

Abstract
Pediatric inflammatory bowel disease (PIBD) is a globally increasing chronic inflammatory disease associated with an imbalanced intestinal microbiota and treated with several treatment options, including anti-tumor necrosis factor alpha (TNF-α), such as infliximab (IFX). Up to half of the patients do not respond to the drug and there are no methods for response prediction. Our aim was to predict IFX response from the gut microbiota composition since this is largely unexplored in PIBD. The gut microbiota of 30 PIBD patients receiving IFX was studied by MiSeq sequencing targeting 16S and ITS region from fecal samples collected before IFX and two and six weeks after the start of treatment. The response to IFX induction was determined by fecal calprotectin value < 100 µg/g at week six. The bacterial microbiota differed significantly between response groups, with higher relative abundance of butyrate-producing bacteria in responders compared to non-responders at baseline, validated by high predictive power (area under curve = 0.892) for baseline Ruminococcus and calprotectin. Additionally, non-responders had higher abundance of Candida, while responders had higher abundance of Saccharomyces at the end of the study. The gut microbiota composition in PIBD patients could predict response to IFX treatment in the future.
Subject terms: Biomarkers, Microbiome
Introduction
Inflammatory bowel diseases (IBD) including Crohn’s disease (CD), Ulcerative Colitis (UC) and IBD unclassified (IBDU) are chronic inflammatory diseases characterized by inflammation in the gastrointestinal tract. The incidence of pediatric IBD (PIBD) is increasing worldwide both in developing and developed countries1. At diagnosis PIBD often presents with similar symptoms to adults’, which may include diarrhea, abdominal pain, bloody stools and fever2. In addition, PIBD (especially CD) may cause malnourishment, leading to growth impairment, delayed puberty or even psychosocial problems2. Effective treatment is crucial in order to avoid these long lasting sequalae in patients with PIBD.
The current hypothesis for the pathogenesis of IBD is that the combination of genetic, environmental and microbial factors leads to altered gut permeability and abnormal and prolonged immune responses with the alteration of gut microbiota composition with predominance of pathobionts that contribute to gut inflammation3. The role of the fungal gut microbiota, the mycobiota, is not as established, but may also be characterized as dysbiotic4, with Candida spp. being possible drivers of the imbalance5–7. Additionally, it has been observed that Saccharomyces species are less abundant in patients with IBD compared to healthy controls (HC)6. Studies concerning the gut mycobiota in PIBD are few, and although the results vary somewhat between the studies, both Candida and Malassezia have been reported as increased in PIBD, as well as the phylum Basidiomycota, as reviewed recently8. Many studies have reported a reduction in the diversity of commensal bacteria, especially in the two most abundant phyla Bacteroidetes and Firmicutes, in adult patients with IBD3. Additionally, a reduced abundance of potentially anti-inflammatory genera such as Roseburia, Bifidobacterium and Faecalibacterium and an increased abundance of Proteobacteria, Veillonellaceae, Pasteurellaceae, Fusobacterium, and Ruminococcus gnavus, some of which have proinflammatory effects, have been reported in adult patients with IBD when compared to HC3. Accordingly, the bacterial microbiota of patients with PIBD has been found to differ from HC with an increased relative abundance of Prevotella9. A recent review regarding gut microbiota profiles in PIBD, encompassing 41 studies, reported an increase in Enterococcus and significant decrease in Anaerostipes, Blautia, Coprococcus, Faecalibacterium, Roseburia, Ruminococcus, and Lachnospira in PIBD compared to HC, and a decrease in alpha-diversity within majority of the included articles10.
There are multiple therapy choices for PIBD, including conventional medications such as 5-ASA, immunomodulators and steroids, and explored for novel treatment options, such as fecal microbiota transplantation11. Recent ECCO-ESPGHAN guideline recommends anti-tumor necrosis factor alpha (TNF-α), such as infliximab (IFX), in pediatric CD with a high risk of poor outcome, if serious growth delay is present, or EEN and corticosteroid treatment do not induce remission12. A recent ESPGHAN guideline for pediatric UC recommends IFX in chronically active of steroid dependent UC, uncontrolled by 5-ASA and thiopurines13. However, up to 50% of patients do not respond to IFX, and loss of response is frequent14. To this day, there are no clinical methods available to predict the response although many have studied the bacterial microbiota with the aim to find biomarkers for prediction of anti-TNF-α treatment response to improve the cost-effectiveness of the therapy15–20.
There are very limited studies on the mycobiota in patients with PIBD receiving IFX therapy, and the study approach differ from this study21. Previous studies regarding the treatment response and disease severity have reported a reduced relative abundance of Faecalibacterium prausnitzii (F. prausnitzii)17,22 and we recently found an increased relative abundance of Candida to correlate with poor treatment response to anti-TNF-α in adult patients with IBD23. It has been discovered that adult patients with IBD have an imbalanced Ascomycota and Basidiomycota ratio24 and a higher abundance of Candida albicans compared to HC4. In children, a low baseline abundance of F. prausnitzii was associated with a lack of mucosal healing and need of surgical treatment9. In our previous study regarding the fecal microbiota and the treatment response to anti-TNF-α in PIBD patients we discovered several bacteria as potential biomarkers predicting treatment response such as higher relative abundances of Bifidobacterium, Clostridium colinum, Eubacterium rectale, uncultured Clostridiales, and Vibrio, and a lower abundance of Streptococcus mitis20.
The aim of this study was to describe the gut microbiota in patients with PIBD introduced to anti-TNF-α medication IFX and to study whether the fecal fungal and bacterial microbiota profiles predict IFX treatment response. Also, we aimed to study the association between the gut fungal and bacterial microbiota. Improved understanding of the complex interactions between the fungi and bacteria of the intestine could provide new insight to dysbiosis seen in IBD, possibly even having impact on the adjustment of therapy.
Results
Finding methods for predicting the response to IFX therapy to avoid unnecessary side effects and high cost, particularly for pediatric patients, is crucial. Here we have investigated the fungal and bacterial gut microbiota profiles with the aim to find predictive markers for IFX response.
