

Abstract
Background
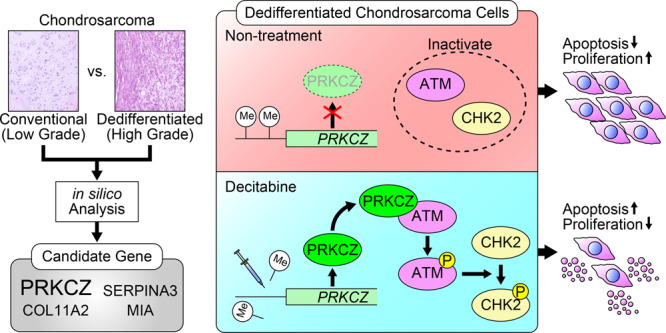
Dedifferentiated chondrosarcoma (DDCS) is an aggressive bone tumour with a poor prognosis and no effective treatment. Because changes in DNA methylation play critical roles in DDCS, we explored the roles that DNA methylation plays in oncogenesis to potentially identify an effective epigenetic treatment.
Methods
We identified genes downregulated in DDCS vs. conventional chondrosarcoma (CCS) due to DNA methylation using in silico analysis. The results were validated in DDCS clinical samples, and the molecular functions of the genes of interest were investigated in multiple chondrosarcoma cell lines (NDCS-1, SW1353, and OUMS-27). The therapeutic effect of decitabine, a DNA methyltransferase inhibitor, was evaluated in vitro and in vivo.
Results
PRKCZ was specifically downregulated by DNA methylation in DDCS. Overexpression of PRKCZ decreased the proliferation of NDCS-1 and SW1353 cells. PRKCZ directly bound to and activated ATM, which was followed by phosphorylation of CHK2 and subsequent apoptosis. Decitabine increased PRKCZ expression through de-methylating the promoter region of PRKCZ, which activated the ATM/CHK2 pathway and inhibited cell proliferation by inducing apoptosis.
Conclusions
Increased DNA methylation and reduced expression of PRKCZ prevents apoptosis via inactivation of the ATM/CHK2 pathway in DDCS. Decitabine-induced expression of PRKCZ represents a promising therapy for DDCS.
Subject terms: Sarcoma, Apoptosis, Chemotherapy, DNA methylation
Background
Chondrosarcoma, the second most common primary sarcoma of bone, is characterised by the production of a cartilage matrix. Chondrosarcoma is divided into several subtypes: conventional (CCS), dedifferentiated (DDCS), mesenchymal, and clear-cell [1]. The genetic characteristics differ for each subtype. Mutations in isocitrate dehydrogenase (IDH) 1 or 2 are characteristic markers for CCS and DDCS, but not for the other subtypes [1, 2]. By contrast, the presence of a fusion gene is common for the mesenchymal subtype, but not for the other subtypes [1]. DDCS, which has a high-grade dedifferentiated component occurring adjacent to a low-grade chondrogenic component, comprises ~10% of chondrosarcoma [3]. The prognosis for patients with DDCS is much poorer than for those with other chondrosarcoma subtypes; the five-year survival rates for patients with DDCS, CCS, mesenchymal, and clear-cell are 0–13%, 70%, 48%, and 100%, respectively [4–6]. Despite the poor prognosis, there is no established and efficacious systemic therapy for DDCS other than clinical resection [2]. Hence, there is an urgent need for more effective systemic treatment for this cancer.
Several retrospective reports describe the activity of a variety of sarcoma-focussed cytotoxic agents, including adriamycin and ifosfamide, in combination with surgical resection of DDCS; however, the prognosis following such approaches remains generally poor [7]. Several candidate molecularly targeted medicines, such as mTOR inhibitor [8], CDK4/6 inhibitor, multi-kinase inhibitor [9], and Hedgehog pathway inhibitor [10, 11], have been proposed for the treatment of chondrosarcoma, including DDCS; however, their clinical efficacy has been unsatisfactory. Consequently, alternative approaches other than cytotoxic agents and molecularly targeted medicines are needed.
Mutation of IDH induces DNA methylation due to excessive accumulation of 2-hydroxyglutarate [12, 13]. Aberrant DNA methylation, which is detected in various types of cancer, regulates the biological behaviour of cancers by suppressing the transcription of specific genes [14]. In addition, a multi-omics molecular profiling study of patients with chondrosarcoma revealed that DNA hypermethylation is associated with a worse prognosis of chondrosarcoma [15]. Accordingly, inhibitors of mutant IDH exert tumour-suppressive activity in chondrosarcoma in vitro and in vivo, which is concomitant with DNA demethylation [16–18]. For the same reason, DNA methyltransferase inhibitors (DNMTis) were thought to suppress chondrosarcoma growth; however, whether DNMTis inhibit or exacerbate oncogenicity remains controversial [19–23]. In addition, no studies have focused on the activity of DNMTis in DDCS.
In this study, by analysing comprehensive datasets from patients with chondrosarcoma, we identified genes downregulated by DNA methylation in DDCS relative to CCS. In particular, we identified PRKCZ, which encodes the atypical PKC isoform protein kinase C zeta [24], as one of the genes of interest. The role PRKCZ plays in oncogenesis varies depending on the tumour histology; [25–32] no previous studies have reported a relationship between PRKCZ and the oncogenesis of DDCS. Hence, we investigated the biological roles of PRKCZ in DDCS. In addition, we evaluated the therapeutic efficacy of decitabine, a well-known DNMTi, on DDCS in vitro and in vivo.
Methods
Bioinformatic analysis
Gene expression, methylation, and clinical data of chondrosarcoma were collected from Gene Expression Omnibus (National Center for Biotechnology Information, https://www.ncbi.nlm.nih.gov/geo/) and ArrayExpress (European Bioinformatics Institute, https://www.ebi.ac.uk/arrayexpress/). Values were subjected to quantile normalisation and divided into two groups: CCS and DDCS. The methylation difference, the fold changes, and standard errors were estimated by fitting a linear model for each gene using the R package limma. When processed data were available, they were analysed with GEO2R (http://www.ncbi.nlm.nih.gov/geo/geo2r/). Thresholds were as follows: adjusted p-value, 0.05; methylation difference 0.2 for hypermethylation; log-fold change −0.5 for hypoexpression.
Whole-genome bisulfite sequencing (WGBS) library construction & sequencing
Genomic DNA was extracted using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). Integrity of genomic DNA was checked by agarose gel electrophoresis, and gDNA was quantified using Quant-IT PicoGreen (Invitrogen, Carlsbad, CA, USA). WGBS libraries were prepared using Accel-NGS Methyl-Seq DNA library kit (Swift BioSciences). Briefly, 200 ng of genomic DNA was fragmented on a Covaris LE220 focused-ultrasonicator (Covaris, Woburn, MA, USA) to a target peak size of 400–700 bp. The fragmented DNA was then subjected to bisulfite conversion using the EZ DNA Methylation-Gold Kit (Zymo Research, Irvine, CA, USA). The bisulfite-treated ssDNA fragments were repaired, and truncated adapters 1 and 2 were ligated to the 3′ and 5′ ends of the fragments, respectively. The truncated adapter-ligated DNA was amplified with an indexed primer to complete the WGBS libraries with full-length adapters. The final libraries were then quantified by qPCR according to the qPCR Quantification Protocol Guide (KAPA Library Quantification kits for Illumina Sequencing platforms), and quality was checked using TapeStation DNA ScreenTape D1000 (Agilent). Sequencing was performed on the NovaSeq6000 platform (Illumina, San Diego, CA, USA).
