

Abstract
Metabolism plays a significant role in the regulation of aging at different levels, and metabolic reprogramming represents a major driving force in aging. Metabolic reprogramming leads to impaired organismal fitness, an age-dependent increase in susceptibility to diseases, decreased ability to mount a stress response, and increased frailty. The complexity of age-dependent metabolic reprogramming comes from the multitude of levels on which metabolic changes can be connected to aging and regulation of lifespan. This is further complicated by the different metabolic requirements of various tissues, cross-organ communication via metabolite secretion, and direct effects of metabolites on epigenetic state and redox regulation; however, not all of these changes are causative to aging. Studies in yeast, flies, worms, and mice have played a crucial role in identifying mechanistic links between observed changes in various metabolic traits and their effects on lifespan. Here, we review how changes in the organismal and organ-specific metabolome are associated with aging and how targeting of any one of over a hundred different targets in specific metabolic pathways can extend lifespan. An important corollary is that restriction or supplementation of different metabolites can change activity of these metabolic pathways in ways that improve healthspan and extend lifespan in different organisms. Due to the high levels of conservation of metabolism in general, translating findings from model systems to human beings will allow for the development of effective strategies for human health- and lifespan extension.
Keywords: Aging, Metabolism, Drosophila, C. elegans, Yeast, Mice
1. Age-dependent metabolic reprogramming
Aging is the primary risk factor for many major human pathologies including cancer, diabetes, cardiovascular disorders, and neurodegenerative diseases (Lopez-Otin et al., 2013). Loss of metabolic homeostasis is a hallmark of aging, and it is characterized by dramatic metabolic reprogramming, as reflected in age-dependent changes in organismal and tissue-specific transcriptomes, proteomes, and metabolomes. Metabolic reprogramming leads to impaired organismal fitness, age-dependent increases in susceptibility to diseases, decreased ability to mount a stress response, and increased frailty. Through an understanding of these processes, the affected metabolic pathways can be specifically targeted, paving the way to a delay or even a reversal of aging in humans. In addition, age-dependent metabolic reprogramming (a ‘metabolic signature’) can be used to predict longevity and development of age-dependent diseases, even before the first symptoms appear. As an example, untargeted metabolomics profiling of 770 metabolites in plasma from 268 healthy individuals including 125 twin pairs ranging in age from 6 months to 82 years determined 52 metabolites that can predict age in subjects over 16 years old (Bunning et al., 2020). Similarly, Robinson et al. developed a metabolic age model based on untargeted metabolic profiling of urine and serum that correlated with chronological age and metabolic age. Aging acceleration was associated with obesity, high alcohol intake, diabetes, and depression (Robinson et al., 2020). Also, Johnson et al. developed plasma metabolic signatures that were associated with biological age and were predictive of an accelerated or delayed rate of aging (Johnson et al., 2019). This global age-dependent metabolic reprogramming has been reflected in multiple studies in different organisms at whole-body and tissue-specific levels. Indeed, studies in worms (Fuchs et al., 2010), mice (Houtkooper et al., 2011; Tomas-Loba et al., 2013), and humans (Chaleckis et al., 2016; Darst et al., 2019; Dunn et al., 2015; Menni et al., 2013; Rist et al., 2017; Yu et al., 2012) have documented changes in the metabolome during the aging process. In addition to simply changing with age, differences in the levels of some metabolites have been associated with delayed aging in different human populations. Cheng et al. quantified 217 plasma metabolites from 2327 participants and found that higher concentrations of a TCA cycle intermediate, isocitrate, and a bile acid, taurocholate, were associated with lower odds of longevity (defined as reaching the age of 80 years), and higher concentrations of isocitrate were also associated with poorer cardiovascular health (Cheng et al., 2015). Deelen et al, identified 14 circulating metabolic biomarkers associated with with all-cause mortality (Deelen et al., 2019). MetaboAge is a repository of metabolomic variations, which hosts ageing-related metabolite changes, occurring in healthy individuals (Bucaciuc Mracica et al., 2020).
Moreover, interventions that extend lifespan often exhibit reversal of age-dependent metabolic reprogramming. Caloric restriction (CR) usually means the amount of calories is reduced, for example, via food dilution or reduction of food intake; whereas dietary restriction (DR) usually refers to a change in dietary content that does not affect the calorie content. However, these terms are often used interchangeably, and we use here whichever term was used in the original publication. CR/DR extends lifespan in most tested models and protects against multiple age-related diseases (Soultoukis and Partridge, 2016). An untargeted metabolomics analysis in flies (Hoffman et al., 2014) has suggested that DR might reverse age-dependent metabolic reprogramming at the level of a specific tissue (Laye et al., 2015) and the whole organism (Avanesov et al., 2014). Similarly, Gao et al. compared transcriptomics and metabolomics data in wild-type worms with data from long-lived daf-2 mutant worms (impaired IlS) and long-lived eat-2 mutant worms (a genetic model for studying DR, in which the eat-2 mutation disrupts pharyngeal pumping and thus limits food intake). Combined analysis of transcriptomes and metabolomes revealed increased amino acid metabolism and upregulation of purine metabolism as a commonality between the two long-lived mutants (Gao et al., 2018).
In addition, using an evolutionary approach, a comparison of metabolomes of different species with variable lifespans allowed for the identification of metabolites that can be linked to differences in longevity between these species. Transcriptional analysis using RNA-seq from the liver, kidney, and brain of 33 diverse species of mammals followed by gene set enrichment analysis (GSEA) revealed central energy metabolism as one of the most relevant biological pathways associated with life histories (Fushan et al., 2015). Metabolomics analysis in the brain, heart, kidney, and liver of 26 mammalian species across 10 taxonomic orders revealed a positive correlation between longevity and the levels of several metabolites, namely sphingomyelins (in the brain), and a negative correlation for amino acids (brain), lysophosphatidylcholines (brain and heart), lysophosphatidylethanolamines (brain and kidney), and triacylglycerols (kidney) (Ma et al., 2015).
In addition to the observation that metabolite levels change with age, these changes appear to be delayed in species that are long-lived or following pro-longevity interventions. Nevertheless, these changes could be simply correlative. To determine if these changes are causative to aging, many have asked if up- or down-regulating these metabolic pathways and preventing the associated metabolic changes extends lifespan. Taking a cross-species approach to the study of aging seems particularly important because changes to metabolic pathways that extend lifespan in different species are more likely to extend lifespan and healthspan in humans. This review examines how targeting any one of over a hundred different targets in specific metabolic pathways (Table 1) can prolong lifespan in different organisms. We also provide examples of manipulations of relevant pathways in humans and potential age-related benefits related to these manipulations.
Table 1.
Manipulations of metabolic enzymes known to affect lifespan.
| # | Acronym | Enzyme / Metabolite / Drug | KEGG ID | Metabolic pathway | Manipulation / Delivery | Effect on LS | Reference |
|---|---|---|---|---|---|---|---|
|
|
|
||||||
| Carbohydrate metabolism | |||||||
| Worms | |||||||
| 2-DG | 2-Deoxyglucose | C00586 | Glycolysis | Feeding | 17 % increase | (Schulz et al., 2007) | |
| gpi-1 | Glucose phosphate isomerase | CELE_Y87G2A.8/ K01810 |
Glycolysis | Downregulation | Increase | (Schulz et al., 2007; Hansen et al., 2005) | |
| fgt-1 | Glucose transporter | CELE_H17B01.1/ K07299 | Glucose transporter | Downregulation | 25 % increase | (Feng et al., 2013) | |
| pgm-1 | Phosphoglycerate mutase | CELE_F57B10.3/K15633 | Glycolysis | Downregulation | Increase | (Lee et al., 2003b) | |
| Fructose | Fructose | C00095 | Feeding | 45 % increase | (Zheng et al., 2017) | ||
| Sorbitol | Sorbitol | C00794 | Feeding | Increase | (Chandler-Brown et al., 2015) | ||
| D-Glucosamine | Hexokinase and glucokinase inhibitor | C00329 | Glycolysis | Feeding | 27 % increase | (Weimer et al., 2014) | |
| gfat-1 | glutamine-fructose 6-phosphate aminotransferase | CELE_F07A11.2/ K00820 | Hexosamine pathway | Gain-of-function mutation | 42 % increase | (Denzel et al., 2014) | |
| GlcNAc | UDP-N-acetylhexosamine | C00043 | Hexosamine pathway | Feeding | 38 % increase | (Denzel et al., 2014) | |
| masl | alpha-1,2-mannosidase I | CELE_D2030.1/ K01230 | N-linked glycosylation | Downregulation | 9% increase | (Liu et al., 2009) | |
| MGO | Methylglyoxal | C00546 | Methylglyoxal pathway | Feeding | Increase | (Ravichandran et al., 2018) | |
| gsy-i | Glycogen synthase | CELE_Y46G5A.31/ K00693 | Glycogen metabolism | Downregulation | 15 % increase | (Gusarov et al., 2017; Seo et al., 2018) | |
| K08E3.5 | UTP-glucose-1-phosphate uridylyltransferase | CELE_K08E3.5/K00963 | Glycogen metabolism | Downregulation | Increase | (Hamilton et al., 2005) | |
| Trehalose | Trehalose | C01083 | Trehalose metabolism | Feeding | 32 % increase | (Honda et al., 2010; Seo et al., 2018) | |
| tre-1 | Trehalase | CELE_F57B10.7/ K01194 | Trehalose metabolism | Downregulation | Increase | (Seo et al., 2018) | |
| tre-3 | Trehalase | CELE_W05E10.4/ K01194 | Trehalose metabolism | Downregulation | Increase | (Seo et al., 2018) | |
| Glucose, galactose, fucose, lactose, arabinose, sorbitol | Glucose, galactose, fucose, lactose, arabinose, sorbitol | C00031, C00124, C01019, C00243, C00259, C00794 | Sugar metabolism | Feeding | 10 % increase | (Brokate-Llanos et al., 2014) | |
| CA | Colanic acid | Polysaccharide | Feeding | Increase | (Han et al., 2017a) | ||
| pck-1/PEPCK-C | Phosphoenolpyruvate carboxykinase | CELE_W05G11.6/ K01596 | Gluconeogenesis | Overexpression | 22 % increase | (Yuan et al., 2012) | |
| tald-1 | Transaldolase | CELE_Y24D9A.8/ K00616 | Pentose Phosphate Pathway | Downregulation | Increase | (Bennett et al., 2017, 2014) | |
| tkt-1 | Transketolase | CELE_F01G10.1/ K00615 | Pentose Phosphate Pathway | Downregulation | Increase | (Bennett et al., 2017, 2014; Kim and Sun, 2007) | |
| 6PGD | 6-phosphogluconate dehydrogenase | CELE_T25B9.9/ K00033 | Pentose Phosphate Pathway | Downregulation | Increase | (Bennett et al., 2017, 2014) | |
| slcf-1 | SLC16 monocarboxylate transporter | CELE_F59F5.1/K08179 | Transporter | Downregulation | 40 % increase | (Mouchiroud et al., 2011) | |
| Sodium pyruvate | Sodium pyruvate | C00022 | Pyruvate metabolism | Feeding | 14 % increase | (Mouchiroud et al., 2011; Butler et al., 2013; Mishur et al., 2016) | |
| pdhk-2/ PDHK | Pyruvate dehydrogenase kinase | CELE_ZK370.5/ K00898 | Pyruvate metabolism | Downregulation | 20 % increase | (Mouchiroud et al., 2011) | |
| DCA | Dichloroacetate | C11149 | Pyruvate metabolism | Feeding | Increase | (Schaffer et al., 2011) | |
| a-lipoic acid | a-lipoic acid | C00725 | Pyruvate metabolism | Feeding | Increase | (Benedetti et al., 2008) | |
| tpk-1 | Thiamine pyrophosphokinase | CELE_ZK637.9/ K00949 | Thiamine metabolism | Mutant | 40 % increase | (de Jong et al., 2004) | |
| aco-2 | Aconitase | CELE_F54H12.1/ K01681 | TCA cycle | Downregulation | Increase | (Hamilton et al., 2005) | |
| IDH3A | Isocitrate dehydrogenase | CELE_F43G9.1/K00030 | TCA cycle | Downregulation | Increase | (Hamilton et al., 2005) | |
| IDH1 | Isocitrate dehydrogenase | CELE_F59B8.2/ K00031 | TCA cycle | Downregulation | Increase | (Hamilton et al., 2005) | |
| Acetic acid | Acetic acid | C00033 | Feeding | 20 % increase | (Chuang et al., 2009) | ||
| Malate | Malate | C00149 | TCA cycle | Feeding | 14 % increase | (Edwards et al., 2013) | |
| Fumarate | Fumarate | C00122 | TCA cycle | Feeding | 16 % increase | (Edwards et al., 2013) | |
| Oxaloacetate | Oxaloacetate | C00036 | TCA cycle | Feeding | 25 % increase | (Williams et al., 2009) | |
| a-ketoglutarate | a-ketoglutarate | C00026 | TCA cycle | Feeding | 32 % increase | (Chin et al., 2014) | |
| (R)-2 H G | 2-hydroxygluatarate | C01087 | TCA cycle | Feeding | 40 % increase | (Fu et al., 2015) | |
| (S)-2 H G | 2-hydroxyglutarate | C03196 | TCA cycle | Feeding | 30 % increase | (Fu et al., 2015) | |
| DLD | Dihydrolipoamide dehydrogenase | CELE_LLC1.3/K00382 | TCA cycle | Downregulation | Increase | (Butler et al., 2013; Mishur et al., 2016) | |
| 3M2OB | 3-methyl-2-oxobutyrate | C00141 | Feeding | Increase | (Butler et al., 2013; Mishur et al., 2016) | ||
| 3M2OV | 3-methyl-2-oxovalerate | C03465 | Feeding | Increase | (Butler et al., 2013; Mishur et al., 2016) | ||
| 4M2OV | 4-methyl-2-oxovalerate | C00233 | Feeding | Increase | (Butler et al., 2013; Mishur et al., 2016) | ||
| 2,4-PDA | a-ketoglutarate mimetic | Feeding | 15 % increase | (Butler et al., 2013; Mishur et al., 2016) | |||
| ETC Complex I | Multiple subunits | CELE_C09H10.3/K03942; CELE_T10E9.7/K03936; CELE_Y57G11C.12/K03950; CELE_K04G7.4/K03954; CELE_Y45G12B.1/K03934; CELE_W01A8.4/K03960; CELE_C18E9.4/K03959; CELE_C25H3.9/K03961; CELE_C33A12.1/K03949; CELE_D2030.4/K03963; CELE_F59C6.5/K03966; CELE_T20H4.5/K03941; CELE_Y53G8AL.2/K03953; CELE_Y56A3A.19/K03955; CELE_Y71H2AM.4/K03968; CELE_ZK809.3/K03962; CELE_T26A5.3/K03935; | ETC | Downregulation | Increase | Reviewed in (Munkacsy and Rea, 2014) | |
| ETC Complex III | Multiple subunits | CELE_C54G4.8/K00413; CELE_E04A4.7/K08738; CELE_F42G8.12/K00411; K00414; CELE_F45H10.2/K00418; CELE_R07E4.3/K00418; CELE_T02H6.11/K00417; CELE_T27E9.2/K00416; | ETC | Downregulation | Increase | Reviewed in (Munkacsy and Rea, 2014) | |
| ETC Complex IV | Multiple subunits | CELE_F26E4.9/K02265; CELE_Y37D8A.14/K02264; CELE_F26E4.6/K02272; CELE_F29C4.2/K02268; CELE_F54D8.2/K02266; CELE_T06D8.5/K02259; CELE_W09C5.8/K02263; CELE_Y71H2AM.5/K02267; | ETC | Downregulation | Increase | Reviewed in (Munkacsy and Rea, 2014) | |
| ETC Complex V | Multiple subunits | CELE_C53B7.4/K02140; CELE_F02E8.1/K02127; CELE_C34E10.6/K02133; CELE_F27C1.7/K02137; K02131; CELE_C06H2.1/K02138; CELE_H28O16.1/K02132 | ETC | Downregulation | Increase | Reviewed in (Munkacsy and Rea, 2014) | |
| antimycin A | ETC complex III inhibitor | C11339 | ETC | Feeding | Increase | (Dillin et al., 2002) | |
| Ethidium bromide | mtDNA transcription/replication inhibitor | C11161 | Feeding | Increase | (Tsang and Lemire, 2002) | ||
| Arsenite | Mitochondrial poison | C06697 | Feeding | Increase | (Schmeisser et al., 2013b) | ||
| F13G3.7 | F13G3.7 | CELE_F13G3.7./K15121 | Mitochondrial transporter | Downregulation | Increase | (Lee et al., 2003b) | |
| K01C8.7 | Slc-25A32 | CELE_K01C8.7/K15115 | Mitochondrial transporter | Downregulation | Increase | (Lee et al., 2003b) | |
| ceNAC-3/ceNaDC2 | ceNAC-3/ceNaDC2 | CELE_K08E5.2/K14445 | Mitochondrial transporter | Downregulation | 15 % increase | (Fei et al., 2003, 2004) | |
| ceNAC-2/NaCT | ceNAC-2/NaCT | Mitochondrial transporter | Downregulation | 19 % increase | (Fei et al., 2003, 2004) | ||
| dk-1 /Coq7 | COQ7 | CELE_ZC395.2/K06134 | Ubiquinone biosynthesis | Mutant | Increase | (Ewbank et al., 1997; Lakowski and Hekimi, 1996 | |
| CoQ8 | CoQ | Ubiquinone biosynthesis | Diet removal | 50 % increase | (Larsen and Clarke, 2002) | ||
| CoQ10 | CoQ | C11378 | Ubiquinone biosynthesis | Feeding | 18 % increase | (Ishii et al., 2004) | |
| CCCP | Mitochondrial uncoupler | C11164 | Uncoupling agent | Feeding | 60 % increase | (Lemire et al., 2009) | |
| FCCP | Mitochondrial uncoupler | Uncoupling agent | Feeding | 22 % increase | (Morcos et al., 2008) | ||
| zUCP2 | Zebrafish uncoupling protein 2 | K15103 | Uncoupling | Overexpression | 40 % increase | (Sagi and Kim, 2012) | |
| Drosophila | |||||||
| Hex-A | Hexokinase-A | Dmel_CG3001/K00844 | Glycolysis | Downregulation | Increase | (Talbert et al., 2015) | |
| Hex-C | Hexokinase-C | Dmel_CG8094/K00844 | Glycolysis | Downregulation | Increase | (Talbert et al., 2015) | |
| masl | alpha-1,2-mannosidase I | Dmel_CG31202/K01230 | N-linked glycosylation | Suppression | 38 % increase | (Liu et al., 2009) | |
| Edeml | ER degradation-enhancing alpha-1,2-mannosidase-like protein | Dmel_CG3810/K10085 | N-linked glycosylation | Suppression | 30 % increase | (Liu et al., 2009) | |
| Tpi + Pgi(Gpi) | Triose phosphate isomerase + Phosphoglucose isomerase | Dmel_CG2171/K01803 + Dmel_CG8251/K01810 | Glycolysis | Upregulation | Increase | (Ma et al., 2018b) | |
| LDH | Lactate dehydrogenase | Dmel_CG10160/K00016 | Glycolysis | Downregulation | Increase | (Long et al., 2020) | |
| GlyS/CG6904 | Glycogen synthase | Dmel_CG6904/K00693 | Glycogen metabolism | Downregulation | 10 % increase | (Sinadinos et al., 2014) | |
| GlyP | Glycogen phosphorylase | Dmel_CG7254/K00688 | Glycogen | Overexpression | Increase | (Post et al., 2018) | |
| CG33138 | 1,4-alpha glucan branching | Dmel_CG33138/K00700 | metabolism Glycogen | EP element | Increase | (Paik et al., 2012) | |
| CA | enzyme Colanic acid | metabolism Polysaccharide | insertion Feeding | Increase | (Han et al., 2017a) | ||
| G6PD | Glucose-6-phosphate dehydrogenase | Dmel_CG7140/K00036 | Pentose Phosphate Pathway | Ubiquitous and neuronal overexpression | 38 % increase | (Legan et al., 2008; Wang et al., 2019) | |
| Rpi | Ribose-5-phosphate isomerase | Dmel_CG30410/K01807 | Pentose Phosphate Pathway | Downregulation | 38 % increase | (Wang et al., 2012) | |
| DCA | Dichloroacetate | C11149 | Pyruvate metabolism | Feeding | 15 % increase | (Pandey et al., 2014) | |
| Men/ME1 | Malic enzyme | Dmel_CG10120/K00029 | Overexpression | 15 % increase | (Kim et al., 2015) | ||
| ATPCL | ATP citrate lyase | Dmel_CG8322/K01648 | Acetyl-CoA metabolism | Heterozygous | Increase | (Peleg et al., 2016) | |
| AcCoAS | Acetyl-CoA Synthase | Dmel_CG9390/K01895 | Acetyl-CoA metabolism | Downregulation | Increase | (Eisenberg et al., 2014) | |
| α-KG | α-ketoglutarate | C00026 | TCA cycle | Feeding | Increase | (Lylyk et al., 2018; Su et al., 2019) | |
| J147 | ATP5A inhibitor | Feeding | Increase | (Goldberg et al., 2018) | |||
| CG9172 | ETC complex I, NDUFS7 subunit | Dmel_CG9172/K03940 | ETC | Downregulation | Increase | (Copeland et al., 2009) | |
| CG9762 | ETC complex I, NDUFB5 subunit | Dmel_CG9762 /K03961 | ETC | Downregulation | Increase | (Copeland et al., 2009) | |
| CG17856 | ETC complex III, UQCRB subunit | Dmel_CG17856/K00417 | ETC | Downregulation | Increase | (Copeland et al., 2009) | |
| CG18809 | ETC complex IV | Dmel_CG18809 | ETC | Downregulation | Increase | (Copeland et al., 2009) | |
| CG5389 | ETC complex V, CG5389/ATP5F1B subunit | Dmel_CG5389/K02133 | ETC | Downregulation | Increase | (Copeland et al., 2009) | |
| ATPsynD | ATP synthase subunit d | Dmel_CG6030/K02138 | ETC | Downregulation | Increase | (Sun et al., 2014) | |
| Indy | Indy | Dmel_CG3979/K14445 | Mitochondrial transporter | Downregulation | Increase | (Rogina et al., 2000) | |
| sbo/Coq2 | COQ2 | Dmel_CG9613/K06125 | Ubiquinone biosynthesis | Heterozygous | 31 % increase | (Liu et al., 2011) | |
| 2,4-dinitrophenol (DNP) | Mitochondrial uncoupler | C02496 | Mitochondrial uncoupling | Feeding | Increase | (Padalko, 2005; Ulgherait et al., 2020) | |
| UCP5 | Mitochondrial uncoupling protein 5 | Dmel_CG7314/K15106 | Mitochondrial uncoupling | Deletion | 30 % increase | (Sanchez-Blanco et al., 2006) | |
| hUCP2 | Human uncoupling protein 2 | 7351/K15103 | Mitochondrial uncoupling | Overexpression | Increase | (Fridell et al., 2005; Fridell et al., 2009) | |
| mUCP1 | Mouse uncoupling protein 1 | K08769 | Mitochondrial uncoupling | Overexpression | Increase | (Fridell et al., 2009) | |
| NDI | Alternative NADH dehydrogenase | Complex I bypass | Overexpression | 40 % increase | (Bahadorani et al., 2010a; Sanz et al., 2010; Hur et al., 2013) | ||
| NDX | Alternative NADH dehydrogenase | Complex III bypass | Overexpression | 50 % increase | (Gospodaryov et al., 2014, 2019) | ||
| Yeast | |||||||
| HXK2 | Hexokinase 2 | YGL253W/K00844 | Glycolysis | Deletion | Increase | (Lin et al., 2000, 2002) | |
| Glucose | Glucose | C00031 | Glycolysis | Deprivation | Increase | (Lin et al., 2000, 2002; Brokate-Llanos et al., 2014) | |
| Xylitol | Xylitol | C00379 | Feeding | Increase | (Kaeberlein et al., 2002) | ||
| Glycerol | Glycerol | C00116 | Feeding | Increase | (Kaeberlein et al., 2002) | ||
| Sorbitol | Sorbitol | C00794 | Feeding | Increase | (Chandler-Brown et al., 2015; Brokate-Llanos et al., 2014) | ||
| TDH2 | Glyceraldehyde-3- phosphate dehydrogenase | YJR009C/K00134 | Glycolysis | Deletion | RLS increase | (Hachinohe et al., 2013) | |
| Latl | Dihydrolipoamide acetyltransferase | YNL071W/K00627 | Pyruvate metabolism | Overexpression | 30 % increase | (Easlon et al., 2007) | |
| 2,4-dinitrophenol (DNP) | Mitochondrial uncoupling agent | C02496 | Mitochondrial uncoupling | Feeding | Increase | (Barros et al., 2004) | |
| Aatl, mdhl, Gut2 | Mitochondrial NADH shuttles | YKL106W/K14455; YKL085W/K00026; YIL155C/K00111 | NADH shuttling | Overexpression | 25 % RLS increase | (Easlon et al., 2008) | |
| Aat2 and Mdh2 | Mitochondrial NADH shuttles | YLR027C/K14454; YOL126C/K00026 | NADH shuttling | Overexpression | 15 % RLS increase | (Easlon et al., 2008) | |
| Mice | |||||||
| Acarbose | Acarbose | C06802 | Feeding | 16 % increase | (Harrison et al., 2019, 2014) | ||
| GlcN | D-Glucosamine | C00329 | Glycolysis | Feeding | Increase | (Weimer et al., 2014) | |
| G6PD | Human glucose-6-phosphate dehydrogenase | 2539/K00036 | Pentose phosphate pathway | Overexpression | 14 % increase | (Nobrega-Pereira et al., 2016) | |
| Mclkl | Dimethyl-Q 7 | 12,850/K06134 | Ubiquinone biosynthesis | Heterozygous | 31 % increase | (Liu et al., 2005) | |
| SURF1 | Cytochrome C oxidase | 20,930/K14998 | Deficiency | Increase | (Dell’agnello et al., 2007) | ||
| 2,4-dinitrophenol (DNP) | Mitochondrial uncoupling agent | C02496 | Mitochondrial uncoupling | Feeding | Increase | (Caldeira da Silva et al., 2008) | |
| UCP1 | Uncoupling protein 1 | 22,227/K08769 | Mitochondrial uncoupling | Skeletal muscle- specific expression | 10 % increase | (Gates et al., 2007) | |
| Amino acid metabolism | |||||||
| Worms | |||||||
| bcat-1 | Branched amino acid transferse-1 | CELE_K02A4.1/K00826 | Amino acid degradation | Downregulation | Increase | (Mansfeld et al., 2015) | |
| leucine | Leucine | C00123 | Amino acid metabolism | Feeding | Increase | (Edwards et al., 2015a; Mansfeld et al., 2015) | |
| valine | Valine | C00183 | Amino acid metabolism | Feeding | Increase | (Edwards et al., 2015a; Mansfeld et al., 2015) | |
| isoleucine | Isoleucine | C00407 | Amino acid metabolism | Feeding | Increase | (Edwards et al., 2015a; Mansfeld et al., 2015) | |
| Gcat | Glycine-C-acetyltransferase | CELE_T25B9.1/K00639 | Threonine metabolism | Downregulation | 22 % increase | (Ravichandran et al., 2018) | |
| CeGly | Glyoxalase-1 | CELE_C16C10.10/no KO assigned | Methylglyoxal pathway | Overexpression | Increase | (Morcos et al., 2008) | |
| Rifampicin | Rifampicin | D00211 | Feeding | 60 % increase | (Golegaonkar et al., 2015) | ||
| Proline | Proline | C00148 | Proline metabolism | Feeding | Increase | (Edwards et al., 2015a; Zarse et al., 2012) | |
| ARGK-1 | Arginine kinase | CELE_F44G3.2/K00934 | Arginine metabolism | Overexpression, downregulation | Increase | (McQuary et al., 2016; Rozanov et al., 2020) | |
| sams-1 | Methionine adenosyltransferase | CELE_C49F5.1/K00789 | Methionine metabolism | Downregulation | Increase | (Hansen et al., 2005) | |
| cbs-1 | Cystathionine beta-synthase | CELE_ZC373.1/K01697 | Transsulfuration pathway | Overexpression | Increase | (Hine et al., 2015) | |
| N-Acetyl-L-cysteine | N-acetyl-L-cysteine | C06809 | Transsulfuration pathway | Feeding | Increase | (Oh et al., 2015) | |
| Spermidine | Spermidine | C00315 | Methionine metabolism | Feeding | 15 % increase | (Eisenberg et al., 2009) | |
| Glycine | Glycine | C00037 | Methionine metabolism | Feeding | Increase | (Liu et al., 2019) | |
| mel-32 | Serine hydroxymethyltransferase | CELE_C05D11.11/K00600 | Glycine metabolism | Downregulation | Increase | (Liu et al., 2019) | |
| Hpd-1 | 4-hydroxyphenylpyruvate dioxygenase | CELE_T21C12.2/K00457 | Tyrosine metabolism | Downregulation | 30 % increase | (Lee et al., 2003a) (Yuan et al., 2012) | |
| tatn-1 | Tyrosine aminotransferase | CELE_F42D1.2/K00815 | Tyrosine metabolism | Mutation | Increase | (Ferguson et al., 2013) | |
| Tdo-2 | Tryptophan 2,3 dioxygenase | CELE_C28H8.11/K00453 | Tryptophan metabolism | Depletion | Increase | (van der Goot et al., 2012) | |
| Tryptophan | Tryptophan | C00078 | Tryptophan metabolism | Feeding | Increase | (Edwards et al., 2015a) (Gebauer et al., 2016) | |
| Ibuprofen | Ibuprofen | D00126 | Tryptophan uptake inhibition | Feeding | Increase | (He et al., 2014) | |
| Acsd-1 | Aminocarboxymuconate- semialdehyde decarboxylase (ACMSD) | CELE_Y71D11A.3/K03392 | NAD + synthesis | Downregulation | Increase | (Katsyuba et al., 2018) | |
| Kynu-1 | Kynureninase | CELE_C15H9.7/K01556 | NAD + synthesis | Downregulation | 23 % increase | (Sutphin et al., 2017) | |
| Nicotinamide | Nicotinamide (NAM) | C00153 | Nicotinamide pathway | Feeding | Increase | (Mouchiroud et al., 2013) (Schmeisser et al., 2013a) | |
| Nicotinamide riboside | Nicotinamide riboside (NR) | C03150 | Nicotinamide pathway | Feeding | Increase | (Mouchiroud et al., 2013) | |
| NAD | NAD | C00003 | Nicotinamide pathway | Feeding | 15 % increase | (Hashimoto et al., 2010) | |
| Nicotinic acid | Nicotinic acid | C00253 | Nicotinamide pathway | Feeding | Increase | (Schmeisser et al., 2013a) | |
| 1- methylnicotinamide | 1-methylnicotinamide (MNA) | C02918 | Nicotinamide pathway | Feeding | Increase | (Schmeisser et al., 2013a) | |
| ANMT-1 | Nicotinamide-N- methyltransferase | CELE_B0303.2/K00541 | Nicotinamide pathway | Overexpression | Increase | (Schmeisser et al., 2013a) | |
| Drosophila | |||||||
| dAhcyLl | Adenosylhomocysteinase like 1 | Dmel_CG9977/K01251 | Methionine metabolism | Downregulation | Increase | (Parkhitko et al., 2016) | |
| dAhcyL2 | Adenosylhomocysteinase like 2 | Dmel_CG8956/K01251 | Methionine metabolism | Downregulation | Increase | (Parkhitko et al., 2016) | |
| GNMT | Glycine N-methyltransferase | Dmel_CG6188/K00552 | Methyltransferase | Overexpression | Increase | (Obata and Miura, 2015) | |
| dCBS | Cystathionine beta synthase | Dmel_CG1753/K01697 | Transsulfuration pathway | Ubiquitous or neuron-specific overexpression | Increase | (Kabil et al., 2011) | |
| GCLc | Glutamate-cysteine ligase catalytic subunit | Dmel_CG2259/K11204 | Glutamatecysteine pathway | Global or neuronal overexpression | Increase | (Orr et al., 2005) | |
| GCLm | Glutamate-cysteine ligase modulatory subunit | Dmel_CG4919/K11205 | Glutamatecysteine pathway | Global or neuronal overexpression | Increase | (Orr et al., 2005) | |
| NAC | N-acetylcysteine | C06809 | Glutamatecysteine pathway | Feeding | Increase | (Brack et al., 1997) | |
| spermidine | Spermidine | C00315 | Methionine pathway | Feeding | 30 % increase | (Eisenberg et al., 2009) | |
| CG1461 | Tyrosine aminotransferase (TAT) | Dmel_CG1461/K00815 | Tyrosine metabolism | Neuronal-specific downregulation | Increase | (Parkhitko et al., unpublished | |
| HPD | 4-hydroxyphenylpyruvate dioxygenase | Dmel_CG11796/K00457 | Tyrosine metabolism | Neuronal-specific downregulation | Increase | (Parkhitko et al., unpublished | |
| HGO | Homogentisate 1,2- dioxygenase | Dmel_CG4779/K00451 | Tyrosine metabolism | Neuronal-specific downregulation | Increase | (Parkhitko et al., unpublished | |
| TDO | Tryptophan 2,3 dioxygenase (vermilion) | Dmel_CG2155/K00453 | Tryptophan metabolism | Depletion | 27 % increase | (Oxenkrug, 2010) | |
| Alpha-methyl tryptophan | TDO inhibitor | Tryptophan metabolism | Feeding | 27 % increase | (Oxenkrug et al., 2011) | ||
| minocycline | Minocycline (tetracycline antibiotic) | D05045 | Tryptophan metabolism | Feeding | Increase | (Oxenkrug et al., 2012) | |
| Ibuprofen | Ibuprofen | D00126 | Tryptophan uptake inhibition | Feeding | Increase | (He et al., 2014) | |
| CG9940 | NAD + synthase (NADSYN) | Dmel_CG9940/K01950 | NAD metabolism | Overexpression | Increase | (Wen et al., 2016) | |
| D-NAAM/NAMase | Nicotinamidase | Dmel_CG31216/no KO assigned | NAD metabolism | Whole-body expression | 30 % increase | (Balan et al., 2008) | |
| CYB5R | Cytochrome b5 reductase 3 | Dmel_CG5946/K00326 | NAD metabolism | Overexpression | 17 % increase | (Martin-Montalvo et al., 2016) | |
| Yeast | |||||||
| Threonine | Threonine | C00188 | Amino acid metabolism | Feeding | CLS increase | (Alvers et al., 2009) | |
| Leucine | Leucine | C00123 | Amino acid metabolism | Feeding | CLS increase | (Alvers et al., 2009) | |
| Isoleucine | Isoleucine | C00407 | Amino acid metabolism | Feeding | CLS increase | (Alvers et al., 2009) | |
| Valine | Valine | C00183 | Amino acid metabolism | Feeding | CLS increase | (Alvers et al., 2009) | |
| Ibuprofen | Ibuprofen | D00126 | Tryptophan uptake inhibition | Feeding | Increase | (He et al., 2014) | |
| PNC1 | Nicotinamidase | YGL037C/K01440 | Nicotinamide pathway | Overexpression | Increase | (Anderson et al., 2003) | |
| MET3 | Sulfate adenylyltransferase | YJR010W/K00958 | Methionine synthesis | Deletion | RLS increase | (McCormick et al., 2015) | |
| SAM1 | Methionine adenosyltransferase | YLR180W/K00789 | Methionine cycle | Deletion | RLS increase | (McCormick et al., 2015) | |
| Methionine | Methionine | C00073 | Methionine metabolism | Feed reduction in strain auxotrophic for his/leu/lys | CLS increase | (Wu et al., 2013) | |
| MET2 | Methionine biosynthesis | YNL277W/K00641 | Methionine metabolism | Deletion | CLS increase | (Johnson and Johnson, 2014; Ruckenstuhl et al., 2014) | |
| MET15 | Methionine biosynthesis | YLR303W/K17069 | Methionine metabolism | Deletion | CLS increase | (Johnson and Johnson, 2014; Ruckenstuhl et al., 2014) | |
| MGL | L-methionine gamma lyase (Methioninase) from Clostridium sporogenes | Methionine metabolism | Overexpression | CLS increase | (Plummer and Johnson, 2019) | ||
| Mice | |||||||
| Methionine | Methionine | C00073 | Methionine metabolism | Feed reduction | Increase | (Miller et al., 2005) | |
| Spermidine | Spermidine | C00315 | Methionine metabolism | Feeding | Increase | (Eisenberg et al., 2016) | |
| Spermine | Spermine | C00750 | Methionine metabolism | Feeding | Increase | (Eisenberg et al., 2016) | |
| Nicotinamide riboside | Nicotinamide riboside (NR) | C03150 | Nicotinamide pathway | Feeding | 5% increase | (Zhang et al., 2016) | |
| Nqo1, Cyb5r3 | NAD(P)H dehydrogenase, cytochrome b5 reductase 3 | 18,104/K00355; 109,754/K00326 | NADPH metabolism | Overexpression | 4% increase | (Diaz-Ruiz et al., 2018) | |
| eNAMPT | Nicotinamide phosphoribosyltransferase | 59,027/K03462 | Nicotinamide metabolism | Overexpression | 8% increase | (Yoshida et al., 2019) | |
| Rats | |||||||
| Methionine | Methionine | C00073 | Methionine metabolism | Feed reduction | 30 % increase | (Orentreich et al., 1993) | |
| Tryptophan | Tryptophan | C00078 | Tryptophan metabolism | Feed reduction | Increase | (Ooka et al., 1988) | |
| Nucleotide metabolism | |||||||
| Worms | |||||||
| Xdh | Xanthine dehydrogenase | CELE_F55B11.1/K00106 | Nucleotide metabolism | Downregulation | Increase | (Hamilton et al., 2005) | |
| B0286.3 | PAICS | CELE_B0286.3/K01587 | Nucleotide metabolism | Downregulation | 15 % increase | (Sutphin et al., 2017) | |
| Hypoxanthine | Purine metabolism intermediate | C00262 | Nucleotide metabolism | Feeding | 5% increase | (Copes et al., 2015) | |
| Uric Acid | Uric Acid | C00366 | Nucleotide metabolism | Feeding | 15 % increase | (Wan et al., 2020) | |
| Allantoin | Oxidation product (UA) | C02348 | Uric Acid metabolism | Feeding | 22 % increase (wildtype) | (Calvert et al., 2016) | |
| Thymine | Pyrimidine metabolism intermediate | C00178 | Nucleotide metabolism | Feeding | 18 % increase | (Wan et al., 2019) | |
| β-aminoisobutyrate | Pyrimidine metabolism intermediate | C03284 | Nucleotide metabolism | Feeding | 10 % increase | (Wan et al., 2019) | |
| Orotate | C00295 | Feeding | (Wan et al., 2019) | ||||
| Pyrimidine metabolism intermediate | Nucleotide metabolism | 15 % increase | |||||
| Uridine | Pyrimidine metabolism intermediate | C00299 | Nucleotide metabolism | Feeding | 10 % increase | (Wan et al., 2019) | |
| Cytidine | Pyrimidine metabolism intermediate | C00475 | Nucleotide metabolism | Feeding | 8% increase | (Wan et al., 2019); Copes et al., 2015) | |
| dpyd-1 | Dihydropyrimidine dehydrogenase | CELE_C25F6.3/K00207 | Nucleotide metabolism | Downregulation | 13 % increase | (Wan et al., 2019) | |
| upp-l | Uridine phosphorylase | CELE_ZK783.2/K00757 | Nucleotide metabolism | Downregulation | 19 % increase | (Wan et al., 2019) | |
| SMX | Sulfamethoxazole | lC-metabolism | Feeding | Increase | (Virk et al., 2012) | ||
| Drosophila | |||||||
| AdSS | Adenylosuccinate synthetase | Dmel_CG17273/K01939 | Nucleotide metabolism | Heterozygous | 20 % increase | (Stenesen et al., 2013) | |
| AdenoK | Adenosine kinase | Dmel_CG11255/K00856 | Nucleotide metabolism | Heterozygous | Increase | Stenesen et al., 2013) | |
| Aprt | Adenine phosphoribosyltransferase | Dmel_CG18315/K00759 | Nucleotide metabolism | Heterozygous | Increase | Stenesen et al., 2013) | |
| Adk2 | Adenylate kinase | Dmel_CG3140/K00939 | Nucleotide metabolism | Heterozygous | Increase | Stenesen et al., 2013) | |
| Nmdmc | NMDMC/MTHFD2 | Dmel_CG18466/K13403 | 1C metabolism | Overexpression | Increase | (Yu et al., 2015) | |
| Mice | |||||||
| Uox | Urate oxidase/uricase | 22,262/K00365 | Uric Acid metabolism | Heterozygous | Increase | Cutler et al., 2019 | |
| Lipid metabolism | |||||||
| Worms | |||||||
| lipl-4 | Lipase | CELE_K04A8.5/K19771 | Lipid metabolism | Intestine overexpression | 24 % increase | (Wang et al., 2008) | |
| Arachidonic acid | ra-6 fatty acid, PUFA | C00219 | Lipid metabolism | Feeding | Increase | (O’Rourke et al., 2013) | |
| di-homo-y-linoleic acid | ra-6 fatty acid, PUFA | C03242 | Lipid metabolism | Feeding | Increase | (O’Rourke et al., 2013) | |
| f061;-linolenic acid | ra-3 fatty acid, PUFA | C06427 | Lipid metabolism | Feeding | 30 % increase | (Qi et al., 2017) | |
| 10-hydroxy-2- decenoic acid | Fatty Acid from Royal Jelly | C02774 | Lipid metabolism | Feeding | 12 % increase | (Honda et al., 2011) | |
| Oleic acid | Monounsaturated ra -9 fatty acid, MUFA | C00712 | Lipid metabolism | Feeding | 15–20% increase | (Han et al., 2017b) | |
| Palmitoleic acid | Monounsaturated ra -7 fatty acid, MUFA | C08362 | Lipid metabolism | Feeding | 15–20% increase | (Han et al., 2017b) | |
| cis-vaccenic acid | Monounsaturated ra -7 fatty acid, MUFA | C21944 | Lipid metabolism | Feeding | 15–20% increase | (Han et al., 2017b) | |
| FAT-7 | Desaturase | CELE_F10D2.9/K00507 | Lipid metabolism | Intestine overexpression | Increase | (Han et al., 2017b) | |
| elo-1 | Elongase | CELE_F56H11.4/K10203 | Lipid metabolism | Downregulation | 11 % increase | (Shmookler Reis et al., 2011) | |
| elo-2 | Elongase | CELE_F11E6.5/K10203 | Lipid metabolism | Downregulation | 8% increase | (Shmookler Reis et al., 2011) | |
| fat-4 | Desaturase | CELE_T13F2.1 | Lipid metabolism | Downregulation | 25 % increase | (Shmookler Reis et al., 2011) | |
| lbp-8 | Fatty acid-binding protein | CELE_T22G5.6/K08752 | Lipid metabolism | Overexpression | 30 % increase | (Folick et al., 2015) | |
| KDS-5104 | Oleoylethanolamide analog | Lipid metabolism | Feeding | Increase | (Folick et al., 2015) | ||
| Acs-2 | Acyl-CoA synthetase | Mitochondrial 1d5d;-oxidation | Overexpression | Increase | (Ramachandran et al., 2019) | ||
| NDG-4 | NDG-4 | CELE_F56F3.2/no KO assigned | Lipid transport pathway | Downregulation | Increase | (Brejning et al., 2014) | |
| NRF-5 | NRF-5 | CELE_F55B12.5 | Lipid transport pathway | Downregulation | Increase | (Brejning et al., 2014) | |
| NRF-6 | NRF-6 | CELE_C08B11.4 | Lipid transport pathway | Downregulation | Increase | (Brejning et al., 2014) | |
| VIT/vitellogenin | Yolk lipoprotein | CELE_K09F5.2; CELE_C42D8.2; CELE_F59D8.1; CELE_F59D8.2; CELE_C04F6.1 | Lipid metabolism | Downregulation | 16–40% Increase | (Murphy et al., 2003; Seah et al., 2016) | |
| Sodium butyrate | HDACS class I and II inhibitor (bacterial product) | C00246 | Lipid metabolism | Feeding | Increase | (Zhang et al., 2009) | |
| D- ß-hydroxybutyrate | D- p-hydroxybutyrate | C01089 | Lipid metabolism | Feeding | 20 % increase | (Edwards et al., 2014) | |
| DAGL/inaE/dagl-1 | Diacylglycerol lipase | CELE_F42G9.6/K13806 | Lipid metabolism | Overexpression | Increase | (Lin et al., 2014) | |
| DGK/rdgA/dgk-5 | Diacylglycerol kinase | CELE_K06A1.6/K00901 | Lipid metabolism | Downregulation | Increase | (Lin et al., 2014) | |
| ISP-1 | SPT inhibitor | Lipid metabolism | Feeding | 31 % Increase | (Cutler et al., 2014) | ||
| SPT (sptl-1) | Serine palmitoyltransferase | CELE_C23H3.4/K00654 | Lipid metabolism | Downregulation | 33 % Increase | (Cutler et al., 2014) | |
| D609 | Sphingomyelin synthase inhibitor | Lipid metabolism | Feeding | 25 % Increase | (Cutler et al., 2014) | ||
| Dihydroceramide desaturase | Dihydroceramide desaturase | CELE_Y54E5A.1/K04712 | Lipid metabolism | Downregulation | 40 % Increase | (Cutler et al., 2014) | |
| PDMP | Glucosylceramide synthase inhibitor | Lipid metabolism | Feeding | 38 % Increase | (Cutler et al., 2014) | ||
| Glucosylceramide synthase | Glucosylceramide synthase | CELE_F20B4.6/K00720 | Lipid metabolism | Downregulation | 40 % Increase | (Cutler et al., 2014) | |
| Epoxyquinone G109 | Neutral sphingomyelinase inhibitor | Lipid metabolism | Feeding | 6% Increase | (Cutler et al., 2014) | ||
| Neutral/acidic ceramidase | Neutral/acidic ceramidase | CELE_F27E5.1/K12348 | Lipid metabolism | Downregulation | 40 % Increase | (Cutler et al., 2014) | |
| ASM-1 | Acid sphingomyelinase | CELE_B0252.2/K12350 | Lipid metabolism | Downregulation | 12 % Increase | (Kim and Sun, 2012) | |
| ASM-2 | Acid sphingomyelinase | CELE_ZK455.4/K12350 | Lipid metabolism | Downregulation | 10 % Increase | (Kim and Sun, 2012) | |
| ASM-3 | Acid sphingomyelinase | CELE_W03G1.7/K12350 | Lipid metabolism | Mutant and downregulation | 14–19% Increase | (Kim and Sun, 2012) | |
| Desipramine | ASM inhibitor | D07791 | Lipid metabolism | Feeding | 24 % Increase | (Kim and Sun, 2012) | |
| Clomipramine | ASM inhibitor | D07727 | Lipid metabolism | Feeding | 14 % Increase | (Kim and Sun, 2012) | |
| HYL-1 and LAGR-1 | Ceramide synthase genes | CELE_C09G4.1/K23727 and CELE_Y6B3B.10/K04710 | Lipid metabolism | Downregulation | 21.4 % Increase | (Mosbech et al., 2013) | |
| HYL-1 | Ceramide synthase gene | CELE_C09G4.1/K23727 | Lipid metabolism | Downregulation | 15 % Increase | (Tedesco et al., 2008) | |
| FAAH | Fatty acid amide hydrolase | CELE_B0218.1/K15528 | Lipid metabolism | Overexpression | Increase | (Lucanic et al., 2011) | |
| Pregnenolone | Pregnenolone | C01953 | Steroid hormone biosynthesis | Feeding | 15–20% increase | (Broue et al., 2007) | |
| Ascr#2 | Ascaroside | Lipid metabolism | Endogenous | 17 % increase | (Ludewig et al., 2013) | ||
| Ascr#3 | Ascaroside | Lipid metabolism | Endogenous production | 21 % increase | (Ludewig et al., 2013) | ||
| Drosophila | |||||||
| CG6783 | Fatty acid-binding protein | Dmel_CG6783/K08752 | Lipid metabolism | Overexpression | Increase | (Lee et al., 2012) | |
| CG13890 | Dodecenoyl-CoA delta- | Dmel_CG13890/K13239 | Lipid metabolism | Overexpression | Increase | (Lee et al., 2012) | |
| Glaz | isomerase Lipid-binding protein | Dmel_CG4604/K03098 | Lipid metabolism | Overexpression | 18 % Increase | (Walker et al., 2006) | |
| human ApoD | Lipid-binding protein | K03098 | Lipid metabolism | Overexpression | 40 % Increase | (Muffat et al., 2008) | |
| Enigma (Egm) | P-oxidation of fatty acids | Dmel_Cg9006/no KO assigned | Lipid metabolism | Heterozygous | 19.5 % increase | (Mourikis et al., 2006) | |
| Sodium butyrate | Short-chain fatty acid (bacterial product) | C00246 | Lipid metabolism | Feeding | Increase | (Vaiserman et al., 2012) | |
| DAGL/inaE/dagl-1 | Diacylglycerol lipase | Dmel_Cg33174/K13806 | Lipid metabolism | Overexpression | Increase | (Lin et al., 2014) | |
| DGK/rdgA/dgk-5 | Diacylglycerol kinase | CELE_K06A1.6/K00901 | Lipid metabolism | Downregulation | Increase | (Lin et al., 2014) | |
| Dacer / bwa | Alkaline ceramidase | Dmel_CG13969/K01441 | Lipid metabolism | Downregulation | 50 % increase | (Yang et al., 2010) | |
| Yeast | |||||||
| Tgl3 | TAG lipase | YMR313C/K14675 | Glycerolipid metabolism | Deletion | CLS increase | (Handee et al., 2016) | |
| Tgl4 | TAG lipase | YKR089C/K14674 | Glycerolipid metabolism | Deletion | CLS increase | (Handee et al., 2016) | |
| Dga1p | Diacylglycerol O- acyltransferase | YOR245C/K14457 | Glycerolipid metabolism | Overexpression | CLS increase | (Handee et al., 2016) | |
| Mice Dgat1 | Diacylglycerol O- acyltransferase-1 | 13,350/K11155 | Lipid metabolism | Deficiency | 25 % increase | (Streeper et al., 2012) | |
| AMP-activated protein kinase (AMPK) | |||||||
| Worms | |||||||
| Aak-2 | AMP-activated protein kinase subunit alpha-1 | CELE_PAR2.3/K07198 | AMPK pathway | Overexpression | 13 % increase | (Apfeld et al., 2004; Greer et al., 2007) | |
| Metformin | Metformin | D04966 | AMPK pathway | Feeding with E. coli co-culture | Increase | (Cabreiro et al., 2013) | |
| Agmatine | Agmatine (bacterial metabolite) | C00179 | AMPK pathway | Feeding | Increase | (Pryor et al., 2019) | |
| Drosophila | |||||||
| AMPK alpha | AMP-activated protein kinase alpha subunit, isoform A | Dmel_CG3051/K07198 | AMPK pathway | Overexpression | Increase | (Stenesen et al., 2013) | |
| Agmatine | Agmatine (bacterial metabolite) | C00179 | AMPK pathway | Feeding | Increase | (Pryor et al., 2019) | |
| Mice | |||||||
| Rapamycin & metformin | Rapamycin & metformin | C07909, D04966 | AMPK pathway | Feeding | Increase | (Strong et al., 2016) | |
2. Metabolic pathways
2.1. Carbohydrate metabolism
Glycolysis is a central carbohydrate metabolism pathway with an important role in cancer and aging. During glycolysis, glucose is broken down into pyruvate, producing ATP, while glycolytic intermediates serve as direct precursors of many cellular building blocks (Fig. 1). Hexokinase (HK) is the first and one of the rate-limiting enzyme of glycolysis, catalyzing the phosphorylation of glucose to form glucose-6-phosphate. Adding glucose to C. elegans feeding media shortened worm lifespan (Schulz et al., 2007) and inhibited activities of the lifespan-extending transcription factors DAF-16 and HSF-1 (Lee et al., 2009). 2-Deoxyglucose (2-DG) is a glucose analog that can be phosphorylated by hexokinase, resulting in the formation of 2-deoxyglucose-phosphate, which lacks the ability to undergo glycolysis. In adult C. elegans, exposure to 5 mM 2-DG led to a specific blockade of glucose metabolism and glycolysis. Worms maintained on food containing 2-DG exhibited a 17 % extension of lifespan. This lifespan extension was possible even under short-term exposure to 2-DG, i.e. when worms were fed on the supplemented media for 6 days in the beginning of their adult stage (Schulz et al., 2007). A similar lifespan extension effect was achieved by inhibition of a glycolytic enzyme, glucose phosphate isomerase (gpi-1). Exposure of worms to 2-DG or gpi-1 inhibition activated aak-2 (C. elegans ortholog of AMPK), and aak-2 was required for lifespan extension. Treatment of worms with 2-DG caused a significant increase in reactive oxygen species (ROS) formation, and pretreatment with N-acetylcysteine (NAC), a membrane-permeable glutathione precursor known to ameliorate the effects of ROS, significantly decreased ROS formation and completely abolished the effects of 2-DG on lifespan extension, whereas NAC alone had no significant effect on lifespan (Schulz et al., 2007). Gpi-1 was also identified as a hit in a C. elegans RNAi library screen for clones that extend lifespan (Hansen et al., 2005). Despite the positive effect of 2-DG in worms, supplementation of male Fischer-344 rats with 2-DG led to increased mortality and caused cardiotoxicity (Minor et al., 2010). A glycolysis inhibitor less potent than 2-DG, glucosamine, extended lifespan in mice; this effect will be discussed later in detail (Weimer et al., 2014).
