

SUMMARY
Metabolic reprogramming is a hallmark of activated T cells. The switch from oxidative phosphorylation to aerobic glycolysis provides energy and intermediary metabolites for the biosynthesis of macromolecules to support clonal expansion and effector function. Here we show that glycolytic reprogramming additionally controls inflammatory gene expression via epigenetic remodeling. We found that the glucose transporter GLUT3 is essential for the effector functions of Th17 cells in models of autoimmune colitis and encephalomyelitis. At the molecular level, we show that GLUT3-dependent glucose uptake controls a metabolic-transcriptional circuit that regulates the pathogenicity of Th17 cells. Metabolomic, epigenetic and transcriptomic analyses linked GLUT3 to mitochondrial glucose oxidation and ACLY-dependent acetyl-CoA generation as a rate-limiting step in the epigenetic regulation of inflammatory gene expression. Our findings are also important from a translational perspective because inhibiting GLUT3-dependent acetyl-CoA generation is a promising metabolic checkpoint to mitigate Th17 cell-mediated inflammatory diseases.
eTOC blurb
Hochrein et al. report that inflammatory T cells express high levels of GLUT3. Ablation of GLUT3 curtailed Th17 cell-mediated immune responses and protected mice from autoimmune colitis and encephalomyelitis. GLUT3-dependent glucose metabolism controls the generation of nucleo-cytosolic acetyl-CoA and the epigenetic regulation of cytokine responses through histone acetylation.
Graphical Abstract
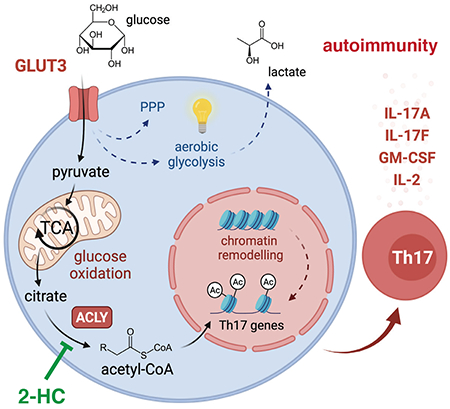
INTRODUCTION
Autoimmune diseases, including multiple sclerosis (MS) and inflammatory bowel disease (IBD), are a heterogeneous group of pathologies with tissue-specific characteristics but common immunological hallmarks, such as T cell-mediated inflammation (Theofilopoulos et al., 2017). T helper 17 (Th17) cells are implicated in the pathogenesis of most autoimmune diseases and targeting their effector cytokines showed promising clinical efficacy in psoriasis, IBD and rheumatoid arthritis (RA) (Hamilton, 2019; Lai and Dong, 2016; Miossec and Kolls, 2012). The molecular mechanisms that promote the pathogenicity of Th17 cells remain incompletely understood but culminating evidence suggest that dietary factors contribute to the development and progression of autoimmune diseases (Manzel et al., 2014; Nobs et al., 2020; Zhang et al., 2019). Nutritional patterns that are characterized by high-calorie, high-salt and excess sugar intake (collectively termed the ‘Western diet’) are correlated with metabolic and inflammatory disorders and are considered as risk factors for autoimmunity (Manzel et al., 2014; Matveeva et al., 2018; Zhang et al., 2019). How changes in systemic metabolism influence local inflammatory processes is complex but recent data suggest that metabolic reprogramming of lymphocytes contributes significantly to the development of autoimmune diseases (Alwarawrah et al., 2018; Teng et al., 2019).
Naive T cells are characterized by a low-rate catabolic metabolism, which generates ATP through oxidation of pyruvate and fatty acids. Following activation, (autoreactive) T cells dramatically increase their bioenergetic and metabolic profile to support cellular growth and clonal expansion. To meet these demands, T cells rewire their metabolic machinery and use primarily glycolysis for ATP production and the biosynthesis of building blocks (O’Neill et al., 2016; Vaeth et al., 2017a; Wang et al., 2020). The seemingly paradoxical phenomenon that proliferative cells rely predominantly on glycolysis even when oxygen levels allow more energy-efficient respiration was first described by Otto Warburg in 1956 (Warburg, 1956). However, lymphocytes prefer aerobic glycolysis over oxidative phosphorylation (OxPhos) not only because glycolysis generates ATP ~ 100 times faster than respiration, but also because glycolysis provides critical intermediary metabolites that serve as building blocks for the biosynthesis of macromolecules (O’Neill et al., 2016; Shen and Shi, 2019). Nonetheless, glycolytic and mitochondrial metabolism are tightly interconnected to precisely meet the metabolic demands of different T cell subsets (Shen and Shi, 2019).
Glucose is the universal fuel for the immune system and can be taken up from the environment by three structurally distinct transporter families (Deng and Yan, 2016). Sodium-glucose linked transporters (SGLTs) require an ATP-dependent sodium gradient at the plasma membrane to import glucose, whereas the SLC2A family of glucose transporters (GLUTs) and the more recently identified SWEET (SLC50A1) proteins promote facilitated diffusion across membranes (Deng and Yan, 2016; Mueckler and Thorens, 2013; Simpson et al., 2008). 14 different GLUTs have been identified in humans with 12 homologues in mice (Deng and Yan, 2016; Mueckler and Thorens, 2013; Simpson et al., 2008). Different tissues express a heterogenous spectrum of glucose transporters but if individual glucose transporters have specific functions remains poorly defined. In the adaptive immune system, only the role of GLUT1 has been investigated to date. T cell-specific inactivation of Slc2a1 (encoding GLUT1) attenuated glucose uptake and glycolysis, which impaired proliferation and differentiation of CD4+ T cells (Macintyre et al., 2014). Surprisingly, GLUT1-deficient cytotoxic and T regulatory (Treg) cells remained phenotypically and functionally unaffected (Macintyre et al., 2014), indicating that other glucose uptake mechanisms exist in these T cell subsets. We and others showed previously that antigen receptor signaling massively upregulated the expression of ‘brain-type’ glucose transporter GLUT3 in T cells (Beckermann et al., 2020; Klein-Hessling et al., 2017; Macintyre et al., 2014; Vaeth et al., 2020; Vaeth et al., 2017a; Wang et al., 2020) but the function of GLUT3 in lymphocytes remained elusive.
We here report that GLUT3 controls the expression of inflammatory cytokines by Th17 cells through glycolytic-epigenetic reprogramming. GLUT3-dependent glucose consumption was largely dispensable for ATP production, T cell activation and clonal expansion. By contrast, GLUT3 was critical for the inflammatory cytokine production by Th17 cells and ablation of GLUT3 protected mice from autoimmune colitis and encephalomyelitis. Mechanistically, we found that GLUT3-mediated glucose metabolism supports the generation of acetyl-CoA and epigenetic remodeling. Inactivation of ATP-citrate lyase (ACLY), which catalyzes the release of cytosolic acetyl-CoA from citrate, phenocopied the defects of GLUT3-deficient Th17 cells, suggesting that glucose-derived acetyl-CoA drives Th17 pathogenicity. Pharmacological and genetic suppression of ACLY-dependent acetyl-CoA generation prevented histone acetylation at inflammatory gene loci and mitigated cytokine responses in murine and human Th17 cells. These findings suggest that ACLY, downstream of GLUT3, is a druggable ‘metabolic checkpoint’ to treat autoimmune diseases, in which Th17 cells play an important pathophysiological role.
RESULTS
Multiple pathways coordinately upregulate GLUT3 in Th17 cells
Activated T cells massively upregulated GLUT3 at mRNA and protein levels (Fig. 1A, B) at the plasma membrane (Fig. S1A, B). We found the highest transcripts of GLUT3 in inflammatory Th1 and Th17 cells, whereas T regulatory (Treg) cells showed only a moderate expression of GLUT3 (Fig. 1B). Because GLUT3 was highly expressed in Th17 cells, we further investigated the signaling pathways that control the upregulation of GLUT3 in this subset (Fig. S1C–J). Ablation of the regulatory subunit Bα of calcineurin (gene name Ppp3r1) prevents the activation of NFAT transcription factors and completely abolished GLUT3 induction (Fig. S1C). Similarly, deletion of the co-stimulatory receptor CD28 (Fig. S1D), the CD28-dependent transcription factor IRF4 (Fig. S1E) or STAT3 (Fig. S1F) also prevented GLUT3 expression in Th17 cells, demonstrating that antigen receptor, co-stimulation and cytokine signaling coordinately regulate the expression of GLUT3 in Th17 cells. When we differentiated T cells under hypoxia, we found even higher GLUT3 expression levels (Fig. S1G). We reasoned that the hypoxia-inducible factor HIF-1α promotes GLUT3 expression and found that HIF-1α-deficient Th17 cells failed to express GLUT3 (Fig. S1H). In silico analyses of the Slc2a3 (GLUT3) gene locus using DNAse I hypersensitivity (Bevington et al., 2016) and ATAC-sequencing data (Mognol et al., 2017) demonstrated open chromatin regions at the promoter region and at two regulatory elements located −16 and −22 kb upstream of the transcription start site (TSS) (Fig. S1I). Analyses of genome-wide chromatin immunoprecipitation followed by sequencing (ChIP-seq) datasets revealed that NFATc1 (Klein-Hessling et al., 2017) and NFATc2 (Martinez et al., 2015) bind to both the Slc2a3 promoter and the −16 kb element, whereas IRF4 (Man et al., 2013) and STAT3 (Hirahara et al., 2015) binding was found at the −16 kb locus (Fig. S1I). Binding of HIF-1α (Ciofani et al., 2012) was enriched at the −22 kb element. Collectively, these data demonstrate that NFAT, IRF4, STAT3 and HIF-1α together control the expression of GLUT3 in Th17 cells by integrating antigen, costimulatory and cytokine signaling (Fig. S1J).
Figure 1. GLUT3 is required for the effector function of Th17 cells.
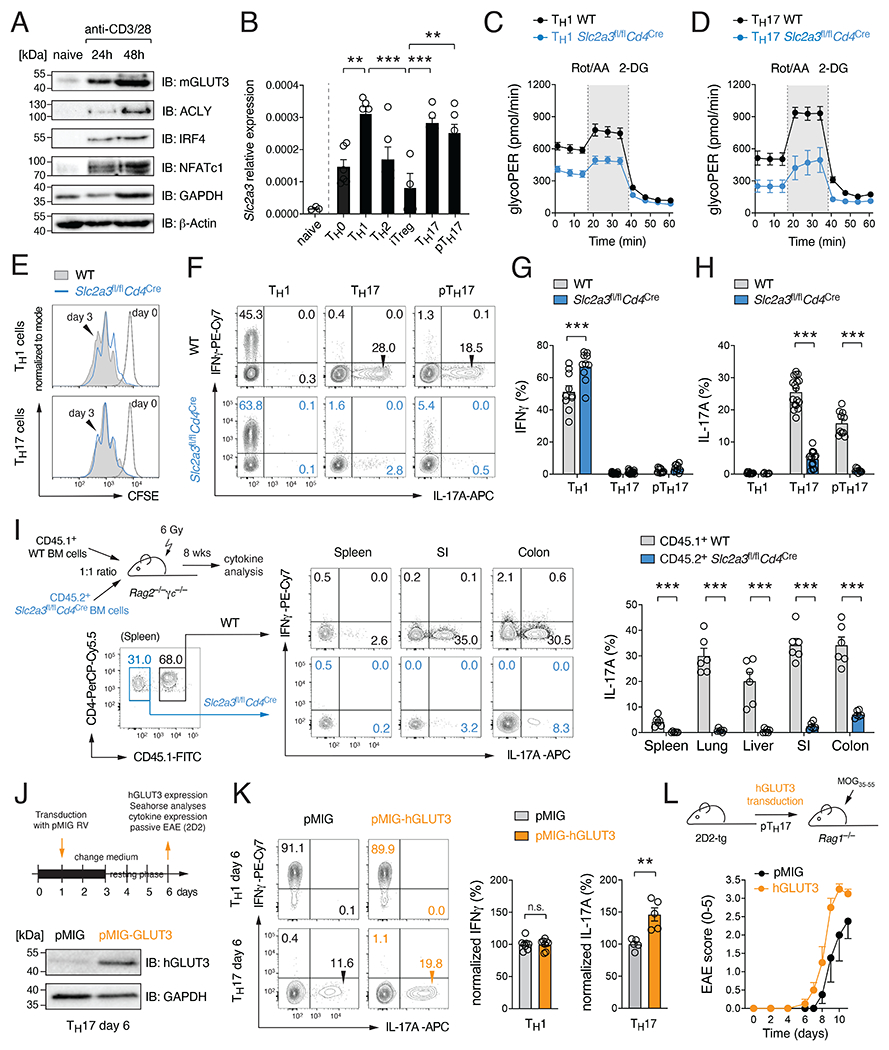
(A) Immunoblot analysis of murine GLUT3, ACLY, IRF4, NFATc1 and GAPDH expression. (B) Analysis of Slc2a3 (GLUT3) gene expression in naïve CD4+ T cells and T helper (Th) cell subsets by qRT-PCR; means ± SEM of 5-6 mice. (C and D) Glycolytic proton efflux rate (glycoPER) analyses of WT and GLUT3-deficient Th1 (C) and Th17 (D) cells using a Seahorse extracellular flux analyzer; means ± SEM of 5 mice. (E) Proliferation analysis of WT and GLUT3-deficient Th1 and Th17 cells. (F-H) Flow cytometric analysis of IFNγ (G) and IL-17 (H) production of WT and GLUT3-deficient Th1, Th17 and pathogenic Th17 (pTh17) cells after re-stimulation with PMA/Iono for 5 h; means ± SEM of 9-15 mice. (I) Generation of mixed BM chimeras using BM from CD45.2+ Slc2a3fl/flCd4Cre and CD45.1+ WT mice at a 1:1 ratio. 8 weeks after reconstitution, the production of IFNγ and IL-17 in CD4+ T cells of WT and GLUT3-deficient BM origin was analyzed; means ± SEM of 6 mice. (J-L) Ectopic expression of GLUT3 in T cells augments Th17 cell effector function. (J) Retroviral transduction of WT T cells with GLUT3 or empty control vectors (EV). Immunoblot analysis of GLUT3 overexpression. (K) Flow cytometric analysis of IFNγ and IL-17 production of GLUT3-transduced Th1 and Th17 cells; means ± SEM of 5-8 mice. (L) Clinical EAE scoring of Rag1−/− mice after transfer of GLUT3-transduced 2D2 T cells; means ± SEM of 4 mice per cohort. **, p<0.01, ***, p<0.001 by unpaired Student’s t-test (B), (G-I) and (K); n.s., non-significant.
GLUT3 controls the effector function of Th17 cells
To investigate the function of GLUT3 in T cells, we generated mice with T cell-specific inactivation of the Slc2a3 gene by crossing Slc2a3fl/fl mice (Fidler et al., 2017) with Cd4Cre animals. We first verified that GLUT3 expression was abolished in CD4+ T cells without a compensatory upregulation of other glucose transporters (Fig. S1K,L). Slc2a3fl/flCd4Cre mice had normal populations of immature, double and single positive thymic lymphocytes (Fig. S1M,N). GLUT3-deficient mice showed ~ 30% reduced conventional and Treg cells in their spleens and lymph nodes (LNs), without changing the composition of naïve, effector and memory T cell subsets (Fig. S1O–R). Uptake of the fluorescent glucose analogue 2-NBDG (Fig. S2A) and tritiated [3H] 2-desoxy-glucose (2-DG) (Fig. S2B) revealed that both Th1 and Th17 cells have an impaired glucose uptake capacity in absence of GLUT3 with the strongest defect in pathogenic Th17 cells. To further test whether GLUT3-deficient T cells have also an impaired glycolytic flux, we analyzed the proton efflux rate (PER) as a measure for lactate production. Both Th1 (Fig. 1C) and Th17 cells (Fig. 1D) showed reduced basal and maximal PER. However, their glycolytic defect neither prevented cell cycle entry (Fig. S2C), cellular growth (Fig. S2D), expression of activation markers (Fig. S2E), nor the proliferation of GLUT3-deficient Th1 and Th17 cells (Fig. 1E and S2F). Ablation of GLUT3 slightly enhanced apoptosis in both subsets (Fig. S2G) and, consequently, attenuated their cellular expansion (Fig. S2H). By analyzing the expression of T-bet and RORγt as the ‘signature’ transcription factors of Th1 and Th17 cells, respectively, we found that GLUT3-deficient Th1 cells express slightly higher levels of T-bet, whereas RORγt expression was reduced in Th17 cells (Fig. S2I). Intriguingly, ablation of GLUT3 in Th1 cells did not impair the expression of IFNγ (Fig. 1F,G), whereas IL-17, IL-2 and GM-CSF expression was almost completely abolished in ‘homeostatic’ and ‘pathogenic’ (p) Th17 cell subsets (Fig. 1F,H and S2J–L). In addition to Th17 cells, the differentiation of in vitro generated Treg and Th2 cells was also affected by GLUT3 inactivation (Fig. S2M,N).
To further demonstrate that GLUT3 is cell-intrinsically required for Th17 cell differentiation, we generated mixed bone marrow (BM) chimeric mice (Fig. 1I). Eight weeks after reconstitution, we analyzed the expression of IL-17 by CD4+ T cells in the spleen and non-lymphoid tissues and found that IL-17 was almost exclusively produced by CD45.1+ WT T cells (Fig. 1I). To further confirm that GLUT3 expression directly supports cytokine expression of Th17 cells, we retrovirally (RV) overexpressed GLUT3 in WT Th1 and Th17 cells (Fig 1J). Ectopic expression of GLUT3 elevated both basal and maximal glycolytic activity in transduced Th17 cells after 6 days in culture (Fig. S2O). GLUT3 overexpression also increased IL-17 and GM-CSF expression in Th17 cells, whereas IFNγ remained unchanged (Fig. 1K and S2P,Q). To investigate whether ectopic GLUT3 expression also augments the pathogenicity of Th17 cells in vivo, we transduced myelin oligodendrocyte glycoprotein (MOG)-specific 2D2 T cells with GLUT3. After transfer into Rag1−/− mice, we immunized the recipient mice with MOG35-55 peptide to induce experimental autoimmune encephalomyelitis (EAE) (Fig. 1L). Mice that received GLUT3-overexpressing 2D2 T cells not only showed a more severe EAE immunopathology (Fig. 1L) but also elevated numbers of cytokine producing T cells in their CNS compared to control animals (Fig. S2R,S). Together, these data demonstrate that GLUT3 promotes the differentiation and effector function of Th17 cells in vitro and in vivo.
