

Abstract
Individuals who maintain cognitive function despite high levels of Alzheimer’s disease (AD)-associated pathology are said to be ‘resilient’ to AD. Identifying mechanisms underlying resilience represents an exciting therapeutic opportunity. Human studies have identified a number of molecular and genetic factors associated with resilience, but the complexity of these cohorts prohibits a complete understanding of which factors are causal or simply correlated with resilience. Genetically and phenotypically diverse mouse models of AD provide new and translationally-relevant opportunities to identify and prioritize new resilience mechanisms for further cross-species investigation. This review will discuss insights into resilience gained from both human and animal studies and highlight future approaches that may help translate these insights into therapeutics designed to prevent or delay AD-related dementia.
Keywords: genetic diversity, mouse models, cognition, pathology, tau, amyloid
Prevalence of resilience to Alzheimer’s neuropathology
Alzheimer’s disease (AD) is characterized clinically by a progressive loss of memory functions that impact daily life[1]. Dementia is preceded pathologically by progressive neurodegeneration, deposition of extracellular beta amyloid (Aβ) aggregates and intracellular accumulation of hyperphosphorylated tau-containing neurofibrillary tangles[2]. These proteinopathies are thought to be drivers of the neuronal dysfunction and cognitive impairment that characterize the disease[3], in part due to the identification of inherited mutations in amyloid precursor protein (APP) that cause familial forms of autosomal dominant AD[4].
Despite the strong genetic link between amyloid and AD, a significant proportion of individuals with brain amyloid deposition remain non-demented throughout their lifespan[5]. These individuals are referred to in the literature as ‘resilient’ to AD pathology and represent a clinically interesting subset of the population, as they are able to cope with the presence of amyloid and/or tau while escaping the deficits typically associated with these pathological hallmarks. In large-scale studies, up to one-third of elderly individuals are estimated to reach this criterion as measured by either cerebrospinal fluid (CSF) biomarker monitoring, amyloid- or tau-specific imaging techniques, or autopsy[6–8]. A better understanding of the mechanisms that confer protection against cognitive decline in these individuals represents an outstanding therapeutic opportunity; information gained from studying these ‘resilient’ individuals could provide the key to preventing or delaying the onset of dementia in susceptible individuals. This review will primarily focus on studies aimed at identifying cellular and molecular mediators of preserved cognition in the face of amyloid and/or tau neuropathology. In addition, we will discuss how animal models can be used to nominate, prioritize, and investigate potential therapeutic targets.
Features of resilient individuals
A number of definitions have been used in recent years to attempt to describe and understand resilient individuals (Box 1, Figure 1). A comprehensive histopathological study comparing pathology-positive non-demented individuals with AD cases and pathology-free controls found a striking preservation of neuron numbers, synaptic markers, and axonal morphology in resilient individuals compared to the demented cases[7] (Table 1). In addition, resilient individuals exhibited a unique cytokine profile consisting of higher levels of anti-inflammatory cytokines and neurotrophins along with lower levels of chemokines associated with microglial recruitment and activation[9]. Spine density was found to be significantly reduced in AD patients relative to both pathology-free controls and pathology-positive non-demented individuals[10], suggesting maintenance of critical brain structures is an important determinant of maintained cognitive function in the face of pathology. Interestingly, studies have found that resilient individuals have lower levels of hyperphosphorylated tau accumulation in the synaptic compartment[7] and neocortical areas[11], despite comparable levels of amyloid. Across studies, tau pathology seems to be more strongly linked to cognitive outcomes than amyloid pathology[12–14], suggesting resilience to tau pathology is less common than resilience to amyloid pathology.
Box 1: Defining AD resilience.
Generally, resilient individuals are those who evade cognitive decline associated with observed levels of amyloid and/or tau neuropathology and/or inheritance of deleterious autosomal dominant AD mutations[74–76] (Figure 1a–b). It has been hypothesized that these individuals exhibit higher levels of ‘cognitive reserve’, where functional brain processes are better able to cope with changes induced by neuropathology, or ‘brain reserve’, where variation in structural characteristics of the brain (e.g. neuron or synapse numbers) increase the ability of an individual’s brain to deal with neuropathological insults[77]. To understand the mechanisms and specific factors that differentiate resilient versus susceptible individuals, individuals can be grouped into bins (e.g. non-demented with AD pathology versus demented with AD pathology). Historically, this has been done post-mortem using classifications from autopsy-based neuropathological scores on standardized scales such as the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) score for neuritic plaques[78] and Braak staging of neurofibrillary tangles[79] in combination with antemortem scores on standardized cognitive tests.
Beyond discrete classification systems, the development of approaches that define resiliency as a continuous metric have been aided by the development of quantitative measurements of pathology, including CSF biomarkers, uptake of amyloid- and tau-specific ligands measured via positron emission tomography (PET) imaging, and structural magnetic resonance imaging (MRI) that quantifies various metrics such as brain volume. The measures are aggregated to calculate a predicted cognitive outcome given an individual’s measured pathology; this predicted value is compared to the same individual’s actual cognitive performance. The discrepancy between the predicted and observed outcome, or the proportion of the cognitive score not explained by neuropathology (i.e. the model residuals), is used to quantify an individual’s level of resilience (Figure 1B). Non-residual-based continuous metrics have also been developed[80], such as the AD Cognitive Resilience (AD-CR) score. This score is computed using a global measure of cognition obtained proximal to death and postmortem AD pathology. Machine learning algorithms trained using this information may eventually be able to predict whether an individual will have an extremely high or low AD-CR score and thus be at extremely high or low risk for progressing into AD dementia. Across these measures, individuals that perform better than predicted given an observed level of pathology are said to have high resilience. The main benefit of these approaches is that they do not exclude individuals that do not fit neatly into discrete bins but allow for interrogation of the entire study population.
Figure 1: Strategies to identify resilient individuals in human populations.
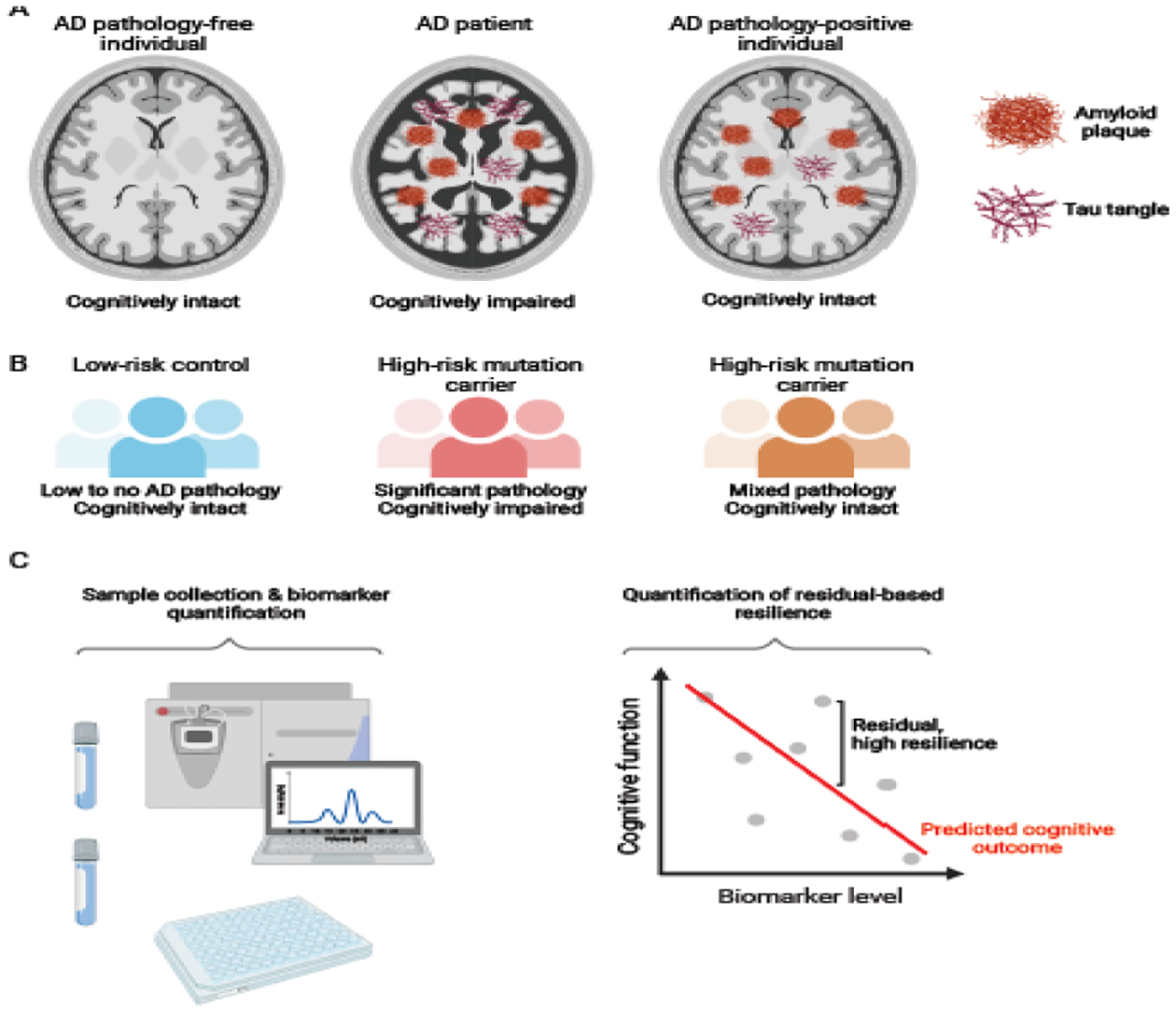
