

Abstract
Activation of the Stimulator of Interferon Gene (STING) pathway within the tumor microenvironment has been shown to generate a strong antitumor response. While local administration of STING agonists has promise for cancer immunotherapy, the dosing regimen needed to achieve efficacy requires frequent intratumoral injections over months. Frequent dosing for cancer treatment is associated with poor patient adherence, with as high as 48% of patients failing to comply. Multiple intratumoral injections also disrupt the tumor microenvironment and vascular networks and therefore increase the risk of metastasis. Here, we developed microfabricated polylactic-co-glycolic acid (PLGA) particles that remain at the site of injection and release encapsulated STING agonist as a programmable sequence of pulses at pre-determined time points that mimic multiple injections over days to weeks. A single intratumoral injection of STING agonist-loaded microparticles triggered potent local and systemic antitumor immune responses, inhibited tumor growth, and prolonged survival as effectively as multiple soluble doses, but with reduced metastasis in several mouse tumor models. STING agonist-loaded microparticles improved the response to immune checkpoint blockade therapy and substantially decreased the tumor recurrence rate from 100% to 25% in mouse models of melanoma when administered during surgical resection. In addition, we demonstrated the therapeutic efficacy of STING microparticles on an orthotopic pancreatic cancer model in mice that does not allow multiple intratumoral injections. These findings could directly benefit current STING agonist therapy by decreasing the number of injections, reducing risk of metastasis, and expanding its applicability to hard-to-reach cancers.
One Sentence Summary:
Microfabricated particles mimic multiple intratumoral injections of STING agonist over weeks to improve ease of use and decrease metastasis.
Introduction
The advent of immune checkpoint blockade (ICB) has had a profound impact on cancer treatment, with several drugs receiving approval from the United States Food and Drug Administration (FDA) (1). Despite great promise, the clinical benefits of ICB remain limited by a low response rate (2). Clinical studies have shown that patients who respond to ICB have higher amounts of tumor-infiltrating lymphocytes (TILs) and display a signature of type I interferon (IFN) producing genes indicative of innate immune system activation (3–5). Therefore, strategies to improve TIL infiltration and innate immune system activation have been proposed as combination therapies to further improve the response rate of ICB. Among many innate immune pathways that are initiated through toll-like receptors (TLRs), mitochondrial antiviral-signaling protein (MAVS), or P2X purinergic receptor 7, activation of stimulator of interferon genes (STING) shows great promise for increasing TILs and improving the antitumor efficacy of ICB (6, 7). STING pathway activation is initiated through recognition of cytoplasmic DNA. Cyclic guanosine monophosphate–adenosine monophosphate (cGAMP) synthase senses cytoplasmic DNA and produces the second messenger cGAMP, which then binds to STING to trigger a signaling cascade through tank-binding kinase 1 (TBK1)/interferon regulatory factor 3 (IRF3) for production of type I IFNs and other cytokines (8, 9). Substantial evidence has shown that intratumoral injections of STING agonist stimulate potent antitumor immunity in clinically relevant tumor models (7, 10–13). As a result, phase I clinical trials (NCT02675439, NCT03172936, and NCT03010176) using STING agonist alone or in combination with ICB are currently under way to treat patients with advanced solid tumors and lymphoma.
The dosing regimen of STING agonist in current clinical trials consists of multiple intratumoral injections administered repeatedly (for example, 3 injections over a 28-day period or 1 injection every week for 9 weeks per treatment cycle) for as long as two years to achieve therapeutic efficacy (14, 15). Such high dosing frequency over a long period of time can cause chronic injection pain, increase the risk of infection, and ultimately result in poor adherence, especially when every dose requires a health care visit (16–18). The adherence rates to cancer treatment are as low as ~52%, with similar rates (~50%) reported for patients with other chronic diseases (19, 20). Poor adherence can result in failed treatment and constitutes a financial burden of approximately $100 billion each year in the United States alone (21). In addition, multiple intratumoral injections limit the scope of STING agonist-based therapies to readily accessible tumor types and introduce the risk of disrupting the tumor microenvironment (TME) and vascular network, potentially promoting cancer cell extravasation and metastases (22–24). Therefore, a delivery system that mimics current clinical dosage regimens within a single injection is an attractive solution to improve patient adherence, decrease risk of metastasis and therapeutic cost, and expand the scope of current STING agonist-based therapies.
Here, we developed such a multidose drug delivery platform through engineering polylactic-co-glycolic acid (PLGA), an FDA-approved and commercially available polymer, into cubic microparticles (Fig. 1A). Unlike commonly used local drug delivery materials, such as hydrogels or microparticles produced using a double emulsion-solvent evaporation technique, which exhibit sustained drug release kinetics (25–28), these microfabricated PLGA particles (PLGA-MPs) released individual doses of encapsulated STING agonist in pulses for up to several months with essentially no leakage. We demonstrated that a single injection containing multiple populations of STING agonist-loaded PLGA-MPs inhibited tumor growth and improved survival as effectively as multiple injections of soluble STING agonist in several mouse tumor models. The ability to combine multiple doses into single injection PLGA-MPs also decreased metastasis and expanded the potential applications of current STING agonist-based therapy to hard-to-reach tumors.
Figure 1. Design and fabrication of PLGA-MPs.
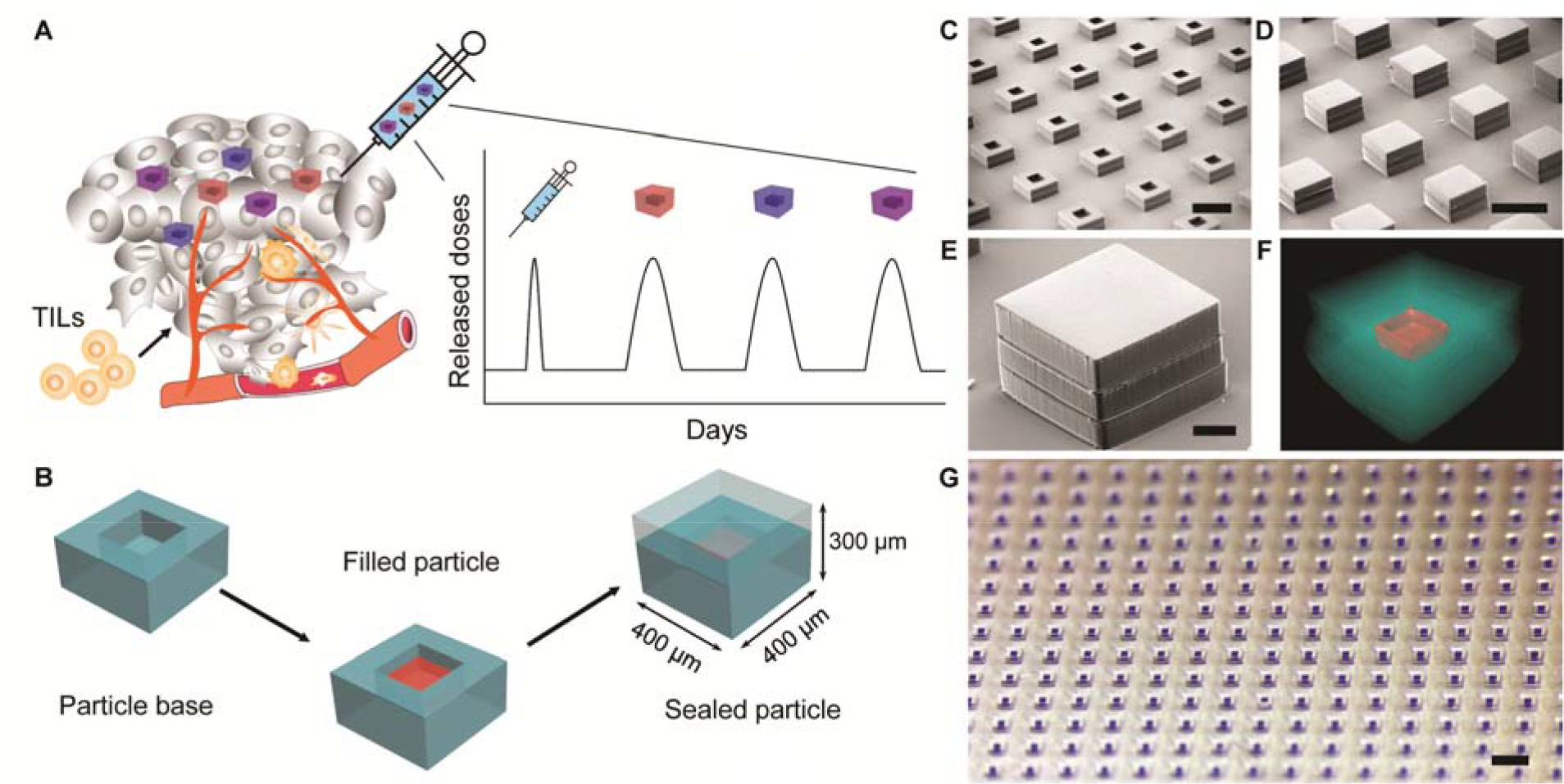
A. Schematics of single injection drug delivery platform for cancer immunotherapy. Different PLGA microparticles reside in the tumor after a single intratumoral injection, release encapsulated STING agonist in pulses at discrete time points, and promote infiltration of tumor-infiltrating lymphocytes (TILs). B. Schematics of the fabrication process of PLGA-MPs, which are prepared by filling cargo of interest into particle bases and then sealing the bases with corresponding particle caps by briefly applying heat. C, D, E. Representative scanning electron microscopy images of empty particle bases (C) and sealed array of particles (D) or an individual particle (E). Scale bars are 500 μm in C and D, and 100 μm in E. F. Representative high resolution X-ray computed tomography image of sealed particle encapsulating 3’3’-cGAMP. Red color represents dried 3’3’-cGAMP. Scale bar is 100 μm. G. Representative optical image of an array of sealed particles encapsulating Alexa Fluor 647-labeled dextran. Scale bar is 1 mm.
Results
Fabrication of PLGA microparticles with different release kinetics
We used soft lithography techniques to fabricate arrays of cubic PLGA microparticles with a fully enclosed cavity for drug loading (Fig. 1B). Briefly, PLGA was heated and pressed into a polydimethylsiloxane (PDMS) mold to form microparticle bases with internal cavities (200 × 200 × 100 μm by length × width × height), which corresponds to a volumetric capacity of 4 nL. We then filled aqueous drug or model drug into these bases using a piezoelectric dispenser that can dispense volumes on the order of 100 picoliter (pL). The water component of the drug solutions evaporated rapidly due to the small volume, which provided space for filling additional cargo. Multiple filling and drying cycles were used to achieve maximum cargo loading. Filled microparticles were then aligned with PLGA caps embedded in a PDMS mold and sealed by heating above the glass transition temperature (approximately 50 °C) of PLGA. Sealed microparticles have external dimensions of 400 × 400 × 300 μm (length × width × height) and wall thickness of 100 μm in each dimension. The loading capacity of each microparticle is 8.4% by volume. Scanning electron microscopy, high resolution X-ray computed tomography (CT), and optical images showed that microparticles can be made in large arrays (over 300 per array) with high fidelity (Figs. 1C to G, and fig. S1).
To study the release kinetics, we filled a fluorescently labeled macromolecule, Alexa Fluor 647-labeled dextran (AF647-dextran), into PLGA microparticles with different polymer properties (table S1). These microparticles were sealed with corresponding caps and incubated in phosphate-buffered saline (PBS, pH = 6.84) at 37 °C to mimic the acidic TME. PLGA microparticles released AF647-dextran in pulses at approximately 1 ± 0, 4 ± 0, 8 ± 0, 11 ± 1, 15 ± 1, 18 ± 1, and 97 ± 2 days without detectable leakage prior to release (fig. S2). To mimic the dosing regimen of four consecutive doses with 3 to 4 days between each dose that has been shown to be effective at inhibiting tumor growth in animal models (7), microparticles that release AF647-dextran at day 4 (PLGA-1), day 8 (PLGA-2), and day 11 (PLGA-3) were selected for further study (Fig. 2A). To validate the release kinetics in vivo, we subcutaneously injected AF647-dextran-loaded PLGA-1, 2, and 3 into hairless mice. The release of AF647-dextran was monitored by in vivo fluorescence imaging (IVIS). Released AF647-dextran showed an over 100-fold increase in fluorescence intensity compared to encapsulated AF647-dextran in particles due to the self-quenching effects of the fluorophore when dry or at an extremely high local concentration (fig. S3). Fig. 2B and fig. S4 showed that microparticles released AF647-dextran in vivo with similar release times as in vitro. The average releasing times of PLGA-1, 2, and 3 in vivo were 3.9 ± 1.1, 8.1 ± 1.5, and 11.5 ± 1.4 days, respectively (fig. S5).
Figure 2. Release kinetics of PLGA-MPs.
