

Abstract
Background:
The blood coagulation factor fibrin(ogen) can modulate inflammation by altering leukocyte activity. Analyses of fibrin(ogen)-mediated proinflammatory activity have largely focused on leukocyte integrin binding activity revealed by conversion of fibrinogen to a stabilized fibrin polymer by blood coagulation enzymes. In addition to coagulation enzymes, fibrinogen is a substrate for tissue transglutaminase-2 (TG2), a widely expressed enzyme that produces unique fibrinogen Aα-γ chain cross-linked products.
Objectives:
We tested the hypothesis that TG2 dependent cross-linking alters the proinflammatory activity of surface-adhered fibrinogen.
Methods:
Mouse bone marrow-derived macrophages (BMDMs) were cultured on tissue culture plates coated with fibrinogen or TG2-cross-linked fibrinogen (10 μg/mL) and then stimulated with lipopolysaccharide (LPS, 1 ng/mL) or vehicle for various times.
Results:
In the absence of LPS stimulation, TG2-cross-linked fibrin(ogen) enhanced inflammatory gene induction (e.g., Tnfα) compared with unmodified fibrinogen. LPS stimulation induced MAP kinase phosphorylation, IκBα degradation, and expression of proinflammatory cytokines (e.g., TNFα) within 60 minutes. This initial cellular activation was unaffected by unmodified or TG2-cross-linked fibrinogen. In contrast, LPS induction of IL-10 mRNA and protein and STAT3 phosphorylation was selectively attenuated by TG2-cross-linked fibrinogen, which was associated with enhanced proinflammatory cytokine secretion by LPS stimulated BMDMs at later time points (6 and 24 h).
Conclusions:
The results indicate that atypical cross-linking by TG2 imparts unique proinflammatory activity to surface-adhered fibrinogen. The results suggest a novel coagulation-independent mechanism controlling fibrinogen-directed macrophage activation.
Keywords: fibrinogen, inflammation, lipopolysaccharide, macrophage, transglutaminase
Introduction:
Cross-linked fibrin polymer plays a key role in both hemostasis and thrombosis and links the hemostatic system to the inflammatory response [1, 2]. In vitro studies suggest that fibrin(ogen) promotes expression of proinflammatory cytokines, cell migration, cell adhesion, and phagocytosis in monocytes, macrophages, and neutrophils [3, 4]. In addition to sites of intravascular coagulation activity, fibrin(ogen) is deposited in the extravascular space in injured tissues, such as the liver, lung, and kidney. Extravascular fibrin(ogen) deposition has been linked to leukocyte-mediated inflammation and tissue repair [5, 6], indicating that fibrin(ogen) plays an important role in regulating the local inflammatory response in varied settings.
Fibrin(ogen) regulates leukocyte inflammatory activity by engaging with heterodimeric cell surface adhesion receptors (integrins) that are expressed on the leukocyte surface. Fibrin(ogen) binds with high affinity to the integrins αMβ2 (CD11b/CD18) and αXβ2 (CD11c/CD18). Leukocyte β2 integrins bind to a cryptic site on the fibrin(ogen) γ-chain, revealed upon conversion of fibrinogen to fibrin [7]. Indeed, soluble plasma fibrinogen is a poor ligand for β2 integrins, whereas polymerized fibrin or surface-adhered fibrinogen (e.g., fibrinogen adhered to damaged surfaces, cells, or foreign bodies) binds leukocyte β2 integrins with high affinity. Thus, surface-adhered fibrinogen is viewed as bona fide β2 integrin ligand. However, it is unclear if adherence of fibrinogen to a surface fully unmasks proinflammatory activity evident in pathological settings such as tissue injury and extravascular fibrinogen accumulation.
Tissue transglutaminase-2 (TG2) is ubiquitously present in tissues and erythrocytes [8, 9] at the protein level, and is capable of cross-linking fibrinogen to produce atypical Aα-γ inter- and intra-molecular cross-links [10, 11]. This contrasts with cross-linking of fibrin polymer by the coagulation transglutaminase factor XIIIa (FXIIIa), which yields γ-γ dimers and α-polymers [10, 12]. Experimental studies indicate that TG2-cross-linked fibrin(ogen) is present in human atherosclerotic aortas [13], deposited in the chronically-injured liver [14], and formed at the surface of primary hepatocytes [15]. Cross-linking of fibrinogen by TG2 modestly delays fibrin polymerization [11] and inhibits degradation of fibrin by plasmin [16]. Moreover, fibrinogen αC regions cross-linked by TG2 promote endothelial cell activation via integrin αVβ3 [17]. Notably, the precise impact of TG2 on fibrinogen-directed leukocyte inflammatory responses has not been examined.
We tested the hypothesis that cross-linking of fibrinogen by TG2 enhances fibrinogen-directed macrophage proinflammatory activity in vitro. We validated atypical cross-linking characteristics of TG2-cross-linked fibrinogen and examined basal and LPS-stimulated inflammatory responses of bone marrow-derived macrophages (BMDMs) plated on a fibrinogen or TG2-cross-linked fibrinogen-coated surface.
Materials and Methods:
Cross-linking of fibrinogen by tissue transglutaminase-2 and coating of cell culture plates:
TG2-cross-linked fibrinogen was prepared as described previously with minor modifications [12]. Human fibrinogen 1 (Enzyme Research Labs, South Bend, IN, 2.3 μM) was incubated with recombinant human TG2 (R&D Systems, Minneapolis, MN, 0.3 μM) in buffer containing 5 mM CaCl2, 50 mM Tris-HCl, and 274 mM NaCl at 37 °C for 1 hour. Unmodified fibrinogen was prepared under the same conditions, by incubating in buffer containing 5 mM CaCl2, 50 mM Tris-HCl, and 274 mM NaCl at 37 °C for 1 hour, in the absence of TG2. In some experiments, the transglutaminase inhibitor cystamine (Cayman Chemical, Ann Arbor, MI, 0–2.5 mM) was added to the reaction. Purified mouse fibrinogen (Enzyme Research Labs) was used in select experiments, where indicated.
To coat tissue culture plates, fibrinogen and TG2-cross-linked fibrinogen were diluted to 10 μg/mL in sterile phosphate-buffered saline (PBS) and 1 mL, 0.5 mL, or 0.2 mL of solution was added to the wells of 6, 12, or 24-well tissue culture plates (Greiner Bio-One, Monroe, NC), respectively. Plates were incubated overnight at 4 °C and rinsed 3 times with PBS before plating cells.
Isolation and culture of mouse bone marrow-derived macrophages:
Mice were maintained in an Association for Assessment and Accreditation of Laboratory Animal Care International-accredited facility at Michigan State University at approximately 22 ± 2 °C with alternating 12-h light/dark cycles and were provided ad libitum access to reverse-osmosis purified drinking water and rodent diet (Teklad 8940, Envigo, Hackensack, NJ, USA). All animal procedures were approved by Michigan State University (MSU) Institutional Animal Care and Use Committee. Bone marrow cells were isolated from male and female C57Bl/6J mice (8–20 weeks old) by flushing femurs and tibias with RPMI-1640 media (Thermo Fisher Scientific, Waltham, MA) containing 1% penicillin/streptomycin (Thermo Fisher Scientific). In select experiments, bone marrow cells were also isolated from CD11b-deficient mice (CD11b−/− mice, B6.129S4-Itgamtm1Myd/J) or age-matched C56Bl6/J mice (The Jackson Laboratory Bar Harbor, ME). Bone marrow cells were differentiated into bone marrow macrophages by culturing in suspension flasks in macrophage medium (RPMI-1640 medium containing 10% fetal bovine serum, 1% penicillin/streptomycin, and 25 mM HEPES) supplemented with 20 ng/mL recombinant mouse macrophage colony stimulating factor (MCSF, BioLegend, San Diego, CA) for 6 days. On day 6, media were replaced with macrophage medium. Cells for experiments were harvested on day 7 by lifting cells from flasks with non-enzymatic cell lifting reagent (CellStripper, Corning, Corning, NY).
BMDMs were plated at a density of 5 × 105 cell/mL on uncoated, fibrinogen-coated, or TG2-cross-linked fibrinogen-coated surface and incubated for various times (1–24 h), with or without the addition of lipopolysaccharide (LPS, 1 ng/mL; Millipore Sigma, St. Louis, MO). In some experiments, cells were treated with the TG2 inhibitor ERW1041E (20 μM, R&D Systems) or vehicle (0.1% DMSO final concentration).
