

Abstract
Targeted sequencing enables sensitive and cost-effective analysis by focusing resources on molecules of interest. Existing methods, however, are limited in enrichment power and target capture length. Here, we present a novel method that uses compound nucleic acid cytometry to achieve million-fold enrichments of molecules >10 kbp in length using minimal prior target information. We demonstrate the approach by sequencing HIV proviruses in infected individuals. Our method is useful for rare target sequencing in research and clinical applications, including for identifying cancer-associated mutations or sequencing viruses infecting cells.
Introduction
Target enrichment focuses valuable sequencing on important molecules and is useful when the sample comprises a large background of uninteresting DNA.1 For instance, characterizing HIV genomic diversity is important for understanding persistent infection, but under treatment viral DNA is outnumbered by human DNA by billions of times.2−4 In metagenomic analyses, organisms of interest may be present at a few percent,5−7 while in a human genetic disease, variants may be present at fractions of a percent.8−10 In instances such as these, sequencing all DNA is wasteful because only a fraction of reads corresponds to the region of interest. The most common target enrichment strategies are based on polymerase chain reaction (PCR) amplification or hybridization capture.1,11,12 PCR methods recover only the amplified portion and miss information beyond primers.13,14 Hybridization capture recovers information extending beyond probes but can require hundreds of probes;15,16 this necessitates considerable prior information for probe design, which is often unavailable, especially when little information is known about the region of interest, such as in the novel microbe or genetic lesion sequencing.17,18
Nucleic acid cytometry (NAC) is a conceptually novel approach to target enrichment based on droplet microfluidics.19 The overarching principle is to physically isolate molecules by hydrodynamic sorting. Target identification is accomplished using droplet PCR, while isolation is accomplished by sorting positive droplets.19−22 The approach is akin to querying a diverse mixture for keyword subsequences and isolating all molecules containing the keyword. The critical factor in NAC enrichment is sensitivity for recovering the target of interest. Sensitivity, in turn, is limited by the number of droplet PCRs that can be sorted which, presently, is ∼10 million. Considering losses in DNA recovery and the need for sufficient material to perform sequencing, current enrichments are capped to ∼30,000, allowing NAC to maximally concentrate the target by this factor.5,8,23,24 This enrichment is insufficient for applications with ultrarare targets below one in a million. To broaden the applicability of NAC, a strategy to increase enrichment power is needed.
In this study, we demonstrate the ability to perform NAC repeatedly on a sample to achieve compound enrichment over multiple rounds. The final enrichment is the product of each round, allowing a ∼6 million-fold enrichment over two rounds. This is ∼200-fold higher than enrichments with the next best technology, and it affords the ability of recovering single virus genomes.8,15,23 To demonstrate the approach, we use it to isolate and sequence single HIV genomes from infected individuals. Because of their limited enrichment power, previous approaches lack the sensitivity to recover and sequence such rare single virus genomes. Compound NAC provides a general platform for recovering long, ultrarare molecules with minimal prior sequence information.
Experimental Section
Microfluidic Device Fabrication
The microfluidic devices were fabricated in polydimethylsiloxane (PDMS) using standard soft lithography. Photomasks designed by AutoCAD were printed on transparencies and the features on the photomask were transferred to a silicon wafer (University Wafer) using a negative photoresist (MicroChem, SU-8 2025) by UV photolithography. A PDMS (Dow Corning, Sylgard 184) prepolymer mixture of polymer and cross-linker at a ratio of 10:1 was poured over the pattered silicon wafer and cured in a 65 °C oven for 2 h. The PDMS replica was peeled off and punched for inlets and outlets by a 0.75 mm biopsy core (World Precision Instruments). The PDMS slab was bound to a clean glass using an oxygen plasma cleaner (Harrick Plasma), followed by baking at 65 °C for 30 min to ensure strong bonding between the PDMS and glass. The microfluidic channels were treated with Aquapel (PPG Industries) and baked at 65 °C overnight for hydrophobicity.
Droplet TaqMan PCR
50 fg of ΦX 174 virion DNA and 500 ng of lambda DNA (New England BioLabs) were added to 150 μL of PCR reagents containing 1× Platinum Multiplex PCR Master Mix (Life Technologies, catalog no. 4464269), 200 nM TaqMan probe (IDT), 1 μM forward primer and 1 μM reverse primer (IDT), 2.5% (w/w) Tween 20 (Fisher Scientific), 2.5% (w/w) poly(ethylene glycol) 6000 (Sigma-Aldrich), and 0.8 M 1,2-propanediol (Sigma-Aldrich). Tween 20 and poly(ethylene glycol) 6000 were used to increase the stability of droplets during thermal cycling.22 1,2-Propanediol was used as a PCR enhancer when low temperature was used for denaturation.25 Two syringes backfilled with HFE-7500 fluorinated oil (3M, catalog no. 98-0212-2928-5) were loaded with (1) TaqMan PCR reaction mix and (2) HFE-7500 oil with a 2% (w/w) PEG–PFPE amphiphilic block copolymer surfactant (RNA Biotechnologies, catalog no. 008-FluoroSurfactant-1G). The aqueous phase and oil phase were injected into a flow-focus droplet maker at controlled flow rates (400 μL/h for the PCR mix and 800 μL/h for the oil phase) sustained by computer-programmed syringe pumps (New Era). ∼3 million monodispersed droplets (diameter ∼40 μm) were generated and collected in PCR tubes via polyethylene tubing. The bottom oil was then removed and replaced with FC-40 fluorinated oil (Sigma-Aldrich, catalog no. 51142-49-5) with a 5% (w/w) PEG–PFPE amphiphilic block copolymer surfactant for better droplet stability before putting the emulsion into a thermal cycler (Bio-Rad, T100 model). Thermal cycling was performed at the following conditions: 2 min 30 s at 86 °C; 35 cycles of 30 s at 86 °C, 1 min 30 s at 60 °C, and 30 s at 72 °C; and a final extension of 5 min at 72 °C. A low denaturation temperature of 86 °C was used to minimize DNA fragmentation. After PCR, a small aliquot of drops was visualized with an EVOS inverted fluorescence microscope. Another small aliquot of drops was taken and broken with a 10% (v/v) solution of perfluoro-octanol (Sigma-Aldrich, catalog no. 370533) and an addition of 10 μL of deionized (DI) water, followed by gentle vortexing for 5 s and centrifuging for 1 min at 500 rpm. The recovered DNA in water, denoted as “unsort”, was saved for later measurement of the enrichment factor.
