

Summary
Human pluripotent stem cell (hPSC)-derived pancreatic progenitors (PPs) can be differentiated into beta-like cells in vitro and in vivo and therefore have therapeutic potential for type 1 diabetes (T1D) treatment. However, the purity of PPs varies across different hPSC lines, differentiation protocols, and laboratories. The uncommitted cells may give rise to non-pancreatic endodermal, mesodermal, or ectodermal derivatives in vivo, hampering the safety of hPSC-derived PPs for clinical applications and their differentiation efficiency in research settings. Recently, proteomics and transcriptomics analyses identified glycoprotein 2 (GP2) as a PP-specific cell surface marker. The GP2-enriched PPs generate higher percentages of beta-like cells in vitro, but their potential in vivo remains to be elucidated. Here, we demonstrate that the GP2-enriched-PPs give rise to all pancreatic cells in vivo, including functional beta-like cells. Remarkably, GP2 enrichment eliminates the risk of teratomas, which establishes GP2 sorting as an effective method for PP purification and safe pancreatic differentiation.
Keywords: human embryonic stem cells, type 1 diabetes, pancreatic progenitors, beta cells, cell replacement therapy, teratoma, cell surface marker, endoderm, mesoderm, ectoderm
Graphical abstract
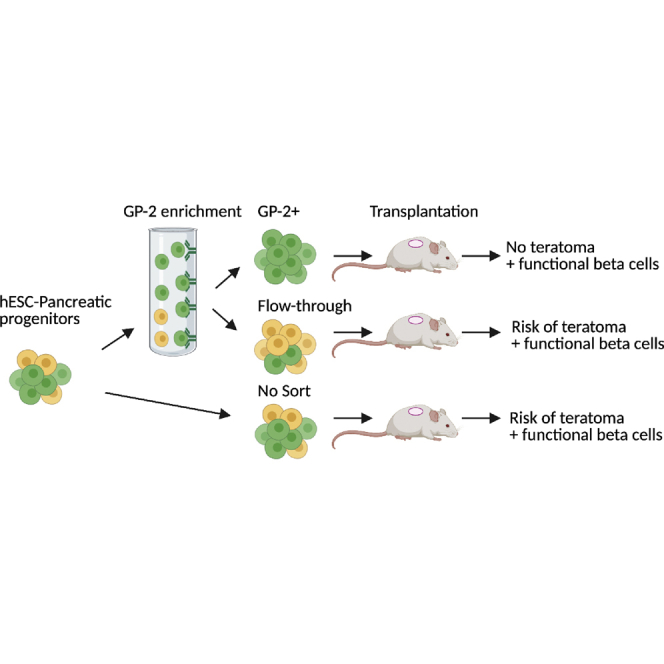
Highlights
-
•
GP2-enriched PPs give rise to pancreatic endocrine and exocrine cells in vivo
-
•
GP2-enriched PPs give rise functional beta cells in vivo
-
•
GP-2 enrichment of hPSC-PPs eliminates teratoma formation in vivo
Aghazadeh et al. established that GP2-enriched pancreatic progenitors give rise to endocrine and exocrine pancreatic cells in vivo, including functional beta cells, while eliminating the risk of teratoma formation. These findings indicate that the enrichment of GP2-expressing pancreatic progenitors can improve the safety of these cells for clinical translation.
Introduction
Type 1 diabetes (T1D) is an autoimmune disease characterized by the destruction of pancreatic beta cells. The standard treatment for people living with T1D is the administration of exogenous insulin with frequent glucose monitoring, which significantly increases life expectancy but fails to fully protect from long-term vascular complications (Aye and Atkin, 2014; Beck and Cogen,2015). Therefore, beta cell replacement strategies that are able to endogenously regulate blood glucose are highly sought after (Odorico et al., 2018; Shapiro et al., 2000, 2016). Indeed, islet transplantation using the Edmonton protocol has succeeded in controlling brittle diabetes, providing protection from hypoglycemic episodes, and in several cases, has led to insulin independence (McCall and Shapiro, 2012). In addition to donor-derived islets, the recent progress in the development of pancreatic cells from human pluripotent stem cells (hPSCs) offers an unprecedented opportunity to generate renewable, readily available, and quality-controlled pancreatic cells for clinical applications (Migliorini et al., 2021; Odorico et al., 2018). hPSCs can be differentiated into pancreatic progenitors (PPs) or pancreatic endodermal cells (PECs), which can give rise to functional beta-like cells both in vitro and in vivo (Kroon et al., 2008; Nostro et al., 2015; Rezania et al., 2012, 2014). However, preclinical studies reported the formation of teratoma in 15%–50% of mice transplanted with hPSC-derived PEC/PP populations (Kelly et al., 2011; Kroon et al., 2008; Rezania et al., 2012). Remarkably, PECs, delivered in vivo via encapsulation devices, have entered clinical trials (NCT02239354 and NCT03163511). Results from the first clinical trials (VC01, NCT02239354) demonstrated that the device designed to prevent vessel penetration into the graft is not a viable option for beta cell replacement therapy, because it led to graft fibrosis and cell death (Henry et al., 2018). In contrast, the results from the trial using an open device (VC02, NCT03163511) showed that, in the absence of a full physical barrier between the graft and the host, PECs can give rise to beta cells that secrete C-peptide in a majority of recipients (Ramzy et al., 2021; Shapiro et al., 2021). These findings are consistent with our studies demonstrating the importance of vascularization for graft survival and beta cell function (Aghazadeh et al., 2021). However, PEC-derived beta cells secreted a 10th of the C-peptide levels required to normalize blood glucose (Ramzy et al., 2021), suggesting that improving vascularization strategies, PEC-generating protocols, and/or the transplantation of higher number of PECs in the open devices will be necessary to treat T1D. Importantly, while no teratomas were formed from PECs in these trials, the need for a higher number of cells in open devices could increase this risk. Therefore, designing strategies to purify the PP population is imperative to ensure the safety of this therapeutic approach.
The pancreatic secretory granule membrane major glycoprotein 2 (GP2) was previously identified as a specific cell surface marker of hPSC-derived PPs and shown to be co-expressed with PTF1A and NKX6-1 in the putative multi-potent PPs in the developing human pancreas (Ameri et al., 2017; Cogger et al., 2017). Purification and in vitro culture of GP2-enriched epithelial cells, obtained from human pancreas at 7 weeks of development, confirmed that these cells give rise to acinar cells, in which GP2 is upregulated, as well as ductal and endocrine cells, in which GP2 is downregulated or silenced (Ramond et al., 2017). Similarly, GP2-expressing hPSC-derived PPs downregulate GP2 expression as they differentiate to beta-like cells in vitro (Ameri et al., 2017; Cogger et al., 2017). Collectively, these studies demonstrate that the GP2-expressing PPs are committed to the pancreatic fate. However, there is a lack of data examining the in vivo commitment of GP2-enriched PPs to pancreatic lineage and, ultimately, to beta cells. In this study, we explore the developmental potential of GP2-enriched PPs and demonstrate that sorted GP2-expressing PPs give rise to all endocrine and exocrine cells in vivo, including functional beta cells. Furthermore, we demonstrate that GP2 sorting does not impact the endocrine-to-acinar ratio within the graft and more importantly prevents teratoma formation in vivo. These findings indicate that GP2-enriched PPs represent a safe cell population with the potential for clinical use.