A) Example of a discrete classification system used to identify resilient individuals. In this classification, each individual is assigned into one of three groups: cognitively unimpaired individuals, free from significant Alzheimer’s disease (AD) pathology (left); Alzheimer’s disease patients, with brain atrophy, amyloid plaques, and hyperphosphorylated tau tangles (middle); and “resilient” individuals, with no apparent cognitive impairment despite the presence of AD pathology (right). Preservation of brain volume and decreased accumulation of tau tangles have been associated with resilience, although specific features of resilient individuals can vary across studies and cohorts. B) Resilient individuals can also be identified based on susceptibility to mutations typically associated with increased risk for AD. A subset of the population is at relatively low genetic risk for AD and will exhibit limited accumulation of AD pathology and has a high likelihood of remaining cognitively intact (left). Another subset will inherit deleterious mutations associated with increased risk for AD: some of these individuals will go on to develop AD pathology and dementia as expected (middle), while a resilient subset will remain cognitively intact throughout the lifespan (right), despite varying levels of pathology. C) Resilience can also be quantified as a continuous metric. In this approach, diagnostic samples are first collected and biomarkers quantified (left). These measures are aggregated with other demographic factors including for instance age, sex, and educational status, to predict an individual’s expected cognitive performance (prediction line in red). Results are then compared to the same individual’s actual cognitive performance, and the discrepancy between these two values (i.e. the residual) is used to quantify resilience. Individuals with better than predicted cognitive function (above red line) are said to exhibit high resilience, while those with worse than predicted cognitive function (below red line) are said to exhibit low resilience.
Table 1:
Select factors correlated with resilience in humans.
| Trait | Cohort | Directionality | Reference |
|---|---|---|---|
| Cortical thickness | Aβ-positive individuals with MCI or dementia; residual metrics used to define resilience | Increased thickness associated with cognitive resilience to tau pathology | [15] |
| Spine density | AD patients vs pathology-free controls vs pathology-positive, non-demented controls | Spine density and numbers of thin and mushroom spines reduced in AD cases only | [10] |
| Neuron numbers, synaptic markers | AD vs pathology-free controls vs pathology-positive, non-demented | Preservation of these features observed in pathology-positive controls | [7] |
| Glial activation | AD vs pathology-free controls vs pathology-positive, non-demented | Lower activation in resilient individuals | [7] |
| Cortical proteins including NRN1, EPHX4, SGTB, CPLX1 | ROSMAP | Higher levels of these proteins associated with greater resilience to several pathologies | [35] |
| REST protein levels | Pathology-positive controls vs AD patients | Nuclear REST levels higher in cognitively intact individuals | [53] |
| CSF levels of VEGF | ADNI | Elevated CSF VEGF associated with better cognitive outcomes in pathology-positive individuals | [86] |
| IGFBP5, HSPB2, AK4, ITPK1 protein in DLPFC | ROSMAP | IGFBP5, HSPB2, AK4 - higher protein levels associated with faster cognitive function, opposite for ITPK1. Associations not explained by neuropathology. | [87] |
| ENC1, UNC5C | ROSMAP | Multi-omics approach (genetic, epigenetic, transcriptomic) prioritized the aforementioned genes; increased UNC5C RNA associated with worse residual-based resilience, and increased ENC1 associated with increased residual-based resilience | [33] |
| Cytokine profiles in entorhinal cortex and superior temporal sulcus | Non-demented individuals with 1) no, 2) moderate, 3) high pathology and AD patients | Resilient cases had unique profile – increased anti-inflammatory cytokines, increased neurotrophic factors, decreased microglial recruitment chemokines | [9] |
| sTREM2 levels in CSF | Subjects with AD defined by CSF biomarkers | Higher CSF sTREM2 associated with attenuated decline in memory and cognition | [30] |
| MEF2C | Pathology-positive subjects with or without AD dementia | Higher MEF2C observed in resilient individuals, specifically within a subset of translationallyactive excitatory neurons | [38] |
| LAP3, MACROD1, SEMA7A protein in synapse | Pathology-positive cognitively intact ‘resilient’ patients vs. AD patients | Resilient subjects had lower levels of these proteins in synapses compared to AD patients | [37] |
| Proteins involved in serotonin release, oxidative phosphorylation, glycolysis, and proteasome function | Pathology-positive cognitively intact ‘resilient’ patients vs. AD patients | Resilient subjects had an enrichment for serotonin- and oxidative phosphorylation-related proteins in the synapse, as well as depletion of glycolysis- and proteasome-related proteins in the synapse | [37] |
| APOE Christchurch mutation | PSEN1 E280A carrier | Homozygous mutation protected against high levels of pathology | [20] |
| BDNF Val66Met mutation | DIAN, among others | Mutation associated with worsened neurodegeneration & cognitive decline | [26, 88–90] |
| Klotho haplotype VS | APOE-e4 positive individuals | One copy of haplotype lowered AD risk (but also reduced Aβ and tau positivity on PET) | [28, 31, 32] |
| SNX25, PDLIM3, SORBS2 | Caribbean Hispanics with PSEN1 p.G206A mutation | Association with age at onset in early- and late-onset AD | [24] |
| Gene variants near CD44, NPHP1, CADPS2, GREM2 | Families carrying PSEN1 E280A mutation | Variants associated with age at onset of AD | [25] |
| Gene variants near CASP7, SERPINA3 | APOE-ε4 carriers | Variants associated with AD risk | Reviewed in [27] |
| Variant upstream ATP8B1 | Four cohorts: A4, ROSMAP, ADNI, ACT | Variant associated with combined (i.e. cognitive and brain structural-based) resilience in cognitively intact subset of patients | [17] |
| Variation at chromosome 8 locus | Mayo Clinic Study of Aging and ADNI | Variant associated with differential cognitive resilience to brain amyloid | [34] |
| Educational attainment | Various | Higher education is associated with increased resilience across cohorts and studies | [15–17, 91], among others |
| Early-life cognitive enrichment | MAP | Cognitive enrichment was associated with less cognitive decline, 80% of the effect was independent of neuropathology | [92] |
Abbreviations: ROSMAP: Religious Orders Study/Memory and Aging Project, DLPFC: Dorsolateral prefrontal cortex, MCI: mild cognitive impairment, GWAS: genome-wide association studies, AD: Alzheimer’s disease, ADNI: Alzheimer’s disease Neuroimaging Initiative, A4: Anti-Amyloid Treatment in Asymptomatic Alzheimer’s study, ACT: Adult Changes in Thought study
Beyond discrete classification systems, a number of studies have used a residual-based approach to identify features common to resilient individuals (Box 1, Table 1). For example, cortical thickness is associated with cognitive resilience to pathological tau[15], while younger age and higher education was well-correlated with resilience to structural changes and CSF/plasma-based biomarker metrics[15–17]. An additional benefit to studies that incorporate relatively non-invasive measurements is their ability to be repeated over time. There is some evidence that resilience capacity can change over time, with overall levels of resilience decreasing with age[18]. While cross-sectional measures of residual-based resilience were indeed associated with cognitive outcomes[19], there is some evidence that change in resilience metrics over time is even more useful for predicting cognitive decline over time and identifying those individuals most at risk of developing dementia[18], which is important for identifying when individuals stand to benefit most from early intervention and preventative-based therapeutics and clinical trials.
