

Abstract
Since its discovery over 100 years ago, insulin has been recognized as a key hormone in control of glucose homeostasis. Deficiencies of insulin signaling are central to diabetes and many other disorders. The brain is among the targets of insulin action, and insulin resistance is a major contributor to many diseases including brain disorders. Here, we summarize key roles of insulin action in the brain and how this involves different brain cell types. Disordered brain insulin signaling can also contribute to neuropsychiatric diseases, affecting brain circuits involved in mood and cognition. Understanding of insulin signaling in different brain cell types/circuits and how these are altered in disease may lead to the development of new therapeutic approaches to these challenging disorders.
Keywords: Diabetes, Insulin resistance, Neurons, Astrocytes, Depression, Alzheimer’s Diseases
The Brain is Insulin-Sensitive
Type 2 Diabetes (T2D, see Glossary), obesity and metabolic syndrome are increasing worldwide at epidemic rates. T2D affects over 450 million people worldwide, and over 2 billion people are overweight or obese [1]. In these disorders, impaired insulin signaling, i.e., insulin resistance, is a defining feature [2, 3]. While metabolic changes associated with insulin resistance have been largely related to alterations in so-called “classical” insulin-sensitive tissues: liver, skeletal muscle, and fat [2, 4], it is now known that most, if not all, tissues of the body, including the brain, express insulin receptors (InsR) and are insulin sensitive (Figure 1) [4]. Accumulating evidence suggests that brain insulin signaling not only plays a key role in regulation of metabolism [5], but also in regulation of mood, behavior, and cognition. Dysregulated insulin signaling has also been implicated in a range of brain disorders (Figure 1). Over the past few decades, epidemiological studies have demonstrated a high rate of comorbidity between T2D and brain disorders such as depression and Alzheimer’s Disease (AD) [6, 7], leading to recommendations that the evaluation of neuropsychiatric comorbidities should be part of the initial assessment of T2D patients [8]. Understanding the role of brain insulin signaling in these disorders may also provide new therapeutics targeting the comorbidity between T2D and brain disorders.
Figure 1.
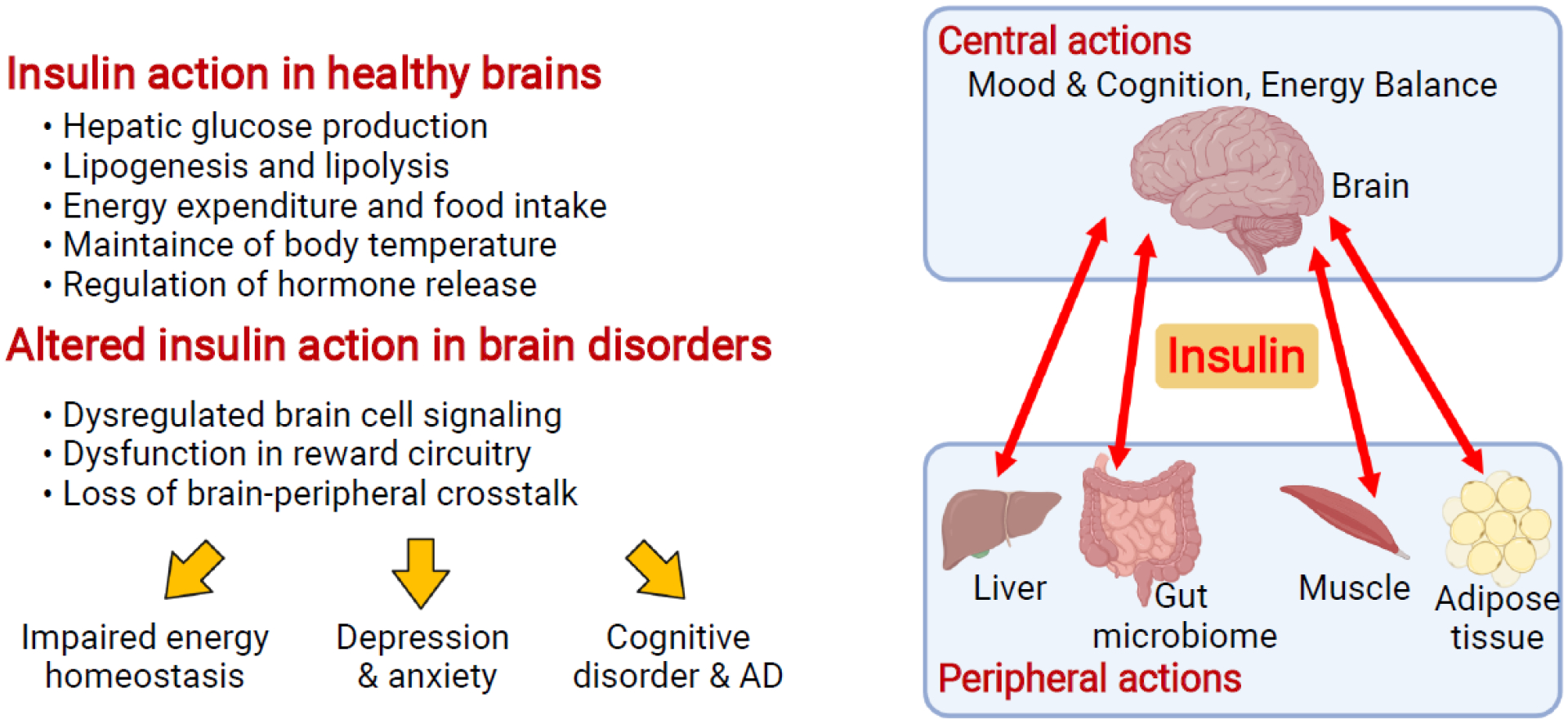
Brain insulin signaling in healthy and disordered brains. Insulin not only serves as a master regulator of systemic metabolism and normal functions within organs such as liver, fat, and muscle, but also regulates key brain functions such as mood and cognition. As a result, altered brain insulin signaling leads to brain disorders such as emotional and cognitive disorders. Created with BioRender.com.
In this review, we summarize the clinical evidence connecting insulin resistance/T2D and brain dysfunction, and explore this link at a mechanistic level with a particular focus on cell type-specific regulation in the brain mediated by insulin signaling. We also connect these cellular insights to brain-wide changes in circuits and to the pathogenesis of brain disorders.
Comorbidity of T2D and Brain Disorders
Growing evidence has shown that T2D is closed associated with mood disorders and neurodegenerative diseases independent of dysglycemia. Thus, patients with type 1 diabetes have similar rates of depression as the general population [9], while the prevalence of depression in T2D is about twice the population average [10] and increases with insulin resistance [11]. Prospective studies have pointed to a bidirectional relationship: the incidence of depression is increased [12], and depressive symptoms are more persistent and remission less stable in patients with T2D [13]. Likewise, a previous diagnosis of depression is associated with increased incidence of T2D by 60% [14] and with more severe long-term diabetic complications [15]. The effect of depression on T2D is partially, but not completely, explained by the increased risk of T2D with antidepressant use, especially noradrenergic and tricyclic antidepressants [16, 17]. Conversely, diabetic patients treated with selective serotonin reuptake inhibitors experience improvements in glycemic control [18] and increased insulin sensitivity [19], although this has not been observed in all studies [20]. Understanding the molecular mechanisms underlying these interactions between diabetes and psychiatric medications should provide new insights into the pathogenesis of depression as well as diabetes, and how to improve treatment of both (see Outstanding questions).
Outstanding questions.
Does insulin signaling in the mesolimbic dopamine reward pathway directly modulate this pathway’s function, and could this play a role in the manifestation of mood and anxiety symptoms in disorders of insulin signaling?
Given the marked heterogeneity of brain cell types, including neurons, astrocytes and microglia, how does InsR or IGF1R signaling in these subtypes change with normal brain development and during disease progression? Are some subtypes more vulnerable than others to systemic metabolic dysfunction? How does insulin resistance in one subtype affect function in other subtypes?
Would targeting dopaminergic or serotonergic neurons modulate brain insulin release and alleviate the comorbidity of T2D and psychiatric disorders? How do antidepressants or cognition-enhancing drugs modify systemic metabolic status?
Are there InsR-independent mechanisms or actions of insulin on other cells which can modulate how insulin enters the brain, and if so, how do they change in disease?
What are the best preclinical animal models to study comorbidity of T2D and brain disorders and the most efficient way to achieve clinical translation of these studies?
Investigations of the relationship between T2D and anxiety have uncovered similar connections. The prevalence of clinically significant anxiety in patients with T2D is ~20% higher than the population average [21]. People with T2D also have increased prevalence of generalized anxiety disorder (GAD), panic disorder, post-traumatic stress disorder, and agoraphobia [22]. Attempts to establish causality, however, yielded mixed results. A meta-analysis demonstrated that patients with higher baseline levels of anxiety have an increased incidence of T2D [23]. In patients with GAD and T2D, treatment with alprazolam has been noted to improve glycemic control in a manner unrelated to the level of improvement in anxiety, suggesting these are occurring through independent mechanisms [24].
It has long been known that schizophrenia is associated with increased levels of hyperglycemia [25], and the prevalence of T2D in patients with schizophrenia is 2–3 times the population norm [26]. Part of this is attributable to medication side effects. All atypical antipsychotics, the first-line treatment for schizophrenia since the 1990s, increase the incidence of diabetes [27]. The co-existence of diabetes, however, is not fully explained by medications. Indeed, prior to the advent of antipsychotics, hospitalized patients with psychosis exhibited abnormal glucose tolerance [28] and hepatic insulin resistance [29]. Intriguingly, the efficacy of current antipsychotics tends to positively correlate with the severity of their metabolic side effects, suggesting that disruptions in insulin signaling and/or metabolism is linked to the therapeutic mechanism [30].
