

Abstract
Hsp100 is an ATP-dependent unfoldase that promotes protein disaggregation or facilitates the unfolding of aggregation-prone polypeptides marked for degradation. Recently, new Hsp100 functions are emerging. In Plasmodium, an Hsp100 drives malaria protein export, presenting a novel drug target. Whether Hsp100 has a similar function in other protists is unknown. We present the 1.06-Å resolution crystal structure of the Hsp100 N-domain from Leishmania spp., the causative agent of leishmaniasis in humans. Our structure reveals a network of methionines and aromatic amino acids that define the putative substrate-binding site and likely evolved to protect Hsp100 from oxidative damage in host immune cells.
Keywords: Leishmania, Molecular chaperone, Protein unfoldase, Hsp100
Introduction.
Leishmaniasis is a vector-borne tropical disease caused by infection with the Leishmania parasite that is transmitted zoonotically through the bite of an infected female sandfly. Approximately 1.3 million new cases are reported annually, including ulcerative skin lesions (cutaneous leishmaniasis) and systemic infections (visceral leishmaniasis) that can be fatal if left untreated. Lack of preventive vaccines and safe drugs calls for additional research.
Leishmania parasites are flagellated protists that cycle between extracellular promastigotes in the insect vector and intracellular amastigotes inside the mammalian host 1. Following host infection, promastigotes rapidly infiltrate neutrophils before parasitizing macrophages where they reside within parasitophorous vacuoles and transform into amastigotes that are responsible for parasite replication and all clinical manifestations of leishmaniasis. To cope with changes in the intracellular lifestyle, the parasite must continually adapt, which depends on the expression of molecular chaperones to confer general stress tolerance, including exposure to reactive oxygen species that are rapidly released from host immune cells to eradicate the engulfed pathogen 1. For instance, it was shown that the Leishmania spp. heat-shock protein 100 (Hsp100) is required for full amastigote development 2,3, supporting a role as an essential virulence factor and potent drug target.
Hsp100 chaperones are ATP-driven protein unfoldases that are ubiquitously found in bacteria, fungi, protozoa, and plants, as well as in mitochondria of animal cells. Hsp100 ATPases possess a multi-domain structure composed of an N-domain (Hsp100N) followed by one or two nucleotide-binding domains (NBD). The N-domain is a hallmark of plant and microbial Hsp100 chaperones, including protozoan Hsp100, and, unlike the NBD, is not shared with any human protein. In addition, Hsp100 unfoldases often feature additional structural elements such as a coiled-coil motif and the ClpP-binding loop that confer specific Hsp100 functions 4. In this regard, Leishmania Hsp100 resembles members of the ClpB/Hsp104 subgroup that function as protein disaggregases 4, although the molecular function of Leishmania Hsp100 remains unknown. It was recently shown that Hsp101 from Plasmodium falciparum, the causative agent of malaria, is an integral component of the Plasmodium Translocon of Exported Proteins (PTEX) complex that is essential for the transport of malarial proteins into host erythrocytes 5. However, other PTEX components are not found outside of Plasmodium spp. 6, arguing against the existence of a homologous PTEX complex in Leishmania and related kinetoplastids. To inform on Leishmania Hsp100 function, we determined the high-resolution crystal structure of the Leishmania mexicana Hsp100N in order that this information might be exploited for rational structure-based drug design to fight off Leishmania infections.
Materials and Methods.
Protein expression.
The L. mexicana Hsp100 N-domain (LmHsp100N) comprising residues 1 to 158 was PCR amplified from genomic DNA and cloned into pProEX-HTb (Invitrogen), which adds a Tobacco Etch Virus (TEV) protease cleavable N-terminal His6-tag. Escherichia coli BL21-CodonPlus (DE3)-RIL cells (Agilent Technologies) transformed with pLmHsp100N were cultured at 37°C in lysogeny broth medium supplemented with 100 μg/mL ampicillin and 34 μg/mL chloramphenicol until mid-log phase. Cells were induced with 0.5 mM isopropyl β-D-1-thiogalactopyranoside and continued to grow at 25°C for an additional 18 hours before harvesting.
