

Abstract
In electrospray ionization mass spectrometry (ESI-MS), analytes are introduced into the mass spectrometer in typically aqueous-organic solvent mixtures, including pH modifiers. One mechanism for improving the signal intensity and simultaneously increasing the generation of higher charge-state ions is the inclusion of small amounts (approx. <0.5% v/v mobile phase solution) of charge-inducing or supercharging reagents, such as m-nitrobenzyl alcohol, o-nitrobenzyl alcohol, m-nitrobenzonitrile, m-(trifluoromethyl)-benzyl alcohol and sulfolane. We explore the direct and indirect (colligative properties) that have been proposed as responsible for their modes of action during ESI. Of the many theorized mechanisms of ESI, we re-visit the three most popular and highlight how they are impacted by supercharging observations on small ions to large molecules including proteins. We then provide a comprehensive list of 34 supercharging reagents that have been demonstrated in ESI experiments. We include an additional 19 potential candidate isomers as supercharging reagents and comment on their broad physico-chemical properties. It is becoming increasingly obvious that advances in technology and improved ion source design, analyzers e.g. the use of ion mobility, ion trap, circular dichroism (CD) spectroscopy, together with computer modeling are increasing the knowledge base and, together with the untested isomers and yet-to-be unearthed ones, offer opportunities for further research and application in other areas of polymer research.
A simple illustration of the positive electrospray ionization (ESI) environment.
1. Introduction
Three main models seek to explain the mechanisms of electrospray ionization (ESI), on which early work began with John Fenn.1 The generation of ions in the dynamic ESI environment which is under an electric field and at elevated temperatures but at ambient pressure is explored within the concept of these three popular models. Fundamentals such as solution chemistry and gas-phase interactions are summarised to explain the ESI processes. The role of supercharging reagents during ESI is discussed and key findings explored. Some of these findings are the direct and more complex interactions between these reagents and macromolecules, especially proteins in the ESI environment. The influence of colligative properties of both ESI solvents and supercharging reagents, including surface tension, polarizability, dipole moments, and acidity/basicity are highlighted. Konermann et al. offer a detailed and rigorous review, which includes experimentation and computational studies on the mechanisms of ESI, dwelling mainly on the interactions between supercharging reagents and proteins.2
In this brief review, we provide a comprehensive list of 34 supercharging reagents that have been used in studies and include an additional 19 potential candidate isomers commenting on their broad physico-chemical properties. These suggested isomers have the potential to further enhance our understanding of supercharging during ESI-MS. This report may be viewed as a first compendium or a ‘one-stop-shop’ of supercharging reagents for fellow ESI mass spectrometrists to peruse.
1.1. Supercharging during ESI
One of the unique advantages during ESI-MS is the formation of multiply charged ions, which enables the observation of molecules of high molecular weights at relatively low m/z values.3,4 Thus, for instance, biological macromolecules including proteins (>100 KDa), nucleic acids, industrial polymers of high molecular weights can be analyzed in a mass spectrometer with a triple quadrupole (QqQ), several types of ion trap (IT) analysers (including orbitraps), and combinations, such as the quadrupole time-of-flight (QToF), quadrupole-ion trap (QIT), and with ion-mobility mass spectrometry.
In ESI, one of the main approaches for increasing the generation and detection of higher charge state in peptide ions (i.e. from [M + nH]n+ to [M + (n + 1)H]n+1)+ and simultaneously improving the signal intensity (ESI response) is the inclusion of very small amounts (approx. <5% v/v of mobile phase solution) charge-inducing compounds or supercharging reagents. Supercharging reagents include glycerol,5m-nitrobenzyl alcohol (mNBA; 3-nitro(phenyl) methanol),4,6,7o-nitrobenzyl alcohol (2-nitro(phenyl)methanol) 8 and sulfolane.8,9 Thus far, the supercharging reagents used in experiments are all small molecules (mol. wt. <180 Da (Table 1).