T cell-specific ablation of GLUT3 protects mice from autoimmunity
Both Th1 and Th17 cells contribute to immunopathology in a variety of autoimmune diseases, including MS and IBD. To investigate the role of GLUT3 in vivo, we employed animal models of T cell-mediated cerebral (Fig. 2A–D) and intestinal (Fig. 2E–L) inflammation. We first induced EAE in Slc2a3fl/flCd4Cre and littermate control mice by immunization with MOG35-55 peptide. Strikingly, mice with T cell-specific deletion of GLUT3 were completely protected from EAE immunopathology, including paralysis of their extremities (Fig 2A), inflammation-induced weight loss (Fig. S3A) and the demyelination of the spinal cord (Fig. 2B). The infiltration of lymphocytes into the spinal cord was reduced in Slc2a3fl/flCd4Cre mice compared to control animals (Fig. 2B and S3B–E). The expression of T-bet (Fig. S3F,G) and IFNγ (Fig. 2C and S3H) were unaltered in GLUT3-deficient T cells, whereas RORγt (Fig. S3F,G), IL-17, GM-CSF and other inflammatory cytokines were markedly reduced in the CNS of Slc2a3fl/flCd4Cre mice (Fig. 2C,D and S3I–K). These findings demonstrate that T cell-specific deletion of GLUT3 prevents EAE immunopathology by attenuating the effector function of encephalitogenic Th17 cells.
Figure 2. Ablation of GLUT3 in T cells prevents autoimmunity.
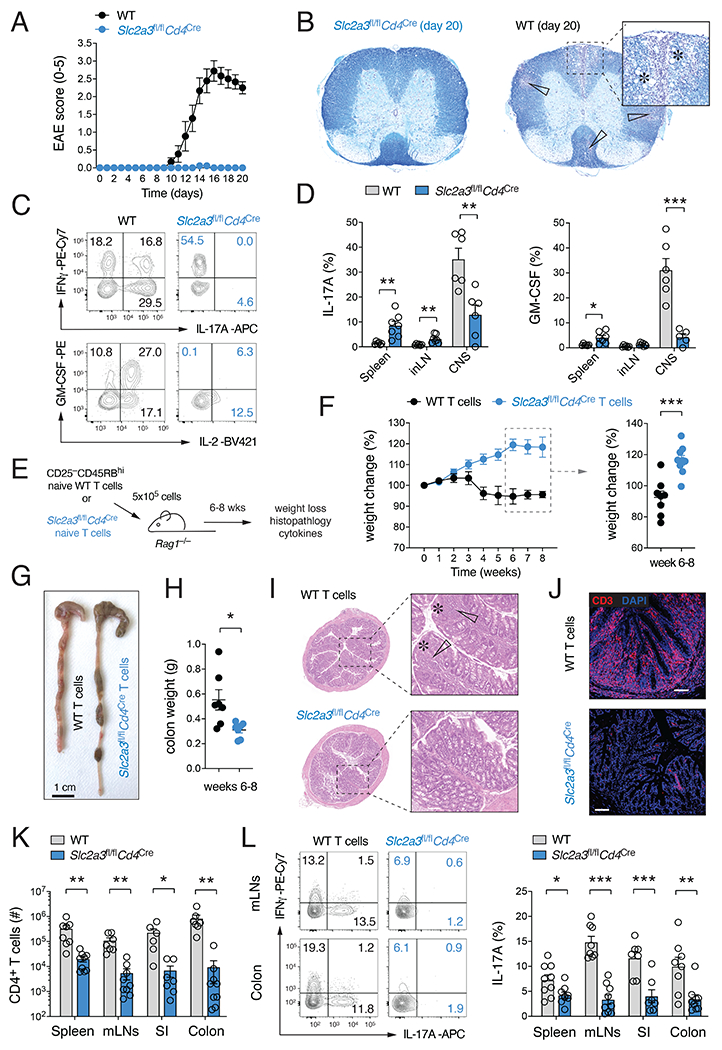
(A-D) Slc2a3fl/flCd4Cre mice are protected from experimental autoimmune encephalomyelitis (EAE). (A) Clinical EAE scores of WT and Slc2a3fl/flCd4Cre mice after immunization with MOG35-55 peptide emulsified in CFA; means ± SEM of 9 mice per cohort. (B) Representative histopathological examination of spinal cord sections of WT and Slc2a3fl/flCd4Cre mice 20 days after MOG35-55 peptide immunization. White arrows and asterisks indicate leukocytic infiltrates and areas of demyelination, respectively. (C and D) Frequencies of IL-17 and GM-CSF-producing CD4+ T cells in the spleen, inLNs and CNS of WT and Slc2a3fl/flCd4Cre mice; means ± SEM of 6-7 mice. (E-L) GLUT3-deficient T cells fail to induce adoptive transfer autoimmune colitis. (F) Weight loss of Rag1−/− host mice after transfer of naive CD4+ T cells from WT or Slc2a3fl/flCd4Cre mice; means ± SEM of 8-9 host mice. (G and H) Representative macroscopic pictures (G) and colon weights (H) 8 weeks after T cell transfer; means ± SEM of 6-7 recipient mice. (I and J) Representative H&E-stained colon sections (I) and anti-CD3 immunofluorescence analysis of inflammatory tissue damage and T cell infiltration (J). (K) T cell numbers in the spleen, mLNs, small intestine (SI) and colon in Rag1−/− recipient mice 6 to 8 weeks after transfer of T cells; means ± SEM of 6-9 host mice. (L) IFNγand IL-17 cytokine production of WT and GLUT3-deficient T cells in the spleen, mLNs, SI and colon 6-8 weeks after T cell transfer; means ± SEM of 7-9 mice. *, p<0.05; **, p<0.01, ***, p<0.001 by unpaired Student’s t-test (D), (F), (H) and (K,L).
Slc2a3fl/flCd4Cre mice had slightly reduced T cell numbers in their peripheral lymphoid organs (Fig. S1O), which could contribute to the ameliorated EAE pathology. To directly probe the cell-intrinsic function of GLUT3 in pathogenic Th17 cells, we transferred naïve CD4+ T cells from WT and Slc2a3fl/flCd4Cre mice into lymphopenic host mice to induce autoimmune colitis (Fig. 2E). Mice that received WT T cells showed significantly reduced body weights compared to animals that were transplanted with GLUT3-deficient lymphocytes (Fig. 2F). Macroscopic inspection of colonic samples showed significant shortening, thickening, edema formation and bleeding in mice that received WT T cells (Fig. 2G,H). In contrast, mice with T cells from Slc2a3fl/flCd4Cre mice showed no obvious colon immunopathology (Fig. 2G). Histopathological examination of colon sections no IBD symptoms, such as lymphocytic infiltration, epithelial hyperplasia, goblet cell depletion and/or ulceration in these mice, whereas mice that received WT T cells showed a pronounced colonic inflammation (Fig. 2I). In line with their ameliorated immunopathology, mice transplanted with GLUT3-deficient T cells showed decreased T cell accumulation in the small intestine and colon (Fig. 2J,K) and markedly reduced IL-17 production (Fig. 2L). We next explored GLUT3’s function in the context of infections. Th1 and Th17 cell responses are both important for the control of Citrobacter rodentium infections (Silberger et al., 2017). TNFα and IFNγ production was unaffected after C. rodentium infection in Slc2a3fl/flCd4Cre mice (Fig. S3L), whereas IL-17 expression and Th17 cell numbers were markedly decreased (Fig. S3L,M). Because the clearance of C. rodentium depends on Th17 cell-mediated immunity, Slc2a3fl/flCd4Cre mice lost more weight than the littermate controls (Fig. S3N) and failed to eliminate the pathogen (Fig. S3O). We next infected WT and Slc2a3fl/flCd4Cre mice with the ‘acute’ Armstrong strain of lymphocytic choriomeningitis virus (LCMV), which promotes the differentiation of Th1 and Tfh cells, but does not support a Th17 cell-mediated immune response. Similar as observed before, IFNγ expression by LCMV-specific CD4+ T cells (identified by NP309-328 tetramers) was comparable to WT T cells (Fig. S3P). Collectively, these data demonstrate that GLUT3 is specifically required for the differentiation and effector function of Th17 cells in both autoimmunity and infection.
GLUT3-dependent metabolism controls a complex gene expression program in Th17 cells
To define the function of GLUT3 in Th1 and Th17 cells at the molecular level, we performed transcriptomics and isotope tracing using RNA-sequencing (RNA-seq) and liquid chromatography with mass spectrometry (LC/MS), respectively. The gene expression profiles of WT and GLUT3-deficient T cell subsets were clearly distinct (Fig. 3A) and revealed 460 and 933 differential expressed genes (DEGs) in Th1 and Th17 cells, respectively (Fig. 3B,C). Only 217 DEGs were shared between Th1 and Th17 cells (Fig. 3D), suggesting that GLUT3 controls the transcriptional program of Th1 and Th17 cells in different ways. Among them was Slc2a3, which was strongly downregulated in both GLUT3-deficient T cell subsets as expected (Fig. 3B–D). Gene set enrichment analyses (GSEA) revealed that the transcriptome of GLUT3-deficient Th17 cells is negatively correlated with Th17 cell-specific gene signatures and was shifted towards Th1 and Treg cell signatures (Fig. 3E). To further explore the biological processes governed by GLUT3 in Th17 cells, we performed pathway enrichment and network analyses (Fig. 3F). Surprisingly, we found that signatures correlating with immune cell activation, such as kinase signaling, immune responses, leukocyte migration and ROS production were enriched in GLUT3-deficient Th17 cells (Fig. 3F), indicating that loss of GLUT3 does not impair T cell activation in general. The expression of the ‘signature’ transcription factors T-bet (Tbx21) and RORγt (Rorc) were unaltered and no compensatory upregulation of other glucose transporters was observed in absence of GLUT3 (Fig. 3G). By contrast, and in line with the leukocyte migration signatures of our network analyses, several genes encoding chemokines and their receptors were dysregulated in GLUT3-deficient T cells (Fig. 3G). Intriguingly, genes that are involved in metabolic processes, such as mitochondrial respiration and amino acid metabolism were markedly downregulated in GLUT3-deficient Th17 cells (Fig. 3F). The notion that GLUT3-dependent glucose uptake affects – in addition to glycolysis – also mitochondrial function was further supported by GSEA using gene ontology signatures, such as mitochondrial gene expression, mitochondrial protein complex and oxidative phosphorylation (Fig. 3H).
Figure 3. GLUT3 controls a complex metabolic-transcriptional network in Th17 cells.
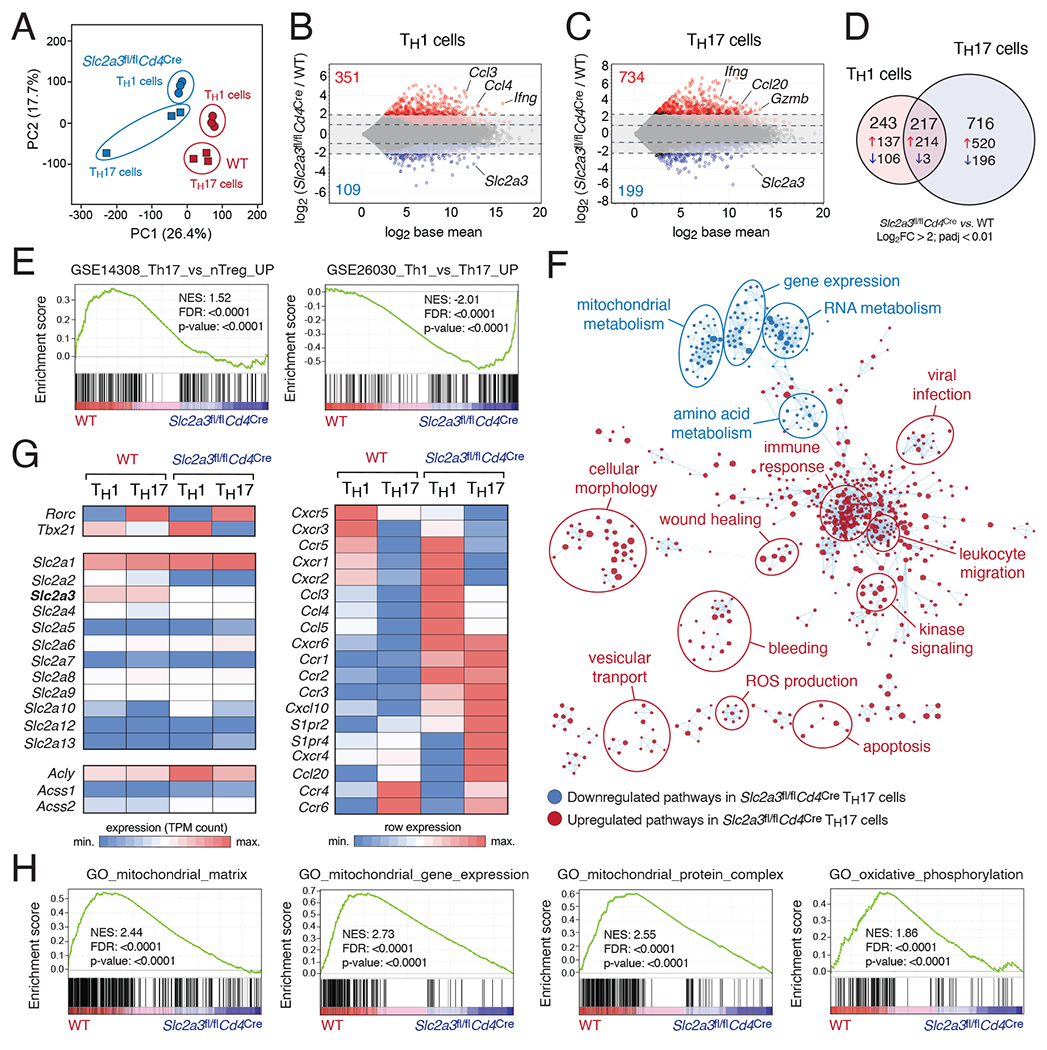
(A) Principal component (PC) analysis of WT and GLUT3-deficient (Slc2a3fl/flCd4Cre) Th1 and Th17 cell RNA-seq data; n=3 biological replicates per T cell subset and genotype. (B and C) MA plots of differentially expressed genes (DEGs) in WT versus GLUT3-deficient Th1 (B) and Th17 cells (C); genes significantly (padj < 0.01) up- and downregulated are depicted in red and blue, respectively. (D) Venn diagram analyses of > 4-fold DEGs (padj < 0.01) of GLUT3-deficient Th1 and Th17 cells. (E) Gene set enrichment analysis (GSEA) of WT versus GLUT3-deficient Th17 cells. (F) Network clustering of significantly (p < 0.005) enriched gene expression signatures to identify dysregulated physiological processes in GLUT3-deficient Th17 cells. Down- and upregulated gene sets in GLUT3-deficient Th17 cells compared to WT are shown in blue and red, respectively. (G) Heatmap analysis of selected genes in GLUT3-deficient and WT Th1 and Th17 cells. (H) GSEAs of WT versus GLUT3-deficient Th17 cells highlight impaired mitochondrial gene expression and function.
To determine how impaired (mitochondrial) gene expression affects the metabolome of GLUT3-deficient T cells, we performed stable isotope tracing of polar metabolites using LC/MS. WT and GLUT3-deficient T cells were incubated with 13C-glucose for 24 h before the samples were analyzed by LC/MS (Fig. S4A). The metabolomic analyses clearly separated Th1 from Th17 cells; the latter showed an enrichment of lipid species, whereas Th1 cells had higher amounts of glycolytic intermediates (Fig. S4B). 13C-glucose-derived metabolites can be identified by an isotopic increase in their specific molecular mass (Fig. 4A), for example m+6 for the six-carbon hexose-6-phosphate and m+3 for the three-carbon metabolites 3-phosphoglycerate and lactate (Fig. S4C). The 13C-labelling pattern downstream of TCA cycle metabolites is more complex due to repeated cycle rounds and the integration of other (unlabeled) metabolites (Fig. 4A and S4D). In addition to intracellular metabolites, we also analyzed the consumption of 13C-glucose from the cell culture supernatant and the secretion of lactate by WT and GLUT3-deficient Th1 and Th17 cells (Fig. 4B). Significant enrichment of intracellular 13C-isotopologues was observed in intermediates of the glycolytic pathway, metabolites of the tricarboxylic acid (TCA) cycle, nucleotides, nucleotide sugars and amino acids that are involved in one-carbon metabolism (serine, glycine) (Fig 4C–E). GLUT3-deficient Th1 and Th17 cells differed only little in their relative 13C-labelling pattern of glycolytic intermediates compared to WT T cells (Fig. 4C and S4C), indicating that the glycolytic flux is not completely inhibited in the absence of GLUT3. By contrast, mitochondrial isotopologues, such as fumarate, malate and acetyl-CoA, were significantly decreased in GLUT3-deficient Th17 cells compared to WT controls (Fig. 4C). This suggests that GLUT3 also fuels not only glycolysis but also supports an oxidative metabolism of glucose. Of note, differential metabolites in WT and GLUT3-deficient T cells were only partially shared between Th1 and Th17 cells (Fig. S4E). ATP levels were not significantly reduced in GLUT3-deficient Th17 cells (Fig. S4F), which suggests that T cells do not experience a bioenergetic crisis in absence of GLUT3. The notion that GLUT3 differentially affects Th1 and Th17 cells was supported by global metabolomic analyses that revealed specific changes in their metabolite profiles (Fig. 4D,E). Among the most downregulated metabolites in GLUT3-deficient Th17 cells was acetyl-CoA, which was only slightly reduced in Th1 cells (Fig. 4D,E). Metabolite set enrichment analysis (MSEA) revealed that GLUT3 not only supports glycolytic pathways, such as Warburg effect, glycolysis and gluconeogenesis, but also mitochondrial signatures (Fig. 4F and S4G). Intriguingly, pyruvate metabolism, citric acid cycle and transfer of acetyl groups into mitochondria were selectively enriched in Th17 cells but not in Th1 cells (Fig. 4F and S4G). The notion that Th17 cells rely more on mitochondrial glucose oxidation than Th1 cells was supported by a higher OCR:ECAR ratio of Th17 cells (Fig. S4H). Inactivation of GLUT3 more severely impaired the oxygen consumption of Th17 than Th1 cells (Fig. 4G,H) without changing their mitochondrial volume, membrane potential or ROS production (Fig. 4I). These data suggest that Th1 cells primarily perform aerobic glycolysis, whereas in Th17 cells GLUT3-dependent glucose uptake supports both glycolytic and mitochondrial metabolism.