Data processing and methylation profile calling
Sequence quality of paired-end sequencing reads (151 bp) generated from WGBS libraries was verified with FastQC. Before analysis, TrimGalore was used to remove adapter sequences. The cleaned reads were aligned to the Homo sapiens genome (hg38) using Bismark. Mapped data (BAM file format) were sorted and indexed using Samtools. Methylation level was called with the R package methylKit. Methylation of the promoter region was called for 1000 bases before and after the transcription start site of each gene. WGBS data are available at GEO (accession number: GSE185124).
Targeted bisulfite sequencing
Genomic DNA was extracted using a QIAamp DNA Mini Kit (Qiagen). Bisulfite treatment of genomic DNA was performed using the EZ DNA Methylation-Gold kit (Zymo Research). After the bisulfite treatment, methylated cytosine (C) of CpG remained as C, whereas non-methylated C was converted into uracil. Polymerase chain reaction (PCR) was performed with TaKaRa EpiTaq™ HS (Takara Bio, Kusatsu, Japan). Primers are listed in Supplementary Table S1. Following agarose gel electrophoresis, the bands of PCR products were cut out of the gel and purified using the QIAquick Gel Extraction Kit (Qiagen). PCR products were inserted into pUC19 DNA (Takara Bio) and cloned using E. coli DH5α Competent Cells (Takara Bio). Sequencing reactions were performed in a BioRad DNA Engine Dyad PTC-220 Peltier Thermal Cycler using an ABI PRISM BigDye Terminator v3.1 Cycle Sequencing Kits (Applied Biosystems). Single-pass sequencing was performed on each template. Fluorescently labelled fragments were purified from unincorporated terminators by ethanol precipitation. The samples were resuspended in distilled water and subjected to electrophoresis on an ABI 3730xl sequencer (Applied Biosystems). Assays were performed in five clones to calculate the methylation percentage.
Reagents
Decitabine was purchased from Tokyo Chemical Industry (Tokyo, Japan). Chloroquine diphosphate was purchased from FUJIFILM Wako (Osaka, Japan). Dulbecco’s phosphate-buffered saline (PBS) was purchased from ThermoFisher Scientific (Waltham, MA, USA). Decitabine and chloroquine diphosphate was dissolved in PBS. For proliferation assays, decitabine or PBS were administered at the time of seeding. For DNA extraction, RNA extraction, protein extraction, and cell cycle assays, decitabine or PBS were administered at 0 and 48 h after seeding. Cells were harvested 96 h after seeding. Chloroquine diphosphate was administered at a concentration of 400 µM for 3 h.
Cell lines and culture
Each cell line was maintained at 37 °C in an atmosphere of 5% CO2. NDCS-1 (CVCL_EJ23) was kindly provided by Dr. Akira Ogose [33] and maintained in Roswell Park Memorial Institute 1640 (RPMI 1640, ThermoFisher Scientific). OUMS-27 (CVCL_3090) was obtained from the Japanese Collection of Research Bioresources Cell Bank (Osaka, Japan). SW1353 (CVCL_0543) was obtained from the American Type Culture Collection (Manassas, Virginia, USA). OUMS-27 and SW1353 were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM; ThermoFisher Scientific). Each medium was supplemented with 10% fetal bovine serum (HyClone Laboratories, Logan, UT, USA), 100 units/ml penicillin, and 100 μg/ml streptomycin. OUMS-27 and SW1353 were confirmed with STR DNA fingerprinting. NDCS-1 was not registered in the database. All cell lines were used after confirmation of non-contamination with Mycoplasma using the EZ-PCR Mycoplasma detection kit (Biological Industries, Kibbutz Beit-Haemek, Israel).
Quantitative reverse transcription-polymerase chain reaction (RT-qPCR)
RNA of the cell line was extracted using the RNeasy Mini Kit (Qiagen, Hilden, Germany), and RNA of frozen tissue was extracted using TRIzol (ThermoFisher Scientific). Complementary DNA (cDNA) was synthesised using a Prime-Script™ RT reagent Kit (Takara Bio). RT-qPCR was performed on a LightCycler 2.0 (Roche, Basel, Switzerland) with TB Green Premix Ex Taq II (Takara Bio). mRNA levels of protein kinase C zeta (PRKCZ), ataxia telangiectasia mutated (ATM), collagen type II alpha 1 chain (COL2A1), aggrecan (ACAN), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were measured; relative expression was determined by the delta-delta Ct method. Assays were performed in triplicate. Primers are listed in Supplementary Table S1.
Western blotting
Whole-cell protein extracts were obtained using CelLytic M (Sigma-Aldrich) containing protease inhibitor cocktail tablets (cOmplete™; Roche) and phosphatase inhibitor cocktail (PhosSTOP™; Roche). Western blot analysis was performed as described previously [34]. Antibodies and their dilutions are listed in Supplementary Table S2. Semi-quantitation was performed on CS Analyzer 3.0 (ATTO, Tokyo, Japan).
Clinical samples
Clinical samples were obtained from 17 DDCS patients and nine CCS patients who underwent surgical resection between 1982 and 2018 at the Graduate School of Medical Science, Kyushu University, Fukuoka, Japan. In each case, a diagnosis was made on the basis of histologic features. All patients provided written informed consent before undergoing the study procedures. The Institutional Review Board at Kyushu University approved the use of human specimens for this study (approval number: 27-420). This study was conducted in accordance with the Declaration of Helsinki. The demographics of patients are listed in Supplementary Table S3.
Immunohistochemistry (IHC) staining
All samples were fixed in 4% paraformaldehyde (FUJIFILM Wako) and embedded in paraffin. After being deparaffinized in xylene and dehydrated in a graded ethanol series, sections were pretreated with TE buffer (10 mM Tris-HCl and 1 mM EDTA) in an autoclave at 98 °C for 25 min, and then incubated with primary antibody at 4 °C overnight. Anti-PRKCZ antibody (HPA021851, Sigma-Aldrich, St. Louis, MO, USA) was used as the primary antibody. Samples were incubated with Dako EnVision Dual Link System-HRP (Agilent Technologies, Santa Clara, CA, USA), visualised using the diaminobenzidine substrate system (FUJIFILM Wako), and counterstained with diluted hematoxylin. All images were captured a BZ-X800 microscope (Keyence, Osaka, Japan) and the percentage of positive cells were calculated by Hybrid Cell Count Software (Keyence). Staining intensity was classified into four classes in each of the cartilaginous and dedifferentiated components. Evaluation of intensity was performed by two bone oncologists who were blinded to the clinical information.
Vector construction and transfection
The full-length cDNA of PRKCZ was generated from cDNA of HEK293 cell line by PCR and gel extraction. Primers are listed in Supplementary Table S1. The PRKCZ overexpression vector was created by inserting full-length PRKCZ cDNA into pCDH-EF1-MCS-IRES-Puro (System Biosciences, Palo Alto, CA, USA). GFP-tagged PRKCZ vector was created by inserting full-length PRKCZ cDNA into pEGFP-N1-FLAG (Addgene, Toddington, UK). Plasmids without inserts were used as control vectors. Plasmids were transfected into cells using Lipofectamine3000 (Invitrogen).
