

Abstract
Spinal muscular atrophy with respiratory distress type I (SMARD1) is a neurodegenerative disease defined by respiratory distress, muscle atrophy and sensory and autonomic nervous system defects. SMARD1 is a result of mutations within the IGHMBP2 gene. We have generated six Ighmbp2 mouse models based on patient-derived mutations that result in SMARD1 and/or Charcot-Marie Tooth Type 2 (CMT2S). Here we describe the characterization of one of these models, Ighmbp2D564N (human D565N). The Ighmbp2D564N/D564N mouse model mimics important aspects of the SMARD1 disease phenotype, including motor neuron degeneration and muscle atrophy. Ighmbp2D564N/D564N is the first SMARD1 mouse model to demonstrate respiratory defects based on quantified plethysmography analyses. SMARD1 disease phenotypes, including the respiratory defects, are significantly diminished by intracerebroventricular (ICV) injection of ssAAV9-IGHMBP2 and the extent of phenotypic restoration is dose-dependent. Collectively, this model provides important biological insight into SMARD1 disease development.
Introduction
Spinal muscular atrophy with respiratory distress type I (SMARD1) is a rare, autosomal recessive motor neuron disease that primarily affects infants. This devastating disease typically manifests between 6 and 12 months of age, with respiratory impairment due to diaphragm paralysis as the primary clinical symptom (1–4). SMARD1 clinical symptoms also include severe muscle atrophy that begins in the distal limbs and progresses proximally, intrauterine growth delay, weak cry, fatty deposits in the phalanges, and sensory and autonomic nervous system defects (1,3–7). The main cause of muscle atrophy and paralysis is the selective loss of alpha motor neurons in the ventral horns of the spinal cord and subsequent denervation of skeletal muscle (8,9). Motor nerve conduction velocities are dramatically reduced in SMARD1 patients (4). Interestingly, analyses in patients and mouse models demonstrate that selective muscle groups are more vulnerable to denervation than others; however, the underlying cause of disease development is still unknown (10,11). Although SMARD1 is similar to spinal muscular atrophy (SMA), it is clinically and genetically distinct (2,3,12). Unlike SMA, there are no SMARD1-specific therapeutic options available and only palliative care measures are used. The majority of patients require permanent artificial mechanical ventilation to support breathing.
SMARD1 is caused by loss-of-function mutations in the immunoglobulin heavy-chain μ-DNA binding protein 2 (IGHMBP2) gene, located on human chromosome 11q13.3 (3,12). Compound heterozygous and homozygous recessive mutations have been identified in SMARD1 patients. Missense, nonsense, splice donor, frameshift, deletion and insertions are distributed throughout IGHMBP2, with mutations clustering within exons 10 and 12 of the helicase domain (3–5,13,14). Currently, there is no clear correlation between the type of mutation and disease severity or disease onset. Interestingly, several SMARD1 compound heterozygous mutations in IGHMBP2 have also been associated with Charcot-Marie Tooth Type 2 (CMT2S), another related neurodegenerative disease (15–17).
IGHMBP2 is composed of 15 exons that encode a 993 amino acid protein with homology to the UPF1-like superfamily I (SF1) helicases that include UPF1, Senataxin and MOV10 (18). IGHMBP2 contains RNA/DNA helicase and ATP-binding domains as well as an AN1 zinc finger motif and a R3H nucleic acid-binding motif (19–21). IGHMBP2 is ubiquitously expressed and is predominantly found within the cytoplasm (10). Several biochemical roles for IGHMBP2 include immunoglobulin class switching (19,22); pre-mRNA processing, and transcription (23,24); however, more recent studies suggest a key role for IGHMBP2 in translation (25,26). IGHMBP2 interacts with translation factors Reptin, Pontin, TFIIIC220 and multiple tRNAs, but the specific cellular mechanism leading to a specific loss of motor neurons remains unclear (25,26).
The first mouse model for SMARD1 arose from a spontaneous mutation and is referred to as the neuromuscular degeneration (nmd) model B6.BKS-Ighmbp2nmd-2J/J, herein referred to as BKS-nmd-2J (27,28). The nmd mutation is an A to G transition in intron 4 of Ighmbp2 that creates a cryptic pre-mRNA splice site, adding an additional 23 nucleotides and generating a premature termination. As a result, there is ~20% wild-type Ighmbp2 mRNA produced and significant reduction in full-length IGHMBP2 protein (28). A well-defined phenotype associated with BKS-nmd-2J mice is hindlimb weakness and contractures beginning around postnatal day (PND) 14 with severe hindlimb paralysis by 5 weeks (10,27,28). The lifespan of BKS-nmd-2J mice is variable, ranging from 8 weeks to 10 months with typical survival 2–3 months. Motor neuron loss is detected at ~PND10 before any disease phenotypes are present. By 5 weeks, when hindlimb paralysis is severe, there is approximately a 40% reduction in motor neurons and by 12–14 weeks only ~28% of motor neurons remain in BKS-nmd-2J mice (10). Neuromuscular junctions (NMJs) located within muscles of the distal appendages were more susceptible to denervation as opposed to NMJs within the trunk and neck muscles of BKS-nmd-2J mice (11). Axonal degeneration was observed in the sciatic and femoral quadricep nerves (10). Differences in axonal myelination were also detected in disease-susceptible muscle populations versus resistant muscles (10,11). Interestingly however, NMJ end plate fragmentation was present in susceptible and resistant muscle populations (11). Recently, our lab generated a mouse model that possesses the same A to G transition in intron 4 of Ighmbp2, but is within the FVB/NJ background (FVB/NJ-Ighmbp2nmd) (29). These mice survive 18–21 days, display similar hindlimb contraction and weakness, and paralysis as the original BKS-nmd-2J mice (29). Although the nmd-2J models are not based on a patient mutation, these models have provided very important insight into SMARD1 biology. As well, multiple pre-clinical gene-replacement studies using AAV9-IGHMBP2 have shown improvement in phenotypic and cellular pathology features in nmd-2J mice (29,30).
SMARD1 patients present with significant differences in disease onset, progression and severity. To better understand the relationship of genotype to disease development, we generated six Ighmbp2 mouse models based on SMARD1 patient mutations; C46X in exon 2, C496X in exon 10, D565N in exon 12, R603C in exon 13, R605X in exon 13 and H924Y in exon 14. Of the six Ighmbp2 models generated, the R602C (R603C in humans) and the C495X (C496X in humans) homozygous mutations appear embryonic lethal upon first examination. The remaining four Ighmbp2 mutations, C46X, D564N (D565N in humans), R604X (R605X in humans) and H922Y (H924Y in humans) generate distinct phenotypes and functional deficits in homozygous mutant mice. Here we describe the generation and characterization of the novel SMARD1 mouse model FVB/NJ-Ighmpb2D564N (D565N in humans); the remaining Ighmbp2 mutants will be reported independently. Previous studies demonstrated that the corresponding human D565N mutation located within exon 12 lies in motif V within the flexible hoop in domain 2A (20). Ighmbp2D565N has been identified in SMARD1 patients as a compound heterozygous mutation that results in respiratory distress in 6 months (D565N/R790X) or 167 days (D565N/L577P) (5,13). In vitro studies demonstrated that this pathogenic mutation maintains nucleic acid binding properties and ATPase activity but does not possess helicase activity (25). Mapping of the D565N mutation using the crystal structure of IGHMBP2 suggests the D565 mutation likely alters the ability of IGHMBP2 to translocate along bound RNA (20).
These studies demonstrate that FVB/NJ-Ighmpb2D564N homozygous mutant mice recapitulate important aspects of SMARD1 patient pathology, including hindlimb weakness and paralysis, decreased lifespan and motor neuron loss. Importantly, these studies show that the Ighmpb2D564N model is the first SMARD1 mouse model that demonstrates quantifiable respiratory deficiencies.
Results
CRISPR/Cas9 generation of FVB/NJ-Ighmbp2+/D564N mice
Although SMA is primarily a result of a homozygous deletion of the SMN1 gene, SMARD1 is defined by many mutations throughout the IGHMBP2 gene. Here we report the generation and characterization of the FVB/NJ-Ighmpb2D564N mouse model (referred to as Ighmbp2D564N or D564N). The D564N missense mutation lies within the IGHMBP2 helicase domain (Fig. 1A). Using CRISPR/Cas9, the Ighmbp2D564N mutation was introduced onto the mouse FVB/NJ background. Founder mice genotypes were verified by Sanger sequencing (Fig. 1B). None of the founders contained off-site mutations from non-specific CRISPR/Cas9 activity, indicating that the sole mutation in each line was in Ighmbp2 (Supplementary Material, Fig. S1). Founders were outcrossed to FVB/NJ animals to establish the colony and to reduce potential undetected off-target CRISPR edits.
Figure 1.
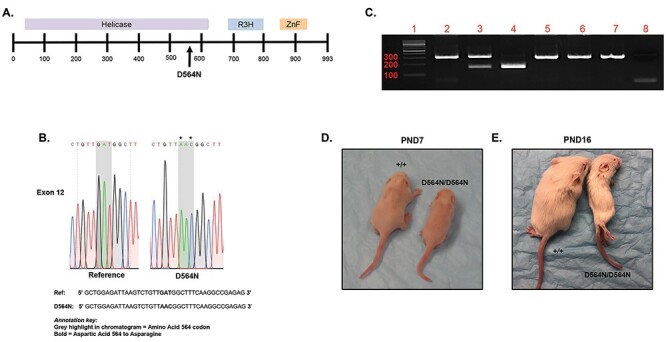
CRISPR/Cas9 generation of the Ighmbp2D564N/+ mouse model. (A) Depiction of murine IGHMBP2 protein and the location of the D564N mutation. (B) Sanger sequencing data from a N1 Ighmbp2+/D564N mouse confirming the D564N mutation. The * indicates the nucleotide changes between the wild-type reference FVB genomic sequence and the N1 Ighmbp2+/D564N genomic sequence. (C) Genotyping of mice. Lanes 2–4 were HpaI digested, whereas lanes 5–7 were undigested controls. Lane 1, 100 bp ladder, lane 2, Ighmbp2+/+ genomic DNA, lane 3, Ighmbp2+/D564N genomic DNA, lane 4, Ighmbp2D564N/D564N genomic DNA, lane 5, Ighmbp2+/+ genomic DNA, lane 6, Ighmbp2+/D564N genomic DNA, lane 7, Ighmbp2D564N/D564N genomic DNA, lane 8, no DNA control. (D) Representative image of PND7 wild-type and Ighmbp2D564N/D564N mice. (E) Representative image of PND16 wild-type and Ighmbp2D564N/D564N mice.