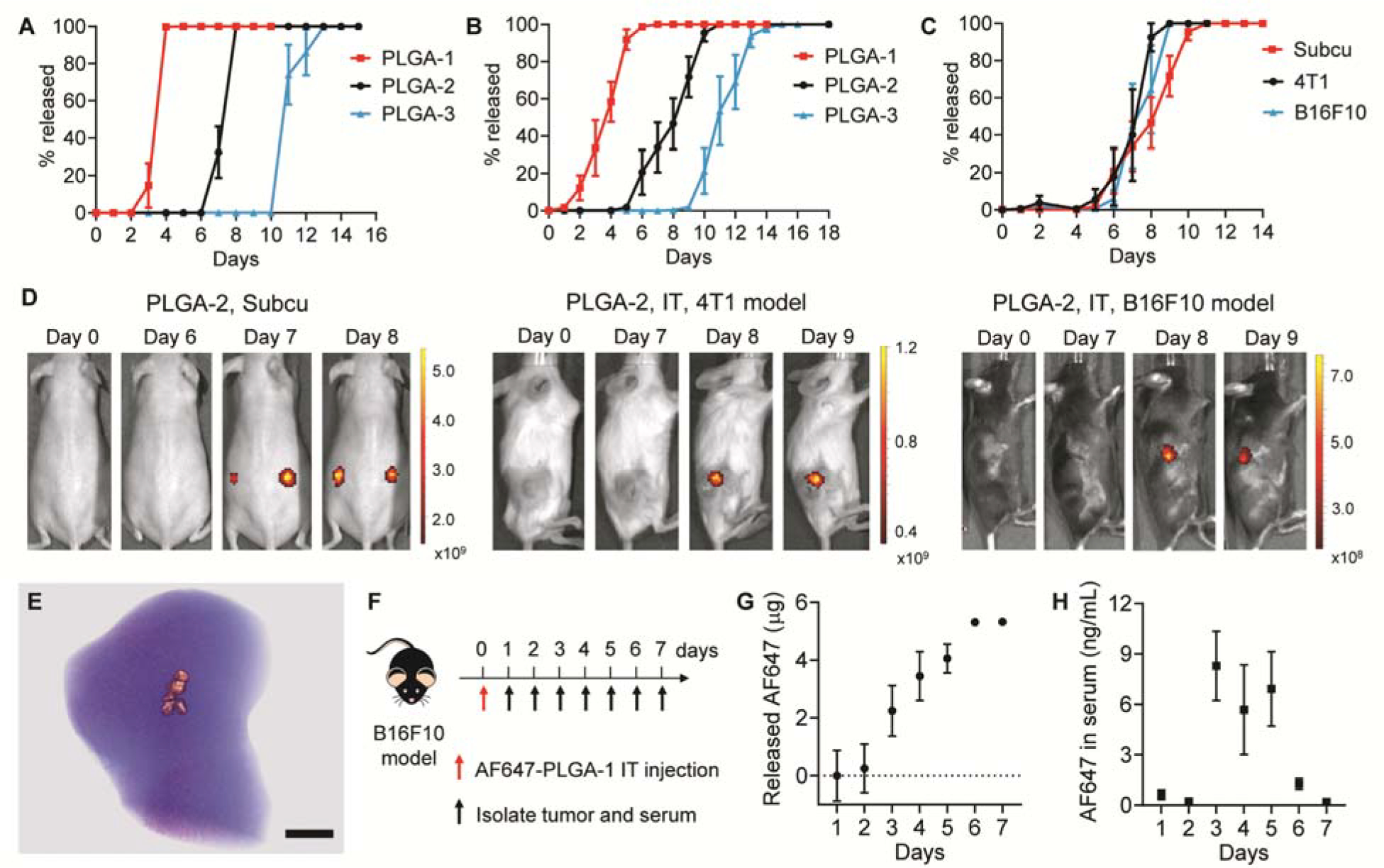
A, B. Cumulative in vitro (A) and in vivo (B) release kinetics of AF647-dextran from PLGA-1, 2, and 3. PLGA-MPs were administered subcutaneously (n = 6–8). Data are shown as mean ± SEM. C, D. Cumulative in vivo release kinetics (C) and representative fluorescence images (D) of AF647-dextran-loaded PLGA-2 that were administered subcutaneously in SKH1E hairless mice (n = 8) or intratumorally in B16F10 melanoma model (n = 4) and 4T1 breast cancer model (n = 4). Data are shown as mean ± SEM. Scale bars represent radiant efficiency. E. MicroCT image of the B16F10 tumor, which was isolated from mice 1 h after injection of 5% phosphotungstic acid-doped PLGA-1. Scale bar is 2 mm. F. Treatment and sampling schedule of B16F10 tumor-bearing mice after intratumoral injection of AF647-loaded PLGA-1. G. Cumulative in vivo release of AF647 from PLGA-1 in B16F10 tumors (n = 4). H. AF647 concentration in serum after intratumoral injection of AF647-loaded PLGA-1 (n = 4). Data are shown as mean ± SEM.
We then evaluated the influence of different TMEs on release kinetics. AF647-dextran-loaded PLGA-2 was intratumorally injected into mice bearing B16F10 melanoma or 4T1 breast tumors. Release kinetics were then monitored by daily IVIS imaging. PLGA-2 showed consistent release kinetics in both tumors and the subcutaneous environment (Figs. 2C and 2D). To study the distribution of microparticles in tumors, we doped 5% phosphotungstic acid (PTA) in PLGA-1 and imaged the tumor using MicroCT. The particles were successfully injected into tumors and aggregated at the injection site due to the low mobility in confined environment (Fig. 2E). To further demonstrate that PLGA-MPs released all encapsulated cargo during the release window, we fabricated AF647-loaded PLGA-1 and intratumorally injected particles into B16F10 tumor-bearing mice. AF647 is a small and hydrophilic molecule with a MW of 753.9, which is similar to that of cGAMP (MW 674.4). Free AF647 was rapidly cleared from tumors after intratumoral injection (>95% within 2.5 hours, fig. S6). We measured the amount of unreleased AF647 in tumors every day and back-calculated the amount of released AF647 (Fig. 2F). Fig. 2G shows that PLGA-1 completely released AF647 from 3 to 6 days in tumors. Some released AF647 also diffused into the blood stream, as demonstrated by increased AF647 concentration in serum from day 3 to day 6 (Fig. 2H). These data demonstrated that PLGA-MPs released all encapsulated cargos at anticipated time points in tumors.
We then fabricated STING agonist-loaded PLGA-1, 2, and 3 with a drug loading of 2 μg per particle. 3’3’-cGAMP, a linkage isomer of naturally produced 2’3’-cGAMP, is used here because of its enhanced stability against ecto-nucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1), which primarily hydrolyzes cGAMP (29). 3’3’-cGAMP was released from PLGA-1, 2, and 3 in pulses at nearly the same times as the fluorescent molecules in vitro (Fig. 3A). The stability of encapsulated cGAMP in physiological conditions is critical for the retention of bioactivity upon release. To study the stability of 3’3’-cGAMP in particles, we incubated cGAMP-loaded PLGA-2 particles in PBS at 37°C and analyzed the structure integrity of cGAMP in the supernatant over time by liquid chromatography-mass spectrometry (LC-MS). Released cGAMP showed identical elution time and molecular mass to those of standard 3’3’-cGAMP (Fig. 3B and fig. S7). The bioactivity of released cGAMP was also tested by an interferon regulatory factor (IRF)-reporter cell line (RAW-Lucia ISG cells). Released cGAMP from PLGA-2 maintained over 95% of bioactivity (Fig. 3C). Collectively, these data demonstrated that encapsulated cGAMP remained stable and could be completely released from PLGA microparticles.
Figure 3. Single injection of cGAMP-MPs inhibited tumor growth and prolonged animal survival.
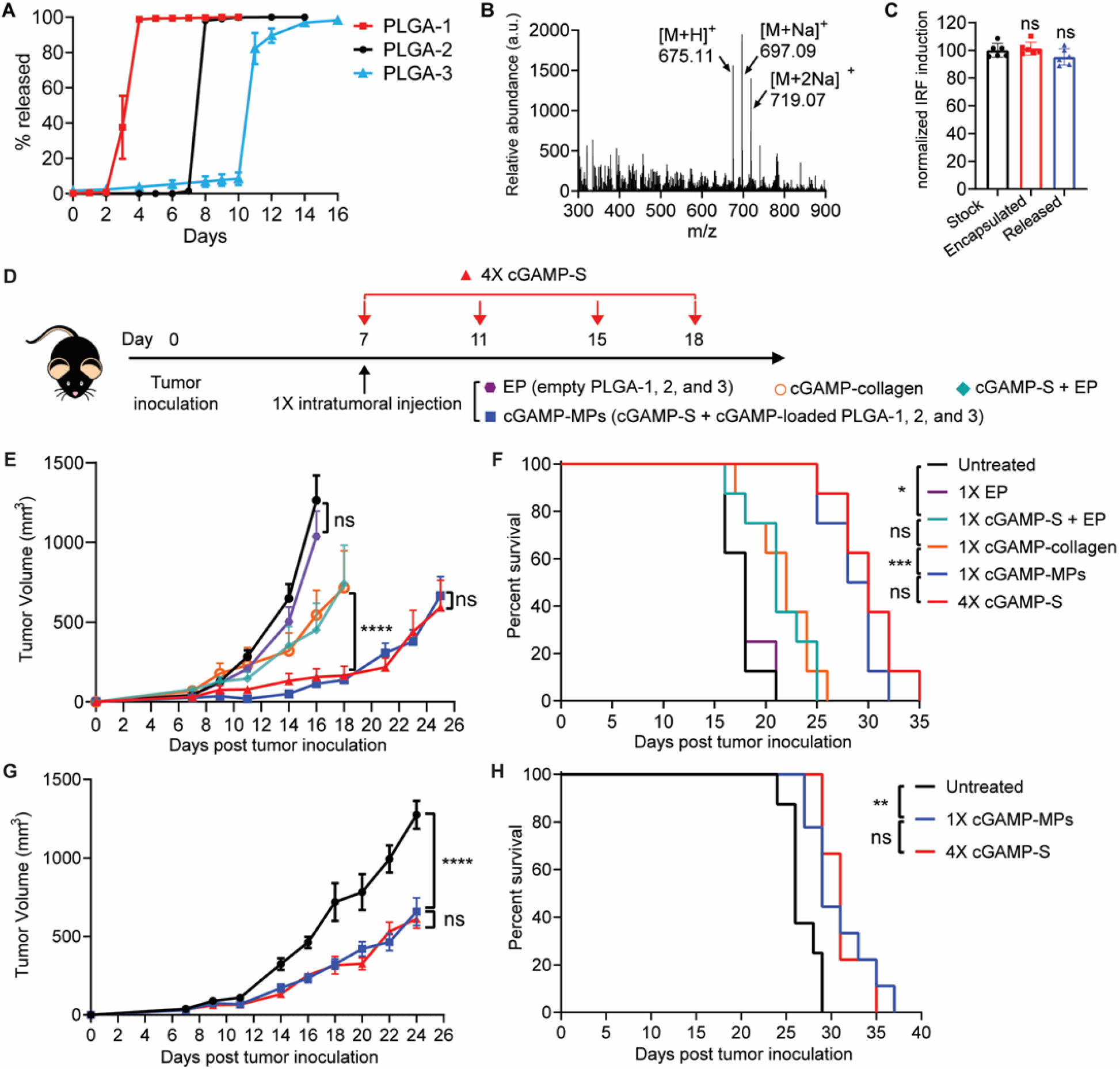
A. Cumulative in vitro release of 3’3’-cGAMP from PLGA-1, 2, and 3 (n = 6–8). Data are shown as mean ± SEM. B. Mass spectrum of 3’3’-cGAMP released from PLGA-2 on day 8 showed molecular ions [M+H]+ = 675.11, [M+Na]+ = 697.09, [M+2Na]+ = 719.07. Particles were incubated at 37 °C in PBS. C. Response of encapsulated cGAMP in PLGA-2 after sealing or released cGAMP from PLGA-2 (incubated at 37 °C in PBS for 8 days) on an interferon regulatory factor (IRF) reporter cell line (n = 6). Stock solution of cGAMP was used as a positive control. Data are shown as mean ± SD. Statistical significance was calculated using one-way analysis of variance (ANOVA). D. Treatment scheme of B16F10 melanoma and orthotopic 4T1 breast tumor models. Tumor-bearing mice were treated with a single intratumoral injection of EP, cGAMP-S+EP (40 μg cGAMP), cGAMP-collagen (40 μg cGAMP), or cGAMP-MPs (40 μg cGAMP: 10 μg in each of cGAMP-S, PLGA-1, 2, and 3) at day 7, or four intratumoral injections of soluble cGAMP at days 7, 11, 15, and 18 (4×cGAMP-S, 10 μg per injection) after tumor inoculation. E, F. Average tumor growth (E) and Kaplan-Meier survival curves (F) of B16F10 melanoma-bearing mice treated with different therapeutic combinations (n = 8 biologically independent samples). G, H. Average tumor growth curve (G) and survival analysis (H) of mice bearing orthotopic 4T1 breast tumors (n = 8 biologically independent samples). Statistical significance was calculated by two-way ANOVA and Tukey’s multiple comparisons test: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Data are shown as mean ± SEM.
Single injection of cGAMP-loaded particles effectively inhibited tumor growth
To test our hypothesis that a single injection of several timed-release populations of cGAMP-loaded PLGA-MPs could stimulate anti-tumor immunity comparable to multiple injections of soluble cGAMP (cGAMP-S), mice bearing poorly immunogenic B16F10 melanoma tumors were treated intratumorally with 1) a single injection of cGAMP-S (10 μg) combined with cGAMP-loaded PLGA-1, 2, and 3 microparticles (cGAMP-MPs, 10 μg cGAMP per formulation) to mimic four doses; 2) four injections of cGAMP-S (10 μg cGAMP per injection) administered at multiple time points corresponding to PLGA release (4×cGAMP-S); 3) a single intratumoral injection of empty PLGA-1, 2, and 3 microparticles (EP); or 4) a single intratumoral injection of high-dose cGAMP-S (40 μg) and EP (cGAMP-S+EP, Fig. 3D). Untreated mice were used as negative controls. To compare the therapeutic efficacy of cGAMP-MPs to other sustained release systems, we fabricated three sustained release formulations including dextran hydrogel, polyvinyl alcohol (PVA) hydrogel, and collagen hydrogel. The in vitro release kinetics study showed that over 99% of cGAMP was released from all three hydrogel formulations within 24 hours (fig. S8), which is consistent with previously reported sustained release systems for cGAMP (30, 31). Collagen gel, which exhibits the slowest release rate among tested gels, was loaded with 40 μg of cGAMP and intratumorally administered into tumor-bearing mice as controls (cGAMP-collagen). Tumors grew rapidly in untreated and EP-treated groups and all mice died within 21 days, indicating that PLGA microparticles alone did not inhibit tumor growth (Figs. 3E and 3F). A single injection of cGAMP-S+EP exhibited an antitumor effect at early time points but failed to achieve sustained tumor inhibition. The survival time was slightly extended from 21 days for untreated mice to 25 days, indicating the necessity of multiple doses for effective tumor inhibition. cGAMP-collagen did not show superior tumor inhibition or survival compared to cGAMP-S+EP. In contrast, cGAMP-MPs effectively inhibited tumor growth and prolonged animal survival with no statistical difference compared to 4×cGAMP-S at equivalent doses. Similar results were also observed in both orthotopic (Figs. 3G and 3H) and subcutaneous (fig. S9) triple-negative breast cancer model (4T1) with the same treatments. The systemic interleukin-6 (IL-6) response of orthotopic 4T1 tumor-bearing mice was evaluated from day 1 to 7. The IL-6 concentrations were increased in both cGAMP-MPs and 4×cGAMP-S treated groups compared to untreated group (fig. S10), suggesting the successful release of cGAMP from PLGA-MPs into tumors as well as into the bloodstream.