Results
GP2 enrichment of hESC-derived PPs
To assess the efficiency of GP2+ PP purification using magnetic-activated cell sorting (MACS), H1 and H9 human embryonic stem cell (hESC) lines were differentiated to PPs using previously published protocols (Korytnikov and Nostro, 2016) (Figure 1A). PP commitment was assessed by flow cytometric analysis of NKX6-1, PDX1, and GP2 expression at the end of stage 4. Both H1 and H9 cell lines generated more than 80% PDX1+/NKX6-1+ cells (Figures 1B–1D), while H1 cells generated higher frequencies of GP2+ cells (∼67%) compared with H9 cells (∼31%) (Figures 1E–1G). These results demonstrated that the percentage of PDX1/NKX6-1-expressing cells may not correlate with that of GP2 (Figures 1D and 1G). This is consistent with previous studies showing that not all NKX6-1+/PDX1+ cells express GP2 at the end of stage 4, in line with its transient expression in the developing pancreas (Cogger et al., 2017; Ramond et al., 2017). GP2 enrichment was performed at the end of stage 4 for both cell lines using MACS (Figure 1H) (Cogger et al., 2017). The purity of the H1-derived GP2-enriched population (henceforth referred to as GP2+) was 88% (n = 6), while the flow-through (FT) and the not sorted (NS) cells retained approximately 67% GP2-expressing cells (Figures 1I and 1J). The quantification of GP2 mean fluorescence intensity (MFI) indicated significant enrichment of GP2high-expressing cells in the GP2+ compared with FT and NS cell populations (Figure 1K). Approximately 76% of GP2+ cells (n = 3) were obtained using H9-derived PPs, which was lower than the H1-derived GP2+ cells (Figures 1L and 1M). The MFI of the GP2+ cells was similar between the two lines (Figures 1K and 1N). The percentage of GP2− cells was significantly lower in the GP2+ compared with NS and FT cell populations in both H1- and H9-derived PPs (Figures 1A and 1B). Overall, these data demonstrate that MACS can effectively enrich GP2+ cells with higher cell surface GP2 expression per cell compared with NS and FT populations (Figures 1I–1J). The recovery yield from MACS column was 16% and 24% in H1 and H9 line, respectively (Table 1).
Figure 1.

Enrichment of GP2+ cells in hESC-derived PPs
(A) Schematic of the hESC differentiation to pancreatic progenitors (PPs).
(B–D) Representative flow cytometry plots and quantification of NKX6-1/PDX1 co-expression in hESC-H1- (B) or H9- (C) derived PPs at the end of stage 4 (D).
(E–G) Representative flow cytometry plots and quantification of GP2 expression in H1- (E) or H9- (F) derived PPs at the end of stage 4 (G). (H1, n = 6 independent experiments; H9, n = 3 independent experiments; unpaired Student’s t test; error bars represent SEM).
(H) Schematic of GP2 enrichment using MACS.
(I, J, L, and M) Representative flow cytometry plots and quantification of the percentage of GP2-expressing cells in not sorted (NS), flow-through (FT), and GP2-enriched (GP2+) fractions of H1- (I and J) or H9- (L and M) derived PPs.
(K and N) Mean fluorescence intensity (MFI) over isotype control in NS, FT, and GP2+ fractions of H1- (K) or H9- (N) derived PPs. (H1, n = 6 independent experiments; H9, n = 3 independent experiments; one-way ANOVA analysis with Tukey’s multiple comparisons; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; error bars represent SEM).
See also Figure S1.
Table 1.
Percent of recovery after MACS, calculated by comparing the number of GP2+ cells obtained from MACS, to total number of cells loaded on the column
| Biological replicate | 1 | 2 | 3 | 4 | 5 | 6 | Mean | SD |
|---|---|---|---|---|---|---|---|---|
| % Cell recovery H1 | 27 | 10.86 | 11.81 | 6.65 | 10.4 | 28.16 | 15.81 | 4.89 |
| % Cell recovery H9 | 14.76 | 33.26 | 22.12 | 23.83 | 7.6 |
Related to Figure 1.
GP2 sorting prevents teratoma formation in vivo
To assess if GP2 sorting at the end of stage 4 could reduce the risk of teratoma formation in vivo, we transplanted GP2+, NS, and FT populations into immunocompromised male NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice. We first examined if the sorted fractions could survive long-term in the subcutis, because this site allows the detection of teratomas with the naked eye. To assess long-term cell survival, an hESC-H1 line that constitutively expresses DsRED Fluorescent Protein (RFP-H1) (Davidson et al., 2012) was used to generate RFP-PPs, which were sorted using MACS to generate GP2+, FT, and NS populations (Figure 2A), which were transplanted subcutaneously in NSG mice (Figure 2B). The RFP signal was monitored at weeks 1, 4, and 12 post-transplantation by live imaging. The background RFP signal was assessed using untransplanted mice (No Tx) and sham mice (Figure S2B). The RFP signal in GP2+-, FT-, and NS-transplanted mice was detected at all time points, confirming that all three populations can survive long-term in the subcutis (Figure 2B). We then utilized this model for the transplantation of MACS H1-derived GP2+, FT, and NS cell populations. The transplanted NSG mice were monitored weekly, and outgrowths visible with the naked eye were detected in NS and FT recipients from week 8 onward (Figure 2C). Mice without visible outgrowths were maintained for 15 weeks, a period required for the development of functional beta cells (Kroon et al., 2008; Nostro et al., 2015; Rezania et al., 2014). The NS- and FT-derived outgrowths were harvested and analyzed by H&E staining, which indicated the presence of mesodermal, ectodermal, and endodermal cell types (Figures 2D and S3A). Further pathological analysis using toluidine blue staining confirmed the presence of cartilage (Figures 2E and S3B), and immunostaining indicated the presence of cells expressing SOX2 (stomach and neuronal marker, Figures 2F and S3C), FOXA2 (endodermal cells, Figures 2G and S3E), CDX2 (intestinal marker, Figures 2H and S3D), and NKX2-1 (lung marker, Figures 2I and S3F) within NS- and FT-derived outgrowths. These data indicate that transplanted NS- and FT-derived PPs give rise to non-pancreatic tissues from multiple germ layers, characteristic of teratoma, in 27% (4/15) of NS recipients and in 18% (3/17) of FT-recipients. Remarkably, none of the 11 mice transplanted with GP2+ cells developed teratomas/outgrowths (Figure 2J). These data strongly demonstrate that GP2+ cell enrichment prevents teratoma formation in vivo.
Figure 2.

Transplantation of GP2-enriched PPs prevents teratoma formation
(A) Schematic of RFP-H1 differentiation to RFP-PPs followed by GP2 enrichment using MACS.
(B) Live imaging of mice transplanted subcutaneously with NS, FT, and GP2+ cell populations, at weeks 1, 4, and 12 post-transplantation. See also Figure S2.
(C) Representative image of outgrowths visible with the naked eye in the mouse transplanted with NS cells (dashed line). No outgrowths detected in mice transplanted with GP2+ cells.
(D–I) Representative images of NS-derived grafts stained with H&E (D), toluidine blue (E), SOX2 (F), FOXA2 (G), CDX2 (H), and NKX2-1 (F) at week 15 post-transplantation ([D] scale bar is 100 μm; [F] scale bar is 200 μm; and [E, G–I] scale bar is 400 μm). See also Figure S3.
(J) Percentage of NS-, FT-, and GP2+-recipient mice that generated teratomas/outgrowths (NS, n = 15 mice from six independent experiments; FT, n = 17 mice from six independent experiments; and GP2+, n = 11 mice from six independent experiments).