Generating Ighmbp2D564N/D564N mutants
SMARD1 is a result of recessive inheritance of mutant IGHMBP2 alleles. Heterozygous Ighmbp2+/D564N mice were bred, and pups were genotyped using D564N primers that amplify a 321 base pair (bp) fragment (Fig. 1C). Amplicons were restriction enzyme digested with HpaI, a unique restriction site located within the mutant allele. Wild-type mice were identified based on a single 321 bp fragment following gel electrophoresis (Fig. 1C, lane 2). Heterozygous mice were identified by the presence of the wild-type 321 bp band and two smaller 156 and 165 base pair bands representing the mutant allele (Fig. 1C, lane 3). Homozygous (Ighmbp2D564N/D564N) animals were identified by the exclusive presence of the 156 and 165 bp products (Fig. 1C, lane 4). Due to the similarity in size, the 156 and 165 bp products could not be independently resolved. As controls, undigested samples were analyzed for wild-type, heterozygous (+/D564N) and homozygous (D564N/D564N) samples (Fig. 1C, lanes 5–7), as well as a no template negative control (Fig. 1C, lane 8).
Ighmbp2 D564N/D564N mice have reduced survival and weight
To assess disease severity of the D564N mutation, FVB/NJ-Ighmbp2+/D564N animals were bred. Fitness, lifespan and total body weight progression were measured. Ighmbp2D564N/D564N mice were indistinguishable from Ighmbp2+/D564N and Ighmbp2+/+ littermates at birth in all parameters measured. By PND7, D564N mutant mice were phenotypically distinct from their healthy littermates; homozygous mutant mice were smaller in size and weighed less (Figs. 1D and2D). By PND16, D564N mutant mice exhibited severely reduced mobility, weight and coat quality (Figs. 1E and2D). Ighmbp2D564N/D564N mice had an average lifespan of ~16–17 days (n = 24, P < 0.0001), ranging from 12 to 22 days (Fig. 2A). Ighmbp2+/D564N and Ighmbp2+/+ animals did not die during the 24-day lifespan evaluation; Ighmbp2+/D564N animals did not demonstrate a detectable phenotype (Fig. 2A). D564N mutant mice were noticeably smaller and only achieved a maximum weight of ~4 g, whereas wild-type mice achieved weights exceeding 10 g (Fig. 2B). To determine whether some or all of the SMARD1 disease characteristics could be diminished, ssAAV9-IGHMBP2 (human IGHMBP2) was intracerebroventricular (ICV) injected at PND2. When ssAAV9-IGHMBP2 was injected at low dose (1 × 1011 viral genomes), lifespan nor weight was significantly increased (Figs. 2A and B). However, when ssAAV9-IGHMBP2 was injected at the high dose (5 × 1011 viral genomes), weight and lifespan were significantly increased (Figs. 2A and B). D564N mutants injected with the high dose of ssAAV9-IGHMBP2 achieved survival over 100 days and gained over 10 g during the lifespan. These results also confirm expression of human IGHMBP2 from the ssAAV9-IGHMBP2 vector. Although not reaching the benchmarks of wild-type mice, improvement of lifespan and weight in D564N mutant mice with human AAV9-IGHMBP2 was dose-dependent and significant.
Figure 2.
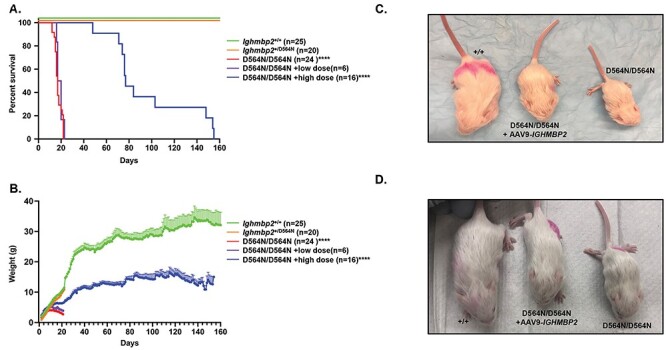
Ighmbp2 D564N/D564N mice have reduced survival and weight that is improved in a dose-dependent manner with ssAAV9-IGHMBP2. (A) Survival of Ighmbp2+/+ mice (green), Ighmbp2+/D564N mice (orange), Ighmbp2D564N/D564N mice (red) and Ighmbp2D564N/D564N mice treated with either a low dose (1 × 1011 viral genomes-purple) or a high dose (5 × 1011 viral genomes-blue) of ssAAV9-IGHMBP2. Statistical analysis performed using the log-rank (Mantel-Cox) test. Median survival of Ighmbp2D564N/D564N mice was 17 days (n = 24). (B) Weight differences between the five cohorts listed above. The average peak weight of Ighmbp2D564N/D564N mice was 3.83 ± 0.1399 g (n = 24). (C) Representative image at PND16 of wild-type, Ighmbp2D564N/D564N and Ighmbp2D564N/D564N mice treated with a low dose (1 × 1011 viral genomes) of ICV injected ssAAV9-IGHMBP2. (D) Representative image at PND16 of wild-type, Ighmbp2D564N/D564N and Ighmbp2D564N/D564N mice treated with a high dose (5 × 1011 viral genomes) of ICV injected ssAAV9-IGHMBP2. Data bars expressed as mean ± SEM ****P < 0.0001, * indicates significance.
Ighmbp2 D564N/D564N mice exhibit hindlimb muscle atrophy and reduced motor function
Two assays used to measure early changes in motor function are time-to-right (TTR) and hindlimb splay (HLS). In homozygous BKS-nmd2J mice, TTR is indistinguishable between wild-type and nmd-2J mutant mice; however, HLS is significantly reduced in nmd-2J mice as SMARD1 disease progresses (31). To assess motor function in homozygous D564N mice, HLS and TTR assays were used. The HLS assay was initiated at PND7. Defects in HLS for D564N mice became apparent as early as PND7 (Fig. 3A). By PND16, severe contractures were present with little to no mobility in the hindlimbs and reduced mobility in the forelimbs (Fig. 3D). Differences in motor function were also assessed by conducting the TTR assay. D564N mutant mice took significantly longer to correct their positions at PND8 compared with heterozygous and wild-type controls (Fig. 3C). The TTR scores for D564N mutants did not improve throughout the remainder of life indicating deficiencies in motor function (Fig. 3C). ICV injection of ssAAV9-IGHMBP2 (high dose) into D564N mutants at PND2 improved HLS and TTR throughout the duration of these studies. These results demonstrate clear improvements in motor function following ssAAV9-IGHMBP2 delivery (Figs. 3C and D). The rotarod assay tests motor coordination, strength and balance. The rotarod assay was initiated at PND46. D564N mutants ICV injected with a high dose of ssAAV9-IGHMBP2 at PND2 performed as well as wild-type mice on the rotarod assay (Fig. 3E). There was no significant difference between wild-type males and females nor ssAAV9-IGHMBP2 treated D564N mutant males and females (Supplementary Material, Fig. S2). D564N mutants could not be evaluated in the rotarod assay due to death. These results suggest that injection of ssAAV9-IGHMBP2 significantly improves motor coordination, strength and balance across the sexes. Grip strength was used to measure forelimb strength. This assay is initiated at PND46; therefore, D564N homozygous mutants could not be evaluated on this assay. Although ssAAV9-IGHMBP2 injected D564N mutants did not achieve wild-type measurements, the ability to perform the assay was significant (Fig. 3F). There was no significant difference in the grip strength assay between sexes (Supplementary Material, Fig. S2).
Figure 3.
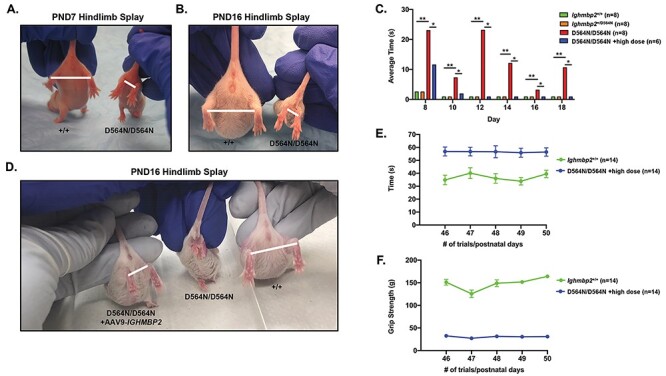
Ighmbp2 D564N/D564N mice exhibit hindlimb atrophy and reduced motor function that can be improved by ssAAV9-IGHMBP2 gene therapy. (A) Representative images of PND7 and (B) PND16 wild-type and D564N mutant mice performing the HLS assay. Bar indicates the distance measured as an evaluation of relative splay. (C) Motor performance was analyzed using the TTR test in seconds (one-way ANOVA P < 0.0001). TTR from PND8 to PND18 in wild-type (n = 8), +/D564N (n = 8), D564N/D564N (n = 8) and D564N/D564N mice ICV injected with a high dose (5 × 1011 viral genomes) of ssAAV9-IGHMBP2 (n = 6). (D) HLS in D564N mutant mice ICV injected with a high dose (5 × 1011 viral genomes) of ssAAV9-IGHMBP2 along with wild type and D564N/D564N animals. (E) Motor strength and coordination assayed by rotarod in wild-type and D564N/D564Nmice ICV injected with a high dose (5 × 1011 viral genomes) of ssAAV9-IGHMBP2 (n = 14). Assay is measured in seconds. (F) Grip strength in wild type and D564N/D564N mice ICV injected with a high dose (5 × 1011 viral genomes) of ssAAV9-IGHMBP2 (n = 14). Data bars expressed as mean ± SEM; *P < 0.05, **P < 0.01, * indicates significance.