Lentiviral vector production and infection
Vectors were co-transfected with Lentivirus Package plasmid mix (System Biosciences) into 293TN cells (System Biosciences) using Lipofectamine3000 (Invitrogen). Culture supernatants containing lentivirus particles were collected 48 h post-transfection. The supernatant was concentrated using PEG-it Virus Precipitation Solution (System Biosciences), and the concentrated supernatant was used to infect target cells. Cells were plated at a density of 5.0 × 104 cells per well in 24-well plates containing culture medium supplemented with 10% FBS, without antibiotics. After incubation for 24 h, the cells were transfected with lentivirus particles using the TransDux Virus Transduction Reagent (System Biosciences). Two days after transfection, puromycin was added to the medium to eliminate uninfected cells, and single clones were picked.
Knockdown
Small interfering RNA (siRNA) and negative control siRNA were purchased from ThermoFisher Scientific. siRNA sequences were as follows: siPRKCZ#1 (#s11129), sense 5′-GGACUUUGACCUAAUCAGA(dTdT)-3′, antisense 5′-UCUGAUUAGGUCAAAGUCC(dTdG)-3′; siPRKCZ#2 (#s11128), sense 5′-CGUUCGACAUCAUCACCGA(dTdT)-3′, antisense 5′-UCGGUGAUGAUGUCGAACG(dGdG)-3; siATM (#s42813), sense 5′-GGAAAUCAGUAGUUUGGUC(dTdT)-3′, antisense 5′-CACCAAACUACUGAUUUCC(dTdG)-3. siRNA (50 nM) was transfected into cells with Lipofectamine3000 (ThermoFisher).
Proliferation assay
Cells were seeded at 1.0 × 103 cells per well in 96-well plates. Four hours after seeding, the cell viability in each well was measured as a standard value. After that, relative cell viability was monitored every 24 h for 72 h using the CellTiter-Glo Luminescent Cell Viability kit (Promega, Madison, WI, USA).
Cell cycle analysis by flow cytometry
Cells were harvested with trypsin, washed with PBS, and fixed in ice-cold 70% ethanol at 4 °C for 30 min. The fixed cells were washed with PBS and incubated with propidium iodide and RNase (Immunostep S.L, Salamanca, Spain) for 15 min. Flow cytometric analyses were performed on a BD Accuri C6 flow cytometer (Becton, Dickinson and Company, Franklin Lakes, NJ, USA), and cell cycle analyses were performed with ModFit (Verity Software House, Topsham, ME, USA).
Colony formation assay
Assays were performed in six-well plates. Cells were seeded at a density of 500 cells/well and cultured for 7–14 days. The cells were stained by the addition of 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (FUJIFILM Wako), followed by incubation for 1 h. Colonies were counted by the naked eye.
Xenograft transplantation experiments
Male 4-week-old BALB/cAJcl-nu/nu mice were purchased from CLEA Japan (Tokyo, Japan) and maintained in a pathogen-free environment with a 12-h dark/light cycle in the animal facility at Kyushu University. Three to five animals were housed per cage, and all animals were fed and watered ad libitum. Animal health was checked at least twice a week to visually assess appearance behaviour, faces, and other conditions. The mice were anaesthetised using a mixed cocktail of midazolam (Fuji Pharma, Tokyo, Japan), butorphanol tartrate (Meiji Seika Pharma, Tokyo, Japan), and medetomidine hydrochloride (Kyoritsu Seiyaku Corp., Tokyo, Japan). Cells (1.0 × 106) were dissolved in 100 μl Matrigel (Corning, Corning, NY, USA) and injected subcutaneously (s.c.) into the trunks of mice on day 0. Tumour size was measured weekly by callipers in a blinded manner, and the tumour volume was calculated using the following formula: (major axis of tumour) × (minor axis of tumour)2/2. When conducting experiments with the drug, 14 days after tumour injection, mice were randomly divided into two groups based on a random number table and injected intraperitoneally (i.p.) twice per week with 100 μl of 200 μg/ml decitabine or 100 μl PBS. The Institutional Review Board at Kyushu University approved the use of xenograft models for this study (approval number: A20-339-0). All experiments met the National Institutes of Health guide for the care and use of laboratory animals. Although sample sizes were calculated using a significance criterion of α = 0.05 to achieve 80% statistical power, the sample sizes were kept as small as possible for the sake of animal welfare.
Antibody array analysis
Each cell line was cultured in a 15-cm culture dish to 80% confluence. Whole-cell protein extracts were obtained using Cell Lysis Buffer (R&D Systems, Minneapolis, MN, USA) containing protease inhibitor cocktail tablets (cOmplete™; Roche). Protein concentration in cell lysates was measured by the Bradford method. Antibody array analysis was performed using Proteome Profiler Human Phospho-Kinase Array Kits (ARY003B; R&D Systems). Semi-quantitation was performed on CS Analyzer 3.0 (ATTO)
Immunoprecipitation
Whole-cell protein extracts were obtained as described in the Western blotting section. Anti-GFP mAb-Magnetic Beads (Medical & Biological Laboratories, Nagoya, Japan) or rat IgG2a-Magnetic Beads (Medical & Biological Laboratories) were added to the extracts and incubated at 4 °C for 1 h. Incubated lysates were placed on a magnetic rack, and the supernatant was removed. The beads were then washed three times with cold wash buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.05% NP-40). Proteins were resuspended with 1× LDS sample buffer (Invitrogen) and boiled at 90 °C for five min.
Statistical analysis
Equality of data variances was assessed by the F-test. When the variances had equality, Student’s t-test was applied; otherwise, the Welch’s t-test was applied for two-group comparisons. p-values < 0.05 were considered significant. Data in graphs are given as means ± standard deviation (S.D.). All statistical analyses were performed with R (The R Foundation for Statistical Computing, Vienna, Austria).
Results
PRKCZ is epigenetically silenced in DDCS
To determine which candidate genes are epigenetically silenced in DDCS, we applied a bioinformatic approach. We began by analysing the E–MTAB–7263 and E–MTAB–7264 datasets from Array Express [15]. To generate this dataset, DNA methylation and gene expression in chondrosarcoma were analysed using array-based assays. We compared methylation status between CCS (n = 73) and DDCS (n = 16). Methylation near the transcription start site was significantly higher for 74 genes, and mRNA levels were significantly lower for 681 genes in DDCS vs. CCS (Fig. 1a, left and middle); 17 of these genes overlapped (Fig. 1b). To further narrow down the candidate genes, we analysed the GSE12532 dataset from Gene Expression Omnibus [35]. To generate this dataset, gene expression in chondrosarcoma was analysed using array-based assays. Again, we compared CCS (n = 12) and DDCS (n = 3). mRNA levels of 54 genes were lower in DDCS than in CCS (Fig. 1a, right). After these processes, the candidates were narrowed down to four genes (Fig. 1b). Of these, two had CpG island near the transcription start sites: PRKCZ and COL11A2 (Supplementary Fig. S1). To determine which genes to investigate, we performed whole-genome bisulfite sequencing (WGBS) for two cell lines: NDCS-1 and OUMS-27. NDCS-1 was established from a DDCS tumour and is characterised by rapid growth and xenografts that present with a dedifferentiation component and a cartilage component. OUMS-27 was established from a CCS tumour and is characterised by relatively slow growth and stable expression of a differentiated chondrocyte phenotype (Supplementary Fig. S2). The detailed data for the WGBS analysis is presented in Supplementary Table S4. PRKCZ was hypermethylated in NDCS-1 relative to OUMS-27 (Fig. 1c). Therefore, we decided to target PRKCZ for further investigation.