A growing body of evidence indicates that neurodegenerative diseases, such as AD, also have strong epidemiological connections with T2D and that T2D is a major risk factor for AD and other dementias [31]. Patients with T2D have a ~50% increased risk of developing mild cognitive impairment [32, 33], and a meta-analysis of prospective studies including more than 1.7 million patients has demonstrated a similar increase in AD risk for patients with T2D [34]. This robust epidemiological connection is supported by analysis of postmortem brain tissue of AD patients. Defects in insulin and IGF-1 signaling or associated signaling proteins have been shown to co-occur with Aβ plaques in the temporal lobe, hippocampus, and cerebellum [35, 36]. A decrease in the primary substrate of these receptors, IRS-1, appears to be a more consistent marker of insulin resistance in the brain [36, 37]. Interestingly, hyperactivation of the downstream targets of IRS-1, including phosphoinositide 3-kinase (PI3K), Akt, and glycogen synthase kinase 3β (GSK-3β), have also been observed in both early- and late-stage AD [38]. Furthermore, patients with AD have increased plasma insulin levels and decreased cerebrospinal fluid insulin levels [39, 40].
Cell Type-Specific Regulation of Insulin Receptor Signaling in the Brain
To decipher the comorbidity of T2D and brain disorders, it is key to understand insulin signaling in the brain. Both the InsR and the closely-related insulin-like growth factor 1 receptor (IGF-1) receptor are highly expressed in the brain [41]. The first direct evidence for a role of the InsR in brain came with the generation of a brain InsR knockout (NIRKO) mouse, produced using the Cre/LoxP system with Cre driven by the Nestin promoter [42]. Nestin targets both neuronal and glial cell precursors in the brain and also to some extent the peripheral nervous system (Table 1 summarizes the use of the Cre/loxP system for probing InsR signaling in different cell types). NIRKO mice exhibited a complete loss of insulin-stimulated signaling in the brain at a molecular level [42]. This was accompanied by slightly increased food intake, mild obesity and systemic insulin resistance [5]. When NIRKO mice were challenged with hypoglycemia, the expected sympathoadrenal response with elevation in epinephrine and norepinephrine levels was significantly blunted, indicating that insulin signaling in the brain is required for a normal counterregulatory response to hypoglycemia [43, 44] (for more information on brain insulin action in regulation of peripheral metabolism, see reference [45]). In addition, NIRKO mice also exhibited increased anxiety- and depression-like behavioral changes [6] and demonstrated some biochemical hallmarks of AD, such as increased Tau phosphorylation [42]. These data indicate the interplay between insulin signaling in brain function and its control of peripheral metabolism, behavior and brain disorders.
Table 1.
A summary of studies using Cre/loxP system to probe physiological roles of InsR signaling in different brain cell types
| Name | Cell-type of interests | Cre-driver | Main findings of the InsR KO phenotypes | Physiological importance | Refs |
|---|---|---|---|---|---|
| NIRKO | brain-specific | Nestin-Cre |
|
Regulation of energy disposal, fuel metabolism, reproduction, brain-liver crosstalk, glucose sensing, brain cholesterol metabolism, mitochondrial function, and dopamine signaling | [5, 6, 42–44, 107, 142] |
| GIRKO | Astrocyte-specific | GFAP-Cre, GFAP-CreERT2, GLAST-CreERT2 |
|
hypothalamic glucose sensing, regulation of systemic glucose metabolism, dopaminergic signaling, ATP exocytosis, HPG axis | [7, 62, 143] |
| EndoIRKO | Vascular endothelial cells-specific | Cdh5-Cre Slcolc1-CreERT2 |
|
Transendothelial insulin delivery; kinetics of insulin signaling | [74, 77, 79] |
Using mice which express cell type specific Cre driven by promoters for unique neuropeptides or their receptors, it has been possible to explore the role of insulin signaling within specific neuronal subtypes (Table 2). Among these, the role of insulin signaling in hypothalamic proopiomelanocortin (POMC) and agouti-related neuropeptide/neuropeptide Y (AgRP/NPY) expressing neurons in terms of food intake and energy expenditure are the best characterized (Figure 2). Although the InsR is expressed in both of these neuronal types [46], insulin signaling in AgRP/NPY neurons acts to suppress hepatic glucose production and decrease energy expenditure [47, 48], whereas in POMC neurons, insulin acts to promote hepatic glucose production and increase energy expenditure, at least in part by activating TRPC5 channels and PI3K [49–51] and regulating neuronal plasticity [47, 51, 52]. These divergent effects occur as the result of signaling within competitive circuits (Figure 2, and see also discussion below and references [47, 48]).
Table 2.
A summary of InsR signaling in different neuronal subtypes
| Neuronal subtypes | Transgenic approaches | Main findings | Physiological role | Ref. |
|---|---|---|---|---|
| ARC POMC neurons | POMC InsR KO POMC InsR KI mice POMC-eGFP POMC InsR/LepR double KO IRS2 KO mice |
|
Regulation of adipose lipolysis, neuronal plasticity, energy expenditure, glucose homeostasis and fertility | [47, 48, 50–52, 144–146] |
| ARC AgRP neurons | AgRP InsR KO AgRP InsR KI mice |
|
Regulation of glucose metabolism | [47, 48] |
| LH MCH neurons | MCH InsR KO MCH-GFP MCH−/− mice |
|
Regulation of body weight, insulin sensitivity, and energy homeostasis | [53, 54, 147] |
| Orexin-A neurons | Orexin−/− mice Orexin-cre (DREADDs, viral ablation) |
|
Prevention of hepatic insulin resistance Regulation of feeding behavior, metabolism | [56, 57, 148–150] |
| HPA axis | InsRNkx2.1 KO (hypothalamus) mice, InsRSim1 KO (PVN), mice InsRAgRP KO (ARH) mice |
This study used separate Cre lines to achieve disruption of InsR signals in selective hypothalamic areas and found:
|
Regulation of HPA axis and stress response; Regulation of mood |
[151] |
| Sensory neurons | Advillin-cre mice (SNIRKO) |
|
Regulation of islet function | [152] |
Figure 2.
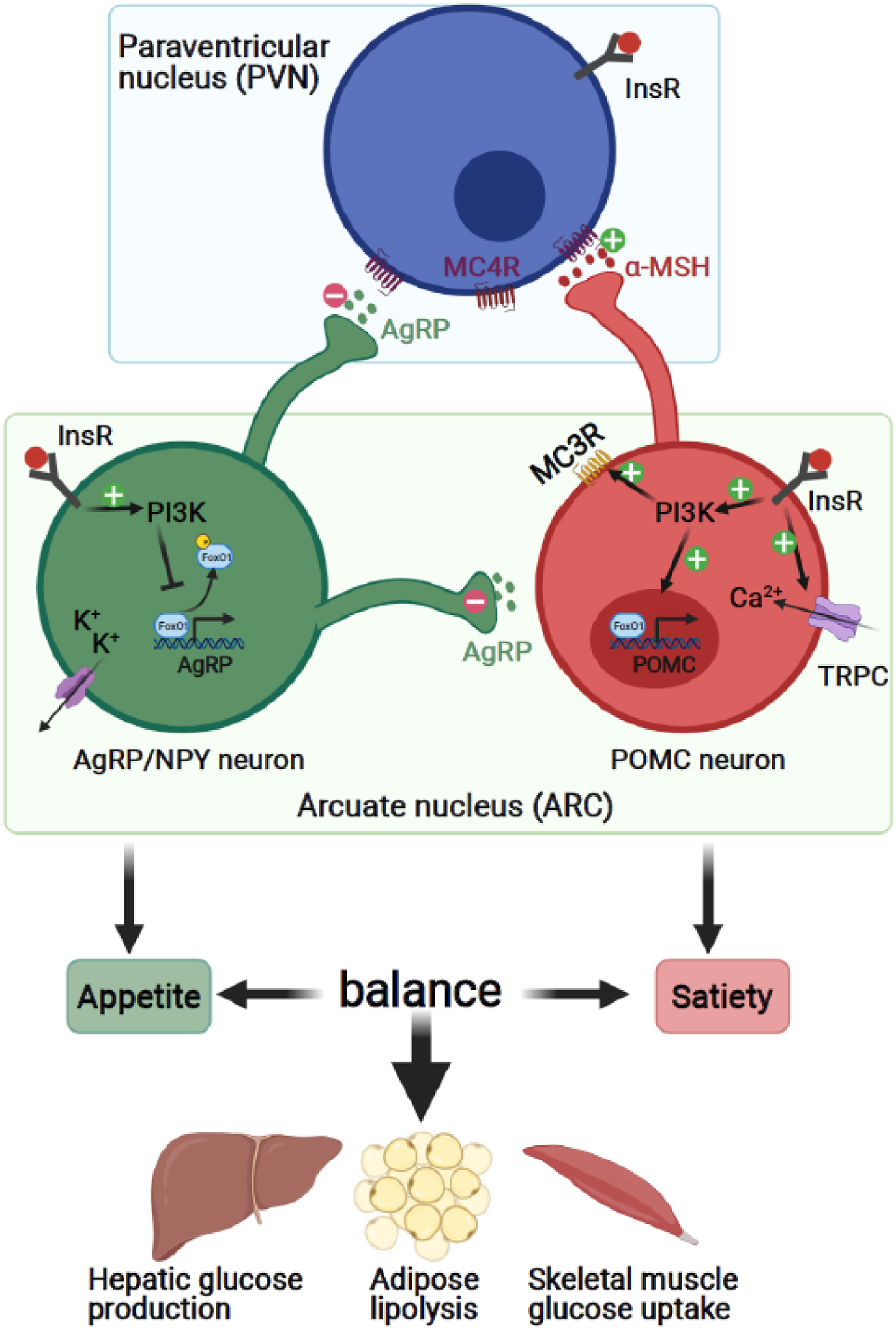
Insulin regulates food intake and energy expenditure through hypothalamic neurons. A prominent role of insulin signaling in the brain is to regulate food intake and energy metabolism through hypothalamic POMC and AgRP/NYP neurons. The interplay across these cell types contributes to the maintenance of the balance between appetite and satiety. Created with BioRender.com.
Abbreviations: AgRP/NPY, agouti-related neuropeptide/neuropeptide Y; α-MSH, alpha-Melanocyte-stimulating hormone; InsR, insulin receptor; MC3R, melanocortin 3 receptor; MC4R, melanocortin 4 receptor; PI3K, Phosphoinositide 3-kinase; POMC, proopiomelanocortin; TRPC, transient receptor potential canonical channels.
