

Abstract
Cancers have been associated with a diverse array of genomic alterations. To help mechanistically understand such alterations in breast invasive carcinoma, we have applied affinity-purification mass spectrometry to delineate comprehensive biophysical interaction networks for 40 frequently altered breast cancer (BC) proteins, with and without relevant mutations, across three human breast cell lines. These networks identify cancer-specific protein-protein interactions (PPIs), interconnect and enrich for common and rare cancer mutations, and are substantially rewired by the introduction of key BC mutations. Our analysis identified PIK3CA-interacting proteins which repress AKT signaling and has uncovered USP28 and UBE2N as functionally relevant interactors of BRCA1. We also show that the PP1 phosphatase regulatory subunit, Spinophilin, interacts with and regulates dephosphorylation of BRCA1 to promote DNA double-strand break repair. Thus, PPI landscapes provide a powerful framework for mechanistically interpreting disease genomic data and can identify valuable new therapeutic targets.
One Sentence Summary
Large-scale protein interaction maps of breast cancer genes provide a framework to recognize previously unidentified oncogenic drivers.
Breast cancer (BC) is the most common malignancy in women and the second leading cause of cancer‐related death in the United States, where an estimated 281,550 women and 2,650 men will be newly diagnosed in 2021 (1). The disease has been divided into different subtypes, based largely on the presence or absence of three key proteins: estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2/ERBB2). Despite this and much additional heterogeneity at the molecular level, the majority of BC patients are treated using untailored chemotherapy or hormone therapies. Therefore, there is an urgent need to develop targeted therapies matched to the specific molecular alterations in a tumor, with the goal of achieving better efficacy and avoiding unnecessary treatment.
Advances in DNA sequencing technology have enabled the widespread analysis of breast tumor genomes, creating a catalog of genetic mutations that may initiate or drive tumor progression (2, 3). In addition to common mutations in well-known cancer genes, such as TP53 and PIK3CA, breast cancers harbor many additional mutations, each of which are seen rarely across the patient population. A key question is how these less common alterations, dispersed across a multitude of genes, elicit pathologic consequences and patient outcomes. An important answer may lie in understanding how individual gene mutations converge on multi-gene functional modules, including the signaling pathways orchestrating cell proliferation and apoptosis and DNA repair complexes (4–15). To broadly enable a pathway-based understanding of cancer, a prerequisite is to generate general and comprehensive maps of cancer molecular networks in relevant malignant and premalignant cell contexts.
Protein-protein interaction mapping of breast cancer drivers
Using the TCGA analysis of BC tumors (2, 3), we collected a panel of genes that are associated with molecular alterations, in terms of cell growth, proliferation, and genome stability in BC and used this list to guide the selection of 40 proteins for generation of protein-protein interaction (PPI) networks. Our selected targets included proteins with well-known roles in BC (e.g., TP53, PIK3CA, CDH1, and BRCA1) as well as less-well characterized proteins with recurrent mutations (e.g., CHEK2, MLH1, SMARCB1, and XPC) (16–19). Importantly, 93% of BC tumors in TCGA harbor an alteration in one or more of these 40 genes (Fig. 1A). Three breast cell lines derived from human mammary epithelium – MCF7 (ER+, luminal A subtype), MDA-MB-231 (ER−, PR−, HER2− triple-negative TN subtype) and MCF10A (non-tumorigenic mammary epithelial cells) – were selected for the PPI analysis because they have been shown to replicate therapeutically relevant responses found in BC tumors (20), their RNA profiles are highly correlated with those of BC tumors (21) and ER+ and TN subtypes together account for approximately 90% of BC patients (22). We reasoned that comparing protein networks among ER+, TN and non-tumorigenic models would allow study of how PPI networks are altered between normal and tumorigenic backgrounds as well as influenced by different mammary epithelial lineages.
Figure 1. Overview of protein-protein interaction mapping in breast epithelial cells.
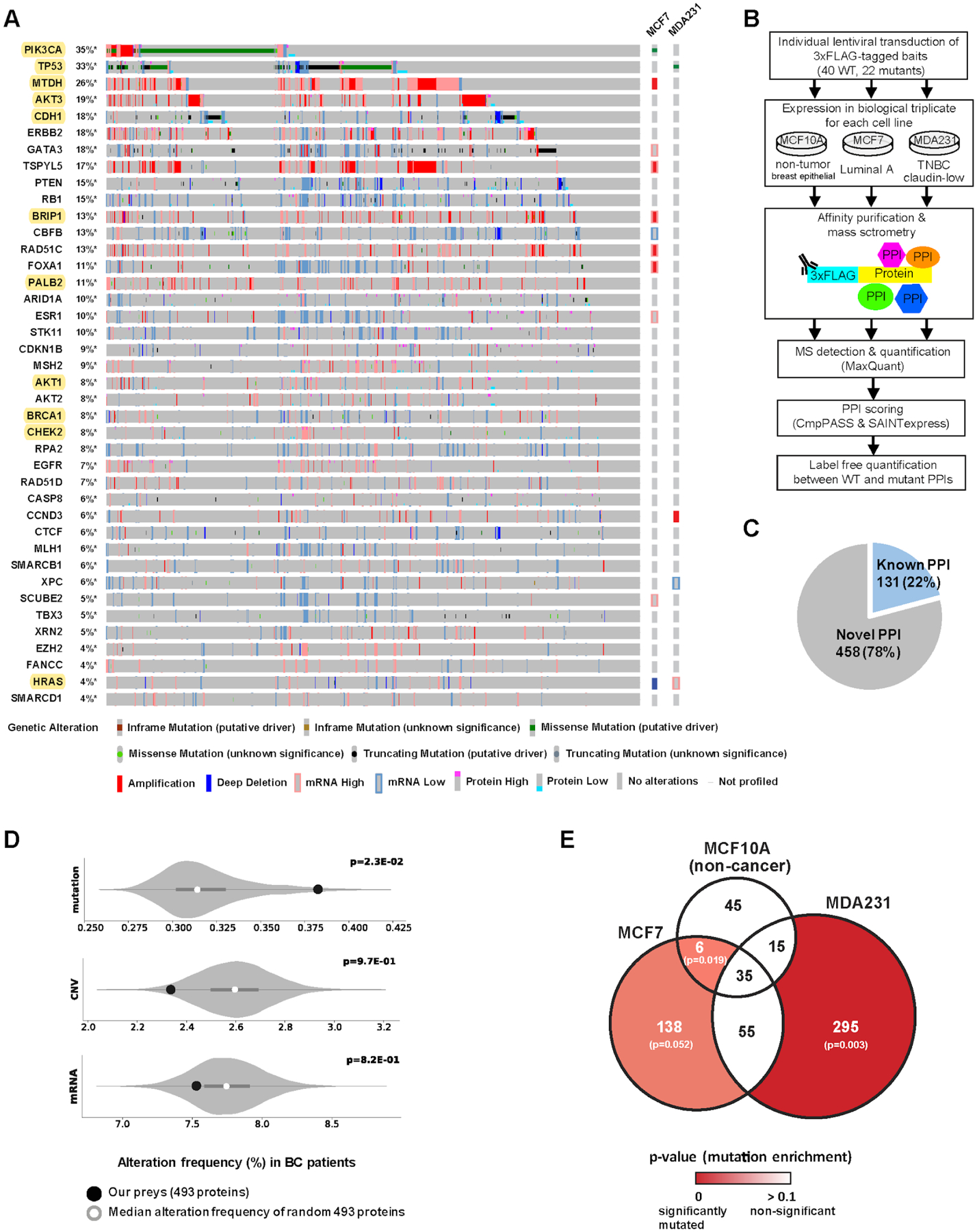
(A) The gene alteration frequencies from the breast invasive carcinoma (TCGA Firehose Legacy) dataset for the 40 genes selected as AP-MS baits in this study. Each vertical grey column represents a patient, such that various genetic alterations of 40 genes in a given patient are indicated as seen at the bottom. In total, 93% (1028 out of 1108) of BC patients have non-synonymous mutation, chromosomal copy-number alteration (CNA), or mRNA/protein expression alteration in one or more of these 40 genes. Genes analyzed for both wild-type and mutant proteins are highlighted in yellow. Existing gene alterations in MCF7 and MDA-MB-231 are shown on the right. (B) The experimental workflow in which each bait was expressed in biological triplicate in 3 cell lines and subjected to AP-MS analysis. (C) Majority (78%) of the high-confidence PPIs identified in this study are not represented in a panel of public PPI databases (CORUM, BioPlex 2.0, IMEx and BioGRID low throughput & multivalidated). (D) The frequency of non-synonymous mutations, chromosomal CNVs, or mRNA expression alterations of 10,000 random size-matched permutations taken from the list of genes detected in the global protein abundance analysis. The white circle indicates the median of the random sampling, and the grey bar represents ±1 standard deviation. The frequency of alterations found in the prey retrieved in our PPI dataset is indicated in the black circle. (E) Venn diagram illustrating the overlap of PPIs (PPI score ≥ 0.9) across the 3 cell lines. PPI score is an average of the PPI confidence scores calculated from compPASS and SAINTexpress (see methods for details). The frequency of non-synonymous mutations of the prey genes in each sector of the Venn diagram was compared to those of 10,000 random size-matched permutations as in (D). The p-values for mutation enrichment in each prey set were shown in a color scale, where a stronger red represents more significant mutation enrichment.
To generate PPI maps, “bait” proteins were cloned into triple FLAG-tagged lentiviral vectors (table S2), individually transduced into each cell line and expressed in biological triplicate via a doxycycline inducible promoter (Fig. 1B). Cells were harvested after approximately 40 hr doxycycline-induction, and anti-FLAG tag-based affinity purification was performed followed by mass spectrometry to detect interacting “prey” proteins in an unbiased manner. We employed two PPI scoring algorithms to quantify high-confidence interacting proteins: compPASS (23, 24) and SAINTexpress (25) (Materials and Methods). Although overexpression of tagged baits may generate possible artifacts, it permits the capture of PPIs in a highly sensitive and reproducible manner with a relatively uniform background signal, which can be removed by the appropriate algorithms. We and many others have used overexpression AP-MS to characterize thousands of physiologically relevant PPIs (26–30). Using this approach, we identified a total of 589 high-confidence PPIs involving 493 prey proteins (Fig. 1C, fig. S1A–B, tables S3 and S4). Collectively, 78% of the BC PPIs we identified were not previously reported in protein-protein interaction databases (CORUM, BioPlex 2.0, IMEx and the low throughput and multivalidated BIOGRID) (Fig. 1C). The high percentage of new interactions may reflect cell type-enriched PPIs as nearly all systematic protein-protein interaction analyses to date have been performed in HEK293T or HeLa cell lines (24, 26, 27, 31, 32).
PPIs often suggest functional relationships among proteins that work together to regulate a specific cellular process. Previously, we and others had found a significant enrichment of frequently mutated proteins in large PPI repositories (8–12, 33–35). Similarly, we investigated whether our BC PPI network showed enrichment for three major types of alterations – non-synonymous mutations, chromosomal CNVs, and mRNA expression alterations – documented in the BC TCGA cohort. Accordingly, we calculated the average frequency of each alteration for prey proteins detected in our PPIs, compared to background expectation (fig. S1C and table S5). We observed that BC-associated mutations were significantly enriched in BC PPIs, but that CNVs and mRNA expression alterations were not (Fig. 1D), a trend that was also observed in head and neck squamous cell carcinoma (Swaney et al., accompanying paper). Furthermore, we found enrichment of tumor mutations in preys detected specifically in either of the two cancer cell lines (MCF7, MDA-MB-231) among BC patient tumors but not in the preys of non-cancerous MCF10A cells (Fig. 1E and table S4). This result supports the notion that the interaction partners of frequently altered cancer proteins are also under positive pressure for mutations. Along this line, in a prior study, we compared the preys of frequently mutated baits to those of non-frequently mutated baits from the BioPlex dataset and found the same trend to emerge for almost all cancer types (33).
Out of 589 PPIs identified, 81% were not shared with other cell lines, reflecting high cell type-specificity of PPIs in different genetic contexts (Fig. 1E). We speculated that differential protein abundance across cell lines might provide one explanation for cell type-enriched PPIs. However, while some changes in interaction could be explained by changes in protein abundance, we also found many cases with the opposite behavior, in which a gain in interaction was observed with a concomitant decrease in protein abundance (table S5 and fig. S1D).
Cell type-enriched interactions
To compare PPIs across cell lines, we defined a cancer-enriched differential interaction score (DIS) as the probability of the PPI being present in a cancer cell line (either MCF7 or MDA-MB-231) but absent in the normal cell line (MCF10A) (Materials and Methods). Given that each PPI has a specific DIS, a continuous color scheme was used to represent cancer versus non-cancer differential interactions. We used the results of this differential scoring analysis to visualize the entire BC PPI network showing PPIs that are (1) enriched in a cancer cell line, (2) enriched in non-cancerous MCF10A cells, or (3) conserved in the two cancer cell lines but absent in the non-cancerous context (Fig. 2 and table S6).
Figure 2. Differential interaction analysis of the BC-enriched interactome.
