

Abstract

We put to the test a recent suggestion [Shee, J., et al. J. Phys. Chem. Lett.2021, 12 (50), 12084–12097] that MP2 regularization might improve the performance of double-hybrid density functionals. Using the very large and chemically diverse GMTKN55 benchmark, we find that κ-regularization is indeed beneficial at lower percentages of Hartree–Fock exchange, especially if spin-component scaling is not applied [such as in B2GP-PLYP or ωB97M(2)]. This benefit dwindles for DSD and DOD functionals and vanishes entirely in the ∼70% HF exchange region optimal for them.
Double-hybrid density functional (DHDF) theory (for reviews, see refs (1−4)) represents a special case of fifth-rung functionals on “Jacob’s Ladder”5 (the fifth rung is where dependence on unoccupied orbitals enters). As such, DHDF theory resides on the seamline between density functional theory (DFT) and wave function approaches. DHDFs are the most accurate DFT methods available to date for main group energetics,6−8 transition metal catalysis,9−13 electronic excitation spectroscopy,14−22 external magnetic23−26 and electric field27,28 induced properties, and vibrational frequencies.29 Its reliance on a second-order perturbation theory term for part of the correlation energy creates an Achilles’ heel for molecules with small band gaps, due to the presence of orbital energy differences in the denominator. Using the spin–orbital notation, the MP2-like Görling–Levy30 nonlocal correlation term (GLPT2) has the following form:
where the indices i and j refer to occupied orbitals and a and b to virtual orbitals, while the energy denominator Δijab = εa + εb – εi – εj.
One remedy that has been proposed in the past, in the context of single-reference or multireference MP2, has been DCPT (degeneracy-corrected perturbation theory31). Another is regularization of the expression presented above to remove the singularity for Δijab → 0. Stück and Head-Gordon32 proposed a simple level-shift regularizer (δ) in the context of ROOMP233−35 (restricted orbital-optimized second-order perturbation theory). They found that this works well for single-bond breaking, but the required level shifts for multiple bonds were so large that they disrupted thermochemistry results.36 Lee and Head-Gordon37 have recently proposed two energy gap-dependent regularizers: σ and κ. Although these forms were developed initially with OOMP2 in mind, Head-Gordon and co-workers38,39 have shown that even without orbital optimization, σ- and κ-MP2 can achieve better accuracy than ordinary (unregularized) MP2 for both main group and transition metal thermochemistry, barrier heights, and noncovalent interactions. They also found that both sets of regularizers performed comparably; hence, we will focus here on only κ-regularized MP2 correlation, which has the following expression:
where κ is a fixed regularization parameter. In the large-κ or large-Δijab limits, the regularization factor approaches unity, while in the small-κ or small-Δijab limits, the corresponding term in the energy summation approaches zero, i.e., the Hartree–Fock energy is recovered.
Reference (37) states that “We are optimistic that this study could pave the way for future development of double-hybrid density functionals based on nonlocal correlation expressions that are more appropriate than conventional MP2 for large dispersion-bound systems and organometallic bonding, yet still free of self-correlation errors. κMP2, σ2MP2, and σMP2 are promising candidates in this regard.”
Such DHDFs as our own minimally empirical dispersion-corrected spin-component scaled families, e.g., revDSD,37 revDOD,37 and xDSD,40 Grimme’s PWPB95,41 or the more heavily parametrized ωB97M(2)42 range-separated DHDF reach accuracies approaching those of composite wave function approaches (see ref (43) for a head-to-head comparison). Yet there might still be room for further improvement, particularly in terms of resilience for systems with small band gaps, and hence significant type A static correlation (also known as absolute near-degeneracy correlation44).
In this letter, we will attempt to confirm or refute the conjecture presented above from ref (37), that is, to determine whether using spin-component-scaled κ-GLPT2 instead of unregularized (i.e., conventional) PT2 correlation can further improve the performance of DSD functionals. For the κ-regularized DSD functionals, the final energy has the following expression:
EN1e stands for the sum of nuclear repulsion and one-electron energy terms. EX,HF represents the exact exchange, and cX,HF the corresponding coefficient. EX,DFT is the exact exchange energy component from the semilocal generalized gradient approximation (GGA), and cX,DFT the corresponding parameter. EC,DFT represents the semilocal GGA correlation component, and cC,DFT the coefficient for that energy part. E2ab,κ-PT2 and E2ss,κ-PT2 are the opposite-spin and same-spin κ-GLPT2 correlation energies, respectively, and their respective linear coefficients are c2ab and c2ss. Finally, Edisp is a dispersion correction such as D3(BJ)45−47 with any associated adjustable parameters: in the work presented here (as in ref (48)), the nonlinear damping-function shape parameters are fixed at a1 = 0 and a2 = 5.5, and the higher-order coefficient is fixed at s8 = 0 (as we have found48 to be appropriate for double hybrids).
We have used the GMTKN55 benchmark suite7 (general main group thermochemistry, kinetics, and noncovalent interactions) throughout. It comprises 55 types of chemical problems, which can be further divided into five major subcategories: thermochemistry of small- and medium-sized molecules, barrier heights, large-molecule reactions, intermolecular interactions, and intramolecular interactions (or conformer energies). WTMAD2 (weighted mean absolute deviation) as defined in eq 2 of ref (7) has been used as our primary metric:
where 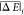 is the mean absolute value of all of the
reference energies from i = 1 to 55, Ni is the number of systems in each subset,
and MADi is the mean absolute difference
between calculated and reference energies for each of the 55 subsets.
For the details of all 55 subsets with proper references, see Table
1 of ref (7).
is the mean absolute value of all of the
reference energies from i = 1 to 55, Ni is the number of systems in each subset,
and MADi is the mean absolute difference
between calculated and reference energies for each of the 55 subsets.
For the details of all 55 subsets with proper references, see Table
1 of ref (7).
All electronic structure calculations were performed using the QCHEM 5.449 package on the ChemFarm HPC cluster in the Faculty of Chemistry at the Weizmann Institute of Science. The Weigend–Ahlrichs def2-QZVPP50 basis set was used for all subsets except seven, for which we used the diffuse-function augmented variant def2-QZVPPD51 instead: the rare gas clusters RG18 and the six anion-containing subsets WATER27, IL16, G21EA, BH76, BH76RC, and AHB21. For the C60ISO and UPU23 subsets (which have comparatively small weights in WTMAD2), we settled for the def2-TZVPP basis set to reduce the computational cost. For the remaining 46 subsets, the def2-QZVPP50 basis set was used. The corresponding standard RI-MP252 and Coulomb exchange fitting (RI-JK)53 basis sets were employed throughout to reduce the computational cost further. The large pruned integration grid SG-3 was used across the board.54 Generally, inner-shell orbitals were frozen, but in subsets where such orbitals come close enough to the valence shell to qualify as “honorary valence orbitals”,55 the same frozen-core settings were used as in refs (48), (56), and (57).