Fig. 1.
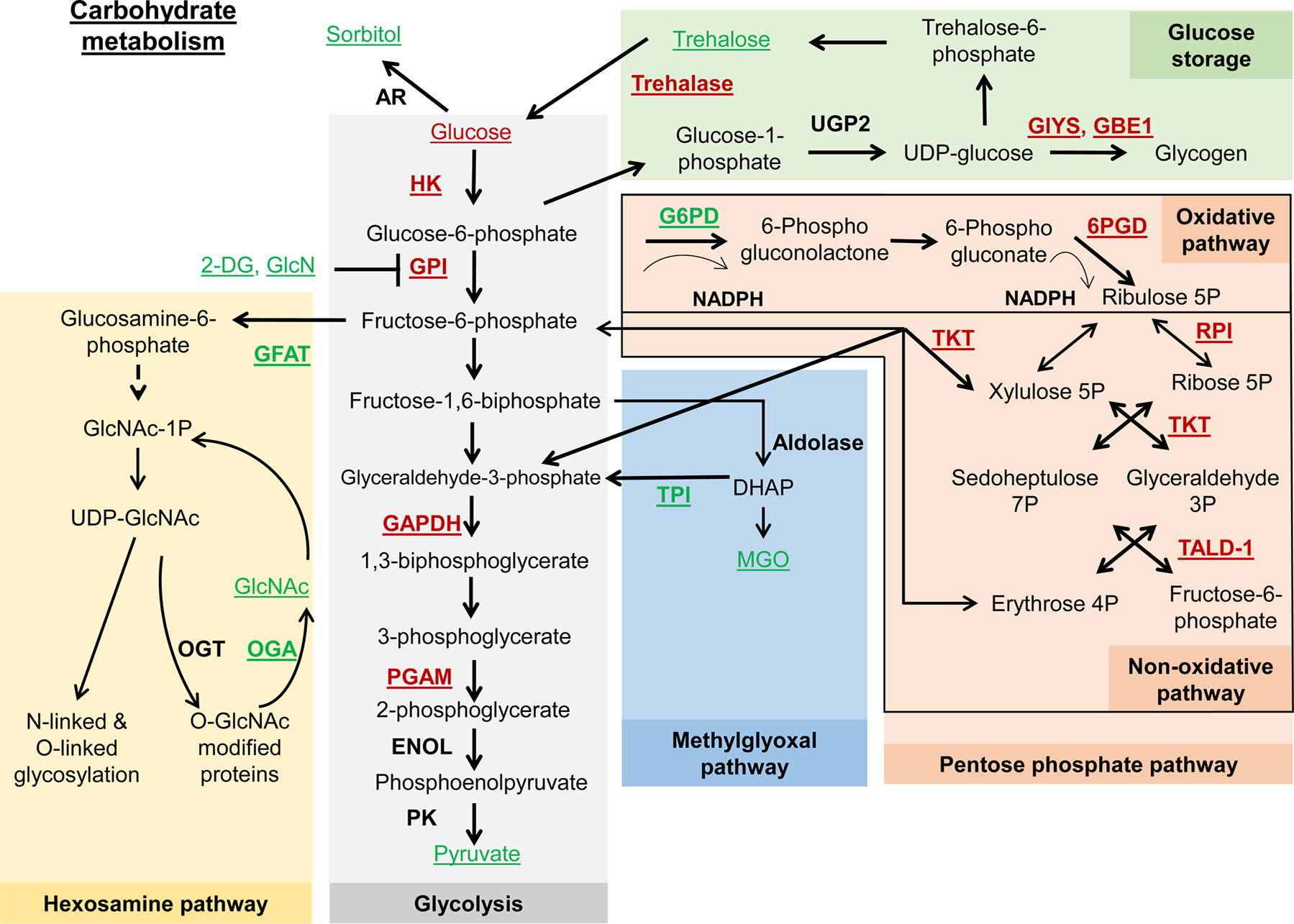
Schematic representation of glycolysis and related metabolic pathways. Underlined are metabolites and enzymes that were associated with lifespan extension. Red font color represents downregulation or depletion from food, while green font color represents overexpression or supplementation. Dashed line represents that multiple steps are involved. In the glycolysis pathway, glucose is broken down into pyruvate, producing ATP. Lifespan extension was associated with glycolysis inhibition through the downregulation of hexokinase (HK), glucose isomerase (GPI), glyceraldehyde 3-phosphate dehydrogenase (GAPDH), or phosphoglycerate mutase (PGAM); or through addition of inhibitors such as 2-Deoxyglycose (2-DG) and D-glucosamine (GlcN). The hexosamine pathway converts fructose-6-phosphate to UDP-N-acetylglucosamine (UDP-GlcNAc). Lifespan extension was associated with an increased expression of glutamine-fructose 6-phosphate aminotransferase (GFAT) and O-GlcNAcase (OGA), as well as with added acetylglucosamine (GlcNAc). The methylglyoxal pathway produces methylglyoxal (MGO) from glyceraldehyde-3-phosphate and dihydroxyacetone phosphate (DHAP). While excessive MGO can disrupt protein function, moderate supplementation was associated with increased lifespan in worms. Increased expression of triosephosphate isomerase (TPI) also increased lifespan. The pentose phosphate pathway (PPP) consists of the oxidative and nonoxidative branches. Lifespan extension was associated with downregulation of 6-phosphogluconate dehydrogenase (6PGD), ribose-5-phosphate isomerase (RPI), transketolase (TKT), and transaldolase (TALD-1), as well as with upregulation of glucose-6-phosphate dehydrogenase (G6PD). Downregulation of enzymes responsible for glycogen synthesis (glycogen synthase, GlyS; and 1,4-alpha-glucan branching enzyme 1, GBE1) extended lifespan. Increased levels of trehalose and downregulation of trehalase was associated with extended lifespan in worms.
Similar to 2-DG, downregulation of fgt-1, the only functional glucose transporter found in worms, decreased total body glucose uptake and led to a lifespan extension of 20–25 % (Feng et al., 2013). In accordance with the beneficial role of glycolysis inhibition, Lee et al. identified phosphoglycerate mutase (F57B10.3/pgm-1) as one of the hits in a systematic RNAi screen of 5690 C. elegans genes for lifespan extension (Lee et al., 2003b). Similarly, flies with lower levels of hexokinase-A (Hex-A) or hexokinase-C (Hex-C) were longer lived under normal diet (Talbert et al., 2015). Furthermore, in yeast, reducing the glucose content of the media from 2% to 0.5 % or deletion of HEXK2, one of three hexokinases that introduce glucose into glycolysis, extended lifespan and caused a metabolic shift toward respiration. This metabolic shift was required for lifespan extension by CR. Activation of this metabolic shift via overexpression of the Hap4 transcription factor resulted in a ~3-fold increase in the respiration rate and was enough to extend lifespan, but did not further extend lifespan under CR (Lin et al., 2000, 2002). However, later it was shown that CR exhibits robust lifespan extension in respiratory-deficient cells (Kaeberlein et al., 2005). There are two aging models in budding yeast: chronological aging (CLS) and replicative aging (RLS). CLS is defined as the length of time that a non-dividing yeast cell survives. RLS is defined as the number of daughter cells produced by a mother cell prior to senescence (Longo et al., 2012). Rho° yeast cells completely lack mitochondrial DNA and are incapable of respiratory metabolism. In mitochondrial DNA-deficient cells, the RLS did not change (BY4742 strain) or was decreased (PSY316 strain). However, these strains responded similarly to wildtype cells under CR, exhibiting a comparable lifespan enhancement (Kaeberlein et al., 2005).
Age-dependent changes in glucose metabolism are also evident in humans. Goyal et al. analyzed cerebral metabolic rate of glucose use (CMRGlc), oxygen consumption (CMRO2), and cerebral blood flow (CBF) in adult humans throughout their lifespans. They found a decrease in CMRGlc during late adulthood, whereas whole-brain CMRO2 and CBF remained unchanged. Based on these measurements, they demonstrated that the level of aerobic glycolysis at the whole-brain level gradually decreases with age, reaching zero levels at the age of 60. In addition, aerobic glycolysis topography significantly changed with age. During young adulthood, regions with high aerobic glycolysis correlate with transcriptional neoteny (i.e. regions with transcriptional characteristics of childhood development). Age-dependent changes in aerobic glycolysis predominantly occurred in the most neotenous regions of the human brain in the absence of amyloid pathology or neurologically evident brain pathology (Goyal et al., 2017). Consistent with this, the 18F-fluorodeoxyglucose ratio declined with advancing age in many regions of the brain, including in most cortical and subcortical regions (Knopman et al., 2014). A prospective cohort study, named PURE (Prospective Urban Rural Epidemiology) analyzed more than 135,335 individuals, 5796 deaths, and 4784 major cardiovascular disease events over ~7.4 years. They found that carbohydrate intake was associated with an increased risk of total mortality. By contrast, the intake of total fat and each type of fat (saturated/mono-unsaturated/poly-unsaturated) reduced the risk of total mortality (Dehghan et al., 2017).
In contrast to glucose, which suppressed lifespan in worms, the addition of fructose at the same concentrations tested for glucose (i.e. 55 mM or 111 mM), increased lifespan by 22 and 45 %, respectively, and addition of a higher concentration of fructose (555 mM) decreased lifespan (Zheng et al., 2017). Similar to fructose, sorbitol (monosaccharide alcohol), which is produced from glucose by aldose reductase, prolonged lifespan in yeast (1 M sorbitol) (Kaeberlein et al., 2002) and worms (275 mM sorbitol) (Chandler-Brown et al., 2015), potentially due to activation of the osmotic response and stress response pathways. In yeast, in addition to sorbitol, supplementation with 1 M xylitol or 1 M glycerol also extended lifespan (Kaeberlein et al., 2002).
D-Glucosamine / 2-amino-2-deoxy-D-glucose (GlcN) is a well-established inhibitor of both hexokinase and glucokinase, which are key players in the first step of glycolysis (Fig. 1). Supplementation of worms with 100 μM of GlcN reduced glucose oxidation rates, decreased ATP content, and extended lifespan by 27 %. Interestingly, supplementation of GlcN increased lifespan independent of increased hexosamine metabolism since downregulation of F21D5.1 (the only C. elegans orthologue of mammalian phospho-acetyl-GlcN-mutase) had no effect on the lifespan-extending capabilities of GlcN. Exposing worms to GlcN increased phosphorylation of AAK-2/AMPK, while GlcN failed to extend lifespan in aak-2-deficient worms (Weimer et al., 2014). Similar to the worm study, supplementation of 10 g/kg of GlcN to C57BL/6NRj mice of both sexes, starting at an age of 100 weeks, increased lifespan (Weimer et al., 2014). In a human population-based prospective cohort study, glucosamine supplementation was associated with lower all-cause mortality and cause-specific mortality associated with cancer and cardiovascular, respiratory, and digestive diseases (Li et al., 2020). Although GlcN extended lifespan independently of increased hexosamine metabolism, Denzel et al. found that the hexosamine pathway metabolites prolonged lifespan in worms via enhanced protein quality control (Denzel et al., 2014). All membrane and secreted proteins undergo N-glycosylation on the amino group of asparagine residues in the ER. This requires UDP-N-acetylglucosamine as a precursor for N-glycosylation. Glutamine-fructose 6-phosphate aminotransferase (Gfat) is the key rate-limiting enzyme of the hexosamine pathway, which synthesizes UDP-N-acetylglucosamine (UDP-GlcNAc). Denzel et al. found that gain-of-function (gof) mutations in the gfat-1 gene induced the ER-associated protein degradation (ERAD) machinery, activated autophagy and prolonged lifespan by 42 %. Moreover, levels of endogenous UDP-N-acetylhexosamines, UDP-GlcNAc and UDP-N-acetylgalactosamine (UDP-GalNAc), decreased with age, and supplementation of wild-type worms with 1–10 mM GlcNAc extended lifespan by 38 % (Denzel et al., 2014). O-GlcNAc transferase (OGT) post-translationally GlcNAcylates proteins, and O-GlcNAcase (OGA) catalyzes the removal of O-GlcNAc from proteins. Mutation of ogt-1 (this strain completely lacks the O-GlcNAc modification) significantly reduced lifespan in wild-type (by ~20 %) and long-lived daf-2 mutant worms, while the oga-1 mutation (elevated levels of O-GlcNAc-modified proteins) significantly extended lifespan in the daf-2 mutants (~12 % extension) but not in wild-type (Love et al., 2010). alpha-1,2-mannosidase I (mas1) is a member of the class I glycosidases and is involved in N-linked glycosylation via mannose removal from permanently unfolded proteins. De-mannosed proteins are recognized by Edem (ER degradation-enhancing alpha-1,2-mannosidase-like protein) and are degraded via ER-associated degradation (ERAD). In flies, suppression of mas1 and Edem1 extended lifespan by 38 % and 30 %, respectively (Liu et al., 2009). In worms, suppression of mas1/D2030.1 extended lifespan by 9% (Liu et al., 2009). McCormick et al. performed a systematic analysis of yeast RLS in 4698 viable single-gene deletion strains and identified 238 long-lived strains. One of the most enriched functional categories was related to protein mannosylation; 9 single-gene deletions that affect this activity extended lifespan (McCormick et al., 2015).
Another pathway that stems from glycolysis is the methylglyoxal pathway. Methylglyoxal (MGO) is a highly reactive carbonyl species that is mainly produced from the glycolytic intermediates glyceraldehyde 3-phosphate (GA3P) and dihydroxyacetone phosphate (DHAP) but can be also generated during the catabolism of threonine and other metabolic processes (Fig. 1). MGO is removed by the glyoxalase system. However, excessive amounts of MGO can react with proteins and generate advanced glycation end-products (AGEs) that alter or disrupt protein function. In addition to amino acids, specific nucleotides can also be modified by MGO (Chaudhuri et al., 2018; Kold-Christensen and Johannsen, 2020). glod-4/GLO1-mutant worms with an impaired glyoxalase system have a dramatic increase in MGO levels and rapidly exhibit several pathogenic phenotypes and early mortality (Chaudhuri et al., 2016). In humans, higher serum levels of carboxymethyl-lysine (CML), a ubiquitous human advanced glycation end-product, are associated with the incidence of disability and the prevalence of frailty (Whitson et al., 2014). Although excessive formation of MGO is detrimental and related to various pathological processes, supplementation of worms with 50 or 100 u M MGO increased worm lifespan (further discussed in the next section) (Ravichandran et al., 2018). An additional player in the regulation of lifespan by MGO is microbiota. Shin et al. performed a genome-wide screen using 3792 E. coli mutants and identified three mutants that extend C. elegans longevity via decreased production of bacterial MGO (Shin et al., 2020).
Whereas in worms, inhibition of glycolysis is beneficial for lifespan extension, in flies, extension of lifespan can be achieved through increased expression of glycolytic genes (Ma et al., 2018b). In Drosophila, levels of the repressive histone mark H3K27me3 increase with age, and reduction of components of Polycomb repressive complex 2 (PRC2) (esc, E(z), Pcl, Su(z)12) and PRC1 (Psc and Su(z)2) promotes lifespan via activation of glycolysis. Expression of two glycolytic genes, Tpi and Pgi(Gpi), was upregulated in long-lived PRC2 mutant flies. Based on metabolomics analysis, glycolysis was one of the most significantly affected metabolic pathways that changed with age. In particular, lactate, a specific indicator of anaerobic glycolysis, was significantly decreased during normal aging in wild-type animals but became elevated in long-lived PRC2 mutants. Combined increased expression of Tpi and Pgi(Gpi) stimulated glycolysis and improved locomotion, resistance to oxidative stress, and lifespan (Ma et al., 2018b). However, it is still not clear whether lactate plays protective or detrimental roles. Lactate dehydrogenase (LDH) catalyzes the conversion of glycolysis-derived pyruvate to lactate. In contrast to the Ma et al. study, where they demonstrated a beneficial role of lactate production; pan-neuronal reduction of Ldh in neurons extended lifespan and delayed age-dependent neurodegeneration, while overexpression of Ldh caused a significant reduction in lifespan and increased brain neurodegeneration (Long et al., 2020). Interestingly, normal aging and premature aging in mtDNA mutator mice exhibit increased brain lactate (Ross et al., 2010), and cerebrospinal fluid lactate is elevated in aging humans (Yesavage et al., 1982). Lin et al. performed transcriptional profiling and simultaneously measured glycogen and metabolites from the gluconeogenic, glycolytic, and glyoxylate pathways in wild-type, long-lived (Snf4-mutant), and short-lived (Sip2-mutant) yeast strains. They found that age-dependent transcriptional and metabolic changes in yeast were associated with a shift from glycolysis towards gluconeogenesis and energy storage. Accordingly, these changes were accompanied by a rise in glycogen levels with age (Lin et al., 2001). Similarly, Hachinohe et al. found that several metabolites from glycolysis and the TCA cycle accumulated with age in yeast, further confirming that with age, yeast cells have enhanced gluconeogenesis and reduced glycolysis. They also found that deletion of the TDH2 gene, which encodes yeast GAPDH, extended the RLS of wild-type cells but not in cells mimicking CR, suggesting that this was happening in a CR-dependent manner (Hachinohe et al., 2013). In mice, administration of acarbose, a glucoamylase inhibitor that reduces the rate of digestion of carbohydrates in the small intestine, at 3 different doses (400, 1,000, and 2500 ppm) significantly extended lifespan in genetically heterogeneous mice, with a stronger effect in males (up to 16 %) (Harrison et al., 2019; Harrison et al., 2014). In humans, acarbose has been used for many years to treat hyperglycemia and type 2 diabetes.
In flies and worms, glucose is stored in two main forms: as the disaccharide trehalose and as the polysaccharide glycogen (Fig. 1). Gusarov et al. found that high levels of glucose, which suppressed lifespan in worms, also increased resistance to oxidative stress. This resistance to oxidative stress was due to increased production of glycogen, while downregulation of glycogen synthase (gsy-1) abolished glycogen accumulation and the antioxidant effect of the high level of glucose. Moreover, the detrimental effect of high levels of glucose on lifespan can be reversed by the addition of exogenous oxidants that also deplete glycogen storage. In wild-type worms, downregulation of gsy-1 inhibited glycogen production and extended lifespan by 15 % via an AMPK-dependent mechanism (Gusarov et al., 2017). In agreement with the detrimental role of glycogen, Hamilton et al. found in a large-scale RNAi screen that downregulation of K08E3.5/UTP-glucose-1-phosphate uridylyltransferase, an enzyme that generates glycogen precursor - UDP-glucose, extended worm lifespan (Hamilton et al., 2005).
Accumulation of glycogen granules was identified in old flies, and inhibition of GlyS/CG6904 in neurons reduced glycogen granule accumulation, improved neurological function with age, and also extended lifespan in male flies by 10 % (Sinadinos et al., 2014). In a Drosophila screen of 45 EP (overexpression) lines for extension of lifespan, Paik et al. identified CG33138, the ortholog of human 1,4-Alpha-Glucan Branching Enzyme 1 (GBE1) that promotes branching and solubility of glycogen and potentially might regulate glycogen granule accumulation (Paik et al., 2012). Drosophila insulin-like peptide 2 (DILP-2) is a hormone made in the insulin-producing cells of the adult Drosophila brain that plays an important role in the regulation of carbohydrate metabolism (Kannan and Fridell, 2013). Post et al. performed phosphoproteomic analysis in S2 cells treated with DILP2 and found that phosphorylation of Glycogen phosphorylase (GlyP), the rate-limiting step in glycogenolysis, at Ser15 was greatly decreased in response to DILP2 treatment. Mutation of dilp2 was sufficient to extend longevity in Drosophila (Bai et al., 2012; Gronke et al., 2010) and also led to the activation of GlyP. Moreover, overexpression of GlyP decreased total glycogen and was sufficient to extend lifespan (Post et al., 2018).
Although glycogen depletion extends worm lifespan, feeding worms with 5 mM trehalose increased reproductive span, retarded age-associated accumulation of lipofuscin, enhanced thermotolerance, reduced polyglutamine accumulation, and extended lifespan by 32 % (Honda et al., 2010). In agreement with this data, Seo et al. also found that when glucose is stored as glycogen it is detrimental, whereas when stored as trehalose, it promotes a longer, healthier worm lifespan. In line with this, downregulation of gsy-1 with RNAi or in worms mutant for gsy-1, glycogen stores were dramatically decreased and these worms exhibited ~20 % lifespan increase, lower levels of AGE (an indication of physiological age), and greater levels of locomotion (Seo et al., 2018). Trehalase is the enzyme that breaks down trehalose to produce two glucose molecules. C. elegans have five trehalase genes (tre-1, tre-2, tre-3, tre-4, and tre-5). As expected, downregulation of two different trehalases (tre-1 or tre-3) or supplementation with 5 mM trehalose led to increased trehalose levels and increased lifespan in a DAF-16 and autophagy-dependent manner (Seo et al., 2018). Interestingly, although mammals do not use trehalose to store carbohydrates, they possess the trehalase enzyme to break down trehalose derived from food (Richards et al., 2002). Moreover, supplementation of trehalose to mammalian cells, mice or humans exerts health benefits (Mizote et al., 2016).
Another level of complexity comes from the intestinal flora. When C. elegans were cultured with live E. coli OP50, the addition of a low concentration of saccharides (0.1 % of glucose, galactose, fucose, lactose, arabinose, or sorbitol) to the medium promoted longevity by ~10 %. However, this effect was abolished when the bacterium could not metabolize the sugar (Brokate-Llanos et al., 2014). In addition, Han et al. performed a screen using 3792 E. coli mutants and identified five mutants that extended C. elegans longevity via increased secretion of the polysaccharide colanic acid (CA). Addition of purified colanic acid was sufficient to extend lifespan of both worms and flies (Han et al., 2017a).
Gluconeogenesis is the reverse pathway of glycolysis that serves to generate glucose from non-carbohydrate carbon substrates (Fig. 2). In addition to glycolytic enzymes, gluconeogenesis utilizes 4 enzymes that are exclusive to this pathway (Wang and Dong, 2019). Phosphoenolpyruvate carboxykinase (PEPCK) converts oxaloacetate (OAA) into phosphoenolpyruvate (PEP) in the second step of gluconeogenesis, which allows the cell to use glutamine, lactate, and TCA cycle intermediates under nutrient starvation (Hanson, 2009; Wang and Dong, 2019). Yuan et al. used quantitative proteomics in C. elegans to compare enzyme expression in wild-type and long-lived eat-2 worms and found that the long-lived worms had decreased levels of multiple enzymes critical for carbohydrate metabolism (ENOL-1/a-enolase, PYK-1 and PYK-2/pyruvate kinase, FBP-1/ fructose-1,6-biphosphatase, PYC-1/pyruvate carboxylase, and pck-1/PEPCK-C). To determine if there was a switch in fuel utilization, they measured the capacity of wild-type and long-lived eat-2 nematodes to oxidize specific radio-labeled substrates (acetate, palmitate, glutamate, or glucose) to CO2. They found much higher rates of acetate, glutamate, and glucose oxidation in eat-2 worms, whereas the rate of palmitate oxidation was not different (Yuan et al., 2012). This suggested that instead of decreasing general metabolic rates, CR leads to metabolic reprogramming and a switch in fuel utilization. Furthermore, they found that downregulation of pck-1/PEPCK-C decreased lifespan, whereas overexpression of pck-1/PEPCK-C significantly extended lifespan of transgenic worms by 22 % (Yuan et al., 2012). In a later publication, these authors found reciprocal changes in locomotor muscle between an age-dependent progressive decrease of PEPCK-C and increase in glycolytic pyruvate kinase (PK), which shunts energy metabolism towards glycolysis and reduces mitochondrial function. In addition, CR could prevent these age-dependent reciprocal changes in PEPCK-C and PK (Yuan et al., 2016). There are two PEPCKs in humans, the cytosolic form PEPCK-C, which is encoded by PCK1, and the mitochondrial form PEPCK2, encoded by PCK2, and there are three forms of PEPCK in worms, two cytosolic forms, encoded by pck-1 and pck-3, and a potentially mitochondrial form, encoded by pck-2. The worm data complements data in rodent studies, in which transgenic mice overexpressing muscle-specific PEPCK-C were seven times more active (running up to 6 km compared to 0.2 km for control mice), ate 60 % more, had half the body weight, and lived longer than controls. Before exercise, both control and PEPCK-C transgenic mice had equal blood lactate concentrations of around 4 mM, while after exercise, at exhaustion, lactate levels in control mice increased by 17 mM but remained unchanged in transgenic mice. It is possible that PEPCK-C transgenic mice rely on fatty acids as a source of energy for muscles during exercise and thus do not generate lactate. The authors generated several lines of PEPCK-C transgenic mice and found that the amount of PEPCK-C activity correlated with muscle triglyceride concentrations. These mice also had a larger number of mitochondria that potentially can fuel their increased activity (Hakimi et al., 2007).
Fig. 2.
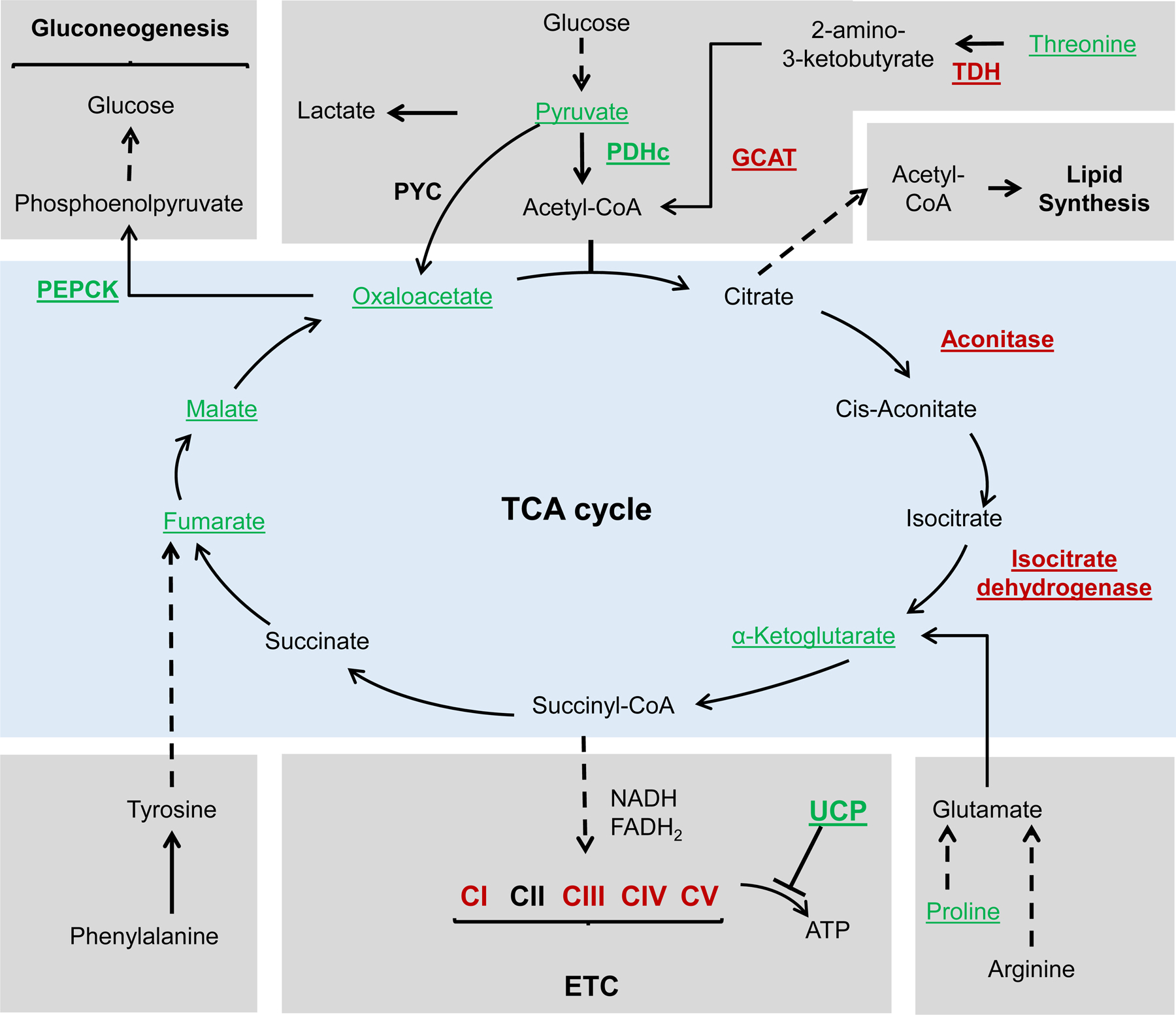
Schematic representation of the TCA cycle and related metabolic pathways. Underlined are metabolites and enzymes that were associated with lifespan extension. Red font color represents downregulation or depletion from food, while green font color represents overexpression or supplementation. Dashed line represents that multiple steps are involved. Before entering the TCA cycle, pyruvate must be converted to acetyl-CoA through the pyruvate dehydrogenase complex (PDHc). Overexpression of the dihydrolipoamide acetyltransferase (E2 component) of the PDHc extended lifespan in yeast. In addition, downregulation of pyruvate dehydrogenase kinase (not shown), an inhibitor of PDHc, extended lifespan in worms. Threonine can be converted to acetyl-CoA through a series of reactions. Threonine supplementation, as well as downregulation of the enzymes l-threonine-3-dehydrogenase (TDH) and glycine-C-acetyltransferase (GCAT), extended lifespan in yeast. Dietary supplementation of several TCA cycle intermediates, including oxaloacetate, α-ketoglutarate, fumarate, and malate, was associated with lifespan extension. Downregulation of aconitase and isocitrate dehydrogenase, two enzymes in the TCA cycle, also extended lifespan. The electron transport chain (ETC) is a series of complexes which ultimately generates ATP through electron transfer and redox reactions. Downregulation of components of complexes I, II, IV and V was associated with lifespan extension in worms. In addition, lifespan was extended through the expression of some mitochondrial uncoupling proteins (UCPs). Gluconeogenesis is a process that allows cells to convert TCA intermediates into glucose under nutrient starvation. Phosphoenolpyruvate carboxykinase (PEPCK) is a key enzyme in this process, and overexpression of PEPCK extends lifespan in worms.