RNA isolation and quantitative reverse-transcription PCR:
Cells were lysed in TRI Reagent (Molecular Research Center, Cincinnati, OH), and RNA was isolated from cells using Direct-Zol RNA miniprep kits (Zymo Research, Irvine, CA) according to the manufacturer’s instructions. Equivalent amounts of total RNA were used to synthesize cDNA using a High Capacity cDNA Reverse Transcription Kit (Life Technologies, Foster City, CA) and a C1000 Thermal Cycler (Bio-Rad Laboratories, Hercules, CA). SYBR Green quantitative real-time PCR (qPCR) amplification was performed using a CFX Connect thermal cycler (Bio-Rad) with primers purchased from IDT (Coralville, IA) and PerfeCTa SYBR Green SuperMix (Quanta Biosciences, Beverly, MA). The expression of each gene was normalized to the geometric mean Ct of two housekeeping genes [18], Hprt and Gapdh, and relative fold change was determined using the ΔΔCt method. Primer sequences are listed in Supplemental Table 1.
Immunoblotting:
Cells were lysed in 2X Laemmli sample buffer (Bio-Rad) containing 5% 2-ME and protease and phosphatase inhibitors (G-Biosciences, St. Louis, MO). Samples were loaded on 4–12% Bis-Tris gels (Bio-Rad Laboratories, Hercules, CA) and proteins separated by SDS-PAGE in XT-MOPS running buffer (Bio-Rad). Proteins were transferred to PVDF membrane (Millipore Sigma Burlington, MA), using a Bio-Rad Transblot Semi-dry Transfer Cell, and the membrane was blocked in 5% BSA in tris-buffered saline + 0.1% Tween-20 pH 7.4 (TBST) for 1 h at room temperature. Following blocking, membranes were incubated in 5% BSA containing antibodies against phosphorylated or total ERK, p38, SAPK/JNK, IκBα, or tubulin overnight at 4 °C. Complete information regarding antibodies is provided in Supplemental Table 2. The membrane was washed in TBST and incubated in 1% BSA in TBST containing HRP-conjugated goat anti-rabbit IgG (Jackson Immuno Research Labs, West Grove, PA). For chemiluminescent detection, all membranes were incubated with EcoBright Pico HRP substrate (Innovative Solutions, Beverly Hills, MI) and exposed to blue-lite autoradiography film (DOT Scientific, Burton, MI).
Enzyme-linked immunosorbent assay (ELISA):
Mouse TNFα, IL1β, and IL-6, and IL-10 concentration in cell-free media supernatants was determined using ELISA MAX Standard Sets (Biolegend) according to the manufacturer’s instructions.
Statistics:
Comparison of 2 groups was performed using a paired Student’s t-test. Comparison of 3 or more groups was performed using a one-way repeated measures analysis of variance (ANOVA) and Tukey’s post-hoc test. Results were considered significant when the p-value was less than 0.05. Analysis was performed using Prism (Version 8, GraphPad Software, La Jolla, CA).
Results
TG2-cross-linked fibrinogen enhances Tnfa gene expression in BMDMs compared to unmodified fibrinogen.
Validating prior studies [10, 12], cross-linking of fibrinogen with TG2 produced a unique cross-linking pattern compared to unmodified fibrinogen or thrombin-polymerized fibrin. Under reducing conditions, unmodified fibrinogen produced characteristic bands representing the Aα, Bβ, and γ fibrinogen polypeptide chains (Fig. 1A, lanes 1 and 2). Thrombin treatment catalyzed removal of fibrinopeptides A and B, evident in decreased molecular weight of fibrinogen α and β chains, and production of characteristic γ-γ dimer (Fig. 1A, lanes 3 and 4). Cross-linking of fibrinogen by TG2 produced Aα-y complexes and high molecular weight species without evidence of fibrinopeptide removal (Fig. 1A, lanes 5 and 6). The same cross-linking pattern was also observed in TG2-cross-linked FXIII-free (peak 1) fibrinogen (Supplemental Figure 1), indicating that FXIIIa is not required for cross-linking of fibrinogen by TG2.
Figure 1: Effect of TG2 cross-linking on fibrinogen structure and Tnfa and Il10 gene expression in BMDMs.
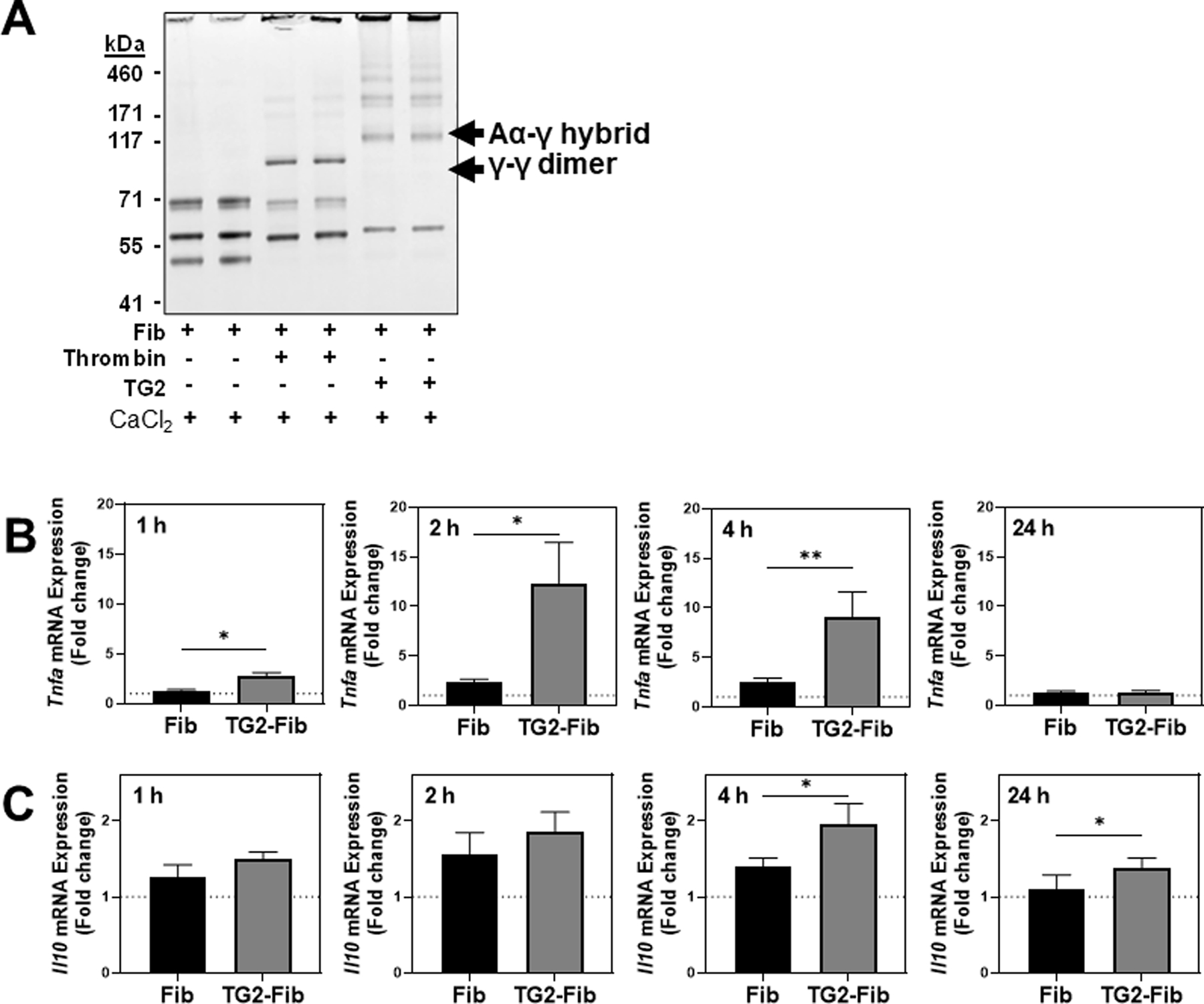
(A) Fibrinogen was polymerized by thrombin or cross-linked by TG2 as described in Materials and Methods. Samples were resolved on a 4–12% Bis-tris gradient gel, and the gel was stained with SimplyBlue Total Protein Stain, scanned, and depicted in grayscale. BMDMs were cultured on an uncoated surface, fibrinogen-coated surface, or TGM2-cross-linked fibrinogen-coated surface for 1, 2, 4, or 24 hours. Gene expression of Tnfa (B) and Il10 (C) was measured via qRT-PCR. Data are expressed as mean fold change + SEM relative to gene expression in macrophages cultured on an uncoated surface (n=4–17). *p<0.05, **p<0.01
To determine the effect of TG2-cross-linked fibrinogen on BMDM proinflammatory activity, mouse BMDMs were cultured on an uncoated surface, fibrinogen-coated surface, or TG2-cross-linked fibrinogen-coated surface for 1, 2, 4, or 24 h. Compared to an uncoated surface (dashed line; Fig 1B), unmodified fibrinogen modestly induced Tnfa expression. Tnfa expression was significantly enhanced in BMDMs plated on TG2 cross-linked fibrinogen after 1, 2, and 4 h. Importantly, TG2-cross-linked mouse fibrinogen also increased Tnfa expression compared to mouse fibrinogen alone (Supplemental Figure 2A). Induction of Tnfa is often balanced by induction of the anti-inflammatory cytokine, interleukin-10 (IL-10) [19]. Compared to an uncoated surface, unmodified fibrinogen did not induce Il10 gene expression; however, a slight but statistically significant increase in Il10 expression was observed in BMDMs plated on TG2-cross-linked fibrinogen compared to unmodified fibrinogen.
