

Abstract
The clustered regularly interspaced short palindromic repeats (CRISPR)-Cas12a system is composed of a Cas12a effector that acts as a DNA-cleaving endonuclease and a crispr RNA (crRNA) that guides the effector to the target DNA. It is considered a key molecule for inducing target-specific gene editing in various living systems. Here, we improved the efficiency and specificity of the CRISPR-Cas12a system through protein and crRNA engineering. In particular, to optimize the CRISPR-Cas12a system at the molecular level, we used a chimeric DNA-RNA guide chemically similar to crRNA to maximize target sequence specificity. Compared with the wild-type (wt)-Cas12a system, when using enhanced Cas12a system (en-Cas12a), the efficiency and target specificity improved on average by 2.58 and 2.77 times, respectively. In our study, when the chimeric DNA-RNA-guided en-Cas12a effector was used, the gene-editing efficiency and accuracy were simultaneously increased. These findings could contribute to highly accurate genome editing, such as human gene therapy, in the near future.
Keywords: RNA/DNA editing, highly specific, efficient, chimeric DNA-RNA, genome editing, en-Cas12a, gene therapy
Graphical abstract
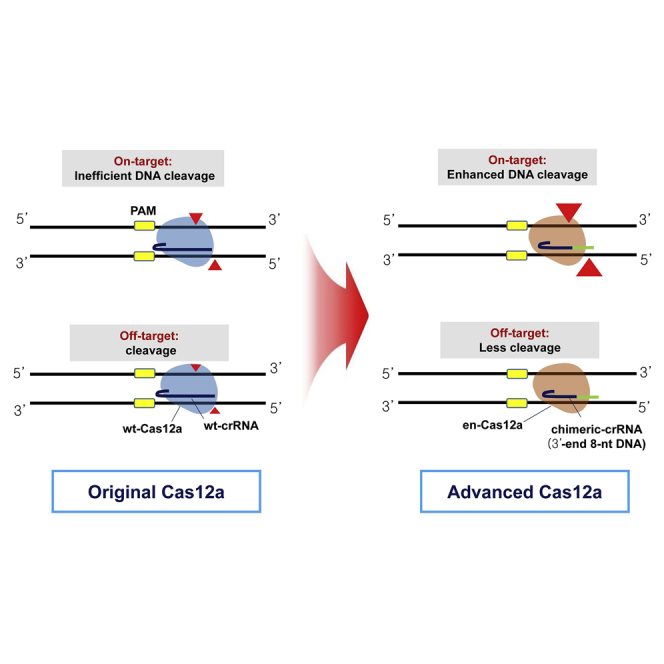
We demonstrated that the chimeric DNA-RNA-guided en-Cas12a-based genome-editing system developed in this paper serves as a versatile tool for highly specific genome editing in living organisms. This is the first study to show that simple engineering of the Cas12a component could simultaneously enhance the efficiency and accuracy of previous Cas12a systems.
Introduction
The clustered regularly interspaced short palindromic repeats (CRISPR)-Cas system, which is known to be a bacterial defense system, is composed of Cas endonuclease and guide RNA; it is known to operate in various living organisms.1, 2, 3 Recently, it has been used as a key tool for in vivo therapeutics because it can be reprogrammed specifically for a target gene. Thus, it is easy to use the CRISPR-Cas system to access genetic diseases.4,5 The field of gene therapy is growing into a large market in which these advanced genome-editing tools are frequently employed; thus, it is important to determine whether the CRISPR-Cas system can accurately induce mutations into a target.6,7 The target sequence specificity of CRISPR occurs because of molecular-level interactions resulting from its intrinsic properties.8, 9, 10 CRISPR-Cas endonuclease recognizes target DNA based on the complementary nucleotide sequence contained in the guide RNA. CRISPR-Cas recognizes the protospacer adjacent motif (PAM) sequence in the target gene through the PAM interaction domain, melts the DNA double helix, and propagates the hybridization of guide RNA and target DNA to form a stable R loop that induces target DNA cleavage.11, 12, 13, 14, 15 It has been reported that the hybridization between the guide RNA and the target DNA, which is formed to aid the CRISPR-Cas system in stably binding to the target DNA, is approximately 20–24 bp; it can have various mismatch tolerances depending on the target sequence.16, 17, 18, 19, 20 Accordingly, the possibility of inducing cleavage to off-targets similar to the target sequence has been reported, and efforts have been made to reduce such errors.9,21, 22, 23
Among CRISPR-Cas endonucleases, the CRISPR-Cas12a system, which belongs to class II and type V, has excellent target specificity.13,24, 25, 26, 27 Therefore, it has attracted much attention as an accurate genome-editing tool for use as a therapeutic agent for humans in the future. Unfortunately, the CRISPR-Cas12a system has also been reported to have a tolerance for mismatches in the intermediate region (8–9 bp) or in the PAM distal region inside the protospacer, which is necessary for target recognition.18 This off-target cleavage effect appears to be more serious for engineered CRISPR-Cas12a, which has enhanced target recognition and improved gene-editing efficiency.28,29 When considering gene therapy for human systems in the future, efficiency and safety will likely be important issues; they must be addressed simultaneously to improve the CRISPR-Cas12a system.