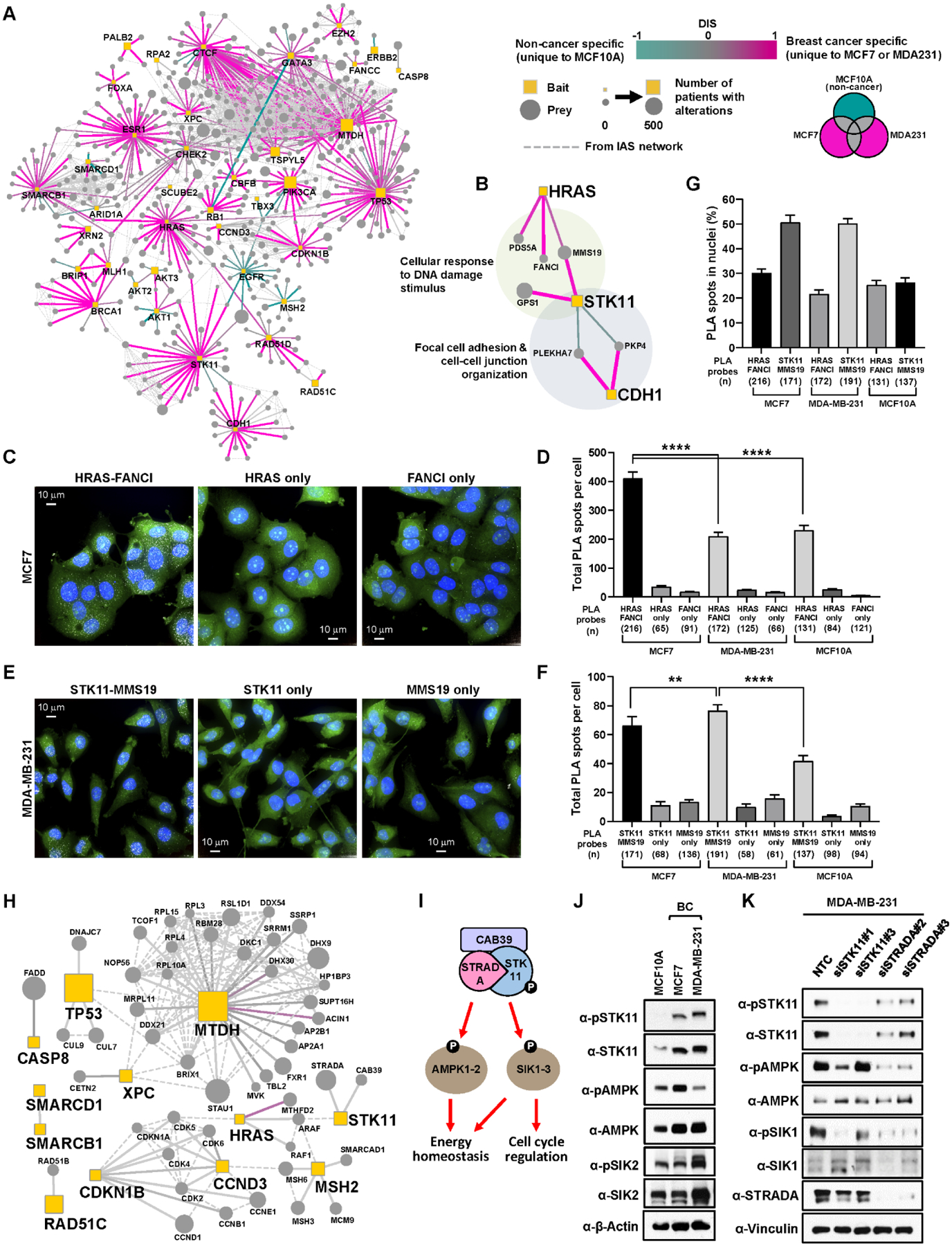
(A) Interactome of the union of all high-confidence PPIs detected across all cell lines. Edges are colored based on their differential interaction, with pink edges representing PPIs that are enriched to BC cell lines (unique to either MDA-MB-231 or MCF7) as compared to MCF10A cells (shown in teal edges). Dotted line represents the physical protein-protein association (validated in other studies) with high Integrated Association Stringency score. (B) PPIs connecting HRAS, STK11, and CDH1. HRAS and STK11 have several interactors including FANCI and MMS19 in BC cells involved in cellular response to DNA damage stimulus. STK11 and CDH1 interact with PKP4 and PLEKHA7 in a cell type-specific manner, implying differential regulation of cell adhesion and cell-cell junction in non-BC and BC cells. (C) Representative images of MCF7 cells from the PLA between HRAS and FANCI antibodies. Maximum intensity projection images are shown to represent total PLA interactions. PLA with only one of the two primary antibodies was performed as negative control. PLA spots (white), HCS CellMask Green stain (Green) and DAPI (blue). Scale bar = 10 μm. (D) Total PLA spots per cell using HRAS and FANCI antibodies were quantified in MCF7, MDA-MB-231, and MCF10A cells. n= total number of cells analyzed in each condition. **** p-value ≤ 1.0×10−4. (E) Representative images of MDA-MB-231 cells from the PLA between STK11 and MMS19 antibodies. (F) Total PLA spots per cell using STK11 and MMS19 antibodies were quantified in MCF7, MDA-MB-231, and MCF10A cells. **** p-value ≤ 1.0×10−4, ** p-value ≤ 1.0×10−2. (G) Percent nuclear PLA spots in each PLA condition. (H) High-confidence PPIs that are commonly detected only in two cancer cell lines (MDA-MB-231 and MCF7) but not in non-cancerous MCF10A cells. Node and edge styles and colors as seen in A. (I) STK11 forms a heterotrimeric complex with CAB39 and STRADA to activate its kinase activity and phosphorylate downstream kinases including AMPK and SIKs for regulating energy homeostasis and cell cycle. (J) STK11 kinase activity was monitored by measuring total and phosphorylation levels of its known downstream substrates (AMPK and SIK2) as well as itself. The following phospho-epitopes were detected by antibodies: pSTK11 (pS428), pAMPKα (pT172), pSIK2 (pT175). (K) Knockdown of STK11 and its interacting protein (STRADA) by two individual small-interfering RNAs reduces phosphorylation of STK11 (S428), AMPK (T172), and SIK1 (T182).
Among interactions enriched to a cancer cell line, we found the HRAS proto-oncogene and the tumor suppressor kinase STK11 (also known as LKB1) to interact with a set of DNA damage response (DDR) proteins (PDS5A, FANCI, MMS19, GPS1) in MCF7 and MDA-MB-231 cells, respectively (Fig. 2B). The HRAS-FANCI and STK11-MMS19 interactions were further assessed using a proximity ligation assay (PLA). Abundant PLA spots were observed when both HRAS and FANCI antibodies were incubated with fixed MCF7 cells, but either antibody alone generated only a background level of signal (Fig. 2C–D and fig. S2A), confirming the interaction seen by AP-MS. Also consistent with the mass spectrometry results, a significantly higher number of HRAS-FANCI PLA spots were detected in MCF7 compared to MDA-MB-231 and MCF10A cells (Fig. 2D). Again consistent with the cell type specificity uncovered via AP-MS, STK11-MMS19 PLA spots were observed with consistently higher numbers in MDA-MB-231 than in MCF7 and MCF10A cells (Fig. 2E–F and fig. S2A). Intriguingly, we also observed the HRAS-FANCI and STK11-MMS19 interactions in two additional BC cell lines (T47D and SKBR3) by PLA (fig. S2B–C). Analysis using 3D segmentation revealed that the PLA spots are present in the cytoplasm as well as the nucleus (Fig. 2G and fig. S2B–C), indicating nuclear roles of HRAS and STK11, potentially involving DNA damage/repair functions based on their PPIs. Consistent with our findings, the Human Protein atlas (www.proteinatlas.org) reveals that HRAS and STK11 are present both in the nucleus and cytosol based on immunofluorescence microscopy (36). Given the previous observations that silencing of HRAS and STK11 lead to defective DNA repair and genome instability (37), these interactions may provide insights into direct effectors by which HRAS and STK11 modulate DDR. STK11 also interacted with cell adhesion factors in MCF10A cells (PLEKHA7 and PKP4, Fig. 2B), consistent with its role in cell autonomous polarization (38) and actin filament assembly at the cellular leading edge (39).
Interestingly, CDH1 but not STK11 was found to interact with cell adhesion factors in MDA-MB-231 cells. CDH1 plays critical roles as a master regulator of cell-cell adhesion via adherens junctions, cell polarity and cell migration (40), and abrogation of CDH1 expression is a hallmark of the epithelial-to-mesenchymal transition (41). The observed interaction patterns suggest that STK11 may contribute to cell polarity and focal adhesion through a physical interaction with PLEKHA7 and PKP4, but that it requires the cellular ability to form adherens junctions. This may explain the lack of interaction of STK11 with PLEKHA7 and PKP4 in MDA-MB-231, which do not express CDH1 due to promoter hypermethylation (42).
We also found that STK11 interacts with STRADA and CAB39 (also known as MO25) preferentially in the two cancer cell lines (Fig. 2H). STRADA and CAB39 form a heterotrimeric complex with STK11 (43) to properly position the activation loop of STK11 in an active conformation (44), enabling STK11 to phosphorylate and activate downstream kinases, including AMP-activated protein kinases (AMPKs) and salt-inducible kinases (SIKs) involved in energy homeostasis and cell cycle regulation (Fig. 2I) (45–47). The increased associations among STK11, CAB39, and STRADA (regardless of protein abundance in these cells) suggested that STK11 activity is generally augmented in cancer. Consistent with this hypothesis, we found that both total and activated STK11 (phosphorylated at Ser428) are more abundant in MCF7 and MDA-MB-231 than in MCF10A cells (Fig. 2J). Furthermore, phosphorylation of STK11 downstream targets, including SIK2 and AMPK, was higher in either of two BC cell lines (Fig. 2J). Knockdown of STRADA by two different individual small-interfering RNAs (siRNAs) significantly reduced phosphorylation of STK11 (S428), AMPK (T172), and SIK1 (T182) in MDA-MB-231 cells (Fig. 2K), providing further evidence the association of STK11 with STRADA contributes to downstream signaling of activated STK11. Increased STK11 activity may support cellular fitness by balancing energy production with anabolic metabolism, as previously seen in hepatocellular carcinoma (48).
Previously unidentified regulators of PIK3CA signaling
PIK3CA and AKT activating mutations and copy-number amplifications are frequently found in many cancer types including BC (49–51), indicating that the PI3K/AKT pathway is a key signaling module for cancer cell proliferation, and thus an attractive target for therapeutic intervention (52). However, given the substantial role of the PI3K pathway in tumorigenesis, mechanisms of regulation or tuning by interacting proteins remains largely unknown as prior research predominantly centers around how mutations and alterations in the PIK3CA and AKT genes themselves regulate pathway activity.
Activation of PIK3CA via receptor tyrosine kinase (RTK) or oncogenic mutations leads to membrane recruitment and activation of AKT (Fig. 3A) (53, 54). In BC, mutations at E545 and H1047 residues are most frequently found (Fig. 3B). Using AP-MS, we identified 20 prey proteins which interact with PIK3CA, 18 of which were observed in MCF7 cells (table S4). Of the 18 proteins, only 4 (IRS1 and PIK3R1/2/3) were previously known interactors (55–58). Many of these new PPIs are significantly decreased, and in some cases completely abolished, by different PIK3CA mutations (Fig. 3C). To determine whether these PIK3CA interactors modulate the PI3K/AKT pathway, we tested whether depletion of each target by siRNAs affects downstream AKT activation in MCF7 cells by measuring cellular phospho-AKT (pS473) levels in an in-cell western assay (59). Four independent siRNAs per target gene were pooled and transfected for knockdown (Materials and Methods). Non-targeting control siRNAs (NTCs) as well as siRNAs targeting PIK3CA (a positive regulator) and PTEN (a negative regulator) were included as controls (60). As expected, knockdown of PIK3CA in MCF7 cells significantly diminished pAKT signal, while knockdown of PTEN augmented it (Fig. 3D, fig. S3A, and table S7). Knockdown of the PIK3CA interactors BPIFA1 and SCGB2A1 (also named PLUNC and Mammaglobin-B, respectively) increased pAKT activity to a degree higher than or the same as the PTEN knockdowns, indicating that these two proteins are negative regulators of the PI3K/AKT pathway (Fig. 3D, fig. S3A, and table S7). To verify these in-cell western results, we performed pooled and individual siRNA-mediated knockdown of BPIFA1 and SCGB2A1 and confirmed the increase of AKT pS473 by standard western blot analysis in MCF7 cells (Fig. 3E and fig. S3B). Although BPIFA1 and SCGB2A1 were below the level of detection by the PIK3CA AP-MS in MDA-MB-231 cells (table S3), pooled and individual siRNA-mediated knockdown of BPIFA1 and SCGB2A1 in MDA-MB-231 cells also led to an increase in AKT pS473 (Fig. 3F and fig. S3C). These results suggest that BPIFA1 and SCGB2A1 may act as regulators of the PI3K/AKT pathway in multiple cellular contexts. Consistent with this, the interaction of BPIFA1 with 3xFLAG-tagged PIK3CA (WT) was confirmed by PLA in not only MCF7 but MDA-MB-231 cells (fig. S3D), albeit at lower levels. Furthermore, abundant PLA spots between BPIFA1 and endogenous PIK3CA were also observed in T47D and SKBR3 cells (fig. S3E). To verify a negative role of BPIFA1 and SCGB2A1 in PIK3CA signaling, we measured their effect on the PIK3CA kinase activity in a reconstituted in vitro assay using recombinant proteins (PIK3CA/PIK3R1, BPIFA1 and SCGB2A1) and lipid substrate (phosphoinositol-4,5-bisphosphate) (Materials and Methods). Consistent with the AP-MS results (Fig. 3C), increasing amounts of recombinant BPIFA1 and SCGB2A1 preferentially inhibit WT PIK3CA kinase activity towards lipid substrate when compared to the two mutant forms of PIK3CA (E545K and H147R) (Fig. 3G–H). These data both confirm the validity of the PPI and provide further verification of the WT vs. mutant specificity revealed by the AP-MS.
Figure 3. Comparative interactome analysis of WT and mutant PIK3CA proteins.