Direct activity of TG2 does not drive the proinflammatory activity of TG2 cross-linked fibrinogen.
TG2 itself has been demonstrated to modulate monocyte/macrophage function [20]. To exclude a contribution of residual TG2 coating the tissue culture surface to the observed macrophage response to TG2-cross-linked fibrinogen, BMDMs were cultured on plates coated with fibrinogen, TG2-cross-linked fibrinogen, or TG2 alone at the concentration present in TG2-cross-linked fibrinogen-coated wells. Each group of cells was incubated for 4 h in the presence or absence of the small molecule TG2 inhibitor ERW1041E. Expression of Tnfa was not induced in BMDMs cultured on surfaces coated with TG2 alone (Fig. 2A). Consistent with our initial studies, TG2 cross-linked fibrinogen enhanced Tnfa expression by BMDMs compared to fibrinogen alone. Notably, enhanced Tnfa induction was unaffected by addition of TG2 inhibitor to the culture medium. Collectively, these studies indicate that TG2 cross-linked fibrinogen, not residual TG2 activity, is driving the response of BMDMs to TG2-cross-linked fibrinogen. Indeed, concentration-dependent inhibition of the initial TG2-mediated fibrinogen cross-linking reaction with the transglutaminase inhibitor cystamine (Figure 2B) markedly reduced the induction of Tnfa by BMDMs (Figure 2C), while addition of cystamine to the reaction mixture containing fibrinogen alone had no effect on Tnfa expression.
Figure 2: Enhanced Tnfa expression in BMDMs is driven by TG2-cross-linked fibrinogen and not independent effects of TG2.
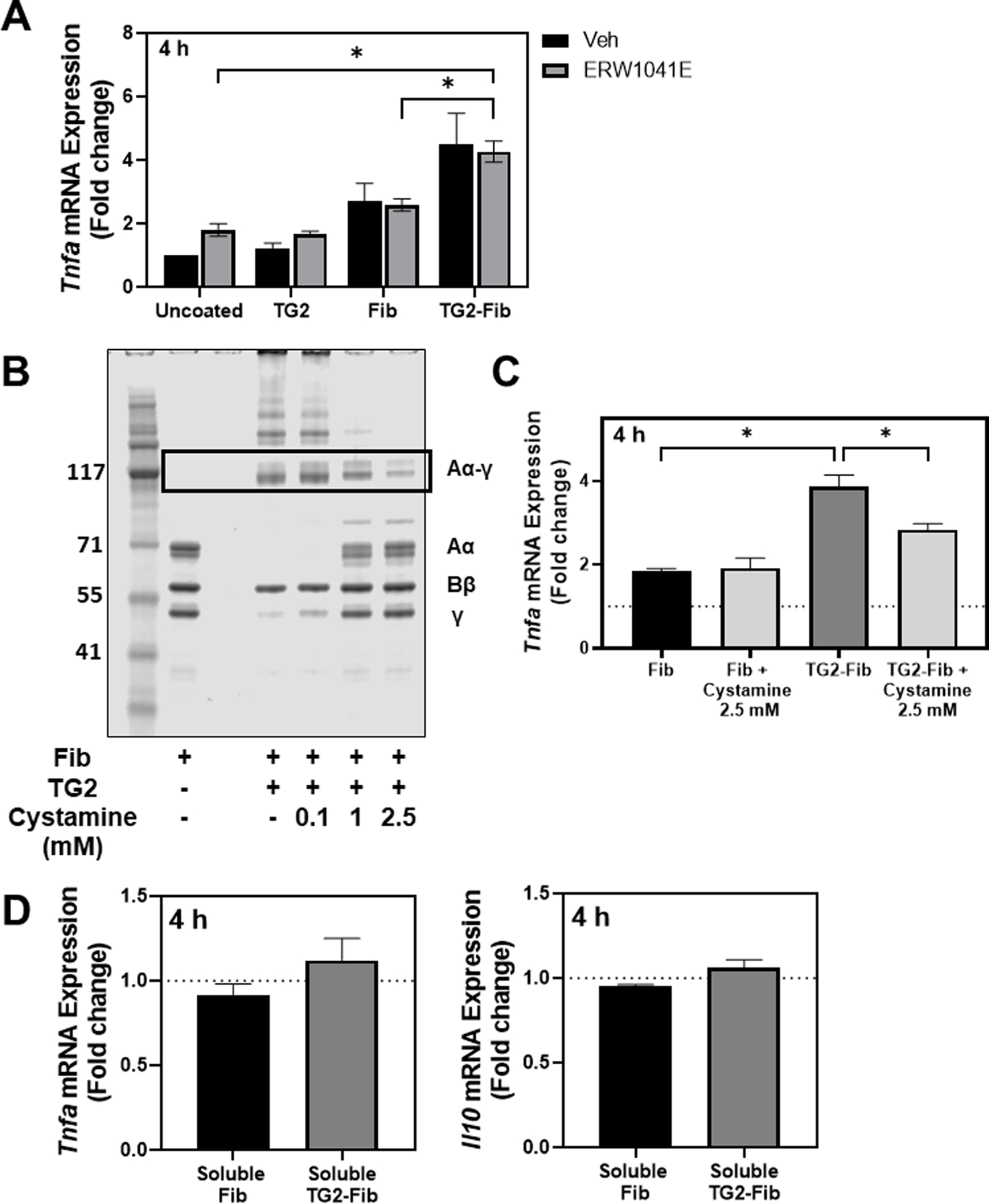
(A) BMDMs were cultured on a fibrinogen- or TG2-cross-linked fibrinogen-coated surface or uncoated surface. Immediately after plating, the TG2-specific inhibitor ERW1041E (20 μM) or vehicle (0.1% DMSO, final concentration) was added. Tnfa expression was measured via qRT-PCR after four hours. (B) Fibrinogen was incubated with TG2 or TG2 + 0.1, 1.0, or 2.5 mM of the transglutaminase inhibitor, cystamine. Cross-linked products were resolved on a 4–12% Bis-tris gel and stained with SimplyBlue total protein stain. Lane 1: Molecular weight marker, Lane 2: fibrinogen, Lane 3: empty, Lanes 4–6: Fibrinogen +TG2 with the indicated concentration of cystamine. (C) BMDMs were cultured on unmodified fibrinogen, fibrinogen incubated with 2.5 mM cystamine, TG2-cross-linked fibrinogen, or fibrinogen incubated with TG2 + 2.5 mM cystamine for 4 hours. Tnfa expression was measured via qRT-PCR. Data are presented as mean fold change + SEM compared to BMDMs on uncoated surface (n=3). *p<0.05. (D) BMDMs were cultured on uncoated plates and treated with either unmodified fibrinogen or TG2-cross-linked fibrinogen (10 μg/mL) or vehicle for 4 hours. Tnfa and Il10 expression were measured via qRT-PCR. Data are presented as mean fold change + SEM compared to vehicle-treated BMDMs (n=3).
