

Abstract
The COVID-19 pandemic is now approaching 2 years old, with more than 440 million people infected and nearly six million dead worldwide, making it the most significant pandemic since the 1918 influenza pandemic. The severity and significance of SARS-CoV-2 was recognized immediately upon discovery, leading to innumerable companies and institutes designing and generating vaccines and therapeutic antibodies literally as soon as recombinant SARS-CoV-2 spike protein sequence was available. Within months of the pandemic start, several antibodies had been generated, tested, and moved into clinical trials, including Eli Lilly’s bamlanivimab and etesevimab, Regeneron’s mixture of imdevimab and casirivimab, Vir’s sotrovimab, Celltrion’s regdanvimab, and Lilly’s bebtelovimab. These antibodies all have now received at least Emergency Use Authorizations (EUAs) and some have received full approval in select countries. To date, more than three dozen antibodies or antibody combinations have been forwarded into clinical trials. These antibodies to SARS-CoV-2 all target the receptor-binding domain (RBD), with some blocking the ability of the RBD to bind human ACE2, while others bind core regions of the RBD to modulate spike stability or ability to fuse to host cell membranes. While these antibodies were being discovered and developed, new variants of SARS-CoV-2 have cropped up in real time, altering the antibody landscape on a moving basis. Over the past year, the search has widened to find antibodies capable of neutralizing the wide array of variants that have arisen, including Alpha, Beta, Gamma, Delta, and Omicron. The recent rise and dominance of the Omicron family of variants, including the rather disparate BA.1 and BA.2 variants, demonstrate the need to continue to find new approaches to neutralize the rapidly evolving SARS-CoV-2 virus. This review highlights both convalescent plasma- and polyclonal antibody-based approaches as well as the top approximately 50 antibodies to SARS-CoV-2, their epitopes, their ability to bind to SARS-CoV-2 variants, and how they are delivered. New approaches to antibody constructs, including single domain antibodies, bispecific antibodies, IgA- and IgM-based antibodies, and modified ACE2-Fc fusion proteins, are also described. Finally, antibodies being developed for palliative care of COVID-19 disease, including the ramifications of cytokine release syndrome (CRS) and acute respiratory distress syndrome (ARDS), are described.
Supplementary Information
The online version contains supplementary material available at 10.1007/s40259-022-00529-7.
Key Points
| Antibodies against SARS-CoV-2 and its variants have become critically important weapons in the arsenal against COVID-19, contributing to the effort to save lives and reduce severe disease and hospitalization. |
| As SARS-CoV-2 has drifted antigenically from the Wuhan virus, to its Alpha, Beta, Gamma, Delta, and Omicron variants, antibodies in development have been brought forward to address each variant, including sometimes revisiting “older” antibodies that may work against new variants that arise. |
| Of all of the antibodies and targets tested for palliative therapy, it appears that only the anti-IL-6 receptor antibodies provide benefit for the immunological effects brought on by COVID-19 disease. |
| For all antibody-based approaches to treat COVID-19, the earliest possible treatment with high doses appear to be required for optimal activity and efficacy. |
Introduction
Historically, the general concept of “antibody” treatment for pathogenic diseases is more than 130 years old, when Behring and Kitasato demonstrated that the transfer of serum from a guinea pig immunized with diphtheria toxin to another guinea pig offered protection against that toxin [1]. Immunized animal serum-based therapy, which was used widely until the 1940s for a variety of infectious diseases ranging from diphtheria to whooping cough to chickenpox [2], eventually gave way to immunized/convalescent human plasma-based and specific (hyperimmune) intravenous immunoglobulin (IVIg) therapy, and finally, to the use of monoclonal antibodies (mAbs) for prevention and/or treatment of infectious diseases [3–5].