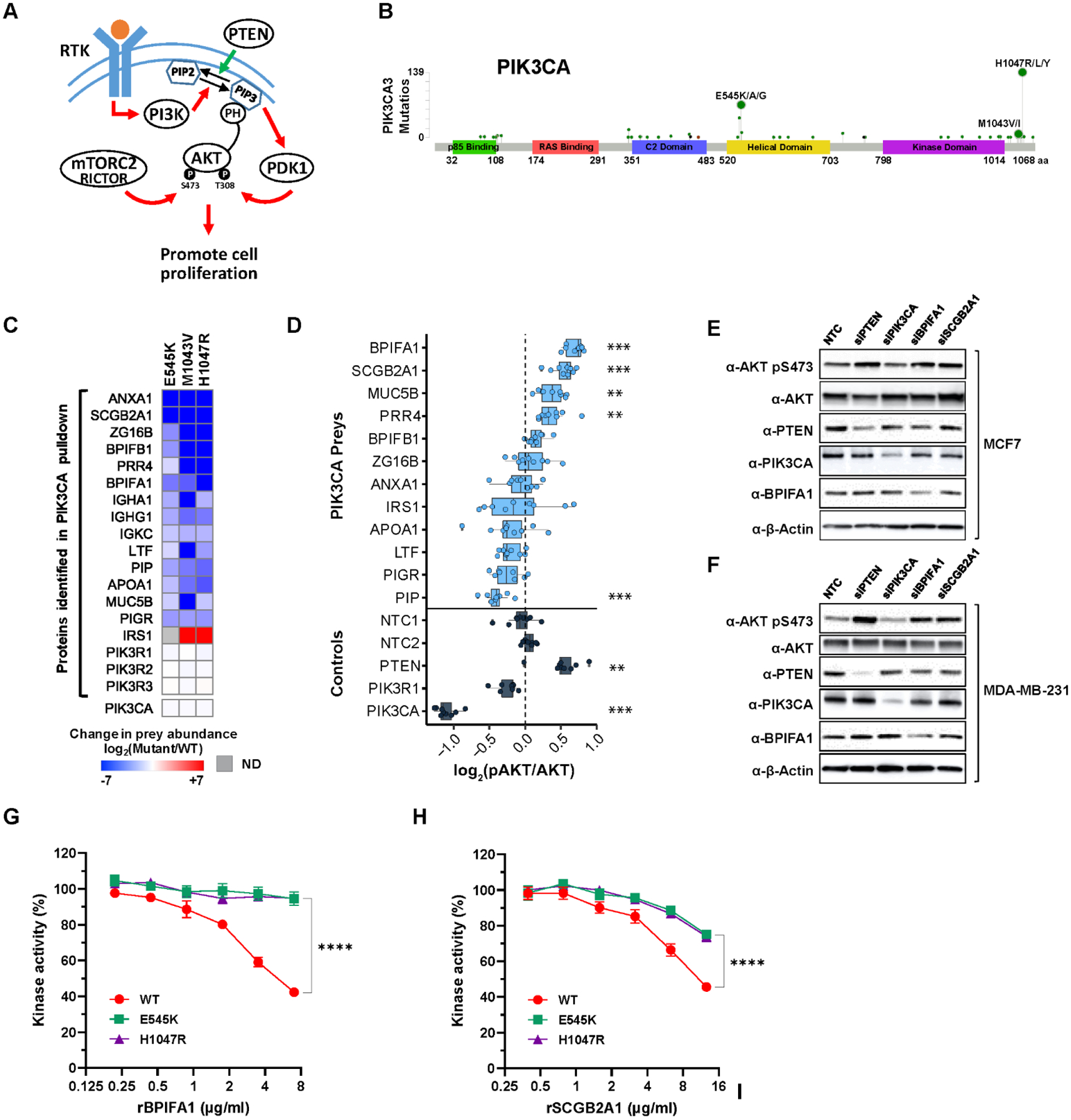
(A) Overview of the receptor tyrosine kinase (RTK)-PI3K signaling cascade leading to the phosphorylation (T308 and S473) and activation of the AKT pathway. (B) A lollipop plot representing the sites of PIK3CA mutations and the number of BC patients bearing a given PIK3CA mutation from TCGA (Firehose Legacy) study. (C) Relative quantification of the abundance of high-confidence preys observed from pull-down of PIK3CA (WT and mutants) in MCF7 cells. Preys detected only in WT are represented in deep blue while preys detected only in mutants are in deep red. All three PIK3CA mutants were expressed and detected at a similar level. ND, not detected. (D) The level of AKT S473 phosphorylation (as proxy of activation) was measured by in-cell western analysis upon siRNA-mediated knockdown of PIK3CA interacting preys and control genes (PTEN, PIK3CA, and PIK3R1) in MCF7 cells. The intensity of AKT pS473 was normalized to total AKT as well as cell viability. *** p-value ≤ 1.0×10−3, ** p-value ≤ 1.0×10−2. (E-F) Increase of AKT S473 phosphorylation upon knockdown of BPIFA1 and SCGB2A1 was confirmed by western blot in both MCF7 and MDA-MB-231 cells, respectively. (G-H) PIK3CA (WT, E545K, and H1047R) kinase activity towards phosphatidylinositol-4, 5-bisphosphate (PIP2) was measured in vitro in the presence of increasing amounts of recombinant BPIFA1 and SCGB2A1, respectively. Results are from at least 3–6 independent experiments. **** p-value ≤ 1.0×10−4.
Effect of pathogenic mutations on the BRCA1 interactome
BRCA1 is a major hereditary cancer susceptibility gene (61, 62) that plays critical roles in DNA repair by homologous recombination (HR) (63) in addition to other processes, such as regulation of transcription, RNA splicing and cell cycle (64, 65). BRCA1 carries out its functions in concert with other proteins (63), leading to many studies of BRCA1-containing complexes and their roles in DNA repair (66, 67). To date, many of these findings have been based on either immunoprecipitation with antibodies against the WT BRCA1 protein or interrogation of pairwise protein interactions with the yeast two-hybrid system. Moreover, these analyses were done mainly using WT BRCA1 protein and did not systematically capture how different mutations in BRCA1 might affect its protein interactions.
To comprehensively catalog the BRCA1 interactome and how pathogenic BRCA1 mutations alter these interaction profiles, we performed AP-MS on WT and pathogenic variants reported in cancer patients, including C61G and R71G in the N-terminal RING domain (68, 69) and S1655F, 5382insC and M1775R in the C-terminal tandem BRCT domain (70, 71) (Fig. 4A). Given that alternative splicing in cancer often generates BRCA1 isoforms lacking exon 11, which confers residual HR activity and therapeutic resistance (72), an isoform (isoform 6; UniProt identifier P38398–6) lacking exon 11 with distinct C-terminus was also included in the analysis. The I26A separation of function mutation in the RING domain, which abrogates E3 ubiquitin ligase activity but retains BARD1 binding, was also analyzed (73). We induced the expression of these BRCA1 proteins in all three breast cell lines; however, only MDA-MB-231 cells (harboring the TP53 R280K mutation) supported ectopic 3xFLAG-BRCA1 expression. These observations were consistent with previous studies which have shown that ectopic overexpression of BRCA1 (both WT and mutants) is not stably maintained without a compensatory TP53 mutation (74). AP-MS experiments in MDA-MB-231 cells identified 128 high-confidence interactions from 8 BRCA1 constructs (WT and 7 mutants, PPI score ≥ 0.65; fig. S4A and table S8); of these interacting proteins, 70 showed ≥ 8-fold change (Fig. 4B and fig. S4B).
Figure 4. Quantitative analysis of the effect of mutations on the BRCA1 interactome.
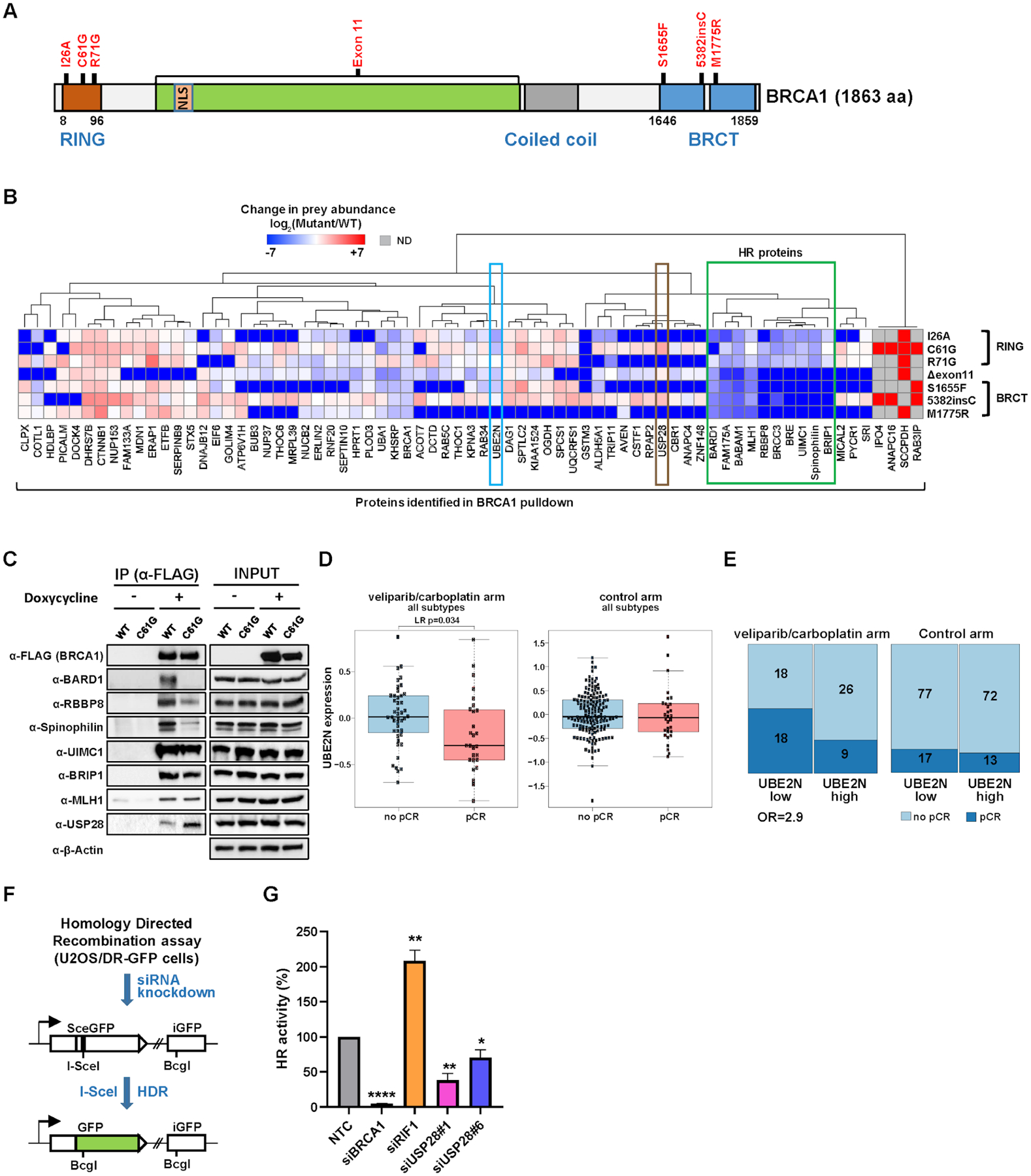
(A) Functional domains in the BRCA1 gene and the location of mutations analyzed by AP-MS. (B) Relative quantification of the abundance of prey proteins (PPI score ≥ 0.65, ≥ 8-fold change) identified by BRCA1 AP-MS in MDA-MB-231 cells. All prey abundance values were normalized by 3xFLAG-tagged BRCA1 levels in their respective AP-MS experiments. Preys detected only in WT are represented in deep blue while preys detected only in mutants are in deep red. A group of proteins involved in HR repair (boxed in green) are clustered together, wherein RING domain and BRCT domain BRCA1 mutants show distinct PPI abundance profiles. Spinophilin has not previously been known to have a function relevant to HR repair. UBE2N is boxed in sky blue. ND, not detected. (C) PPIs of selected proteins with BRCA1 (WT or C61G mutant) were confirmed by co-immunoprecipitation with anti-FLAG antibody followed by western blot analysis. (D) Box plot shows that the patient group (enrolled in the I-SPY 2 clinical trial) with pCR to veliparib (PARP inhibitor) and carboplatin (VC) had pre-treatment tumors with significantly lower UBE2N mRNA expression (LR p-value = 0.034) than those of non-responding patients. In contrast, BC patient tumors in the control arm did not show any significant difference in UBE2N expression between pCR and no pCR groups. (E) Mosaic plot shows that BC patients who did pCR to VC in addition to standard chemotherapy had 2.9 times more likely had lower mRNA expression of UBE2N in their pre-treatment tumors (Odds Ratio = 2.9). In the control arm, there is no significant difference in pCR between low and high UBE2N expression groups. Numbers in each block represent the patient sample size. Column width indicates the relative proportion of the UBE2N low and high expression group on the patient population. (F) A schematic of the HR reporter assay. The DR-GFP reporter contains two defective copies of the GFP gene, one disrupted by an I-SceI site and the other lacking a promoter. I-SceI cutting of the first copy generates a DSB, and repair by HR with the second copy as a template leads to restoration of a functional GFP gene. siRNA-mediated knockdown of HR-related genes leads to reduction of GFP+ cells (a proxy of HR activity) compared to NTC experiments. (G) HR activities upon depletion of USP28 relative to NTC (set to 100%). Depletion of BRCA1 and RIF1 was included and analyzed as controls. Data shown are the means from 3–6 independent experiments for each siRNA. Error bars represent standard deviations (SDs). **** p-value ≤ 1.0×10−5, ** p-value ≤ 0.01, * p-value ≤ 0.05.
These data revealed a number of previously unidentified BRCA1-interacting proteins, along with known interactors, many of which were differentially affected by mutations in different domains of BRCA1. For example, HR proteins previously known to interact with BRCA1 (including BRIP1 and RBBP8) (71, 75) had a similar pattern of interaction loss (boxed in green in Fig. 4B) associated with BRCT domain mutants (S1655F, 5382insC, M1775R), whereas RING domain mutants (I26A, C61G, R71G) maintained these interactions. In a separate pattern, we found that the C61G RING domain mutant abolishes interaction with BARD1 (Fig. 4B), as previously reported (66). Several interactions could be confirmed by co-IP/western blot analysis (Fig. 4C). These results suggest that RING domain mutants are hypomorphic and may retain some BRCA1 functions, which could explain at least in part why the BRCA1 C61G variant is only moderately sensitive to cisplatin and poly (ADP-ribose) polymerase (PARP) inhibitors and becomes readily resistant to these drugs (76, 77).
A ubiquitin E2-conjugating enzyme, UBE2N (also known as UBC13), was found to interact with WT BRCA1, but to a lesser degree with mutant forms of BRCA1 (PPI score < 0.6) (boxed in sky blue in Fig. 4B). For example, consistent with reports from yeast two-hybrid studies (78), we found a six-fold reduction in UBE2N associated with the I26A mutant compared to WT, suggesting that UBE2N interacts with BRCA1 through the RING domain. Notably, the M1775R mutation in the BRCT domain also dramatically reduced the interaction with UBE2N (Fig. 4B), suggesting that M1775 residue in the BRCTs domain may also contribute to the interaction with UBE2N, although the underlying mechanism is currently unclear. Depletion of UBE2N shows HR defects including altered RAD51 filament formation and E3 Ub ligase function of BRCA1 (79), indicating a critical role of UBE2N in HR repair. Consistent with the cell line models, we found that baseline UBE2N mRNA expression was significantly lower in patients who achieved pathologic complete response (pCR) to the PARP-inhibitor (veliparib)/carboplatin (80) in comparison to non-responsive patients (p = 0.034, Fig. 4D) in I-SPY 2, a neoadjuvant, adaptive clinical platform trial for high risk early stage breast cancer (81). In contrast, BC tumors in the control arm did not show any significant difference in UBE2N expression between pCR and no pCR groups. Although the average expression of UBE2N is numerically lower in TN than HR+HER2-tumors, the difference is not statistically significant (p=0.1, fig. S4C). Moreover, the association of low UBE2N expression to pCR is seen more strongly in the HR+HER2- subset (p=0.0012, fig. S4D) than in the population as a whole (p=0.034, Fig. 4D), suggesting that the association is not merely a proxy for the TN subtype. BRCA1/2 germline mutation status is available for 112 out of 115 patients in the VC and concurrent control arms, and 15 out of 112 (13%) are BRCA1/2-. UBE2N trends toward lower expression levels in BRCA1/2 mutation carriers (p=0.07, fig. S4E). However, in the HR+HER2- subset (n=55 with BRCA1/2 mutation status data), where lower UBE2N levels associate with response to VC, there are only 4 mutation carriers and no difference in UBE2N levels by BRCA1/2 status (p=1). Furthermore, only 2 HR+HER2- BRCA1/2 mutation carriers were treated by VC (only one of these achieved pCR), implying that association between UBE2N and pCR in this subset may not be driven by ‘spurious’ association with germline BRCA1/2 mutation (fig. S4F). These results indicate that expression of UBE2N may serve as a biomarker of response to PARP inhibitors and other DNA repair targeted therapies (Odds Ratio = 2.9, Fig. 4E). Importantly, there are several observations that not all of BRCA1-interacting HR proteins show a response to PARPi upon depletion (by CRISPR or siRNA) when tested in multiple cell line models, as seen in a recent CRISPR screen (82). Furthermore, the clinical response of many BRCA1-interacting proteins to PARP inhibition has never been explicitly tested in a clinical setting. Therefore, identification of UBE2N as a potential biomarker for PARPi response could be clinically valuable to help stratify patients with UBE2N alteration for targeted therapy.