Targeted investigation of genetic and molecular mediators of resilience
Identification of the specific genetic factors that promote resilience to AD pathology or autosomal dominant familial AD mutations may provide a starting point for uncovering novel therapeutic targets. Specific populations at high genetic risk for AD represent an opportunity to interrogate mechanisms that enable some mutation carriers to ‘escape’ the predicted effect of the mutation on cognitive outcomes. For example, a recent case study reported a carrier of a PSEN1 mutation (PSEN1 E280A) who did not develop mild cognitive impairment until her seventies despite high levels of amyloid[20]. Carriers of the PSEN1 E280A mutation exhibit an average age of dementia onset at 49 years[21, 22], so this individual seemed highly resilient to the typically deleterious effects of the mutation[20]. This specific incidence of resilience was attributed to the presence of a homozygous APOE variant known as the Christchurch mutation[23] that confers reductions in low density lipoprotein receptor and heparin binding[20], implicating these mechanisms as putative drivers of resilience in the face of high amyloid deposition. The value of these pathways as therapeutic targets remains to be elucidated, as the Christchurch mutation was also linked to reduced tau pathology in the PSEN1 E280A carrier[20], which suggests some of the benefit of the mutation may be conferred through resistance to developing primary pathologies associated with AD rather than true resilience as defined earlier.
Populations at high genetic risk for AD provide several other important opportunities for the investigation of resilience. One approach, for instance, involved genome-wide searches for the modifiers of the age at onset of cognitive decline in families carrying what are typically considered highly penetrant mutations. These studies have identified several putatively involved genes[24, 25] (Table 1). In addition, these populations provide the opportunity for targeted investigation into hypothesized resilience factors such as BDNF. For example, it was found that a single nucleotide polymorphism (SNP) in BDNF (BDNFVal66Met) was associated with worsened neurodegeneration and cognitive decline given amyloid pathology across individuals with autosomal dominant AD, studied as part of the Dominant Inherited Alzheimer Network (DIAN) cohort[26]. Individuals with APOE-ε4 genotype provide a similar opportunity. Although APOE- ε4 has been identified as the most significant genetic risk factor for AD, some ε4 homozygous individuals do not develop AD as expected. Several candidate genes (Table 1) have been associated with reduced AD risk in APOE-ε4-positive individuals[27, 28], highlighting these genes as potential therapeutic intervention points. For example, levels of the soluble form of the AD risk gene TREM2 (sTREM2) found in the CSF of biomarker-positive AD patients were found to significantly modify the effect of an APOE-ε4 allele independent of primary AD pathology[29]. Higher CSF levels of sTREM2 were also found to be associated with slower decline in memory and cognition in biomarker-positive AD patients[30]. It is worth noting that some of the ‘resilience’ genes identified have also been associated with reduced levels of pathology[31, 32], highlighting the need to disentangle effects associated with true resilience to pathology from resistance to developing these pathologies.
Genome-wide approaches to discover genetic and molecular mediators of resilience
With the advent of more affordable genetic profiling, higher-throughput monitoring of relevant biomarker metrics including CSF-, plasma-, and imaging-based metrics, as well as quantitative, continuous metrics of resilience, whole-genome discovery-based studies of resilience are now feasible. Here, residual resilience metrics are calculated across a population and treated as a quantitative variable for association studies in order to identify specific variants, genes, proteins and/or pathways associated with the resilience phenotype[18]. As sample sizes are generally still small compared to traditional case-control GWAS, relatively few genome-wide significant associations have been found[17, 33, 34]. Hits have been prioritized by combining genetic data with various additional functional –omics datasets, including methylation data and dorsolateral prefrontal cortex (DLPFC) RNA expression[33]. At the pathway level, associations with resilience included amino acid metabolism, prolactin receptor signaling, and the dehydrogenase pathway[17] as well as integrin-related cell adhesion and T-cell factor signaling related to the Wnt/B-catenin pathway[34], nominating relatively broad biological categories for therapeutic intervention rather than specific therapeutic targets. Beyond genetic mediators of resilience, unbiased proteomics has identified cortical proteins strongly associated with levels of resilience[35], including NRN1 (Table 1), which, similar to BDNF, is a neurotrophic factor known to play important roles in synaptic function and plasticity as well as maintenance of axonal morphology[36]. An additional study uniquely focused on synaptic proteins identified decreased levels of proteins including MAP3 and SEMA7A in synapses from resilient individuals relative to AD patients, as well as an overall enrichment for proteins involved in the serotonin release cycle and oxidative phosphorylation and depletion of protein involved in glycolysis and proteasome-related processes[37] (Table 1). Single-nuclear sequencing has also been used to identify a resilience-related increase in MEF2C in a specific subpopulation of translationally active excitatory neurons[38] (Table 1), demonstrating the utility of these high-dimensional and unbiased assays to identify specific molecular mediators of resilience to AD pathology.
Using animal models to study resilience
While human-focused studies of resilience are crucial, drawbacks to these studies exist (Box 2), calling for complementary research approaches. Model organism studies provide an important resource to begin to address the outstanding questions surrounding resilience mechanisms through longitudinal, mechanistic, and/or interventional studies with controlled environmental factors. Mice have emerged as a particularly useful model organisms due to their affordability, shared genetic features with humans, short generation time, and the vast array of mouse-specific genetic engineering resources. As mice do not naturally develop AD, genetic engineering is necessary to introduce human genes harboring mutations that lead to early-onset AD. These mutations typically drive the overproduction of amyloid, which accumulates in the brain of these animals and induces AD-relevant symptoms including cognitive decline, synaptic dysfunction, and neurodegeneration.
Box 2: Potential drawbacks to human-based studies of resilience.
While both targeted and genome-wide studies identified correlates with resilience in the human population, it is important to note several limitations of these types of analyses. Because brain tissue from individuals in these cohorts is not accessible during their lives, many studies use postmortem tissue to identify molecules (e.g. RNA, protein) that are associated with performance on cognitive tests prior to death. However, in these studies, brain regions used to examine the molecular mediators of resilience are typically selected based on already-known involvement in disease risk (e.g. hippocampus and prefrontal cortex). It is possible that molecular changes that confer resilience originate in brain regions outside those classically affected in AD and thus not typically selected for analyses. In addition, mechanisms and molecules important for resilience are likely expressed and act well before the time at which these tissues can be accessed. This lack of access to brain tissue early in the disease course is a significant barrier to understanding the molecules most closely associated with the onset of resilience (and/or dementia). In addition, the ability to test mechanistic hypotheses is generally limited in human populations, as the identification of molecules associated with cognitive outcomes is largely correlative[81]. Other potential caveats and considerations, such as the limitations associated with retrospective group assignments, have been highlighted elsewhere[81]. Together, these factors have precluded a clear understanding of when in disease course and where in the brain differences between resilient and susceptible individuals are first detectable, a key piece of information that will enable the eventual development of targeted therapeutics. These limitations create a critical need for innovative approaches to synergize the power of animal AD models with the wealth of medically relevant human data presently available to parse out the spatial and temporal origins of resilience, as well as to establish causation and screen actionable resilience interventions.
Historically, model organism studies have varied one condition in genetically identical individuals (e.g. inbred mice) to evaluate the impact on disease outcomes. While many important insights into disease pathogenesis have been achieved using this approach, recently the importance of expanding model organism studies to include additional unique genotypes has been receiving growing recognition. Specifically, genetic reference panels (GRPs) have emerged as a useful tool to incorporate genetic diversity into mouse studies in a systematic way (Box 3). Another notable development is that in addition to various forms of inbred lines, studies have begun to incorporate genetic diversity in the form of outbred mice, particularly the Diversity Outbred (DO) panel population of systematically outbred mice originating from the same 8 founder strains as the Collaborative Cross (CC) GRP[39].
Box 3: Mouse genetic reference panels.