GP2 enrichment eliminates non-pancreatic cells
To evaluate how GP2 enrichment prevented teratoma formation, we quantified the expression levels of neuroectodermal marker ZIC3 (Bernal and Arranz, 2018) (Figure S4A), mesodermal marker PDGFRA (Farahani and Xaymardan, 2015) (Figure S4B), and pluripotency marker POU5F1 (Pan et al., 2002) (Figure S4C) in the NS, FT, and GP2+ fractions by RT-PCR and compared them with hPSC-derived neuroectoderm, mesoderm, or undifferentiated hESCs, respectively. These data indicated significantly lower expression of all markers in the NS, FT, and GP2+ fractions compared to undifferentiated mesoderm, neuroectoderm, or hESC controls, but no significant differences were detected by RT-PCR between the three PP fractions (Figures S4A–S4C), which is likely due to robust endodermal commitment of H1 line (∼99.2%) at stage 1 of differentiation (Figures S4D and S4E). Quantification of GP2 in NS, FT, and GP2+ fractions was performed for quality control (Figure S4F). Next the presence of non-pancreatic endodermal cells was assessed in the three fractions by analyzing the expression levels of the anterior foregut markers SOX2 and NKX2-1 and hindgut marker CDX2 (Figures 3A–3C). No significant differences were observed in NKX2-1 expression between the three fractions, but SOX2 and CDX2 expression was significantly lower in GP2+ compared with NS and FT fractions (Figures 3A–3C). To confirm these data, cell sorting using fluorescence-activated cell sorting (FACS) was performed to purify GP2+ and GP2− populations, which is not feasible by MACS. The frequencies of SOX2+ and CDX2+ cells were quantified in NS cells, as well as GP2− and GP2+ populations. Less than 1% of the cells at stage 4 expressed SOX2 (0.24% ± 0.05%) (Figures 3D and 3E) and all SOX2+ cells were GP2− (Figures 3D and 3E). Surprisingly, a majority of stage 4 cells expressed CDX2 (76.1% ± 6.1%) (Figures 3F and 3G). This could be due to the fact that the loss of CDX2 in the pre-pancreatic domain occurs gradually in developing mice, while the pancreas-stomach boundary is defined as early as embryonic day 8.75, with very few cells co-expressing SOX2 and PDX1 (Shih et al., 2015). Co-staining of the NS population for GP2 and CDX2 indicated that 66.64% ± 5% of the CDX2+ cells lacked GP2, which was greatly reduced in the GP2+ fraction after FACS (Figures 3F and 3G). Collectively, these data indicate that sorting for GP2 significantly eliminates SOX2+/GP2− and CDX2+/GP2− non-pancreatic cells from stage 4 populations.
Figure 3.

GP2-enriched PPs have lower expression of endoderm-derived non-pancreatic cell markers
(A–C) RT-PCR quantification of SOX2, NKX2-1, and CDX2 normalized to RPL-19 housekeeping gene, in H1-derived NS, FT, and GP2+ PP fractions (n = 3 independent experiments, NS, FT, and GP2+; n = 1, human stomach, lung, and intestine; one-way ANOVA analysis with Tukey’s multiple comparisons; ∗p < 0.05, ∗∗∗p < 0.01; error bars represent SEM).
(D–G) Representative flow cytometry plots indicating SOX2 and CDX2 expression in NS, GP2−, and GP2+ populations and quantification of cell percentages (n = 7 independent experiments; one-way ANOVA analysis with Tukey’s multiple comparisons; ∗p < 0.05, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, error bars represent SEM).
See also Figures S4 and S5.
GP2-enriched PPs are less proliferative than GP2− cells
To assess the developmental potential of GP2+ PPs, all grafts from mice transplanted with GP2+ cells and those transplanted with NS and FT recipients that did not form visible outgrowths were explanted at 15 weeks post-transplantation and histologically analyzed. Results indicated that significantly larger grafts were generated from the same number of transplanted NS population compared with the GP2+ population (Figures 4A and 4B). This observation was confirmed by quantifying the number of human cells within the graft by KU80 immunostaining, a marker of human nuclei, which demonstrated significantly higher number of human cells in the NS-derived grafts compared with the GP2+-derived grafts (Figures 4C and 4D). Because previous studies indicated a negative correlation between GP2 expression and cell proliferation (Ameri et al., 2017), we next quantified the expression of MKI67 and TOP2A proliferation markers in the MAC-Sorted populations to evaluate whether the larger size of grafts is due to higher proliferative potential of NS and FT compared with the GP2+ population. RT-PCR analysis showed significantly lower expression of MKI67 in the GP2+ compared NS and FT fractions and lower TOP2A expression in the GP2+ compared with the FT fraction (Figures 4E and 4F). These results were confirmed by flow cytometric quantification of the frequency of KI67+ cells in FAC-Sorted GP2+ and GP2− fractions., indicating that GP2+ cells are significantly less proliferative than GP2− cells, which may explain the formation of smaller grafts (Figures 4G and 4H). Therefore, we next investigated whether a larger graft area translates to higher number of off-target cells or to higher number of pancreatic cells. Non-teratoma grafts were analyzed by immunostaining of SOX2, NKX2-1, and CDX2 at week 15 post-transplantation. SOX2 expression was not detected in any of the grafts (data not shown), while NS-, FT-, and GP2+-derived grafts contained a small population of CDX2+ and NKX2-1+ cells (Figures 4I–4L). While the number of NKX2-1+ cells did not vary between the different fractions (Figures 4K and 4L), CDX2+ cells were significantly lower in the GP2+ compared with the NS-derived grafts (Figures 4I and 4J). These results are consistent with the analysis of cell populations before transplantation (Figures 3D–3G), which indicated negligible SOX2 expression in all three cell populations (Figures 3D and 3E), significantly lower CDX2 expression in GP2+ cells compared with NS and FT fractions (Figures 3C and 3F), and no differences in NKX2-1 levels between the three cell populations (Figure 3B). We then analyzed the proportion of pancreatic cells in NS-, FT-, and GP2+-derived grafts by immunostaining for the endocrine pancreatic marker chromogranin A (CHGA), pancreatic ductal marker cytokeratin 19 (KRT19), and pancreatic acinar marker trypsin (PRSS1) (Figures 4M–4Q). Interestingly, while no significant differences were detected in the total number of pancreatic endocrine cells (CHGA+) (Figures 4M and 4N), significantly lower acinar cells (PRSS1+) were observed in the GP2+ compared with the FT-derived grafts (Figures 4P and 4Q). The ratio of endocrine to acinar cells was comparable between the three conditions (Figure 4R), demonstrating that GP2 enrichment of PPs does not affect endocrine versus acinar commitment post-transplantation. This is not surprising because stage 4 PPs have not yet committed to endocrine or exocrine cell fates. RT-PCR analysis confirmed that GP2 sorting does not enrich for endocrine- or exocrine-committed cells, because no significant differences were detected in CELA3A (acinar marker), HNF1B (ductal marker), and NEUROG3 and NEUROD1 (endocrine markers) between NS, FT, and GP2+ populations (Figures 5A–5D). Indeed, endocrine commitment is initiated in vitro at stage 5 (Figure S5E), and it is marked by the expression of NEUROD1 and CHGA (Figures S5F and S5G), while additional factors would be required to induce exocrine commitment in vitro (Breunig et al., 2021; Huang et al., 2021). Collectively, these data demonstrated that GP2+ PPs are less proliferative than GP2− and give rise to fewer trypsin+ and CDX2+ cells post-transplantation, while generating endocrine cells with similar efficacy as unsorted cells.
Figure 4.

GP2-enriched PPs are less proliferative and give rise to smaller grafts with lower CDX2+ and PRSS1+ cells
(A–D) Representative images and quantification of H&E and KU80 staining of the non-teratoma grafts retrieved from mice 15 weeks post-transplantation (scale bar is 400 μm; n = 3 independent experiments; one-way ANOVA analysis with Tukey’s multiple comparisons; ∗p < 0.05, ∗∗p < 0.01; error bars are SEM).
(E and F) RT-PCR quantification of MKI67 and TOP2A normalized to RPL19 housekeeping gene in NS, FT, and GP2+ populations before transplantation. (n = 3 independent experiments; one-way ANOVA analysis with Tukey’s multiple comparisons; ∗p < 0.05, ∗∗p < 0.01; error bars represent SEM).