Insulin signaling is also critical in other neuronal cell types. While mice with whole-body knockout of melanin-concentrating hormone (MCH) exhibit hypophagia and reduced body weight [53], deletion of InsR from MCH-expressing neurons produces no metabolic phenotype in lean mice but increases locomotor activity and improves insulin sensitivity in obese mice [54]. Orexin is another peptide that plays a key role in the CNS regulation of glucose and energy homeostasis [55], and deletion of orexin-producing neurons or manipulation of orexin-dependent neuronal activity leads to hepatic insulin resistance [56] and alters feeding behavior and metabolism [57]. For additional information on insulin signaling in neuronal subpopulations, see Table 2 and other excellent reviews [58–60].
Investigations into the roles that insulin signaling plays in non-neuronal cell types in the brain are relatively recent. Astrocytes play an important role in regulating neuronal functions, such as synaptic plasticity and neurotransmission [61]. InsR KO in astrocytes guided by the glial fibrillary acidic protein+ (GFAP+) Cre has been achieved using both constitutive and inducible Cre/LoxP-mediated recombination (GIRKO/iGIRKO mice, Table 1). These studies have revealed that in astrocytes, insulin stimulates tyrosine phosphorylation of Munc18c which regulates SNARE complex formation, leading to exocytosis of ATP, which in turn modulates synaptic plasticity at dopaminergic axonal terminals by acting through P2Y receptor (Figure 3) [7]. Strikingly, loss of insulin signaling in astrocytes reduces evoked dopamine (DA) release in dorsal striatum and nucleus accumbens (NAc) by almost 50%, leading to anxiety- and depression-like behaviors in GIRKO mice [7].
Figure 3.
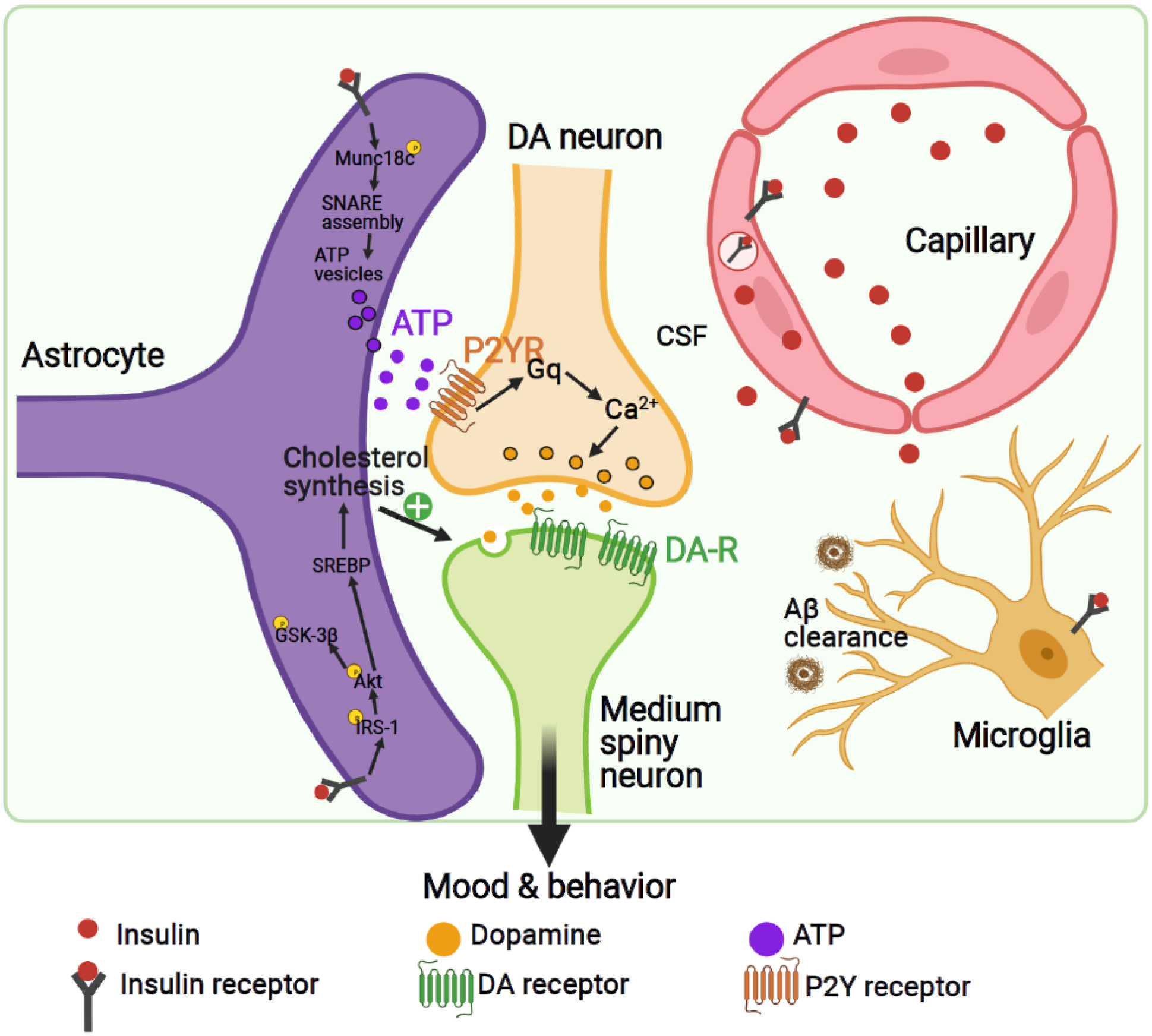
Cell type-specific regulation of insulin signaling in the brain. Insulin regulates a wide range of brain cell types, including neurons, astrocytes, endothelial cells, and microglia. Created with BioRender.com.
Abbreviations: ATP, adenosine triphosphate; CSF, cerebrospinal fluid; DA, dopamine; DA-R, dopamine receptors; P2YR, P2Y receptors; SREBP, sterol regulatory element-binding protein.
Astrocytic insulin signaling also regulates hypothalamic glucose sensing. In GIRKO mice, there is a reduction of glucose-induced activation of POMC neurons and an impairment of the physiological response to altered glucose availability, indicating that astrocytic insulin signaling plays an important role in systemic glucose homeostasis [62]. However, GFAP+ astrocytes represent only one subpopulation of astrocytes in brain. Thus, going forward, it will be important to utilize other astrocyte markers, such as GLAST [62] or ALDH1L1 [61, 63] or to target disease-associated astrocytic subpopulations [64] (see Outstanding questions).
Studies on the role of insulin signaling in microglia, the resident immune cells in the brain, are still at a nascent stage. In obesity and metabolic syndrome, increases in inflammatory cells are observed in many organs, including adipose tissue, liver and even brain [65, 66], therefore it seems plausible that microglial responses could be altered during states of insulin resistance. In microglial cell lines, insulin inhibits Aβ42 clearance induced by isoproterenol (Figure 3) [67]. This seems contradictory to the finding that intranasal insulin treatment has been shown to reduce Aβ levels and microglia activation and restore impaired insulin signaling in an AD mouse model [68]. It is possible that the beneficial effects of intranasal insulin are related to an impact on inflammation gene expression in the hippocampus [69, 70], suggesting these as potential targets of insulin in the AD brain. Microglial overactivation had also been observed in rats made insulin-resistant by high fructose diet, and this effect can be reduced with the antidiabetic PPARγ-agonist pioglitazone or the GLP-1 analogue exenatide associated with enhanced cognition [71]. These studies, together with others, suggest that microglia activation and inflammation occur in the brain in both AD and states of insulin resistance, and that insulin and other antidiabetic drugs might prevent microglia activation via reduction of inflammation.
Compared to astrocytes and microglia, little is known about insulin action on oligodendrocytes, but clinical clues are pointing to their potential involvement. In patients with multiple system atrophy (MSA), a neurodegenerative disorder characterized by α-synuclein aggregation in oligodendrocytes, there is increased phosphorylation of IRS-1 at serine 312, a site associated with insulin resistance [72]. Studies also suggest that both neurons and oligodendrocytes, but not microglia and astrocytes, are insulin resistant in MSA, implicating the potential for differentially regulated insulin signaling in different brain cell types [73]. Further studies are needed to determine the role of insulin signaling in oligodendrocyte function and survival.
Brain vascular endothelial cells (BVECs) also express InsR (Figure 3). Conditional InsR KO in vascular endothelium delays the onset of insulin signaling in brain areas such as the hippocampus, certain areas of the hypothalamus, and prefrontal cortex (Figure 4) [74]. In addition, loss of endothelial InsR impairs blood-brain barrier (BBB) function and causes systemic insulin resistance and mild obesity [74]. While early studies showed that insulin enters the brain via InsR-mediated transcytosis across BVECs [75], recent studies suggest that insulin can also be transported across BVECs via InsR-independent routes [76, 77]. While these alternative routes remain to be revealed, identification of insulin transport-related protein(s) [77] may provide crucial insights into understanding how brain insulin signaling contributes to the pathogenesis of brain disorders.
Figure 4.