We also sought to investigate the possibility that TG2 cross-linking increases inflammatory activity of soluble (i.e., not surface-adhered) fibrinogen. Accordingly, we treated BMDMs with either fibrinogen, TG2-cross-linked fibrinogen (10 μg/mL) or vehicle. Neither soluble fibrinogen or TG2-cross-linked soluble fibrinogen induced expression of Tnfa or Il10 (Figure 2D), indicating that TG2-cross-linked fibrinogen required adherence to a surface to exert proinflammatory effects. Notably, these results largely exclude the possibility that enhanced proinflammatory activity of TG2-cross-linked fibrinogen is attributable to contaminants in the reaction mixture (e.g., LPS).
TG2-cross-linked fibrinogen selectively suppresses LPS induction of IL-10 in BMDMs.
Next, we sought to determine the effect of fibrinogen or TG2-cross-linked fibrinogen on the response of BMDMs to LPS. BMDMs were plated on wells coated with fibrinogen or TG2-cross-linked fibrinogen or uncoated wells for 4 h, then stimulated with LPS (1 ng/mL) for various times. Tnfa gene expression was robustly induced in BMDMs 30, 60, and 120 min after LPS stimulation (Figure 3A). Expression of Tnfa after LPS stimulation was not significantly affected by either fibrinogen coating. Similarly, LPS stimulation induced Il10 gene expression compared to untreated cells at each time point (Figure 3B). Interestingly, compared to cells in uncoated wells or cells on unmodified fibrinogen, TG2-cross-linked fibrinogen significantly suppressed Il10 induction by 120 minutes after LPS stimulation. Similar results were observed using mouse fibrinogen (Supplemental Figure 2B). The impact of TG2-cross-linked fibrinogen on Tnfa and Il10 gene expression was reflected by changes in the concentration of each cytokine in the cell culture supernatant 120 min after LPS stimulation. Indeed, we observed similar levels of TNFα produced by LPS-stimulated BMDMs on fibrinogen and TG2-cross-linked fibrinogen as well as uncoated wells (Figure 3C). Supernatant levels of a second pro-inflammatory mediator, interleukin-6 (IL-6), were also not affected by fibrinogen or TG2-cross-linked fibrinogen at this time point (Fig. 3D). In contrast, IL-10 concentration was significantly reduced in supernatants from LPS-stimulated cells cultured on TG2-cross-linked fibrinogen compared to cells on unmodified fibrinogen or uncoated wells (Fig. 3E).
Figure 3: TG2-cross-linked fibrin(ogen) selectively attenuates early LPS-induced IL-10 expression.
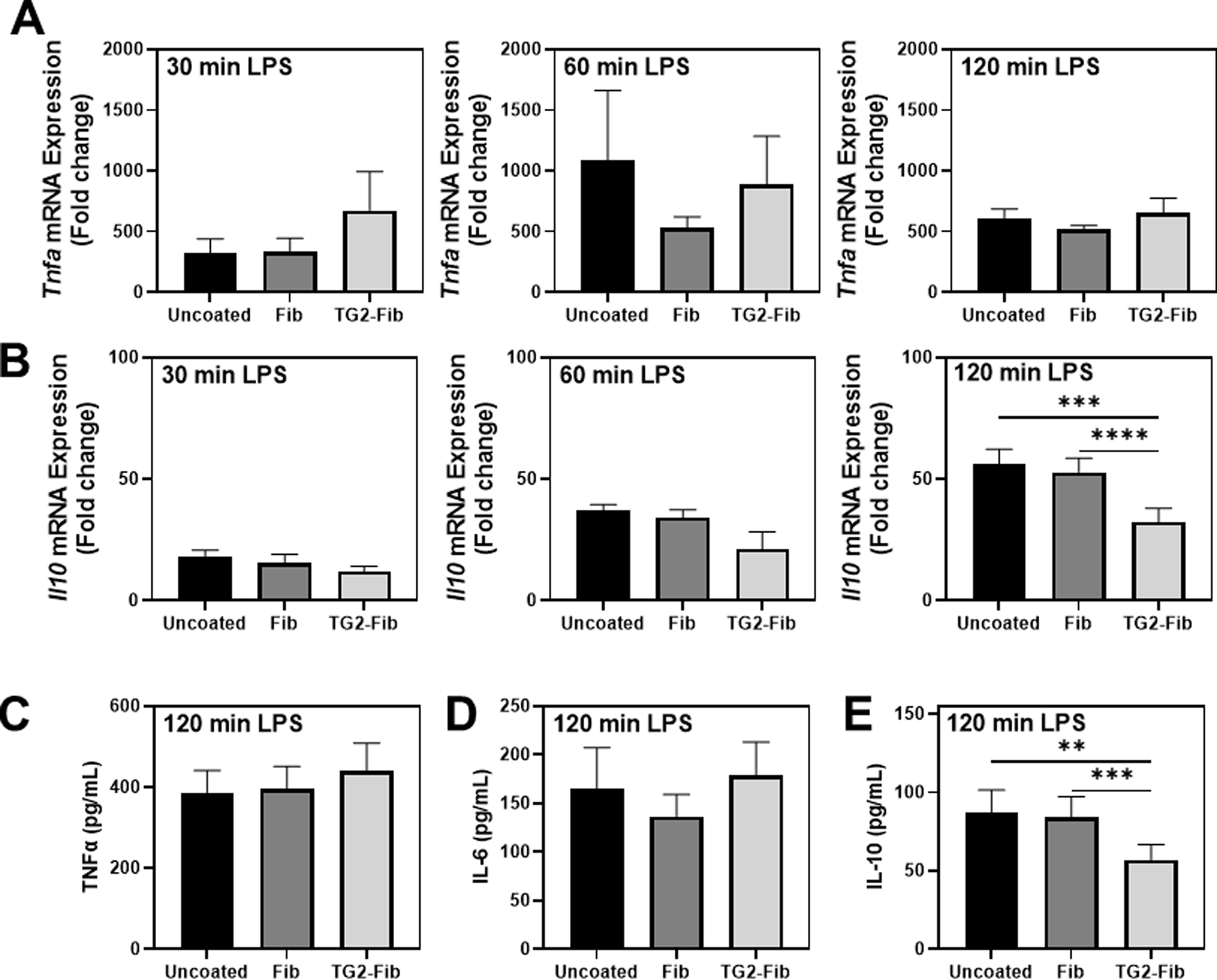
BMDMs were cultured on an uncoated surface, fibrinogen-coated surface, or TG2-cross-linked fibrinogen-coated surface for 4 hours then stimulated with lipopolysaccharide (LPS) for the indicated time points. (A) Tnfa and (B) Il10 expression was measured in BMDMs treated with 1 ng/mL LPS for 30, 60, or 120 min via qRT-PCR. Data are presented as mean fold induction + SEM compared to vehicle-treated BMDMs on an uncoated surface at t=0 (not shown) (n=3–11). (C) TNFα, (D) IL-6, and (E) IL-10 protein levels were measured in supernatant following stimulation of BMDMs with 1 ng/mL LPS for two hours (n=7–13). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001
TG2-cross-linked fibrinogen does not alter early LPS-induced proinflammatory signaling in BMDMs but suppresses STAT3 phosphorylation.
LPS coordinates inflammatory gene induction through rapid activation of multiple pathways including the mitogen-activated protein kinases (MAPKs) ERK1/2, JNK 1/2, and p38MAPK and the transcription factor NFκB. Phosphorylation of MAPKs was near undetectable levels in vehicle-treated cells, and neither fibrinogen nor TG2-cross-linked fibrinogen had any detectable effect on MAPK phosphorylation in unstimulated cells (Fig. 3A). The impact of TG2-cross-linked fibrinogen on LPS activation of these pathways was determined in BMDMs plated on fibrinogen, TG2-cross-linked fibrinogen or uncoated wells. LPS activation of Erk1/2, SAPK/JNK, and p38MAPK, evident as an increase in the phosphorylated form of these kinases, occurred within 30 minutes and was not affected by fibrinogen or TG2-cross-linked fibrinogen (Figure 4A–D). Likewise, degradation of IκBα, which permits translocation of NFκB to the nucleus, was not significantly affected by fibrinogen or TG2-cross-linked fibrinogen (Figure 4A, E).
Figure 4: Effect of TG2-cross-linked fibrinogen on LPS-induced cell signaling in BMDMs.