Protein purification.
The cell pellet was resuspended in lysis buffer (25 mM Tris-HCl pH 8.8, 0.3 M NaCl, 30 mM imidazole, 5% glycerol, and 5 mM β-mercaptoethanol) supplemented with 1 mM phenylmethylsulfonyl fluoride and 1 mM benzamidine. After cell lysis, the clarified supernatant was loaded onto a 20 mL Ni Sepharose 6 Fast Flow (Cytiva) column that was then washed with 50 column volumes of lysis buffer. His-tagged LmHsp100N was eluted in lysis buffer using a linear gradient of 30–500 mM imidazole. His-TEV protease was added to the eluted sample, which was dialyzed against 25 mM Tris-HCl pH 8.8, 50 mM NaCl, 5% glycerol, and 5 mM β-mercaptoethanol for 16 hours at 4°C. The sample was reapplied onto a Ni Sepharose 6 Fast Flow column to remove the liberated His-tag, the His-TEV protease, and any uncleaved protein. The flow-through was further purified on a 10 ml Q Sepharose Fast Flow column (Cytiva) and eluted in 25 mM Tris-HCl pH 8.8, 5% glycerol, and 1 mM dithiothreitol using a linear gradient of 0.05–1.0 M NaCl. Protein concentration was determined by denaturing the protein in 6 M guanidine-HCl and using a calculated molar extinction coefficient of 9,970 M−1cm−1.
Crystallization.
Crystals of LmHsp100N were grown at 14°C using the hanging drop vapor diffusion method by mixing 1 μL of protein (110 mg/mL) with an equal volume of reservoir solution consisting of 5% polyethylene glycol (PEG) 2000, 5% PEG 3350, 5% PEG 4000, 5% PEG methyl ether 5000, 0.1 M KCl, 0.1 M MgCl2, 0.1 M PIPES pH 7.0 and setup over a 100 μL reservoir. Crystals were harvested in reservoir solution supplemented with 15.63% (v/v) glycerol and flash frozen in liquid nitrogen. Diffraction data were collected at 100 K using a Pilatus3 X 6M detector at the APS SBC ID19 beamline at Argonne National Laboratory (Lemont, IL). Data were processed and scaled using the HKL3000 software suite 7.
Structure solution and refinement.
The structure of LmHsp100N was determined by molecular replacement with PHASER 8 using the X-ray structure of an N-terminal fragment of Saccharomyces cerevisiae Hsp104 comprising residues 6–149 (PDB: 5WBW_A) 9 as search model. A single solution was found despite a sequence identity of only 24%. The structure was refined with PHENIX 10, which was interspersed by manual rebuilding of the model. The final structure has excellent stereochemical properties with 99.29% and 0.71% of residues in favored and allowed regions, and none of the residues in disallowed regions of the Ramachandran plot (Table 1).
Table 1:
Summary of data collection and refinement statistics.
| LmHsp100N | |
|---|---|
|
| |
| Data collection | |
|
| |
| Space group | P 41 21 2 |
| Unit cell dimensions | |
| a, b, c (Å) | 60.971, 60.971, 71.807 |
| α, β, γ (°) | 90.0, 90.0, 90.0 |
| Wavelength (Å) | 0.9794 |
| Resolution (Å) | 50.00–1.05 (1.07–1.05) |
| Unique reflections | 61,983 |
| Rsym or Rmerge | 0.071 (0.528) |
| R pim | 0.023 (0.259) |
| I / σI | 10.9 |
| Completeness (%) | 97.1 (92.6) |
| Redundancy | 9.1 (4.8) |
| Refinement | |
| Resolution (Å) | 36.96–1.06 |
| No. reflections | 41,615 / 2,515 |
| Rwork / Rfree | 0.1339 / 0.1528 |
| No. atoms | 1,349 |
| Protein | 1,145 |
| Ligand/ion | 6 |
| Water | 198 |
| B-factors (Å2) | |
| Protein | 19.83 |
| Ligand/ion | 19.35 |
| Water | 30.46 |
| RMS deviation | |
| Bond lengths (Å) | 0.004 |
| Bond angles (°) | 0.721 |
| Ramachandran | |
| Outliers | 0.00% |
| Allowed | 0.71% |
| Favored | 99.29% |
Values in parentheses are for highest-resolution shell.