A list of supercharging reagents and some of their physico-chemical propertiesa.
| Common name/[IUPAC name]α | Molecular structureα | Ave. mass,α Da | Boiling ptα °C at 760 mmHg | Vapour press.αβ mmHg at 25 °C | Surface tensionαβ mN m−1 at 25 °C | Densityαβ (specific gravity) 25 °C | Acidity pKa 25 °C | Dipole momentδ μ(D)/μD* | Ref. | |
|---|---|---|---|---|---|---|---|---|---|---|
| 3- to 4-membered & planar molecules | ||||||||||
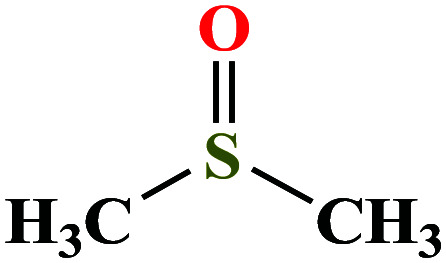
| 1 | Dimethyl sulfoxide (DMSO) [(Methylsulfinyl)methane] |
|
78.133 | 189 | 0.60 | 43.54 | 1.101 (ref. 65) | 35.1 | 3.96/4.44 | 3 and 25 |
| 2 | Ethylene glycol [1,2-ethandiol] |

|
62.068 | 196–198 | 0.06 | 47.99 | 1.113 | 14.22 | 2.747/— | 3 |
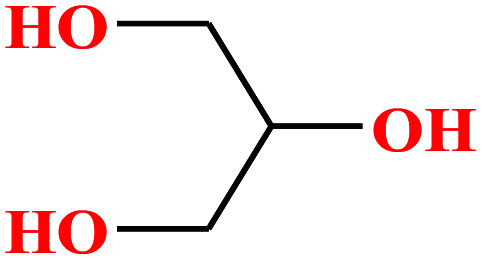
| 3 | Glycerol [1,2,3-propanetriol] |
|
92.094 | 182 | 1.68 × 10−4 | 72.6 | 1.26 (ref. 65) | 14.4 | 2.68/— | 5 and 8 |
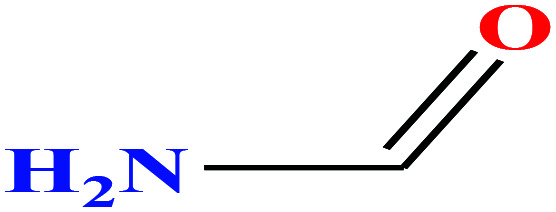
| 4 | Formamide |
|
45.041 | 210 | 6.10 × 10−2 (ref. 66) | 58.35 (ref. 67) | 1.13 | 23.5 (in DMSO) | — | 3 |
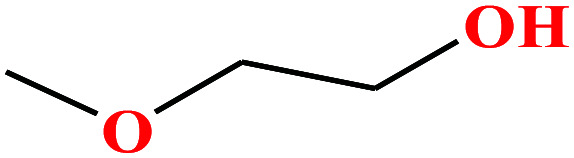
| 5 | 2-Methoxyethanol |
|
76.094 | 124–125 | 6.2 (ref. 68) | 30.84 (ref. 69) | 0.97 (ref. 65 and 70) | 14.8 | — | 3 |
| 6ε | Methoxypropanol [1-methoxypropan-2-ol] |
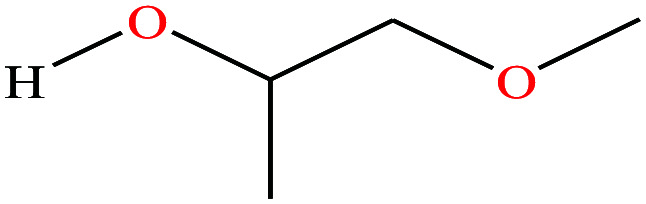
|
90.122 | 120 | 10.9 | 2.64 × 10−3 | 0.926 | 14.49 ± 0.20 (ref. 70 and 71) | — | |
| 7 | Crotononitrile[(2E)-2-butenenitrile] |

|
67.089 | 120–121 | 31.95 | — | — | — | 4.3/— | 15 |
| Heterocyclic 4- & 5-membered ring structures, including organosulfur cpd (sulfones) | ||||||||||
| 8 | Dimethyl sulfone [methylsulfonylmethane] |
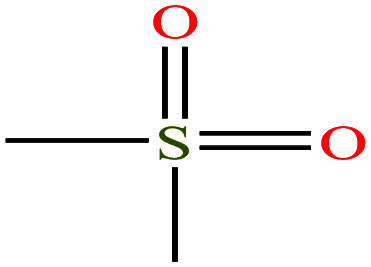
|
94.133 | 238 | — | — | 1.45 | 31 | 4.5/— | 15 |
| 9 | N,N,N′N′-Tetraethylsulfamide (TES) [N-(diethylsulfamoyl)-N-ethylethanamine] |
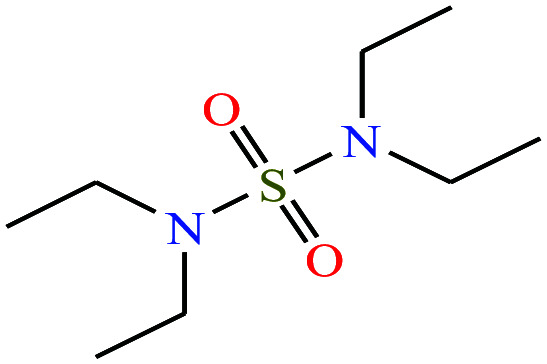
|
208.32 | 125 | — | — | 1.283 | — | — | 16 |
| 10 | 3-Chlorothiete-1,1-dioxide |
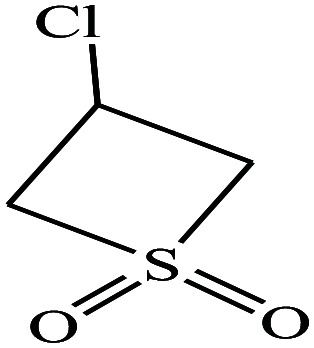
|
140.59 | 328.9 | — | — | 1.64 | — | —/3.38 | 9 |
| 11ε | 3-Chloro-2H-thiete 1,1-dioxide |
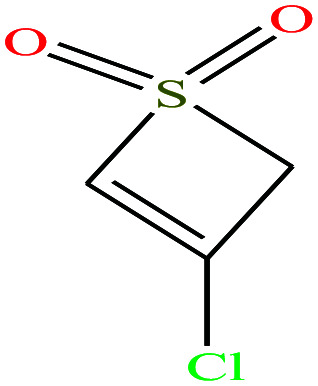
|
138.573 | 328.9 ± 41.0 | 3.2 ± 0.3 | — | 1.6 ± 0.1 | — | — | |
| 12 | 2-Thiophenone [2H-thiophen-5-one] |
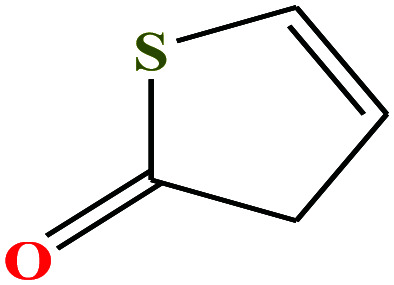
|
100.139 | 197.4 | 0.4 ± 0.3 | 38.7 ± 3.0 | 1.24 | 49 | ||
| 13ε | 4-Butyrothiolactone [thiolan-2-one] |
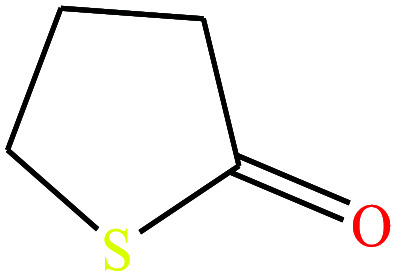
|
102.16 | 76.5429 | — | — | 1.18 | Poorly soluble in water | — | |
| 14 | Sulfolane [2,3,4,5-tetrahydrothiophene-1,1-dioxide] |
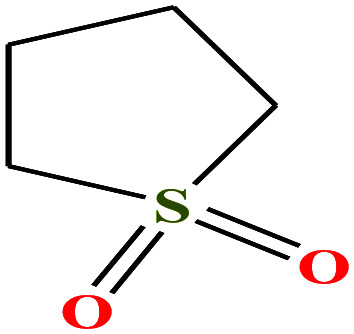
|
120.170 | 285 | 6.2 × 10−3 (27.6 °C) | 35.5 (ref. 72) | 1.26 (ref. 65) | 12.9 | 4.35/5.68 | 7, 8, 52 and 53 |
| 15 | Sulfolene [2,5-dihydrothiophene 1,1-dioxide] |
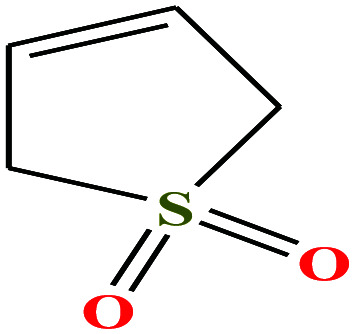
|
118.154 | 64–65.5 °C mpt (ref. 73) | — | 41.0 | 1.3 (ref. 68) | — | —/5.69 | 47 |
| 16 | 1,3-Propanesultone [1,2-oxathiolane 2,2-dioxide] |
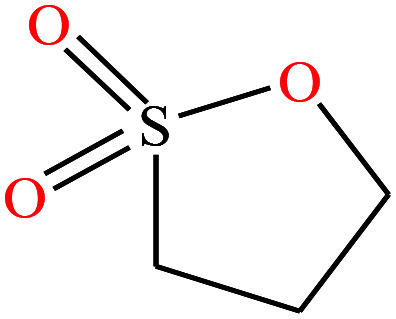
|
122.143 | 315.87 | — | — | 1.392 | — | — | 15 |
| 17 | 1,4-Butanesultone [1,2-oxathiane 2,2-dioxide] |
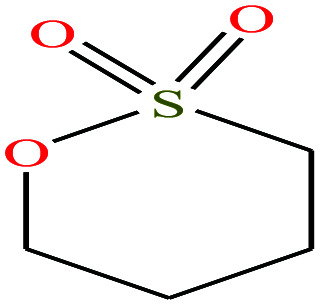
|
136.169 | 304.047 | — | — | 1.335 | — | — | 15 |
| 18 | 3-Methyl-2-oxazolidone (MOZ) [3-methyl-1,3-oxazolidin-2-one] |
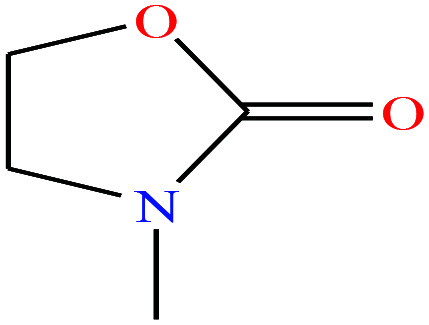
|
101.1 | 88 | — | — | 1.17 | — | — | 16 |
| Heterocyclic 4- & 5-membered ring structures (including the heterocyclic acetals) | ||||||||||
| 19 | Glycerol carbonate [4-hydroxymethyl-1,3-dioxolan-2-one] |
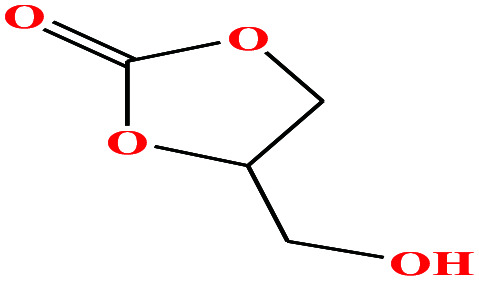
|
118.088 | 353.9 ± 15 | 0.0 ± 1.8 | 41.1 | 1.375 | — | — | 48 |
| 20 | Propylene carbonate [4-methyl-1,3-dioxol-2-one] |
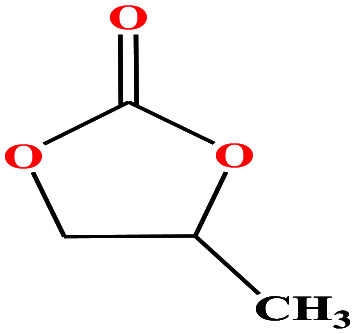
|
102.089 | 240 | 0.045 (ref. 66) | 45 | 1.2 (ref. 69) | — | 4.9 | 48 and 49 |
| 21 | Ethylene carbonate [1,3-dioxolan-2-one] |
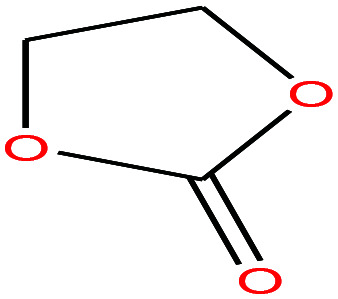
|
88.062 | 244–245 | — | 54 (30 °C) | 1.32 | — | 4.9/— | 15 |
| 22 | Butylene carbonate [4-ethyl-1,3-dioxolan-2-one] |
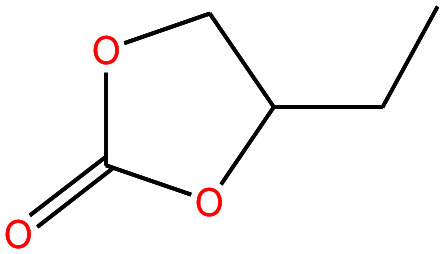
|
116.115 | 281.18 | — | — | 1.141 | — | — | 15 |
| 23 | Vinylethylene carbonate [4-vinyl-1,3-dioxolan-2-one] |
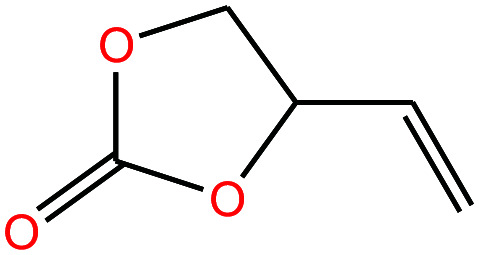
|
114.099 | 238.72 | — | — | 1.188 | — | — | 15 |
| Benzene-ring containing molecules | ||||||||||
| 24 | Nitrobenzene |
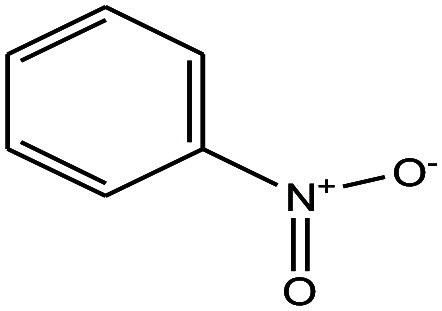