Fig. 1. Bioinformatic analysis identified epigenetic silencing of PRKCZ in DDCS.

a Volcano plots illustrating differences in DNA methylation (left) or mRNA expression (middle and right). Red dashed lines indicate thresholds: 0.05, 0.2, and −0.5 for adjusted p-value, Δβ value of methylation in DDCS vs. CCS, and log-fold change of mRNA expression in DDCS vs. CCS, respectively. Hypermethylated genes are indicated by red dots. Hypoexpressed genes are indicated by blue dots. b Venn diagram illustrating the number of genes detected by analysis of differential DNA methylation or mRNA expression. c Bisulfite sequencing to detect differences in DNA methylation levels in the promoters of the candidate genes in NDCS-1 and OUMS-27. Methylation rates of each C in CpGs are indicated by dots. Curved lines indicate smooth local regression of methylation rates in each cell line. CpG island are indicated by blue bands. The locus was defined by GRCh38 Reference Sequence.
PRKCZ expression levels correlate with the prognosis for DDCS patients
Methylation and expression levels of PRKCZ in DDCS and CCS were validated with the datasets used in silico analysis (Fig. 2a). We observed no differences in the methylation or expression of PRKCZ amongst the histological grades of CCS (Fig. 2b). To verify the correlation between PRKCZ expression and the prognosis of chondrosarcoma, we examined the difference in overall survival between patients with high and low PRKCZ expression using the E–MTAB–7264 dataset described above. DDCS patients with lower-than-median PRKCZ had a significantly lower survival rate (Fig. 2c). By contrast, CCS patients exhibited no differences in survival rate as a function of PRKCZ expression (Fig. 2d). These results suggest that a lower expression level of PRKCZ correlates with a poor prognosis for patients with DDCS, but the expression level of PRKCZ does not correlate with the tumour histological grade or prognosis for patients with CCS.
Fig. 2. Validation of DNA methylation and expression of PRKCZ.

a Comparison of DNA methylation level and expression level of PRKCZ between DDCS and CCS. Methylation levels of all probes near the transcription starting site of PRKCZ are shown (left). b Comparison of DNA methylation level and expression level of PRKCZ among the histological grades of CCS. In E-MTAB-7263 and 7264 datasets, n = 73 in CCS; 17 in Grade 1, 39 in Grade 2, and 17 in Grade 3; n = 16 in DDCS. In GSE12532 dataset, n = 12 in CCS; one in Grade 1, 10 in Grade 2, one in Grade 3; n = 3 in DDCS. c, d Comparison of survival rates in chondrosarcoma. c DDCS. d CCS. Log-rank test.
Immunohistological analysis of PRKCZ in DDCS
To determine whether protein expression of PRKCZ was reduced in DDCS, we performed IHC staining of clinical samples of DDCS. The semi-quantitative assessment was performed using immunoreactive scores (IRS) [36]. The dedifferentiated components had significantly lower IRS relative to the cartilaginous components (Fig. 3a, b). The expression levels of PRKCZ in clinical samples were validated using RT-qPCR (Supplementary Fig. S3). These results suggest that PRKCZ protein expression decreased in the dedifferentiated components in DDCS.
Fig. 3. PRKCZ is suppressed in dedifferentiated component of DDCS relative to the cartilaginous component.
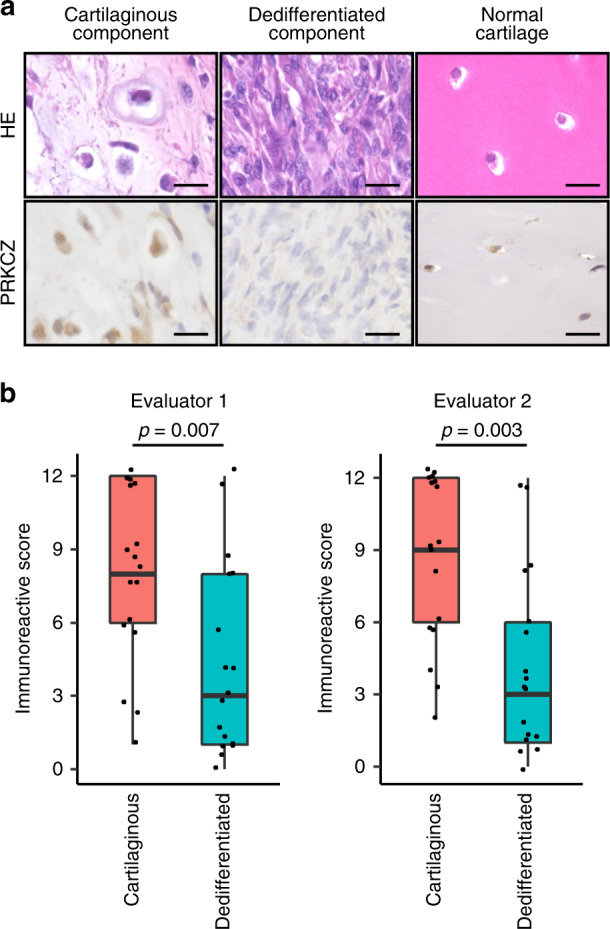
a Representative images of HE staining and PRKCZ IHC staining in DDCS. Normal cartilage is for comparison. Scale bars, 20 µm. b Evaluation of staining in each component of DDCS using immunoreactive score. Wilcoxon signed-rank test.
PRKCZ decreases the proliferation rate in a DDCS cell line
For in vitro analysis, we used the SW1353 cell line in addition to the NDCS-1 and OUMS-27 cell lines used for WGBS analysis. SW1353 was characterised by rapid growth, no expression of differentiated chondrocyte genes, and xenografts that presented with a dedifferentiated tissue structure despite the cell line being derived from a CCS tumour (Supplementary Fig. S2). Therefore, we concluded that the SW1353 phenotype more closely matches that observed in DDCS tumours. PRKCZ was expressed at a lower level in NDCS-1 and SW1353 relative to the level observed in OUMS-27 (Fig. 4a, b).
Fig. 4. PRKCZ overexpression decreases proliferation and induces apoptosis in NDCS-1.

a PRKCZ expression in each cell line is determined by RT-qPCR. b PRKCZ expression in each cell line determined by western blotting. C28/I2, human chondrocyte cell line for comparison. In c–h PRKCZ-overexpressing NDCS-1 vs. mock-transfected NDCS-1. c PRKCZ expression determined by RT-qPCR. d PRKCZ expression determined by western blotting. e Proliferation assay. f Sub-G1 fraction rate determined by cell cycle assay. g Levels of apoptosis-related proteins expression determined by western blotting. h Time course of tumour volume of xenograft transplantation. Images were harvested at the sacrifice; representative images are shown. In i–l siPRKCZ-transfected OUMS-27 vs. scrambled siRNA-transfected OUMS-27. i. PRKCZ expression determined by qRT-PCR. j PRKCZ expression determined by western blotting. k Proliferation assay. l Sub-G1 fraction rate determined by cell cycle assay. In a, c, e, f, h, i, k, and l data are means ± SD; n = 3. Two-tailed t-tests. *p < 0.05. NS not significant.