Patient characteristics and response to IFX
In this study, a total of 30 patients with PIBD introduced to TNF-α antagonist IFX were included with a total of 85 samples collected before the start of IFX treatment and at two and six weeks of induction therapy. Out of the 30 patients, 27 patients with 68 samples were available for 16S sequencing, while 85 samples of 30 patients were available for ITS sequencing. 25 were diagnosed with CD and 5 with UC/IBDU. The background characteristics of the patients are shown in Table 1. At six weeks three patients were transferred to adult care. Therefore, the IFX response for three patients was determined by the fecal calprotectin value at week two since these patients terminated the study before week six (lack of third sample). One patient discontinued at week 2 due to loss of response and was considered a non-responder. In total 18 patients (60%) were NRs and 12 Rs. The number of patients and samples at each timepoint and the response to IFX of these for both the ITS and 16S data is presented in Fig. 1. IFX trough levels were available from 15 patients (9 NRs and 6 Rs) at 6 weeks post treatment. The median value was 4.6 mg/L (min–max = 0–11) for NRs and 11.05 mg/L (min–max = 0.79–10.1) for Rs. The trough levels did not differ significantly between the response groups.
Table 1.
Patient characteristics.
| Characteristics | n (%) or Median (min–max) | |
|---|---|---|
| No. of patients | 30 | |
| 12 R | 18 NR | |
| Male | 8 (67) | 13 (72) |
| CD | 11 (92) | 14 (78) |
| UC | 1 (8) | 1 (6) |
| IBD unclassified | 0 (0) | 3 (17) |
| Age at diagnosis, years | 13 (6–16) | 13 (6–16) |
| Age at IFX initiation, years | 15 (9–18) | 14 (6–17) |
| Anti-TNF-α naïve | 9 (75) | 17 (94) |
| Previous exposure to IFX* | 3 | 1 |
| Disease duration at recruitment, years | 1.4 (0–4) | 0.3 (0–6) |
| IBD surgery | 0 (0) | 2 (11) |
| Baseline fecal calprotectin value (µg/g)** | 360 (7–1317) | 769 (55–6293) |
| 2-week fecal calprotectin value (µg/g) | 25 (< 5–496) | 456 (36–1876) |
| 6-week fecal calprotectin value (µg/g) | 41 (5–89) | 399 (162–2142) |
| Baseline symptom index score38 | 1 (1–4) | 1 (1–8) |
| Baseline VAS (disease impact on QOL)38 | 2 (1–4) | 2 (1–6) |
| Baseline physicians’ global assessment39 | 2 (1–3) | 2 (1–3) |
| Concomitant medication at IFX initiation | ||
| Steroid | 6 (50) | 12 (67) |
| 5-aminosalicylic acid | 5 (42) | 7 (58) |
| Azathioprine | 7 (58) | 3 (17) |
| Methotrexate | 1 (8) | 0 (0) |
| Ursodeoxycholic acid | 1 (8) | 1 (6) |
| Antibiotics*** | 5 (42) | 9 (50) |
| Saccharomyces boulardii supplement**** | 0 (0) | 2 (11) |
| Lactic acid bacteria supplement (regular use) | 9 (75) | 9 (50) |
CD Crohn's disease, UC ulcerative colitis, IBD inflammatory bowel disease, IFX infliximab, TNF tumor necrosis factor.
*Time since exposure at baseline 10–26 months.
**Three patients had baseline fecal calprotectin value < 100 µg/g.
***Metronidazole, cephalosporin or amoxicillin in 12, other = 1.
****Two weeks prior to the study.
Figure 1.

Overview of the study outline presenting the number of patients and samples at the three different timepoints (before start of treatment and 2- and 6 weeks post-treatment), response to infliximab (IFX) and reason of exclusion. R = responder, and NR = non-responder to IFX therapy.
We had three patients with fecal calprotectin value < 100 at the start of the study (Table 1). Of the three patients with calprotectin < 100 at baseline the indication to start IFX treatment were steroid-dependency (n = 2) and perianal disease (n = 1).
Overview of fungal and bacterial gut microbiota in patients with PIBD
Fungal gut microbiota
For fungal analyses, successful annotation with read count above 100 was achieved for 47 samples from 23 patients. Across all samples, the gut mycobiota was composed of Ascomycota (83%) and Basidiomycota (10%) and uncultured Eukaryota (7%). Altogether 33 genera were present in the gut mycobiota, and the most abundant genera were Candida (64%), followed by Saccharomyces (51%), uncultured fungus (20%) and Cladosporium (14%), presented in Fig. 2A. The prevalence for all genera is presented in Supplementary Table 1. At species level [Candida albicans] (38%) and [Saccharomyces cerevisiae] (22%) were most prevalent.
Figure 2.

Overview of the fecal (A) fungal mycobiota and (B) bacterial microbiota composition before start of infliximab (IFX) therapy (baseline), two weeks and six weeks after treatment stratified by response to induction therapy to IFX. The plots present the most abundant genera, which are color-coded and shown on the right-side panel. (C) The standardized confounder-adjusted relative abundance of the fungal genera that significantly differed between response groups presented throughout the study and (D) the relative abundance of the bacterial genera and (E) classes that differed significantly at baseline between response groups presented throughout the study. R presents responders and NR presents non-responders. (*p FDR).
Bacterial gut microbiota
For bacterial analyses, a total of 68 samples from 27 patients were available. The fecal microbiota of the samples was composed of the phyla Firmicutes (68%), Actinobacteria (14%), Bacteroidetes (12%), and Proteobacteria (5%). The most abundant genera across all samples were Faecalibacterium (9.1%). Bifidobacterium (5.5%), Blautia (4.2%) and Bacteroides (4%). The most prevalent genera at different timepoints are presented in Fig. 2B.
Fungal and bacterial microbiota composition as markers for prediction of IFX response
Differences in fecal fungal microbiota according to IFX therapy response
The fecal mycobiota did not differ significantly between response groups at baseline. At two weeks after induction the order Saccharomycetales was more abundant in Rs (by 1.03-fold, P FDR < 0.001). At six weeks the genus Candida [C. albicans] was more abundant in NRs (by 22-fold, P FDR < 0.001) and Saccharomyces [S. cerevisiae] in Rs (by 1.9-fold, P FDR < 0.001). This is presented in Fig. 2C where the standardized confounder-adjusted relative abundance of the genera Candida and Saccharomyces is plotted throughout the study. The fungal diversity and richness at the different timepoints stratified by response groups is presented in Supplementary Fig. 1A, although there were no significant differences between response groups.