Figure 4. GLUT3 supports mitochondrial acetyl-CoA generation.
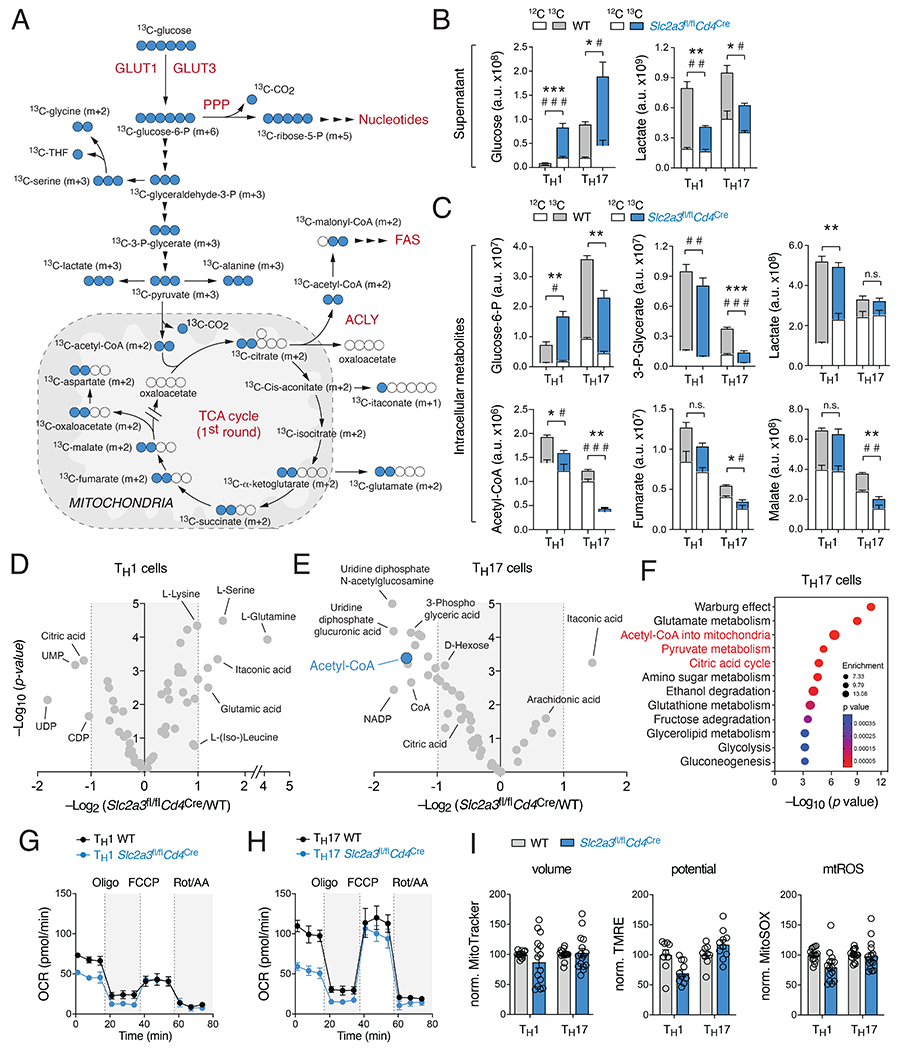
(A) Isotope tracing of glucose-derived metabolites in WT and GLUT3-deficient T cells by liquid chromatography and mass spectrometry (LC/MS). (B) Analysis of 13C-labelled glucose and lactate levels in cell culture supernatants of WT and GLUT3-deficient Th1 and Th17 cells by LC/MS. Asterisks and hash signs indicate significant differences in 13C and 12C-metabolites, respectively. (C) Fractional enrichment of 13C-glucose-derived glycolytic and TCA cycle intracellular metabolites in WT and GLUT3-deficient Th1 and Th17 cells; means ± SEM of 4 biological replicates. (D and E) Volcano plots of differential metabolite concentrations between WT and GLUT3-deficient Th1 (D) and Th17 (E) cells; 4 biological replicates per group. (F) Metabolite set enrichment analysis (MSEA) of differential metabolite concentrations (p < 0.05) between WT and GLUT3-deficient Th17 cells. (G and H) Oxygen consumption rate (OCR) measurements of WT and GLUT3-deficient Th1 (G) and Th17 cells (H) using a Seahorse extracellular flux analyzer; means ± SEM of 5 mice. (I) Analysis of mitochondrial volume (MitoTracker), membrane potential (TMRE) and mitochondrial ROS production (MitoSOX) in WT and GLUT3-deficient T cells; means ± SEM of 10-17 mice. *, p<0.05; ***, p<0.001 by unpaired Student’s t-test (B and C).
ACLY promotes GLUT3-mediated acetyl-CoA generation and Th17 cell effector function
To further elucidate the source of the reduced acetyl-CoA levels in the absence of GLUT3, we directly measured citrate and acetyl-CoA in fractionated WT and GLUT3-deficient Th17 cells (Fig. 5A,B). In whole cell lysates (WCL) and in cytosolic fractions, both citrate and acetyl-CoA levels were significantly reduced, confirming the results of the LC/MS analyses (Fig. 4C). No differences within the mitochondrial fractions were detected (Fig. 5A,B), indicating that reduced acetyl-CoA levels in the LC/MS analyses were mainly due to the lower levels in the cytosol. Because the mitochondrial membrane is impermeable to acetyl-CoA, the concentration of cytosolic acetyl-CoA is regulated by the release of citrate from mitochondrial stores and the subsequent conversion to acetyl-CoA by the enzyme ATP-citrate lyase (ACLY) (Zhao et al., 2016). Independently from mitochondrial citrate, cytosolic acetyl-CoA can be also generated from acetate by acetyl-CoA synthetase (ACSS) (Zhao et al., 2016). The provision of exogenous acetate during Th17 cell differentiation almost completely rescued the defective IL-17, GM-CSF and IL-2 production of GLUT3-deficient T cells (Fig. 5C). Because lactate, glutamine or α-ketoglutarate and lipids had little or no effect (Fig. S4I–R), this data suggest that defective cytokine production of GLUT3-deficient Th17 cells is mainly due to an impaired generation of acetyl-CoA from mitochondrial citrate.
Figure 5. ACLY controls acetyl-CoA production and Th17 cell effector function.
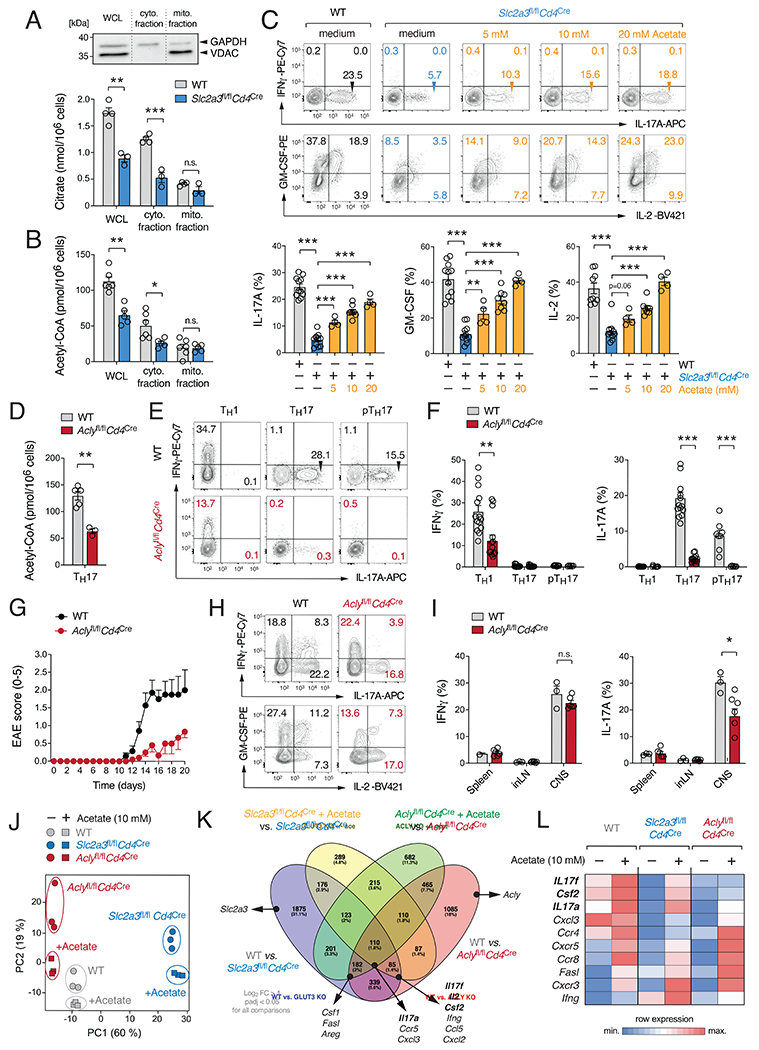
(A and B) Analysis of subcellular citrate (A) and acetyl-CoA (B) in whole cell lysates (WCL) and isolated cytosolic and mitochondrial fractions; means ± SEM of 5-6 mice. Immunoblot analysis of cytosolic (GAPDH) and mitochondrial (VDAC) proteins. (C) Exogenous acetate rescues impaired cytokine production of GLUT3-deficient Th17 cells. Flow cytometric analyses of IFNγ, IL-17, IL-2 and GM-CSF expression of WT and Slc2a3fl/flCd4Cre Th17 cells treated with acetate; means ± SEM of 4-11 mice. (D-I) ACLY controls Th17 cell effector function. (D) Quantification of acetyl-CoA in WCL of WT and Aclyfl/flCd4Cre Th17 cells; means ± SEM of 3-5 mice. (E and F) Flow cytometric analysis of IFNγ and IL-17 production by WT and ACLY-deficient (Aclyfl/flCd4Cre) T cells; means ± SEM of 6-14 mice. (G–I) Inactivation of ACLY in T cells prevents autoimmune encephalomyelitis. (G) Clinical EAE scores of WT and Aclyfl/flCd4Cre mice after MOG35-55 immunization; means ± SEM of 7-10 mice per cohort. (H and I) Flow cytometric analyses of IFNγ and IL-17 production of CD4+ T cells from WT and Aclyfl/flCd4Cre mice; means ± SEM of 3-6 mice. (J-L) Global gene expression analysis of WT, GLUT3-deficient (Slc2a3fl/flCd4Cre) and ACLY-deficient (Aclyfl/flCd4Cre) Th17 cells in the presence and absence of acetate. (J) Principal component (PC) analysis of WT, GLUT3-deficient and ACLY-deficient Th17 cell gene expression data. (K) Venn diagram analyses comparing GLUT3-deficient, ACLY-deficient and WT Th17 cells in the presence or absence of 10 mM acetate; > 2-fold expression change and padj < 0.05. (L) Heatmap analysis of selected gene expression in WT, GLUT3- and ACLY-deficient Th17 cells. *, p<0.05; **, p<0.01, ***, p<0.001 by unpaired Student’s t-test (A-D), (F) and (I).
The induction of ACLY and GLUT3 protein expression followed a similar kinetic in activated T cells (Fig. 1A), indicating that glucose uptake and ACLY-dependent acetyl-CoA generation are co-regulated in Th17 cells. To directly probe the role of ACLY in Th17 cells, we inactivated ACLY protein expression using CRISPR/Cas9 genome editing (Fig. S5A,B). We transduced Cas9-expressing Th1 and Th17 cells with gRNAs targeting the second exon of the Acly gene. At least three of the four gRNAs completely abolished ACLY protein expression in Th17 cells three days after RV transduction (Fig. S5A). In Th1 cells, CRISR/Cas9-mediated ACLY inactivation only marginally affected IFNγ expression, whereas the IL-17 production by Th17 cells was significantly reduced (Fig. S5B). We next generated mice with T cell-specific ablation of ACLY by crossing Aclyfl/fl mice (Zhao et al., 2016) with Cd4Cre animals. As expected, Acly gene expression was completely abolished in T cells isolated from Aclyfl/flCd4Cre mice (Fig. S5C). Aclyfl/flCd4Cre mice were indistinguishable from their littermates and showed a normal number and composition of immature, DP and SP thymic T cell populations (Fig. S5D,E). The frequencies of peripheral conventional and regulatory T cell subsets were unaltered and Aclyfl/flCd4Cre mice showed no obvious immune dysregulation (Fig. S5F–I). The total acetyl-CoA levels were reduced by ~ 60% in ACLY-deficient Th17 cells (Fig. 5D), indicating that – in absence of ACLY – a fraction of the cellular acetyl-CoA pool is generated from citrate-independent sources (e.g. from acetate, amino acids, ketone bodies and/or lipids). Like GLUT3-deficient T cells, ablation of ACLY did neither prevent cell cycle entry (Fig. S5J) nor the expression of activation markers (Fig. S5K) but slightly impaired the proliferation of Th17 cells (Fig. S5L). The expression of T-bet and RORγt was comparable between WT and ACLY-deficient T cells (Fig. S5M). IFNγ expression was reduced by ~50% in in ACLY-deficient Th1 cells, whereas IL-17 and GM-CSF were completely abolished Th17 cells (Fig. 5E,F and S5N). To investigate the role of ACLY in T cell-mediated immune responses in vivo, we next induced EAE in Aclyfl/flCd4Cre mice and littermate controls (Fig 5G–I and S5O–R). Clinical EAE scoring (Fig. 5G) and the demyelination of the spinal cord (Fig. S5O) was significantly lower in ACLY-deficient mice, although infiltration of lymphocytes into the spinal cord was comparable in Aclyfl/flCd4Cre mice and littermate controls (Fig. S5P). Importantly, the frequencies and absolute numbers of IL-17 and GM-CSF-producing T cells were markedly reduced in the CNS of Aclyfl/flCd4Cre mice, whereas IFNγ expression was unaltered (Fig. 5H,I and S5Q,R). These data suggest that the generation of cytosolic acetyl-CoA downstream of GLUT3 is a critical metabolic checkpoint for (pathogenic) Th17 cell immune responses.
To further explore the specific functions of GLUT3 and ACLY in Th17 cells and to elucidate the role of acetyl-CoA in both genotypes, we performed RNA-seq analyses (Fig. 5J–L) of WT, GLUT3 and ACLY-deficient Th17 cells. Although the gene expression profiles of all genotypes were distinct, ACLY-deficient Th17 cells clustered closer to WT T cells, indicating that the loss of GLUT3 has a broader effect on gene expression (Fig. 5J). Comparing untreated WT and GLUT3-deficient Th17 cells revealed ~ 3000 DEGs that were significantly up- or downregulated, whereas ~ 2400 genes were different between WT and ACLY-deficient cells. 716 DEGs were shared by GLUT3 and ACLY-deficient Th17 cells, including Il17a, Il17f, Il22, Csf2 (GM-CSF) and Il2 (Fig 5K). Importantly, 110 genes of the 716 DEGs in both genotypes were restored by the treatment with acetate, including Il17a (Fig 5K,L). Collectively, these data demonstrate that many effector molecules of Th17 cells, including cytokines and chemokines, are dependent on both GLUT3 and ACLY in an acetyl-CoA-dependent fashion.
GLUT3-dependent epigenetic reprogramming controls the pathogenicity of Th17 cells
Acetyl-CoA levels are an important molecular rheostat connecting metabolic activity with cellular function (Pietrocola et al., 2015). Cytosolic acetyl-CoA is the substrate for fatty acid and sterol biosynthesis, thereby promoting the generation of lipids. In addition, cytosolic acetyl-CoA is required for protein acetylation, including the modification of histones.