Intriguingly, a deubiquitinase USP28 was found to interact more strongly with the BRCA1 mutants C61G and 5382insC than WT by AP-MS (boxed in brown in Fig. 4B), and this differential interaction was confirmed by co-IP/western blot analysis (Fig. 4C). USP28 was previously known to stabilize multiple DDR proteins (e.g. CHEK2, CLSPN, NBS1, and 53BP1) in response to DNA damage (83, 84), and its recruitment to DNA double-strand breaks (DSBs) was shown to be dependent on the tandem BRCT domain of TP53BP1 (85). To further delineate the role of USP28 in DNA DSB repair, we analyzed the effect of USP28 knockdown on HR activity. In this assay, DNA DSBs were induced by I-SceI endonuclease, which cleaves non-functional GFP cassettes engineered in the genome of U2OS reporter cell lines (DR-GFP) (86). DSB repair that depends on the HR mechanism restores a functional GFP gene, yielding a readout tied to fluorescent signal intensity (Fig. 4F). Upon USP28 knockdown by siRNA, HR activity was significantly reduced when compared to NTC (Fig. 4G and fig. S5A), suggesting USP28 is regulating proper HR repair. In the same assay, knockdown of BRCA1 greatly decreased HR as expected (87), while depletion of RIF1, a protein functioning in an alternative non-homologous end joining DNA repair pathway did not. To identify proteins potentially regulated by USP28 via deubiquitination, we performed a global ubiquitome analysis upon knockout of USP28 in MDA-MB-231 cells by CRISPR/Cas9 ribonucleoprotein (RNP) strategy (88). Polyclonal USP28 knockout cells with ~85% knockout efficiency (crRNA#3, fig. S5B) as well as control (NTC) cells were lysed and ubiquitinated peptides were subsequently enriched and analyzed by mass spectrometry. Functional enrichment analysis of 275 ub-enriched proteins observed in USP28 knockout cells compared to control cells (log2FC ≥ 1, p-value ≤ 0.05; table S9) revealed that 19 proteins (7%) belong to the cellular response to stress category (GO: 0033554) (fig. S5C), and subsequent analysis further categorized proteins involved in cellular response to DNA damage stimulus (8 proteins) (fig. S5D–E). Notably, these include ubiquitylation-dependent DDR signaling proteins (UBA1, RAD18, DDB2) (89–91) and DNA replication helicases (MCM3, MCM6) (92), providing further evidence for a role of USP28 in responding to DNA damage and replication stress in cells.
Spinophilin is a previously unidentified BRCA1-interacting protein
Another protein interacting with BRCA1 in a mutation-dependent manner is Spinophilin (encoded by PPP1R9B), a neuronal scaffolding protein that regulates synaptic transmission through its ability to target protein phosphatase 1 (PP1) to dendritic spines where it inactivates glutamate receptors (93). Binding of Spinophilin to BRCA1 was abolished by BRCT domain mutations similar to the pattern observed earlier for HR proteins (Fig. 4B). Reciprocal AP-MS was performed using 3xFLAG-tagged Spinophilin in MDA-MB-231 cells, which confirmed the interaction of Spinophilin with BRCA1 as well as with PP1 catalytic subunits (PPP1CA, PPP1CB, PPP1CC) (Fig. 5A). MSstats analysis of differential interactions between BRCA1 WT and BRCT domain mutants demonstrated that an intact BRCT domain is required for the BRCA1-Spinophilin interaction (fig. S6A). In fact, Spinophilin was previously observed but unexplored in a systematic analysis of proteins interacting with the BRCT domain of BRCA1 (94). The BRCA1 BRCT domain binds to a pS-x-x-F motif on target proteins (95), and such recognition has been reported for BRCA1 interactions with FAM175A, BRIP1, and RBBP8 (71, 75, 96). Importantly, Spinophilin has three potential pS-x-x-F recognition motifs at S212, S248, and S635. To map the BRCT domain-binding motif on Spinophilin, we mutagenized Serine residues into Alanine at each pS-x-x-F site and transfected each Spinophilin (HA-tagged) construct individually along with 3xFLAG-BRCA1 DNA construct into HEK293T cells. The association of each Spinophilin mutant with BRCA1 was monitored after immunoprecipitation of Spinophilin using anti-HA beads followed by western blotting with anti-FLAG antibody. This experiment showed that the S635A mutant interacted significantly less with BRCA1, demonstrating that Spinophilin requires the pS635PTF motif for binding the BRCT domain of BRCA1 (Fig. 5B and fig. S6B). The F451A mutation, which resides in the interface with PP1 RVXF binding groove, also significantly diminished the interaction of Spinophilin with PP1 (PPP1CA) (Fig. 5B and fig. S6B), as observed previously (97).
Figure 5. Spinophilin interacts with BRCA1 and regulates DNA damage response via dephosphorylation.
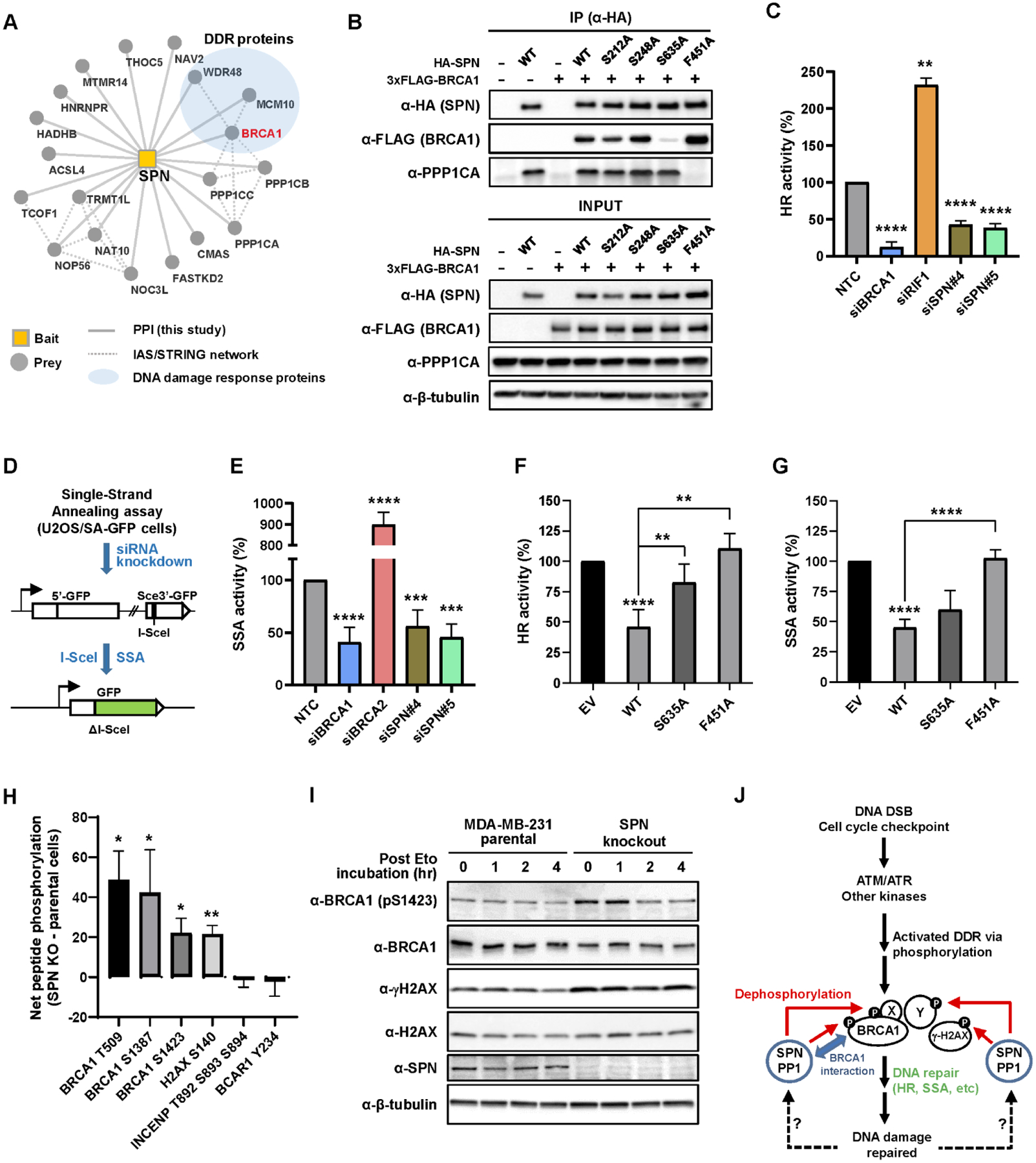
(A) AP-MS of 3xFLAG-tagged Spinophilin (SPN, encoded by PPP1R9B) identifies BRCA1 (highlighted in a red edge) and other DDR-related proteins as well as PP1 catalytic subunits (PPP1CA, PPP1CB, and PPP1CC) in MDA-MB-231 cells. (B) HA-tagged SPN (WT, S212A, S248A, S635A, or F451A) was transfected with 3xFLAG-BRCA1 into HEK293T cells. After pulldown with anti-HA magnetic beads, co-associated 3xFLAG-BRCA1 was monitored. S635A mutation significantly diminished BRCA1 pulldown, while F451A mutation abolished the association with PP1 catalytic subunit (PPP1CA). Empty vectors were used as negative control. (C) HR activities upon depletion of SPN relative to NTC (set to 100%) were measured as in Fig. 4G. Data shown are the means from 3–9 independent experiments for each siRNA. Error bars represent standard deviations (SDs). **** p-value ≤ 1.0×10−5, ** p-value ≤ 1.0×10−2. (D) A schematic of the SA-GFP reporter assay. The SA-GFP reporter contains a 5’-fragment of GFP (5’-GFP) and a 3’-fragment of GFP (Sce3’-GFP) that contains an I-SceI site. Repair of the DSB in Sce3’-GFP using 266 nt homology by single-strand annealing (SSA) restores a functional GFP gene. (E) SSA activities upon depletion of SPN relative to NTC (set to 100%). Depletion of BRCA1 and BRCA2 was included and analyzed as controls. Data shown are the means ± SDs from six independent experiments for each siRNA. **** p-value ≤ 1.0×10−4, *** p-value ≤ 1.0×10−3. (F-G) CMV promoter-driven SPN (WT, S635A, or F451A) expression DNA construct was transfected into U2OS DR and SA-GFP reporter cells, and the effect of SPN overexpression on HR and SSA activities was monitored, respectively. **** p-value ≤ 1.0×10−4, ** p-value ≤ 1.0×10−2. (H) Selective peptides derived from various proteins including BRCA1 and H2AX as well as non-DNA repair proteins (INCENP and BCAR1) were individually mixed with lysates from either SPN KO or parental cells and subsequently monitored for phosphorylation by measuring ATP consumption in each reaction. Net peptide phosphorylation values are net changes in ATP concentrations between SPN knockout cells and parental control cells (subtraction of parental runs from SPN KO runs). Mean value of two independent runs was shown in the y-axis. Units are arbitrary. Error bars represent standard deviations (SDs). * p-value ≤ 0.05, ** p-value ≤ 1.0×10−2. (I) SPN KO and parental cells were treated with 2.5 μM etoposide (Eto) for 16 hr and changes in the phosphorylation level of BRCA1 S1423 (pS1423) and H2AX S140 (γ-H2AX) were monitored at 0, 1, 2, and 4 hr post Eto treatment with fresh medium. (J) Model for the role of SPN in regulating DDR. See text for details.
The AP-MS experiment found that Spinophilin interacts with additional proteins involved in DNA replication and repair, including MCM10 and WDR48 (Fig. 5A). These results suggest that Spinophilin may participate in and/or regulate DNA repair by interacting with various DNA repair and replication proteins, including BRCA1. To explore this hypothesis, we analyzed the effect of Spinophilin knockdown on DNA repair by HR and single-strand annealing (SSA). Upon Spinophilin knockdown, HR activity was significantly reduced compared to NTC siRNA (Fig. 5C and fig. S6C). Similarly, knockdown of Spinophilin significantly reduced SSA activity, comparable to BRCA1 depletion, while BRCA2 depletion dramatically increased SSA as seen previously (87) (Fig. 5D–E and fig. S6D), implying that Spinophilin promotes both HR and SSA-mediated DSB repair.
According to the breast invasive carcinoma patient cohort study by TCGA (2), there is no clear evidence that the Spinophilin (PPP1R9B) gene is recurrently silenced by deep DNA copy loss or mutation. Using the interface to the TCGA methylation data (http://maplab.imppc.org/wanderer/), we do not observe evidence for methylation of the Spinophilin (PPP1R9B) promoter, either. However, among the sequenced TCGA BC patient tumors that have alterations in the Spinophilin (PPP1R9B) gene (73 out of 1084), the majority (69 out of 73) of the alterations are amplification, suggesting that higher expression of Spinophilin may be a pathogenic driver of breast cancer. Thus, we also analyzed the effect of Spinophilin overexpression (mimicking amplification) on HR and SSA DSB repair. CMV promoter-driven overexpression of WT Spinophilin in the U2OS reporter cell lines led to significant reduction in both HR and SSA activities, compared to empty vector control (Fig. 5F–G). Consistent with these observations, a recent study identified Spinophilin as a BC oncogene (98). However, the effect of overexpression of the S635A mutant (incapable of interacting with BRCA1) on HR was significantly mitigated, and more importantly, overexpression of the F451A mutant (incapable of interacting with PP1) did not affect HR or SSA at all (Fig. 5F–G and fig. S6E–F), implying that the inhibitory effect of WT Spinophilin overexpression is likely exerted by PP1-mediated dephosphorylation and/or BRCA1 interaction (at least for HR).