Ighmbp2 D564N/D564N express similar levels of IGHMBP2
Ighmbp2 D564N/D564N mice demonstrated more severe phenotypes than previously characterized BKS-Ighmbp2nmd-2J and FVB-Ighmbp2nmd-2J mice. To determine whether the disease phenotypes were associated with changes in IGHMBP2 protein, Ighmbp2D564N/D564N whole brain and whole spinal cord extracts were analyzed by western blot. IGHMBP2 protein in PND12 Ighmbp2+/+ and Ighmbp2D564N/D564N mice were similar and not statistically different in either brain or spinal cord (Fig. 4). D564N mutants did not demonstrate decreased intracellular protein levels in contrast to nmd-2J mice. These results suggest that the biochemical activity, not the amount of IGHMBP2 protein, is deficient in D564N mutants. The amount of total protein in D564N mutants injected with ssAAV9-IGHMBP2 is similar to wild-type animals (Fig. 4). This result is not surprising, as it is likely that IGHMBP2 protein is tightly regulated such that the amount of protein whether endogenous or from the transgene is static.
Figure 4.
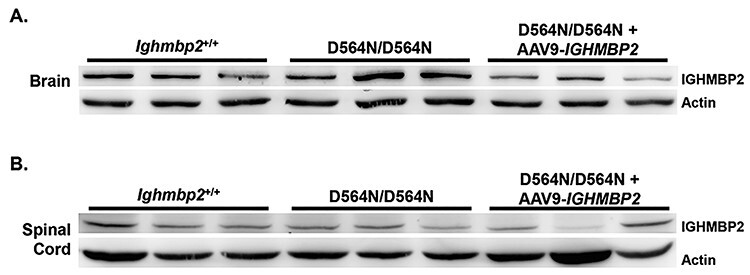
D564N mutant phenotypes are not due to reduced IGHMBP2 protein. (A) Western blot of brain extracts from PND12 Ighmbp2+/+ (n = 3), Ighmbp2D564N/D564N mice (n = 3) and Ighmbp2D564N/D564N mice ICV injected with a high dose of ssAAV9-IGHMBP2 (n = 3). Actin was used as a loading control. (B) Western blot of spinal cord extracts from PND12 Ighmbp2+/+ (n = 3), Ighmbp2D564N/D564N mice (n = 3) and Ighmbp2D564N/D564N mice ICV injected with a high dose of ssAAV9-IGHMBP2 (n = 3). Actin was used as a loading control.
Ighmbp2 D564N/D564N mice display reduced motor neuron numbers and size
SMARD1 patients as well as nmd-2J mice demonstrate progressive loss of motor neurons. Motor neuron loss precedes detection of mutant phenotypes with a 40% reduction in motor neurons by 10 days in BKS-nmd-2J mice. A plateau period follows and then there is a reduction in motor neurons to 28% at 12 weeks (10). To evaluate motor neuron loss in D564N animals, the L3–L5 lumbar spinal cord region was analyzed at PND15, a late symptomatic stage. As predicted, the number of motor neurons were significantly reduced in D564N mutant mice compared with wild-type (Figs. 5A and B). Wild-type mice had an average of ~11 motor neurons per section, whereas D564N homozygous mutants had an average of ~5 motor neurons per section (n = 78 motor neuron sections, P < 0.0001) (Fig. 5). Motor neuron measurements were also taken as an indication of progressive motor neuron degeneration. Wild-type motor neurons had an average area of 651.6 μm2 and while the area of D564N mutant motor neurons was significantly reduced to an average of 606.6 μm2 (n = 675 total motor neurons, P = 0.0201) (Fig. 5C). Motor neuron perimeter was also reduced in D564N homozygous mutants with an average of 93.78 μm compared with the wild-type animals with an average perimeter of 100.2 μm (n = 675 total motor neurons, P < 0.0001) (Fig. 5D). To determine whether reduced motor neuron number, area and perimeter could be corrected, ssAAV9-IGHMBP2 was ICV injected at the high dose into D564N mutants. Under these conditions, there was an increase in motor neuron number, area and perimeter (Figs. 5A and D). Collectively, these data indicate that Ighmbp2D564N/D564N mice display motor neuron degeneration that can be reduced or delayed with ICV injection of ssAAV9-IGHMBP2.
Figure 5.
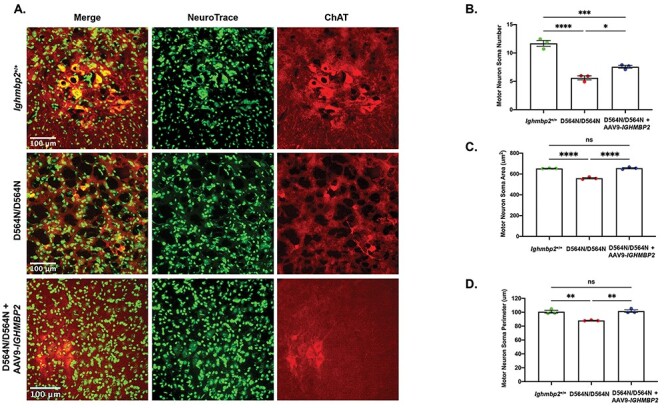
Motor neuron number and size is reduced in Ighmbp2D564N/D564N mice and is partially rescued with ssAAV9-IGHMBP2 gene therapy. (A) Representative images of cross sections from PND15 L3–L5 spinal cord motor neurons of Ighmbp2+/+ mice (top panels), Ighmbp2D564N/D564N mice (middle panels) and Ighmbp2D564N/D564N ssAAV9-IGHMBP2 injected mice (bottom panels). Mice were injected with a high dose (5 × 1011 viral genomes) of ssAAV9-IGHMBP2. Sections are immunostained with anti-ChAT antibody and Nissl stain (Neurotrace). Fluorescent microscope images are taken at 40x magnification. (B-D) Motor neuron somas in Ighmbp2+/+ mice (green), Ighmbp2D564N/D564N mice (red) and Ighmbp2D564N/D564N ssAAV9-IGHMBP2 injected mice (blue). (B) Quantification of motor neuron soma number. (C) Area analysis of motor neuron somas. (D) Quantification of motor neuron soma perimeter. Data analyzed as a one-way ANOVA with Tukey’s post-hoc test for multiple comparisons. Data bars expressed as mean ± SEM. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, n = 3 animals per cohort in which >400 cells were measured per cohort and averaged; ns = not significant.
Ighmbp2 D564N/D564N mice have selective muscle vulnerability to denervation
One of the hallmarks of motor neuron disease is the disruption of the NMJ. Antibodies against neurofilament-heavy (NF-H) and synaptophysin were used to visualize the axon and the presynaptic axon termini, and fluorochrome-tagged alpha-bungarotoxin was used to visualize the postsynaptic end plates. NMJ defects were quantified based on (1) denervated, (2) partially denervated or (3) fully innervated. NMJ innervation was scored based upon the overlap between the axon terminal and the NMJ end plate. Complete overlap between the terminal and end plate was considered fully innervated, whereas incomplete overlap was considered partially denervated. The absence of axon terminal over an end plate was scored as fully denervated.
To evaluate NMJ denervation, the gastrocnemius, a muscle previously shown to be extremely susceptible to denervation in BKS-nmd-2J mutants, was analyzed in wild-type and Ighmbp2D564N/D564N animals. NMJ denervation was analyzed at PND7 and as anticipated, D564N homozygous mutant mice displayed denervation in the gastrocnemius as compared with wild-type controls with either partially innervated or fully denervated end plates (Fig. 6) (n > 300 total end plates, P = 0.0374). NMJ denervation could be prevented in D564N mutants following ICV injection of the high dose ssAAV9-IGHMBP2 (Fig. 6). To determine whether NMJ denervation progressed throughout disease development, the gastrocnemius was examined in PND15 animals. As expected, the gastrocnemius in D564N homozygous mutant mice showed significant denervation defects with 60% of NMJs denervated compared to the unaffected wild-type animals (n > 300 total end plates, P < 0.0001) (Figs. 7A and B). These results demonstrate progressive denervation of NMJs within the gastrocnemius from PND7 to PND15. Importantly, despite the significant denervation observed at PND15 in D564N mutants, these defects could be significantly improved or at least delayed with ICV injection of a high dose of ssAAV9-IGHMBP2 (Figs. 7A and B).
Figure 6.
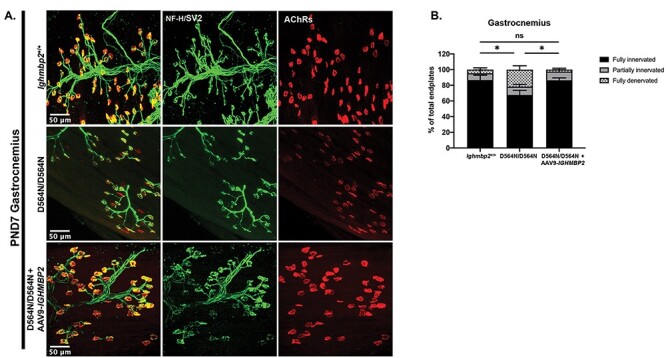
Ighmbp2 D564N/D564N mice demonstrate decreased number of fully innervated end plates at PND7 that is restored with ssAAV9-IGHMBP2 gene therapy. (A) Representative images of gastrocnemius muscle of PND7 Ighmbp2+/+ mice (top panels), Ighmbp2D564N/D564N mice (middle panels) and Ighmbp2D564N/D564N ssAAV9-IGHMBP2 injected mice (bottom panels). Mice were injected with a high dose (5 × 1011 viral genomes) of ssAAV9-IGHMBP2. Muscles were labeled with anti-neurofilament heavy polypeptide (NF-H) antibody, anti-synaptic vesicle 2 (SV2) antibody and α-bungarotoxin (α-BTX) that labels the acetylcholine receptors (AChRs). Fluorescent images were taken at 40x magnification. (B) Quantification of fully innervated (black), partially innervated (gray) and fully denervated end plates (hatched). Data bars expressed as mean ± SEM; *P < 0.05, n = 3 animals per cohort (>100 end plates per cohort were counted and averaged); ns = not significant.
Figure 7.