The use of mAbs as therapeutic drugs to treat viral infections has a long history prior to the recent COVID-19 pandemic. In 1998, the sixth mAb ever approved by the US Food and Drug Administration (FDA) was palivizumab (Synagis®), for prophylaxis against respiratory syncytial virus (RSV) in premature infants, demonstrating early on the potential importance for use of mAbs against viral pathogens [6]. Not including the multitude of antibodies targeting severe acute respiratory syndrome (SARS)-coronavirus-2 (CoV-2) (SARS-CoV-2) described herein, there are currently at least 40 different mAbs recently or currently in clinical trials targeting a wide variety of viruses, including RSV (e.g., NCT03979313; [7, 8]), human immunodeficiency virus (HIV) (e.g., NCT03707977) [7, 9], influenza virus (e.g., NCT02623322) [7, 10], Middle East respiratory syndrome (MERS) virus (e.g., NCT03301090) [7, 11], Ebola virus (e.g., NCT03576690) [12], zika virus (e.g., NCT03776695) [13], dengue virus (e.g., NCT04273217), chikungunya virus (e.g., NCT04441905; an mRNA encoded antibody) [14], herpes simplex virus (e.g., NCT04539483), hepatitis B virus (e.g., NCT04856085) [15], rabies virus (e.g., NCT04644484) [16], and cytomegalovirus (e.g., sevirumab, NCT00001061) [17]. Across the virus landscape, therapeutic antibodies have been shown to neutralize and kill viruses via a wide variety of mechanisms of action (MOAs) [3], including inhibition of virus binding to receptor [3], complement mediated killing [3, 18, 19], antibody-dependent cellular cytotoxicity (ADCC) [3, 18–21], and opsonization and phagocytosis [22, 23].
While some of these antibodies are being tested individually, many are dosed as mixtures of two or more individual mAbs to provide broader protection against a wider variety of viral strains or serotypes [24] and/or against viral mutations leading to antigenic drift [25]. One such mixture of antiviral antibodies, Inmazeb®, a combination of the anti-Ebola mAbs atoltivimab, maftivimab, odesivimab-ebgn, was fully approved for use by the FDA in 2020 [12].
When considering antibody therapeutics for infectious diseases, the dosing paradigm is for treatment of a rapidly developing, acute disease, which is very different from the use of therapeutics mAbs for chronic diseases such as rheumatoid arthritis and cancer. Thus, the two critical lessons learned through the long history of anti-viral mAb therapy have been that timing (i.e., earliest possible administration after diagnosis) and dosage (i.e., providing a high enough dose to result in therapeutic mAb concentrations in targeted tissues) are critical factors for successful therapy [4]. These lessons are equally as true today for the use of mAbs, mixtures of mAbs, hyper-immune intravenous immunoglobulin (IVIg), and convalescent plasma therapy to treat or prevent disease caused by SARS-CoV-2 [26].
A little over 2 years ago, in 4Q2019, the world had no clue about what was to come, and what would dominate societies from one end of the earth to the other. SARS-CoV-2 has swept through our populations, now in at least four separate waves, perhaps forever changing how we interact, conduct business, and deal with one another. To date (2 March 2022), about 440 million people have been infected and nearly six million people worldwide have died from coronavirus disease 2019 (COVID-19), the disease caused by SARS-CoV-2 [27]. Countries have gone into “lock-down,” sometimes for extended periods of time, to avoid rampant infection rates as the virus has mutated to become even more infectious and transmissible over time. Had this pandemic occurred 100 years ago, as the H1N1 Spanish flu virus did [28, 29], the results could have been even more devastating than they have been and continue to be. Because the pandemic happened now, incredible new vaccine and antibody discovery and development technologies were available to move from validated sequence to product launch under Emergency Use Authorizations (EUAs) in less than a year. Antibodies in many formats and from many processes have played, or are being developed to play, a critical role in saving lives, including prophylactic antibodies, convalescent plasma therapy, and therapeutic antibodies targeting SARS-CoV-2, as well as antibodies for use in palliative care to modulate the immune responses to the viral infection that can lead to severe disease and potentially death. The timing for the use of each different type of antibody treatment depends on the goal and disease status, as noted in Fig. 1, but in any case, antibody treatment of SARS-CoV-2 infection and/or the disease it causes, COVID-19, requires as early intervention as possible. This review tries to capture the salient aspects of the antibodies discovered and developed both to fight the SARS-CoV-2 virus and the disease it causes, COVID-19. The supplemental section accompanying this manuscript provides an historical context for development of antibodies against SARS-CoV-2 as well as details on mutations to the spike protein that driver differences amongst the variants.
Fig. 1.