(G and H) Representative flow cytometry plots indicating KI67 expression in NS, GP2−, and GP2+ populations (n = 7 independent experiments; one-way ANOVA analysis with Tukey’s multiple comparisons; ∗p < 0.05, error bars represent SEM).
(I–Q) Representative immunostaining staining of grafts at week 15 post-transplantation, indicating the presence of cells expressing CDX2 (intestinal marker), NKX2-1 (lung marker), chromogranin A (CHGA, pancreatic endocrine marker), cytokeratin 19 (KRT19, ductal cell marker), and trypsin (PRSS1, acinar cell marker) (scale bar is 200μm), and quantification of cell numbers (n = 3 independent experiments; one-way ANOVA analysis with Tukey’s multiple comparisons; ∗p < 0.05; error bars represent SEM).
(R) Ratio of endocrine to acinar cells (n = 3 independent experiments; error bars are SEM).
Figure 5.

GP2+-enriched PPs give rise to functional beta cells in vivo
(A and B) Glucose-stimulated C-peptide secretion assay at 15 weeks post-transplantation of H1- (A) or H9- (B) derived NS, FT, and GP2+ fractions ([A] NS, n = 15 mice from 6 independent experiments; FT, n = 14 mice from six independent experiments; GP2+, n = 9 mice from 6 independent experiments; [B] NS, n = 6 mice from three independent experiments; FT, n = 7 mice from 3 independent experiments; GP2+, n = 4 mice from three independent experiments; two-way ANOVA analysis with Tukey’s multiple comparisons; ∗p < 0.05, ∗∗p < 0.01; error bars represent SEM).
(C and D) Representative immunostaining staining of grafts without teratoma retrieved from mice at week 15 post-transplantation indicating the presence of cells expressing C-peptide (Cp, beta cell marker), glucagon (GCG, alpha cell marker), somatostatin (SST, delta cell marker), and SLC18A1 (enterochromaffin cell marker). DAPI marks cell nuclei. (Scale bar is 200μm).
GP2-enriched PPs give rise to functional beta cells in vivo
We next examined whether GP2+ cells could differentiate in vivo to functional beta cells by performing a glucose-stimulated C-peptide secretion assay at week 15 post-transplantation, which indicated significantly higher C-peptide secretion in response to glucose in NS-, FT-, and GP2+-transplanted mice (Figure 5A). Importantly, the small size of the grafts derived from GP2+ cells did not jeopardize the concentration of secreted human C-peptide in response to glucose challenge (Figure 5A). To assess reproducibility across stem cell lines, this experiment was replicated using H9-derived NS, FT, and GP2+ fractions(Figure 5B). Interestingly, using the H9 line, GP2+ recipients were the only group that secreted significantly higher C-peptide levels in response to glucose challenge, which indicates that GP2 enrichment can be highly beneficial for PPs derived from hESC lines, such as H9, that have suboptimal GP2 expression at stage 4 (Figure 1G). The grafts were further analyzed by immunofluorescence staining which identified beta cells (C-peptide+ [Cp]), alpha cells (glucagon [GCG]), and delta cells (somatostatin [SST]) in grafts derived from NS, FT, and GP2+ fractions (Figure 5C). It is noteworthy that, in the grafts generated by all three populations, we detected the expression of SLC18A1, a marker of enterochromaffin cells (Figure 5D), which are generated as a by-product of beta cell differentiation in vitro or after transplantation of PPs or beta-like cells (Aghazadeh et al., 2021; Augsornworawat et al., 2020; Veres et al., 2019). Therefore, GP2 sorting does not eliminate enterochromaffin cells (Figure 5D). Collectively, these data demonstrate that GP2 enrichment of PPs prevents teratoma formation without jeopardizing the developmental potential of PPs for generating functional beta cells in vivo.
Discussion
The frequency of PPs generated from hPSC-directed differentiation varies between cell lines, protocols, and laboratories, and, despite optimization of the culture conditions, the presence of uncommitted cells cannot be ruled out. These contaminants reduce beta cell differentiation efficacy and pose risks for teratoma formation in transplant settings (Kelly et al., 2011; Kroon et al., 2008; Rezania et al., 2012). The latter could be an obstacle for clinical translation if these cells were to be transplanted without encapsulation to maximize vascularization required for engraftment. To purify the hESC-derived PP population and eliminate uncommitted cells, we and others have identified PP cell surface markers, such as GP2, CD142, and CD24 (Ameri et al., 2017; Cogger et al., 2017; Jiang et al., 2011; Kelly et al., 2011; Ramond et al., 2017). However, because CD24 and CD142 are expressed by polyhormonal cells and undifferentiated hESCs, they are not as PP-specific as GP2 (Cogger et al., 2017; Jiang et al., 2011). Building on in vitro studies showing that GP2-sorted cells can give rise to beta cells, here we demonstrate that GP2+ PPs have the potential to give rise to both exocrine and endocrine cells in vivo, including functional beta cells, while eliminating the risk of teratoma. In line with previous reports (Ameri et al., 2017), our data indicated that the GP2+ PPs are less proliferative than unsorted PPs and give rise to smaller grafts in vivo without jeopardizing graft efficacy in responding to glucose challenge. As such, the GP2+ PPs show great promise for clinical use as well as studying pancreas development from stem cells in vivo and in vitro. It is important to note, however, that GP2 enrichment using MACS resulted in significant cell loss (16% cell recovery), which may limit the use of MACS in large-scale manufacturing and research settings. This approach could be improved by developing better antibodies against GP2, introducing a lineage depletion step prior to GP2 sorting, and improving the magnetic separation steps. Using a negative selection of uncommitted cells prior to sorting for GP2 is an approach that is widely used in the hematopoietic field to sort for rare stem and progenitor populations (Spangrude et al., 1988). Lineage depletion could be achieved by using a selection of antibodies for proteins known to not be expressed by the PPs, which could be selected from a proteomic dataset that we have previously generated (Cogger et al., 2017). Even with improvements, loss of target cells is inevitable with sorting strategies, and improving the differentiation protocols may be a more cost-effective alternative to cell sorting. However, we show that despite robust differentiation of the H1 cell line to definitive endoderm at stage 1 and PPs at stage 4, the unsorted cells can give rise to teratomas, which could render the use of enrichment strategies imperative for cell therapies without the use of encapsulation. Alternatively, because we show that GP2 expression negatively correlates with the risk of tumorigenesis, the assessment of GP2 levels, together with other markers, could be used for quality control prior to PP transplantation. Currently, NKX6-1 is prominently measured to assess pancreatic commitment at stage 4. This is based on mouse developmental studies that showed that NKX6-1 is the master regulator of beta cell fate (Sander et al., 2000; Schaffer et al., 2010; Taylor et al., 2013) and critical for the upregulation of insulin biosynthesis and glucose-sensing genes (Aigha and Abdelalim, 2020; Schaffer et al., 2013). However, modeling pancreas development with hPSCs could generate aberrant cell types that co-express NKX6-1 and non-pancreatic transcription factors, such as CDX2 and SOX2 (Veres et al., 2019; Wesolowska-Andersen et al., 2020). Such aberrant phenotypes are not unique to hPSC-derived pancreatic cultures and were reported in hPSC-derived lung and kidney populations (Hurley et al., 2020; Wu et al., 2018). Indeed, mapping hPSC-derived cell fate trajectories elucidated that early multi-potent/progenitor cells could exhibit lineage plasticity (Petersen et al., 2017; Shih et al., 2015), suggesting that assessing the expression of a single cell marker does not accurately represent cell commitment. Here we show that sorting for GP2 reduces the frequency of SOX2+/GP2− and CDX2+/GP2− cells and prevents teratoma formations. Accordingly, SOX2+ cells were only detected in teratomas, while CDX2+ cells were significantly reduced in GP2+-derived grafts compared with unsorted and FT-derived grafts. Interestingly, CDX2 staining appeared to be cytoplasmic, which is associated with aberrant pancreatic or intestinal pathologies (Bai et al., 2002; Hinoi et al., 2002; Ishikawa et al., 2004). Therefore, in addition to NKX6-1, measuring GP2 expression could serve as an indicator of PP purity and commitment to pancreatic cell fate. Additional cell surface markers, such as CD177, which is expressed in endoderm at stage 1, could provide further indication of successful differentiations (Mahaddalkar et al., 2020). The CD177-enriched endoderm exhibited improved efficiency for pancreatic specification and differentiated to a homogeneous NKX6-1+/PDX1+ at stage 4 in vitro (Mahaddalkar et al., 2020); however, whether transplantation of CD177+-derived PPs eliminates teratoma formation in vivo remains to be explored. Because teratoma formation from the transplanted hPSC-derived beta-like cells (that include non-beta cells) has not been reported (Hogrebe et al., 2020; Pagliuca et al., 2014; Rezania et al., 2014; Velazco-Cruz et al., 2019; Veres et al., 2019), beta-like cells could be a safer alternative to PPs for clinical translation. The lack of teratomas could be attributed to the low proliferation rate of cells after endocrine commitment (McDonald et al., 2012; Oakie and Nostro, 2021; Rosado-Olivieri et al., 2020). However, generation of beta-like cells is more costly compared with PP production, due to lower differentiation yield (Cogger et al., 2017; Pagliuca et al., 2014; Rezania et al., 2014; Wesolowska-Andersen et al., 2020), which challenges scalability. Furthermore, beta cells have a high oxygen consumption rate, which could negatively impact survival post-transplantation (Papas et al., 2015; Pepper et al., 2012). It is noteworthy that previous studies indicated that the in vivo microenvironment can also influence cell fate specification and cell growth, because the same cell population transplanted into different sites developed teratomas with various compositions and at different rates (Hentze et al., 2009; Lawrenz et al., 2004). Thus far, teratoma formation from unsorted PPs has been reported in the subrenal capsule (Kroon et al., 2008; Rezania et al., 2012) and epidydimal fat pad (Kelly et al., 2011; Kroon et al., 2008). In this study, we show teratoma formation in the subcutis, which is a clinically relevant site. However, it should be considered that encapsulation devices or other sites of transplantation expose the grafts to distinct microenvironments, which could alter the risk of teratoma formation.