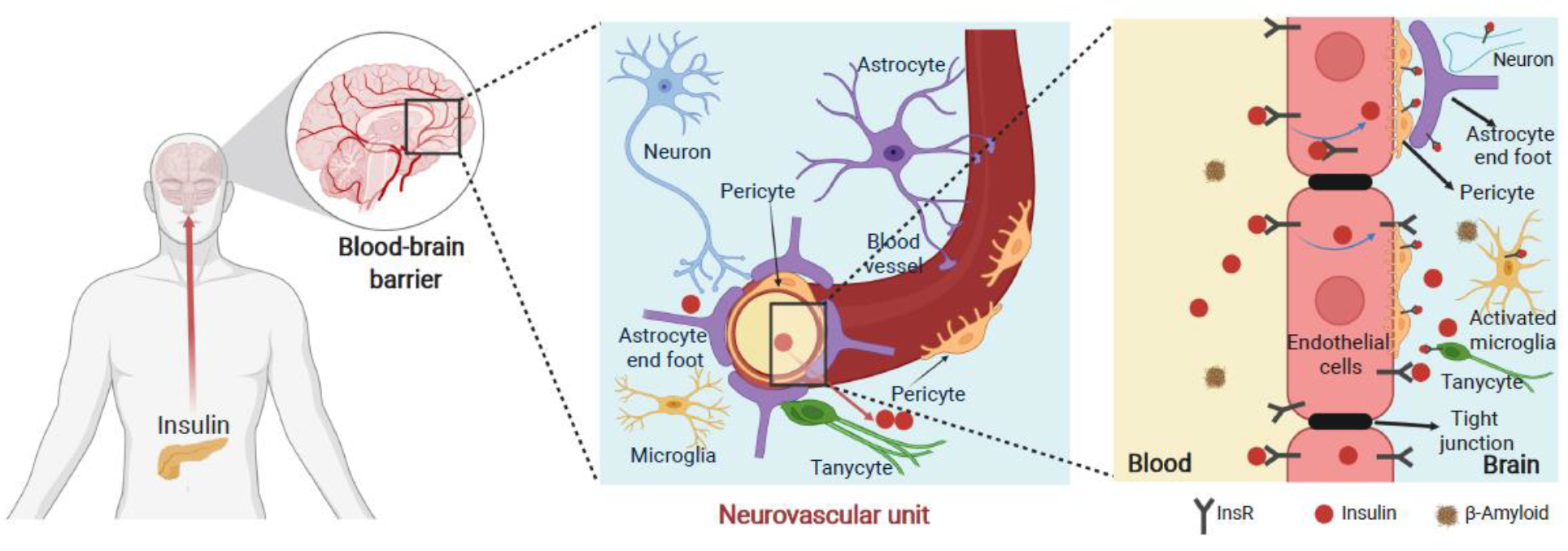
Insulin signaling in brain vascular endothelial system. The blood-brain barrier (BBB) is a highly selective barrier that dynamically maintains a stable milieu for the brain. Endothelial InsR regulates transendothelial insulin delivery thus differentially modulating the kinetics of insulin signaling across brain regions. Altered BBB function plays a role in brain insulin resistance and contributes to the comorbidity of T2D and AD. Blue lines within the endothelial cells (in the right panel) denote the direction of insulin transport across the brain capillary. Created with BioRender.com.
Recently, it has been shown that tanycytes, a form of non-neuronal cells that share some features with astrocytes and have processes extending into the hypothalamus, may play an important role in both transcytosis of peripheral hormones to the arcuate nucleus (ARC) and in nutrient sensing and regulation of energy homeostasis (Figure 4) [78, 79]. Comparison of mice with InsR deletion in endothelial cells versus tanycytes showed that InsR in tanycytes maybe rate limiting in regulation of insulin transport into the ARC [79]. This study also suggested that insulin action in tanycytes has an important role in gating systemic insulin sensitivity, ghrelin action and AgRP neuronal activity [79].
In addition to insulin’s actions in the periphery and in the brain, it remains debated whether some cells in the brain may actually produce insulin, at least under some conditions. Insulin-like peptides are produced in Drosophila brain [80], and comparison of mice with conditional targeting of Ins1 versus Ins2 gene have suggested that Ins2 (which is expressed in the brain during development) may have a unique role in brain [81–83]. A recent study showed that insulin can also be released from the choroid plexus (ChP), and this release is modulated by serotonin, but not glucose [84]. This has been further supported by single-cell RNA-sequencing (scRNA-seq) showing expression of insulin in the ChP [85]. Future studies should further investigate whether serotonergic antidepressants modulate insulin release from the ChP (see Outstanding questions). For further reading on insulin signaling in BVECs, we refer readers to references [86, 87].
Insulin Action in Brain Circuits
The mesolimbic dopaminergic pathway projects from the ventral tegmental area (VTA) to the NAc and plays a key role in circuits involving (among other brain regions) the hippocampus and amygdala, and that are critical to the regulation of mood, cognition, and reward-based behaviors. Insulin can modulate mesolimbic circuitry to alter feeding and reward behaviors by acting at the NAc and VTA, key brain regions in regulation of motivational salience and emotional valence. Direct infusion of insulin into the VTA decreases food ingestion [88], whereas loss of InsR in VTA dopaminergic neurons leads to hyperphagia and obesity [89], consistent with a role for insulin in satiety. Insulin modulates the excitability of VTA dopaminergic neurons by two mechanisms. First, insulin elicits long-term depression (LTD) of the presynaptic glutamatergic inputs to VTA dopaminergic neurons [90]. Second, insulin enhances spontaneous tonic activity of dopaminergic neurons in a cell-autonomous fashion [89]. The net effect of insulin on dopaminergic neuronal activity represents an integration of the negative presynaptic and positive cell-autonomous effects (Figure 5). Complicating the picture, insulin, acting via the PI3K-Akt pathway, also promotes the surface expression of the DA transporter [91], which counterbalances the increased DA release.
Figure 5.

Insulin regulates key brain functions through multiple mechanisms involving different cell types and neural circuits. Insulin not only regulates functions of various brain cell types, including neurons, astrocytes, microglia, and tanycytes, but also modulates key brain pathways such as the mesolimbic dopaminergic pathway and hypothalamic circuits. In addition, insulin modulates global brain processes such as synaptic transmission and brain metabolism. Dysregulated insulin signaling in the brain contributes to brain disorders such as depression and AD. Created with BioRender.com.
Abbreviations: AgRP/NPY, agouti-related neuropeptide/neuropeptide Y; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; ATP, adenosine triphosphate; GABA, γ-aminobutyric acid; GSK-3β, glycogen synthase kinase 3β; NMDA, N-Methyl-d-aspartic acid; POMC, proopiomelanocortin.
Insulin modulates reward behaviors by acting on the mesolimbic dopaminergic circuitry in the NAc (Figure 5), a primary target of VTA dopaminergic neurons. Insulin indirectly enhances evoked DA release in both dorsal striatum and the NAc by increasing the excitability of the cholinergic interneurons and enhancing astrocyte-derived ATP release [7, 92]. Thus, in NAc, insulin indirectly potentiates DA release through astrocyte-neuron and interneuron-neuron communication. Functionally, the potentiation of DA release in NAc by insulin is important for reward valuation, and ablating insulin action in NAc leads to impairment of food preference and development of anhedonic behavior [7]. In line with this, insulin resistant obese animals lose insulin-enhanced DA release in NAc [92], which contributes to the anhedonic behavior partly resulting from deficient reward processing. The mechanism by which impairment of insulin action in NAc contributes to depressive symptoms, however, has yet to be fully elucidated (see below discussion and Outstanding questions).
Insulin, in addition to leptin and ghrelin, is a primary modulator of an important hypothalamic circuit projecting from the ARC to the paraventricular nucleus (PVN) involved in regulation of food intake and satiety. As noted above, insulin binds to its receptors on both POMC and AgRP neurons in the ARC. These cells project to PVN neurons that express melanocortin 4 receptors (MC4R+). POMC and AgRP are released and competitively bind to MC4R with POMC potentiating downstream firing of these neurons, while AgRP inhibits firing. This competitive action results in POMC neurons suppressing appetite while AgRP neurons promote hunger and eating behavior (Figure 2). Insulin inhibits GABA release to AgRP neurons and hence alleviates the inhibitory constraint on POMC neurons [52, 93–95], resulting in reduced food intake. Electrophysiological recording of glucose-inhibited hypothalamic neurons from brain slices of NIRKO mice show a significant reduction in membrane potential and input resistance in response to a decrease of glucose concentration [43], i.e., impaired glucose sensing. The exact links between these changes in glucose sensing and altered InsR signaling remain to be determined.
The roles of gut microbiota in modulating mood and behavior, as well as brain insulin sensitivity [66], have been receiving growing attention, and represent one of the rapidly advancing areas in the field of brain-metabolism interactions. Modification of gut microbiota, due to high-fat diet feeding, is associated with altered insulin signaling and inflammation in key brain reward processing centers such as the NAc and amygdala [66]. Concordantly, gut microbiome transfer between mice on high-fat diet and high-fat diet plus antibiotics results in changes in many basic behaviors in recipient germ-free mice [66]. Further investigations should help reveal key metabolites or other mediators involved in modulating insulin sensitivity through the gut-brain axis.
Insulin Action on Synaptic Transmission
Beyond modulation of distinct circuits, insulin can modulate global brain processes. In mice, increases of circulating insulin levels within a physiological range under conditions when glucose levels are clamped elicits robust transcriptional regulation of genes in brain, especially in hypothalamus, where the number of transcriptional changes exceeds those in classic insulin target tissues, such as liver and muscle [96]. Many of the regulated genes are involved in neurotransmission (Figure 5). Insulin regulates synaptic plasticity by upregulating GABAA receptor subunits and SNARE proteins responsible for neurotransmitter release, while altering expression of different glutamate receptor subunits (Figure 5). In addition, in hippocampus, insulin not only enhances phosphorylation of NMDA receptor subunits and promotes NMDA receptor membrane localization [97, 98], but also stimulates GluR1-containing AMPA receptor surface expression [99]. With chronic loss of insulin/IGF-1 signaling in hippocampus, a dramatic reduction of GluR1 expression is observed [100]. Insulin also stimulates endocytosis of GluR2-containing AMPA receptors [101] and triggers internalization of AMPA receptors in hippocampus [102], contributing to induction of LTD. Thus, through transcriptional and post-translational regulation affecting NMDA and AMPA receptors, insulin modulates hippocampal long-term plasticity, a cellular component of learning and memory (Figure 5). Consistent with this, hippocampal deletion of InsR results in impairment of spatial and contextual learning [100, 103]. To add to this picture, a recent study demonstrated that insulin produces bidirectional effects on synaptic transmission in NAc involving a disinhibition mechanism mediated by opioid receptor [104]. These studies open an avenue for future studies of brain insulin dysfunction in cognitive deficits and mood disturbance. In fact, restoration of brain insulin signaling via intranasal insulin treatment has been suggested as a potential treatment for cognitive decline [105] and mood disorders [106] (see discussion below).