To investigate the biological functions of PRKCZ in DDCS, we forced the expression of PRKCZ in NDCS-1. Elevated expression levels of PRKCZ were confirmed by RT-qPCR and western blotting in three independent clones (Fig. 4c, d). Overexpression of PRKCZ significantly inhibited proliferation (Fig. 4e). To determine why the proliferation rate was reduced, we performed cell cycle analysis. We found that overexpression of PRKCZ resulted in the expansion of a sub-G1 fraction relative to control cells (Fig. 4f and Supplementary Fig. S4a), and the expression levels of apoptosis-related proteins such as cleaved caspase 3 and cleaved poly (ADP-ribose) polymerase (PARP) increased (Fig. 4g). These results suggest that overexpression of PRKCZ in NDCS-1 induced apoptosis, which was followed by inhibition of cell proliferation. Overexpression of PRKCZ in NDCS-1 also inhibited colony-forming ability (Supplementary Fig. S4b). To confirm the effects from the overexpression of PRKCZ in NDCS-1 cells in vivo, we injected PRKCZ-overexpressing and control NDCS-1 cells into the trunk of BALB/cAJcl-nu/nu mice and periodically measured the volume of the resultant tumours (n = 3 per group). All PRKCZ-overexpressing NDCS-1 cells grew significantly more slowly than control NDCS-1 cells, consistent with the results in vitro (Fig. 4h). Forced expression of PRKCZ in SW1353 also produced the same results; that is, overexpression of PRKCZ induced apoptosis, which was followed by the inhibition of cell proliferation in vitro, and the tumour exhibited slow growth in vivo (Supplementary Fig. S5). Because OUMS-27 expressed PRKCZ endogenously, we knocked down PRKCZ in this cell line using siRNA. The effects of siRNA on PRKCZ expression were confirmed by RT-qPCR and western blotting 48 h after siRNA transfection (Fig. 4i, j). Notably, PRKCZ-knockdown cells exhibited no difference in proliferation rates relative to cells transfected with scrambled control siRNA (Fig. 4k). In addition, cell cycle analysis revealed no difference in the sub-G1 fraction between knockdown and control cells (Fig. 4l and Supplementary Fig. S4c).
PRKCZ induces apoptosis by activating ATM/CHK2 pathway
We next investigated the molecular mechanism of apoptosis induced by overexpression of PRKCZ. Because PRKCZ has kinase activity, we used a human phospho-antibody array (Human Phospho–Kinase Array, Proteome Profiler Array Kit) to semi-quantitatively determine the phosphorylations of many proteins in parallel. Compared to control NDCS-1, all PRKCZ-overexpressing NDCS-1 clones had higher phosphorylation of CHK2 (Fig. 5a), a well-known activator of the apoptosis pathway [37]. Therefore, we further investigated the relationship between PRKCZ and CHK2. The upstream pathway of apoptosis induction by phosphorylation of CHK2 is as follows: once the MRE11–RAD50–NBS1 (MRN) complex senses double-strand breaks of DNA, ATM is recruited to chromatin, where it is activated; there, it promotes activation of CHK2, which can lead to apoptosis depending on the level of stress [37]. Hence, we investigated the protein and phosphorylation levels of the factors upstream of CHK2. The phosphorylation levels of ATM and CHK2 were higher, with no change in total protein expression, in the PRKCZ-overexpressing NDCS-1 cells relative to the mock-transfected cells (Fig. 5b and Supplementary Fig. S6a). In the SW1353 cells, PRKCZ-overexpressing clones also exhibited higher phosphorylation levels of ATM and CHK2 relative to the control cells (Supplementary Fig. S6b). To confirm whether phosphorylation of ATM induced apoptosis in chondrosarcoma, we administered chloroquine diphosphate, which induces the phosphorylation of ATM [38]. Consistent with reports using other cell lines, chloroquine induced the phosphorylation of ATM, which was followed by apoptosis (Supplementary Fig. S6c).
Fig. 5. PRKCZ induces apoptosis through activation of the ATM/CHK2 pathway.

a Phospho-antibody array showing that PRKCZ overexpression induces CHK2 phosphorylation. Red squares indicate CHK2 spots (left). Bar plots indicate semi-quantitative intensity of representative proteins in PRKCZ-overexpressing or mock-transfected NDCS-1 (right). Data are means ± SD; n = 2. b Expression of ATM/CHK2-related proteins in PRKCZ-overexpressing NDCS-1 vs. mock-transfected NDCS-1. Band intensities were quantified and normalised using actin levels. The relative intensities compared to mock are shown under each band. c Microscopic images of GFP-tagged PRKCZ-transfected NDCS-1 and mock-transfected NDCS-1 (left) and HeLa (right). Scale bars, 100 µm. d Immunoprecipitation assay to detect the interaction between PRKCZ and ATM/CHK2-related proteins in NDCS-1 (left) and HeLa (right).
To elucidate the physical interaction between PRKCZ and proteins involved in ATM/CHK2 pathway, we constructed a GFP-tagged PRKCZ vector, transfected it into NDCS-1, and established a single clone (Fig. 5c, left). Immunoprecipitation with anti-GFP antibody confirmed that PRKCZ interacted with ATM, RAD50, and MRE11 (Fig. 5d, left). To further confirm these interactions in other cells, we transiently transfected GFP-tagged PRKCZ into HeLa cells (Fig. 5c, right) and performed immunoprecipitation as described above for NDCS-1. The results were consistent with those obtained with NDCS-1 (Fig. 5d, right). Moreover, a computational prediction of PRKCZ phosphorylation sites identified several predicted sites in ATM (Supplementary Fig. S7). Together, these results suggest that PRKCZ phosphorylated ATM by physical interaction.
DNA methylation inhibitor increases PRKCZ expression
Our results strongly implied that in DDCS, the expression of PRKCZ is suppressed by DNA methylation, and that this reduction in PRKCZ expression enhances cell proliferation. We then tested the effect of decitabine, a DNMTi, on PRKCZ expression, proliferation, and apoptosis. We confirmed the effects of decitabine on demethylation using bisulfite sequencing (Fig. 6a) and decided to increase the concentration of decitabine to 10 µM because the methylation rate was low. As expected, decitabine-treated NDCS-1 cells expressed higher levels of PRKCZ relative to PBS-treated cells (Fig. 6b, c). Furthermore, in decitabine-treated NDCS-1, proliferation was slower, and more apoptosis was observed (Fig. 6d, e and Supplementary Fig. S8a). Consistent with our observations in PRKCZ-overexpressing NDCS-1, western blotting of decitabine-treated NDCS-1 revealed more phosphorylation of ATM and CHK2, and higher expression of apoptosis-related proteins (Fig. 6c). Colony formation was inhibited by decitabine (Supplementary Fig. S8b). To confirm the effects of decitabine on NDCS-1 in vivo, we injected NDCS-1 cells into the trunk of BALB/cAJcl-nu/nu mice. Starting 2 weeks after injection, mice were injected i.p. twice per week with 100 μl of 200 μg/ml decitabine or 100 μL of PBS (n = 6 per group) (Fig. 6f). The tumour volume was significantly smaller in the decitabine-treated mice relative to the PBS-treated mice, which was consistent with our observations from the PRKCZ-overexpressing NDCS-1 cells (Fig. 6g, h). We confirmed that PRKCZ was expressed at higher levels in the tumours from decitabine-treated mice relative to the PBS-treated mice (Supplementary Fig. S8c). Decitabine also induced PRKCZ expression, which was followed by ATM/CHK2 activation and apoptosis in SW1353 cells (Supplementary Fig. S9a–h). By contrast, decitabine did not induce PRKCZ expression or inhibit the growth of OUMS-27 cells (Supplementary Fig. S10).
