

ABSTRACT
Osteogenesis imperfecta (OI) is a genetically heterogenous disorder most often due to heterozygosity for mutations in the type I procollagen genes, COL1A1 or COL1A2. The disorder is characterized by bone fragility leading to increased fracture incidence and long‐bone deformities. Although multiple mechanisms underlie OI, endoplasmic reticulum (ER) stress as a cellular response to defective collagen trafficking is emerging as a contributor to OI pathogenesis. Herein, we used 4‐phenylbutiric acid (4‐PBA), an established chemical chaperone, to determine if treatment of Aga2 +/− mice, a model for moderately severe OI due to a Col1a1 structural mutation, could attenuate the phenotype. In vitro, Aga2 +/− osteoblasts show increased protein kinase RNA‐like endoplasmic reticulum kinase (PERK) activation protein levels, which improved upon treatment with 4‐PBA. The in vivo data demonstrate that a postweaning 5‐week 4‐PBA treatment increased total body length and weight, decreased fracture incidence, increased femoral bone volume fraction (BV/TV), and increased cortical thickness. These findings were associated with in vivo evidence of decreased bone‐derived protein levels of the ER stress markers binding immunoglobulin protein (BiP), CCAAT/−enhancer‐binding protein homologous protein (CHOP), and activating transcription factor 4 (ATF4) as well as increased levels of the autophagosome marker light chain 3A/B (LC3A/B). Genetic ablation of CHOP in Aga2 +/− mice resulted in increased severity of the Aga2 +/− phenotype, suggesting that the reduction in CHOP observed in vitro after treatment is a consequence rather than a cause of reduced ER stress. These findings suggest the potential use of chemical chaperones as an adjunct treatment for forms of OI associated with ER stress. © 2022 The Authors. Journal of Bone and Mineral Research published by Wiley Periodicals LLC on behalf of American Society for Bone and Mineral Research (ASBMR).
Keywords: osteogenesis imperfecta, Aga2 , bone, 4‐PBA, ER stress, Chop −/− , Bip +/−
Introduction
Osteogenesis Imperfecta (OI) is a genetically heterogenous disorder characterized by brittle bones and is one of the most common osteochondrodysplasia phenotypes.( 1 , 2 ) OI varies in severity from perinatal lethality to milder phenotypes characterized by an increased number of low impact fractures, particularly in childhood. Severe or progressive deforming OI is associated with recurrent fractures, poor fracture healing, long‐bone deformities, vertebral compression, and kyphoscoliosis. Although poor bone quality is of principal importance in OI, other manifestations include short stature, dentinogenesis imperfecta, deafness, and cardiopulmonary compromise.( 1 , 2 , 3 ) Approximately 80%–85% of OI cases are caused by dominantly inherited mutations in the genes encoding type I procollagen (COL1A1 and COL1A2). Most of the mutations in the remaining 19 genes encode proteins involved in the synthesis, posttranslational processing, and cellular trafficking of type I procollagen, or proteins involved in osteoblast function.( 1 , 4 , 5 ) Based on the genes and the associated mutations, several pathogenetic mechanisms have been proposed to produce OI. For mutations that lead to qualitatively abnormal collagen, due to either structural mutations in the type I procollagen genes and/or altered posttranslational modification, the defective collagen accumulates in the extracellular matrix (ECM), altering fibril organization, mineralization, and the biomechanical properties of the bones. Conversely, for mutations that lead to reduced type I procollagen synthesis, primarily due to haploinsufficiency for COL1A1, there is reduced abundance in the ECM and altered stoichiometry relative to other matrix proteins. Additionally, abnormal osteoblast function due to altered signaling pathway activity, including WNT and transforming growth factor β (TGFβ), have also been shown to contribute to disease.( 4 , 5 , 6 ) Although these mechanisms have all contributed to our understanding of bone fragility, both OI and skeletal formation are complex, and additional mechanisms are likely to contribute to the underlying pathology.
During the last decade, endoplasmic reticulum (ER) stress has emerged as a common cellular pathologic process for a number of diseases (reviewed in Oyadomari and Mori( 7 )). The ER is the primary site for synthesis, posttranslational modifications, folding, transport, and secretion of proteins.( 8 , 9 , 10 ) Structurally abnormal proteins, absence of proper posttranslational modifications, and insufficient chaperone activity can lead to accumulation of unfolded or misfolded proteins in the ER lumen, inducing ER stress.( 11 ) These perturbations initiate evolutionarily conserved, complex signal transduction cascades referred to as the unfolded protein response (UPR). Central to this process is binding immunoglobulin protein (BiP) (also known as GRP78 or HSPA5), a multifunctional protein that has a role in the activation of ER stress pathways. BiP binds to hydrophobic residues of misfolded proteins after dissociating from the UPR pathway receptor regulators protein kinase RNA‐like endoplasmic reticulum kinase (PERK), activating transcription factor 6 (ATF6), and inositol‐requiring enzyme‐1 (IRE1),( 12 ) thereby activating these mediators of the three major branches of the UPR.( 13 ) PERK is an important sensing element for ER stress( 14 ) that, when activated, induces a downstream pathway to inhibit protein translation in order to restore ER homeostasis.( 15 ) However, if ER stress persists and is severe, PERK activates activating transcription factor 4 (ATF4),( 16 ) which then induces the transcription of the CCAAT/−enhancer‐binding protein homologous protein (CHOP). High levels and persistent expression of CHOP induce cellular apoptosis.( 17 ) Bone cells, particularly osteoblasts, are especially at risk for detrimental levels of ER stress due to their abundant secretion of ECM proteins.
There is an increasing literature on the effects of the chemical chaperone 4‐PBA or the related sodium phenylbutyrate in various biological systems.( 18 , 19 , 20 , 21 ) 4‐PBA has been shown to have multiple mechanisms of action. It acts as an ammonia scavenger and, in this context, has been employed in humans to treat the hyperammonemia in urea cycle defects.( 22 , 23 , 24 ) Additionally, 4‐PBA acts as a weak histone deacetylase inhibitor and may affect mitochondrial and peroxisome biogenesis.( 25 , 26 ) Several studies suggest that 4‐PBA acts as a molecular chaperone, aiding in protein folding and preventing ER aggregation of proteins.( 27 , 28 , 29 ) Numerous studies have demonstrated that ER stress in osteoblasts is an important factor in OI and other low‐bone‐mass disorders.( 30 , 31 , 32 , 33 ) Moreover, varying degrees of ER stress have been demonstrated in several OI mouse models.( 30 , 34 , 35 ) 4‐PBA has been reported to relieve ER stress in human OI fibroblasts with mutations in COL1A1, COL1A2, CRTAP, P3H1, and PPIB.( 32 , 36 ) In vivo, 4‐PBA treatment of Chihuahua, a zebrafish model of dominant OI with a type I collagen structural mutation, improved bone mineralization and decreased adult skeletal deformities.( 37 ) These published data imply that a chemical chaperone has the potential to attenuate the OI phenotype through the relief of ER stress.
This study describes a potential molecular treatment that targets the intrinsic cellular pathology of OI caused by type I procollagen gene mutations. We tested the effects of 4‐PBA treatment in an OI mouse model, Aga2 +/− , that manifests moderate to severe OI.( 30 , 34 , 38 ) The Aga2 +/− mouse has a dominant frameshift mutation in the Col1a1 C‐propeptide domain and exhibits decreased bone mass with increased bone brittleness and fracture incidence.( 30 ) The Aga2 +/− mutation has been shown to induce ER stress in fibroblasts, osteoblasts, and cortical bone through the retention of type I procollagen in the ER.( 30 ) Our findings reveal that the upregulated ER stress response observed in Aga2 +/− osteoblasts and femoral bone lysates can be reduced by 4‐PBA treatment. 4‐PBA treatment increased the weight and length of Aga2 +/− mice, indicating a positive effect on overall postnatal bone growth. Importantly, 4‐PBA treatment of Aga2 +/− mice resulted in increased bone strength and reduced fracture incidence. However, genetic reduction of the ER stress response by either ablation of CHOP or reduced expression of BiP, worsened or did not improve, respectively, the Aga2 +/− OI phenotype, indicating that these proteins are important in managing the deleterious effects of OI mutations. Collectively, this study demonstrates that improving chaperone function reduced ER stress and improved bone parameters in one mouse model of OI, and suggests this strategy could be effective in patients with OI.