Differences in response groups to IFX therapy in fecal bacterial microbiota
At baseline the bacterial microbiota composition differed between Rs and NRs. The classes Clostridia (1.2-fold, P FDR < 0.001) and Bacilli (1.2-fold, P FDR < 0.001) were more abundant in Rs, whereas the class Gammaproteobacteria (4.4-fold, P FDR < 0.001) was more abundant in NRs. At genus level, the Rs had an increased relative abundance of Faecalibacterium (1.4-fold, P FDR < 0.001) and Subdoligranulum (1.7-fold, P FDR < 0.001), whereas the NRs had an increased abundance of Dialister (1.2-fold, P FDR < 0.001) and Anaerostipes (2.3-fold, P FDR < 0.001) when compared to the Rs. The statistically significantly differing genera are presented in Fig. 2D at all timepoints, and classes Bacilli and Clostridia are depicted in Fig. 2E. All statistically significantly differing bacterial taxa of other taxonomic levels are listed in Supplementary Table 2.
At six weeks after start of IFX treatment the Rs had an increased relative abundance of classes Actinobacteria (by 1.5-fold, P FDR = 0.041) and Erysipelotrichia (by 3.3-fold, P FDR = 0.024). At genus level, the Rs had decreased relative abundances of Blautia (by 5.6-fold, P FDR < 0.001), Coprococcus (by 6.3-fold, P FDR = 0.024), Lachnospiraceae (Insertae Sedis) (by 4.7-fold, P FDR = 0.0031) and Dialister (by 11-fold, P FDR = 0.048), but an increased relative abundance of Bifidobacterium (by 2.0-fold, P FDR = 0.048). All statistically significantly differing bacterial taxa of other taxonomic levels are listed in Supplementary Table 3.
The bacterial diversity and richness at the different timepoints stratified by response groups is presented in Supplementary Fig. 1B. Neither bacterial diversity nor richness differed significantly between treatment response groups at any timepoints. The response to IFX therapy was predictable by the bacterial genus Ruminococcus combined with baseline fecal calprotectin values before start of treatment with AUC 0.892 respectively (Fig. 3). Additionally, the response was predicted by baseline Ruminococcus relative abundance alone (AUC 0.790).
Figure 3.
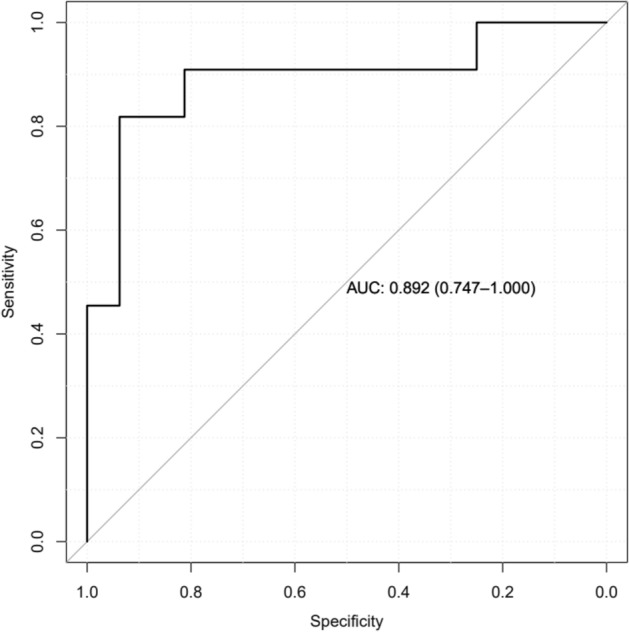
Receiver operating characteristic (ROC) curve to predict the response to infliximab therapy in patients with pediatric inflammatory bowel disease at baseline. The bacterial genus Ruminococcus and baseline fecal calprotectin values were included in the model. The area under curve (AUC) is indicated.
Disease duration, age, sex and antibiotics
Gut microbiota composition and disease duration before the start of IFX
Disease duration was defined as the time from diagnosis to the start of IFX treatment. In fecal mycobiota we observed no difference in the composition in relation to disease duration.
Regarding fecal bacterial microbiota, the patients with disease duration for more than a year (n = 14) had a lower relative abundance of the class Clostridia (by 1.2-fold, P FDR < 0.001) at baseline. At genus level, this group of patients had a higher relative abundance of the genera Faecalibacterium (1.1-fold, P FDR < 0.001) and Subdoligranulum (1.3-fold P FDR < 0.001) when compared to patients with a disease duration less than a year (n = 14). All statistically significantly differing bacterial taxa of other taxonomic levels are listed in Supplementary Table 4.
Gut microbiota composition in age groups
The age of the patients varied from 6 to 18 years (median = 14) at the initiation of IFX therapy. To investigate the gut microbiota composition according to age stratification we divided the patients into older and younger than 12 years (an arbitrary cut-off for prepubertal and pubertal patients), with 8 patients being younger than 12 years at IFX therapy initiation. The analyses were done at baseline.
In the fungal microbiota we observed that the genus Saccharomyces [S. cerevisiae] was significantly more abundant (P FDR < 0.001, by twofold) in patients younger than 12 years of age.
In the bacterial microbiota, we discovered that the patients older than 12 years had an increased relative abundance of the class Clostridia (by 1.1-fold, P FDR < 0.001), but a decrease in Bacilli (by 2.8-fold, P FDR < 0.001). At genus-level, the patients older than 12 years had a lower relative abundance of Dialister (by 1.9-fold, P FDR < 0.001). All statistically significantly differing bacterial taxa of other taxonomic levels are listed in Supplementary Table 5.
Gut microbiota composition difference between sex
There was a significant difference in mycobiota composition between sex with a higher abundance of the class Saccharomycetes (by 2.9-fold, P FDR = 0.04) and a lower abundance of the genus Saccharomyces [S. cerevisiae] (by 2.1-fold, P FDR < 0.001) in females compared to males at baseline.
At baseline we observed a higher relative abundance of the class Clostridia (by 1.1-fold, P FDR < 0.001) in males, when compared to females. At genus level, females had significantly higher relative abundances of Faecalibacterium (2.1-fold, P FDR < 0.001), but a lower relative abundance of Dialister (by 1.5-fold, P FDR < 0.001). All statistically significantly differing bacterial taxa of other taxonomic levels are listed in Supplementary Table 6.