Fig. 6. Decitabine increases PRKCZ expression and induces apoptosis in NDCS-1.

a DNA methylation levels on the PRKCZ promoter in decitabine-treated vs. PBS-treated NDCS-1, as determined by bisulfite sequencing. Methylation rates of each C in CpG islands are indicated by dots (left). Curved lines indicate smooth local regression of methylation rates in each group (left). Average methylation rates (right). n = 5. In b–e 10 μM of decitabine-treated NDCS-1 vs. PBS-treated NDCS-1. b PRKCZ expression determined by RT-qPCR. c Expression of ATM/CHK2-related proteins and apoptosis-related proteins determined by western blotting. d Proliferation assay. e Sub-G1 fraction rate determined by cell cycle assay. f Schedule of tumour implantation and decitabine treatment in the xenograft assay. g Time course of tumour volume of xenografts in decitabine-treated NDCS-1 vs. PBS-treated NDCS-1. Images were obtained at the time of sacrifice; representative images are shown. n = 6. In h–j 10 μM of decitabine-treated NDCS-1 with or without siPRKCZ. h PRKCZ expression determined by RT-qPCR. i PRKCZ expression determined by western blotting. j Proliferation assay. In b, d, e, h, and j n = 3. In a, b, d, e, g, h, and j data are means ± SD. Two-tailed t-tests. *p < 0.05. NS not significant. DAC decitabine.
To determine whether the effect of decitabine was due to the expression of PRKCZ, we administered decitabine to NDCS-1 along with siRNA against PRKCZ. Twenty-four hours after transfection with 50 nM siRNA, the medium was changed, and 10 μM decitabine was administered for another two days. The effect of knockdown of PRKCZ was confirmed by RT-qPCR and western blotting (Fig. 6h, i and Supplementary Fig. S8d). Interestingly, the inhibitory effect of decitabine on cell proliferation was decreased by PRKCZ-knockdown (Fig. 6j and Supplementary Fig. S8e). The same result was not observed in SW1353 cells because siRNA could not sufficiently reduce the expression of PRKCZ (Supplementary Fig. S9i–k). Furthermore, a siRNA against ATM produced the same results as a siRNA against PRKCZ (Supplementary Fig. S11). These results indicated that growth inhibition by administration of decitabine of NDCS-1 might be mediated predominantly through an expression of PRKCZ and subsequent activation of the ATM/CHK2 pathway.
Discussion
This study demonstrates that increased DNA methylation and reduced expression of PRKCZ prevent apoptosis via inactivation of the ATM/CHK2 pathway in DDCS. Our results also show that decitabine induces apoptosis in DDCS by increasing PRKCZ expression levels.
The relationships between certain genes, including RUNX3, p16, FHIT, and E-cadherin [39–41], with methylation in high-grade chondrosarcomas are known. In our study, we adopted a bioinformatic strategy to identify PRKCZ as a target gene by analysing multiple datasets; this approach could be used to discover candidate genes that have not been previously examined and would strengthen the reliability of the results.
We then investigated the biological significance of PRKCZ in the oncogenesis of DDCS. PRKCZ plays an oncosuppressive role in other cancers [25, 30, 32]. Consistent with these studies, forced expression of PRKCZ also produced an oncosuppressive effect in DDCS by inducing apoptosis; however, our study showed that PRKCZ activated ATM/CHK2, which was not activated by PRKCZ in other studies that reported PRKCZ-mediated activation of ADAR2 [25], c-Myc [30], p38 MAPK [42], STAT3, Akt, and Erk [43, 44]. Interestingly, PRKCZ induced apoptosis exclusively in DDCS tumours, but not in the CCS tumours with low-grade histology in our study, implying that the presence of ATM/CHK2 activation by PRKCZ followed by the induction of apoptosis depends on the cellular context. Although the mechanism of this cellular context-dependence remains unclear, our results strongly suggest that reduced expression of PRKCZ helps DDCS escape from apoptosis.
We then used decitabine to express PRKCZ in DDCS tumours, keeping in mind future clinical applications, because it is used as a treatment for myelodysplastic syndromes. Decitabine produced oncosuppressive effects following PRKCZ expression in only DDCS, which may explain the controversies over whether DNMTi suppresses oncogenicity or not in chondrosarcoma; that is, DNMTi cannot induce PRKCZ expression, and PRKCZ does not induce apoptosis even if PRKCZ is expressed in CCS tumours. Hence, in malignant cartilaginous tumours, the use of decitabine as an effective treatment may be limited to the treatment of patients with DDCS tumours.
There were several limitations in this study. First, although we showed that PRKCZ could bind ATM directly, it is not clear whether PRKCZ phosphorylates ATM. In vitro kinase assays may solve this problem, and we are currently working to overcome the technical difficulties of that experiment. Second, although the promoter region of PRKCZ was hypermethylated, there are no IDH mutations in NDCS-1, and the reason for hypermethylation is unclear. A multi-omics study revealed several DDCS cases that exhibited hypermethylation without IDH mutation [15]. Thus, unknown mechanisms of hypermethylation might exist in DDCS. In addition, the mechanism by which decitabine inhibits oncogenicity in DDCS was not clearly elucidated, as decitabine is a non-specific DNMTi that causes global demethylation of DNA. To solve this problem, we are considering carrying out targeted demethylation of the PRKCZ promoter region using the CRISPR–dCas9 system [45]. Finally, because of the rarity of the disease, we examined only a small number of clinical DDCS cases. Therefore, in order to validate the clinical significance of PRKCZ in the biology of DDCS, it will be necessary to examine more clinical samples.
Supplementary information
Acknowledgements
We appreciate the technical assistance from The Research Support Center, Research Center for Human Disease Modeling, Kyushu University Graduate School of Medical Sciences.
Author contributions
YM and YN supervised the study; YM, MN, KY, AK, TH, MK, and RO provided ideas for the research design; ES performed the in silico, in vitro, and in vivo experiments and analysed the data; YS and YO collected clinical samples of chondrosarcoma; ES wrote the original draft; YM, ME, NS, TF, KI, and AN reviewed and revised the manuscript; YM, MN, and ME acquired funding All authors approved the final version of the manuscript.
Funding
This work was supported in part by AMED (#JP20ck0106523 (MN)), Grant-in-Aid for Young Scientists (#JP18K16627), Grant-in-Aid for Scientific Research (#JP21K09325), and Grant-in-Aid for Research Activity Start-up (#JP21K20838) from the Japan Society for the Promotion of Science, Grant of the Clinical Research Promotion Foundation (2021).
Data availability
Data used in the study are available from the corresponding author upon reasonable request.