Similar to the unregularized (x)DSD functionals, one fully optimized κ-DSD also has six adjustable linear parameters: cX,HF, cC,DFT, c2ab, c2ss (=c2aa + c2bb), and for the D3(BJ) dispersion correction one prefactor s6 and one parameter a2 for the damping function (like in refs (58) and (59), we constrain a1 and s8 to zero). In addition, our new κ(x)DSD functionals also contain the PT2 regularization parameter κ.
Powell’s BOBYQA60 (bound optimization by quadratic approximation) derivative-free constrained optimizer together with in-house written scripts and Fortran programs were used to optimize all parameters.
To find our bearings, we first explore the effect of using κ-GLPT2 energies in the revDSD-PBEP86-D3BJ and xDSD75-PBEP86-D3BJ double hybrids using the “Diet-100” statistical reduction61 version of GMTKN55. The unregularized same-spin and opposite-spin PT2 energies were replaced by the corresponding κ-PT2 terms, evaluated for a fixed κ value, and then all four linear parameters were reoptimized to obtain the regularized double hybrids, κDSD-PBEP86-D3BJ and κxDSD75-PBEP86-D3BJ. Including κ = ∞ (i.e., unregularized GLPT2), we calculated WTMAD2 for 17 κ values ranging from 0.9 to 10. For both κDSD-PBEP86-D3BJ and κxDSD75-PBEP86-D3BJ, WTMAD2 decreases with an increase in κ, and beyond κ = 4.0, WTMAD2 approaches WTMAD2κ=∞ (see Figure S1). Interestingly, for κDSD-PBEP86-D3BJ, we observe a very shallow dip near κ = 4.0, but WTMAD2κ=4.0 is only 0.03 kcal/mol lower than WTMAD2κ=∞.
Having initially scanned κ-space for one gDH and one xDH, we then used full GMTKN55 for the final parametrization of the κ-regularized DSD functionals. For this purpose, we selected nine κ values (i.e., κ = 1.1, 1.45, 2.0, 2.2, 2.5, 3.0, 3.5, 4.0, and ∞) and three different exchange correlation (XC) combinations: PBE-P86, PBE-PBE, and B88-LYP. Among the three regularized DSD (i.e., κDSD-PBEP86-D3BJ, κDSD-PBEPBE-D3BJ, and κDSD-BLYP-D3BJ) and one regularized xDSD (i.e., κxDSD75-PBBEP86-D3BJ) functionals, only κDSD-BLYP-D3BJ marginally benefits from the PT2 regularization. We obtained the lowest WTMAD2 (2.34 kcal/mol) for κDSD-BLYP-D3BJ at κ = 3.0, which is just 0.09 kcal/mol lower than the WTMAD2 of the unregularized counterpart, revDSD-BLYP-D3BJ (see Figure 1 and Table S1). [By way of perspective, in the Supporting Information of ref (57), we applied the Bayesian information criterion62,63 to see what reduction in WTMAD2 could be considered “decisive” and found 3.5%, or (in this context) ∼0.09 kcal/mol.] Now, partitioning each WTMAD2 into five major subcategories, we found that small-molecule thermochemistry, barrier heights, and intramolecular interactions do not benefit at all from PT2 regularization and κ = ∞ always offers the best performance (see Table S1). However, for the large-molecule reactions, all four κDSD functionals benefit from regularization, most prominently for κDSD-BLYP-D3BJ. Finally, for the intermolecular interactions, κ-regularization improved performance for κxDSD75-PBEP86-D3BJ, κDSD-PBEP86-D3BJ, and κDSD-PBEPBE-D3BJ. We obtained the best results at κ = 2.5 while using κxDSD75-PBEP86-D3BJ and at κ = 1.45 for κDSD-PBEP86-D3BJ and κDSD-PBEPBE-D3BJ. However, the performance of those three functionals deteriorates with an increase in κ. The unregularized variant wins the race for this subset when κDSD-BLYP-D3BJ is considered (see Figure S2 and Table S1).
Figure 1.
Dependence of total WTMAD2 (kcal/mol) on reciprocal κ for three variants of each of the regularized double-hybrid functionals (A) κxDSD75-PBBEP86-D3BJ, (B) κDSD-PBEPBE-D3BJ, (C) κDSD-BLYP-D3BJ, and (D) κDSD-PBEP86-D3BJ. Green solid lines represent the functionals where same- and opposite-spin coefficients are both optimized independently. The black lines are for the DOD variants, while the red lines represent the special case in which c2ss = c2ab (i.e., κDH-XC-D3BJ forms).
What if we allow only one degree of freedom between c2ab and c2ss but self-consistently reoptimize the remaining three parameters (i.e., cC,DFT, c2ab, and s6)? We considered two possibilities. The first is imposing the condition c2ab = c2ss, i.e., simple double hybrids like B2PLYP,64 B2GPPLYP,65 PBE0-2,66 etc., as well as the combinatorially optimized ωB97M(2).42 The nomenclature, “DSD”, is no longer appropriate in this case and will be replaced by κDH-XC-D3BJ. The second option is to exclude same-spin correlation entirely (c2ss = 0), also known as the DOD forms. These are of interest because they are amenable to reduced-scaling opposite-spin MP2 techniques like Laplace transform MP2 of Häser and Almlöf,67 which scales as Ο(N4) with system size, or the tensor hypercontraction approach of Song and Martinez,68 which scales as Ο(N3). As expected, constraining c2ab = c2ss always increases WTMAD2 over the κDSD forms. For both κDH-PBEPBE-D3BJ and κDH-PBEP86-D3BJ, we found the lowest WTMAD2 at κmin = 2.5, corresponding to decreases of 0.11 and 0.10 kcal/mol, respectively, compared to their unregularized forms. For κDH-BLYP-D3BJ, the WTMAD2 gap between κmin (κ value for which we obtain the minimum WTMAD2) and κ = ∞ is ∼0.08 kcal/mol. Interestingly, for the κDOD functionals, GLPT2 regularization does more harm than good, and the unregularized forms always offer the best performance (see Figure 1).