The timing of COVID-19 in four stages, pre-disease (Stage 0), viral infection and amplification in the respiratory tract (Stage 1), viral expansion to other organs and initiation of immune response (Stage 2), and, in more severe cases, excessive inflammation in response to viral infection (Stage 3). A Prophylactic antibodies are provided at Stage 0 to protect the uninfected or recently exposed from being infected, similar to how a vaccine would work; B antiviral antibody therapy is provided as soon as possible after infections and/or symptoms appear to thwart the viral expansion phase; and C palliative care antibodies are those given in Stage 2 or Stage 3 to reduce the out-of-control immune response to the virus. This figure was modified and redrawn from Patel et al. [30]
Evolution of SARS-CoV-2 in Humans—Variants of Concern
SARS-CoV-2 has provided researchers with an incredibly difficult challenge due to its ability to rapidly mutate and form novel variants with potentially improved transmissibility and/or virulence characteristics. The essence of this review is the tug of war between those developing antibody-based therapeutics or prophylactics against the rapidly mutating and adapting SARS-CoV-2. Starting in March 2021, the WHO and US Centers for Disease Control (CDC) provided labels for SARS-CoV-2 variants according to their potential for both increased transmissibility and pathogenesis [31, 32]. The variant labels come in two major forms, “Variant of Interest” (VOI) and the more serious “Variant of Concern” (VOC) (Fig. 2). Additionally, the WHO has identified Variants under Monitoring (VUMs), i.e., those variants observed, but not rising to the importance of VOIs. Individual key mutations and the variants of lesser significance, VOIs, are described in the Online Supplemental Material (OSM), Sect. S2.
Fig. 2.

Locations of mutations in SARS-CoV-2 spike protein. A Linear representation of SARS-CoV-2 spike protein showing some of the key subunits involved with mutations as well as antibodies, including the N-terminal domain (NTD), the receptor binding domain (RBD), receptor binding motif (RBM), S1/S2 region around the furin protease cleavage site, the S2 domain, and the transmembrane (TM) region at the C-terminus of S2. Notes: X, residues 980-1006 are an epitope for S2-targeting neutralizing antibody 3A3; Y, residues 1140–1164 in stem-helix region targeted by antibody CC40.8; Z, residues 1229–1243 in stem-helix region targeted by neutralizing antibody 28D9. B A complete list of all the World Health Organization (WHO) designated Variants of Concern (VOC) and the Variants of Interest (VOI) as well as the “original variant” D614G, and the mutations each variant carries in each of the major domains. All amino acids are noted by their single letter designation. NC not categorized. Deletions are noted by Δ followed by the deleted amino acids. aD614G was found in many sequences very soon after sequencing efforts began in early 2020; b the VOIs Epsilon, Theta, Eta, Kappa, Iota, and Zeta, were declassified as VOIs, so those names are provided parenthetically; cmutant positions in parentheses (e.g., (S13I)) indicate mutations that are only sometimes associated with the variant listed. Data are from the WHO [31] and US Centers for Disease Control (US CDC) [32]. Data for Omicron are from the US CDC [33]
VOC Alpha (B.1.1.7)
Alpha was initially detected in Kent, England on 20 September 2020 and, with a reproductive number (R0, “R naught”; described in OSM section S2.3) of 4.0, which is about double that of the Wuhan SARS-CoV-2 virus [34], quickly spread across the UK [35]. It was designated as a VOC in December 2020 [36], about the same time it was first observed in the USA. Alpha replicates in cells around four to ten times faster than wild-type virus (WA-1/2020) [37]. As a result, the Alpha SARS-CoV-2 variant rapidly spread and represented 70% of USA cases by 8 May 2020 [33], before declining to its currently level of virtually undetectable US cases. Alpha generally was associated with more severe disease, including increased mortality, than other variants in multiple studies [38].
Alpha has the following mutations in the spike protein: Δ69-70 and Δ144-145 in the N-terminal domain (NTD), E484K*, S494P*, and N501Y in the receptor binding domain (RBD), A570D, D614G, P681H, and T716I in the S1/S2 region, and S982A, D1118H, and K1191N* in the S2 domain (Fig. 2B). Here and throughout this section, the asterisk indicates mutations that are only sometimes present in the variant. Recent studies have shown that the deletion of residues H69-V70 have arisen multiple times in the SARS epidemic [39]. The Δ69-70 spike confers increased ease of S1/S2 cleavage, spike incorporation and fusion, rapid syncytium formation, resulting in enhanced infectivity [39]. As noted above, N501Y dramatically increases the affinity of the RBD to human ACE2, and for the sub-population of Alpha variants that possess E484K as well, the affinity is even further enhanced [40]. Additionally, the combination of A570D, D614G, and S982A are thought to enhance cleavage into S1 and S2 [37] and, as noted above, P681H in the furin cleavage site is expected to enhance cleavage of spike into S1 and S2 over wild-type virus.