To further unravel the biological function of Spinophilin, we knocked out Spinophilin in MDA-MB-231 cells using a CRISPR/Cas9 RNP method (88). Using genomic PCR/sequencing and western blot analyses, at least two independent Spinophilin knockout clones were identified (fig. S7). We hypothesized that Spinophilin targets PP1 to specific DNA repair proteins for dephosphorylation. To uncover potential dephosphorylation targets under this model, we employed a high-throughput peptide phosphorylation assay platform (99). This system uses a collection of peptide sequences derived from biological targets of multiple kinases, which serves as phosphorylatable probes in a large-scale ATP-consumption assay (100). In this assay, we measured changes in phosphorylation (i.e., ATP-consumption) of peptide substrates derived from various proteins, including BRCA1 and the DSB-associated histone H2AX as well as proteins unrelated to DNA repair (e.g., INCENP, BCAR1), in Spinophilin knockout (KO) and parental cells. We found that BRCA1 residues at T509, S1387, and S1423, as well as H2AX at S140 (γ-H2AX), were significantly increased in phosphorylation in Spinophilin KO cell lysates compared to lysates from parental cells, and, in fact, were among the top 20 most increased sites (Fig. 5H and fig. S8A). BRCA1 pT509 enhances nuclear localization and transcriptional activity of BRCA1 (101), and pS1387 and pS1423 sites in the BRCA1 SQ-cluster region are critical for HR repair and cell-cycle checkpoint functions (102–104). γ-H2AX is a hallmark of DNA DSB (105) and initiates a signaling cascade to recruit various DSB repair proteins to properly repair the DNA damage (106). These results were in contrast to phosphorylation of the INCENP and BCAR1 peptides which were not significantly changed by Spinophilin disruption. To validate these results, Spinophilin KO and parental cells were treated with 2.5 μM etoposide (Eto) for 16 hr to induce BRCA1 pS1423 and γ-H2AX, and persistence of these phosphosites was monitored by western blot at 0, 1, 2, 4 hr post Eto treatment. Indeed, these phosphosites remained at a significantly higher level and were maintained for longer in Spinophilin KO cells compared to parental cells (Fig. 5I). Consistently, we also observed higher levels of BRCA1 pS1423 and γ-H2AX after Eto treatment in Spinophilin knockdown U2OS cells (fig. S8B).
To unbiasedly identify targets that are potentially dephosphorylated by Spinophilin, we performed a phospho-proteomic analysis using the two Spinophilin KO clones and parental MDA-MB-231 cells. In this experiment, we found that 473 phospho-peptides (from 407 proteins) were more than 4-fold enriched (log2FC ≥ 2, p-value ≤ 0.05) in both Spinophilin KO clones compared to parental cells, while 328 phospho-peptides (from 280 proteins) were more than 4-fold depleted (log2FC ≤ −2, p-value ≤ 0.05) (fig. S9A–B and table S10). Notably, proteins known to be directly connected to BRCA1 via physical and/or functional interactions were significantly enriched in the group of up-regulated phospho-proteins seen in Spinophilin KO cells (fig. S9C). Functional enrichment analysis of these 407 up-regulated phospho-proteins revealed that 32 proteins (8%) belong to the DNA repair category (GO: 0006281) (fig. S9D), and subsequent analysis further categorized 15 proteins involved in double-strand break repair (fig. S9E). Importantly, the 32 DNA repair proteins include not only BRCA1-interacting proteins (e.g. BRIP1) but also contain key regulators of DNA repair pathways (e.g. MDC1, TP53BP1), and helicase/nuclease (e.g. MRE11, WRN), suggesting broad roles of Spinophilin in modulating DNA repair and genome integrity (fig. S9F–G). Taken together with the previous data, these results indicate that Spinophilin regulates BRCA1 and DDR signaling by dephosphorylation (Fig. 5J).
DISCUSSION
The cell is comprised of a series of protein complexes, or “machines” (107), that function together in an elaborate network of pathways. Mutations, such as seen in cancer, perturb the machines and therefore the network of pathways (108). Understanding the machines and networks in healthy and diseased states is crucial for a deeper understanding of disease biology and ultimately the discovery of new therapeutic strategies and the application of precision medicine (109). Using this premise as an underlying motivation, we generated comprehensive interaction maps for 40 frequently altered BC proteins. This large-scale study of biophysical interactions in BC and across three cell lines of human breast tissue origin provides a PPI resource to study BC biology and to contextualize uncharacterized mutations within signaling pathways and protein complexes. Approximately 78% of PPIs we identified have not been previously reported (Fig. 1C), and 81% are not shared across cell lines (Fig. 1E). The cell line PPI specificity we observe in this study and our accompanying manuscript (Swaney et al.), as well as in a recent large-scale AP-MS study (27), speaks to the complexity of PPI networks and their heterogeneity among different cell types, calling for more studies to deeply characterize these networks in a wider range of cellular contexts. Given that each BC subtype arises by distinct tumorigenic mechanisms due to different genomic alterations, PPIs from cell lines representing other BC subtypes will most likely capture additional sets of interactors reflecting their distinct tumorigenic mechanisms. Our results also suggest that protein abundance in a cell line is not the sole mechanism for PPI specificity (fig. S1D). Presumably other features, such as differential posttranslational modifications (PTM), cellular compartmentalization, and/or mutational status of proteins may contribute to cell type-specificity. Notably, prey proteins enriched to either of the two BC cell lines are more frequently mutated in breast tumors than preys from non-tumorigenic cells (Fig. 1E), implying that proteins interacting with cancer drivers may also contribute to the onset of cancer.
Using the systematic proteomic approach described in the manuscript, we have identified previously unidentified interactors of PIK3CA that negatively regulate the PI3K/AKT pathway (Fig. 3D–H). Interestingly, RNA expressions of several of these interactors (e.g. BPIFA1, BPIFB1, and MUC5B) were found to be correlated in human airway epithelial cells (110), implying that they may be controlled under the same transcriptional regulatory program and/or functionally related. Our results reveal that they play a role in the PI3K/AKT signaling cascade in BC context. BPIFA1 is a lipid-binding protein with antimicrobial and immunomodulatory functions (111). It is significantly downregulated in nasopharyngeal carcinoma (112) and its single-nucleotide polymorphisms are associated with increased susceptibility to this tumor type (113). BPIFA1 is known to increase the expression of PTEN via downregulating the miR-141 oncogene (114), thus knockdown of BPIFA1 could indirectly activate PI3K/AKT. The BPIFA1-PIK3CA interaction we identified and the inhibition of WT PIK3CA kinase activity by BPIFA1 in vitro suggests that BPIFA1 may also directly modulate PI3K/AKT via PPI, which warrants a structural study of the complex. Another PIK3CA interactor, SCGB2A1, is a small secreted protein highly differentially expressed in multiple types of cancer including breast and endometrium (115, 116). Previous studies have shown that SCGB2A1 is expressed at lower levels in luminal BC compared to histologically normal breast epithelium (117), and that decreased SCGB2A1 expression in endometrial tumors is significantly correlated with higher grade, advanced stage, and worse overall survival (118). Neither the BPIFA1 or SCGB2A1 gene harbors recurrent focal deep deletions, missense mutations, or truncating mutations in their coding regions among 996 breast tumors in TCGA analysis (2). However, SCGB2A1 harbored loss of heterozygosity (LOH) in 10% of breast tumors (7% LOH in all tumors) according to a recent study (119), which suggests that its allelic imbalance may contribute to the development of tumors.
We also uncovered a number of previously unidentified interactors of the well-studied BRCA1, which is tightly connected to DNA repair processes, including Spinophilin. Interestingly, both knockdown and overexpression of Spinophilin led to significant impairment in DSB repair by both HR and SSA pathways (Fig. 5C, E–G), establishing that this protein has a defined role in DNA repair. An intriguing question is how the alterations of Spinophilin expression interfere with HR and SSA repair activity. Given the role of Spinophilin in dephosphorylation, one plausible explanation is that prolonged phosphorylation or prevailed dephosphorylation of BRCA1 and other DDR proteins is inhibitory to multiple steps during DNA repair, including DSB-end resection which is a prerequisite for HR and SSA. In agreement with this hypothesis, continuous DNA damage signaling and phosphorylation of several DDR proteins (including H2AX, NBN, RPA2, and CHEK2) induced by short double-stranded DNA molecules (mimicking DNA DSB) was shown to disorganize the cellular DNA repair system and inhibit DSB repair (120). Alternatively, but not exclusively, Spinophilin may play a role in initiating the DSB repair process by removing constitutive phosphorylations that inhibit the function of DDR proteins. Supporting this scenario, a phospho-proteomic study revealed that over one-third of the captured phospho-peptides were dephosphorylated within minutes of DNA damage (121). Additionally, Spinophilin may be involved in counteracting DSB-induced phosphorylation events, thus promoting recycling of DDR proteins as DNA damage is being repaired, possibly through its interaction with BRCA1.
Finally, while the approach we described in this study was applied to BC, it is equally powerful against other cancers, including head and neck squamous cell carcinoma (HNSCC) (Swaney et al., accompanying manuscript). Efforts such as this will ultimately lead to hierarchical maps of protein complexes and systems in both healthy and diseased cells (Zheng et al., accompanying manuscript), which, based on the mutational landscape, can be predictive for use of known treatments and can also be used to uncover previously unidentified therapeutic strategies across a multitude of disease areas.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Jeremy Stark for the kind gift of U2OS GFP reporter cell lines. We also thank Christina Moelders for technical support running the peptide phosphorylation assay, David Quigley for comments on the genomic alterations in the BPIFA1, SCGB2A1, and PPP1R9B genes, and Dexter Pratt for deposition of PPI network data to the NDEx.
Funding:
This work was supported by the NIH U54 CA209891 (NJK, TI, LV, and AA), U24 CA184427 (TI), P41 GM103504 (TI), R50 CA243885 (JFK) and R01 CA138804 (BX). Additional support was received from the Martha and Bruce Atwater Breast Cancer Research Program via UCSF Helen Diller Family Comprehensive Cancer Center (MK), UCSF Prostate Cancer Program Research Pilot Funding (MK), Benioff Initiative for Prostate Cancer Research (MK) and the V foundation and BRCA foundation (AA). PLA Data for this study were acquired at the Center for Advanced Light Microscopy-Nikon Imaging Center at UCSF on instruments obtained using grants from the UCSF Program for Breakthrough Biomedical Research funded in part by the Sandler Foundation and the UCSF Research Resource Fund Award. MB is a recipient of NIH F32 CA239333. BT is a recipient of NIH F32 CA239336.
Competing interests:
LV is a co-founder, stockholder, and part-time employee of Agendia NV. AA is a co-founder of Tango Therapeutics, Azkarra Therapeutics, Ovibio Corporation and Kytarro; a consultant for SPARC, Bluestar, GenVivo, Earli, Cura, ProLynx and GSK; a member of the Scientific Advisory Board (SAB) of Genentech and GLAdiator; receives grant/research support from SPARC and AstraZeneca; holds patents on the use of PARP inhibitors held jointly with AstraZeneca which he has benefited financially (and may do so in the future). TI is a co-founder of Data4Cure, Inc., is on the SAB, and has an equity interest. TI is on the SAB of Ideaya BioSciences, Inc., has an equity interest, and receives sponsored research funding. The terms of these arrangements have been reviewed and approved by the University of California San Diego in accordance with its conflict of interest policies. NJK is a shareholder of Tenaya Therapeutics and has received stocks from Maze Therapeutics and Interline Therapeutics; has consulting agreements with the Icahn School of Medicine at Mount Sinai, New York, Maze Therapeutics and Interline Therapeutics. The laboratory of NJK has received research support from Vir Biotechnology and F. Hoffmann-La Roche.
Footnotes
Data and materials availability:
All raw data files and search results are available from the Pride partner ProteomeXchange repository (122). For AP-MS and protein abundance data from MCF10A, MCF7, and MDA-MB-231 cells, these can be accessed under the PXD019639 identifier. For PTM (ub and ph) analysis data, these are available under the PXD025931 identifier. PPI networks can be accessed on the NDEx platform (https://bit.ly/3y10JFY). DOIs and links for each PPI network are in table S1. Expression vectors used in this study are readily available from the authors for biomedical researchers and educators in the nonprofit sector under MTA. Further information and requests for resources and reagents should be directed to and will be fulfilled by NJK (nevan.krogan@ucsf.edu).