Single injection of cGAMP-MPs stimulated potent antitumor immunity
Next, we investigated the activation of STING pathway and antitumor immunity within the TME of B16F10 melanoma tumors. A combination of cGAMP-S, cGAMP-loaded PLGA-1, and cGAMP-loaded PLGA-2 microparticles (cGAMP-MPs) was injected intratumorally at day 7 after tumor inoculation to mimic a total of three doses (Fig. 4A). Mice treated with a single intratumoral injection of EP or multiple intratumoral injections of cGAMP-S at day 7, 11, and 15 were used as controls. cGAMP-MPs substantially inhibited tumor growth (fig. S11), which is consistent with our tumor inhibition findings. Tumors were isolated one day after the third cGAMP-S injection and analyzed by western blot and quantitative polymerase chain reaction (qPCR). cGAMP-MPs increased messenger RNA (mRNA) expression of interferon-stimulated genes (ISGs) Cxcl10 (6.8-fold over untreated) and Irf7 (58.5-fold over untreated), which was comparable to 3×cGAMP-S treated mice (7.2- and 66.5-fold increase of Cxcl10 and Irf7, Fig. 4B). Additionally, tumors treated with cGAMP-MPs and 3×cGAMP-S showed high phosphorylation of TBK1 (p-TBK1) and IRF3 (p-IRF3, Fig. 4C). Untreated and EP treated tumors did not exhibit detectable p-TBK1 and p-IRF3. These data demonstrate that cGAMP-MPs successfully activated the STING pathway and induced ISG production to an extent similar to that of multiple injections (32, 33). In contrast, empty particles did not trigger production of p-TBK1, p-IRF3, and ISGs.
Figure 4. A single injection of cGAMP-MPs activates STING pathway and stimulates an immunogenic TME.
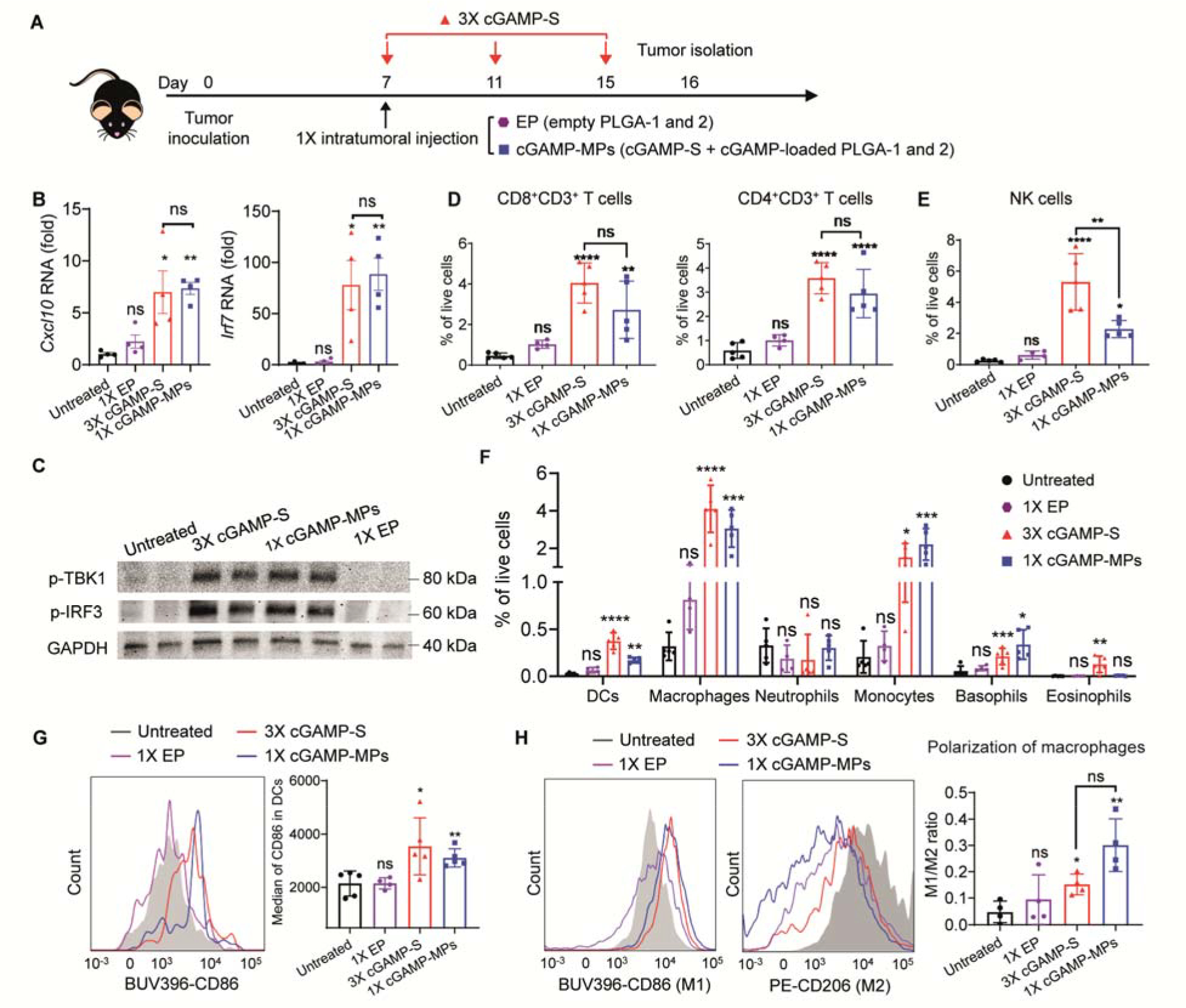
A. Treatment scheme of B16F10 tumor-bearing mice receiving a single intratumoral injection of EP or cGAMP-MPs (30 μg cGAMP: 10 μg in each of cGAMP-S, PLGA-1, and PLGA-2) at day 7, or three intratumoral injections of soluble cGAMP at days 7, 11, and 15 (3×cGAMP-S, 10 μg per injection) after tumor inoculation. Tumors were isolated on day 16. B. qPCR analysis of Cxcl10 and Irf7 mRNA expression in tumors (n = 4). Data are shown as mean ± SEM. C. Western blot analysis of p-TBK1 and p-IRF3. GAPDH is used as an internal reference (n = 2). D, E, F. Percentages of infiltrating lymphocytes (D, CD8+CD3+ T cells and CD4+CD3+ T cells; E, NK1.1+CD3− NK cells), CD11b−CD11c+ dendritic cells (DCs, F), and myeloid cells (F, CD11b+F4/80+ macrophages, CD11b+F4/80−Ly6c+Ly6g+ neutrophils, CD11b+F4/80−Ly6c+Ly6g− monocytes, CD11b+Gr-1−CD200R3+ basophils, and CD11b+Gr-1−CD170+ eosinophils) in TME among all live cells (n = 4 to 5). Data are shown as mean ± SD. G. Representative flow cytometry measurements of activated DCs (CD86+CD11c+CD11b−) in tumors treated with different therapeutic combinations (n = 4 to 5). Quantitative analysis was shown on the right. BUV396 and PE represent BD Horizon Brilliant Ultraviolet 396 and phycoerythrin, respectively. Data are shown as mean ± SD. H. Representative flow cytometry measurements of M1 (CD86+CD11b+F4/80+) and M2 (CD206+ CD11b+F4/80+) macrophages in tumors treated with different therapeutic combinations. The ratio of M1/M2 macrophages was calculated and presented on the right (n = 4). Data are shown as mean ± SD. Statistical significance was calculated by one-way ANOVA or Student’s t test when comparing multiple or two groups, respectively. Data were compared with untreated group unless indicated otherwise. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Activation of the STING pathway in the TME has been shown to promote lymphocyte infiltration, which is the major mediator for effective cancer immunotherapy (34). Flow cytometry analysis of tumors showed that 3×cGAMP-S and cGAMP-MPs increased TILs by approximately 23.5- and 17.6-fold compared to the untreated group (figs. S12 and S13). Among these TILs, tumor-infiltrating CD8+ and CD4+ T cells had been increased by 24.4- and 23.6-fold for 3×cGAMP-S treated group, and 16.2- and 22.1- fold for cGAMP-MPs treated group, respectively (Fig. 4D and fig. S14). The amounts of CD8+ and CD4+ T cells in the cGAMP-S treated group were not significantly different from those in the cGAMP-MP treated group. 3×cGAMP-S and cGAMP-MPs treatments also showed 1.2- and 1.5-fold increase of CD8+/CD4+ T cell ratio compared to untreated group, respectively (fig. S13), which is a commonly reported positive prognostic indicator of immunotherapy (10, 35). In agreement with this enriched CD8+ T cell infiltration and enhanced antitumor activity, terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL) showed a greater abundance of apoptotic cells for the cGAMP-MPs and 3×cGAMP-S treated groups than untreated and EP treated groups (fig. S15). Additionally, 3×cGAMP-S and cGAMP-MPs increased infiltrating natural killer (NK) cells (Fig. 4E), another important group of cytotoxic lymphocytes that shape the adaptive immune response and were found to be effective for spontaneous STING-mediated protection against B16F10 tumors (36). Conversely, the group receiving empty particles did not increase TILs within the TME, confirming its inability to activate the STING pathway. We did not observe differences in regulatory T cells between any of the groups (fig. S16).
We next evaluated the changes in dendritic cells (DCs) and myeloid composition in B16F10 melanoma TME after treatments. Both 3×cGAMP-S and cGAMP-MPs promoted the influx of DCs (CD11b−CD11c+), basophils (CD11b+Gr-1−CD200R3+), monocytes (CD11b+F4/80−Ly6c+Ly6g−), and macrophages (CD11b+F4/80+), creating an innate inflammatory niche with the potential to prime adaptive immunity (Fig. 4F) (37). In contrast, empty particles did not increase the myeloid cell population, suggesting the low immunogenicity of PLGA. Additionally, surface expression of CD86, which is a maturation marker overexpressed on activated tumor-infiltrating DCs (38), was increased by 1.7- and 1.5-fold for 3×cGAMP-S and cGAMP-MPs treated groups, respectively (Fig. 4G). The maturation of DCs in combination with enriched TILs and increased intratumoral ISGs suggest potential activation of adaptive immunity (10, 39, 40). We next evaluated the polarization of macrophages within the TME, another key function associated with cGAMP (34, 41). After 3×cGAMP or 1×cGAMP-MPs treatments, repolarization of M2-like macrophages in the tumor to M1-like phenotype was observed, which is consistent with previous studies of STING agonist-treated tumors and suggests a less immunosuppressive TME (34, 42, 43). cGAMP-MPs downregulated the canonical M2 surface marker (CD206) and upregulated M1 surface markers (CD86, Fig. 4H). Quantitative analysis showed that cGAMP-MPs induced approximately a 2-fold greater M1/M2 ratio than did the 3×cGAMP-S treated group. Empty particles had no significant effect on the M1/M2 ratio compared to the untreated group.
Single injection of cGAMP-MPs triggered potent systemic antitumor immunity
To study whether activation of STING in the TME triggers systemic antitumor immunity, mouse serum was collected from the antitumor efficacy study of B16F10 melanoma model (Fig. 3D) 21 and 28 days after tumor inoculation and analyzed by flow cytometry. cGAMP-MPs and 4×cGAMP-S treatments increased IFNγ+CD8+ T cells in serum at day 21 by 5.1- and 4.9-fold, respectively, compared to the untreated group (Fig. 5A and fig. S17). The number of IFNγ+CD8+ T cells remained unchanged at day 28 (Fig. 5A), demonstrating long-lasting systemic immune response. In addition, cGAMP-MPs also increased the number of memory CD62L−CD44+CD4+ T cells (~6.2-fold over untreated group) and CD62L−CD44+CD8+ T cells (~5.4-fold over untreated group) in TME (Fig. 5B) (44, 45). Collectively, a single injection of cGAMP-MPs generated long-lived systemic antitumor immunity and local immunological memory, which may be able to prevent tumor recurrence and metastases.
Figure 5. Single injection of cGAMP-MPs induced systemic antitumor immunity and inhibited metastasis.