Results.
The structure of LmHsp100N consists of one molecule in the asymmetric unit and was determined to 1.06-Å resolution, which allowed the unequivocal fitting of side-chains and ordered water molecules. To our knowledge, our structure represents the highest resolution crystal structure of a microbial Hsp100N domain solved to date. After iterative structure refinement and rebuilding of the atomic model, the final structure consists of residues 3–144 of LmHsp100, 198 water molecules, and one glycerol molecule (Fig. 1A).
Figure 1.
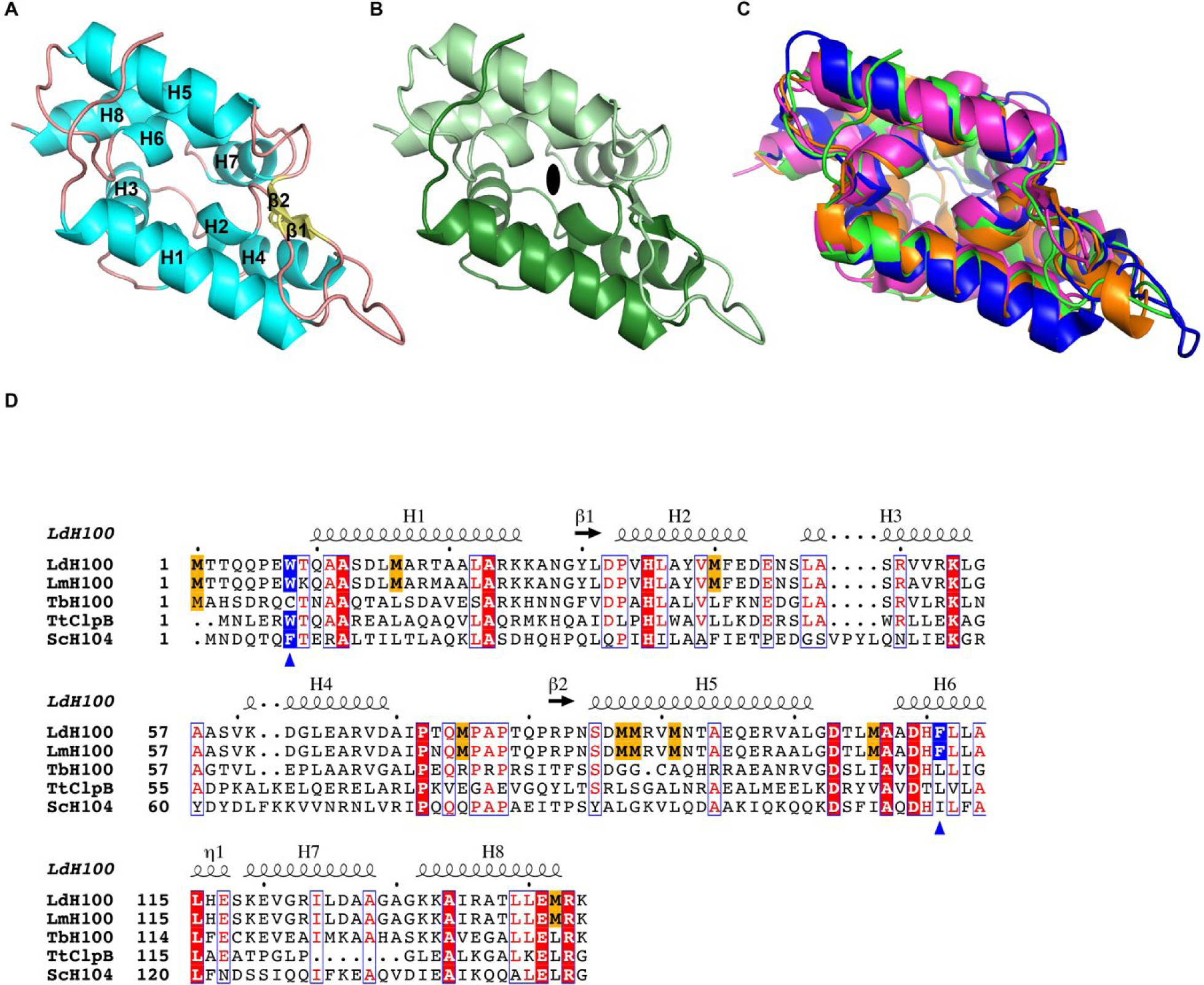
Crystal structure of the L. mexicana Hsp100N. (A) Atomic structure of LmHsp100N shown as ribbon diagram with α-helices (H1-H8) and β-strands (β1-β2) labelled sequentially. (B) View along the pseudo two-fold symmetry axis (symbol) with each Clp repeat subdomain shown in different hues. (C) Superposition of the N-domain from L. mexicana Hsp100 (green), E. coli ClpA (PDB: 1K6K; blue), T. thermophilus ClpB (PDB: 1QVR_A; orange), and B. subtilis ClpC (PDB: 2Y1Q; magenta). (D) Structural elements and sequence alignment of Hsp100 N-domains from L. donovani (LdH100), L. major (LmH100), T. brucei (TbH100), T. thermophilus (TtClpB), and S. cerevisiae (ScH104). The amino acid sequence for Hsp100N from L. mexicana (New World species) and L. donovani (Old World species) are identical. Conserved methionines in Leishmania spp. Hsp100 are highlighted in gold. The blue triangles mark the critical aromatic/hydrophobic amino acid. The dot above the sequence marks every tenth residue.
LmHsp100N is mostly α-helical consisting of eight α-helices (H1-H8), which can be grouped into two repeats related by pseudo two-fold symmetry (Fig. 1B). The two repeats are held together by a pair of parallel β-strands that precede H2 and H5, and give rise to the characteristic N-domain structure, also known as the Clp repeat domain (PROSITE: PS51903). Pair-wise superposition of LmHsp100N with the N-domains of bacterial ClpA, ClpB, and ClpC shows that the overall structure is highly similar and superposes with a root-mean-square-deviation of 1.84-Å for E. coli ClpA (410 out of 555 atoms), 1.30-Å for Thermus thermophilus ClpB (598 out of 747 atoms), and 2.20-Å for Bacillus subtilis ClpC (576 out of 732 atoms) (Fig. 1C). However, despite sharing the same fold, it