Two of the most well-characterized GRPs are 1) the BXDs, named for the parental common inbred strains C57BL6J (B) and DBA2J (D) from which it is derived, and 2) the Collaborative Cross (CC), derived from a systematic crossing scheme incorporating five common inbred strains and three wild-derived inbred strains [82]. In these genetic reference panels, mice with unique combinations of each parental genotype are inbred until a new stable inbred line is created. The result is a large number of related inbred strains (the BXD GRP is currently comprised of 140 unique strains while the CC GRP contains ~70 strains), each of which can be reproducibly studied across time, laboratories, and under different environmental conditions [72]. In addition, functional data such as transcriptional, proteomic, and behavioral profiles for each strain can be collected, combined, and shared in order to better prioritize nominated candidates as described in the main text for human data. GRPs allow for intense community collaboration, and a wealth of resources exist for each system (e.g. GeneNetwork.org[83]iii, Systems-Genetics.org[84]iv, and the Mouse Phenome Database [85]v allow for data mining, visualization, and investigations of relationships between measures of interest). These resources enable investigation into the downstream effects of prioritized candidate genes and SNPs associated with traits of interest and facilitate the identification of relevant SNPs and molecular mechanisms from a large number of putative candidates.
Hypothesis-driven approaches to understanding mechanisms of resilience in mouse models
Animal models offer opportunities to test the causality of resilience factors identified in human studies. As mentioned above, historically, this has often been performed in a single inbred line where the intervention or treatment is the only factor being manipulated to assess causality of the putative resilience factor. For example, viral delivery or overexpression of BDNF in various animal models of AD has been shown to improve outcomes, as assessed via various markers of disease including hippocampal connectivity and cognitive tests, independent of any effects on amyloid or tau neuropathology[40, 41] (Table 2). Similarly, overexpression of Mef2-family transcription factors in neurons of a mouse model of tauopathy was sufficient to improve cognitive outcomes and rescue pathology-induced neuronal hyperexcitability without altering tau pathology[38] (Table 2).
Table 2:
Select factors associated with resilience in mouse models
| Trait | Cohort | Directionality | Reference |
|---|---|---|---|
| Intrinsic neuronal excitability | B6SJL 5XFAD | Decreased neuronal excitability in ‘weak-learners’ | [50] |
| Pla2g4e expression and genotype | Tg2576 | Increased Pla2g4e observed in ‘resilient’ mice | [51] |
| Synaptic protein levels | Tg2576 | Increased in ‘resilient’ mice | [51] |
| Several gene networks, including immune- and amyloid degradation-related networks | Tg2576 | Results suggest reduced CD4+ T-cell infiltration in resilient mice, | [49] |
| BDNF | J20 (amyloid transgenic) P301L (tau transgenic) mouse models | Overexpression improved AD-related outcomes independent of pathological changes | [40, 41] |
| Mef2 family of transcription factors | P301S tauopathy model | Overexpression of Mef2a/c improved cognitive outcomes and rescued neuronal hyperexcitability without changing tau pathology | [38] |
| Apoe genotype | AD-BXDs | D allele associated with worsened cognitive outcomes | [56] |
| Genetic risk score derived from common AD risk genes | AD-BXDs | Genotype at common AD risk genes associated with late-life cognitive outcomes | [56] |
| Several gene networks, including vascular- and immune-related networks | AD-BXDs | Higher expression of immune-enriched networks early in life correlated with worse cognitive outcomes later in life, reverse was true for expression of vascular-related networks | [57] |
| Variation at chromosome 2 locus | AD-BXDs | Genotype at identified locus regulated immune-related network expression | [57] |
| X chromosome, putatively Kdm6a | Transgenically modified mice with altered numbers of sex chromosomes and hAPP model mice | Additional X chromosome in either males or females improved Aβ-related mortality, cognitive deficits, and cellular viability. Overexpression of Kdm6a, which escapes X inactivation, was sufficient to recapitulate this effect in cell and animal models | [46] |
Abbreviations: B6SJL: mixed background strain derived from the inbred strains C57BL6/J and SJL/J, 5XFAD: common AD transgene incorporating 5 mutations known to cause familial AD in humans, BDNF: Brain-derived neurotrophic factor, AD-BXDs: F1 genetic reference panel generated by introducing the 5XFAD transgene into the BXD panel (see Box 3 for details), AD: Alzheimer’s disease
Beyond the testing of specific gene candidates, animal models also enable targeted experimental investigation into pathways hypothesized to mediate resilience. In resilient patients, preservation of dendritic spines is consistently observed, raising the question of what are the mechanisms and pathways that preserve spines. Using cell and animal models, Aβ42-induced spine loss was identified to be mediated, at least in part, through Rho-associated kinase ROCK2 and downstream activation of the kinase LIMK1[42]. Treatment of AD model mice with a LIMK1 inhibitor rescued Aβ-induced hippocampal spine loss and morphologic aberrations[42]. In addition, an intriguing observation across human populations is that despite similar levels of Aβ and tau, men show worse cognition[43], greater cognitive decline[44], increased neurodegeneration[45], and increased mortality[46] compared to women, suggesting an effect of sex on resilience and reduced resilience in men. A study in transgenic animal models found that the presence of an additional X-chromosome either in males or females consistently improved Aβ-related mortality, cognitive deficits, and cellular viability[46]. The authors localized the protective effect to the gene Kdm6a, a histone demethylase that escapes X-chromosome inactivation and is enriched in the brain. Overexpression of Kdm6a attenuated Aβ-induced neuotoxicity and cognitive impairment in both cell and animal models[46], nominating Kdm6a as a putative mechanistic driver of resilience.
Exploiting phenotypic diversity to identify resilience mechanisms
Beyond mechanistic investigation of previous-identified resilience factors[40, 41, 47], mouse models also provide a powerful tool to investigate differential susceptibility to typically highly deleterious AD mutations – similar to the approach described above using families harboring autosomal dominant AD mutations (Figure 2). In animal model studies, various studies have shown that even within a single inbred strain, mice are not uniformly susceptible to the cognitive effects of amyloid and/or tau accumulation[48–51]. For example, two independent groups found that when mice are subjected to paradigms meant to measure learning and memory, age-matched AD mice could be divided into either ‘weak-learner’ or ‘strong-learner’ subsets[49–51]. Across studies, whether a particular mouse exhibited cognitive deficits was not significantly associated with amyloid plaque density, levels of tau hyperphosphorylation, or markers of glial activation, suggesting the ‘strong-learners’ were exhibiting resilience to neuropathology[50, 51]. The identification of these independent subsets provides an opportunity to investigate causal mechanisms driving resilience. In the case of Tg2576 transgenic mice, resilient animals exhibited higher levels of synaptic proteins, suggesting preserved synaptic function[51] (Table 2). Similarly, resilient mice carrying the 5XFAD transgene exhibited a higher degree of learning-related intrinsic neuronal plasticity relative to 5XFAD weak-learners[50] (Table 2). Together, these studies highlight maintenance of synaptic structure and neuronal excitability as an important mechanism mediating resilience to AD pathology. Transcriptomic analysis comparing Tg2576 resilient and susceptible mice identified the gene Pla2g4e (phospholipase A2 group IVE) as a putative driver of resilience[51]. Subsequent overexpression of Pla2g4e in hippocampal neurons of AD mice was sufficient to completely restore cognitive function and increase spine number without affecting amyloid or tau pathology[51]. In addition, PLA2G4E was found to be decreased in the brains of late-stage AD human patients, suggesting this approach identified a translationally relevant therapeutic target. A similar transcriptomic analysis also identified several additional pathways putatively involved in resilience and predicted a reduced infiltration of peripheral immune cells into the brains of resilient mice[49]. A previous investigation into the hippocampal membrane proteome of 5XFAD strong-learners and weak-learners identified several putative drivers of resilience, including the epigenetic modifier REST[52] previously associated with resilience to pathology in human populations[53] (Table 2). Together, these findings demonstrate the powerful translational potential of harnessing variable AD susceptibly in animal models.
Figure 2: Exploiting phenotypic or genetic diversity in mouse populations to investigate molecular and genetic mediators of resilience.