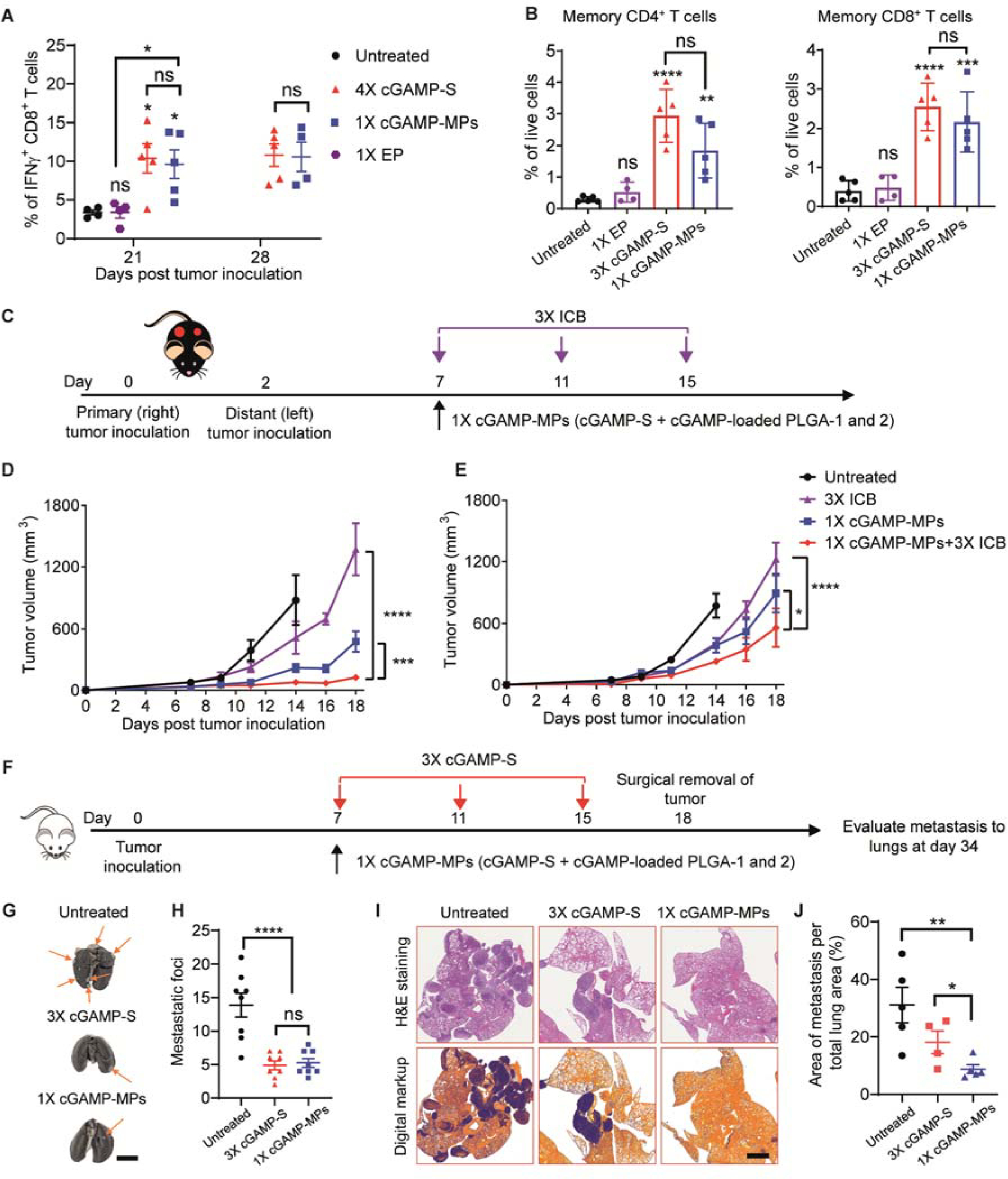
A. Quantitative analysis of IFNγ+CD8+ T cells in the serum collected at days 21 and 28 (n = 4 to 5, treatment scheme shown in Fig. 3D). Untreated and 1×EP-treated mice did not survive to day 28. Data are shown as mean ± SEM. B. Numbers of effective memory CL62L−CD44+CD4+CD3+ and CL62L−CD44+CD8+CD3+ T cells in the TME (treatment scheme shown in Fig. 4A). C. Schematic of treatment regimen on a contralateral B16F10 model. Tumors were inoculated on the right (primary) and left (distant) rear flanks of mice at days 0 and 2, respectively. The mice were treated with a single intratumoral injection of cGAMP-MPs (30 μg cGAMP: 10 μg cGAMP in each of cGAMP-S, PLGA-1, PLGA-2) on the primary tumor, three intraperitoneal injections of anti-PD-1 (3×ICB, 100 μg per injection), or the combination of both cGAMP-MPs and 3×ICB. The distant tumor did not receive any treatments. D, E. Average tumor growth curves of treated (D) and distant tumors (E, n = 8). Data are shown as mean ± SEM. F. Schematic of treatment regimen on a metastatic 4T1 model. Mice were treated with a single intratumoral injection of cGAMP-MPs (30 μg cGAMP: 10 μg cGAMP in each of cGAMP-S, PLGA-1, PLGA-2) or three intratumoral injections of cGAMP-S (3×cGAMP-S). G, H. Representative lung photographs (G) and number of metastatic foci (H) on lung surfaces after treatments (n = 8). Arrows point to metastatic tumors. Scale bar is 0.5 cm. I. Representative H&E stained lung sections and digitally processed images used for quantifying metastatic tumor cells. Scale bar is 2 mm. J. Percentage of tumor area within total lung area after treatments (n = 4 to 5). Statistical significance was calculated by Student’s t test or two-way ANOVA: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
To study whether cGAMP-MPs could inhibit the growth of distant tumors, we used a contralateral B16F10 tumor model. The primary tumor was treated by a single intratumoral injection of cGAMP-MPs (cGAMP-S, cGAMP-loaded PLGA-1, and cGAMP-loaded PLGA-2) to achieve overall three doses at days 7, 11, and 15 after tumor inoculation. The distant tumor did not receive any treatment (Fig. 5C). cGAMP-MPs effectively inhibited the growth of both the primary and distant tumors compared with the untreated group, thereby demonstrating strong systemic antitumor immunity (Fig. 5D and 5E). To evaluate whether cGAMP-MPs could improve the antitumor efficacy of ICB, we tested a combination therapy of cGAMP-MPs with anti-programmed death 1 (PD-1) in the same contralateral B16F10 tumor model. The combination of cGAMP-MPs and ICB showed greater inhibition of primary and distant tumor growth than individual therapies by themselves (Figs. 5D and 5E), demonstrating the potential of combining cGAMP-MPs with ICB to enhance the therapeutic effect.
We then studied the effect of cGAMP-MPs on metastasis using an orthotopic 4T1 breast cancer model. We treated the primary tumor with a single intratumoral injection of cGAMP-MPs at day 7 or three intratumoral injections of cGAMP-S at days 7, 11, and 15 (3×cGAMP-S) after tumor inoculation. The primary tumors were surgically removed at day 18 to extend survival time, which was necessary to allow the development of metastasis. We then isolated lungs and analyzed metastasis at day 34 (Fig. 5F). cGAMP-MPs treatment significantly decreased the number of metastatic foci on lung surfaces (Figs. 5G and 5H, p<0.0001) and reduced the relative area of tumors in the lungs compared to untreated groups (Figs. 5I and 5J, P<0.01). cGAMP-MPs also exhibited a greater ability to decrease the percentage of metastatic tumor cells within lungs compared to 3×cGAMP-S (Fig. 5J, P<0.5), suggesting the benefits of a single injection for decreasing metastasis.
cGAMP-MPs could expand the scope of current STING agonist-based therapies
In the clinic, patients often develop recurrent tumors after surgery because of residual micro-tumors and circulating tumor cells (46–48). To expand the clinical applications of cGAMP-MPs, we adopted a surgical resection melanoma model to evaluate the efficacy of cGAMP-MPs on inhibiting tumor recurrence (49, 50). Six days after tumor inoculation, approximately 99% of each B16F10 tumor was surgically removed. We then directly deposited cGAMP-MPs (cGAMP-S, cGAMP-loaded PLGA-1 and 2) at the fresh surgical bed to achieve three doses overall at days 0, 4, and 8. Mice treated with 3×cGAMP-S or 1×cGAMP-S+EP were used as controls (Fig. 6A). cGAMP-MPs and 3×cGAMP-S treatments improved tumor inhibition (Fig. 6B) and enhanced survival (Fig. 6C) compared to untreated and cGAMP-S+EP treated mice. Six out of eight cGAMP-MPs and 3×cGAMP-S treated mice were tumor-free and survived for over 60 days after inoculation (Fig. 6C). The tumor recurrence rates of cGAMP-MPs and 3×cGAMP-S treated groups were both 25% (2 out of 8 mice), which was lower than that of the untreated group (100%, 8 out of 8 mice) and 1×cGAMPs+EP treated group (87.5%, 7 out of 8 mice. Fig. 6C). We then rechallenged these tumor-free mice through subcutaneous injection of B16F10 cells at day 60. The tumors grew significantly slower in cGAMP-MPs and 3×cGAMP-S treated mice than in naïve mice (Fig. 6D, p<0.001). Survival analysis also showed prolonged survival time for treated groups (Fig. 6E), suggesting cGAMP-MPs and 3×cGAMP-S can provide protective immunity.
Figure 6. cGAMP-MPs prevented tumor recurrence after surgery and inhibited growth of hard-to-reach tumors.
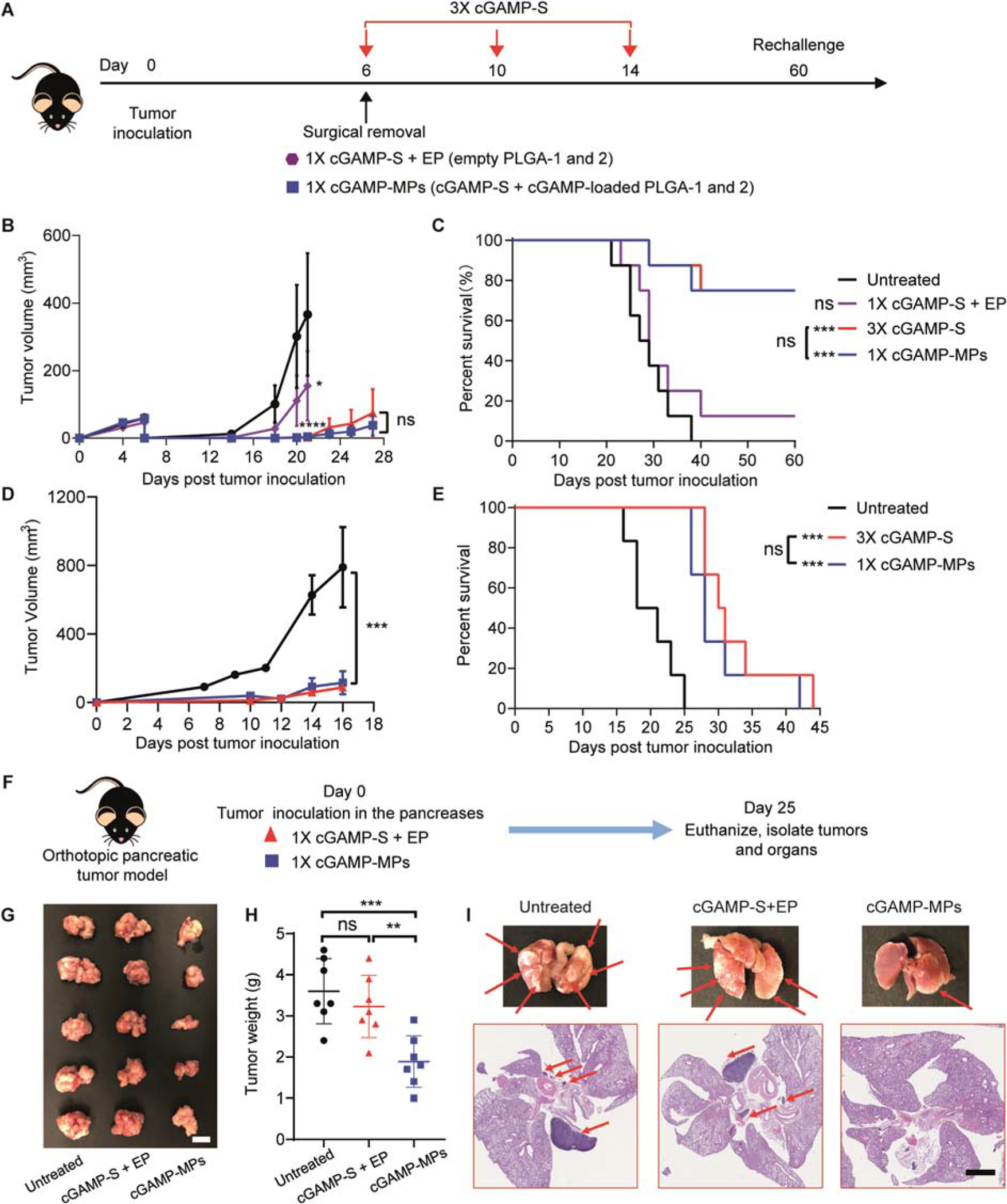
A. Schematic of treatment regimen in a surgical removal B16F10 model. Approximately 99% of the tumor mass was surgically removed at day 6 after tumor inoculation. cGAMP-MPs (30 μg cGAMP: 10 μg cGAMP in each of cGAMP-S, PLGA-1, PLGA-2) or 1×cGAMP-S+EP (10 μg cGAMP with empty PLGA-1 and 2) were directly deposited at the surgical bed. For 3×cGAMP-S treatment, cGAMP-S was administered intratumorally at days 6, 10, and 14 (10 μg per injection). B, C. Average tumor growth curve (B) and survival analysis (C) of treated mice (n = 8). Numbers of tumor-free mice out of treated mice for each group are indicated in C. D, E. Tumor-free mice after cGAMP-MPs and 3×cGAMP-S treatments were rechallenged with B16F10 cells at day 60 after tumor inoculation. Naïve mice were challenged as negative controls. Tumor growth (D) and survival (E) were monitored over time (n = 6). Data are shown as means ± SEM. F. Schematic of treatment regimen on an orthotopic pancreatic tumor model. cGAMP-MPs or 1×cGAMP-S+EP were injected into the pancreases immediately after tumor inoculation (n = 7). G, H. Representative images (G) and weights (H) of isolated pancreatic tumors. Scale bar is 1 cm. I. Representative photographs of lungs and H&E stained lung sections. Red arrows point to metastatic tumors. Scale bar is 2 mm. Statistical significance was calculated by one way or two-way ANOVA and Tukey’s multiple comparisons test: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
We further tested the therapeutic efficacy of cGAMP-MPs on an allograft model of pancreatic cancer (KPC model). We injected cGAMP-MPs (10 μg each of cGAMP-S, cGAMP-loaded PLGA-1 and 2) after tumor inoculation in the pancreas to achieve overall three doses at days 0, 4, and 8 (Fig. 6F). Multiple intratumoral injections of soluble cGAMP are extremely difficult on such hard-to-reach tumors. Therefore, we performed a single injection of a high dose of cGAMP-S (30 μg)+EP at day 0 (Fig. 6F). Untreated mice were used as negative controls. We then analyzed the tumor growth and metastasis 25 days after treatments. cGAMP-MPs significantly inhibited primary tumor growth in the pancreas and metastasis to lungs compared with untreated group (Figs. 6G, 6H, and 6I, p<0.001). In contrast, cGAMP-S+EP did not show benefits on tumor growth or metastasis. Collectively, these data demonstrate cGAMP-MPs could be useful for hard-to-reach tumors and suggest the necessity of multiple doses at extended time points for effective therapy.