Dielectrophoretic Sorting
The thermocycled drops were transferred to a 1 mL syringe and reinjected to a microfluidic dielectrophoretic (DEP) sorter (Figure 2) at 50 μL/h.8,20 The syringe was placed vertically so that the drops remained at the top and closely packed. Individual drops were separated after entering the sorter by a spacer oil of HFE-7500 with a flow rate of 950 μL/h. Another stream of HFE-7500 oil at 1000 μL/h was introduced at the sorting junction to drive the drops to waste collection when the DEP force was off. A syringe at −1000 μL/h was used to produce a negative pressure at the waste collection to further ensure that unsorted drops flowed to waste. The salt water electrodes and moat shielding were filled with 2 M NaCl solution. A laser of 100 mW, 532 nm was focused upstream of the sorting junction to excite droplet fluorescence. Photomultiplier tubes (PMTs, Thorlabs, PMM01 model) were focused on the same spot to measure the emission fluorescence. A data acquisition card (FPGA card) and a LabVIEW program (available at GitHub: https://github.com/AbateLab/sorter-code) (National Instruments) were used to collect PMT outputs and activate the salt electrode when the emission fluorescence intensity is higher than a preset threshold. A high-voltage amplifier (Trek) was used to amplify the electrode pulse to 0.8–1 kV for DEP sorting. The sorted drops were collected into a 1.5 mL Eppendorf DNA LoBind tube.
Figure 2.
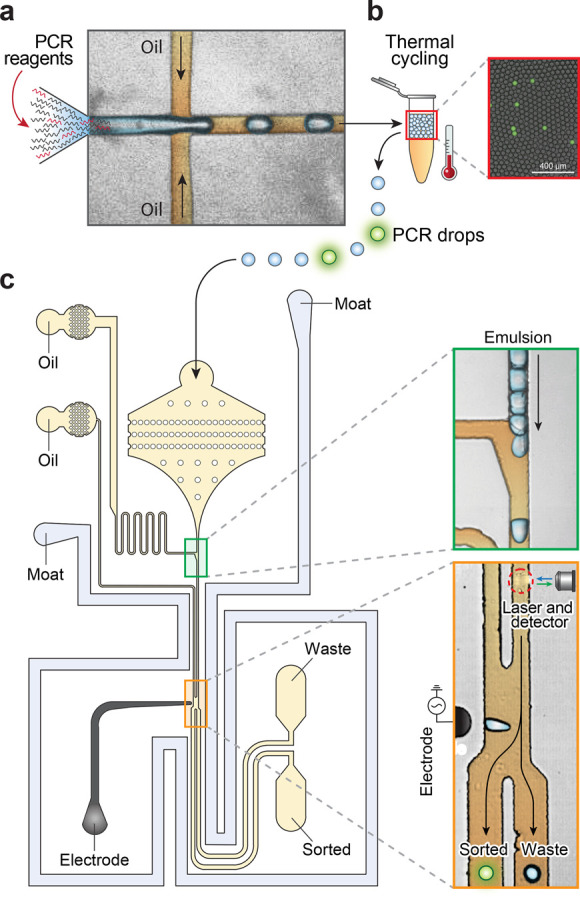
Microfluidic devices of NAC. (a) Droplet encapsulation of DNA and PCR reagents. (b) In-drop PCR to generate fluorescence when the target is present. The merged bright field/fluorescence image shows a representative sample post thermocycling. (c) Fluorescence-activated sorting selects droplets containing target sequences. Scale bars: 400 μm.
DNA Recovery and Second Round of Enrichment
DNA from sorted drops was recovered by breaking the emulsion with 10% (v/v) solution of perfluoro-octanol (Sigma-Aldrich, catalog no. 370533) and the addition of 20 μL of DI water, followed by gentle vortexing for 5 s and centrifugation for 1 min at 500 rpm. 2 μL of the recovered DNA, denoted as “single sort”, was saved for later measurement of the enrichment factor by qPCR. The remaining 18 μL recovered DNA was processed with a second round of droplet TaqMan PCR and DEP sorting as described above. After sorting, the sorted drops were broken and the recovered DNA, denoted as “double sort”, was used to measure the degree of enrichment.