In this study, we devised a technology that dramatically lowers the induction of off-target mutations while efficiently inducing on-target mutations by effectively recognizing various target nucleotide sequences in human-derived cell lines. When using this enhanced Cas12a system (en-Cas12a) with strong target recognition, the average efficiency of inducing mutations in the target sequence increased (1.7- to 16.9-fold) when guided by a chimeric DNA-RNA guide in comparison with the wild-type Cas12a (wt-Cas12a) system. In addition, the average (0.5%–10.6%) of the mutation induction efficiency of off-target nucleotide sequences was reduced (0.1%–3.6%) by using a chimeric DNA-RNA guide, which increased the target specificity 2.8-fold on average. Using the chimeric DNA-RNA guide-based en-Cas12a system developed in this study, it is possible to induce target-specific, high-efficiency gene editing. Therefore, our proof-of-concept study could contribute in the near future to the fundamental treatment of various incurable human diseases resulting from genetic mutations.
Results
Comparison of target DNA cleavage activity of chimeric DNA-RNA-guided en-AsCas12a and wt-AsCas12a
The CRISPR-Cas12a system uses single-stranded crispr RNA (crRNA) to hybridize target DNA with 20 bases, form a stable R loop, and induce target DNA cleavage. When the amino acid residues (Lys548, Ser542, and Glu174) interacting near the PAM (TTTN) sequence were changed to positive residues for the interaction between Cas12a and the target DNA (Figure 1A, left inset), the target-induced indel ratio (%) was improved for various genes.28 From these results, we speculate that PAM recognition contributes to the kinetics of the entire Cas12a target recognition and DNA cleavage process, and that it can eventually affect stable R-loop formation through the hybridization of DNA and crRNA. The target specificity (on-target editing/off-target editing) of the Cas12a system has previously been optimized by substituting DNA for the 3′ end of the crRNA to change the hybridization energy between crRNA and target DNA (Figure 1A, right inset).30 Based on this system, here we attempted to maximize the target specificity and genome-editing efficiency using en-AsCas12a (Acidaminococcus sp. Cas12a), which has enhanced target recognition. First, to improve target specificity by changing the binding energy of the target DNA-crRNA hybridization region, we gradually substituted the crRNA with DNA; we then confirmed the influence of this substitution on the target DNA cleavage for wt-AsCas12a and en-AsCas12a effectors (Figure 1B). Amplicon cleavage assays were performed on the target nucleotide sequences of both genes (DNMT1 and CCR5-site2) (Figure S1). When DNA was gradually substituted from the 3′ end of the crRNA (recognized by AsCas12a), en-AsCas12a showed improved target recognition compared with wt-AsCas12a; it demonstrated more tolerance to the DNA substitution of crRNA (Figures 1B and S2). As previously reported,30 when 12 or more DNAs are substituted from the 3′ end of Cas12a, the cleavage activity of the DNA amplicon is reduced, and almost no activity is shown in substitutions over 16 nt DNAs. However, en-AsCas12a showed robust cleavage activity after 12 nt DNA substitutions at the 3′ end of crRNA, but showed a decrease in activity by more than half following 16 nt or more DNA substitutions. This indicates that en-AsCas12a is more tolerant than wt-AsCas12a to DNA substitution at the 3′ end of crRNA, which is advantageous for target DNA cleavage.
Figure 1.

Comparison of target DNA cleavage activity of chimeric DNA-RNA guided en-Cas12a and wt-Cas12a
(A) Structure of the target-strand DNA-crRNA-AsCas12a complex (PDB: 5B43). Left inset: amino acids in AsCas12a interacting with around the PAM sequence in the target DNA. Right inset: amino acid (Lys414) interacting with the target-strand DNA-crRNA duplex in AsCas12a (hydrogen bonding with the 2′-OH group on the crRNA 3′ end region). (B) Comparison of cleavage efficiency of DNA amplicons using chimeric DNA-RNA guided wt-AsCas12a (light purple) and en-AsCas12a (dark purple). Each chimeric DNA-RNA includes target nucleotide sequences (DNMT1, CCR5-site2) with gradual DNA substitution from the 3′ end of crRNA. NC, negative control; WT, wild-type crRNA was treated with wt- or en-AsCas12a. The RNA region of the (cr)RNA is shown in blue, and the substituted DNA region is shown in red (8DNA to 44DNA indicates number of substituted DNA nucleotides in (cr)RNA). The x axis indicates the efficiency of the target gene (DNMT1-site1, CCR5-site2) cleavage by wt- and en-AsCas12a using various chimeric DNA-RNA guides (gradual DNA substitution from the 3′ end of the (cr)RNA). All cleavage efficiencies were calculated from band intensity, which was separated on 2% agarose gel (cleaved fragment intensity [%]/total fragment intensity [%]). All calculated values were normalized to wild-type (cr)RNA (Figure S2). The histograms represent means ± SEM from three independent experimental values. p values were calculated using two-way ANOVA with Sidak’s multiple comparisons test (ns, not significant; ∗p < 0.0332, ∗∗p < 0.0021, ∗∗∗p < 0.0002, ∗∗∗∗p < 0.0001).