VOC Beta (B.1.351)
The Beta variant was initially detected in South Africa in May 2020, and was subsequently identified in the USA late in January 2021. Along with Alpha, Beta was designated a VOC in December 2020. The B.1.351 variant has mutations in the spike protein including L18F*, D80A, D215G, Δ241-243 and R246I* in the NTD, K417N, E484K, and N501Y in the RBD, and D614 and A701V in the S1/S2 region (Fig. 2B). In this case, the approximately 15-fold improved affinity contributed by the mutations N501Y/E484K is significantly offset by the decrease in binding due to K417N, resulting in Beta RBD having an approximately threefold higher affinity to ACE2 than wild-type RBD [40]. Beta has an R0 of 3.8, significantly higher than the Wuhan SARS-CoV-2 virus or the seasonal influenza virus [33], and has a moderate level of immune evasiveness due to the mutations, particularly E484K [41, 42].
VOC Gamma (P.1, B.1.1.28.1)
The Gamma variant was initially identified in travelers from Brazil, who were tested during routine screening at an airport in Japan, in November 2020. This variant was subsequently detected in the USA in January 2021. The Gamma variant contains L18F, T20N, P26S, D138Y, and R190S mutations in NTD, K417T, E484K, and N501Y in the RBD, and D614G and H655Y in the S1/S2 region, and T1027I and V1176F* in the S2 domain of the spike protein (Fig. 2). Similar to Beta, the enhanced affinity conferred by N501Y/E484K is substantially counterbalanced by the K417T mutation, resulting in a combined 5.5-fold higher affinity for Gamma RBD to ACE2 than wildtype RBD [40]. Gamma has an R0 of 5.0, more than twice the transmissibility of the Wuhan SARS-CoV-2 virus [43], and has been associated with reduced neutralization by mAbs, or plasma from convalescent patients or from vaccinated individuals [44].
VOC Delta (B.1.617.2)
The Delta variant was initially identified in India in October 2020, and subsequently detected in the USA in March 2021. Due to its very high transmissibility rate, Delta quickly became the predominant SARS-CoV-2 virus worldwide. In the USA, as of 6 November 2021, the Delta variant made up 99.9% of all sequenced variants [32], although it has since been completely supplanted by the Omicron variant (see next section). The Delta variant contains the following spike mutations: T19R, G142D*, D156-157, R158G in the NTD, L452R, T478K, and E484Q in the RBD, and D614G and P681R in the S1/S2 region, and D950N in the S2 domain. The L452R and T478K mutations in the Delta RBD are thought to be important for stabilizing the RBD:ACE2 complex [45, 46], while L452R/E484Q combined to increase the affinity of the Delta RBD more than fivefold over wild-type RBD [47]. Additionally, as noted in OSM section S2, the P681R mutation in the furin cleavage site enhances the cleavage of full-length spike to S1 and S2 [48], which increases the transmissibility and fitness of Delta over wild-type virus, as well as Alpha, which has the less effective mutation P681H [33, 49].
In August 2021, the United States Centers for Disease Control (US-CDC) announced that they estimated R0 for the Delta variant to be 8.5 [50], three- to fourfold over the R0 value of the Wuhan virus, which provides one explanation why it became the dominant variant in the USA and the world so quickly. In fact, it has been estimated that the SARS-CoV-2 Delta variant is nearly as contagious as chickenpox, but not as transmissible as measles [50]. This played out in the real world as the Delta variant increased from 1.3 to 94.4% incidence in the USA during just a 3-month period (2 May to 31 July 2021), supplanting the Alpha variant, which decreased over the same period from 70 to 2.4% [33].