The pentose phosphate pathway (PPP), also known as the hexose monophosphate shunt, is an alternative pathway for glucose oxidation. It consists of two parts: an oxidative, nonreversible branch that allows NADP+ to be reduced to NADPH while converting glucose-6-phosphate to a pentose phosphate and CO2, and a non-oxidative, reversible branch that connects pentose phosphates to glycolytic intermediates (Fig. 1). PPP is a cytosolic pathway that has two major outcomes: production of NADPH (the source of reducing equivalents in multiple pathways and a key player in the oxidative stress response), and synthesis of ribose-5-phosphate, a precursor for nucleotide synthesis (Stincone et al., 2015; Wamelink et al., 2008). Glucose-6-phosphate dehydrogenase (G6PD) is the first and rate-limiting enzyme that catalyzes the conversion of glucose-6-phosphate into 6-phosphogluconolactone and reduces one molecule of NADP+ to NADPH, redirecting glucose from glycolysis into the PPP (Stincone et al., 2015; Wamelink et al., 2008).
Superoxide radicals and hydrogen peroxide are generated in mitochondria and peroxisomes under normal physiological conditions. Hydrogen peroxide is detoxified by catalase and peroxidases. The activity of peroxidases depends on the availability of reduced forms of glutathione (GSH) or thioredoxin, and peroxidase activity results in the oxidation of GSH to GSSG and reduction of thioredoxin to oxidized thioredoxin. NADPH is an indirectly acting antioxidant that participates as an electron donor. NADPH supplies the reducing equivalents for the reduction of GSSG by glutathione reductase and oxidized thioredoxin by thioredoxin reductase (Bradshaw, 2019). In Drosophila, the NADP+/NADPH ratio increased with age, while the NAD+/NADH ratio declined (Sohal et al., 1990). Drosophila long-lived strains have higher levels of G6PD activity (Luckinbill et al., 1990). Ubiquitous overexpression of G6PD in flies increased G6PD enzymatic activity and extended lifespan by 38 %, whereas neuronal overexpression of G6PD extended lifespan by 18 %, all without significant effects on fertility. G6PD overexpression also resulted in increased resistance to oxidative stress (Legan et al., 2008). Wang et al. found that neuronal activation of JNK in long-lived pucE69 heterozygous flies induced G6PD expression and shifted carbon flux into the pentose phosphate pathway, increasing NADPH production and resistance to oxidative stress. Moreover, neuronal overexpression of G6PD extended lifespan in wild-type flies but not in puc mutant flies (Wang et al., 2019). Similarly, transgenic mice overexpressing the entire human G6PD gene, including upstream and downstream regulatory sequences, had significantly higher levels of NADPH in the liver and brain, and a 13.7 % increase in lifespan was observed for female mice but not in males (Nobrega-Pereira et al., 2016). Downregulation of ribose-5-phosphate isomerase (RPI), a downstream enzyme from the non-oxidative branch of the PPP that catalyzes the isomerization of ribulose-5-phosphate to ribose-5-phosphate, increased levels of G6PD and NADPH and increased lifespan by 38 % (Wang et al., 2012). In worms, tald-1/Transaldolase and tkt-1/Transketolase, enzymes in the non-oxidative branch of the PPP, and 6PGD, an enzyme in the oxidative PPP branch, have been identified as negative regulators of mitochondrial unfolded protein response (UPRmt), and their downregulation extended worm lifespan; however, the effect on the lifespan was independent of UPRmt induction (Bennett et al., 2017, 2014). Downregulation of tald-1/Transaldolase led to decreased cellular NADPH levels, higher endogenous levels of oxidative stress, the appearance of smaller and thinner mitochondria, a reduction in oxygen consumption, and a dramatic reduction in intestinal fat levels (Bennett et al., 2017). Transketolase was also identified in an RNAi screen in C. elegans for genes that promote both resistance to paraquat and lifespan extension (Kim and Sun, 2007). The activity of the PPP is tightly linked to the activity of glycolysis (Stincone et al., 2015) and whether the lifespan benefits associated with glycolysis inhibition are caused by upregulation of the PPP remains to be determined.
In summary, manipulations of glycolysis and related pathways are tightly linked to lifespan extension across different species; however, in some cases, both down- and upregulation of the same enzyme can be beneficial or detrimental, depending on species. In addition, many of these manipulations are linked to AMPK activation, which is an attractive target in the anti-aging field and can be achieved by metformin treatment.
2.2. Mitochondrial energy metabolism
2.2.1. Tricarboxylic acid (TCA) cycle
Mitochondria act as a platform for metabolic pathways, including the TCA cycle, the urea cycle, β-oxidation, and lipid synthesis. The citric acid cycle (also known as the Krebs cycle or TCA cycle) takes place in mitochondria and is an integral part of energy metabolism, macromolecule synthesis, and redox balance (Fig. 2). The TCA cycle begins when the two-carbon acetyl CoA (generated from fatty acids, amino acids, or pyruvate) and the four-carbon oxaloacetate (OAA), combine through the action of citrate synthase to form the six-carbon citrate. Next, citrate is converted to isocitrate by aconitase, isocitrate is converted to five-carbon α-ketoglutarate by isocitrate dehydrogenase, and a-ketoglutarate is converted to four-carbon succinyl-CoA by α-ketoglutarate dehydrogenase, ultimately releasing two molecules of CO2 and generating two NADH molecules. Succinyl-CoA is then converted to succinate by succinyl-CoA-synthetase, generating GTP. Succinate is oxidized to the four-carbon fumarate by succinic dehydrogenase, producing FADH2. Finally, fumarate is converted to malate by fumarase and malate is converted into OAA by malate dehydrogenase with the generation of one molecule of NADH, finishing the cycle. The products of the TCA cycle, NADH and FADH2, feed the ETC complex I and complex II, respectively (Martinez-Reyes and Chandel, 2020). In the yeast screen by McCormick et al. measuring RLS in 4698 viable single-gene deletion strains, one of the most enriched functional categories among 238 identified long-lived gene deletions was the TCA cycle, for which 7 different genes were identified (McCormick et al., 2015).
The pyruvate dehydrogenase complex (PDHc) functionally links glycolysis in the cytoplasm with oxidative phosphorylation (OXPHOS) in mitochondria. The PDHc is composed of three separate enzymes: pyruvate dehydrogenase (E1), dihydrolipoamide acetyltransferase (E2), and lipoamide dehydrogenase (E3). The PDHc catalyzes the irreversible oxidation of pyruvate to acetyl-CoA and plays an important role in aging and various age-dependent pathologies (Fig. 2). Aging is associated with decreased mitochondrial PDHc activity (Stacpoole, 2012). Downregulation of slcf-1, which encodes a predicted SLC16 monocarboxylate transporter, extended C. elegans lifespan by 40 %. Metabolomics analysis revealed significantly increased pyruvate levels in mutant worms. Feeding with 2.5 mM pyruvate (sodium pyruvate) increased lifespan by 14 % in wild-type worms but did not affect lifespan of mutant worms (Mouchiroud et al., 2011). Pyruvate dehydrogenase kinase (PDHK, encoded by pdhk-2 in C. elegans) inhibits the activity of the PDHc. Downregulation of pdhk-2 (which would activate the PDC) extended the lifespan of wild-type worms by 20 % but did not further increase the lifespan of slcf-1 RNAi-treated worms. slcf-1 mutation and pyruvate treatment induced hydrogen peroxide accumulation and treating worms with the antioxidant N-acetyl cysteine (NAC) extended lifespan in slcf-1-mutant worms (Mouchiroud et al., 2011). Although both 2-DG supplementation and slcf-1 downregulation were associated with increased ROS production and extended lifespan, 2-DG was expected to decrease pyruvate levels while slcf-1 downregulation increased them. Dichloroacetate (DCA) promotes pyruvate entry into the TCA cycle by inhibiting pyruvate dehydrogenase kinase (PDHK). Feeding worms with 50 μg/mL of DCA moderately extended worm lifespan (Schaffer et al., 2011). Similarly, treatment of flies with 20 μg/mL of DCA increased lifespan by 15 % (Pandey et al., 2014).
Lat1 (dihydrolipoamide acetyltransferase) is an E2 component of the PDHc in S. cerevisiae. Lat1 deletion abrogated lifespan extension induced by CR but did not affect the lengthened lifespan of mutants with deleted hexokinase. Conversely, Lat1 overexpression extended lifespan by 30 %, and this lifespan extension was not further increased by CR. Lifespan extension by Lat1 overexpression required a functional respiratory chain. Interestingly, overexpression of the E1 or E3 components of PDHc, Pda1 and Lpd1, did not extend lifespan (Easlon et al., 2007). Similarly, feeding lipoic acid/lipoamide (an essential cofactor for E2 activity) to old rats improved several age-associated phenotypes and partially restored mitochondrial structure and function (Liu et al., 2002). Similarly, supplementation of 1 mM α-lipoic acid significantly extended worm lifespan (Benedetti et al., 2008).
Thiamine pyrophosphokinase (tpk-1) catalyzes the formation of thiamine pyrophosphate (TPP) from thiamine (Vitamin B1). TPP is necessary for oxidative phosphorylation and the pentose phosphate pathway by acting as a cofactor for pyruvate dehydrogenase (PDH), a-ketoglutarate dehydrogenase (KGDH), branched-chain-ketoacid dehydrogenase, and transketolase. Tpk-1 mutant worms were identified in a screen for mutations that resulted in slow development and behavior, both phenotypes that are reminiscent of long-lived clk-1 mutant worms. Similar to clk-1 mutants, tpk-1 mutants had a significant lifespan increase of 40 % (de Jong et al., 2004). Malic enzyme (ME1) catalyzes the conversion of the TCA cycle intermediate malate to pyruvate and NADPH, linking glycolysis and the TCA cycle. In mammalian cells, three isoforms of MEs have been identified: a cytosolic NADP+-dependent isoform, ME1; a mitochondrial NAD+-dependent isoform, ME2; and a mitochondrial NADP+-dependent isoform, ME 3. In flies, overexpression of Men (the fly ortholog of ME1) increased pyruvate content and the NADPH/NADP+ ratio, and significantly extended lifespan (Kim et al., 2015; Paik et al., 2012).
In agreement with the fact that activation of the upstream pathways feeding into the TCA cycle is beneficial for lifespan extension, feeding organisms with TCA cycle intermediates and manipulating TCA cycle enzymes are important in regulating lifespan. In a large-scale RNAi screen, Hamilton et al. identified 89 new genes that extend lifespan in C. elegans. Among them were the TCA cycle enzymes aconitase/F54H12.1 and isocitrate dehydrogenase (F43G9.1/IDH3A and F59B8.2/IDH1) (Hamilton et al., 2005).
Acetyl-CoA synthetases (mitochondria-localized acetyl-CoA synthetase 1 (ACSS1) and nucleocytosol-localized (ACSS2)) catalyze the ATP-dependent ligation of acetate and CoA to produce acetyl-CoA. Cytoplasmic acetyl-CoA is used for fatty acid biosynthesis, whereas mitochondrial acetyl-CoA is used in the TCA cycle. Acetyl-CoA hydrolases catalyze the hydrolysis of acetyl-CoA and generate acetate. Acetyl-CoA is a central metabolite between glycolysis and the TCA cycle and serves as an important substrate for the synthesis of sterols, hexosamines and ketones (Schug et al., 2016). Feeding worms with acetic acid significantly extended lifespan by 20 % (Chuang et al., 2009). Acetyl-CoA also serves as a cofactor for the acetylation of lysine residues that is a critical regulatory mechanism for histone proteins, metabolic enzymes, and many other proteins. ATP citrate lyase (ACLY/ ATPCL) catalyzes the formation of acetyl-CoA and oxaloacetate from citrate and CoA with concomitant hydrolysis of ATP to ADP and phosphate. Aging in flies is characterized by increased levels of acetyl-CoA and citrate/isocitrate, and increased proteome acetylation. Flies carrying one hypomorphic atpcl allele displayed reduced ATPCL activity and increased lifespan (Peleg et al., 2016). In yeast, de novo synthesis of Acetyl-CoA is facilitated by two metabolic routes, the mitochondrial (ACS1-, ACH1-, or MPC1-dependent) and the nucleocytosolic (ACS2--dependent) pathways. In addition, nucleocytosolic Acetyl-CoA production in yeast is a metabolic repressor of autophagy during aging. Autophagy is a conserved process that catabolizes intracellular components to maintain energy homeostasis and to protect cells against stress and a key pro-longevity pathway (Parkhitko et al., 2013). Suppression of the mitochondrial branch of acetyl-CoA production by deletion of ACH1 leads to cytosolic accumulation of the Acetyl-CoA precursor acetate, activates nucleocytosolic Acetyl-CoA synthase Acs2p, which triggers histone acetylation, inhibits autophagy, and reduces lifespan. Brain-specific downregulation of Acetyl-CoA Synthase in flies extended lifespan and induced autophagy (Eisenberg et al., 2014).
Edwards et al. demonstrated that supplementing the growth medium of C. elegans with TCA cycle intermediates, 10 mM malate and 10 mM fumarate, increased lifespan by 14 % and 16 %, respectively, whereas adding 10 mM succinate or 10 mM of α-ketoglutarate had no effect on lifespan (Edwards et al., 2013). The lifespan extension by malate did not require malic enzyme, which catalyzes the conversion of malate to pyruvate, or malate dehydrogenase, which catalyzes the reversible conversion of malate to oxaloacetate, but was abrogated by downregulation of fumarate hydratase/fumarase (fum-1), which catalyzes the conversion of fumarate to malate. However, fumarate addition was able to extend lifespan by 18 % in fum-1 RNAi knockdown worms (Edwards et al., 2013). C. elegans possess the glyoxylate shunt, which is composed of two enzymes, isocitrate lyase and malate synthase (in worms both enzymes are fused into one bifunctional protein named GEI-7/ICL-1). The glyoxylate shunt, which reversibly converts isocitrate and acetyl-CoA to succinate, malate, and CoA, was essential for lifespan extension by both malate and fumarate. The authors of the study suggested that malate has to be converted into fumarate, and then fumarate must be reduced to succinate by soluble fumarate reductase and the mitochondrial ETC complex II (Edwards et al., 2013). Interestingly, gei-7 was upregulated in long-lived daf-2-mutant worms and its downregulation shortened the lifespan of daf-2 mutants substantially, while shortening wild-type lifespan only slightly (Murphy et al., 2003). Upregulation of glyoxylate shunt activity is predicted to increase NAD levels, therefore activating AMP kinase (Rafaeloff-Phail et al., 2004). Knockdown of two enzymes in the NAD synthesis pathway (qns-1 or nmnat-2) abrogated lifespan extension by malate (Edwards et al., 2013). In agreement with the positive effect of fumarate on lifespan observed for worms, augmentation of fumarate levels in mouse hearts, either via cardiac-specific fumarate hydratase (Fh1) inactivation or via dimethylfumarate supplementation, protected hearts from ischemia-reperfusion injury and upregulated protective antioxidant response element genes (Ashrafian et al., 2012). Moreover, in humans, treatment of relapsing-remitting multiple sclerosis patients with fumarate (BG00012, the oral formulation of dimethyl fumarate) was safe, well-tolerated and had a neuroprotective effect (Kappos et al., 2008).
Williams et al. found that supplementation of worms with 8 mM oxaloacetate resulted in a 25 % lifespan extension. Although the conversion of oxaloacetate to malate promotes the conversion of NADH to NAD+, oxaloacetate-induced lifespan extension was independent of Sir-2 but abrogated in daf-16- and aak-2/AMPK mutant worms (Williams et al., 2009). However, as observed in the NIA Interventions Testing Program, treatment of mice with oxaloacetic acid beginning at 4 months of age did not have a statistically significant effect on lifespan of male or female mice (Strong et al., 2013).
Chin et al. found that α-ketoglutarate (α-KG) binds ATP synthase subunit β (ATP5B) of complex V ETC and inhibits TOR. Supplementation of worms with 8 mM α-KG extended lifespan by 32 % (Chin et al., 2014). 2-hydroxyglutarate (2 H G) is an oncometabolite that is produced from α-KG either by mutant IDH1/2 or by the promiscuous activity of phosphoglycerate dehydrogenase (PHGDH). 2-HG competitively inhibits α-KG-dependent enzymes. Similar to α-KG, both enantiomers of 2 H G, (R)-2 H G and (S)-2 H G, inhibited ATP synthase, suppressed TOR, and extended worm lifespan by 40 % and 30 %, respectively (Fu et al., 2015). In addition, supplementation of flies with 5–10 uM of α-KG activated AMPK inhibited TOR and significantly extended lifespan (Lylyk et al., 2018; Su et al., 2019). Moreover, supplementation of α-KG in the form of a calcium salt (CaAKG) significantly extended lifespan and healthspan of C57BL/6 mice (Asadi Shahmirzadi et al., 2020).
The exometabolome of long-lived mit mutants is enriched in several compounds that are normally metabolized by mitochondrial 1d6fc;-ketoacid dehydrogenase complexes, including 1d6fc; -ketobutyrate (2-oxobutyrate, 2OB), 1d6fc; -ketoisocaproate (4-methyl-2-oxovalerate, 4M2OV), 1d6fc; -ketoisovalerate (3-methyl-2-oxobutyrate, 3M2OB), 1d6fc; -keto-b-methylvalerate (3-methyl-2-oxovalerate, 3M2OV), and 1d6fc; -ketopropionate (pyruvate). Downregulation of dihydrolipoamide dehydrogenase (DLD), which is a pivotal control point in the production of these compounds, significantly upregulated worm lifespan. In addition, supplementation of pyruvate and the three branched-chain 1d6fc; -ketoacids—3M2OB, 3M2OV, and 4M2OV—significantly increased worm lifespan. Moreover, feeding an 1d6fc; -ketoglutarate mimetic, 2,4-PDA, which inhibits multiple 1d6fc; -ketoglutarate-dependent hydroxylases, extended the adult lifespan of C. elegans by up to 15 % (Butler et al., 2013; Mishur et al., 2016). Goldberg et al. found that the Alzheimer’s disease (AD) drug candidate J147 targeted the mitochondrial 1d6fc; -F1-ATP synthase (ATP5A), extended lifespan in Drosophila by 13 %, and prevented age-associated changes in the plasma metabolome of mice (Goldberg et al., 2018).
2.2.2. OXPHOS
Mitochondria function decline is a hallmark of aging in different organisms and is associated with accumulation of mtDNA mutations and decreased OXPHOS (Bratic and Larsson, 2013). Mitochondrial respiration progressively declines in adult C. elegans, and the rate of this decline is slower in long-lived daf-2 mutant worms (Brys et al., 2010). However, mitochondrial perturbations can cause increased longevity (Copeland et al., 2009; Dillin et al., 2002; Feng et al., 2001). Members of the mitochondrial electron transport chain (ETC) provide the stepwise transfer of electrons from reducing equivalents (NADH and FADH2) to molecular oxygen, ultimately resulting in the synthesis of ATP. In C. elegans, RNAi knockdown of several subunits of mitochondrial ETC and ATP synthase has been shown to extend lifespan. Downregulation of ETC genes must occur during a specific stage of development to promote longevity. These genes include components of complex I (NADH/Ubiquinone oxidoreductase), complex III (cytochrome c reductase), complex IV (cytochrome c oxidase), and complex V. A full list of the components can be found in Table 1 (Curran and Ruvkun, 2007; Dillin et al., 2002; Feng et al., 2001; Hamilton et al., 2005; Hansen et al., 2005; Hartman et al., 2001; Kim and Sun, 2007; Lee et al., 2003b; Munkacsy and Rea, 2014; Rea et al., 2007; Tsang et al., 2001; Yang and Hekimi, 2010; Zuryn et al., 2010). In addition, treatment of wild-type worms with antimycin A, a complex III inhibitor (Dillin et al., 2002); ethidium bromide, a DNA-intercalating dye that functions as an inhibitor of mtDNA transcription/replication, leading to the depletion or elimination of mtDNA (Tsang and Lemire, 2002); or arsenite, a mitochondrial poison (Schmeisser et al., 2013b) also increased lifespan. Accordingly, partial pharmacological inhibition of complex I with 15 pM rotenone reversed age-related transcriptional signatures and extended lifespan by 15 % in the short-lived killifish N. furzeri (Baumgart et al., 2016). Similarly in flies, downregulation of components of complexes I (CG9172/NDUFS7 and CG9762/NDUFB5), III (CG17856/UQCRB), IV (CG18809) or V (CG5389/ATP5F1B) led to increased lifespan. Downregulation of fly CG9762 and CG9172 from the onset of adulthood was able to increase lifespan; by contrast, in worms, downregulation of mETC subunits had to occur during the larval stages to increase longevity (Copeland et al., 2009). Zid et al. assayed genome-wide translational changes in Drosophila under DR and found increased ribosomal loading and enhanced activity of nuclear-encoded mitochondrial genes encoding subunits of Complex I and IV of the ETC. Downregulation of components of Complex I (CG9762) and IV (CG11015) of the ETC abolished lifespan extension by DR (Zid et al., 2009). While both DR and downregulation of CG5389 (Complex V) extended Drosophila lifespan, downregulation of CG5389 completely abolished lifespan extension by DR (Bahadorani et al., 2010b). Also, in adult flies, downregulation of ATP synthase subunit d (ATPsyn-d), a component of ATP synthase, ETC complex V, extended lifespan of female but not male flies and increased resistance to oxidative stress (Sun et al., 2014). In addition to the ETC components mentioned above, in a systematic RNAi screen of 5690 C. elegans genes, Lee et al. identified several other genes (mrpl-47/B0261.4, cchl-1/T06D8.6, F13G3.7, slc-25A32/K01C8.7) that are important for mitochondrial function. Two of these genes (F13G3.7 and slc-25A32/K01C8.7) are mitochondrial transporters (Lee et al., 2003b).
Similarly, reduced levels of Indy, which functions as a cation-independent electroneutral transporter for a variety of di/tricarboxylic acid-cycle intermediates, extended lifespan in Drosophila (Rogina et al., 2000). Downregulation of worm ortholog of Indy - ceNAC-1/ceNaDC1 (corresponding to mammalian NaDC1/NaC1) did not affect worm lifespan, while downregulation of ceNAC-3/ceNaDC2 (corresponding to mammalian NaDC3/NaC3) increased worm lifespan by 15 %, and downregulation of ceNAC-2/NaCT (mammalian NaC2/NaC2) increased worm lifespan by 19 % (Fei et al., 2003, 2004). Moreover, a knockout mouse model of the mammalian Indy (mIndy) ortholog, SLC13A5, phenocopies the DR-like phenotype and protects mINDY-deficient mice from the adiposity and insulin resistance that normally result from a high-fat diet and aging (Birkenfeld et al., 2011).
Clk-1/Coq7 is a mitochondrial diiron-containing monooxygenase that catalyzes the hydroxylation of 5-demethoxyubiquinone, a critical step in the biosynthesis of ubiquinone (Coenzyme Q (CoQ)). In mitochondria, CoQ acts as a carrier of electrons from respiratory complexes I and II to complex III, but it can also accept electrons from other donors including dihydroorotate dehydrogenase and acyl-CoA dehydrogenase. In non-mitochondrial membranes, CoQ accepts electrons from cytosolic NAD(P)H. CoQ also functions as an antioxidant, either by quenching ROS or via regeneration of other antioxidants (Varela-Lopez et al., 2016), and is mainly reduced by membrane NADH-cytochrome b5 reductase and NAD(P)H:quinone reductase 1 (NQO1). CoQ also plays roles in plasma membrane electron transport, regulation of the mitochondrial permeability transition pore, and pyrimidine nucleotide biosynthesis. There are species-specific differences in CoQ isoforms that vary in the number of isoprene side-chain units. CoQ8, CoQ9 and CoQ10 predominate in E. coli, C. elegans and humans, respectively. Based on its importance in respiration and as an antioxidant, CoQ is a popular dietary supplement and it has been tested to treat neurodegenerative and cardiovascular diseases in humans. Worms mutant for clk-1/Coq7 gene are longer lived (Ewbank et al., 1997; Lakowski and Hekimi, 1996). 2, 4-dihydroxybenzoate (DHB) can serve as an unnatural precursor for ubiquinone synthesis. Restoring ubiquinone synthesis with DHB in long-lived clk-1 mutant worms completely suppressed the extended longevity (Liu et al., 2017). Also, withdrawal of CoQ8 from the diet of wild-type worms via feeding with Q-less E. coli extended adult lifespan by 60 % (Larsen and Clarke, 2002). These results conflict with another study, which found that feeding C. elegans with 150 μg/mL CoQ10 (the form of CoQ predominant in humans) increased lifespan in wild-type C. elegans by 18 % (Ishii et al., 2004). Saiki et al. developed a water-soluble formulation of CoQ10 that was able to restore the phenotype associated with CoQ-deficiency in worms but could not rescue the phenotype of CoQ-deficient bacteria. Surprisingly, the long lifespan of C. elegans fed with Q-less E. coli was not dependent on the absence of CoQ because its supplementation did not rescue the long lifespan. It is very likely that loss of ability to produce CoQ in bacteria causes secondary effects that play a role in extension of lifespan in worms (Saiki et al., 2008). Moreover, feeding flies with dietary yeast deficient for CoQ did not benefit their survival; rather their survival decreased (Palmer and Sackton, 2003). Interestingly, the product of the clk-1 gene, COQ7, can be localized in mitochondria or nuclei. The nuclear pool of COQ7 regulates ROS metabolism and could partially rescue the lifespan extension of clk-1 mutant worms independent of its role in ubiquinone biosynthesis (Monaghan et al., 2015). Flies heterozygous for sbo/Coq2, which potentially catalyzes the prenylation of p-hydroxybenzoate with the isoprenoid chain during the process of CoQ synthesis, have an extended lifespan of up to 12.5 % in male and 31 % in female flies (Liu et al., 2011). In mice, homozygous inactivation of the mouse ortholog of clk-1 (mclk) protected ES cells against oxidative stress and DNA damage. Mice heterozygous for mclk1 had up to a 31 % increased lifespan in different genetic backgrounds (Liu et al., 2005). SURF1 is a putative Cytochrome c oxidase (COX) assembly factor that is necessary for the correct assembly of COX, which consists of 13 protein subunits. Mice deficient for SURF1 had significantly lower COX activity in several tissues and significantly extended lifespan (Dell’agnello et al., 2007).
On the other hand, interventions that delay aging may also lead to increased mitochondrial function. Dietary restriction increases mitochondrial activity in worms (Bishop and Guarente, 2007), flies (Zid et al., 2009), and mice (Nisoli et al., 2005). This increased mitochondrial activity may be due to an increased NAD/NADH ratio, which stimulates mitochondrial TCA cycle dehydrogenases utilizing NAD as a cofactor. Uncoupling increases the permeability of the mitochondrial inner membrane to protons not coupled to ATP synthesis, dissipates mitochondrial membrane potential, and protects cells from ROS damage. Multiple mutations or RNAi treatments that extend lifespan also decrease mitochondrial membrane potential, and treatment of wild-type worms with the uncoupler CCCP extended lifespan by 60 % (Lemire et al., 2009). Similarly, treatment of wild-type worms with the uncoupler FCCP extended lifespan by 22 % (Morcos et al., 2008). Moreover, the uncoupling agent 2,4-dinitrophenol (DNP), which acts as a protonophore, increases both respiration activity and longevity in yeast (Barros et al., 2004), flies (Padalko, 2005, Ulgherait et al., 2020), and mice (Caldeira da Silva et al., 2008).
Mitochondrial uncoupling proteins (UCPs) reduce the amount of ATP that can be produced via oxidative metabolism. They are located at the inner mitochondrial membrane and cause proton leakage into the matrix, thus disrupting the proton gradient generated by the ETC and uncoupling substrate oxidation from ATP phosphorylation. Mild uncoupling may lead to the attenuation of oxidative stress and lifespan extension. Drosophila contains 4 UCPs—UCP4a, UCP4b, UCP4c, and UCP5—that are the counterparts of 5 mammalian UCPs—UCP1, UCP2, UCP3, UCP4, and UCP5. Flies lacking UCP5 had a 30 % increase in lifespan as compared to controls on low-calorie diets (Sanchez-Blanco et al., 2006). Similar to DNP treatment, expression of human uncoupling protein 2 (hUCP2) also increased longevity in flies (Fridell et al., 2005). Moreover, expression of mUCP1 and hUCP2 specifically in fly insulin-producing cells (IPCs) reduced systemic insulin signaling, extended lifespan by 19 %, and increased resistance to oxidative stress and starvation without any changes in egg production (Fridell et al., 2009). Ubiquitous expression of human UCP3 (hUCP3) in flies did not increase lifespan, while adult-specific neuronal expression caused a marginal lifespan extension only in males. However, higher neuronal expression of hUCP3 expression, which led to measurable increases in proton conductance, resulted in increased DILP protein levels in head samples and dramatically shortened lifespan (Humphrey et al., 2009). Also, overexpression of UCP4C in intestinal stem cells and enteroblasts was sufficient to extend Drosophila lifespan and preserve proliferative homeostasis in the gut with age. Similarly, two mitochondrial uncouplers, 2,4-dinitrophenol and beta-hydroxytoluene (BHT), extended lifespan of wild-type male flies and reduced age-dependent over-proliferation of intestinal stem cells (Ulgherait et al., 2020). Interestingly, in worms, expression of worm ucp-4 did not affect lifespan, but expression of zebrafish ucp2 extended lifespan by 40 %, implying the efficacy or level of uncoupling are important for lifespan extension (Sagi and Kim, 2012). Moreover, primary dermal fibroblasts from long-lived (small) dog breeds had more uncoupled mitochondria, reduced electron escape, greater respiration, and a capacity for respiration comparable to that of short-lived (large) dog breeds (Nicholatos et al., 2019). Mice with skeletal muscle-specific UCP1 expression had 0.5 °C higher core body temperatures than WT mice, ~10 % increased lifespan in both males and females, and lower frequency of lymphomas (the most common probable cause of death for this strain of mice) (Gates et al., 2007). For Ucp2-deficient mice, lifespan was shortened by ~35 %, but hUCP2 transgenic mice did not show any difference in lifespan as compared to control mice (Andrews and Horvath, 2009).
Another plausible endogenous anti-aging mechanism based on mild uncoupling was recently reported by Vyssokikh et al. (Vyssokikh et al., 2020). Using extensive comparative bioenergetic characterization of purified mitochondria from F1 C57Bl/6/CBA hybrid mice, naked mole rats, and bats, the authors demonstrated mild depolarization in long-lived animals (naked mole rats and bats) compared to short-lived mice. This uncoupling mechanism was suggested to substantially inhibit mROS generation, which can account for lifespan differences (Vyssokikh et al., 2020).
Many organisms possess alternative enzymes that can bypass or replace the proton-translocating complexes of the mETC. These enzymes can provide an alternative respiratory chain that allows the maintenance of redox homeostasis and intermediary metabolism when the mETC is restrained. Alternative ubiquinol oxidases (AOX) can bypass complexes III and IV, and alternative NADH dehydrogenases, Nde (NADH dehydrogenase external) or Ndi (NADH dehydrogenase internal), can bypass complex I. Alternative NADH dehydrogenases reduce ubiquinone, taking electrons from NADH, but do not pump protons across the inner mitochondrial membrane. Mammalian mitochondrial complex I (NADH: ubiquinone oxidoreductase) consists of 45 subunits (44 distinct subunits and one subunit appears twice) (Rhooms et al., 2019). Alternative NADH:ubiquinone oxidoreductase (Ndi1) from S. cerevisiae consists of a single polypeptide (Luttik et al., 1998). Two groups generated transgenic flies expressing Ndi1 and demonstrated that Ndi1 expression stimulated rotenone-resistant respiration, rescued the lethality of Complex I subunit knockdown, and increased the NAD+/NADH ratio (Bahadorani et al., 2010a; Sanz et al., 2010). Interestingly, one group demonstrated that ubiquitous expression of Ndi1 in flies during development and in the adult stage prolonged lifespan both in male (30–40 %) and female (10–20 %) flies, and decreased mitochondrial ROS production (Sanz et al., 2010). Another group demonstrated that ubiquitous inducible expression of Ndi1 in flies during development and the adult stage did not significantly affect the lifespan of either male or female flies, and did not affect mitochondrial ROS production. However, expression of Ndi1 in adipose tissue decreased both male and female lifespan, whereas neuronal expression of Ndi1 increased lifespan in both male (by 7%) and female (by 21 %) flies (Bahadorani et al., 2010a). The differences in the effect on lifespan extension might be explained by the fact that the first group used a ubiquitous non-inducible driver (da-Gal4) and another group used a ubiquitous inducible driver (tubulin-GeneSwitch-Gal4), and these can result in different levels of induction of NDI expression. Furthermore, Ndi1 overexpression reduced the CoQ pool and increased ROS production via reverse electron transport (RET) through ETC complex I. The increased ROS production was required for the lifespan extension by Ndi1 (Scialo et al., 2016). Expression of Ndi1 in Drosophila intestinal stem and progenitor cells delayed the onset of multiple markers of intestinal aging and was sufficient to extend lifespan (Hur et al., 2013). Expression of alternative NADH dehydrogenase (NDX) of Ciona intestinalis in Drosophila also increased resistance to different stresses and extended lifespan by 50 % (Gospodaryov et al., 2014, 2019).
The NADH shuttle systems move permeable NAD and NADH across the mitochondrial membrane, which is impermeable to NAD and NADH, thereby balancing the NAD/NADH ratio between the mitochondrial and cytosolic/nuclear pool. There are several mitochondrial NADH shuttles in yeast, including the malate/aspartate shuttle, which consists of aspartate amino transferase (Aat1/Aat2) and malate dehydrogenase (Mdh1/Mdh2); the ethanol/acetaldehyde shuttle, which consists of mitochondrial alcohol dehydrogenase (Adh3) and cytosolic alcohol dehydrogenase (Adh1/Adh2); and the glycerol-3-phosphate shuttle, which oxidizes cytosolic NADH and transfers electrons directly into the respiratory chain. The glycerol-3-phosphate shuttle consists of two components—the cytosolic glycerol-3-phosphate dehydrogenase (Gpd1/Gpd2) and the mitochondrial glycerol-3-phosphate dehydrogenase (Gut2)—that are localized to the inner mitochondrial membrane (Easlon et al., 2008). Overexpression of Aat1, Mdh1, and Gut2 increased the NAD/NADH ratio and caused ~25 % extension of yeast RLS, comparable to lifespan extension by CR, whereas overexpression of Aat2 and Mdh2 caused ~15 % lifespan extension, and overexpression of Adh2 and Adh3 did not significantly affect yeast lifespan. Overexpression of Aat1, Mdh1, and Gut2 did not further extend lifespan under CR, and their deletion partially suppressed CR-induced lifespan extension, while their deletion did not suppress lifespan under normal conditions. Moreover, similar to CR, lifespan extension by Aat1 and Mdh1 overexpression required Sir2, whereas lifespan extension by Gut2 overexpression was not affected by Sir2 deletion. In addition, double deletion of mdh1 and aat1 suppressed lifespan extension induced by other manipulations reminiscent of CR, such as overexpression of Hap4, which induces a metabolic shift toward respiration, or Lat1, an E2 component of PDC (see above) (Easlon et al., 2008).
In summary, manipulating enzymes related to the TCA cycle and OXPHOS, feeding of metabolites associated with these processes, increasing uncoupling, and introducing bacterial enzymes that can rescue age-dependent defects of the ETC are all strategies that have shown potential to extend lifespan across different species.
2.3. Amino acids and NAD metabolism
Responses to calorie restriction or dietary restriction (CR/DR) are evolutionary conserved, and CR/DR provides one of the most robust mechanisms of lifespan extension (Soultoukis and Partridge, 2016). Adding back essential amino acids to the DR diet in flies abrogates the beneficial effects of DR on lifespan (Grandison et al., 2009). In mice, the effects of protein restriction on lifespan extension were stronger than carbohydrate or fat restriction (Solon-Biet et al., 2014). Similarly, in Drosophila, reduction of yeast (the protein component of fly food) has a much stronger effect on lifespan extension compared to restriction of carbohydrates or total calories (Mair et al., 2005). All of this would suggest a detrimental effect of amino acids on lifespan. However, studies in model organisms demonstrate that supplementation of specific amino acids may instead extend lifespan and delay aging (Canfield and Bradshaw, 2019). Individual supplementation of 18 out of 20 amino acids (except phenylalanine and aspartate) extended worm lifespan, with serine and proline supplementation showing the largest effects (Edwards et al., 2015a). Moreover, association studies have revealed positive associations between levels of amino acids and lifespan. Ma et al. found that levels of 11 out of 20 amino acids including arginine, glutamate, histidine, leucine, lysine, methionine, phenylalanine, proline, tryptophan, tyrosine, and valine in fibroblasts from 15 primate species, 33 bird species, and 13 rodent species had a positive association with species lifespan (Ma et al., 2016). Although effects of manipulations of ratios between different food components such as carbohydrates, lipids, and amino acids were recently reviewed (Fontana and Partridge, 2015; Soultoukis and Partridge, 2016; Tatar et al., 2014) (Le Couteur et al., 2016), we will discuss how manipulations of metabolism of single amino acids affect aging and lifespan.
2.3.1. Leucine, isoleucine, and valine
Branched-chain amino acids (BCAAs) leucine, isoleucine or valine have been linked to aging and age-related disease in multiple species (Green and Lamming, 2019; Valerio et al., 2011). Long-term dietary supplementation with a specific branched-chain amino acid (BCAA)-enriched amino acid mixture (BCAAem) beginning at 9 months increased lifespan in male mice, and this effect was abrogated in eNOS-mutant mice. BCAAem-supplemented middle-aged mice demonstrated enhanced mitochondrial biogenesis and function in cardiac and skeletal muscles but not in adipose tissue or liver. Moreover, BCAAem preserved muscle fiber size and improved physical endurance in middle-aged mice (D’Antona et al., 2010). These data were consistent with findings in yeast, where threonine, leucine, isoleucine, and valine extended CLS (Alvers et al., 2009). In worms, downregulation of BCAA transferase-1 (bcat-1) (Fig. 3) led to excessive levels of BCAAs and extension of lifespan, whereas overexpression of bcat-1 shortened lifespan (Mansfeld et al., 2015). In addition, leucine, valine, or isoleucine supplementation led to an increase in lifespan in C. elegans (Edwards et al., 2015a; Mansfeld et al., 2015). Long-lived daf-2 or ife-2 mutant worms, in which the eukaryotic translation initiation factor eIF4E is disrupted, are characterized by a striking increase in levels of BCAAs—isoleucine, leucine and valine—and this increase was abrogated in daf-2,daf16 double-mutant worms (Fuchs et al., 2010). BCAA supplementation improved cognitive performance in dogs with greater benefit to senior dogs (Fretwell et al., 2006). In a randomized trial in elderly humans with sarcopenia aged 66–84 years, supplementation with a special mixture of amino acids including BCAAs (leucine, lysine, isoleucine, valine, threonine, cysteine, histidine, phenylalanine, methionine, tyrosine, and tryptophan) increased whole-body lean mass, reduced levels of tumor necrosis factor-α, and improved insulin sensitivity (Solerte et al., 2008). Various beneficial effects of BCAA supplementation were recently reviewed by Valerio et al. (Valerio et al., 2011).
Fig. 3.
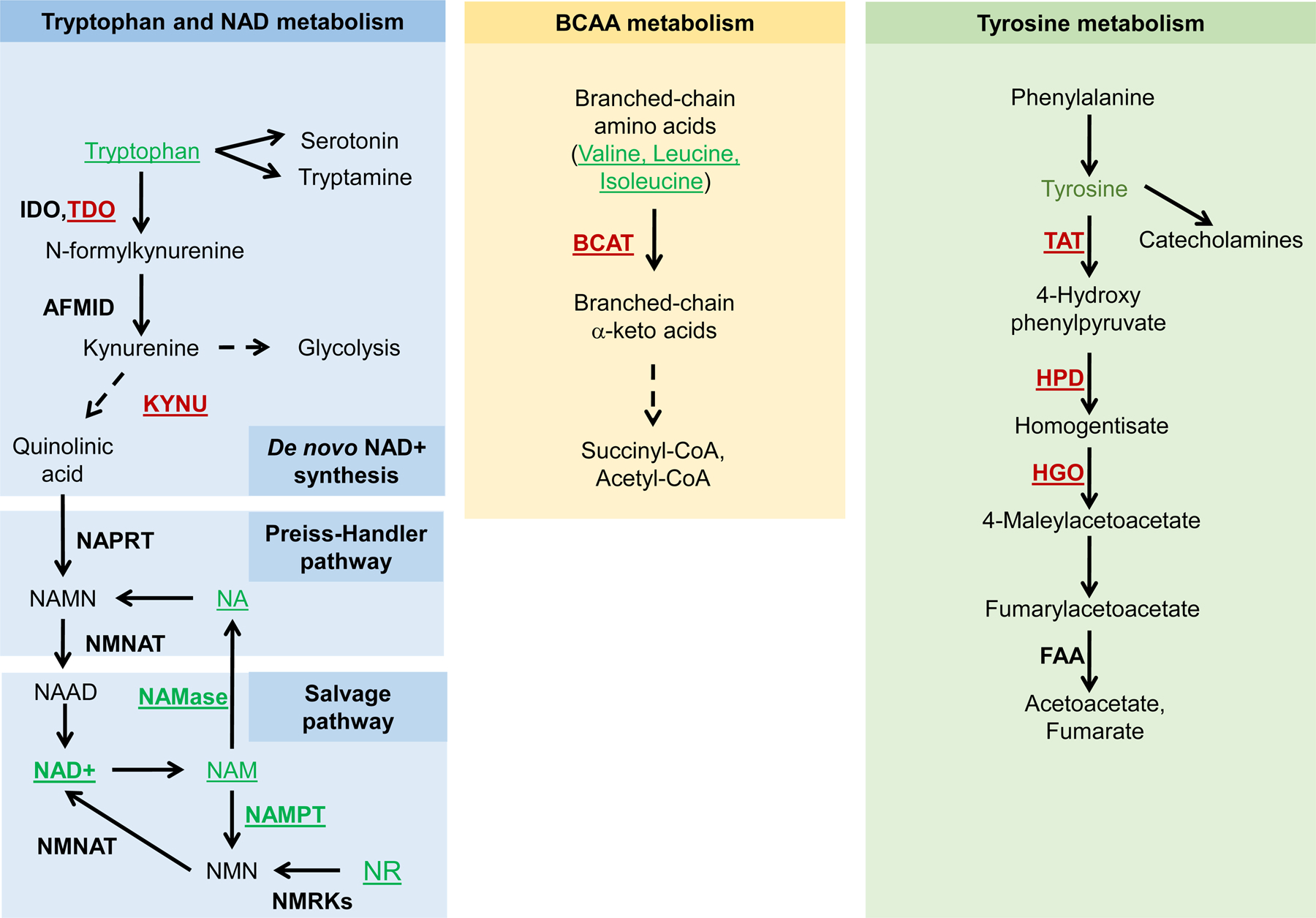
Schematic representation of tryptophan and NAD metabolism (A), BCAAs metabolism (B), and tyrosine metabolism (C). Underlined are metabolites and enzymes that were associated with lifespan extension. Red font color represents downregulation or depletion from food, while green font color represents overexpression or supplementation. Dashed line represents that multiple steps are involved. (A) Supplementation of tryptophan, as well as downregulation of the enzymes tryptophan 2,3-dioxygenase (TDO) and kynureninase (KYNU) was associated with lifespan increase. The Preiss-Handler and salvage pathways can synthesize NAD + from pyridine bases. Increased levels of nicotinic acid (NA), nicotinamide (NAM), nicotinamide riboside (NR) and NAD+, as well as expression of nicotinamidase (NAMase) and nicotinamide phosphoribosyltransferase (NAMPT) were associated with lifespan extension. (B) Branched-chain amino acids (BCAAs) are degraded through a series of reactions, resulting in succinyl-CoA (valine) or acetyl-CoA (leucine, isoleucine). Valine, leucine, and isoleucine supplementation was associated with lifespan extension in mice, yeast, and worms, while downregulation of the enzyme branched-chain amino acid transferase (BCAT) extended lifespan in worms. (C) The tyrosine degradation pathway converts tyrosine into fumarate and acetoacetate. Supplementation of tyrosine and downregulation of the tyrosine degradation enzymes tyrosine aminotransferase (TAT), 4-hydroxyphenylpyruvate dioxygenase (HPD), and homogentisate 1,2 dioxygenase (HGO) resulted in lifespan extension in flies.