The use of the more modern D469,70 dispersion correction instead of D3BJ for the regularized functionals does not affect any trends with respect to the regularizer (κ). For the PBE-PBE and PBE-P86 exchange-correlation combinations, the WTMAD2 gaps between the D3BJ and D4 corrected forms increase gradually with an increase in κ. However, switching from D3BJ to D4 has no significant effect on the performance of the κxDSD75-PBEP86-D4 and κDSD-BLYP-D4 functionals (see Table S2). The same story is repeated for the κDOD and κxDOD functionals with D4 dispersion correction (see Table S3).
Thus far, for the κDSD functionals, we have used the same parameter for exact exchange as for their unregularized forms.40,48 Our next objective is to check whether the Hartree–Fock exchange prefactors of the unregularized forms are still optimal for the new regularized functionals. To answer this question, we have considered seven cX,HF values ranging from 0.5 to 1.0, and the κDSDX-PBEP86-D3BJ functional together with its κDH, κDOD, and dispersion uncorrected variants (X represents the percentage of HF exchange used; only one joint set of electronic structure calculations is required for all variants).
It turns out that regularization in κDSD-PBEP86-D3BJ becomes gradually more beneficial as cX,HF is decreased, with κmin decreasing concomitantly. For example, we obtain the lowest WTMAD2 near κ = 1.67 when cX,HF = 0.55, but when cX,HF = 0.5, the κmin decreases to 1.45 (see Figure 2). Now, splitting each WTMAD2 into five major subcategories, we found that regularization does more harm than good across the board for small-molecule thermochemistry. However, the extent of performance deterioration with respect to the κ values becomes less prominent as cX,HF is decreased. Using a small κ value can severely harm the performance for barrier heights at higher percentages of HF exchange, but much less so at lower percentages. When κ = 1.1, the κDSD50-PBEP86-D3BJ functional marginally outperforms the unregularized variant. For large-molecule reactions, the κDSD-PBEP86-D3BJ functional is a better choice across the board compared to the revDSD-PBEP86-DBJ functional, and κmin decreases gradually with an increase in cX,HF. The regularized forms with 69% and 66% Hartree–Fock exchange offer the best performance near κ = 3.0 and 2.0, respectively. For intramolecular interactions, the trends are largely the same as what we obtained for the small-molecule thermochemistry subsets. Finally, for intermolecular interactions, regularized forms always outperform the unregularized alternatives, and κmin decreases with an increase in cX,HF. For this subset, κDSD69-PBEP86-D3BJ and κDSD63-PBEP86-D3BJ are the two best picks at κ = 1.45 and 1.33, respectively (for optimized parameters, the total WTMAD2 for full GMTKN55, and its decomposition into five major subsets, see Table S4).
Figure 2.
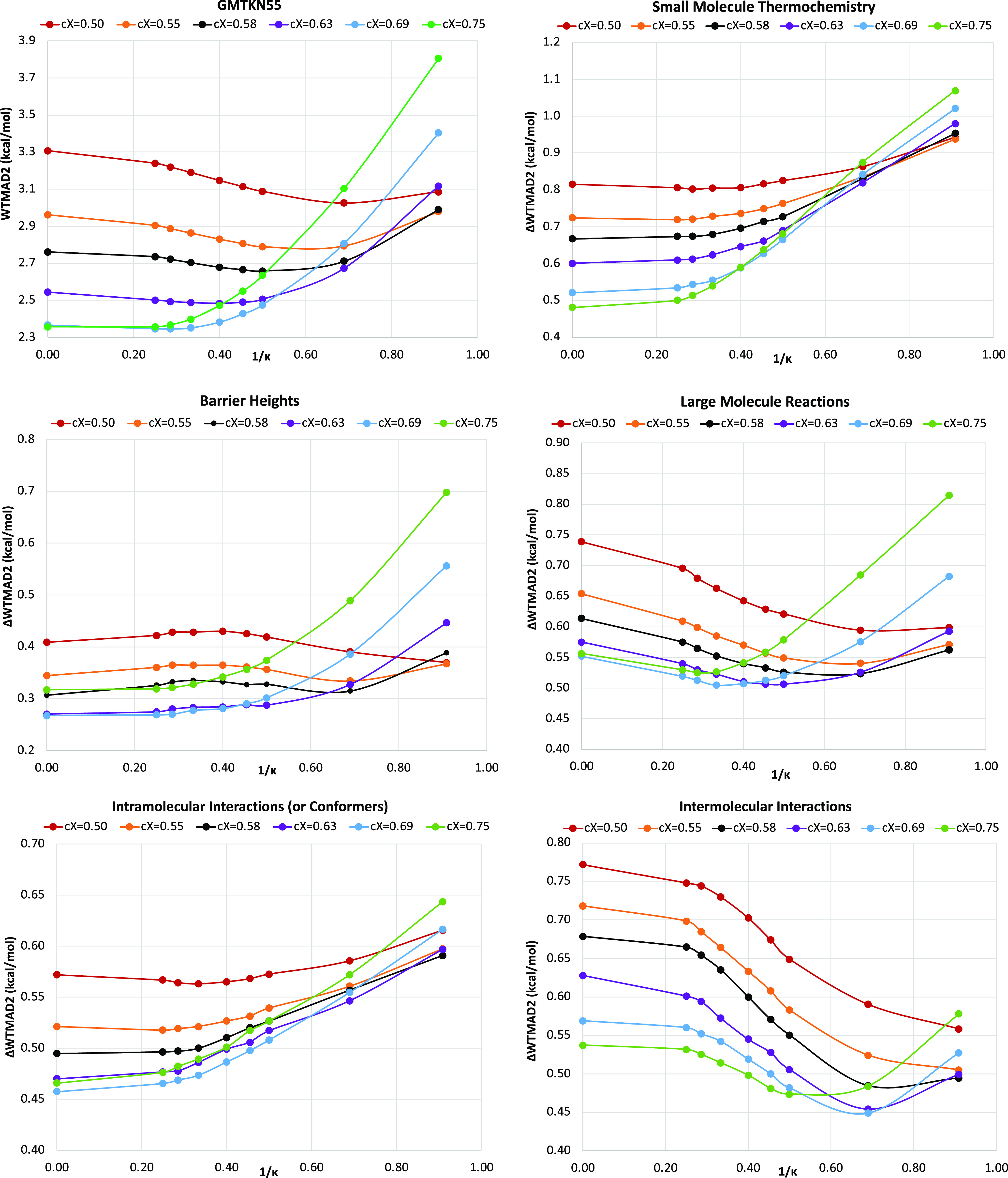
Dependence of total WTMAD2 (kcal/mol) and contribution (ΔWTMAD2 in kcal/mol) from five major subcategories on reciprocal κ for κDSDX-PBEP86-D3BJ. Six colors represent six different fractions of exact exchange (cX) ranging from 0.75 to 0.50.