The effect of antibiotics on the microbiota composition
We discovered that patients who had received antibiotics during the prior month to sampling had a higher relative abundance of the fungal genus Saccharomyces [S. cerevisiae] (by twofold, P FDR < 0.001) at baseline. Additionally, the antibiotic-treated patients had a lower abundance of Clostridia (by 1.2-fold, P FDR < 0.001), but an increased abundance of Bacilli (by threefold, P FDR < 0.001). At genus-level, patients receiving antibiotics had a significantly reduced abundance of Faecalibacterium (by 4.2-fold, P FDR < 0.001) compared to those who did not receive antibiotics one month prior to sampling. All statistically significantly differing bacterial taxa of other taxonomic levels are listed in Supplementary Table 7.
Correlation between fungal and bacterial microbiota composition
Correlations between fungal and bacterial microbiota were eligible to be calculated for 5 Rs and 13 NRs at baseline and the correlations differed between the response groups, values at baseline are presented in Supplementary Tables 8 and 9.
In Rs Saccharomyces correlated positively with Akkermansia (r = 0.89, P = 0.04) and Candida correlated positively with Lactococcus (r = 0.92, P = 0.03) at baseline.
Spearman correlations between fungal and bacterial genera in NRs are presented in Table 2.
Table 2.
Statistical tests assessing Spearman correlations between relative abundance of fungal and bacterial genera were performed at baseline (introduction to infliximab) in pediatric patients with inflammatory bowel disease who turned out to be non-responders to infliximab at the end of induction therapy (week six).
| Bacterial genus | Fungal genus | Spearman correlation (r) | P value |
|---|---|---|---|
| Bacteroides | Botrytis | 0.63 | 0.02 |
| Barnesiella | Uncultured fungus | 0.57 | 0.04 |
| Blautia | Uncultured fungus | 0.68 | 0.01 |
| Collinsella | Candida | 0.60 | 0.03 |
| Parasutterella | Botrytis | − 0.59 | 0.03 |
| Peptostreptococcus | Saccharomyces | − 0.59 | 0.03 |
| Prevotella | Saccharomyces | − 0.58 | 0.04 |
| IncertaeSedis | Uncultured Cladosporium | − 0.71 | 0.006 |
| IncertaeSedis | Uncultured Penicillium | 0.56 | 0.047 |
| Lactobacillus | Uncultured Cladosporium | 0.706 | 0.007 |
| Enterococcus | Uncultured Penicillium | 0.56 | 0.047 |
| Phascolarctobacterium | Uncultured Penicillium | 0.60 | 0.03 |
| Klebsiella | Uncultured Saccharomyces | 0.63 | 0.02 |
Discussion
In this study, we explored the fungal and bacterial gut microbiota in pediatric patients with IBD, who received anti-TNF-α medication IFX with the aim to find microbiota markers for prediction of treatment response. We demonstrate differences in the gut microbiota compositions between response groups to IFX therapy in PIBD, further validated by high predictive power for the bacterial microbiota. Additionally, we found differences in the interkingdom interactions between the response groups. Together these results suggest that intestinal microbes have potential as biomarkers for prediction of treatment response in patients with PIBD, that previously have been investigated from the bacterial microbiota profile but are largely unexplored for the fungal microbiota.
Although being less abundant than bacteria25, the fungal species seem to have a significant role in IBD, with a composition characterized by a higher abundance of Basidiomycota and lower abundance of Ascomycota compared to HC24. This is observed as an increase of Candida and a decrease of Saccharomyces and Malassezia in IBD24. The gut mycobiota of patients with PIBD consisted of the phyla Ascomycota and Basidiomycota across all samples, as previously observed8,24. The most abundant genus was Candida, followed by Saccharomyces, also agreeing with previous studies24. In pediatric studies, Candida has been observed to associate with PIBD and with disease intensity5,24,26.
In younger, in male and in antibiotic-treated patients, the relative abundance of Saccharomyces was higher. The number of female patients was low (less than 30% of all patients), and most were prepubertal. Using age-grouping below and above 12 years is somewhat arbitrary due to not considering factors such as females and males reaching puberty at different ages, and the impact of IBD on the age of reaching puberty. The species S. boulardii is characterized with anti-inflammatory properties and is used as a probiotic27, however only two of our patients used such probiotics two weeks prior to the study, of which both had Saccharomyces present in the mycobiota. Antibiotics cause a decrease in bacteria, which in turn causes less competition to fungal species28. If fungi play a role in IBD disease progression, the use of antibiotics may have unpredicted consequences as fungi will benefit from it.
The bacterial microbiota consisted of the phyla Firmicutes, Actinobacteria, Bacteroidetes and Proteobacteria, accounting up to 99% of all bacteria. Similar bacterial composition has been discovered in adult studies, both within HC and IBD patients, where these four phyla accounted from 95 to 98% of all reads15.
Longer disease duration associated with a reduced relative abundance of the class Clostridia, but an increase in Clostridia genera, namely Faecalibacterium and Subdoligranulum. Faecalibacterium and Subdoligranulum both produce butyrate which has an anti-inflammatory effect by upregulating the secretion of IL-10, an anti-inflammatory cytokine, and strengthening the intestinal barrier26,29–31. This suggests a decreased inflammation in patients for whom the disease duration was more than a year. Active inflammation in the gut has been noted to alter the intestinal microbiota and change during remission in response to anti-TNF-α7,15,16,22.
Finally, patients who received antibiotics two weeks prior to the study had a significantly reduced abundance of Faecalibacterium, an important anti-inflammatory bacterium, as stated above. Either the disease is more severe and perhaps fistulizing and therefore antibiotics have been used as a treatment, or the use of antibiotics has severely disturbed the intestinal homeostasis towards a more inflammatory state.
Few have investigated the mycobiota in PIBD7,8,26, and to our knowledge there is only one study investigating it for prediction of response to IFX treatment21, in which a metagenomic approach was used to investigate the microbiome, making it complicated to compare to this study. Here, the mycobiota was comparable between response groups at baseline, agreeing with the previous study21. However, Candida was significantly elevated in NRs while Saccharomyces was elevated in Rs at six weeks after therapy. This could indicate that the gut mycobiota of Rs shifts towards a healthier composition, since it has been observed that in HC Saccharomyces is increased24, while Candida is associated with disease progression in both adult and pediatric patients with IBD4,23,26. In adult IBD, we recently found that Candida was elevated in adult NRs to IFX prior to therapy initiation23. IBD is different in and pediatric patients, and in our adult study (median age 31 years) there were more UC patients compared to CD, different drugs were used in addition to IFX, and the male to female ratio was different; all of which might cause the differing results23.