The results of other studies cloud the picture with respect to the potential beneficial effects of BCAAs on lifespan. Solon-Biet et al. demonstrated that long-term exposure of mice to high BCAA diets led to hyperphagia, obesity and reduced lifespan (Solon-Biet et al., 2019). Accordingly, a diet low in BCAAs (Cummings et al., 2018; Fontana et al., 2016) or only in leucine (Xiao et al., 2011) improved metabolic health in mice. Similarly, in flies, a diet low in BCAAs extended lifespan as did a diet low in threonine, histidine and lysine (THK) (Juricic et al., 2020). Moreover, similar to the seemingly incongruous effects of BCAAs on the extension of lifespan, the effects of glutamine on yeast lifespan were opposite in two studies. Wu et al. found that high glutamate levels increased CLS in yeast (Wu et al., 2013), whereas Powers et al. found that removal of glutamate from the media or inhibition of glutamine synthesis extended CLS (Powers et al., 2006).
2.3.2. Threonine
Mansfeld et al. performed RNA-seq analysis on skin samples from zebrafish, C57BL/6 J mice, and whole worms and identified 13 genes upregulated during aging and 16 genes downregulated during aging in all three species. Subsequent screening of these genes in C. elegans identified glycine-C-acetyltransferase (Gcat)/T25B9.1, which was downregulated during physiological aging in different species, as a new regulator of lifespan (Mansfeld et al., 2015). RNAi against gcat/T25B9.1 extended C. elegans lifespan by 22 % and improved healthspan-related parameters such as movement, speed, and accumulation of aging pigments without reducing fertility or food uptake (Ravichandran et al., 2018). L-threonine dehydrogenase converts L-threonine to 2-amino-3--ketobutyrate and GCAT converts 2-amino-3-ketobutyrate to l-glycine and acetyl-CoA. 2-amino-3-ketobutyrate is highly unstable and spontaneously decarboxylates into aminoacetone, which can further undergo enzymatic oxidation by amine oxidases to produce methylglyoxal (MGO), a highly reactive dicarbonyl aldehyde, and additionally produces reactive oxygen species (ROS) in the form of hydrogen peroxide (Schmidt et al., 2001). Lifespan extension by suppression of gcat required amine oxidases (amx-1, amx-2, and amx-3 in worms) and was dependent on MGO production, as supplementation of worms with 50 and 100 u M MGO increased worm lifespan (Ravichandran et al., 2018). Gcat/T25B9.1 downregulation and MGO supplementation activated SKN-1 and HSF-1 transcription factors, and lifespan extension was completely abrogated in skn-1 and hsf-1 mutant worms (Ravichandran et al., 2018). In contrast to the positive role of MGO in the regulation of lifespan, Morcos et al. found that the activity of glyoxalase-1, an enzyme that detoxifies MGO, is strongly reduced with age. Overexpression of the C. elegans glyoxalase-1 ortholog, CeGly, decreased MGO production and extended lifespan (Morcos et al., 2008). Similarly, treatment of worms with rifampicin reduced glycation of cellular proteins in vivo and increased lifespan by 60 % (Golegaonkar et al., 2015).
2.3.3. Proline
Zarse and colleagues demonstrated that chronic impairment of insulin/IGF-1 signaling (IIS) caused an induction of mitochondrial activity in daf-2-mutant worms and mouse embryonic fibroblasts (Irs-1−/− and Insr+/−). Interestingly, acute downregulation of daf-2 caused a transient increase in ROS levels, while long-term daf-2 downregulation was accompanied by reduction of ROS levels and increased levels of the antioxidant enzymes SOD and CAT. This transient increase in ROS levels (mitohormetic role) was required for the later induction of SOD and CAT and extension of lifespan. Unexpectedly, chronic impairment of IIS led to decreased glucose uptake despite increased mitochondrial activity. Transcriptome analysis revealed upregulation of L-proline dehydrogenase/prodh (B0513.5 in worms), an enzyme that is essential for proline catabolism and can serve an anaplerotic role. Downregulation of B0513.5 did not affect the lifespan of wild-type worms but decreased lifespan extension induced by daf-2 downregulation. In accordance with a positive role of fueling proline into the TCA cycle, feeding worms with 5 u M proline extended lifespan (Edwards et al., 2015a; Zarse et al., 2012).
2.3.4. Arginine
Arginine is a semi-essential amino acid that plays an important role in the synthesis of nitric oxide, polyamines, proline, glutamate, creatine, agmatine and urea, which are involved in the regulation of aging and lifespan (Gad, 2010). In yeast, deletion of CAN1, which encodes a plasma membrane-localized arginine amino acid transporter, extended RLS, and this effect depended on the transcription factors GCN4 and Hac1 (Beaupere et al., 2017). McQuary et al. identified the arginine kinase ARGK-1/F44G3.2 as the most significantly enriched protein in long-lived rsks-1/S6K mutants and levels of its mammalian ortholog, creatinine kinase, were increased in the brains of S6K1 knockout mice. Arginine kinases are similar to mammalian creatine kinases and function to maintain intracellular ATP levels by catalyzing the reversible reaction of ATP and arginine to produce ADP and phospho-arginine. In worms, overexpression of ARGK-1 phenocopied the rsks-1/S6K mutant phenotype and extended worm lifespan, whereas downregulation of argk-1 suppressed lifespan of rsks-1/S6K mutants but had no effect in wild-type worms (McQuary et al., 2016). Rozanov et al. identified several pro-aging transcription factors in C. elegans. One of them, HLH-2, impacts metabolism and lifespan via regulation of argk-1. In contrast to the findings reported by McQuary et al.; however, Rozanov et al. found that downregulation of argk-1 extended worm lifespan (Rozanov et al., 2020).
2.3.5. Methionine/Cysteine
Methionine restriction (MetR) extends lifespan across different species and exerts beneficial effects on metabolic health and inflammatory responses (Parkhitko et al., 2019). Methionine feeds into complex metabolic pathways including the methionine cycle, the transsulfuration pathway, and polyamine biosynthesis (Fig. 4). Manipulation of each of these pathways extends lifespan.
Fig. 4.
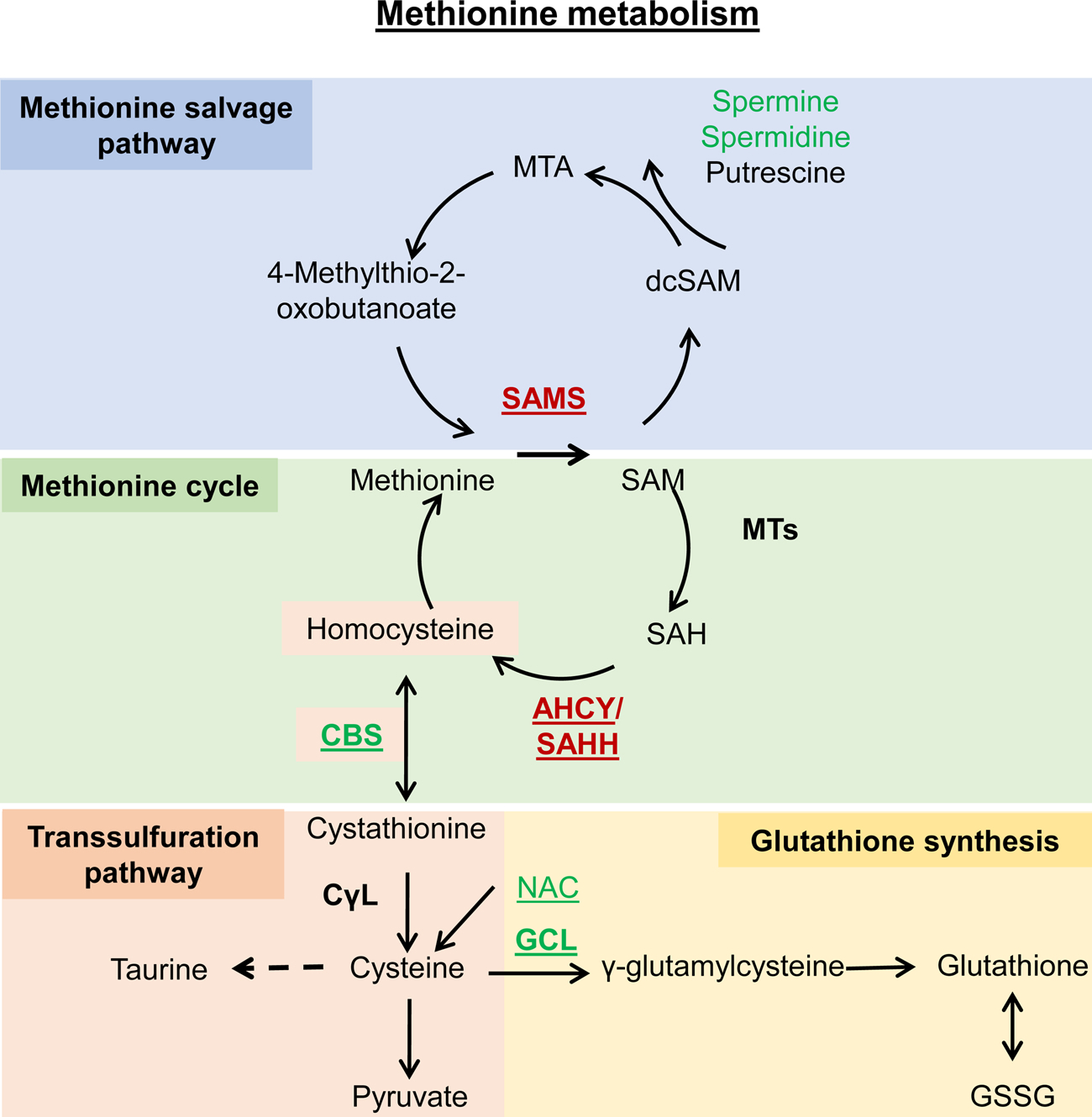
Schematic representation of methionine metabolism. Underlined are metabolites and enzymes that were associated with lifespan extension. Red font color represents downregulation or depletion from food, while green font color represents overexpression or supplementation. In the methionine cycle, methionine is converted to S-adenosyl-l-methionine (SAM), which acts as a methyl donor for methyltransferases, forming S-adenosyl-l-homocysteine (SAH), and finally, homocysteine. Downregulation of methionine adenosyltransferase (SAMS) and S-adenosyl-l-homocysteine hydrolase (ACHY), and overexpression of Glycine N- methyltransferase (GNMT) were associated with lifespan extension. In the methionine salvage pathway, methionine can be regenerated from SAM, forming polyamines during the cycle. Supplementation of the polyamines spermine and spermidine was associated with lifespan extension in several species. In the transsulfuration pathway, homocysteine is converted into cysteine, which can then be metabolized into taurine, pyruvate, and glutathione. Upregulation of cystathionine-β-synthase (CBS) and glutamate-cysteine ligase (GCL), as well as supplementation with the cysteine donor N-acetylcysteine (NAC) were associated with lifespan increase.
The first step in methionine metabolism is performed by methionine adenosyltransferase (MAT), an enzyme conserved from E. coli to humans that catalyzes the biosynthesis of S-adenosyl-l-methionine (SAM) from methionine and ATP. During substrate methylation, SAM donates its methyl group to acceptor molecules, e.g. DNA, RNA, proteins, or other cellular metabolites, generating S-adenosyl-l-homocysteine (SAH). SAH hydrolase (SAHH/AHCY) catalyzes the reversible hydrolysis of SAH to adenosine and l-homocysteine. Homocysteine can be remethylated to form methionine and retained in the methylation cycle, or converted to cysteine via the transsulfuration pathway and thus withdrawn from the methylation cycle. Remethylation of homocysteine to form methionine completes the methionine cycle.
In an unbiased RNAi screen, Hansen et al. identified sams-1/ C49F5.1 as encoding a MAT that catalyzes the biosynthesis of SAM, the first step in methionine metabolism. Downregulation of sams-1 extended lifespan in a daf-16-independent manner but failed to extend the lifespan of eat-2/ad1116 mutants (Hansen et al., 2005). Consistent with this, overexpression of sams-1 partially suppressed lifespan extension of DR worms, and RNAi knockdown of sams-1 reduced the global translation rate (Ching et al., 2010). In flies, restriction of amino acids increased lifespan at any concentration of methionine, whereas MetR extended lifespan only when the levels of amino acids were reduced (Lee et al., 2014). We found that naturally-selected long-lived flies, which have twice the lifespan of wild-type strains, have higher levels of endogenous methionine, suggesting that high levels of methionine are not detrimental to lifespan and that flux via methionine metabolism is more critical than the level of methionine itself (Parkhitko et al., 2016). In agreement with this hypothesis, downregulation of dAhcyL1/dAhcyL2 activates Ahcy13, which in turn promotes SAH and homocysteine processing, resulting in an increase in methionine flux and lifespan extension (Parkhitko et al., 2016). Similarly, Obata et al. showed that overexpression of GNMT, which converts glycine to sarcosine (N-methyl-glycine) by methyl group transfer using SAM and functions as a regulator of SAM levels in metabolic organs, suppresses age-dependent SAM increase and extends lifespan (Obata and Miura, 2015). Drosophila microbiota may also play an important role in the response of flies to MetR. Rearing flies with Escherichia coli mutants for distinct genes relevant to methionine metabolism alters the Drosophila starvation response and longevity (Judd et al., 2018; Matthews et al., 2020). Moreover, in yeast, significant CLS extension could be achieved by restricting methionine in the S. cerevisiae wild-type BY4742 strain, which is auxotrophic for his/leu/lys (Wu et al., 2013), by genetically limiting methionine biosynthesis (Δmet15 or Δmet2) (Johnson and Johnson, 2014; Ruckenstuhl et al., 2014), or by enzymatically degrading methionine (Plummer and Johnson, 2019). In addition, an unbiased genetic screening of 4698 viable single-gene deletion strains for replicative lifespan in S. cerevisiae identified that deletion of MET3, which encodes an enzyme important for sulfate assimilation and methionine synthesis, or SAM1, which encodes methionine adenosyltransferase, significantly extended replicative lifespan (McCormick et al., 2015). Interestingly, glucose restriction in yeast was associated with decreased expression of methionine biosynthetic enzymes and transporters and lifespan extension by glucose restriction was blocked by methionine supplementation (Zou et al., 2020).
The lifelong reduction of a single dietary component—methionine—from 0.86 % to 0.17 % in the diet of Fisher 344 rats resulted in a 30 % increase of male rat lifespan (Orentreich et al., 1993). In female CB6F1 mice, a decrease of methionine from 0.43 % by weight to 0.1–0.15% increased lifespan, slowed immune and lens aging, and decreased levels of serum IGF-I, insulin, glucose, and thyroid hormone, despite an increase in food uptake (Miller et al., 2005). In human diploid fibroblasts, reduction of methionine from 30 mg/L to 1 mg/L had no significant effect on the rate of cell proliferation in early-passage cells but significantly extended their replicative lifespan, postponing cellular senescence. Extended lifespan was associated with reduced oxygen consumption (Koziel et al., 2014). In humans, a vegan diet is associated with decreased methionine content (McCarty et al., 2009).
Homocysteine from the methionine cycle can also be utilized in the transsulfuration pathway to produce cysteine. Cystathionine-β-synthase is the first and rate-limiting enzyme of the transsulfuration pathway, the primary metabolic pathway for the synthesis of cysteine. Cystathionine-β-synthase synthesizes cystathionine from the condensation of homocysteine and serine. Cystathionine is hydrolyzed by cystathionine-γ-lyase to produce cysteine, which is further used in the synthesis of proteins, glutathione, and taurine. Cystathionine-γ-lyase and cystathionine-β-synthase also catalyze the production of hydrogen sulfide (H2S) from cysteine and homocysteine. Long-lived eat-2 mutant worms produced more H2S than wild-type worms. CBS-1 is a worm ortholog of cystathionine-β-synthase. RNAi knockdown of cbs-1 decreased the lifespan extension normally associated with eat-2 mutants, and overexpression of CBS-1 in wild-type worms prolonged lifespan (Hine et al., 2015). Supplementing the diet with the product of the transsulfuration pathway, N-acetyl-l-cysteine, significantly extended lifespan and significantly increased resistance to oxidative stress, heat stress, and UV irradiation in C. elegans (Oh et al., 2015).
In Drosophila, Kabil et al. demonstrated that the activity of dCBS and the transsulfuration pathway is increased under DR; inhibiting the second enzyme in the transsulfuration pathway, γ-cystathionase using propargylglycine caused robust suppression of lifespan extension by DR but not in fully fed flies. In agreement with this, either ubiquitous adult-specific overexpression of dCBS or neuronal overexpression of dCBS was sufficient to increase longevity (Kabil et al., 2011). Moreover, consistent with the positive role of the transsulfuration pathway in lifespan extension, maximal H2S production of flies subjected to various forms of Met and DR correlated with maximal lifespan extension (Hine et al., 2015).
Glutamate-cysteine ligase (GCL) is the rate-limiting enzyme that conjugates glutamate and cysteine to create γ-glutamylcysteine. GSH synthase (GS) links glycine to γ-glutamylcysteine to form GSH. Glutamate-cysteine ligase (GCL) is a heterodimeric enzyme consisting of a catalytic subunit, GCLc, and a modulatory subunit, GCLm. Orr et al. showed that the overexpression of GCLc or GCLm in flies using either global or neuronal drivers of expression led to an increase in the glutathione content observed in fly homogenates and extended lifespan (Orr et al., 2005). In agreement with these findings, feeding flies with N-acetylcysteine (NAC), a cysteine donor for GSH, resulted in a dose-dependent increase in lifespan (Brack et al., 1997). Similar to worms and flies, a heterogeneous stock of mice (NIA Interventions Testing Program) fed with N-acetyl-l-cysteine (NAC) had a significantly extended lifespan. Notably, the effect was sex-specific. In females, NAC treatment did not significantly affect total lifespan whereas in males both high (1200 mg/kg/d) and low (600 mg/kg/d) NAC doses increased total lifespan. However, both doses of NAC caused a sudden drop in body weight, followed by a further slow decline (Flurkey et al., 2010).
The methionine salvage pathway, or 5′-methylthioadenosine (MTA) cycle, regenerates methionine from SAM and is responsible for the production of polyamines (Minois et al., 2011; Pegg, 2016). In the methionine salvage pathway, SAM is decarboxylated by AdoMet decarboxylase into decarboxylated SAM (dcSAM), which serves as an aminopropyl group donor. In parallel, arginase converts arginine into ornithine, which is then decarboxylated by ornithine decarboxylase (ODC) to produce putrescine. Putrescine is further converted to spermidine and spermine through the consecutive action of two distinct aminopropyl transferases, spermidine synthase and spermine synthase, which use dcSAM as an aminopropyl donor (Minois et al., 2011; Pegg, 2016). Supplementing food with spermidine in C. elegans induced autophagy and prolonged lifespan by up to 15 %, whereas knockdown of Beclin-1, a gene essential for autophagy, abolished the spermidine-mediated increase in lifespan (Eisenberg et al., 2009). Similar to worms, supplementation of regular food with 1 mM spermidine was shown to prolong lifespan in flies by up to 30 %. Exogenous supplementation of spermidine also increased CLS and RLS in yeast (Eisenberg et al., 2009). In C57BL/6 J female mice, supplementation with spermidine and spermine (but not putrescine) significantly extended lifespan. Spermidine also significantly extended lifespan when supplementation was started late in life (in 18 months old mice). Spermidine supplementation had cardioprotective effects, resulting in reduced cardiac hypertrophy and preserved diastolic function in old mice. These findings were associated with enhanced cardiac cell autophagy, mitophagy, and mitochondrial respiration and spermidine failed to provide cardioprotection in mice that lack Atg5, which is essential for autophagy in cardiomyocytes (Eisenberg et al., 2016).
Altogether, the results of these methionine metabolism studies imply that strategies such as increasing methionine flux, promoting the transsulfuration pathway, and increasing exposure to polyamines could have positive lifespan and healthspan effects across many species.
2.3.6. Glycine
Supplementation with glycine significantly prolongs C. elegans lifespan via feeding into the methionine cycle, and mutations in components of the methionine cycle—methionine synthase (metr-1) or S-adenosylmethionine synthetase (sams-1)—completely abrogated glycine-induced lifespan extension (Liu et al., 2019). Moreover, downregulation of mel-32 (C. elegans ortholog of SHMT1/2) prevented glycine conversion to serine and extended lifespan (Liu et al., 2019). In rats, glycine supplementation phenocopies lifespan extension by dietary methionine restriction through the clearance of hepatic methionine (Joel Brind et al., 2011). Similarly, glycine supplementation extended lifespan of male and female mice (Miller et al., 2019).
2.3.7. Tyrosine
Tyrosine is a non-essential amino acid that can be produced from phenylalanine. Tyrosine is a precursor for the biogenic amines dopamine, adrenaline (analogous to octopamine in invertebrates), and noradrenaline (analogous to tyramine in invertebrates). Tyrosine also can be degraded via the tyrosine degradation pathway and generate two fragments, each of which can enter the TCA cycle. Four of the nine carbon atoms of tyrosine generate free acetoacetate, which is converted into acetoacetyl-CoA, and the second four-carbon fragment is recovered as fumarate. Eight of the nine carbon atoms of these two amino acids thus enter the citric acid cycle, and the remaining carbon is lost as CO2 (Fig. 3). Tyrosine aminotransferase/TAT is the first and rate-limiting enzyme in the tyrosine catabolic pathway and catalyzes the conversion of tyrosine to 4-hydroxyphenylpyruvate. 4-hydroxyphenylpyruvate dioxygenase (HPD) catalyzes the conversion of 4-hydroxyphenylpyruvate to homogentisate, the second step in the tyrosine degradation pathway, and homogentisate 1,2-dioxygenase (HGO) catalyzes the conversion of homogentisate to 4-maleylacetoacetate.
Lee et al. identified 17 orthologous genes from C. elegans and Drosophila that carry a DAF-16 binding site in the promoter region. The mRNA levels of T21C12.2/hpd-1 were downregulated in long-lived daf-2 mutant worms, and its downregulation caused 30 % increases in lifespan (Lee et al., 2003a). Also, Yuan et al. found that the level of hpd-1 is decreased in long-lived eat-2 mutant worms and downregulation of hpd-1 increased worm lifespan (Yuan et al., 2012). Moreover, the C. elegans ortholog of TAT, tatn-1, influenced insulin signaling, development, and lifespan via modulation of aak-2/AMPK signaling (Ferguson et al., 2013). We found that levels of tyrosine were increased in long-lived flies. We further demonstrated that the levels of enzymes in the tyrosine degradation pathway were increased with age and that whole-body and neuronal-specific downregulation of enzymes in the tyrosine degradation pathway (including TAT, HPD, and HGO) significantly extended Drosophila lifespan and upregulated the levels of tyrosine-derived neuromediators (Parkhitko et al., unpublished). In agreement with the positive role of tyrosine-derived neuromediators in prolonging lifespan, intermittently feeding octopamine to adult flies could substitute for exercise in sedentary flies, providing a number of pro-healthspan benefits (Sujkowski et al., 2017). Also, in worms, AMPK/calcineurin-mediated longevity was regulated cell-nonautonomously via regulation of octopamine (Burkewitz et al., 2015). Together, these studies demonstrate that by shifting the metabolism of tyrosine away from degradation and increasing exposure to tyrosine-derived biogenic amines, multiple lifespan and health benefits could be observed across different species.
2.3.8. Tryptophan and NAD metabolism
Upregulation of the tryptophan/kynurenine metabolic pathway has been suggested to be a mechanism of aging and age-associated medical and psychiatric disorders (van der Goot and Nollen, 2013). Tryptophan is an essential amino acid and a precursor for the synthesis of methoxyindoles (serotonin and melatonin). The major nonproteinogenic function of tryptophan is the production of N-formylkynurenine, which is further converted into kynurenine. Kynurenine metabolism is the major catabolic route for ingested tryptophan, and it has two major branches leading to the production of the neuroactive metabolite kynurenic acid (KA) or nicotinamide adenine dinucleotide (NAD+). Conversion of tryptophan into N-formylkynurenine is catalyzed by indoleamine 2, 3-dioxygenase 1 (IDO) or tryptophan 2,3-dioxygenase (TDO/TDO2). N-formylkynurenine is degraded by arylformamidase (AFMID) to yield kynurenine (KYN) (Schwarcz et al., 2012) (Fig. 3). In humans, aging is accompanied by an increased kynurenine/tryptophan ratio, reflecting changes in the tryptophan degradation rate and suggesting that aging is accompanied by accelerated degradation of tryptophan through the kynurenine pathway (Frick et al., 2004; Pertovaara et al., 2006). In rats, a low tryptophan diet (30 and 40 % of control diet) increased mortality early in life but substantially prolonged lifespan at late ages (Ooka et al., 1988). Depletion of TDO (encoded by vermilion in flies) or treating flies with 18.3 mM of alpha-methyl tryptophan, a TDO inhibitor, increased lifespan by 27 % (Oxenkrug, 2010; Oxenkrug et al., 2011). Similarly, depletion of TDO (tdo-2) extends lifespan in worms, suppresses α-synuclein toxicity and suppresses the age-related decline in motility (van der Goot et al., 2012). Inhibition of the tryptophan/kynurenine pathway may exert beneficial effects either via decrease accumulation of downstream products or via accumulation of tryptophan. Knockdown of tdo-2 in worms strongly increased the level of tryptophan, and supplementation of worms with increasing amounts of tryptophan but not threonine caused a dose-dependent suppression of α-synuclein toxicity (van der Goot et al., 2012). In line with this, in a screen of 20 proteogenic amino acids that were individually supplemented to determine their effects on C. elegans lifespan, tryptophan supplementation increased lifespan and thermo tolerance, and induced the UPRmt and an ER stress response (Edwards et al., 2015a). Minocycline, a tetracycline derivative with antibacterial, anti-inflammatory, antioxidant, and neuroprotective properties, also inhibits kynurenine formation from tryptophan (Henry et al., 2008). Oxenkrug et al. demonstrated that 0.87 mM of minocycline prolonged lifespan of wild-type flies (Oxenkrug et al., 2012). However, later it was shown that minocycline can extend lifespan not only in a wild-type strain but also in a w1118 strain, which is defective (at least partially) in intracellular transport of tryptophan (Mackenzie et al., 1999), and this finding does not support the hypothesis that minocycline acts via the tryptophan/kynurenine pathway (Lee et al., 2017). Ibuprofen, a common non-steroidal anti-inflammatory drug, increased lifespan in yeast, worms, and flies. In yeast, ibuprofen destabilized Tat2p, a high-affinity tryptophan permease, and inhibited tryptophan uptake. Furthermore, loss of Tat2p significantly extended RLS, and the effect of ibuprofen in Tat2p-deficient cells was attenuated (He et al., 2014).
One branch of the tryptophan/kynurenine pathway leads to de novo synthesis of nicotinamide adenine dinucleotide (NAD+) (Fig. 3). NAD+ is a pyridine nucleotide that is present in all living cells and is an essential cofactor that plays a critical role in many enzymatic redox reactions, mitochondrial energy production, and regulation of aging and lifespan. Kynurenine (KYN) can be converted to 3-hydroxykynurenine (3 H K) by kynurenine 3-monooxygenase (KMO). 3-hydroxykynurenine (3 H K) is further converted to 3-hydroxyanthranilic acid (3HAA) by kynureninase (KYNU). 3-hydroxyanthranilic acid (3HAA) is converted to 2-amino-3-carboxymuconic semialdehyde (ACMSA) by 3-hydroxyanthranilate 3,4-dioxygenase (HAAO), and ACMSA can be spontaneously converted to quinolinic acid (QA). QA is processed by quinolinate phosphoribosyl transferase (QPRT) to an NAD+ precursor, nicotinic acid mononucleotide (NAMN). Alternatively, 2-amino-3-carboxymuconic semialdehyde (ACMSA) can be converted to 2-aminomuconic semialdehyde (AMSA) by aminocarboxymuconate-semialdehyde decarboxylase (ACMSD) and further to picolinic acid (PA) and glutaryl-coenzyme A (Glutaryl-CoA). Nicotinic acid mononucleotide (NAMN) is converted to nicotinic acid adenine dinucleotide (NAAD) by NAMN adenylyltransferases (NMNATs). Finally, nicotinic acid adenine dinucleotide (NAAD) is converted to NAD+ by the glutamine-dependent NAD+ synthetase (NADSYN) (Canto et al., 2015; Castro-Portuguez and Sutphin, 2020; Lautrup et al., 2019; Verdin, 2015).
In worms, increasing levels of NAD+ is known to extend lifespan (Hashimoto et al., 2010; Katsyuba et al., 2018; Mouchiroud et al., 2013). Dose-dependent tryptophan supplementation and downregulation of acsd-1, which encodes ACMSD, increased NAD+ levels (Katsyuba et al., 2018) and extended worm lifespan in a sir-2.1-dependent manner (Edwards et al., 2015a; Katsyuba et al., 2018). Interestingly, Gebauer et al. created a genome-scale reconstruction of C. elegans metabolism assembled from genomic, transcriptomic, metabolomic, and literature data. Their metabolic network reconstruction predicted the lack of the enzyme NNDPR, which converts QA to nicotinate D-ribonucleotide in the tryptophan conversion to NAD+. They confirmed that feeding of tryptophan at the concentrations of 0.5 and 5 u M extended worm lifespan, while at higher concentrations it decreased lifespan. Supplementation of tryptophan led to a dramatic increase of QA, but there was no increase in the NAD+ concentration, suggesting that the conversion of tryptophan into NAD+ is not possible in C. elegans and that tryptophan extends lifespan by mechanisms independent of NAD+ (Gebauer et al., 2016). In mice, pharmacological inhibition of ACMSD with TES-991 and TES-1025 dose-dependently increased NAD+ levels in the liver, kidneys and brain of C57BL/6 J mice (Katsyuba et al., 2018). Suthpin et al. reported a systematic RNAi longevity screen of 82 C. elegans genes selected based on orthology to human genes differentially expressed with age. Among five genes with the greatest impact on lifespan, one encoded the enzyme kynureninase (kynu-1/ KYNU). Downregulation of kynu-1 increased levels of tryptophan and 3-hydroxykynurenin, and extended healthspan and lifespan by ~23 % (Sutphin et al., 2017). The Drosophila CG9940 gene encodes the NADSYN, which catalyzes the final step in de novo NAD+ biosynthesis. Overexpression of CG9940/NADSYN improved age-related cardiac function and extended Drosophila lifespan (Wen et al., 2016).
There are three major metabolic pathways for NAD+ production. The kynurenine pathway is the only route for de novo NAD+ biosynthesis from food-derived tryptophan. The Preiss-Handler pathway and the salvage pathway synthesize NAD+ from pyridine bases. In the Preiss-Handler pathway, cells generate NAD+ from nicotinic acid (NA). NA is converted by nicotinate phosphoribosyltransferase (NAPRT) to nicotinic acid mononucleotide (NAMN), where it converges with de novo synthesis. In the salvage pathway, NAD+ is produced from nicotinamide riboside (NR). Nicotinamide riboside (NR) is converted by nicotinamide riboside kinases (NMRKs) to nicotinamide mononucleotide (NMN) and then by NAMN adenylyltransferases (NMNATs) to NAD+. The Preiss-Handler pathway and the salvage pathway recycle NAD+ from the nicotinamide (NAM) when NAD+ is consumed by NAD-dependent enzymes. C. elegans and D. melanogaster genomes do not have nicotinamide phosphoribosyltransferase (NAMPT), which converts nicotinamide (NAM) to nicotinamide mononucleotide (NMN), but they do have nicotinamides (NAMase), which convert NAM to NA. Thus, invertebrates utilize the Preiss-Handler pathway to recycle NAD+ from NAM. By contrast, the mammalian genome does not encode NAMase, but does encode NAMPT. Thus, mammals use the salvage pathway to recycle NAD+ from nicotinamide (NAM) (Canto et al., 2015; Castro-Portuguez and Sutphin, 2020; Lautrup et al., 2019; Verdin, 2015).
Whole-body expression of Drosophila nicotinamidase (D-NAAM/NAMase) increased NAD+/NADH levels and extended lifespan by 30 %, an effect on lifespan that was abrogated in Sir2 mutant flies. This effect on lifespan can be recapitulated by neuronal-specific expression of D-NAAM (Balan et al., 2008). In S. cerevisiae, constitutive overexpression of PNC1, the yeast ortholog of D-NAAM, extended lifespan and increased resistance to stress (Anderson et al., 2003). In worms, a null mutation in pnc-1 dramatically reduced the lifespan increase associated with either DR or dietary deprivation, but it did not abrogate healthspan benefits associated with DR (Moroz et al., 2014). Supplementing worms with either 200 μM NAM or 500 μM nicotinamide riboside (NR) extended lifespan in sir-2.1-dependent manner, increased respiration, increased levels of ATP, and improved metabolic state. Moreover, feeding worms with NR induced UPRmt, and its suppression abrogated lifespan extension (Mouchiroud et al., 2013). Consistent with this, feeding worms with 100 μM of NAD extended lifespan by 15 %, and this lifespan extension depended on sir-2.1 and daf-16 (Hashimoto et al., 2010). Similarly, feeding worms with 1 mM NA also extended worm lifespan in a sir-2.1-dependent manner (Schmeisser et al., 2013a). Besides recycling of NAM to NAD+, NAM can be methylated by nicotinamide N-methyltransferase (NNMT, encoded by anmt-1 in C. elegans) to 1-methylnicotinamide (MNA), which may promote formation of ROS by inhibiting ETC complex I. Feeding worms with 100 μM NAM or 1 μM of MNA, or ANMT-1 overexpression, extended worm lifespan; however, in contrast to what was found for NAD and NA, this lifespan extension was independent of sir-2.1. Specifically, lifespan extension by MNA was dependent on its oxidation by aldehyde oxidase (AOX1, encoded by gad-3 in C. elegans) to 1-methyl-2-pyridone-5-carboxamide (PYR-2) and 1-methyl-4-pyridone-5-carboxamide (PYR-4) due to the formation of ROS as a byproduct during oxidation (Schmeisser et al., 2013a). In mice, chronic NAM supplementation improved healthspan without extending lifespan (Mitchell et al., 2018). Supplementation with NR starting at 24 months old in C57BL/6 mice increased mouse lifespan by ~5%; this supplementation with NR also rejuvenated muscle stem cells and attenuated senescence of neural and melanocyte stem cells in aged mice (Zhang et al., 2016). Interestingly, gut microbiota substantially contributes to the NAD+-boosting effect of oral NAM and NR supplementation in mice. This effect is mediated by bacterial nicotinamidase (PncA), which converts NAM to NA, a precursor in the alternative deamidated NAD salvage pathway (Shats et al., 2020).
In mice, CR rescues the age-dependent decrease of the function of 2 NADH-dehydrogenases, cytochrome b5 reductase 3 (CYB5R3) and NAD (P)H:quinone oxidoreductase 1 (NQO1), which function as detoxifying enzymes and intracellular generators of NAD+. Transgenic mice overexpressing rat Nqo1 and Cyb5r3 genes exhibited significantly higher levels of NAD+ and NADP+, a ~4% increase in lifespan and performed significantly better on the wire hang and rotarod tests, suggesting enhanced overall physical fitness (Diaz-Ruiz et al., 2018). Nqr1p, which encodes NADH-Coenzyme Q reductase 1 (NQR1), is a yeast plasma membrane-associated cytochrome b5 reductase induced by CR. In S. cerevisiae, overexpression of NQR1 extended both RLS and CLS via shifting yeast from fermentative to respiratory metabolism and modulation of the NAD+/NADH ratio (Jimenez-Hidalgo et al., 2009). Overexpression of the single Drosophila ortholog of cytochrome b5 reductases, CYB5R, extended lifespan by 17 % and improved lipid metabolism in flies (Martin-Montalvo et al., 2016). Similarly, transgenic mice overexpressing the rat CYB5R3 gene in the C57BL/6 J background had an increased lifespan (Martin-Montalvo et al., 2016).
To facilitate in vivo studies, Zhu et al. developed a magnetic resonance (MR)-based in vivo NAD assay that is capable of noninvasively assessing NAD+ and NADH content and the NAD+/NADH redox state in intact human brains. They found an age-dependent increase in intracellular NADH and age-dependent reductions in NAD+, total NAD content, and NAD+/NADH redox potential of the healthy human brain (Zhu et al., 2015). In humans, chronic supplementation with 1000 mg per day of NR for 6 weeks was well tolerated and effectively stimulated NAD+ metabolism in healthy 55- to 79-year-old adults (Martens et al., 2018). Several approaches have been implemented in humans to boost NAD levels and these have been reviewed recently (Rajman et al., 2018; Yoshino et al., 2018). Overexpression of nicotinamide phophoribosyltransferase (NAMPT) in adipose tissue elevated NAD+ levels and increased lifespan of female mice by 8%. Supplementation of mice with extracellular nicotinamide phophoribosyltransferase (eNAMPT) contained in extracellular vesicles starting at 26 months of age elevated NAD+ levels, improved physical activity, and increased lifespan by 10 % (Yoshida et al., 2019). Muscle-specific depleting of Nampt in mice led to a dramatic decline in intramuscular NAD content that was accompanied by fiber degeneration and progressive loss of both muscle strength and treadmill endurance, while administration of NR rapidly ameliorated functional deficits and restored muscle function. Moreover, lifelong overexpression of Nampt preserved muscle NAD+ levels and exercise capacity in aged mice (Frederick et al., 2016).
In summary, NAD metabolism is one of the most studied metabolic pathways related to lifespan extension in all species, and it can be effectively targeted at different levels in humans.
2.4. Nucleotide metabolism
Unbiased large-scale ‘omics’ analyses have led researchers to look at nucleotide metabolism as another metabolic process that might impact lifespan. To identify the global age-dependent metabolic changes in worms, Copes et al. employed a mass spectroscopy-based approach to discover endogenous metabolite differences between young (4 d) and aged (10 d) control and long-lived glp-4 mutant worms. Among multiple changes detected in different metabolic pathways, levels of purine metabolites (adenine, guanine, adenosine, adenosine monophosphate, ribose, ribose 5-phosphate, hypoxanthine, and inosine) exhibited the largest total decrease with age. Pyrimidine metabolite levels were also largely decreased with age (Copes et al., 2015). In a different study, Gao et al. compared transcriptomics and metabolomics data in wild-type and long-lived daf-2 (impaired IlS) and eat-2 (CR model) mutant worms. Both long-lived mutants shared the metabolic signature associated with upregulated purine metabolism (Gao et al., 2018). By highlighting nucleotide metabolism changes in aging worms, as well as metabolic differences in long-lived mutants, these two studies show that nucleotide metabolism may play an important role in regulating aging and lifespan.
2.4.1. Purine metabolism
Adenine and guanine nucleotides are derived from a common precursor, inosine monophosphate (IMP), which consists of ribose phosphate, and a purine derivative known as hypoxanthine. The de novo biosynthesis of AMP from IMP occurs in two steps. First, adenylosuccinate synthetase (AdSS) catalyzes the conversion of IMP to adenylosuccinate; then, adenylosuccinate lyase (AdSL) acts to create AMP by removing fumarate. In addition to the de novo pathway, AMP can also be produced via a salvage pathway. In the salvage pathway, adenine phosphoribosyltransferase (Aprt) converts adenine into AMP; alternatively, adenosine kinase (AdenoK) converts adenosine into AMP. Finally, AMP can be generated when adenylate kinase (Adk) catalyzes the conversion of two molecules of ADP into AMP and ATP (Fig. 5). Stenesen et al. found that heterozygous mutations of AMP biosynthetic enzymes extended Drosophila lifespan (Stenesen et al., 2013). The lifespan of isogenic male and female AdSS heterozygous mutants was approximately 20 % longer than in sibling controls. Similarly, flies heterozygous for AdSL (de novo AMP biosynthesis), AdenoK, Aprt (salvage pathway), and Adk2 were long-lived. All long-lived heterozygous mutant flies exhibited an increased AMP/ATP ratio and increased AMPK activity; when dominant-negative AMPK was expressed, the lifespan extension of AdSS heterozygous mutants was suppressed. Moreover, muscle- and fat body-specific AMPK overexpression alone increased lifespan, while muscle- and fat body-specific AMPK downregulation reduced lifespan. Stenesen et al. also demonstrated that the AMP/ATP ratio was also increased under DR. Dietary supplementation of adenine, a substrate for the AMP biosynthesis salvage pathway, rescued the lifespan extension in AdSS heterozygous mutants and reversed the longevity benefit associated with DR (Stenesen et al., 2013).