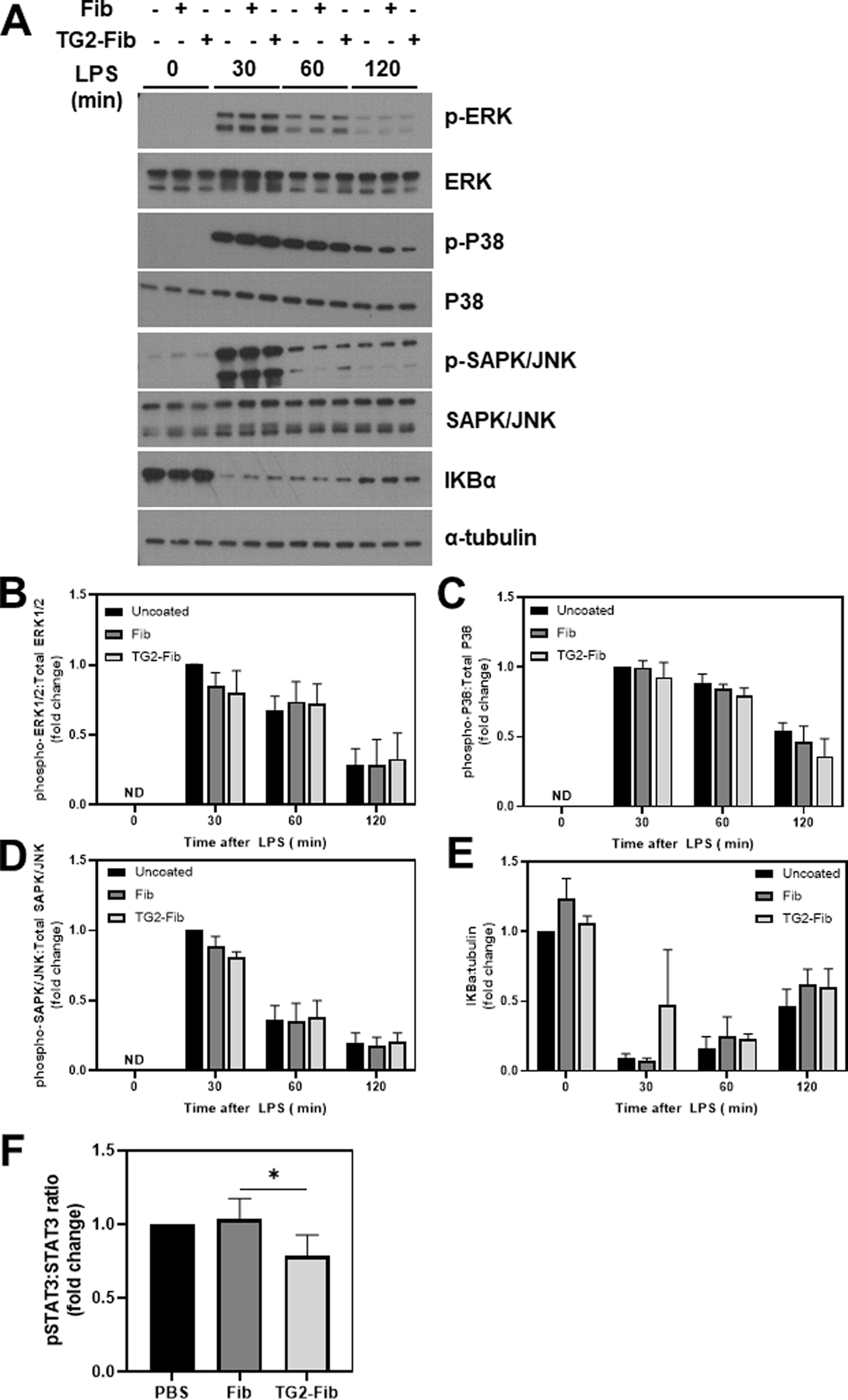
BMDMs were cultured on an uncoated surface, fibrinogen-coated surface, or TG2-cross-linked fibrinogen coated surface for 4 hours then stimulated with 1 ng/mL lipopolysaccharide (LPS) for the indicated time points. Representative western blots (A) show phosphorylation of ERK (B), P38 (C), and SAPK/JNK (D), and degradation of IKBα in BMDMs 0–2 hours after LPS stimulation. Data are presented as mean ratio of phospho-protein to total or IKBα to tubulin + SEM compared to vehicle-treated BMDMs on an uncoated surface (n=4–5). *p<0.05
Initial LPS-induced proinflammatory signaling is balanced by induction of anti-inflammatory signaling to attenuate the initial proinflammatory response. Anti-inflammatory signaling is mediated in part by STAT3 phosphorylation, driven by paracrine and autocrine IL-10 signaling [21]. STAT3 phosphorylation was increased in BMDMs challenged with LPS for 2 h compared to vehicle-treated cells (not shown). In agreement with inhibition of LPS-induced IL-10 expression TG2-cross-linked fibrinogen, STAT3 phosphorylation was significantly reduced in LPS-challenged BMDMs, with BMDMs in 4 out of 5 independent experiments displaying a ~10–50% reduction in STAT3 phosphorylation compared to cells in uncoated wells (Figure 4F, Supplemental Figure 3).
TG2-cross-linked fibrinogen enhances late proinflammatory cytokine secretion in LPS-challenged BMDMs.
IL-10 mediates its anti-inflammatory effects via STAT3 signaling, which ultimately inhibits expression of proinflammatory mediators such as TNFα, IL-1, and IL-6 [21, 22]. Because TG2-cross-linked fibrinogen suppressed IL-10 induction and STAT3 phosphorylation in LPS-stimulated BMDMs, we determined the impact of TG2-cross-linked fibrinogen on inflammatory cytokine secretion following extended LPS stimulation. BMDMs were plated on fibrinogen or TG2-cross-linked fibrinogen or in uncoated wells for 4 h, then stimulated with LPS (1 ng/mL) for 6 or 24 h. LPS treatment caused robust secretion of IL-10 (Figure 5A), TNFα (Figure 5B), IL-1β (Figure 5C), and IL-6 (Figure 5D) in BMDMs. These cytokines were below the limit of detection in supernatant from vehicle-treated cells on each surface (not shown). LPS-induced IL-10 secretion remained suppressed 6 hours after LPS challenge in BMDMs plated on TG2-cross-linked fibrinogen (Figure 5A). Furthermore, supernatant levels of TNFα were significantly elevated in BMDMs plated on TG2-cross-linked fibrinogen compared to unmodified fibrinogen, both 6 and 24 h after LPS stimulation. Notably, similar results were observed for IL-1β and IL-6 24 h after LPS stimulation.
Figure 5: Effect of TG2-cross-linked fibrinogen on prolonged LPS-induced inflammatory cytokine release.