|
123.109 | 210–211 | 4.2 | 0.245 | 1.201 | 1.204 slightly soluble in water | 4.2/— | 15 |
| 25 | Benzyl alcohol [(hydroxymethyl)benzene] |
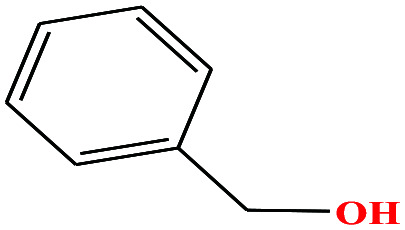
|
108.138 | 205 | 0.094 (ref. 66) | 39.0 (ref. 70) | 1.0 (ref. 65) | 15.4 | 1.71/1.79 | 8 |
| 26 | m-Chlorophenol [3-chlorophenol] |
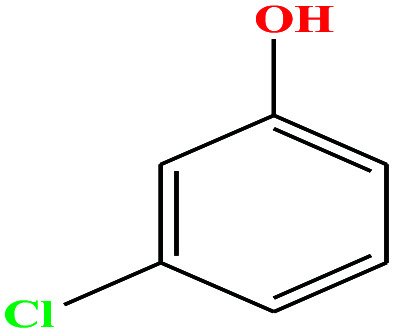
|
128.556 | 214 | 0.125 | 44.7 | 1.3 (ref. 68) | 9.12 | 1.03 ± 1.08 | 4 |
| 27ε | o-Chlorophenol [2-chlorophenol] |
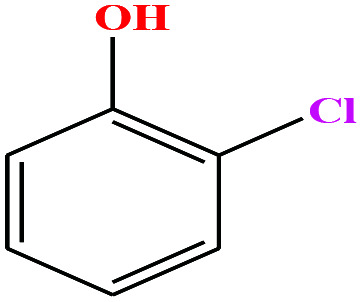
|
128.556 | 175–176 | 2.53 (ref. 66) | 40.50 | 1.26 (ref. 65) | 8.56 | — | |
| 28ε | p-Chlorophenol [4-chlorophenol] |
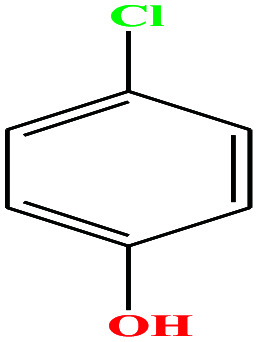
|
128.556 | 217–220 | 1 (ref. 74) | 8.7 × 10−2 mm Hg | 1.306 (ref. 65) | 9.41 | 2.10/— | |
| 29 | 2-Nitrochlorobenzene [1-chloro-2-nitrobenzene] |
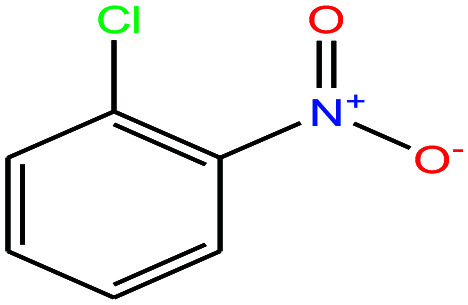
|
157.555 | 245–246 | — | 48.4 ± 3.0 | 1.348 | 0.6 | 4.6/— | 15 |
| 30ε | 3-Nitrochlorobenzene [1-chloro-3-nitrobenzene] |
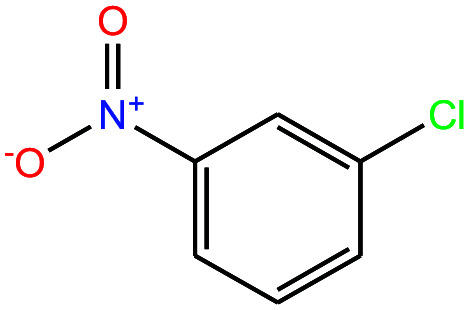
|
157.555 | 235–236 | 0.097 (ref. 66) | 4.37 × 10−2 | 1.3 | — | — | |
| 31ε | 4-Nitrochlorobenzene [1-chloro-4-nitrobenzene] |
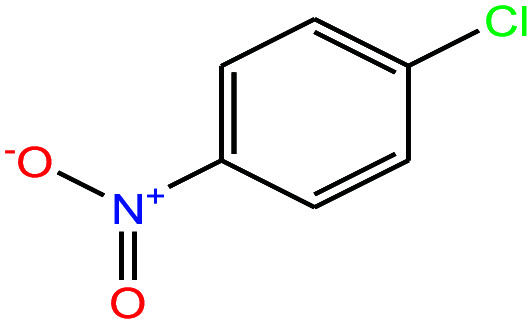
|
157.555 | 241.66 | 2.19 × 10−2 (ref. 66) | 3.71 × 10−2 | 1.520 | n/a | — | |
| 32 | o-Nitrobenzyl alcohol (oNBA) [2-nitro(phenyl)methanol] |
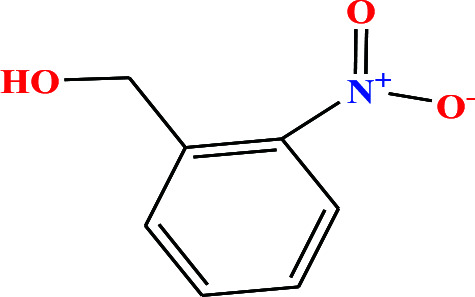
|
153.135 | 270 (solid at RT) | 0.0 ± 0.5 | — | 1.3 | — | — | 8 |
| 33 | m-Nitrobenzyl alcohol (mNBA) [3-nitro(phenyl) methanol] |
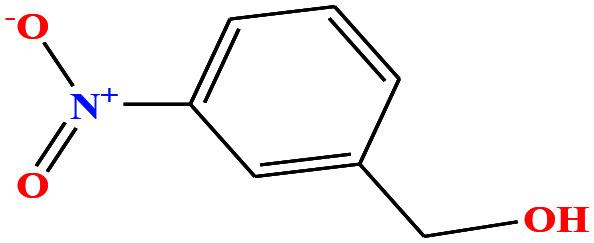
|
153.135 | 349.8 | 0.0 ± 0.5 | 50 ± 5 | 1.29 | — | — | 4, 6, 7, 8, 48, 49 and 75 |
| 34 | p-Nitrobenzyl alcohol (pNBA) [4-nitro(phenyl)methanol] |
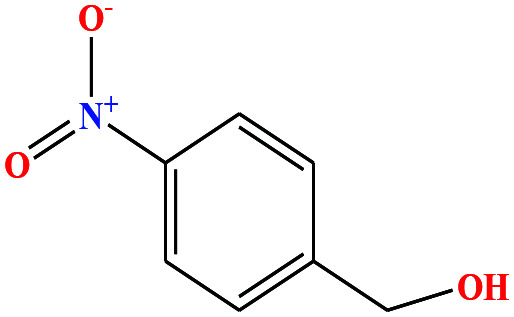
|
153.135 | 185 (solid at RT) | Negligible | — | — | 7.15 | — | 8 |
| 35 | m-Nitroacetophenone [1-(3-nitrophenyl)ethanone] |
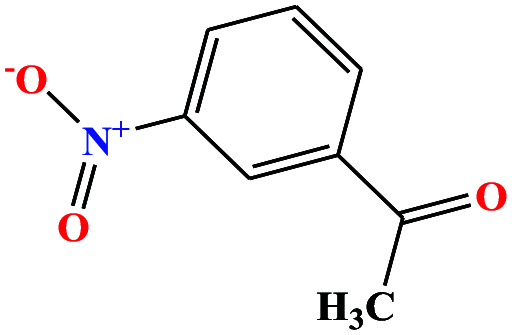
|
165.146 | 202 (solid at RT) | 3.86 × 10−5 | — | 1.4 | — | — | 8 and 27 |
| 36 | m-Nitrobenzonitrile [3-benzonitronitrile] |
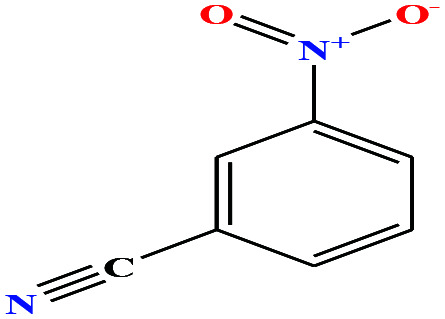
|
148.119 | 165 | 0.0 ± 0.5 | — | 1.3 ± 0.1 | — | — | 6, 8 and 27 |
| 37ε | o-Nitrobenzonitrile [2-benzonitronitrile] |
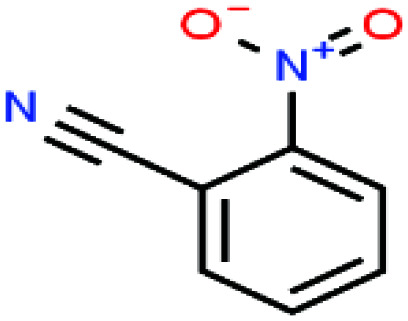
|
148.119 | 321.78 165.0 °C (16.0 mmHg) | Insoluble in water | — | — | — | — | |
| 38ε | p-Nitrobenzonitrile [4-benzonitronitrile] |
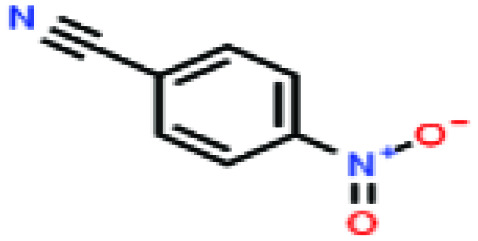
|
148.119 | — | — | — | — | — | — | |
| 39 | m-nitrophenyl ethanol [1-(3-nitrophenyl)ethanol] |
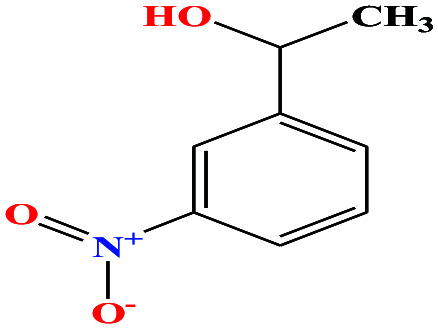
|
167.162 | 281.3 ± 23.0 | 0.0 ± 0.6 | — | 1.3 ± 0.1 | — | — | 6 and 8 |
| 40ε | o-Nitrophenyl ethanol [1-(2-nitrophenyl)ethanol) |
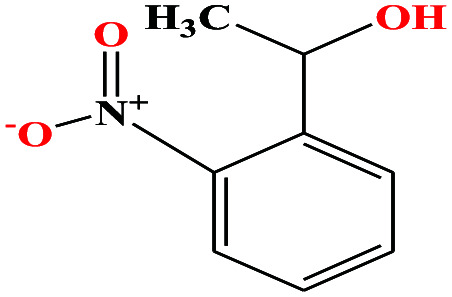
|
167.162 | 319.0 ± 17.0 | 0.0 ± 0.6 | — | 1.3 ± 0.1 | — | — | |
| 41ε | p-Nitrophenyl ethanol (1-(4-nitrophenyl)ethanol] |
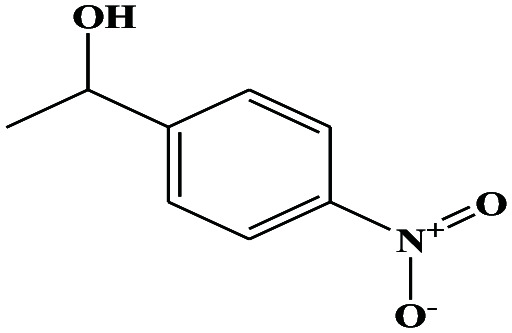
|
167.162 | 290.68 | 0.0 ± 0.7 g | — | 1.3 ± 0.1 | — | — | |
| 42 | o-Nitroanisole [1-methoxy-2-nitrobenzene] |
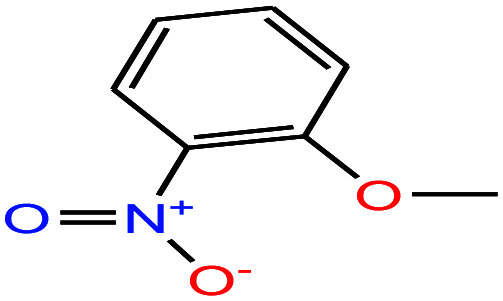
|
153.135 | 272–273 | — | 48 | 1.254 | — | 5.0/— | 15 |
| 43ε | m-Nitroanisole [1-methoxy-3-nitrobenzene] |
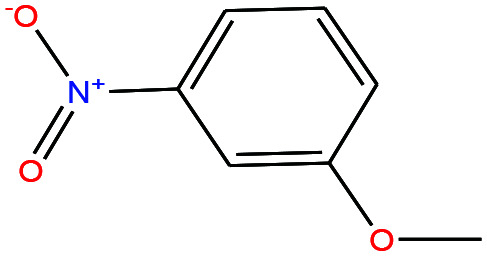
|
153.135 | 256.3 ± 13.0 | 0.0 ± 0.5 | — | 1.2 ± 0.1 | — | — | |
| 44 | p-Nitroanisole [1-methoxy-4-nitrobenzene] |
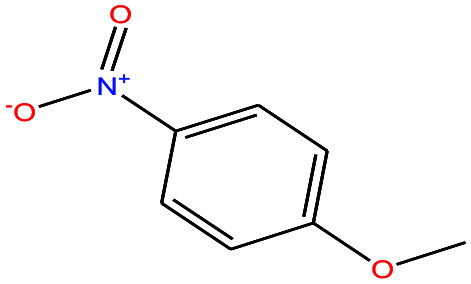
|
153.135 | 260 | — | — | 1.233 | — | 5.3/— | 15 |
| 45 | 3-Nitrophenethyl alcohol [2-(3-nitrophenyl)ethanol] |
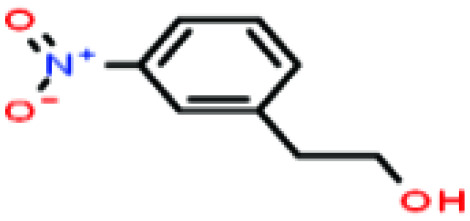
|
167.162 | 341.7–349.0 | — | — | — | — | — | 8 |
| 46ε | 2-Nitrophenethyl alcohol [2-(2-nitrophenyl)ethanol] |
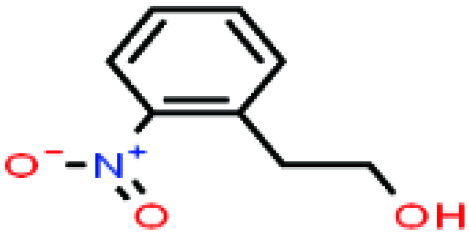
|
167.162 | 267 | — | — | 1.19 | — | — | |
| 47ε | 4-Nitrophenethyl alcohol [2-(4-nitrophenyl)ethanol] |
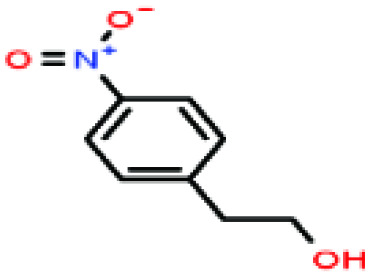
|
167.162 | 337 | — | — | — | — | — | |
| 48 | m-(Trifluoromethyl)-benzyl alcohol [[3-(trifluoromethyl)phenylmethanol] |
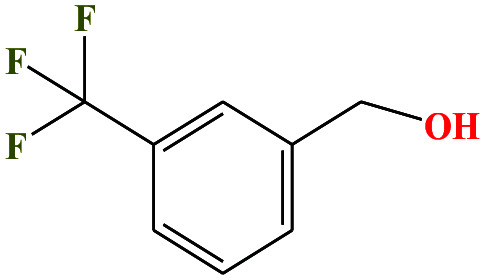
|
176.136 | 257–261 | 0.1 ± 0.4 | — | 1.3 ± 0.1 | — | — | 6 |
| 49ε | o-(Trifluoromethyl)-benzyl alcohol [2-(trifluoromethyl)phenylmethanol] |
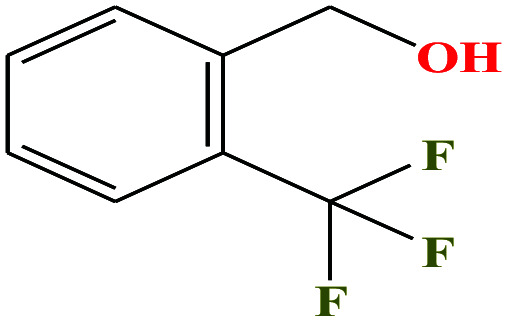
|
176.136 | 214–262 | — | — | 1.3 ± 0.1 | — | — | |
| 50ε | p-(Trifluoromethyl)-benzyl alcohol [4-(trifluoromethyl)benzyl alcohol] |
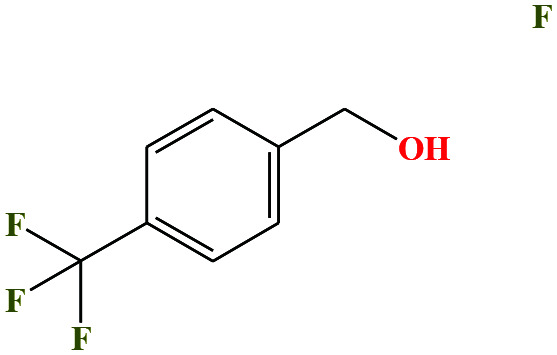
|
176.136 | 250 | 0.1 ± 0.4 | — | 1.28 (ref. 74) | — | — | |