Optimization of the genome-editing activity of en-AsCas12a based on a chimeric DNA-RNA guide to a target nucleotide sequence on the intracellular genome
To check whether the engineered en-AsCas12a effector, based on this chimeric DNA-RNA guide, could effectively induce target-specific gene mutations in human cells, we used various chimeric DNA-RNA guides to induce mutations and analyzed the efficiency in comparison with wt-AsCas12a. Comparative analysis of mutation induction efficiencies for the target nucleotide sequences of three genes (DNMT1, IL2A-AS1, and CCR5-site1) revealed that engineered en-AsCas12a outperformed wt-AsCas12a in terms of editing efficiency (Figures 2 and S1). In particular, the induction of gene mutations targeting intracellular loci showed a different trend from that of amplicon cleavage (Figure 1B). Unlike wt-AsCas12a, which exhibited a significantly lower operating efficiency based on chimeric DNA-RNA guides for the target sequence in the genome, engineered en-AsCas12a allowed up to 8 nt DNA substitutions (8DNA) from the 3′ end of the crRNA while maintaining the editing activity (Figures 2, S3, S4, and S8). Surprisingly, engineered en-AsCas12a showed 1.7- to 16.9-fold improvements in mutation induction efficiency compared with wt-AsCas12a when the 3′ end of crRNA was substituted with 8 nt DNA to increase target specificity. This effect was universally confirmed in various genes (CCR5-site2, FANCF); on average, a 7.3-fold higher recovery was achieved (Figures S3, S4, and S8). Previous studies have reported that when AsCas12a targets the intracellular genome and operates on the basis of a chimeric DNA-RNA guide it is difficult to induce mutations in the target nucleotide sequence, owing to many restrictions on the topology of the intracellular genome.30 Accordingly, in this study we attempted to change the genome topology near the target sequence by using nickase. We compared the resulting changes in the genome-editing efficiency of the target sequence for wt-AsCas12a and en-AsCas12a (Figures S1, S3, S4, and S8). In the case of wt-AsCas12a, the mutation induction efficiency, which was reduced by the use of an 8 nt DNA-substituted chimeric DNA-RNA guide (8DNA), was completely recovered by co-treatment with nickase. In the case of en-AsCas12a, the mutation induction efficiency was maintained at a level similar to that of wt-crRNA using chimeric DNA-RNA (8DNA); it was not significantly affected by nickase (Figures 2, S3, S4, and S8). Therefore, these data indicate that using the en-AsCas12a effector, based on the chimeric DNA-RNA guide (8DNA), enables more effective genome editing than wt-AsCas12a when inducing mutations on the target DNA, without the help of nickase.
Figure 2.

Comparison and optimization of genome-editing efficiency of en-Cas12a and wt-Cas12a based on chimeric DNA-RNA guide targeting endogenous locus in cell lines (HEK293FT)
Comparison of genome-editing efficiency (%) of wt-AsCas12a (light color) and en-AsCas12a (dark color) using a chimeric DNA-RNA guide for human-derived cell line (HEK293FT). Comparison of the indel induction efficiency (%) in intracellular genomic target sequences DNMT1-site1 (A), IL2A-AS1 (B), and CCR5-site1 (C) by gradual DNA substitution of the 3′ end of crRNA. All the indel ratios (%) were calculated from targeted amplicon sequencing (indel ratio [%] = mutant DNA read number/total DNA read number). Data are shown as means ± SEM from three independent experiments. p values were calculated using two-way ANOVA with Sidak’s multiple comparisons test (ns: not significant; ∗p < 0.0332, ∗∗p < 0.0021, ∗∗∗p < 0.0002, ∗∗∗∗p < 0.0001). NC, negative control; WT, wild-type crRNA was treated with wt- or en-AsCas12a, 8DNA to 44DNA. Chimeric crRNA (sequential 8DNA to 44DNA substitution at 3′ end of crRNA) was treated with wt- or en-AsCas12a.
Improving target specificity for inducing genetic mutations in the intracellular genome using chimeric DNA-RNA-guided engineered en-AsCas12a
Next, we compared the chimeric DNA-RNA guide-based engineered en-AsCas12a and wt-AsCas12a effectors regarding their target specificities under optimized conditions (3′ end 8DNA substitution of crRNA) that effectively induced mutations in the target sequences (Figures 3, S1, S5, S6, and S9). We performed mutation induction experiments with the Cas12a system for various genes in advance and then selected gene sites (CCR5, AAVS1, DNMT1) with a high off-target indel ratio. For these gene sites, mutations were induced using wt-AsCas12a and en-AsCas12a effectors, after which the on-/off-target activity was directly compared by performing sequencing-based analysis. In the case of wt-Cas12a-based targeting of the CCR5-site1, the target mutation induction efficiency was greatly reduced by the use of chimeric DNA-RNA (Figure 3A). Unlike wt-AsCas12a, in which mutation induction efficiency was recovered in a nickase-dependent manner, for en-AsCas12a the target mutation efficiency was maintained in a nickase-independent manner by using chimeric DNA-RNA, in which the 3′ end was substituted with 8 nt DNA (Figure 3B). The nickase dependency was lowered by 11.25 times (Figure 3C) and target specificity was increased by 2.79 times (Figure 3D) using en-AsCas12a. In addition, when the chromosome topology near the target sequence was changed using nickase, the target specificity using chimeric DNA-RNA was further increased by 3.45-fold compared with that of wt-AsCas12a (Figure 3D). We further compared the target specificities of the engineered en-AsCas12a and wt-AsCas12a effectors in the target sequences of two other genes (AAVS1-site1 and DNMT1-site2) (Figures S5 and S6). When a chimeric DNA-RNA guide was used for AAVS1-site1, neither en-AsCas12a nor wt-AsCas12a effectors showed nickase dependence, and gene mutations were induced with similar efficiencies to conditions using wt-crRNA (Figure S5). However, when using the en-AsCas12a effector to target the AAVS1-site1 sequence, the indel ratio (%) was significantly higher (3.9-fold) than that of wt-Cas12a. However, there were also more unintentional mutations in the off-target sequence (off-target1) (Figures S5A and S5B). We confirmed that the number of mutations induced in the off-target1 sequence was dramatically reduced by the use of chimeric DNA-RNA, in which the 3′ end was substituted with 8 nt DNA. Therefore, the overall target specificity was increased 3.5-fold compared with that of wt-AsCas12a when chimeric DNA-RNA (8DNA)-guided en-AsCas12a was used (Figure S5D). The DNMT1-site2 showed the same trend as the AAVS1-site1 locus targeted by the Cas12a system (Figure S6). In the case of the en-AsCas12a effector based on the chimeric DNA-RNA guide with 8 nt DNA substitution at the 3′ end, the indel ratio (%) was significantly increased (1.9-fold) compared with that of wt-AsCas12a. Furthermore, the off-target nucleotide sequence (off-target1, 2)-induced mutations were dramatically reduced (Figures S6A and S6B). As a result, the target specificity was increased 2-fold, regardless of the use of nickase (Figures S6C and S6D). We summarized the data to see at a glance the effect of inducing mutations targeting various genes using wt- or en-AsCas12a. These results show that when mutations are induced in target DNA using chimeric-crRNA-based en-AsCas12a, the off-targeting effect can be reduced while maintaining target mutagenesis activity for various genes in different cell lines (Figures 4, S7, and S10). In conclusion, by inducing mutations in multiple genes using chimeric DNA-RNA-guided en-AsCas12a, the targeted indel ratio (%) was improved 2.6-fold on average without the help of nickase. The off-target mutation induction efficiency was also reduced. Eventually, the target specificity was improved 2.8-fold compared with that of wt-AsCas12a.