Subjects and Methods
Cell culture
For Figs. 1 and 2, mouse osteoblast (mOB) cultures were established from P5 collagenase‐digested calvaria of both Aga2 +/− and wild‐type (WT) mice and plated in six‐well plates at 200,000 cells/well, cultured for 48 hours in FBS supplemented media (Dulbecco‐Vogt Modified Eagle Medium [DMEM] supplemented with 10% fetal bovine serum [FBS] and then cultured in osteogenic media DMEM supplemented with 10% FBS, 50 μg/mL ascorbic acid, 10mM β‐glycerophosphate, 10nM dexamethasone) for a total of 7 days with media changes every 2 days. Cells were treated with 5nM 4‐PBA after plating for a total of 7 days with media changes every 2 days. For protein analyses, cells were collected in Lysis Buffer (Thermo Fisher Scientific, Waltham, MA, USA; 87787) supplemented with proteinase inhibitors; media was collected for collagen analysis 72 hours after the final media change and supplemented with proteinase inhibitors prior to concentration.
Fig. 1.
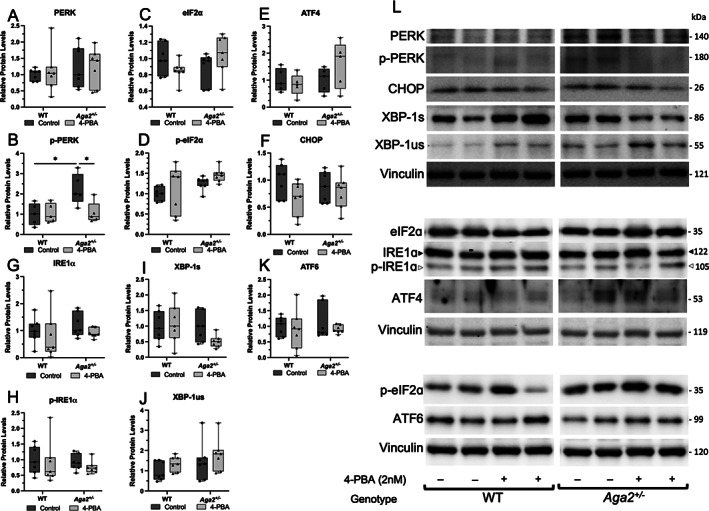
Acute 4‐PBA treatment reduces UPR activation in short‐term primary osteoblast culture. (A–K) Quantification of Western blots probed for UPR receptors and downstream transducers in primary calvarial osteoblasts cultured and treated with 5nM 4‐PBA for 7 days. n = 7 per treatment group. Quantifications are displayed with median and interquartile range. Two‐way ANOVAs were performed, *p < 0.05 was considered statistically significant. (L) Representative Western blots of relative protein levels.
Fig. 2.
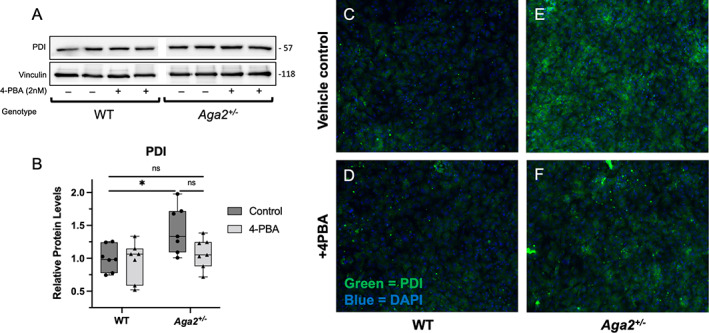
4‐PBA treatment reduces PDI levels and localization in Aga2 +/− primary osteoblast culture. (A,B) Representative Western blot and quantification of relative PDI levels in primary calvarial osteoblasts cultured and treated with 5nM 4‐PBA for 7 days. n = 7 per treatment group. A two‐way ANOVA was performed, *p < 0.05 was considered statistically significant. (C–F) Immunofluorescent images of primary osteoblasts probed with a conjugated antibody against PDI. Green = PDI, blue = DAPI, n = 6.
To concentrate media, 593 μL of ice‐cold 95% ethanol was added to 1.1 mL of media, collected, and kept on ice. Each tube was inverted to mix, then precipitated on ice for 60 minutes. Samples were centrifuged at 18,000 g and 0°C for 5 minutes, then the supernatant was removed. With the pellet remaining, the sample was left open and inverted to dry at room temperature for 60 minutes, then resuspended in 100 μL radioimmunoprecipitation assay (RIPA) buffer (EMD Millipore, Burlington, MA, USA; 20‐188) supplemented with Halt Protease and Phosphatase Inhibitor (Thermo Fisher Scientific; 1861281).
Immunofluorescence
Immunofluorescence experiments were performed using an Echo Revolution fluorescence microscope (ECHO, San Diego, CA, USA). Cultured cells were fixed in 4% paraformaldehyde (PFA) in phosphate buffered saline (PBS), then washed and permeabilized with 0.1% Triton X100 for 5 minutes, followed by blocking in 10% goat serum for 1 hour. The conjugated primary antibody (protein disulfide isomerase [PDI]; Cell Signaling Technology, Danvers, MA, USA; 5051; 1:100) was incubated overnight at 4°C. 4′,6‐Diamidino‐2‐phenylindole (DAPI) at a 1:1000 dilution for 10 minutes at room temperature was applied before mounting. Experiments in Fig. 3 were repeated with at least six biological replicates in each genotype.
Fig. 3.
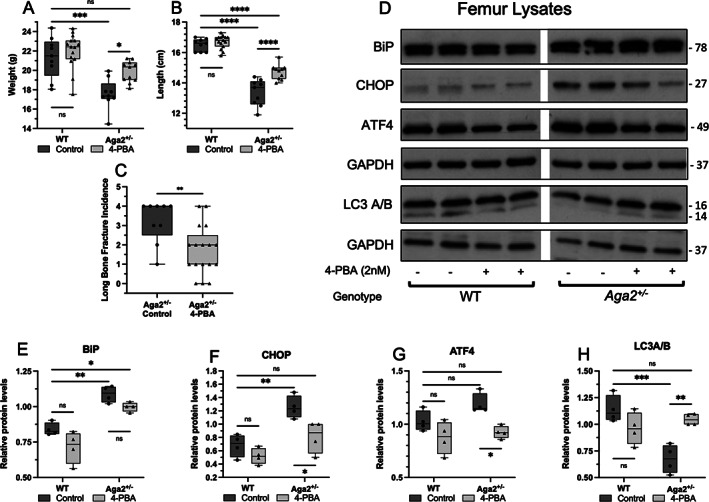
Improvement of the OI phenotype and ER stress levels in male Aga2 +/− mice treated with 4‐PBA. (A,B) Quantification of total body length and weight in WT and Aga2 +/− male mice treated with 4‐PBA or untreated controls, n = 9 (WT control), 15 (WT treated), 5 (Aga2 +/− control), 11 (Aga2 +/− treated). Two‐way ANOVAs were performed, *p < 0.05 was considered statistically significant. (C) Quantification of long‐bone fracture incidence in 4‐PBA treated and untreated 2‐month‐old male Aga2 +/− mice, n = 11 (untreated) and 17 (treated). Significance was determined by t test. (D–H) Representative western blot and quantification of relative protein levels in femoral lysates of WT and Aga2 +/− mice treated with 4‐PBA or untreated control animals. n = 4/per group. Quantifications are displayed with median and interquartile range. Two‐way ANOVAs were performed, *p < 0.05 was considered statistically significant. *p < 0.05, **p < 0.01, ***p < 0.001, ns = not significant.
Animal studies
The animal studies were performed under an approved by the University of California at Los Angeles (UCLA) Research Safety and Animal Welfare Committee (ARC Committee). The Animal Research Committee (ARC) is an independent research review committee mandated by the Animal Welfare Act the PHS Policy on Humane Care and Use of Laboratory Animals. Aga2 +/− animals were received as a gift from the Jacobsen laboratory at Harvard Medical School and maintained on a C57BL/6J background.( 30 ) The Aga2 +/− colony was maintained through mutant alleles only passed through males. All models were housed in the UCLA vivarium under a 12‐hour light/dark cycle and provided water and standard chow ad libitum. Both male and female animals were studied. Treatment with 4‐PBA was performed as follows; 2.5 g/kg/d of 4‐PBA was placed in the drinking water and changed three times per week over a 5‐week treatment period (postweaning, days 21 to 60). A maximum of two Aga2 +/− and two WT animals were housed per cage (only the same genotypes were housed together) where water consumption was observed between 4 and 6 mL per day (2–3 mL per day per animal from 21 days to 2 months of age). Thus, 4‐PBA was offered in drinking water at 25 mg/mL concentration, calculating that each mouse consumes about 50 mg per day per mouse. There was no correlation observed between weight and water consumption in Aga2 +/− and WT mice. No adverse events were reported with treatment. All animal studies were terminated at 2 months of age and euthanasia was performed via isoflurane inhalation and cervical dislocation according to Association for Assessment and Accreditation of Laboratory Animal Care International (AALAC) protocols. At this point animals were measured, X‐rayed, and dissected for collection of calvaria (for osteoblast extraction) and femurs (for protein analyses, micro–computed tomography [μCT] scanning, and biomechanical studies). Fracture incidence was calculated from the anterior/posterior radiographs and the number of fractures counted in the long bones (humeri, radii, ulnas, femurs, tibias and fibulas). Whole bones were immediately cleaned and processed for protein lysis, fixed in 4% PFA for histology, fixed and partially dehydrated in 70% ethanol for μCT, and frozen in saline‐embedded gauze for fracture studies.