| 51ε | o-Nitrobenzoic acid [2-nitrobenzoic acid] |
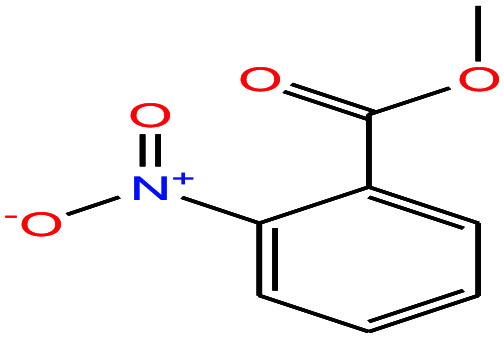
|
167.12 | 295.67 | 0.0 ± 0.8 (ref. 75) | 66.4 ± 3.0 | 1.575 | 2.17 | 4.07/— | |
| 52ε | m-Nitrobenzoic acid [3-nitrobenzoinc acid] |
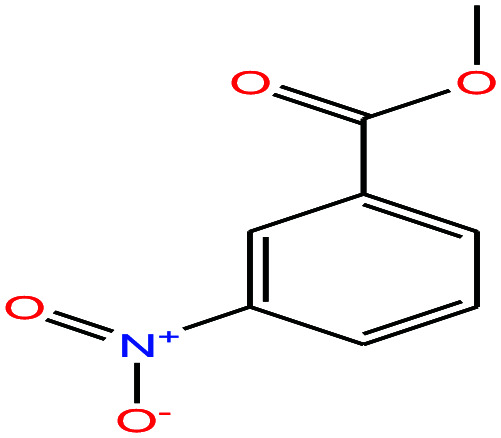
|
167.12 | 340.7 ± 25.0 | 3.71 × 10−5 (ref. 75) | 66.4 ± 3.0 | 1.5 ± 0.1 | 3.47 | 4.03/— | |
| 53ε | p-Nitrobenzoic acid [4-nitrobenzoic acid] |
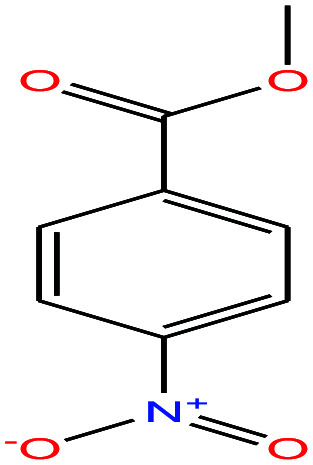
|
167.12 | It sublimes | — | — | 1.58 | 3.41 | 4.05/— | |
| Typical solvents used in ESI | ||||||||||
| Common name/IUPAC name | Molecular structure | Ave. mass, Da | Boiling pt °C at 760 mmHg | Vap. press. mmHg at 25 °C (ref. 80) | Surface tension mN m−1 at 25 °C | Density (specific gravity) | Acididty pKa 25 °C | Dipole moment μ(D)/μD* | 66 and 69 | |
| 1 | Water |
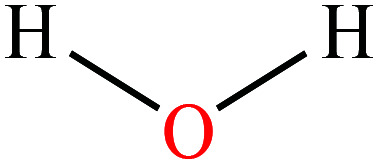
|
18.02 | 100 | 17.44 | 72.8 | 1.00 | 14 | 1.85/2.16 | |
| 2 | Methanol |
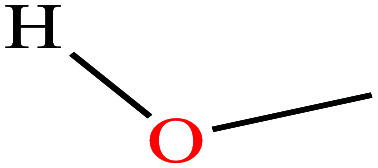
|
32.04 | 64.7 | 127 | 22.07 | 0.792 | 15.5 | 1.70/ | |
| 3 | Ethanol |
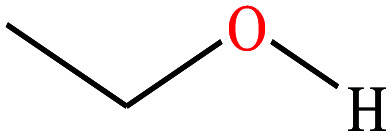
|
46.07 | 78.2 | 59.3 | 21.97 | 0.789 | 15.9 | 1.69/ | |
| 4 | Acetonitrile |
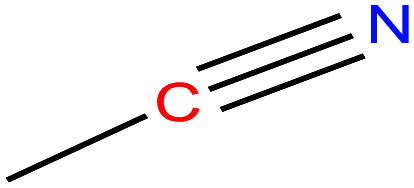
|
41.05 | 81.3–82.1 | 88.8 | 29.04 | 0.786 | 25 | 3.92/ | |
| 5 | Ethyl acetate |
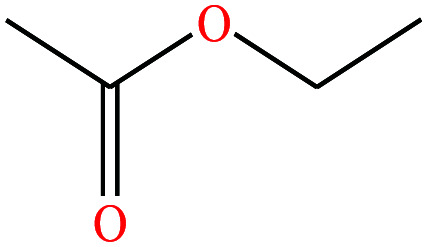
|
88.106 | 77.1 | 93.2 | 24 | 0.902 | 25 | 1.78/ | |
| 6 | Acetone |
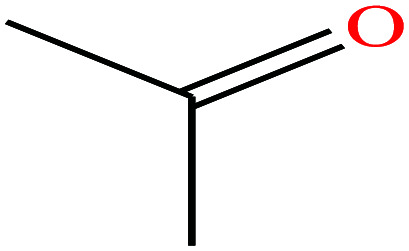
|
58.08 | 56.05 | 231 | 23.7 | 0.785 | 19.16 | 2.91/ | |
Key: α chemspider: http://www.chemspider.com/, γ pKaε candidate isomers proposed supercharging reagents, β pubchem: https://pubchem.ncbi.nlm.nih.gov/, δ dipole moment μ(D)/μD*: determined by experiment/calculated*. We acknowledge that some values in Table 1 are incomplete.
1.1.1. Proposed modes of action of supercharging reagents: colligative properties
In ESI, supercharging reagents assist in surmounting deficiencies such as ion suppression and improve the linear dynamic range, that is, in the latter case, they can decrease the detection limit across a wide sample concentration linear range. Some colligative properties have been indicated as responsible for the supercharging phenomena. These are boiling points, surface tension, polarizability, dipole moments, and acidity/basicity.
Boiling points and surface tension
One of the fundamental properties of these reagents which promote higher ESI charging is that they all have much higher boiling points than both water and the organic solvents in the mobile phase mixture. They are therefore less volatile than the solvents. Although opposing arguments exist, the reason for their action has been attributed to two phenomena:
(1) Their ability to raise the solution surface tension.10 Due to the low volatility of these compounds compared with that of common mobile-phase compositions (e.g. water/methanol or water/acetonitrile), their concentration increases as evaporation of the more volatile components from the droplets occurs. This enrichment of the supercharging reagents raises the surface tension which results in a requirement for a higher degree of surface charging to reach the Rayleigh limit for a spherical droplet. This, in turn, increases the boiling point of the supercharging reagent-enriched environment10 However, Samalikova and Grandori11 reported that their results were not easily reconciled by considering only charge availability of the precursor droplets at the Rayleigh limit as determined by surface tension.
(2) Protein denaturation: Williams and co-workers argued that protein unfolding induced by chemical and or thermal denaturation appears to be the primary origin of the enhanced charging observed in the protein complexes investigated.7 In their experiments, the arrival time distributions obtained from traveling wave ion mobility spectrometry showed that the higher charge state ions that are formed with m-NBA and sulfolane are significantly more unfolded than lower charge state ions. Sterling et al.12 argued that droplet heating, owing to the high boiling point of m-NBA, resulted in thermal denaturation. However, unlike Sterling et al.12 Hogan et al.13 reported that supercharging reagents do not cause structural protein modifications in solution and that the modest mobility decrease observed could be partly attributed to trapping of the sulfolane within the protein ions.