Fig. 5.
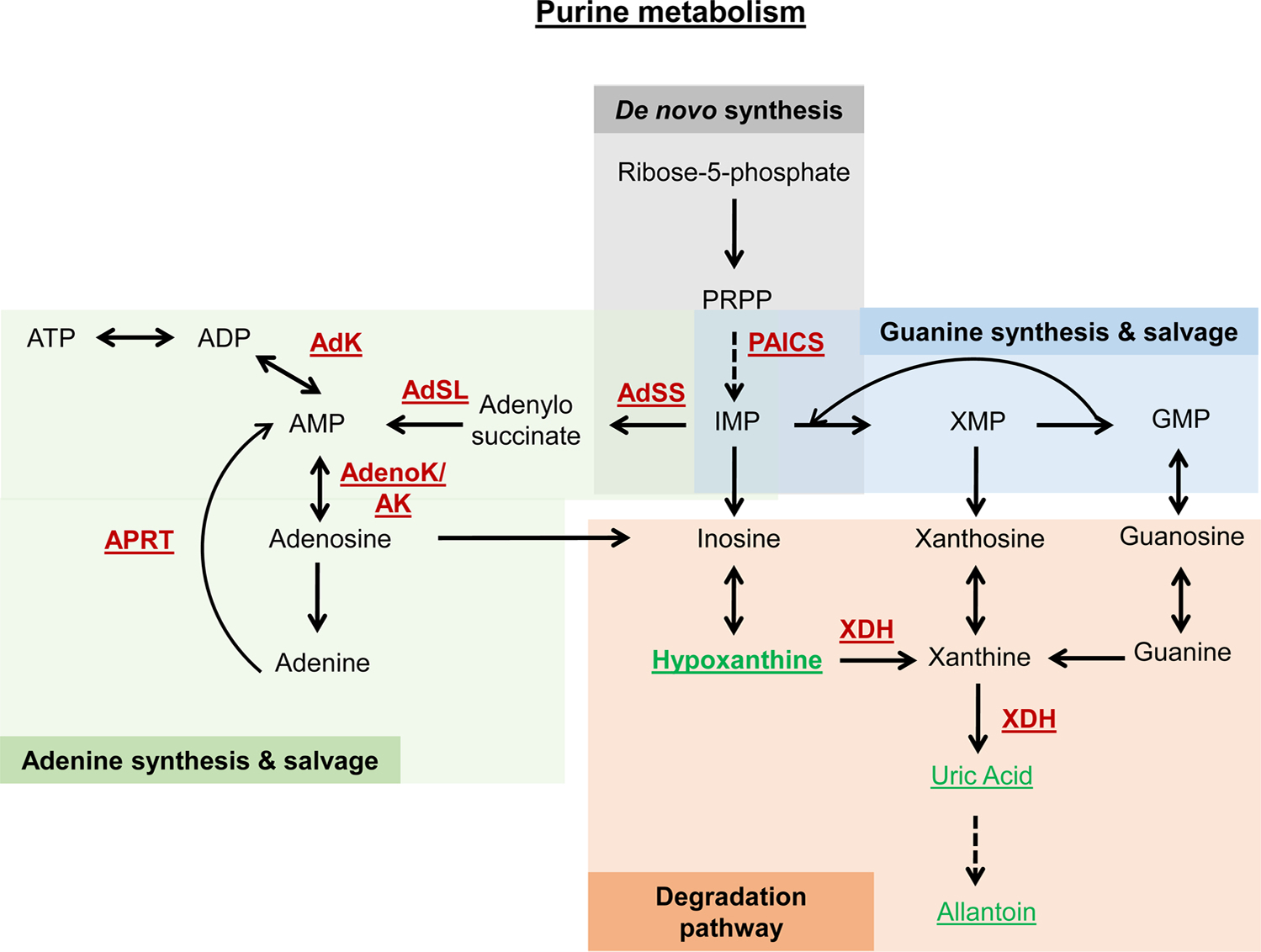
Schematic representation of purine metabolism. Underlined are metabolites and enzymes that were associated with lifespan extension. Red font color represents downregulation or depletion from food, while green font color represents overexpression or supplementation. Dashed line represents that multiple steps are involved. During de novo synthesis, ribose-5-phosphate is converted to inosine monophosphate (IMP), which can then be converted to adenine and guanine nucleotides. In worms, downregulation of PAICS increased lifespan. In flies, heterozygous mutations of adenylosuccinate synthetase (AdSS), adenylosuccinate lyase (AdSL), adenine phosphoribosyltransferase (Aprt), adenosine kinase (AdenoK), and adenylate kinase (AdK) increased lifespan. The degradation pathway converts the purine nucleotides into xanthine, which can then be metabolized to uric acid. Downregulation of xanthine dehydrogenase increased lifespan in flies, while supplementation with hypoxanthine, uric acid and allantoin increased lifespan in worms.
In agreement with the fly data, a large-scale RNAi screen conducted by Hamilton et al. identified 89 new genes that extend lifespan in C. elegans, including Xanthine Dehydrogenase (XDH)/F55B11.1, a key enzyme in purine degradation (Hamilton et al., 2005). Similarly, in a systematic RNAi longevity screen in worms, Suthpin et al. found that downregulation of B0286.3, the worm ortholog of phosphoribosylaminoimidazole carboxylase and phosphoribosylaminoimidazolesuccinocarboxamide synthase (PAICS), an enzyme involved in de novo biosynthesis of purine nucleotides, extended worm lifespan by ~15 % (Sutphin et al., 2017). Copes et al. tested whether an age-dependent decrease in purine levels might limit lifespan and found that supplementation with the purine derivative 10 mM hypoxanthine increased worm lifespan by 5% (Copes et al., 2015).
Uric acid (UA), an end-product of purine metabolism, is one of the most abundant antioxidant molecules and may also affect lifespan. A common metabolite in mammalian serum, it can scavenge peroxynitrite, nitric oxide, and hydroxyl radicals to prevent lipid peroxidation. Feeding worms with 2 mM of UA extended worm lifespan by ~15 % and enhanced the worms’ resistance to oxidative stress (Wan et al., 2020). Urate oxidase (UOX)/uricase, encoded by the Uox gene in mice, catalyzes the oxidation of UA to 5-hydroxyisourate. Mice lacking both copies of Uox had more than a 10-fold increase in UA levels and died early, while mice heterozygous for Uox had increased UA levels (the level of UA increase differed between young and old mice, with increases of 25 % and 500 %, respectively). Female mice heterozygous for Uox exhibited extended lifespan (Cutler et al., 2019). Humans have high levels of UA because they lack a functional urate oxidase (UOX) enzyme. Allantoin is a product of oxidation of uric acid (UA). Because humans lack UOX, the presence of allantoin in their plasma results from non-enzymatic oxidation. Using gene expression signatures, Calvert et al. identified allantoin as a CR mimetic. Feeding 250 μM allantoin to wild-type worms extended lifespan by ~22 %, but no lifespan effects were observed in long-lived eat-2 (model of CR) mutant worms (Calvert et al., 2016).
One-carbon (1C) / folate metabolism provides 1C units (methyl groups) for biosynthetic processes including purine and thymidine synthesis and homocysteine remethylation (Ducker and Rabinowitz, 2017). NAD-dependent methylenetetrahydrofolate dehydrogenase-methenyltetrahydrofolate cyclohydrolase (NMDMC/MTHFD2) is involved in the generation of both glycine and one-carbon units. Nmdmc overexpression enhanced lifespan and oxidative stress resistance in flies. Moreover, fat body-specific Nmdmc overexpression was enough for lifespan extension (Yu et al., 2015). In worms, inhibition of folate synthesis either with sulfamethoxazole (SMX), a sulfonamide drug that blocks folate synthesis, or by supplementing with mutant bacteria in which aroD had been deleted significantly extended lifespan (Virk et al., 2012).
2.4.2. Pyrimidine metabolism
In de novo pyrimidine synthesis, glutamine is converted to orotate through a series of four steps. Orotate can then be converted into ribonucleotides including UDP and CTP, which in turn can be converted through a series of reactions into thymine and cytidine, respectively (Fig. 6). To test the effects of intermediate metabolites from pyrimidine metabolism on the regulation of lifespan, Wan et al. fed C. elegans on a diet of heat-inactive E. coli supplemented with 0.5 mM thymine, β-aminoisobutyrate, orotate, uridine, cytidine, uracil or thymidine. Feeding worms with thymine, β-aminoisobutyrate, orotate, uridine or cytidine extended lifespan by 15.2, 9.69, 14.8, 9.83, or 7.87 %, respectively, whereas treatment with uracil or thymidine had no effect on lifespan (Wan et al., 2019). In the range from 0.05 mM to 10 mM, 2 mM thymine had the strongest effect, extending lifespan by 18 %. Interestingly, thymine, β-aminoisobutyrate, uridine, cytidine, or orotate failed to extend the lifespan of long-lived glp-1 worms. Wild-type worms treated with these pyrimidine metabolism intermediates had increased lipid storage (Wan et al., 2019). Similarly, Copes et al. found that supplementation with 10 mM cytidine increased worm lifespan by 11 % (Copes et al., 2015). In agreement with the lifespan extension by feeding metabolites from the pyrimidine metabolism pathway, manipulations of rate-limiting enzymes in the pyrimidine metabolism pathway were also found to affect worm lifespan. RNAi against dpyd-1, an ortholog of human dihydropyrimidine dehydrogenase (DPYD), the first and rate-limiting enzyme for the metabolism of thymine to 5,6-dihydrothymine, extended the lifespan by 13 %. Similarly, downregulation of upp-1, a uridine phosphorylase that catalyzes the reversible phosphorylation of uridine to uracil and ribose-1-phosphate, extended lifespan by 19 % (Wan et al., 2019). Further studies across different organisms could elucidate how pyrimidine metabolism might affect lifespan.
Fig. 6.
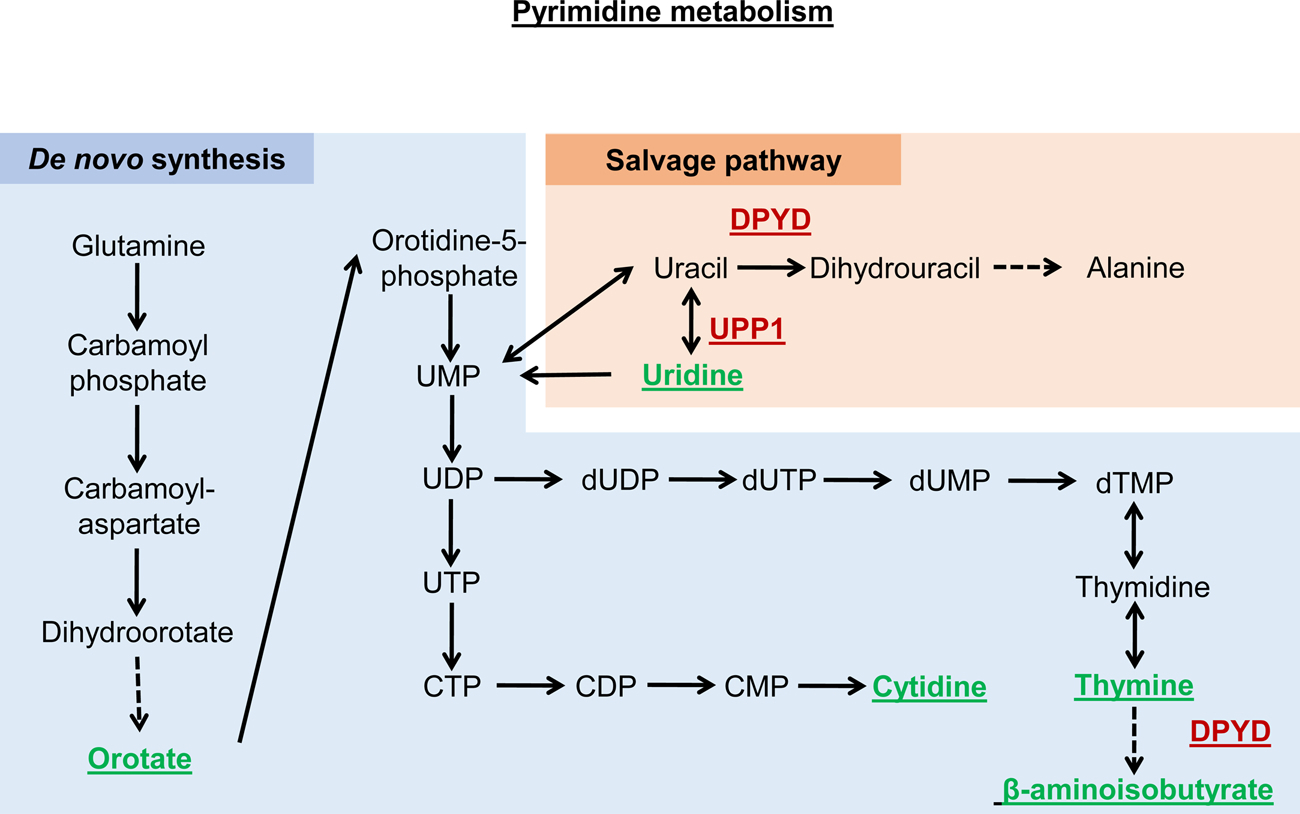
Schematic representation of pyrimidine metabolism. Underlined are metabolites and enzymes that were associated with lifespan extension. Red font color represents downregulation or depletion from food, while green font color represents overexpression or supplementation. Dashed line represents that multiple steps are involved. In de novo pyrimidine synthesis, glutamine is converted to orotate, which can then be converted to uridine nucleotides. Feeding worms with pyrimidine synthesis intermediates including orotate, uridine, thymine, cytidine, and β-aminoisobutyrate increased lifespan, while downregulating dihydropyrimidine dehydrogenase (DPYD) and uridine phosphorylase (UPP1) also increased lifespan.
2.5. Lipid metabolism
2.5.1. Overview of lipid metabolism
Lipids are a diverse class of molecules (fatty acyls, glycerolipids, glycerophospholipids, sphingolipids, sterol lipids, and others) important for energy storage, membrane formation, and signaling. Lipid metabolism plays a key role in the regulation of aging and longevity (Bustos and Partridge, 2017; Hou and Taubert, 2012; Johnson and Stolzing, 2019) (Fig. 7). Aging in humans is associated with a dramatic change in fat mass and tissue distribution; in particular, with age, lipids accumulate in non-adipose tissue like muscle. These changes are associated with multiple health complications (Cartwright et al., 2007). Too little or too much fat is associated with early mortality in rodents and humans, whereas leanness, or an optimal amount of fat, is associated with a longer lifespan. The plasma lipidomic profile of 11 mammalian species, with maximum lifespans ranging from 3.5–120 years, revealed species-specific lipid composition signatures that could accurately predict animal longevity (Jove et al., 2013). Similarly, analysis of transcriptional longevity signatures from 14 Drosophila species with lifespans ranging from 8 to 16 days (shorter-lived) to 52–62 days (longer-lived) identified that longer-lived flies were characterized by upregulated fatty acid metabolism (Ma et al., 2018a). Moreover, a lipid signature from a small subset of 20 lipid species in human plasma could discriminate between adult, aged and centenarian subjects (Jove et al., 2017). Bozek et al. measured more than 20,000 lipid compounds from 6 tissues of 35 species representing three mammalian clades: primates, rodents and bats. They determined lipid predictors of long lifespan that differed between neuronal and non-neuronal tissues and found higher content of saturated lipids in cell membranes of long-lived species (Bozek et al., 2017). Jobson et al. compared the nonsynonymous and synonymous evolution of −5.7 million codon sites across 25 species and found that genes involved in lipid composition were under increased selective pressure in long-lived species (Jobson et al., 2010). Gonzalez-Covarrubias et al. performed plasma lipidome analysis measuring 128 lipid species in 1526 middle-age offspring of nonagenarians (who were shown to have a life-long survival advantage) and 675 controls from the Leiden Longevity Study. Although they did not observe significant differences in males, they did observe 19 lipid species in females that were associated with familial longevity, and the longevity-associated lipid profile was characterized by a higher ratio of MUFA (monounsaturated) over PUFA (polyunsaturated) lipid species (Gonzalez-Covarrubias et al., 2013).
Fig. 7.

Schematic representation of lipid metabolism. Underlined are metabolites and enzymes that were associated with lifespan extension. Red font color represents downregulation or depletion from food, while green font color represents overexpression or supplementation. Dashed line represents that multiple steps are involved. (A) In fatty acid metabolism, triglycerides can form saturated or unsaturated fatty acids. Higher levels of mono-unsaturated fatty acids (MUFAs) were associated with increased lifespan. Downregulation of diacylglycerol O-acetyltransferase (DGAT), elongases (ELO), and a desaturase converting MUFAs to PUFAs (FAT), as well as upregulation of the lipase LIPL was associated with lifespan extension. (B) In sphingolipid metabolism, palmitoyl-CoA is converted to ceramides, which can then form sphingomyelins. Downregulation of several enzymes, including serine palmitoyltransferase (SPT), sphingomyelinase (SMase), sphingomyelin synthase (Smsynthase), glucosylceramide synthase (PDMP), ceramidase, and ceramide synthase was associated with lifespan increase. (C) In diacylglycerol metabolism, diacylglycerol can be converted to 2-arachidonoyl-sn-glycerol (2-AG) or phosphatidic acid (PA). PA promotes TOR activity. Diacylglycerol lipase (DAGL-1) overexpression and diacylglycerol kinase (DGK) downregulation were associated with lifespan extension in worms.
Fatty acids are important energy fuels for the cell; they can be degraded via β–oxidation to generate acetyl-CoA and subsequently used to generate ATP via the TCA cycle (Houten and Wanders, 2010). The main regulators of lifespan and aging, such as AMPK, daf-2 deficiency, and DR, have been also shown to regulate lipid metabolism. However, these interventions can have opposite effects on lipid metabolism. Long-lived daf-2 (Perez and Van Gilst, 2008) or glp-1 (O’Rourke et al., 2009) mutant worms have increased lipid levels, whereas long-lived eat-2 mutant worms (CR model) have decreased lipid levels (Heestand et al., 2013).
AMPK is one of the central regulators of aging. One of the first identified AMPK targets was acetyl-CoA carboxylase (ACC), the enzyme that converts acetyl-CoA to malonyl-CoA. Malonyl-CoA is a critical precursor of fatty acid biosynthesis and can also inhibit carnitine palmitoyl-transferase-1, an enzyme required in mitochondrial fatty acid uptake. AMPK inhibits ACC and lowers the concentration of malonyl-CoA, which results in increased fatty acid oxidation (FAO) capacity and inhibited de novo lipogenesis. AMPK is therefore expected to coordinate the partitioning, or redistribution, of fatty acids between oxidative and biosynthetic pathways (Houten and Wanders, 2010). In addition, CR and inhibition of insulin signaling promote FAO (Bruss et al., 2010; Xu et al., 2012). Neutral lipids such as sterol esters and triacylglycerols (TAGs) are stored in lipid droplets (LDs). TAG is composed of a glycerol backbone esterified to three fatty acids. Intestinal monoacylglycerides (MAG), derived from the hydrolysis of dietary fats, can serve as substrates for the synthesis of triglycerides. Diacylglycerol O-acyltransferase-1 (Dgat1) catalyzes the conversion of diacylglycerol and fatty acyl-CoA to TAGs. Female Dgat1-deficient mice were protected from an age-related increase in body fat, tissue TAGs, and inflammation in white adipose tissue, and were characterized by a −25 % increased lifespan (Streeper et al., 2012). In contrast, yeast cells that are genetically engineered to store more TAG (via deletion of lipases Tgl3 and Tgl4 alone or in combination, or overexpression of TAG biosynthetic acyltransferase Dga1p), live significantly longer (CLS), whereas diminishing TAG synthesis (via a simultaneous deletion of the two TAG biosynthetic acyltransferases, Dga1p and Lro1p) shortened lifespan (Handee et al., 2016).
2.5.2. Lipolysis
When cells need lipids to generate energy or synthesize membranes, two pathways are activated to mobilize substrates from LDs: lipolysis or lipophagy. Lipases cleave individual fatty acyl chains from TAG, DAG, MAG, and phospholipids (Farese and Walther, 2009). A triglyceride lipase, LIPS-7, is activated in the long-lived ctbp-1(ok498)-mutant worms and its downregulation results in complete suppression of lifespan extension associated with ctbp1(ok498), whereas its downregulation does not affect lifespan in wild-type worms (Chen et al., 2009).
In C. elegans, ablation of germline stem cells (GSCs) either by laser or genetic mutation (glp-1 mutant worms) protects worms against stress and extends lifespan (Hsin and Kenyon, 1999). A lysosomal triglyceride lipase, K04A8.5 / LIPL-4, is activated in the long-lived glp-1 mutants and is required for its longevity, but downregulation of LIPL-4 does not reduce lifespan in wild-type worms. However, constitutive expression of K04A8.5 / LIPL-4 specifically in the intestine led to 24 % longer lifespan than in control siblings (Wang et al., 2008). Similar to LIPL-4, lips-17 (triacylglycerol lipase) and fard-1 (fatty acyl reductase) were upregulated in long-lived glp-1 mutant worms, and their downregulation shortened the lifespan to that of wild-type worms (McCormick et al., 2012). Steinbaugh et al. found that SKN-1/Nrf is required for increased stress resistance and lifespan extension caused by ablation of GSCs (via either glp-1 mutation or laser ablation) and activated diverse lipid metabolism genes, including lipl-3, acs-10, cpt-3, and ech-9. Ablation of GSCs led to the accumulation of high levels of yolk lipoproteins and activation of SKN-1/Nrf prevented excessive lipid accumulation. Interestingly, addition of oleic acid was enough to induce SKN-1/Nrf (Steinbaugh et al., 2015). LIPL-4 has also been shown to be transcriptionally upregulated in worms. This physiological response to fasting led to an enrichment of ω-3/6, i.e. 20-carbon ω-3 - eicosapentaenoic acid (EPA), 20-carbon ω-6 - arachidonic acid (AA) polyunsaturated fatty acids (PUFAs). Unlike mammals, which require dietary PUFAs to maintain health, nematodes have all of the enzymes necessary for PUFA biosynthesis. Dietary supplementation with ω-6, i.e. arachidonic acid or its precursor di-homo-γ-linoleic acid (DGLA), activated autophagy in worms and human cells, and increased worm lifespan in well-fed animals. Moreover, this lifespan extension was dependent on the essential autophagy genes bec-1, atg-16.2, and lgg-1 (O’Rourke et al., 2013). In addition to EPA, the ω-3 fatty acids also include α-linolenic acid (ALA) and docosahexaenoic acid (DHA). ω −3 fatty acids are essential in humans and mainly received from plants (ALA) and fish/fish oil (EPA and DHA).
In accordance with the significance of ω-3 fatty acids, greater fish intake is linked to a greatly reduced rate of hypertension and atherosclerotic cardiovascular disease. However, in the NIA Interventions Testing Program, the treatment of mice with fish oil did not extend mouse lifespan (Strong et al., 2016). Qi et al. demonstrated that the treatment of worms with the 18-carbon ω-3 fatty acid α-linolenic acid (ALA) increases the lifespan of treated worms by 30 % and this lifespan increase was dependent on the activation of NHR-49/PPARα, SKN-1/Nrf2, and increased β-oxidation (Qi et al., 2017). ω-3 fatty acids can undergo both spontaneous and enzymatically mediated oxidation reactions that lead to the production of many distinct oxylipin molecules. Oxylipins represent biologically active molecules that often act via unique receptors. The beneficial effects of ω-3 fatty acids in humans can be associated with the production of oxylipins (Gabbs et al., 2015). Qi et al. demonstrated that the product of nonenzymatic oxidation of ALA, 9(S)-HpOTrE, enhanced the effect of ALA on lifespan extension (Qi et al., 2017). In addition to AA and ALA, treatment with the fatty acid 10-hydroxy-2-decenoic acid (10-HDA), a component of Royal Jelly that is produced by glands of worker honeybees Apis mellifera L., extended worm lifespan by −12 % (Honda et al., 2011).
In different organisms, longevity is tightly linked to the epigenetic state and changes of epigenetic state are a hallmark of aging (Lopez-Otin et al., 2013). In worms, a deficiency in H3K4me3 methyltransferase promotes fat accumulation with specific enrichment of mono-unsaturated fatty acids (MUFAs)—palmitoleic acid, cis-vaccenic acid, and oleic acid—while SFAs and PUFAs remained mostly unchanged. Deficiency of the COMPASS chromatin complex, which trimethylates lysine 4 on histone H3 (H3K4me3), leads to lifespan extension (Greer et al., 2010). Moreover, dietary supplementation of MUFAs was sufficient to extend worm lifespan by 15–20 % and increased total fat accumulation, while dietary supplementation of the PUFAs linoleic and alpha-linolenic acid did not significantly affect lifespan. Additionally, overexpression of the oleic acid-synthesizing enzyme FAT-7 in the intestine increased fat accumulation and extended lifespan, which was not further changed by oleic acid supplementation (Han et al., 2017b). Interestingly, fat-7 together with other lipid metabolism-related genes such as dod-9/acs-17, dod-12/acdh-1, gpd-2 and dod-10/stdh-2 were upregulated in long-lived daf-2-mutant worms, and their downregulation shortened the lifespan of daf-2 mutants more significantly than the lifespan of wild-type worms (Murphy et al., 2003).
2.5.3. Lipid synthesis
Synthesis of FAs de novo involves acetyl-CoA carboxylase (ACC) and fatty acid synthase (FAS). The fatty acid product released from FAS is palmitate (16 carbons, saturated). FA elongase enzymes function to elongate the FA chain. Elongation of fatty acids involves condensation of fatty acyl-CoA groups with either acetyl-CoA or malonyl-CoA, which results in FAs that are longer by two carbons. FA desaturase enzymes catalyze the conversion of saturated FAs (SFA) into MUFAs. In C. elegans, knockdown of either elo-1 or elo-2 FA elongases but not of elo-5 elicited moderate and significant extensions of lifespan by 11 % and 8%, respectively. Moreover, the combined knockdown of both elongases was more effective in lifespan extension (by 15 %) than the depletion of either elongase gene alone. In addition, downregulation of FA desaturase fat-4 led to a lifespan extension of 25 % (Shmookler Reis et al., 2011). As discussed earlier, K04A8.5 / LIPL-4, is activated in the glp-1 mutant and is required for its longevity (Wang et al., 2008). Goudeau et al. found that the extended lifespan of glp-1 mutants also required the activation of FAT-6/SCD1 via the activation of NHR-80/HNF4, independent of the activation of K04A8.5 / LIPL-4. FAT-6/SCD1 is a stearoyl-CoA-Δ9-desaturase that converts stearic acid to OA. In agreement with the activation of FAT-6/SCD1 observed in glp-1 mutant worms, the levels of OA, as well as the stearic/oleic acid ratio, were also specifically increased (Goudeau et al., 2011). In contrast to Han et al. study (Han et al., 2017b), dietary supplementation of wild-type worms with OA did not extend lifespan (Goudeau et al., 2011).
2.5.4. Lipid transport
Fatty acid-binding proteins (FABPs) are lipid chaperones that shuttle lipid molecules between cellular compartments for different functions. The amount of C. elegans FABP LBP-8 was increased in the intestine of K04A8.5/lipl-4 transgenic worms and RNAi-mediated depletion of lbp-8 or lbp-8 deletion mutant suppressed the lifespan extension in lipl-4 transgenic animals without affecting lifespan in wild-type worms (Folick et al., 2015). Moreover, worms constitutively expressing lbp-8 had a 30 % increase in lifespan and improved maintenance of physical activity in old age compared to wild-type worms. The lifespan extension by lbp-8 required a NLS motif, suggesting that LBP-8 may function as a lysosomal lipid chaperone transferring lipid signals to the nucleus. Nuclear hormone receptors NHR-49 and NHR-80 were both required for lipl-4– and lbp-8–mediated longevity. Moreover, LBP-8 has been found to bind the C20 fatty acid oleoylethanolamide (OEA), which is an N-acylethanolamine fatty acid derivative, and the OEA analog KDS-5104. Direct treatment of worms with KDS-5104 prolonged lifespan and improved physical activity maintenance in aged animals (Folick et al., 2015). Ramachandran et al. further demonstrated that LIPL-4/LBP-8 lysosomal signaling actively promoted mitochondrial β-oxidation, reduced complex II activity, and promoted the mtROS production and activation of JNK signaling that were necessary for LIPL-4/LBP-8 longevity-promoting effects. In addition, activation of mitochondrial β-oxidation via acyl-CoA synthetase (acs-2) overexpression in the intestine was sufficient to prolong worm lifespan (Ramachandran et al., 2019).
Direct modulation of FAO via the overexpression of CG6783/fabp and CG13890 extended Drosophila lifespan (Lee et al., 2012). CG6783/fabp encodes the fatty-acid-binding protein, which facilitates the intracellular movement of fatty acids and promotes FAO (Smathers and Petersen, 2011). CG13890 encodes dodecenoyl-CoA delta-isomerase (DCI), which is localized in the mitochondria and catalyzes the degradation of long-chain fatty acids during FAO (Houten and Wanders, 2010). In addition to increased lifespan, overexpression of CG6783/fabp and CG13890 increased resistance to oxidative stress and starvation (Lee et al., 2012). In mice, combined deficiency of Fabp4 and Fabp5 promoted metabolic healthspan via attenuation of age-related body weight gain and deterioration of glucose tolerance, insulin sensitivity and hepatosteatosis, but without any extension of lifespan (Charles et al., 2017).
Similar to CG6783/fabp, another lipid-binding protein that is responsible for transporting lipids, apolipoprotein D (ApoD), is involved in the regulation of aging and stress response in flies (Muffat et al., 2008). Human ApoD is elevated during aging and a variety of pathological conditions, including Alzheimer’s disease (AD), stroke, schizophrenia, and bipolar disorder (Kalman et al., 2000; Thomas et al., 2001). Elevation of ApoD during aging and neurodegeneration can be mediated by a compensatory response. ApoD can carry membrane lipids, such as arachidonic acid and sterols, and may be involved in the clearance and/or repair of damaged membranes as well as quenching harmful material released by neurons and glial cells in response to damage (Morais Cabral et al., 1995; Muffat et al., 2008). The Drosophila genome contains three Lipocalin genes: NLaz, GLaz, and karl. Overexpression of a fly ortholog of ApoD, Glial Lazarillo (Glaz), protected against hyperoxia and starvation, and extended lifespan at 29 °C by 18 % (Walker et al., 2006). Similarly, overexpression of human ApoD (hApoD) in flies extended lifespan by 40 % and also protected flies against hyperoxia, paraquat treatment (oxidative stress), and heat stress. Aging is accompanied by the accumulation of lipid peroxides, which are formed when free radicals react with membrane and storage lipids. In addition to lifespan extension, hApoD overexpression in flies prevented the age-dependent increase of lipid peroxide burden (Muffat et al., 2008). NLaz transcription is induced by oxidative stress and by JNK signaling in the fat body, and its induction mediates the antagonistic interaction between JNK and insulin signaling in flies. Overexpression of NLaz both ubiquitously and in the fat body increased resistance to paraquat, and ubiquitous NLaz overexpression increased lifespan (Hull-Thompson et al., 2009). In mice, apolipoprotein E (apoE) deficiency is characterized by altered lipoprotein metabolism, advanced atherosclerosis, and reduced lifespan (Moghadasian et al., 2001). Several genome-wide association (GWAS) and whole-genome sequencing (WGS) studies linked polymorphisms in ApoE genes with human lifespan (Abondio et al., 2019). Polymorphism in the apolipoprotein C3 gene (APOC3) has been associated with favorable lipoprotein profile, outcomes of age-related disease, and lifespan (Atzmon et al., 2006). Brejning et al. identified that downregulation of NDG-4, NRF-5, and NRF-6 genes that function in the lipid transport pathway increased resistance to stress and extended lifespan. Moreover, when ndg-4 was downregulated together with insulin/IGF-1 signaling, the lifespan was increased almost fivefold (Brejning et al., 2014). While downregulation of ndg-4 in worms extends lifespan, flies mutant for the drop-dead gene (fly ortholog of ndg-4) are characterized by striking early death (Rogina et al., 1997).
Yolk lipoproteins are lipid transfer complexes for transporting phospholipids and cholesterol to oocytes to provide lipids used during embryonic development. In C. elegans, six isoforms of the yolk lipoprotein VIT/vitellogenin (vit-1 to −6) are synthesized in the intestine and then transported to oocytes; of these, vit-1 to vit-5 are the most similar to human APOB (apolipoprotein B) (Seah et al., 2016). Murphy et al. analyzed gene expression changes regulated by DAF-2 and DAF-16 and found that vit-2 and vit-5 were downregulated under daf-2(−) conditions and upregulated in daf-16(−) animals. Moreover, downregulation of vit-2 or vit-5 increased the lifespan of daf-2(+) worms (Murphy et al., 2003). A study from a different group also found that transcription of the complete vitellogenin (vit) gene family (vit-1 to vit-6) was decreased in daf-2 mutant worms (Halaschek-Wiener et al., 2005). Seah et al. found that overexpression of VIT/vitellogenin (vit-2) had no effect on the lifespan of wild-type worms but it reduced the lifespan of long-lived glp-1 and daf-2 mutant worms, decreased their intestinal lipid storage and inhibited expression of autophagy-related and lysosomal acid lipase genes. The suppression of vitellogenesis using RNAi against vit genes (vit-1/2, −3, −4, and −5) increased lifespan by 16–40%, increased lipid storage, and stimulated autophagy and lysosomal lipolysis. Moreover, inhibition of autophagy (Atg18), inhibition of NHR-49 and NHR-80, and inhibition of lipogenic genes (lipl-3 and lipl-4) prevented lifespan extension conferred by vit RNAi (Seah et al., 2016).
In agreement with the role for lipid metabolism in regulation of lifespan, flies heterozygous for a null allele of Enigma (Egm), the mitochondrial enzyme responsible for β-oxidation of fatty acids, had a lifespan on average 19.5 % greater than control flies without a loss in reproductive potential. Similar to CG6783/fabp, CG13890, and ApoD, Enigma mutant flies demonstrated a significantly increased resistance to paraquat (oxidative stress) (Mourikis et al., 2006). Mitochondrial β-oxidation produces acetyl-CoA by degrading fatty acids via a cascade of four reactions: dehydration, hydration, oxidation, and thiolysis. Mitochondrial trifunctional protein (MTP) consists of four MTP1d6fc; and four MTP1d6fd; subunits and catalyzes the last three steps in the β-oxidation of long-chain fatty acids. Flies deficient for either Mtp1d6fc; or Mtp1d6fd; are viable but had a shortened lifespan, defective locomotor activity, reduced fecundity, and abnormal lipid catabolism (Kishita et al., 2012). Upon dietary restriction, flies shift their metabolism toward increasing fatty-acid synthesis and breakdown, and inhibition of this shift via downregulation of Acetyl-CoA Carboxylase (dACC) in muscle tissue inhibited lifespan extension upon DR. In addition, overexpression of adipokinetic hormone (dAKH), the functional ortholog of glucagon, enhanced fat metabolism and lifespan (Katewa et al., 2012).
2.5.5. Butyrate and ketone bodies
Butyrate (butanoate), a natural product of bacteria, is a short-chain fatty acid (SCFA) that functions as an HDACs class I and class II inhibitor. Feeding flies (Kang et al., 2002; Vaiserman et al., 2012; Zhao et al., 2005) or worms (Zhang et al., 2009) with sodium butyrate increases their lifespan. Feeding butyrate to mice from 16 to 26 months of age reduced percent fat mass and prevented hind limb muscle loss (Walsh et al., 2015). Dietary restriction in the form of calorie or carbohydrate deprivation leads to ketogenesis and serum ketone elevation. Under ketogenic diet conditions, the liver is the main site of fatty acid β-oxidation of serum-derived fatty acids, which generates ketone bodies (acetoacetate, acetone, and β-hydroxybutyrate) that can subsequently serve as energy substrates for the body as a whole and, most importantly, for the brain. In the liver, fatty acids are first metabolized to acetyl-CoA via mitochondrial β-oxidation; then, acetyl-CoA condenses with acetoacetyl-CoA by HMGCS2 to form HMG-CoA from which acetoacetate is liberated by HMG-CoA lyase (HMGCL). Acetoacetate is the precursor of the two other circulating ketone bodies, acetone and β-hydroxybutyrate. In target tissues, β-hydroxybutyrate is broken down in mitochondria into acetoacetate by β-hydroxybutyrate dehydrogenase 1 (BDH1), generating NADH. Acetoacetate is further metabolized to acetyl-CoA via acetoacetyl-CoA, which is subsequently metabolized in the TCA cycle to produce NADH and FADH2 (Newman and Verdin, 2017).
Similar to butyrate, β-hydroxybutyrate is an inhibitor of histone deacetylases (HDACs). Dietary restriction benefits can be potentially linked to the elevated levels of ketone bodies (Veech et al., 2017). Addition of 20 mM D-β-hydroxybutyrate to cultures of C. elegans extended lifespan by 20 %, delayed Alzheimer’s amyloid-beta toxicity, and decreased Parkinson’s alpha-synuclein aggregation; by contrast, L-β-hydroxybutyrate did not have any effects on lifespan. D-β-hydroxybutyrate lifespan extension was dependent on SIR-2.1, AMPK, DAF16, and SKN-1. Downregulation of HDACs hda-2 or hda-3 also increased lifespan and prevented lifespan extension by β-hydroxybutyrate (Edwards et al., 2014, 2015b). In mice, a cyclic isoprotein ketogenic diet started in middle age and alternated weekly with a control diet generated plasma beta-hydroxybutyrate levels similar to fasting (1–2 mM), reduced midlife mortality, and improved healthspan, but did not affect maximum lifespan (Newman et al., 2017). Similarly, starting a ketogenic diet (89 % kcal from fat) in C57BL/6 mice at 12 months of age significantly increased levels of circulating β-hydroxybutyrate, increased lifespan by 13.6 % compared to control diet (65 % kcal from carbohydrate), preserved physiological function, and decreased incidence of tumors; while mice on a low-carbohydrate diet (70 % kcal from fat) had intermediate lifespan (Roberts et al., 2017). In humans, food supplementation with β-hydroxybutyrate or sodium butyrate is associated with multiple health benefits (Cavaleri and Bashar, 2018). In a randomized clinical trial, 4 weeks of alternate-day fasting in humans improved markers of general health in healthy middle-aged humans such as reduced body weight, reduced lean and fat mass, improved cardiovascular markers, and increased levels of β-hydroxybutyrate (Stekovic et al., 2019).
Diacylglycerol (DAG) is an important lipid metabolic intermediate that can be hydrolyzed by DAG lipase (DAGL) to become 2-arachidonoyl-sn-glycerol (2-AG) or modified by DAG kinase (DGK), resulting in its conversion to phosphatidic acid (PA). Overexpression of diacylglycerol lipase (DAGL/inaE/dagl-1) or knockdown of diacylglycerol kinase (DGK/rdgA/dgk-5) in Drosophila or C. elegans extended lifespan and enhanced responses to oxidative stress (Lin et al., 2014). The mechanistic Target of Rapamycin (mTOR) is a serine/threonine protein kinase, which phosphorylates a diverse set of substrates to regulate numerous cellular and physiological processes serving as a central regulator of growth, metabolism, and aging (Parkhitko et al., 2014). Manipulation of DAGL and DGK activity resulted in altered levels of PA that in turn modulate the activity of TOR signaling (Lin et al., 2014).
2.5.6. Sphingolipids
Sphingolipids are a class of lipids with important roles as structural entities of biological membranes. In addition to their structural roles, sphingolipids, and in particular ceramide, are important bioactive signaling molecules (Hannun and Obeid, 2018). Sphingolipids have been shown to play an important role in aging and lifespan in multiple species including yeast, worms, and flies (Huang et al., 2014). The enzyme serine palmitoyltransferase (SPT) catalyzes the initial and rate-limiting step of sphingolipid synthesis. SPT uses pyridoxal phosphate (vitamin B6) as a cofactor in the decarboxylating transfer of palmitoyl-CoA onto serine to form sphingosine, which is converted into biologically inert dihydroceramides. Dihydroceramides can either be transformed into dihydrosphingomyelin or converted to biologically active ceramides by sphingolipid desaturase. Sphingomyelin is produced by de novo synthesis from ceramide or by desaturation of dihydrosphingomyelin. Sphingomyelinases (SMases) cleave sphingomyelin to generate ceramides and other bioactive metabolites including sphingosine-1-phosphate and gangliosides (Cutler et al., 2014).
A variety of studies point to the relevance of sphingolipids to aging. Cutler et al. found that the sphingomyelin composition was remodeled with age in worms with levels of C18:1, C22:0, and C24:0 being significantly higher in 11-day-old worms compared to young worms. They further showed that inhibitors of SPT (ISP-1), sphingomyelin synthase (D609), glucosylceramide synthase (PDMP), or neutral sphingomyelinase (Epoxyquinone G109) increased worm lifespan by 31 %, 25 %, 38 %, and 6%, respectively. In addition to taking a pharmacological approach, these authors further showed that downregulation of SPT (C23H3.4), dihydroceramide desaturase (Y54E5A.1), neutral/acidic ceramidase (W02F12.2), and glucosylceramide synthase (F20B4.6) resulted in a significant extension of lifespan by 33 %, 40 %, 40 %, and 40 %, respectively (Cutler et al., 2014). Acid sphingomyelinase (ASM) is a phosphodiesterase that hydrolyzes sphingomyelin to produce ceramide and phosphorylcholine. The C. elegans genome encodes three ASM homologs, asm-1, asm-2, and asm-3. Kim et al. found that asm-3--mutant worms or worms treated with asm-3 RNAi were 14–19% longer-lived. In addition, worms fed with asm-1 and asm-2 RNAi were 12 % and 10 % longer-lived, respectively. Combined inactivation of asm-3 and asm-1 or asm-3 and asm-2 further extended lifespan by 30 % and 28 %, respectively. Furthermore, chemical inhibition of ASM with FDA-approved drugs desipramine or clomipramine extended the lifespan of wild-type animals by 24 % or 14 %, respectively (Kim and Sun, 2012). Asm-3 was also identified in an RNAi screen in C. elegans for genes that promote resistance to paraquat and extend lifespan (Kim and Sun, 2007).