Genetic crosses were performed with Aga2 +/− and Bip +/− mice to generate animals with the Aga2 +/− ;BiP +/− genotype. Similarly, Aga2 +/− mice were crossed with Chop −/− mice to generate mice with the Aga2 +/− ;Chop −/− genotype. Animal studies were terminated at 2 months of age and the animals were measured, X‐rayed, and femurs from the Aga2 +/− ;BiP +/− mice underwent μCT analyses. The Aga2 +/− ;Chop −/− mice could not undergo μCT due to the fragility and deformity of the femurs. BiP +/− mice were generously shared by Dr. Amy Lee at the University of Southern California.( 39 ) Chop −/− mice were originally generated by Dr. David Ron, NYU School of Medicine and were deposited for commercial use at Jackson Laboratory (Bar Harbor, ME, USA; B6.129S(Cg)‐Ddit3tm2.1Dron/J; Stock No: 005530).
Western blots
For Western blot analyses, protein lysates were separated by electrophoresis on either 5%, 7.5%, 10%, 12%, or gradient (Bio‐Rad Laboratories, Hercules, CA, USA; 4% to 20%, catalog no. 4568093) sodium dodecylsulfate (SDS)‐polyacrylamide gels or TGX Stain‐Free polyacrylamide gels (Bio‐Rad Laboratories; catalog no. 1610180), transferred to polyvinylidene difluoride membranes, blocked in 5% nonfat milk or 5% bovine serum albumin (BSA) in Tris‐buffered saline with 0.1% Tween 20 (TBST) and probed overnight with primary antibody (anti‐BiP antibody [Cell Signaling Technology; 1:1000; milk, catalog no. 3177], anti‐CHOP [Cell Signaling Technology; 1:1000; milk, catalog no. 5554], anti‐ATF4 [Cell Signaling Technology; 1:1000; milk, catalog no. 11815], anti–light chain 3A/B [LC3A/B] [Cell Signaling Technology; 1:1000; milk; catalog no. 4108], anti‐GAPDH [Cell Signaling Technology; 1:2000; milk, catalog no. 2118], anti‐PERK [Cell Signaling Technology; 1:1000; milk, catalog no. 5683], anti‐phospho‐PERK [Cell Signaling Technology; 1:1000; BSA, catalog no. 3179], anti‐XBP‐1 [Cell Signaling Technology; 1:1000; BSA, catalog no. 40435], anti‐Vinculin [Abcam; 1:10,000; milk, catalog no. ab129002], anti‐eIF2ɑ [Cell Signaling Technology; 1:1000; milk, catalog no. 5324], anti‐phospho‐eIF2ɑ [Cell Signaling Technology; 1:1000; BSA, catalog no. 3398], anti‐IRE1ɑ [Cell Signaling Technology; 1:1000; BSA, catalog no. 3294], anti‐phospho‐IRE1ɑ [Invitrogen; 1:1000; BSA, catalog no. PA1‐16927], anti‐ATF6 [Cell Signaling Technology; 1:1000; milk, catalog no. 65880], anti‐PDI [Cell Signaling Technology; 1:1000; milk, catalog no. 3501], anti‐β‐Actin [Cell Signaling Technology; 1:1000; milk, catalog no. 4967]). Peroxidase‐conjugated secondary antibodies (Cell Signaling Technology; 1:2000, catalog nos. 7071 and 7072) were used, and immunocomplexes were identified using the enhanced chemiluminescence (ECL) Detection Reagent (Cell Signaling Technology; catalog no. 7003) or West Femto Maximum Sensitivity Substrate (Thermo Fisher; catalog no. 34095). Protein band quantification was performed using either ImageLab 6.1.0 (BioRad; Figs. 2 and 3) or ImageJ (Fiji; Fig. 1) following recommendations in (http://rsb.info.nih.gov/ij/docs/menus/analyze.html#gels). In Figs. 1 and 2, experiments were repeated at least six times from six independent animals.
CT and fracture studies
μCT was conducted on the right femur with the Scanco μCT‐40 system (SCANCO Medical AG, Brüttisellen, Switzerland; 55‐peak kilovoltage and 145‐μA X‐ray source). A standardized region of the distal femoral metaphysis was scanned at 16‐μm resolution. Biomechanical testing by three‐point bending: the contralateral left femurs of the same mouse were tested by three‐point bending with a span of 6 mm, using an Instron 5848 device (Instron Inc., Grove City, PA, USA). Femurs were tested wet at room temperature using an Instron 5848 microtester (Instron Inc.). The femurs were tested to failure in three‐point bending at a rate of 0.1 mm/s and were oriented in the test fixture such that the anterior surface was in compression and the posterior surface was in tension. The test fixture span was about 6.5 mm. A 100‐N load cell was used to collect data, and load and displacement data was captured at rate of 40 Hz by using BLUEHILL Software (Instron Inc.; Instron 5848).
The maximum load was determined by finding the highest load value recorded before the specimen fractured. The region of the load–displacement curve between 1 N and the maximum load was separated into five segments and the fitted line of the segment with greatest slope was defined as the stiffness. A line representing 0.2% offset of this stiffness was used to define the yield point. The elastic region was identified as the region from the completion of the preload to the yield point. The postyield (plastic) region was identified as the region from the yield point until point of specimen fracture. Using a trapezoidal numerical integration method, the energy (elastic, plastic, or total) was calculated as the area under the load–displacement curve. The cross‐sectional geometry of each bone as determined by μCT image analysis was used to convert the maximum load and stiffness data into ultimate stress and elastic modulus values using beam theory.( 40 ) Toughness values were calculated as areas (elastic, plastic, and total) under the stress/strain curve in the same way as energy for the load/displacement curve.
Statistical analysis
GraphPad Prism (GraphPad Software, Inc., La Jolla, CA, USA) was used for statistical analysis. All values are shown as box plots with median and interquartile range, as indicated in the figure legends. Comparisons in the study of two groups with n = 6 or greater were performed using the Student's t test. Comparisons in the study with n < 6 and multiple groups were performed using a two‐way analysis of variance (ANOVA) with a Tukey test as a correction for multiple comparisons.
Results
4‐PBA treatment reduced ER stress markers in Aga2 +/− osteoblasts
To determine the effects of 4‐PBA on endogenous levels of ER stress and specific UPR components in Aga2 +/− osteoblasts, we cultured calvarial osteoblasts and we performed short‐term primary osteoblast culture experiments with a 5nM 4‐PBA treatment for 7 days while in osteogenic media. Cell layer lysates from these cultures were collected and probed for proteins representing the three main signaling receptors of the UPR (PERK, IRE1a, ATF6) as well as their downstream transducers (Fig. 1). At baseline, we observed increased p‐PERK in Aga2 +/− osteoblasts versus WT and this increase was significantly diminished with 4‐PBA treatment while total PERK levels were unchanged (Fig. 1A,B ). However, we did not see significant changes in ATF4 and eIF2α, downstream targets of activated PERK (Fig. 1C–E,L ). Further, CHOP is transcribed upon ATF4 binding to nuclear DNA and although we observed increases in CHOP in our in vivo data, we did not see changes in CHOP in shorter‐term culture (Fig. 1F,L ).( 14 , 16 , 41 ) Increases in ATF4 and CHOP transcription are generally seen in long‐term ER stress conditions; therefore, it is likely our short‐term treatment protocol was not sufficient to induce downstream activation. To address the IRE1a‐activated arm of the UPR, we probed for IRE1a and p‐IRE1a levels and found no significant changes by genotype or treatment condition (Fig. 1G,H,L ). We did, however, show a slight though insignificant decrease in X‐box binding protein 1 (XBP1) splicing (a downstream event of IRE1a activation) in Aga2 +/− mice following 4‐PBA treatment but no change in WT osteoblasts. The short‐term culture/treatment protocol may not be sensitive enough to detect changes in IRE1a activation; however, the decrease in XBP‐1 splicing seen only in Aga2 +/− osteoblasts indicates a differential response and increased sensitivity of the UPR to 4‐PBA treatment in Aga2 +/− compared to WT (Fig. 1I,J,L ). ATF6, the third arm of the UPR, did not show any significant changes in the levels between Aga2 and WT (Fig. 1K,L ). To further delineate the effect of 4‐PBA on stress within the ER, we probed primary osteoblasts derived for our short‐term culture/treatment protocol cell lysates as well as performed immunofluorescence for levels of PDI, an abundant folding chaperone whose expression is induced by ER stress.( 42 , 43 ) Lysates showed increased levels of PDI in Aga2 +/− versus WT and these levels were diminished with 4‐PBA treatment to a level more similar to WT (Fig. 2A,B ). Immunofluorescent images also reflected these results, indicating that Aga2 +/− osteoblasts exhibit increased PDI localization and amelioration with 4‐PBA treatment (Fig. 2C–F ). Overall, this data indicates that specific elements of the UPR, but not all, are activated endogenously in Aga2 +/− primary calvarial osteoblasts and can be diminished via 4‐PBA treatment, highlighting the complexity of the UPR.