Toxicity analysis
PLGA has been used in many FDA-approved medical devices owing to its biodegradability and biocompatibility (51). Flow cytometry analysis of immune cells in TME indicated that intratumorally administered empty PLGA-MPs did not induce any detectable inflammation in situ (Figs. 4D to G). In addition, we did not observe weight loss or behavior changes in any of the animals throughout the treatment period for all in vivo studies (fig. S18). Hematoxylin and eosin (H&E) staining of histological sections of major organs (heart, liver, spleen, lung, and kidney) showed no obvious change in morphology (fig. S19). We further studied the biodegradability of PLGA-MPs by subcutaneously injecting empty PLGA-2 into immunocompetent mice and performed H&E staining of skin tissues at days 2, 8, and 30 after injection. At day 2, PLGA-MPs exhibited cubic morphology under the skin with few lymphocytes and leukocytes around the particle, suggesting the presence of minimal inflammation. The particles then deformed to an ellipse shape due to hydrolysis of PLGA with diminished immune cells at day 8, just around the release window, which is consistent with flow cytometry analysis. We did not observe any particles left in mice at day 30, which suggests complete degradation and clearance (fig. S20). Collectively, these data demonstrate that microfabricated PLGA-MPs did not show toxicity and can be completely degraded and cleared in vivo.
PLGA-MPs could be a platform technology to deliver various cancer therapeutics
Loading drugs into PLGA-MPs is an independent step during particle fabrication, thus allowing us to fill other types of cancer therapeutics into PLGA-MPs. We performed preliminary studies of loading either a chemotherapeutic drug (pemetrexed) or a TLR agonist (CpG DNA) into PLGA-2. Pemetrexed and CpG DNA exhibited nearly the same in vitro release kinetics from PLGA-2 as cGAMP (fig. S21), indicating the potential applicability of PLGA-MPs for these cancer therapeutics. The drug loading capacity of PLGA-MPs could also be tuned by changing the particle design. We fabricated a different particle base with a bigger cavity (300 × 300 × 200 μm length × width × height) and wall thickness of 50 μm to increase the loading capacity. We could load ~10 μg of cGAMP into each of these particle bases (fig. S22).
Discussion
Adherence to current STING agonist-based therapy is challenging because of the frequent injections over a long period of time and the need of trained healthcare professionals for each injection. Poor patient adherence represents a notable challenge resulting in treatment failures and large financial costs (19, 52). Frequent injections for cancer treatment also cause a substantial burden to daily lives of patients (53). Previous research efforts have mainly focused on improving the cellular uptake of STING agonist and tumor targeting efficacy after systemic administration (10, 34, 54). Here, we describe an approach of improving the overall effectiveness of STING agonist therapy by ensuring that patients receive every required dose at the correct time.
To replace multiple intratumoral injections of soluble STING agonist, PLGA-MPs need to stay inside tumors and release biologically active cargo at anticipated times. MicroCT analysis showed that these microparticles aggregated at the tumor injection site due to low mobility. LC-MS and in vitro analysis in cells showed that cGAMP maintained >95% of bioactivity after release from PLGA-MPs. We then extensively studied the release kinetics of different cargos in vitro and in vivo. The release kinetics of PLGA-MPs were independent of the cargo released (AF647, AF647-dextran, and STING agonist) and the in vivo microenvironment (subcutaneous, B16F10, and 4T1 tumors). These observations are expected because the degradation of PLGA is predominantly driven by hydrolysis. It has been previously reported that enzymatic activity has negligible effect on PLGA degradation (51). The slightly acidic tumor microenvironment did not accelerate the release of low MW PLGAs, but may have effects on the long-term release of PLGA-MPs because of the acid-catalyzed hydrolysis of PLGA. Therefore, testing in vitro release in an acidic environment is needed to predict release kinetics in tumors.
Pulsatile release over long periods of time is usually achieved by implantable drug delivery devices, which require invasive surgery to administer and remove (55). One benefit of PLGA-MPs is that they could be injected using a regular needle and completely degraded over time, thus improving patient compliance. Other injectable long-term drug release systems, such as emulsion-based microparticles (including PLGA formulations) or hydrogels, often show an initial phase of burst release and then a second phase of sustained release for hydrophilic drugs (26, 56, 57). Such release kinetics result in an initial high dose, which could cause toxic side effects. Additionally, achieving sustained release of small/hydrophilic drugs, such as STING agonists, over weeks is extremely challenging. Drug encapsulation efficacy is also relatively low for emulsion-based microparticles (58). Our engineered PLGA microparticles could achieve essentially 100% drug encapsulation efficacy and be combined to exhibit multiple burst release events at time points up to months. We tested the release kinetics of several low MW PLGAs in this study to achieve release time within the previously reported treatment schedules using cGAMP in animal models. Expanding the release kinetics library to cover pulsatile release over months or even a year should be possible by tuning the MW, chain-end functionality, and copolymer ratio of PLGA. Therefore, we could potentially administer customizable doses by physically mixing PLGA-MPs with different release profiles within one injection.
We demonstrated that the antitumor efficacy of single-administered PLGA-MPs is comparable with multiple injections of cGAMP solutions in multiple mouse models. PLGA-MP-treated B16F10 tumors showed high amounts of ISGs and phosphorylated TBK-1 and IRF-3 proteins, suggesting the successful activation of the STING pathway by a sequence of pulsatile releases of cGAMP. PLGA-MPs induced an immunogenic TME, as demonstrated by increased tumor-infiltrating CD8+ T cells, NK cells, DCs, and the shift from an M2 to an M1 macrophage phenotype. Loading cGAMP in PLGA-MPs improved M1 polarization as compared to free cGAMP, possibly due to the acidic degradation products of PLGA, which have been shown to stimulate proinflammatory macrophages (59–61). Further studies on macrophage polarization kinetics are required to fully elucidate the function of PLGA-MPs on macrophage polarization in the TME. We also observed an increased number of memory T cells in tumors and increased circulating IFNγ+CD8+ T cells, which contributed to the inhibition of distant tumor growth, reduction in metastasis, and protective immunity against rechallenge. Neither cGAMP-MPs- nor 3×cGAMP-S-treated mice completely rejected tumor rechallenge. This observation is consistent with a recent study showing that repetitive intratumoral injections of STING-agonist attenuated systemic T cell responses (62). Nonetheless, multiple doses showed a better tumor inhibition effect than a single dose of soluble STING agonist in previous publications and in the current study (34, 63, 64). Identifying the best dosing regimen of STING agonist to maximize therapeutic efficacy and systemic T cell response will be needed in future studies. Lastly, PLGA-MPs do not exhibit apparent toxicity and could be completely degraded, as supported by body weight and histology analysis, respectively. Collectively, these results demonstrated the efficacy and safety of PLGA-MPs in comparison to multiple soluble injections.
Current STING-agonist therapies in clinical trials focus on easily accessible tumors. Intratumoral injection of therapeutics into major organs in the clinic is usually achieved under CT or ultrasound guidance (65). Therefore, applying STING-agonist therapy to hard-to-reach tumors is challenging because of the complexity and high financial cost of multiple imaging-guided injections. We have demonstrated that cGAMP-MPs could be administered into orthotopic pancreatic tumors and effectively inhibit tumor growth and metastasis with one injection. These data suggest that cGAMP-MPs could not only benefit readily accessible tumors (for example, melanoma) but also open up the possibility of treating major organ cancers. Additionally, cGAMP-MPs could be used after surgical resection of tumors that are not compatible with multiple intratumoral injections, to prevent tumor recurrence. Collectively, cGAMP-MPs hold great potential to broaden the scope of STING agonist-based therapies.
Despite the encouraging efficacy and safety profile of PLGA-MPs, several aspects could be improved before clinical translation. The drug loading of PLGA-MPs (currently at 8.4% by volume) could be increased in order to maximize the achievable number of doses within a single injection. One solution to increase drug loading is to reduce particle wall thickness while maintaining the outer dimensions. For example, decreasing wall thickness from 100 μm to 50 μm would increase drug loading by 450% and allow loading 10 μg of cGAMP into each particle. The current dose of STING agonist in clinical trials is 100 μg/injection, which requires ~10 particles to match the same dose. The volume of 10 particles is 4.8×10−4 cm3. The overall volume of PLGA-MPs for 20 doses is 9.6×10−3 cm3, which is less than 1% of the volume of a 1 cm3 tumor. Additionally, the size and geometry of PLGA-MPs could also be optimized to further increase drug loading and enable injections using smaller needles. Scale-up production also needs to be investigated for translation. The fabrication process for PLGA-MPs uses a combination of photo lithography, soft lithography, and ultralow volume dispensing technologies. These technologies have been widely used in microelectronic and microfluidics industries (66); however, integrating different fabrication steps into an automatic process needs to be investigated in future studies to increase the throughput of production.
In summary, by engineering PLGA into a core-shell microstructure, we developed a fully degradable delivery system for STING agonist that could improve patient adherence and lower financial costs by eliminating repeated injections and doctor visits, decrease the risk of metastasis, and ultimately lead to better effectiveness of STING agonist-based cancer immunotherapy. PLGA-MPs also expand the scope of STING agonist-based therapy to hard-to-reach tumors and as an adjuvant therapy to prevent tumor recurrence after surgery. This single injection delivery system contains only PLGA, which is widely used in FDA-approved drug products, thus holding great promise for rapid clinical translation. The modularity of this platform makes it easily compatible with loading any hydrophilic drug and many hydrophobic drugs (for example, pemetrexed and CpG DNA), or even delivering various drugs at different times for combination cancer therapies. This platform also opens up opportunities for many other diseases that require frequent or repeated local administration of therapeutics.
Materials and Methods
Study design
The objective of this study was to develop an injectable delivery platform that could release multiple doses of STING agonist at distinct time points within one injection. We first screened the release kinetics of different PLGA-MPs in vitro to find desired releasing time points for treating tumor-bearing animal models. We further verified the release kinetics of PLGA microparticles in vivo using fluorescence imaging. The antitumor efficacy was evaluated in mouse B16F10 melanoma, 4T1 breast cancer, contralateral B16F10 melanoma, orthotopic pancreatic cancer, and incomplete surgical removal tumor models. To demonstrate the activation of STING pathway, we analyzed STING-related mRNA expression and protein abundance using qPCR and western blots, respectively. The local and systemic immune responses of microparticles were also examined using flow cytometry and immunofluorescence staining. The toxicity and biodegradability of PLGA microparticles were studied by body weight analysis and histology. Mice were randomly divided into different treatment groups for all studies. The researchers were not blinded in this study. Power analysis was not used to predetermine sample size. Sample sizes were determined on the basis of our previous studies. The replicate numbers for each experiment and statistical analysis methods are indicated in the figure legends. Raw data are shown in data file S1.
Fabrication of PLGA microparticles
PLGA were purchased from Evonik and PolySciTech. PLGA microparticles were fabricated through the Stamp Assembly of Polymer Layers (SEAL) process (67). Photomasks with microscale patterns of bases and caps were made by Front Range Photomask. Positive master molds of microparticle base and cap were created by SU-8 lithography on silicon wafers. The mixture of PDMS base and curing agent (Sylgard 184, Dow Corning) was poured onto the silicon master mold, then exposed to high vacuum for 1 h. A glass slide with two cover slips at each end was then pushed against the silicon mold while curing in the oven for 2 h at 150°C to yield a thin PDMS mold. The obtained PDMS molds were then used as negative molds to press desired microparticles. PLGA films were prepared by solvent casting 60% weight/volume PLGA in acetone solution. The thickness of PLGA films was ~1650–1750 μm. For molding the caps of the microparticles, a small piece of PLGA film was placed between the PDMS cap mold and a Teflon film, and covered with a glass slide. A pair of spring-loaded clamps was then used to fix and compress the microparticles in a 120 oC vacuum oven for 2 h. PLGA film melted and flowed in the cap PDMS mold during heating. The setup was then allowed to cool down to room temperature and separated to yield the PLGA caps in the PDMS mold. For molding the base of the microparticles, the above steps were repeated without using the Teflon film. Therefore, the PLGA base would separate from PDMS base mold and stick to the covering glass slide. The cargos of interest were filled into the PLGA microparticles using a BioJet Ultra picoliter dispensing instrument (BioDot). The aqueous solutions of cargos were dispensed for multiple 15-drop cycles of 180–200 pL drops. Filled particles were then aligned and sintered with corresponding PLGA caps using a photomask aligner (MA4, Karl SUSS) retrofitted with a Peltier heater to enable simultaneous alignment and sealing. Sealed particles were then separated from glass slides using a razor blade. Scanning electron microscopy images were collected using a JSM-5600LV scanning electron microscope (JEOL) with an acceleration voltage of 5 kV. High resolution X-ray CT images were collected at the Biotechnology Resource Center of Cornell University.
In vitro release kinetics
To study the in vitro release kinetics of AF647-dextran (Life Technologies), 3’3’-cGAMP (InvivoGen), Cy5-CpG DNA (5’-TCC ATG ACG TTC CTG ACG TT-Cy5–3’, IDT Inc.), and pemetrexed (Sigma-Aldrich), PLGA microparticles were separately filled with 1 μg of AF647-dextran, 2 μg of 3’3-cGMAP, 1 μg of Cy5-CpG, or 2 μg of pemetrexed, respectively. To determine the cargo loading in particles, filled particles were suspended in 200 μL of PBS buffer individually, vortexed for 15 seconds, and centrifuged at 14000 rcf for 1 minute. The supernatants were then analyzed by a microplate reader (AF647, AF647-dextran, Cy5-CpG), HPLC (pemetrexed), and Nanodrop (3’3’-cGAMP, absorbance at 260 nm). The results were quantified using a standard curve of a serial dilution of stock solutions. Filled particles were then sealed with corresponding PLGA caps. Each particle was placed into 200 μL of PBS (pH = 6.84) in a 0.5 mL microcentrifuge tube (Eppendorf) and incubated on an orbital shaker at 37 °C. The supernatant of each centrifuge tube was collected at predetermined time points. The supernatants of AF-647 dextran and Cy5-CpG groups were analyzed by a microplate reader (Tecan Infinite M200 spectrophotometer, excitation/emission = 640/680 nm). The supernatants of 3’3’-cGAMP and pemetrexed were analyzed via HPLC (Alliance HPLC systems, Waters Co.) using a C-18 column (Acclaim PolarAdvantage II, 3 μm, 4.6 × 150 mm) and a photodiode detector at 260 nm for 3’3’-cGAMP and 254 nm for pemetrexed. Water and acetonitrile were used as mobile phases. The results were quantified using a standard curve of a serial dilution of stock solutions and normalized to total cumulative release (n = 6–8). The actual release day of each PLGA was determined as the day at which more than 50% of cargo was released.