To define whether de novo fatty acid synthesis or protein acetylation controls cytokine expression of Th17 cells, we treated WT T cells with 2-hydroxycitrate (2-HC), 5-tetradecyloxy-2-furonic acid (TOFA) or soraphen A (SorA). 2-HC is a competitive inhibitor of ACLY, whereas SorA and TOFA target acetyl-CoA carboxylases 1 (ACC1) and ACC2, which catalyze lipid biosynthesis (Fig. 6A). Consistent with previous reports, both SorA (Berod et al., 2014) and TOFA (Endo et al., 2015) prevented IL-17 expression when present during Th17 cell differentiation (Fig. 6B). By contrast, when these inhibitors were added acutely during the restimulation of differentiated Th17 cells, only 2-HC attenuated the expression of IL-17 (Fig. 6B). Because the generation of complex lipids such as sphingolipids, including sphingomyelins and ceramides, was not affected in GLUT3-deficient Th17 cells (Fig. S4N) and the provision of exogenous lipids had only a negligible effect on the IL-17 expression (Fig. S4O–R), we hypothesized that GLUT3-dependent acetyl-CoA generation is primarily required for the epigenetic reprogramming of Th17 cells. Intriguingly, GSEA also indicated dysregulated protein and histone acetylation in GLUT3-deficient Th17 cells (Fig. S6A). When we analyzed global histone 3 (H3) acetylation at lysins K9/14 and K27, marks that are associated with chromatin opening and transcription, we observed reduced H3 acetylation in GLUT3-deficient T cells compared to control cells (Fig. 6C and 6D). By contrast, H3 tri-methylation at lysin K9 and K27 was unaltered (Fig. S6B) or elevated (Fig. S6C), respectively. To substantiate the notion that GLUT3 and ACLY-derived acetyl-CoA is utilized for epigenetic remodeling, we measured the incorporation of glucose-derived, radio-labelled [14C] carbons into the post-translational modifications of histones. Significantly less [14C] counts in the histone fraction was observed in both genotypes, demonstrating that GLUT3 and ACLY participate in glucose-dependent epigenetic modification (Fig. 6E). Inhibition of histone de-acetylation using the pan-HDAC inhibitor Panobinostat partially restored IL-17 expression in GLUT3-deficient Th17 cells (Fig. 6F), corroborating the concept that GLUT3-dependent histone acetylation controls cytokine responses. To examine if changes in histone acetylation are directly associated with attenuated gene expression, we performed genome-wide analysis of H3 K9/14 acetylation using ChIP-seq (Fig. 6G–K). Global H3 acetylation (scaled to 1kb up- and downstream of TSS with an extension of 0.2 kb) was clearly distinct between WT and GLUT3-deficient Th17 cells containing ~ 14.000 differential acetylated regions, with 11.000 being lower acetylated compared to input samples (Fig. 6G). Differentially acetylated CpG-rich elements mapped to both intergenic regions and gene bodies, including upstream regulatory sites, promoters and coding regions (Fig. S6D). To define the regulatory elements at different gene loci, we aligned our H3 K9/14 ChIP-seq data with published ATAC-seq datasets of in vitro generated Th17 cells and IL-17-expressing T cells isolated from homeostatic ileum and the spinal cord of EAE mice (Qiu et al., 2020). Accessible chromatin regions were used to demarcate promoters (green shading) and regulatory elements, such as enhancers and other conserved noncoding sequences (CNS) (orange shading) (Fig. 6H,K and S6E–G). Intriguingly, no obvious differences in H3 acetylation were observed within the loci of ‘housekeeping’ genes, such as Actb, Pgk1, Hprt and B2m (Fig. S6E), or ‘activation marker’ genes, including Mki67 (encoding Ki-67), Il2ra (CD25) and Cd44 (Fig. S6F). By contrast, acetylation around the Il17a and Il17f genes were clearly reduced in GLUT3-deficient Th17 cells compared to WT control cells (Fig. 6H), demonstrating that GLUT3-dependent acetyl-CoA controls epigenetic remodeling in a locus-specific manner. The acetylated histone marks aligned closely with open chromatin within the Il17 locus that have been described as regulatory elements (originally termed CNS 1-8) that control the accessibility to the Il17a and Il17f promoters (Akimzhanov et al., 2007). We further confirmed impaired H3 acetylation at the CNS-2 and the Il17 promoter regions using ChIP-qPCR assays with antibodies against acetylated K9/14 (Fig. 6I) and K27 residues (Fig. 6J). Of note, impaired H3 modifications were not limited to the Il17 locus in GLUT3-deficient Th17 cells but were also observed at other cytokine loci, including Il2 (Fig. 6K), whose expression was also markedly downregulated in GLUT3-deficient T cells (Fig. 2C and S2L). We also identified several ‘non-cytokine’ genes with impaired K9/14 acetylation marks. One of these genes was Tfrc (encoding the transferrin receptor CD71) that promotes iron-dependent cytokine expression and Th17 cell-mediated autoimmunity (Wang et al., 2018). K9/14 acetylation was strongly reduced at the Tfrc promoter and an −15kb upstream element (Fig. S6G), which correlated with an impaired expression of CD71 at the plasma membrane (Fig. S6H). In line with our RNA-seq data (Fig. 3F), pathway enrichment analysis of differentially acetylated genes predicted alterations in signaling pathways and mitochondrial signatures, such as electron transport chain and oxidative phosphorylation (Fig. S6I). These data suggest that GLUT3-dependent histone acetylation controls the effector responses of Th17 cells through epigenetic remodeling.
Figure 6. GLUT3-dependent histone acetylation promotes Th17 cell effector function.
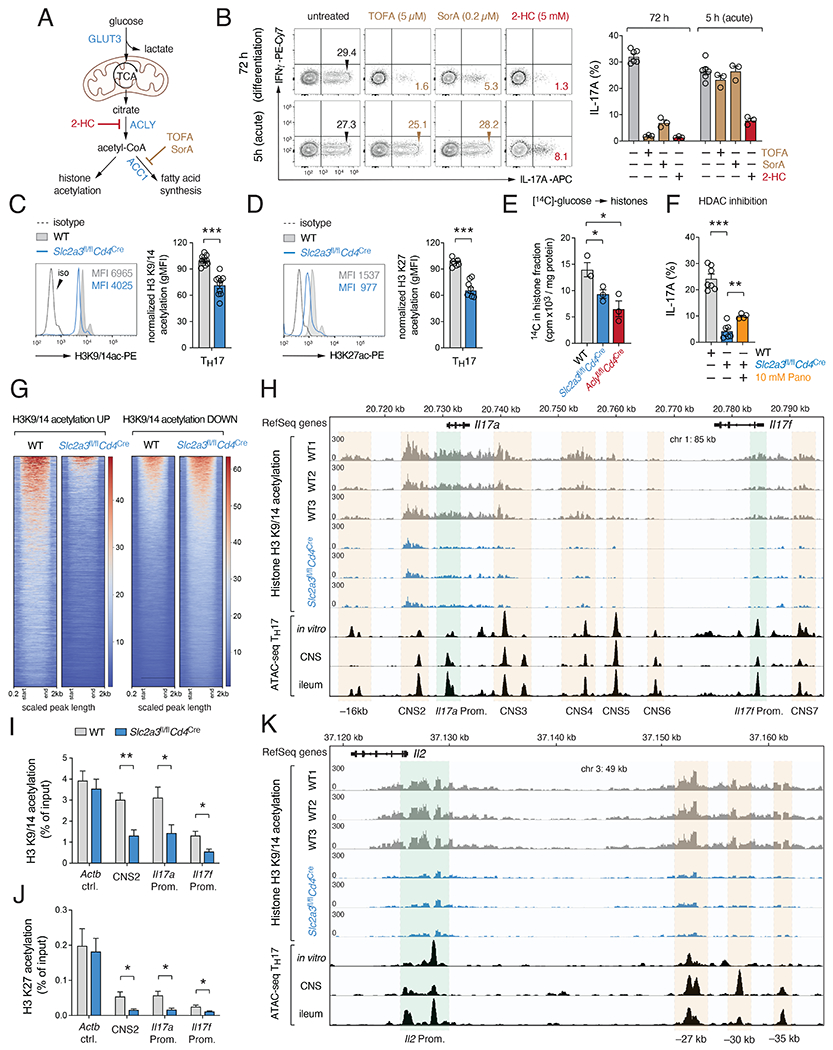
(A) Effects of 2-Hydroxycitrate (2-HC), Soraphen A (SorA) or 5-Tetradecyloxy-2-furonic acid (TOFA) to inhibit ATP-citrate lyase (ACLY), acetyl-CoA carboxylases (ACC1/2) or fatty acid synthase (FAS), respectively. (B) Analysis of IL-17 expression in Th17 cells treated with 2-HC, SorA or TOFA during differentiation (72 h) or re-stimulation (5 h). (C-J) GLUT3-dependent acetyl-CoA controls the epigenetic re-programming of Th17 cells. (C and D) Analysis of global histone 3 (H3) acetylation at lysins K9/14 (C) and K27 (D) in WT and GLUT3-deficient Th17 cells; means ± SEM of 5=9 mice. (E) Glucose-derived incorporation of [14C] carbons into histones of WT, GLUT3- and ACLY-deficient Th17 cells; means ± SEM of 3 mice. (F) Analysis of IL-17 expression in GLUT3-deficient Th17 cells treated with 10 mM Panobinostat for 24 h; means ± SEM of 4-7 mice. (G-K) Genome-wide analysis of H3 K9/14 acetylation in WT and GLUT3-deficient Th17 cells by chromatin immunoprecipitation followed by DNA-sequencing (ChIP-seq). (G) Heatmaps using depth-normalized coverages of global K9/14 histone acetylation of WT and GLUT3-deficient Th17 cells relative to input samples. (H) Analysis of H3 K9/14 acetylation at the Il17a and Il17f gene cluster in WT and GLUT3-deficient Th17. Promoters (green shading), enhancers and other conserved noncoding regions (CNS, orange shading) were determined using ATAC-seq datasets of Th17 cells (Qiu et al., 2020). (I and J) Quantification of K9/14 (I) and K27 (J) acetylation in WT and GLUT3-deficient Th17 cells at the CNS-2, Il17a and Il17f promoter using ChIP-qPCR; n=5-6 mice. (K) Analysis of H3 K9/14 acetylation at the Il2 locus in WT and GLUT3-deficient Th17 cells. *, p<0.05; **, p<0.01, ***, p<0.001 by unpaired Student’s t-test (C-F), (I) and (J).
Pharmacological inhibition of ACLY ameliorates T cell-mediated autoimmunity
2-hydroxycitrate (2-HC) is a competitive inhibitor of ACLY and found naturally in fruits of the Garcinia cambogia tree (Chuah et al., 2013). Garcinia preparations are traditionally used to treat infections and rheumatism, highlighting an anti-inflammatory effect of 2-HC (Chuah et al., 2013). Treatment of Th17 cells with 5 mM 2-HC reduced their total acetyl-CoA levels by ~ 50% (Fig. 7A), comparable to the reduction observed in GLUT3 (Fig. 5B) and ACLY-deficient T cells (Fig. 5D). 2-HC also decreased histone H3 acetylation at K9/14 (Fig. 7B) without impairing the proliferation of Th17 cells (Fig. S7A). By contrast, addition of 2-HC strongly attenuated their cytokine production (Fig. 7C). IL-17 expression was more severely reduced after 2-HC treatment compared to IFNγ, indicating that Th17 cells are more sensitive to ACLY inhibition. To explore if ACLY inhibition can be exploited as a novel therapeutic strategy to treat autoimmune diseases, we immunized WT mice with MOG35-55 peptide and treated these mice with 2-HC, starting when the first EAE symptoms became apparent. Therapeutic 2-HC treatment significantly ameliorated the clinical symptoms of EAE (Fig. 7D and S7B–F), without affecting the overall numbers and composition of leukocytes in the CNS (Fig. S7C,D). However, cytokine-producing Th17 cells in the CNS were markedly reduced after 2-HC treatment (Fig. 7E and S7E), suggesting that 2-HC inhibits primarily the effector function of encephalitogenic T cells. To exclude that the effects of 2-HC are due to a shift in the T cell repertoire or the deletion of pathogenic T cell clones, we adoptively transferred in vitro-generated MOG-specific Th17 cells from 2D2 mice into lymphopenic host mice, with or without 2-HC treatment (Fig. 7F). Similar as in active EAE, 2-HC also ameliorated infiltration of mononuclear cells into the CNS and the demyelination of the spinal cord (Fig. S7G). To verify that the effects of ACLY inhibition also applicable to human T cells, we stimulated healthy donor PBMCs in presence of 2-HC (Fig. 7G and S7H–K). As observed in murine T cells, 2-HC did not affect cell cycle entry (Fig. S7H), proliferation (Fig. S7I) or the survival of human T cells (Fig. S7J). By contrast, treatment of PBMCs with 2-HC during stimulation dramatically decreased the expression of IL-17 in CD45RO+CCR6+ memory T cells (Fig. 7G). The levels of IFNγ, IL-2 and GM-CSF were also reduced in CD45RO+CCR6+ and CD45RO+CCR6−T cells after treatment with 2-HC, albeit to a lesser extent compared to IL-17 (Fig. 7G and S7K).
Figure 7. Pharmacological inhibition of ACLY ameliorates autoimmunity.
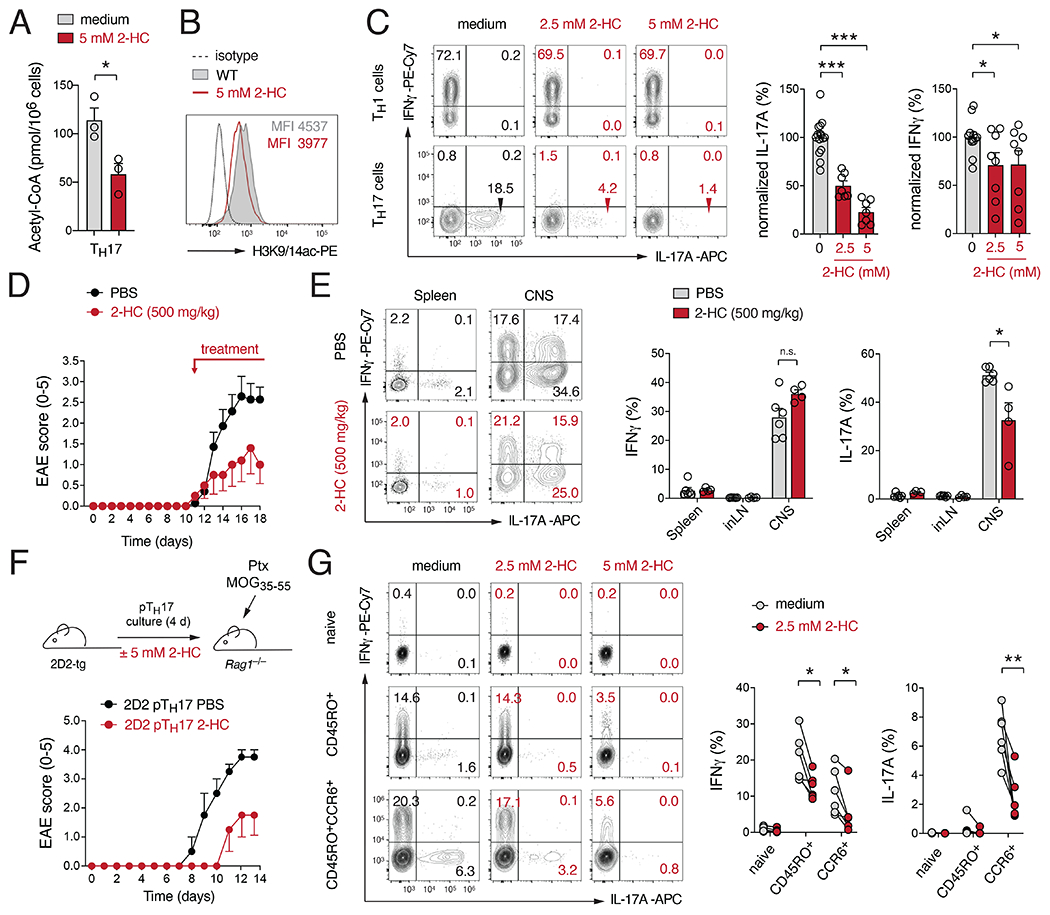
(A) Quantification of acetyl-CoA levels in Th17 cells treated with 5 mM 2-hydroxycitrate (2-HC) for 24 h; means ± SEM of 3 mice. (B) Analysis of global histone H3 acetylation at lysins K9/14 in Th17 cells treated with 5 mM 2-HC for 24 h. (C) Analysis of IFNγ and IL-17 expression in T cells after treatment with 2-HC for 24 h; means ± SEM of 7-15 mice. (D) Clinical EAE scores of WT mice immunized with MOG35-55 peptide and treated therapeutically with 500 mg/kg 2-HC; means ± SEM of 5-6 mice per cohort. (E) Flow cytometric analysis of IL-17 and IFNγ production in T cells isolated from mice treated with or without 2-HC; means ± SEM of 4-6 mice. (F) Transfer of in vitro differentiated 2D2 Th17 cells into Rag1−/− recipient mice with and without 2-HC treatment. Clinical EAE scores are means ± SEM of 2-4 mice. (G) 2-HC inhibits cytokine production of human CD4+ T cells. Flow cytometric analysis of IFNγ and IL-17 expression in naïve, CD45RO+ and CD45RO+CCR6+ memory CD4+ T cell subsets from healthy donors after stimulation with PMA/Iono in presence or absence of 2-HC; n=6 donors. *, p<0.05; **, p<0.01, ***, p<0.001 by unpaired Student’s t-test (A), (C), (E) and (G).
Collectively, these findings demonstrate that GLUT3 and ACLY-dependent acetyl-CoA generation is a ‘metabolic checkpoint’ in inflammatory T cells that can be pharmacologically exploited as a novel strategy to treat inflammatory and autoimmune diseases.
DISCUSSION
We here report a novel function of the glucose transporter GLUT3 in the adaptive immune system. Although largely dispensable for the activation, cell cycle entry and clonal expansion of Th17 cells, GLUT3 was critical for their effector function in vitro and in animal models of colitis and encephalomyelitis. At the molecular level, we found that GLUT3-dependent glucose metabolism controls a complex metabolic-transcriptional network in Th17 cells governed by epigenetic remodeling. Transcriptomic and metabolomic data linked GLUT3 to ACLY-dependent acetyl-CoA generation, which promotes inflammatory cytokine expression in Th17 cells through locus-specific histone acetylation. Our work highlights a mechanism through which glycolytic metabolism directly controls the effector function of pathogenic T cells at the epigenetic level. These findings also have important translational implications because inhibition of glucose-derived acetyl-CoA generation is a promising pharmacological strategy to treat T cell-mediated (auto-) inflammatory diseases.
We found that GLUT3-dependent glucose uptake not only supports aerobic glycolysis but also the generation of acetyl-CoA in Th17 cells through mitochondrial oxidation of glucose to citrate. T cell-specific inactivation of ACLY, which catalyzes the release of acetyl-CoA from mitochondrial citrate, phenocopied many defects of GLUT3-deficient Th17 cells, suggesting that GLUT3-mediated glucose consumption, mitochondrial oxidation of pyruvate to citrate and its recycling back to acetyl-CoA in the cytosol is a hardwired metabolic pathway that controls the effector function of Th17 cells. Acetyl-CoA is a fundamental metabolic intermediate for numerous biochemical reactions but is generally associated with the biosynthesis of fatty acids (Pietrocola et al., 2015). Besides lipogenesis, acetyl-CoA also regulates the activity of proteins through posttranslational modification, including those of histones. Covalent addition of acetyl groups disrupts the ionic interaction between DNA and histones, thus, promoting euchromatin formation and gene expression. Because clonal expansion requires fatty acids, phospholipids and cholesterols, activation of T cells correlates with elevated lipid metabolism (Wang et al., 2011). Previous studies showed that Th17 cells primarily depend on fatty acid and sterol biosynthesis from glucose rather than utilizing extracellular lipids (Berod et al., 2014; Hu et al., 2015). Pharmacological and genetic suppression of ACC1, the rate-limiting enzyme for de novo fatty acid synthesis, abrogated the differentiation of Th17 cells in vitro (Berod et al., 2014; Endo et al., 2015; Wang et al., 2011). Consistent with these reports, we also found that permanent inhibition of lipid synthesis prevented the proliferation of T cells, thereby curtailing their differentiation into Th17 cells. However, ablation of GLUT3 or ACLY neither abolished T cell proliferation nor the differentiation of Th17 cells but mitigated the expression of their ‘signature’ cytokines IL-17A, IL-17F and GM-CSF. The generation of complex lipids was not perturbed in GLUT3-deficient Th17 cells, suggesting that glycolytic acetyl-CoA controls cytokine expression independently of de novo fatty acid synthesis. The provision of exogenous acetate, which can be converted intracellularly to acetyl-CoA, almost completely restored the defective cytokine responses in both GLUT3 and ACLY-deficient Th17 cells. These data suggest that the GLUT3-dependent effector function of Th17 cells does not rely on lipogenesis but is, instead, mediated by an alternative, acetyl-CoA-dependent mechanism.