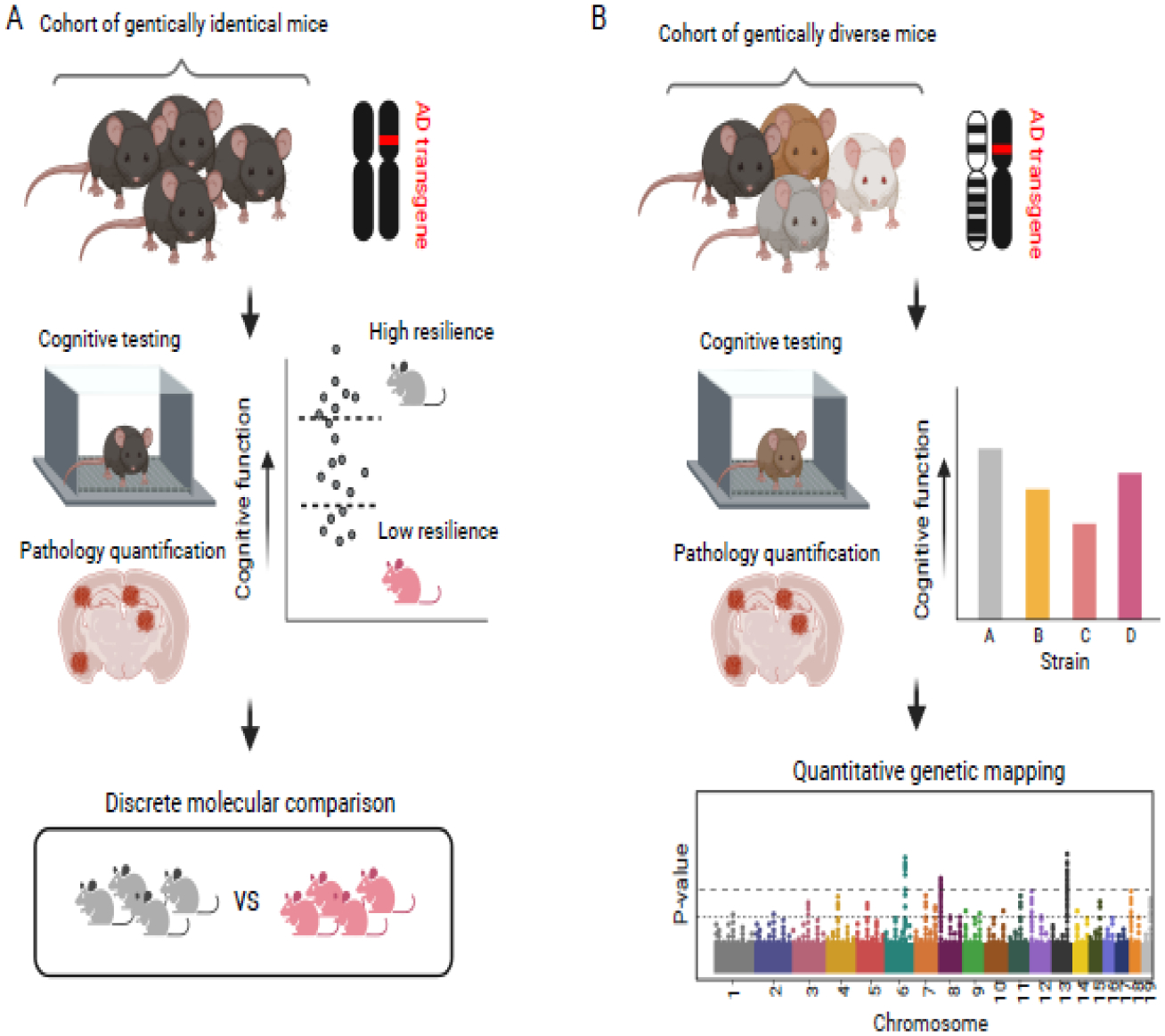
A) Phenotypic diversity among genetically homogenous strains of mice harboring Alzheimer’s disease (AD) transgenes can be harnessed to classify individuals into discrete ‘high resilience’ (i.e. strong-learner) or ‘low resilience’ (i.e. weak-learner) categories. Pathology can be quantified using a range of techniques including immunohistochemistry to account for differences in amyloid and/or hyperphosphorylated tau accumulation. B) Traditional animal models do not incorporate genetic diversity, which in human populations contributes to differing levels of resilience. By introducing AD transgenes into diverse models including the BXDs, Collaborative Cross, or Diversity Outbred panels, strain-by-strain differences in cognitive outcomes and pathology can be quantified and used for genetic mapping in order to identify specific genetic mediators of resilience.
Exploiting genetic diversity to identify resilience mechanisms
Traditionally, animal model studies of resilience to AD have often relied on experiments within a single strain. While variable susceptibility within a single strain provides important mechanistic insight into resilience, focusing on a single strain leads to a scenario that is distant from the degree of genetic diversity of human populations. Comparison of genetically distinct mouse strains harboring identical amyloid- or tau- overexpressing transgenes has confirmed the presence of variable susceptibility across strains to disease outcomes including pathological burden and overall survival [48, 54, 55]. To specifically use GRPs (Box 3) to identify resilience factors, a new F1 mouse population was generated by introducing the well-established 5XFAD transgene into almost 30 BXD strains[56]. This panel, referred to as the AD-BXDs, was thoroughly characterized for a variety of disease-relevant traits, including amyloid deposition and cognitive decline. As hypothesized, a wide range of cognitive outcomes was observed across the strains despite similar levels of amyloid pathology[56], demonstrating differing levels of cognitive resilience depending on an individual strain’s genetic makeup. Notably, genotype at well-defined late-onset AD risk factors including Apoe, Bin1, and Clu, among others, influenced cognitive outcomes in the AD-BXDs, suggesting this novel panel represents a resource with high translational potential[56] (Table 2). Of note, the single Apoe mutation segregating across the AD-BXDs occurs near the receptor binding region of the protein, where the highly protective Christchurch mutation occurs in human populations. This suggests that the AD-BXDs, in addition to their utility in identifying modifiers of familial AD, could be useful for identifying more general modifiers of late-onset AD susceptibility.
The reproducible nature of an F1 GRP design provides an opportunity to understand genetic and molecular drivers of resilience. Specifically, transcriptional networks present early in life, at a time point at which the AD-BXDs were not impaired as a population relative to their wild-type littermates, were related to strain-specific cognitive outcomes in later life[57]. Networks related to neuroinflammation, brain vasculature, extracellular matrix organization, and synaptic signaling were associated with cognitive resilience in this population[57], mirroring some of the pathways (e.g. vascular risk, synaptic structure) important in regulating human resilience (Table 2) and highlighting these as mechanisms that may drive resilience. Due to the genetic structure of the AD-BXDs, genetic mapping can be used to identify specific regions associated with traits of interest. A region on chromosome two was identified as containing variants that directly modulate expression of identified resilience networks in AD-BXD mice[57]. Interestingly, the human equivalent of this this region was also associated with age at AD onset in familial AD studies[58, 59], further demonstrating the translational relevance of this panel for identifying drivers of resilience that may be harnessed for future therapeutic development. Notably, beyond specific resilience to AD pathology, the AD-BXDs can also be combined with their wild-type or non-transgenic littermates (i.e. the Ntg-BXDs) to understand and identify mechanisms that promote cognitive longevity across disease and normal aging[60, 61].
Future directions for animal studies of resilience
Translating findings from animal models to the human condition is not without challenges. For example, microglia, the resident immune cells of the brain, have been identified as cells involved in disease progression[62]. However, significant differences exist between the mouse and human immune systems, and these could translate into marked differences in the microglial response to amyloid[63]. In addition, most transgenes used to study AD in mouse models overexpress human mutant APP and PSEN1 in a cell-type specific manner. In humans, mutations are presumably expressed in all cell types so the expression of transgenes only in a single cell type, typically neurons, prohibits the identification of how these mutations may drive disease through effects on inter-cell communication or non-neuronal mechanisms. Newer models incorporating AD mutations directly into the mouse orthologs have not replicated all findings in earlier transgenic models, raising the concern that some aspects of disease in these mice are driven purely by overexpression artifacts rather than mutation status[64]. Finally, most of the widely used animal models of AD produce no detectable tau pathology. This is primarily because variants in the gene encoding for tau, MAPT, have not been genetically associated with AD and were therefore thought not to be a valid way to genetically model AD in the mouse. However, because tau pathology is part of the diagnostic criteria for AD, models that incorporate some degree of tau pathology likely come closer to recapitulating the human disease. A recent study identified the MEF2 family of transcription factors as potential mediator(s) of resilience to tau pathology with high translational relevance, demonstrating the utility and potential of this approach[38]. As such, incorporating tau pathology into future studies of resilience to AD will be critical. For example, the AD-BXD reference panel could be regenerated using alternative AD models harboring tau mutations. This approach could provide important insight into the overlap or discordance between the genetic mediators of resilience to amyloid or tau pathology. Importantly, as tau pathology appears to be more intimately coupled to cognitive outcomes than amyloid in human populations, understanding the genetic mechanisms that enable resilience to tau pathology may provide the most broadly applicable therapeutic targets.