Insulin Action on Global Brain Metabolism
The brain has high energy demands and relies on glucose as its main fuel. In addition, the brain is the most cholesterol-rich organ in the body, containing approximately 25% of total body cholesterol. In brain, cholesterol is important not only in myelin formation, but in neuronal membrane function. Since cholesterol-containing lipoproteins in the blood cannot cross the BBB, the brain relies on local production of cholesterol for myelination and synaptic formation and remodeling. Peripheral infusion of insulin under euglycemic conditions in mice induces expression of genes involved in cholesterol and long-chain fatty acid biosynthesis while suppressing expression of many genes involved in glycolysis and the pentose phosphate pathway [96]. The overall effect is to reroute carbon substrates from glucose metabolism to lipid metabolism. Thus, although insulin does not regulate brain glucose uptake, insulin can regulate substrate flux and brain metabolism, which affects many neural functions including cognition (Figure 5). Consistent with this, in streptozotocin-induced diabetic mice with low circulating insulin levels, genes involved in cholesterol biosynthesis in brain are downregulated, and brain cholesterol synthesis is low [107]. Likewise, in the hypothalamus of NIRKO mice, there is downregulation of SREBP2, the master regulator of cholesterol metabolism [107]. Reduced brain cholesterol synthesis impairs LTP and contextual learning in mice [108], which in the context of the other findings discussed earlier indicates that the anabolic effect of insulin on brain cholesterol synthesis is necessary for normal memory formation.
Mitochondria are central regulators of cellular metabolism and energetics. Mitochondrial dysfunction in multiple peripheral tissues has been associated with diabetes and insulin resistance [109]. It is therefore not surprising that, in brain, insulin can regulate mitochondrial biogenesis, morphology, and function, which in turn can modulate metabolism and higher-order brain functions and cognition (Figure 5). Insulin also mediates the innate stress-response machinery in mitochondria by regulating chaperone proteins, including Hsp60 and Hsp10 [89, 90]. Thus, in NIRKO mice, there are fewer and smaller mitochondria in brain and decreased expression of mitochondrial oxidative phosphorylation complexes [6]. In addition, normal insulin signaling prevents mitochondrial swelling and accumulation of reactive oxygen species in hippocampus [110]. Intranasal insulin treatment increases mitochondrial ATP production in heathy mouse brain, demonstrating that insulin direct regulating brain mitochondrial function [111]. However, increased mitochondrial ATP release could also increase oxidative stress, which might contribute to AD (recently reviewed in [112]). Given the vulnerability of neurons to oxidative stress, a question that requires further investigation is how mitochondrial dysfunction related to abnormal insulin signaling contributes to neurodegenerative disorders.
Cell Type-Specific Regulation of IGF-1 Receptor Signaling in the Brain
Insulin-like growth factors (IGFs) bind with high affinity to the IGF1 receptor (IGF1R) on target cells and modulate multiple processes, including cell growth and differentiation [113]. Insulin and IGF-1 receptors are expressed in overlapping neuroanatomical patterns, but at different levels in different regions [45]. Despite many similarities in signaling, these two receptors have preferential pathways of signaling and exert distinct downstream biological effects [114]. Both ligands can also cross-react with each other’s receptors, albeit with lower affinity.
The neuroprotective effects of IGF-1 signaling have been studied for decades [115]. In mouse models of neurodegeneration, IGF-1 administration has been shown to improve the neurologic phenotype [116–118], suggesting that IGF-1 might serve as a treatment for these disorders. Conversely, dysfunction of IGF-1 signaling leads to poor outcomes after traumatic brain injury [119, 120]. Not all effects of IGF-1, however, are positive. For instance, a recent study found that microglia-produced IGF-1 may serve as a tumor-supporting factor [121]. Thus, a balance of homeostasis of IGF-1 signaling is essential.
In contrast to insulin which is almost exclusively produced in pancreatic β-cells, IGF-1 is produced by many cell types. scRNA-seq has revealed that IGF-1 expression is enriched in specific subsets of inhibitory neurons [122], and this might form a bridge between neuronal activity and IGF-1-related cognitive function [123]. While IGF1Rs are present on both neurons and glial cells, IGF-1 signaling in astrocytes appears largely responsible for its neuroprotective role. Consistent with this, mice with an astrocyte-specific IGF1R KO have impaired hippocampal-dependent learning [124]. In addition, studies have shown that loss of IGF-1 signaling in astrocytes impairs the astrocytic protection of neurons during oxidative stress in a human astrocyte-neuron co-culture system [125] and in mice subjected to ischemic brain injury [126].
Microglia-derived IGF-1 also plays a role in protection of neurons from damage and apoptosis. In contrast to astrocytes, microglial IGF-1 signaling mainly protects from focal ischemic injury [127, 128]. Following ischemic brain injury, Igf1 expression is increased in reactive microglia, but not in astrocytes [127], indicating a cell-type-specific response. Thus, insulin/IGF-1 action in different glia cell types play distinct roles in the CNS response to injury.
New Technologies to Probe Insulin and IGF-1 Signaling in the Brain
New technologies are enabling a better characterization of individual brain cell types and may help elucidate the roles of insulin/IGF-1 signaling in the brain. As discussed above, distinct VTA dopaminergic subpopulations exhibit different responses to insulin. Thus, insulin increases the spontaneous firing rates of only ~50% of midbrain dopaminergic neurons [89]. Going forward, technologies such as scRNA-seq should help identify these phenotypically distinct neuronal subsets and help better understand the CNS response to insulin and IGF-1. Additionally, in vivo Ca2+ imaging via a fiber or lens implanted in the brain enables simultaneous recording of activity of hundreds of brain cells while animals are engaged in behavioral tasks [129]. A recent study has applied a fiber photometry-based Ca2+ imaging in freely moving mice with tanycytic InsR deletion and discovered a role of insulin signaling in tanycytes in regulation of AgRP neuronal activity [79]. Although it is clear that fiber photometry lacks single-cell resolution, this has already offered significant insights into tanycyte’s regulatory effects upon application of stimuli such as gut-secreted hormones and feeding. Future studies should also investigate the dynamic neuronal response in real-time to different metabolic states, such as insulin resistance and diabetes (see Outstanding Questions).
A Translational View
While translational studies are limited, preliminary clinical trials in healthy subjects [130] and small trialsi,ii in AD patients [131–133] suggested that intranasal insulin therapy might produce modest cognitive improvement. In AD, patients without the Apoe4 risk allele showed a more profound response to intranasal insulin than patients carrying Apoe4 [134]. However, results from the larger multicenter trial have been mixed. While one clinical trialiii found no cognitive benefits of 12-month intranasal insulin treatment [135], a recent follow-up analysis of brain imaging of the study participants demonstrated a reduction in white matter hyperintensity with insulin therapy [136]. Intranasal insulin treatment of patients with mood disorders has also shown mixed results. An early clinical trialiv found a significant improvement of neurocognitive performance in bipolar patients [106], however, the same research group failed to demonstrate beneficial effects of intranasal insulin on overall mood or neurocognition in patients with major depressive disordersv [137]. Interestingly, animal studies and pilot clinical trials of other antidiabetic medications, including liraglutide (a GLP-1 analogue)vi and pioglitazonevii,viii, have shown promising effects in AD [138, 139], but this effect was not observed with rosiglitazoneix,x, another thiazolidinedione [140].
Investigations of the basic mechanisms of insulin action in the brain have also raised the potential of identifying new therapeutic targets. Cholesterol synthesis provides a promising example. Thus, acute insulin signaling regulates de novo cholesterol synthesis in neurons and glia, whereas chronic hyperinsulinemia in diabetes and insulin resistance leads to brain insulin resistance and a loss of the normal dynamic regulation of brain cholesterol production. This could contribute to the acceleration of AD progression, possibly through altered apoE metabolism, which is strongly genetically linked to development of AD [141]. Further advances to our understanding of brain insulin and IGF-1 signaling should help pinpoint the precise site where insulin acts on different subtypes of astrocytes and neurons in regulating cholesterol and long-chain fatty acid biosynthesis and understand how these might contribute to the pathogenesis of brain disorders such as depression and AD (see Outstanding Questions). These results raise hope for new therapeutic approaches to brain disorders centered around insulin signaling and metabolism, but also underscore the need for better understanding of the multiple roles of these pathways in brain.
Concluding Remarks
Insulin signaling regulates a broad range of physiological processes at the systemic level by modulating substrate uptake and metabolism in classic target organs - liver, fat and muscle. Insulin also plays important roles in brain, modulating both neurons and glial cells through changes in gene expression and cell function at multiple levels. Accumulating evidence demonstrates that dysregulated insulin signaling plays a role in psychiatric disorders and neurodegenerative diseases. In animal models, manipulation of brain InsR or IGF1R using transgenic approaches, as well as regional manipulation, has increased our understanding of insulin action and its role in brain functions. However, the brain has an incomparable level of cellular heterogeneity, and a deep cell-type-specific understanding of insulin action remains an important goal for future research. New tools, such as scRNA-seq, optogenetics, chemogenetics, and neuronal tracing techniques, as well as ways to modify insulin action in specific cell populations, should provide deeper insights and better understanding of insulin signaling in the brain and hopefully provide new targets for the treatment of both metabolic and brain disorders.
Highlights.
Insulin is a key hormone in regulation of energy metabolism. Insulin resistance, a state when cells become unresponsive to insulin, is a key feature of type 2 diabetes (T2D), obesity, and many other metabolic disorders.
The brain is an insulin sensitive tissue. Insulin signaling plays a key role in different brain cell types.
Insulin modulates neuronal and glial function, resulting in changes in mood, cognition, and behavior.
Dysfunction in brain insulin signaling underlies comorbidity of T2D and disorders such as depression and Alzheimer’s disease.
Better understanding of brain insulin actions may lead to new therapeutic targets for the treatment of brain disorders.
Acknowledgements
This work was supported by grants from the US National Institutes of Health (R01DK031036 to CRK and K01DK120740, R01MH125903 to WKC). Figures were created with BioRender.com.