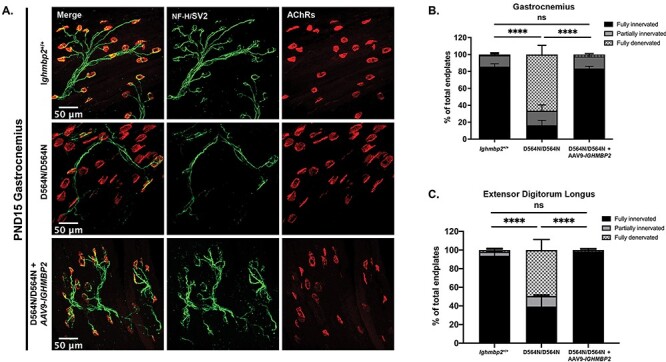
Ighmbp2 D564N/D564N mice demonstrate progressive denervation that can be restored with ssAAV9-IGHMBP2. (A) Representative images of the gastrocnemius muscle of PND15 Ighmbp2+/+ mice (top panels), Ighmbp2D564N/D564N mice (middle panels) and Ighmbp2D564N/D564N ssAAV9-IGHMBP2 injected mice (bottom panels). Muscles were labeled with anti-neurofilament heavy polypeptide (NF-H) antibody, anti-synaptic vesicle 2 (SV2) antibody and α-bungarotoxin (α-BTX) that labels the AChRs. Fluorescent images were taken at 40x magnification. (B) Quantification of fully innervated (black), partially innervated (gray) and full denervated (hatched) NMJs in the gastrocnemius muscle. (C) Quantification of fully innervated (black), partially innervated (gray) and full denervated (hatched) NMJs in the EDL muscle. Data analyzed by a two-way ANOVA with Tukey’s post-hoc test for multiple comparisons. Data bars expressed as mean ± SEM. ****P < 0.0001; n = 3 animals per cohort (n > 100 end plates were analyzed and averaged per cohort); ns = not significant
Previous studies in BKS-nmd-2J mice demonstrated certain muscle groups were differentially susceptible to denervation. The extensor digitorum longus (EDL), plantaris, soleus, tibialis anterior (TA), diaphragm, rectus abdominis (RA) and transverse abdominis (TVA) were analyzed at PND15 in wild-type and D564N homozygous mutants to examine denervation. In addition to the gastrocnemius, the EDL and TA are also required in limb movement and show varying degrees of denervation in D564N mutant animals (Fig. 7C, Supplementary Material, Fig. S3C). Interestingly, NMJ denervation in the EDL was more than the TA in D564N homozygous mutant mice, which is in contrast to BKS-nmd-2J mice. The denervation defects in D564N mutants were significantly reduced or delayed with ICV injection of ssAAV9-IGHMBP2 at the high dose (Fig. 7C, Supplementary Material, Fig. S3C). The plantaris, soleus and gastrocnemius muscles form the triceps surae that functions in plantar flexion. Significant NMJ denervation was detected in the plantaris and soleus muscles to the extent that most NMJs were partially or fully denervated in Ighmbp2D564N/D564N mice (Supplementary Material, Fig. S3A and B). Importantly, NMJ denervation could be prevented with ssAAV9-IGHMBP2 delivered at the high dose in D564N mutants (Supplementary Material, Fig. S3A and B). Lastly, the TVA, RA and diaphragm were analyzed (Supplementary Material, Fig. S3D-F). The TVA and RA muscles are abdominal wall muscles that stabilize the spine and support movement and respiration. The diaphragm muscle is also required for respiration. NMJs within each of these muscles were largely resistant to denervation in D564N mutant mice (Supplementary Material, Fig. S3D-F). The RA muscle had ~10% of the NMJs denervated (n > 300 total end plates, P = 0.0071) (Supplementary Material, Fig. S3E). These data suggest that NMJ denervation begins early and becomes progressively more severe in susceptible motor units of Ighmbp2D564N/D564N mice; however, these defects can be largely prevented or at least delayed with ssAAV9-IGHMBP2 delivered at the high dose.
Ighmbp2 D564N/D564N mice exhibit reduced muscle fiber size
Muscle atrophy has been observed in SMARD1 patients and BKS-nmd-2J mice. To determine if this pathology is present in D564N homozygous mutant mice, gastrocnemius and diaphragm muscle fiber cross sections were analyzed at PND15, a late-symptomatic stage (Fig. 8). Muscle fiber quantification of the gastrocnemius showed a significant reduction in mean muscle fiber area of Ighmbp2D564N/D564N mice (~147.0 μm2) compared with Ighmbp2+/+ mice (~414.7 μm2) (P < 0.0001) (Figs. 8A and B). Muscle fiber perimeter showed similar results with Ighmbp2D564N/D564N mice (~46.60 μm) compared with Ighmbp2+/+ mice (~79.58 μm) (P < 0.0001). ICV injection of ssAAV-IGHMBP2 into Ighmbp2D564N/D564N mice slightly improved muscle fiber size (Figs. 8A-C). Interestingly, diaphragm muscle fiber was significantly reduced (~151.2 μm2) in D564N mutant mice compared with wild-type mice (~232.9 μm2) (P < 0.0001) (Fig. 8D and E). The diaphragm muscle fiber perimeter revealed similar results with Ighmbp2D564N/D564N and Ighmbp2+/+ averaging 47.78 and 61.01 μm, respectively (P < 0.0001) (Fig. 8E). Diaphragm muscle fiber could be improved following delivery of ssAAV9-IGHMBP2 in D564N mutants (Fig. 8D and E). Interestingly, although the gastrocnemius had severe NMJ denervation and reduced muscle fiber size in Ighmbp2D564N/D564N mice, the diaphragm was largely resistant to NMJ denervation.
Figure 8.
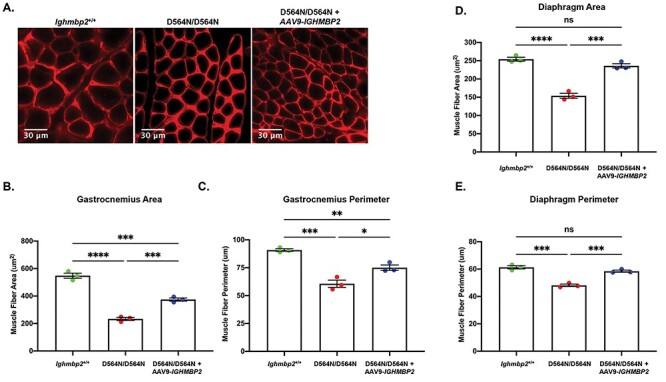
Ighmbp2 D564N/D564N mice exhibit reduced muscle fiber size that is improved with ssAAV9-IGHMBP2 gene therapy. (A) Representative images of PND15 cross-sections of the gastrocnemius muscle from Ighmbp2+/+ mice, Ighmbp2D564N/D564N mice and Ighmbp2D564N/D564N ssAAV9-IGHMBP2 injected mice. Muscles were immunostained with anti-laminin antibody and images were taken at 40x magnification. Analysis of gastrocnemius muscle fiber area (B) and perimeter (C) from Ighmbp2+/+ mice (green), Ighmbp2D564N/D564N mice (red) and Ighmbp2D564N/D564N ssAAV9-IGHMBP2 injected mice (blue). Quantification of diaphragm muscle fiber area (D) and perimeter (E) from Ighmbp2+/+ mice (green), Ighmbp2D564N/D564N mice (red) and Ighmbp2D564N/D564N ssAAV9-IGHMBP2 injected mice (blue). Data bars expressed as mean ± SEM. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, n = 3 animals per cohort (n > 200 muscle fibers were analyzed and averaged per cohort).
Ighmbp2 D564N/D564N mice demonstrate respiratory abnormalities
Respiratory abnormalities are a defining clinical symptom of SMARD1; however, respiratory insufficiency is not reported as a phenotype in BKS-nmd-2J mice. To determine whether respiratory abnormalities were detectable in D564N mutants, a whole-body plethysmography analysis was performed on PND12 Ighmbp2D564N/D564N and Ighmbp2+/+ mice, as well as Ighmbp2D564N/D564N mice ICV injected with the high dose of ssAAV9-IGHMBP2. Mice were subjected to two conditions: normoxia (21% O2, balance N2) and hypercapnia + hypoxia (10.5% O2, 7% CO2). Hypercapnia with hypoxia conditions were used to evaluate the response to a respiratory challenge; under these conditions wild-type mice respond with an increase in respiratory parameters. Measurements taken were respiratory frequency (breaths per minute), tidal volume (volume of air moved between normal inhalation and exhalation), minute ventilation (volume of gas inhaled from the lungs per minute) and mean inspiratory flow (amount of air inspired in a given time).
Under normoxia conditions, wild-type mice had an average of 343.1 breaths per minute (bpm), whereas D564N mutant mice had a significantly decreased respiratory frequency of 258.9 bpm (n = 5, P < 0.0001) (Fig. 9A). The respiratory frequency decreased further in D564N mutants when challenged (253.5 bpm, n = 5, P < 0.0001). These results are in contrast to wild-type animals where the respiratory frequency increased to 392.7 bpm as a response to the challenge (hypercapnia + hypoxia conditions) (Fig. 9). Minute ventilation and mean inspiratory flow were equivalent in wild-type mice and D564N mutants under normoxia conditions (Fig. 9B and C). However, when challenged, the D564N mutants did not respond by increasing the minute ventilation and mean inspiratory flow as observed in wild-type mice (Fig. 9B and C). Interestingly, D564N homozygous mutants demonstrated a higher tidal volume compared with wild-type mice under normoxia conditions that was increased further under hypercapnia + hypoxia conditions (Fig. 9D). These studies demonstrate that Ighmbp2D564N/D564N mice fail to respond to challenge conditions by increasing the respiratory frequency, minute ventilation or mean inspiratory flow as compared with wild-type animals; instead, D564N mutants appear to over-compensate by increasing tidal volume under normoxia and challenge conditions. Additionally, the number of apneas is significantly increased in Ighmbp2D564N/D564N mice versus wild-type mice under normoxia conditions but not during hypercapnia + hypoxia conditions (Fig. 9E). The percent of time D564N mutants spent in erratic breathing was significantly increased under normoxia and hypercapnia + hypoxia conditions (Fig. 9F). ssAAV9-IGHMBP2 delivery to Ighmbp2D564N/D564N mice improved the respiratory frequency, number of apneas and erratic breathing to nearly wild-type under normoxia and hypercapnia + hypoxia conditions (Fig. 9). These results are significant in that they demonstrate that D564N mutants are capable of responding to challenge conditions following ssAAV9-IGHMBP2 delivery. Although there was not a significant difference between minute ventilation and mean inspiratory flow in wild-type and Ighmbp2D564N/D564N mice under normoxia conditions, ssAAV9-IGHMBP2 delivery to D564N mutants substantially increased minute volume and mean inspiratory flow well above wild-type animals following challenge conditions (Fig. 9B and C). Tidal volume was also further increased under challenge conditions following ssAAV9-IGHMBP2 delivery to Ighmbp2D564N/D564N mice (Fig. 9D). These results suggest that although ssAAV9-IGHMBP2 delivery to Ighmbp2D564N/D564N mice improved the respiratory frequency to wild-type; minute volume, mean inspiratory flow nor tidal volume were corrected to wild-type under hypercapnia + hypoxia conditions and were substantially elevated above wild-type (Fig. 9). These data demonstrate that D564N mutant mice exhibit reduced respiratory function. Although treatment does significantly improve many aspects of respiration, it is noteworthy that ssAAV9-IGHMBP2 delivery to D564N mutant mice did not improve all respiratory deficits.