is immediately apparent that LmHsp100N is enriched in methionines, featuring eight methionines in addition to the initiating methionine and comprising 5.6% of the amino acid sequence. This compares to 2.4% for the remainder of the protein, and 0–1.5% for the isolated Hsp100N domain from other microbes (Fig. 1D). Notably, while the eight methionines are conserved across Leishmania spp., they are not found outside the Leishmania genus, including the closely related trypanosomes, arguing for an important function specific to the Leishmania life cycle.
The structure shows that the side-chains of six methionines (Met16, Met40, Met87, Met88, Met91, Met106) are exposed on the surface of a hydrophobic grove formed by the two Clp repeat subdomains (Fig. 2A). It was previously shown that this surface facilitates substrate interaction in bacterial and fungal Hsp100 11–13 (Fig. S1). Notably, the sulfur atoms of Met16, Met40, and Met106 form hydrophobic interactions with the aromatic side-chain of both Trp8 and Phe111, with the latter featuring an additional hydrophobic interaction with Met91. These interactions are characteristic of the methionine-aromatic amino acid motif that is widely found in protein structures and was proposed to provide additional stabilization 14. Because methionine oxidation not only increases the stability of the methionine-aromatic interaction 14 but also is reversible and is not known to adversely affect enzymatic activity 15, methionine serves as an important in vivo anti-oxidant that prevents damage to other residues critical to protein function. Although Trp8 and Phe111 are not strictly conserved across species and are often substituted for another aromatic or aliphatic amino acid, it nevertheless underscores the importance for a hydrophobic amino acid at this position. Phe111 is of particular interest because this residue is buried deep inside the hydrophobic groove and coordinates multiple side chain interactions that define the hydrophobic cleft (Fig. 2 A,B) and how substrates might bind to its surface (Fig. S1). Consistent with the critical role of this residue, we previously showed that an alanine substitution for the equivalent hydrophobic residue in yeast Hsp104 (Ile116) caused a destabilization of protein structure, which irreversibly inhibited Hsp104 function 12.