Glossary
- Comorbidity
the existence of two or more medical conditions in an individual, which is often associated with worse outcomes
- Cre/LoxP system
a technique enabling precise genetic modification based on the recognition of Cre recombinase of the 34 bp LoxP sites inserted around a gene or exon
- Cre/LoxP system
a technique enabling precise genetic modification based on the recognition of Cre recombinase of the 34 bp LoxP sites inserted around a gene or exo
- Emotional valence
a term broadly defining emotional state as positive or negative
- Glucose homeostasis
the tightly regulated process to maintain the blood glucose within a narrow range
- Insulin-like growth factor 1 receptor (IGF1R)
a closely related tyrosine kinase receptor activated primarily by IGF-1 and IGF-2, and to a lesser extent by insulin
- Insulin receptor (InsR)
a member of the class of tyrosine kinase receptors activated primarily by insulin, and to a lesser extent by IGF-1 and IGF-2. The InsR plays a key role in maintaining glucose homeostasis
- Insulin resistance
a state when cells become unresponsive to insulin. In individuals in which hyperinsulinemia cannot overcome the insulin resistance, the result is elevated level of blood glucose
- Motivational salience
a cognitive process regulating behavior to pursue a reward (incentive salience) or to avoid a punishment (aversive salience)
- Synaptic plasticity
the ability of synapses to modify the strength, efficacy, or target of synaptic transmission
- Type 1 diabetes
a chronic autoimmune condition in which the β cells in the pancreas are destroyed resulting in low insulin levels. While historically this was viewed as a form of diabetes which appeared in childhood, it is now clear that it can develop at any age
- Type 2 diabetes (T2D)
a chronic condition in which body cells do not respond appropriately to insulin; as a result, glucose remains in the blood and cannot be utilized as fuel. Circulating insulin levels may be high, low, or normal
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
CRK is a consultant to Kaleido Biosciences, Sana Biotechnology, ERX Pharmaceuticals, and CohBar, but none of these are related to the contents of this manuscript. The other authors declare no conflicts of interests in relation to this work.
References
- 1.Cho NH et al. (2018) IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res Clin Pract 138, 271–281. [DOI] [PubMed] [Google Scholar]
- 2.Rask-Madsen C and Kahn CR (2012) Tissue-specific insulin signaling, metabolic syndrome, and cardiovascular disease. Arterioscler Thromb Vasc Biol 32 (9), 2052–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kahn CR et al. (2019) Altered adipose tissue and adipocyte function in the pathogenesis of metabolic syndrome. J Clin Invest 129 (10), 3990–4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boucher J et al. (2014) Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb Perspect Biol 6 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bruning JC et al. (2000) Role of brain insulin receptor in control of body weight and reproduction. Science 289 (5487), 2122–5. [DOI] [PubMed] [Google Scholar]
- 6.Kleinridders A et al. (2015) Insulin resistance in brain alters dopamine turnover and causes behavioral disorders. Proc Natl Acad Sci U S A 112 (11), 3463–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cai W et al. (2018) Insulin regulates astrocyte gliotransmission and modulates behavior. J Clin Invest 128 (7), 2914–2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.American Diabetes A (2019) 4. Comprehensive Medical Evaluation and Assessment of Comorbidities: Standards of Medical Care in Diabetes-2019. Diabetes Care 42 (Suppl 1), S34–S45. [DOI] [PubMed] [Google Scholar]
- 9.Johnson B et al. (2013) Prevalence of depression among young people with Type 1 diabetes: a systematic review. Diabet Med 30 (2), 199–208. [DOI] [PubMed] [Google Scholar]
- 10.Anderson RJ et al. (2001) The prevalence of comorbid depression in adults with diabetes: a meta-analysis. Diabetes Care 24 (6), 1069–78. [DOI] [PubMed] [Google Scholar]
- 11.Kan C et al. (2013) A systematic review and meta-analysis of the association between depression and insulin resistance. Diabetes Care 36 (2), 480–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nouwen A et al. (2010) Type 2 diabetes mellitus as a risk factor for the onset of depression: a systematic review and meta-analysis. Diabetologia 53 (12), 2480–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peyrot M and Rubin RR (1999) Persistence of depressive symptoms in diabetic adults. Diabetes Care 22 (3), 448–52. [DOI] [PubMed] [Google Scholar]
- 14.Mezuk B et al. (2008) Depression and type 2 diabetes over the lifespan: a meta-analysis. Diabetes Care 31 (12), 2383–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Groot M et al. (2001) Association of depression and diabetes complications: a meta-analysis. Psychosom Med 63 (4), 619–30. [DOI] [PubMed] [Google Scholar]
- 16.Pan A et al. (2012) Bidirectional association between depression and metabolic syndrome: a systematic review and meta-analysis of epidemiological studies. Diabetes Care 35 (5), 1171–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khoza S et al. (2012) Use of antidepressant agents and the risk of type 2 diabetes. Eur J Clin Pharmacol 68 (9), 1295–302. [DOI] [PubMed] [Google Scholar]
- 18.Brieler JA et al. (2016) Antidepressant medication use and glycaemic control in co-morbid type 2 diabetes and depression. Fam Pract 33 (1), 30–6. [DOI] [PubMed] [Google Scholar]
- 19.McIntyre RS et al. (2006) The effect of antidepressants on glucose homeostasis and insulin sensitivity: synthesis and mechanisms. Expert Opin Drug Saf 5 (1), 157–68. [DOI] [PubMed] [Google Scholar]
- 20.Nicol GE et al. (2018) Metabolic Effects of Antipsychotics on Adiposity and Insulin Sensitivity in Youths: A Randomized Clinical Trial. JAMA Psychiatry 75 (8), 788–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith KJ et al. (2013) Association of diabetes with anxiety: a systematic review and meta-analysis. J Psychosom Res 74 (2), 89–99. [DOI] [PubMed] [Google Scholar]
- 22.Lin EH et al. (2008) Mental disorders among persons with diabetes--results from the World Mental Health Surveys. J Psychosom Res 65 (6), 571–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith KJ et al. (2018) Investigating the longitudinal association between diabetes and anxiety: a systematic review and meta-analysis. Diabet Med 35 (6), 677–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lustman PJ et al. (1995) Effects of alprazolam on glucose regulation in diabetes. Results of double-blind, placebo-controlled trial. Diabetes Care 18 (8), 1133–9. [DOI] [PubMed] [Google Scholar]
- 25.Kasanin J (1926) The blood sugar curve in mental disease: II. The schizophrenic (dementia praecox) groups. Archives of Neurology & Psychiatry 16 (4), 414–419. [Google Scholar]
- 26.Mitchell AJ et al. (2013) Prevalence of metabolic syndrome and metabolic abnormalities in schizophrenia and related disorders--a systematic review and meta-analysis. Schizophr Bull 39 (2), 306–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kessing LV et al. (2010) Treatment with antipsychotics and the risk of diabetes in clinical practice. Br J Psychiatry 197 (4), 266–71. [DOI] [PubMed] [Google Scholar]
- 28.Freeman H, Looney JM, Hoskins RG, & Dyer CG (1943) Results of insulin and epinephrine tolerance tests in schizophrenic patients and in normal subjects. Archives of Neurology & Psychiatry 49 (2), 195–203. [Google Scholar]
- 29.van Nimwegen LJ et al. (2008) Hepatic insulin resistance in antipsychotic naive schizophrenic patients: stable isotope studies of glucose metabolism. J Clin Endocrinol Metab 93 (2), 572–7. [DOI] [PubMed] [Google Scholar]
- 30.Girgis RR et al. (2008) Antipsychotic drug mechanisms: links between therapeutic effects, metabolic side effects and the insulin signaling pathway. Mol Psychiatry 13 (10), 918–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arnold SE et al. (2018) Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat Rev Neurol 14 (3), 168–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cooper C et al. (2015) Modifiable predictors of dementia in mild cognitive impairment: a systematic review and meta-analysis. Am J Psychiatry 172 (4), 323–34. [DOI] [PubMed] [Google Scholar]
- 33.Gudala K et al. (2013) Diabetes mellitus and risk of dementia: A meta-analysis of prospective observational studies. J Diabetes Investig 4 (6), 640–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang J et al. (2017) An updated meta-analysis of cohort studies: Diabetes and risk of Alzheimer’s disease. Diabetes Res Clin Pract 124, 41–47. [DOI] [PubMed] [Google Scholar]
- 35.Frolich L et al. (1998) Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J Neural Transm (Vienna) 105 (4–5), 423–38. [DOI] [PubMed] [Google Scholar]
- 36.Talbot K et al. (2012) Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest 122 (4), 1316–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moloney AM et al. (2010) Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol Aging 31 (2), 224–43. [DOI] [PubMed] [Google Scholar]
- 38.Tramutola A et al. (2015) Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD. J Neurochem 133 (5), 739–49. [DOI] [PubMed] [Google Scholar]
- 39.Steen E et al. (2005) Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease--is this type 3 diabetes? J Alzheimers Dis 7 (1), 63–80. [DOI] [PubMed] [Google Scholar]
- 40.Craft S et al. (1998) Cerebrospinal fluid and plasma insulin levels in Alzheimer’s disease: relationship to severity of dementia and apolipoprotein E genotype. Neurology 50 (1), 164–8. [DOI] [PubMed] [Google Scholar]
- 41.Goldstein BJ et al. (1987) Variation in insulin receptor messenger ribonucleic acid expression in human and rodent tissues. Mol Endocrinol 1 (11), 759–66. [DOI] [PubMed] [Google Scholar]
- 42.Schubert M et al. (2004) Role for neuronal insulin resistance in neurodegenerative diseases. Proc Natl Acad Sci U S A 101 (9), 3100–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Diggs-Andrews KA et al. (2010) Brain insulin action regulates hypothalamic glucose sensing and the counterregulatory response to hypoglycemia. Diabetes 59 (9), 2271–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fisher SJ et al. (2005) Insulin signaling in the central nervous system is critical for the normal sympathoadrenal response to hypoglycemia. Diabetes 54 (5), 1447–51. [DOI] [PubMed] [Google Scholar]