4‐PBA treatment reduces ER stress and improves OI bone quality in vivo
To determine the effect of 4‐PBA treatment in vivo, we measured body length and weight in the untreated and 4‐PBA treated 2‐month‐old mice. At this time point, untreated Aga2 +/− mice were smaller and weighed less than WT controls (Fig. 3A,B ). Male Aga2 +/− animals treated with 4‐PBA showed increased body length and weight (Fig. 3A,B ). Similar results were observed in female animals (Fig. S1). Further, male Aga2 +/− animals exhibited reduced long‐bone fracture incidence determined by radiographic analyses (Fig. 3C ). These results suggest that 4‐PBA treatment had a positive effect on postnatal growth and increased bone resistance to fracture in the Aga2 +/− model, with no changes seen in WT animals.
Analysis of ER stress markers by Western blot was performed on bone lysates showing that relative to WT animals, there were increased levels of BiP, CHOP, and ATF4 in Aga2 +/− mice at baseline in vivo (Fig. 3D–H ). By contrast, the level of LC3A/B, a marker of autophagocytosis, was decreased in the Aga2 +/− bone lysates. In previous studies, mutant collagen has been shown to be eliminated by autophagy and not by proteasomal degradation, and previous in vitro studies have reported increased levels of autophagy in OI cell models.( 32 , 44 ) However, our in vivo data showed the opposite, indicating either a decrease in the ability of the cells to initiate autophagy or an increase in autophagosome degradation. Bone lysates from Aga2 +/− mice treated with 4‐PBA showed a slight decrease in BiP, significant decreases in CHOP and ATF4, as well as increased LC3A/B levels, indicating suppression of the UPR and a positive effect on autophagy, bringing these values closer to WT levels (Fig. 3D–H ). These results show that 4‐PBA treatment reduced ER stress‐related molecular changes in OI mice in vivo. Although these results agree with the increased ER stress levels observed in vitro, they also highlight important differences when analyzing ER stress in vivo versus in vitro, again revealing the sensitivity of the UPR to cell and tissue conditions.
To determine the effects of 4‐PBA treatment on the material properties of bones, we studied femoral bone quality and strength of the untreated and treated 2‐month‐old mice using μCT and biomechanical studies. 4‐PBA treated Aga2 +/− femurs show a trend toward increased relative bone volume (BV/TV; Fig. 4A ), indicating improved density. Further, levels of bone connectivity density (Conn‐Dens; Fig. 4B ), and bone trabecular number (Tb.N; Fig. 4C ), with an indirect correlation with decreased trabecular space (Tb‐Sp; Fig. 4D ) were increased to levels closer to WT, implying significant improvements in bone quality in OI mice treated with 4‐PBA. Average cortical thickness (Cort.Th; Fig. 4E ) also significantly increased in treated animals. Visual representations of these improvements are displayed in Fig. 4I–L . Additional μCT measurements of trabecular and cortical bone in Fig. S2 showed no significant changes in trabecular thickness or geometry and the proportional change in bone surface with the increased bone. Biomechanical testing also showed an improvement in bone strength as measured by resistance to fracture via three‐point bending at the femur. We observed increased values in the ultimate load of Aga2 +/− 4‐PBA‐treated femurs that were more similar to WT control levels (Fig. 4F ) and a trend toward increased yield load necessary to deform the bone (Fig. 4G ). We did not, however, see significant changes in ultimate strength necessary to fracture (Fig. 4H ), or bone stiffness or ductility via elastic, plastic, and total displacement measures, indicating no effect on the brittleness that is commonly observed in OI bone (Fig. S3). Altogether, these results revealed that treatment with 4‐PBA during stages of growth led to a reduction in the ER stress response, increased expression of markers for autophagy, and improved OI bone mass and strength.
Fig. 4.
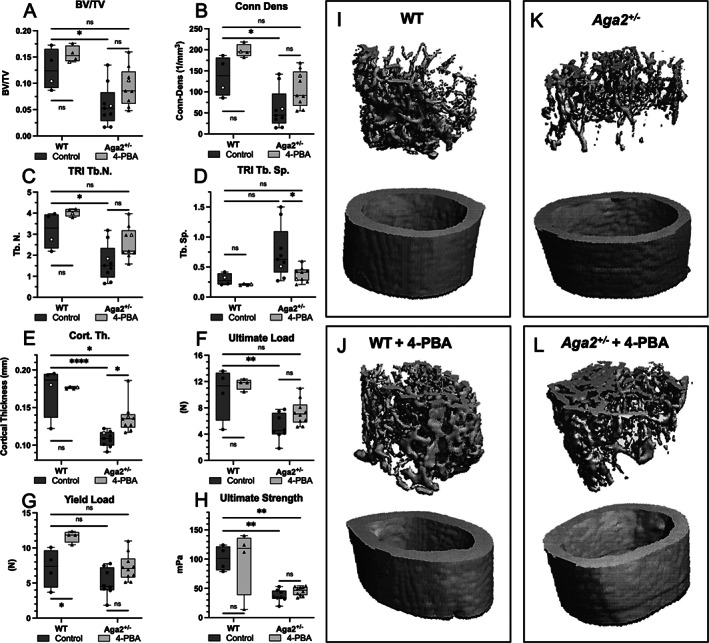
4‐PBA treatment improves bone mineralization, trabecular bone formation, and bone strength in Aga2 +/− mice. (A–E) Trabecular and bone parameter quantification of 4‐PBA treated and untreated Aga2 +/− 2‐months‐old male mice. Bars represent median and interquartile range. n = 8. (F–H) Quantification of biomechanical analyses via three‐point‐bending in 4‐PBA‐treated and untreated Aga2 +/− 2‐month‐old male mice. Bars represent median and interquartile range. n = 8. Open circles correspond to the animals represented in I,J. Two‐way ANOVAs were performed, *p < 0.05 was considered statistically significant. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns = not significant. (I–L) Representative visual images of trabecular (top) and cortical bone (bottom) μCT analysis in 4‐PBA–treated and untreated Aga2 +/− mice.
Genetic ablation of the key ER stress responders, BiP and CHOP, on the Aga2 +/− phenotype
As shown herein and previously by others,( 30 , 31 , 32 , 33 ) ER stress and UPR activation are increased in OI. We have also shown that, in vivo, 4‐PBA treatment reduced bone levels of BiP and CHOP. However, it is unclear whether the reduction in the abundance of these ER stress–responsive proteins directly improved the OI phenotype, or if they indirectly reflect improved intracellular dynamics. To determine if the genetic reduction of UPR pathway activation has an effect on the OI phenotype, we analyzed the phenotypes of the Aga2 +/− OI mouse models with either reduced BiP or absent CHOP. Radiographs of representative mice with each genotype are shown in Fig. 5A–H . Because BiP −/− animals are embryonic lethal at embryonic day 3.5 (E3.5),( 45 ) and BiP +/− mice are viable without a skeletal phenotype, we generated Aga2 +/− ;BiP +/− mice and found that with the genetic reduction of BiP expression, aspects of Aga2 +/− bone were not improved. Aga2 +/− ;BiP +/− mice showed no change in either body length or weight (Fig. S4 A,B), and femoral μCT analysis from 2‐month‐old mice showed no significant changes in trabecular or cortical parameters when compared to Aga2 +/− mice (Figs. S4 C–F, S5). Chop −/− mice were previously shown to be born at the expected Mendelian frequencies, appeared phenotypically normal, and had normal fertility and reproductive behavior, though the absence of CHOP had an effect on osteoblastic function based on decreased expression of type I collagen and osteocalcin mRNA expression.( 46 , 47 ) Generation of Aga2 +/− ;Chop −/− animals showed strikingly increased severity of the Aga2 +/− (OI) phenotype (Fig. 5). When compared to Aga2 +/− mice, Aga2 +/− ;Chop −/− animals showed markedly increased fracture incidence and deformity in long bones (Fig. 5B,D,F,H,K ), as well as decreased body length and weight (Fig. 5I,J ). Due to the severity of the phenotype and the extreme fragility of the long bones, we were unable to perform μCT analysis. Aga2 +/− ;Chop −/− mice also showed early mortality, with 13% dead before 2 months of age and scoliosis in 26% of the animals (Fig. 5D , blue arrow). Neither mortality before 2 months of age nor scoliosis was observed in our Aga2 +/− colony. These results signify that the UPR is necessary for managing the deleterious effects of defective type I procollagen molecules within the cell and suggests that although reduction of BiP and CHOP were seen with 4‐PBA treatment, the reduction was likely a consequence of an improved cellular environment. This is reflected in our in vitro experiments and illustrates the critical and necessary role of these molecules in responding to the abnormal ER environment in OI.