For the sustained release systems, fast-dextran hydrogel kits (Part No. TURE2–1KT), fast-PVA hydrogel kits (Part No. TRUE4–1KT), and 3D collagen Kit (Part No. ECM675) were purchased from Millipore Sigma. 40 μg of cGAMP was loaded into 30 μL hydrogels according to manufacturers’ instructions. To study the in vitro release kinetics, cGAMP-loaded hydrogels were incubated on an orbital shaker at 37 °C. The supernatant of each centrifuge tube was collected at predetermined time points and analyzed by Nanodrop.
Animals and cell lines
All animal procedures were approved by Massachusetts Institute of Technology Committee on Animal Care. Six- to eight-week-old SKH1-E, C57BL/6, and BALB/c female mice were purchased from Charles River Laboratories Inc. The mouse breast cancer cell line 4T1 and melanoma cell line B16F10 were purchased from American Type Culture Collection. The RAW-Lucia ISG cell line was purchased from InvivoGen Inc. KPC (LSL-KrasG12D/+; LSL-Trp53R172H/+; Pdx-1-Cre) pancreatic cancer cells were kindly given by Dr. Serguei Kozlov (Frederick National Laboratory of Cancer Research). Cells were cultured in Dulbecco’s modified Eagle medium (DMEM, B16F10), DMEM/F12 (KPC), and RPMI 1640 (4T1) supplemented with 10% fetal bovine serum (FBS), penicillin (100 units/mL), and streptomycin (100 μg/mL) at 37 °C and with 5% CO2. Cell culture media and antibiotics were purchased from Invitrogen. RAW-Lucia ISG cells were cultured in DMEM supplemented with 2 mM L-glutamine, 10% FBS, 100 μg/ml Normocin (InvivoGen), and 200 μg/ml Zeocin (InvivoGen).
In vivo release kinetics
One PLGA microparticle encapsulating AF647-dextran (1 μg) was tip-loaded into an 18-gauge Monoject filter needle (Covidien) in approximately 20 μl of 15 mg/ml methyl cellulose (MC, Sigma Aldrich) used as a viscosity enhancer. Particles were then injected subcutaneously into the left and rear flanks (1 particle per side) of hairless mice (SKH1-E) or intratumorally into tumor-bearing mice. Mice were imaged every 1–2 days using a PerkinElmer Spectrum In Vivo Imaging System (excitation/emission=640/700 nm, IVIS). Cumulative release was normalized to the maximum and minimum total fluorescence in the region of interest corresponding to a particular particle’s complete release and background signal, respectively. Because fluorescence dropped after release due to biological clearance, values after the highest signal were set to 100% in Fig. 2. Release timing was considered to be the day on which fluorescence achieved half of its final maximum value above background.
To evaluate the amount of released cargo in tumors, we intratumorally injected 10 AF647-loaded PLGA-1 (0.5 μg AF647 per particle) into B16F10 tumor-bearing mice at day 0. Free cGAMP were also administered intratumorally at days 0 and 4 to control tumor growth. We euthanized 4 mice and isolated tumors and serum every day until day 7. The tumors were homogenized in PBS buffer. Unreleased AF647-PLGA-1 was physically broken to release AF647 during homogenization. The supernatant containing unreleased AF647 and serum samples were analyzed by a microplate reader (excitation/emission = 640/680 nm).
Micro-CT analysis of particle distribution
5% of phosphotungstic acid (PTA) was doped into PLGA-1 while making PLGA-1 films to increase the contrast for microCT imaging. PTA-doped PLGA-1 were intratumorally injected into B16F10 tumor-bearing mice. Mice were then euthanized 1 h after injection. The tumors were isolated and imaged by a Bruker Skyscan 1276 microCT imaging system. The reconstructed images were analyzed by MicroView.
Bioactivity of released 3’3’-cGAMP
To evaluate the activity of 3’3’-cGAMP after particle fabrication, cGAMP-loaded PLGA-2 were placed in PBS buffer and mechanically broken by a scalpel to release encapsulated cargo. To evaluate the activity of cGAMP after release, cGAMP-loaded PLGA-2 were incubated on an orbital shaker in PBS buffer at 37 °C. Supernatant was collected at release window and quantified by Nanodrop. 5×104 RAW-Lucia ISG cells were plated in a 96-well plate. A serial dilution of cGAMP stock solutions, dissolved cGAMP after particle fabrication, and released cGAMP were incubated with cells for 24 h before adding QUANTI-Luc solution. The plate was then analyzed by a microplate reader. The results were quantified using a standard curve of a serial dilution of stock solutions.
Treatment of B16F10 and 4T1 tumors
2×105 4T1 or 2×105 B16F10 cells were subcutaneously injected into the right rear flank of BALB/c or C57BL/6 female mice, respectively. For orthotopic 4T1 model, 2×105 4T1 cells were injected into the mammary fat pad of BALB/c female mice. Seven days after tumor injection, B16F10 tumor-bearing mice were divided into 6 experimental groups (n = 8 for each group): untreated, 1×empty particles (EP), 1×cGAMP-S+EP, 1×cGAMP-collagen, 4×cGAMP-S, and cGAMP-MPs. For the 4×cGAMP-S group, mice were intratumorally administered 10 μg of cGAMP in 50 μL of 15 mg/mL methyl cellulose solution (MC, Sigma Aldrich) at days 7, 11, 15, and 18. The overall dose of cGAMP was 40 μg per mouse throughout the treatment period. For the cGAMP-MP group, mice were given a single intratumoral injection of a mixture of 10 μg cGAMP-S, 5 PLGA-1 particles containing a total of 10 μg cGAMP, 5 PLGA-2 particles containing a total of 10 μg cGAMP, and 5 PLGA-3 particles containing a total of 10 μg cGAMP in 50 μL MC solution via an 18G filter needle. The overall dose of cGAMP is 40 μg. For 1×EP and 1×cGAMP-S+EP groups, 5 each of empty PLGA-1, PLGA-2, and PLGA-3 particles with or without 40 μg cGAMP-S were injected intratumorally in 50 μL MC solution. Mice from untreated groups were injected intratumorally with 50 μL MC solution at day 7 after tumor inoculation. In the subcutaneous and orthotopic 4T1 models, tumor-bearing mice were subjected to MC solution (untreated group), 4×cGAMP-S, or 1×cGAMP-MPs treatments at day 7 after tumor inoculation (n = 8 for each group). Tumor size was measured every other day started at day 7 after tumor inoculation with a digital caliper. Tumor volume was calculated using the following formula: length (mm) × width2 (mm) × 0.5. Animals were euthanized when they showed signs of poor health or when the tumor size exceeded 1500 mm3.
Western blot and quantitative polymerase chain reaction (qPCR)
Mice bearing B16F10 tumors were divided into the following 4 experimental groups (n = 5): untreated, 1×EP (5 each of empty PLGA-1 and 2), 3×cGAMP-S, and 1×cGAMP-MPs (10 μg of cGAMP-S, 5 PLGA-1 particles containing a total of 10 μg of cGAMP, and 5 PLGA-2 particles containing a total of 10 μg of cGAMP). For the 3×cGAMP-S group, mice were intratumorally administered 10 μg of cGAMP in 50 μL of MC solution at days 7, 11, and 15 after tumor inoculation. Tumors were collected at day 16 after tumor inoculation and cut into 50–100 mg pieces in a 1.5 mL microcentrifuge tube. The tumors were lysed in radioimmunoprecipitation assay (RIPA) buffer (Sigma-Aldrich), homogenized, and centrifuged at 20130 rcf for 10 min. The protein content in the supernatant was quantified using a bicinchoninic acid protein assay kit (Thermo Fisher Scientific). Equal amounts of proteins (20 μg) were separated on 4 to 15% gradient SDS–polyacrylamide gels (Bio-Rad) and electro-transferred to nitrocellulose membranes. The membranes were then blocked with 5% milk in tris-buffered saline supplemented with 0.05% Tween 20 and further incubated with GAPDH monoclonal antibody (Invitrogen, cat. no. MA5–27912), phospho-TBK1/NAK (Ser172) (D52C2) rabbit mAb (Cell Signaling Technology, cat. no. 5483S), or phospho-IRF-3 (S396) rabbit mAb (Cell Signaling Technology, cat. no. 4947S) at 4 °C overnight. The membranes were then incubated with goat anti-rabbit IgG (H+L) Secondary Antibody, HRP (Invitrogen, cat. no. TG266717) for 1 h at room temperature. Protein bands were visualized by chemiluminescence using the ECL Western Blotting Substrate (Thermo Fisher Scientific).
For qPCR experiments, total RNAs were extracted from tumors by RNeasy Kit (Qiagen, Inc.) according to manufacturer’s protocol. Total RNA was then reversed-transcribed to cDNA using a high-capacity cDNA reverse transcription kit (Thermo Fisher Scientific). The obtained cDNA was amplified with TaqMan Gene Expression Master mix (Thermo Fisher Scientific) using a 384-well LightCycler 480 (Roche). We used primers for Irf7 (Thermo Fisher, assay Id. Mm00516793_g1), Cxcl10 (Thermo Fisher, assay Id. Mm00445235_m1), and Gapdh (Thermo Fisher, assay Id. Mm99999915_g1). Samples were analyzed in triplicates.
Flow cytometry
To stain the cell surface markers for flow cytometry analysis, cells were pre-treated with anti-CD16/32-Fc blocker (Biolegend, cat. no. 101319) and stained with fluorophore-conjugated antibody solution according to manufacturer-suggested dilutions on ice for 1 h. To stain each intracellular marker, for example IFN-γ, cells were pre-stimulated with cell stimulation cocktail (eBioscience, cat. no. 00–4970-93) for 4–6 h, fixed and permeabilized using fixation/permeabilization solution kit (BD, cat. no. 554714), and then stained with both anti-IFN-γ and other surface antibodies. Antibodies used for flow cytometry studies were anti-CD86-BUV395 (BD, cat. no. 564199), anti-CD45-BUV737 (BD, cat. no. 564880), anti-TCRβ-BV421 (Biolegend, cat. no. 109229), anti-NK1.1-BV605 (Biolegend, cat. no. 108739), anti-NK1.1-BV605 (Biolegend, cat. no. 108739), anti-CD8a-FITC (BD, cat. no. 553030), anti-CD4-PerCP/Cy5.5 (BD, cat. no. 550954), anti-CD62L-PE (Biolegend, cat. no. 104407), anti-CD19-PE/Cy7 (eBioscience, cat. no. 25–0193-81), anti-CD3-PE/594 (Biolegend, cat. no. 100245), anti-FOXP3-APC (eBioscience, cat. no. 17–5773-80), anti-CD11b-AF700 (Biolegend, cat. no. 201222), anti-CD8a-BV421 (Biolegend, cat. no. 100737), anti-Ly6g-BV510 (Biolegend, cat. no. 127633), anti-Siglec F-BV605 (BD, cat. no. 740388), anti-MHC II-BV786 (BD, cat. no. 743875), anti-Ly6c-AF488 (Biolegend, cat. no. 128021), anti-CD11c-PerCP/Cy5.5 (Biolegend, cat. no. 117327), anti-CD206-PE (Biolegend, cat. no. 141705), anti-CD197-PE/594 (Biolegend, cat. no. 120121), anti-F4/80-PE/Cy7 (Biolegend, cat. no. 123113), anti-CD200R3-APC (Biolegend, cat. no. 142207), and anti-CD11b-AF700 (Biolegend, cat. no. 101222), as well as viability dye eFluor 780 (eBiosciecne, cat. no. 65–0865-14). Flow cytometry data were acquired on an LSRFortessa cell analyzer (BD) and analyzed using FlowJo software.
Immunofluorescence staining
Tumor sections (5 um) were fixed with 4% paraformaldehyde, blocked with 3% bovine serum albumin, and permeabilized with 0.1% Triton X-100 in PBS. The tumor sections were then incubated with anti-CD8 alpha antibody (1:200, Abcam, cat. no. ab217344) at 4 °C overnight and goat anti-rabbit IgG H&L (Alexa Fluor 488) (1:1000, Abcam, cat. no. ab150077) secondary antibody at room temperature for 1 h. Apoptotic tumor cells were stained using an in situ cell death detection kit (Roche) according to the manufacturer’s instructions. Images were acquired on a Nikon A1R Ultra-Fast Spectral Scanning Confocal Microscope.
Treatment of contralateral B16F10 tumors
2×105 B16F10 cells were subcutaneously injected into the right rear flank of C57BL/6 female mice at day 0. Another 2×105 B16F10 cells were subcutaneously injected into the left rear flank at day 2 to mimic metastatic tumor. Seven days after primary tumor inoculation, B16F10 tumor-bearing mice were divided into 4 experimental groups (n = 8 for each group): untreated, 1×cGAMP-MPs, 3×ICB (anti-PD-1), and 1×cGAMP-MPs+3×ICB. cGAMP-MPs (10 μg of cGAMP-S, 5 PLGA-1 particles containing a total of 10 μg of cGAMP, and 5 PLGA-2 particles containing a total of 10 μg of cGAMP) in 50 μL MC solution were intratumorally injected into the primary tumor (on the right side). For 3×ICB and 1×cGAMP-MPs+3×ICB treated groups, 100 μg of anti-PD-1 antibody (Biolegend, cat. no. 114114) was intraperitoneally injected at days 7, 11, and 15 after primary tumor inoculation. The distant tumor (left side) did not receive any treatments. Tumor size was measured every other day starting at day 7 after tumor inoculation with a digital caliper. Tumor volume was calculated using the following formula: length (mm) × width2 (mm) × 0.5. Animals were euthanized when they showed signs of poor health or when the tumor size on either side exceeded 1500 mm3.