REFERENCES AND NOTES
- 1.Siegel RL, Miller KD, Fuchs HE, Jemal A, Cancer Statistics, 2021. CA: A Cancer Journal for Clinicians. 71 (2021), pp. 7–33. [DOI] [PubMed] [Google Scholar]
- 2.Cancer Genome Atlas, Network, Comprehensive molecular portraits of human breast tumours. Nature. 490, 61–70 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, Yates LR, Papaemmanuil E, Beare D, Butler A, Cheverton A, Gamble J, Hinton J, Jia M, Jayakumar A, Jones D, Latimer C, Lau KW, McLaren S, McBride DJ, Menzies A, Mudie L, Raine K, Rad R, Chapman MS, Teague J, Easton D, Langerod A, Oslo Breast Cancer, Consortium, Lee MT, Shen CY, Tee BT, Huimin BW, Broeks A, Vargas AC, Turashvili G, Martens J, Fatima A, Miron P, Chin SF, Thomas G, Boyault S, Mariani O, Lakhani SR, van de Vijver M, van ‘t Veer L, Foekens J, Desmedt C, Sotiriou C, Tutt A, Caldas C, Reis-Filho JS, Aparicio SA, Salomon AV, Borresen-Dale AL, Richardson AL, Campbell PJ, Futreal PA, Stratton MR, The landscape of cancer genes and mutational processes in breast cancer. Nature. 486, 400–404 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wood LD, Parsons DW, Jones S, Lin J, Sjöblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, Silliman N, Szabo S, Dezso Z, Ustyanksky V, Nikolskaya T, Nikolsky Y, Karchin R, Wilson PA, Kaminker JS, Zhang Z, Croshaw R, Willis J, Dawson D, Shipitsin M, Willson JKV, Sukumar S, Polyak K, Park BH, Pethiyagoda CL, Pant PVK, Ballinger DG, Sparks AB, Hartigan J, Smith DR, Suh E, Papadopoulos N, Buckhaults P, Markowitz SD, Parmigiani G, Kinzler KW, Velculescu VE, Vogelstein B, The genomic landscapes of human breast and colorectal cancers. Science. 318, 1108–1113 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Cho A, Shim JE, Kim E, Supek F, Lehner B, Lee I, MUFFINN: cancer gene discovery via network analysis of somatic mutation data. Genome Biology. 17 (2016), , doi: 10.1186/s13059-016-0989-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Knijnenburg TA, Wang L, Zimmermann MT, Chambwe N, Gao GF, Cherniack AD, Fan H, Shen H, Way GP, Greene CS, Liu Y, Akbani R, Feng B, Donehower LA, Miller C, Shen Y, Karimi M, Chen H, Kim P, Jia P, Shinbrot E, Zhang S, Liu J, Hu H, Bailey MH, Yau C, Wolf D, Zhao Z, Weinstein JN, Li L, Ding L, Mills GB, Laird PW, Wheeler DA, Shmulevich I, Cancer Genome Atlas Research, Network, Monnat RJ Jr., Xiao Y, Wang C, Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 23, 239–254 e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, Dimitriadoy S, Liu DL, Kantheti HS, Saghafinia S, Chakravarty D, Daian F, Gao Q, Bailey MH, Liang W-W, Foltz SM, Shmulevich I, Ding L, Heins Z, Ochoa A, Gross B, Gao J, Zhang H, Kundra R, Kandoth C, Bahceci I, Dervishi L, Dogrusoz U, Zhou W, Shen H, Laird PW, Way GP, Greene CS, Liang H, Xiao Y, Wang C, Iavarone A, Berger AH, Bivona TG, Lazar AJ, Hammer GD, Giordano T, Kwong LN, McArthur G, Huang C, Tward AD, Frederick MJ, McCormick F, Meyerson M, Cancer Genome Atlas Research Network, Van Allen EM, Cherniack AD, Ciriello G, Sander C, Schultz N, Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell. 173, 321–337.e10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Creixell P, Reimand J, Haider S, Wu G, Shibata T, Vazquez M, Mustonen V, Gonzalez-Perez A, Pearson J, Sander C, Raphael BJ, Marks DS, Ouellette BFF, Valencia A, Bader GD, Boutros PC, Stuart JM, Linding R, Lopez-Bigas N, Stein LD, Mutation Consequences and Pathway Analysis Working Group of the International Cancer Genome Consortium, Pathway and network analysis of cancer genomes. Nat. Methods 12, 615–621 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hofree M, Shen JP, Carter H, Gross A, Ideker T, Network-based stratification of tumor mutations. Nat. Methods 10, 1108–1115 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leiserson MDM, Vandin F, Wu H-T, Dobson JR, Eldridge JV, Thomas JL, Papoutsaki A, Kim Y, Niu B, McLellan M, Lawrence MS, Gonzalez-Perez A, Tamborero D, Cheng Y, Ryslik GA, Lopez-Bigas N, Getz G, Ding L, Raphael BJ, Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat. Genet 47, 106–114 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paczkowska M, Barenboim J, Sintupisut N, Fox NS, Zhu H, Abd-Rabbo D, Mee MW, Boutros PC, PCAWG Drivers and Functional Interpretation Working Group, Reimand J, PCAWG Consortium, Integrative pathway enrichment analysis of multivariate omics data. Nat. Commun 11, 735 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reyna MA, Haan D, Paczkowska M, Verbeke LPC, Vazquez M, Kahraman A, Pulido-Tamayo S, Barenboim J, Wadi L, Dhingra P, Shrestha R, Getz G, Lawrence MS, Pedersen JS, Rubin MA, Wheeler DA, Brunak S, Izarzugaza JMG, Khurana E, Marchal K, von Mering C, Sahinalp SC, Valencia A, PCAWG Drivers and Functional Interpretation Working Group, Reimand J, Stuart JM, Raphael BJ, PCAWG Consortium, Pathway and network analysis of more than 2500 whole cancer genomes. Nat. Commun 11, 729 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taylor IW, Linding R, Warde-Farley D, Liu Y, Pesquita C, Faria D, Bull S, Pawson T, Morris Q, Wrana JL, Dynamic modularity in protein interaction networks predicts breast cancer outcome. Nat. Biotechnol 27, 199–204 (2009). [DOI] [PubMed] [Google Scholar]
- 14.Kennedy SA, Jarboui M-A, Srihari S, Raso C, Bryan K, Dernayka L, Charitou T, Bernal-Llinares M, Herrera-Montavez C, Krstic A, Matallanas D, Kotlyar M, Jurisica I, Curak J, Wong V, Stagljar I, LeBihan T, Imrie L, Pillai P, Lynn MA, Fasterius E, Al-Khalili Szigyarto C, Breen J, Kiel C, Serrano L, Rauch N, Rukhlenko O, Kholodenko BN, Iglesias-Martinez LF, Ryan CJ, Pilkington R, Cammareri P, Sansom O, Shave S, Auer M, Horn N, Klose F, Ueffing M, Boldt K, Lynn DJ, Kolch W, Extensive rewiring of the EGFR network in colorectal cancer cells expressing transforming levels of KRASG13D. Nat. Commun 11, 499 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gill MK, Christova T, Zhang YY, Gregorieff A, Zhang L, Narimatsu M, Song S, Xiong S, Couzens AL, Tong J, Krieger JR, Moran MF, Zlotta AR, van der Kwast TH, Gingras A-C, Sicheri F, Wrana JL, Attisano L, A feed forward loop enforces YAP/TAZ signaling during tumorigenesis. Nat. Commun 9, 3510 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harkness EF, Barrow E, Newton K, Green K, Clancy T, Lalloo F, Hill J, Evans DG, Lynch syndrome caused by MLH1 mutations is associated with an increased risk of breast cancer: a cohort study. J. Med. Genet 52, 553–556 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Mimori K, Inoue H, Shiraishi T, Ueo H, Mafune K-I, Tanaka Y, Mori M, A single-nucleotide polymorphism of SMARCB1 in human breast cancers. Genomics. 80, 254–258 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Zheng W, Cong X-F, Cai W-H, Yang S, Mao C, Zou H-W, Current evidences on XPC polymorphisms and breast cancer susceptibility: a meta-analysis. Breast Cancer Res. Treat 128, 811–815 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Hoenerhoff MJ, Chu I, Barkan D, Liu Z-Y, Datta S, Dimri GP, Green JE, BMI1 cooperates with H-RAS to induce an aggressive breast cancer phenotype with brain metastases. Oncogene. 28, 3022–3032 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iorio F, Knijnenburg TA, Vis DJ, Bignell GR, Menden MP, Schubert M, Aben N, Gonçalves E, Barthorpe S, Lightfoot H, Cokelaer T, Greninger P, van Dyk E, Chang H, de Silva H, Heyn H, Deng X, Egan RK, Liu Q, Mironenko T, Mitropoulos X, Richardson L, Wang J, Zhang T, Moran S, Sayols S, Soleimani M, Tamborero D, Lopez-Bigas N, Ross-Macdonald P, Esteller M, Gray NS, Haber DA, Stratton MR, Benes CH, Wessels LFA, Saez-Rodriguez J, McDermott U, Garnett MJ, A Landscape of Pharmacogenomic Interactions in Cancer. Cell. 166, 740–754 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu K, Chen B, Aran D, Charalel J, Yau C, Wolf DM, van ‘t Veer LJ, Butte AJ, Goldstein T, Sirota M, Comprehensive transcriptomic analysis of cell lines as models of primary tumors across 22 tumor types. Nature Communications. 10 (2019), , doi: 10.1038/s41467-019-11415-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Santagata S, Thakkar A, Ergonul A, Wang B, Woo T, Hu R, Harrell JC, McNamara G, Schwede M, Culhane AC, Kindelberger D, Rodig S, Richardson A, Schnitt SJ, Tamimi RM, Ince TA, Taxonomy of breast cancer based on normal cell phenotype predicts outcome (2014), , doi: 10.1172/JCI70941. [DOI] [PMC free article] [PubMed]
- 23.Sowa ME, Bennett EJ, Gygi SP, Harper JW, Defining the human deubiquitinating enzyme interaction landscape. Cell. 138, 389–403 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K, Dong R, Guarani V, Vaites LP, Ordureau A, Rad R, Erickson BK, Wuhr M, Chick J, Zhai B, Kolippakkam D, Mintseris J, Obar RA, Harris T, Artavanis-Tsakonas S, Sowa ME, De Camilli P, Paulo JA, Harper JW, Gygi SP, The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell. 162, 425–440 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Teo G, Liu G, Zhang J, Nesvizhskii AI, Gingras AC, Choi H, SAINTexpress: improvements and additional features in Significance Analysis of INTeractome software. J. Proteomics 100, 37–43 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huttlin EL, Bruckner RJ, Paulo JA, Cannon JR, Ting L, Baltier K, Colby G, Gebreab F, Gygi MP, Parzen H, Szpyt J, Tam S, Zarraga G, Pontano-Vaites L, Swarup S, White AE, Schweppe DK, Rad R, Erickson BK, Obar RA, Guruharsha KG, Li K, Artavanis-Tsakonas S, Gygi SP, Harper JW, Architecture of the human interactome defines protein communities and disease networks. Nature. 545, 505–509 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huttlin EL, Bruckner RJ, Navarrete-Perea J, Cannon JR, Baltier K, Gebreab F, Gygi MP, Thornock A, Zarraga G, Tam S, Szpyt J, Panov A, Parzen H, Fu S, Golbazi A, Maenpaa E, Stricker K, Thakurta SG, Rad R, Pan J, Nusinow DP, Paulo JA, Schweppe DK, Vaites LP, Wade Harper J, Gygi SP, Dual Proteome-scale Networks Reveal Cell-specific Remodeling of the Human Interactome. bioRxiv (2020), p. 2020.01.19.905109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shah PS, Link N, Jang GM, Sharp PP, Zhu T, Swaney DL, Johnson JR, Von Dollen J, Ramage HR, Satkamp L, Newton B, Hüttenhain R, Petit MJ, Baum T, Everitt A, Laufman O, Tassetto M, Shales M, Stevenson E, Iglesias GN, Shokat L, Tripathi S, Balasubramaniam V, Webb LG, Aguirre S, Willsey AJ, Garcia-Sastre A, Pollard KS, Cherry S, Gamarnik AV, Marazzi I, Taunton J, Fernandez-Sesma A, Bellen HJ, Andino R, Krogan NJ, Comparative Flavivirus-Host Protein Interaction Mapping Reveals Mechanisms of Dengue and Zika Virus Pathogenesis. Cell. 175, 1931–1945.e18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Batra J, Hultquist JF, Liu D, Shtanko O, Von Dollen J, Satkamp L, Jang GM, Luthra P, Schwarz TM, Small GI, Arnett E, Anantpadma M, Reyes A, Leung DW, Kaake R, Haas P, Schmidt CB, Schlesinger LS, LaCount DJ, Davey RA, Amarasinghe GK, Basler CF, Krogan NJ, Protein Interaction Mapping Identifies RBBP6 as a Negative Regulator of Ebola Virus Replication. Cell. 175, 1917–1930 e13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, O’Meara MJ, Rezelj VV, Guo JZ, Swaney DL, Tummino TA, Huettenhain R, Kaake RM, Richards AL, Tutuncuoglu B, Foussard H, Batra J, Haas K, Modak M, Kim M, Haas P, Polacco BJ, Braberg H, Fabius JM, Eckhardt M, Soucheray M, Bennett MJ, Cakir M, McGregor MJ, Li Q, Meyer B, Roesch F, Vallet T, Mac Kain A, Miorin L, Moreno E, Naing ZZC, Zhou Y, Peng S, Shi Y, Zhang Z, Shen W, Kirby IT, Melnyk JE, Chorba JS, Lou K, Dai SA, Barrio-Hernandez I, Memon D, Hernandez-Armenta C, Lyu J, Mathy CJP, Perica T, Pilla KB, Ganesan SJ, Saltzberg DJ, Rakesh R, Liu X, Rosenthal SB, Calviello L, Venkataramanan S, Liboy-Lugo J, Lin Y, Huang X-P, Liu Y, Wankowicz SA, Bohn M, Safari M, Ugur FS, Koh C, Savar NS, Tran QD, Shengjuler D, Fletcher SJ, O’Neal MC, Cai Y, Chang JCJ, Broadhurst DJ, Klippsten S, Sharp PP, Wenzell NA, Kuzuoglu D, Wang H-Y, Trenker R, Young JM, Cavero DA, Hiatt J, Roth TL, Rathore U, Subramanian A, Noack J, Hubert M, Stroud RM, Frankel AD, Rosenberg OS, Verba KA, Agard DA, Ott M, Emerman M, Jura N, von Zastrow M, Verdin E, Ashworth A, Schwartz O, d’Enfert C, Mukherjee S, Jacobson M, Malik HS, Fujimori DG, Ideker T, Craik CS, Floor SN, Fraser JS, Gross JD, Sali A, Roth BL, Ruggero D, Taunton J, Kortemme T, Beltrao P, Vignuzzi M, García-Sastre A, Shokat KM, Shoichet BK, Krogan NJ, A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature (2020), doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hein MY, Hubner NC, Poser I, Cox J, Nagaraj N, Toyoda Y, Gak IA, Weisswange I, Mansfeld J, Buchholz F, Hyman AA, Mann M, A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell. 163, 712–723 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Enfield KSS, Marshall EA, Anderson C, Ng KW, Rahmati S, Xu Z, Fuller M, Milne K, Lu D, Shi R, Rowbotham DA, Becker-Santos DD, Johnson FD, English JC, MacAulay CE, Lam S, Lockwood WW, Chari R, Karsan A, Jurisica I, Lam WL, Epithelial tumor suppressor ELF3 is a lineage-specific amplified oncogene in lung adenocarcinoma. Nat. Commun 10, 5438 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bouhaddou M, Eckhardt M, Chi Naing ZZ, Kim M, Ideker T, Krogan NJ, Mapping the protein–protein and genetic interactions of cancer to guide precision medicine. Curr. Opin. Genet. Dev 54, 110–117 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eckhardt M, Zhang W, Gross AM, Von Dollen J, Johnson JR, Franks-Skiba KE, Swaney DL, Johnson TL, Jang GM, Shah PS, Brand TM, Archambault J, Kreisberg JF, Grandis JR, Ideker T, Krogan NJ, Multiple Routes to Oncogenesis Are Promoted by the Human Papillomavirus–Host Protein Network. Cancer Discovery. 8 (2018), pp. 1474–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.IMEx Consortium Curators, Del-Toro N, Duesbury M, Koch M, Perfetto L, Shrivastava A, Ochoa D, Wagih O, Piñero J, Kotlyar M, Pastrello C, Beltrao P, Furlong LI, Jurisica I, Hermjakob H, Orchard S, Porras P, Capturing variation impact on molecular interactions in the IMEx Consortium mutations data set. Nat. Commun 10, 10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thul PJ, Åkesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, Alm T, Asplund A, Björk L, Breckels LM, Bäckström A, Danielsson F, Fagerberg L, Fall J, Gatto L, Gnann C, Hober S, Hjelmare M, Johansson F, Lee S, Lindskog C, Mulder J, Mulvey CM, Nilsson P, Oksvold P, Rockberg J, Schutten R, Schwenk JM, Sivertsson Å, Sjöstedt E, Skogs M, Stadler C, Sullivan DP, Tegel H, Winsnes C, Zhang C, Zwahlen M, Mardinoglu A, Pontén F, von Feilitzen K, Lilley KS, Uhlén M, Lundberg E, A subcellular map of the human proteome. Science. 356 (2017), doi: 10.1126/science.aal3321. [DOI] [PubMed] [Google Scholar]