Figure 3.

Comparison of genome-editing specificity of en-AsCas12a and wt-AsCas12a based on chimeric DNA-RNA guide targeting endogenous locus in cell lines (HEK293FT)
Upper table shows the on-target nucleotide sequence (On) for target gene (CCR5-site1) and the corresponding off-target nucleotide sequence (OT1, OT2) predicted from in silico analysis (Bae et al. 31). Underlining indicates the PAM (TTTN) nucleotide sequence, and the nucleotides mismatched with the target sequence in the off-target are indicated in red. (A and B) Indel ratio (%) of the wt-AsCas12a-based (A) or en-AsCas12a-based (B) editing on the endogenous target sequences (on-/off-target sites for CCR5-site1) using wt-crRNA (WT) and 3′ end 8 nt DNA-substituted crRNA (8DNA). NC, negative control; Only_Cas12a, only protein treated; nCas9, nickase Cas9 (D10A). (C and D) Nickase dependency (C) and target specificity (D) were calculated from next-generation sequencing results. Nickase dependency = (without [w/o] nCas9 editing [%]/with [w/] nCas9 editing [%]); Target specificity = (on-target editing [%]/off-target editing [%]). Histograms represent means ± SEM from three independent experimental values. p values were calculated using two-way ANOVA with Sidak’s multiple comparisons test (ns, not significant; ∗p < 0.0332, ∗∗p < 0.0021, ∗∗∗p < 0.0002, ∗∗∗∗p < 0.0001).
Figure 4.

Comparison of the indel ratio (%) of on-target and off-target sites for various genes (AAVS1, CCR5, DNMT1, FANCF, IL12A_AS1) when wt-AsCas12a- or en-AsCas12a-based editing was attempted in cell lines (HEK293FT)
After inducing genetic mutations based on wt- or en-AsCas12a, the results of targeted amplicon sequencing for various targets (AAVS1, CCR5, DNMT1, FANCF, IL12A_AS1) and predicted corresponding off-target sites were analyzed and plotted. WT, wild-type crRNA was treated with wt- or en-AsCas12a; 8DNA, chimeric crRNA (sequential 8 nt DNA substitution at 3′ end of crRNA) was treated with wt- or en-AsCas12a. Each horizontal bar indicates the mean value of indel ratio (%). p values were calculated using two-tailed Student’s t test (ns, not significant; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001).
A model for improving target specificity and genome-editing efficiency of en-Cas12a, based on chimeric DNA-RNA guides
Combining all of the above findings, the results of the working mechanism of en-Cas12a, compared with the existing wt-Cas12a, are presented in Figure 5, based on the chimeric DNA-RNA guide. When wt-Cas12a was used to cleave the target sequence in the genome of the cell, there was tolerance for mismatches between the protospacer (20 bp) middle part and the PAM (TTTN) distal region, so there was a possibility that Cas12a could recognize and cleave off-target sequences. When wt-Cas12a, based on the chimeric DNA-RNA guide, was used (in which the 3′ end was substituted with 8 DNAs), the off-target effect could be reduced by destabilizing the binding of Cas12a to DNA due to crRNA-target strand DNA distortion.32 For the off-target seqeunce, DNA cleavage property is more severely hampered by mismatch incorporation in the protospacer region than perfectly matched on-target sequence. However, when chimeric DNA-RNA-based cleavage was performed on the genome in the cell, the mutation-inducing effect on the target nucleotide sequence was largely decreased. In this case, the target specificity was increased only by changing the topology of the genome with the simultaneous use of nickase (Figures 3A–3C). For the en-Cas12a, in which target sequence recognition is reinforced by engineering the interacting amino acids of the PAM sequence recognition part, its efficiency in inducing target sequence mutations for various genes was increased compared with that of Cas12a, but its unintended off-target effects also increased greatly. Using the chimeric DNA-RNA-based en-Cas12a effector to target the intracellular genome can dramatically reduce the effects of off-target mutations. In general, en-Cas12a shows a largely enhanced DNA-targeting property under a chimeric DNA-RNA (3′ end 8 nt DNA substitution) guided condition than wt-Cas12a. Without the help of nickase, it is thus possible to increase the target sequence editing efficiency and dramatically lower the off-target sequence editing efficiency. Regarding the efficient induction of gene mutations with respect to the use of chimeric guides, accurate and high-efficiency gene targeting is possible when using chimeric DNA-RNA-based en-Cas12a.