Treatment of metastatic 4T1 model
2×105 4T1 cells were injected into the mammary fat pad. Seven days after injection, tumor-bearing mice were divided into 3 experimental groups: untreated, 3×cGAMP-S, and 1×cGAMP-MPs (10 μg of cGAMP-S, 5 PLGA-1 particles containing a total of 10 μg of cGAMP, and 5 PLGA-2 particles containing a total of 10 μg of cGAMP). For the 3×cGAMP-S group, mice were intratumorally administered 10 μg of cGAMP in 50 μL of MC solution at days 7, 11, and 15 after tumor inoculation. The primary tumor was surgically removed at day 18 to extend survival. Mice were euthanized at day 34. Lung tissues were stained with India ink and fixed in Fekete’s solution. Metastatic foci on the lung were counted under a microscope. Unstained lung tissues were fixed in formalin and stained by H&E. Quantitation of metastatic tumor cells in H&E stained sections was performed using an Aperio ImageScope using tuned positive pixel count algorithm. Briefly, we tuned input hue value in the positive pixel count algorithm to positively select normal lung tissues in red to orange range, whereas the tumors were negatively selected in purple. Percentage area of tumor metastasis per total lung area was calculated as the number of negative counts (purple)/the number of total counts (purple, orange, and red) × 100%. Three H&E sections per lung at different depths were analyzed and averaged to obtain the percentage of tumor over the lung for one mouse. Four or five mice were analyzed for each group.
Treatment of surgically removed B16F10 tumor
2×105 B16F10 cells were subcutaneously injected into the right rear flank of C57BL/6 female mice. Six days after tumor inoculation, B16F10 tumor-bearing mice were randomly divided into 4 experimental groups (n=8 for each group): untreated, 1×cGAMP-S+EP, 3×cGAMP-S, and 1× cGAMP-MPs. ~99% of tumor volume was surgically removed, leaving ~1% residual tumor to mimic residual microtumors. Upon the removal of tumors, cGAMP-MPs (10 μg of cGAMP-S, 5 PLGA-1 particles containing a total of 10 μg of cGAMP, and 5 PLGA-2 particles containing a total of 10 μg of cGAMP) or 1×cGAMP-S+EPs (10 μg of cGAMP-S, 5 each of empty PLGA-1 and PLGA-2 particles) in 50 μL MC solution were directly applied to the surgical bed through a micropipette. For 3×cGAMP-S treated group, 10 μg of cGAMP-S was applied to the surgical bed after surgery followed by intratumoral injections of 10 μg of cGAMP at days 4 and 8 post-surgery. The wound was closed by an autoclip wound clip system. Tumor size were measured every other day started at day 7 after tumor inoculation with a digital caliper. For the rechallenge experiment, 2×105 B16F10 cells were subcutaneously injected into the left rear flank of treated mice with complete responses or naïve mice. Tumor size was measured every other day with a digital caliper.
Treatment of an orthotopic pancreatic tumor model
A small incision was made to exteriorize the spleen and pancreas of C57BL/6 female mice. 5×105 KPC cells in 50 μL of PBS and Matrigel (1:1 mixture by volume) were injected into the tail of the pancreas. cGAMP-MPs (10 μg of cGAMP-S, 5 PLGA-1 particles containing a total of 10 μg of cGAMP, and 5 PLGA-2 particles containing a total of 10 μg of cGAMP) or 1×cGAMP-S+EP (30 μg of cGAMP-S, 5 each of empty PLGA-1 and PLGA-2 particles) in 50 μL MC solution were also injected into the tail of the pancreas. The wound was then closed by an autoclip wound clip system. Mice were euthanized 25 days after tumor inoculation. The tumors were isolated and weighed by a balance. Metastasis to lungs was evaluated by H&E staining of lung sections.
Biodegradation of PLGA-MPs
Five empty PLGA-2 microparticles were subcutaneously injected into the rear flank of SKH1-E mouse. Mice were euthanized at days 2, 8, and 30 after injection. The skin and subdermal tissue were collected and fixed in formalin-free fixative (Sigma-Aldrich) for 24 h. Tissues were then embedded in paraffin, cut into 5 μm sections, stained with H&E, and imaged using an Aperio AT2 Slide Scanner (Leica Biosystems).
Statistical analysis
All statistical analyses were performed using the GraphPad Prism software package (PRISM 8.0.2; GraphPad Software). Biological replicates were used in all experiments unless otherwise stated. Survival benefit was determined using a log-rank test. All experimental results were indicated as the means ± SD or the means ± SEM. One-way or two-way analysis of variance (ANOVA) were used when there were multiple comparisons. Student’s t test was used for single comparations. The specific statistical methods are indicated in figure legends.
Supplementary Material
Fig. S1. Additional optical and scanning electron microscopy images of microparticles.
Fig. S2. Cumulative in vitro release kinetics of AF647-dextran from PLGA-MPs.
Fig. S3. Fluorescence of AF647-dextran was quenched before release from microparticles.
Fig. S4. Representative IVIS images of mice that received AF647-dextran-loaded PLGA-MPs.
Fig. S5. Averaged release time of AF647-dextran from subcutaneously injected PLGA-MPs.
Fig. S6. Free AF647 was rapidly cleared from tumors.
Fig. S7. HPLC analysis of cGAMP before and after release.
Fig. S8. Cumulative in vitro release of 3’3’-cGAMP from hydrogels.
Fig. S9. Tumor growth and survival analysis of subcutaneous 4T1 breast tumor-bearing mice treated with cGAMP-MPs.
Fig. S10. Systemic IL-6 concentrations in orthotopic 4T1 tumor-bearing mice after treatments.
Fig. S11. Representative image of B16F10 tumors and average tumor growth curve after treatments.
Fig. S12. Gating scheme for flow cytometry measurements of myeloid and lymphoid cells in TME.
Fig. S13. Quantification of lymphocytes in the TME by flow cytometry.
Fig. S14. Immunofluorescence images of histological sections of B16F10 tumors after treatments.
Fig. S15. TUNEL staining of histological sections of B16F10 tumors after treatments.
Fig. S16. Analysis of regulatory T cells in the TME after treatments.
Fig. S17. Flow cytometry measurements of T cells in systemic circulation.
Fig. S18. Body weight analysis of mice bearing B16F10 melanoma and 4T1 breast tumors after treatments.
Fig. S19. H&E-stained sections of major organs from B16F10 tumor-bearing mice after different treatments.
Fig. S20. H&E staining images showing PLGA microparticles deformed and completely degraded over time in vivo.
Fig. S21. Cumulative in vitro release kinetics of pemetrexed and Cy5-labeled CpG DNA from PLGA-2.
Fig. S22. Representative scanning electron microscopy image of a thinner walled microparticle and cGAMP loading per particle.
Table S1. PLGA compositions and in vitro release time points of AF-647 dextran from different PLGA-MPs.
Data file S1. Individual subject-level data.
Acknowledgments:
We thank Lauren Milling from Dr. Darrell Irvine lab for her advice on orthotopic 4T1 tumor models. We thank Dr. Serguei Kozlov for providing KPC cells. We thank the Koch Institute Swanson Biotechnology Center for technical support, specifically Microscopy, Histology, Flow cytometry, and Animal Imaging & Preclinical Testing facilities.
Funding:
X. L. was supported by a Ludwig Postdoctoral Fellowship. L. M. was supported by a Misrock Postdoctoral Fellowship. K. M. was supported by an NIH Ruth L. Kirschstein National Research Service Award (F32EB022416). K. S. was supported by a Ruth L. Kirschstein NRSA Postdoctoral Fellowship (1F32EB025688-0A1). This work is supported in part by the Cancer Center Support (core) (grant no. P30-CA14051) from the National Institutes of Health.
Footnotes
Competing interests: D.G.A. is a consultant with Translate Bio, Sigilon Therapeutics, and Verseau Therapeutics. X. L., R. L., and A. J. are inventors on patent application (Micromolded particles for cancer treatment, 21543) held by Massachusetts Institute of Technology that covers the use of core-shell technology for treating cancer. K. M., A. J., and R. L. are inventors on patent applications (US 62/558,172 and US 14/572,631) submitted by Massachusetts Institute of Technology that cover the core-shell particle fabrication technology. For a list of entities with which R.L. is involved, compensated or uncompensated, see www.dropbox.com/s/yc3xqb5s8s94v7x/Rev%20Langer%20COI.pdf?dl=0. All other authors declare that they have no competing interests.
Data and materials availability: All data associated with this study are present in the paper and supplementary materials.
References and notes
- 1.Postow MA, Callahan MK, Wolchok JD, Immune checkpoint blockade in cancer therapy, J. Clin. Oncol. 33, 1974–1982 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sharma P, Allison JP, The future of immune checkpoint therapy, Science (80-. ). 348, 56–51 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, McKee M, Gajewski TF, Chemokine Expression in Melanoma Metastases Associated with CD8+ T-Cell Recruitment, Cancer Res. 69, 3077–3085 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gajewski TF, Woo S-R, Zha Y, Spaapen R, Zheng Y, Corrales L, Spranger S, Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment, Curr. Opin. Immunol. 25, 268–276 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Galon J, Pagès F, Marincola FM, Angell HK, Thurin M, Lugli A, Zlobec I, Berger A, Bifulco C, Botti G, Tatangelo F, Britten CM, Kreiter S, Chouchane L, Delrio P, Arndt H, Asslaber M, Maio M, V Masucci G, Mihm M, Vidal-Vanaclocha F, Allison JP, Gnjatic S, Hakansson L, Huber C, Singh-Jasuja H, Ottensmeier C, Zwierzina H, Laghi L, Grizzi F, Ohashi PS, Shaw PA, Clarke BA, Wouters BG, Kawakami Y, Hazama S, Okuno K, Wang E, O’Donnell-Tormey J, Lagorce C, Pawelec G, Nishimura MI, Hawkins R, Lapointe R, Lundqvist A, Khleif SN, Ogino S, Gibbs P, Waring P, Sato N, Torigoe T, Itoh K, Patel PS, Shukla SN, Palmqvist R, Nagtegaal ID, Wang Y, D’Arrigo C, Kopetz S, Sinicrope FA, Trinchieri G, Gajewski TF, Ascierto PA, Fox BA, Cancer classification using the Immunoscore: a worldwide task force, J. Transl. Med. 10, 205 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang H, Hu S, Chen X, Shi H, Chen C, Sun L, Chen ZJ, cGAS is essential for the antitumor effect of immune checkpoint blockade, Proc. Natl. Acad. Sci. 114, 1637–1642 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, Woo S-RR, Lemmens E, Banda T, Leong JJ, Metchette K, Dubensky TW, Gajewski TF, Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity, Cell Rep. 11, 1018–1030 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Q, Sun L, Chen ZJ, Regulation and function of the cGAS–STING pathway of cytosolic DNA sensing, Nat. Immunol. 17, 1142–1149 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Röhl I, Hopfner K-P, Ludwig J, Hornung V, cGAS produces a 2′−5′-linked cyclic dinucleotide second messenger that activates STING, Nature 498, 380–384 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shae D, Becker KW, Christov P, Yun DS, Lytton-Jean AKR, Sevimli S, Ascano M, Kelley M, Johnson DB, Balko JM, Wilson JT, Endosomolytic polymersomes increase the activity of cyclic dinucleotide STING agonists to enhance cancer immunotherapy, Nat. Nanotechnol. (2019), doi: 10.1038/s41565-018-0342-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E, Mechette K, Leong JJ, Lauer P, Liu W, Sivick KE, Zeng Q, Soares KC, Zheng L, Portnoy DA, Woodward JJ, Pardoll DM, Dubensky TW, Kim Y, STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade, Sci. Transl. Med. 7 (2015), doi: 10.1126/scitranslmed.aaa4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Curran E, Chen X, Corrales L, Kline DEE, Dubensky TWW, Duttagupta P, Kortylewski M, Kline J, STING Pathway Activation Stimulates Potent Immunity against Acute Myeloid Leukemia, Cell Rep. 15, 2357–2366 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luo M, Wang H, Wang Z, Cai H, Lu Z, Li Y, Du M, Huang G, Wang C, Chen X, Porembka MR, Lea J, Frankel AE, Fu YX, Chen ZJ, Gao J, A STING-activating nanovaccine for cancer immunotherapy, Nat. Nanotechnol. 12, 648–654 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.ClinicalTrails.gov, Study of MK-1454 Alone or in Combination With Pembrolizumab in Participants With Advanced/Metastatic Solid Tumors or Lymphomas (MK-1454–001) (available at https://clinicaltrials.gov/ct2/show/NCT03010176).
- 15.I. Aduro Biotech, Safety and Efficacy of MIW815 (ADU-S100) +/− Ipilimumab in Patients With Advanced/Metastatic Solid Tumors or Lymphomas (available at https://clinicaltrials.gov/ct2/show/NCT02675439).