Figure 2.
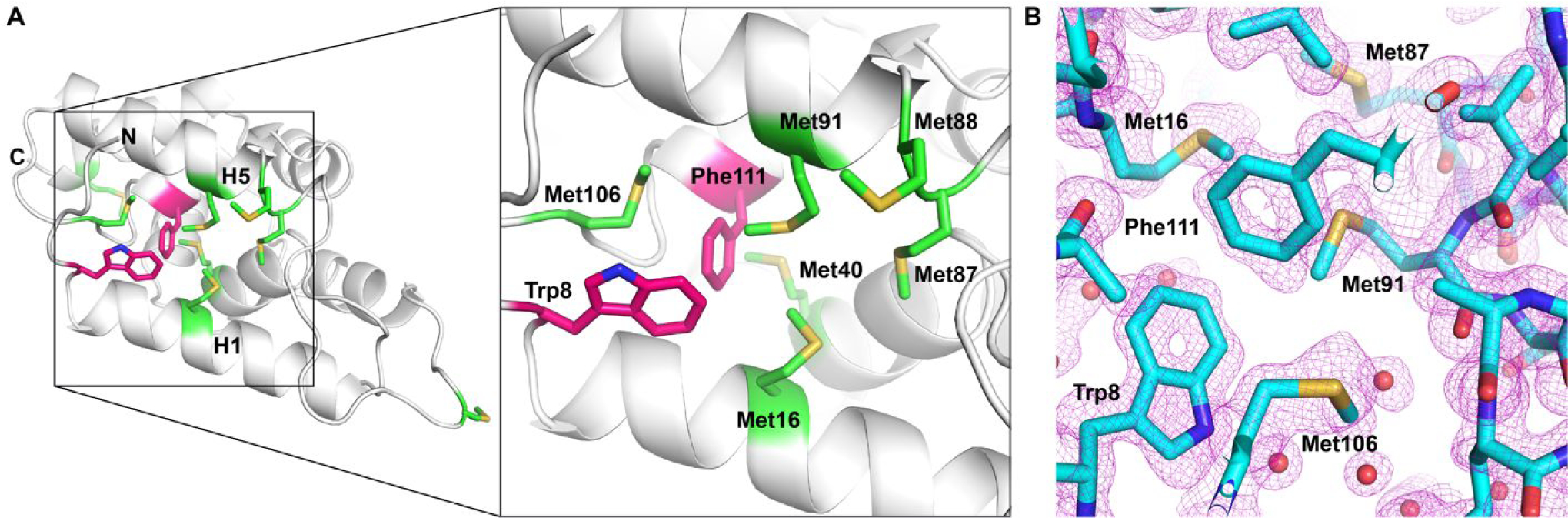
LmHsp100N features the methionine-aromatic amino acid motif that protects the N-domain from oxidative damage. (A) Enlarged view of the hydrophobic cleft showing the location of the six methionines (green) as well as of Trp8 and Phe111 (purple), with the tryptophan side chain being solvent exposed. (B) Phe111 is surrounded by methionine residues that protect this critical residue from oxidative damage. Four out of five methionines are shown for clarity. A section of the composite 2Fo-Fc electron density map is shown as purple mesh and contoured at the 1.0 σ level.
Discussion.