Figure 9.
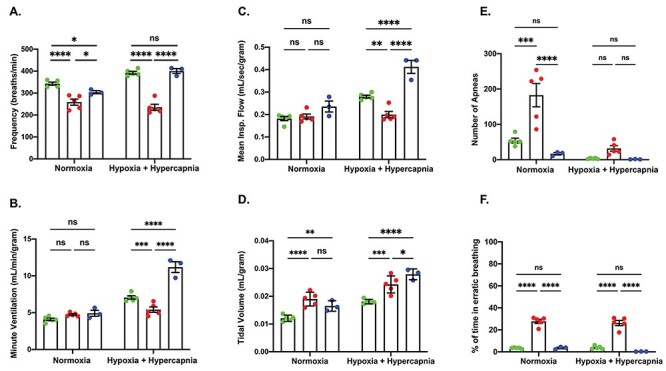
Ighmbp2 D564N/D564N mice have respiratory abnormalities that can be improved with ssAAV9-IGHMBP2 delivery. (A-F) PND13 Ighmbp2+/+ mice (green), Ighmbp2D564N/D564N mice (red) and Ighmbp2D564N/D564N ssAAV9-IGHMBP2 injected mice (blue) treated under normoxia and hypoxia + hypercapnia conditions. (A) Respiratory frequency of mice under normoxia and hypoxia + hypercapnia conditions. (B) Minute volume of the three cohorts under normoxia and hypoxia + hypercapnia conditions. (C) Mean inspiratory flow of mice under normoxia and hypoxia + hypercapnia conditions. (D) Tidal volume of the three cohorts under normoxia and hypoxia + hypercapnia conditions. (E) Number of apneas of mice under normoxia and hypoxia + hypercapnia conditions. (F) Percent time spent in erratic breathing of three cohorts under normoxia and hypoxia + hypercapnia conditions. Statistical test used is a two-way ANOVA with a Tukey’s post-hoc test for multiple comparisons. Data bars expressed as mean ± SEM. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, n = 5 animals per cohort; ns = not significant.
Discussion
SMARD1 is a complex genetic disease defined by homozygous recessive and compound heterozygous mutations throughout the IGHMBP2 gene. To further understand the relationship between IGHMBP2 mutations and disease progression, our laboratory generated six FVB/NJ-Ighmbp2 mouse models based on mutations that correspond to SMARD1 and CMT2S patients. In this study, we characterized the Ighmbp2D564N mutation in mice (D565N in humans). The D565N mutation lies within the helicase domain of IGHMBP2 and patients with compound heterozygous mutations including D565N develop SMARD1. D564N mutant mice develop SMARD1 phenotypes and this is the first SMARD1 mouse model with quantifiable respiratory deficiencies as well as SMARD1 pathology.
Several hallmark measurements of disease severity: lifespan, weight, TTR and HLS were significantly reduced in the D564N homozygous mutant mice compared with the BKS-Ighmbp2nmd-2J model. Lifespan is measured in several months and is highly variable in BKS-Ighmbp2nmd-2J mice in contrast to Ighmbp2D564N/D564N mice (16–17 days). Interestingly, the lifespan and weight of D564N mutant mice is more consistent with the newly derived FVB/NJ-Ighmbp2nmd mice that have a lifespan of 18–21 days. Although the lifespan is shortened in D564N mutants, there remains a sufficient window for therapeutic delivery and improvement of SMARD1 phenotypes.
Hindlimb contractures and impaired mobility developed earlier (PND7) and progressed rapidly in D564N mutant mice as compared with BKS-nmd-2J (21 days) or FVB-nmd-2J (severe by 17 days) mice. BKS-nmd-2J mice show no defects in time to right assays, whereas D564N mutant mice have significant time to right differences observed as early as PND8 when testing is initiated. BKS-nmd-2J mice were also capable of completing rotarod assays until ~14 weeks of age suggesting at least moderate motor function is present despite significant motor neuron loss (10). Disease progression was more rapid in D564N mutants and loss of motor function and death ensued before rotarod testing could be initiated. The significant differences in lifespan, weight and motor function between nmd-2J and D564N homozygous mutant mice are likely attributed to the amount of functional IGHMBP2 protein present in each of these mutants. In nmd-2J mice, there is a reduction in full-length, functional IGHMBP2 protein. In contrast, D564N mutant mice do not significantly differ in the amount of IGHMBP2 as compared with wild-type mice suggesting that the disease phenotype is likely associated with lack of functional IGHMBP2 protein. Interestingly, AAV9-IGHMBP2 delivery at the low dose (1e11 viral genomes) did not impact survival nor weight in Ighmbp2D564N/D564N mice, whereas clear differences were observed at the high dose (5e11 viral genomes). These results are in contrast to those observed in BKS-nmd-2J and FVB-nmd-2J mice. (30–32) One possible explanation could be available IGHMBP2 functional protein. Nmd-2J mice already have 20–25% functional protein; therefore, any additional functional protein pushes these mice over the threshold of IGHMBP2 protein needed for normal cellular function. In contrast, D564N mutant mice produce no wild-type and exclusively produce full-length D564N IGHMBP2 protein. In vitro studies demonstrate D564N protein lacks helicase activity (25) and wild-type IGHMBP2 forms oligomers (26). Therefore, it is possible that if a heterooligomeric complex forms consisting of wild-type and mutant IGHMBP2, it is less functional than a homo-oligomeric complex consisting of wild-type IGHMBP2. The threshold is overcome by increasing to a higher viral titer for ICV delivery of human IGHMBP2 and mutant phenotypes are reduced. Biochemical analyses of the D564N mutant protein is currently being investigated.
Ighmbp2 D564N/D564N mice demonstrate significant motor neuron loss very early and these results are consistent with what is observed in nmd-2J mice. The presence of fully denervated end plates (~25%) in the gastrocnemius was significant by PND7 and substantially increased (>60%) by PND15 in D564N mutants. In other muscle groups used for limb movement, the plantaris and the soleus revealed only 10% or less of fully innervated end plates. In D564N mutants, the EDL and TA demonstrated significant but less severe denervation of 60% and 50%, respectively. These differences are likely not attributed due to fiber type alone in that the gastrocnemius and plantaris are considered primarily fast-twitch muscle fiber muscles, whereas the soleus is composed of primarily slow-twitch muscle fibers. These results are also in contrast to Amyotrophic Lateral Sclerosis (ALS) disease progression where motor neurons that innervate fast-twitch fibers are more vulnerable to denervation than slow twitch (33). Although there was significant denervation in muscles for limb movement in D564N mutants, it was surprising that muscles used in respiration (diaphragm, rectus abdominis, transverse abdominis) were largely spared from denervation. This result is even more interesting when taking into account the significant respiratory deficiencies measured by plethysmography.
Motor neuron loss could be reduced following ICV delivery of ssAAV9-IGHMBP2; however, motor neuron numbers did not achieve wild-type. Although motor neuron numbers were not completely restored, lifespan and motor function assays suggest that motor function was significantly improved well past ICV delivery at PND2 (46+ days). ssAAV9-IGHMBP2 delivery did, however, significantly preserve end plate innervation. These results suggest that motor neuron loss likely occurs early in Ighmbp2D564N/D564N mice and PND2 delivery of ssAAV9-IGHMBP2 preserves motor neurons that are still present. In contrast, ssAAV9-IGHMBP2 delivery maintained end plate innervation to wild-type likely either by stimulating branching and/or by sprouting.
Respiratory defects are a primary clinical symptom in SMARD1 patients; however, previous studies have not reported respiratory defects in Ighmbp2nmd-2J mice. Ighmbp2D564N/D564N mice demonstrate significant respiratory impairment. Ighmbp2D564N/D564N mice have a lower respiratory frequency compared with Ighmbp2+/+ mice under normoxia conditions that is further reduced under hypoxia + hypercapnia conditions. The lower respiratory frequency could be a result of a combination of different defects, including within the respiratory circuit. When wild-type mice are placed under conditions of hypoxia + hypercapnia the brain responds by increasing the respiratory frequency; however, the opposite occurs in D564N mutants as they fail to compensate in response to the respiratory challenge. Interestingly, the tidal volume was significantly increased in Ighmbp2D564N/D564N mice under normoxia and hypoxia with hypercapnia conditions relative to Ighmbp2+/+ mice. These results suggest that the Ighmbp2D564N/D564N mutant mice may be compensating for reduced respiratory frequency by generating increased tidal volume either through the diaphragm alone or by recruiting accessory inspiratory muscles such as the external intercostals. Recruitment of accessory breathing muscles is observed in several diseases including ALS. (34–36) Under hypoxia and hypercapnia conditions, minute volume and mean inspiratory flow was significantly decreased in Ighmbp2D564N/D564N mice suggesting the compensation, through whatever mechanism, was not sufficient when exposed to a respiratory challenge. Although NMJ denervation was not observed within the diaphragm of D564N mutants, there was reduction in myofiber area and perimeter. As the diaphragm is the primary muscle for inspiration, myofiber defects as well as motor neuron defects within the respiratory circuit could lead to some of the respiratory deficiencies detected in the D564N mutants. We cannot exclude however that there are additional abnormalities, not examined in this manuscript, that result in the respiratory defects observed. Importantly, the Ighmbp2D564N/D564N mice recapitulate a critical aspect of SMARD1 clinical symptoms, respiratory deficiencies, not observed in other SMARD1 animal models to date.