The bacterial fecal microbiota in PIBD patients has been studied previously7,9,15–17,20,21. The methodologies used in these studies vary, which must be considered when comparing the results. We found that treatment response was associated with a microbiota characterized by bacteria capable of anti-inflammatory butyrate production, which might help reach and maintain remission. Rs had a higher relative abundance of the classes Clostridia and Bacilli, belonging to the phylum Firmicutes and the order Clostridiales at baseline. These results are supported by studies on both pediatric and adult cohorts investigating the response to biological therapy in the fecal microbiota15,20,23. In a previous study on adults, the relative abundance of Clostridiales increased during IFX treatment, resulting in a microbiota resembling HC, indicating that Clostridiales re-enrichment correlated with remission15. In PIBD, we previously discovered that the relative abundance of uncultured Clostridiales predicted treatment response to IFX28. Differences between Rs and NRs at such a high taxonomic level indicate a deeper dysbiosis in NRs (compared to Rs), since the relative abundance of Clostridiales is reduced in IBD7,15,20.
An increased baseline abundance of Faecalibacterium has been associated with response to biological therapy in adult and pediatric IBD9,15,17,22. Additionally, an increase in Faecalibacterium and another butyrate producing genus, Roseburia, has been associated with response to anti-TNF treatment7,15,16,22. The higher relative abundance of Clostridiales, Faecalibacterium and Subdoligranulum, in Rs reflects gained similarity to microbiota in HC along therapeutic response7,9,16,20. In Rs, we also discovered an increase in the class Bacilli and order Lactobacillales and a reduced relative abundances of the genus Dialister. In our previous study on adults, we observed Dialister to be decreased in UC in Rs to IFX before the start of treatment23.
In most studies the microbiota of Rs to anti-TNF-α has been associated with a baseline microbiota closer to HC and shifting during treatment even more towards that of HC7,9,15–17. Unexpectedly, we discovered that the Rs had a decreased relative abundance of Dialister, Blautia and Coprococcus at six weeks after the start of IFX treatment and all of these have been reported decreased in PIBD7,16,20, especially in those patients with a more profound dysbiosis7. However, we observed Bifidobacterium to be increased in the Rs at six weeks potentially balancing the dysbiosis caused by the lack of the likely beneficial Dialister, Blautia and Coprococcus.
A higher baseline diversity and an increase in microbial diversity during treatment has been associated with response to anti-TNF-α treatment in PIBD7,20. However, we did not discover statistically significant differences in fungal or bacterial diversities between treatment response groups at any timepoints.
The predictive power of the microbiota composition was further studied by the performance of the models created from the genera that we determined to differ significantly between response groups by the PathModel function. The AUC indicated high predictive power for the bacterial model. Ruminococcus, a bacterial genus that we used in the model, has previously been associated with response to anti-TNF-α medication22 with a decrease in Rs, in line with our results. In our previous study done on adult patients with IBD we found that the family unknown Ruminococcaceae was predicting the response at baseline with high AUC23. In the model for ROC curve analyses baseline fecal calprotectin value together with Ruminococcus was also predicting response. Fecal calprotectin is a neutrophil-derived biomarker of intestinal inflammation that is used in the evaluation of therapeutic responses particularly in pediatric patients where invasive methods such as biopsy are not preferable32. Candida predicted response in our previous study done on adult IBD patients23. Although a significant difference was not observed between response groups at baseline here, Candida was significantly more abundant in NRs at the end of the study.
The interaction between fungi and bacteria in the gut may have a significant role in the pathology of IBD23,24. Interestingly, we found that the interkingdom correlations differed between response groups. This difference was characterized by bacteria that previously have been shown to have beneficial roles in patients with IBD that correlated with Candida and Saccharomyces in Rs, while bacteria that have been associated with dysbiosis in IBD correlated with Candida and Saccharomyces in NRs. In Rs we found positive correlations between the abundance of Candida and Lactococcus, and between Saccharomyces and Akkermansia. Lactococcus has previously been associated with response to anti-TNF-α7 and lower abundances of Akkermansia has been associated with more severe inflammation in adult patients with UC33. In NRs we found Candida to correlate positively with Collinsella. As previously stated, Candida has been associated with non-response to anti-TNF-α23, while Collinsella has been associated with penetrating disease in PIBD34. Further, the abundance of Saccharomyces correlated negatively with Peptostreptococcus and Prevotella while unknown Saccharomyces correlated positively with Klebsiella in NRs. In adult patients with IBD, Peptostreptococcus was increased in NRs to anti-TNF-α19. High Prevotella and Klebsiella relative abundance is associated with dysbiosis35. Improved understanding of the interplay and relation of myco- and microbiota is needed to find out whether it is possible to modify the intestinal balance of species towards a more anti-inflammatory form to improve therapeutic responses.
The strengths of this study are that the samples were immediately frozen after sampling, ensuring good quality and exceptionally low loss of follow up. As a limitation, we had a relatively limited number of patients, as in most pediatric studies. However, this cohort is, to our knowledge, the largest one studying both the fungal and bacterial gut microbiota as predictive markers for IFX response in PIBD patients thus far. The problem with zero-inflation considering the ITS data is one not only present in this study, but in most studies concerning ITS data. Here it is evident in the number of samples with no detectable fungi or unsuccessful annotation. This could be either biological or methodological. Finally, the predictive taxa of fungi and bacteria found need to be validated in an independent, larger cohort to be applicable in clinical settings.
To conclude, we found significant differences between response groups to IFX medication in the fungal and bacterial microbiota composition of patients with PIBD, seen in the bacterial microbiota already before the start of IFX therapy. Non-responders had lower abundance of butyrate-producers such as bacteria in the Clostridia class, particularly the genera Faecalibacterium and Subdoligranulum before start of IFX therapy. The response was predictable at baseline by Ruminococcus (belonging to class Clostridia) and baseline fecal calprotectin values. Additionally, Candida was more abundant in NRs, while Saccharomyces was more abundant in Rs after IFX therapy. Together with previously published data, our results indicate that gut microbiota composition is a valuable resource in prediction of anti-TNF-α treatment response.
Methods
Study design
Patients with IBD introduced to IFX therapy at the Children´s Hospital, University of Helsinki were recruited to the study between the years 2011 and 2016. The participants were requested to collect a stool sample for study purposes before the start of IFX infusion and at two- and six-weeks infusions concurrent to obtaining the routine sample for fecal calprotectin. Samples were immediately frozen at home in –20 °C and transported to laboratory where they were stored at − 80 °C until processing.