The C. elegans genome contains three ceramide synthase genes; hyl-1, hyl-2, and lagr-1. HYL-1 is required for the synthesis of ceramides and sphingolipids containing very long acyl-chains (≥C24), whereas HYL-2 is required for the synthesis of ceramides and sphingolipids with shorter acyl-chains (≤C22). Functional loss of HYL-2 decreased lifespan by 16.7 %, whereas the loss of HYL-1 or LAGR-1 did not affect lifespan. However, simultaneous loss of HYL-1 and LAGR-1 functions increased lifespan by 21.4 %, increased resistance to heat, and reduced feeding and reproduction. Furthermore, this lifespan extension required a functional autophagy pathway, was dependent on DAF-16 and SKN-1, and was partly dependent on PHA-4 (Mosbech et al., 2013). Mosbech et al. did not observe an effect of hyl-1 downregulation on lifespan, and Tedesco et al. demonstrated that although RNAi against hyl-1 extended worm lifespan by 15 %, deletion mutation of hyl-1 had no effect on lifespan (Tedesco et al., 2008). Interestingly, similar to worms, both genetic and pharmacological inhibition of sphingolipid synthesis increased yeast lifespan (D’Mello et al., 1994; Huang et al., 2012).
Ceramidases catalyze the hydrolysis of ceramides to generate sphingosine and fatty acids. Dacer, encoded by the bwa (brainwashing) gene, is a Drosophila alkaline ceramidase. Inactivation of Dacer has been shown to elevate levels of most ceramide species, increase lifespan by 50 % and increase resistance to oxidative stress (Yang et al., 2010). Interestingly, although inactivation of bwa/Dacer elevates levels of ceramide and causes an abnormality in the structure of the mushroom body in Drosophila, it also extended lifespan. Serum lipidomics in centenarians revealed unique changes in lipids biosynthesis and revealed phospho/sphingolipids as putative markers and biological modulators of healthy aging in humans (Montoliu et al., 2014).
2.5.7. N-acylethanolamines
N-acylethanolamines (NAEs) are lipid-derived signaling molecules that participate in endocannabinoid signaling. In worms, NAEs are degraded by fatty acid amide hydrolase (FAAH), which is encoded by faah-1, and synthesized by N-acyl-phosphatidylethanolamine-specific phospholipase D (NAPE-PLD). Lucanic et al. found that manipulation of both enzymes affect lifespan in worms. Specifically, they found that DR reduces NAEs and that worms overexpressing faah-1 in the pharynx showed increased adult lifespan in the presence of abundant food but not under dietary restriction. Further, fat-4 mutants show reduced NAE levels and extended lifespan, consistent with its opposing effects on NAE levels. Their data suggest that NAEs could provide a signal of nutrient availability and energy balance in worms further supported by the fact that supplementation with NAEs inhibits dietary restriction-induced lifespan extension in wild-type worms and suppresses lifespan extension in a TOR pathway mutant (Lucanic et al., 2011).
2.5.8. Sterol metabolites
Another class of lipids, sterol metabolites (oxysterols, bile acids, and steroids), are critical signaling molecules that regulate metabolism, development, and lifespan (Wollam and Antebi, 2011). The extended lifespan of worms with ablated GSCs requires DAF-16, DAF-12, and a lipophilic hormone that activates DAF-12. DAF-12 is a worm ortholog of vertebrate vitamin D and liver X receptors and controls the choice between reproductive growth and dauer diapause during development, while also regulating longevity in response to signals from the gonad during adulthood. Longevity is regulated by DAF-12 and its endogenous ligands, 3-keto bile acid-like steroids called dafachronic acids (Gerisch et al., 2007; Motola et al., 2006; Yamawaki et al., 2010). Dafachronic acid, DAF-12 and biosynthetic enzymes such as daf-36/Rieske-like oxygenase (Wollam et al., 2011) and dhs-16/3-hydroxysteroid dehydrogenase (Wollam et al., 2012) that are required for the production of dafachronic acid, facilitate DAF-16 nuclear localization, and contribute to the extended lifespan of germline-less worms.
Another hormonal steroid linked to the regulation of lifespan—pregnenolone (3β-hydroxy-pregn-5-en-20-one; PREG), which is present in both worms and humans—was upregulated in long-lived glp-1 mutant worms. Moreover, supplementation of pregnenolone to worms extended lifespan by 15–20%, but it could not further extend lifespan of glp-1 mutant worms (Broue et al., 2007).
Bile acids are endogenous products of cholesterol catabolism. Although treatment with the bile acid UDCA exhibited some beneficial effects on healthspan in mice and humans; in a study by the NIA Interventions Testing Program, treatment of mice with UDCA did not significantly extend lifespan (Strong et al., 2016).
2.5.9. Ascarosides
Ascarosides are derivatives of the dideoxysugar ascarylose, which is linked to fatty acid-like side chains of varying lengths. C. elegans excrete a variety of ascarosides, which regulate different aspects of worm biology, including but not limited to the development, mating and olfactory learning (Ludewig and Schroeder, 2013). Feeding with either of two endogenously produced ascarosides, ascr#2 and ascr#3, increased worm lifespan by 17 % and 21 %, improved survival under oxidative stress, and required SIR-2.1 for these effects (Ludewig et al., 2013).
2.6. AMP-activated protein kinase (AMPK) and Metformin
AMP-activated protein kinase (AMPK) is the principle energy sensor that functions to maintain cellular energy homeostasis. When cellular energy is low, AMPK is activated and targets a variety of physiological processes to increase energy production and coordinately decrease ATP usage, thereby transforming the metabolic state from energy-consuming to energy-generating and energy-storing. AMPK also integrates key energetic metabolic pathways related to longevity (Burkewitz et al., 2014; Hardie et al., 2016). Increased gene dosage of aak-2 extends worm lifespan and contributes to lifespan extension by insulin/IGF-1 signaling (Apfeld et al., 2004). DR in worms significantly increases the AMP/ATP ratio and lifespan extension by DR requires aak-2 (C. elegans catalytic subunits of AMPK). Increasing expression of aak-2 in C. elegans increases lifespan by 13 % and mimics DR in well-fed animals (Greer et al., 2007). Both AMPK and DR promote longevity in worms via maintaining mitochondrial metabolism and network homeostasis and blocking of mitochondria fusion/fission blocks AMPK- and DR-extended lifespan (Burkewitz et al., 2015; Weir et al., 2017). As mentioned above, muscle- and fat body-specific AMPK overexpression increased Drosophila lifespan, while muscle- and fat body-specific AMPK RNAi reduced lifespan (Stenesen et al., 2013). Furthermore, upregulation of AMPK in the adult intestine and nervous system also prolonged Drosophila lifespan and caused an organism-wide response to tissue-specific AMPK activation (Ulgherait et al., 2014). Besides manipulations of AMPK itself, AMPK is also frequently activated as a result of different pro-longevity manipulations, such as treatment of worms with 2-DG (Schulz et al., 2007), while AMPK downregulation abolishes these lifespan extension effects. Other examples have been extensively discussed throughout the review.
Metformin is a widely prescribed FDA-approved oral antidiabetic drug that has been found to target several molecular mechanisms of aging (Kulkarni et al., 2020). One of the main effects of metformin is inhibition of mitochondrial complex I, which leads to a change in the AMP/ATP ratio and AMPK activation. Metformin demonstrated protective effects against several age-related diseases in humans and is currently in the Targeting Aging with Metformin (TAME) trial, which is aimed at delaying aging in humans (Barzilai et al., 2016).
Interestingly, although feeding metformin to adult flies resulted in robust activation of AMPK and reduced lipid stores, it did not increase lifespan in either male or female flies (Slack et al., 2012). One explanation for why metformin does not extend lifespan in Drosophila could be complications attributable to host-microbiota interactions. Indeed, results from worms support the idea that metformin in food impacts microbiota. C. elegans co-cultured with Escherichia coli as a food source after treatment with metformin had an extended lifespan. However, metformin treatment did not extend C. elegans lifespan in the absence of bacteria, when bacteria are metabolically impaired, or when bacteria develop resistance to the growth-inhibitory effects of metformin. Metformin inhibited folate production and methionine metabolism in bacteria, leading to changes in methionine metabolism and a decrease in SAM levels in the worms (Cabreiro et al., 2013). Pryor et al. identified two transcriptional regulators of metformin-E. coli interactions: Crp, a master regulator of carbon metabolism, and ArgR, a master regulator of nitrogen metabolism. Both Crp and ArgR were required for the increased worm longevity induced by metformin. Moreover, Crp overexpression in E. coli extended worm lifespan in a dose-dependent manner. Similarly, in flies, they showed that metformin extended Drosophila lifespan in a dose-dependent manner when the fly gut was colonized with control OP50 E. coli but not in germ-free flies or when flies were colonized with Crp-mutant OP50 E. coli. They further identified a bacterial metabolite, agmatine, and the enzyme agmatinase, which is encoded by SpeB, as regulated by Crp, and found that feeding agmatine to worms or Drosophila extended lifespan of both in a dose-dependent manner (Pryor et al., 2019).
With regards to rodent studies, metformin has been shown to extend the lifespan of short-lived and tumor-prone HER2/neu mice by 8% (Anisimov et al., 2005). Also, long-term treatment with metformin (0.1 % w/w in diet) starting at middle age extended lifespan by ~6% and extended healthspan in male C57BL/6 mice (Martin-Montalvo et al., 2013). In the NIA Interventions Testing Program, treatment of mice with metformin alone at a dose of 0.1 % in the diet did not significantly extend lifespan; however, when combined with rapamycin, metformin robustly extended lifespan and exhibited an added benefit to rapamycin as compared with rapamycin alone (Strong et al., 2016).
In summary, AMPK activation was associated with the majority of manipulations of metabolic pathways that led to an extension of lifespan across different species, and AMPK downregulation abrogated lifespan benefits of these manipulations. The TAME trial will pave a road for future investigation of prospective small molecule inhibitors and metabolites for delaying aging in humans.
In summary, lipids represent a wide class of diverse molecules that are associated with aging and have been associated with lifespan regulation across different species.
3. Conclusion
Although several dozen enzymes and metabolites belonging to different metabolic pathways have been discovered to play important roles in the regulation of lifespan, there are still multiple open questions that will have to be addressed to gain a full picture of the interplay between metabolism and aging.
How do we predict which lifespan-extending manipulations performed in different model organisms would be beneficial in humans?
For example, while mutations in different subunits of the ETC extend lifespan in various organisms including worms, flies, and mice, mutations in these genes result in devastating diseases in humans.
What interplay exists between metabolic pathways and microbiota, and how does this impact lifespan?
Above, we discussed an example of the dramatic effects that microbiota can play in lifespan extension by metformin in worms and flies.
How can we differentiate between direct effects of metabolic pathway manipulation on lifespan versus indirect effects, for example, via altering proliferation, epigenetics or redox state?
Altering metabolism in one organ can prolong lifespan; however, this lifespan extension may be caused by the improved function of a specific organ rather than by altered metabolism of an organism as a whole. An example is the lifespan extension by preserving proliferative homeostasis in the Drosophila gut (Biteau et al., 2010); specific metabolic manipulations may restrict age-dependent proliferation of ISCs and indirectly extend organismal lifespan.
How can we predict which combinations of metabolic genes and/or pathway manipulations will have additive or synergistic effects on lifespan extension?
Simultaneous inactivation of two different subunits (nuo-6 and isp-1) of mitochondrial respiratory complexes has an additive effect on lifespan extension (Yang and Hekimi, 2010). The combination of daf-2 and rsks-1 mutations produces a 5-fold increase in longevity, which is much greater than the sum of the effects of the single mutations (Chen et al., 2013). Alternatively, could we use a metabolic signature of aging to predict additive/synergistic effects of different drugs on the lifespan extension? In flies, the combination of trametinib (MEK inhibitor), rapamycin (mTORC1 inhibitor), and lithium (GSK-3 inhibitor) additively increased longevity in Drosophila (Castillo-Quan et al., 2019). It would be interesting to know whether each of these drugs has a distinctive metabolic signature that can predict its effects on lifespan.
Would it be possible to expand the toolbox and create tools that might be used for a compartment-specific manipulation of metabolites in model organisms for testing their effects on the regulation of lifespan?
Metabolites play important and different roles in different subcellular compartments. Manipulations of rate-limiting enzymes within a given metabolic pathway would most likely directly or indirectly affect the levels of a specific metabolite in all compartments. Genetic tools that can directly manipulate levels of specific metabolites in specific compartments will open doors to new types of studies. Titov et al. utilized a NADH oxidase from L. brevis as a genetic tool for inducing a compartment-specific increase of the NAD+/NADH ratio in human cells (Titov et al., 2016). It would be interesting to investigate roles of organelle-specific functions of specific metabolites in the regulation of aging.
Would it be possible to create universal cross-species ‘metabolic clocks’?
A set of CpG sites, the so-called ‘epigenetic clocks’, have been developed to predict chronological and biological age in humans (Horvath, 2013) and is currently used for an early prediction of the effects of specific manipulations on lifespan. Development of comparable ‘metabolic clocks’ potentially may serve a similar role and complement or substitute epigenetic clocks and improve the precision with which we can predict biological age in various organisms.
Is there a unifying principle of lifespan extension among all manipulations of metabolic enzymes that result in lifespan extension?
We have reviewed how changes in organismal and organ-specific metabolomes are associated with aging and discussed over a hundred different perturbations of metabolic pathways that can result in lifespan extension (Table 1). The differences in experimental strategies applied in different organisms, as well as the complexity and interconnection of processes implicated in lifespan extension, make it difficult to identify a single unifying principle of lifespan extension. Also, different interventions may cooperate in lifespan extension, assuming they extend lifespan via different mechanisms, and we have very little information on the effects of combined inactivation of different genes. What is clear, however, is that much has been learned, and that common pathways relevant to lifespan extension are beginning to emerge that would further require systematic testing of all pro-longevity interventions in order to reveal common downstream players, their interactions between each other, and organismal functional response to these interventions.
Acknowledgments
We thank Damir Khabibullin (Brigham and Women’s Hospital and Harvard Medical School), Patrick Jouandin (Harvard Medical School), and Valentin Cracan (the Scintillon Institute and the Scripps Research Institute) for critical reading of the manuscript. This work was supported by NIAR00AG057792 (A.P.) and AFAR (N.P.). N.P. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Declaration of Competing Interest
The authors report no declarations of interest.
References
- Abondio P, Sazzini M, Garagnani P, Boattini A, Monti D, Franceschi C, Luiselli D, Giuliani C, 2019. The genetic variability of APOE in different human populations and its implications for longevity. Genes (Basel) 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvers AL, Fishwick LK, Wood MS, Hu D, Chung HS, Dunn WA Jr., Aris JP, 2009. Autophagy and amino acid homeostasis are required for chronological longevity in Saccharomyces cerevisiae. Aging Cell 8, 353–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RM, Bitterman KJ, Wood JG, Medvedik O, Sinclair DA, 2003. Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature 423, 181–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews ZB, Horvath TL, 2009. Uncoupling protein-2 regulates lifespan in mice. Am. J. Physiol. 296, E621–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anisimov VN, Berstein LM, Egormin PA, Piskunova TS, Popovich IG, Zabezhinski MA, Kovalenko IG, Poroshina TE, Semenchenko AV, Provinciali M, Re F, Franceschi C, 2005. Effect of metformin on life span and on the development of spontaneous mammary tumors in HER-2/neu transgenic mice. Exp. Gerontol. 40, 685–693. [DOI] [PubMed] [Google Scholar]
- Apfeld J, O’Connor G, McDonagh T, DiStefano PS, Curtis R, 2004. The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to lifespan in C. Elegans. Genes Dev. 18, 3004–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asadi Shahmirzadi A, Edgar D, Liao CY, Hsu YM, Lucanic M, Asadi Shahmirzadi A, Wiley CD, Gan G, Kim DE, Kasler HG, Kuehnemann C, Kaplowitz B, Bhaumik D, Riley RR, Kennedy BK, Lithgow GJ, 2020. Alpha-Ketoglutarate, an Endogenous Metabolite, Extends Lifespan and Compresses Morbidity in Aging Mice. Cell metabolism 32, 447–456 e446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafian H, Czibik G, Bellahcene M, Aksentijevic D, Smith AC, Mitchell SJ, Dodd MS, Kirwan J, Byrne JJ, Ludwig C, Isackson H, Yavari A, Stottrup NB, Contractor H, Cahill TJ, Sahgal N, Ball DR, Birkler RI, Hargreaves I, Tennant DA, Land J, Lygate CA, Johannsen M, Kharbanda RK, Neubauer S, Redwood C, de Cabo R, Ahmet I, Talan M, Gunther UL, Robinson AJ, Viant MR, Pollard PJ, Tyler DJ, Watkins H, 2012. Fumarate is cardioprotective via activation of the Nrf2 antioxidant pathway. Cell Metab. 15, 361–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atzmon G, Rincon M, Schechter CB, Shuldiner AR, Lipton RB, Bergman A, Barzilai N, 2006. Lipoprotein genotype and conserved pathway for exceptional longevity in humans. PLoS Biol. 4, e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avanesov AS, Ma S, Pierce KA, Yim SH, Lee BC, Clish CB, Gladyshev VN, 2014. Age- and diet-associated metabolome remodeling characterizes the aging process driven by damage accumulation. eLife 3, e02077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahadorani S, Cho J, Lo T, Contreras H, Lawal HO, Krantz DE, Bradley TJ, Walker DW, 2010a. Neuronal expression of a single-subunit yeast NADH-ubiquinone oxidoreductase (Ndi1) extends Drosophila lifespan. Aging Cell 9, 191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahadorani S, Hur JH, Lo T Jr., Vu K, Walker DW, 2010b. Perturbation of mitochondrial complex V alters the response to dietary restriction in Drosophila. Aging Cell 9, 100–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai H, Kang P, Tatar M, 2012. Drosophila insulin-like peptide-6 (dilp6) expression from fat body extends lifespan and represses secretion of Drosophila insulin-like peptide-2 from the brain. Aging Cell 11, 978–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balan V, Miller GS, Kaplun L, Balan K, Chong ZZ, Li F, Kaplun A, VanBerkum MF, Arking R, Freeman DC, Maiese K, Tzivion G, 2008. LiF.e span extension and neuronal cell protection by Drosophila nicotinamidase. J. Biol. Chem. 283, 27810–27819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros MH, Bandy B, Tahara EB, Kowaltowski AJ, 2004. Higher respiratory activity decreases mitochondrial reactive oxygen release and increases life span in Saccharomyces cerevisiae. J. Biol. Chem. 279, 49883–49888. [DOI] [PubMed] [Google Scholar]
- Barzilai N, Crandall JP, Kritchevsky SB, Espeland MA, 2016. Metformin as a tool to target aging. Cell Metab. 23, 1060–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgart M, Priebe S, Groth M, Hartmann N, Menzel U, Pandolfini L, Koch P, Felder M, Ristow M, Englert C, Guthke R, Platzer M, Cellerino A, 2016. Longitudinal RNA-Seq analysis of vertebrate aging identifies mitochondrial complex I as a small-molecule-Sensitive modifier of lifespan. Cell Syst. 2, 122–132. [DOI] [PubMed] [Google Scholar]
- Beaupere C, Wasko BM, Lorusso J, Kennedy BK, Kaeberlein M, Labunskyy VM, 2017. CAN1 arginine permease deficiency extends yeast replicative lifespan via translational activation of stress response genes. Cell Rep. 18, 1884–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti MG, Foster AL, Vantipalli MC, White MP, Sampayo JN, Gill MS, Olsen A, Lithgow GJ, 2008. Compounds that confer thermal stress resistance and extended lifespan. Exp. Gerontol. 43, 882–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett CF, Vander Wende H, Simko M, Klum S, Barfield S, Choi H, Pineda VV, Kaeberlein M, 2014. Activation of the mitochondrial unfolded protein response does not predict longevity in Caenorhabditis elegans. Nat. Commun. 5, 3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett CF, Kwon JJ, Chen C, Russell J, Acosta K, Burnaevskiy N, Crane MM, Bitto A, Vander Wende H, Simko M, Pineda V, Rossner R, Wasko BM, Choi H, Chen S, Park S, Jafari G, Sands B, Perez Olsen C, Mendenhall AR, Morgan PG, Kaeberlein M, 2017. Transaldolase inhibition impairs mitochondrial respiration and induces a starvation-like longevity response in Caenorhabditis elegans. PLoS Genet. 13, e1006695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkenfeld AL, Lee HY, Guebre-Egziabher F, Alves TC, Jurczak MJ, Jornayvaz FR, Zhang D, Hsiao JJ, Martin-Montalvo A, Fischer-Rosinsky A, Spranger J, Pfeiffer AF, Jordan J, Fromm MF, Konig J, Lieske S, Carmean CM, Frederick DW, Weismann D, Knauf F, Irusta PM, De Cabo R, Helfand SL, Samuel VT, Shulman GI, 2011. Deletion of the mammalian INDY homolog mimics aspects of dietary restriction and protects against adiposity and insulin resistance in mice. Cell Metab. 14, 184–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop NA, Guarente L, 2007. Two neurons mediate diet-restriction-induced longevity in C. Elegans. Nature 447, 545–549. [DOI] [PubMed] [Google Scholar]
- Biteau B, Karpac J, Supoyo S, Degennaro M, Lehmann R, Jasper H, 2010. Lifespan extension by preserving proliferative homeostasis in Drosophila. PLoS Genet. 6, e1001159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozek K, Khrameeva EE, Reznick J, Omerbasic D, Bennett NC, Lewin GR, Azpurua J, Gorbunova V, Seluanov A, Regnard P, Wanert F, Marchal J, Pifferi F, Aujard F, Liu Z, Shi P, Paabo S, Schroeder F, Willmitzer L, Giavalisco P, Khaitovich P, 2017. Lipidome determinants of maximal lifespan in mammals. Sci. Rep. 7, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brack C, Bechter-Thuring E, Labuhn M, 1997. N-acetylcysteine slows down ageing and increases the life span of Drosophila melanogaster. Cell. Mol. Life Sci. 53, 960–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw PC, 2019. Cytoplasmic and mitochondrial NADPH-Coupled redox systems in the regulation of aging. Nutrients 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratic A, Larsson NG, 2013. The role of mitochondria in aging. J. Clin. Invest. 123, 951–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brejning J, Norgaard S, Scholer L, Morthorst TH, Jakobsen H, Lithgow GJ, Jensen LT, Olsen A, 2014. Loss of NDG-4 extends lifespan and stress resistance in Caenorhabditis elegans. Aging Cell 13, 156–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brokate-Llanos AM, Garzon A, Munoz MJ, 2014. Escherichia coli carbon source metabolism affects longevity of its predator Caenorhabditis elegans. Mech. Ageing Dev. 141–142, 22–25. [DOI] [PubMed] [Google Scholar]
- Broue F, Liere P, Kenyon C, Baulieu EE, 2007. A steroid hormone that extends the lifespan of Caenorhabditis elegans. Aging Cell 6, 87–94. [DOI] [PubMed] [Google Scholar]
- Bruss MD, Khambatta CF, Ruby MA, Aggarwal I, Hellerstein MK, 2010. Calorie restriction increases fatty acid synthesis and whole body fat oxidation rates. Am. J. Physiol. 298, E108–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brys K, Castelein N, Matthijssens F, Vanfleteren JR, Braeckman BP, 2010. Disruption of insulin signalling preserves bioenergetic competence of mitochondria in ageing Caenorhabditis elegans. BMC Biol. 8, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucaciuc Mracica T, Anghel A, Ion CF, Moraru CV, Tacutu R, Lazar GA, 2020. MetaboAge DB: a repository of known ageing-related changes in the human metabolome. Biogerontology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunning BJ, Contrepois K, Lee-McMullen B, Dhondalay GKR, Zhang W, Tupa D, Raeber O, Desai M, Nadeau KC, Snyder MP, Andorf S, 2020. Global metabolic profiling to model biological processes of aging in twins. Aging Cell 19, e13073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkewitz K, Zhang Y, Mair WB, 2014. AMPK at the nexus of energetics and aging. Cell Metab. 20, 10–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkewitz K, Morantte I, Weir HJM, Yeo R, Zhang Y, Huynh FK, Ilkayeva OR, Hirschey MD, Grant AR, Mair WB, 2015. Neuronal CRTC-1 governs systemic mitochondrial metabolism and lifespan via a catecholamine signal. Cell 160, 842–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustos V, Partridge L, 2017. Good Ol’ fat: links between lipid signaling and longevity. Trends Biochem. Sci. 42, 812–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler JA, Mishur RJ, Bhaskaran S, Rea SL, 2013. A metabolic signature for long life in the Caenorhabditis elegans Mit mutants. Aging Cell 12, 130–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabreiro F, Au C, Leung KY, Vergara-Irigaray N, Cocheme HM, Noori T, Weinkove D, Schuster E, Greene ND, Gems D, 2013. Metformin retards aging in C. Elegans by altering microbial folate and methionine metabolism. Cell 153, 228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldeira da Silva CC, Cerqueira FM, Barbosa LF, Medeiros MH, Kowaltowski AJ, 2008. Mild mitochondrial uncoupling in mice affects energy metabolism, redox balance and longevity. Aging Cell 7, 552–560. [DOI] [PubMed] [Google Scholar]
- Calvert S, Tacutu R, Sharifi S, Teixeira R, Ghosh P, de Magalhaes JP, 2016. A network pharmacology approach reveals new candidate caloric restriction mimetics in C. Elegans. Aging Cell 15, 256–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canfield C, Bradshaw P, 2019. Amino acids in the regulation of aging and aging-related diseases. Transl. Med. Aging 70–89. [Google Scholar]
- Canto C, Menzies KJ, Auwerx J, 2015. NAD(+) metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. 22, 31–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartwright MJ, Tchkonia T, Kirkland JL, 2007. Aging in adipocytes: potential impact of inherent, depot-specific mechanisms. Exp. Gerontol. 42, 463–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo-Quan JI, Tain LS, Kinghorn KJ, Li L, Gronke S, Hinze Y, Blackwell TK, Bjedov I, Partridge L, 2019. A triple drug combination targeting components of the nutrient-sensing network maximizes longevity. Proc. Natl. Acad. Sci. U.S.A. 116, 20817–20819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Portuguez R, Sutphin GL, 2020. Kynurenine pathway, NAD(+) synthesis, and mitochondrial function: targeting tryptophan metabolism to promote longevity and healthspan. Exp. Gerontol. 132, 110841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaleri F, Bashar E, 2018. Potential synergies of beta-hydroxybutyrate and butyrate on the modulation of metabolism, inflammation, cognition, and general health. J. Nutr. Metab. 2018, 7195760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaleckis R, Murakami I, Takada J, Kondoh H, Yanagida M, 2016. Individual variability in human blood metabolites identifies age-related differences. Proc. Natl. Acad. Sci. U.S.A. 113, 4252–4259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler-Brown D, Choi H, Park S, Ocampo BR, Chen S, Le A, Sutphin GL, Shamieh LS, Smith ED, Kaeberlein M, 2015. Sorbitol treatment extends lifespan and induces the osmotic stress response in Caenorhabditis elegans. Front. Genet. 6, 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles KN, Li MD, Engin F, Arruda AP, Inouye K, Hotamisligil GS, 2017. Uncoupling of metabolic health from longevity through genetic alteration of adipose tissue lipid-binding proteins. Cell Rep. 21, 393–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri J, Bose N, Gong J, Hall D, Rifkind A, Bhaumik D, Peiris TH, Chamoli M, Le CH, Liu J, Lithgow GJ, Ramanathan A, Xu XZS, Kapahi P, 2016. A Caenorhabditis elegans model elucidates a conserved role for TRPA1-Nrf signaling in reactive alpha-dicarbonyl detoxification. Curr. Biol. 26, 3014–3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri J, Bains Y, Guha S, Kahn A, Hall D, Bose N, Gugliucci A, Kapahi P, 2018. The role of advanced glycation end products in aging and metabolic diseases: bridging association and causality. Cell Metab. 28, 337–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Whetstine JR, Ghosh S, Hanover JA, Gali RR, Grosu P, Shi Y, 2009. The conserved NAD(H)-dependent corepressor CTBP-1 regulates Caenorhabditis elegans life span. Proc. Natl. Acad. Sci. U.S.A. 106, 1496–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Li PW, Goldstein BA, Cai W, Thomas EL, Chen F, Hubbard AE, Melov S, Kapahi P, 2013. Germline signaling mediates the synergistically prolonged longevity produced by double mutations in daf-2 and rsks-1 in C. Elegans. Cell Rep. 5, 1600–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S, Larson MG, McCabe EL, Murabito JM, Rhee EP, Ho JE, Jacques PF, Ghorbani A, Magnusson M, Souza AL, Deik AA, Pierce KA, Bullock K, O’Donnell CJ, Melander O, Clish CB, Vasan RS, Gerszten RE, Wang TJ, 2015. Distinct metabolomic signatures are associated with longevity in humans. Nat. Commun. 6, 6791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin RM, Fu X, Pai MY, Vergnes L, Hwang H, Deng G, Diep S, Lomenick B, Meli VS, Monsalve GC, Hu E, Whelan SA, Wang JX, Jung G, Solis GM, Fazlollahi F, Kaweeteerawat C, Quach A, Nili M, Krall AS, Godwin HA, Chang HR, Faull KF, Guo F, Jiang M, Trauger SA, Saghatelian A, Braas D, Christofk HR, Clarke CF, Teitell MA, Petrascheck M, Reue K, Jung ME, Frand AR, Huang J, 2014. The metabolite alpha-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature 510, 397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching TT, Paal AB, Mehta A, Zhong L, Hsu AL, 2010. drr-2 encodes an eIF4H that acts downstream of TOR in diet-restriction-induced longevity of C. Elegans. Aging Cell 9, 545–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang MH, Chiou SH, Huang CH, Yang WB, Wong CH, 2009. The lifespan-promoting effect of acetic acid and Reishi polysaccharide. Bioorg. Med. Chem. 17, 7831–7840. [DOI] [PubMed] [Google Scholar]
- Copeland JM, Cho J, Lo T Jr., Hur JH, Bahadorani S, Arabyan T, Rabie J, Soh J, Walker DW, 2009. Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr. Biol. 19, 1591–1598. [DOI] [PubMed] [Google Scholar]
- Copes N, Edwards C, Chaput D, Saifee M, Barjuca I, Nelson D, Paraggio A, Saad P, Lipps D, Stevens SM Jr., Bradshaw PC, 2015. Metabolome and proteome changes with aging in Caenorhabditis elegans. Exp. Gerontol. 72, 67–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings NE, Williams EM, Kasza I, Konon EN, Schaid MD, Schmidt BA, Poudel C, Sherman DS, Yu D, Arriola Apelo SI, Cottrell SE, Geiger G, Barnes ME, Wisinski JA, Fenske RJ, Matkowskyj KA, Kimple ME, Alexander CM, Merrins MJ, Lamming DW, 2018. Restoration of metabolic health by decreased consumption of branched-chain amino acids. J. Physiol. 596, 623–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran SP, Ruvkun G, 2007. Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet. 3, e56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler RG, Thompson KW, Camandola S, Mack KT, Mattson MP, 2014. Sphingolipid metabolism regulates development and lifespan in Caenorhabditis elegans. Mech. Ageing Dev. 143–144, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler RG, Camandola S, Feldman NH, Yoon JS, Haran JB, Arguelles S, Mattson MP, 2019. Uric acid enhances longevity and endurance and protects the brain against ischemia. Neurobiol. Aging 75, 159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Antona G, Ragni M, Cardile A, Tedesco L, Dossena M, Bruttini F, Caliaro F, Corsetti G, Bottinelli R, Carruba MO, Valerio A, Nisoli E, 2010. Branched-chain amino acid supplementation promotes survival and supports cardiac and skeletal muscle mitochondrial biogenesis in middle-aged mice. Cell Metab. 12, 362–372. [DOI] [PubMed] [Google Scholar]
- D’Mello NP, Childress AM, Franklin DS, Kale SP, Pinswasdi C, Jazwinski SM, 1994. Cloning and characterization of LAG1, a longevity-assurance gene in yeast. J. Biol. Chem. 269, 15451–15459. [PubMed] [Google Scholar]
- Darst BF, Koscik RL, Hogan KJ, Johnson SC, Engelman CD, 2019. Longitudinal plasma metabolomics of aging and sex. Aging (Albany NY) 11, 1262–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong L, Meng Y, Dent J, Hekimi S, 2004. Thiamine pyrophosphate biosynthesis and transport in the nematode Caenorhabditis elegans. Genetics 168, 845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deelen J, Kettunen J, Fischer K, van der Spek A, Trompet S, Kastenmuller G, Boyd A, Zierer J, van den Akker EB, Ala-Korpela M, Amin N, Demirkan A, Ghanbari M, van Heemst D, Ikram MA, van Klinken JB, Mooijaart SP, Peters A, Salomaa V, Sattar N, Spector TD, Tiemeier H, Verhoeven A, Waldenberger M, Wurtz P, Davey Smith G, Metspalu A, Perola M, Menni C, Geleijnse JM, Drenos F, Beekman M, Jukema JW, van Duijn CM, Slagboom PE, 2019. A metabolic profile of all-cause mortality risk identified in an observational study of 44,168 individuals. Nature communications 10, 3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehghan M, Mente A, Zhang X, Swaminathan S, Li W, Mohan V, Iqbal R, Kumar R, Wentzel-Viljoen E, Rosengren A, Amma LI, Avezum A, Chifamba J, Diaz R, Khatib R, Lear S, Lopez-Jaramillo P, Liu X, Gupta R, Mohammadifard N, Gao N, Oguz A, Ramli AS, Seron P, Sun Y, Szuba A, Tsolekile L, Wielgosz A, Yusuf R, Hussein Yusufali A, Teo KK, Rangarajan S, Dagenais G, Bangdiwala SI, Islam S, Anand SS, Yusuf S, Prospective Urban Rural Epidemiology study, i, 2017. Associations of fats and carbohydrate intake with cardiovascular disease and mortality in 18 countries from five continents (PURE): a prospective cohort study. Lancet 390, 2050–2062. [DOI] [PubMed] [Google Scholar]
- Dell’agnello C, Leo S, Agostino A, Szabadkai G, Tiveron C, Zulian A, Prelle A, Roubertoux P, Rizzuto R, Zeviani M, 2007. Increased longevity and refractoriness to Ca(2+)-dependent neurodegeneration in Surf1 knockout mice. Hum. Mol. Genet. 16, 431–444. [DOI] [PubMed] [Google Scholar]
- Denzel MS, Storm NJ, Gutschmidt A, Baddi R, Hinze Y, Jarosch E, Sommer T, Hoppe T, Antebi A, 2014. Hexosamine pathway metabolites enhance protein quality control and prolong life. Cell 156, 1167–1178. [DOI] [PubMed] [Google Scholar]
- Diaz-Ruiz A, Lanasa M, Garcia J, Mora H, Fan F, Martin-Montalvo A, Di Francesco A, Calvo-Rubio M, Salvador-Pascual A, Aon MA, Fishbein KW, Pearson KJ, Villalba JM, Navas P, Bernier M, de Cabo R, 2018. Overexpression of CYB5R3 and NQO1, two NAD(+) -producing enzymes, mimics aspects of caloric restriction. Aging Cell 17, e12767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillin A, Hsu AL, Arantes-Oliveira N, Lehrer-Graiwer J, Hsin H, Fraser AG, Kamath RS, Ahringer J, Kenyon C, 2002. Rates of behavior and aging specified by mitochondrial function during development. Science (New York, N.Y 298, 2398–2401. [DOI] [PubMed] [Google Scholar]
- Ducker GS, Rabinowitz JD, 2017. One-carbon metabolism in health and disease. Cell Metab. 25, 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn WB, Lin W, Broadhurst D, Begley P, Brown M, Zelena E, Vaughan AA, Halsall A, Harding N, Knowles JD, Francis-McIntyre S, Tseng A, Ellis DI, O’Hagan S, Aarons G, Benjamin B, Chew-Graham S, Moseley C, Potter P, Winder CL, Potts C, Thornton P, McWhirter C, Zubair M, Pan M, Burns A, Cruickshank JK, Jayson GC, Purandare N, Wu FC, Finn JD, Haselden JN, Nicholls AW, Wilson ID, Goodacre R, Kell DB, 2015. Molecular phenotyping of a UK population: defining the human serum metabolome. Metabolomics 11, 9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easlon E, Tsang F, Dilova I, Wang C, Lu SP, Skinner C, Lin SJ, 2007. The dihydrolipoamide acetyltransferase is a novel metabolic longevity factor and is required for calorie restriction-mediated life span extension. J. Biol. Chem. 282, 6161–6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easlon E, Tsang F, Skinner C, Wang C, Lin SJ, 2008. The malate-aspartate NADH shuttle components are novel metabolic longevity regulators required for calorie restriction-mediated life span extension in yeast. Genes Dev. 22, 931–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards CB, Copes N, Brito AG, Canfield J, Bradshaw PC, 2013. Malate and fumarate extend lifespan in Caenorhabditis elegans. PLoS One 8, e58345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards C, Canfield J, Copes N, Rehan M, Lipps D, Bradshaw PC, 2014. D-beta-hydroxybutyrate extends lifespan in C. Elegans. Aging (Albany NY) 6, 621–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards C, Canfield J, Copes N, Brito A, Rehan M, Lipps D, Brunquell J, Westerheide SD, Bradshaw PC, 2015a. Mechanisms of amino acid-mediated lifespan extension in Caenorhabditis elegans. BMC Genet. 16, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards C, Copes N, Bradshaw PC, 2015b. D-ss-hydroxybutyrate: an anti-aging ketone body. Oncotarget 6, 3477–3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg T, Knauer H, Schauer A, Buttner S, Ruckenstuhl C, Carmona-Gutierrez D, Ring J, Schroeder S, Magnes C, Antonacci L, Fussi H, Deszcz L, Hartl R, Schraml E, Criollo A, Megalou E, Weiskopf D, Laun P, Heeren G, Breitenbach M, Grubeck-Loebenstein B, Herker E, Fahrenkrog B, Frohlich KU, Sinner F, Tavernarakis N, Minois N, Kroemer G, Madeo F, 2009. Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 11, 1305–1314. [DOI] [PubMed] [Google Scholar]
- Eisenberg T, Schroeder S, Andryushkova A, Pendl T, Kuttner V, Bhukel A, Marino G, Pietrocola F, Harger A, Zimmermann A, Moustafa T, Sprenger A, Jany E, Buttner S, Carmona-Gutierrez D, Ruckenstuhl C, Ring J, Reichelt W, Schimmel K, Leeb T, Moser C, Schatz S, Kamolz LP, Magnes C, Sinner F, Sedej S, Frohlich KU, Juhasz G, Pieber TR, Dengjel J, Sigrist SJ, Kroemer G, Madeo F, 2014. Nucleocytosolic depletion of the energy metabolite acetyl-coenzyme a stimulates autophagy and prolongs lifespan. Cell Metab. 19, 431–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg T, Abdellatif M, Schroeder S, Primessnig U, Stekovic S, Pendl T, Harger A, Schipke J, Zimmermann A, Schmidt A, Tong M, Ruckenstuhl C, Dammbrueck C, Gross AS, Herbst V, Magnes C, Trausinger G, Narath S, Meinitzer A, Hu Z, Kirsch A, Eller K, Carmona-Gutierrez D, Buttner S, Pietrocola F, Knittelfelder O, Schrepfer E, Rockenfeller P, Simonini C, Rahn A, Horsch M, Moreth K, Beckers J, Fuchs H, Gailus-Durner V, Neff F, Janik D, Rathkolb B, Rozman J, de Angelis MH, Moustafa T, Haemmerle G, Mayr M, Willeit P, von Frieling-Salewsky M, Pieske B, Scorrano L, Pieber T, Pechlaner R, Willeit J, Sigrist SJ, Linke WA, Muhlfeld C, Sadoshima J, Dengjel J, Kiechl S, Kroemer G, Sedej S, Madeo F, 2016. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat. Med. 22, 1428–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewbank JJ, Barnes TM, Lakowski B, Lussier M, Bussey H, Hekimi S, 1997. Structural and functional conservation of the Caenorhabditis elegans timing gene clk-1. Science (New York, N.Y 275, 980–983. [DOI] [PubMed] [Google Scholar]