The benefit in noncovalent interactions is not as prominent as that found by Shee et al.39 for pure MP2, which is expected because MP2 correlation in their case has a coefficient of unity while in a DHDF functional the MP2-like correlation (or PT2 correlation) is scaled down by a factor in the range of 0.3–0.5; hence, regularization will impact overall performance less. Additionally, HF exchange and PT2 correlation in a basis of KS orbitals are a different proposition from the same in a basis of HF orbitals.
As we found no material change from D3BJ to D4 in the previous section, we have decided not to explore that avenue for different cX,HF values.
Now, what happens if we impose c2ab = c2ss, i.e., a simple double hybrid rather than a DSD or DOD form? (As always, parameters are reoptimized self-consistently.) The results can be found in Figure S3 and Table S5. Even when cX,HF = 0.75, we find a shallow WTMAD2 reduction (0.04 kcal/mol) at κmin; as cX,HF is decreased, this “well” is deepened until it reaches 0.36 kcal/mol at cX,HF = 0.50 (κmin decreases in tandem with cX,HF). Among the five major subsets, at low cX,HF values, the WTMAD2 component from noncovalent interactions (NCI) decreases as κmin decreases, while this is detrimental to small-molecule thermochemistry and (at κmin = 1.1–1.45) for barrier heights: the former tendency grows weaker, and the latter stronger, as cX,HF is increased. We note that the ωB97M(2) combinatorially optimized range-separated double hybrid of Mardirossian and Head-Gordon42 has c2ab = c2ss and might hence benefit. (The way spin-component-scaled MP2 behaves differently from standard MP2 has been rationalized to some degree as approximate higher-order effects.71,72)
Interestingly, the behavior seen for the κDODX-PBEP86-D3BJ functionals (i.e., when c2ss = 0) is fairly similar, with lower percentages of HF exchange significantly benefiting from PT2 regularization. For example, the WTMAD2 “well” for κDOD50-PBEP86-D3BJ is 0.39 kcal/mol, but for κDOD63-PBEP86-D3BJ, this shrinks to just 0.04 kcal/mol. Except for small-molecule thermochemistry, we found the same trend as κDSDX-PBEP86-D3BJ for the remaining subsets. At small cX,HF values, regularization seems to be slightly beneficial for the small-molecule thermochemistry subsets (see Figure S3 and Table S6).
For the sake of completeness, for dispersion-uncorrected κnoDispSDX-PBEP86 functionals, we likewise found that at higher fractions of HF exchange, κmin approaches infinity (see Figure S3 and Table S7). In this case, obviously the balance is tipped more strongly toward high percentages of PT2 correlation (and of HF exchange, as the two are well-known65 to be linearly related) as long-range dispersion is not covered by anything else.
Thus far, we have used the same percentage of HF and semilocal exchange for orbital generation and final energy calculation. What happens when we use a different fraction of HF and DFT exchange for orbitals and final energies, e.g., PBE0P86 (i.e., 0.25HFx + 0.75PBEx + 1.0P86c) orbitals in κDSD69-PBEP86-D3BJ? These new DSD functionals are found not to benefit from PT2 regularization (see Table S8 and Figure S4). When WTMAD2 = 2.28 kcal/mol, the unregularized version, revDSD-PBEP86-D3BJ@PBE0P86, slightly outperforms the original revDSD-PBEP86-D3BJ (WTMAD2 = 2.38 kcal/mol): nearly all of that gain comes from RSE43 (radical-stabilization energies), due to substantially reduced spin contamination thanks to the smaller cX,HF in the orbitals. If we constrain c2ab = c2ss, the WTMAD2 gap between κmin and κ = ∞ is 0.13 kcal/mol, marginally larger than what we obtained for κDH69-PBEP86-D3BJ (0.10 kcal/mol). κmin also increases from 2.0 (for κDH69-PBEP86-D3BJ) to 3.0. For the κDOD variants of these new functionals, we saw the same trend that we did for the κDSD forms.
Next, we check the effect of PT2 regularization in three nonempirical double hybrids: SOS0-PBE0-2-D3BJ,73,74 SOS1-PBE-QIDH-D3BJ,75,76 and PBE0-DH-D3BJ.1,77 Benchmarking against GMTKN55, Mehta et al.74 reported that the first two among these three functionals are the best performers among the nonempirical DHDFs. Similar to what we found for κDODX-PBEP86-D3BJ, employing regularization in the PT2 term does more harm than good for SOS0-PBE0-2-D3BJ and SOS1-PBE-QIDH-D3BJ (see Table S9). Unlike κDH50-PBEP86-D3BJ, regularized PT2 correlation offers no benefit for PBE0-DH-D3BJ. One reason the nonempirical double hybrid with 50% HF exchange sees no benefit may well be that the parameter for the PT2 correlation is 1/8 only; hence, it would not matter enough in the total WTMAD2.
Summing up an extensive survey of regularized DHDFs using the large and chemically diverse GMTKN55, we can conclude the following.
The benefits of PT2 regularization for intermolecular interactions and large-molecule reactions are negated by the losses for small-molecule thermochemistry, barrier heights, and conformer energies. Hence, κ-GLPT2 correlation causes no significant reduction in WTMAD2 compared to the unregularized revDSD-PBEP86-D3BJ, revDSD-PBE-D3BJ, and xDSD75-PBEP86-D3BJ functionals. However, the significantly better performance of κDSD-BLYP-D3BJ when κ = 2.2 for large-molecule reactions has enough impact on WTMAD2 that, overall, it marginally outperforms revDSD-BLYP-D3BJ. Replacing D3BJ with D4 dispersion does not affect those trends.
If we eliminate spin-component scaling (i.e., c2ab = c2ss), the WTMAD2 gap between the κmin and κ = ∞ (i.e., unregularized) forms of κ(x)DH-XC-D3BJ is more significant than that we obtained for the κ(x)DSD functionals. In contrast, for the κ(x)DOD forms, unregularized functionals always perform better.
Regularization of the GLPT2 terms in double hybrids is most helpful at lower percentages (e.g., 50%) of HF exchange. At higher percentages of HF exchange, the benefits for intermolecular interactions and large-molecule reactions are outweighed by the deterioration in the remaining three subsets. At lower percentages of HF exchange, the benefits are heightened and the deterioration is mitigated, hence, an overall beneficial effect. In special cases in which DHs with a small fraction of HF exchange might be more resilient (e.g., systems with strong static correlation or prone to severe spin contamination error), κ-regularized double hybrids will offer advantages over their unregularized counterparts.