IFX response
The response to IFX was evaluated by fecal calprotectin value at 6 weeks after IFX therapy initiation. IFX response (R) was determined by fecal calprotectin value below 100 µg/g and non-response (NR) with a value above 100 µg/g36. The cut-off value of < 100 µg/g has a specificity of 0.92 and negative prediction value of 95%36 and predicts transmural healing with high accuracy37. We assessed clinical disease activity using a validated tool with symptom score and visual analogy scale (VAS) from 1 to 7, measuring the impact of disease to quality of life (QOL)38. Symptom score contained 5 questions (total score 0–15 points, higher score being worse) regarding general well-being, abdominal pain, nocturnal and daily bowel movements, and the presence of blood in the stools. This index is applicable to adult and pediatric patients with IBD and in UC as well as in Crhon’s disease38. We also did physicians global assessment (PGA) of the general activity of the disease ranging from 1 to 3, with 3 being the most active disease39. All measurements of disease activity are listed in Table 1.
DNA extraction from fecal samples and preparation of sequencing libraries
The samples were thawed, and DNA was extracted by using a repeated bead beating method as previously described40. The DNA from fungal and bacterial organisms was amplified in PCR reactions targeting the 16S rRNA gene and ITS1 regions in separate reactions as previously23. The fecal bacterial composition was determined by sequencing the V3-V4 region of the 16S rRNA gene with primers 341FWD 5′-CCTACGGGNGGCWGCAG-3′ and 785REV 5′-GACTACHVGGGTATCTAATCC-3′41. The fecal fungal composition was analyzed by sequencing the ITS1 region using primer pair ITS1F (FWD, CTTGGTCATTTAGAGGAAGTAA) and ITS2 (REV, GCTGCGTTCTTCATCGATGC)38,42. The libraries for sequencing were prepared as previously described23. Illumina MiSeq paired-end sequencing was performed in Functional Genomics Unit, University of Helsinki, Helsinki, Finland in separate runs. The ITS and 16S rDNA sequences are available at the ENA database (ITS accession number: PRJEB50351, 16S accession number: PRJEB50380).
Analysis of sequencing data
The 16S MiSeq sequencing data was analyzed using the R package mare43. The median number of reads obtained per sample was 38 037 (range 21 743–94 758) for bacteria. Default parameters were used in processing of only forward reads truncated to 150 bases by using the function ProcessReads in mare. Reads below the abundance of 0.002% were discarded. After the processing the median read count was 29 254 (range 16 051-73 478). All samples from the 27 patients included were annotated successfully and included in further analyses. Annotation to bacterial taxa was done using USEARCH44 and the reads were annotated to the database SILVA v3v445, restricted to gut-specific bacteria.
The ITS data was processed according to the ITS pipeline included in DADA246. Annotation was done as described previously23 by annotating the amplicon sequence variants (ASVs) with BLAST47. The median number of reads per sample was 58,700 before pre-processing. After annotation the average read count was 4000 reads. Successful annotation to fungal taxa was obtained from 59 samples (69%) from 27 patients (out of 30 patients available for ITS sequencing). Annotations at species-level were the best hits based on the annotations to BLAST47 and are included in brackets for fungi.
Statistical analysis
Both for the 16S rRNA gene and ITS relative abundance data, the package mare43 was used for analysis with the program R using the packages vegan48, MASS49, nmle50, and pROC51,52. P-values for taxon-specific differences were corrected for false discovery rate (FDR; Benjamini–Hochberg53). The data-based selection of background variables was based on PERMANOVA and all background variables presented in Table 1 were evaluated. Those that were statistically significantly associated with the fungal and bacterial microbiota composition were used as co-variates to adjust the analyses at the three different timepoints. Generalized linear models with negative binomial distribution (glm.nb) from the MASS package49 and Generalized Least Squares (gls) from the nlme package50 were used to analyze differences in response groups and IBD subtypes for both the fungal and bacterial microbiota data. In all bacterial analyses at the three timepoints, IBD subtype, sex, previous antibiotics (less than one month prior to sample), the use of corticosteroid-medication and age (grouped to below and over 12 years) were used as confounders. In bacterial analyses 0.1 was used as minimum prevalence and 0.01 as minimum abundance. In all fungal analyses at the three different timepoints, age, sex, IBD subtype and antibiotics were used as confounders. Due to zero-inflation in the fungal data, only taxa with a prevalence above 50% were included in the analysis. In all analyses the IBDU patients were included with the UC patients. The diversity was calculated as the inverse Simpson diversity index and richness as the number of operational taxonomic units.
PathModel function in R package mare43 was used to identify the genera that together predicted response and to find the ideal glm model49 to fit the data. The performance of the model was tested and visualized by performing receiver operating characteristic (ROC) analysis with the pROC package51,52.
The association between fecal fungal and bacterial genera was done by calculating Spearman correlations and p values, with phyloseq objects created for both ITS and 16S rRNA gene amplicon data54. To obtain a list of uniquely annotated ASVs, the tax_glom function from phyloseq54 was used for agglomeration over the "genus" taxonomic rank. The resulting list was filtered to select taxa with over 100 reads across the samples. No co-variates were used to adjust the correlations.
Ethics statement
The study was approved by the ethical committee of the Hospital District of Helsinki and Uusimaa (extension approved in 2014 to study, approved with a diary number 183/13/03/03/2011). All participants (or their guardians) signed an informed consent. All methods were performed in accordance with relevant guidelines and regulations.
Supplementary Information
Acknowledgements
We would like to acknowledge the excellent work by Tinja Kanerva on DNA extraction and library preparation.
Author contributions
R.V.H., M.H.: data analysis and interpretation, design of study and revising and drafting the manuscript. SS: data analysis. A.S., W.V.: micro- and mycobiota data acquisition, supervision of sample processing. K.L.K.: designing the study and participating in data interpretation. A.N.: recruiting patients, collected samples and demographics. S.S., A.N., A.S., W.V., K.L.K.: revising the manuscript. All authors approved the final version of the manuscript. We did not use any writing assistance.
Funding
This work was supported by a grant from Pediatric Research Foundation (Finland) [Grant not numbered] and Helsinki University Hospital Grant [Grant number TYH2018212] to KLK. Sweet Crosstalk ITN Funded by EU H2020 programme [Grant number 814102] to AS.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Rebecka Ventin-Holmberg and Miikka Höyhtyä.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-022-10548-7.