Polarizability and dipole moments
The addition of the supercharging reagent sulfolane on cytochrome introduced another dimension to the debate, in that, while supercharging was observed in the positive ionization mode, no change in the charge state distribution was observed in the negative mode.9 This, therefore, eliminates polarity-independent factors such as conformational changes or surface tension effects. The authors also reported the formation of adducts between cytochrome and sulfolane thus supporting the report of Hogan et al.13
Further, the study by Douglass and Venter demonstrated that supercharging was shown to increase with increasing dipole moment for the supercharging reagents sulfolane and sulfolene.9 Finally, if an increase in surface tension is the cause of supercharging, then it should occur in both the positive and negative modes since the effect of surface tension on the Rayleigh limit is independent of the polarity of the charge. Like many other identified supercharging reagents, sulfolane is highly polar with a dipole moment of 4.35 D, much higher than that of water (1.85 D) or methanol (1.70 D) (Table 1).
The interaction of supercharging reagents with charged basic sites would depend on the molecular structure, such as the type and location of functional groups. Thus, they exhibit low solution-phase basicities and relatively low gas-phase basicities.8 For example, the most effective supercharging reagents have one or more carbonyl, sulfonyl, or nitro groups present in their structure (Table 1), which may be important for intermolecular interactions between the supercharging reagent and the charged basic site. Lomeli et al. demonstrated that supercharging occurs not only when liquid m-NBA is present, but also with solid o- or p-isomers. This provides some evidence against increased surface tension causing supercharging.8
1.1.2. Direct interaction
Adduct formation
Examination of the mass spectra generated from the addition of these reagents show adduct formation on the higher charges. This suggests that there is a direct interaction between these reagents and fragments of unfolding polypeptides and proteins.8,9 Circular dichroism measurements while titrating myoglobin with 6 M guanidinium hydrochloride in the presence of 0%, 2.5%, 5.0%, or 7.5% sulfolane established that sulfolane destabilized the protein by ∼1.5 kcal mol−1 (moles L−1 of GuHCl)−1, which Sterling and co-workers12 argued was evidence that chemical denaturation caused the charge increases observed when sulfolane is added to native-like solutions. However, they also found that without the added GuHCl, the helicity of myoglobin was lower, even at the highest sulfolane concentrations tested.
Chemical denaturation
Sterling's circular dichroism experiment indicates a chemical denaturation activity of sulfolane.12 As supercharging reagents have low vapor pressures (higher surface tension), therefore it has been suggested that aqueous droplets are preferentially enriched in these reagents as evaporation occurs in the ESI environment. Sterling et al. reasoned that less evaporative cooling will occur after the droplets are substantially enriched in the low volatility supercharging reagent (i.e. the organic solvent is evaporated first), and the droplet temperature should be higher compared with when these reagents are not present, in the more aqueous environment.12 Furthermore, Sterling et al. rationalized that protein unfolding induced by chemical and/or thermal denaturation in the electrospray droplet appeared to be the primary origin of the enhanced charging observed for noncovalent protein complexes formed from aqueous solutions that contain these supercharging reagents, although other factors most certainly influence the extent of charging. However, some studies are not in agreement with this. For example, Yao et al.14 reported that supercharging of lysozyme by sulfolane was not related to protein unfolding during the ESI process.
A report evaluating the potential of low volatility of supercharging reagents demonstrated that there was not a strong correlation between the extent of analyte (protein) charging with either surface tension, dipole moment, or dielectric constant of the additives.15 That is, there was no single physical property of an additive that significantly determined the extent of protein charging when formed from a solution mixture. What is clear, however, was that most of the supercharging reagents have higher boiling points, higher surface tension values, high dielectric constants, and higher dipole moments than the ESI solvents. Thus, a combination of these physicochemical properties together with other factors including adduct formation between analyte and supercharger, basicity at the solution, and gaseous phases, may all influence charging, although the extent of charging may depend on the experimental conditions.
A comprehensive list of 53 molecules of which 34 have been shown to have supercharging ability in experiments and 19 potential candidate isomers, together with some of their physico-chemical properties are given in Table 1. The molecules are grouped conveniently in terms of molecular structure and functional group given rise to four categories:
(1) Acyclic 3- to 4-membered and/or planar structures.
(2) Heterocyclic 4- to 5-membered molecules including the sulfones and S-containing ones. Dimethyl sulfone and N,N,N′N′-tetraethylsulfamide (TES) are acyclic molecules but are included in this category because they contain the sulfone functional group.
(3) Heterocyclic 4- to 5-membered molecules including the heterocyclic acetals.
(4) Molecules containing the benzene structure with at least two functional group attachments which may have opposite charges e.g. m-nitrobenzyl alcohol.
Recently, two new supercharging reagents N,N,N′,N′-tetraethylsulfamide (TES), and 3-methyl-2-oxazolidone (MOZ) were shown to counter the suppression activity of trifluoroacetic acid (TFA).16 The study also demonstrated that the two molecules increased charge states of peptides and proteins and improved separation efficiency during reverse-phase LC-MS determinations.
We suggest that the following 19 additives (denoted by ε and in italics, Table 1) are potential supercharging reagents and could be evaluated:
(1) Based on the fact that some of their isomers have successfully been demonstrated; methoxypropanol, 3-chloro-2H-thiete 1,1-dioxide, 4-butyrothiolactone, o-chlorophenol, p-chlorophenol, 3-nitrochlorobenzene, 4-nitrochlorobenzene, o-nitrophenylethanol, p-nitrophenyl ethanol, m-nitroanisole, o-(trifluoromethyl)-benzyl alcohol, and p-(trifluoromethyl)-benzyl alcohol, and,
(2) Molecules having a benzene ring structure and with two or more attached functional groups having opposing charges (‘dual polarity’ isomers); e.g. the isomers o-, m-, and p- of nitrobenzoic acid, nitrobenzonitrile and nitrophenethyl alcohol.
ESI mobile phase solvents
Six solvents commonly used during ESI-MS are also included for comparison. The list in Table 1 includes the following physico-chemical properties: molecular structure, molecular weight (Da), boiling point (or melting point, °C), vapor pressure (mm Hg), surface tension (mN m−1), acidity (pKa)/basicity (pKb), dipole moment (D), and references of key publications in which the superchargers were employed. The sources of the properties are also indicated. We concede that some of the properties of the reagents are either not determined or could not be found in the literature.
1.2. The ESI mechanism
During ESI-MS operation and for samples extracted from a complex and an electrolyte-rich biological matrice, such as saliva, serum, cerebrospinal fluid, or microbial extract, resolution and detector signal response can be hindered. A clean-up procedure, for example, solid-phase extraction (SPE) or desalting steps is necessary to decrease the concentration of or remove these usually confounding electrolytes, detergents, or other compounds. In (positive) ESI-MS, analytes including peptides are introduced into the mass spectrometer in typically aqueous-organic solvent mixtures, together with other additives including pH modifiers like formic acid (≤0.3% v/v). Following the application of an electric charge at the capillary tip, there follows the simultaneous processes of solution-phase ion generation, solvent evaporation, a plume of charged ions in residual solvent, charge transfer, and/or concentration, and finally, gas phase ion generation (Fig. 1). These processes typically occur under an electric field and in a heated (approx. 90° to 120 °C) environment in the ion source compartment of the mass spectrometer. And finally, the generated highly charged gas-phase ions are then differentially drawn into the first stage of the mass spectrometer under negative pressure. The uncharged gaseous solutes, molecules, are also removed under negative pressure into a separate exhaust stream.
Fig. 1. A simple illustration of the ESI environment. For simplicity, droplets are presented with positive charges. According to the model presented by Loo et al.27 opposing charges may also be present in the gas phase ion. Under an electric field, elevated temperature but at ambient pressure, the sprayed analyte solution undergoes the simultaneous processes of the droplet splitting, generation of highly charged droplets, repeated splitting and shrinkage of the droplets, solvent evaporation, and removal under a directed gas (typically N2) stream. These progeny droplets themselves undergo subsequent and repeated evaporation and splitting, and this process is repeated until only gas-phase ions are generated.76 The ions are then drawn under negative pressure into the mass spectrometer.

Observations show that many parameters e.g. solvent composition,3,4,17,18 analyte composition and concentration,19–21 pH, flow rate,18,21 denaturing solutions,22,23 and non-denaturing solutions,24–26 solution- & gas-phase basicity, solution-phase conformation,26,27 instrument settings: source voltage,28,29 sprayer orifice diameter,30 gas pressures,31 and, ion source type (laser ESI-LEMS vrs conventional ESI)32 affect charge state distributions (CSD) especially, in large analytes such as proteins. Further, sub-ambient pressure ESI source combined with nano-flow significantly improves ion yield and sensitivity33
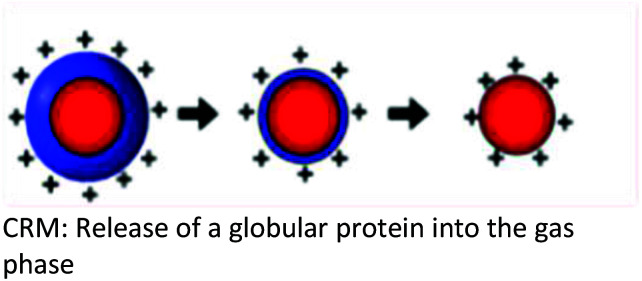
Several models have been put forward to explain the mechanisms of action of the processes of generating gas-phase ions from the liquid mobile phase. Among these, three are popular: the charge residue model (CRM),34,35 the ion evaporation model (IEM)36–40 and the third postulate, chain ejection model (CEM)41,42 which was recently described for unfolded proteins.2,43,44 These models are summarized in Table 2.