In line with this notion, we show that GLUT3 and ACLY directly control the epigenetic program of (pathogenic) Th17 cells through locus-specific histone acetylation. The interrelation between nutrient consumption and gene regulation by posttranslational protein modifications, including the acetylation of histones, was first demonstrated in yeast and cancer cells (Etchegaray and Mostoslavsky, 2016; Wellen et al., 2009). More recently, and in line with our findings, a direct link between the glycolytic activity and the epigenetic remodeling of T cells has been established (Bailis et al., 2019; Vodnala et al., 2019; Xu et al., 2021a; Xu et al., 2021b). In cytotoxic T cells and Th1 cells, defective nutrient uptake in response to elevated extracellular potassium levels or T cell-specific deletion of lactate dehydrogenase A (LDHA), the rate limiting enzyme for converting pyruvate to lactate, diminished cytosolic acetyl-CoA levels and histone acetylation (Peng et al., 2016; Vodnala et al., 2019). Ablation of LDHA also abolished the proliferation and effector differentiation of Th17 cells, which was – at least in part – due to altered epigenetic regulation of the Il17 locus (Xu et al., 2021a). However, LDHA deletion almost completely abolished aerobic glycolysis and the differentiation of all T cell subsets, whereas T cell-specific inactivation of GLUT3 affected primarily the effector functions of Th17 cells. These data suggest that quantitative and/or qualitative thresholds exist that metabolically uncouple T cell activation from effector function, thus providing a therapeutic window to target specifically the pathogenic functions of Th17 cells. In line with this notion, we here show that the natural compound 2-HC, an over-the-counter dietary supplement for weight loss therapy, ameliorates EAE in mice and abolishes inflammatory cytokine responses in murine and human Th17 cells. These proof-of-concept experiments establish that GLUT3 and ACLY are promising metabolic checkpoints in lymphocytes that can be targeted to treat inflammatory diseases, in which Th17 cells play an important pathophysiological role.
LIMITATIONS OF THE STUDY
In this study, we demonstrate that GLUT3 in controls the effector function of Th17 cells – at least in part – through epigenetic reprogramming. This does, however, not exclude additional roles of GLUT3 in other metabolic processes, such as the pentose phosphate pathway, amino acid synthesis and redox regulation. We focused on Th17 cells because their ‘signature’ cytokines IL-17A, IL-17F and GM-CSF were almost completely abolished in absence of GLUT3, whereas IFNγ and TNFα were unaffected. However, ablation of GLUT3 also impaired the differentiation of Th2 and Treg cells, which we have not investigated in detail. Because Treg cells do not produce inflammatory cytokines, it is tempting to speculate that GLUT3 fulfills cell type-specific (metabolic) functions in other T cell subsets. Finally, our findings in GLUT3-deficient Th17 cells deviated from the previously reported phenotypes of GLUT1-deficient T cells, indicating that individual glucose transporters have non-redundant roles in lymphocytes.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Additional information and requests for reagents and resources should be directed to and will be fulfilled by the Lead Contact, Dr. Martin Vaeth (martin.vaeth1@uni-wuerzburg.de).
Materials availability
Plasmids, cell lines and other materials generated in this study are available upon reasonable request to the lead contact.
Data and code availability
The RNA-seq and ChIP-seq datasets have been deposited at GEO under accession numbers GSE192659, GSE192663 and GSE193515. Original Western blots and values for creating the graphs in the paper are provided in Data S1.
This study did not generate any code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All mice were bred maintained under specific pathogen free conditions at the Center for Experimental Medicine (ZEMM) or the Institute for Systems Immunology at the Julius-Maximilians University of Würzburg. Mice were maintained on a 12/12 h light/dark cycle at between 20-24 °C in individually ventilated cages. Mice had access to standard chow (Ssniff; cat# V1534) and autoclaved water ad libitum and health status of the animals was inspected daily by the responsible animal caretakers. Hygiene status of the sentinel mice was monitored quarterly according to the FELASA guidelines. Both male and female mice between 8 and 16 weeks old at the time of the experiment were used in this study. All animal protocols were approved by government of Lower Franconia, Germany. CD45.1+ (strain 002014), Rag2−/−γc−/− (strain 014593), Rag1−/− (strain 002216), Cd28−/− (strain 002666), 2D2 (strain 006912), Aclyfl/fl (strain 43555), Irf4fl/fl (strain 009380), Stat3fl/fl (strain 016923), Hif1afl/fl (strain 007561), Rosa26LSL-Cas9 (strain 026175) and Cd4Cre mice (strain 017336) have been described previously and were purchased from the Jackson Laboratories (JAX) or kindly provided by Kathryn E. Wellen (University of Pennsylvania, USA), Elisabeth Sock (University of Erlangen, Germany), Alma Zernecke-Madsen, Friederike Berberich-Siebelt, Niklas Beyersdorf and Kristen Rak (University of Würzburg, Germany). Slc2a3fl/fl mice were generated with the knockout mouse project (KOMP) at UC Davis (stock number 049702-UCD) and have been described before (Fidler et al., 2017a; Fidler et al., 2017b). All animals used in this study were on a pure C57BL/6J genetic background, except for the Ppp3r1fl/fl mice that were backcrossed onto C57BL/6J for 4 generations from a mixed CBA:B6 background.
In vitro T cell cultures and cell lines
For in vitro cultures, murine CD4+T cells from male and female mice (see above) were isolated from single cell suspension of lymph nodes and spleen by negative selection using the MojoSort Mouse CD4 T cell isolation kit (BioLegend). T cells were cultured in modified RPMI 1640 medium with physiological glucose concentration (i.e. 100 mg/dL) by diluting standard RPMI 1640 medium (Gibco) with glucose-free RPMI medium (Roth). The medium was supplemented with 10% FBS (Sigma), 50 μM 2-mercaptoethanol (β-ME), 1% penicillin/streptomycin and 1% GlutaMAX-I (all Gibco), unless otherwise stated. Human PBMCs were isolated from buffy coats of fresh blood samples or anonymous donors of unknown sex and age. Donors provided written informed consent to participate in research protocols approved by the Institutional Review Board of the University of Würzburg. PBMCs were isolated by gradient centrifugation using the SepMate-15 kit (StemCell technologies) and cultivated in fully supplemented RPMI 1640 without β-ME. Human and murine T cells were cultured at 37°C with 5% CO2. Platinum-E retroviral packaging cell line (Cell Biolabs Inc.) was cultured in standard DMEM with 10% FBS (Sigma) and 1% penicillin and streptomycin (Gibco) at 37°C with 5% CO2. After thawing, Platinum-E cells were selected for 7 days using 1 μg/ml puromycin and 10 μg/ml blasticidin S (both Gibco) and cultured afterwards in standard DMEM medium.
METHOD DETAILS
T cell activation and differentiation
For T helper (Th) cell differentiation, delta-surface plates (Nunc) were pre-coated with 12 μg/ml polyclonal anti-hamster IgG (MP Biomedicals) for 2 h and washed once with PBS. In 24-well plates, 1 ×106 cells were activated with 0.25 μg/ml (for Th17 and pTh17) or 0,5 μg/ml (for Th1, Th2 and iTreg subsets) of anti-CD3 (clone 145-2C1) together with 1 μg/ml anti-CD28 (clone 37.51, both Bio X Cell) and polarized into different Th subsets using the following cytokines and antibodies. For Th1 cells: 2,5 μg/ml anti-IL-4 (clone 11B11, Bio X Cell), 10 ng/ml rhIL-2 and 10 ng/ml rmIL-12 (both Peprotech). For Th2 cells: 5 μg/ml anti-IFNg (clone XMG1.2), 10 ng/ml rhIL-2 and 50 ng/ml rmIL-4 (both Preprotech). For iTreg cells: 2,5 μg/ml anti-IL-4 (clone 11B11), 2,5 μg/ml anti-IFNg (clone XMG 1.2, both Bio X Cell), 10 ng/ml rhIL-2 and 5 ng/ml rhTGFb1 (both Peprotech). To differentiate naïve CD4+ T cells into ‘pathogenic’ and ‘non-pathogenic’ Th17 cell subsets, different cytokine cocktails were used as described before (Kaufmann et al., 2019; Lee et al., 2012; Sharma et al., 2013). For ‘non-pathogenic’ Th17 cells: 2,5 μg/ml anti-IL-4 (clone 11B11), 2,5 μg/ml anti-IFNg (clone XMG1.2, both Bio X Cell), 20 ng/ml rmIL-6 and 0.5 ng/ml rhTGFβ1 (both Peprotech). For ‘pathogenic’ (p)Th17 cells: 2,5 μg/ml anti-IL-4 (clone 11B11), 2,5 μg/ml anti-IFNg (clone XMG1.2, both Bio X Cell), 20 ng/ml rmIL-6, 20 ng/ml rmIL-1β (both Peprotech) and 20 ng/ml rmIL-23 (Invitrogen). In some experiments, sodium acetate, sodium pyruvate, sodium lactate, dimethyl-α-ketoglutarate (DMK), palmitic acid, sodium oleate, 2-hydroxycitrate (2-HC), 5-tetradecyloxy-2-furonic acid (TOFA, all Sigma), L-glutamine (Gibco) or soraphen A (kindly provided by Rolf Möller, Helmholtz Center for Infection Research, Braunschweig, Germany) were added to the cell culture at the indicated concentrations and time points. Palmitic acid was firstly dissolved in ethanol at 5 mM concentration and then complexed with 4 volumes of 4% fatty acid-free BSA (Sigma) in medium (1 mM final concentration) and incubated for 1 h at 37°C. Sodium oleate (Sigma) was firstly diluted at a concentration of 100 mM in hot distilled water and then complexed to fatty acid-free BSA at a molar ratio of 8:1 (fatty acid:albumin) to obtain a final concentration of 1 mM oleate. All other exogenous metabolites were dissolved in medium stock solutions based on the manufacture’s recommendation.
Flow cytometry
Flow cytometric staining was performed as previously described (Vaeth et al., 2019). Briefly, cells were stained with Fixable Viability Dye eFluor 780 (eBioscience) for 10 min in PBS at RT together with an anti-FcgRII/FcgRIII antibody (clone 2.4G2; Bio X Cell) to prevent unspecific binding. After washing, surface antigens were stained with fluorophore-conjugated antibodies (see Key Resource Table) in PBS containing 0.5% BSA for 20 min at RT in the dark. For intracellular cytokine staining, cells were stimulated with 1 μM ionomycin (BioMol) and 30 nM phorbol-12-myristat-13-acetate (PMA, Sigma) in the presence of 2 μg/ml brefeldin A and/or 2 μM monensin (both eBioscience) for 4-5 h at 37°C. After surface staining, cells were fixed with IC-fixation buffer (eBioscience) and intracellular cytokines were stained using 1x permeabilization buffer (both eBioscience) for 40 min at RT. For detection of intranuclear antigens, cells were fixed using Foxp3/TF staining buffer set according to manufacturer’s recommendations (eBioscience). To analyze T cell proliferation, CD4+ T cells were loaded with 2,5 μM CFSE (BioLegend) or 5 μM CellTrace Violet (Invitrogen) according to manufacturer’s instructions. To assess histone modifications, stimulated T cells were fixed with the Foxp3/TF staining buffer set (eBioscience) for 30 min at RT and stained intranuclearly with rabbit polyclonal anti-mouse acetyl-histone H3K9/14ac (Cell Signaling), rabbit monoclonal anti-mouse acetyl-histone H3K27ac (Cell Signaling, clone D5E4), rabbit monoclonal anti-mouse H3K9me3 (Cell Signaling, clone D4W1U) or rabbit monoclonal anti-mouse H3K27me3 (Cell Signaling, clone C36B11) for 30 min in permeabilization buffer (eBioscience). After washing, anti-histone antibodies were detected using a donkey polyclonal anti-rabbit IgG secondary antibody conjugated to Alexa Fluor 546 (Invitrogen) diluted 1:1000 in permeabilization buffer for 30 min. Neutral lipid content of cultivated Th cells was measured using the BODIPY 493/503 reagent (Invitrogen). For the quantification of the mitochondrial volume, membrane potential and ROS production, T cells were loaded with 500 nM MitoTracker deep red or 2 nM Tetramethylrhodamin-Ethylester (TMRE) and 2.5. μM MitoSOX, respecitvely (all Invitrogen). As background control for TMRE, T cells were pre-treated with 20 μM Trifluoromethoxy carbonylcyanide phenylhydrazone (FCCP, Cayman Chemicals) for 15 min to depolarize the membrane potential. All sample acquisition was performed with a BD Celesta flow cytometer (BD Biosciences) or an Aurora Flow Cytometer (Cytek) and further analyzed with the FlowJo software (Tree Star).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat polyclonal anti-hamster IgG | MP Biomedicals | Cat#: 56984 |
| Hamster monoclonal anti-mouse CD3 | Bio X Cell | Cat#: BE0001-1; Clone 145-2C1 |
| Hamster monoclonal anti-mouse CD28 | Bio X Cell | Cat#: BE00015-1; Clone 37.51 |
| Hamster monoclonal anti-mouse IFN-gamma | Bio X Cell | Cat#: BE0055; Clone XMG1.2 |
| Rat monoclonal anti-mouse IL-4 | Bio X Cell | Cat#: BE0045; Clone 11B11 |
| Rat monoclonal anti-mouse CD16/CD32 | Bio X Cell | Cat#: BE0307; Clone 2.4G2 |
| Hamster monoclonal anti-mouse CD3 | BioLegend | Clone 145-2C11 |
| Rat monoclonal anti-mouse CD4 | BioLegend | Clone GK1.5 |
| Rat monoclonal anti-mouse CD8a | BioLegend | Clone 53-6.7 |
| Rat monoclonal anti-mouse CD11b | BioLegend | Clone M1/70 |
| Rat monoclonal anti-mouse CD45 | BioLegend | Clone 30-F11 |
| Rat monoclonal anti-mouse CD45RB | BioLegend | Clone C363-16A |
| Mouse monoclonal anti-mouse CD45.1 | BioLegend | Clone A20 |
| Rat monoclonal anti-mouse IL-17a | BioLegend | Clone C11-18H10.1 |
| Rat monoclonal anti-mouse IFN-gamma | BioLegend | Clone XMG1.2 |
| Rat monoclonal anti-mouse GM-CSF | BioLegend | Clone MP1-22E9 |
| Rat monoclonal anti-mouse TNF-alpha | BioLegend | Clone MP6-XT22 |
| Rat monoclonal anti-mouse IL-2 | BioLegend | Clone JES6-5H4 |
| Rat monoclonal anti-mouse Foxp3 | Thermo | Clone FJK-16s |
| Mouse monoclonal anti-mouse Tbet | BioLegend | Clone 4B10 |
| Rat monoclonal anti-mouse/human GATA3 | Thermo Fisher | Clone TWAJ |
| Mouse monoclonal anti-mouse RORgamma t | BD | Clone Q31-378 |
| Rat monoclonal anti-mouse CD25 | BioLegend | Clone PC61 |
| Mouse monoclonal anti-mouse Ki-67 | BD | Clone B56 |
| Rat monoclonal anti-mouse CD62L | BioLegend | Clone MEL-14 |
| Hamster monoclonal anti-mouse CD69 | BD | Clone H1.2F3 |
| Rat monoclonal anti-mouse CD44 | BioLegend | Clone IM7 |
| Rat monoclonal anti-mouse OX-40 | BioLegend | Clone OX-86 |
| Hamster monoclonal anti-mouse ICOS | BioLegend | Clone C398.4A |
| Rat monoclonal anti-mouse CD38 | BioLegend | Clone 90 |
| Rat monoclonal anti-mouse CD279 | BioLegend | Clone 29F.1A12 |
| Rat monoclonal anti-mouse CD98 | BioLegend | Clone 4F2 |
| Rat monoclonal anti-mouse CD71 | BioLegend | Clone RI7217 |
| Rat monoclonal anti-mouse CD11b | BioLegend | Clone M1/70 |
| anti-FcgRII/FcgRIII antibody | eBioscience | Clone 2.4G2 |
| Rabbit polyclonal anti-mouse acetyl-Histone H3 (Lys9/14) | Cell Signaling | Cat#: 9677 |
| Rabbit monoclonal anti-mouse acetyl-Histone H3 (Lys27) | Cell Signaling | Cat#: 8173; Clone D5E4 |