- 45.Kleinridders A et al. (2014) Insulin action in brain regulates systemic metabolism and brain function. Diabetes 63 (7), 2232–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lam BYH et al. (2017) Heterogeneity of hypothalamic pro-opiomelanocortin-expressing neurons revealed by single-cell RNA sequencing. Mol Metab 6 (5), 383–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin HV et al. (2010) Divergent regulation of energy expenditure and hepatic glucose production by insulin receptor in agouti-related protein and POMC neurons. Diabetes 59 (2), 337–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shin AC et al. (2017) Insulin Receptor Signaling in POMC, but Not AgRP, Neurons Controls Adipose Tissue Insulin Action. Diabetes 66 (6), 1560–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Qiu J et al. (2018) Insulin and leptin excite anorexigenic pro-opiomelanocortin neurones via activation of TRPC5 channels. J Neuroendocrinol 30 (2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qiu J et al. (2014) Insulin excites anorexigenic proopiomelanocortin neurons via activation of canonical transient receptor potential channels. Cell Metab 19 (4), 682–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Williams KW et al. (2010) Segregation of acute leptin and insulin effects in distinct populations of arcuate proopiomelanocortin neurons. J Neurosci 30 (7), 2472–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dodd GT et al. (2018) Insulin regulates POMC neuronal plasticity to control glucose metabolism. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shimada M et al. (1998) Mice lacking melanin-concentrating hormone are hypophagic and lean. Nature 396 (6712), 670–4. [DOI] [PubMed] [Google Scholar]
- 54.Hausen AC et al. (2016) Insulin-Dependent Activation of MCH Neurons Impairs Locomotor Activity and Insulin Sensitivity in Obesity. Cell Rep 17 (10), 2512–2521. [DOI] [PubMed] [Google Scholar]
- 55.Sakurai T et al. (1998) Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell 92 (4), 573–85. [DOI] [PubMed] [Google Scholar]
- 56.Tsuneki H et al. (2013) Hypothalamic orexin prevents hepatic insulin resistance induced by social defeat stress in mice. Neuropeptides 47 (3), 213–9. [DOI] [PubMed] [Google Scholar]
- 57.Inutsuka A et al. (2014) Concurrent and robust regulation of feeding behaviors and metabolism by orexin neurons. Neuropharmacology 85, 451–60. [DOI] [PubMed] [Google Scholar]
- 58.Baldini G and Phelan KD (2019) The melanocortin pathway and control of appetite-progress and therapeutic implications. J Endocrinol 241 (1), R1–R33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Valassi E et al. (2008) Neuroendocrine control of food intake. Nutr Metab Cardiovasc Dis 18 (2), 158–68. [DOI] [PubMed] [Google Scholar]
- 60.Belgardt BF et al. (2009) Hormone and glucose signalling in POMC and AgRP neurons. J Physiol 587 (Pt 22), 5305–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Khakh BS and Deneen B (2019) The Emerging Nature of Astrocyte Diversity. Annu Rev Neurosci 42, 187–207. [DOI] [PubMed] [Google Scholar]
- 62.Garcia-Caceres C et al. (2016) Astrocytic Insulin Signaling Couples Brain Glucose Uptake with Nutrient Availability. Cell 166 (4), 867–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Srinivasan R et al. (2016) New Transgenic Mouse Lines for Selectively Targeting Astrocytes and Studying Calcium Signals in Astrocyte Processes In Situ and In Vivo. Neuron 92 (6), 1181–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Habib N et al. (2020) Disease-associated astrocytes in Alzheimer’s disease and aging. Nat Neurosci 23 (6), 701–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Saltiel AR and Olefsky JM (2017) Inflammatory mechanisms linking obesity and metabolic disease. J Clin Invest 127 (1), 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Soto M et al. (2018) Gut microbiota modulate neurobehavior through changes in brain insulin sensitivity and metabolism. Mol Psychiatry 23 (12), 2287–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kong Y et al. (2010) Norepinephrine promotes microglia to uptake and degrade amyloid beta peptide through upregulation of mouse formyl peptide receptor 2 and induction of insulin-degrading enzyme. J Neurosci 30 (35), 11848–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen Y et al. (2014) Intranasal insulin restores insulin signaling, increases synaptic proteins, and reduces Abeta level and microglia activation in the brains of 3xTg-AD mice. Exp Neurol 261, 610–9. [DOI] [PubMed] [Google Scholar]
- 69.Rhea EM et al. (2019) Molecular Mechanisms of Intranasal Insulin in SAMP8 Mice. J Alzheimers Dis 71 (4), 1361–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Frazier HN et al. (2020) Long-Term Intranasal Insulin Aspart: A Profile of Gene Expression, Memory, and Insulin Receptors in Aged F344 Rats. J Gerontol A Biol Sci Med Sci 75 (6), 1021–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gad ES et al. (2016) Pioglitazone and exenatide enhance cognition and downregulate hippocampal beta amyloid oligomer and microglia expression in insulin-resistant rats. Can J Physiol Pharmacol 94 (8), 819–28. [DOI] [PubMed] [Google Scholar]
- 72.Bassil F et al. (2017) Insulin resistance and exendin-4 treatment for multiple system atrophy. Brain 140 (5), 1420–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.O’Grady JP et al. (2019) Elevated Insulin and Insulin Resistance are Associated with Altered Myelin in Cognitively Unimpaired Middle-Aged Adults. Obesity (Silver Spring) 27 (9), 1464–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Konishi M et al. (2017) Endothelial insulin receptors differentially control insulin signaling kinetics in peripheral tissues and brain of mice. Proc Natl Acad Sci U S A 114 (40), E8478–E8487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.King GL and Johnson SM (1985) Receptor-mediated transport of insulin across endothelial cells. Science 227 (4694), 1583–6. [DOI] [PubMed] [Google Scholar]
- 76.Hersom M et al. (2018) The insulin receptor is expressed and functional in cultured blood-brain barrier endothelial cells but does not mediate insulin entry from blood to brain. Am J Physiol Endocrinol Metab 315 (4), E531–E542. [DOI] [PubMed] [Google Scholar]
- 77.Rhea EM et al. (2018) Insulin transport across the blood-brain barrier can occur independently of the insulin receptor. J Physiol 596 (19), 4753–4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Elizondo-Vega RJ et al. (2019) Nutrient Sensing by Hypothalamic Tanycytes. Front Endocrinol (Lausanne) 10, 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Porniece Kumar M et al. (2021) Insulin signalling in tanycytes gates hypothalamic insulin uptake and regulation of AgRP neuron activity. Nat Metab 3 (12), 1662–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Broughton SJ et al. (2005) Longer lifespan, altered metabolism, and stress resistance in Drosophila from ablation of cells making insulin-like ligands. Proc Natl Acad Sci U S A 102 (8), 3105–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Song J et al. (2010) Brain expression of Cre recombinase driven by pancreas-specific promoters. Genesis 48 (11), 628–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wicksteed B et al. (2010) Conditional gene targeting in mouse pancreatic ss-Cells: analysis of ectopic Cre transgene expression in the brain. Diabetes 59 (12), 3090–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tamarina NA et al. (2014) Characterization of mice expressing Ins1 gene promoter driven CreERT recombinase for conditional gene deletion in pancreatic beta-cells. Islets 6 (1), e27685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mazucanti CH et al. (2019) Release of insulin produced by the choroid plexis is regulated by serotonergic signaling. JCI Insight 4 (23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dani N et al. (2021) A cellular and spatial map of the choroid plexus across brain ventricles and ages. Cell 184 (11), 3056–3074 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rhea EM and Banks WA (2019) Role of the Blood-Brain Barrier in Central Nervous System Insulin Resistance. Front Neurosci 13, 521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rhea EM and Banks WA (2021) A historical perspective on the interactions of insulin at the blood-brain barrier. J Neuroendocrinol 33 (4), e12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu S et al. (2016) Consumption of palatable food primes food approach behavior by rapidly increasing synaptic density in the VTA. Proc Natl Acad Sci U S A 113 (9), 2520–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Konner AC et al. (2011) Role for insulin signaling in catecholaminergic neurons in control of energy homeostasis. Cell Metab 13 (6), 720–728. [DOI] [PubMed] [Google Scholar]
- 90.Labouebe G et al. (2013) Insulin induces long-term depression of ventral tegmental area dopamine neurons via endocannabinoids. Nat Neurosci 16 (3), 300–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Carvelli L et al. (2002) PI 3-kinase regulation of dopamine uptake. J Neurochem 81 (4), 859–69. [DOI] [PubMed] [Google Scholar]
- 92.Stouffer MA et al. (2015) Insulin enhances striatal dopamine release by activating cholinergic interneurons and thereby signals reward. Nat Commun 6, 8543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rau AR and Hentges ST (2019) GABAergic Inputs to POMC Neurons Originating from the Dorsomedial Hypothalamus Are Regulated by Energy State. J Neurosci 39 (33), 6449–6459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Aponte Y et al. (2011) AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. Nat Neurosci 14 (3), 351–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen Y et al. (2015) Sensory detection of food rapidly modulates arcuate feeding circuits. Cell 160 (5), 829–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cai W et al. (2021) Peripheral Insulin Regulates a Broad Network of Gene Expression in Hypothalamus, Hippocampus, and Nucleus Accumbens. Diabetes 70 (8), 1857–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Skeberdis VA et al. (2001) Insulin promotes rapid delivery of N-methyl-D- aspartate receptors to the cell surface by exocytosis. Proc Natl Acad Sci U S A 98 (6), 3561–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Christie JM et al. (1999) Insulin causes a transient tyrosine phosphorylation of NR2A and NR2B NMDA receptor subunits in rat hippocampus. J Neurochem 72 (4), 1523–8. [DOI] [PubMed] [Google Scholar]
- 99.Passafaro M et al. (2001) Subunit-specific temporal and spatial patterns of AMPA receptor exocytosis in hippocampal neurons. Nat Neurosci 4 (9), 917–26. [DOI] [PubMed] [Google Scholar]
- 100.Soto M et al. (2019) Insulin signaling in the hippocampus and amygdala regulates metabolism and neurobehavior. Proc Natl Acad Sci U S A 116 (13), 6379–6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Man HY et al. (2000) Regulation of AMPA receptor-mediated synaptic transmission by clathrin-dependent receptor internalization. Neuron 25 (3), 649–62. [DOI] [PubMed] [Google Scholar]
- 102.Beattie EC et al. (2000) Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat Neurosci 3 (12), 1291–300. [DOI] [PubMed] [Google Scholar]