- 37.Grabocka E, Pylayeva-Gupta Y, Jones MJK, Lubkov V, Yemanaberhan E, Taylor L, Jeng HH, Bar-Sagi D, Wild-type H- and N-Ras promote mutant K-Ras-driven tumorigenesis by modulating the DNA damage response. Cancer Cell. 25, 243–256 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Forcet C, Etienne-Manneville S, Gaude H, Fournier L, Debilly S, Salmi M, Baas A, Olschwang S, Clevers H, Billaud M, Functional analysis of Peutz–Jeghers mutations reveals that the LKB1 C-terminal region exerts a crucial role in regulating both the AMPK pathway and the cell polarity. Hum. Mol. Genet 14, 1283–1292 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Xu X, Omelchenko T, Hall A, LKB1 tumor suppressor protein regulates actin filament assembly through Rho and its exchange factor Dbl independently of kinase activity. BMC Cell Biol. 11, 77 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brieher WM, Yap AS, Cadherin junctions and their cytoskeleton(s). Current Opinion in Cell Biology. 25 (2013), pp. 39–46. [DOI] [PubMed] [Google Scholar]
- 41.Canel M, Serrels A, Frame MC, Brunton VG, E-cadherin-integrin crosstalk in cancer invasion and metastasis. Journal of Cell Science. 126 (2013), pp. 393–401. [DOI] [PubMed] [Google Scholar]
- 42.Lombaerts M, van Wezel T, Philippo K, Dierssen JWF, Zimmerman RME, Oosting J, van Eijk R, Eilers PH, van de Water B, Cornelisse CJ, Cleton-Jansen A-M, E-cadherin transcriptional downregulation by promoter methylation but not mutation is related to epithelial-to-mesenchymal transition in breast cancer cell lines. Br. J. Cancer 94, 661–671 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zeqiraj E, Filippi BM, Goldie S, Navratilova I, Boudeau J, Deak M, Alessi DR, van Aalten DMF, ATP and MO25alpha regulate the conformational state of the STRADalpha pseudokinase and activation of the LKB1 tumour suppressor. PLoS Biol. 7, e1000126 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zeqiraj E, Filippi BM, Deak M, Alessi DR, van Aalten DMF, Structure of the LKB1-STRAD-MO25 complex reveals an allosteric mechanism of kinase activation. Science. 326, 1707–1711 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hardie DG, Grahame Hardie D, Alessi DR, LKB1 and AMPK and the cancer-metabolism link - ten years after. BMC Biology. 11 (2013), , doi: 10.1186/1741-7007-11-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hollstein PE, Eichner LJ, Brun SN, Kamireddy A, Svensson RU, Vera LI, Ross DS, Rymoff TJ, Hutchins A, Galvez HM, Williams AE, Shokhirev MN, Screaton RA, Berdeaux R, Shaw RJ, The AMPK-Related Kinases SIK1 and SIK3 Mediate Key Tumor-Suppressive Effects of LKB1 in NSCLC. Cancer Discov. 9, 1606–1627 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lizcano JM, Göransson O, Toth R, Deak M, Morrice NA, Boudeau J, Hawley SA, Udd L, Mäkelä TP, Hardie DG, Alessi DR, LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 23, 833–843 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee S-W, Li C-F, Jin G, Cai Z, Han F, Chan C-H, Yang W-L, Li B-K, Rezaeian AH, Li H-Y, Huang H-Y, Lin H-K, Skp2-dependent ubiquitination and activation of LKB1 is essential for cancer cell survival under energy stress. Mol. Cell 57, 1022–1033 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, Hostetter G, Boguslawski S, Moses TY, Savage S, Uhlik M, Lin A, Du J, Qian Y-W, Zeckner DJ, Tucker-Kellogg G, Touchman J, Patel K, Mousses S, Bittner M, Schevitz R, Lai M-HT, Blanchard KL, Thomas JE, A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 448, 439–444 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Vivanco I, Sawyers CL, The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer 2, 489–501 (2002). [DOI] [PubMed] [Google Scholar]
- 51.Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT, The PI3K Pathway in Human Disease. Cell. 170, 605–635 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yap TA, Yan L, Patnaik A, Fearen I, Olmos D, Papadopoulos K, Baird RD, Delgado L, Taylor A, Lupinacci L, Riisnaes R, Pope LL, Heaton SP, Thomas G, Garrett MD, Sullivan DM, de Bono JS, Tolcher AW, First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J. Clin. Oncol 29, 4688–4695 (2011). [DOI] [PubMed] [Google Scholar]
- 53.Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P, Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr. Biol 7, 261–269 (1997). [DOI] [PubMed] [Google Scholar]
- 54.Stokoe D, Stephens LR, Copeland T, Gaffney PRJ, Reese CB, Painter GF, Holmes AB, McCormick F, Hawkins PT, Dual Role of Phosphatidylinositol-3,4,5-trisphosphate in the Activation of Protein Kinase B. Science. 277, 567–570 (1997). [DOI] [PubMed] [Google Scholar]
- 55.Tzatsos A, Kandror KV, Nutrients suppress phosphatidylinositol 3-kinase/Akt signaling via raptor-dependent mTOR-mediated insulin receptor substrate 1 phosphorylation. Mol. Cell. Biol 26, 63–76 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Woscholski R, Dhand R, Fry MJ, Waterfield MD, Parker PJ, Biochemical characterization of the free catalytic p110 alpha and the complexed heterodimeric p110 alpha.p85 alpha forms of the mammalian phosphatidylinositol 3-kinase. J. Biol. Chem 269, 25067–25072 (1994). [PubMed] [Google Scholar]
- 57.Kuchay S, Duan S, Schenkein E, Peschiaroli A, Saraf A, Florens L, Washburn MP, Pagano M, FBXL2- and PTPL1-mediated degradation of p110-free p85β regulatory subunit controls the PI(3)K signalling cascade. Nat. Cell Biol 15, 472–480 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mothe I, Delahaye L, Filloux C, Pons S, White MF, Van Obberghen E, Interaction of wild type and dominant-negative p55PIK regulatory subunit of phosphatidylinositol 3-kinase with insulin-like growth factor-1 signaling proteins. Mol. Endocrinol 11, 1911–1923 (1997). [DOI] [PubMed] [Google Scholar]
- 59.Chen H, Kovar J, Sissons S, Cox K, Matter W, Chadwell F, Luan P, Vlahos CJ, Schutz-Geschwender A, Olive DM, A cell-based immunocytochemical assay for monitoring kinase signaling pathways and drug efficacy. Anal. Biochem 338, 136–142 (2005). [DOI] [PubMed] [Google Scholar]
- 60.Cantley LC, Neel BG, New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc. Natl. Acad. Sci. U. S. A 96, 4240–4245 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Futreal PA, Liu Q, Shattuck-Eidens D, Cochran C, Harshman K, Tavtigian S, Bennett LM, Haugen-Strano A, Swensen J, Miki Y, BRCA1 mutations in primary breast and ovarian carcinomas. Science. 266, 120–122 (1994). [DOI] [PubMed] [Google Scholar]
- 62.Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W, A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 266, 66–71 (1994). [DOI] [PubMed] [Google Scholar]
- 63.Prakash R, Zhang Y, Feng W, Jasin M, Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol. 2015; 7: a016600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mullan PB, Quinn JE, Harkin DP, The role of BRCA1 in transcriptional regulation and cell cycle control. Oncogene. 25, 5854–5863 (2006). [DOI] [PubMed] [Google Scholar]
- 65.Hill SJ, Rolland T, Adelmant G, Xia X, Owen MS, Dricot A, Zack TI, Sahni N, Jacob Y, Hao T, McKinney KM, Clark AP, Reyon D, Tsai SQ, Joung JK, Beroukhim R, Marto JA, Vidal M, Gaudet S, Hill DE, Livingston DM, Systematic screening reveals a role for BRCA1 in the response to transcription-associated DNA damage. Genes Dev. 28, 1957–1975 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu LC, Wang ZW, Tsan JT, Spillman MA, Phung A, Xu XL, Yang MC, Hwang LY, Bowcock AM, Baer R, Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat. Genet 14, 430–440 (1996). [DOI] [PubMed] [Google Scholar]
- 67.Yu X, Chini CC, He M, Mer G, Chen J, The BRCT domain is a phospho-protein binding domain. Science. 302, 639–642 (2003). [DOI] [PubMed] [Google Scholar]
- 68.Górski B, Byrski T, Huzarski T, Jakubowska A, Menkiszak J, Gronwald J, Płużańska A, Bębenek M, Fischer-Maliszewska Ł, Grzybowska E, Narod SA, Lubiński J, Founder Mutations in the BRCA1 Gene in Polish Families with Breast-Ovarian Cancer. Am. J. Hum. Genet 66, 1963–1968 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vega A, Campos B, Bressac-de-Paillerets B, Bond PM, Janin N, Douglas FS, Domènech M, Baena M, Pericay C, Alonso C, Carracedo A, Baiget M, Diez O, The R71GBRCA1is a founder Spanish mutation and leads to aberrant splicing of the transcript. Human Mutation. 17 (2001), pp. 520–521. [DOI] [PubMed] [Google Scholar]
- 70.Levy-Lahad E, Catane R, Eisenberg S, Kaufman B, Hornreich G, Lishinsky E, Shohat M, Weber BL, Beller U, Lahad A, Halle D, Founder BRCA1 and BRCA2 mutations in Ashkenazi Jews in Israel: frequency and differential penetrance in ovarian cancer and in breast-ovarian cancer families. Am. J. Hum. Genet 60, 1059–1067 (1997). [PMC free article] [PubMed] [Google Scholar]
- 71.Clapperton JA, Manke IA, Lowery DM, Ho T, Haire LF, Yaffe MB, Smerdon SJ, Structure and mechanism of BRCA1 BRCT domain recognition of phosphorylated BACH1 with implications for cancer. Nat. Struct. Mol. Biol 11, 512–518 (2004). [DOI] [PubMed] [Google Scholar]
- 72.Wang Y, Bernhardy AJ, Cruz C, Krais JJ, Nacson J, Nicolas E, Peri S, van der Gulden H, van der Heijden I, O’Brien SW, Zhang Y, Harrell MI, Johnson SF, Candido Dos Reis FJ, Pharoah PDP, Karlan B, Gourley C, Lambrechts D, Chenevix-Trench G, Olsson H, Benitez JJ, Greene MH, Gore M, Nussbaum R, Sadetzki S, Gayther SA, Kjaer SK, kConFab Investigators, D’Andrea AD, Shapiro GI, Wiest DL, Connolly DC, Daly MB, Swisher EM, Bouwman P, Jonkers J, Balmaña J, Serra V, Johnson N, The BRCA1-Δ11q Alternative Splice Isoform Bypasses Germline Mutations and Promotes Therapeutic Resistance to PARP Inhibition and Cisplatin. Cancer Res. 76, 2778–2790 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shakya R, Reid LJ, Reczek CR, Cole F, Egli D, Lin C-S, deRooij DG, Hirsch S, Ravi K, Hicks JB, Szabolcs M, Jasin M, Baer R, Ludwig T, BRCA1 tumor suppression depends on BRCT phosphoprotein binding, but not its E3 ligase activity. Science. 334, 525–528 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Holstege H, Joosse SA, van Oostrom CTM, Nederlof PM, de Vries A, Jonkers J, High incidence of protein-truncating TP53 mutations in BRCA1-related breast cancer. Cancer Res. 69, 3625–3633 (2009). [DOI] [PubMed] [Google Scholar]
- 75.Yu X, Chen J, DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol. Cell. Biol 24, 9478–9486 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang Y, Krais JJ, Bernhardy AJ, Nicolas E, Cai KQ, Harrell MI, Kim HH, George E, Swisher EM, Simpkins F, Johnson N, RING domain-deficient BRCA1 promotes PARP inhibitor and platinum resistance. J. Clin. Invest 126, 3145–3157 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Drost R, Dhillon KK, van der Gulden H, van der Heijden I, Brandsma I, Cruz C, Chondronasiou D, Castroviejo-Bermejo M, Boon U, Schut E, van der Burg E, Wientjens E, Pieterse M, Klijn C, Klarenbeek S, Loayza-Puch F, Elkon R, van Deemter L, Rottenberg S, van de Ven M, Dekkers DH, Demmers JA, van Gent DC, Agami R, Balmana J, Serra V, Taniguchi T, Bouwman P, Jonkers J, BRCA1185delAG tumors may acquire therapy resistance through expression of RING-less BRCA1. J. Clin. Invest 126, 2903–2918 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Christensen DE, Brzovic PS, Klevit RE, E2–BRCA1 RING interactions dictate synthesis of mono- or specific polyubiquitin chain linkages. Nat. Struct. Mol. Biol 14, 941–948 (2007). [DOI] [PubMed] [Google Scholar]
- 79.Zhao GY, Sonoda E, Barber LJ, Oka H, Murakawa Y, Yamada K, Ikura T, Wang X, Kobayashi M, Yamamoto K, Boulton SJ, Takeda S, A critical role for the ubiquitin-conjugating enzyme Ubc13 in initiating homologous recombination. Mol. Cell 25, 663–675 (2007). [DOI] [PubMed] [Google Scholar]
- 80.Rugo HS, Olopade OI, DeMichele A, Yau C, van ‘t Veer LJ, Buxton MB, Hogarth M, Hylton NM, Paoloni M, Perlmutter J, Symmans WF, Yee D, Chien AJ, Wallace AM, Kaplan HG, Boughey JC, Haddad TC, Albain KS, Liu MC, Isaacs C, Khan QJ, Lang JE, Viscusi RK, Pusztai L, Moulder SL, Chui SY, Kemmer KA, Elias AD, Edmiston KK, Euhus DM, Haley BB, Nanda R, Northfelt DW, Tripathy D, Wood WC, Ewing C, Schwab R, Lyandres J, Davis SE, Hirst GL, Sanil A, Berry DA, Esserman LJ, I-SPY 2 Investigators, Adaptive Randomization of Veliparib-Carboplatin Treatment in Breast Cancer. N. Engl. J. Med 375, 23–34 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Barker AD, Sigman CC, Kelloff GJ, Hylton NM, Berry DA, Esserman LJ, I-SPY 2: An Adaptive Breast Cancer Trial Design in the Setting of Neoadjuvant Chemotherapy. Clinical Pharmacology & Therapeutics. 86 (2009), pp. 97–100. [DOI] [PubMed] [Google Scholar]
- 82.Zimmermann M, Murina O, Reijns MAM, Agathanggelou A, Challis R, Tarnauskaitė Ž, Muir M, Fluteau A, Aregger M, McEwan A, Yuan W, Clarke M, Lambros MB, Paneesha S, Moss P, Chandrashekhar M, Angers S, Moffat J, Brunton VG, Hart T, de Bono J, Stankovic T, Jackson AP, Durocher D, CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature. 559, 285–289 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang D, Zaugg K, Mak TW, Elledge SJ, A role for the deubiquitinating enzyme USP28 in control of the DNA-damage response. Cell. 126, 529–542 (2006). [DOI] [PubMed] [Google Scholar]