Acknowledgments
G.S. acknowledges a doctoral fellowship from the Feinberg Graduate School (WIS). The authors thank Dr. James Shee for helpful tips regarding the Q-Chem inputs required. This research was funded by the Israel Science Foundation (Grant 1969/20) and the Minerva Foundation (Grant 2020/05).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpclett.2c00718.
Optimized parameters, total WTMAD2 and its division into major subsets for κDSD-PBEP86-D3BJ, κDSD-PBEPBE-D3BJ, κDSD-BLYP-D3BJ, κxDSD75-PBEP86-D3BJ, κDSD-PBEP86-D4, κDSD-PBEPBE-D4, κDSD-BLYP-D4, κxDSD75-PBEP86-D4, κDOD-PBEP86-D4, κDOD-PBEPBE-D4, κDOD-BLYP-D4, and κxDOD75-PBEP86-D4; optimized parameters, total WTMAD2, and its division into major subcategories for κDSDX-PBEP86-D3BJ, κDHX-PBEP86-D3BJ, κDODX-PBEP86-D3BJ, and κnoDispSDX-PBEP86, where seven different fractions of exact exchange (cX,HF) ranging from 1.0 to 0.5 are considered; optimized parameters, total WTMAD2, and its decomposition into major subcategories for κDSD69-PBEP86-D3BJ and κDSD69-PBEPBE-D3BJ@PBE0P86; dependence of WTMAD2 (kcal/mol) on reciprocal κ for κDSD-PBEP86-D3BJ and κxDSD75-PBEP86-D3BJ; dependence of total WTMAD2 (kcal/mol) and contribution (ΔWTMAD2 in kcal/mol) from five major subcategories on reciprocal κ for κxDSD75-PBEP86-D3BJ, κDSD-PBEP86-D3BJ, κDSD-PBEPBE-D3BJ, κDSD-BLYP-D3BJ, and κxDSD75-PBEP86-D3BJ; dependence of WTMAD2 (kcal/mol) on reciprocal κ for κDSD-PBEP86-D3BJ, κDOD-PBEP86-D3BJ, κDH-PBEP86-D3BJ, and κnoDispSD-PBEP86 functionals [the six colors represent six different fractions of exact exchange (cX) ranging from 0.75 to 0.50]; dependence of total WTMAD2 (kcal/mol) on reciprocal κ for κDSD69-PBEP86-D3BJ@PBE0P86 and κDSD69-PBEP86-D3BJ (A), their κDH counterparts (B), κDOD variants (C), and dispersion-uncorrected forms (D); total WTMAD2 and its decomposition into major subcategories for selected nonempirical DHDFs; and QCHEM sample input for κDSD-BLYP-D3BJ with κ = 2.2 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Goerigk L.; Grimme S. Double-Hybrid Density Functionals. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4 (6), 576–600. 10.1002/wcms.1193. [DOI] [Google Scholar]
- Brémond E.; Ciofini I.; Sancho-García J. C.; Adamo C. Nonempirical Double-Hybrid Functionals: An Effective Tool for Chemists. Acc. Chem. Res. 2016, 49 (8), 1503–1513. 10.1021/acs.accounts.6b00232. [DOI] [PubMed] [Google Scholar]
- Martin J. M. L.; Santra G. Empirical Double-Hybrid Density Functional Theory: A ‘Third Way’ in Between WFT and DFT. Isr. J. Chem. 2020, 60 (8–9), 787–804. 10.1002/ijch.201900114. [DOI] [Google Scholar]
- Zhang I. Y.; Xu X. Doubly Hybrid Density Functional for Accurate Description of Thermochemistry, Thermochemical Kinetics and Nonbonded Interactions. Int. Rev. Phys. Chem. 2011, 30 (1), 115–160. 10.1080/0144235X.2010.542618. [DOI] [Google Scholar]
- Perdew J. P.; Schmidt K. Jacob’s Ladder of Density Functional Approximations for the Exchange-Correlation Energy. AIP Conf. Proc. 2001, 577, 1–20. 10.1063/1.1390175. [DOI] [Google Scholar]
- Mardirossian N.; Head-Gordon M. Thirty Years of Density Functional Theory in Computational Chemistry: An Overview and Extensive Assessment of 200 Density Functionals. Mol. Phys. 2017, 115 (19), 2315–2372. 10.1080/00268976.2017.1333644. [DOI] [Google Scholar]
- Goerigk L.; Hansen A.; Bauer C.; Ehrlich S.; Najibi A.; Grimme S. A Look at the Density Functional Theory Zoo with the Advanced GMTKN55 Database for General Main Group Thermochemistry, Kinetics and Noncovalent Interactions. Phys. Chem. Chem. Phys. 2017, 19 (48), 32184–32215. 10.1039/C7CP04913G. [DOI] [PubMed] [Google Scholar]
- Goerigk L.; Mehta N. A Trip to the Density Functional Theory Zoo: Warnings and Recommendations for the User. Aust. J. Chem. 2019, 72 (8), 563. 10.1071/CH19023. [DOI] [Google Scholar]
- Dohm S.; Hansen A.; Steinmetz M.; Grimme S.; Checinski M. P. Comprehensive Thermochemical Benchmark Set of Realistic Closed-Shell Metal Organic Reactions. J. Chem. Theory Comput. 2018, 14 (5), 2596–2608. 10.1021/acs.jctc.7b01183. [DOI] [PubMed] [Google Scholar]
- Iron M. A.; Janes T. Evaluating Transition Metal Barrier Heights with the Latest Density Functional Theory Exchange–Correlation Functionals: The MOBH35 Benchmark Database. J. Phys. Chem. A 2019, 123 (17), 3761–3781. 10.1021/acs.jpca.9b01546. [DOI] [PubMed] [Google Scholar]
- Efremenko I.; Martin J. M. L. Coupled Cluster Benchmark of New Density Functionals and of Domain Pair Natural Orbital Methods: Mechanisms of Hydroarylation and Oxidative Coupling Catalyzed by Ru(II) Chloride Carbonyls. AIP Conf. Proc. 2019, 2186, 030005. 10.1063/1.5137916. [DOI] [Google Scholar]
- Maurer L. R.; Bursch M.; Grimme S.; Hansen A. Assessing Density Functional Theory for Chemically Relevant Open-Shell Transition Metal Reactions. J. Chem. Theory Comput. 2021, 17 (10), 6134–6151. 10.1021/acs.jctc.1c00659. [DOI] [PubMed] [Google Scholar]
- Efremenko I.; Martin J. M. L. Coupled Cluster Benchmark of New DFT and Local Correlation Methods: Mechanisms of Hydroarylation and Oxidative Coupling Catalyzed by Ru(II, III) Chloride Carbonyls. J. Phys. Chem. A 2021, 125 (40), 8987–8999. 10.1021/acs.jpca.1c05124. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Neese F. Double-Hybrid Density Functional Theory for Excited Electronic States of Molecules. J. Chem. Phys. 2007, 127 (15), 154116. 10.1063/1.2772854. [DOI] [PubMed] [Google Scholar]