- 103.Grillo CA et al. (2015) Hippocampal Insulin Resistance Impairs Spatial Learning and Synaptic Plasticity. Diabetes 64 (11), 3927–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fetterly TL et al. (2021) Insulin Bidirectionally Alters NAc Glutamatergic Transmission: Interactions between Insulin Receptor Activation, Endogenous Opioids, and Glutamate Release. J Neurosci 41 (11), 2360–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Erichsen JM et al. (2021) Intranasal insulin and orexins to treat age-related cognitive decline. Physiol Behav 234, 113370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.McIntyre RS et al. (2012) A randomized, double-blind, controlled trial evaluating the effect of intranasal insulin on neurocognitive function in euthymic patients with bipolar disorder. Bipolar Disord 14 (7), 697–706. [DOI] [PubMed] [Google Scholar]
- 107.Suzuki R et al. (2010) Diabetes and insulin in regulation of brain cholesterol metabolism. Cell Metab 12 (6), 567–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Suzuki R et al. (2013) Reduction of the cholesterol sensor SCAP in the brains of mice causes impaired synaptic transmission and altered cognitive function. PLoS Biol 11 (4), e1001532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ruegsegger GN et al. (2018) Altered mitochondrial function in insulin-deficient and insulin-resistant states. J Clin Invest 128 (9), 3671–3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pipatpiboon N et al. (2012) PPARgamma agonist improves neuronal insulin receptor function in hippocampus and brain mitochondria function in rats with insulin resistance induced by long term high-fat diets. Endocrinology 153 (1), 329–38. [DOI] [PubMed] [Google Scholar]
- 111.Ruegsegger GN et al. (2019) Insulin deficiency and intranasal insulin alter brain mitochondrial function: a potential factor for dementia in diabetes. FASEB J 33 (3), 4458–4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Banks WA and Rhea EM (2021) The Blood-Brain Barrier, Oxidative Stress, and Insulin Resistance. Antioxidants (Basel) 10 (11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fernandez AM and Torres-Aleman I (2012) The many faces of insulin-like peptide signalling in the brain. Nat Rev Neurosci 13 (4), 225–39. [DOI] [PubMed] [Google Scholar]
- 114.Cai W et al. (2017) Domain-dependent effects of insulin and IGF-1 receptors on signalling and gene expression. Nat Commun 8, 14892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Linseman DA et al. (2002) Insulin-like growth factor-I blocks Bcl-2 interacting mediator of cell death (Bim) induction and intrinsic death signaling in cerebellar granule neurons. J Neurosci 22 (21), 9287–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tsai LK et al. (2014) Systemic administration of a recombinant AAV1 vector encoding IGF-1 improves disease manifestations in SMA mice. Mol Ther 22 (8), 1450–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Banerjee A et al. (2016) Jointly reduced inhibition and excitation underlies circuit-wide changes in cortical processing in Rett syndrome. Proc Natl Acad Sci U S A 113 (46), E7287–E7296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shcheglovitov A et al. (2013) SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature 503 (7475), 267–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Santi A et al. (2018) Circulating insulin-like growth factor I modulates mood and is a biomarker of vulnerability to stress: from mouse to man. Transl Psychiatry 8 (1), 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Guan J et al. (2014) Cyclic glycine-proline regulates IGF-1 homeostasis by altering the binding of IGFBP-3 to IGF-1. Sci Rep 4, 4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yao M et al. (2020) Astrocytic trans-Differentiation Completes a Multicellular Paracrine Feedback Loop Required for Medulloblastoma Tumor Growth. Cell 180 (3), 502–520 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bhattacherjee A et al. (2019) Cell type-specific transcriptional programs in mouse prefrontal cortex during adolescence and addiction. Nat Commun 10 (1), 4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yap EL and Greenberg ME (2018) Activity-Regulated Transcription: Bridging the Gap between Neural Activity and Behavior. Neuron 100 (2), 330–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Logan S et al. (2018) Insulin-like growth factor receptor signaling regulates working memory, mitochondrial metabolism, and amyloid-beta uptake in astrocytes. Mol Metab 9, 141–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ratcliffe LE et al. (2018) Loss of IGF1R in Human Astrocytes Alters Complex I Activity and Support for Neurons. Neuroscience 390, 46–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Genis L et al. (2014) Astrocytes require insulin-like growth factor I to protect neurons against oxidative injury. F1000Res 3, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Beilharz EJ et al. (1998) Co-ordinated and cellular specific induction of the components of the IGF/IGFBP axis in the rat brain following hypoxic-ischemic injury. Brain Res Mol Brain Res 59 (2), 119–34. [DOI] [PubMed] [Google Scholar]
- 128.Dempsey RJ et al. (2003) Stroke-induced progenitor cell proliferation in adult spontaneously hypertensive rat brain: effect of exogenous IGF-1 and GDNF. J Neurochem 87 (3), 586–97. [DOI] [PubMed] [Google Scholar]
- 129.Parker JG et al. (2018) Diametric neural ensemble dynamics in parkinsonian and dyskinetic states. Nature 557 (7704), 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Reger MA et al. (2008) Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-beta in memory-impaired older adults. J Alzheimers Dis 13 (3), 323–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Stein MS et al. (2011) A randomized controlled trial of high-dose vitamin D2 followed by intranasal insulin in Alzheimer’s disease. J Alzheimers Dis 26 (3), 477–84. [DOI] [PubMed] [Google Scholar]
- 132.Craft S et al. (2012) Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol 69 (1), 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Craft S et al. (2017) Effects of Regular and Long-Acting Insulin on Cognition and Alzheimer’s Disease Biomarkers: A Pilot Clinical Trial. J Alzheimers Dis 57 (4), 1325–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Avgerinos KI et al. (2018) Intranasal insulin in Alzheimer’s dementia or mild cognitive impairment: a systematic review. J Neurol 265 (7), 1497–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Craft S et al. (2020) Safety, Efficacy, and Feasibility of Intranasal Insulin for the Treatment of Mild Cognitive Impairment and Alzheimer Disease Dementia: A Randomized Clinical Trial. JAMA Neurol 77 (9), 1099–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kellar D et al. (2021) Intranasal Insulin Reduces White Matter Hyperintensity Progression in Association with Improvements in Cognition and CSF Biomarker Profiles in Mild Cognitive Impairment and Alzheimer’s Disease. J Prev Alzheimers Dis 8 (3), 240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Cha DS et al. (2017) A randomized, double-blind, placebo-controlled, crossover trial evaluating the effect of intranasal insulin on cognition and mood in individuals with treatment-resistant major depressive disorder. J Affect Disord 210, 57–65. [DOI] [PubMed] [Google Scholar]
- 138.Galimberti D and Scarpini E (2017) Pioglitazone for the treatment of Alzheimer’s disease. Expert Opin Investig Drugs 26 (1), 97–101. [DOI] [PubMed] [Google Scholar]
- 139.Gejl M et al. (2016) In Alzheimer’s Disease, 6-Month Treatment with GLP-1 Analog Prevents Decline of Brain Glucose Metabolism: Randomized, Placebo-Controlled, Double-Blind Clinical Trial. Front Aging Neurosci 8, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gold M et al. (2010) Rosiglitazone monotherapy in mild-to-moderate Alzheimer’s disease: results from a randomized, double-blind, placebo-controlled phase III study. Dement Geriatr Cogn Disord 30 (2), 131–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Husain MA et al. (2021) APOE and Alzheimer’s Disease: From Lipid Transport to Physiopathology and Therapeutics. Front Neurosci 15, 630502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Inoue H et al. (2006) Role of hepatic STAT3 in brain-insulin action on hepatic glucose production. Cell Metab 3 (4), 267–75. [DOI] [PubMed] [Google Scholar]
- 143.Manaserh IH et al. (2019) Ablating astrocyte insulin receptors leads to delayed puberty and hypogonadism in mice. PLoS Biol 17 (3), e3000189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Qiu J et al. (2018) Estradiol Protects Proopiomelanocortin Neurons Against Insulin Resistance. Endocrinology 159 (2), 647–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.He Z et al. (2019) Acute effects of zinc and insulin on arcuate anorexigenic proopiomelanocortin neurons. Br J Pharmacol 176 (5), 725–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hill JW et al. (2010) Direct insulin and leptin action on pro-opiomelanocortin neurons is required for normal glucose homeostasis and fertility. Cell Metab 11 (4), 286–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Jeon JY et al. (2006) MCH−/− mice are resistant to aging-associated increases in body weight and insulin resistance. Diabetes 55 (2), 428–34. [DOI] [PubMed] [Google Scholar]
- 148.Kale AY et al. (2006) Type II glucocorticoid receptor involvement in habituated activation of lateral hypothalamic area orexin-A-immunopositive neurons during recurring insulin-induced hypoglycemia. Neurosci Res 56 (3), 309–13. [DOI] [PubMed] [Google Scholar]
- 149.Moriguchi T et al. (1999) Neurons containing orexin in the lateral hypothalamic area of the adult rat brain are activated by insulin-induced acute hypoglycemia. Neurosci Lett 264 (1–3), 101–4. [DOI] [PubMed] [Google Scholar]
- 150.Park JH et al. (2015) Orexin A regulates plasma insulin and leptin levels in a time-dependent manner following a glucose load in mice. Diabetologia 58 (7), 1542–50. [DOI] [PubMed] [Google Scholar]
- 151.Chong AC et al. (2015) Central insulin signaling modulates hypothalamus-pituitary-adrenal axis responsiveness. Mol Metab 4 (2), 83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Grote CW et al. (2018) Deletion of the insulin receptor in sensory neurons increases pancreatic insulin levels. Exp Neurol 305, 97–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Resources
- i. https://clinicaltrials.gov/ct2/show/NCT00438568 .
- ii. https://clinicaltrials.gov/ct2/show/NCT01595646 .
- iii. https://clinicaltrials.gov/ct2/show/NCT01767909 .
- iv. https://clinicaltrials.gov/ct2/show/NCT00314314 .
- v. https://clinicaltrials.gov/ct2/show/NCT00570050 .
- vi. https://clinicaltrials.gov/ct2/show/NCT01469351 .
- vii. https://clinicaltrials.gov/ct2/show/NCT00982202 .
- viii. https://clinicaltrials.gov/ct2/show/NCT02284906 .
- ix. https://clinicaltrials.gov/ct2/show/NCT00428090 .
- x. https://clinicaltrials.gov/ct2/show/NCT00550420 .