References
- 1.Benchimol EI, Fortinsky KJ, Gozdyra P, et al. Epidemiology of pediatric inflammatory bowel disease: A systematic review of international trends. Inflamm. Bowel Dis. 2011;17:423–439. doi: 10.1002/ibd.21349. [DOI] [PubMed] [Google Scholar]
- 2.Kelsen J, Baldassano RN. Inflammatory bowel disease: The difference between children and adults. Inflamm. Bowel Dis. 2008;14:S9–11. doi: 10.1002/ibd.20560. [DOI] [PubMed] [Google Scholar]
- 3.Nishida A, Inoue R, Inatomi O, et al. Gut microbiota in the pathogenesis of inflammatory bowel disease. J. Clin. Gastroenterol. 2018;11:1–10. doi: 10.1007/s12328-017-0813-5. [DOI] [PubMed] [Google Scholar]
- 4.Standaert-Vitse A, Sendid B, Joossens M, et al. Candida albicans colonization and ASCA in familial Crohn's disease. Am. J. Gastroenterol. 2009;104:1745–1753. doi: 10.1128/mBio.01250-16. [DOI] [PubMed] [Google Scholar]
- 5.Nelson A, Stewart CJ, Kennedy NA, et al. The impact of NOD2 genetic variants on the gut mycobiota in Crohn’s disease patients in remission and individuals without gastrointestinal inflammation. J. Crohns Colitis. 2020 doi: 10.1093/ecco-jcc/jjaa220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoarau G, Mukherjee PK, Gower-Rousseau C, et al. Bacteriome and mycobiome interactions underscore microbial dysbiosis in familial Crohn's disease. mBio. 2016;7:e01250–e0125016. doi: 10.1128/mBio.01250-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lewis JD, Chen EZ, Baldassano RN, et al. Inflammation, antibiotics, and diet as environmental stressors of the gut microbiome in pediatric Crohn's disease. Cell Host Microbe. 2015;18:489–500. doi: 10.1016/j.chom.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fitzgerald RS, Sanderson IR, Claesson MJ. Paediatric inflammatory bowel disease and its relationship with the microbiome. Microb. Ecol. 2021 doi: 10.1007/s00248-021-01697-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olbjørn C, Småstuen MC, Thiis-Evensen E, et al. Fecal microbiota profiles in treatment-naïve pediatric inflammatory bowel disease-associations with disease phenotype, treatment, and outcome. Clin. Exp. Gastroenterol. 2019;12:37–49. doi: 10.2147/CEG.S186235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhuang X, Liu C, Zhan S, et al. Gut microbiota profile in pediatric patients with inflammatory bowel disease: A systematic review. Front. Pediatr. 2021;9:626232. doi: 10.3389/fped.2021.626232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borody TJ, Eslick GD, Clancy RL. Fecal microbiota transplantation as a new therapy: From Clostridioides difficile infection to inflammatory bowel disease, irritable bowel syndrome, and colon cancer. Curr. Opin. Pharmacol. 2019;49:43–51. doi: 10.1016/j.coph.2019.04.017. [DOI] [PubMed] [Google Scholar]
- 12.Van Rheenen PF, Aloi M, Assa A, et al. The medical management of paediatric Crohn’s disease: An ECCO-ESPGHAN guideline update. J. Crohns Colitis. 2021;15:171–194. doi: 10.1093/ecco-jcc/jjaa161. [DOI] [PubMed] [Google Scholar]
- 13.Turner D, Ruemmele FM, Orlanski-Meyer E, et al. Management of paediatric ulcerative colitis, part 1: Ambulatory care-an evidence-based guideline from European Crohn’s and Colitis Organization and European society of paediatric gastroenterology, hepatology and nutrition. J. Pediatr. Gastroenterol. Nutr. 2018;67:257–291. doi: 10.1097/MPG.0000000000002035. [DOI] [PubMed] [Google Scholar]
- 14.Nielsen OH, Ainsworth MA. Tumor necrosis factor inhibitors for inflammatory bowel disease. N. Engl. J. Med. 2013;369:754–762. doi: 10.1056/NEJMct1209614. [DOI] [PubMed] [Google Scholar]
- 15.Zhou Y, Xu ZZ, He Y, et al. Gut microbiota offers universal biomarkers across ethnicity in inflammatory bowel disease diagnosis and infliximab response prediction. mSystems. 2018;30:3e00188. doi: 10.1128/mSystems.00188-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y, Gao X, Ghozlane A, et al. Characteristics of faecal microbiota in paediatric Crohn's disease and their dynamic changes during infliximab therapy. J. Crohns Colitis. 2018;12:337–346. doi: 10.1093/ecco-jcc/jjx153. [DOI] [PubMed] [Google Scholar]
- 17.Rajca S, Grondin V, Louis E, et al. Alterations in the intestinal microbiome (dysbiosis) as a predictor of relapse after infliximab withdrawal in Crohn’s disease. Inflamm. Bowel Dis. 2014;20:978–986. doi: 10.1097/MIB.0000000000000036. [DOI] [PubMed] [Google Scholar]
- 18.Dovrolis N, Michalopoulos G, Theodoropoulos GE, et al. The interplay between mucosal microbiota composition and host gene-expression is linked with infliximab response in inflammatory bowel diseases. Microorganisms. 2020;8:438. doi: 10.3390/microorganisms8030438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding NS, McDonald JAK, Perdones-Montero A, et al. Metabonomics and the gut microbiome associated with primary response to anti-TNF therapy in Crohn’s disease. J. Crohns Colitis. 2020 doi: 10.1093/ecco-jcc/jjaa039. [DOI] [PubMed] [Google Scholar]
- 20.Kolho K-L, Korpela K, Jaakkola T, et al. Fecal microbiota in pediatric inflammatory bowel disease and its relation to inflammation. Am. J. Gastroenterol. 2015;110:921–930. doi: 10.1038/ajg.2015.149. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y, Gao X, Zhang X, et al. Microbial and metabolic features associated with outcome of infliximab therapy in pediatric Crohn’s disease. Gut Microbes. 2021;13:1–18. doi: 10.1080/19490976.2021.1900996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Magnusson MK, Strid H, Sapnara M, et al. Anti-TNF therapy response in patients with ulcerative colitis is associated with colonic antimicrobial peptide expression and microbiota composition. J. Crohns Colitis. 2016;10:943–952. doi: 10.1093/ecco-jcc/jjw051. [DOI] [PubMed] [Google Scholar]
- 23.Ventin-Holmberg R, Eberl A, Saqib S, et al. Bacterial and fungal profiles as markers of infliximab drug response in inflammatory bowel disease. J. Crohns Colitis. 2020 doi: 10.1093/ecco-jcc/jjaa252. [DOI] [PubMed] [Google Scholar]