Material and Methods
Animal use and procedures
All experimental procedures were approved by the University of Missouri’s Institutional Animal Care and Use Committee and were performed according to the guidelines set forth in the Guide for the Use and Care of Laboratory Animals.
CRISPR/Cas9
To reduce off-target effects, an enhanced-specificity Cas9 (eSPCas9) protein was utilized. This variant has been shown to significantly reduce CRISPR off-targeting (37). The method used for examining CRISPR off-target sites utilized the CRISPR RGEN Tools website maintained by the Center for Genome Engineering Institute (Korea) and the CCTop website maintained by the Centre for Organismal Studies (Heidelberg) to calculate off-target scores. Any predicted off-target site with less than a 2 bp mismatch (including DNA or RNA bulges) or with less than 3 bp mismatches if no mismatches are in the 12 bp seed region of the sgRNA was PCR amplified and sequenced to ensure no erroneous edits were made. The sequence was aligned with the reference FVB_NJ_v1 genomic assembly.
D564N sgRNA and repair template
sgRNAs were ordered as chemically modified synthetic sgRNAs through Synthego. The chemical modifications on each sgRNA were 2′-O-methyl analogs and 3′ phosphorothioate internucleotide linkages at the first three 5′ and 3′ terminal RNA residues. The repair template was chemically synthesized by IDT as a 200 bp single-stranded DNA oligo (ssODN). The ssODN was complementary to the non-target strand and contained symmetrical homology arms. An additional silent mutation was introduced for genotyping purposes.
sgRNA sequence: 5′-CTGGAGATTAAGTCTGTTGA-3′.
Repair template sequence (sgRNA sequence disrupted by desired mutation): 5′-ATGATATCTTGGCCCTGCTTTCTGTTTCAGGTGGATCTGCTCAGACAGAGCCTCTCCAACAAGCACCCTGAGCTGGAGATTAAGTCTGTTAACGGCTTTCAAGGCCGAGAGAAAGAGGCTGTGCTCCTGACCTTTGTCAGGTCCAATAGGAAAGGTATGTGGCACAGCCCGTTTGAGCCTTGGGTTCAGCCTGGGAGTGT-3′. The boxed area represents the sgRNA target sequence and the bold letter indicates mutations engineered in the DNA repair template.
Zygote electroporation and embryo transfer
A mix containing a final concentration of 3.0 μM sgRNA, 2.0 μM enhanced specificity Cas9 protein (Sigma) and 1.6 μM ssODN was made immediately prior to electroporation. CRISPR sgRNA/Cas9 RNP complexes were formed by gently mixing the sgRNA and Cas9 protein together and incubating at room temperature for 10 min. After RNP formation, the ssODN repair template was added to the mixture. Using a NepaGene21 electroporator along with a 1.5-mm gap glass slide electrode, zygotes were electroporated under the following conditions: Poring pulse: 40 V, 3.5 ms length, 50 ms interval, 10% decay rate, positive polarity (x4 pulses) Transfer pulse: 5 V, 50 ms length, 50 ms interval, 40% decay rate, alternating polarity (x5 pulses). Electroporated zygotes were surgically transferred to pseudo pregnant surrogate females the same day as the electroporation. FVB/NJ embryo donor females (3 weeks of age) and stud males (10 weeks of age) were purchased from Jackson Laboratory. CD1 surrogate females (8 weeks of age) were purchased from Charles River.
Genotyping
Genotyping of neonatal pups was performed at PND1. Genomic DNA was obtained using the DNA isolation protocol from Jackson labs. The FVB/NJ-Ighmbp2D564N line was genotyped using FWD 5’-TGGTCACTGAAAGGTACAGTGCAG-3′ and REV 5’-GCTCCCTGCCCAGACTTAGTCTTC-3′ primers and Go Taq Green Master Mix (Promega). PCR conditions were 94°C denaturing for 3:00 min followed by 39 cycles of 94°C denaturing for 30 s, 57°C annealing for 30 s and 72°C extension for 1:00 min, the final extension was 72°C for 5 min. Amplicons were digested with HpaI and separated on a 4% gel to differentiate wild-type from mutant alleles.
Motor function tests
Motor function was measured by the TTR assay from PND8, a standardized time that unaffected controls begin to correct their position to upright after being placed on their backs. Each pup was placed on its backside and the time it takes for them to turn over and stabilize on all four paws was recorded. The maximum attempted time was 30 s. The TTR assay was conducted every other day from PND8 to PND20, and the averages were calculated for each cohort every other day. Hindlimb function was assessed measuring HLS, a spontaneous reflex reaction of mice to spread out their hind legs when they are held by their tail. HLS was initiated at PND7 daily. To administer the test, mice were held ~0.5″ from the base of their tail over the wire top of the cage for ~2 s. The relative distance between the splayed hindlimbs was recorded as a qualitative measurement. The rotarod assay measures the ability of the mouse to walk forward on a rotating rod in time. Mice were trained for the rotarod assay at PND40 for 5 days before data collection at PND46. The time that the mouse walked forward on the rotating rod was measured in seconds and recorded. Mice were trained on the grip strength apparatus beginning at PND40 for 5 days before data collection at PND46.
Western blot
Three mice per cohort (Ighmbp2+/+, Ighmbp2D564N/D564N, Ighmbp2D564N/D564N injected with AAV9-IGHMBP2) were sacrificed using 2.5% isoflurane. Whole spinal cord and whole brain tissue samples were freshly harvested from the mouse and immediately flash-frozen in liquid nitrogen. Protein was extracted with JLB buffer (50 mM Tris, pH = 8.0, 150 mM NaCl, 10% Glycerol, 20 mM NaH2PO4, 50 mm NaF, 2 mM EDTA) supplied with Complete Mini Protease Inhibitor Cocktail (Roche). 20 μg of total protein was separated by gel electrophoresis using a 4–12% SDS-PAGE gel (catalog M00653, GenScript). Proteins were transferred to Immobilon-P membrane (Millipore Sigma). The membrane was blocked with 5% BSA (Bovine Serum Albumin) for 1 h at room temperature. Membranes were incubated with anti-IGHMBP2 (1:1000; catalog PA5-68960; Invitrogen) and anti-Actin (11 000; Sigma) primary antibodies at 4°C overnight followed by incubating with horseradish peroxidase-conjugated anti-Rabbit secondary antibody to detect IGHMBP2 protein and horseradish peroxidase anti-Mouse to detect Actin protein (Jackson ImmunoResearch). The specificity of the IGHMBP2 antibody to IGHMBP2 protein was determined by multiple methods including confirmation using several IGHMBP2 antibodies, and recognition of the IGHMBP2 antibody to protein isolated from HEK293T cells transfected with pAAV-IGHMBP2. Immunoblots were visualized and images were acquired using a BioSpectrum 816 Imaging System (UVP, LLC). Densitometry quantification of the band of interest was determined by Image Studio software (v5.2.5, LI-COR) with a rectangle box drawn around the band and background density subtracted. Densitometry data were analyzed by PROC GLM of Statistical Analysis Systems (v9.4). F-test was used to determine treatment effects and Duncan-adjusted multiple range test for differences between groups.
Generation of ssAAV9-IGHMBP2 virus and ICV injections
The single-stranded AAV9-IGHMBP2 viral vector utilized has been previously described (31). Viral particles were generated in HEK293T cells (ATCC® CRL-3216™) and purified using three CsCl density-gradient ultracentrifugation steps followed by dialysis against HEPES buffer. Number of viral genomes were determined by qPCR using SYBR Green. The animals were injected via ICV injection, (38) at ~1 × 1011 viral genomes (low dose) or ~5 × 1011 viral genomes (high dose) on PND2.
Motor neuron immunohistochemistry
Animals were sacrificed by perfusion with ice-cold 4% paraformaldehyde (PFA) followed by a subsequent post-fixing at 4% PFA for 24 h at 4°C. Lumbar (L3–L5) regions from spinal cord tissue were dissected and cryoprotected in 30% sucrose solution overnight before embedded in optimal cutting temperature (OCT) media. Embedded tissues were cryosectioned at 16 μm thickness with every 10th section from the spinal cord tissue collected for immunohistochemistry. Sections were stained with choline acetyltransferase (ChAT) primary antibody (1:100; catalog AB144P; Millipore Sigma), Donkey anti-Goat Alexa Fluor-594 secondary antibody, (1:250; Jackson ImmunoResearch) and NeuroTrace Green Fluorescent Nissl (1:100; catalog N21480; ThermoFisher Scientific) for motor neuron identification. Images were collected using a Leica DM5500 B fluorescent microscope (Leica Microsystem Inc.) under 20x magnification. Motor neuron counts, cell body perimeter and area measurements were performed manually with Fiji Software (NIH) in a blinded manner from 14 sections per mouse.
NMJ immunohistochemistry
Whole mount preparations were post-fixed in 4% PFA following perfusion of each mouse. Anti-Neurofilament Heavy Chain (NF-H) (1:2000; catalog AB5539, Chemicon, EMD Millipore) and anti-Synaptic Vesicle 2 (SV2) (1:200; catalog YE269, Life Technologies) primary antibodies followed by Donkey anti-Chicken Alexa Fluor 488 (1:400; Jackson ImmunoResearch) and Goat anti-Rabbit Alexa Fluor 488 (1:200; Jackson ImmunoResearch) secondary antibodies were used to label the axon and synaptic terminal. Acetylcholine receptors were labeled with Alexa Fluor 594-conjugated α-Bungarotoxin (1:200; Life Technologies). Representative images were obtained using a laser scanning confocal microscope at 40x magnification (Leica TCS SP8, Leica Microsystems Inc.). NMJ analyses was performed in a blinded manner on at least three randomly selected fields of view per muscle at 20x magnification (Leica DM5500 B, Leica Microsystems Inc.). Images were analyzed based on the end plate overlap with the synaptic terminal. End plates with missing overlapping terminal were considered fully denervated, end plates with partial overlap were considered partially denervated and end plates with complete overlap were considered fully innervated using Fiji Software (NIH).