Fig. 5.
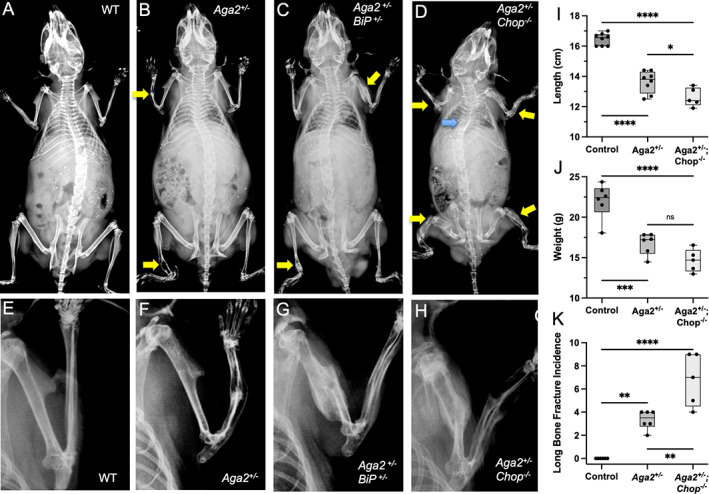
Genetic deletion of CHOP results in increased bone fragility and impaired bone development. (A–H) X‐ray images of WT, Aga2 +/−, Aga2 +/− ;BiP +/− , and Aga2 +/− ;Chop −/− male 2‐month‐old mice with insets showing closer imaging of forearm. Yellow arrows point to long‐bone fractures, blue arrow points to kyphoscoliosis. (I–K) Quantification of body length, body weight, and fracture incidence in WT (n = 6), Aga2 +/− (n = 6), and Aga +/− ;Chop −/− (n = 6) male mice. Bars represent median and interquartile range. Two‐way ANOVAs were performed, *p < 0.05 was considered statistically significant. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns = not significant.
Discussion
OI is a genetically heterogenous skeletal disorder that encompasses a broad range of phenotypic severity from mildly increased fracture rates over a lifetime, to severe ongoing progressive skeletal deformity due to recurrent fractures and undermineralized bone. Presently, the most commonly employed treatment is the use of bisphosphonates, stable derivatives of inorganic pyrophosphate (PPi).( 48 ) Multiple studies have shown that bisphosphonate therapy can preserve vertebral morphometry and decrease fracture incidence; however, this reduction in fracture incidence is generally only about 20%.( 49 ) Newer therapies that include neutralizing antibodies directed against sclerostin, receptor activator of nuclear factor κB ligand (RANKL), or TGFβ, and daily parathyroid hormone (PTH) administration( 50 , 51 , 52 , 53 , 54 , 55 , 56 ) are in preclinical and clinical trials for patients with OI. These treatments focus on improving bone quality by increasing collagen synthesis or influencing bone mineralization through modulation of osteoblast or osteoclast functions. They do not, however, address the molecular consequences of ER stress in the context of the effect of mutant type I collagen on cellular homeostasis.
It is important to note that the Aga2 +/− mutation results in a frameshift within the C‐propeptide region that alters a splice acceptor site and results in an extension of the COL1A1 peptide, differentiating it from the more common OI triple helical mutations.( 30 ) This may result in the inability of the peptide to accomplish early assembly and therefore induce a UPR that is different from the more common triple helical mutations. ER stress, however, has been shown to be a unifying molecular phenotype as a result of many differing types of OI mutations.( 32 , 36 , 44 , 57 ) Cultured fibroblasts derived from patients with type I procollagen gene structural mutations result in misfolded proteins that are retained in the ER and induce a UPR response.( 32 ) These structural mutations include those in triple helical domains, and affected human cultured fibroblasts show enlarged ERs as well as increased activity of multiple UPR pathways, with PERK being the most often upregulated.( 32 ) Similarly, cultured fibroblasts derived from OI patients with mutations in the genes encoding FKBP10 or HSP47, members of the FKBP65‐HSP47‐LH2‐BIP ER chaperone complex, demonstrated abnormally dilated ER with intracellular protein aggregates.( 58 , 59 , 60 , 61 ) Fibroblasts derived from individuals with recessively inherited mutations in CRTAP, P3H1, and PPIB (an ER complex responsible for the hydroxylation of specific proline residues in type I procollagen) produced overmodified type I procollagen, showed enlarged ER with retention of type I procollagen, presence of protein aggregates, activation of the PERK/ATF4 branch of the UPR, and apoptotic cell death.( 36 ) In our study, we addressed whether an in vivo treatment that reduces ER stress and the UPR response could have an effect on bone development and postnatal bone homeostasis.
Chemical ER stress inhibitors such as 4‐PBA have been shown to have increasing relevance in clinical treatment settings. 4‐PBA has several functions, including acting as an ammonia scavenger, histone deacetylase (HDAC) inhibitor, and protein folding chaperone (reviewed in Kolb and colleagues( 62 )). More specifically, 4‐PBA functions as a chaperone through its ability to bind to the exposed hydrophobic residues of misfolded proteins.( 63 ) Our short‐term treatment/culture protocol in Aga2 +/− osteoblasts revealed increased levels of PERK activation, reductions in XBP‐1 splicing, and increased levels of PDI, indications of increased ER stress that were then reduced with 4‐PBA treatment. This falls in line with previous studies( 37 , 63 ) showing a reduction in the ER stress response in both 4‐PBA treated OI patient fibroblasts through reduced PERK activation and in the zebrafish OI model, reducing ER cisternae size.
In vivo, 4‐PBA–treated Aga2 +/− mice showed significantly decreased fracture incidence as well as increased length and weight compared to mice that received vehicle alone. μCT analysis revealed bone volumes, cortical bone volumes, and trabecular number that were either improved compared to untreated Aga2 +/− bone or no longer significantly different from WT values. Thus 4‐PBA treatment improved trabecular bone parameters as well as cortical thickness, an indication of increased bone formation. Importantly, biomechanical testing of femurs via three‐point bending revealed improvements in the ultimate load and yield load of treated Aga2 +/− long bones to where they were no longer significantly different from WT samples. Notably, 4‐PBA treatment did not have a significant effect on the stiffness and ductility of Aga2 +/− bone. This may be because Aga2 +/− bone still contains mutated type I collagen proteins that likely disturb collagen fibril formation and therefore bone ECM structure. In this study we did not measure mineralization in 4‐PBA–treated femurs, which could contribute to the improved bone parameters. Further, Lisse and colleagues( 30 ) showed increased mineralization in Aga2 +/− cultured osteoblasts at baseline, making it unlikely that mineralization plays a major role in the reduced fracture incidence following 4‐PBA treatment. Finally, our experiments demonstrated not only improved OI bone strength but also postnatal bone development that included increased linear growth. This indicates an effect on the cartilage growth plate and supports evidence from Scheiber and colleagues( 31 ) that growth plate hypertrophic chondrocytes in OI exhibit increased ER stress.