- Goerigk L.; Grimme S. Calculation of Electronic Circular Dichroism Spectra with Time-Dependent Double-Hybrid Density Functional Theory. J. Phys. Chem. A 2009, 113 (4), 767–776. 10.1021/jp807366r. [DOI] [PubMed] [Google Scholar]
- Goerigk L.; Moellmann J.; Grimme S. Computation of Accurate Excitation Energies for Large Organic Molecules with Double-Hybrid Density Functionals. Phys. Chem. Chem. Phys. 2009, 11 (22), 4611. 10.1039/b902315a. [DOI] [PubMed] [Google Scholar]
- Goerigk L.; Kruse H.; Grimme S.. Theoretical Electronic Circular Dichroism Spectroscopy of Large Organic and Supramolecular Systems. In Comprehensive Chiroptical Spectroscopy; John Wiley & Sons, Inc.: Hoboken, NJ, 2012; Vol. 1, pp 643–673. 10.1002/9781118120187.ch22 [DOI] [Google Scholar]
- Schwabe T.; Goerigk L. Time-Dependent Double-Hybrid Density Functionals with Spin-Component and Spin-Opposite Scaling. J. Chem. Theory Comput. 2017, 13 (9), 4307–4323. 10.1021/acs.jctc.7b00386. [DOI] [PubMed] [Google Scholar]
- Casanova-Páez M.; Goerigk L. Time-Dependent Long-Range-Corrected Double-Hybrid Density Functionals with Spin-Component and Spin-Opposite Scaling: A Comprehensive Analysis of Singlet–Singlet and Singlet–Triplet Excitation Energies. J. Chem. Theory Comput. 2021, 17 (8), 5165–5186. 10.1021/acs.jctc.1c00535. [DOI] [PubMed] [Google Scholar]
- Casanova-Páez M.; Goerigk L. Assessing the Tamm–Dancoff Approximation, Singlet–Singlet, and Singlet–Triplet Excitations with the Latest Long-Range Corrected Double-Hybrid Density Functionals. J. Chem. Phys. 2020, 153 (6), 064106. 10.1063/5.0018354. [DOI] [PubMed] [Google Scholar]
- Goerigk L.; Casanova-Paéz M. The Trip to the Density Functional Theory Zoo Continues: Making a Case for Time-Dependent Double Hybrids for Excited-State Problems. Aust. J. Chem. 2021, 74 (1), 3–15. 10.1071/CH20093. [DOI] [Google Scholar]
- Mester D.; Kállay M. A Simple Range-Separated Double-Hybrid Density Functional Theory for Excited States. J. Chem. Theory Comput. 2021, 17 (2), 927–942. 10.1021/acs.jctc.0c01135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoychev G. L.; Auer A. A.; Neese F. Efficient and Accurate Prediction of Nuclear Magnetic Resonance Shielding Tensors with Double-Hybrid Density Functional Theory. J. Chem. Theory Comput. 2018, 14 (9), 4756–4771. 10.1021/acs.jctc.8b00624. [DOI] [PubMed] [Google Scholar]
- Tran V. A.; Neese F. Double-Hybrid Density Functional Theory for g-Tensor Calculations Using Gauge Including Atomic Orbitals. J. Chem. Phys. 2020, 153 (5), 054105. 10.1063/5.0013799. [DOI] [PubMed] [Google Scholar]
- Dittmer A.; Stoychev G. L.; Maganas D.; Auer A. A.; Neese F. Computation of NMR Shielding Constants for Solids Using an Embedded Cluster Approach with DFT, Double-Hybrid DFT, and MP2. J. Chem. Theory Comput. 2020, 16 (11), 6950–6967. 10.1021/acs.jctc.0c00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bursch M.; Gasevic T.; Stückrath J. B.; Grimme S. Comprehensive Benchmark Study on the Calculation of 29 Si NMR Chemical Shifts. Inorg. Chem. 2021, 60 (1), 272–285. 10.1021/acs.inorgchem.0c02907. [DOI] [PubMed] [Google Scholar]
- Alipour M. How Well Can Parametrized and Parameter-Free Double-Hybrid Approximations Predict Response Properties of Hydrogen-Bonded Systems? Dipole Polarizabilities of Water Nanoclusters as a Working Model. J. Phys. Chem. A 2013, 117 (21), 4506–4513. 10.1021/jp402659w. [DOI] [PubMed] [Google Scholar]
- Alipour M. Novel Recipe for Double-Hybrid Density Functional Computations of Linear and Nonlinear Polarizabilities of Molecules and Nanoclusters. J. Phys. Chem. A 2014, 118 (28), 5333–5342. 10.1021/jp503959w. [DOI] [PubMed] [Google Scholar]
- Kesharwani M. K.; Brauer B.; Martin J. M. L. Frequency and Zero-Point Vibrational Energy Scale Factors for Double-Hybrid Density Functionals (and Other Selected Methods): Can Anharmonic Force Fields Be Avoided?. J. Phys. Chem. A 2015, 119 (9), 1701–1714. 10.1021/jp508422u. [DOI] [PubMed] [Google Scholar]
- Görling A.; Levy M. Exact Kohn-Sham Scheme Based on Perturbation Theory. Phys. Rev. A 1994, 50 (1), 196–204. 10.1103/PhysRevA.50.196. [DOI] [PubMed] [Google Scholar]
- Assfeld X.; Almlöf J. E.; Truhlar D. G. Degeneracy-Corrected Perturbation Theory for Electronic Structure Calculations. Chem. Phys. Lett. 1995, 241 (4), 438–444. 10.1016/0009-2614(95)00650-S. [DOI] [Google Scholar]
- Stück D.; Head-Gordon M. Regularized Orbital-Optimized Second-Order Perturbation Theory. J. Chem. Phys. 2013, 139 (24), 244109. 10.1063/1.4851816. [DOI] [PubMed] [Google Scholar]
- Lochan R. C.; Head-Gordon M. Orbital-Optimized Opposite-Spin Scaled Second-Order Correlation: An Economical Method to Improve the Description of Open-Shell Molecules. J. Chem. Phys. 2007, 126 (16), 164101. 10.1063/1.2718952. [DOI] [PubMed] [Google Scholar]
- Neese F.; Schwabe T.; Kossmann S.; Schirmer B.; Grimme S. Assessment of Orbital-Optimized, Spin-Component Scaled Second-Order Many-Body Perturbation Theory for Thermochemistry and Kinetics. J. Chem. Theory Comput. 2009, 5 (11), 3060–3073. 10.1021/ct9003299. [DOI] [PubMed] [Google Scholar]