While the BXD family contains 6 million single nucleotide polymorphisms[65], this degree of genetic diversity does not reach what is observed across the human population. In contrast, the wild-derived strains used to generate the CC and DO panels harbor almost 40 million SNPs relative to the reference C57BL6J genome[39]. Introduction of AD transgenes into these wild-derived strains themselves produces a wide variation in amyloid deposition, neuronal cell loss, and microglial morphology and activation[66, 67]. Similar to the approach used to generate the AD-BXDs discussed earlier, future studies incorporating AD transgenes into CC strains could provide insights into which, if any, CC strains exhibit cognitive resilience to amyloid deposition, and could offer opportunities to identify novel resilience factors that may not exhibit genetic variation in the BXD GRP. The areas of oncology and immunology offer some precedents in that regard: The introduction of disease-causative transgenes into the DO population have proven useful in identifying mediators of prostate cancer[68] and immune dysfunction[69, 70]. Possibly, the DO panel may prove similarly useful in nominating specific genetic resilience candidates. Encouraging data pointing in this direction are the differential resilience to normal cognitive aging in the DO panel, driven by genotype at Dlgap2 and presumably subsequent alterations in spine morphology[71].
Laboratory mice also provide a unique opportunity to begin to understand the mechanisms by which various lifestyle factors may contribute to resilience[72]. Because of the well-defined environment in which laboratory mice are housed, researchers can systematically vary a single lifestyle factor, such as diet, physical activity, or cognitive enrichment. The effect of each of these well-defined manipulations on cognitive outcomes can then be interrogated and mechanisms investigated, providing insight into how each of these interventions may influence disease outcomes. There is abundant evidence that environmental enrichment and physical activity improve disease outcomes in animal models of AD[73], but most of these studies have been conducted only in single inbred strains. Given that genetic heterogeneity may influence the effects of these interventions, it would be valuable to pair these approaches with genetically diverse models[72]. These approaches may also help identify specific human populations that stand to benefit most from certain lifestyle interventions, providing a pathway to personalized recommendations to promote resilience.
Finally, future studies utilizing genetically diverse animal models should continually strive to complement their findings with human data in order to maximize their translational relevance. Due to the combination of increased availability of large-scale human datasets and increased collaboration through consortia such as the Collaboratory on Research Definitions for Reserve and Resilience in Cognitive Aging and Dementiai, and Resilience-ADii, it has become increasingly possible to perform cross-species analyses. Often, human studies that aim to identify resilience mediators by testing many genes or variants for association with cognitive function simultaneously suffer from low power due to low sample size and high statistical burden. Using prior knowledge gained from animal studies to test specific variants or genes for association with cognitive function in humans, rather than performing genome-wide searches, enables researchers to nominate new resilience mediators that may have been previously overlooked in these complex studies. It is worth noting that the approaches described here to understand resilience to AD, including incorporating genetic and phenotypic diversity across animal models of disease, are broadly applicable to a range of complex diseases. As these studies require large numbers of animals and phenotyping tests, and also generate large amounts of data, the continued development of high-throughput tools (e.g. automated behavioral assays, imaging, and biomarker analysis platforms) will be essential for effective progress.
Concluding remarks
Understanding how some individuals can retain cognitive function despite high levels of neuropathology is critical for developing therapeutics to prevent or delay the onset of dementia in the larger population. Initial studies have identified several factors that contribute to resilience to AD pathology, including the ability to maintain brain synapse structure and functional connectivity, potentially through the maintenance of neurotrophic factors including NRN1 and BDNF. Recent advances in the ability to quantitatively define resilience across large populations, in combination with relatively non-invasive CSF/plasma biomarkers and structural imaging measurements, have facilitated the search for genetic contributors to resilience.
To facilitate the identification of actionable resilience factors, a major goal for future work is to better understand which brain regions are involved, and at what time point in disease progression resilience mechanisms act (see Outstanding Questions). This will necessitate brain-wide assessment at various time points throughout the lifespan, in contrast to profiling a single region with a single technology (e.g. PFC- and hippocampus-specific RNA sequencing). Understanding where in the brain (or body) resilience originates, when in the disease course it emerges, and how it progresses with age will provide critical insight for the development of effective therapies. Each of these questions can be systematically addressed when paired with both comprehensive human data and innovative mouse models that harness genetic complexity under well-defined environmental conditions. With these techniques, the field is hopefully well-positioned to identify new avenues for therapeutic development in the coming years.
Outstanding Questions.
Quantifying resilience as a continuous metric eliminates the need to ‘discard’ data from patients that do not fit into discrete classification bins. This perspective, combined with non-invasive techniques to evaluate AD pathology, allows performing large-scale analyses of the genetic and molecular mediators of resilience. Will the use of larger cohorts result in the identification and prioritization of novel mediators of resilience to AD pathology?
Previous studies identified candidate features of resilience. Can these various metrics be integrated to identify individuals who may have low levels of innate resilience and thus, be at a higher risk for developing dementia due to AD? Can this information be used to begin early intervention and/or optimize clinical trial enrollment? Is early intervention necessary, or can treatments which intervene after the onset of cognitive decline be effective?
While the use of the BXD genetic reference panel improved translational relevance of AD mouse models, the genetic diversity present in this panel does not approach the diversity present in the human population. Will the introduction of AD transgenes into more complex animal models of genetic diversity (i.e. CC, DO – Box 3) yield new insights into mediators of resilience?
As tau pathology is more closely linked to cognitive outcomes than amyloid and is a diagnostic criterion for AD, is inclusion of tau pathology necessary to identify translationally-relevant mediators of resilience? What is the best translationally relevant model(s) for preclinical studies of therapeutic targets?
The exact brain region(s) and molecular mediator(s) for conferring enhanced resilience to AD pathology remain unclear. Can brain-wide studies designed to survey putative mediators at multiple levels (i.e DNA, RNA, protein) help pinpoint which brain regions or molecules may be most highly associated with resilience? In addition, when do ‘resilient’ and ‘susceptible’ individuals first start to deviate?
Highlights.
Some individuals retain cognitive function despite high levels of Alzheimer’s disease (AD) pathology. Understanding the biology behind this form of ‘AD resilience’ may lead to opportunities for therapeutic development.
Human genetic studies and postmortem analyses have identified a number of factors associated with cognitive resilience to AD pathology, including overall maintenance of synapse structure and candidate molecular mediators (e.g. REST, NRN1, among others).
Genetically and phenotypically diverse mouse models of AD provide new and translationally-relevant opportunities to identify and prioritize new resilience mechanisms.
Approaches integrating results from both human and mouse studies will provide an effective path toward the development of therapeutics promoting resilience in susceptible human populations.
Acknowledgements:
This work was funded by the National Institutes of Health, National Institute on Aging (R01AG057914, R01AG054180, and RF1AG063755 to CCK, F32AG069503 to SMN, R01AG066831 to VM, R01AG059716, R01AG061518, and R01AG074012 to TJH, RF1AG059778 to KMSO), the BrightFocus Foundation (A2016397S to CCK), and the Zenith Fellows Award (ZEN-21-846037 to CCK and MT). All figures were made in BioRender (www.biorender.com) with publication license to MT.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests:
Dr. Hohman serves on the Scientific Advisory Board for Vivid Genomics. All other authors have no conflicts of interest to declare in relation to this work.