Figure 5.

A model for enhancing target specificity and editing efficiency of en-Cas12a based on chimeric DNA-RNA-guided engineering
(Upper left) wt-crRNA guided wt-Cas12a system. In general, the wt-Cas12a effector can induce genetic mutations in target sequences but can also induce mutations in similar off-target sequences. (Bottom left) wt-crRNA-guided en-Cas12a system. Recognition of a target sequence is enhanced by an effector engineered by amino acid substitution, and thus the efficiency of inducing gene mutations is increased compared with that of the wt-Cas12a effector. However, due to the same principle as target sequence recognition, there is a problem in that the mutation induction efficiency of off-target sequences is also increased. (Upper right) Chimeric-crRNA-guided wt-Cas12a system. Effectively reduced off-target mutation induction efficiency when using a chimeric DNA-RNA guide with 8 nt DNA substituted at the 3′ end. However, the efficiency of mutation induction for the target nucleotide sequence in the genome is also reduced, and the efficiency is restored only when there is the action of nickase on the nucleotide sequence near the target. (Bottom right) Chimeric-crRNA-guided en-Cas12a system. This maximizes the target sequence indel ratio (%) and minimizes the off-target indel ratio (%) when using a chimeric DNA-RNA (8DNA)-guided en-Cas12a effector. It can induce more accurate and high-efficiency gene editing than wt-Cas12a on genomic DNA. PAM (TTTN) sequence for Cas12a effector is indicated by the yellow box. DNA cleavage points are indicated by red arrowheads, and the degree of cleavage is indicated by arrowhesd size according to the Cas12a activity. The wt-Cas12a and en-Cas12a effectors are shown in blue and brown, respectively. In the wt-crRNA and chimeric DNA-RNA guides, RNA is indicated in dark blue and nucleotides replaced with DNA in green.
Discussion
The CRISPR-Cas12a effector is attracting attention as a potential future target-specific genome-editing tool as it is known to be capable of inducing mutations in a target sequence on a desired gene; it also has the highest target specificity among previously known CRISPR systems. However, the Cas12a system has been reported to have lower activity than Cas9 in general, and there remains room to improve the properties of the endonuclease itself for applications in various in vivo conditions. Efforts have been made to more effectively recognize the target DNA sequence and induce cleavage by engineering the Cas12a system.28,29 These studies have shown overall improved activity compared with wt-Cas12a in various genes, through enhanced binding, by changing amino acids around the domain, within Cas12a, and by recognizing the PAM sequence in the target DNA. However, most CRISPR endonucleases induce double-stranded cleavage by forming an R loop through the complementary binding of target-strand DNA and crRNA. In general, as the tolerance of mismatch recognition increases due to enhanced binding affinity, the chance of off-target binding also increases, and we believe that the effects of off-target binding would be maximized when using engineered Cas12a systems. In the future, if a genome-editing technology is directly applied to the human body, the target specificity of the CRISPR system can be a very important issue. For the gene-editing system to work efficiently in the human body, safety issues must also be considered in parallel with editing efficiency. Therefore, a method for increasing target specificity is also required, in parallel with methods to enhance activity.
Previous studies have improved target specificity by applying crRNA engineering to the Cas12a system; target-specific gene mutations have been effectively induced by optimizing the length of DNA substitution. Mismatch tolerance has been reduced by changing the complementary binding energy of target-strand DNA and crRNA through sequential 8 nt DNA substitutions at the 3′ end of crRNA. This principle confirms that the induction of off-target mutations can be reduced while maintaining the efficiency of inducing target mutations. Based on this, here we used chimeric DNA-RNA crRNA to engineer en-AsCas12a, which displayed maximized target sequence recognition and improved mutation induction efficiency in various target nucleotide sequences in comparison with wt-AsCas12a. Surprisingly, the induction of off-target mutations was dramatically decreased, and eventually the target specificity was largely improved. Interestingly, the observed discrepancy between amplicon cleavage (Figure 1B) and genome editing inside the cell (Figure 2) showed that the cleavage activity of Cas12a endonuclease is greatly influenced by DNA topology. Chimeric DNA-RNA crRNA-based en-AsCas12a and wt-AsCas12a displayed differing sensitivities to the intracellular genome, so en-AsCas12a showed higher gene mutation induction efficiency than wt-AsCas12a. These results suggest that en-AsCas12a can, to some extent, overcome structures that are unfavorable to target DNA cleavage due to unstable R-loop formation (due to the DNA substitution of crRNA in the genomic sequence). This is achievable by enhancing PAM recognition through protein engineering. In general, the low operating efficiency induced in the intracellular genome by wt-Cas12a (based on the DNA substitution of crRNA to improve target specificity) could be recovered by changing the genome topology by using nickase around the target sequence. However, in the case of en-Cas12a, it was possible to induce a significant amount of mutation following up to eight DNA substitutions at the 3′ end of the crRNA, without the help of nickase. Therefore, when using chimeric DNA-RNA guide-based en-AsCas12a, it was possible to simultaneously improve the genome-editing efficiency (%) and the target specificity (on-target editing [%]/off-target editing [%]) compared with those of wt-AsCas12a by changing the hybridization energy of the target DNA strand and crRNA. In addition, when analyzing the results in our study, although weak compared with wt-Cas12a, the editing function of en-Cas12a is likely to be slightly increased by the topology change by nickase treatment. Although the engineered Cas12a system shows improved target specificity while maintaining high efficiency, genome editing was only effectively conducted by sequential delivery of an en-Cas12a expression plasmid into human-derived cell lines and a synthesized chimeric DNA-RNA guide rather than delivery with mixture of purified protein and chimeric DNA-RNA guide. To enable effective gene editing in the future by applying this technology to living tissues, it is thought that the technology such as the mRNA form expression system of the en-Cas12a and the highly efficient lipid carrier must be fused for more effective delivery.