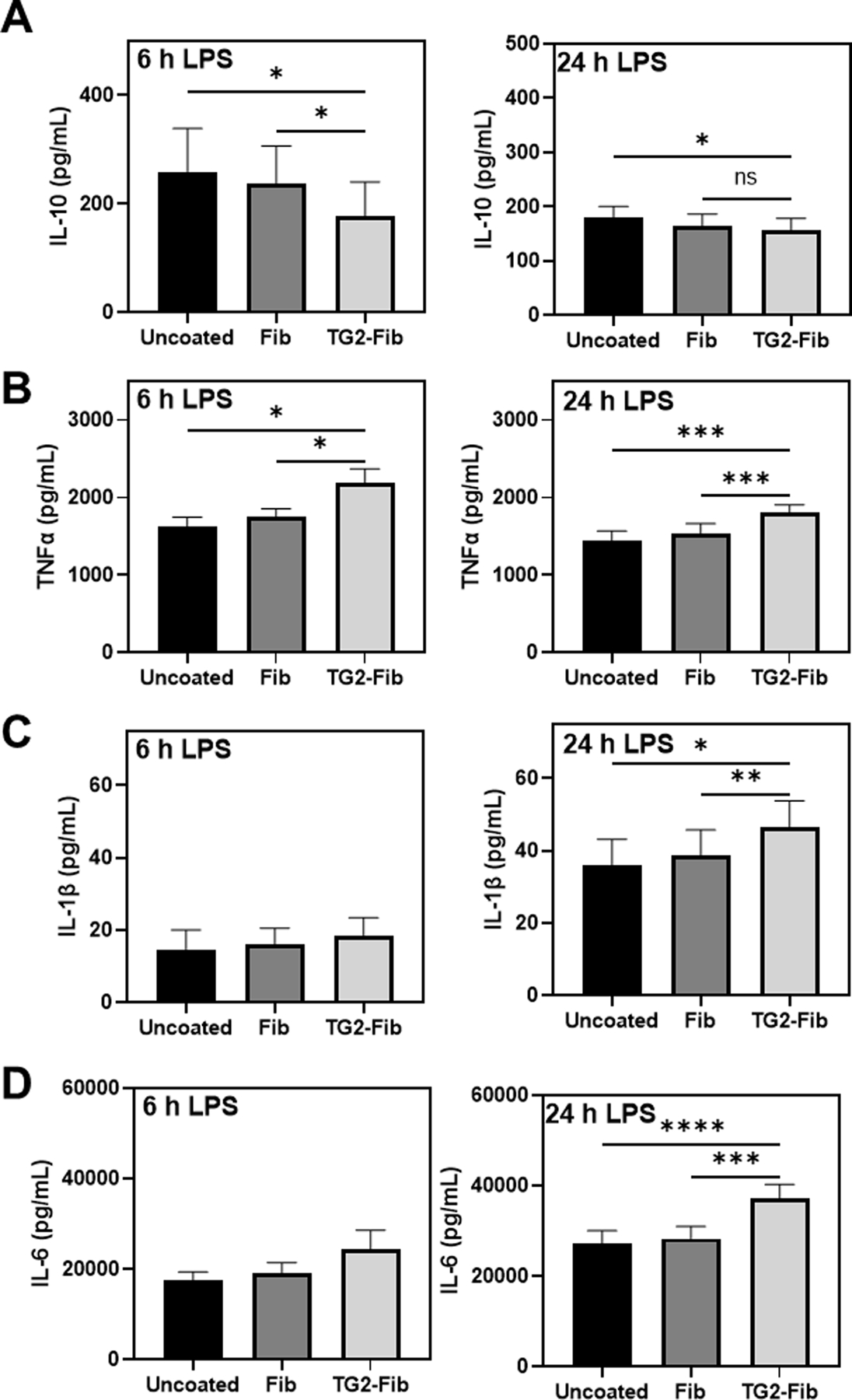
BMDMs were cultured on an uncoated surface, fibrinogen-coated surface, or TG2-cross-linked fibrinogen coated surface for 4 hours then stimulated with 1 ng/mL lipopolysaccharide (LPS) for 6 or 24 h. IL-10 (A), TNFα (B), IL-1β (C), and IL-6 (D) were measured in supernatant using enzyme-linked immunosorbent assay (ELISA), n=4–8. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001
Discussion:
Fibrin(ogen) bridges the hemostatic and inflammatory responses by engaging with leukocyte β2 integrins. Conversion of fibrinogen to a fibrin polymer during clot formation and/or immobilization of fibrinogen to a surface enables leukocyte β2 integrin binding and drives inflammatory activity. However, it is possible that thrombin-independent modifications of fibrin(ogen) structure may create a β2 integrin ligand uniquely driving cellular responses. In the current study, we found that the proinflammatory activity of surface-adhered fibrinogen, already documented as an effective β2 integrin ligand [7], can be further enhanced by TG2 cross-linking. Notably, our results uncover a coagulation-independent mechanism altering the proinflammatory activity of surface-adhered fibrin(ogen). Indeed, the results suggest that modification of extravascular fibrin(ogen) deposits by TG2 could form a novel mechanism whereby products of tissue injury could exacerbate extravascular fibrinogen proinflammatory activity.
Prior studies have examined the deposition of ‘non-traditional’ extravascular fibrin(ogen) deposits [14] in injured tissue, including fibrin(ogen) deposition driven by TG2. Here, we validated results of prior studies documenting unique TG2-dependent formation of Aα-γ hybrid cross-linked fibrinogen [10, 12]. Importantly, the cellular response to TG2-cross-linked fibrinogen was not impacted by TG2 inhibitors in the culture medium, excluding a fibrinogen-independent effect of TG2. Rather, pharmacologic inhibition of the cross-linking reaction prior to coating cell culture plates reduced cytokine induction in BMDMs. It is not clear whether the proinflammatory effects of TG2-cross-linked fibrinogen are unique, or whether cross-linking by FXIIIa would similarly amplify the cellular response to fibrin(ogen). Unlike TG2, FXIIIa does not efficiently cross-link non-polymerized fibrinogen in physiological conditions [23]. An alternative approach to make this comparison could include comparing the proinflammatory effects of TG2-cross-linked fibrinogen with that of polymerized fibrin gels, although this is complicated by inherent differences in the response of macrophages to different surface properties (e.g., stiffness). Interestingly, prior studies found that cross-linking of fibrinogen with TG2 delayed fibrin polymerization [11], whereas subjecting polymerized fibrin to secondary cross-linking by TG2 delayed fibrinolysis [16]. As these reactions may be paired in injured tissues, it would certainly be interesting to determine if TG2 also amplifies the proinflammatory properties of polymerized fibrin.
The mechanism whereby TG2 cross-linking of fibrinogen exacerbates cytokine expression by BMDMs is not fully understood but is thought to be mediated by interaction of fibrin(ogen) with various cell surface receptors, such as toll-like receptor-4 (TLR4) or integrin receptors. One study suggested that soluble fibrinogen enhances chemokine secretion via a TLR4-dependent mechanism in murine thioglycolate-elicited peritoneal exudate cells (PECs) [24]. In our study, TG2-cross-linked fibrinogen in solution did not display inflammatory activity, and surface-adhered TG2-cross-linked fibrinogen did not affect primary TLR4 signaling pathways in the presence or absence of LPS treatment. This suggests that TLR4 is not a key receptor for TG2-cross-linked fibrinogen in BMDMs, at least in conditions investigated used for these studies. The precise mechanisms whereby TLR4 may regulate the macrophage response to fibrinogen remain unclear. For example, it is unclear whether fibrinogen interacts with TLR4 directly, or whether TLR4-mediated signaling events prime macrophages for response to fibrinogen via a different receptor, such as a β2 integrin receptor.
Fibrin(ogen) interaction with integrins, such as αMβ2 (CD11b/CD18), may also mediate fibrinogen’s proinflammatory effects. Mouse BMDMs express high levels of αMβ2, which binds fibrin(ogen) via a cryptic binding domain in the fibrin(ogen) γ-chain that is revealed upon conversion of fibrinogen to fibrin or immobilization of fibrinogen to a surface [7]. In multiple types of leukocytes, including monocytes [25–27] and neutrophils [28], blockade or deletion of β2 integrins has been demonstrated to reduce the proinflammatory effects of fibrinogen. We therefore tested the hypothesis that αMβ2 mediates the pro-inflammatory effects of TG2-cross-linked fibrinogen in this setting. Interestingly, in our studies, we observed a similar reduction in LPS-induced Il10 expression in BMDMs plated on TG2-cross-linked fibrinogen compared to unmodified fibrinogen in both wildtype and CD11b-deficient BMDMs. These findings suggest that the pro-inflammatory effects of TG2-cross-linked fibrinogen are mediated by a novel, αMβ2-independent mechanism.
Apart from αMβ2, it is possible that TG2-cross-linking of fibrinogen promotes binding to a different β2 integrin receptor, such as αXβ2. Bone marrow-derived macrophages do not readily express αXβ2, but this receptor may become up-regulated in proinflammatory conditions [29]. Previous studies indicate that in monocytes, CD11b and not CD11c is the integrin receptor which mediates the proinflammatory effects of fibrinogen [27], but importantly, this mechanism has not been investigated in BMDMs. It is possible that TG2-cross-linking of fibrinogen may cause BMDMs to upregulate CD11c as they polarize to a more proinflammatory phenotype. Finally, it is possible that TG2-cross-linked fibrinogen mediates macrophage proinflammatory activity independently β2 integrins. Indeed, BMDMs also express integrin αVβ3 [30], which binds fibrinogen via its RGD domain [31]. Cross-linking of fibrinogen by TG2 has been demonstrated to promote binding of the fibrinogen α-C domain to endothelial cell αVβ3. It is possible that by enhancing fibrinogen’s ability to bind to multiple macrophage integrins, TG2-directed cross-linking may enhance expression of inflammatory cytokines.