- 24.Sokol H, Leducq V, Aschard H, et al. Fungal microbiota dysbiosis in IBD. Gut. 2017;66:1039–1048. doi: 10.1136/gutjnl-2015-310746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. PLOS Biol. 2016;14:e1002533. doi: 10.1371/journal.pbio.1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chehoud C, Albenberg LG, Judge C, et al. Fungal signature in the Gut microbiota of pediatric patients with inflammatory bowel disease. Inflamm Bowel Dis. 2015;21:1948–1956. doi: 10.1097/MIB.0000000000000454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McFarland LV. Systematic review and meta-analysis of Saccharomyces boulardii in adult patients. World J. Gastroenterol. 2010;16:2202–2222. doi: 10.3748/wjg.v16.i18.2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seelbinder B, Chen J, Brunke S, et al. Antibiotics create a shift from mutualism to competition in human gut communities with a longer-lasting impact on fungi than bacteria. Microbiome. 2020;8:1–20. doi: 10.1186/s40168-020-00899-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng L, Kelly CJ, Battista KD, et al. Microbial-derived butyrate promotes epithelial barrier function through IL-10 receptor-dependent repression of claudin-2. J. Immunol. 2017;199:2976–2984. doi: 10.4049/jimmunol.1700105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kasubuchi M, Hasegawa S, Hiramatsu T, et al. Dietary gut microbial metabolites, short-chain fatty acids, and host metabolic regulation. Nutrients. 2015;7:2839–2849. doi: 10.3390/nu7042839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Furusawa Y, Obata Y, Fukuda S, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504:446–450. doi: 10.1038/nature12721. [DOI] [PubMed] [Google Scholar]
- 32.Hämäläinen A, Sipponen T, Kolho K-L. Infliximab in pediatric inflammatory Bowel disease rapidly decreases fecal calprotectin levels. World J. Gastroenterol. 2011;17:5166. doi: 10.3748/wjg.v17.i47.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Earley H, Lennon G, Balfe Á, et al. The abundance of Akkermansia muciniphila and its relationship with sulphated colonic mucins in health and ulcerative colitis. Sci. Rep. 2019;9:1–9. doi: 10.1038/s41598-019-51878-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kugathasan S, Denson LA, Walters TD, et al. Prediction of complicated disease course for children newly diagnosed with Crohn’s disease: A multicentre inception cohort study. Lancet. 2017;389:1710–1718. doi: 10.1016/S0140-6736(17)30317-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pope JL, Yang Y, Newsome RC, et al. Microbial colonization coordinates the pathogenesis of a Klebsiella pneumoniae infant isolate. Sci. Rep. 2019;9:1–13. doi: 10.1038/s41598-019-39887-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sipponen T, Kolho K-L. Fecal calprotectin in diagnosis and clinical assessment of inflammatory bowel disease. Scand J. Gastronterol. 2015;50:74–80. doi: 10.3109/00365521.2014.987809. [DOI] [PubMed] [Google Scholar]
- 37.Weinstein-Nakar I, Focht G, Church P, et al. Associations among mucosal and transmural healing and fecal level of calprotectin in children with Crohn’s disease. Clin. Gastroenterol. Hepatol. 2018;16:1089–1097. doi: 10.1016/j.cgh.2018.01.024. [DOI] [PubMed] [Google Scholar]
- 38.Puolanne AM, Kolho K-L, Alfthan H, et al. Rapid fecal calprotectin test and symptom index in monitoring the disease activity in colonic inflammatory bowel disease. Dig. Dis. Sci. 2017;62:3123–3130. doi: 10.1007/s10620-017-4770-0. [DOI] [PubMed] [Google Scholar]
- 39.Haapamäki J, Roine RP, Sintonen H, Kolho KL. Health-related quality of life in paediatric patients with inflammatory bowel disease related to disease activity. J. Paediatr. Child Health. 2011;47:832–837. doi: 10.1111/j.1440-1754.2011.02034.x. [DOI] [PubMed] [Google Scholar]
- 40.Salonen A, Nikkilä J, Jalanka-Tuovinen J, et al. Comparative analysis of fecal DNA extraction methods with phylogenetic microarray: Effective recovery of bacterial and archaeal DNA using mechanical cell lysis. J. Microbiol. Methods. 2010;81:127–134. doi: 10.1016/j.mimet.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 41.Klindworth A, Pruesse E, Schweer T, et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013;41:e1. doi: 10.1093/nar/gks808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.White TJ, Bruns T, Lee S, Taylor JW. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protocols: A Guide to Methods and Applications. Cambridge: Elsevier; 1990. pp. 315–22. [Google Scholar]
- 43.Korpela, K. Mare: microbiota analysis in R easily. R package version 1.0. 2016. https://github.com/katrikorpela/mare.
- 44.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 45.Quast C, Pruesse E, Yilmaz P, et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012;4:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Callahan BJ, McMurdie PJ, Rosen MJ, et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods. 2016;13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Altschul SF, Gish W, Miller W, et al. BLAST PROGRAMS. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 48.Oksanen J, Blanchet FG, Kindt R, et al. Package ‘vegan’. Commun. Ecol. 2013;9:1–295. [Google Scholar]
- 49.Venables W, Ripley B. Modern Appl. Stat. 2002 doi: 10.1007/b97626. [DOI] [Google Scholar]
- 50.Pinheiro, J., Bates, D., DebRoy, S., Sarkar, D. R. nlme: Linear and Nonlinear Mixed Effects Models. R package version 3.1–14. Core T. https://svn.r-project.org/R-packages/trunk/nlme/.
- 51.Robin X, Turck N, Hainard A, et al. pROC: anopen-source package for R and S+ to analyze and compare ROC curves. BMC Bioinf. 2011;12:77. doi: 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Burnham KP, Anderson DR. Multimodel inference-understanding AIC and BIC in model selection. Sociol Methods Res. 2014;33:261–304. doi: 10.1177/0049124104268644. [DOI] [Google Scholar]
- 53.Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B. 1995;57:289–300. [Google Scholar]
- 54.McMurdie PJ, Holmes S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE. 2013;8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.