Quantitative PCR Analysis of Sorted Droplets
We used a multiplex TaqMan PCR, with one FAM-based probe targeting ΦX 174 DNA and one Cy5-based probe targeting lambda DNA to quantify ΦX 174 and lambda DNA in “unsort”, “single sort”, and “double sort”. The PCR was set as follows: 1× Platinum Multiplex PCR Master Mix, 200 nM TaqMan probes, 1 μM forward primers and 1 μM reverse primers (IDT), recovered DNA, and DNAse-free water to bring the volume to 25 μL. The PCR was performed in a QuantStudio 5 Real-Time PCR System (Thermo Fisher Scientific) using the following parameters: 95 °C for 2 min; 40 cycles of 95 °C for 30 s, 60 °C for 90 s, and 72 °C for 30 s. Ct values for each sample were obtained and used to compute the enrichment factor. All primer and TaqMan probe sequences are listed in Table S2. The TaqMan assays were tested for specificity and linearity by constructing a serial dilution of the ΦX 174 DNA with a fixed concentration of lambda DNA. We obtained two Ct values for ΦX 174 (FAM) and lambda (Cy5) for each of the “unsort”, “single sort”, and “double sort” samples to compute the enrichment factors for each round of sorting.
HIV-Associated DNA Sample Preparation
The HIV-infected cells were prepared by plating resting CD4 T cells from an ART-treated person at ∼1 infected cell per 5 wells (∼100 total cells per well), followed by stimulation and a period of in vitro culture to allow proliferation.26 Non-HIV-infected Jurkat cells (ATCC TIB-152) were cultured following the provided protocol. DNA was extracted from clonally expanded cells (from one well of the culture plate) and from Jurkat cells using Quick-DNA Miniprep Plus Kits (Zymo research, catalog no. D4068) according to the manufacturer’s instructions and mixed at a 1:30 ratio.
Compound Enrichment of Single HIV Genomes
The DNA mixture was processed with droplet TaqMan PCR and 1st DEP sorting as described above using the HIV pol specific TaqMan probe and biotinylated primers. All primer and TaqMan probe sequences for HIV are listed in Table S3. The sorted emulsions were broken using perfluoro-1-octanol, and the aqueous fraction was diluted in 5 μL of H2O. The aqueous layer containing sorted DNA was then added to streptavidin-conjugated magnetic beads (Dynabeads MyOne Streptavidin C, Thermo Fisher Scientific) and incubated for 15 min. D1 buffer from the REPLI-g single cell kit (Qiagen, catalog no. 150343) was added to denature the DNA. Biotinylated primers and amplicons were attached to the magnetic beads and removed after transferring the supernatant to a fresh tube. The multiple-displacement amplification (MDA) reaction mixture was then prepared with a REPLI-g single cell kit by following the manufacturer’s protocol and emulsified by a flow-focus droplet maker (diameter ∼20 μm) as described above. The emulsion was collected in a 1 mL syringe and incubated at 30 °C for 20 h. After incubation, MDA droplets and 2nd TaqMan PCR reagents were injected into a microfluidic merger device.27 PCR reagent drops were formed on a chip and merged with MDA drops pairwise. Merging was achieved at a salt electrode connected to a cold cathode fluorescent inverter and a DC power supply (Mastech) to generate a ∼2 kV AC signal from a 2 V input voltage. The merged drops (diameter: 40 μm) were collected to PCR tubes. The bottom oil layer was removed and replaced with FC-40 fluorinated oil with a 5% (w/w) PEG–PFPE surfactant for thermal cycling: 3 min at 86 °C; 35 cycles of 30 s at 86 °C, 90 s at 60 °C, and 30 s at 72 °C; and finally, 5 min at 72 °C. After PCR, the drops were reinjected into a DEP sorter for the second sorting round as described above.
Library Preparation and Sequencing from Sorted Droplets
The sorted single droplets with their carrier oil were collected into individual PCR tubes and dried out in a vacuum chamber. 1 μL of DI H2O was added to dissolve the sorted DNA. The dissolved DNA was then tagmented using 0.6 μL of the TD Tagmentation buffer and 0.3 μL of the ATM Tagmentation enzyme from the Nextera DNA Library Prep Kit (Illumina, catalog no. FC-121-1030) for 5 min at 55 °C. 1 μL of the NT buffer was added to neutralize the tagmentation. The tagmented DNA was then mixed with a PCR solution containing 1.5 μL of the NPM PCR master mix, 0.5 μL of each index primers i5 and i7 from the Nextera Index Kit (Illumina, catalog no. FC-121-1011), and 1.5 μL of H2O and placed on a thermal cycler with the following program: 3 min at 72 °C; 30 s at 95 °C; 20 cycles of 10 s at 95 °C, 30 s at 55 °C, and 30 s at 72 °C; and finally, 5 min at 72 °C. The DNA library was purified using a DNA Clean & Concentrator-5 kit (Zymo Research, catalog no. D4004), size-selected for 200–600 bp fragments using Agencourt AMPure XP beads (Beckman Coulter), and quantified using the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific) and the High Sensitivity DNA Bioanalyzer chip (Agilent). The library was sequenced using Illumina Miseq and ∼1 million paired-end reads of 150 bp were used for each sorted droplet or unsorted starting sample. Sequencing reads were mapped to the HIV reference genome (HXB2) using Bowtie 2.28 Genomic coverage as a function of genome position was generated using SAMtools.29 HIV genome average coverage with or without duplicates removing by Picard (MarkDuplicates) (http://broadinstitute.github.io/picard/) was calculated using SAMtools.29 TaqMan amplicon regions were excluded in the average coverage calculation. The non-HIV regions of chimeric reads were extracted using extractSoftclipped (https://github.com/dpryan79/SE-MEI) and analyzed by a web-based tool for integration sites (https://indra.mullins.microbiol.washington.edu/integrationsites/).