In vivo, we found that 4‐PBA treatment reduced elevated BiP, CHOP, and ATF4 levels in Aga2 +/− mice. We do not discount the possibility that the relief of ER stress may have an effect on osteoblast differentiation or progenitor availability; this would be an important topic for future study. Similar to previous publications, we also saw the modulation of autophagosome markers in Aga2 +/− femur lysates compared to WT.( 30 , 32 ) Besio and colleagues( 32 ) showed increased levels of LC3A/B in OI human fibroblasts in vitro before and after chloroquine treatment, indicating an increase in autophagosome formation. However, whereas previous publications demonstrated increased autophagy in either Aga2 +/− fibroblasts or OI patient fibroblasts in vitro,( 30 , 32 ) we detected decreased LC3A/B levels in Aga2 +/− bone in vivo. This could be interpreted as either a decrease in autophagosome formation or an increase in autophagosome degradation/flux. The possibility of altered autophagy in OI bone cells in vivo requires further experimental study. Nevertheless, our results indicate that 4‐PBA treatment also had an effect on autophagy in bone in vivo and restored autophagy (LC3A/B) activity to WT levels. This falls in line with a publication showing increased LC3A/B levels upon 4‐PBA treatment in vitro.( 32 ) Further, a recent publication using a PC12 neuroblastic cell line showed enhancement of autophagy upon 4‐PBA treatment following prolonged ER stress.( 64 ) This effect occurred via the phosphatidylinositol‐3′‐kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) signaling pathway. Ishida and colleagues( 44 ) showed that mutated collagens in OI are mainly cleared through autophagy, making mTOR signaling and the autophagosome an ideal target for treatment. However, another recent study showed that inhibition of mTOR signaling in the OI G610C model resulted in improved trabecular bone formation but no change in biomechanical properties, and negative effects on longitudinal bone growth.( 65 ) Altogether, this implies that the improved autophagy observed in our 4‐PBA–treated Aga2 +/− mice likely contributes to bone formation improvements, whereas the upstream effects of 4‐PBA on UPR activation improves bone density, biomechanical parameters, and longitudinal bone growth. Additionally, it should be noted that our study showed that protein analysis of OI bone in vivo leads to different results than observed in in vitro cultured cells and care should be taken in drawing conclusions solely from in vitro data.
The UPR and ER stress pathway involves several major components that could be targeted for therapy. Because we observed significantly increased BiP and CHOP levels in Aga2 +/− mice in vivo, we performed genetic crosses in order to gain insight into the role of these ER stress responses in OI and their effect on Aga2 +/− bone quality, strength, and development. Genetic reduction of BiP expression in Aga2 +/− mice had no effect on trabecular bone parameters, cortical bone width, overall mouse length, or mouse weight when compared to Aga2 +/− mice. It is possible that 50% reduction of BiP expression is not sufficient to affect the Aga2 +/− phenotype. When CHOP levels were ablated in Aga2 +/− mice, fracture incidence was significantly increased, mouse overall length and weight were significantly decreased, and the Aga2 +/− OI phenotype was notably worsened. This may result from the normal role of CHOP in osteoblasts( 47 , 66 , 67 ) that includes effects on osteoblast differentiation when diminished and increased osteoblast apoptosis when overexpressed. The data suggest that a certain baseline expression of CHOP is needed in osteoblasts, and our findings indicate that the downstream ER stress response is at some level necessary to maintain OI bone integrity. Reduction of the response alone is unable to rescue the OI phenotype, but worsens it.
Although there are several ER stress inhibitors used as clinical treatments for cancer, the goal of these treatments is to sensitize the cells to ER stress and thereby induce cell toxicity (reviewed in Li and colleagues( 68 ) and Almanza and colleagues( 69 )), which would likely not be effective in treating OI. Furthermore, because our study demonstrated that the genetic reduction of the ER stress response did not rescue the OI phenotype, therapeutic strategies targeting the downstream components of the UPR are also unlikely to be effective in OI. However, molecules that aid in or increase protein chaperone activity, and lead to improved folding and/or secretion of type I procollagen, may improve the OI phenotype. Overall, our study presents novel data showing that the UPR is part of the mechanism of disease of OI and that in vivo alleviation of ER stress via 4‐PBA treatment in an OI mouse model diminishes its cellular consequences and improves multiple bone parameters, including bone strength, partly due to increasing autophagy and reduction of varying components of the UPR.
Author Contributions
Ivan Duran: Conceptualization, Investigation, Methodology, Visualization, Writing‐original draft; Jennifer Zieba: Conceptualization, Investigation, Methodology, Visualization, Writing‐original draft, Writing‐review & editing; Fabiana Csukasi: Investigation; Jorge H. Martin: Investigation; Davis Wachtell: Investigation, Writing‐original draft; Maya Barad: Investigation; Brian Dawson: Investigation; Bohumil Fafilek: Investigation; Christina M. Jacobsen: Resources; Catherine G. Ambrose: Investigation; Pavel Krejci: Conceptualization, Resources, Writing‐review & editing; Brendan H. Lee: Funding acquisition; Supervision, Writing‐review & editing; Deborah Krakow: Conceptualization, Funding acquisition, Supervision, Writing‐original draft, Writing‐review & editing.
Conflict of Interests
None of the authors have any potential conflicts of interest to disclose.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1002/jbmr.4501.
Supporting information
Fig. S1 Improvement of the OI phenotype and ER stress levels in female Aga2 +/− mice treated with 4‐PBA. Quantification of body length, body weight, and long bone fracture incidence in 4‐PBA treated and untreated Aga2 +/− female mice. Bars represent median and interquartile range. For length and weight, 2‐way ANOVAs were performed, * = p < 0.05 was considered statistically significant. * = p < 0.05, ** = p < 0.01, *** = p < 0.001, **** = p < 0.0001, ns = not significant. For fracture incidence, significance was determined via t test.
Fig. S2 Supplemental μCT trabecular data for 4‐PBA treated Aga2 +/− mice. Trabecular and bone parameter quantification of 4‐PBA treated and untreated Aga2 +/− mice taken from femoral trabecular and midshaft μCT analysis. Bars represent median and interquartile range. 2‐way ANOVAs were performed, * = p < 0.05 was considered statistically significant. * = p < 0.05, ** = p < 0.01, ns = not significant.
Fig. S3 Supplemental biomechanical data of 4‐PBA treated Aga2 +/− mice. Quantification of biomechanical analyses via three‐point‐bending in 4‐PBA treated and untreated Aga2 +/− mice. Bars represent median and interquartile range. 2‐way ANOVAs were performed, * = p < 0.05 was considered statistically significant. * = p < 0.05, ** = p < 0.01, *** = p < 0.001, ns = not significant.
Fig. S4 Aga2 +/− ;BiP +/− mice show no change in trabecular bone formation. (A,B) Quantification of body length and weight in WT (n = 6), Aga2 +/− (n = 6), and Aga2 +/− ;Chop −/− (n = 6) mice. 2‐way ANOVAs were performed, * = p < 0.05 was considered statistically significant. * = p < 0.05, ** = p < 0.01, *** = p < 0.001, **** = p < 0.0001, ns = not significant. (C–F) Trabecular and bone parameter quantification of Aga2 +/− (n = 6) and Aga2 +/− ;BiP +/− (n = 7). Bars represent median and interquartile range. Significance was determined via t test. (G,H) Representative visual images of trabecular (top) and cortical bone (bone) μCT analysis of Aga2 +/− and Aga2 +/− ;BiP +/− mice.
Fig. S5 Supplemental μCT data of Aga2 +/− ;BiP +/− mouse femurs. Trabecular and bone parameter quantification of Aga2 +/− and Aga2 +/− ;BiP +/− male mice. Bars represent median and interquartile range. Significance was determined via t test.
Acknowledgments
DK, DHC, and BHL are supported by NIH P01 HD070394. DK and DHC are also supported by NIH R01 AR066124. ID is supported by a Geisman Award from the Osteogenesis Imperfecta Foundation, Junta de Andalucia FEDER funds UMA18‐FEDERJA‐177, and the AHUCE foundation. PK was supported by Agency for Healthcare Research of the Czech Republic (Grant NV18‐08‐00567), and the Czech Science Foundation (Grants GA17‐09525S, GA19‐20123S). We appreciate the sharing of Aga2 mice by Dr. Martin Hrabé de Angelis to Dr. Christina M. Jacobson.