Collectively, our findings validate GP2 as a marker of committed PPs devoid of contaminants, which can give rise to functional beta-like cells in vivo and can be used to improve the safety of cell therapies.
Experimental procedures
Cell culture
H1 and H9 hESCs (WiCell) or RFP-H1 (gift from Dr. Gordon Keller, McEwen Stem Cell Institute, University Health Network) were expanded in vitro to reach 70%–80% confluency, as previously described (Kennedy et al., 2007). The differentiation of hESC was carried out in 4 stages, yielding definitive endoderm (days 0–3), primitive gut tube (days 3–5), posterior foregut (days 6–8), and PPs (days 8–13), as previously described (Korytnikov and Nostro, 2016). Neuroectoderm differentiation was adapted from previously described protocols (Chambers et al., 2009; Tchieu et al., 2017) and performed with the H1 cell line. Cells were cultured in serum-free DMEM media (Gibco) and a mixture of DMEM (Gibco) with 15% KnockOut Serum Replacement (KSR; Gibco), 1 mM glutamine, and 1× non-essential amino acids (Gibco). Media changes were performed using the following base media: day 0, 100% DMEM/KSR; day 2, 100% DMEM/KSR; day 4, 75% DMEM/KSR and 25% serum-free differentiation media (SFD) (Korytnikov and Nostro, 2016); day 6, 50% DMEM/KSR and 50% SFD; day 8, 25% DMEM/KSR and 75% SFD; and day 10, 100% SFD. All media changes were supplemented with 10μM SB 431542 (Sigma) and 500 ng/mL Noggin (R&D Systems) to promote neuroectoderm differentiation. All RNA samples were collected at day 11. Neuronal differentiation was confirmed with immunofluorescence antibody staining for βIII-tubulin (Chemicon, dilution: 1:1000,) and by RT-PCR for neuronal markers (data not shown). Mesodermal cells were a gift from Dr. Stephanie Protze (McEwen Stem Cell Institute, University Health Network) and differentiated according to published protocol (Lee et al., 2017). Adult human stomach, intestine, and lung mRNAs were purchased form Takara.
Magnetic-activated cell sorting (MACS)
Stage 4 PPs were dissociated to form single cells using TrypLE (Gibco) for 5 min at 37°C, and washed with PBS without calcium and magnesium supplemented with 10% fetal bovine serum (FACS buffer), DNase 1 bovine pancreas (10 μl/mL, Millipore), and 10 uM rock inhibitor Y27632 (Tocris). For cell surface GP2 staining, live cells were incubated with anti-GP2 antibody (1/10000 dilution, MBL International) in FACS buffer for 20 min at room temperature. Cells were washed with FACS buffer, followed by an incubation with anti-mouse phycoerythrin (PE)-conjugated secondary antibody for 20 min at room temperature. Another wash with MACS buffer (Miltenyi Biotech) was performed using MACS buffer (75 μL/1 × 107 cells) and PE magnetic beads were added (20 μL/1 × 107 cells). Cells and beads were incubated for 15 min at 4°C followed by a wash with MACS buffer. Cells were counted and NS fraction was used for further experiments. The remaining cells were loaded onto LS-positive selection columns (Miltenyi Biotech), FT fraction was collected and treated with Y27632 prior to transplantation. The columns were washed twice with MACS buffer and cells were eluted in FACS buffer with Y27632. These cells constituted the GP2+-enriched fraction. The recovery yield from MACS column was assessed by dividing the total number of GP2+ cells eluted from the MACS columns to the total cells loaded onto the columns (Table 1). The 3 fractions NS, FT, and GP2+ were then analyzed for GP2 expression by flow cytometry and then cultured overnight at a plating density of 5 × 106 cells/dish in stage 5 media (+Y27632 and DNase1) to allow spontaneous aggregation before transplant. Based on the cell counts on day 13 of differentiation, 5 × 106 cells were encapsulated in collagen hydrogels at day 14 for transplantation.
Fluorescence-activated cell sorting (FACS)
Stage 4 PPs were dissociated from the monolayer using TrypLE Express (Gibco) and incubated at 37°C to generate a single-cell suspension. Live cells were incubated for 20 min at 4°C with GP2 antibody conjugated to PE in FACS buffer. Cells were sorted using the BD AriaII-RITT or AriaIII Fusion cell sorters.
Flow cytometry
Stage 1, 4, 5, or 6 cells were dissociated to form single cells using TrypLE (Gibco) for 5 min at 37°C. Live cell staining was performed using Zombie Violet (Biolegend) diluted in PBS for 20 min at room temperature in the dark. For GP2 (stage 4), and KIT and CXCR4 (stage 1) staining, cells were stained live using antibodies in Table S1. For NKX6-1 and PDX1 (stage 4) or NEUROD1 and CHGA (stages 4, 5, and 6), cells were fixed in Cytofix/Cytoperm (BD Bioscience) for 24 h at 4°C, washed twice with Perm/Wash (BD Bioscience), and resuspended either in unconjugated primary antibodies, listed in Table S1, and incubated overnight at 4°C, or in conjugated antibodies, incubated for 30–60 min at room temperature in the dark After washing two times with Perm/Wash, secondary antibodies were added if needed, as described in Table S1, and the cells were incubated for 20–30 min at room temperature. Cells were washed in Perm/Wash; flow cytometric analysis was performed on a BD LSR Fortessa flow cytometer and Flowjo v. 10.1 was used for data analysis.