Results and Discussion
NAC isolates molecules of interest from a mixed population based on specific sequence biomarkers.19 This is achieved by combining the droplet TaqMan PCR identification and microfluidic droplet sorting to physically isolate molecules based on the TaqMan signal (Figure 1a). The DNA mixture is partitioned at the limiting dilution such that individual droplets rarely contain more than one target. The purity of the target sequence before sorting Pbefore and after sorting Pafter
| 1 |
| 2 |
where NT is the number of target molecules (positively sorted drops), NO is the total number of off-target molecules, D is the total number of droplets, and f is the assay false positive rate. Thus, the enrichment power E is (derivation in Supporting Information)
| 3 |
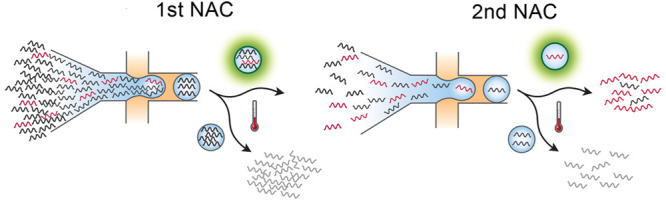
Figure 1.

Schematic of compound NAC workflow. (a) A mixed DNA sample is sorted in the first NAC round using TaqMan targeting a desired sequence biomarker. Each NAC round comprises (i) DNA encapsulation with TaqMan reagents; (ii) in-droplet PCR to generate fluorescence when the target is present; (iii) sorting to select positive drops; and (iv) recovery of sorted DNA by droplet demulsification. (b) DNA recovered from the first NAC round is diluted and processed through another round consisting of the same steps (v–viii). (c) The double-enriched DNA is analyzed by qPCR to estimate enrichment and sequenced.
Because the false positive rate is generally small (∼10–4) and difficult to reduce,30 the best way to increase the enrichment power is to encapsulate the sample into more droplets, which thus delivers fewer co-encapsulated off-target molecules per sorted positive. However, the number of droplets that can be sorted is limited to ∼10 million.8,19 It is possible to increase D in several ways, including implementing faster droplet sorting with gapped dividers, batched sorting, and novel sorting mechanisms.31 However, while promising, none have yet been demonstrated for this purpose and doing so would introduce risk and require additional development. Consequently, the maximum practical enrichment that can be achieved per NAC round is ∼104.
Similar to hybridization capture, NAC does not fragment the original target molecules. However, in contrast to hybridization capture, NAC can recover long intact targets (>100 kbp) present in a sample over a wide range of DNA concentrations.8 These features allow it to be performed repeatedly on a sample such that compound enrichment is achieved (Figure 1b). In such a strategy, the overall enrichment with two rounds Ecompound is
| 4 |
when using D1 drops in the first round and D2 drops in the second. Compound enrichment thus allows marked increases to enrichment compared to sorting more drops in a single round. For example, for a total of ∼107 drops sorted, one round typically achieves ∼103 enrichment of 10,000 target molecules, while two consecutive rounds achieve ∼106. Obtaining such an enrichment with a single-round of NAC would require sorting over a billion droplets, which is impractical. The resultant concentrated DNA is intact and readily amenable to qPCR or sequencing analysis (Figure 1c).
Microfluidic Workflow for Compound Enrichment
NAC uses ultrahigh-throughput microfluidics to perform, analyze, and sort millions of PCRs. Flow focusing loads sample DNA with TaqMan reagents in ∼45 μm droplets at ∼2.5 kHz, partitioning the entire 150 μL reaction in ∼3 million drops in ∼20 min (Figure 2a). The drops are thermocycled, generating TaqMan fluorescence when the target is present (Figure 2b). The drops are analyzed and sorted using a laser-induced fluorescence detector and a DEP droplet deflector27,32 (Figure 2c). We operate this integrated device at ∼400 Hz to ensure accurate sorting and efficient positive recovery, screening ∼3 million drops in ∼2 h. The sorted target molecules are recovered by droplet demulsification with perfluoro-octanol32 and diluted into new TaqMan reagents for the next round of NAC.
Compound Enrichment of the ΦX 174 Virus
To demonstrate the power of compound NAC, we apply it to enrich ΦX 174 viral genomes from a 107-fold greater background of lambda DNA. The TaqMan set used in each round detects a different region of the ΦX 174 genome, preventing amplicons carried over from the first round generating false positives in the second (Figure 3a). Both sets reliably detect ΦX 174 DNA (Figure S1). We set the target concentration such that ∼0.3% droplets are expected to be positive and observe an actual positive rate of 0.24% in the first round (Figure 3b(i)), yielding an enrichment of ∼400. This input concentration also generates less than 0.001% multiplets, in accordance with Poisson statistics. We screen a total of ∼3 million droplets, collecting ∼7000 positives. The recovered DNA is diluted into a fresh reaction buffer again to achieve another 400-fold enrichment and subjected to another round of NAC, collecting ∼5000 positives (Figure 3b(ii)). Because the method is nondestructive (Figure S2), the number of positive droplets should be equal for both rounds, but sample loss during transfer and preparation for the second round results in a ∼30% positive reduction (Figure 3b(iii)). To confirm the enrichment, we use qPCR to measure the fractions of ΦX 174 and lambda DNA in the sorted samples. After a single round, the qPCR curve for ΦX 174 shifts to lower cycles by ∼5 (concentrated), while that for lambda shifts to higher cycles by ∼3 (diluted), illustrating enrichment (Figure 3c(i)). For two rounds compounded, the shifts are greater (−2 for ΦX 174 and +12 for lambda) (Figure 3c(ii)). To quantify the enrichments, we calculate the enrichment factor E based on the cross-threshold values of the qPCR curves (Table S1).20 For one round of sorting ∼3 million droplets, the estimated enrichment is ∼150. For two rounds of sorting comprising a total of ∼6 million droplets, the enrichment is ∼16,000. Achieving this enrichment in one round would require sorting ∼300 million droplets, totaling 16 mL of PCR reagent, and a week of nonstop sorting.