TG2-cross-linked fibrinogen displayed direct proinflammatory activity compared to unmodified fibrinogen and enhanced the macrophage response to LPS by selectively suppressing anti-inflammatory signaling. Indeed, TG2-cross-linked fibrinogen inhibited LPS induction of IL-10 expression and the activation of STAT3. IL-10 suppresses induction of proinflammatory cytokines, including TNFα, via activation of STAT3 [21, 22]. Reflecting this reduction in anti-inflammatory signaling, proinflammatory cytokine release at later time points (6–24 h after LPS stimulation) was increased in BMDMs cultured on TG2-cross-linked fibrinogen. The mechanism whereby TG2-cross-linked fibrinogen inhibits LPS induction of IL-10 is unclear. Induction of IL-10 gene expression in LPS-treated BMDMs is mediated by expression of type I interferons (e.g., IFN-α, IFNβ), which signal through the IFN-α receptor [32]. IFNαR signaling also induces expression of the anti-inflammatory cytokine IL-27, which further drives IL-10 gene expression [33]. It is possible that initial type I interferon signaling is disrupted by TG2-cross-linked fibrinogen. Interestingly, a prior study found that IFNα-induced STAT1 and STAT3 phosphorylation was suppressed in human monocyte-derived macrophages plated on fibrinogen [34]. Although we did not observe an impact of unmodified fibrinogen on LPS induction of IL-10, this could be attributed to differences in cell type, concentration of fibrinogen, or concentration of LPS in the cell culture system. TG2-modified fibrin(ogen) may be a unique modifier of these pathways, ultimately enhancing proinflammatory cytokine induction.
The results of these fundamental studies have multiple implications. Fibrin(ogen)-leukocyte interactions contribute to disease pathogenesis in multiple settings, including peritonitis [5], colitis-associated cancer [35], Duchenne muscular dystrophy [36], bile duct fibrosis [37], acute liver injury [6], obesity [38], and arthritis [39]. The extent to which TG2 plays a role in fibrin(ogen) cross-linking in these various pathologies is understudied. TG2 could potentially cross-link extravascular fibrinogen or further modify fibrin deposits, tailoring the local inflammatory response to tissue injury. There is some precedent for this concept. Fibrin(ogen) cross-linking in the acutely-injured liver is largely mediated by FXIII [40]. However, residual fibrin(ogen) cross-linking is evident even in FXIIIA-deficient mice. Interestingly, hepatic fibrin(ogen) deposition in a widely-used model of hepatic fibrosis is driven by TG2 [14], and TG2-cross-linked fibrinogen may also play an important role in defining macrophage phenotype in other disease processes, including vascular diseases such as atherosclerosis. For example, TG2 is highly expressed in atherosclerotic plaques, which are also rich in fibrin(ogen), in close proximity to macrophages [41]. Cross-linking of fibrin(ogen) by TG2 in these lesions may promote proinflammatory foam cell activity and disease progression. Although fibrin formation is certainly a key component of the inflammatory response in multiple diseases, these studies and our in vitro observations suggest the proinflammatory properties of TG2-directed fibrin(ogen) cross-linking in vivo should not be overlooked. Current experimental tools (TG2-knockout mice and conditional knockout mice) may be insufficient to identify the role of TG2-cross-linked fibrinogen in the inflammatory response in vivo. Indeed, leukocyte TG2 is involved in key cellular functions in the setting of inflammation, including phagocytosis [20]. To isolate the precise role of TG2-cross-linked fibrinogen in disease settings, future studies could include generation of mice expressing fibrinogen in which the residues required for TG2 cross-linking are mutated, as has recently been generated for the FXIIIa cross-linking sites [42].
In summary, we identified a novel mechanism whereby coagulation-independent modifications of fibrinogen enhance its proinflammatory properties. Specifically, TG2-cross-linked fibrinogen enhanced inflammatory gene expression in macrophages and modified the balance of proinflammatory and anti-inflammatory cytokines produced after LPS stimulation. Because TG2 is ubiquitously expressed, the unique cross-linking of extravascular fibrin(ogen) deposits by TG2 may constitute a highly relevant but underrecognized mechanism linking fibrinogen to the progression of multiple diseases.
Supplementary Material
Essentials:
Fibrinogen links the hemostatic and inflammatory responses by interacting with leukocytes.
Fibrinogen can be cross-linked by tissue transglutaminase (TG2) independent of polymerization.
TG2-cross-linked fibrinogen enhances macrophage proinflammatory responses in vitro.
TG2-cross-linking is a novel mechanism whereby fibrinogen may attain proinflammatory function.
Funding Information:
This work was supported by grants from the National Institutes of Health (R01ES017537 and R01DK120289 to JPL and K99DK129710 and F32DK121423 to LGP), and R01DK112778 (to MJF), as well as additional support from the US Department of Agriculture (USDA) National Institute of Food and Agriculture (to JPL). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institute of Environmental Health Sciences, or the USDA.
Footnotes
Conflicts of Interest:
L.G. Poole, A. K. Kopec, M. J. Flick, and J.P. Luyendyk express no conflicts of interest regarding this work.
Bibliography and References Cited
- 1.Flick MJ, Du X, Degen JL. Fibrin(ogen)-alpha M beta 2 interactions regulate leukocyte function and innate immunity in vivo. Exp Biol Med (Maywood). 2004;229(11):1105–10. [DOI] [PubMed] [Google Scholar]
- 2.Luyendyk JP, Schoenecker JG, Flick MJ. The multifaceted role of fibrinogen in tissue injury and inflammation. Blood. 2019;133(6):511–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rubel C, Fernandez GC, Dran G, Bompadre MB, Isturiz MA, Palermo MS. Fibrinogen promotes neutrophil activation and delays apoptosis. J Immunol. 2001;166(3):2002–10. [DOI] [PubMed] [Google Scholar]
- 4.Smiley ST, King JA, Hancock WW. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J Immunol. 2001;167(5):2887–94. [DOI] [PubMed] [Google Scholar]
- 5.Flick MJ, Du X, Witte DP, Jiroušková M, Soloviev DA, Busuttil SJ, Plow EF, Degen JL. Leukocyte engagement of fibrin(ogen) via the integrin receptor α(M)β(2)/Mac-1 is critical for host inflammatory response in vivo. Journal of Clinical Investigation. 2004;113(11):1596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kopec AK, Joshi N, Cline-Fedewa H, Wojcicki AV, Ray JL, Sullivan BP, Froehlich JE, Johnson BF, Flick MJ, Luyendyk JP. Fibrin(ogen) drives repair after acetaminophen-induced liver injury via leukocyte alphaMbeta2 integrin-dependent upregulation of Mmp12. J Hepatol. 2017;66(4):787–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lishko VK, Kudryk B, Yakubenko VP, Yee VC, Ugarova TP. Regulated Unmasking of the Cryptic Binding Site for Integrin αMβ2 in the γC-Domain of Fibrinogen. Biochemistry. 2002;41(43):12942–51. [DOI] [PubMed] [Google Scholar]
- 8.Breve JJ, Drukarch B, van Strien M, van Dam AM. Validated sandwich ELISA for the quantification of tissue transglutaminase in tissue homogenates and cell lysates of multiple species. J Immunol Methods. 2008;332(1–2):142–50. [DOI] [PubMed] [Google Scholar]
- 9.Wolf J, Lachmann I, Wagner U, Osman AA, Mothes T. Quantification of human tissue transglutaminase by a luminescence sandwich enzyme-linked immunosorbent assay. Anal Biochem. 2011;419(2):153–60. [DOI] [PubMed] [Google Scholar]
- 10.Murthy SN, Lorand L. Cross-linked A alpha.gamma chain hybrids serve as unique markers for fibrinogen polymerized by tissue transglutaminase. Proc Natl Acad Sci U S A. 1990;87(24):9679–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murthy SNP, Wilson JH, Lukas TJ, Veklich Y, Weisel JW, Lorand L. Transglutaminase-catalyzed crosslinking of the Aα and γ constituent chains in fibrinogen. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(1):44–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murthy SN, Wilson J, Guy SL, Lorand L. Intramolecular crosslinking of monomeric fibrinogen by tissue transglutaminase. Proc Natl Acad Sci U S A. 1991;88(23):10601–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shainoff JR, Valenzuela R, Urbanic DA, DiBello PM, Lucas FV, Graor R. Fibrinogen A alpha and gamma-chain dimers as potential differential indicators of atherosclerotic and thrombotic vascular disease. Blood Coagul Fibrinolysis. 1990;1(4–5):499–503. [PubMed] [Google Scholar]