| Rabbit monoclonal anti-mouse H3K9me3 | Cell Signaling | Cat#:13969; Clone D4W1U |
| Rabbit monoclonal anti-mouse H3K27me3 | Cell Signaling | Cat#: 9733; Clone C36B11 |
| Rabbit polyclonal anti-mouse acetyl-Histone H3 (Lys9/14) | Diagenode | Cat#: C15410005 |
| Rabbit polyclonal anti-mouse acetyl-Histone H3 (Lys27) | Diagenode | Cat#: C15410174 |
| Isotype rabbit IgG | Cell Signaling | Cat#: 2729 |
| Mouse monoclonal anti-mouse/human/rat GLUT1 | Abcam | Cat#: ab40084; Clone SPM498 |
| Rabbit polyclonal anti-mouse/human IRF4 | Cell Signaling | Cat#: 4948S |
| Rabbit polyclonal anti-human GLUT3 | BioRad | Cat#: VPA00651 |
| Rabbit polyclonal anti-mouse/rat GLUT3 | Merck Millipore | Cat#: AB1344 |
| Mouse monoclonal anti-mouse/human/rat NFATc1 | Abcam | Cat#: ab2796; Clone 7A6 |
| Rabbit polyclonal anti-mouse/human/rat ACLY | Cell Signaling | Cat#: 4332 |
| Mouse monoclonal anti-mouse/human GAPDH | SCBT | Cat#: sc-3223; Clone 6C5 |
| Mouse monoclonal anti-mouse/human/rat beta-Actin-HRP | BioLegend | Cat#: 643807; Clone 2F1-1 |
| Mouse monoclonal anti-mouse/human/rat VDAC | Biolegend | Cat#: 820702; Clone N152B/23 |
| Goat polyclonal anti-mouse-HRP | Bio-Rad | Cat#: STAR207P |
| Goat anti-rabbit-HRP | Bio-Rad | Cat#: STAR208P |
| Rabbit polyclonal anti-human/mouse CD3 | Dako | Cat#: A0452 |
| Donkey polyclonal anti-rabbit IgG, Alexa Fluor 546 | Thermo Fisher | Cat#: A10040 |
| Rabbit monoclonal anti-human/mouse/rat/monkey TOM20 | CellSignalling | Cat#: D8T4N |
| Rabbit polyclonal anti-human Slc2a3 (GLUT3) | Sigma | Cat#:HPA006539 |
| Chemicals,Reagents, Peptides, and Recombinant Proteins | ||
| RPMI 1640 GlutaMAX | Gibco | Cat#: 61870-010 |
| RPMI 1640 w/o glucose | Roth | Cat#: 9094.1 |
| Penicillin-streptomycin | Gibco | Cat#: 15140122 |
| Puromycin | Gibco | Cat#:A1113802 |
| Plasticidin | Gibco | Cat#:A1113902 |
| 2-mercaptoethanol | Gibco | Cat#: 21985023 |
| FCS | Sigma | Cat#: 12133 |
| EDTA | Thermo | Cat#: 15575020 |
| HEPES | Gibco | Cat#: 11560496 |
| Bovine Serum Albumin (BSA) fatty acid free | Sigma | Cat#: A8806 |
| Seahorse XF RPMI medium, pH 7.4 | Agilent | Cat#: 103576-100 |
| Seahorse XF Calibrant Solution, pH 7.4 | Agilent | Cat#: 100840-000 |
| Prolong Gold Antifade Mounting Medium | Molecular Probes | Cat#: P10144 |
| Recombinant human TGF-beta | Peprotech | Cat#: 100-21 |
| Recombinant murine IL-6 | Peprotech | Cat#: 216-16 |
| Recombinant murine IL-1-beta | Peprotech | Cat#: AF-211-11B |
| Recombinant murine IL-12 p70 | Peprotech | Cat#: 210-12 |
| Recombinant human IL-2 | Peprotech | Cat#: 200-02 |
| Recombinant murine IL-4 | Peprotech | Cat#: AF-214-14 |
| Recombinant murine IL-23 | Invitrogen | Cat#: 14-8231-63 |
| Phorbol 12-myristate 13-acetate (PMA) | Sigma | Cat#: P8139-5MG |
| Ionomycin | Biomol | Cat#: AG-CN2-0418 |
| Brefeldin A | eBioscience | Cat#: 00-4506-51 |
| Monensin | eBioscience | Cat#: 00-4505-51 |
| Fixable Viability Dye eFluor 780 | eBioscience | Cat#: 65-0865-18 |
| Annexin A5 | BioLegend | Cat#: 640920 |
| 4’, 6-Diamidino-Phenylindole (DAPI) | Thermo | Cat#: D1306 |
| Propidium iodide (PI) | BioLegend | Cat#: 421301 |
| MitoTracker Deep Red | Invitrogen | Cat#: M22426 |
| Red Mitochondrial Superoxide Indicator (MitoSOX) | Invitrogen | Cat#: M36008 |
| Tetramethzlyrhodamine, Ethyl Ester, Percholorate (TMRE) | Invitrogen | Cat#: T669 |
| Trifluoromethoxy carbonylcyanide phenylhydrazone (Fccp) | Cayman Chemical | Cat#: Cay15218-50 |
| 4,4-Difluoro-1,3,5,7,8-Pentamethyl-4-Bora-3a,4a-Diaza-s-Indacene (BODIPY) | Invitrogen | Cat#: D3922 |
| 2-deoxy-2-[(7-nitro-2,1,3-benzoxadiazol-4-yl)amino]-D-glucose (2-NBDG) | Cayman Chemical | Cat#: Cay11046 |
| 123count eBeads Counting Beads | Invitrogen | 01-1234-42 |
| Permeabilization Buffer (10X) | eBioscience | Cat#: 00-8333-56 |
| IC Fixation Buffer | eBioscience | Cat#: 00-8222-49 |
| Foxp3/TF Staining Buffer | eBioscience | Cat#: 00-5523-00 |
| Oligomycine | Cayman Chemical | Cat#: Cay11341-5 |
| Rotenone | AdipoGen Life Sciences | Cat#: AG-CN2-0516-G001 |
| Antimycin A | Sigma | Cat#: A8674 |
| 2-Deoxy-D-glucose (2-DG) | Sigma | Cat#: D8375 |
| Cell-Tak | Corning | Cat#: 354240 |
| L-Glutamine | Gibco | Cat#: 25030081 |
| D-Glucose | Sigma | Cat#: G7021 |
| Sodium Pyruvate | Sigma | Cat#: P2256-5G |
| Formaldehyde 16%, methanol-free | ThermoFischer | Cat#: 28906 |
| Luxol Fast Blue (Solvent Blue) | Roth | Cat#: 7709.1 |
| Cresyl Violet | Sigma | Cat#: C5042 |
| MOG35-55 peptide | Synpeptide | Cat#: MOG35-55 |
| Pertussis Toxin | Enzo | Cat#: BML-G100 |
| Mycobacterium tuberculosis H37Ra | FisherScientific | Cat#: 10218823 |
| Incomplete Freund’s Adjuvant | BD | Cat#: 263910 |
| Phenol-Chloroform-Isoamylalkohole | Roth | Cat#: A156.1 |
| Potassium Hydroxycitrate Tribasic Monohydrate (2-HC) | Sigma | Cat#: 59847 |
| Sodium Acetate | Sigma | Cat#: S5636 |
| Sodium Lactate | Sigma | Cat#: 71718 |
| Palmitic Acid | Sigma | Cat#: P0500-10G |
| Sodium Oleate | Sigma | Cat#: O7501-250mg |
| Soraphen A | Rolf Möller | Berod, Friedrich et al. 2014 |
| 5-Tetradecyloxy-2-furonic acid (TOFA) | Sigma | Cat#: T6575-5MG |
| 2-Oxoglutarsäure-dimethylester (DMK) | Sigma | Cat#: 349631 |
| Polybrene | SantaCruz | Cat#: sc-134220 |
| GeneJet Tranfection reagent | Thermo Fisher | Cat#: SL100488 |
| Collagenase D | Roche | Cat#: 11088858001 |
| DNase 1 | Thermo Fisher | Cat#: EN0521 |
| Dispase | BD | Cat#: 354235 |
| RIPA lysis buffer | SantaCruz | Cat#: sc-24948 |
| complete Mini, EDTA-free | Roche | Cat#: 11836170001 |
| ROTI Load 1 | Roth | Cat#: K929.1 |
| RNase A | Thermo Scientific | Cat#: EN0531 |
| Proteinase K | Thermo Scientific | Cat#: EO0491 |
| [3H] 2-Deoxy-D-glucose | Perkin Elmer | Cat#: NET328A250UC |
| ROTISZINT®eco plus LSC-universal cocktail | Roth | Cat#: 0016.3 |
| [14C(U)]-D-glucose | Hartman Analytic | Cat#: 20562910 |
| Critical Commercial Assays | ||
| MojoSortTM Mouse CD4 T Cell Isolation Kit | BioLegend | Cat#: 480033 |
| CellTrace Violet Cell Proliferation Kit | Invitrogen | Cat#: C34557 |
| Acetyl-Coenzym A Assay Kit | Sigma | Cat#: MAK039-1KT |
| Seahorse Xfe FluxPak | Agilent | Cat#: 102601-100 |
| Pierce 660nm Protein Assay | Thermo Fisher | Cat#: 22662 |
| Protein A/G Magnetic Beads | Pierce | Cat#: 88802 |
| RNeasy Plus Micro Kit | Quiagen | Cat#: 74034 |
| RNAprotect Cell Reagent | Quiagen | Cat#: 76526 |
| Roti-Prep RNA Mini | Roth | Cat#: 8485.2 |
| iScript cDNA Synthesis Kit | Bio-Rad | Cat#: 1708891 |
| iTaq Universal SYBR Green Supermix | Bio-Rad | Cat#: 1725124 |
| CHIP DNA Clean & Concentrator Kit | Zymo | Cat#: D5205 |
| Deposited Data | ||
| Data S1 | This study | |
| RNA-seq data 1; Th1 and Th17 cells differentiated from WT and Slc2a3fl/flCd4Cre mice | This study | GSE192659 |
| RNA-seq data 2; Th17 cells differentiated from WT, Slc2a3fl/flCd4Cre and Aclyfl/flCd4Cre mice with and without 10 mM acetate treatment | This study | GSE192663 |
| ChIP-seq data (H3 K9/14ac) | This study | GSE193515 |
| ChIP-seq data (NFATc1) | Klein Hessling et al. 2017 | GSE98726 |
| ChIP-seq data (STAT3) | Hirahara et al 2015 | GSE65621 |
| ChIP-seq data (HIF-1a) | Ciofani et al 2012 | GSE40918 |
| ChIP-seq data (IRF4) | Man et al 2013 | GSE49931 |
| ChIP-seq data (NFATc2) | Martinez et al 2015 | GSE64409 |
| DNaseI-seq data | Bevington et al 2016 | GSE67465 |
| ATAC-seq data | Mognol et al 2017 | GSE88987 |
| ATAC-seq data | Qui et al 2020 | GSE127768 |
| Experimental Models: Cell Lines/bacteria/viruses | ||
| Platinum-E retroviral packaging cell line | Cell Biolabs Inc. | Cat# RV-101 |
| Citrobacter rodentium (strain ICC180) | Friedrich et al. 2017 | N/A |
| LCMV (Armstrong strain) | Vaeth et al. 2017 | N/A |
| Experimental Models: Mouse Strain | ||
| Mouse: Cd4Cre | Jackson Laboratories | Cat#: 017336 |
| Mouse: Slc2a3fl/fl | Provided by E. Dale Abel (Iowa) | Trevor P. Fidler et al., 2017 |
| Mouse: Aclyfl/fl | Jackson Laboratories | Cat#: 43555-JAX |
| Mouse: Irf4fl/fl | Jackson Laboratories | Cat#: 009380 |
| Mouse: Stat3fl/fl | Jackson Laboratories | Cat#: 016923 |
| Mouse: Cd28−/− | Jackson Laboratories | Cat#: 002666 |
| Mouse: Hif1afl/fl | Jackson Laboratories | Cat#: 007561 |
| Mouse: Rag2−/−γc−/− | Jackson Laboratories | Cat#: 014593 |
| Mouse: Rag1−/− | Jackson Laboratories | Cat#: 002216 |
| Mouse: Rosa26LSL-Cas9 | Jackson Laboratories | Cat#: 026175 |
| Mouse: CD45.1+ C57BL/6 | Jackson Laboratories | Cat#: 002014 |
| Oligonucleotides | ||
| See Table S1 for gRNAs | Sigma | This study |
| See Table S2 for primers for qRT-PCR | Sigma | This study |
| See Table S3 for primers for ChIP-qPCR | Sigma | This study |
| Recombinant DNA | ||
| pMIG | Addgene | Cat#: 9044 |
| pMIG-GLUT3 | This study | N/A |
| retro-gRNA-eGFP | Addgene | Cat#: 116926 |
| retro-gAcly-eGFP | This study | N/A |
| Software and Algorithms | ||
| FACSDiva | BD Bioscience | N/A |
| FlowJo software (Tree Star, Inc.) | FlowJo LLC | https://www.flowjo.com/ |
| GraphPad Prism V9 | GraphPad Software | https://www.graphpad.com/ |
| MetaboAnalyst 4.0 | Xia Lab, McGill University | https://www.metaboanalyst.ca |
| Illustrator CS5 | Adobe Systems Inc. | https://www.adobe.com/de/creativecloud/membership.html |
| Integrative Genome Viewer (IGV) | Broad Institute | https://software.broadinstitute.org/software/igv/ |
| LCS software | Leica | N/A |
| ImageJ | NIH | N/A |
| Icy | Institut Pasteur, France BioImaging | http://icy.bioimageanalysis.org |
| Xcalibur software | Thermo Scientific | https://www.thermofisher.com/order/catalog/product/OPTON-30965#/OPTON-30965 |
| ChopChop | Labun et al., 2019 | https://chopchop.cbu.uib.no |
| Other | ||
| XFe96 Extracellular Flux Analyzer | Seahorse Bioscience | N/A |
| ChemiDoc imaging system | BioRad | N/A |
| DMi8 automated laser-scanning confocal microscope | Leica | N/A |
| FlexStation 3 multi-mode microplate reader | Molecular Devices | N/A |
| Celesta Flow Cytometer | BD Bioscience | N/A |
| Aurora Flow Cytometer | Cytek | N/A |
| Biorupter Plus Sonication System | Diagenode | N/A |
| E220 sonicator | Covaris | N/A |
| Dionex UltiMate 3000 LC System | Thermo Scientific | N/A |
| Q Exactive Orbitrap mass spectrometer | Thermo Scientific | N/A |
| SX-8G Compact IP-Star | Diagenode | N/A |
| NovaSeq Illumina sequencer | Illumina | N/A |
| BD FACSAria III cell sorter | BD | N/A |
| QubitTM 3.0 Fluometer | Thermo Scientific | N/A |
| 2100 Bioanalyzer | Agilent | N/A |
| NextSeq 500 platform | Illumina | N/A |
Retroviral transduction
Retroviral infection of primary T cells was performed as described previously (Vaeth et al., 2017a). Briefly, Platinum-E retroviral packaging cells were transfected transiently with modified pMIG (Addgene, #9044) or retro-gRNA-eGFP retroviral plasmids (Addgene, #116926) using the GeneJet reagent (SignaGene). The transfection medium was replaced 24 h later with standard DMEM medium and the supernatant containing retroviral particles was collected 2 and 3 days after transfection. CD4+ T cells were isolated by negative selection using the MojoSort Mouse CD4 T cell isolation kit (BioLegend) and polarized into Th1 or Th17 cells as described above. 24 h after activation, the medium of the T cells was replaced by retroviral supernatant the T cells were transduced by spin-infection (2.500 rpm, 30°C, 90 min) in the presence of 10 mg/ml polybrene (SantaCruz). After transduction, cells were incubated at 37°C for 4 h before the viral supernatant was removed and replaced by fresh medium. After 3 days of stimulation, transduced T cells were rested o/n and GFP+ cells and purified using a FACSAria III cell sorter (BD Pharmingen) before resting for additional 2 days in fresh medium containing Th1 or Th17 cell-polarizing cytokines.
Genome editing using CRISPR/Cas9
All gRNAs were designed as 20nt oligomers with the ChopChop software (Labun et al., 2019) and synthesized by Sigma (see Table S1). gRNAs were annealed with the respective complement-reverse sequences with the following overhangs: phospho-5’-CACCG-gRNA-3’(forward) and 5’-AAAC-gRNA-C-3’ (reverse). The annealed oligos were cloned into Bbs I-linearized retro-gRNA-eGFP retroviral plasmids (Addgene#116926) using a standard T4 ligase protocol (NEB). The correct integration of the gRNA was verified by DNA sequencing (Eurofins) using the following primer: 5’-TGGACTATCATATGCTTACCGTAACTTGAA-3’. Th1 and Th17 cells were retrovirally transduced with retro-gRNA-eGFP plasmids as described above and ablation of the target protein was tested by Western blotting.
Glucose uptake and utilization measurements
Glucose uptake by T cells in vitro was monitored using the fluorescent glucose analog 2-NBDG (Cayman Chemicals). After starving for 15 min in glucose-free medium, T cells were incubated with 300 μM 2-NBDG for 30 min at 37°C, washed and processed for flow cytometric analysis. Alternatively, glucose uptake was measured directly using tritiated 2-desoxy-glucose ([3H]-2-DG) (Perkin Elmer). CD4+T cells were differentiated into Th1 and Th17 cells for 3 days, before 5x105 T cells were incubated with 200 μl glucose-free medium (with 10% FBS) containing 1 μCi/ml [3H]-2-DG in 96-well plates. After 6 h of incubation, T cells were washed twice with ice-cold PBS, resuspended in 40 μl H2O before lysing in 160 μl scintillation cocktail (Roth). Intracellular [3H] counts per minute (cpm) were measured using a MicroBeta2 microplate scintillation counter (Perkin Elmer) and normalized to cell number. To measure the incorporation of glucose-derived carbons into histones, CD4+ T cells were differentiated into Th17 cells in complete RPMI 1640 medium (Gibco) containing 0.5 g/l D-glucose and 1 μCi/ml radio-labelled [14C(U)]-D-glucose (Hartmann Analytic). After 72 h of incubation, 8×106 cells were harvested, washed twice with ice-cold PBS and histones were extracted as described before (Shechter et al., 2007). Briefly, nuclei were isolated using a hypotonic buffer containing 10 mM HEPES pH 7.9, 10 mM KCl, 1.5 mM MgCl, 0.34 M sucrose, 10% glycerol and 0.2% NP-40. PMSF and complete protease inhibitor cocktail (Roche) were added prior to use. Nuclei were washed in extraction buffer without NP-40 and further lysed in salt-free buffer containing 3 mM EDTA and 0.2 mM EGTA. After 30 min of incubation on a rotator at 4°C, the nucleoplasm was removed by centrifugation and the chromatin was resuspended in 150 μl of high-salt solubilization buffer containing 10 mM Tris-Cl pH 8, 2.5 mM NaCl and 0.05% NP-40. After 30 min incubation on a rotator at 4°C, samples were centrifuged at 16.000 g and the supernatant containing the histones was collected. 40 μl of histone extract was mixed with 160 μl scintillation cocktail (Roth) and [14C] incorporation was measured using a MicroBeta2 microplate scintillation counter (Perkin Elmer). [14C] counts per minute (cpm) were normalized to histone protein concentration.