References
- 1.Selkoe DJ, The molecular pathology of Alzheimer’s disease. Neuron, 1991. 6(4): p. 487–98. [DOI] [PubMed] [Google Scholar]
- 2.Gao Y, et al. , Tau in Alzheimer’s Disease: Mechanisms and Therapeutic Strategies. Curr Alzheimer Res, 2018. 15(3): p. 283–300. [DOI] [PubMed] [Google Scholar]
- 3.Hardy JA and Higgins GA, Alzheimer’s disease: the amyloid cascade hypothesis. Science, 1992. 256(5054): p. 184–5. [DOI] [PubMed] [Google Scholar]
- 4.Goate A, et al. , Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature, 1991. 349(6311): p. 704–6. [DOI] [PubMed] [Google Scholar]
- 5.Rahimi J and Kovacs GG, Prevalence of mixed pathologies in the aging brain. Alzheimers Res Ther, 2014. 6(9): p. 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aizenstein HJ, et al. , Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol, 2008. 65(11): p. 1509–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perez-Nievas BG, et al. , Dissecting phenotypic traits linked to human resilience to Alzheimer’s pathology. Brain, 2013. 136(Pt 8): p. 2510–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schneider JA, et al. , The neuropathology of older persons with and without dementia from community versus clinic cohorts. J Alzheimers Dis, 2009. 18(3): p. 691–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barroeta-Espar I, et al. , Distinct cytokine profiles in human brains resilient to Alzheimer’s pathology. Neurobiol Dis, 2019. 121: p. 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boros BD, et al. , Dendritic spines provide cognitive resilience against Alzheimer’s disease. Ann Neurol, 2017. 82(4): p. 602–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Latimer CS, et al. , Resistance and resilience to Alzheimer’s disease pathology are associated with reduced cortical pTau and absence of limbic-predominant age-related TDP-43 encephalopathy in a community-based cohort. Acta Neuropathol Commun, 2019. 7(1): p. 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Rossum IA, et al. , Injury markers but not amyloid markers are associated with rapid progression from mild cognitive impairment to dementia in Alzheimer’s disease. J Alzheimers Dis, 2012. 29(2): p. 319–27. [DOI] [PubMed] [Google Scholar]
- 13.Arriagada PV, et al. , Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology, 1992. 42(3 Pt 1): p. 631–9. [DOI] [PubMed] [Google Scholar]
- 14.Hyman BT, et al. , National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement, 2012. 8(1): p. 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ossenkoppele R, et al. , Assessment of Demographic, Genetic, and Imaging Variables Associated With Brain Resilience and Cognitive Resilience to Pathological Tau in Patients With Alzheimer Disease. JAMA Neurol, 2020. 77(5): p. 632–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin L, et al. , Resilience to Plasma and Cerebrospinal Fluid Amyloid-beta in Cognitively Normal Individuals: Findings From Two Cohort Studies. Front Aging Neurosci, 2021. 13: p. 610755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dumitrescu L, et al. , Genetic variants and functional pathways associated with resilience to Alzheimer’s disease. Brain, 2020. 143(8): p. 2561–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bocancea DI, et al. , Measuring Resilience and Resistance in Aging and Alzheimer Disease Using Residual Methods: A Systematic Review and Meta-analysis. Neurology, 2021. 97(10): p. 474–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hohman TJ, et al. , Asymptomatic Alzheimer disease: Defining resilience. Neurology, 2016. 87(23): p. 2443–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arboleda-Velasquez JF, et al. , Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: a case report. Nat Med, 2019. 25(11): p. 1680–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quiroz YT, et al. , Association Between Amyloid and Tau Accumulation in Young Adults With Autosomal Dominant Alzheimer Disease. JAMA Neurol, 2018. 75(5): p. 548–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Acosta-Baena N, et al. , Pre-dementia clinical stages in presenilin 1 E280A familial early-onset Alzheimer’s disease: a retrospective cohort study. Lancet Neurol, 2011. 10(3): p. 213–20. [DOI] [PubMed] [Google Scholar]
- 23.Wardell MR, et al. , Apolipoprotein E2-Christchurch (136 Arg----Ser). New variant of human apolipoprotein E in a patient with type III hyperlipoproteinemia. J Clin Invest, 1987. 80(2): p. 483–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee JH, et al. , Genetic Modifiers of Age at Onset in Carriers of the G206A Mutation in PSEN1 With Familial Alzheimer Disease Among Caribbean Hispanics. JAMA Neurol, 2015. 72(9): p. 1043–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Velez JI, et al. , Pooling/bootstrap-based GWAS (pbGWAS) identifies new loci modifying the age of onset in PSEN1 p.Glu280Ala Alzheimer’s disease. Mol Psychiatry, 2013. 18(5): p. 568–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Franzmeier N, et al. , The BDNFVal66Met SNP modulates the association between beta-amyloid and hippocampal disconnection in Alzheimer’s disease. Mol Psychiatry, 2021. 26(2): p. 614–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huq AJ, et al. , Genetic resilience to Alzheimer’s disease in APOE epsilon4 homozygotes: A systematic review. Alzheimers Dement, 2019. 15(12): p. 1612–1623. [DOI] [PubMed] [Google Scholar]
- 28.Belloy ME, et al. , Association of Klotho-VS Heterozygosity With Risk of Alzheimer Disease in Individuals Who Carry APOE4. JAMA Neurol, 2020. 77(7): p. 849–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Franzmeier N, et al. , Higher CSF sTREM2 attenuates ApoE4-related risk for cognitive decline and neurodegeneration. Mol Neurodegener, 2020. 15(1): p. 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ewers M, et al. , Increased soluble TREM2 in cerebrospinal fluid is associated with reduced cognitive and clinical decline in Alzheimer’s disease. Sci Transl Med, 2019. 11(507). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neitzel J, et al. , KL-VS heterozygosity is associated with lower amyloid-dependent tau accumulation and memory impairment in Alzheimer’s disease. Nat Commun, 2021. 12(1): p. 3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Belloy ME, et al. , KL *VS heterozygosity reduces brain amyloid in asymptomatic at-risk APOE *4 carriers. Neurobiol Aging, 2021. 101: p. 123–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.White CC, et al. , Identification of genes associated with dissociation of cognitive performance and neuropathological burden: Multistep analysis of genetic, epigenetic, and transcriptional data. PLoS Med, 2017. 14(4): p. e1002287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramanan VK, et al. , Coping with brain amyloid: genetic heterogeneity and cognitive resilience to Alzheimer’s pathophysiology. Acta Neuropathol Commun, 2021. 9(1): p. 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu L, et al. , Cortical Proteins Associated With Cognitive Resilience in Community-Dwelling Older Persons. JAMA Psychiatry, 2020. 77(11): p. 1172–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yao JJ, et al. , Functions and the related signaling pathways of the neurotrophic factor neuritin. Acta Pharmacol Sin, 2018. 39(9): p. 1414–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carlyle BC, et al. , Synaptic proteins associated with cognitive performance and neuropathology in older humans revealed by multiplexed fractionated proteomics. Neurobiol Aging, 2021. 105: p. 99–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barker SJ, et al. , MEF2 is a key regulator of cognitive potential and confers resilience to neurodegeneration. Sci Transl Med, 2021. 13(618): p. eabd7695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Churchill GA, et al. , The Diversity Outbred mouse population. Mamm Genome, 2012. 23(9–10): p. 713–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nagahara AH, et al. , Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat Med, 2009. 15(3): p. 331–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiao SS, et al. , Brain-derived neurotrophic factor protects against tau-related neurodegeneration of Alzheimer’s disease. Transl Psychiatry, 2016. 6(10): p. e907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Henderson BW, et al. , Pharmacologic inhibition of LIMK1 provides dendritic spine resilience against beta-amyloid. Sci Signal, 2019. 12(587). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buckley RF, et al. , Sex, amyloid, and APOE epsilon4 and risk of cognitive decline in preclinical Alzheimer’s disease: Findings from three well-characterized cohorts. Alzheimers Dement, 2018. 14(9): p. 1193–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jack CR Jr., et al. , Age, Sex, and APOE epsilon4 Effects on Memory, Brain Structure, and beta-Amyloid Across the Adult Life Span. JAMA Neurol, 2015. 72(5): p. 511–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jack CR Jr., et al. , Transition rates between amyloid and neurodegeneration biomarker states and to dementia: a population-based, longitudinal cohort study. Lancet Neurol, 2016. 15(1): p. 56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davis EJ, et al. , A second X chromosome contributes to resilience in a mouse model of Alzheimer’s disease. Sci Transl Med, 2020. 12(558). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dubal DB, et al. , Life extension factor klotho prevents mortality and enhances cognition in hAPP transgenic mice. J Neurosci, 2015. 35(6): p. 2358–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Swarup V, et al. , Identification of evolutionarily conserved gene networks mediating neurodegenerative dementia. Nat Med, 2019. 25(1): p. 152–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Perez-Gonzalez M, et al. , Identifying the Main Functional Pathways Associated with Cognitive Resilience to Alzheimer’s Disease. Int J Mol Sci, 2021. 22(17). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kaczorowski CC, et al. , Mechanisms underlying basal and learning-related intrinsic excitability in a mouse model of Alzheimer’s disease. Neurobiol Aging, 2011. 32(8): p. 1452–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Perez-Gonzalez M, et al. , PLA2G4E, a candidate gene for resilience in Alzheimer s disease and a new target for dementia treatment. Prog Neurobiol, 2020. 191: p. 101818. [DOI] [PubMed] [Google Scholar]