A recent study has demonstrated how Delta achieved its very high person-to-person transmission rate, which is due to the rapid increase in viral load, particularly in the upper respiratory tract [51]. Before Delta, infected individuals took an average of 5.5 days after initial infection to test positive for virus by the polymerase chain reaction (PCR) viral RNA test, but developed symptoms by an average of 6.3 days after infection, leaving very little time (0.8 day) to shed virus while asymptomatic [52]. With the Delta variant, however, patients tested RNA-positive within an average of four days after infection and developed a high nasal load of virus before symptoms emerged at an average of 5.8 days [52], leaving an average of 1.8 days to shed virus while asymptomatic [53]. This more rapid viral replication was shown to lead to more than 1000-fold increased Delta virus titer in nasopharyngeal swabs compared with the initial SARS-CoV-2 [51]. Importantly, the nasopharyngeal viral load for Delta appears similar for vaccinated and unvaccinated individuals, as well as asymptomatic individuals irrespective of vaccination status [54, 55], which explains why vaccinated and/or asymptomatic individuals may still infect others at high R0 numbers.
Additionally, B.1.617.1, a variant closely related to the Delta variant, was shown to be more pathogenic in hamsters than the B.1 variant [56]. Whether that translates to humans is still not certain, but data also suggest that some of the VOCs may have a more significant disease impact on individuals. The two-dose mRNA-based vaccines have been shown to provide about 88% protection against the Delta variant [57]. Nevertheless, a recent publication demonstrated that the Delta variant was sixfold and eightfold less sensitive to antibodies induced by vaccination and previous infection, respectively [58]. This resistance to antibody neutralization appears to be primarily focused on mutated RBD residues E484Q, T478K, and L452R [48], but also effects both NTD targeted antibodies, which, when combined, likely explains the higher breakthrough rates associated with Delta [58]. The issue going forward, however, is the percentage of population that is fully vaccinated, which in some US states and some countries is below 50%. The burden then shifts to therapeutic treatment options, of which therapeutic antibodies represent a significant option.
In July 2021, a variation of Delta now known as AY.4.2 was discovered in the United Kingdom. This variant increased to 11–12% in populations previously saturated with Delta [59], suggesting that the additional mutations it possesses (Y145H, A222V) may improve its transmissibility over that of “normal” Delta. Mutant Y145H has been implicated as a spike trimer destabilizing mutation, interestingly, by itself to lower interaction of RBD with ACE2 [60]. The combination of mutations, however, may increase interaction of variant AY.4.2 with ACE2, allowing it to outpace Delta in some populations [58]. Both Delta and its subvariant, AY.4.2, have now become entirely supplanted by Omicron, as described below.
VOC Omicron BA.1 (B.1.1.529)
Omicron was first detected in a sample taken on 8 November 2021, in Botswana, and was identified as a new variant in South Africa in late November 2021 [61] (Fig. 2B). It was given the PANGO lineage designation B.1.1.529 on 24 November 2021 and then designated as a VOC on 26 November 2021. The Omicron variant is apparently descended from the original B.1.1. lineage rather than evolving from any of the major variants such as Alpha, Beta, Gamma or Delta. It has a very long branch [62], suggesting that it had been evolving undetected for perhaps up to a year in countries with poor surveillance. Interestingly, phylogenetic analysis shows that Omicron is most closely related to Gamma and Alpha [63, 64], with Delta as the phylogenetic outlier [63, 64].
After Omicron was discovered, it was eventually split into three major subtypes, designated as BA.1 (Pango B.1.1.529.1), BA.2 (Pango B.1.1.529.2), and BA.3 (Pango B.1.1.529.3) [65–67]. BA.1 is the variant widely recognized as “Omicron.” An additional subvariant, named BA.1.1, is BA.1 with the additional mutation of R346K [66, 67]. BA.2, which was initially expected to die out, has emerged as a significant variant on its own, as described in the next section. Importantly, BA.1 and BA.2 are antigenically distinct from all other variants, and are antigenically distinct from each other [68]. BA.3, which so far is a minor variant in terms of numbers of total cases worldwide, has a mutation profile that is more of a combination of mutations in BA.1 and BA.2 than having its own unique profile [66, 67, 69].