- 14.Poole LG, Pant A, Baker KS, Kopec AK, Cline-Fedewa HM, Iismaa SE, Flick MJ, Luyendyk JP. Chronic liver injury drives non-traditional intrahepatic fibrin(ogen) crosslinking via tissue transglutaminase. Journal of Thrombosis and Haemostasis. 2019;17(1):113–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barsigian C, Stern AM, Martinez J. Tissue (type II) transglutaminase covalently incorporates itself, fibrinogen, or fibronectin into high molecular weight complexes on the extracellular surface of isolated hepatocytes. Use of 2-[(2-oxopropyl)thio] imidazolium derivatives as cellular transglutaminase inactivators. Journal of Biological Chemistry. 1991;266(33):22501–9. [PubMed] [Google Scholar]
- 16.Mutch NJ, Koikkalainen JS, Fraser SR, Duthie KM, Griffin M, Mitchell J, Watson HG, Booth NA. Model thrombi formed under flow reveal the role of factor XIII-mediated cross-linking in resistance to fibrinolysis. Journal of thrombosis and haemostasis : JTH. 2010;8(9):2017–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Belkin AM, Tsurupa G, Zemskov E, Veklich Y, Weisel JW, Medved L. Transglutaminase-mediated oligomerization of the fibrin(ogen) alphaC domains promotes integrin-dependent cell adhesion and signaling. Blood. 2005;105(9):3561–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3(7):RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Platzer C, Meisel C, Vogt K, Platzer M, Volk HD. Up-regulation of monocytic IL-10 by tumor necrosis factor-alpha and cAMP elevating drugs. Int Immunol. 1995;7(4):517–23. [DOI] [PubMed] [Google Scholar]
- 20.Chrobok NL, Sestito C, Wilhelmus MM, Drukarch B, van Dam AM. Is monocyte- and macrophage-derived tissue transglutaminase involved in inflammatory processes? Amino Acids. 2017;49(3):441–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williams L, Bradley L, Smith A, Foxwell B. Signal transducer and activator of transcription 3 is the dominant mediator of the anti-inflammatory effects of IL-10 in human macrophages. J Immunol. 2004;172(1):567–76. [DOI] [PubMed] [Google Scholar]
- 22.Berlato C, Cassatella MA, Kinjyo I, Gatto L, Yoshimura A, Bazzoni F. Involvement of suppressor of cytokine signaling-3 as a mediator of the inhibitory effects of IL-10 on lipopolysaccharide-induced macrophage activation. J Immunol. 2002;168(12):6404–11. [DOI] [PubMed] [Google Scholar]
- 23.Siebenlist KR, Meh DA, Mosesson MW. Protransglutaminase (factor XIII) mediated crosslinking of fibrinogen and fibrin. Thromb Haemost. 2001;86(5):1221–8. [PubMed] [Google Scholar]
- 24.Smiley ST, King JA, Hancock WW. Fibrinogen Stimulates Macrophage Chemokine Secretion Through Toll-Like Receptor 4. The Journal of Immunology. 2001;167(5):2887–94. [DOI] [PubMed] [Google Scholar]
- 25.Perez RL, Ritzenthaler JD, Roman J. Transcriptional regulation of the interleukin-1beta promoter via fibrinogen engagement of the CD18 integrin receptor. Am J Respir Cell Mol Biol. 1999;20(5):1059–66. [DOI] [PubMed] [Google Scholar]
- 26.Perez RL, Roman J. Fibrin enhances the expression of IL-1 beta by human peripheral blood mononuclear cells. Implications in pulmonary inflammation. J Immunol. 1995;154(4):1879–87. [PubMed] [Google Scholar]
- 27.Sitrin RG, Pan PM, Srikanth S, Todd RF 3rd. Fibrinogen activates NF-kappa B transcription factors in mononuclear phagocytes. J Immunol. 1998;161(3):1462–70. [PubMed] [Google Scholar]
- 28.Walzog B, Weinmann P, Jeblonski F, Scharffetter-Kochanek K, Bommert K, Gaehtgens P. A role for beta(2) integrins (CD11/CD18) in the regulation of cytokine gene expression of polymorphonuclear neutrophils during the inflammatory response. FASEB J. 1999;13(13):1855–65. [DOI] [PubMed] [Google Scholar]
- 29.Ying W, Cheruku PS, Bazer FW, Safe SH, Zhou B. Investigation of Macrophage Polarization Using Bone Marrow Derived Macrophages. Journal of Visualized Experiments : JoVE. 2013(76):50323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Erwig LP, Gordon S, Walsh GM, Rees AJ. Previous uptake of apoptotic neutrophils or ligation of integrin receptors downmodulates the ability of macrophages to ingest apoptotic neutrophils. Blood. 1999;93(4):1406–12. [PubMed] [Google Scholar]
- 31.Yokoyama K, Zhang XP, Medved L, Takada Y. Specific binding of integrin alpha v beta 3 to the fibrinogen gamma and alpha E chain C-terminal domains. Biochemistry. 1999;38(18):5872–7. [DOI] [PubMed] [Google Scholar]
- 32.Chang EY, Guo B, Doyle SE, Cheng G. Cutting edge: involvement of the type I IFN production and signaling pathway in lipopolysaccharide-induced IL-10 production. J Immunol. 2007;178(11):6705–9. [DOI] [PubMed] [Google Scholar]
- 33.Iyer SS, Ghaffari AA, Cheng G. Lipopolysaccharide-mediated IL-10 transcriptional regulation requires sequential induction of type I IFNs and IL-27 in macrophages. J Immunol. 2010;185(11):6599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang L, Gordon RA, Huynh L, Su X, Park Min KH, Han J, Arthur JS, Kalliolias GD, Ivashkiv LB. Indirect inhibition of Toll-like receptor and type I interferon responses by ITAM-coupled receptors and integrins. Immunity. 2010;32(4):518–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Steinbrecher KA, Horowitz N, Blevins EA, Barney KA, Shaw MA, Harmel-Laws E, Finkelman FD, Flick MJ, Pinkerton MD, Talmage KE, Kombrinck KW, Witte DP, Palumbo JS. Colitis-associated cancer is dependent on the interplay between the hemostatic and inflammatory systems and supported by integrin α(M)β(2) engagement of fibrinogen. Cancer research. 2010;70(7):2634–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vidal B, Ardite E, Suelves M, Ruiz-Bonilla V, Janue A, Flick MJ, Degen JL, Serrano AL, Munoz-Canoves P. Amelioration of Duchenne muscular dystrophy in mdx mice by elimination of matrix-associated fibrin-driven inflammation coupled to the alphaMbeta2 leukocyte integrin receptor. Hum Mol Genet. 2012;21(9):1989–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Joshi N, Kopec AK, Ray JL, Cline-Fedewa H, Nawabi A, Schmitt T, Nault R, Zacharewski TR, Rockwell CE, Flick MJ, Luyendyk JP. Fibrin deposition following bile duct injury limits fibrosis through an α(M)β(2)-dependent mechanism. Blood. 2016;127(22):2751–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kopec AK, Abrahams SR, Thornton S, Palumbo JS, Mullins ES, Divanovic S, Weiler H, Owens AP 3rd, Mackman N, Goss A, van Ryn J, Luyendyk JP, Flick MJ. Thrombin promotes diet-induced obesity through fibrin-driven inflammation. J Clin Invest. 2017;127(8):3152–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Flick MJ, LaJeunesse CM, Talmage KE, Witte DP, Palumbo JS, Pinkerton MD, Thornton S, Degen JL. Fibrin(ogen) exacerbates inflammatory joint disease through a mechanism linked to the integrin alphaMbeta2 binding motif. J Clin Invest. 2007;117(11):3224–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Poole LG, Kopec AK, Groeneveld DJ, Pant A, Baker KS, Cline-Fedewa HM, Flick MJ, Luyendyk JP. Factor XIII cross-links fibrin(ogen) independent of fibrin polymerization in experimental acute liver injury. Blood. 2021;137(18):2520–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Auld GC, Ritchie H, Robbie LA, Booth NA. Thrombin upregulates tissue transglutaminase in endothelial cells: a potential role for tissue transglutaminase in stability of atherosclerotic plaque. Arterioscler Thromb Vasc Biol. 2001;21(10):1689–94. [DOI] [PubMed] [Google Scholar]
- 42.Duval C, Baranauskas A, Feller T, Ali M, Cheah LT, Yuldasheva NY, Baker SR, McPherson HR, Raslan Z, Bailey MA, Cubbon RM, Connell SD, Ajjan RA, Philippou H, Naseem KM, Ridger VC, Ariens RAS. Elimination of fibrin gamma-chain cross-linking by FXIIIa increases pulmonary embolism arising from murine inferior vena cava thrombi. Proc Natl Acad Sci U S A. 2021;118(27). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.