Animal transplantation-
After MACS and aggregation overnight, 5 × 106 GP2+, NS, or FT cells were embedded in a collagen hydrogel composed of 3 mg/mL rat tail collagen type I (Corning) and diluted in cell culture grade water (Gibco), low glucose DMEM powder (Life Technologies, 0.4 mg/mL), sodium bicarbonate (Sigma, 0.015 g/mL), and HEPES sodium salt (Sigma, 0.02 g/mL). All animal use was approved by the Animal Care Committee of University Health Network ; 4- to 6-week-old male NSG mice were anesthetized using isoflurane. A subcutaneous incision was formed in the dorsal area. Solidified hydrogels were picked up using forceps and inserted through the incision, and the incision site was sutured.
Glucose-stimulated C-peptide secretion and ELISA assay
At week 15 post-transplantation, the animals were fasted overnight. The next day, blood was collected though saphenous vein. Each mouse was weighed, and 3 g/kg glucose was injected intraperitoneally per mouse; 60 min later, a second blood collection was performed from the saphenous vein. High-speed centrifugation was performed to separate the serum fraction. Ultra-sensitive C-peptide ELISA assay (Mercodia) was performed to measure the levels of human C-peptide in the sera.
Live animal imaging
Mice were anesthetized using isoflurane, and RFP signal was visualized using live imaging by PerkinElmer Xenogen IVIS Spectrum Imaging System.
Immunostaining and quantifications
Immunostaining was performed as previously described (Cogger et al., 2017). Briefly, grafts were embedded in agarose and paraffin, and 3 μm sections were cut by the Toronto General Hospital, Pathology Research Program Laboratory. Sections were de-paraffinized using xylene and rehydrated in a serial dilution of absolute alcohol. Antigen retrieval was performed, and the sections were blocked using 10% non-immune donkey serum (Jackson ImmunoResearch Laboratories) in PBS. Primary antibodies listed in Table S1 were diluted in 1× PBS supplemented with 0.3% Triton X-100 (Sigma) and 0.25% BSA (Sigma) (PBS-Triton-BSA), and incubation was conducted at 4°C overnight. After washing, the sections were incubated with secondary antibodies, listed in Table S1, and diluted in PBS-Triton-BSA for 45 min at room temperature. Slides were counterstained with 4′,6-diamidino-2-phenylindole (DAPI,Biotium) for 1 min and mounted with Dako fluorescence mounting media. Digital images were acquired using the EVOS FL Cell Imaging System (Thermo Fisher). The graft area was measured using ImageJ (version: https://imagej.net). Automated cell count was used on KU80+ cells in ImageJ, all other cell counts were performed manually using cell counter function of ImageJ.
RT-PCR cells were harvested and the RNA was extracted using Ambion PureLink RNA Mini Kit (Invitrogen). RT-PCR was performed to obtain cDNA using SuperScript III Reverse Transcriptase and RNaseOUT Recombinant Ribonuclease Inhibitor (Invitrogen). qPCR was further performed using Bio-Rad SsoAdvanced Universal SYBR Green Supermix on the Biorad CFX Connect Real-Time PCR system. Relative gene expression was calculated by normalizing mRNA levels of the gene of interest to the housekeeping gene RPL19. The sequences of RT-PCR primers used are listed in Table S2.
Author contributions
Y.A. designed and conducted experiments, analyzed and interpreted the result, and prepared the manuscript. F.S. designed and conducted experiments and analyzed data. F.P. assisted with RT-PCR and GSIS. B.N. and S.S.N. performed KU80 staining. E.C.M. contributed to PP differentiation. M.C.N. designed the experiments, analyzed and interpreted the results, and prepared the manuscript.
Conflicts of interest
M.C.N. has filed a patent related to the selection of PPs (US patent application 16/043,782). M.C.N. has a sponsored research agreement with Universal Cells, has a patent (WO2013163739A1) licensed to Sernova, and is a scientific consultant for Sigilon Therapeutics.
Acknowledgments
This work was supported by funding from the Toronto General and Western Hospital Foundation and a Canadian Institute of Health Research project grant to M.C.N. Y.A. was supported by post-doctoral fellowships from the JDRF Canadian Clinical Trial Network and Toronto General Hospital Research Institute as well as Medicine by Design. F.P. was supported by scholarships from Ontario Graduate Scholarship and the Banting & Best Diabetes Centre, University of Toronto. We acknowledge Dr. Amanda Oakie for neurectoderm differentiation, Dr. Stephanie Protze for mesoderm differentiation, and Dr. Rangarajan Sambathkumar and Angel Sing for technical support. Schematics in Figures 1, 2, and S5 were generated using Biorender.com.
Published: March 31, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.stemcr.2022.03.004.
Supplemental information
References
- Aghazadeh Y., Poon F., Sarangi F., Wong F.T.M., Khan S.T., Sun X., Hatkar R., Cox B.J., Nunes S.S., Nostro M.C. Microvessels support engraftment and functionality of human islets and hESC-derived pancreatic progenitors in diabetes models. Cell Stem Cell. 2021;28:1936–1949.e1938. doi: 10.1016/j.stem.2021.08.001. [DOI] [PubMed] [Google Scholar]
- Aigha I.I., Abdelalim E.M. NKX6.1 transcription factor: a crucial regulator of pancreatic beta cell development, identity, and proliferation. Stem Cell Res Ther. 2020;11:459. doi: 10.1186/s13287-020-01977-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameri J., Borup R., Prawiro C., Ramond C., Schachter K.A., Scharfmann R., Semb H. Efficient generation of glucose-responsive beta cells from isolated GP2+ human pancreatic progenitors. Cell Rep. 2017;19:36–49. doi: 10.1016/j.celrep.2017.03.032. [DOI] [PubMed] [Google Scholar]
- Augsornworawat P., Maxwell K.G., Velazco-Cruz L., Millman J.R. Single-cell transcriptome profiling reveals beta cell maturation in stem cell-derived islets after transplantation. Cell Rep. 2020;32:108067. doi: 10.1016/j.celrep.2020.108067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aye M.M., Atkin S.L. Patient safety and minimizing risk with insulin administration - role of insulin degludec. Drug Healthc Patient Saf. 2014;6:55–67. doi: 10.2147/DHPS.S59566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y.Q., Yamamoto H., Akiyama Y., Tanaka H., Takizawa T., Koike M., Kenji Yagi O., Saitoh K., Takeshita K., Iwai T., et al. Ectopic expression of homeodomain protein CDX2 in intestinal metaplasia and carcinomas of the stomach. Cancer Lett. 2002;176:47–55. doi: 10.1016/s0304-3835(01)00753-4. [DOI] [PubMed] [Google Scholar]