The Omicron variants are the most transmissible SARS-CoV-2 variants to date [67, 70]. The initial estimates of transmissibility of Omicron BA.1 suggest an intrinsic R0 number for BA.1 of approximately twofold greater than Delta [71], putting the Omicron R0 in the range of 7–14, which would make it approximately equivalent to mumps (R0 of 7) as the second most transmissible virus ever known, behind measles R0 of 12–18 [72]. Additionally, other analyses suggest that Omicron spread across the population at a rate three- to fivefold greater than Delta [71–74]. Part of the discrepancy between intrinsic and observed spread rates for Omicron versus Delta may come from Omicron’s superior ability to escape antibodies from vaccination or previous infection [71, 73, 74]. As an example of the transmissibility of Omicron in South Africa, 80% of all SARS-COV-2 samples sequenced in October 2021, were Delta, but by November 2021, Delta was found in only 22% of sequenced samples whereas Omicron was found in 75%—a marked change in just 1 month [75]. Similarly, the US-CDC data indicated that as of 26 February 2022, the Omicron family of variants makes up virtually 100% of all COVID-19 cases in the USA, BA.1 and BA.2 comprising 91.8% and 8.3% of the cases, respectively [76]. Thus, between 1 December 2021, when the first case of Omicron was detected in the USA, to the last week of January 2022, i.e., less than 2 months, Omicron went from first detection to 100% of cases, completely replacing Delta [76]. Unfortunately, the BA.2 data represent a doubling of BA.2 in one reporting week (2/19/22 to 2/26/22), a potential harbinger for a next wave dominated by BA.2.
As shown in Fig. 2B, Omicron has four mutations, three deletions, and an insert in the NTD, 15 mutations in the RBD, five mutations in the S1/S2 region, and six mutations in the S2 domain [77], making it one of the most heavily mutated variants thus far observed. The combination of RBD mutations found in Omicron is intriguing (Fig. S2B). While the K417N mutation typically lowers affinity to ACE2, it has been shown to be offset (as noted above and in the OSM) by N501Y. The Q498R mutation in the Omicron RBD is unique amongst VOCs and VOIs (Fig. 2). In an in vitro phage display-based evolution study of SARS-CoV-2 mutations, Zahradnik et al. [78] found that the combination of Q498R and N501Y resulted in an “epistatic” effect, yielding the highest affinity for ACE2 amongst the evolved mutations found. Moreover, they found that mutants S477N, Q498R, and N501Y, all three of which are in Omicron, formed new contacts with ACE2 [78]. As such, Zahradnik et al. [78], with no fore-knowledge of Omicron, predicted that Q498R, which had not been observed previously in VOI or VOC variants, would eventually emerge as a partner to the N501Y and E484K (in Omicron it is E484A) mutations.
It appears that despite the N501Y, Q493K/R, and T478K mutations, Omicron BA.1 has an affinity for human ACE2 that is lower than other variants such as Alpha, Beta, and Gamma [79], and approximately in the range of 24-30 nM, similar to that of ancestral SARS-CoV-2 [79–81]. Moreover, the mutations in Omicron BA.1 have provided it with the ability to bind ACE2 from additional species, giving it a broader species tropism including mice, rats, and domestic poultry, which is potentially concerning as reservoirs for future infections [82]. The spike of Omicron BA.1 is found exclusively in the one RBD-up (or open) conformation [83]. Additionally, the RBDs were shown to have modified local conformations resulting in significant remodeling of the ACE2 binding domain, which helps to explain why it evades antibody binding so well [83–85].
To infect cells, SARS-CoV-2 fuses with host cell membranes via one of two mechanisms, a cell surface-based fusion, largely mediated by the protease TMPRSS2, and an endosomal fusion process, in which fusion only takes place after pinocytosis and formation of an endosome containing virus [82, 86]. This latter process is mediated by endosomal cathepsin, which cleaves S2 and allow for maturation of the viral entry fusogenic mechanism [82, 86]. SARS-CoV-2 variant Delta largely prefers the cell surface fusion mode of entry and Delta entry kinetics and cell tropism are largely correlated to TMPRSS2 expression by the target cells [86]. Cell types that strongly express TMPRSS2, such as lung, alveolar, and gut epithelial cells [82, 86–88], favor Delta entry [86, 89]. On the other hand, Omicron has mutations in the S1/S2 furin cleavage site region that impair its ability to use TMPRSS2, and lung and alveolar epithelial cells that strongly express TMPRSS2 suppress, albeit not eliminate, Omicron entry and replication [85–88]. Omicron, however, strongly utilizes the cathepsin-dependent endosomal fusion pathway, which results in a modified cell tropism towards nasal airway epithelial cells which exhibit poor TMPRSS2 expression [82, 86–88, 90]. Thus, while Delta is fourfold more efficient than Omicron at using TMPRSS2 to enter cells via the cell surface entry MOA, Omicron is tenfold more efficient than Delta at utilizing the endosomal cell entry MOA [91].