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All patients provided written informed consent before undergoing the study procedures. The Institutional Review Board at Kyushu University approved the use of human specimens for this study (approval number: 27-420). This study was conducted in accordance with the Declaration of Helsinki.
Consent to publish
All patients provided written informed consent for publication.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41416-021-01695-1.
References
- 1.Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F (eds.) WHO classification of tumours of soft tissue & bone. Geneva, Switzerland: World Health Organization; 2013.
- 2.Chow, WA. Chondrosarcoma: biology, genetics, and epigenetics. F1000Res. 2018;7:F1000. [DOI] [PMC free article] [PubMed]
- 3.Bovee JVMG, Cleton-Jansen A-M, Rosenberg C, Taminiau AHM, Cornelisse CJ, Hogendoorn PCW. Molecular genetic characterization of both components of a dedifferentiated chondrosarcoma, with implications for its histogenesis. J Pathol. 1999;189:454–62. doi: 10.1002/(SICI)1096-9896(199912)189:4<454::AID-PATH467>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 4.Evans HL, Ayala AG, Romsdahl MM. Prognostic factors in chondrosarcoma of bone. Cancer. 1977;40:818–31. doi: 10.1002/1097-0142(197708)40:2<818::AID-CNCR2820400234>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 5.Giuffrida AY, Burgueno JE, Koniaris LG, Gutierrez JC, Duncan R, Scully SP. Chondrosarcoma in the United States (1973 to 2003): an analysis of 2890 cases from the SEER database. J Bone Jt Surg Am. 2009;91:1063–72. doi: 10.2106/JBJS.H.00416. [DOI] [PubMed] [Google Scholar]
- 6.Yokota K, Sakamoto A, Matsumoto Y, Matsuda S, Harimaya K, Oda Y, et al. Clinical outcome for patients with dedifferentiated chondrosarcoma: a report of 9 cases at a single institute. J Orthop Surg Res. 2012;7:38. doi: 10.1186/1749-799X-7-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kawaguchi S, Sun T, Lin PP, Deavers M, Harun N, Lewis VO. Does ifosfamide therapy improve survival of patients with dedifferentiated chondrosarcoma? Clin Orthop Relat Res. 2014;472:983–9. doi: 10.1007/s11999-013-3360-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dienstmann R, Rodon J, Serra V, Tabernero J. Picking the point of inhibition: a comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol Cancer Ther. 2014;13:1021–31. doi: 10.1158/1535-7163.MCT-13-0639. [DOI] [PubMed] [Google Scholar]
- 9.Duffaud F, Italiano A, Bompas E, Rios M, Penel N, Mir O, et al. Efficacy and safety of regorafenib in patients with metastatic or locally advanced chondrosarcoma: results of a non-comparative, randomised, double-blind, placebo controlled, multicentre phase II study. Eur J Cancer. 2021;150:108–18. doi: 10.1016/j.ejca.2021.03.039. [DOI] [PubMed] [Google Scholar]
- 10.Italiano A, Le Cesne A, Bellera C, Piperno-Neumann S, Duffaud F, Penel N, et al. GDC-0449 in patients with advanced chondrosarcomas: a French Sarcoma Group/US and French National Cancer Institute Single-Arm Phase II Collaborative Study. Ann Oncol. 2013;24:2922–6. doi: 10.1093/annonc/mdt391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Campbell VT, Nadesan P, Ali SA, Wang CY, Whetstone H, Poon R, et al. Hedgehog pathway inhibition in chondrosarcoma using the smoothened inhibitor IPI-926 directly inhibits sarcoma cell growth. Mol Cancer Ther. 2014;13:1259–69. doi: 10.1158/1535-7163.MCT-13-0731. [DOI] [PubMed] [Google Scholar]
- 12.L MG, Boulay K, Topisirovic I, Huot ME, Mallette FA. Oncogenic activities of IDH1/2 mutations: from epigenetics to cellular signaling. Trends Cell Biol. 2017;27:738–52. doi: 10.1016/j.tcb.2017.06.002. [DOI] [PubMed] [Google Scholar]
- 13.Lu C, Venneti S, Akalin A, Fang F, Ward PS, Dematteo RG, et al. Induction of sarcomas by mutant IDH2. Genes Dev. 2013;27:1986–98. doi: 10.1101/gad.226753.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klutstein M, Nejman D, Greenfield R, Cedar H. DNA methylation in cancer and aging. Cancer Res. 2016;76:3446–50. doi: 10.1158/0008-5472.CAN-15-3278. [DOI] [PubMed] [Google Scholar]
- 15.Nicolle R, Ayadi M, Gomez-Brouchet A, Armenoult L, Banneau G, Elarouci N, et al. Integrated molecular characterization of chondrosarcoma reveals critical determinants of disease progression. Nat Commun. 2019;10:4622. doi: 10.1038/s41467-019-12525-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakagawa M, Nakatani F, Matsunaga H, Seki T, Endo M, Ogawara Y, et al. Selective inhibition of mutant IDH1 by DS-1001b ameliorates aberrant histone modifications and impairs tumor activity in chondrosarcoma. Oncogene. 2019;38:6835–49. doi: 10.1038/s41388-019-0929-9. [DOI] [PubMed] [Google Scholar]
- 17.Sujiker J, Oosting J, Koornneef A, Struys EA, Salomons GS, Schaap FG, et al. Inhibition of mutant IDH1 decreases D-2-HG levels without affecting tumorigenic properties of chondrosarcoma cell lines. Oncotarget. 2015;6:12505–19. doi: 10.18632/oncotarget.3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li L, Paz AC, Wilky BA, Johnson B, Galoian K, Rosenberg A, et al. Treatment with a small molecule mutant IDH1 inhibitor suppresses tumorigenic activity and decreases production of the oncometabolite 2-hydroxyglutarate in human chondrosarcoma cells. PLoS ONE. 2015;10:e0133813. doi: 10.1371/journal.pone.0133813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bui C, Ouzzine M, Talhaoui I, Sharp S, Prydz K, Coughtrie MW, et al. Epigenetics: methylation-associated repression of heparan sulfate 3-O-sulfotransferase gene expression contributes to the invasive phenotype of H-EMC-SS chondrosarcoma cells. FASEB J. 2010;24:436–50. doi: 10.1096/fj.09-136291. [DOI] [PubMed] [Google Scholar]
- 20.Sheng W, Zhang ZC, Shi DY, Wang BC, Wu Q, Shao ZW, et al. Epigenetic silencing of SFRP5 promotes the metastasis and invasion of chondrosarcoma by expression inhibition and Wnt signaling pathway activation. Chem Biol Interact. 2018;296:1–8. doi: 10.1016/j.cbi.2018.08.020. [DOI] [PubMed] [Google Scholar]
- 21.Hamm CA, Xie H, Costa FF, Vanin EF, Seftor EA, Sredni ST, et al. Global demethylation of rat chondrosarcoma cells after treatment with 5-aza-2’-deoxycytidine results in increased tumorigenicity. PLoS ONE. 2009;4:e8340. doi: 10.1371/journal.pone.0008340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Venneker S, Kruisselbrink AB, Baranski Z, Palubeckaite I, Briaire-de Bruijn IH, Oosting J, et al. Beyond the influence of IDH mutations: exploring epigenetic vulnerabilities in chondrosarcoma. Cancers. 12;2020:3589. [DOI] [PMC free article] [PubMed]