Figure 3.

Enrichment of the ΦX 174 DNA from a background of lambda DNA with compound NAC. (a) TaqMan assays detect droplets containing ΦX 174 (green) and lambda (red) DNA. (b) The microfluidic sorter interrogates the droplets for fluorescence and sorts PCR positives. (i) Scatter plot of fluorescence vs size of drops from the first NAC round, with 0.24% positive. (ii) DNA from the first round is recovered, diluted, and processed again. (iii) Scatter plot of fluorescence vs size of drops from the second NAC round, with 0.17% positive. (c) qPCR plots for (i) single- and (ii) double-enriched DNA; based on curve shifts, single-round sorting enriches ΦX 174 by ∼150-fold and double-round sorting by ∼16,000-fold. Inset in (i) shows ΦX 174 and lambda standard curves.
Single Genome Sequencing of Ultrarare HIV Proviruses
During effective antiretroviral therapy, HIV persists in a latent state and circulates at extremely low levels, with human DNA outnumbering it by over a billion-fold.2,3,26 Under such circumstances, unbiased sequencing would recover a minute fraction of one viral genome per human genome sequenced. To obtain comprehensive information on the genetics of HIV under such circumstances, potent enrichment of the virus is needed. Due to limited information on HIV genomes in a specific sample, capture probe design is challenging. In addition, hybridization methods require considerable input DNA and do not provide single provirus information. Long-range PCR methods are only applicable when specific primers can be designed to target known conserved sequences within the virus or host; this information is often not available for novel virus integrations and precludes the recovery of unknown integrations. The only effective strategy presently available is terminal dilution PCR in well plates.3,4,33 This brute force approach aliquots thousands of cells in hundreds of microwells using long-ranged multiprimer amplification to obtain near full-length HIV genomes. However, in addition to often generating artifacts that can confound analysis, the approach does not obtain the crucial virus–host junction with the complete virus genome in a single contig. Without this dual information, specific proviruses cannot be related to host insertion sites and thereby the combination associated to disease behavior.3,34 Consequently, other strategies must be employed to infer viral genome and host junction relationships.33,34
Due to the potent enrichment enabled by compound NAC and its ability to recover intact DNA fragments spanning integrated HIV genomes, such analyses are possible. To demonstrate this, we use compound NAC to isolate and sequence HIV proviruses from a patient-infected cell expansion.26 This clonal lineage of infected cells contains one HIV provirus per ∼100 cells, each bearing the same provirus. To demonstrate the enrichment power of compound NAC, we dilute the sample with non-HIV cell gDNA at a 1:30 ratio. This dilution models the concentration of the latent infection. Due to the extreme rarity of HIV DNA in this sample, we use a multiplexed TaqMan PCR targeting multiple conserved regions of HIV in the second cycle for accurate detection and isolation before sequencing (Figure 4a). The DNA mixture is encapsulated in droplets and the ones containing HIV genomes are isolated. Microfluidic enrichment and pooled sequencing of DNA collected from many droplets have been used to characterize rare genome sequences, but isolation and sequencing of single viruses are impossible with this approach.5,23 By incorporating in-droplet MDA before the second round,35 each sorted droplet yields ∼3 pg DNA, just enough for sequencing; this enables the linkage of a specific HIV genome to its integration site (Figure 4b). This novel workflow affords superior enrichment and single drop sequencing, allowing recovery of integrated provirus genomes (Figure 4c, first to third row). We thus identify the integration site in the host gene ARIH2 by extracting virus–human chimeric reads from the sequencing data. Shearing during PCR and DNA preparation results in a partial genome dropout. Thus, by assembling reads from three droplets, we obtain complete coverage of the full-length viral genome (Figure 4c, fourth row). In total, sequencing after two rounds detects ∼6 million times more proviral reads compared to the initial sample, with an average coverage of ∼200× for the targeted proviral genome and ∼0.1× for the untargeted human genome. Discarding duplicates reduces the coverage of the targeted region to 60× but is not always necessary.36 These results illustrate that compound NAC enables the sequencing of extremely rare HIV proviruses and that the enriched molecules retain information on the genetic context of the integration.
Figure 4.