The ESI mechanism: gas-phase ion generation.
| The three popular models that seek to explain the ESI mechanism | |||
|---|---|---|---|
| Charge residue model (CRM) | Ion evaporation model (IEM) | Chain ejection model (CEM) | |
| Background | The CRM was proposed based on the ability of the SCRs to raise the surface tension of the mobile phase solvent, a requirement for a higher degree of surface charging to reach the Rayleigh limit which, results in a coulombic fission event | Following the repeated splitting and shrinkage of droplets, the size of droplets reach a certain critical radius (10−6 cm = 10−8 m), the field strength at the surface of the droplet becomes large enough to assist the field desorption of solvated ions | Neither the CRM nor IEM mechanisms explain quantitatively the extent of macromolecular multiple charging, especially in unfolded proteins |
| Key points in the mechanism | Millions of small, highly charged progeny droplets containing the analyte leave the parent droplet with a disproportionately large fraction of the surface charge | Analyte ions desorb directly from charged nanodroplets, driven by the large electric field at the droplet surface | The inclusion of SCRs has opened up new ways to modulate protein charge states, thereby challenging the existing ESI models |
| Progeny droplets containing single analytes are charged through a charge transfer process between the charge carriers on the surface of the droplet and the analyte molecule upon final droplet evaporation | The ion evaporation process then becomes operative for a highly electrified cloud of droplets at low solute concentrations | ESI of unfolded proteins yields [M + (z + 1)H]z+ ions that are much more highly charged than their folded counterparts | |
| That is, analyte ions desorb directly from charged nanodroplets, driven by the large electric field at the droplet surface | Solvent properties such as dipole moment and protein–SCR adduct formation have been shown to play prominent roles in supercharging in proteins | ||
| Analytes | Larger ions (e.g. from folded proteins under non-denaturing conditions) are formed by CRM | It has been demonstrated that small ions (i.e. Na+), generated from small molecules, are liberated into the gas phase through the ion evaporation mechanism | This proposed model accounts for the protein ESI behavior under such non-native conditions and recently has been proposed to apply to unfolded proteins |
| Illustration |
|
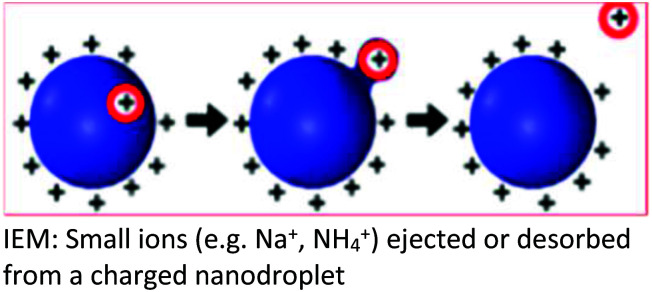
|
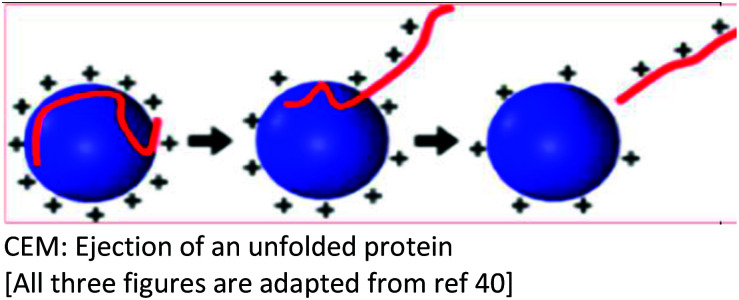
|
| References | 34, 77 and 78 | 10, 36–40 and 79 | 2, 41–44, 52 and 53 |
The use of nano-ESI, conventionally a non-pneumatic operation with very low mobile flow rates (∼<1 μL min−1) and much small samples sizes which then provides improved desolvation, greater salt tolerance, and a higher ion yield29,33 has added another dimension to the debate although the generated final ions are believed to be the same for both conventional and nano-ESI.45
Hogan et al. proposed that both the IEM and CRM are in play, in which macromolecules are charged residues but, in negative ion mode, carry less charge because highly-charged droplets field-emit anions. Why positively charged droplets would not also field-emit small ions has not yet been explained.46,47 Excellent discussions are also given.9,27,48,49
1.3. The impact of supercharging reagents on ESI
After extensive investigations of ESI of macromolecules, neither model (CRM or IEM) adequately and quantitatively explain the extent of macromolecular multiple charging.50,51 Quite recently, with increasing interest in using ESI-MS as a technology for top-down proteomics and for studying protein interactions, the expanding application of ion mobility mass spectrometry (IM-MS), and the use of supercharging reagents, most of the reported supercharging research has been on proteins in the native state, that is, with solution pH around neutral and the low voltage conditions to preserve protein structure. Thus, the role of additive properties such as dipole moment and adduct formation (between analyte and supercharger) have been shown to play prominent roles in supercharging in proteins. Therefore, a third model, the chain ejection model (CEM) has been proposed42 where the ESI of unfolded proteins yields M + (z + 1)H]z+ ions that are much more highly charged than their folded counterparts. This model, it has been suggested, accounts for the protein ESI behavior under such non-native conditions and has been proposed to apply for unfolded proteins.2,43,44,52,53
Proteins that are unfolded in solution produce higher charge states during ESI than their natively folded counterparts. Building on the work by Douglass and Venter,9 who showed experimentally the formation of adducts between sulfolane and the most highly charged protein ions, Peters et al. recently, reported that the CEM involves the partitioning of mobile H+ (e.g. from formic acid) between the droplet and the departing protein.44 Their results indicate that the supercharging of unfolded proteins is caused by residual sulfolane that stabilizes protonated sites on the protruding chains. This, therefore, promotes H+ retention on the protein. Their report suggested that charge stabilization on the sites of projecting chains is due to charge–dipole interactions which are mediated by the large dipole moment and the low volatility of sulfolane.
Denaturing ESI follows the CEM, where protein ions are gradually expelled from the droplet surface. Proton (H+) equilibration between the droplets and the protruding chains culminates in highly charged gaseous proteins.2 The presence of internal disulfide (S–S) bonds on the extent of supercharging was determined in three proteins each containing multiple internal disulfide bonds; bovine serum albumin, β-lactoglobulin, and lysozyme.43 Reduction of the disulfide bonds led to a marked increase in charge state following the addition of sulfolane without significantly altering folding in solution. This evidence supports a supercharging mechanism in which these proteins unfold before or during evaporation of the electrospray droplet and ionization would therefore, occur by the CEM.
Ogorzalek Loo et al. proposed a three-regime view of ESI, i.e. with solution, intermediate, and gas-phase regimes in the ESI environment.27 The intermediate phase was introduced to explain the charge transfer and other accommodations that analytes especially protein ions undergo when exiting the bulk solution, this concept rationalizes observations that ion intensities vary little over pH 3–11, despite huge changes in solution compositions.17,54,55 This kinetic model is unique in treating the evaporating droplets as charge-polarized and uses their decompositions and the variation in solution-to-gas phase properties to explain charge enrichment in ESI.
The 3-phase ESI environment could, in principle, be imagined for the IEM, CRM, and other models, particularly as distinctions between them blur in attempts to explain non-conforming observations.29,56 Thus far, the IEM and the CRM appear to represent the foundations of any discussion related to the mechanism of ESI. Molecular dynamics (MD) simulations52,57 suggest that small ions such as Na+ are ejected from the surface of an aqueous ESI droplet (IEM), while folded proteins in native ESI could be released by water evaporation to dryness (CRM).43,52,53,58
Other factors that increase the charge state during ESI are the diameter of the emitter opening: proteins (native and denatured) charge state distributions (CSDs) undergo a small, but reproducible shift to higher charge when delivered by nano-spray versus standard electrospray59,60 and source voltage.26,61 Ogorzalek Loo, et al.27 noted that this relationship between the extent of charging and initial droplet diameter could not be observed for ions released as charge residues, thus, these observations argue against CRM being the primary source of native-like ions.
Improvements in ESI source design including sub-ambient pressure with nano-flow,33,62,63 chemically-etched emitters with sheath gas capillaries and nano-flow64 are reported to enhance signal intensity, increase desolvated ion transmission to the mass spectrometer proper and thus increasing overall sensitivity. The use of these improvements together with supercharging reagents should further enhance our understanding of the ESI mechanism and how it is impacted by the supercharging reagents.
2. Summary
2.1. The following are the main highlights on supercharging during ESI
(1) Konermann et al.2 indicated that the ion evaporation model (IEM) and the charged residue model (CRM) represent the foundations underpinning the mechanism of ESI. Simulations of molecular dynamics (MD) supported by classical solution chemistry demonstrate that small ions such as Na+ or NH4+ are ejected from the surface of aqueous ESI droplets (IEM), while folded proteins in the native ESI are released by water evaporation to dryness (CRM).
(2) The application of ESI-MS in protein analysis and the inclusion of supercharging reagents (SCR) has been the main driver in the exploration of the mechanism of the ESI process and the dynamics governing proteins in the native and unfolded state.
(3) The Konermann team2,44,52,53 and Donor et al.43 indicated that the CEM largely explains the CSD in proteins as it accounts for the dipole-moment of solvents, charge distribution from the folded to the unfolded state in proteins, and also the presence of intramolecular di-sulfide bonds within proteins.
(4) SCRs have expanded and increased our knowledge of the ESI environment and protein unfolding. These molecules have boiling points and higher surface tension than the solvent mixtures used in ESI,10 although studies by Samalikova and Grandori11 reported a diminished role of surface tension. The vast majority of supercharging molecules have densities greater than that of water and other ESI solvents (Table 1). William's and Loo's teams observed that SCRs also have low solution-phase basicities (Brønsted bases weaker than H2O) and relatively low gas-phase basicities.26–28 They also have higher dipole moments (except for acetonitrile (3.92 D)). In solution, SCRs are ionized, and highly polarized.
(5) Loo's team8 suggested that for dual-polarity superchargers, the molecule should both be a weak Brønsted base and a weak Brønsted acid.
(6) Venter's team9 demonstrated supercharging in cytochrome in the positive ionization mode, however, no change in the CSD was observed in the negative mode. This, therefore, diminishes the role of polarity-independent factors such as conformational changes or surface tension effects as key vehicles for supercharging.
(7) Venter's team9 also demonstrated that when a SCR is added in concentration about equal to or greater than the proteins concentration in solution, during ESI, there is adduct formation between the proteins and SCR.
(8) Using SCRs such as nitrobenzyl alcohol, where there are two functional groups (hydroxyl and nitro), supercharging increases in the order of the para, meta, and ortho isomers. The same principle could perhaps be applied to the isomers of nitrochlorobenzene, nitroanisole, nitrobenzoic acid, nitrobenzonitrile, and nitrophenethyl alcohol.
(9) Improvements in ion source design to include operating at sub-ambient pressure combined with nano-flow increases desolvation of charged species and elevates the ion transmission rate into the analyser section.33,62–64
It should be pointed out that all these different supercharging attributes have been proposed to matter, but not all are of them are relevant. Experimental conditions would increase the prominence of one or several of them over the others depending on the aims of the researchers.
3. Conclusions
In summary, we presented a brief review of supercharging and the attributed factors which influence supercharging during ESI. We highlighted the three most popular models, IEM, CRM and CEM that seek to explain the ESI process. The use of supercharging reagents have unraveled and expanded on the ESI mechanisms. Advances in technology including improved source design, the applications of IM-MS, nano-ESI, detectors (e.g. ion traps), circular dichroism spectroscopy, and computer software to explore molecular dynamics coupled with the use of supercharging reagents have expanded the knowledge base of the ESI mechanism, especially as applied to proteins. The list of 19 untested candidate isomers and, many more to be unearthed, offers opportunities for further study. The use of extreme supercharging reagents should be very useful for maximizing MS and MS/MS performance. A combination of these tools offers avenues for further research and applications in other areas of polymer research.