Authors' roles: Conceptualization, ID, JZ, PK, and DK; Investigation, ID, JZ, FC, JHM, MB, DW, BD, BF, and CGA; Writing – Original Draft, ID, JZ, and DK; Writing – Review & Editing, DHC, PK, MLW, BHL, JZ, DW, and DK; Funding Acquisition, DK, DHC, and BHL; Resources, CMJ, PK, and MLW; Supervision, DK, DHC, and BHL.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Forlino A, Cabral WA, Barnes AM, Marini JC. New perspectives on osteogenesis imperfecta. Nat Rev Endocrinol. 2011;7(9):540‐557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rauch F, Glorieux FH. Osteogenesis imperfecta. Lancet. 2004;363(9418):1377‐1385. [DOI] [PubMed] [Google Scholar]
- 3. Marini JC, Forlino A, Bachinger HP, et al. Osteogenesis imperfecta. Nat Rev Dis Primers. 2017;3:17052. [DOI] [PubMed] [Google Scholar]
- 4. Jacobsen CM, Schwartz MA, Roberts HJ, et al. Enhanced Wnt signaling improves bone mass and strength, but not brittleness, in the Col1a1(+/mov13) mouse model of type I osteogenesis imperfecta. Bone. 2016;90:127‐132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kaupp S, Horan DJ, Lim KE, et al. Combination therapy in the Col1a2(G610C) mouse model of osteogenesis imperfecta reveals an additive effect of enhancing LRP5 signaling and inhibiting TGFbeta signaling on trabecular bone but not on cortical bone. Bone. 2020;131:115084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zimmerman SM, Dimori M, Heard‐Lipsmeyer ME, Morello R. The osteocyte transcriptome is extensively dysregulated in mouse models of osteogenesis imperfecta. JBMR Plus. 2019;3(7):e10171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11(4):381‐389. [DOI] [PubMed] [Google Scholar]
- 8. Marciniak SJ, Ron D. Endoplasmic reticulum stress signaling in disease. Physiol Rev. 2006;86(4):1133‐1149. [DOI] [PubMed] [Google Scholar]
- 9. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8(7):519‐529. [DOI] [PubMed] [Google Scholar]
- 10. Nakayama H, Hamada M, Fujikake N, et al. ER stress is the initial response to polyglutamine toxicity in PC12 cells. Biochem Biophys Res Commun. 2008;377(2):550‐555. [DOI] [PubMed] [Google Scholar]
- 11. Boyce M, Yuan J. Cellular response to endoplasmic reticulum stress: a matter of life or death. Cell Death Differ. 2006;13(3):363‐373. [DOI] [PubMed] [Google Scholar]
- 12. Pobre KFR, Poet GJ, Hendershot LM. The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: getting by with a little help from ERdj friends. J Biol Chem. 2019;294(6):2098‐2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Adams CJ, Kopp MC, Larburu N, Nowak PR, Ali MMU. Structure and molecular mechanism of ER stress signaling by the unfolded protein response signal activator IRE1. Front Mol Biosci. 2019;6:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13(2):89‐102. [DOI] [PubMed] [Google Scholar]
- 15. Ron D, Harding HP. Protein‐folding homeostasis in the endoplasmic reticulum and nutritional regulation. Cold Spring Harb Perspect Biol. 2012;4(12):a013177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harding HP, Novoa I, Zhang Y, et al. Regulated translation initiation controls stress‐induced gene expression in mammalian cells. Mol Cell. 2000;6(5):1099‐1108. [DOI] [PubMed] [Google Scholar]
- 17. Yang Y, Liu L, Naik I, Braunstein Z, Zhong J, Ren B. Transcription factor C/EBP homologous protein in health and diseases. Front Immunol. 2017;8:1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guo Q, Xu L, Li H, Sun H, Wu S, Zhou B. 4‐PBA reverses autophagic dysfunction and improves insulin sensitivity in adipose tissue of obese mice via Akt/mTOR signaling. Biochem Biophys Res Commun. 2017;484(3):529‐535. [DOI] [PubMed] [Google Scholar]
- 19. Ricobaraza A, Cuadrado‐Tejedor M, Perez‐Mediavilla A, Frechilla D, Del Rio J, Garcia‐Osta A. Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in an Alzheimer's disease mouse model. Neuropsychopharmacology. 2009;34(7):1721‐1732. [DOI] [PubMed] [Google Scholar]
- 20. Zeng M, Sang W, Chen S, et al. 4‐PBA inhibits LPS‐induced inflammation through regulating ER stress and autophagy in acute lung injury models. Toxicol Lett. 2017;271:26‐37. [DOI] [PubMed] [Google Scholar]
- 21. Andersen E, Chollet ME, Baroni M, et al. The effect of the chemical chaperone 4‐phenylbutyrate on secretion and activity of the p.Q160R missense variant of coagulation factor FVII. Cell Biosci. 2019;9:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pena‐Quintana L, Llarena M, Reyes‐Suarez D, Aldamiz‐Echevarria L. Profile of sodium phenylbutyrate granules for the treatment of urea‐cycle disorders: patient perspectives. Patient Prefer Adherence. 2017;11:1489‐1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lichter‐Konecki U, Diaz GA, Merritt JL 2nd, et al. Ammonia control in children with urea cycle disorders (UCDs); phase 2 comparison of sodium phenylbutyrate and glycerol phenylbutyrate. Mol Genet Metab. 2011;103(4):323‐329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xiao C, Giacca A, Lewis GF. Sodium phenylbutyrate, a drug with known capacity to reduce endoplasmic reticulum stress, partially alleviates lipid‐induced insulin resistance and beta‐cell dysfunction in humans. Diabetes. 2011;60(3):918‐924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brose RD, Shin G, McGuinness MC, et al. Activation of the stress proteome as a mechanism for small molecule therapeutics. Hum Mol Genet. 2012;21(19):4237‐4252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Miller AC, Cohen S, Stewart M, Rivas R, Lison P. Radioprotection by the histone deacetylase inhibitor phenylbutyrate. Radiat Environ Biophys. 2011;50(4):585‐596. [DOI] [PubMed] [Google Scholar]
- 27. Koyama M, Furuhashi M, Ishimura S, et al. Reduction of endoplasmic reticulum stress by 4‐phenylbutyric acid prevents the development of hypoxia‐induced pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol. 2014;306(9):H1314‐H1323. [DOI] [PubMed] [Google Scholar]
- 28. Zhu M, Guo M, Fei L, Pan XQ, Liu QQ. 4‐phenylbutyric acid attenuates endoplasmic reticulum stress‐mediated pancreatic beta‐cell apoptosis in rats with streptozotocin‐induced diabetes. Endocrine. 2014;47(1):129‐137. [DOI] [PubMed] [Google Scholar]
- 29. Mimori S, Ohtaka H, Koshikawa Y, et al. 4‐Phenylbutyric acid protects against neuronal cell death by primarily acting as a chemical chaperone rather than histone deacetylase inhibitor. Bioorg Med Chem Lett. 2013;23(21):6015‐6018. [DOI] [PubMed] [Google Scholar]
- 30. Lisse TS, Thiele F, Fuchs H, et al. ER stress‐mediated apoptosis in a new mouse model of osteogenesis imperfecta. PLoS Genet. 2008;4(2):e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Scheiber AL, Guess AJ, Kaito T, et al. Endoplasmic reticulum stress is induced in growth plate hypertrophic chondrocytes in G610C mouse model of osteogenesis imperfecta. Biochem Biophys Res Commun. 2019;509(1):235‐240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Besio R, Iula G, Garibaldi N, et al. 4‐PBA ameliorates cellular homeostasis in fibroblasts from osteogenesis imperfecta patients by enhancing autophagy and stimulating protein secretion. Biochim Biophys Acta Mol Basis Dis. 2018;1864(5 Pt A):1642‐1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lindert U, Weis MA, Rai J, et al. Molecular consequences of the SERPINH1/HSP47 mutation in the dachshund natural model of osteogenesis imperfecta. J Biol Chem. 2015;290(29):17679‐17689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Forlino A, Tani C, Rossi A, et al. Differential expression of both extracellular and intracellular proteins is involved in the lethal or nonlethal phenotypic variation of BrtlIV, a murine model for osteogenesis imperfecta. Proteomics. 2007;7(11):1877‐1891. [DOI] [PubMed] [Google Scholar]
- 35. Kasamatsu A, Uzawa K, Hayashi F, et al. Deficiency of lysyl hydroxylase 2 in mice causes systemic endoplasmic reticulum stress leading to early embryonic lethality. Biochem Biophys Res Commun. 2019;512(3):486‐491. [DOI] [PubMed] [Google Scholar]
- 36. Besio R, Garibaldi N, Leoni L, et al. Cellular stress due to impairment of collagen prolyl hydroxylation complex is rescued by the chaperone 4‐phenylbutyrate. Dis Model Mech. 2019;12(6):dmm038521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gioia R, Tonelli F, Ceppi I, et al. The chaperone activity of 4PBA ameliorates the skeletal phenotype of Chihuahua, a zebrafish model for dominant osteogenesis imperfecta. Hum Mol Genet. 2017;26(15):2897‐2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hrabe de Angelis MH, Flaswinkel H, Fuchs H, et al. Genome‐wide, large‐scale production of mutant mice by ENU mutagenesis. Nat Genet. 2000;25(4):444‐447. [DOI] [PubMed] [Google Scholar]