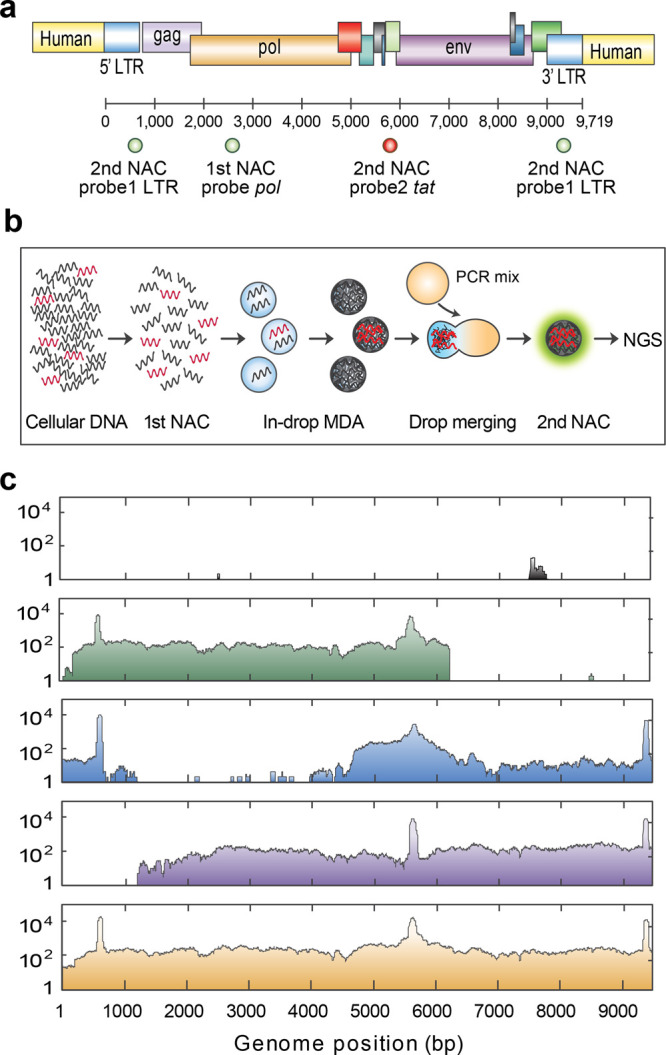
Compound enrichment and sequencing of HIV proviruses in infected individuals. (a) The first round of NAC uses a TaqMan assay targeting the pol gene (FAM). The second round of NAC uses a degenerate TaqMan assay targeting the long terminal repeat (LTR, FAM) and tat gene (Cy5). (b) Implementation of in-droplet MDA before the second round generates sufficient material for single droplet sequencing. The MDA droplet is merged with a droplet containing PCR reagents for TaqMan detection in the second round. (c) HIV genome and integration site coverage maps for nonenriched sample (top panel) and three sorted individual drops. Aggregating all data (bottom panel) assembles the full-length HIV genome including the human integration site. The peaks at LTR and tat are the TaqMan PCR amplicons.
By leveraging droplet microfluidics, NAC enables enrichment of target molecules containing sequence biomarkers. By processing the sample repeatedly, feeding the output of the first round into the input of the second, compound enrichment is achieved, allowing the recovery of ultrarare targets. In single-round NAC, maximum enrichment is limited by the false positive droplet rate. Partitioning the sample into more droplets enhances enrichment in a linear fashion but does not allow the marked increases required for ultrarare targets. Moreover, such brute force also increases cost and processing time and becomes impractical beyond enrichments of 30,000.8,23 Our method eliminates the need for large numbers of droplets and increases the maximum enrichment to 109 fold, allowing highly specific target recovery. A drawback of the multiround NAC is sample loss during transfer and processing. We observe ∼30% loss between the two NAC rounds, which may result in the loss of low-abundant variants. Nevertheless, recovery of these variants by single-round NAC is essentially impossible, so compound enrichment is still the best approach for analysis of rare sequences.
Conclusions
Compound NAC allows the isolation of long molecules with minimal prior sequence information, opening new avenues in target enrichment. For example, million-fold enrichment of >100 kbp molecules is useful for a variety of ultrarare target applications, including characterizing novel human genetic mutations or natural product gene clusters in metagenomic samples.5,8 Additionally, the approach is generalizable to other targets because it uses TaqMan PCR to define the sequence biomarker of capture and thus can be applied to any nucleic acid detectable by this assay, including RNA by adding a reverse transcription step; this would allow sequencing of fusion genes or low-abundant variants. Finally, as we have shown, implementation of in-droplet MDA allows sequencing of compound-enriched single molecules, making it a powerful tool for single virus genomics.
Acknowledgments
We thank James I. Mullins at the University of Washington for providing HIV-infected cells. We also thank members of the Abate lab, in particular Leqian Liu, Cyrus Modavi, David J. Sukovich, and Samuel C. Kim, for helpful discussions. This work was supported by the Chan Zuckerberg Biohub and the National Institutes of Health (NIH) (grant nos. R01-EB019453-01 and R01-HG008978-01).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.0c04749.