Author contributions
DAA and BVN developed the original concept. All authors contributed to the writing of the original draft, and all authors contributed to the writing of and approved the final draft.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
The manuscript was prepared using a laptop purchased from a grant received from Royal Society of Chemistry Research Fund (Ref: 2625).
References
- Fenn F. B. Mann M. Meng C. K. Wong S. F. Whitehouse C. M. Electrospray ionization for mass spectrometry of large biomolecules. Science. 1989;246:64–71. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- Konermann L. Metwally H. Duez Q. Peters I. Charging and supercharging of proteins for mass spectrometry: recent insights into the mechanisms of electrospray ionization. Analyst. 2019;144(21):6157–6171. doi: 10.1039/C9AN01201J. [DOI] [PubMed] [Google Scholar]
- Iavarone A. T. Williams E. R. Supercharging in electrospray ionization: effects on signal and charge. Int. J. Mass Spectrom. 2002;219:63–72. doi: 10.1016/S1387-3806(02)00587-0. [DOI] [Google Scholar]
- Iavarone A. T. Williams E. R. Mechanism of charging and supercharging molecules in electrospray ionization. J. Am. Chem. Soc. 2003;125:2319–2327. doi: 10.1021/ja021202t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iavarone A. T. Jurchen J. C. Williams E. R. Supercharged protein and peptide ions formed by electrospray ionization. Anal. Chem. 2001;73:1455–1460. doi: 10.1021/ac001251t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomeli S. H. Yin S. Ogorzalek Loo R. R. Loo J. A. Increasing charge while preserving noncovalent protein complexes for ESI-MS. J. Am. Soc. Mass Spectrom. 2009;20:593–596. doi: 10.1016/j.jasms.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterling H. J. Williams E. R. Origin of supercharging in electrospray ionization of non-covalent complexes from aqueous solution. J. Am. Soc. Mass Spectrom. 2009;20:1933–1943. doi: 10.1016/j.jasms.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomeli S. H. Peng I. X. Yin S. Ogorzalek Loo R. R. Loo A. J. New reagents for increasing ESI multiple charging of proteins and protein complexes. J. Am. Soc. Mass Spectrom. 2010;21:127–131. doi: 10.1016/j.jasms.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglass K. A. Venter A. R. Investigating the role of adducts in protein supercharging with sulfolane. J. Am. Soc. Mass Spectrom. 2012;23:489–497. doi: 10.1007/s13361-011-0319-1. [DOI] [PubMed] [Google Scholar]
- Gamero-Castaño M. Fernández de la Mora J. Mechanisms of electrospray ionization of singly and multiply charged salt clusters. Anal. Chim. Acta. 2000;406:67–91. doi: 10.1016/S0003-2670(99)00596-6. [DOI] [Google Scholar]
- Samalikova M. Grandori R. Testing the role of solvent surface tension in protein ionization by electrospray. J. Mass Spectrom. 2005;40(4):503. doi: 10.1002/jms.821. doi: 10.1002/jms.821. [DOI] [PubMed] [Google Scholar]
- Sterling H. J. Daly M. Feld G. Thoren K. Kintzer A. Krantz B. Williams E. R. Effects of supercharging reagents on noncovalent complex structure in electrospray ionization from aqueous solutions. J. Am. Soc. Mass Spectrom. 2010;21:1762–1774. doi: 10.1016/j.jasms.2010.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan C. J. Loo R. R. O. Loo J. A. de la Mora J. F. Ion mobility-mass spectrometry of phosphorylase B ions generated with supercharging reagents but in chargereducing buffer. Phys. Chem. Chem. Phys. 2010;12(41):13476. doi: 10.1039/c0cp01208d. doi: 10.1039/c0cp01208d. [DOI] [PubMed] [Google Scholar]
- Yao Y. Richards M. R. Kitova E. N. Klassen J. S. Influence of sulfolane on ESI-MS measurements of protein-ligand affinities. J. Am. Soc. Mass Spectrom. 2016;27(3):498–506. doi: 10.1007/s13361-015-1312-x. [DOI] [PubMed] [Google Scholar]
- Chen A. T. Donald W. A. Solution additives for supercharging proteins beyond the theoretical maximum proton-transfer limit in electrospray ionization mass spectrometry. Anal. Chem. 2014;86:4455–4462. doi: 10.1021/ac500304r. [DOI] [PubMed] [Google Scholar]
- Nshanian M. Lakshmanan R. Chen H. Ogorzalek Loo R. R. Loo J. A. Enhancing sensitivity of liquid chromatography–mass spectrometry of peptides and proteins using supercharging agents. Int. J. Mass Spectrom. 2018;427:157–164. doi: 10.1016/j.ijms.2017.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G. Cole R. B. Effect of solution ionic strength on analyte charge state distributions in positive and negative ion electrospray mass spectrometry. Anal. Chem. 1994;66(21):3702–3708. doi: 10.1021/ac00093a026. [DOI] [Google Scholar]
- Karas M. Bahr U. Dulcks T. Nano-electrospray ionization mass spectrometry: addressing analytical problems beyond routine. Fresenius. J. Anal. Chem. 2000;366:669–676. doi: 10.1007/s002160051561. [DOI] [PubMed] [Google Scholar]
- Wang G. Cole R. B. Mechanistic interpretation of the dependence of charge state distributions on analyte concentrations in electrospray ionization mass spectrometry. Anal. Chem. 1995;67:2892–2900. doi: 10.1021/ac00113a025. [DOI] [Google Scholar]
- Allard E. Troger R. A. Arvidsson B. Sjoberg P. J. R. Quantitative aspects of analyzing small molecules - monitoring singly or doubly charged ions? A case study of ximelagatran. Rapid Commun. Mass Spectrom. 2010;24:429. doi: 10.1002/rcm.4414. [DOI] [PubMed] [Google Scholar]
- Abaye D. A. Pullen F. S. Nielsen B. V. Practical considerations in analysing neuropeptides, calcitonin gene-related peptide and vasoactive intestinal peptide, by nano-electrospray ionisation and quadrupole time-of-flight mass spectrometry: monitoring multiple protonations. Rapid Commun. Mass Spectrom. 2011;25:1107–1116. doi: 10.1002/rcm.4961. [DOI] [PubMed] [Google Scholar]
- Loo J. A. Edmonds C. G. Udseh H. R. Smith R. D. Effect of reducing disulfide-containing proteins on electrospray ionization mass spectra. Anal. Chem. 1990;62:693–698. doi: 10.1021/ac00206a009. [DOI] [PubMed] [Google Scholar]
- Dobo A. Kaltashov I. A. Detection of multiple protein conformational ensembles in solution via deconvolution of charge-state distributions in ESI MS. Anal. Chem. 2001;73:4763–4773. doi: 10.1021/ac010713f. [DOI] [PubMed] [Google Scholar]
- Katta V. Chait B. T. Observation of the heme-globin complex in native myoglobin by electrospray-ionization mass spectrometry. J. Am. Chem. Soc. 1991;113:8534–8535. doi: 10.1021/ja00022a058. [DOI] [Google Scholar]
- Kitova E. N. El-Hawiet A. Schnier P. D. Klassen J. S. Reliable determinations of protein–ligand interactions by direct ESI-MS measurements. Are we there yet? J. Am. Soc. Mass Spectrom. 2012;23:431–441. doi: 10.1007/s13361-011-0311-9. [DOI] [PubMed] [Google Scholar]
- Sterling H. J. Prell J. S. Cassou C. A. Williams E. R. Protein conformation and supercharging with DMSO from aqueous solution. J. Am. Soc. Mass Spectrom. 2011;22(7):1178–1186. doi: 10.1007/s13361-011-0116-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogorzalek Loo R. R. Lakshmanan R. Loo J. A. What protein charging (and supercharging) reveal about the mechanism of electrospray ionization. J. Am. Soc. Mass Spectrom. 2014;25(10):1675–1693. doi: 10.1007/s13361-014-0965-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo J. A. Udseth H. R. Smith R. D. Futrell J. H. Collisional effects on the charge distribution of ions from large molecules, formed by electrospray-ionization mass spectrometry. Rapid Commun. Mass Spectrom. 1988;2:207–210. doi: 10.1002/rcm.1290021006. [DOI] [Google Scholar]
- Juraschek R. Dülcks T. Karas M. Nanoelectrospray—More than just a minimized-flow electrospray ionization source. J. Am. Soc. Mass Spectrom. 1999;10:300–308. doi: 10.1016/S1044-0305(98)00157-3. [DOI] [PubMed] [Google Scholar]
- Li Y. Cole R. B. Shifts in peptide and protein charge state distributions with varying spray tip orifice diameter in nanoelectrospray Fourier transform ion cyclotron resonance mass spectrometry. Anal. Chem. 2003;75:5739–5746. doi: 10.1021/ac0301402. [DOI] [PubMed] [Google Scholar]
- Samalikova M. Matecko I. Muller N. Grandori R. Interpreting conformational effects in protein nano-ESI-MS spectra. Anal. Bioanal. Chem. 2004;378:1112–1123. doi: 10.1007/s00216-003-2339-6. [DOI] [PubMed] [Google Scholar]
- Karki S. Flanigan IV P. M. Perez J. J. Archer J. J. Levis R. J. Increasing protein charge state when using laser electrospray mass spectrometry. J. Am. Soc. Mass Spectrom. 2015;26:706–715. doi: 10.1007/s13361-015-1084-3. [DOI] [PubMed] [Google Scholar]
- Marginean I. Page J. S. Tolmachev A. V. Tang K. Q. Smith R. D. Achieving 50% ionization efficiency in sub-ambient pressure ionization with nanoelectrospray. Anal. Chem. 2010;82:9344–9349. doi: 10.1021/ac1019123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dole M. Mack L. L. Hines R. L. Mobley R. C. Ferguson L. D. Alice M. B. Molecular beams of macroions. J. Chem. Phys. 1968;49(5):2240–2249. doi: 10.1063/1.1670391. [DOI] [Google Scholar]
- Blades A. T. Ikonomou M. G. Kebarle P. Mechanism of electrospray mass spectrometry. Electrospray as an electrolysis cell. Anal. Chem. 1991;63:2109–2114. doi: 10.1021/ac00019a009. [DOI] [Google Scholar]
- Iribarne J. V. Thomson B. A. On the evaporation of small ions from charged droplets. J. Chem. Phys. 1976;64(6):2287–2294. doi: 10.1063/1.432536. [DOI] [Google Scholar]
- Thomson B. A. Iribarne J. V. Field induced ion evaporation from liquid surfaces at atmospheric pressure. J. Chem. Phys. 1979;71:4451–4463. doi: 10.1063/1.438198. [DOI] [Google Scholar]
- Kebarle P. Tang L. From ions in solution to ions in the gas phase - the mechanism of electrospray mass spectrometry. Anal. Chem. 1993;65(22):A972–A986. [Google Scholar]