An additional hallmark of Delta and other variants infection is the strong ability to form TMPRSS2 cleavage-dependent syncytia between cells, allowing for efficient cell-cell transmission [87, 92]. Omicron, on the other hand, due to its inability to use TMPRSS2 efficiently, does not form syncytia between cells, eliminating cell-cell direct transmission [87]. This modified cell tropism could explain two significant factors related to Omicron: (i) significantly increased transmissibility over other VOCs, likely driven by increased replication in the upper respiratory tract where it can easily be shed into the environment and (ii) somewhat attenuated disease, driven by poorer ability to infect lung epithelial cells and lack of cell-cell direct transmission, as compared with Delta and other variants [86, 88, 90].
In a separate approach to compare Omicron with Delta and ancestral virus, Lamers et al. measured rate of infections and virus shedding in a 2D organoid-based air-liquid interface airway model [87]. They demonstrated a significant increase in competitive infectivity of Omicron over Delta in the first 5 days, followed thereafter by Delta becoming dominant. Since humans are most infectious within the first few days of infection, this initial competitiveness by Omicron in culture may help to explain its ability to outcompete Delta in several populations across the world [87].
Due to its significantly altered antigenicity, Omicron has demonstrated resistance against human antibodies generated as a result of infection with earlier variants [68, 93–95] as well as immunization [93–98]. This makes sense, considering that seven out of 17 SARS-CoV-2 RBD contact residues for binding ACE2 are mutated in Omicron (Fig. 2). The immune escape index, I-index, measures the predicted ability of SARS-CoV-2 variant to escape detection and neutralization by antibodies as compared with the ancestral virus [91, 92]. Thus, the escape index for ancestral virus is one (1), alpha (~ 1.2), beta (~ 2.6), gamma (~ 2.8), and delta (3.1) all are less than or around 3, and Omicron has an I-index of 5.8, a reflection of its mutational pattern, which is substantially different from the other VOCs and is both predicted and observed to make Omicron resistant to most antibodies generated by infection to previous variants as well as standard two-dose vaccination regimens. Luckily, immune evasion of primed or prime/boost vaccines, which is more significant for ChAdOx-1 than for the BioNtech/Pfizer mRNA vaccine [85], can be overcome with a third vaccine boost, which increased titers [98], potentially improved T-cell epitope activity [97–99], and/or may lead to epitope spreading, as observed with other vaccines [100], that could help to cover the antigenic drift exhibited by Omicron [85]. It should be noted, however, that a third vaccine boost, or even more effective, the combination of vaccination and previous infection [101], provided protection against Omicron BA.1 [93, 94, 98], albeit at neutralizing titer levels ranging six- to 23-fold lower than anti-Delta titers [102].
Thus, while Omicron BA.1 has demonstrated reduced virulence compared with Delta and other SARS-CoV-2 variants in both rodents [103, 104] and the human population [88, 90, 105, 106], it has a significantly increased ability over other VOCs including Delta to spread through the population due both to its significant antibody resistance as well as increased transmission rate [70–72, 107], especially early in the infectious period [87]. As noted by Suzuki et al. [107], pathogenicity is on a linear scale with respect to increase in hospital admissions, morbidity and mortality, whereas Omicron population spread rate is exponential with respect to those outcomes. Additionally, with increased species tropism, the potential for non-human reservoirs is potentially increased, which could broaden the ability of omicron-like CoVs to re-enter and spread in humans. Thus, as recently summarized by Bhattacharyya and Hanage [108], the intrinsic severity of Omicron infection to the world population as a whole remains significantly high.
A subvariant of Omicron BA.1, called BA.1.1 (Pango B.1.1.529.1.1), is BA.1 plus the R346K mutation [67, 68], which by February 2022, comprised about 30% of that total “BA.1+BA.1.1” infections globally [109]. It is thought that BA.1.1. has a slightly higher transmission rate than BA.1, and this is borne out by the current rate of Omicron infections in the USA, in which BA.1.1 subvariant comprises about 81% of all “BA.1+BA.1.1” COVID-19 cases [76].
A very recent analysis suggests that approximately 73% (range 63–81%) of Americans have antibodies against Omicron, due either to infection, vaccination and boosting, or both [110]. This number is expected to rise to the 80% range by March, although with the expectation of new variants and around 35% of the US population being vaccine-hesitant, achieving true herd immunity is unlikely [110]. This is likely also the case in other countries in which Omicron has spread widely. Nevertheless, with so many people having at least some immunity to SARS-CoV-2, it is expected that future variants may not cause as much mortality or burden on the healthcare system [103, 110].