Seahorse extracellular flux analysis
Mitochondrial respiration and lactate secretion of T cells was measured as their oxygen consumption rate (OCR) and glycolytic proton efflux rate (glycoPER), respectively, using an oxygen-controlled XFe96 extracellular flux analyzer (Seahorse Bioscience). XFe96 cell culture microplates (Agilent) were pre-coated with 22 μg/ml Cell-Tak (Corning) and 1.5x105 T cells per well were attached in 5-8 replicates in Seahorse XF RPMI medium (Agilent) supplemented with 2 mM L-glutamine (Gibco), 1 mM sodium pyruvate (Sigma) and 10 mM D-glucose (Sigma). After incubation for 1 h in a CO2-free incubator at 37°C, glycolytic and mitochondrial stress tests were performed. For assessing glycolysis, basal extracellular acidification rate (ECAR) was measured followed by addition of 0.5 μM rotenone (AdipoGen) and 0.5 μM antimycin A (Sigma) to inhibit mitochondrial complex 1 and 3, respectively. At the end of the measurement, 50 mM 2-DG (Sigma) was added to completely block glycolysis. To analyze mitochondrial respiration, basal oxygen consumption was measured followed by the addition of 2 μM oligomycin (Cayman Chemicals), an ATP synthase inhibitor, 1 μM of the protonophore carbonyl cyanide-4-(trifluoromethoxy)-phenylhydrazone (FCCP, Cayman Chemical) and 0.5 μM rotenone (AdipoGen) together with 0.5 μM antimycin A (Sigma). The basal oxygen consumption was calculated by subtracting the OCR after rotenone and antimycin A treatment from the OCR before oligomycin treatment. The maximal OCR was calculated by subtracting the OCR after rotenone and antimycin A treatment from the OCR measured after addition of FCCP.
Metabolomic profiling
To analyze polar intracellular and secreted metabolites, Th1 and Th17 cells were differentiated in RPMI containing 1 g/l 12C-glucose as described above. After 3 days, T cell were labelled in modified RPMI 1640 containing 1 g/l 13C6-glucose (Sima) for 24 h. After washing with pre-warmed PBS, metabolite extraction was performed in ice-cold 80% methanol containing 1 μM of 13C5-d5-15N glutamic acid, 1 μM d7-15N4 arginine, 1 μM d27 myristic acid and 1 μM d12 glucose as internal standards. Following centrifugation with 20.000 g for 10 min at 4°C, the supernatant containing polar metabolites was transferred to a new tube and stored at −80°C. The pellet was used for protein quantification as an internal normalization method. Alternatively, cell-free culture supernatant was used for analysis. Liquid chromatography followed by mass spectrometry (LC/MS) was performed by the VIB Metabolomics Expertise Center (MEC) at the KU Leuven, Belgium. 10 μl sample lysate was loaded into a Dionex UltiMate 3000 LC System (Thermo Scientific) equipped with a C-18 chromatography column (Acquity UPLC -HSS T3 1. 8 μm; 2.1 x 150 mm, Waters) coupled to a Q Exactive Orbitrap mass spectrometer (Thermo Scientific) operating in negative ion mode. A step gradient was carried out using solvent A (10 mM TBA and 15 mM acetic acid) and solvent B (100% methanol). The gradient started with 0% of solvent B and 100% solvent A and remained at 0% B until 2 min post injection. A linear gradient to 37% B was carried out until 7 min and increased to 41% until 14 min. Between 14 and 26 minutes the gradient increased to 100% of B and remained at 100% B for 4 minutes. At 30 min the gradient returned to 0% B. The chromatography stopped at 40 min. The flow was kept constant at 250 μl/min and the column was placed at 25°C throughout the analysis. The MS operated in full scan mode (m/z range: [70–1050]) using a spray voltage of 3.2 kV, capillary temperature of 320 °C, sheath gas at 40.0, auxiliary gas at 10.0. The AGC target was set at 3e6 using a resolution of 140.000, with a maximum IT fill time of 512 ms. Data collection was performed using the Xcalibur software (Thermo Scientific) and analyzed by integrating the peak areas using the El-Maven to Polly data processing engine (Elucidata). Cellular sphingolipids were analyzed after extraction with methanol:chloroform (2:1) using a 1290 Infinity II HPLC coupled with a 6495C triple-quadrupole mass spectrometer (Agilent Technologies) as previously described (Naser et al., 2020).
Detection of subcellular acetyl-CoA, ATP and citrate levels
Acetyl-CoA, ATP and citrate concentrations in different subcellular fractions were determined using the acetyl-coenzym A assay kit (Sigma), CellTiter-Glo Viability assay kit (Promega) and citrate assay kit (Sigma), respectively. 5x106 Th17 cells were harvested in 100 μl fractionation buffer, containing 20 mM HEPES, 10 mM KCl, 2 mM MgCl2, 1 mM EDTA, 1 mM EGTA and 1 M dithiothreitol (DTT). Samples were incubated on ice for 20 min, followed by centrifugation at 10.000 g at 4°C for 5 min to separate nucleo-cytosolic fractions in the supernatant from the intact mitochondria in the pellet. Mitochondrial lysates were obtained by washing the pellet once with ice-cold PBS and permeabilization with 80% methanol for 10 min on ice followed by centrifugation at 13.000 g for 2 min at 4°C. For the quantification of total cellular acetyl-CoA, ATP or citrate levels, T cells were directly harvested in 80% methanol, permeabilized for 20 min on ice following centrifugation at 13.000 g for 2 min at 4 °C. The cellular fractions were deproteinized by perchloric acid precipitation following KOH neutralization and acetyl-CoA, ATP and citrate levels were quantified according to the manufacturer’s instructions.
Immunofluorescence and histochemistry
Tissue samples were fixed in 4% PFA for 24 h, processed for paraffin embedding and cut into 5 μM sections for immunofluorescence or hematoxylin and eosin staining as described previously (Vaeth et al., 2015). EAE mice were perfused with PBS before the spinal cord of the lumbar region was dissected and directly fixed with 4% PFA for 24 h. Spinal cord samples were processed as described above and stained with Luxol Fast Blue (Roth) and Cresyl Violet (Sigma) to detect demyelination and leukocytic infiltration. For immunofluorescence staining, Jurkat T cells were spun on Superfrost slides (Roth), dried and fixed with 4% PFA. After an 1 h incubation at RT with mouse-anti-Tom20 (Abcam, clone EPR15581-39) and rabbit-anti-human GLUT3 (BioRad), the samples were washed and stained for 1 h with secondary donkey anti-rabbit IgG (H+L) Alexa Fluor 488 and donkey anti-mouse IgG (H+L) Alexa Fluor 647 antibodies (both Invitrogen). To detect T cell infiltration in tissues, sections were deparaffinized and re-hydrated before antigen retrieval was achieved by heating in 10 mM citric acid buffer (pH 6.0) in a high-pressure cooker (Deni) for 20 min, followed by blocking with antibody diluent (Dako) for 1 h. After an overnight incubation with a rabbit anti-mouse CD3 (1:1000, Dako) at 4 °C, the slides were washed and stained for 1 h with a donkey anti-rabbit IgG (H+L) Alexa Fluor 546 (1:800; Invitrogen). The slides were embedded in Prolong Gold Antifade Mounting Medium containing DAPI (Invitrogen). Imaging acquisition was performed with a DMi8 automated laser-scanning confocal microscope (Leica) and analyzed with the Icy software.
ChIP-qPCR and ChIP-sequencing assays
For chromatin immunoprecipitation (ChIP) followed by qPCR, Th17 cells were differentiated for 3 days and re-stimulated for 3 h with PMA and ionomycin. After fixation in 1% formaldehyde (Pierrce) for 10 min at RT, 125 mM glycine was added for 10 min at RT to quench the reaction. After 3 times washing with cold PBS, nuclei were isolated in lysis buffer containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 5 mM EDTA (pH 8.0), 0.5% NP-40 and 1% Triton-X-100. After 20 min on ice, cells were washed twice in PBS and nuclei were lysed in 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 2 mM EDTA (pH 8.0), 1% NP-40 and 20% SDS for 30 min on ice. Nuclear lysates were sonicated using a Biorupter Plus Sonication system (Diagenode) for 25 cycles, high power, 20 sec on, 20 sec off). Histone-DNA complexes were isolated using polyclonal anti-mouse antibodies against acetylated histone H3 lysine 9/14 (anti-H3K9/14ac) and acetylated histone H3 lysine 27 (anti-H3K27ac, both Diagenode). Briefly, magnetic beads (Pierce) were coupled with 2 μg of the precipitating antibody for 2 h while rotating at 4°C. The supernatant was removed and the pre-coupled beads were rotated with 400 μl of the nuclear lysate o/n at 4°C. The precipitate was washed several times with washing buffer, and the protein-DNA complexes were de-crosslinked o/n with 250 mM NaCl at 65°C, before RNA and proteins were digested with 80 μg RNase A and proteinase K (both Thermo Scientific). The precipitated DNA was extracted with a mixture of phenol:chloroform:isoamylalcohol (25:24:1 ratio) followed by concentration of the DNA with ChIP DNA Clean and Concentrator Kit (Zymo). Quantification of the precipitated DNA was performed by qPCR using primers listed in Table S3. The results were normalized to both input and isotype control. For ChIP followed by genome wide DNA-sequencing (ChIP-seq), Th17 cells were re-stimulated with PMA and ionomycin for 3 h and fixed with formaldehyde for 15 min at RT to perform the RELACS protocol as previously described (Arrigoni et al., 2018). Briefly, cells were resuspended in lysis buffer and nuclei were extracted by sonication using the E220 sonicator (Covaris, 75 W peak incident power, 2% duty factor, 200 cycles/burst, 90 seconds of treatment). 1×106 nuclei per sample were digested overnight at 20°C using 50 units of CviKi-1 (NEB). Nuclei were washed once, quantified and normalized using buffer EB (Qiagen) to concentration of 500,000 nuclei per 25 μi. Sample were barcoded using the RELACS procedure, which includes intra-nuclei A-tailing and ligation of barcoded hairpin sequencing adaptors to the digested chromatin. Six samples were barcoded and pooled together, resuspended in 300 μl of shearing buffer and lysed. A third of the volume of chromatin has been used for ChIP, corresponding to about 2x106 nuclei per ChIP total (~ 330,000 per each pooled sample). Chromatin was mixed 1:1 with the ChIP buffer H from the Auto Histone ChIP-seq Kit (Diagenode), supplemented with protease inhibitor cocktail and 5 μg of anti-H3K9/14ac (Diagenode). A fraction of original chromatin (~ 0.1%) was set aside as input. The immunoprecipitation, washing and elution of immunocomplexes from the beads was performed using solutions and protocols from the Auto Histone ChIP-seq Kit and the automatic liquid handler SX-8G Compact IP-Star (Diagenode) following these parameters: indirect method, 200 μl sample volume of ChIP reactions, 10 h antibody incubation, 3 h beads incubation, 5 min washes (all steps at 4°C). Eluates were de-crosslinked for 2 h at 65 °C and DNA was purified using MinElute columns (Qiagen). Samples were USER-treated and amplified with 10 PCR cycles. For these steps components of NEBNext Ultra II DNA Library Prep kit for Illumina (NEB) kit were used as described before (Arrigoni et al., 2018). Libraries were quality controlled and sequenced on a NovaSeq Illumina sequencer, using 50 bp paired-end read length. The raw fastq data files were processed using the snakePipes pipeline (version 2.5.0) for DNA Mapping and ChIP-Seq (https://snakepipes.readthedocs.io). Fastq files were pre-processed using cutadapt 2.8 and then mapped to the GRCm38 genome assembly (mm10) using Bowtie2 (version 2.3.5.1) with default parameters. The bam files were de-duplicated using sambamba markdup and umitools dedup. The resulting bam files were then processed using the ChIP-seq command from the snakePipes pipeline by providing sample information (WT or Slc2a3fl/flCd4Cre) under the --sampleSheet flag and by specifying the corresponding input sample and information about the expected peak signal (broad: False) in the config file. Briefly, we used the deeptools package (3.3.2) (http://deeptools.ie-freiburg.mpg.de) for comparing bam files and plotting mapped regions. In addition, peaks were called using MACS2 (version 2.2.6) with macs2 callpeak and the flags: -f BAM --mfold 0 50 -g 2652783500 --nomodel --extsize228 --keep-dup all --qvalue 0.001. Differential binding on the MACS2 peak files was performed using the CSAW R package (version 1.20.0) as part of the snakePipes pipeline.
qRT-PCR and RNA-sequencing
Total RNA was extracted using the Roti-Prep Mini Kit (Roth) and cDNA was synthesized using the iScript cDNA synthesis kit (Bio-Rad). Quantitative real-time PCR was performed using the SYBR Green qPCR Master Mix (Bio-Rad) and specific primers (see Table S2). The relative abundance of transcripts was normalized to the expression of housekeeping genes (18S) using the 2−ΔCT method. For bulk RNA-sequencing (RNA-seq), naïve T cells of Slc2a3fl/flCd4Cre or WT mice were differentiated for 3 days into Th1 and Th17 cells and 1×106 cells were directly harvested in RNAprotect Cell reagent and stored at −80°C o/n before total RNA was extracted using the RNeasy Plus Micro Kit (both Quiagen). Alternatively, WT, Slc2a3fl/flCd4Cre and Aclyfl/flCd4Cre CD4+ T cells were differentiated for 3 days into Th17 cells. 1×106 cells were treated for 24 h with or without the addition of 10 mM acetate before 5 h re-stimulation with PMA/Iono. Cell were directly harvested into RNAprotect Cell Reagent and total RNA was extracted using the RNeasy Plus Micro Kit (both Quiagen). RNA quality and quantity were checked using a 2100 Bioanalyzer with the RNA 6000 Nano kit (Agilent Technologies) and RNA Pico kit depending on the sample concentration. The RIN for all samples was >7. cDNA libraries suitable for sequencing were prepared from 5 ng of total RNA with SMART-Seq v4 Ultra Low Input RNA Kit (Takara) according to manufacturer’s instructions (1/4 volume) or 500 ng of total RNA with TruSeq mRNA Stranded Library Prep Kit from Illumina according to manufacturer’s instructions (1/2 volume). Libraries were quantified by QubitTM 3.0 Fluometer (Thermo Scientific) and quality was checked using 2100 Bioanalyzer (Agilent) with High Sensitivity DNA kit (Agilent). 0.5 ng of each library was subjected to a tagmentation-based protocol (Nextera XT, Illumina) using a quarter of the recommended reagent volumes. Libraries were quantified again by QubitTM 3.0 Fluometer (Thermo Scientific) and quality was checked using 2100 Bioanalyzer with High Sensitivity DNA kit (Agilent) before pooling. In both experiments, sequencing of pooled libraries, spiked with 1% PhiX control library, was performed at 19-36 million reads/sample in single-end mode with 75 nt read length on the NextSeq 500 platform (Illumina) with 1 High Output Kit v2.5. Demultiplexed FASTQ files were generated with bcl2fastq2 v2.20.0.422 (Illumina). To assure high sequence quality, Illumina reads were quality- and adapter-trimmed via Cutadapt [1] version 2.5 using a cutoff Phred score of 20 in NextSeq mode and reads without any remaining bases were discarded (command line parameters: -- nextseq-trim=20 -m 1 -a CTGTCTCTTATACACATCT). Processed reads were subsequently mapped to the mouse genome (GRCm38.p6 primary assembly and mitochondrion) using STAR v2.7.2b with default parameters based on RefSeq annotation version 108.20200622 for GRCm38.p6 [2]. Read counts on exon level summarized for each gene were generated using featureCounts v1.6.4 from the Subread package [3]. Multi-mapping and multi-overlapping reads were counted non-strand-specific with a fractional count for each alignment and overlapping feature (command line parameters: -s 0 -t exon -M - O --fraction). The count output was utilized to identify differentially expressed genes using DESeq2 [4] version 1.24.0. Read counts were normalized by DESeq2 and fold-change shrinkage was applied by setting the parameter “betaPrior=TRUE”. Differences in gene expression were considered significant if padj < 0.05. The DEseq2 data was further analyzed by gene set enrichment analysis (GSEA) and visualized in Cytoscape using the plugin ErichmentMap in edge cutoff 0.5. Pathways were filtered and displayed when p value < 0.005 and Q value < 0.1.
Immunoblotting
Cell lysates for immunoblotting were directly lysed in Laemmli buffer (Roth) and sonicated for 30 min on ice or in RIPA buffer (SantaCruz) supplemented with complete protease-inhibitor cocktail (Roche). Protein quantification was performed with the Pierce 660 nm Protein Assay (Pierce). Alternatively, samples were prepared in RIPA buffer and mixed in equal amounts with Laemmli buffer (Roth) before denaturation at 98°C for 5 min. Protein samples were separated via SDS-PAGE, blotted onto nitrocellulose membranes, blocked with 5% BSA and incubated with the primary antibodies (see Key Resources Table). Detection was carried out with goat anti-mouse or anti-rabbit horse radish peroxidase (HRP) conjugated secondary antibodies (BioRad), visualized with the enhanced chemiluminescent SuperSignal reagent (Pierce) and detected using a digital ChemiDoc imaging system (BioRad).