Derivation of equations for enrichment power, images and plots showing that TaqMan sets reliably detect ΦX 174 DNA in compound NAC, gel analysis showing minimal DNA fragmentation during thermo cycling, Ct values from the qPCR plots for single and double enrichment, and primers and probes for the compound enrichment of ΦX 174 virus DNA and HIV provirus DNA (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Mamanova L.; Coffey A. J.; Scott C. E.; Kozarewa I.; Turner E. H.; Kumar A.; Howard E.; Shendure J.; Turner D. J. Target-enrichment strategies for next-generation sequencing. Nat. Methods 2010, 7, 111–118. 10.1038/nmeth.1419. [DOI] [PubMed] [Google Scholar]
- Finzi D.; Blankson J.; Siliciano J. D.; Margolick J. B.; Chadwick K.; Pierson T.; Smith K.; Lisziewicz J.; Lori F.; Flexner C.; Quinn T. C.; Chaisson R. E.; Rosenberg E.; Walker B.; Gange S.; Gallant J.; Siliciano R. F. Latent infection of CD4(+) T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 1999, 5, 512–517. 10.1038/8394. [DOI] [PubMed] [Google Scholar]
- Einkauf K. B.; Lee G. Q.; Gao C.; Sharaf R.; Sun X.; Hua S.; Chen S. M. Y.; Jiang C.; Lian X.; Chowdhury F. Z.; Rosenberg E. S.; Chun T.-W.; Li J. Z.; Yu X. G.; Lichterfeld M. Intact HIV-1 proviruses accumulate at distinct chromosomal positions during prolonged antiretroviral therapy. J. Clin. Invest. 2019, 129, 988–998. 10.1172/jci124291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiener B.; Horsburgh B. A.; Eden J.-S.; Barton K.; Schlub T. E.; Lee E.; von Stockenstrom S.; Odevall L.; Milush J. M.; Liegler T.; Sinclair E.; Hoh R.; Boritz E. A.; Douek D.; Fromentin R.; Chomont N.; Deeks S. G.; Hecht F. M.; Palmer S. Identification of Genetically Intact HIV-1 Proviruses in Specific CD4(+) T Cells from Effectively Treated Participants. Cell Rep. 2017, 21, 813–822. 10.1016/j.celrep.2017.09.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P.; Modavi C.; Demaree B.; Twigg F.; Liang B.; Sun C.; Zhang W.; Abate A. R. Microfluidic automated plasmid library enrichment for biosynthetic gene cluster discovery. Nucleic Acids Res. 2020, 48, e48 10.1093/nar/gkaa131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suenaga H. Targeted metagenomics: a high-resolution metagenomics approach for specific gene clusters in complex microbial communities. Environ. Microbiol. 2012, 14, 13–22. 10.1111/j.1462-2920.2011.02438.x. [DOI] [PubMed] [Google Scholar]
- Sharon I.; Banfield J. F. Genomes from Metagenomics. Science 2013, 342, 1057–1058. 10.1126/science.1247023. [DOI] [PubMed] [Google Scholar]
- Eastburn D. J.; Huang Y.; Pellegrino M.; Sciambi A.; Ptacek L. J.; Abate A. R. Microfluidic droplet enrichment for targeted sequencing. Nucleic Acids Res. 2015, 43, e86 10.1093/nar/gkv297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X.; Ju X. C.; Yi X.; Zhu Q.; Qu N.; Liu T. F.; Chen Y.; Jiang H.; Yang G. H.; Zhen R.; Lan Z. Z.; Qi M.; Wang J. M.; Yang Y.; Chu Y. X.; Li X. Y.; Guang Y. F.; Huang J. Identification of Sequence Variants in Genetic Disease-Causing Genes Using Targeted Next-Generation Sequencing. PLoS One 2011, 6, e29500 10.1371/journal.pone.0029500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomba L.; Walter K.; Soranzo N. The impact of rare and low-frequency genetic variants in common disease. Genome Biol. 2017, 18, 77. 10.1186/s13059-017-1212-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houldcroft C. J.; Beale M. A.; Breuer J. Clinical and biological insights from viral genome sequencing. Nat. Rev. Microbiol. 2017, 15, 183–192. 10.1038/nrmicro.2016.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertes F.; ElSharawy A.; Sauer S.; van Helvoort J. M. L. M.; van der Zaag P. J.; Franke A.; Nilsson M.; Lehrach H.; Brookes A. J. Targeted enrichment of genomic DNA regions for next-generation sequencing. J. Brief. Funct. Genom. 2011, 10, 374–386. 10.1093/bfgp/elr033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnakumar S.; Zheng J.; Wilhelmy J.; Faham M.; Mindrinos M.; Davis R. A comprehensive assay for targeted multiplex amplification of human DNA sequences. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 9296–9301. 10.1073/pnas.0803240105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewhey R.; Warner J. B.; Nakano M.; Libby B.; Medkova M.; David P. H.; Kotsopoulos S. K.; Samuels M. L.; Hutchison J. B.; Larson J. W.; Topol E. J.; Weiner M. P.; Harismendy O.; Olson J.; Link D. R.; Frazer K. A. Microdroplet-based PCR enrichment for large-scale targeted sequencing. Nat. Biotechnol. 2009, 27, 1025–1031. 10.1038/nbt.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwase S. C.; Miyazato P.; Katsuya H.; Islam S.; Yang B. T. J.; Ito J.; Matsuo M.; Takeuchi H.; Ishida T.; Matsuda K.; Maeda K.; Satou Y. HIV-1 DNA-capture-seq is a useful tool for the comprehensive characterization of HIV-1 provirus. Sci. Rep. 2019, 9, 12326. 10.1038/s41598-019-48681-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodi K.; Perera A. G.; Adams P. S.; Bintzler D.; Dewar K.; Grove D. S.; Kieleczawa J.; Lyons R. H.; Neubert T. A.; Noll A. C.; Singh S.; Steen R.; Zianni M. Comparison of commercially available target enrichment methods for next-generation sequencing. J. Biomol. Tech. 2013, 24, 73–86. 10.7171/jbt.13-2402-002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl J.; Ding Y.; Fleming A. M.; Burrows C. J. Identification of DNA lesions using a third base pair for amplification and nanopore sequencing. Nat. Commun. 2015, 6, 8807. 10.1038/ncomms9807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petti C. A. Detection and identification of microorganisms by gene amplification and sequencing. Clin. Infect. Dis. 2007, 44, 1108. 10.1086/512818. [DOI] [PubMed] [Google Scholar]