In this study, we developed a technology that maximizes the safety and efficiency of genome editing using chimeric DNA-RNA crRNA-based en-Cas12a. In the future, many improvements are needed in terms of efficiency and safety regarding the application of gene therapy to humans using the CRISPR system or for precise gene editing in in vivo systems. Through this technology, it is expected that the safety and efficacy of various CRISPR endonucleases can be optimized in a similar way when applied in vivo.
Materials and methods
Preparation of the CRISPR-Cas12a recombinant protein and chimeric guides
wt- and en-AsCas12a recombinant proteins were prepared for the in vitro DNA cleavage assay. Codon-optimized AsCas12a (Acidaminococcus sp. Cas12a) coding sequence was cloned into a pET28a bacterial expression vector and then transformed into BL21 (DE3) Escherichia coli cells. Transformed bacterial colonies were cultured at 37°C until the optical density reached 0.6, after which isopropylthio-β-galactoside (IPTG) inoculation was performed. After 48 h, E. coli cells were precipitated at 4°C and 5,000 rpm, following which the culture medium of the upper layer was removed. The precipitated E. coli cell pellet was resuspended in lysis buffer (10 mM β-mercaptoethanol, 300 mM NaCl, 20 mM Tris-HCl [pH 8.0], 1 mM PMSF, and 1% Triton X-100). To disturb the bacterial cell membrane, we performed sonication on ice water for 3 min, following which the cell lysate was precipitated for 10 min at 5,000 rpm at 4°C. Next, the nitrilotriacetic acid (Ni-NTA) resin was pre-washed with wash buffer (20 mM Tris-HCl [pH 8.0], 300 mM NaCl), and the precipitated cell lysate was stirred at 4°C for 90 min. Washing was performed with 10× the volume of wash buffer to remove non-specific binding components in the mixed cell lysate solution. For the elution of AsCas12a protein, an elution buffer (20 mM Tris-HCl [pH 8.0], 300 mM NaCl, 200 mM imidazole) was used and finally exchanged against the storage buffer (200 mM NaCl, 50 mM HEPES [pH 7.5], 1 mM dithiothreitol [DTT], 40% glycerol) using a Centricon (Millipore, Amicon Ultra-15), and stored at −80°C. Chimeric DNA-RNA oligonucleotides (Bioneer) were synthesized for each target gene sequence and dissolved in diethyl pyrocarbonate (DEPC) water, then stored at −80°C (Table S1).
Preparation of the guide RNA for Cas12a and nCas9 (D10A)
Guide RNAs for Cas12a and nCas9 (D10A) were generated by in vitro transcription (Tables S1 and S2). A DNA template for in vitro transcription was constructed using annealing or extension PCR with sense and antisense DNA oligonucleotides (Macrogen) containing the target DNA sequence (Table S3). DNA templates were mixed with T7 RNA polymerase (New England Biolabs, M0251L) and reaction buffer mixture (50 mM MgCl2, 100 mM ribonucleoside triphosphate [Jena Bio, NU-1014], 10× RNA polymerase reaction buffer, 100 mM DTT, RNase inhibitor Murine, DEPC), and incubated at 37°C. After 16 h, to remove the original DNA template, DNase I was added and the mixture was incubated for another 1 h at 37°C. The transcribed RNA was purified using a column (GENECLEAN Turbo Kit; MP Biomedicals). The purified RNA was concentrated through lyophilization (2,000 rpm) at −55°C for 1 h.
In vitro DNA cleavage assay
On-/off-target site PCR amplicons of each gene (DNMT1, CCR5, IL2A-AS1, and AAVS1) were obtained from purified human genomic DNA using DNA primers (Table S3). The purified target PCR amplicon was incubated with purified recombinant wt- or en-Cas12a protein and crRNA (RNA guides or chimeric DNA-RNA guides) in 10× buffer (NEBuffer3.1; New England Biolabs) for 1 h. After adding a stop buffer (100 mM EDTA, 1.2% SDS) to stop the reaction, the cleaved fragment was separated using 2% agarose gel electrophoresis. DNA cleavage efficiency (cleaved fragment intensity [%]/total fragment intensity [%]) was measured using ImageJ software (NIH).
Cell culture and transfection
The HEK293FT (ATCC) cell line was cultured in Dulbecco’s modified Eagle medium (DMEM; Gibco) with 10% fetal bovine serum (Gibco) at 37°C and in 5% CO2. To ensure efficient chimeric DNA-RNA guide delivery, we performed sequential transfection with wt- or en-Cas12a expression vectors and crRNAs. For the primary transfection, 105 cells were mixed with a plasmid vector (AsCas12a, n-SpCas9 (D10A, H840A)) and 20 μL of electroporation buffer (Lonza, V4XC-2032) and were nucleofected according to the manufacturer’s instructions (program: CM-137). The transfected cells were transferred to a 24-well plate with 500 μL of medium and incubated at 37°C in 5% CO2. Twenty-four hours after primary transfection, for secondary transfection crRNA (200 pmol), single guide RNA (30 pmol), 1 μL of P3000, and 1.5 μL of Lipofectamine 3000 reagent (Thermo Fisher Scientific) were mixed in 50 μL of Opti-MEM (Gibco), incubated for 10 min, and added to DMEM medium. Forty-eight hours after the second transfection, cells were harvested and genomic DNA was extracted using a genomic DNA purification kit (DNeasy Blood & Tissue Kit; Qiagen).