- Beck J.K., Cogen F.R. Outpatient management of pediatric type 1 diabetes. J. Pediatr Pharmacol Ther. 2015;20:344–357. doi: 10.5863/1551-6776-20.5.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernal A., Arranz L. Nestin-expressing progenitor cells: function, identity and therapeutic implications. Cell Mol Life Sci. 2018;75:2177–2195. doi: 10.1007/s00018-018-2794-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breunig M., Merkle J., Wagner M., Melzer M.K., Barth T.F.E., Engleitner T., Krumm J., Wiedenmann S., Cohrs C.M., Perkhofer L., et al. Modeling plasticity and dysplasia of pancreatic ductal organoids derived from human pluripotent stem cells. Cell Stem Cell. 2021;28:1105–1124.e1119. doi: 10.1016/j.stem.2021.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers S.M., Fasano C.A., Papapetrou E.P., Tomishima M., Sadelain M., Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009;27:275–280. doi: 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogger K.F., Sinha A., Sarangi F., McGaugh E.C., Saunders D., Dorrell C., Mejia-Guerrero S., Aghazadeh Y., Rourke J.L., Screaton R.A., et al. Glycoprotein 2 is a specific cell surface marker of human pancreatic progenitors. Nat. Commun. 2017;8:331. doi: 10.1038/s41467-017-00561-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson K.C., Adams A.M., Goodson J.M., McDonald C.E., Potter J.C., Berndt J.D., Biechele T.L., Taylor R.J., Moon R.T. Wnt/beta-catenin signaling promotes differentiation, not self-renewal, of human embryonic stem cells and is repressed by Oct4. Proc. Natl. Acad Sci. U S A. 2012;109:4485–4490. doi: 10.1073/pnas.1118777109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farahani R.M., Xaymardan M. Platelet-derived growth factor receptor alpha as a marker of mesenchymal stem cells in development and stem cell biology. Stem Cells Int. 2015;2015:362753. doi: 10.1155/2015/362753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry R.R., Pettus J., Wilensky J., SHAPIRO A.J., Senior P.A., Roep B., Wang R., Kroon E.J., Scott M., D'AMOUR K. Am Diabetes Assoc; 2018. Initial Clinical Evaluation of VC-01TM Combination Product—A Stem Cell–Derived Islet Replacement for Type 1 Diabetes (T1D) [Google Scholar]
- Hentze H., Soong P.L., Wang S.T., Phillips B.W., Putti T.C., Dunn N.R. Teratoma formation by human embryonic stem cells: evaluation of essential parameters for future safety studies. Stem Cell Res. 2009;2:198–210. doi: 10.1016/j.scr.2009.02.002. [DOI] [PubMed] [Google Scholar]
- Hinoi T., Lucas P.C., Kuick R., Hanash S., Cho K.R., Fearon E.R. CDX2 regulates liver intestine-cadherin expression in normal and malignant colon epithelium and intestinal metaplasia. Gastroenterology. 2002;123:1565–1577. doi: 10.1053/gast.2002.36598. [DOI] [PubMed] [Google Scholar]
- Hogrebe N.J., Augsornworawat P., Maxwell K.G., Velazco-Cruz L., Millman J.R. Targeting the cytoskeleton to direct pancreatic differentiation of human pluripotent stem cells. Nat. Biotechnol. 2020;38:460–470. doi: 10.1038/s41587-020-0430-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L., Desai R., Conrad D.N., Leite N.C., Akshinthala D., Lim C.M., Gonzalez R., Muthuswamy L.B., Gartner Z., Muthuswamy S.K. Commitment and oncogene-induced plasticity of human stem cell-derived pancreatic acinar and ductal organoids. Cell Stem Cell. 2021;28:1090–1104.e1096. doi: 10.1016/j.stem.2021.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley K., Ding J., Villacorta-Martin C., Herriges M.J., Jacob A., Vedaie M., Alysandratos K.D., Sun Y.L., Lin C., Werder R.B., et al. Reconstructed single-cell fate trajectories define lineage plasticity windows during differentiation of human PSC-derived distal lung progenitors. Cell Stem Cell. 2020;26:593–608.e598. doi: 10.1016/j.stem.2019.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa A., Sasaki M., Ohira S., Ohta T., Oda K., Nimura Y., Chen M.F., Jan Y.Y., Yeh T.S., Nakanuma Y. Aberrant expression of CDX2 is closely related to the intestinal metaplasia and MUC2 expression in intraductal papillary neoplasm of the liver in hepatolithiasis. Lab. Invest. 2004;84:629–638. doi: 10.1038/labinvest.3700087. [DOI] [PubMed] [Google Scholar]
- Jiang W., Sui X., Zhang D., Liu M., Ding M., Shi Y., Deng H. CD24: a novel surface marker for PDX1-positive pancreatic progenitors derived from human embryonic stem cells. Stem Cells. 2011;29:609–617. doi: 10.1002/stem.608. [DOI] [PubMed] [Google Scholar]
- Kelly O.G., Chan M.Y., Martinson L.A., Kadoya K., Ostertag T.M., Ross K.G., Richardson M., Carpenter M.K., D'Amour K.A., Kroon E., et al. Cell-surface markers for the isolation of pancreatic cell types derived from human embryonic stem cells. Nat. Biotechnol. 2011;29:750–756. doi: 10.1038/nbt.1931. [DOI] [PubMed] [Google Scholar]
- Kennedy M., D'Souza S.L., Lynch-Kattman M., Schwantz S., Keller G. Development of the hemangioblast defines the onset of hematopoiesis in human ES cell differentiation cultures. Blood. 2007;109:2679–2687. doi: 10.1182/blood-2006-09-047704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korytnikov R., Nostro M.C. Generation of polyhormonal and multipotent pancreatic progenitor lineages from human pluripotent stem cells. Methods. 2016;101:56–64. doi: 10.1016/j.ymeth.2015.10.017. [DOI] [PubMed] [Google Scholar]
- Kroon E., Martinson L.A., Kadoya K., Bang A.G., Kelly O.G., Eliazer S., Young H., Richardson M., Smart N.G., Cunningham J., et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat. Biotechnol. 2008;26:443–452. doi: 10.1038/nbt1393. [DOI] [PubMed] [Google Scholar]
- Lawrenz B., Schiller H., Willbold E., Ruediger M., Muhs A., Esser S. Highly sensitive biosafety model for stem-cell-derived grafts. Cytotherapy. 2004;6:212–222. doi: 10.1080/14653240410006031. [DOI] [PubMed] [Google Scholar]
- Lee J.H., Protze S.I., Laksman Z., Backx P.H., Keller G.M. Human pluripotent stem cell-derived atrial and ventricular cardiomyocytes develop from distinct mesoderm populations. Cell Stem Cell. 2017;21:179–194.e174. doi: 10.1016/j.stem.2017.07.003. [DOI] [PubMed] [Google Scholar]
- Mahaddalkar P.U., Scheibner K., Pfluger S., Ansarullah, Sterr M., Beckenbauer J., Irmler M., Beckers J., Knobel S., Lickert H. Generation of pancreatic beta cells from CD177(+) anterior definitive endoderm. Nat. Biotechnol. 2020;38:1061–1072. doi: 10.1038/s41587-020-0492-5. [DOI] [PubMed] [Google Scholar]
- McCall M., Shapiro A.M. Update on islet transplantation. Cold Spring Harb. Perspect. Med. 2012;2:a007823. doi: 10.1101/cshperspect.a007823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald E., Li J., Krishnamurthy M., Fellows G.F., Goodyer C.G., Wang R. SOX9 regulates endocrine cell differentiation during human fetal pancreas development. Int. J. Biochem. Cell Biol. 2012;44:72–83. doi: 10.1016/j.biocel.2011.09.008. [DOI] [PubMed] [Google Scholar]
- Migliorini A., Nostro M.C., Sneddon J.B. Human pluripotent stem cell-derived insulin-producing cells: a regenerative medicine perspective. Cell Metab. 2021;33:721–731. doi: 10.1016/j.cmet.2021.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nostro M.C., Sarangi F., Yang C., Holland A., Elefanty A.G., Stanley E.G., Greiner D.L., Keller G. Efficient generation of NKX6-1+ pancreatic progenitors from multiple human pluripotent stem cell lines. Stem Cell Rep. 2015;4:591–604. doi: 10.1016/j.stemcr.2015.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakie A., Nostro M.C. Harnessing proliferation for the expansion of stem cell-derived pancreatic cells: advantages and limitations. Front. Endocrinol. 2021;12:636182. doi: 10.3389/fendo.2021.636182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odorico J., Markmann J., Melton D., Greenstein J., Hwa A., Nostro C., Rezania A., Oberholzer J., Pipeleers D., Yang L., et al. Report of the key opinion leaders meeting on stem cell-derived beta cells. Transplantation. 2018;102:1223–1229. doi: 10.1097/TP.0000000000002217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliuca F.W., Millman J.R., Gurtler M., Segel M., Van Dervort A., Ryu J.H., Peterson Q.P., Greiner D., Melton D.A. Generation of functional human pancreatic beta cells in vitro. Cell. 2014;159:428–439. doi: 10.1016/j.cell.2014.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan G.J., Chang Z.Y., Scholer H.R., Pei D. Stem cell pluripotency and transcription factor Oct4. Cell Res. 2002;12:321–329. doi: 10.1038/sj.cr.7290134. [DOI] [PubMed] [Google Scholar]