- Bozkaya U.; Turney J. M.; Yamaguchi Y.; Schaefer H. F.; Sherrill C. D. Quadratically Convergent Algorithm for Orbital Optimization in the Orbital-Optimized Coupled-Cluster Doubles Method and in Orbital-Optimized Second-Order Møller-Plesset Perturbation Theory. J. Chem. Phys. 2011, 135 (10), 104103. 10.1063/1.3631129. [DOI] [PubMed] [Google Scholar]
- Razban R. M.; Stück D.; Head-Gordon M. Addressing First Derivative Discontinuities in Orbital-Optimised Opposite-Spin Scaled Second-Order Perturbation Theory with Regularisation. Mol. Phys. 2017, 115 (17–18), 2102–2109. 10.1080/00268976.2017.1284355. [DOI] [Google Scholar]
- Lee J.; Head-Gordon M. Regularized Orbital-Optimized Second-Order Møller–Plesset Perturbation Theory: A Reliable Fifth-Order-Scaling Electron Correlation Model with Orbital Energy Dependent Regularizers. J. Chem. Theory Comput. 2018, 14 (10), 5203–5219. 10.1021/acs.jctc.8b00731. [DOI] [PubMed] [Google Scholar]
- Loipersberger M.; Bertels L. W.; Lee J.; Head-Gordon M. Exploring the Limits of Second- and Third-Order Møller–Plesset Perturbation Theories for Noncovalent Interactions: Revisiting MP2.5 and Assessing the Importance of Regularization and Reference Orbitals. J. Chem. Theory Comput. 2021, 17 (9), 5582–5599. 10.1021/acs.jctc.1c00469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shee J.; Loipersberger M.; Rettig A.; Lee J.; Head-Gordon M. Regularized Second-Order Møller–Plesset Theory: A More Accurate Alternative to Conventional MP2 for Noncovalent Interactions and Transition Metal Thermochemistry for the Same Computational Cost. J. Phys. Chem. Lett. 2021, 12 (50), 12084–12097. 10.1021/acs.jpclett.1c03468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santra G.; Cho M.; Martin J. M. L. Exploring Avenues beyond Revised DSD Functionals: I. Range Separation, with x DSD as a Special Case. J. Phys. Chem. A 2021, 125 (21), 4614–4627. 10.1021/acs.jpca.1c01294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goerigk L.; Grimme S. Efficient and Accurate Double-Hybrid-Meta-GGA Density Functionals—Evaluation with the Extended GMTKN30 Database for General Main Group Thermochemistry, Kinetics, and Noncovalent Interactions. J. Chem. Theory Comput. 2011, 7 (2), 291–309. 10.1021/ct100466k. [DOI] [PubMed] [Google Scholar]
- Mardirossian N.; Head-Gordon M. Survival of the Most Transferable at the Top of Jacob’s Ladder: Defining and Testing the ωB97M(2) Double Hybrid Density Functional. J. Chem. Phys. 2018, 148 (24), 241736. 10.1063/1.5025226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semidalas E.; Martin J. M. L. Canonical and DLPNO-Based G4(MP2)XK-Inspired Composite Wave Function Methods Parametrized against Large and Chemically Diverse Training Sets: Are They More Accurate and/or Robust than Double-Hybrid DFT?. J. Chem. Theory Comput. 2020, 16 (7), 4238–4255. 10.1021/acs.jctc.0c00189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollett J. W.; Gill P. M. W. The Two Faces of Static Correlation. J. Chem. Phys. 2011, 134 (11), 114111. 10.1063/1.3570574. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132 (15), 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Goerigk L.A Comprehensive Overview of the DFT-D3 London-Dispersion Correction. In Non-Covalent Interactions in Quantum Chemistry and Physics: Theory and Applications; Elsevier, 2017; pp 195–219. 10.1016/B978-0-12-809835-6.00007-4 [DOI] [Google Scholar]
- Grimme S.; Ehrlich S.; Goerigk L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32 (7), 1456–1465. 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- Santra G.; Sylvetsky N.; Martin J. M. L. Minimally Empirical Double-Hybrid Functionals Trained against the GMTKN55 Database: RevDSD-PBEP86-D4, RevDOD-PBE-D4, and DOD-SCAN-D4. J. Phys. Chem. A 2019, 123 (24), 5129–5143. 10.1021/acs.jpca.9b03157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epifanovsky E.; Gilbert A. T. B.; Feng X.; Lee J.; Mao Y.; Mardirossian N.; Pokhilko P.; White A. F.; Coons M. P.; Dempwolff A. L.; et al. Software for the Frontiers of Quantum Chemistry: An Overview of Developments in the Q-Chem 5 Package. J. Chem. Phys. 2021, 155 (8), 084801. 10.1063/5.0055522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigend F.; Ahlrichs R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7 (18), 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Rappoport D.; Furche F. Property-Optimized Gaussian Basis Sets for Molecular Response Calculations. J. Chem. Phys. 2010, 133 (13), 134105. 10.1063/1.3484283. [DOI] [PubMed] [Google Scholar]
- Hättig C. Optimization of Auxiliary Basis Sets for RI-MP2 and RI-CC2 Calculations: Core-Valence and Quintuple-ζ Basis Sets for H to Ar and QZVPP Basis Sets for Li to Kr. Phys. Chem. Chem. Phys. 2005, 7 (1), 59–66. 10.1039/B415208E. [DOI] [Google Scholar]
- Weigend F. Hartree–Fock Exchange Fitting Basis Sets for H to Rn. J. Comput. Chem. 2008, 29 (2), 167–175. 10.1002/jcc.20702. [DOI] [PubMed] [Google Scholar]
- Dasgupta S.; Herbert J. M. Standard Grids for High-Precision Integration of Modern Density Functionals: SG-2 and SG-3. J. Comput. Chem. 2017, 38 (12), 869–882. 10.1002/jcc.24761. [DOI] [PubMed] [Google Scholar]