- 52.Neuner SM, et al. , Hippocampal proteomics defines pathways associated with memory decline and resilience in normal aging and Alzheimer’s disease mouse models. Behav Brain Res, 2017. 322(Pt B): p. 288–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lu T, et al. , REST and stress resistance in ageing and Alzheimer’s disease. Nature, 2014. 507(7493): p. 448–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jackson HM, et al. , DBA/2J genetic background exacerbates spontaneous lethal seizures but lessens amyloid deposition in a mouse model of Alzheimer’s disease. PLoS One, 2015. 10(5): p. e0125897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sipe JD, et al. , Characterization of the inbred CE/J mouse strain as amyloid resistant. Am J Pathol, 1993. 143(5): p. 1480–5. [PMC free article] [PubMed] [Google Scholar]
- 56.Neuner SM, et al. , Harnessing Genetic Complexity to Enhance Translatability of Alzheimer’s Disease Mouse Models: A Path toward Precision Medicine. Neuron, 2019. 101(3): p. 399–411 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Neuner SM, et al. , Identification of Pre-symptomatic Gene Signatures That Predict Resilience to Cognitive Decline in the Genetically Diverse AD-BXD Model. Front Genet, 2019. 10: p. 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kehoe P, et al. , A full genome scan for late onset Alzheimer’s disease. Hum Mol Genet, 1999. 8(2): p. 237–45. [DOI] [PubMed] [Google Scholar]
- 59.Pericak-Vance MA, et al. , Identification of novel genes in late-onset Alzheimer’s disease. Exp Gerontol, 2000. 35(9–10): p. 1343–52. [DOI] [PubMed] [Google Scholar]
- 60.Heuer SE, et al. , Identifying the molecular systems that influence cognitive resilience to Alzheimer’s disease in genetically diverse mice. Learn Mem, 2020. 27(9): p. 355–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dunn AR, et al. , Identifying Mechanisms of Normal Cognitive Aging Using a Novel Mouse Genetic Reference Panel. Front Cell Dev Biol, 2020. 8: p. 562662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Novikova G, et al. , Integration of Alzheimer’s disease genetics and myeloid genomics identifies disease risk regulatory elements and genes. Nat Commun, 2021. 12(1): p. 1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhou Y, et al. , Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat Med, 2020. 26(1): p. 131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sasaguri H, et al. , APP mouse models for Alzheimer’s disease preclinical studies. EMBO J, 2017. 36(17): p. 2473–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ashbrook DG, et al. , A platform for experimental precision medicine: The extended BXD mouse family. Cell Syst, 2021. 12(3): p. 235–247 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Onos KD, et al. , Enhancing face validity of mouse models of Alzheimer’s disease with natural genetic variation. PLoS Genet, 2019. 15(5): p. e1008155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yang HS, et al. , Natural genetic variation determines microglia heterogeneity in wild-derived mouse models of Alzheimer’s disease. Cell Rep, 2021. 34(6): p. 108739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Winter JM, et al. , Mapping Complex Traits in a Diversity Outbred F1 Mouse Population Identifies Germline Modifiers of Metastasis in Human Prostate Cancer. Cell Syst, 2017. 4(1): p. 31–45 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wei WZ, et al. , Diversity Outbred Mice Reveal the Quantitative Trait Locus and Regulatory Cells of HER2 Immunity. J Immunol, 2020. 205(6): p. 1554–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hackett JB, et al. , A Diversity Outbred F1 mouse model identifies host-intrinsic genetic regulators of response to immune checkpoint inhibitors bioRxiv, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ouellette AR, et al. , Cross-Species Analyses Identify Dlgap2 as a Regulator of Age-Related Cognitive Decline and Alzheimer’s Dementia. Cell Rep, 2020. 32(9): p. 108091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dunn AR, O’Connell KMS, and Kaczorowski CC, Gene-by-environment interactions in Alzheimer’s disease and Parkinson’s disease. Neurosci Biobehav Rev, 2019. 103: p. 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shepherd A, et al. , Transgenic Mouse Models as Tools for Understanding How Increased Cognitive and Physical Stimulation Can Improve Cognition in Alzheimer’s Disease. Brain Plast, 2018. 4(1): p. 127–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bekris LM, et al. , Genetics of Alzheimer disease. J Geriatr Psychiatry Neurol, 2010. 23(4): p. 213–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Reitz C, Rogaeva E, and Beecham GW, Late-onset vs nonmendelian early-onset Alzheimer disease: A distinction without a difference? Neurol Genet, 2020. 6(5): p. e512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cruchaga C, et al. , Rare variants in APP, PSEN1 and PSEN2 increase risk for AD in late-onset Alzheimer’s disease families. PLoS One, 2012. 7(2): p. e31039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stern Y, et al. , Whitepaper: Defining and investigating cognitive reserve, brain reserve, and brain maintenance. Alzheimers Dement, 2020. 16(9): p. 1305–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fillenbaum GG, et al. , Consortium to Establish a Registry for Alzheimer’s Disease (CERAD): the first twenty years. Alzheimers Dement, 2008. 4(2): p. 96–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Braak H, et al. , Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol, 2006. 112(4): p. 389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yao T, et al. , Quantifying cognitive resilience in Alzheimer’s Disease: The Alzheimer’s Disease Cognitive Resilience Score. PLoS One, 2020. 15(11): p. e0241707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Arenaza-Urquijo EM and Vemuri P, Resistance vs resilience to Alzheimer disease: Clarifying terminology for preclinical studies. Neurology, 2018. 90(15): p. 695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Churchill GA, et al. , The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat Genet, 2004. 36(11): p. 1133–7. [DOI] [PubMed] [Google Scholar]
- 83.Mulligan MK, et al. , GeneNetwork: A Toolbox for Systems Genetics. Methods Mol Biol, 2017. 1488: p. 75–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li H, et al. , An Integrated Systems Genetics and Omics Toolkit to Probe Gene Function. Cell Syst, 2018. 6(1): p. 90–102 e4. [DOI] [PubMed] [Google Scholar]
- 85.Bogue MA, et al. , Mouse Phenome Database: a data repository and analysis suite for curated primary mouse phenotype data. Nucleic Acids Res, 2020. 48(D1): p. D716–D723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hohman TJ, et al. , The role of vascular endothelial growth factor in neurodegeneration and cognitive decline: exploring interactions with biomarkers of Alzheimer disease. JAMA Neurol, 2015. 72(5): p. 520–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yu L, et al. , Targeted brain proteomics uncover multiple pathways to Alzheimer’s dementia. Ann Neurol, 2018. 84(1): p. 78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lim YY, et al. , BDNF Val66Met moderates memory impairment, hippocampal function and tau in preclinical autosomal dominant Alzheimer’s disease. Brain, 2016. 139(Pt 10): p. 2766–2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lim YY, et al. , APOE and BDNF polymorphisms moderate amyloid beta-related cognitive decline in preclinical Alzheimer’s disease. Mol Psychiatry, 2015. 20(11): p. 1322–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lim YY, et al. , Effect of BDNFVal66Met on disease markers in dominantly inherited Alzheimer’s disease. Ann Neurol, 2018. 84(3): p. 424–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Busatto GF, et al. , In vivo imaging evidence of poor cognitive resilience to Alzheimer’s disease pathology in subjects with very low cognitive reserve from a low-middle income environment. Alzheimers Dement (Amst), 2020. 12(1): p. e12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Oveisgharan S, et al. , Association of Early-Life Cognitive Enrichment With Alzheimer Disease Pathological Changes and Cognitive Decline. JAMA Neurol, 2020. 77(10): p. 1217–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]