Skeletal muscle immunohistochemistry
Animals were sacrificed and perfused with 4% PFA followed by a post-fixing in 4% PFA for 24 h at 4°C. Skeletal muscle was dissected and cryoprotected in 30% sucrose overnight. Cryoprotected muscles were embedded in OCT media and cryosectioned at 16 μm. Sections were stained with anti-Laminin primary antibody (1:200; catalog L9393; Millipore Sigma) and Donkey anti-Rabbit Alexa Fluor-594 secondary antibody (1:400; Jackson ImmunoResearch). Imaging was obtained using a Leica DM5500 B fluorescent microscope (Leica Microsystem Inc.) under 40x magnification. Muscle fiber area and perimeter measurements were analyzed manually using Fiji Software (NIH) in a blinded manner.
Whole-body plethysmography
Mice were placed in a whole-body plethysmography chamber (Data Sciences International) to allow for quantitative measurement of ventilation through different gas concentrations (gas concentrations controlled by Flow Commander, Therapeutiq Research). Ventilation was assessed at PND12. The mice were acclimated to the chamber while breathing room air (21% O2, balance N2) for 5 min before ventilatory measurements were recorded for baseline conditions for an additional 30 min. Ventilatory measurements were then made during exposure to a hypoxic and hypercapnic gas mixture (10.5% O2, 7% CO2) for 5 min. The pressure calibration signal, ambient and chamber pressures, and mouse body mass were used to calculate the respiratory frequency (f), tidal volume (VT), minute ventilation (VE), mean inspiratory flow (VT/Ti), using Buxco FinePointe Software (Data Sciences International). In addition, the apnea detection function within FinePointe software was used to identify the percentage of erratic breathing (defined as any breathing that was not classified as a normal breath, sigh, apnea or sniff; Data Sciences International), and apneas that were defined as the absence of at least two inspirations (i.e. a pause in breathing 2x the normalized breath duration threshold). VT, VE and VT/Ti are reported normalized to mouse body weight (per g). Data were rejected if there was evidence of pressure fluctuations caused by gross body movements (39,40).
Statistical analysis
The statistical significance was compared for the experimental groups: Ighmbp2+/+, Ighmbp2D564N/D564N and Ighmbp2D564N/D564N + ssAAV9-IGHMBP2 (high or low dose). Survival analysis was determined with a log-ranked (Mantel-Cox) test. Percentages were calculated as values from unaffected and were standardized as 100%. Motor neurons, skeletal muscle fiber, peak weight, TTR and whole-body plethysmography statistical significance was used with a one-way ANOVA with a Tukey’s multiple comparison post-hoc test. NMJs were analyzed by a two-way ANOVA with a Tukey’s multiple comparison post-hoc test. Analyses were all performed with GraphPad Prism software. Error bars represent mean ± standard error of the mean (SEM). Data points on graph represent the average per animal with statistical analysis comparing the average of each animal.
Abbreviations
- SMARD1
spinal muscular atrophy with respiratory distress type I
- SMA
spinal muscular atrophy
- Ighmbp2
immunoglobulin mu DNA binding protein 2
- SEM
standard error of the mean
- CMT
Charcot-Marie Tooth disease
- ICV
intracerebroventricular
- NMJ
neuromuscular junction
- ssAAV
single stranded adeno-associated virus
- EDL
extensor digitorum longus
- TA
tibialis anterior
- RA
rectus abdominis
- TVA
transverse abdominis
- TTR
time to right
Conflict of Interest statement
C.L.L. is the co-founder and chief scientific officer for Shift Pharmaceuticals. M.A.L. has interests in Shift Pharmaceuticals.
Funding
National Institutes of Health/National Institute of Neurological Disorders and Stroke (R01NS113765 to M.A.L. and C.L.L.); National Institutes of Health/National Institute of General Medical Sciences (Training Grant T32 GM008396 to C.E.S. and S.M.R.H.); National Institutes of Health Post-baccalaureate Research Education Program (R25GM064120 to J.M.); National Institutes of Health/National Institute of General Medical Sciences (T34 GM136493 to Z.A.R.) and Missouri Spinal Cord Injury/Disease Research Program (R01 HL153612 to N.L.N.).
Supplementary Material
Contributor Information
Caley E Smith, Department of Veterinary Pathobiology, College of Veterinary Medicine, University of Missouri, Columbia, MO 65211, USA; Bond Life Sciences Center, University of Missouri, Columbia, MO 65211, USA.
Monique A Lorson, Department of Veterinary Pathobiology, College of Veterinary Medicine, University of Missouri, Columbia, MO 65211, USA; Bond Life Sciences Center, University of Missouri, Columbia, MO 65211, USA.
Sara M Ricardez Hernandez, Department of Veterinary Pathobiology, College of Veterinary Medicine, University of Missouri, Columbia, MO 65211, USA; Bond Life Sciences Center, University of Missouri, Columbia, MO 65211, USA.
Zayd Al Rawi, Department of Veterinary Pathobiology, College of Veterinary Medicine, University of Missouri, Columbia, MO 65211, USA; Bond Life Sciences Center, University of Missouri, Columbia, MO 65211, USA.
Jiude Mao, Department of Veterinary Pathobiology, College of Veterinary Medicine, University of Missouri, Columbia, MO 65211, USA; Bond Life Sciences Center, University of Missouri, Columbia, MO 65211, USA.
Jose Marquez, Department of Veterinary Pathobiology, College of Veterinary Medicine, University of Missouri, Columbia, MO 65211, USA; Bond Life Sciences Center, University of Missouri, Columbia, MO 65211, USA.
Eric Villalón, Department of Veterinary Pathobiology, College of Veterinary Medicine, University of Missouri, Columbia, MO 65211, USA; Bond Life Sciences Center, University of Missouri, Columbia, MO 65211, USA.
Amy N Keilholz, Department of Biomedical Sciences, College of Veterinary Medicine, University of Missouri, Columbia, MO 65211, USA.
Catherine L Smith, Department of Biomedical Sciences, College of Veterinary Medicine, University of Missouri, Columbia, MO 65211, USA.
Mona O Garro-Kacher, Department of Veterinary Pathobiology, College of Veterinary Medicine, University of Missouri, Columbia, MO 65211, USA; Bond Life Sciences Center, University of Missouri, Columbia, MO 65211, USA.
Toni Morcos, Department of Veterinary Pathobiology, College of Veterinary Medicine, University of Missouri, Columbia, MO 65211, USA; Bond Life Sciences Center, University of Missouri, Columbia, MO 65211, USA.
Daniel J Davis, Animal Modeling Core, University of Missouri, Columbia, MO 65211, USA.
Elizabeth C Bryda, Department of Veterinary Pathobiology, College of Veterinary Medicine, University of Missouri, Columbia, MO 65211, USA; Animal Modeling Core, University of Missouri, Columbia, MO 65211, USA.
Nicole L Nichols, Department of Biomedical Sciences, College of Veterinary Medicine, University of Missouri, Columbia, MO 65211, USA.
Christian L Lorson, Department of Veterinary Pathobiology, College of Veterinary Medicine, University of Missouri, Columbia, MO 65211, USA; Bond Life Sciences Center, University of Missouri, Columbia, MO 65211, USA.