- Clark I. C.; Abate A. R. Finding a helix in a haystack: nucleic acid cytometry with droplet microfluidics. Lab Chip 2017, 17, 2032–2045. 10.1039/c7lc00241f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S. W.; Tran T. M.; Abate A. R. PCR-Activated Cell Sorting for Cultivation-Free Enrichment and Sequencing of Rare Microbes. PLoS One 2015, 10, e0113549 10.1371/journal.pone.0113549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lance S. T.; Sukovich D. J.; Stedman K. M.; Abate A. R. Peering below the diffraction limit: robust and specific sorting of viruses with flow cytometry. Virol. J. 2016, 13, 201. 10.1186/s12985-016-0655-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukovich D. J.; Lance S. T.; Abate A. R. Sequence specific sorting of DNA molecules with FACS using 3dPCR. Sci. Rep. 2017, 7, 39385. 10.1038/srep39385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han H.-S.; Cantalupo P. G.; Rotem A.; Cockrell S. K.; Carbonnaux M.; Pipas J. M.; Weitz D. A. Whole-Genome Sequencing of a Single Viral Species from a Highly Heterogeneous Sample. Angew. Chem., Int. Ed. 2015, 54, 13985–13988. 10.1002/anie.201507047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y.; Rotem A.; Zhang H.; Cockrell S. K.; Koehler S. A.; Chang C. B.; Ung L. W.; Cantalupo P. G.; Ren Y.; Lin J. S.; Feldman A. B.; Wobus C. E.; Pipas J. M.; Weitz D. A. Artifact-Free Quantification and Sequencing of Rare Recombinant Viruses by Using Drop-Based Microfluidics. ChemBioChem 2015, 16, 2167–2171. 10.1002/cbic.201500384. [DOI] [PubMed] [Google Scholar]
- Mousavian Z.; Sadeghi H. M. M.; Sabzghabaee A. M.; Moazen F. Polymerase chain reaction amplification of a GC rich region by adding 1,2 propanediol. Adv. Biomed. Res. 2014, 3, 65. 10.4103/2277-9175.125846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruner K. M.; Wang Z.; Simonetti F. R.; Bender A. M.; Kwon K. J.; Sengupta S.; Fray E. J.; Beg S. A.; Antar A. A. R.; Jenike K. M.; Bertagnolli L. N.; Capoferri A. A.; Kufera J. T.; Timmons A.; Nobles C.; Gregg J.; Wada N.; Ho Y.-C.; Zhang H.; Margolick J. B.; Blankson J. N.; Deeks S. G.; Bushman F. D.; Siliciano J. D.; Laird G. M.; Siliciano R. F. A quantitative approach for measuring the reservoir of latent HIV-1 proviruses. Nature 2019, 566, 120–125. 10.1038/s41586-019-0898-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciambi A.; Abate A. R. Generating electric fields in PDMS microfluidic devices with salt water electrodes. Lab Chip 2014, 14, 2605–2609. 10.1039/c4lc00078a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B.; Salzberg S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Handsaker B.; Wysoker A.; Fennell T.; Ruan J.; Homer N.; Marth G.; Abecasis G.; Durbin R.; The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowlands V.; Rutkowski A. J.; Meuse E.; Carr T. H.; Harrington E. A.; Barrett J. C. Optimisation of robust singleplex and multiplex droplet digital PCR assays for high confidence mutation detection in circulating tumour DNA. Sci. Rep. 2019, 9, 12620. 10.1038/s41598-019-49043-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi H.-D.; Zheng H.; Guo W.; Gañán-Calvo A. M.; Ai Y.; Tsao C.-W.; Zhou J.; Li W.; Huang Y.; Nguyen N.-T.; Tan S. H. Active droplet sorting in microfluidics: a review. Lab Chip 2017, 17, 751–771. 10.1039/c6lc01435f. [DOI] [PubMed] [Google Scholar]
- Mazutis L.; Gilbert J.; Ung W. L.; Weitz D. A.; Griffiths A. D.; Heyman J. A. Single-cell analysis and sorting using droplet-based microfluidics. Nat. Protoc. 2013, 8, 870–891. 10.1038/nprot.2013.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro S. C.; Brandt L. D.; Bale M. J.; Halvas E. K.; Joseph K. W.; Shao W.; Wu X.; Guo S.; Murrell B.; Wiegand A.; Spindler J.; Raley C.; Hautman C.; Sobolewski M.; Fennessey C. M.; Hu W.-S.; Luke B.; Hasson J. M.; Niyongabo A.; Capoferri A. A.; Keele B. F.; Milush J.; Hoh R.; Deeks S. G.; Maldarelli F.; Hughes S. H.; Coffin J. M.; Rausch J. W.; Mellors J. W.; Kearney M. F. Combined HIV-1 sequence and integration site analysis informs viral dynamics and allows reconstruction of replicating viral ancestors. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 25891–25899. 10.1073/pnas.1910334116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegand A.; Spindler J.; Hong F. F.; Shao W.; Cyktor J. C.; Cillo A. R.; Halvas E. K.; Coffin J. M.; Mellors J. W.; Kearney M. F. Single-cell analysis of HIV-1 transcriptional activity reveals expression of proviruses in expanded clones during ART. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, E3659–E3668. 10.1073/pnas.1617961114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidore A. M.; Lan F.; Lim S. W.; Abate A. R. Enhanced sequencing coverage with digital droplet multiple displacement amplification. Nucleic Acids Res. 2016, 44, e66 10.1093/nar/gkv1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebbert M. T. W.; Wadsworth M. E.; Staley L. A.; Hoyt K. L.; Pickett B.; Miller J.; Duce J.; Kauwe J. S. K.; Ridge P. G. Alzheimer’s Dis Neuroimaging, I., Evaluating the necessity of PCR duplicate removal from next-generation sequencing data and a comparison of approaches. BMC Bioinf. 2016, 17, 239. 10.1186/s12859-016-1097-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.