- Kebarle P. Peschke M. On the mechanisms by which the charged droplets produced by electrospray lead to gas phase ions. Anal. Chim. Acta. 2000;406:11–35. doi: 10.1016/S0003-2670(99)00598-X. [DOI] [Google Scholar]
- Nguyen S. Fenn F. B. Gas-phase ions of solute species from charged droplets of solutions. Proc. Natl. Acad. Sci. U. S. A. 2007;104(4):1111–1117. doi: 10.1073/pnas.0609969104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann L. Rodriguez A. D. Liu J. On the formation of highly charged gaseous ions from unfolded proteins by electrospray ionization. Anal. Chem. 2012;84:6798–6804. doi: 10.1021/ac301298g. [DOI] [PubMed] [Google Scholar]
- Konermann L. Ahadi E. Rodriguez A. D. Vahidi S. Unraveling the mechanism of electrospray ionization. Anal. Chem. 2013;85:2–9. doi: 10.1021/ac302789c. [DOI] [PubMed] [Google Scholar]
- Donor M. T. Ewing S. A. Zenaidee M. A. Donald W. A. Prell J. S. Extended protein ions are formed by the chain ejection model in chemical supercharging electrospray ionization. Anal. Chem. 2017;89(9):5107–5114. doi: 10.1021/acs.analchem.7b00673. [DOI] [PubMed] [Google Scholar]
- Peters I. Metwally H. Konermann L. mechanism of electrospray supercharging for unfolded proteins: solvent-mediated stabilization of protonated sites during chain ejection. Anal. Chem. 2019;91(10):6943–6952. doi: 10.1021/acs.analchem.9b01470. [DOI] [PubMed] [Google Scholar]
- Kebarle P. Verkerk U. H. Electrospray: from ions in solution to ions in the gas phase, what we know now. Mass Spectrom. Rev. 2009;28:898–917. doi: 10.1002/mas.20247. [DOI] [PubMed] [Google Scholar]
- Hogan C. J. Carroll J. A. Rohrs H. W. Biswas P. Gross M. L. Charge carrier field emission determines the number of charges on native state proteins in electrospray ionization. J. Am. Chem. Soc. 2008;130:6926–6927. doi: 10.1021/ja801280c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan C. J. Carroll J. A. Rohrs H. W. Biswas P. Gross M. L. Combined charged residue-field emission model of macromolecular electrospray ionization. Anal. Chem. 2009;81(1):369–377. doi: 10.1021/ac8016532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Going C. C. Williams E. R. Supercharging with m-nitrobenzyl alcohol and propylene carbonate: forming highly charged ions with extended, near-linear conformations. Anal. Chem. 2015;87(7):3973–3980. doi: 10.1021/acs.analchem.5b00071. [DOI] [PubMed] [Google Scholar]
- Going C. C. Xia Z. Williams E. R. New supercharging reagents produce highly charged protein ions in native mass spectrometry. Analyst. 2015;140(21):7184–7194. doi: 10.1039/C5AN01710F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschke M. Blades A. Kebarle P. Charged states of proteins. Reactions of doubly protonated alkyldiamines with NH3: solvation or deprotonation. Extension of two proton cases to multiply protonated globular proteins observed in the gas phase. J. Am. Chem. Soc. 2002;124:11519–11530. doi: 10.1021/ja012591e. [DOI] [PubMed] [Google Scholar]
- Felitsyn N. Kitova E. N. Klassen J. S. Thermal decomposition of a gaseous multiprotein complex studied by blackbody infrared radiative dissociation. Investigating the origin of the asymmetric dissociation behaviour. Anal. Chem. 2001;73:4647–4661. doi: 10.1021/ac0103975. [DOI] [PubMed] [Google Scholar]
- Metwally H H. Konermann L L. Crown ether effects on the location of charge carriers in electrospray droplets: Implications for the mechanism of protein charging and supercharging. Anal. Chem. 2018;90(6):4126–4134. doi: 10.1021/acs.analchem.8b00099. [DOI] [PubMed] [Google Scholar]
- Metwally H. Duez Q. Konermann L. Chain ejection model for electrospray ionization of unfolded proteins: Evidence from atomistic simulations and ion mobility spectrometry. Anal. Chem. 2018;90(16):10069–10077. doi: 10.1021/acs.analchem.8b02926. [DOI] [PubMed] [Google Scholar]
- Hiraoka K. Murata K. Kudaka I. Do the electrospray mass spectra reflect the ion concentrations in sample solution? J. Mass Spectrom. Soc. Jpn. 1995;43:127–138. doi: 10.5702/massspec.43.127. [DOI] [Google Scholar]
- Mansoori B. A. Volmer D. A. Boyd R. K. ‘Wrong-Way-Round’ electrospray ionization of amino acids. Rapid Commun. Mass Spectrom. 1997;11:1120–1130. doi: 10.1002/(SICI)1097-0231(19970630)11:10<1120::AID-RCM976>3.0.CO;2-Q. [DOI] [Google Scholar]
- Verkerk U. H. Kebarle P. Ion-Ion and Ion-Molecule reactions at the surface of proteins produced by nanospray. Information on the number of acidic residues and control of the number of ionized acidic and basic residues. J. Am. Soc. Mass Spectrom. 2005;16:1325–1341. doi: 10.1016/j.jasms.2005.03.018. [DOI] [PubMed] [Google Scholar]
- Caleman C. van der Spoel D. Evaporation from water clusters containing singly charged ions. Phys. Chem. Chem. Phys. 2007;9:5105–5111. doi: 10.1039/B706243E. [DOI] [PubMed] [Google Scholar]
- Consta S. Oh M. I. Sharawy M. Malevanets A. Macroion-Solvent Interactions in Charged Droplets. J. Phys. Chem. A. 2018;122:5239–5250. doi: 10.1021/acs.jpca.8b01404. [DOI] [PubMed] [Google Scholar]
- Fenselau C. Szilágyi Z. Williams T. J. Intercharge Distances in Zn7-metallothionein analyzed by nanospray on a quadrupole ion trap and molecular modeling. J. Mass Spectrom. Soc. Jpn. 2000;48:23–25. doi: 10.5702/massspec.48.23. [DOI] [Google Scholar]
- Nesatyy V. J. Mass spectrometry evaluation of the solution and gas-phase binding properties of noncovalent protein complexes. Int. J. Mass Spectrom. 2002;221:147–161. doi: 10.1016/S1387-3806(02)00956-9. [DOI] [Google Scholar]
- Sterling H. J. Cassou C. A. Susa A. C. Williams E. R. Electrothermal supercharging of proteins in native electrospray ionization. Anal. Chem. 2012;84:3795–3801. doi: 10.1021/ac300468a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page J. S. Tang K. Kelly R. T. Smith R. D. Subambient pressure ionization with nanoelectrospray source and interface for improved sensitivity in mass spectrometry. Anal. Chem. 2008;80(5):1800–1805. doi: 10.1021/ac702354b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang K. Page J. S. Marginean I. Kelly R. T. Smith R. D. Improving liquid chromatography-mass spectrometry sensitivity using a subambient pressure ionization with nanoelectrospray (SPIN) interface. J. Am. Soc. Mass Spectrom. 2011;22(8):1318–1325. doi: 10.1007/s13361-011-0135-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J. T. Marginean I. Kelly R. T. Smith R. D. Tang K. Improving the sensitivity of mass spectrometry by using a new sheath flow electrospray emitter array at subambient pressures. J. Am. Soc. Mass Spectrom. 2014;25(12):2028–2037. doi: 10.1007/s13361-014-0856-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. Coast Guard, Department of Transportation. CHRIS – Hazardous Chemical Data, Volume II, Government Printing Office, Washington, D.C. U.S, 1984-5 [Google Scholar]
- Daubert T. E. and Danner R. P., Physical and Thermodynamic Properties of Pure Chemicals: Data Compilation, Taylor and Francis, Washington, D.C, 1989 [Google Scholar]
- The Merck Index – An Encyclopedia of Chemicals, Drugs, and Biologicals, ed. S. Budavari, Merck and Co., Inc., Whitehouse Station, NJ, 1996, p. 718 [Google Scholar]
- National Toxicology Program, Institute of Environmental Health Sciences, National Institutes of Health (NTP), National Toxicology Program Chemical Repository Database, Research Triangle Park, North Carolina, USA, 1992 [Google Scholar]
- CRC Handbook of Chemistry and Physics, ed. W. M. Haynes, CRC Press LLC, Boca Raton: FL, 95th edn, 2014-2015, pp. 3–468 [Google Scholar]
- Mookerjee B. D. and Wilson R. A., Benzyl Alcohol and beta-Phenethyl Alcohol, Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, New York, NY, 1999-2014 [Google Scholar]
- Chemical book: https://www.chemicalbook.com/CASDetailList_0_EN.htm, accessed 12 Oct 2020
- Kirk-Othmer Encyclopedia of Chemical Technology, ed. I. J. Kroschwitz and M. Howe-Grant, John Wiley & Sons Inc., New York, NY, 1991–1998, p. V27:135 [Google Scholar]
- The Merck Index – Encyclopedia of Chemicals, Drugs and Biologicals, ed. S. Budavari, Merck and Co., Inc., Rahway, NJ, 1989, p. 1415 [Google Scholar]
- Lewis R. J., Sax's Dangerous Properties of Industrial Materials, John Wiley & Sons Inc., New York, NY, 10th edn, 1999, vol. 1–3, p. V2: 871 [Google Scholar]
- Nielsen B. V. Abaye D. A. Influence of electrolytes and a supercharging reagent on charge state distribution and response of neuropeptide ions generated during positive electrospray ionisation mass spectrometry. Eur. J. Mass Spectrom. 2013;19(5):335–344. doi: 10.1255/ejms.1246. [DOI] [PubMed] [Google Scholar]
- Cech N. B. Enke C. G. Practical implications of some recent studies in electrospray ionization fundamentals. Mass Spectrom. Rev. 2001;20:362–387. doi: 10.1002/mas.10008. [DOI] [PubMed] [Google Scholar]
- Cole R. B. Some tenets pertaining to electrospray ionization mass spectrometry. J. Mass Spectrom. 2000;35:763–772. doi: 10.1002/1096-9888(200007)35:7<763::AID-JMS16>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Taflin D. C. Ward T. L. J Davis E. Electrified droplet fission and the Rayleigh limit. Langmuir. 1989;5:376–384. doi: 10.1021/la00086a016. [DOI] [Google Scholar]
- Gamero-Castaño M. Fernández de la Mora J. Kinetics of small ion evaporation from the charge and mass distribution of multiply charged clusters in electrosprays. J. Mass Spectrom. 2000;35:790–803. doi: 10.1002/1096-9888(200007)35:7<790::AID-JMS21>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Boublik T., Fried V. and Hala E., The Vapour Pressures of Pure Substances, Elsevier, Amsterdam, 2nd revised edn, 1984, p. 57, https://searchworks.stanford.edu/view/954536, accessed 1 Nov 2020 [Google Scholar]