- 84.Bassermann F, Frescas D, Guardavaccaro D, Busino L, Peschiaroli A, Pagano M, The Cdc14B-Cdh1-Plk1 axis controls the G2 DNA-damage-response checkpoint. Cell. 134, 256–267 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Knobel PA, Belotserkovskaya R, Galanty Y, Schmidt CK, Jackson SP, Stracker TH, USP28 is recruited to sites of DNA damage by the tandem BRCT domains of 53BP1 but plays a minor role in double-strand break metabolism. Mol. Cell. Biol 34, 2062–2074 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gunn A, Stark JM, I-SceI-based assays to examine distinct repair outcomes of mammalian chromosomal double strand breaks. Methods Mol. Biol 920, 379–391 (2012). [DOI] [PubMed] [Google Scholar]
- 87.Anantha RW, Simhadri S, Foo TK, Miao S, Liu J, Shen Z, Ganesan S, Xia B, Functional and mutational landscapes of BRCA1 for homology-directed repair and therapy resistance. Elife. 6 (2017), doi: 10.7554/eLife.21350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hultquist JF, Hiatt J, Schumann K, McGregor MJ, Roth TL, Haas P, Doudna JA, Marson A, Krogan NJ, CRISPR-Cas9 genome engineering of primary CD4+ T cells for the interrogation of HIV-host factor interactions. Nat. Protoc 14, 1–27 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Moudry P, Lukas C, Macurek L, Hanzlikova H, Hodny Z, Lukas J, Bartek J, Ubiquitin-activating enzyme UBA1 is required for cellular response to DNA damage. Cell Cycle. 11, 1573–1582 (2012). [DOI] [PubMed] [Google Scholar]
- 90.Unk I, Hajdu I, Fatyol K, Szakal B, Blastyak A, Bermudez V, Hurwitz J, Prakash L, Prakash S, Haracska L, Human SHPRH is a ubiquitin ligase for Mms2-Ubc13-dependent polyubiquitylation of proliferating cell nuclear antigen. Proceedings of the National Academy of Sciences. 103 (2006), pp. 18107–18112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang H, Zhai L, Xu J, Joo H-Y, Jackson S, Erdjument-Bromage H, Tempst P, Xiong Y, Zhang Y, Histone H3 and H4 ubiquitylation by the CUL4-DDB-ROC1 ubiquitin ligase facilitates cellular response to DNA damage. Mol. Cell 22, 383–394 (2006). [DOI] [PubMed] [Google Scholar]
- 92.Chen Y, Weng C, Zhang H, Sun J, Yuan Y, A Direct Interaction Between P53-Binding Protein 1 and Minichromosome Maintenance Complex in Hepg2 Cells. Cell. Physiol. Biochem 47, 2350–2359 (2018). [DOI] [PubMed] [Google Scholar]
- 93.Sarrouilhe D, di Tommaso A, Métayé T, Ladeveze V, Spinophilin: from partners to functions. Biochimie. 88, 1099–1113 (2006). [DOI] [PubMed] [Google Scholar]
- 94.Woods NT, Mesquita RD, Sweet M, Carvalho MA, Li X, Liu Y, Nguyen H, Thomas CE, Iversen ES, Marsillac S, Karchin R, Koomen J, Monteiro ANA, Charting the Landscape of Tandem BRCT Domain-Mediated Protein Interactions. Science Signaling. 5 (2012), pp. rs6–rs6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wu Q, Jubb H, Blundell TL, Phosphopeptide interactions with BRCA1 BRCT domains: More than just a motif. Prog. Biophys. Mol. Biol 117, 143–148 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wu Q, Paul A, Su D, Mehmood S, Foo TK, Ochi T, Bunting EL, Xia B, Robinson CV, Wang B, Blundell TL, Structure of BRCA1-BRCT/Abraxas Complex Reveals Phosphorylation-Dependent BRCT Dimerization at DNA Damage Sites. Mol. Cell 61, 434–448 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ragusa MJ, Dancheck B, Critton DA, Nairn AC, Page R, Peti W, Spinophilin directs protein phosphatase 1 specificity by blocking substrate binding sites. Nat. Struct. Mol. Biol 17, 459–464 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sack LM, Davoli T, Li MZ, Li Y, Xu Q, Naxerova K, Wooten EC, Bernardi RJ, Martin TD, Chen T, Leng Y, Liang AC, Scorsone KA, Westbrook TF, Wong K-K, Elledge SJ, Profound Tissue Specificity in Proliferation Control Underlies Cancer Drivers and Aneuploidy Patterns. Cell. 173, 499–514.e23 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Coppe JP, Mori M, Pan B, Yau C, Wolf DM, Ruiz-Saenz A, Brunen D, Prahallad A, Cornelissen-Steijger P, Kemper K, Posch C, Wang C, Dreyer CA, Krijgsman O, Lee PRE, Chen Z, Peeper DS, Moasser MM, Bernards R, van ‘t Veer LJ, Mapping phospho-catalytic dependencies of therapy-resistant tumours reveals actionable vulnerabilities. Nat. Cell Biol 21, 778–790 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Olow A, Chen Z, Niedner RH, Wolf DM, Yau C, Pankov A, Lee EP, Brown-Swigart L, van ‘t Veer LJ, Coppe JP, An Atlas of the Human Kinome Reveals the Mutational Landscape Underlying Dysregulated Phosphorylation Cascades in Cancer. Cancer Res. 76, 1733–1745 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hinton CV, Fitzgerald LD, Thompson ME, Phosphatidylinositol 3-kinase/Akt signaling enhances nuclear localization and transcriptional activity of BRCA1. Exp. Cell Res 313, 1735–1744 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Beckta JM, Dever SM, Gnawali N, Khalil A, Sule A, Golding SE, Rosenberg E, Narayanan A, Kehn-Hall K, Xu B, Povirk LF, Valerie K, Mutation of the BRCA1 SQ-cluster results in aberrant mitosis, reduced homologous recombination, and a compensatory increase in non-homologous end joining. Oncotarget. 6 (2015), , doi: 10.18632/oncotarget.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Xu B, O’Donnell AH, Kim S-T, Kastan MB, Phosphorylation of serine 1387 in Brca1 is specifically required for the Atm-mediated S-phase checkpoint after ionizing irradiation. Cancer Res. 62, 4588–4591 (2002). [PubMed] [Google Scholar]
- 104.Cortez D, Wang Y, Qin J, Elledge SJ, Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science. 286, 1162–1166 (1999). [DOI] [PubMed] [Google Scholar]
- 105.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM, DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem 273, 5858–5868 (1998). [DOI] [PubMed] [Google Scholar]
- 106.Lukas J, Lukas C, Bartek J, More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat. Cell Biol 13, 1161–1169 (2011). [DOI] [PubMed] [Google Scholar]
- 107.Alberts B, The cell as a collection of protein machines: preparing the next generation of molecular biologists. Cell. 92, 291–294 (1998). [DOI] [PubMed] [Google Scholar]
- 108.Hanahan D, Weinberg RA, The hallmarks of cancer. Cell. 100, 57–70 (2000). [DOI] [PubMed] [Google Scholar]
- 109.Krogan NJ, Lippman S, Agard DA, Ashworth A, Ideker T, The cancer cell map initiative: defining the hallmark networks of cancer. Mol. Cell 58, 690–698 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Goldfarbmuren KC, Jackson ND, Sajuthi SP, Dyjack N, Li KS, Rios CL, Plender EG, Montgomery MT, Everman JL, Bratcher PE, Vladar EK, Seibold MA, Dissecting the cellular specificity of smoking effects and reconstructing lineages in the human airway epithelium. Nat. Commun 11, 2485 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Britto CJ, Cohn L, Bactericidal/Permeability-Increasing Protein Fold–Containing Family Member A1 in Airway Host Protection and Respiratory Disease. Am. J. Respir. Cell Mol. Biol 52, 525–534 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bingle L, Bingle CD, Distribution of human PLUNC/BPI fold-containing (BPIF) proteins. Biochem. Soc. Trans 39, 1023–1027 (2011). [DOI] [PubMed] [Google Scholar]
- 113.He Y, Zhou G, Zhai Y, Dong X, Lv L, He F, Yao K, Association of PLUNC gene polymorphisms with susceptibility to nasopharyngeal carcinoma in a Chinese population. J. Med. Genet 42, 172–176 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chen P, Guo X, Zhou H, Zhang W, Zeng Z, Liao Q, Li X, Xiang B, Yang J, Ma J, Zhou M, Peng S, Xiang J, Li X, L E CW, Xiong W, McCarthy JB, Li G, SPLUNC1 regulates cell progression and apoptosis through the miR-141-PTEN/p27 pathway, but is hindered by LMP1. PLoS One. 8, e56929 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Aihara T, Fujiwara Y, Ooka M, Sakita I, Tamaki Y, Monden M, Mammaglobin B as a novel marker for detection of breast cancer micrometastases in axillary lymph nodes by reverse transcription-polymerase chain reaction. Breast Cancer Res. Treat 58, 137–140 (1999). [DOI] [PubMed] [Google Scholar]
- 116.Tassi RA, Bignotti E, Falchetti M, Calza S, Ravaggi A, Rossi E, Martinelli F, Bandiera E, Pecorelli S, Santin AD, Mammaglobin B expression in human endometrial cancer. Int. J. Gynecol. Cancer 18, 1090–1096 (2008). [DOI] [PubMed] [Google Scholar]
- 117.Zubor P, Hatok J, Moricova P, Kajo K, Kapustova I, Mendelova A, Racay P, Danko J, Gene expression abnormalities in histologically normal breast epithelium from patients with luminal type of breast cancer. Mol. Biol. Rep 42, 977–988 (2015). [DOI] [PubMed] [Google Scholar]
- 118.Zhou H, Zou X, Li H, Li T, Chen L, Cheng X, Decreased secretoglobin family 2A member 1expression is associated with poor outcomes in endometrial cancer. Oncol. Lett 20, 24 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Park S, Supek F, Lehner B, Systematic discovery of germline cancer predisposition genes through the identification of somatic second hits. Nat. Commun 9, 2601 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Quanz M, Chassoux D, Berthault N, Agrario C, Sun J-S, Dutreix M, Hyperactivation of DNA-PK by double-strand break mimicking molecules disorganizes DNA damage response. PLoS One. 4, e6298 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bensimon A, Schmidt A, Ziv Y, Elkon R, Wang S-Y, Chen DJ, Aebersold R, Shiloh Y, ATM-Dependent and -Independent Dynamics of the Nuclear Phosphoproteome After DNA Damage. Science Signaling. 3 (2010), pp. rs3–rs3. [DOI] [PubMed] [Google Scholar]
- 122.Vizcaíno JA, Deutsch EW, Wang R, Csordas A, Reisinger F, Ríos D, Dianes JA, Sun Z, Farrah T, Bandeira N, Binz P-A, Xenarios I, Eisenacher M, Mayer G, Gatto L, Campos A, Chalkley RJ, Kraus H-J, Albar JP, Martinez-Bartolomé S, Apweiler R, Omenn GS, Martens L, Jones AR, Hermjakob H, ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol 32, 223–226 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cox J, Mann M, MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 26, 1367–1372 (2008). [DOI] [PubMed] [Google Scholar]
- 124.Choi M, Chang C-Y, Clough T, Broudy D, Killeen T, MacLean B, Vitek O, MSstats: an R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics. 30, 2524–2526 (2014). [DOI] [PubMed] [Google Scholar]
- 125.Behrends C, Sowa ME, Gygi SP, Harper JW, Network organization of the human autophagy system. Nature. 466, 68–76 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mi H, Ebert D, Muruganujan A, Mills C, Albou L-P, Mushayamaha T, Thomas PD, PANTHER version 16: a revised family classification, tree-based classification tool, enhancer regions and extensive API. Nucleic Acids Res. 49, D394–D403 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, Jensen LJ, von Mering C, The STRING database in 2017: quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 45, D362–D368 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chen Z, Coppé J-P, Method and System for Building and Using a Centralized and Harmonized Relational Database. US Patent (2012), (available at https://patentimages.storage.googleapis.com/af/b1/85/846490ba6d0844/US20120296880A1.pdf). [Google Scholar]
- 129.Chien AJ, Tripathy D, Albain KS, Symmans WF, Rugo HS, Melisko ME, Wallace AM, Schwab R, Helsten T, Forero-Torres A, Stringer-Reasor E, Ellis ED, Kaplan HG, Nanda R, Jaskowiak N, Murthy R, Godellas C, Boughey JC, Elias AD, Haley BB, Kemmer K, Isaacs C, Clark AS, Lang JE, Lu J, Korde L, Edmiston KK, Northfelt DW, Viscusi RK, Yee D, Perlmutter J, Hylton NM, Van’t Veer LJ, DeMichele A, Wilson A, Peterson G, Buxton MB, Paoloni M, Clennell J, Berry S, Matthews JB, Steeg K, Singhrao R, Hirst GL, Sanil A, Yau C, Asare SM, Berry DA, Esserman LJ, I-SPY 2 Consortium, MK-2206 and Standard Neoadjuvant Chemotherapy Improves Response in Patients With Human Epidermal Growth Factor Receptor 2-Positive and/or Hormone Receptor-Negative Breast Cancers in the I-SPY 2 Trial. J. Clin. Oncol 38, 1059–1069 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Johnson WE, Li C, Rabinovic A, Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 8, 118–127 (2007). [DOI] [PubMed] [Google Scholar]
- 131.Wulfkuhle JD, Yau C, Wolf DM, Vis DJ, Gallagher RI, Brown-Swigart L, Hirst G, Voest EE, DeMichele A, Hylton N, Symmans F, Yee D, Esserman L, Berry D, Liu M, Park JW, Wessels LFA, Veer LV, Petricoin EF, Evaluation of the HER/PI3K/AKT Family Signaling Network as a Predictive Biomarker of Pathologic Complete Response for Patients With Breast Cancer Treated With Neratinib in the I-SPY 2 TRIAL. JCO Precision Oncology (2018), pp. 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wolf DM, Yau C, Sanil A, Glas A, Petricoin E, Wulfkuhle J, Severson TM, Linn S, Brown-Swigart L, Hirst G, Buxton M, DeMichele A, Hylton N, Symmans F, Yee D, Paoloni M, Esserman L, Berry D, Rugo H, Olopade O, van ‘t Veer L, DNA repair deficiency biomarkers and the 70-gene ultra-high risk signature as predictors of veliparib/carboplatin response in the I-SPY 2 breast cancer trial. NPJ Breast Cancer. 3, 31 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw data files and search results are available from the Pride partner ProteomeXchange repository (122). For AP-MS and protein abundance data from MCF10A, MCF7, and MDA-MB-231 cells, these can be accessed under the PXD019639 identifier. For PTM (ub and ph) analysis data, these are available under the PXD025931 identifier. PPI networks can be accessed on the NDEx platform (https://bit.ly/3y10JFY). DOIs and links for each PPI network are in table S1. Expression vectors used in this study are readily available from the authors for biomedical researchers and educators in the nonprofit sector under MTA. Further information and requests for resources and reagents should be directed to and will be fulfilled by NJK (nevan.krogan@ucsf.edu).