Targeted amplicon sequencing and data analysis
To analyze the indel frequency of the on-/off-target locus of each gene, we performed targeted deep sequencing using PCR amplicons. The Cas-OFFinder (http://www.rgenome.net/cas-offinder/) web tool was used to select potential off-target sites corresponding to each on-target site. For the preparation of PCR amplicons, PCR amplification was performed using DNA primers corresponding to each endogenous locus (Table S3). To add adapter and index sequences to each 5′ and 3′ end, we performed nested PCR using Phusion High-Fidelity DNA Polymerase (Thermo Fisher). After index tagging, the PCR amplicon mixture was analyzed using a Mini-Seq (Illumina, SY-420-1001) according to the manufacturer’s guidelines. Sequencing read fatstq files were analyzed using Cas-Analyzer (http://www.rgenome.net/cas-analyzer/), and the indel ratio (mutant DNA read number/total DNA read number) was calculated from the read number.
Data and code availability
CRISPR RGEN Tools is an open-source collaborative initiative available in the repository (http://www.rgenome.net/). Targeted deep-sequencing data are available at the NCBI Sequence Read Archive under accession number SRP334002.
Acknowledgments
The authors thank the members of the National Primate Research Center for their helpful discussions. This research was supported by grants from the National Research Foundation funded by the Korean Ministry of Education, Science and Technology (NRF-2019R1C1C1006603, NRF-2022R1A2C4001609) and the Korean Ministry of Science and ICT program (NRF-2017M3A9D5A01072797) through the National Research Foundation and the Korea Medical Device Development Fund grant funded by the Korean government (the Ministry of Science and ICT, Ministry of Trade, Industry and Energy, Ministry of Health & Welfare, Ministry of Food and Drug Safety) (project number: 9991006929, KMDF_PR_20200901_0264). The study was also supported by grants from the Korea Research Institute of Bioscience and Biotechnology (Research Initiative Program KGM5282113, KGM4562121, KGM5382113).
Author contributions
Conceptualization, H.K., W.L., and S.H.L.; methodology, H.K., W.L., and S.H.L.; software, H.K., W.L., and S.H.L.; validation and formal analysis, H.K., W.L., C.H.K., Y.O., L.W.G., H.L., W.J.S., and J.K.H.; investigation, H.K., W.L., and S.H.L.; resources, K.-S.L., K.J.J., K.-H.N., Y.-S.W., K.-R.L., Y.L., Y.-H.K., J.-W.H., B.-H.J., and S.H.L.; data curation, H.K., W.L., and S.H.L.; writing –original draft, H.K., W.L., B.-H.J., D.-S.L., and S.H.L.; writing – review and editing, H.K., W.L., B.-H.J., D.-S.L., and S.H.L.; visualization, H.K., W.L., and S.H.L.; supervision, D.-S.L. and S.H.L.; project administration, B.-H.J., D.-S.L., and S.H.L.; funding acquisition, Y.-H.K., Y.L., J.-W.H., and S.H.L.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2022.03.021.
Contributor Information
Bong-Hyun Jun, Email: bjun@konkuk.ac.kr.
Dong-Seok Lee, Email: lee1@knu.ac.kr.
Seung Hwan Lee, Email: lsh080390@cau.ac.kr.
Supplemental information
References
- 1.Barrangou R., Fremaux C., Deveau H., Richards M., Boyaval P., Moineau S., Romero D., Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 2.Doudna J.A., Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- 3.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gillmore J.D., Gane E., Taubel J., Kao J., Fontana M., Maitland M.L., Seitzer J., O'Connell D., Walsh K.R., Wood K., et al. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N. Engl. J. Med. 2021;385:493–502. doi: 10.1056/NEJMoa2107454. [DOI] [PubMed] [Google Scholar]
- 5.Frangoul H., Altshuler D., Cappellini M.D., Chen Y.S., Domm J., Eustace B.K., Foell J., Fuente J., Grupp S., Handgretinger R., et al. CRISPR-Cas9 gene editing for sickle cell disease and beta-thalassemia. N. Engl. J. Med. 2021;384:252–260. doi: 10.1056/NEJMoa2031054. [DOI] [PubMed] [Google Scholar]
- 6.Han H.A., Pang J.K.S., Soh B.S. Mitigating off-target effects in CRISPR/Cas9-mediated in vivo gene editing. J. Mol. Med. 2020;98:615–632. doi: 10.1007/s00109-020-01893-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang X.H., Tee L.Y., Wang X.G., Huang Q.S., Yang S.H. Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol. Ther. Nucleic Acids. 2015;4:e264. doi: 10.1038/mtna.2015.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cofsky J.C., Karandur D., Huang C.J., Witte I.P., Kuriyan J., Doudna J.A. CRISPR-Cas12a exploits R-loop asymmetry to form double-strand breaks. Elife. 2020;9:e55143. doi: 10.7554/eLife.55143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsai S.Q., Joung J.K. Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nat. Rev. Genet. 2016;17:300–312. doi: 10.1038/nrg.2016.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee S.H., Park Y.H., Jin Y.B., Kim S.U., Hur J.K. CRISPR diagnosis and therapeutics with single base pair precision. Trends Mol. Med. 2020;26:337–350. doi: 10.1016/j.molmed.2019.09.008. [DOI] [PubMed] [Google Scholar]