References
- 1. Kaindl, A.M., Guenther, U.P., Rudnik-Schoneborn, S., Varon, R., Zerres, K., Schuelke, M., Hubner, C. and von Au, K. (2008) Spinal muscular atrophy with respiratory distress type 1 (SMARD1). J. Child Neurol., 23, 199–204. [DOI] [PubMed] [Google Scholar]
- 2. Rudnik-Schoneborn, S., Forkert, R., Hahnen, E., Wirth, B. and Zerres, K. (1996) Clinical spectrum and diagnostic criteria of infantile spinal muscular atrophy: further delineation on the basis of SMN gene deletion findings. Neuropediatrics, 27, 8–15. [DOI] [PubMed] [Google Scholar]
- 3. Grohmann, K., Schuelke, M., Diers, A., Hoffmann, K., Lucke, B., Adams, C., Bertini, E., Leonhardt-Horti, H., Muntoni, F., Ouvrier, R. et al. (2001) Mutations in the gene encoding immunoglobulin mu-binding protein 2 cause spinal muscular atrophy with respiratory distress type 1. Nat. Genet., 29, 75–77. [DOI] [PubMed] [Google Scholar]
- 4. Pitt, M., Houlden, H., Jacobs, J., Mok, Q., Harding, B., Reilly, M. and Surtees, R. (2003) Severe infantile neuropathy with diaphragmatic weakness and its relationship to SMARD1. Brain, 126, 2682–2692. [DOI] [PubMed] [Google Scholar]
- 5. Grohmann, K., Varon, R., Stolz, P., Schuelke, M., Janetzki, C., Bertini, E., Bushby, K., Muntoni, F., Ouvrier, R., Van Maldergem, L. et al. (2003) Infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1). Ann. Neurol., 54, 719–724. [DOI] [PubMed] [Google Scholar]
- 6. Rudnik-Schoneborn, S., Stolz, P., Varon, R., Grohmann, K., Schachtele, M., Ketelsen, U.P., Stavrou, D., Kurz, H., Hubner, C. and Zerres, K. (2004) Long-term observations of patients with infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1). Neuropediatrics, 35, 174–182. [DOI] [PubMed] [Google Scholar]
- 7. Porro, F., Rinchetti, P., Magri, F., Riboldi, G., Nizzardo, M., Simone, C., Zanetta, C., Faravelli, I. and Corti, S. (2014) The wide spectrum of clinical phenotypes of spinal muscular atrophy with respiratory distress type 1: a systematic review. J. Neurol. Sci., 346, 35–42. [DOI] [PubMed] [Google Scholar]
- 8. Diers, A., Kaczinski, M., Grohmann, K., Hubner, C. and Stoltenburg-Didinger, G. (2005) The ultrastructure of peripheral nerve, motor end-plate and skeletal muscle in patients suffering from spinal muscular atrophy with respiratory distress type 1 (SMARD1). Acta Neuropathol., 110, 289–297. [DOI] [PubMed] [Google Scholar]
- 9. Jedrzejowska, M., Madej-Pilarczyk, A., Fidzianska, A., Mierzewska, H., Pronicka, E., Obersztyn, E., Gos, M., Pronicki, M., Kmiec, T., Migdal, M. et al. (2014) Severe phenotypes of SMARD1 associated with novel mutations of the IGHMBP2 gene and nuclear degeneration of muscle and Schwann cells. Eur. J. Paediatr. Neurol., 18, 183–192. [DOI] [PubMed] [Google Scholar]
- 10. Grohmann, K., Rossoll, W., Kobsar, I., Holtmann, B., Jablonka, S., Wessig, C., Stoltenburg-Didinger, G., Fischer, U., Hubner, C., Martini, R. et al. (2004) Characterization of Ighmbp2 in motor neurons and implications for the pathomechanism in a mouse model of human spinal muscular atrophy with respiratory distress type 1 (SMARD1). Hum. Mol. Genet., 13, 2031–2042. [DOI] [PubMed] [Google Scholar]
- 11. Villalon, E., Shababi, M., Kline, R., Lorson, Z.C., Florea, K.M. and Lorson, C.L. (2018) Selective vulnerability in neuronal populations in nmd/SMARD1 mice. Hum. Mol. Genet., 27, 679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Viollet, L., Barois, A., Rebeiz, J.G., Rifai, Z., Burlet, P., Zarhrate, M., Vial, E., Dessainte, M., Estournet, B., Kleinknecht, B. et al. (2002) Mapping of autosomal recessive chronic distal spinal muscular atrophy to chromosome 11q13. Ann. Neurol., 51, 585–592. [DOI] [PubMed] [Google Scholar]
- 13. Maystadt, I., Zarhrate, M., Landrieu, P., Boespflug-Tanguy, O., Sukno, S., Collignon, P., Melki, J., Verellen-Dumoulin, C., Munnich, A. and Viollet, L. (2004) Allelic heterogeneity of SMARD1 at the IGHMBP2 locus. Hum. Mutat., 23, 525–526. [DOI] [PubMed] [Google Scholar]
- 14. Guenther, U.P., Varon, R., Schlicke, M., Dutrannoy, V., Volk, A., Hubner, C., von Au, K. and Schuelke, M. (2007) Clinical and mutational profile in spinal muscular atrophy with respiratory distress (SMARD): defining novel phenotypes through hierarchical cluster analysis. Hum. Mutat., 28, 808–815. [DOI] [PubMed] [Google Scholar]
- 15. Cottenie, E., Kochanski, A., Jordanova, A., Bansagi, B., Zimon, M., Horga, A., Jaunmuktane, Z., Saveri, P., Rasic, V.M., Baets, J. et al. (2014) Truncating and missense mutations in IGHMBP2 cause Charcot-Marie Tooth disease type 2. Am. J. Hum. Genet., 95, 590–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yuan, J.-H., Hashiguchi, A., Yoshimura, A., Yaguchi, H., Tsuzaki, K., Ikeda, A., Wada-Isoe, K., Ando, M., Nakamura, T., Higuchi, Y. et al. (2017) Clinical diversity caused by novel IGHMBP2 variants. J. Hum. Genet., 62, 599–604. [DOI] [PubMed] [Google Scholar]
- 17. Kulshrestha, R., Forrester, N., Antoniadi, T., Willis, T., Sethuraman, S.K. and Samuels, M. (2018) Charcot Marie Tooth disease type 2S with late onset diaphragmatic weakness: an atypical case. Neuromuscul. Disord., 28, 1016–1021. [DOI] [PubMed] [Google Scholar]
- 18. Kanaan, J., Raj, S., Decourty, L., Saveanu, C., Croquette, V. and Le Hir, H. (2018) UPF1-like helicase grip on nucleic acids dictates processivity. Nat. Commun., 9, 3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mizuta, T.R., Fukita, Y., Miyoshi, T., Shimizu, A. and Honjo, T. (1993) Isolation of cDNA encoding a binding protein specific to 5′-phosphorylated single-stranded DNA with G-rich sequences. Nucleic Acids Res., 21, 1761–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lim, S.C., Bowler, M.W., Lai, T.F. and Song, H. (2012) The Ighmbp2 helicase structure reveals the molecular basis for disease-causing mutations in DMSA1. Nucleic Acids Res., 40, 11009–11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Perego, M.G.L., Galli, N., Nizzardo, M., Govoni, A., Taiana, M., Bresolin, N., Comi, G.P. and Corti, S. (2020) Current understanding of and emerging treatment options for spinal muscular atrophy with respiratory distress type 1 (SMARD1). Cell. Mol. Life Sci., 77, 3351–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fukita, Y., Mizuta, T.R., Shirozu, M., Ozawa, K., Shimizu, A. and Honjo, T. (1993) The human S mu bp-2, a DNA-binding protein specific to the single-stranded guanine-rich sequence related to the immunoglobulin mu chain switch region. J. Biol. Chem., 268, 17463–17470. [PubMed] [Google Scholar]
- 23. Chen, N.N., Kerr, D., Chang, C.F., Honjo, T. and Khalili, K. (1997) Evidence for regulation of transcription and replication of the human neurotropic virus JCV genome by the human S(mu)bp-2 protein in glial cells. Gene, 185, 55–62. [DOI] [PubMed] [Google Scholar]
- 24. Zhang, Q., Wang, Y.C. and Montalvo, E.A. (1999) Smubp-2 represses the Epstein-Barr virus lytic switch promoter. Virology, 255, 160–170. [DOI] [PubMed] [Google Scholar]
- 25. Guenther, U.P., Handoko, L., Laggerbauer, B., Jablonka, S., Chari, A., Alzheimer, M., Ohmer, J., Plottner, O., Gehring, N., Sickmann, A. et al. (2009) IGHMBP2 is a ribosome-associated helicase inactive in the neuromuscular disorder distal SMA type 1 (DSMA1). Hum. Mol. Genet., 18, 1288–1300. [DOI] [PubMed] [Google Scholar]
- 26. de Planell-Saguer, M., Schroeder, D.G., Rodicio, M.C., Cox, G.A. and Mourelatos, Z. (2009) Biochemical and genetic evidence for a role of IGHMBP2 in the translational machinery. Hum. Mol. Genet., 18, 2115–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cook, S.A., Johnson, K.R., Bronson, R.T. and Davisson, M.T. (1995) Neuromuscular degeneration (nmd): a mutation on mouse chromosome 19 that causes motor neuron degeneration. Mamm. Genome, 6, 187–191. [DOI] [PubMed] [Google Scholar]
- 28. Cox, G.A., Mahaffey, C.L. and Frankel, W.N. (1998) Identification of the mouse neuromuscular degeneration gene and mapping of a second site suppressor allele. Neuron, 21, 1327–1337. [DOI] [PubMed] [Google Scholar]
- 29. Shababi, M., Smith, C.E., Kacher, M., Alrawi, Z., Villalon, E., Davis, D., Bryda, E.C. and Lorson, C.L. (2019) Development of a novel severe mouse model of spinal muscular atrophy with respiratory distress type 1: FVB-nmd. Biochem. Biophys. Res. Commun., 520, 341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nizzardo, M., Simone, C., Rizzo, F., Salani, S., Dametti, S., Rinchetti, P., Del Bo, R., Foust, K., Kaspar, B.K., Bresolin, N. et al. (2015) Gene therapy rescues disease phenotype in a spinal muscular atrophy with respiratory distress type 1 (SMARD1) mouse model. Sci. Adv., 1, e1500078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shababi, M., Feng, Z., Villalon, E., Sibigtroth, C.M., Osman, E.Y., Miller, M.R., Williams-Simon, P.A., Lombardi, A., Sass, T.H., Atkinson, A.K. et al. (2016) Rescue of a mouse model of spinal muscular atrophy with respiratory distress type 1 by AAV9-IGHMBP2 is dose dependent. Mol. Ther., 24, 855–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Maddatu, T.P., Garvey, S.M., Schroeder, D.G., Hampton, T.G. and Cox, G.A. (2004) Transgenic rescue of neurogenic atrophy in the nmd mouse reveals a role for Ighmbp2 in dilated cardiomyopathy. Hum. Mol. Genet., 13, 1105–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Atkin, J.D., Scott, R.L., West, J.M., Lopes, E., Quah, A.K. and Cheema, S.S. (2005) Properties of slow- and fast-twitch muscle fibres in a mouse model of amyotrophic lateral sclerosis. Neuromuscul. Disord., 15, 377–388. [DOI] [PubMed] [Google Scholar]
- 34. Johnson, R.A. and Mitchell, G.S. (2013) Common mechanisms of compensatory respiratory plasticity in spinal neurological disorders. Respir. Physiol. Neurobiol., 189, 419–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Romer, S.H., Seedle, K., Turner, S.M., Li, J., Baccei, M.L. and Crone, S.A. (2017) Accessory respiratory muscles enhance ventilation in ALS model mice and are activated by excitatory V2a neurons. Exp. Neurol., 287, 192–204. [DOI] [PubMed] [Google Scholar]
- 36. Seven, Y.B., Nichols, N.L., Kelly, M.N., Hobson, O.R., Satriotomo, I. and Mitchell, G.S. (2018) Compensatory plasticity in diaphragm and intercostal muscle utilization in a rat model of ALS. Exp. Neurol., 299, 148–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Anderson, K.R., Haeussler, M., Watanabe, C., Janakiraman, V., Lund, J., Modrusan, Z., Stinson, J., Bei, Q., Buechler, A., Yu, C. et al. (2018) CRISPR off-target analysis in genetically engineered rats and mice. Nat. Methods, 15, 512–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Coady, T.H., Baughan, T.D., Shababi, M., Passini, M.A. and Lorson, C.L. (2008) Development of a single vector system that enhances trans-splicing of SMN2 transcripts. PLoS One, 3, e3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Drorbaugh, J.E. and Fenn, W.O. (1955) A barometric method for measuring ventilation in newborn infants. Pediatrics, 16, 81–87. [PubMed] [Google Scholar]
- 40. Jacky, J.P. (1978) A plethysmograph for long-term measurements of ventilation in unrestrained animals. J. Appl. Physiol. Respir. Environ. Exerc. Physiol., 45, 644–647. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.