- Martin J. M. L. Electron Correlation: Nature’s Weird and Wonderful Chemical Glue. Isr. J. Chem. 2022, 62 (1–2), e202100111 10.1002/ijch.202100111. [DOI] [Google Scholar]
- Santra G.; Semidalas E.; Martin J. M. L. Exploring Avenues beyond Revised DSD Functionals: II. Random-Phase Approximation and Scaled MP3 Corrections. J. Phys. Chem. A 2021, 125 (21), 4628–4638. 10.1021/acs.jpca.1c01295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santra G.; Semidalas E.; Martin J. M. L. Surprisingly Good Performance of XYG3 Family Functionals Using a Scaled KS-MP3 Correlation. J. Phys. Chem. Lett. 2021, 12 (38), 9368–9376. 10.1021/acs.jpclett.1c02838. [DOI] [PubMed] [Google Scholar]
- Kozuch S.; Martin J. M. L. Spin-Component-Scaled Double Hybrids: An Extensive Search for the Best Fifth-Rung Functionals Blending DFT and Perturbation Theory. J. Comput. Chem. 2013, 34 (27), 2327–2344. 10.1002/jcc.23391. [DOI] [PubMed] [Google Scholar]
- Kozuch S.; Martin J. M. L. DSD-PBEP86: In Search of the Best Double-Hybrid DFT with Spin-Component Scaled MP2 and Dispersion Corrections. Phys. Chem. Chem. Phys. 2011, 13 (45), 20104–20107. 10.1039/c1cp22592h. [DOI] [PubMed] [Google Scholar]
- Powell M.The BOBYQA Algorithm for Bound Constrained Optimization without Derivatives (DAMPT Report 2009/NA06); Department of Applied Mathematics and Theoretical Physics, University of Cambridge: Cambridge, U.K., 2009. [Google Scholar]
- Gould T. ‘Diet GMTKN55’ Offers Accelerated Benchmarking through a Representative Subset Approach. Phys. Chem. Chem. Phys. 2018, 20 (44), 27735–27739. 10.1039/C8CP05554H. [DOI] [PubMed] [Google Scholar]
- Schwarz G. Estimating the Dimension of a Model. Ann. Stat. 1978, 6 (2), 461–464. 10.1214/aos/1176344136. [DOI] [Google Scholar]
- Kass R. E.; Raftery A. E. Bayes Factors. J. Am. Stat. Assoc. 1995, 90 (430), 773–795. 10.1080/01621459.1995.10476572. [DOI] [Google Scholar]
- Grimme S. Semiempirical Hybrid Density Functional with Perturbative Second-Order Correlation. J. Chem. Phys. 2006, 124 (3), 034108. 10.1063/1.2148954. [DOI] [PubMed] [Google Scholar]
- Karton A.; Tarnopolsky A.; Lamère J.-F.; Schatz G. C.; Martin J. M. L. Highly Accurate First-Principles Benchmark Data Sets for the Parametrization and Validation of Density Functional and Other Approximate Methods. Derivation of a Robust, Generally Applicable, Double-Hybrid Functional for Thermochemistry and Thermochemical. J. Phys. Chem. A 2008, 112 (50), 12868–12886. 10.1021/jp801805p. [DOI] [PubMed] [Google Scholar]
- Chai J. Da; Mao S. P. Seeking for Reliable Double-Hybrid Density Functionals without Fitting Parameters: The PBE0–2 Functional. Chem. Phys. Lett. 2012, 538, 121–125. 10.1016/j.cplett.2012.04.045. [DOI] [Google Scholar]
- Häser M.; Almlöf J. Laplace Transform Techniques in Møller–Plesset Perturbation Theory. J. Chem. Phys. 1992, 96 (1), 489–494. 10.1063/1.462485. [DOI] [Google Scholar]
- Song C.; Martínez T. J. Atomic Orbital-Based SOS-MP2 with Tensor Hypercontraction. II. Local Tensor Hypercontraction. J. Chem. Phys. 2017, 146 (3), 034104. 10.1063/1.4973840. [DOI] [PubMed] [Google Scholar]
- Caldeweyher E.; Bannwarth C.; Grimme S. Extension of the D3 Dispersion Coefficient Model. J. Chem. Phys. 2017, 147 (3), 034112. 10.1063/1.4993215. [DOI] [PubMed] [Google Scholar]
- Caldeweyher E.; Ehlert S.; Hansen A.; Neugebauer H.; Spicher S.; Bannwarth C.; Grimme S. A Generally Applicable Atomic-Charge Dependent London Dispersion Correction. J. Chem. Phys. 2019, 150 (15), 154122. 10.1063/1.5090222. [DOI] [PubMed] [Google Scholar]
- Szabados Á. Theoretical Interpretation of Grimme’s Spin-Component-Scaled Second Order Møller-Plesset Theory. J. Chem. Phys. 2006, 125 (21), 214105. 10.1063/1.2404660. [DOI] [PubMed] [Google Scholar]
- Fink R. F. Spin-Component-Scaled Møller-Plesset (SCS-MP) Perturbation Theory: A Generalization of the MP Approach with Improved Properties. J. Chem. Phys. 2010, 133 (17), 174113. 10.1063/1.3503041. [DOI] [PubMed] [Google Scholar]
- Alipour M. Seeking for Spin-Opposite-Scaled Double-Hybrid Models Free of Fitted Parameters. J. Phys. Chem. A 2016, 120 (20), 3726–3730. 10.1021/acs.jpca.6b03406. [DOI] [PubMed] [Google Scholar]
- Mehta N.; Casanova-Páez M.; Goerigk L. Semi-Empirical or Non-Empirical Double-Hybrid Density Functionals: Which Are More Robust?. Phys. Chem. Chem. Phys. 2018, 20 (36), 23175–23194. 10.1039/C8CP03852J. [DOI] [PubMed] [Google Scholar]
- Brémond É.; Savarese M.; Sancho-García J. C.; Pérez-Jiménez Á. J.; Adamo C. Quadratic Integrand Double-Hybrid Made Spin-Component-Scaled. J. Chem. Phys. 2016, 144 (12), 124104. 10.1063/1.4944465. [DOI] [PubMed] [Google Scholar]
- Sancho-García J. C.; Brémond É.; Savarese M.; Pérez-Jiménez A. J.; Adamo C. Partnering Dispersion Corrections with Modern Parameter-Free Double-Hybrid Density Functionals. Phys. Chem. Chem. Phys. 2017, 19 (21), 13481–13487. 10.1039/C7CP00709D. [DOI] [PubMed] [Google Scholar]
- Brémond E.; Adamo C. Seeking for Parameter-Free Double-Hybrid Functionals: The PBE0-DH Model. J. Chem. Phys. 2011, 135 (2), 024106. 10.1063/1.3604569. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.