- 11.Globyte V., Lee S.H., Bae T., Kim J.S., Joo C. CRISPR/Cas9 searches for a protospacer adjacent motif by lateral diffusion. EMBO J. 2019;38:e99466. doi: 10.15252/embj.201899466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Swarts D.C., van der Oost J., Jinek M. Structural basis for guide RNA processing and seed-dependent DNA targeting by CRISPR-cas12a. Mol. Cell. 2017;66:221–233.e4. doi: 10.1016/j.molcel.2017.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stella S., Alcon P., Montoya G. Structure of the Cpf1 endonuclease R-loop complex after target DNA cleavage. Nature. 2017;546:559–563. doi: 10.1038/nature22398. [DOI] [PubMed] [Google Scholar]
- 14.Jiang F., Taylor D.W., Chen J.S., Kornfeld J.E., Zhou K., Thompson A.J., Nogales E., Doudna J.A. Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science. 2016;351:867–871. doi: 10.1126/science.aad8282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stella S., Mesa P., Thomsen J., Paul B., Alcon P., Jensen S.B., Saligram B., Moses M.E., Hatzakis N.S., Montoya G. Conformational activation promotes CRISPR-cas12a catalysis and resetting of the endonuclease activity. Cell. 2018;175:1856–1871.e21. doi: 10.1016/j.cell.2018.10.045. [DOI] [PubMed] [Google Scholar]
- 16.Cho S.W., Kim S., Kim Y., Kweon J., Kim H.S., Bae S., Kim J.S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014;24:132–141. doi: 10.1101/gr.162339.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K., Sander J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kleinstiver B.P., Tsai S.Q., Prew M.S., Nguyen N.T., Welch M.M., Lopez J.M., McCaw Z.R., Aryee M.J., Joung J.K. Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat. Biotechnol. 2016;34:869–874. doi: 10.1038/nbt.3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pattanayak V., Lin S., Guilinger J.P., Ma E., Doudna J.A., Liu D.R. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013;31:839–843. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsai S.Q., Zheng Z., Nguyen N.T., Liebers M., Topkar V.V., Thapar V., Wyvekens N., Khayter C., John lafrate A., Le L.P., et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015;33:187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmid-Burgk J.L., Gao L., Li D., Gardner Z., Strecker J., Lash B., Zhang F. Highly parallel profiling of Cas9 variant specificity. Mol. Cell. 2020;78:794–800.e8. doi: 10.1016/j.molcel.2020.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kocak D.D., Josephs E.A., Bhandarkar V., Adkar S.S., Kwon J.B., Gersbach C.A. Increasing the specificity of CRISPR systems with engineered RNA secondary structures. Nat. Biotechnol. 2019;37:657–666. doi: 10.1038/s41587-019-0095-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fu Y., Sander J.D., Reyon D., Cascio V.M., Joung J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014;32:279. doi: 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamano T., Nishimasu H., Zetsche B., Hirano H., Slaymaker I.M., Li Y., Fedorova I., Nakane T., Makarova K.S., Koonin E.V., et al. Crystal structure of Cpf1 in complex with guide RNA and target DNA. Cell. 2016;165:949–962. doi: 10.1016/j.cell.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zetsche B., Gootenberg J.S., Abudayyeh O.O., Slaymaker I.M., Makarova K.S., Essletzbichler P., Volz S.E., Joung J., Oost J., et al. Cpf1 is a single RNA-guided endonuclease of a Class 2 CRISPR-cas system. Cell. 2015;163:759–771. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Safari F., Zare K., Negahdaripour M., Barekati-Mowahed M., Ghasemi Y. CRISPR Cpf1 proteins: structure, function and implications for genome editing. Cell Biosci. 2019;9:36. doi: 10.1186/s13578-019-0298-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dong D., Ren K., Qiu X., Zheng J., Guo M., Guan X., Liu H., Li N., Zhang B., Yang D., et al. The crystal structure of Cpf1 in complex with CRISPR RNA. Nature. 2016;532:522–526. doi: 10.1038/nature17944. [DOI] [PubMed] [Google Scholar]
- 28.Kleinstiver B.P., Sousa A.A., Walton R.T., Tak Y.E., Hsu J.Y., Clement K., Welch M.M., Horng J.E., Lopez J.M., Scarfo I., et al. Engineered CRISPR-Cas12a variants with increased activities and improved targeting ranges for gene, epigenetic and base editing. Nat. Biotechnol. 2019;37:276–282. doi: 10.1038/s41587-018-0011-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang L., Zuris J.A., Viswanathan R., Edelstein J.N., Turk R., Thommandru B., Rube H.T., Glenn S.E., Collingwood M.A., Bode N.M., et al. AsCas12a ultra nuclease facilitates the rapid generation of therapeutic cell medicines. Nat. Commun. 2021;12:3908. doi: 10.1038/s41467-021-24017-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim H., Lee W.J., Oh Y., Kang S.H., Hur J.K., Lee H., Song W.J., Lim K.S., Park Y.H., Song B.S., et al. Enhancement of target specificity of CRISPR-Cas12a by using a chimeric DNA-RNA guide. Nucleic Acids Res. 2020;48:8601–8616. doi: 10.1093/nar/gkaa605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bae S., Park J., Kim J.S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Donohoue P.D., Pacesa M., Lau E., Vidal B., Irby M.J., Nyer D.B., Rotstein T., Banh L., Toh M.S., Gibson J., et al. Conformational control of Cas9 by CRISPR hybrid RNA-DNA guides mitigates off-target activity in T cells. Mol. Cell. 2021;81:3637–3649.e5. doi: 10.1016/j.molcel.2021.07.035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
CRISPR RGEN Tools is an open-source collaborative initiative available in the repository (http://www.rgenome.net/). Targeted deep-sequencing data are available at the NCBI Sequence Read Archive under accession number SRP334002.