- 39. Luo S, Mao C, Lee B, Lee AS. GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol Cell Biol. 2006;26(15):5688‐5697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Turner CH, Burr DB. Basic biomechanical measurements of bone: a tutorial. Bone. 1993;14(4):595‐608. [DOI] [PubMed] [Google Scholar]
- 41. Wang XZ, Lawson B, Brewer JW, et al. Signals from the stressed endoplasmic reticulum induce C/EBP‐homologous protein (CHOP/GADD153). Mol Cell Biol. 1996;16(8):4273‐4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wilkinson B, Gilbert HF. Protein disulfide isomerase. Biochim Biophys Acta. 2004;1699(1‐2):35‐44. [DOI] [PubMed] [Google Scholar]
- 43. Goldberger RF, Epstein CJ, Anfinsen CB. Acceleration of reactivation of reduced bovine pancreatic ribonuclease by a microsomal system from rat liver. J Biol Chem. 1963;238:628‐635. [PubMed] [Google Scholar]
- 44. Ishida Y, Yamamoto A, Kitamura A, et al. Autophagic elimination of misfolded procollagen aggregates in the endoplasmic reticulum as a means of cell protection. Mol Biol Cell. 2009;20(11):2744‐2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Luo S, Baumeister P, Yang S, Abcouwer SF, Lee AS. Induction of Grp78/BiP by translational block: activation of the Grp78 promoter by ATF4 through and upstream ATF/CRE site independent of the endoplasmic reticulum stress elements. J Biol Chem. 2003;278(39):37375‐37385. [DOI] [PubMed] [Google Scholar]
- 46. Zinszner H, Kuroda M, Wang X, et al. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12(7):982‐995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pereira RC, Delany AM, Canalis E. CCAAT/enhancer binding protein homologous protein (DDIT3) induces osteoblastic cell differentiation. Endocrinology. 2004;145(4):1952‐1960. [DOI] [PubMed] [Google Scholar]
- 48. Drake MT, Clarke BL, Khosla S. Bisphosphonates: mechanism of action and role in clinical practice. Mayo Clin Proc. 2008;83(9):1032‐1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hasegawa K, Inoue M, Seino Y, Morishima T, Tanaka H. Growth of infants with osteogenesis imperfecta treated with bisphosphonate. Pediatr Int. 2009;51(1):54‐58. [DOI] [PubMed] [Google Scholar]
- 50. Orwoll ES, Shapiro J, Veith S, et al. Evaluation of teriparatide treatment in adults with osteogenesis imperfecta. J Clin Invest. 2014;124(2):491‐498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. McClung MR, Lewiecki EM, Cohen SB, et al. Denosumab in postmenopausal women with low bone mineral density. N Engl J Med. 2006;354(8):821‐831. [DOI] [PubMed] [Google Scholar]
- 52. Lacey DL, Tan HL, Lu J, et al. Osteoprotegerin ligand modulates murine osteoclast survival in vitro and in vivo. Am J Pathol. 2000;157(2):435‐448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yasuda H, Shima N, Nakagawa N, et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis‐inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci U S A. 1998;95(7):3597‐3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Glorieux FH, Devogelaer JP, Durigova M, et al. BPS804 anti‐sclerostin antibody in adults with moderate osteogenesis imperfecta: results of a randomized phase 2a trial. J Bone Miner Res. 2017;32(7):1496‐1504. [DOI] [PubMed] [Google Scholar]
- 55. Tauer JT, Abdullah S, Rauch F. Effect of anti‐TGF‐beta treatment in a mouse model of severe osteogenesis imperfecta. J Bone Miner Res. 2019;34(2):207‐214. [DOI] [PubMed] [Google Scholar]
- 56. Grafe I, Yang T, Alexander S, et al. Excessive transforming growth factor‐beta signaling is a common mechanism in osteogenesis imperfecta. Nat Med. 2014;20(6):670‐675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mirigian LS, Makareeva E, Mertz EL, et al. Osteoblast malfunction caused by cell stress response to procollagen misfolding in alpha2(I)‐G610C mouse model of osteogenesis imperfecta. J Bone Miner Res. 2016;31(8):1608‐1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Alanay Y, Avaygan H, Camacho N, et al. Mutations in the gene encoding the RER protein FKBP65 cause autosomal‐recessive osteogenesis imperfecta. Am J Hum Genet. 2010;86(4):551‐559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Christiansen HE, Schwarze U, Pyott SM, et al. Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta. Am J Hum Genet. 2010;86(3):389‐398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Duran I, Nevarez L, Sarukhanov A, et al. HSP47 and FKBP65 cooperate in the synthesis of type I procollagen. Hum Mol Genet. 2015;24(7):1918‐1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kelley BP, Malfait F, Bonafe L, et al. Mutations in FKBP10 cause recessive osteogenesis imperfecta and Bruck syndrome. J Bone Miner Res. 2011;26(3):666‐672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kolb PS, Ayaub EA, Zhou W, Yum V, Dickhout JG, Ask K. The therapeutic effects of 4‐phenylbutyric acid in maintaining proteostasis. Int J Biochem Cell Biol. 2015;61:45‐52. [DOI] [PubMed] [Google Scholar]
- 63. Iannitti T, Palmieri B. Clinical and experimental applications of sodium phenylbutyrate. Drugs R D. 2011;11(3):227‐249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wang Z, Zheng S, Gu Y, Zhou L, Lin B, Liu W. 4‐PBA enhances autophagy by inhibiting endoplasmic reticulum stress in recombinant human Beta nerve growth factor‐induced PC12 cells after mechanical injury via PI3K/AKT/mTOR signaling pathway. World Neurosurg. 2020;138:e659‐e664. [DOI] [PubMed] [Google Scholar]
- 65. Bateman JF, Sampurno L, Maurizi A, et al. Effect of rapamycin on bone mass and strength in the alpha2(I)‐G610C mouse model of osteogenesis imperfecta. J Cell Mol Med. 2019;23(3):1735‐1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pereira RC, Stadmeyer L, Marciniak SJ, Ron D, Canalis E. C/EBP homologous protein is necessary for normal osteoblastic function. J Cell Biochem. 2006;97(3):633‐640. [DOI] [PubMed] [Google Scholar]
- 67. Pereira RC, Stadmeyer LE, Smith DL, Rydziel S, Canalis E. CCAAT/enhancer‐binding protein homologous protein (CHOP) decreases bone formation and causes osteopenia. Bone. 2007;40(3):619‐626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Li A, Song NJ, Riesenberg BP, Li Z. The emerging roles of endoplasmic reticulum stress in balancing immunity and tolerance in health and diseases: mechanisms and opportunities. Front Immunol. 2019;10:3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Almanza A, Carlesso A, Chintha C, et al. Endoplasmic reticulum stress signalling ‐ from basic mechanisms to clinical applications. FEBS J. 2019;286(2):241‐278. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Improvement of the OI phenotype and ER stress levels in female Aga2 +/− mice treated with 4‐PBA. Quantification of body length, body weight, and long bone fracture incidence in 4‐PBA treated and untreated Aga2 +/− female mice. Bars represent median and interquartile range. For length and weight, 2‐way ANOVAs were performed, * = p < 0.05 was considered statistically significant. * = p < 0.05, ** = p < 0.01, *** = p < 0.001, **** = p < 0.0001, ns = not significant. For fracture incidence, significance was determined via t test.
Fig. S2 Supplemental μCT trabecular data for 4‐PBA treated Aga2 +/− mice. Trabecular and bone parameter quantification of 4‐PBA treated and untreated Aga2 +/− mice taken from femoral trabecular and midshaft μCT analysis. Bars represent median and interquartile range. 2‐way ANOVAs were performed, * = p < 0.05 was considered statistically significant. * = p < 0.05, ** = p < 0.01, ns = not significant.
Fig. S3 Supplemental biomechanical data of 4‐PBA treated Aga2 +/− mice. Quantification of biomechanical analyses via three‐point‐bending in 4‐PBA treated and untreated Aga2 +/− mice. Bars represent median and interquartile range. 2‐way ANOVAs were performed, * = p < 0.05 was considered statistically significant. * = p < 0.05, ** = p < 0.01, *** = p < 0.001, ns = not significant.
Fig. S4 Aga2 +/− ;BiP +/− mice show no change in trabecular bone formation. (A,B) Quantification of body length and weight in WT (n = 6), Aga2 +/− (n = 6), and Aga2 +/− ;Chop −/− (n = 6) mice. 2‐way ANOVAs were performed, * = p < 0.05 was considered statistically significant. * = p < 0.05, ** = p < 0.01, *** = p < 0.001, **** = p < 0.0001, ns = not significant. (C–F) Trabecular and bone parameter quantification of Aga2 +/− (n = 6) and Aga2 +/− ;BiP +/− (n = 7). Bars represent median and interquartile range. Significance was determined via t test. (G,H) Representative visual images of trabecular (top) and cortical bone (bone) μCT analysis of Aga2 +/− and Aga2 +/− ;BiP +/− mice.
Fig. S5 Supplemental μCT data of Aga2 +/− ;BiP +/− mouse femurs. Trabecular and bone parameter quantification of Aga2 +/− and Aga2 +/− ;BiP +/− male mice. Bars represent median and interquartile range. Significance was determined via t test.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.