- Farese RV Jr., Walther TC, 2009. Lipid droplets finally get a little R-E-S-P-E-C-T. Cell 139, 855–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei YJ, Inoue K, Ganapathy V, 2003. Structural and functional characteristics of two sodium-coupled dicarboxylate transporters (ceNaDC1 and ceNaDC2) from Caenorhabditis elegans and their relevance to life span. J. Biol. Chem. 278, 6136–6144. [DOI] [PubMed] [Google Scholar]
- Fei YJ, Liu JC, Inoue K, Zhuang L, Miyake K, Miyauchi S, Ganapathy V, 2004. Relevance of NAC-2, an Na+-coupled citrate transporter, to life span, body size and fat content in Caenorhabditis elegans. Biochem. J. 379, 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Bussiere F, Hekimi S, 2001. Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Dev. Cell 1, 633–644. [DOI] [PubMed] [Google Scholar]
- Feng Y, Williams BG, Koumanov F, Wolstenholme AJ, Holman GD, 2013. FGT-1 is the major glucose transporter in C. Elegans and is central to aging pathways. Biochem. J. 456, 219–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson AA, Roy S, Kormanik KN, Kim Y, Dumas KJ, Ritov VB, Matern D, Hu PJ, Fisher AL, 2013. TATN-1 mutations reveal a novel role for tyrosine as a metabolic signal that influences developmental decisions and longevity in Caenorhabditis elegans. PLoS Genet. 9, e1004020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flurkey K, Astle CM, Harrison DE, 2010. Life extension by diet restriction and N-acetyl-L-cysteine in genetically heterogeneous mice. J. Gerontol. A Biol. Sci. Med. Sci. 65, 1275–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folick A, Oakley HD, Yu Y, Armstrong EH, Kumari M, Sanor L, Moore DD, Ortlund EA, Zechner R, Wang MC, 2015. Aging. Lysosomal signaling molecules regulate longevity in Caenorhabditis elegans. Science (New York, N.Y 347, 83–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana L, Partridge L, 2015. Promoting health and longevity through diet: from model organisms to humans. Cell 161, 106–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana L, Cummings NE, Arriola Apelo SI, Neuman JC, Kasza I, Schmidt BA, Cava E, Spelta F, Tosti V, Syed FA, Baar EL, Veronese N, Cottrell SE, Fenske RJ, Bertozzi B, Brar HK, Pietka T, Bullock AD, Figenshau RS, Andriole GL, Merrins MJ, Alexander CM, Kimple ME, Lamming DW, 2016. Decreased consumption of branched-chain amino acids improves metabolic health. Cell Rep. 16, 520–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederick DW, Loro E, Liu L, Davila A Jr., Chellappa K, Silverman IM, Quinn WJ 3rd, Gosai SJ, Tichy ED, Davis JG, Mourkioti F, Gregory BD, Dellinger RW, Redpath P, Migaud ME, Nakamaru-Ogiso E, Rabinowitz JD, Khurana TS, Baur JA, 2016. Loss of NAD homeostasis leads to progressive and reversible degeneration of skeletal muscle. Cell Metab. 24, 269–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fretwell LK, McCune S, Fone JV, Yates DJ, 2006. The effect of supplementation with branched-chain amino acids on cognitive function in active dogs. J. Nutr. 136, 2069S–2071S. [DOI] [PubMed] [Google Scholar]
- Frick B, Schroecksnadel K, Neurauter G, Leblhuber F, Fuchs D, 2004. Increasing production of homocysteine and neopterin and degradation of tryptophan with older age. Clin. Biochem. 37, 684–687. [DOI] [PubMed] [Google Scholar]
- Fridell YW, Sanchez-Blanco A, Silvia BA, Helfand SL, 2005. Targeted expression of the human uncoupling protein 2 (hUCP2) to adult neurons extends life span in the fly. Cell Metab. 1, 145–152. [DOI] [PubMed] [Google Scholar]
- Fridell YW, Hoh M, Kreneisz O, Hosier S, Chang C, Scantling D, Mulkey DK, Helfand SL, 2009. Increased uncoupling protein (UCP) activity in Drosophila insulin-producing neurons attenuates insulin signaling and extends lifespan. Aging (Albany NY) 1, 699–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X, Chin RM, Vergnes L, Hwang H, Deng G, Xing Y, Pai MY, Li S, Ta L, Fazlollahi F, Chen C, Prins RM, Teitell MA, Nathanson DA, Lai A, Faull KF, Jiang M, Clarke SG, Cloughesy TF, Graeber TG, Braas D, Christofk HR, Jung ME, Reue K, Huang J, 2015. 2-hydroxyglutarate inhibits ATP synthase and mTOR signaling. Cell Metab. 22, 508–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs S, Bundy JG, Davies SK, Viney JM, Swire JS, Leroi AM, 2010. A metabolic signature of long life in Caenorhabditis elegans. BMC Biol. 8, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fushan AA, Turanov AA, Lee SG, Kim EB, Lobanov AV, Yim SH, Buffenstein R, Lee SR, Chang KT, Rhee H, Kim JS, Yang KS, Gladyshev VN, 2015. Gene expression defines natural changes in mammalian lifespan. Aging Cell 14, 352–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbs M, Leng S, Devassy JG, Monirujjaman M, Aukema HM, 2015. Advances in our understanding of oxylipins derived from dietary PUFAs. Adv. Nutr. 6, 513–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gad MZ, 2010. Anti-aging effects of l-arginine. J. Adv. Res. 1, 169–177. [Google Scholar]
- Gao AW, Smith RL, van Weeghel M, Kamble R, Janssens GE, Houtkooper RH, 2018. Identification of key pathways and metabolic fingerprints of longevity in C. Elegans. Exp. Gerontol. 113, 128–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gates AC, Bernal-Mizrachi C, Chinault SL, Feng C, Schneider JG, Coleman T, Malone JP, Townsend RR, Chakravarthy MV, Semenkovich CF, 2007. Respiratory uncoupling in skeletal muscle delays death and diminishes age-related disease. Cell Metab. 6, 497–505. [DOI] [PubMed] [Google Scholar]
- Gebauer J, Gentsch C, Mansfeld J, Schmeisser K, Waschina S, Brandes S, Klimmasch L, Zamboni N, Zarse K, Schuster S, Ristow M, Schauble S, Kaleta C, 2016. A genome-scale database and reconstruction of Caenorhabditis elegans metabolism. Cell Syst. 2, 312–322. [DOI] [PubMed] [Google Scholar]
- Gerisch B, Rottiers V, Li D, Motola DL, Cummins CL, Lehrach H, Mangelsdorf DJ, Antebi A, 2007. A bile acid-like steroid modulates Caenorhabditis elegans lifespan through nuclear receptor signaling. Proc. Natl. Acad. Sci. U.S.A. 104, 5014–5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg J, Currais A, Prior M, Fischer W, Chiruta C, Ratliff E, Daugherty D, Dargusch R, Finley K, Esparza-Molto PB, Cuezva JM, Maher P, Petrascheck M, Schubert D, 2018. The mitochondrial ATP synthase is a shared drug target for aging and dementia. Aging Cell 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golegaonkar S, Tabrez SS, Pandit A, Sethurathinam S, Jagadeeshaprasad MG, Bansode S, Sampathkumar SG, Kulkarni MJ, Mukhopadhyay A, 2015. Rifampicin reduces advanced glycation end products and activates DAF-16 to increase lifespan in Caenorhabditis elegans. Aging Cell 14, 463–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Covarrubias V, Beekman M, Uh HW, Dane A, Troost J, Paliukhovich I, van der Kloet FM, Houwing-Duistermaat J, Vreeken RJ, Hankemeier T, Slagboom EP, 2013. Lipidomics of familial longevity. Aging Cell 12, 426–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gospodaryov DV, Lushchak OV, Rovenko BM, Perkhulyn NV, Gerards M, Tuomela T, Jacobs HT, 2014. Ciona intestinalis NADH dehydrogenase NDX confers stress-resistance and extended lifespan on Drosophila. Biochim. Biophys. Acta 1837, 1861–1869. [DOI] [PubMed] [Google Scholar]
- Gospodaryov DV, Strilbytska OM, Semaniuk UV, Perkhulyn NV, Rovenko BM, Yurkevych IS, Barata AG, Dick TP, Lushchak OV, Jacobs HT, 2019. Alternative NADH dehydrogenase extends lifespan and increases resistance to xenobiotics in Drosophila. Biogerontology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudeau J, Bellemin S, Toselli-Mollereau E, Shamalnasab M, Chen Y, Aguilaniu H, 2011. Fatty acid desaturation links germ cell loss to longevity through NHR-80/HNF4 in C. Elegans. PLoS Biol. 9, e1000599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal MS, Vlassenko AG, Blazey TM, Su Y, Couture LE, Durbin TJ, Bateman RJ, Benzinger TL, Morris JC, Raichle ME, 2017. Loss of brain aerobic glycolysis in normal human aging. Cell Metab. 26 (353–360), e353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandison RC, Piper MD, Partridge L, 2009. Amino-acid imbalance explains extension of lifespan by dietary restriction in Drosophila. Nature 462, 1061–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green CL, Lamming DW, 2019. Regulation of metabolic health by essential dietary amino acids. Mech. Ageing Dev. 177, 186–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Dowlatshahi D, Banko MR, Villen J, Hoang K, Blanchard D, Gygi SP, Brunet A, 2007. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. Elegans. Curr. Biol. 17, 1646–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Maures TJ, Hauswirth AG, Green EM, Leeman DS, Maro GS, Han S, Banko MR, Gozani O, Brunet A, 2010. Members of the H3K4 trimethylation complex regulate lifespan in a germline-dependent manner in C. Elegans. Nature 466, 383–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronke S, Clarke DF, Broughton S, Andrews TD, Partridge L, 2010. Molecular evolution and functional characterization of Drosophila insulin-like peptides. PLoS Genet. 6, e1000857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusarov I, Pani B, Gautier L, Smolentseva O, Eremina S, Shamovsky I, Katkova-Zhukotskaya O, Mironov A, Nudler E, 2017. Glycogen controls Caenorhabditis elegans lifespan and resistance to oxidative stress. Nat. Commun. 8, 15868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hachinohe M, Yamane M, Akazawa D, Ohsawa K, Ohno M, Terashita Y, Masumoto H, 2013. A reduction in age-enhanced gluconeogenesis extends lifespan. PLoS One 8, e54011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakimi P, Yang J, Casadesus G, Massillon D, Tolentino-Silva F, Nye CK, Cabrera ME, Hagen DR, Utter CB, Baghdy Y, Johnson DH, Wilson DL, Kirwan JP, Kalhan SC, Hanson RW, 2007. Overexpression of the cytosolic form of phosphoenolpyruvate carboxykinase (GTP) in skeletal muscle repatterns energy metabolism in the mouse. J. Biol. Chem. 282, 32844–32855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halaschek-Wiener J, Khattra JS, McKay S, Pouzyrev A, Stott JM, Yang GS, Holt RA, Jones SJ, Marra MA, Brooks-Wilson AR, Riddle DL, 2005. Analysis of long-lived C. Elegans daf-2 mutants using serial analysis of gene expression. Genome Res. 15, 603–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton B, Dong Y, Shindo M, Liu W, Odell I, Ruvkun G, Lee SS, 2005. A systematic RNAi screen for longevity genes in C. Elegans. Genes Dev. 19, 1544–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han B, Sivaramakrishnan P, Lin CJ, Neve IAA, He J, Tay LWR, Sowa JN, Sizovs A, Du G, Wang J, Herman C, Wang MC, 2017a. Microbial genetic composition tunes host longevity. Cell 169 (1249–1262), e1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Schroeder EA, Silva-Garcia CG, Hebestreit K, Mair WB, Brunet A, 2017b. Mono-unsaturated fatty acids link H3K4me3 modifiers to C. Elegans lifespan. Nature 544, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handee W, Li X, Hall KW, Deng X, Li P, Benning C, Williams BL, Kuo MH, 2016. An energy-independent pro-longevity function of triacylglycerol in yeast. PLoS Genet. 12, e1005878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannun YA, Obeid LM, 2018. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 19, 175–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen M, Hsu AL, Dillin A, Kenyon C, 2005. New genes tied to endocrine, metabolic, and dietary regulation of lifespan from a Caenorhabditis elegans genomic RNAi screen. PLoS Genet. 1, 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson RW, 2009. Thematic minireview series: a perspective on the biology of phosphoenolpyruvate carboxykinase 55 years after its discovery. J. Biol. Chem. 284, 27021–27023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Schaffer BE, Brunet A, 2016. AMPK: an energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol. 26, 190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison DE, Strong R, Allison DB, Ames BN, Astle CM, Atamna H, Fernandez E, Flurkey K, Javors MA, Nadon NL, Nelson JF, Pletcher S, Simpkins JW, Smith D, Wilkinson JE, Miller RA, 2014. Acarbose, 17-alpha-estradiol, and nordihydroguaiaretic acid extend mouse lifespan preferentially in males. Aging Cell 13, 273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison DE, Strong R, Alavez S, Astle CM, DiGiovanni J, Fernandez E, Flurkey K, Garratt M, Gelfond JAL, Javors MA, Levi M, Lithgow GJ, Macchiarini F, Nelson JF, Sukoff Rizzo SJ, Slaga TJ, Stearns T, Wilkinson JE, Miller RA, 2019. Acarbose improves health and lifespan in aging HET3 mice. Aging Cell 18, e12898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman PS, Ishii N, Kayser EB, Morgan PG, Sedensky MM, 2001. Mitochondrial mutations differentially affect aging, mutability and anesthetic sensitivity in Caenorhabditis elegans. Mech. Ageing Dev. 122, 1187–1201. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Horikawa M, Nomura T, Sakamoto K, 2010. Nicotinamide adenine dinucleotide extends the lifespan of Caenorhabditis elegans mediated by sir-2.1 and daf-16. Biogerontology 11, 31–43. [DOI] [PubMed] [Google Scholar]
- He C, TsuchiyaM.a, SK, Nguyen QT, Plyusnina EN, Terrill SR, Sahibzada S, Patel B, Faulkner AR, Shaposhnikov MV, Tian R, Tsuchiya M, Kaeberlein M, Moskalev AA, Kennedy BK, Polymenis M, 2014. Enhanced longevity by ibuprofen, conserved in multiple species, occurs in yeast through inhibition of tryptophan import. PLoS Genet. 10, e1004860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heestand BN, Shen Y, Liu W, Magner DB, Storm N, Meharg C, Habermann B, Antebi A, 2013. Dietary restriction induced longevity is mediated by nuclear receptor NHR-62 in Caenorhabditis elegans. PLoS Genet. 9, e1003651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry CJ, Huang Y, Wynne A, Hanke M, Himler J, Bailey MT, Sheridan JF, Godbout JP, 2008. Minocycline attenuates lipopolysaccharide (LPS)-induced neuroinflammation, sickness behavior, and anhedonia. J. Neuroinflammation 5, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hine C, Harputlugil E, Zhang Y, Ruckenstuhl C, Lee BC, Brace L, Longchamp A, Trevino-Villarreal JH, Mejia P, Ozaki CK, Wang R, Gladyshev VN, Madeo F, Mair WB, Mitchell JR, 2015. Endogenous hydrogen sulfide production is essential for dietary restriction benefits. Cell 160, 132–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman JM, Soltow QA, Li S, Sidik A, Jones DP, Promislow DE, 2014. Effects of age, sex, and genotype on high-sensitivity metabolomic profiles in the fruit fly, Drosophila melanogaster. Aging Cell 13, 596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda Y, Tanaka M, Honda S, 2010. Trehalose extends longevity in the nematode Caenorhabditis elegans. Aging Cell 9, 558–569. [DOI] [PubMed] [Google Scholar]
- Honda Y, Fujita Y, Maruyama H, Araki Y, Ichihara K, Sato A, Kojima T, Tanaka M, Nozawa Y, Ito M, Honda S, 2011. Lifespan-extending effects of royal jelly and its related substances on the nematode Caenorhabditis elegans. PLoS One 6, e23527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, 2013. DNA methylation age of human tissues and cell types. Genome Biol. 14, R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou NS, Taubert S, 2012. Function and regulation of lipid biology in Caenorhabditis elegans aging. Front. Physiol. 3, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houten SM, Wanders RJ, 2010. A general introduction to the biochemistry of mitochondrial fatty acid beta-oxidation. J. Inherit. Metab. Dis. 33, 469–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Argmann C, Houten SM, Canto C, Jeninga EH, Andreux PA, Thomas C, Doenlen R, Schoonjans K, Auwerx J, 2011. The metabolic footprint of aging in mice. Sci. Rep. 1, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsin H, Kenyon C, 1999. Signals from the reproductive system regulate the lifespan of C. Elegans. Nature 399, 362–366. [DOI] [PubMed] [Google Scholar]
- Huang X, Liu J, Dickson RC, 2012. Down-regulating sphingolipid synthesis increases yeast lifespan. PLoS Genet. 8, e1002493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Withers BR, Dickson RC, 2014. Sphingolipids and lifespan regulation. Biochim. Biophys. Acta 1841, 657–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull-Thompson J, Muffat J, Sanchez D, Walker DW, Benzer S, Ganfornina MD, Jasper H, 2009. Control of metabolic homeostasis by stress signaling is mediated by the lipocalin NLaz. PLoS Genet. 5, e1000460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey DM, Toivonen JM, Giannakou M, Partridge L, Brand MD, 2009. Expression of human uncoupling protein-3 in Drosophila insulin-producing cells increases insulin-like peptide (DILP) levels and shortens lifespan. Exp. Gerontol. 44, 316–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur JH, Bahadorani S, Graniel J, Koehler CL, Ulgherait M, Rera M, Jones DL, Walker DW, 2013. Increased longevity mediated by yeast NDI1 expression in Drosophila intestinal stem and progenitor cells. Aging (Albany NY) 5, 662–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Hidalgo M, Santos-Ocana C, Padilla S, Villalba JM, Lopez-Lluch G, Martin-Montalvo A, Minor RK, Sinclair DA, de Cabo R, Navas P, 2009. NQR1 controls lifespan by regulating the promotion of respiratory metabolism in yeast. Aging Cell 8, 140–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobson RW, Nabholz B, Galtier N, 2010. An evolutionary genome scan for longevity-related natural selection in mammals. Mol. Biol. Evol. 27, 840–847. [DOI] [PubMed] [Google Scholar]
- Joel Brind VM, Augie, Ines Caliendo, Nicholas Vogelman, Joseph H, Zimmerman Jay A., Orentreich Norman, 2011. Dietary glycine supplementation mimics lifespan extension by dietary methionine restriction in Fisher 344 rats. Faseb J. [Google Scholar]
- Johnson JE, Johnson FB, 2014. Methionine restriction activates the retrograde response and confers both stress tolerance and lifespan extension to yeast, mouse and human cells. PLoS One 9, e97729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AA, Stolzing A, 2019. The role of lipid metabolism in aging, lifespan regulation, and age-related disease. Aging Cell 18, e13048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LC, Parker K, Aguirre BF, Nemkov TG, D’Alessandro A, Johnson SA, Seals DR, Martens CR, 2019. The plasma metabolome as a predictor of biological aging in humans. Geroscience 41, 895–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jove M, Naudi A, Aledo JC, Cabre R, Ayala V, Portero-Otin M, Barja G, Pamplona R, 2013. Plasma long-chain free fatty acids predict mammalian longevity. Sci. Rep. 3, 3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jove M, Naudi A, Gambini J, Borras C, Cabre R, Portero-Otin M, Vina J, Pamplona R, 2017. A stress-resistant lipidomic signature confers extreme longevity to humans. J. Gerontol. A Biol. Sci. Med. Sci. 72, 30–37. [DOI] [PubMed] [Google Scholar]
- Judd AM, Matthews MK, Hughes R, Veloz M, Sexton CE, Chaston JM, 2018. Bacterial methionine metabolism genes influence Drosophila melanogaster starvation resistance. Appl. Environ. Microbiol. 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juricic P, Gronke S, Partridge L, 2020. Branched-chain amino acids have equivalent effects to other essential amino acids on lifespan and aging-related traits in Drosophila. J. Gerontol. A Biol. Sci. Med. Sci. 75, 24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabil H, Kabil O, Banerjee R, Harshman LG, Pletcher SD, 2011. Increased transsulfuration mediates longevity and dietary restriction in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 108, 16831–16836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M, Andalis AA, Fink GR, Guarente L, 2002. High osmolarity extends life span in Saccharomyces cerevisiae by a mechanism related to calorie restriction. Mol. Cell. Biol. 22, 8056–8066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M, Hu D, Kerr EO, Tsuchiya M, Westman EA, Dang N, Fields S, Kennedy BK, 2005. Increased life span due to calorie restriction in respiratory-deficient yeast. PLoS Genet. 1, e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalman J, McConathy W, Araoz C, Kasa P, Lacko AG, 2000. Apolipoprotein D in the aging brain and in Alzheimer’s dementia. Neurol. Res. 22, 330–336. [DOI] [PubMed] [Google Scholar]
- Kang HL, Benzer S, Min KT, 2002. Life extension in Drosophila by feeding a drug. Proc. Natl. Acad. Sci. U.S.A. 99, 838–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan K, Fridell YW, 2013. Functional implications of Drosophila insulin-like peptides in metabolism, aging, and dietary restriction. Front. Physiol. 4, 288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappos L, Gold R, Miller DH, Macmanus DG, Havrdova E, Limmroth V, Polman CH, Schmierer K, Yousry TA, Yang M, Eraksoy M, Meluzinova E, Rektor I, Dawson KT, Sandrock AW, O’Neill GN, Investigators BGPIS, 2008. Efficacy and safety of oral fumarate in patients with relapsing-remitting multiple sclerosis: a multicentre, randomised, double-blind, placebo-controlled phase IIb study. Lancet 372, 1463–1472. [DOI] [PubMed] [Google Scholar]
- Katewa SD, Demontis F, Kolipinski M, Hubbard A, Gill MS, Perrimon N, Melov S, Kapahi P, 2012. Intramyocellular fatty-acid metabolism plays a critical role in mediating responses to dietary restriction in Drosophila melanogaster. Cell Metab. 16, 97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsyuba E, Mottis A, Zietak M, De Franco F, van der Velpen V, Gariani K, Ryu D, Cialabrini L, Matilainen O, Liscio P, Giacche N, Stokar-Regenscheit N, Legouis D, de Seigneux S, Ivanisevic J, Raffaelli N, Schoonjans K, Pellicciari R, Auwerx J, 2018. De novo NAD(+) synthesis enhances mitochondrial function and improves health. Nature 563, 354–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Sun H, 2007. Functional genomic approach to identify novel genes involved in the regulation of oxidative stress resistance and animal lifespan. Aging Cell 6, 489–503. [DOI] [PubMed] [Google Scholar]
- Kim Y, Sun H, 2012. ASM-3 acid sphingomyelinase functions as a positive regulator of the DAF-2/AGE-1 signaling pathway and serves as a novel anti-aging target. PLoS One 7, e45890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim GH, Lee YE, Lee GH, Cho YH, Lee YN, Jang Y, Paik D, Park JJ, 2015. Overexpression of malic enzyme in the larval stage extends Drosophila lifespan. Biochem. Biophys. Res. Commun. 456, 676–682. [DOI] [PubMed] [Google Scholar]
- Kishita Y, Tsuda M, Aigaki T, 2012. Impaired fatty acid oxidation in a Drosophila model of mitochondrial trifunctional protein (MTP) deficiency. Biochem. Biophys. Res. Commun. 419, 344–349. [DOI] [PubMed] [Google Scholar]
- Knopman DS, Jack CR Jr., Wiste HJ, Lundt ES, Weigand SD, Vemuri P, Lowe VJ, Kantarci K, Gunter JL, Senjem ML, Mielke MM, Roberts RO, Boeve BF, Petersen RC, 2014. 18F-fluorodeoxyglucose positron emission tomography, aging, and apolipoprotein E genotype in cognitively normal persons. Neurobiol. Aging 35, 2096–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kold-Christensen R, Johannsen M, 2020. Methylglyoxal metabolism and aging-related disease: moving from correlation toward causation. Trends Endocrinol. Metab. 31, 81–92. [DOI] [PubMed] [Google Scholar]
- Koziel R, Ruckenstuhl C, Albertini E, Neuhaus M, Netzberger C, Bust M, Madeo F, Wiesner RJ, Jansen-Durr P, 2014. Methionine restriction slows down senescence in human diploid fibroblasts. Aging Cell 13, 1038–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni AS, Gubbi S, Barzilai N, 2020. Benefits of metformin in attenuating the hallmarks of aging. Cell Metab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowski B, Hekimi S, 1996. Determination of life-span in Caenorhabditis elegans by four clock genes. Science (New York, N.Y 272, 1010–1013. [DOI] [PubMed] [Google Scholar]
- Larsen PL, Clarke CF, 2002. Extension of life-span in Caenorhabditis elegans by a diet lacking coenzyme Q. Science (New York, N.Y 295, 120–123. [DOI] [PubMed] [Google Scholar]
- Lautrup S, Sinclair DA, Mattson MP, Fang EF, 2019. NAD(+) in brain aging and neurodegenerative disorders. Cell Metab. 30, 630–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laye MJ, Tran V, Jones DP, Kapahi P, Promislow DE, 2015. The effects of age and dietary restriction on the tissue-specific metabolome of Drosophila. Aging Cell 14, 797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Couteur DG, Solon-Biet S, Cogger VC, Mitchell SJ, Senior A, de Cabo R, Raubenheimer D, Simpson SJ, 2016. The impact of low-protein high-carbohydrate diets on aging and lifespan. Cell. Mol. Life Sci. 73, 1237–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SS, Kennedy S, Tolonen AC, Ruvkun G, 2003a. DAF-16 target genes that control C. Elegans life-span and metabolism. Science (New York, N.Y 300, 644–647. [DOI] [PubMed] [Google Scholar]
- Lee SS, Lee RY, Fraser AG, Kamath RS, Ahringer J, Ruvkun G, 2003b. A systematic RNAi screen identifies a critical role for mitochondria in C. Elegans longevity. Nat. Genet. 33, 40–48. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Murphy CT, Kenyon C, 2009. Glucose shortens the life span of C. Elegans by downregulating DAF-16/FOXO activity and aquaporin gene expression. Cell Metab. 10, 379–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Lee SK, Paik D, Min KJ, 2012. Overexpression of fatty-acid-beta-oxidation-related genes extends the lifespan of Drosophila melanogaster. Oxid. Med. Cell. Longev. 2012, 854502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BC, Kaya A, Ma S, Kim G, Gerashchenko MV, Yim SH, Hu Z, Harshman LG, Gladyshev VN, 2014. Methionine restriction extends lifespan of Drosophila melanogaster under conditions of low amino-acid status. Nat. Commun. 5, 3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee GJ, Lim JJ, Hyun S, 2017. Minocycline treatment increases resistance to oxidative stress and extends lifespan in Drosophila via FOXO. Oncotarget 8, 87878–87890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legan SK, Rebrin I, Mockett RJ, Radyuk SN, Klichko VI, Sohal RS, Orr WC, 2008. Overexpression of glucose-6-phosphate dehydrogenase extends the life span of Drosophila melanogaster. J. Biol. Chem. 283, 32492–32499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemire BD, Behrendt M, DeCorby A, Gaskova D, 2009. C. Elegans longevity pathways converge to decrease mitochondrial membrane potential. Mech. Ageing Dev. 130, 461–465. [DOI] [PubMed] [Google Scholar]
- Li ZH, Gao X, Chung VC, Zhong WF, Fu Q, Lv YB, Wang ZH, Shen D, Zhang XR, Zhang PD, Li FR, Huang QM, Chen Q, Song WQ, Wu XB, Shi XM, Kraus VB, Yang X, Mao C, 2020. Associations of regular glucosamine use with all-cause and cause-specific mortality: a large prospective cohort study. Ann. Rheum. Dis. 79, 829–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SJ, Defossez PA, Guarente L, 2000. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science (New York, N.Y 289, 2126–2128. [DOI] [PubMed] [Google Scholar]
- Lin SS, Manchester JK, Gordon JI, 2001. Enhanced gluconeogenesis and increased energy storage as hallmarks of aging in Saccharomyces cerevisiae. J. Biol. Chem. 276, 36000–36007. [DOI] [PubMed] [Google Scholar]
- Lin SJ, Kaeberlein M, Andalis AA, Sturtz LA, Defossez PA, Culotta VC, Fink GR, Guarente L, 2002. Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature 418, 344–348. [DOI] [PubMed] [Google Scholar]
- Lin YH, Chen YC, Kao TY, Lin YC, Hsu TE, Wu YC, Ja WW, Brummel TJ, Kapahi P, Yuh CH, Yu LK, Lin ZH, You RJ, Jhong YT, Wang HD, 2014. Diacylglycerol lipase regulates lifespan and oxidative stress response by inversely modulating TOR signaling in Drosophila and C. Elegans. Aging Cell 13, 755–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Atamna H, Kuratsune H, Ames BN, 2002. Delaying brain mitochondrial decay and aging with mitochondrial antioxidants and metabolites. Ann. N. Y. Acad. Sci. 959, 133–166. [DOI] [PubMed] [Google Scholar]
- Liu X, Jiang N, Hughes B, Bigras E, Shoubridge E, Hekimi S, 2005. Evolutionary conservation of the clk-1-dependent mechanism of longevity: loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev. 19, 2424–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YL, Lu WC, Brummel TJ, Yuh CH, Lin PT, Kao TY, Li FY, Liao PC, Benzer S, Wang HD, 2009. Reduced expression of alpha-1,2-mannosidase I extends lifespan in Drosophila melanogaster and Caenorhabditis elegans. Aging Cell 8, 370–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Wu Q, He D, Ma T, Du L, Dui W, Guo X, Jiao R, 2011. Drosophila sbo regulates lifespan through its function in the synthesis of coenzyme Q in vivo. J. Genet. Genomics 38, 225–234. [DOI] [PubMed] [Google Scholar]
- Liu JL, Yee C, Wang Y, Hekimi S, 2017. A single biochemical activity underlies the pleiotropy of the aging-related protein CLK-1. Sci. Rep. 7, 859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YJ, Janssens GE, McIntyre RL, Molenaars M, Kamble R, Gao AW, Jongejan A, Weeghel MV, MacInnes AW, Houtkooper RH, 2019. Glycine promotes longevity in Caenorhabditis elegans in a methionine cycle-dependent fashion. PLoS Genet. 15, e1007633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long DM, Frame AK, Reardon PN, Cumming RC, Hendrix DA, Kretzschmar D, Giebultowicz JM, 2020. Lactate dehydrogenase expression modulates longevity and neurodegeneration in Drosophila melanogaster. Aging (Albany NY) 12, 10041–10058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo VD, Shadel GS, Kaeberlein M, Kennedy B, 2012. Replicative and chronological aging in Saccharomyces cerevisiae. Cell Metab. 16, 18–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G, 2013. The hallmarks of aging. Cell 153, 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love DC, Ghosh S, Mondoux MA, Fukushige T, Wang P, Wilson MA, Iser WB, Wolkow CA, Krause MW, Hanover JA, 2010. Dynamic O-GlcNAc cycling at promoters of Caenorhabditis elegans genes regulating longevity, stress, and immunity. Proc. Natl. Acad. Sci. U.S.A. 107, 7413–7418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucanic M, Held JM, Vantipalli MC, Klang IM, Graham JB, Gibson BW, Lithgow GJ, Gill MS, 2011. N-acylethanolamine signalling mediates the effect of diet on lifespan in Caenorhabditis elegans. Nature 473, 226–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luckinbill LS, Riha V, Rhine S, Grudzien TA, 1990. The role of glucose-6-phosphate dehydrogenase in the evolution of longevity in Drosophila melanogaster. Heredity (Edinb) 65 (Pt 1), 29–38. [DOI] [PubMed] [Google Scholar]
- Ludewig AH, Schroeder FC, 2013. Ascaroside signaling in C. elegans. WormBook, pp. 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludewig AH, Izrayelit Y, Park D, Malik RU, Zimmermann A, Mahanti P, Fox BW, Bethke A, Doering F, Riddle DL, Schroeder FC, 2013. Pheromone sensing regulates Caenorhabditis elegans lifespan and stress resistance via the deacetylase SIR-2.1. Proc. Natl. Acad. Sci. U.S.A. 110, 5522–5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttik MA, Overkamp KM, Kotter P, de Vries S, van Dijken JP, Pronk JT, 1998. The Saccharomyces cerevisiae NDE1 and NDE2 genes encode separate mitochondrial NADH dehydrogenases catalyzing the oxidation of cytosolic NADH. J. Biol. Chem. 273, 24529–24534. [DOI] [PubMed] [Google Scholar]
- Lylyk MP, Bayliak MM, Shmihel HV, Storey JM, Storey KB, Lushchak VI, 2018. Effects of alpha-ketoglutarate on lifespan and functional aging of Drosophila melanogaster flies. Ukr. Biochem. J. 90, 49–61. [Google Scholar]
- Ma S, Yim SH, Lee SG, Kim EB, Lee SR, Chang KT, Buffenstein R, Lewis KN, Park TJ, Miller RA, Clish CB, Gladyshev VN, 2015. Organization of the mammalian metabolome according to organ function, lineage specialization, and longevity. Cell Metab. 22, 332–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, Upneja A, Galecki A, Tsai YM, Burant CF, Raskind S, Zhang Q, Zhang ZD, Seluanov A, Gorbunova V, Clish CB, Miller RA, Gladyshev VN, 2016. Cell culture-based profiling across mammals reveals DNA repair and metabolism as determinants of species longevity. eLife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, Avanesov AS, Porter E, Lee BC, Mariotti M, Zemskaya N, Guigo R, Moskalev AA, Gladyshev VN, 2018a. Comparative transcriptomics across 14 Drosophila species reveals signatures of longevity. Aging Cell 17, e12740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Wang H, Cai Y, Wang H, Niu K, Wu X, Ma H, Yang Y, Tong W, Liu F, Liu Z, Zhang Y, Liu R, Zhu ZJ, Liu N, 2018b. Epigenetic drift of H3K27me3 in aging links glycolysis to healthy longevity in Drosophila. eLife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie SM, Brooker MR, Gill TR, Cox GB, Howells AJ, Ewart GD, 1999. Mutations in the white gene of Drosophila melanogaster affecting ABC transporters that determine eye colouration. Biochim. Biophys. Acta 1419, 173–185. [DOI] [PubMed] [Google Scholar]
- Mair W, Piper MD, Partridge L, 2005. Calories do not explain extension of life span by dietary restriction in Drosophila. PLoS Biol. 3, e223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansfeld J, Urban N, Priebe S, Groth M, Frahm C, Hartmann N, Gebauer J, Ravichandran M, Dommaschk A, Schmeisser S, Kuhlow D, Monajembashi S, Bremer-Streck S, Hemmerich P, Kiehntopf M, Zamboni N, Englert C, Guthke R, Kaleta C, Platzer M, Suhnel J, Witte OW, Zarse K, Ristow M, 2015. Branched-chain amino acid catabolism is a conserved regulator of physiological ageing. Nat. Commun. 6, 10043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens CR, Denman BA, Mazzo MR, Armstrong ML, Reisdorph N, McQueen MB, Chonchol M, Seals DR, 2018. Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD(+) in healthy middle-aged and older adults. Nat. Commun. 9, 1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Reyes I, Chandel NS, 2020. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 11, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Montalvo A, Mercken EM, Mitchell SJ, Palacios HH, Mote PL, Scheibye-Knudsen M, Gomes AP, Ward TM, Minor RK, Blouin MJ, Schwab M, Pollak M, Zhang Y, Yu Y, Becker KG, Bohr VA, Ingram DK, Sinclair DA, Wolf NS, Spindler SR, Bernier M, de Cabo R, 2013. Metformin improves healthspan and lifespan in mice. Nat. Commun. 4, 2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Montalvo A, Sun Y, Diaz-Ruiz A, Ali A, Gutierrez V, Palacios HH, Curtis J, Siendones E, Ariza J, Abulwerdi GA, Sun X, Wang AX, Pearson KJ, Fishbein KW, Spencer RG, Wang M, Han X, Scheibye-Knudsen M, Baur JA, Shertzer HG, Navas P, Villalba JM, Zou S, Bernier M, de Cabo R, 2016. Cytochrome b5 reductase and the control of lipid metabolism and healthspan. NPJ Aging Mech. Dis. 2, 16006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews MK, Wilcox H, Hughes R, Veloz M, Hammer A, Banks B, Walters A, Schneider KJ, Sexton CE, Chaston JM, 2020. Genetic influences of the microbiota on the life span of Drosophila melanogaster. Appl. Environ. Microbiol. 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty MF, Barroso-Aranda J, Contreras F, 2009. The low-methionine content of vegan diets may make methionine restriction feasible as a life extension strategy. Med. Hypotheses 72, 125–128. [DOI] [PubMed] [Google Scholar]
- McCormick M, Chen K, Ramaswamy P, Kenyon C, 2012. New genes that extend Caenorhabditis elegans’ lifespan in response to reproductive signals. Aging Cell 11, 192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick MA, Delaney JR, Tsuchiya M, TsuchiyaM.a S, Shemorry A, Sim S, Chou AC, Ahmed U, Carr D, Murakami CJ, Schleit J, Sutphin GL, Wasko BM, Bennett CF, Wang AM, Olsen B, Beyer RP, Bammler TK, Prunkard D, Johnson SC, Pennypacker JK, An E, Anies A, Castanza AS, Choi E, Dang N, Enerio S, Fletcher M, Fox L, Goswami S, Higgins SA, Holmberg MA, Hu D, Hui J, Jelic M, Jeong KS, Johnston E, Kerr EO, Kim J, Kim D, Kirkland K, Klum S, Kotireddy S, Liao E, Lim M, Lin MS, Lo WC, Lockshon D, Miller HA, Moller RM, Muller B, Oakes J, Pak DN, Peng ZJ, Pham KM, Pollard TG, Pradeep P, Pruett D, Rai D, Robison B, Rodriguez AA, Ros B, Sage M, Singh MK, Smith ED, Snead K, Solanky A, Spector BL, Steffen KK, Tchao BN, Ting MK, Vander Wende H, Wang D, Welton KL, Westman EA, Brem RB, Liu XG, Suh Y, Zhou Z, Kaeberlein M, Kennedy BK, 2015. A comprehensive analysis of replicative lifespan in 4,698 single-gene deletion strains uncovers conserved mechanisms of aging. Cell Metab. 22, 895–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuary PR, Liao CY, Chang JT, Kumsta C, She X, Davis A, Chu CC, Gelino S, Gomez-Amaro RL, Petrascheck M, Brill LM, Ladiges WC, Kennedy BK, Hansen M, 2016. C. Elegans S6K mutants require a creatine-kinase-like effector for lifespan extension. Cell Rep. 14, 2059–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menni C, Kastenmuller G, Petersen AK, Bell JT, Psatha M, Tsai PC, Gieger C, Schulz H, Erte I, John S, Brosnan MJ, Wilson SG, Tsaprouni L, Lim EM, Stuckey B, Deloukas P, Mohney R, Suhre K, Spector TD, Valdes AM, 2013. Metabolomic markers reveal novel pathways of ageing and early development in human populations. Int. J. Epidemiol. 42, 1111–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RA, Buehner G, Chang Y, Harper JM, Sigler R, Smith-Wheelock M, 2005. Methionine-deficient diet extends mouse lifespan, slows immune and lens aging, alters glucose, T4, IGF-I and insulin levels, and increases hepatocyte MIF levels and stress resistance. Aging Cell 4, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RA, Harrison DE, Astle CM, Bogue MA, Brind J, Fernandez E, Flurkey K, Javors M, Ladiges W, Leeuwenburgh C, Macchiarini F, Nelson J, Ryazanov AG, Snyder J, Stearns TM, Vaughan DE, Strong R, 2019. Glycine supplementation extends lifespan of male and female mice. Aging Cell 18, e12953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minois N, Carmona-Gutierrez D, Madeo F, 2011. Polyamines in aging and disease. Aging (Albany NY) 3, 716–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor RK, Smith DL Jr., Sossong AM, Kaushik S, Poosala S, Spangler EL, Roth GS, Lane M, Allison DB, de Cabo R, Ingram DK, Mattison JA, 2010. Chronic ingestion of 2-deoxy-D-glucose induces cardiac vacuolization and increases mortality in rats. Toxicol. Appl. Pharmacol. 243, 332–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishur RJ, Khan M, Munkacsy E, Sharma L, Bokov A, Beam H, Radetskaya O, Borror M, Lane R, Bai Y, Rea SL, 2016. Mitochondrial metabolites extend lifespan. Aging Cell 15, 336–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell SJ, Bernier M, Aon MA, Cortassa S, Kim EY, Fang EF, Palacios HH, Ali A, Navas-Enamorado I, Di Francesco A, Kaiser TA, Waltz TB, Zhang N, Ellis JL, Elliott PJ, Frederick DW, Bohr VA, Schmidt MS, Brenner C, Sinclair DA, Sauve AA, Baur JA, de Cabo R, 2018. Nicotinamide improves aspects of healthspan, but not lifespan, in mice. Cell Metab. 27 (667–676), e664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizote A, Yamada M, Yoshizane C, Arai N, Maruta K, Arai S, Endo S, Ogawa R, Mitsuzumi H, Ariyasu T, Fukuda S, 2016. Daily intake of trehalose is effective in the prevention of lifestyle-related diseases in individuals with risk factors for metabolic syndrome. J. Nutr. Sci. Vitaminol (Tokyo) 62, 380–387. [DOI] [PubMed] [Google Scholar]
- Moghadasian MH, McManus BM, Nguyen LB, Shefer S, Nadji M, Godin DV, Green TJ, Hill J, Yang Y, Scudamore CH, Frohlich JJ, 2001. Pathophysiology of apolipoprotein E deficiency in mice: relevance to apo E-related disorders in humans. FASEB J. 15, 2623–2630. [DOI] [PubMed] [Google Scholar]