- 16.Mathes T, Pieper D, Antoine S-L, Eikermann M, Adherence influencing factors in patients taking oral anticancer agents: A systematic review, Cancer Epidemiol. 38, 214–226 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Claxton AJ, Cramer J, Pierce C, A systematic review of the associations between dose regimens and medication compliance, Clin. Ther. 23, 1296–1310 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Puts MTE, Santos B, Hardt J, Monette J, Girre V, Atenafu EG, Springall E, Alibhai SMH, An update on a systematic review of the use of geriatric assessment for older adults in oncology Ann. Oncol. (2014), doi: 10.1093/annonc/mdt386. [DOI] [PubMed] [Google Scholar]
- 19.Osterberg L, Blaschke T, Adherence to Medication N Engl. J. Med. 353, 487–497 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Puts MTE, Tu HA, Tourangeau A, Howell D, Fitch M, Springall E, Alibhai SMH, Factors influencing adherence to cancer treatment in older adults with cancer: a systematic review., Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 25, 564–77 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tan H, Cai Q, Agarwal S, Stephenson JJ, Kamat S, Impact of adherence to disease-modifying therapies on clinical and economic outcomes among patients with multiple sclerosis, Adv. Ther. 28, 51–61 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Hobson J, Gummadidala P, Silverstrim B, Grier D, Bunn J, James T, Rincon M, Acute inflammation induced by the biopsy of mouse mammary tumors promotes the development of metastasis, Breast Cancer Res. Treat. 139, 391–401 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hansen NM, Ye X, Grube BJ, Giuliano AE, Manipulation of the Primary Breast Tumor and the Incidence of Sentinel Node Metastases From Invasive Breast Cancer, Arch. Surg. 139, 634 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Estourgie SH, Nieweg OE, Kroon BBR, High incidence of in-transit metastases after sentinel node biopsy in patients with melanoma, Br. J. Surg. 91, 1370–1371 (2004). [DOI] [PubMed] [Google Scholar]
- 25.Kamaly N, Yameen B, Wu J, Farokhzad OC, Degradable controlled-release polymers and polymeric nanoparticles: Mechanisms of controlling drug release, Chem. Rev. 116, 2602–2663 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li J, Mooney DJ, Designing hydrogels for controlled drug delivery, Nat. Rev. Mater. 1, 16071 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin C-CC, Anseth KS, PEG hydrogels for the controlled release of biomolecules in regenerative medicine, Pharm. Res. 26, 631–643 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang H, Mooney DJ, Biomaterial-assisted targeted modulation of immune cells in cancer treatment, Nat. Mater. 17, 761–772 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Kato K, Nishimasu H, Oikawa D, Hirano S, Hirano H, Kasuya G, Ishitani R, Tokunaga F, Nureki O, Structural insights into cGAMP degradation by Ecto-nucleotide pyrophosphatase phosphodiesterase 1, Nat. Commun. 9, 4424 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leach DG, Dharmaraj N, Piotrowski SL, Lopez-Silva TL, Lei YL, Sikora AG, Young S, Hartgerink JD, STINGel: Controlled release of a cyclic dinucleotide for enhanced cancer immunotherapy, Biomaterials 163, 67–75 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Junkins RD, Gallovic MD, Johnson BM, Collier MA, Watkins-Schulz R, Cheng N, David CN, McGee CE, Sempowski GD, Shterev I, McKinnon K, Bachelder EM, Ainslie KM, Ting JPY, A robust microparticle platform for a STING-targeted adjuvant that enhances both humoral and cellular immunity during vaccination, J. Control. Release 270, 1–13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE, STING is a direct innate immune sensor of cyclic di-GMP, Nature 478, 515–518 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Corrales L, McWhirter SM, Dubensky TW, Gajewski TF, The host STING pathway at the interface of cancer and immunity, J. Clin. Invest. 126, 2404–2411 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng N, Watkins-Schulz R, Junkins RD, David CN, Johnson BM, Montgomery SA, Peine KJ, Darr DB, Yuan H, McKinnon KP, Liu Q, Miao L, Huang L, Bachelder EM, Ainslie KM, Ting JP-Y, A nanoparticle-incorporated STING activator enhances antitumor immunity in PD-L1–insensitive models of triple-negative breast cancer, JCI Insight 3 (2018), doi: 10.1172/jci.insight.120638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rudqvist N-P, Pilones KA, Lhuillier C, Wennerberg E, Sidhom J-W, Emerson RO, Robins HS, Schneck J, Formenti SC, Demaria S, Radiotherapy and CTLA-4 Blockade Shape the TCR Repertoire of Tumor-Infiltrating T Cells, Cancer Immunol. Res. 6, 139–150 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marcus A, Mao AJ, Lensink-Vasan M, Wang L, Vance RE, Raulet DH, Tumor-Derived cGAMP Triggers a STING-Mediated Interferon Response in Non-tumor Cells to Activate the NK Cell Response, Immunity 49, 754–763.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iwasaki A, Medzhitov R, Control of adaptive immunity by the innate immune system, Nat. Immunol. 16, 343–353 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Han TH, Jin P, Ren J, Slezak S, Marincola FM, Stroncek DF, Evaluation of 3 Clinical Dendritic Cell Maturation Protocols Containing Lipopolysaccharide and Interferon-γ, J. Immunother. 32, 399–407 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bose D, Bose, Debojit, cGAS/STING Pathway in Cancer: Jekyll and Hyde Story of Cancer Immune Response, Int. J. Mol. Sci. 18, 2456 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vatner RE, Janssen EM, STING, DCs and the link between innate and adaptive tumor immunity, Mol. Immunol. (2017), doi: 10.1016/J.MOLIMM.2017.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ohkuri T, Kosaka A, Ishibashi K, Kumai T, Hirata Y, Ohara K, Nagato T, Oikawa K, Aoki N, Harabuchi Y, Celis E, Kobayashi H, Intratumoral administration of cGAMP transiently accumulates potent macrophages for anti-tumor immunity at a mouse tumor site, Cancer Immunol. Immunother. 66, 705–716 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shae D, Becker KW, Christov P, Yun DS, Lytton-Jean AKR, Sevimli S, Ascano M, Kelley M, Johnson DB, Balko JM, Wilson JT, Endosomolytic polymersomes increase the activity of cyclic dinucleotide STING agonists to enhance cancer immunotherapy, Nat. Nanotechnol. 14, 269–278 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jing W, McAllister D, Vonderhaar EP, Palen K, Riese MJ, Gershan J, Johnson BD, Dwinell MB, STING agonist inflames the pancreatic cancer immune microenvironment and reduces tumor burden in mouse models, J. Immunother. Cancer 7, 1–18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Berard M, Brandt K, Bulfone-Paus S, Tough DF, IL-15 promotes the survival of naive and memory phenotype CD8+ T cells., J. Immunol. 170, 5018–26 (2003). [DOI] [PubMed] [Google Scholar]
- 45.Hengel RL, Thaker V, V Pavlick M, Metcalf JA, Dennis G, Yang J, Lempicki RA, Sereti I, Lane HC, Cutting edge: L-selectin (CD62L) expression distinguishes small resting memory CD4+ T cells that preferentially respond to recall antigen., J. Immunol. 170, 28–32 (2003). [DOI] [PubMed] [Google Scholar]
- 46.Demicheli R, Retsky MW, Hrushesky WJM, Baum M, Gukas ID, The effects of surgery on tumor growth: a century of investigations, Ann. Oncol. 19, 1821–1828 (2008). [DOI] [PubMed] [Google Scholar]
- 47.Alieva M, van Rheenen J, Broekman MLD, Potential impact of invasive surgical procedures on primary tumor growth and metastasis., Clin. Exp. Metastasis 35, 319–331 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Al-Sahaf O, Wang JH, Browne TJ, Cotter TG, Redmond HP, Surgical Injury Enhances the Expression of Genes That Mediate Breast Cancer Metastasis to the Lung, Ann. Surg. 252, 1037–1043 (2010). [DOI] [PubMed] [Google Scholar]
- 49.Wang C, Sun W, Ye Y, Hu Q, Bomba HN, Gu Z, In situ activation of platelets with checkpoint inhibitors for post-surgical cancer immunotherapy, Nat. Biomed. Eng. 1 (2017), doi: 10.1038/s41551-016-0011. [DOI] [Google Scholar]
- 50.Chen Q, Wang C, Zhang X, Chen G, Hu Q, Li H, Wang J, Wen D, Zhang Y, Lu Y, Yang G, Jiang C, Wang J, Dotti G, Gu Z, In situ sprayed bioresponsive immunotherapeutic gel for post-surgical cancer treatment, Nat. Nanotechnol. (2018), doi: 10.1038/s41565-018-0319-4. [DOI] [PubMed] [Google Scholar]
- 51.Makadia HK, Siegel SJ, Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier., Polymers (Basel). 3, 1377–1397 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brown MT, Bussell JK, Medication Adherence: WHO Cares?, Mayo Clin. Proc. 86, 304–314 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haithcox S, R Ramnes C, Lee H, Lu J, Lyman GH, The impact of frequent injections for hematopoietic growth factor support on patients receiving chemotherapy: an observational study, BMC Nurs. 2, 2 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Koshy ST, Cheung AS, Gu L, Graveline AR, Mooney DJ, Liposomal Delivery Enhances Immune Activation by STING Agonists for Cancer Immunotherapy, Adv. Biosyst 1, 1600013 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Farra R, Sheppard NF, McCabe L, Neer RM, Anderson JM, Santini JT, Cima MJ, Langer R, First-in-Human Testing of a Wirelessly Controlled Drug Delivery Microchip, Sci. Transl. Med. 4, 122ra21–122ra21 (2012). [DOI] [PubMed] [Google Scholar]
- 56.Formiga FR, Pelacho B, Garbayo E, Abizanda G, Gavira JJ, Simon-Yarza T, Mazo M, Tamayo E, Jauquicoa C, Ortiz-de-Solorzano C, Prósper F, Blanco-Prieto MJ, Sustained release of VEGF through PLGA microparticles improves vasculogenesis and tissue remodeling in an acute myocardial ischemia–reperfusion model, J. Control. Release 147, 30–37 (2010). [DOI] [PubMed] [Google Scholar]
- 57.Shahani K, Panyam J, Highly Loaded, Sustained-Release Microparticles of Curcumin for Chemoprevention, J. Pharm. Sci. 100, 2599–2609 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yeo Y, Park K, Control of encapsulation efficiency and initial burst in polymeric microparticle systems Arch. Pharm. Res. 27, 1–12 (2004). [DOI] [PubMed] [Google Scholar]
- 59.Nilsson B, Ekdahl KN, Mollnes TE, Lambris JD, The role of complement in biomaterial-induced inflammation, Mol. Immunol. 44, 82–94 (2007). [DOI] [PubMed] [Google Scholar]
- 60.Amini AR, Wallace JS, Nukavarapu SP, Short-term and long-term effects of orthopedic biodegradable implants., J. Long. Term. Eff. Med. Implants 21, 93–122 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ceonzo K, Gaynor A, Shaffer L, Kojima K, Vacanti CA, Stahl GL, Polyglycolic Acid-Induced Inflammation: Role of Hydrolysis and Resulting Complement Activation, Tissue Eng. 12, 301–308 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sivick KE, Desbien AL, Glickman LH, Reiner GL, Corrales L, Surh NH, Hudson TE, Vu UT, Francica BJ, Banda T, Katibah GE, Kanne DB, Leong JJ, Metchette K, Bruml JR, Ndubaku CO, McKenna JM, Feng Y, Zheng L, Bender SL, Cho CY, Leong ML, van Elsas A, Dubensky TW, McWhirter SM, Magnitude of Therapeutic STING Activation Determines CD8 + T Cell-Mediated Anti-tumor Immunity, Cell Rep. 25, 3074–3085.e5 (2018). [DOI] [PubMed] [Google Scholar]
- 63.Sivick KE, Desbien AL, Glickman LH, Reiner GL, Corrales L, Surh NH, Hudson TE, Vu UT, Francica BJ, Banda T, Katibah GE, Kanne DB, Leong JJ, Metchette K, Bruml JR, Ndubaku CO, McKenna JM, Feng Y, Zheng L, Bender SL, Cho CY, Leong ML, van Elsas A, Dubensky TW, McWhirter SM, Magnitude of Therapeutic STING Activation Determines CD8+ T Cell-Mediated Anti-tumor Immunity, Cell Rep 25, 3074–3085.e5 (2018). [DOI] [PubMed] [Google Scholar]
- 64.Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, Woo SR, Lemmens E, Banda T, Leong JJ, Metchette K, Dubensky TW, Gajewski TF, Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity, Cell Rep. 11, 1018–1030 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Aznar MA, Tinari N, Rullán AJ, Sánchez-Paulete AR, Rodriguez-Ruiz ME, Melero I, Intratumoral Delivery of Immunotherapy—Act Locally, Think Globally, J. Immunol. 198, 31–39 (2017). [DOI] [PubMed] [Google Scholar]
- 66.Convery N, Gadegaard N, 30 years of microfluidicsMicro Nano Eng. 2, 76–91 (2019). [Google Scholar]
- 67.McHugh KJ, Nguyen TD, Linehan AR, Yang D, Behrens AM, Rose S, Tochka ZL, Tzeng SY, Norman JJ, Anselmo AC, Xu X, Tomasic S, Taylor MA, Lu J, Guarecuco R, Langer R, Jaklenec A, Fabrication of fillable microparticles and other complex 3D microstructures, Science (80-. ). 357, 1138–1142 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Additional optical and scanning electron microscopy images of microparticles.
Fig. S2. Cumulative in vitro release kinetics of AF647-dextran from PLGA-MPs.
Fig. S3. Fluorescence of AF647-dextran was quenched before release from microparticles.
Fig. S4. Representative IVIS images of mice that received AF647-dextran-loaded PLGA-MPs.
Fig. S5. Averaged release time of AF647-dextran from subcutaneously injected PLGA-MPs.
Fig. S6. Free AF647 was rapidly cleared from tumors.
Fig. S7. HPLC analysis of cGAMP before and after release.
Fig. S8. Cumulative in vitro release of 3’3’-cGAMP from hydrogels.
Fig. S9. Tumor growth and survival analysis of subcutaneous 4T1 breast tumor-bearing mice treated with cGAMP-MPs.
Fig. S10. Systemic IL-6 concentrations in orthotopic 4T1 tumor-bearing mice after treatments.
Fig. S11. Representative image of B16F10 tumors and average tumor growth curve after treatments.
Fig. S12. Gating scheme for flow cytometry measurements of myeloid and lymphoid cells in TME.
Fig. S13. Quantification of lymphocytes in the TME by flow cytometry.
Fig. S14. Immunofluorescence images of histological sections of B16F10 tumors after treatments.
Fig. S15. TUNEL staining of histological sections of B16F10 tumors after treatments.
Fig. S16. Analysis of regulatory T cells in the TME after treatments.
Fig. S17. Flow cytometry measurements of T cells in systemic circulation.
Fig. S18. Body weight analysis of mice bearing B16F10 melanoma and 4T1 breast tumors after treatments.
Fig. S19. H&E-stained sections of major organs from B16F10 tumor-bearing mice after different treatments.
Fig. S20. H&E staining images showing PLGA microparticles deformed and completely degraded over time in vivo.
Fig. S21. Cumulative in vitro release kinetics of pemetrexed and Cy5-labeled CpG DNA from PLGA-2.
Fig. S22. Representative scanning electron microscopy image of a thinner walled microparticle and cGAMP loading per particle.
Table S1. PLGA compositions and in vitro release time points of AF-647 dextran from different PLGA-MPs.
Data file S1. Individual subject-level data.