- 23.Pollack SM, Li Y, Blaisdell MJ, Farrar EA, Chou J, Hoch BL, et al. NYESO-1/LAGE-1s and PRAME are targets for antigen specific T cells in chondrosarcoma following treatment with 5-Aza-2-deoxycitabine. PLoS ONE. 2012;7:e32165. doi: 10.1371/journal.pone.0032165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reina-Campos M, Diaz-Meco MT, Moscat J. The dual roles of the atypical protein kinase Cs in cancer. Cancer Cell. 2019;36:218–35. doi: 10.1016/j.ccell.2019.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shelton PM, Duran A, Nakanishi Y, Reina-Campos M, Kasashima H, Llado V, et al. The secretion of miR-200s by a PKCzeta/ADAR2 signaling axis promotes liver metastasis in colorectal cancer. Cell Rep. 2018;23:1178–91. doi: 10.1016/j.celrep.2018.03.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deevi RK, Javadi A, McClements J, Vohhodina J, Savage K, Loughrey MB, et al. Protein kinase C zeta suppresses low- or high-grade colorectal cancer (CRC) phenotypes by interphase centrosome anchoring. J Pathol. 2018;244:445–59. doi: 10.1002/path.5035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seto KK, Andrulis IL. Atypical protein kinase C zeta: potential player in cell survival and cell migration of ovarian cancer. PLoS ONE. 2015;10:e0123528. doi: 10.1371/journal.pone.0123528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Llado V, Nakanishi Y, Duran A, Reina-Campos M, Shelton PM, Linares JF, et al. Repression of Intestinal stem cell function and tumorigenesis through direct phosphorylation of beta-catenin and Yap by PKCzeta. Cell Rep. 2015;10:740–54. doi: 10.1016/j.celrep.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma L, Tao Y, Duran A, Llado V, Galvez A, Barger JF, et al. Control of nutrient stress-induced metabolic reprogramming by PKCzeta in tumorigenesis. Cell. 2013;152:599–611. doi: 10.1016/j.cell.2012.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim JY, Valencia T, Abu-Baker S, Linares J, Lee SJ, Yajima T, et al. c-Myc phosphorylation by PKCzeta represses prostate tumorigenesis. Proc Natl Acad Sci USA. 2013;110:6418–23. doi: 10.1073/pnas.1221799110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Valkov A, Sorbye SW, Kilvaer TK, Donnem T, Smeland E, Bremnes RM, et al. The prognostic impact of TGF-beta1, fascin, NF-kappaB and PKC-zeta expression in soft tissue sarcomas. PLoS ONE. 2011;6:e17507. doi: 10.1371/journal.pone.0017507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Galvez AS, Duran A, Linares JF, Pathrose P, Castilla EA, Abu-Baker S, et al. Protein kinase Czeta represses the interleukin-6 promoter and impairs tumorigenesis in vivo. Mol Cell Biol. 2009;29:104–15. doi: 10.1128/MCB.01294-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kudo N, Ogose A, Hotta T, Kawashima H, Gu W, Umezu H, et al. Establishment of novel human dedifferentiated chondrosarcoma cell line with osteoblastic differentiation. Virchows Arch. 2007;451:691–9. doi: 10.1007/s00428-007-0426-3. [DOI] [PubMed] [Google Scholar]
- 34.Iida K, Fukushi J, Matsumoto Y, Oda Y, Takahashi Y, Fujiwara T, et al. miR-125b develops chemoresistance in Ewing sarcoma/primitive neuroectodermal tumor. Cancer Cell Int. 2013;13:21. doi: 10.1186/1475-2867-13-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hallor KH, Staaf J, Bovee JV, Hogendoorn PC, Cleton-Jansen AM, Knuutila S, et al. Genomic profiling of chondrosarcoma: chromosomal patterns in central and peripheral tumors. Clin Cancer Res. 2009;15:2685–94. doi: 10.1158/1078-0432.CCR-08-2330. [DOI] [PubMed] [Google Scholar]
- 36.W R, HE S. Recommendation for uniform definition of an immunoreactive score (IRS) for immunohistochemical estrogen receptor detection (ER-ICA) in breast cancer tissue. Pathologe. 1987;8:138–40. [PubMed] [Google Scholar]
- 37.Kantidze OL, Velichko AK, Luzhin AV, Petrova NV, Razin SV. Synthetically lethal interactions of ATM, ATR, and DNA-PKcs. Trends Cancer. 2018;4:755–68. doi: 10.1016/j.trecan.2018.09.007. [DOI] [PubMed] [Google Scholar]
- 38.Loehberg CR, Thompson T, Kastan MB, Maclean KH, Edwards DG, Kittrell FS, et al. Ataxia telangiectasia-mutated and p53 are potential mediators of chloroquine-induced resistance to mammary carcinogenesis. Cancer Res. 2007;67:12026–33. doi: 10.1158/0008-5472.CAN-07-3058. [DOI] [PubMed] [Google Scholar]
- 39.Jin Z, Han YX, Han XR. Loss of RUNX3 expression may contribute to poor prognosis in patients with chondrosarcoma. J Mol Histol. 2013;44:645–52. doi: 10.1007/s10735-013-9511-x. [DOI] [PubMed] [Google Scholar]
- 40.Asp J, Sangiorgi L, Inerot SE, Lindahl A, Molendini L, Benassi MS, et al. Changes of the p16 gene but not the p53 gene in human chondrosarcoma tissues. Int J Cancer. 2000;85:782–6. doi: 10.1002/(SICI)1097-0215(20000315)85:6<782::AID-IJC7>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 41.Ropke M, Boltze C, Neumann HW, Roessner A, Schneider-Stock R. Genetic and epigenetic alterations in tumor progression in a dedifferentiated chondrosarcoma. Pathol Res Pract. 2003;199:437–44. doi: 10.1078/0344-0338-00443. [DOI] [PubMed] [Google Scholar]
- 42.Mazumder S, De R, Debsharma S, Bindu S, Maity P, Sarkar S, et al. Indomethacin impairs mitochondrial dynamics by activating the PKCzeta-p38-DRP1 pathway and inducing apoptosis in gastric cancer and normal mucosal cells. J Biol Chem. 2019;294:8238–58. doi: 10.1074/jbc.RA118.004415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Butler AM, Scotti Buzhardt ML, Li S, Smith KE, Fields AP, Murray NR. Protein kinase C zeta regulates human pancreatic cancer cell transformed growth and invasion through a STAT3-dependent mechanism. PLoS ONE. 2013;8:e72061. doi: 10.1371/journal.pone.0072061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zang G, Mu Y, Gao L, Bergh A, Landstrom M. PKCzeta facilitates lymphatic metastatic spread of prostate cancer cells in a mice xenograft model. Oncogene. 2019;38:4215–31. doi: 10.1038/s41388-019-0722-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morita S, Noguchi H, Horii T, Nakabayashi K, Kimura M, Okamura K, et al. Targeted DNA demethylation in vivo using dCas9-peptide repeat and scFv-TET1 catalytic domain fusions. Nat Biotechnol. 2016;34:1060–5. doi: 10.1038/nbt.3658. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data used in the study are available from the corresponding author upon reasonable request.