- Papas K.K., Bellin M.D., Sutherland D.E., Suszynski T.M., Kitzmann J.P., Avgoustiniatos E.S., Gruessner A.C., Mueller K.R., Beilman G.J., Balamurugan A.N., et al. Islet oxygen consumption rate (OCR) dose predicts insulin independence in clinical islet autotransplantation. PLoS One. 2015;10:e0134428. doi: 10.1371/journal.pone.0134428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepper A.R., Hasilo C.P., Melling C.W., Mazzuca D.M., Vilk G., Zou G., White D.J. The islet size to oxygen consumption ratio reliably predicts reversal of diabetes posttransplant. Cell Transpl. 2012;21:2797–2804. doi: 10.3727/096368912X653273. [DOI] [PubMed] [Google Scholar]
- Petersen M.B.K., Azad A., Ingvorsen C., Hess K., Hansson M., Grapin-Botton A., Honore C. Single-cell gene expression analysis of a human ESC model of pancreatic endocrine development reveals different paths to beta-cell differentiation. Stem Cell Rep. 2017;9:1246–1261. doi: 10.1016/j.stemcr.2017.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramond C., Glaser N., Berthault C., Ameri J., Kirkegaard J.S., Hansson M., Honore C., Semb H., Scharfmann R. Reconstructing human pancreatic differentiation by mapping specific cell populations during development. Elife. 2017;6:e27564. doi: 10.7554/eLife.27564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramzy A., Thompson D.M., Ward-Hartstonge K.A., Ivison S., Cook L., Garcia R.V., Loyal J., Kim P.T.W., Warnock G.L., Levings M.K., et al. Implanted pluripotent stem-cell-derived pancreatic endoderm cells secrete glucose-responsive C-peptide in patients with type 1 diabetes. Cell Stem Cell. 2021;28:2047–2061.e2045. doi: 10.1016/j.stem.2021.10.003. [DOI] [PubMed] [Google Scholar]
- Rezania A., Bruin J.E., Arora P., Rubin A., Batushansky I., Asadi A., O'Dwyer S., Quiskamp N., Mojibian M., Albrecht T., et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat. Biotechnol. 2014;32:1121–1133. doi: 10.1038/nbt.3033. [DOI] [PubMed] [Google Scholar]
- Rezania A., Bruin J.E., Riedel M.J., Mojibian M., Asadi A., Xu J., Gauvin R., Narayan K., Karanu F., O'Neil J.J., et al. Maturation of human embryonic stem cell-derived pancreatic progenitors into functional islets capable of treating pre-existing diabetes in mice. Diabetes. 2012;61:2016–2029. doi: 10.2337/db11-1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosado-Olivieri E.A., Aigha I.I., Kenty J.H., Melton D.A. Identification of a LIF-responsive, replication-competent subpopulation of human beta cells. Cell Metab. 2020;31:327–338.e326. doi: 10.1016/j.cmet.2019.12.009. [DOI] [PubMed] [Google Scholar]
- Sander M., Sussel L., Conners J., Scheel D., Kalamaras J., Dela Cruz F., Schwitzgebel V., Hayes-Jordan A., German M. Homeobox gene Nkx6.1 lies downstream of Nkx2.2 in the major pathway of beta-cell formation in the pancreas. Development. 2000;127:5533–5540. doi: 10.1242/dev.127.24.5533. [DOI] [PubMed] [Google Scholar]
- Schaffer A.E., Freude K.K., Nelson S.B., Sander M. Nkx6 transcription factors and Ptf1a function as antagonistic lineage determinants in multipotent pancreatic progenitors. Dev. Cell. 2010;18:1022–1029. doi: 10.1016/j.devcel.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer A.E., Taylor B.L., Benthuysen J.R., Liu J., Thorel F., Yuan W., Jiao Y., Kaestner K.H., Herrera P.L., Magnuson M.A., et al. Nkx6.1 controls a gene regulatory network required for establishing and maintaining pancreatic Beta cell identity. PLoS Genet. 2013;9:e1003274. doi: 10.1371/journal.pgen.1003274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro A.M., Lakey J.R., Ryan E.A., Korbutt G.S., Toth E., Warnock G.L., Kneteman N.M., Rajotte R.V. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N. Engl. J. Med. 2000;343:230–238. doi: 10.1056/NEJM200007273430401. [DOI] [PubMed] [Google Scholar]
- Shapiro A.M., Pokrywczynska M., Ricordi C. Clinical pancreatic islet transplantation. Nat. Rev. Endocrinol. 2016;13:268–277. doi: 10.1038/nrendo.2016.178. [DOI] [PubMed] [Google Scholar]
- Shapiro A.M.J., Thompson D., Donner T.W., Bellin M.D., Hsueh W., Pettus J., Wilensky J., Daniels M., Wang R.M., Brandon E.P., et al. Insulin expression and C-peptide in type 1 diabetes subjects implanted with stem cell-derived pancreatic endoderm cells in an encapsulation device. Cell Rep. Med. 2021;2:100466. doi: 10.1016/j.xcrm.2021.100466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih H.P., Seymour P.A., Patel N.A., Xie R., Wang A., Liu P.P., Yeo G.W., Magnuson M.A., Sander M. A gene regulatory network cooperatively controlled by Pdx1 and Sox9 governs lineage allocation of foregut progenitor cells. Cell Rep. 2015;13:326–336. doi: 10.1016/j.celrep.2015.08.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangrude G.J., Heimfeld S., Weissman I.L. Purification and characterization of mouse hematopoietic stem cells. Science. 1988;241:58–62. doi: 10.1126/science.2898810. [DOI] [PubMed] [Google Scholar]
- Taylor B.L., Liu F.F., Sander M. Nkx6.1 is essential for maintaining the functional state of pancreatic beta cells. Cell Rep. 2013;4:1262–1275. doi: 10.1016/j.celrep.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchieu J., Zimmer B., Fattahi F., Amin S., Zeltner N., Chen S., Studer L. A modular platform for differentiation of human PSCs into all major ectodermal lineages. Cell Stem Cell. 2017;21:399–410.e397. doi: 10.1016/j.stem.2017.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velazco-Cruz L., Song J., Maxwell K.G., Goedegebuure M.M., Augsornworawat P., Hogrebe N.J., Millman J.R. Acquisition of dynamic function in human stem cell-derived beta cells. Stem Cell Rep. 2019;12:351–365. doi: 10.1016/j.stemcr.2018.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veres A., Faust A.L., Bushnell H.L., Engquist E.N., Kenty J.H., Harb G., Poh Y.C., Sintov E., Gurtler M., Pagliuca F.W., et al. Charting cellular identity during human in vitro beta-cell differentiation. Nature. 2019;569:368–373. doi: 10.1038/s41586-019-1168-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesolowska-Andersen A., Jensen R.R., Alcantara M.P., Beer N.L., Duff C., Nylander V., Gosden M., Witty L., Bowden R., McCarthy M.I., et al. Analysis of differentiation protocols defines a common pancreatic progenitor molecular signature and guides refinement of endocrine differentiation. Stem Cell Rep. 2020;14:138–153. doi: 10.1016/j.stemcr.2019.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H., Uchimura K., Donnelly E.L., Kirita Y., Morris S.A., Humphreys B.D. Comparative analysis and refinement of human PSC-derived kidney organoid differentiation with single-cell transcriptomics. Cell Stem Cell. 2018;23:869–881.e868. doi: 10.1016/j.stem.2018.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.