Molecular chaperones, such as Hsp100, are essential to the microbial stress-response and confer the ability to adapt to different environments. Recently, new Hsp100 functions are emerging, establishing their role as virulence factors 5,6. Although the N-domain is dispensable for protein unfolding, essential roles in substrate recognition have been reported for several Hsp100 members 4. The structure of LmHsp100N suggests that the structural integrity of the N-domain is important and presents a potent drug target. Although the molecular function of LmHsp100 remains unknown, it is tempting to speculate that the N-domain of LmHsp100 mediates substrate binding on the surface of the hydrophobic grove that necessitates a high structural integrity including guarding Phe111 against oxidative damage.
Our structure provides an improved understanding of how Leishmania parasites have evolved the Hsp100 unfoldase that is essential for promastigote-to-amastigote transformation inside host cells. Concomitantly, our work suggests that disrupting the structural integrity of Hsp100N could potentially abolish LmHsp100 function and may be exploited for the development of new anti-leishmanial drugs.
Supplementary Material
Acknowledgements.
We thank J. Lee for help with cloning. This work was supported by the Welch Foundation (Q-1530-20190330), the National Institutes of Health (R01-GM104980, R01-GM142143, R01-AI130126, and R01-DK115454), and the Cancer Prevention and Research Institute of Texas (RR140038). Use of the Macromolecular X-ray Crystallography Core at Baylor College of Medicine is supported in part by NIH grant S10-OD030246. Use of the SBC beamlines at the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. J.M.M. was supported by a training fellowship from the Gulf Coast Consortia on the Houston Area Molecular Biophysics Program (T32-GM008280).
Footnotes
Accession number.
Atomic coordinates and accompanying structure factors have been deposited with the Research Collaboratory for Structural Bioinformatics Protein Data Bank under accession code PDB: 7TFM.
Declaration of interests.
The authors declare that they have no competing financial interests.
References.
- 1.Walker DM, Oghumu S, Gupta G, McGwire BS, Drew ME, Satoskar AR. Mechanisms of cellular invasion by intracellular parasites. Cell Mol Life Sci. 2014;71(7):1245–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hübel A, Krobitsch S, Hörauf A, Clos J. Leishmania major Hsp100 is required chiefly in the mammalian stage of the parasite. Mol Cell Biol. 1997;17:5987–5995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krobitsch S, Clos J. A novel role for 100 kD heat shock proteins in the parasite Leishmania donovani. Cell Stress Chaperones. 1999;4:191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Katikaridis P, Bohl V, Mogk A. Resisting the heat: bacterial disaggregases rescue cells from devastating protein aggregation. Front Mol Biosci. 2021;8:681439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Koning-Ward TF, Gilson PR, Boddey JA, et al. A newly discovered protein export machine in malaria parasites. Nature. 2009;459:945–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ho CM, Beck JR, Lai M, et al. Malaria parasite translocon structure and mechanism of effector export. Nature. 2018;561:70–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr. 2006;62:859–866. [DOI] [PubMed] [Google Scholar]
- 8.McCoy AJ. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D Biol Crystallogr. 2007;63:32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee J, Sung N, Yeo L, Chang C, Lee S, Tsai FTF. Structural determinants for protein unfolding and translocation by the Hsp104 protein disaggregase. Biosci Rep 2017;37:BSR20171399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adams PD, Afonine PV, Bunkóczi G, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosenzweig R, Farber P, Velyvis A, Rennella E, Latham MP, Kay LE. ClpB N-terminal domain plays a regulatory role in protein disaggregation. Proc Natl Acad Sci USA. 2015;112:E6872–E6881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee J, Sung N, Mercado JM, et al. Overlapping and specific functions of the Hsp104 N domain define its role in protein disaggregation. Sci Rep. 2017;7:11184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rizo AN, Lin J, Gates SN, et al. Structural basis for substrate gripping and translocation by the ClpB AAA+ disaggregase. Nat Commun. 2019;10(1):2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lewis AK, Dunleavy KM, Senkow TL, et al. Oxidation increases the strength of the methionine-aromatic interaction. Nat Chem Biol. 2016;12(10):860–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levine RL, Mosoni L, Berlett BS, Stadtman ER. Methionine residues as endogenous antioxidants in proteins. Proc Natl Acad Sci USA. 1996;93(26):15036–15040. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.