- Monaghan RM, Barnes RG, Fisher K, Andreou T, Rooney N, Poulin GB, Whitmarsh AJ, 2015. A nuclear role for the respiratory enzyme CLK-1 in regulating mitochondrial stress responses and longevity. Nat. Cell Biol. 17, 782–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montoliu I, Scherer M, Beguelin F, DaSilva L, Mari D, Salvioli S, Martin FP, Capri M, Bucci L, Ostan R, Garagnani P, Monti D, Biagi E, Brigidi P, Kussmann M, Rezzi S, Franceschi C, Collino S, 2014. Serum profiling of healthy aging identifies phospho- and sphingolipid species as markers of human longevity. Aging (Albany NY) 6, 9–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morais Cabral JH, Atkins GL, Sanchez LM, Lopez-Boado YS, Lopez-Otin C, Sawyer L, 1995. Arachidonic acid binds to apolipoprotein D: implications for the protein’s function. FEBS Lett. 366, 53–56. [DOI] [PubMed] [Google Scholar]
- Morcos M, Du X, Pfisterer F, Hutter H, Sayed AA, Thornalley P, Ahmed N, Baynes J, Thorpe S, Kukudov G, Schlotterer A, Bozorgmehr F, El Baki RA, Stern D, Moehrlen F, Ibrahim Y, Oikonomou D, Hamann A, Becker C, Zeier M, Schwenger V, Miftari N, Humpert P, Hammes HP, Buechler M, Bierhaus A, Brownlee M, Nawroth PP, 2008. Glyoxalase-1 prevents mitochondrial protein modification and enhances lifespan in Caenorhabditis elegans. Aging Cell 7, 260–269. [DOI] [PubMed] [Google Scholar]
- Moroz N, Carmona JJ, Anderson E, Hart AC, Sinclair DA, Blackwell TK, 2014. Dietary restriction involves NAD(+) -dependent mechanisms and a shift toward oxidative metabolism. Aging Cell 13, 1075–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosbech MB, Kruse R, Harvald EB, Olsen AS, Gallego SF, Hannibal-Bach HK, Ejsing CS, Faergeman NJ, 2013. Functional loss of two ceramide synthases elicits autophagy-dependent lifespan extension in C. Elegans. PLoS One 8, e70087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motola DL, Cummins CL, Rottiers V, Sharma KK, Li T, Li Y, Suino-Powell K, Xu HE, Auchus RJ, Antebi A, Mangelsdorf DJ, 2006. Identification of ligands for DAF-12 that govern dauer formation and reproduction in C. Elegans. Cell 124, 1209–1223. [DOI] [PubMed] [Google Scholar]
- Mouchiroud L, Molin L, Kasturi P, Triba MN, Dumas ME, Wilson MC, Halestrap AP, Roussel D, Masse I, Dalliere N, Segalat L, Billaud M, Solari F, 2011. Pyruvate imbalance mediates metabolic reprogramming and mimics lifespan extension by dietary restriction in Caenorhabditis elegans. Aging Cell 10, 39–54. [DOI] [PubMed] [Google Scholar]
- Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Canto C, Mottis A, Jo YS, Viswanathan M, Schoonjans K, Guarente L, Auwerx J, 2013. The NAD(+)/Sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 154, 430–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourikis P, Hurlbut GD, Artavanis-Tsakonas S, 2006. Enigma, a mitochondrial protein affecting lifespan and oxidative stress response in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 103, 1307–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muffat J, Walker DW, Benzer S, 2008. Human ApoD, an apolipoprotein up-regulated in neurodegenerative diseases, extends lifespan and increases stress resistance in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 105, 7088–7093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munkacsy E, Rea SL, 2014. The paradox of mitochondrial dysfunction and extended longevity. Exp. Gerontol. 56, 221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, Li H, Kenyon C, 2003. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 424, 277–283. [DOI] [PubMed] [Google Scholar]
- Newman JC, Verdin E, 2017. Beta-hydroxybutyrate: a signaling metabolite. Annu. Rev. Nutr. 37, 51–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman JC, Covarrubias AJ, Zhao M, Yu X, Gut P, Ng CP, Huang Y, Haldar S, Verdin E, 2017. Ketogenic diet reduces midlife mortality and improves memory in aging mice. Cell Metab. 26 (547–557), e548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholatos JW, Robinette TM, Tata SVP, Yordy JD, Francisco AB, Platov M, Yeh TK, Ilkayeva OR, Huynh FK, Dokukin M, Volkov D, Weinstein MA, Boyko AR, Miller RA, Sokolov I, Hirschey MD, Libert S, 2019. Cellular energetics and mitochondrial uncoupling in canine aging. Geroscience 41, 229–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisoli E, Tonello C, Cardile A, Cozzi V, Bracale R, Tedesco L, Falcone S, Valerio A, Cantoni O, Clementi E, Moncada S, Carruba MO, 2005. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science (New York, N.Y 310, 314–317. [DOI] [PubMed] [Google Scholar]
- Nobrega-Pereira S, Fernandez-Marcos PJ, Brioche T, Gomez-Cabrera MC, Salvador-Pascual A, Flores JM, Vina J, Serrano M, 2016. G6PD protects from oxidative damage and improves healthspan in mice. Nat. Commun. 7, 10894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Rourke EJ, Soukas AA, Carr CE, Ruvkun G, 2009. C. Elegans major fats are stored in vesicles distinct from lysosome-related organelles. Cell Metab. 10, 430–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Rourke EJ, Kuballa P, Xavier R, Ruvkun G, 2013. omega-6 Polyunsaturated fatty acids extend life span through the activation of autophagy. Genes Dev. 27, 429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata F, Miura M, 2015. Enhancing S-adenosyl-methionine catabolism extends Drosophila lifespan. Nat. Commun. 6, 8332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SI, Park JK, Park SK, 2015. Lifespan extension and increased resistance to environmental stressors by N-acetyl-L-cysteine in Caenorhabditis elegans. Clinics (Sao Paulo) 70, 380–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooka H, Segall PE, Timiras PS, 1988. Histology and survival in age-delayed low-tryptophan-fed rats. Mech. Ageing Dev. 43, 79–98. [DOI] [PubMed] [Google Scholar]
- Orentreich N, Matias JR, DeFelice A, Zimmerman JA, 1993. Low methionine ingestion by rats extends life span. J. Nutr. 123, 269–274. [DOI] [PubMed] [Google Scholar]
- Orr WC, Radyuk SN, Prabhudesai L, Toroser D, Benes JJ, Luchak JM, Mockett RJ, Rebrin I, Hubbard JG, Sohal RS, 2005. Overexpression of glutamate-cysteine ligase extends life span in Drosophila melanogaster. J. Biol. Chem. 280, 37331–37338. [DOI] [PubMed] [Google Scholar]
- Oxenkrug GF, 2010. The extended life span of Drosophila melanogaster eye-color (white and vermilion) mutants with impaired formation of kynurenine. J Neural Transm (Vienna) 117, 23–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxenkrug GF, Navrotskaya V, Voroboyva L, Summergrad P, 2011. Extension of life span of Drosophila melanogaster by the inhibitors of tryptophan-kynurenine metabolism. Fly 5, 307–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxenkrug G, Navrotskaya V, Vorobyova L, Summergrad P, 2012. Minocycline effect on life and health span of Drosophila melanogaster. Aging Dis. 3, 352–359. [PMC free article] [PubMed] [Google Scholar]
- Padalko VI, 2005. Uncoupler of oxidative phosphorylation prolongs the lifespan of Drosophila. Biochemistry Mosc. 70, 986–989. [DOI] [PubMed] [Google Scholar]
- Paik D, Jang YG, Lee YE, Lee YN, Yamamoto R, Gee HY, Yoo S, Bae E, Min KJ, Tatar M, Park JJ, 2012. Misexpression screen delineates novel genes controlling Drosophila lifespan. Mech. Ageing Dev. 133, 234–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer MR, Sackton TB, 2003. The effects of dietary coenzyme Q on Drosophila life span. Aging Cell 2, 335–339. [DOI] [PubMed] [Google Scholar]
- Pandey A, Vimal D, Chandra S, Saini S, Narayan G, Kar Chowdhuri D, 2014. Long-term dietary exposure to low concentration of dichloroacetic acid promoted longevity and attenuated cellular and functional declines in aged Drosophila melanogaster. Age (Dordr) 36, 9628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhitko AA, Favorova OO, Henske EP, 2013. Autophagy: mechanisms, regulation, and its role in tumorigenesis. Biochemistry. Biokhimiia 78, 355–367. [DOI] [PubMed] [Google Scholar]
- Parkhitko AA, Favorova OO, Khabibullin DI, Anisimov VN, Henske EP, 2014. Kinase mTOR: regulation and role in maintenance of cellular homeostasis, tumor development, and aging. Biochemistry. Biokhimiia 79, 88–101. [DOI] [PubMed] [Google Scholar]
- Parkhitko AA, Binari R, Zhang N, Asara JM, Demontis F, Perrimon N, 2016. Tissue-specific down-regulation of S-adenosyl-homocysteine via suppression of dAhcyL1/dAhcyL2 extends health span and life span in Drosophila. Genes Dev. 30, 1409–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhitko AA, Jouandin P, Mohr SE, Perrimon N, 2019. Methionine metabolism and methyltransferases in the regulation of aging and lifespan extension across species. Aging Cell 18, e13034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegg AE, 2016. Functions of Polyamines in mammals. J. Biol. Chem. 291, 14904–14912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peleg S, Feller C, Forne I, Schiller E, Sevin DC, Schauer T, Regnard C, Straub T, Prestel M, Klima C, Schmitt Nogueira M, Becker L, Klopstock T, Sauer U, Becker PB, Imhof A, Ladurner AG, 2016. Life span extension by targeting a link between metabolism and histone acetylation in Drosophila. EMBO Rep. 17, 455–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez CL, Van Gilst MR, 2008. A 13C isotope labeling strategy reveals the influence of insulin signaling on lipogenesis in C. Elegans. Cell Metab. 8, 266–274. [DOI] [PubMed] [Google Scholar]
- Pertovaara M, Raitala A, Lehtimaki T, Karhunen PJ, Oja SS, Jylha M, Hervonen A, Hurme M, 2006. Indoleamine 2,3-dioxygenase activity in nonagenarians is markedly increased and predicts mortality. Mech. Ageing Dev. 127, 497–499. [DOI] [PubMed] [Google Scholar]
- Plummer JD, Johnson JE, 2019. Extension of cellular lifespan by methionine restriction involves alterations in central carbon metabolism and is mitophagy-dependent. Front. Cell Dev. Biol. 7, 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Post S, Karashchuk G, Wade JD, Sajid W, De Meyts P, Tatar M, 2018. Drosophila insulin-like peptides DILP2 and DILP5 differentially stimulate cell signaling and Glycogen Phosphorylase to regulate longevity. Front. Endocrinol. (Lausanne) 9, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers RW 3rd, Kaeberlein M, Caldwell SD, Kennedy BK, Fields S, 2006. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 20, 174–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryor R, Norvaisas P, Marinos G, Best L, Thingholm LB, Quintaneiro LM, De Haes W, Esser D, Waschina S, Lujan C, Smith RL, Scott TA, Martinez-Martinez D, Woodward O, Bryson K, Laudes M, Lieb W, Houtkooper RH, Franke A, Temmerman L, Bjedov I, Cocheme HM, Kaleta C, Cabreiro F, 2019. Host-microbe-Drug-Nutrient screen identifies bacterial effectors of metformin therapy. Cell 178 (1299–1312), e1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi W, Gutierrez GE, Gao X, Dixon H, McDonough JA, Marini AM, Fisher AL, 2017. The omega-3 fatty acid alpha-linolenic acid extends Caenorhabditis elegans lifespan via NHR-49/PPARalpha and oxidation to oxylipins. Aging Cell 16, 1125–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafaeloff-Phail R, Ding L, Conner L, Yeh WK, McClure D, Guo H, Emerson K, Brooks H, 2004. Biochemical regulation of mammalian AMP-activated protein kinase activity by NAD and NADH. J. Biol. Chem. 279, 52934–52939. [DOI] [PubMed] [Google Scholar]
- Rajman L, Chwalek K, Sinclair DA, 2018. Therapeutic potential of NAD-Boosting molecules: the in vivo evidence. Cell Metab. 27, 529–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran PV, Savini M, Folick AK, Hu K, Masand R, Graham BH, Wang MC, 2019. Lysosomal signaling promotes longevity by adjusting mitochondrial activity. Dev. Cell 48 (685–696), e685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravichandran M, Priebe S, Grigolon G, Rozanov L, Groth M, Laube B, Guthke R, Platzer M, Zarse K, Ristow M, 2018. Impairing L-Threonine catabolism promotes healthspan through methylglyoxal-mediated proteohormesis. Cell Metab. 27 (914–925), e915. [DOI] [PubMed] [Google Scholar]
- Rea SL, Ventura N, Johnson TE, 2007. Relationship between mitochondrial electron transport chain dysfunction, development, and life extension in Caenorhabditis elegans. PLoS Biol. 5, e259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhooms SK, Murari A, Goparaju NSV, Vilanueva M, Owusu-Ansah E, 2019. Insights from Drosophila on mitochondrial complex I. Cell. Mol. Life Sci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards AB, Krakowka S, Dexter LB, Schmid H, Wolterbeek AP, Waalkens-Berendsen DH, Shigoyuki A, Kurimoto M, 2002. Trehalose: a review of properties, history of use and human tolerance, and results of multiple safety studies. Food Chem. Toxicol. 40, 871–898. [DOI] [PubMed] [Google Scholar]
- Rist MJ, Roth A, Frommherz L, Weinert CH, Kruger R, Merz B, Bunzel D, Mack C, Egert B, Bub A, Gorling B, Tzvetkova P, Luy B, Hoffmann I, Kulling SE, Watzl B, 2017. Metabolite patterns predicting sex and age in participants of the Karlsruhe Metabolomics and Nutrition (KarMeN) study. PLoS One 12, e0183228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts MN, Wallace MA, Tomilov AA, Zhou Z, Marcotte GR, Tran D, Perez G, Gutierrez-Casado E, Koike S, Knotts TA, Imai DM, Griffey SM, Kim K, Hagopian K, McMackin MZ, Haj FG, Baar K, Cortopassi GA, Ramsey JJ, Lopez-Dominguez JA, 2017. A ketogenic diet extends longevity and healthspan in adult mice. Cell Metab. 26 (539–546), e535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson O, Chadeau Hyam M, Karaman I, Climaco Pinto R, Ala-Korpela M, Handakas E, Fiorito G, Gao H, Heard A, Jarvelin MR, Lewis M, Pazoki R, Polidoro S, Tzoulaki I, Wielscher M, Elliott P, Vineis P, 2020. Determinants of accelerated metabolomic and epigenetic aging in a UK cohort. Aging Cell 19, e13149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogina B, Benzer S, Helfand SL, 1997. Drosophila drop-dead mutations accelerate the time course of age-related markers. Proc. Natl. Acad. Sci. U.S.A. 94, 6303–6306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogina B, Reenan RA, Nilsen SP, Helfand SL, 2000. Extended life-span conferred by cotransporter gene mutations in Drosophila. Science (New York, N.Y 290, 2137–2140. [DOI] [PubMed] [Google Scholar]
- Ross JM, Oberg J, Brene S, Coppotelli G, Terzioglu M, Pernold K, Goiny M, Sitnikov R, Kehr J, Trifunovic A, Larsson NG, Hoffer BJ, Olson L, 2010. High brain lactate is a hallmark of aging and caused by a shift in the lactate dehydrogenase A/B ratio. Proc. Natl. Acad. Sci. U.S.A. 107, 20087–20092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozanov L, Ravichandran M, Grigolon G, Zanellati MC, Mansfeld J, Zarse K, Barzilai N, Atzmon G, Fischer F, Ristow M, 2020. Redox-mediated regulation of aging and healthspan by an evolutionarily conserved transcription factor HLH-2/Tcf3/E2A. Redox Biol. 32, 101448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruckenstuhl C, Netzberger C, Entfellner I, Carmona-Gutierrez D, Kickenweiz T, Stekovic S, Gleixner C, Schmid C, Klug L, Sorgo AG, Eisenberg T, Buttner S, Marino G, Koziel R, Jansen-Durr P, Frohlich KU, Kroemer G, Madeo F, 2014. Lifespan extension by methionine restriction requires autophagy-dependent vacuolar acidification. PLoS Genet. 10, e1004347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagi D, Kim SK, 2012. An engineering approach to extending lifespan in C. Elegans. PLoS Genet. 8, e1002780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiki R, Lunceford AL, Bixler T, Dang P, Lee W, Furukawa S, Larsen PL, Clarke CF, 2008. Altered bacterial metabolism, not coenzyme Q content, is responsible for the lifespan extension in Caenorhabditis elegans fed an Escherichia coli diet lacking coenzyme Q. Aging Cell 7, 291–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Blanco A, Fridell YW, Helfand SL, 2006. Involvement of Drosophila uncoupling protein 5 in metabolism and aging. Genetics 172, 1699–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz A, Soikkeli M, Portero-Otin M, Wilson A, Kemppainen E, McIlroy G, Ellila S, Kemppainen KK, Tuomela T, Lakanmaa M, Kiviranta E, Stefanatos R, Dufour E, Hutz B, Naudi A, Jove M, Zeb A, Vartiainen S, Matsuno-Yagi A, Yagi T, Rustin P, Pamplona R, Jacobs HT, 2010. Expression of the yeast NADH dehydrogenase Ndi1 in Drosophila confers increased lifespan independently of dietary restriction. Proc. Natl. Acad. Sci. U.S.A. 107, 9105–9110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer S, Gruber J, Ng LF, Fong S, Wong YT, Tang SY, Halliwell B, 2011. The effect of dichloroacetate on health- and lifespan in C. Elegans. Biogerontology 12, 195–209. [DOI] [PubMed] [Google Scholar]
- Schmeisser K, Mansfeld J, Kuhlow D, Weimer S, Priebe S, Heiland I, Birringer M, Groth M, Segref A, Kanfi Y, Price NL, Schmeisser S, Schuster S, Pfeiffer AF, Guthke R, Platzer M, Hoppe T, Cohen HY, Zarse K, Sinclair DA, Ristow M, 2013a. Role of sirtuins in lifespan regulation is linked to methylation of nicotinamide. Nat. Chem. Biol. 9, 693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeisser S, Schmeisser K, Weimer S, Groth M, Priebe S, Fazius E, Kuhlow D, Pick D, Einax JW, Guthke R, Platzer M, Zarse K, Ristow M, 2013b. Mitochondrial hormesis links low-dose arsenite exposure to lifespan extension. Aging Cell 12, 508–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A, Sivaraman J, Li Y, Larocque R, Barbosa JA, Smith C, Matte A, Schrag JD, Cygler M, 2001. Three-dimensional structure of 2-amino-3-ketobutyrate CoA ligase from Escherichia coli complexed with a PLP-substrate intermediate: inferred reaction mechanism. Biochemistry 40, 5151–5160. [DOI] [PubMed] [Google Scholar]
- Schug ZT, Vande Voorde J, Gottlieb E, 2016. The metabolic fate of acetate in cancer. Nature reviews 16, 708–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M, 2007. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 6, 280–293. [DOI] [PubMed] [Google Scholar]
- Schwarcz R, Bruno JP, Muchowski PJ, Wu HQ, 2012. Kynurenines in the mammalian brain: when physiology meets pathology. Nat. Rev. Neurosci. 13, 465–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scialo F, Sriram A, Fernandez-Ayala D, Gubina N, Lohmus M, Nelson G, Logan A, Cooper HM, Navas P, Enriquez JA, Murphy MP, Sanz A, 2016. Mitochondrial ROS produced via reverse Electron transport extend animal lifespan. Cell Metab. 23, 725–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seah NE, de Magalhaes Filho CD, Petrashen AP, Henderson HR, Laguer J, Gonzalez J, Dillin A, Hansen M, Lapierre LR, 2016. Autophagy-mediated longevity is modulated by lipoprotein biogenesis. Autophagy 12, 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo Y, Kingsley S, Walker G, Mondoux MA, Tissenbaum HA, 2018. Metabolic shift from glycogen to trehalose promotes lifespan and healthspan in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 115, E2791–E2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shats I, Williams JG, Liu J, Makarov MV, Wu X, Lih FB, Deterding LJ, Lim C, Xu X, Randall TA, Lee E, Li W, Fan W, Li JL, Sokolsky M, Kabanov AV, Li L, Migaud ME, Locasale JW, Li X, 2020. Bacteria boost mammalian host NAD metabolism by engaging the deamidated biosynthesis pathway. Cell Metab. 31 (564–579), e567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin MG, Lee JW, Han JS, Lee B, Jeong JH, Park SH, Kim JH, Jang S, Park M, Kim SY, Kim S, Yang YR, Kim JY, Hoe KL, Park C, Lee KP, Kwon KS, Kwon ES, 2020. Bacteria-derived metabolite, methylglyoxal, modulates the longevity of C. Elegans through TORC2/SGK-1/DAF-16 signaling. Proc. Natl. Acad. Sci. U.S.A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmookler Reis RJ, Xu L, Lee H, Chae M, Thaden JJ, Bharill P, Tazearslan C, Siegel E, Alla R, Zimniak P, Ayyadevara S, 2011. Modulation of lipid biosynthesis contributes to stress resistance and longevity of C. Elegans mutants. Aging (Albany NY) 3, 125–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinadinos C, Valles-Ortega J, Boulan L, Solsona E, Tevy MF, Marquez M, Duran J, Lopez-Iglesias C, Calbo J, Blasco E, Pumarola M, Milan M, Guinovart JJ, 2014. Neuronal glycogen synthesis contributes to physiological aging. Aging Cell 13, 935–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack C, Foley A, Partridge L, 2012. Activation of AMPK by the putative dietary restriction mimetic metformin is insufficient to extend lifespan in Drosophila. PLoS One 7, e47699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smathers RL, Petersen DR, 2011. The human fatty acid-binding protein family: evolutionary divergences and functions. Hum. Genomics 5, 170–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal RS, Arnold L, Orr WC, 1990. Effect of age on superoxide dismutase, catalase, glutathione reductase, inorganic peroxides, TBA-reactive material, GSH/GSSG, NADPH/NADP+ and NADH/NAD+ in Drosophila melanogaster. Mech. Ageing Dev. 56, 223–235. [DOI] [PubMed] [Google Scholar]
- Solerte SB, Gazzaruso C, Bonacasa R, Rondanelli M, Zamboni M, Basso C, Locatelli E, Schifino N, Giustina A, Fioravanti M, 2008. Nutritional supplements with oral amino acid mixtures increases whole-body lean mass and insulin sensitivity in elderly subjects with sarcopenia. Am. J. Cardiol. 101, 69E–77E. [DOI] [PubMed] [Google Scholar]
- Solon-Biet SM, McMahon AC, Ballard JW, Ruohonen K, Wu LE, Cogger VC, Warren A, Huang X, Pichaud N, Melvin RG, Gokarn R, Khalil M, Turner N, Cooney GJ, Sinclair DA, Raubenheimer D, Le Couteur DG, Simpson SJ, 2014. The ratio of macronutrients, not caloric intake, dictates cardiometabolic health, aging, and longevity in ad libitum-fed mice. Cell Metab. 19, 418–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solon-Biet SM, Cogger VC, Pulpitel T, Wahl D, Clark X, Bagley E, Gregoriou GC, Senior AM, Wang QP, Brandon AE, Perks R, O’Sullivan J, Koay YC, Bell-Anderson K, Kebede M, Yau B, Atkinson C, Svineng G, Dodgson T, Wali JA, Piper MDW, Juricic P, Partridge L, Rose AJ, Raubenheimer D, Cooney GJ, Le Couteur DG, Simpson SJ, 2019. Branched chain amino acids impact health and lifespan indirectly via amino acid balance and appetite control. Nat Metab 1, 532–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soultoukis GA, Partridge L, 2016. Dietary protein, metabolism, and aging. Annu. Rev. Biochem. 85, 5–34. [DOI] [PubMed] [Google Scholar]
- Stacpoole PW, 2012. The pyruvate dehydrogenase complex as a therapeutic target for age-related diseases. Aging Cell 11, 371–377. [DOI] [PubMed] [Google Scholar]
- Steinbaugh MJ, Narasimhan SD, Robida-Stubbs S, Moronetti Mazzeo LE, Dreyfuss JM, Hourihan JM, Raghavan P, Operana TN, Esmaillie R, Blackwell TK, 2015. Lipid-mediated regulation of SKN-1/Nrf in response to germ cell absence. eLife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stekovic S, Hofer SJ, Tripolt N, Aon MA, Royer P, Pein L, Stadler JT, Pendl T, Prietl B, Url J, Schroeder S, Tadic J, Eisenberg T, Magnes C, Stumpe M, Zuegner E, Bordag N, Riedl R, Schmidt A, Kolesnik E, Verheyen N, Springer A, Madl T, Sinner F, de Cabo R, Kroemer G, Obermayer-Pietsch B, Dengjel J, Sourij H, Pieber TR, Madeo F, 2019. Alternate day fasting improves physiological and molecular markers of aging in healthy, non-obese humans. Cell Metab. 30 (462–476), e466. [DOI] [PubMed] [Google Scholar]
- Stenesen D, Suh JM, Seo J, Yu K, Lee KS, Kim JS, Min KJ, Graff JM, 2013. Adenosine nucleotide biosynthesis and AMPK regulate adult life span and mediate the longevity benefit of caloric restriction in flies. Cell Metab. 17, 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stincone A, Prigione A, Cramer T, Wamelink MM, Campbell K, Cheung E, Olin-Sandoval V, Gruning NM, Kruger A, Tauqeer Alam M, Keller MA, Breitenbach M, Brindle KM, Rabinowitz JD, Ralser M, 2015. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 90, 927–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streeper RS, Grueter CA, Salomonis N, Cases S, Levin MC, Koliwad SK, Zhou P, Hirschey MD, Verdin E, Farese RV Jr., 2012. Deficiency of the lipid synthesis enzyme, DGAT1, extends longevity in mice. Aging (Albany NY) 4, 13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strong R, Miller RA, Astle CM, Baur JA, de Cabo R, Fernandez E, Guo W, Javors M, Kirkland JL, Nelson JF, Sinclair DA, Teter B, Williams D, Zaveri N, Nadon NL, Harrison DE, 2013. Evaluation of resveratrol, green tea extract, curcumin, oxaloacetic acid, and medium-chain triglyceride oil on life span of genetically heterogeneous mice. J. Gerontol. A Biol. Sci. Med. Sci. 68, 6–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strong R, Miller RA, Antebi A, Astle CM, Bogue M, Denzel MS, Fernandez E, Flurkey K, Hamilton KL, Lamming DW, Javors MA, de Magalhaes JP, Martinez PA, McCord JM, Miller BF, Muller M, Nelson JF, Ndukum J, Rainger GE, Richardson A, Sabatini DM, Salmon AB, Simpkins JW, Steegenga WT, Nadon NL, Harrison DE, 2016. Longer lifespan in male mice treated with a weakly estrogenic agonist, an antioxidant, an alpha-glucosidase inhibitor or a Nrf2-inducer. Aging Cell 15, 872–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Wang T, Wu N, Li D, Fan X, Xu Z, Mishra SK, Yang M, 2019. Alpha-ketoglutarate extends Drosophila lifespan by inhibiting mTOR and activating AMPK. Aging (Albany NY) 11, 4183–4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sujkowski A, Ramesh D, Brockmann A, Wessells R, 2017. Octopamine drives endurance exercise adaptations in Drosophila. Cell Rep. 21, 1809–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Wheeler CT, Yolitz J, Laslo M, Alberico T, Sun Y, Song Q, Zou S, 2014. A mitochondrial ATP synthase subunit interacts with TOR signaling to modulate protein homeostasis and lifespan in Drosophila. Cell Rep. 8, 1781–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutphin GL, Backer G, Sheehan S, Bean S, Corban C, Liu T, Peters MJ, van Meurs JBJ, Murabito JM, Johnson AD, Korstanje R, Cohorts for H, Aging Research in Genomic Epidemiology Consortium Gene Expression Working, G, 2017. Caenorhabditis elegans orthologs of human genes differentially expressed with age are enriched for determinants of longevity. Aging Cell 16, 672–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbert ME, Barnett B, Hoff R, Amella M, Kuczynski K, Lavington E, Koury S, Brud E, Eanes WF, 2015. Genetic perturbation of key central metabolic genes extends lifespan in Drosophila and affects response to dietary restriction. Proc. Biol. Sci. 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatar M, Post S, Yu K, 2014. Nutrient control of Drosophila longevity. Trends Endocrinol. Metab. 25, 509–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedesco P, Jiang J, Wang J, Jazwinski SM, Johnson TE, 2008. Genetic analysis of hyl-1, the C. Elegans homolog of LAG1/LASS1. Age (Dordr) 30, 43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas EA, Dean B, Pavey G, Sutcliffe JG, 2001. Increased CNS levels of apolipoprotein D in schizophrenic and bipolar subjects: implications for the pathophysiology of psychiatric disorders. Proc. Natl. Acad. Sci. U.S.A. 98, 4066–4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titov DV, Cracan V, Goodman RP, Peng J, Grabarek Z, Mootha VK, 2016. Complementation of mitochondrial electron transport chain by manipulation of the NAD+/NADH ratio. Science (New York, N.Y 352, 231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomas-Loba A, Bernardes de Jesus B, Mato JM, Blasco MA, 2013. A metabolic signature predicts biological age in mice. Aging Cell 12, 93–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang WY, Lemire BD, 2002. Mitochondrial genome content is regulated during nematode development. Biochem. Biophys. Res. Commun. 291, 8–16. [DOI] [PubMed] [Google Scholar]
- Tsang WY, Sayles LC, Grad LI, Pilgrim DB, Lemire BD, 2001. Mitochondrial respiratory chain deficiency in Caenorhabditis elegans results in developmental arrest and increased life span. J. Biol. Chem. 276, 32240–32246. [DOI] [PubMed] [Google Scholar]
- Ulgherait M, Rana A, Rera M, Graniel J, Walker DW, 2014. AMPK modulates tissue and organismal aging in a non-cell-autonomous manner. Cell Rep. 8, 1767–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulgherait M, Chen A, McAllister SF, Kim HX, Delventhal R, Wayne CR, Garcia CJ, Recinos Y, Oliva M, Canman JC, Picard M, Owusu-Ansah E, Shirasu-Hiza M, 2020. Circadian regulation of mitochondrial uncoupling and lifespan. Nat. Commun. 11, 1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaiserman AM, Koliada AK, Koshel NM, Simonenko AV, Pasiukova EG, 2012. [Effect of the histone deacetylase inhibitor sodium butyrate on the viability and life span in Drosophila melanogaster]. Adv. Gerontol. 25, 126–131. [PubMed] [Google Scholar]
- Valerio A, D’Antona G, Nisoli E, 2011. Branched-chain amino acids, mitochondrial biogenesis, and healthspan: an evolutionary perspective. Aging (Albany NY) 3, 464–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Goot AT, Nollen EA, 2013. Tryptophan metabolism: entering the field of aging and age-related pathologies. Trends Mol. Med. 19, 336–344. [DOI] [PubMed] [Google Scholar]
- van der Goot AT, Zhu W, Vazquez-Manrique RP, Seinstra RI, Dettmer K, Michels H, Farina F, Krijnen J, Melki R, Buijsman RC, Ruiz Silva M, Thijssen KL, Kema IP, Neri C, Oefner PJ, Nollen EA, 2012. Delaying aging and the aging-associated decline in protein homeostasis by inhibition of tryptophan degradation. Proc. Natl. Acad. Sci. U.S.A. 109, 14912–14917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela-Lopez A, Giampieri F, Battino M, Quiles JL, 2016. Coenzyme q and its role in the dietary therapy against aging. Molecules 21, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veech RL, Bradshaw PC, Clarke K, Curtis W, Pawlosky R, King MT, 2017. Ketone bodies mimic the life span extending properties of caloric restriction. IUBMB Life 69, 305–314. [DOI] [PubMed] [Google Scholar]
- Verdin E, 2015. NAD(+) in aging, metabolism, and neurodegeneration. Science (New York, N.Y 350, 1208–1213. [DOI] [PubMed] [Google Scholar]
- Virk B, Correia G, Dixon DP, Feyst I, Jia J, Oberleitner N, Briggs Z, Hodge E, Edwards R, Ward J, Gems D, Weinkove D, 2012. Excessive folate synthesis limits lifespan in the C. elegans: E. Coli aging model. BMC Biol. 10, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyssokikh MY, Holtze S, Averina OA, Lyamzaev KG, Panteleeva AA, Marey MV, Zinovkin RA, Severin FF, Skulachev MV, Fasel N, Hildebrandt TB, Skulachev VP, 2020. Mild depolarization of the inner mitochondrial membrane is a crucial component of an anti-aging program. Proc. Natl. Acad. Sci. U.S.A. 117, 6491–6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker DW, Muffat J, Rundel C, Benzer S, 2006. Overexpression of a Drosophila homolog of apolipoprotein D leads to increased stress resistance and extended lifespan. Curr. Biol. 16, 674–679. [DOI] [PubMed] [Google Scholar]
- Walsh ME, Bhattacharya A, Sataranatarajan K, Qaisar R, Sloane L, Rahman MM, Kinter M, Van Remmen H, 2015. The histone deacetylase inhibitor butyrate improves metabolism and reduces muscle atrophy during aging. Aging Cell 14, 957–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wamelink MM, Struys EA, Jakobs C, 2008. The biochemistry, metabolism and inherited defects of the pentose phosphate pathway: a review. J. Inherit. Metab. Dis. 31, 703–717. [DOI] [PubMed] [Google Scholar]
- Wan QL, Meng X, Fu X, Chen B, Yang J, Yang H, Zhou Q, 2019. Intermediate metabolites of the pyrimidine metabolism pathway extend the lifespan of C. Elegans through regulating reproductive signals. Aging (Albany NY) 11, 3993–4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan QL, Fu X, Dai W, Yang J, Luo Z, Meng X, Liu X, Zhong R, Yang H, Zhou Q, 2020. Uric acid induces stress resistance and extends the life span through activating the stress response factor DAF-16/FOXO and SKN-1/NRF2. Aging (Albany NY) 12, 2840–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Dong C, 2019. Gluconeogenesis in Cancer: function and regulation of PEPCK, FBPase, and G6Pase. Trends Cancer 5, 30–45. [DOI] [PubMed] [Google Scholar]
- Wang MC, O’Rourke EJ, Ruvkun G, 2008. Fat metabolism links germline stem cells and longevity in C. Elegans. Science (New York, N.Y 322, 957–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CT, Chen YC, Wang YY, Huang MH, Yen TL, Li H, Liang CJ, Sang TK, Ciou SC, Yuh CH, Wang CY, Brummel TJ, Wang HD, 2012. Reduced neuronal expression of ribose-5-phosphate isomerase enhances tolerance to oxidative stress, extends lifespan, and attenuates polyglutamine toxicity in Drosophila. Aging Cell 11, 93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Davis SS, Borch Jensen M, Rodriguez-Fernandez IA, Apaydin C, Juhasz G, Gibson BW, Schilling B, Ramanathan A, Ghaemmaghami S, Jasper H, 2019. JNK modifies neuronal metabolism to promote proteostasis and longevity. Aging Cell 18, e12849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weimer S, Priebs J, Kuhlow D, Groth M, Priebe S, Mansfeld J, Merry TL, Dubuis S, Laube B, Pfeiffer AF, Schulz TJ, Guthke R, Platzer M, Zamboni N, Zarse K, Ristow M, 2014. D-Glucosamine supplementation extends life span of nematodes and of ageing mice. Nat. Commun. 5, 3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir HJ, Yao P, Huynh FK, Escoubas CC, Goncalves RL, Burkewitz K, Laboy R, Hirschey MD, Mair WB, 2017. Dietary restriction and AMPK increase lifespan via mitochondrial network and peroxisome remodeling. Cell Metab. 26 (884–896), e885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen DT, Zheng L, Ni L, Wang H, Feng Y, Zhang M, 2016. The expression of CG9940 affects the adaptation of cardiac function, mobility, and lifespan to exercise in aging Drosophila. Exp. Gerontol. 83, 6–14. [DOI] [PubMed] [Google Scholar]
- Whitson HE, Arnold AM, Yee LM, Mukamal KJ, Kizer JR, Djousse L, Ix JH, Siscovick D, Tracy RP, Thielke SM, Hirsch C, Newman AB, Zieman S, 2014. Serum carboxymethyl-lysine, disability, and frailty in older persons: the Cardiovascular Health Study. J. Gerontol. A Biol. Sci. Med. Sci. 69, 710–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DS, Cash A, Hamadani L, Diemer T, 2009. Oxaloacetate supplementation increases lifespan in Caenorhabditis elegans through an AMPK/FOXO-dependent pathway. Aging Cell 8, 765–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollam J, Antebi A, 2011. Sterol regulation of metabolism, homeostasis, and development. Annu. Rev. Biochem. 80, 885–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollam J, Magomedova L, Magner DB, Shen Y, Rottiers V, Motola DL, Mangelsdorf DJ, Cummins CL, Antebi A, 2011. The Rieske oxygenase DAF-36 functions as a cholesterol 7-desaturase in steroidogenic pathways governing longevity. Aging Cell 10, 879–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollam J, Magner DB, Magomedova L, Rass E, Shen Y, Rottiers V, Habermann B, Cummins CL, Antebi A, 2012. A novel 3-hydroxysteroid dehydrogenase that regulates reproductive development and longevity. PLoS Biol. 10, e1001305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Song L, Liu SQ, Huang D, 2013. Independent and additive effects of glutamic acid and methionine on yeast longevity. PLoS One 8, e79319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao F, Huang Z, Li H, Yu J, Wang C, Chen S, Meng Q, Cheng Y, Gao X, Li J, Liu Y, Guo F, 2011. Leucine deprivation increases hepatic insulin sensitivity via GCN2/mTOR/S6K1 and AMPK pathways. Diabetes 60, 746–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Gopalacharyulu P, Seppanen-Laakso T, Ruskeepaa AL, Aye CC, Carson BP, Mora S, Oresic M, Teleman AA, 2012. Insulin signaling regulates fatty acid catabolism at the level of CoA activation. PLoS Genet. 8, e1002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamawaki TM, Berman JR, Suchanek-Kavipurapu M, McCormick M, Gaglia MM, Lee SJ, Kenyon C, 2010. The somatic reproductive tissues of C. Elegans promote longevity through steroid hormone signaling. PLoS Biol. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Hekimi S, 2010. Two modes of mitochondrial dysfunction lead independently to lifespan extension in Caenorhabditis elegans. Aging Cell 9, 433–447. [DOI] [PubMed] [Google Scholar]
- Yang Q, Gong ZJ, Zhou Y, Yuan JQ, Cheng J, Tian L, Li S, Lin XD, Xu R, Zhu ZR, Mao C, 2010. Role of Drosophila alkaline ceramidase (Dacer) in Drosophila development and longevity. Cell. Mol. Life Sci. 67, 1477–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yesavage JA, Holman CA, Sarnquist FH, Berger PA, 1982. Elevation of cerebrospinal fluid lactate with aging in subjects with normal blood oxygen saturations. J. Gerontol. 37, 313–315. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Satoh A, Lin JB, Mills KF, Sasaki Y, Rensing N, Wong M, Apte RS, Imai SI, 2019. Extracellular vesicle-contained eNAMPT delays aging and extends lifespan in mice. Cell Metab. 30 (329–342), e325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino J, Baur JA, Imai SI, 2018. NAD(+) intermediates: the biology and therapeutic potential of NMN and NR. Cell Metab. 27, 513–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Zhai G, Singmann P, He Y, Xu T, Prehn C, Romisch-Margl W, Lattka E, Gieger C, Soranzo N, Heinrich J, Standl M, Thiering E, Mittelstrass K, Wichmann HE, Peters A, Suhre K, Li Y, Adamski J, Spector TD, Illig T, Wang-Sattler R, 2012. Human serum metabolic profiles are age dependent. Aging Cell 11, 960–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Jang Y, Paik D, Lee E, Park JJ, 2015. Nmdmc overexpression extends Drosophila lifespan and reduces levels of mitochondrial reactive oxygen species. Biochem. Biophys. Res. Commun. 465, 845–850. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Kadiyala CS, Ching TT, Hakimi P, Saha S, Xu H, Yuan C, Mullangi V, Wang L, Fivenson E, Hanson RW, Ewing R, Hsu AL, Miyagi M, Feng Z, 2012. Enhanced energy metabolism contributes to the extended life span of calorie-restricted Caenorhabditis elegans. J. Biol. Chem. 287, 31414–31426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Hakimi P, Kao C, Kao A, Liu R, Janocha A, Boyd-Tressler A, Hang X, Alhoraibi H, Slater E, Xia K, Cao P, Shue Q, Ching TT, Hsu AL, Erzurum SC, Dubyak GR, Berger NA, Hanson RW, Feng Z, 2016. Reciprocal changes in Phosphoenolpyruvate Carboxykinase and pyruvate kinase with age are a determinant of aging in Caenorhabditis elegans. J. Biol. Chem. 291, 1307–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarse K, Schmeisser S, Groth M, Priebe S, Beuster G, Kuhlow D, Guthke R, Platzer M, Kahn CR, Ristow M, 2012. Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial L-proline catabolism to induce a transient ROS signal. Cell Metab. 15, 451–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Poplawski M, Yen K, Cheng H, Bloss E, Zhu X, Patel H, Mobbs CV, 2009. Role of CBP and SATB-1 in aging, dietary restriction, and insulin-like signaling. PLoS Biol. 7, e1000245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Ryu D, Wu Y, Gariani K, Wang X, Luan P, D’Amico D, Ropelle ER, Lutolf MP, Aebersold R, Schoonjans K, Menzies KJ, Auwerx J, 2016. NAD(+) repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 352, 1436–1443. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Sun H, Lu J, Li X, Chen X, Tao D, Huang W, Huang B, 2005. Lifespan extension and elevated hsp gene expression in Drosophila caused by histone deacetylase inhibitors. J. Exp. Biol. 208, 697–705. [DOI] [PubMed] [Google Scholar]
- Zheng J, Gao C, Wang M, Tran P, Mai N, Finley JW, Heymsfield SB, Greenway FL, Li Z, Heber D, Burton JH, Johnson WD, Laine RA, 2017. Lower doses of fructose extend lifespan in Caenorhabditis elegans. J. Diet. Suppl. 14, 264–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu XH, Lu M, Lee BY, Ugurbil K, Chen W, 2015. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc. Natl. Acad. Sci. U.S.A. 112, 2876–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zid BM, Rogers AN, Katewa SD, Vargas MA, Kolipinski MC, Lu TA, Benzer S, Kapahi P, 2009. 4E-BP extends lifespan upon dietary restriction by enhancing mitochondrial activity in Drosophila. Cell 139, 149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou K, Rouskin S, Dervishi K, McCormick MA, Sasikumar A, Deng C, Chen Z, Kaeberlein M, Brem RB, Polymenis M, Kennedy BK, Weissman JS, Zheng J, Ouyang Q, Li H, 2020. Life span extension by glucose restriction is abrogated by methionine supplementation: cross-talk between glucose and methionine and implication of methionine as a key regulator of life span. Sci. Adv. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuryn S, Kuang J, Tuck A, Ebert PR, 2010. Mitochondrial dysfunction in Caenorhabditis elegans causes metabolic restructuring, but this is not linked to longevity. Mech. Ageing Dev. 131, 554–561. [DOI] [PubMed] [Google Scholar]