Experimental autoimmune encephalomyelitis (EAE)
To induce EAE in mice, 2 mg/ml MOG35–55 peptide (Synpeptide) was emulsified in IFA (BD) supplemented with 5 mg/ml M. tuberculosis H37Ra (Fisher Scientific) by syringe extrusion. In total 200 mg MOG35–55 peptide was subcutaneously (s.c.) injected into two different sites on the lower flanks of the mice, followed by intraperitoneal injection of 250 ng pertussis toxin (Enzo) on day 0 and 2. Mice were monitored daily for clinical signs of EAE and weight loss. The scores were assigned as described before (Stromnes and Goverman, 2006). In brief, score 0: no paralysis, score 0,5: partially limp tail, score 1: paralyzed tail, score 2: uncoordinated movement, score 2,5: paralysis of one hind limb, score 3: paralysis of both hind limbs, score 3,5: weakness in forelimbs, score 4: paralysis of forelimbs and score 5: moribund. Where indicated, mice were injected daily with 500 mg/kg 2-HC, starting after the disease onset. At the end of the experiment, mice were sacrificed and cell suspensions from the spleen, LNs and the spinal cords were prepared. Briefly, spinal cords were isolated, mined into small pieces and digested with 1 mg/ml collagenase D (Roche) and 20 μg/ml DNase I (Thermo Scientific) for 40 min at 37°C. After filtering through a 70 μm cell strainer, CNS-infiltrating lymphocytes were isolated by percoll gradient (30:70) centrifugation. To induce passive EAE, CD4+ T cells from 2D2 mice were differentiated into pathogenic Th17 cells with or without 5 mM 2-HC for 4 days. 1×105 Th17 cells were injected i.p. into Rag1−/− host mice and injected s.c. with MOG35–55 peptide and 250 ng pertussis toxin i.p. at day 0 and 2 (Enzo).
Adoptive transfer colitis
For the induction of colitis, naïve CD4+CD25−CD45RBhigh T cells from were isolated from the spleens of WT or Slc2a3fl/flCd4Cre littermate mice using a BD FACSAria III cell sorter as previously described (Vaeth et al., 2017b). 5x105 FACS-sorted T cells were transferred intraperitoneally into lymphopenic Rag1−/− recipient mice and initiation of autoimmune colitis was monitored over the course of 8 weeks by analyzing weight loss of the recipient animals, stool samples and clinical signs of colonic inflammation, such as rectal prolapses. At the end of the experiment, mice were sacrificed before spleen, mesenteric LNs, small intestine and colon were extracted. For the isolation of lamina propria lymphocytes from the small intestine and the colon, tissues were washed with PBS and the intraepithelial fraction was dissociated by adding twice Ca2+ and Mg2+-free HBSS (Sigma) supplemented with 5 mM EDTA (Thermo Scientific) and 10 mM HEPES (Gibco) for 20 min at 37°C shaking. After rinsing with PBS, both tissues were minced into small pieces and digested with HBSS containing 500 μg/ml collagenase D (Roche), 20 μg/ml DNase I (Thermo Scientific) and 0.5 U/ml Dispase (BD) for 45 min at 37 °C shaking. Samples were filtered through 70 μm cell strainers and lymphocyte fractions were concentrated by percoll gradient (40% vs. 80%) centrifugation. Cell suspensions of spleen, mesenteric LNs, small intestine and colon were stimulated with PMA and ionomycin for 4 h and analyzed by flow cytometry to analyze surface marker, transcription factors and cytokine expression.
Mixed bone marrow chimeras
To generate mixed BM chimeras, sub-lethally irradiated Rag2−/−μc−/− host mice were injected retro-orbitally with 5x106 CD45.1+ WT together with 5x106 CD45.2+ Slc2a3fl/flCd4Cre BM cells. Eight weeks after BM reconstitution, lymphocytes from the spleen, LNs, lungs, liver small intestine and colon were enriched as described before (Vaeth et al., 2017b), stimulated with PMA and ionomycin and analyzed by flow cytometry as described above.
Citrobacter rodentium and LCMV infections
Single colonies were generated from a glycerol stock of C. rodentium (strain ICC180) grown on kanamycin LB agar plates. A single colony was picked and cultured in 5 ml LB medium shaking at 37 °C o/n. The next day, 45 ml fresh LB medium was added, and bacteria were grown until they reached an OD600 nm between 0.8 and 1. Bacteria were washed once with PBS and 5x109 CFU in 200 μl PBS were injected i.p. into WT and Slc2a3fl/flCd4Cre mice. The weight change of the mice and the bacterial load of the feces were monitored over 35 days. Cytokine expression of T cells was analyzed by flow cytometry at day 10 post infection. Lymphocytic choriomeningitis virus (LCMV) infections of WT and Slc2a3fl/flCd4Cre mice were performed as previously described (Vaeth et al., 2017b).
QUANTIFICATION AND STATISTICAL ANALYSIS
The results are shown as mean ± standard error of the means (SEM). To determine the statistical significance of the differences between the experimental groups unpaired Student’s t tests were performed using the Prism 9 software (GraphPad). Sample sizes were based on experience and experimental complexity but no methods were used to determine normal distribution of the samples. Differences reached significance with p values < 0.05 (noted in figures as *), p < 0.01 (**) and p < 0.001 (***). The figure legends contain the number of independent experiments or mice per group that were used in the respective experiments.
Supplementary Material
Data S1. Unprocessed source data underlying all blots and graphs. Related to Figures 1–7 and Supplemental Figures 1–7.
HIGHLIGHTS.
GLUT3 controls the effector function of pathogenic Th17 cells
Ablation of GLUT3 in T cells prevents Th17 cell-mediated autoimmunity
GLUT3-dependent acetyl-CoA production controls the epigenetic program of T cells
Pharmacological inhibition of acetyl-CoA generation ameliorates autoimmunity
ACKNOWLEDGEMENTS
We thank Kathryn E. Wellen (University of Pennsylvania, USA), Elisabeth Sock (University of Erlangen, Germany), Alma Zernecke-Madsen, Friederike Berberich-Siebelt, Niklas Beyersdorf and Kristen Rak (all University of Würzburg, Germany) for providing Aclyfl/fl, Ppp3r1fl/fl, Hif1afl/fl, Irf4fl/fl, Cd28−/− and Stat3fl/fl mice, respectively. SorA was kindly provided by Rolf Müller (Helmholtz Center for Infection Research, Germany). This work was supported by the Deutsche Forschungsgemeinschaft (DFG) SFB-TR 124/3 2013, project 210879364, SFB-TR 338/1 2021, project 452881907, SFB 1526/1 2022, project 413535489 and project grant VA882/2-1 (to M.V.), NIH U54 HL112311 (to E.D.A.) and the Interdisciplinary Center for Clinical Research (IZKF, supporting the Core Units FACS and SysMed). We would like to thank Camila Takeno Cologna, Wesley Vermaelen and Anton Willems (VIB Metabolomics Expertise Center), Sheyda Hajiesmaeili, Thorsten Bischler and Christian Linden (IZKF Würzburg) for excellent technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERRSTS
The authors declare no competing interests.
REFERENCES
- Akimzhanov AM, Yang XO, and Dong C (2007). Chromatin remodeling of interleukin-17 (IL-17)-IL-17F cytokine gene locus during inflammatory helper T cell differentiation. J Biol Chem 282, 5969–5972. [DOI] [PubMed] [Google Scholar]
- Alwarawrah Y, Kiernan K, and MacIver NJ (2018). Changes in Nutritional Status Impact Immune Cell Metabolism and Function. Front Immunol 9, 1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailis W, Shyer JA, Zhao J, Canaveras JCG, Al Khazal FJ, Qu R, Steach HR, Bielecki P, Khan O, Jackson R, et al. (2019). Distinct modes of mitochondrial metabolism uncouple T cell differentiation and function. Nature 571, 403–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckermann KE, Hongo R, Ye X, Young K, Carbonell K, Healey DCC, Siska PJ, Barone S, Roe CE, Smith CC, et al. (2020). CD28 costimulation drives tumor-infiltrating T cell glycolysis to promote inflammation. JCI Insight 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, Sandouk A, Hesse C, Castro CN, Bahre H, et al. (2014). De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med 20, 1327–1333. [DOI] [PubMed] [Google Scholar]
- Bevington SL, Cauchy P, Piper J, Bertrand E, Lalli N, Jarvis RC, Gilding LN, Ott S, Bonifer C, and Cockerill PN (2016). Inducible chromatin priming is associated with the establishment of immunological memory in T cells. EMBO J 35, 515–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuah LO, Ho WY, Beh BK, and Yeap SK (2013). Updates on Antiobesity Effect of Garcinia Origin (−)-HCA. Evid Based Complement Alternat Med 2013, 751658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciofani M, Madar A, Galan C, Sellars M, Mace K, Pauli F, Agarwal A, Huang W, Parkhurst CN, Muratet M, et al. (2012). A validated regulatory network for Th17 cell specification. Cell 151, 289–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Custodio TF, Paulsen PA, Frain KM, and Pedersen BP (2021). Structural comparison of GLUT1 to GLUT3 reveal transport regulation mechanism in sugar porter family. Life Sci Alliance 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng D, and Yan N (2016). GLUT, SGLT, and SWEET: Structural and mechanistic investigations of the glucose transporters. Protein Sci 25, 546–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo Y, Asou HK, Matsugae N, Hirahara K, Shinoda K, Tumes DJ, Tokuyama H, Yokote K, and Nakayama T (2015). Obesity Drives Th17 Cell Differentiation by Inducing the Lipid Metabolic Kinase, ACC1. Cell Rep 12, 1042–1055. [DOI] [PubMed] [Google Scholar]
- Etchegaray JP, and Mostoslavsky R (2016). Interplay between Metabolism and Epigenetics: A Nuclear Adaptation to Environmental Changes. Mol Cell 62, 695–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidler TP, Middleton EA, Rowley JW, Boudreau LH, Campbell RA, Souvenir R, Funari T, Tessandier N, Boilard E, Weyrich AS, et al. (2017). Glucose Transporter 3 Potentiates Degranulation and Is Required for Platelet Activation. Arterioscler Thromb Vasc Biol 37, 1628–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton JA (2019). GM-CSF-Dependent Inflammatory Pathways. Front Immunol 10, 2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirahara K, Onodera A, Villarino AV, Bonelli M, Sciume G, Laurence A, Sun HW, Brooks SR, Vahedi G, Shih HY, et al. (2015). Asymmetric Action of STAT Transcription Factors Drives Transcriptional Outputs and Cytokine Specificity. Immunity 42, 877–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Wang Y, Hao LY, Liu X, Lesch CA, Sanchez BM, Wendling JM, Morgan RW, Aicher TD, Carter LL, et al. (2015). Sterol metabolism controls T(H)17 differentiation by generating endogenous RORgamma agonists. Nat Chem Biol 11, 141–147. [DOI] [PubMed] [Google Scholar]
- Kaufmann U, Kahlfuss S, Yang J, Ivanova E, Koralov SB, and Feske S (2019). Calcium Signaling Controls Pathogenic Th17 Cell-Mediated Inflammation by Regulating Mitochondrial Function. Cell Metab 29, 1104–1118 e1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein-Hessling S, Muhammad K, Klein M, Pusch T, Rudolf R, Floter J, Qureischi M, Beilhack A, Vaeth M, Kummerow C, et al. (2017). NFATc1 controls the cytotoxicity of CD8(+) T cells. Nat Commun 8, 511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, and Dong C (2016). Therapeutic antibodies that target inflammatory cytokines in autoimmune diseases. Int Immunol 28, 181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, Wu C, Kleinewietfeld M, Kunder S, Hafler DA, et al. (2012). Induction and molecular signature of pathogenic TH17 cells. Nat Immunol 13, 991–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, Anderson SM, Abel ED, Chen BJ, Hale LP, et al. (2014). The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab 20, 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man K, Miasari M, Shi W, Xin A, Henstridge DC, Preston S, Pellegrini M, Belz GT, Smyth GK, Febbraio MA, et al. (2013). The transcription factor IRF4 is essential for TCR affinity-mediated metabolic programming and clonal expansion of T cells. Nat Immunol 14, 1155–1165. [DOI] [PubMed] [Google Scholar]
- Manzel A, Muller DN, Hafler DA, Erdman SE, Linker RA, and Kleinewietfeld M (2014). Role of “Western diet” in inflammatory autoimmune diseases. Curr Allergy Asthma Rep 14, 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez GJ, Pereira RM, Aijo T, Kim EY, Marangoni F, Pipkin ME, Togher S, Heissmeyer V, Zhang YC, Crotty S, et al. (2015). The transcription factor NFAT promotes exhaustion of activated CD8(+) T cells. Immunity 42, 265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matveeva O, Bogie JFJ, Hendriks JJA, Linker RA, Haghikia A, and Kleinewietfeld M (2018). Western lifestyle and immunopathology of multiple sclerosis. Ann N Y Acad Sci 1417, 71–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miossec P, and Kolls JK (2012). Targeting IL-17 and TH17 cells in chronic inflammation. Nat Rev Drug Discov 11, 763–776. [DOI] [PubMed] [Google Scholar]
- Mognol GP, Spreafico R, Wong V, Scott-Browne JP, Togher S, Hoffmann A, Hogan PG, Rao A, and Trifari S (2017). Exhaustion-associated regulatory regions in CD8(+) tumor-infiltrating T cells. Proc Natl Acad Sci U S A 114, E2776–E2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueckler M, and Thorens B (2013). The SLC2 (GLUT) family of membrane transporters. Mol Aspects Med 34, 121–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobs SP, Zmora N, and Elinav E (2020). Nutrition Regulates Innate Immunity in Health and Disease. Annu Rev Nutr 40, 189–219. [DOI] [PubMed] [Google Scholar]
- O’Neill LA, Kishton RJ, and Rathmell J (2016). A guide to immunometabolism for immunologists. Nat Rev Immunol 16, 553–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, and Li MO (2016). Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 354, 481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, and Kroemer G (2015). Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab 21, 805–821. [DOI] [PubMed] [Google Scholar]
- Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M, et al. (2014). CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159, 440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu R, Yu X, Wang L, Han Z, Yao C, Cui Y, Hou G, Dai D, Jin W, and Shen N (2020). Inhibition of Glycolysis in Pathogenic TH17 Cells through Targeting a miR -21-Peli1-c-Rel Pathway Prevents Autoimmunity. J Immunol 204, 3160–3170. [DOI] [PubMed] [Google Scholar]
- Sharma M, Kaveri SV, and Bayry J (2013). Th17 cells, pathogenic or not? TGF-beta3 imposes the embargo. Cell Mol Immunol 10, 101–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter D, Dormann HL, Allis CD, and Hake SB (2007). Extraction, purification and analysis of histones. Nat Protoc 2, 1445–1457. [DOI] [PubMed] [Google Scholar]
- Shen H, and Shi LZ (2019). Metabolic regulation of TH17 cells. Mol Immunol 109, 81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberger DJ, Zindl CL, and Weaver CT (2017). Citrobacter rodentium: a model enteropathogen for understanding the interplay of innate and adaptive components of type 3 immunity. Mucosal Immunol 10, 1108–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson IA, Dwyer D, Malide D, Moley KH, Travis A, and Vannucci SJ (2008). The facilitative glucose transporter GLUT3: 20 years of distinction. Am J Physiol Endocrinol Metab 295, E242–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng X, Li W, Cornaby C, and Morel L (2019). Immune cell metabolism in autoimmunity. Clin Exp Immunol 197, 181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theofilopoulos AN, Kono DH, and Baccala R (2017). The multiple pathways to autoimmunity. Nat Immunol 18, 716–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaeth M, Kahlfuss S, and Feske S (2020). CRAC Channels and Calcium Signaling in T Cell-Mediated Immunity. Trends Immunol 41, 878–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaeth M, Maus M, Klein-Hessling S, Freinkman E, Yang J, Eckstein M, Cameron S, Turvey SE, Serfling E, Berberich-Siebelt F, et al. (2017a). Store-Operated Ca(2+) Entry Controls Clonal Expansion of T Cells through Metabolic Reprogramming. Immunity 47, 664–679 e666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaeth M, Yang J, Yamashita M, Zee I, Eckstein M, Knosp C, Kaufmann U, Karoly Jani P, Lacruz RS, Flockerzi V, et al. (2017b). ORAI2 modulates store-operated calcium entry and T cell-mediated immunity. Nat Commun 8, 14714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vodnala SK, Eil R, Kishton RJ, Sukumar M, Yamamoto TN, Ha NH, Lee PH, Shin M, Patel SJ, Yu Z, et al. (2019). T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, et al. (2011). The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Tao A, Vaeth M, and Feske S (2020). Calcium regulation of T cell metabolism. Curr Opin Physiol 17, 207–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Yin W, Zhu L, Li J, Yao Y, Chen F, Sun M, Zhang J, Shen N, Song Y, et al. (2018). Iron Drives T Helper Cell Pathogenicity by Promoting RNA-Binding Protein PCBP1-Mediated Proinflammatory Cytokine Production. Immunity 49, 80–92 e87. [DOI] [PubMed] [Google Scholar]
- Warburg O (1956). On the origin of cancer cells. Science 123, 309–314. [DOI] [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, and Thompson CB (2009). ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Yin N, Peng M, Stamatiades EG, Chhangawala S, Shyu A, Li P, Zhang X, Do MH, Capistrano KJ, et al. (2021a). Glycolytic ATP fuels phosphoinositide 3-kinase signaling to support effector T helper 17 cell responses. Immunity 54, 976–987 e977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Yin N, Peng M, Stamatiades EG, Shyu A, Li P, Zhang X, Do MH, Wang Z, Capistrano KJ, et al. (2021b). Glycolysis fuels phosphoinositide 3-kinase signaling to bolster T cell immunity. Science 371, 405–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Jin W, Wu R, Li J, Park SA, Tu E, Zanvit P, Xu J, Liu O, Cain A, et al. (2019). High Glucose Intake Exacerbates Autoimmunity through Reactive-Oxygen-Species-Mediated TGF-beta Cytokine Activation. Immunity 51, 671–681 e675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Torres A, Henry RA, Trefely S, Wallace M, Lee JV, Carrer A, Sengupta A, Campbell SL, Kuo YM, et al. (2016). ATP-Citrate Lyase Controls a Glucose-to-Acetate Metabolic Switch. Cell Rep 17, 1037–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Unprocessed source data underlying all blots and graphs. Related to Figures 1–7 and Supplemental Figures 1–7.
Data Availability Statement
The RNA-seq and ChIP-seq datasets have been deposited at GEO under accession numbers GSE192659, GSE192663 and GSE193515. Original Western blots and values for creating the graphs in the paper are provided in Data S1.
This study did not generate any code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request.