VOC Omicron Subvariants BA.2 and BA.3
Variant BA.2 was first detected in South Africa on 17 November 2021 [61], and was designated VUI-22JAN01 (“variant under investigation”) in the UK on 19 January 2022. While BA.2 is considered a sub-lineage of Omicron, it actually has about 40 amino acid differences in sequence from Omicron BA.1 (Fig. 2B) and it is antigenically distinct, making it a very different virus than BA.1 [67, 68]. There are at least five genetic subgroups of BA.2 that have arisen in different geographical areas, suggesting continued antigenic drift within this subvariant [111].
Omicron BA.2 is sometimes referred to as the “stealth Omicron” because it lacks the D69-70 deletion found in Omicron BA.1. This short deletion causes a phenomenon during polymerase chain reaction (PCR)-based assays called S gene target failure (SGTF), which has become a signature in rapid PCR determination of BA.1 [112]. Thus, BA.2 can only be confirmed after sequencing.
BA.2 has an apparent affinity to ACE2 similar to that of BA.1 [113]. In its cell tropism, BA.2 is more like previous SARS-CoV-2 variants such as Delta in that it uses TMPRSS2-based cell entry mechanisms better than BA.1, so it has more potential for cell-cell fusion and ability to infect lung epithelial cells [113]. In the Delta variant, increased fusogenicity is correlated with S1/S2 cleavage, but that does not appear to be the case with BA.2 [113]. These properties are thought to contribute to potentially higher pathogenicity of BA.2 over BA.1 [113], although this has not been confirmed yet with real-world data.
It was recently demonstrated via surveillance and secondary infection rates in Danish households that BA.2 has a substantially higher transmission rate than Omicron BA.1 [113, 114]. Yamasoba et al. [113] calculated that the effective reproduction rate for BA.2 is 1.4-fold higher than for BA.1. Additionally, it appears that BA.2 is more resistant to antibodies generated as a result of vaccination or previous infection than is Omicron BA.1 [113, 114]. In general, two vaccine doses (prime/boost) provide approximately 10% effectiveness against BA.2-caused symptomatic COVID-19 disease, but a third dose (booster approximately 6 months later) increased the effectiveness to 70% against BA.2 [115], albeit with an approximately eightfold reduced neutralizing titers as compared with titers against ancestral virus [102].
As compared with BA.1, BA.2 has a very different set of mutations, especially in the spike protein (Fig. 2B) [67, 68], is more transmissible, uses TMPRSS2 better resulting in the higher ability to form syncytia and spread via cell-cell fusion, is more resistant to vaccine-induced antibodies, and is more pathogenic [113]. Moreover, patients infected with BA.1 were later re-infected with BA.2, indicating that antibody responses generated against BA.1 were not strongly neutralizing for BA.2 [116]. These differentiating characteristics led Yamasoba et al. [113] to propose that BA.2 be given its own Greek letter to accentuate its differences from BA.1.
As of 26 February 2022, BA.2 has now been detected in over 50 countries, including the several African countries, Denmark, UK, India, Philippines [117], and now makes up approximately 21.5% of all cases worldwide, 86% of all cases in South Africa [117], about 45% of case in Southeast Asia [117], and 8.2% of cases in the USA, up from 3.8% the previous week [76]. So far, BA.2 seems to spread in localized clusters in areas such as Denmark [111, 113, 114], where it now makes up approximately 90% of all Omicron infections [118]. What is not understood is why BA.2, which was discovered in the same time period as BA.1, took longer to establish infections in large populations, and why it has overtaken BA.1 only in isolated circumstances such as Denmark, South Africa, and Southeast Asia [111, 113, 114, 117, 118]. Chen and Wei [116] make a strong case that BA.2 may yet be the next dominating variant, and the recent doubling of BA.2 in the US population [76] is worrying along those lines.
As noted previously, BA.3 is comprised of mutations found in BA.1 and BA.2. BA.3 has 33 mutations identical to those found in BA.1, but lacks six key BA.1 mutations (ins214EPE, S371L, G496S, T547K, N856K, and L981F) as well as picking up two mutations (S371F, D405N) from BA.2 [69]. Thus far, BA.3 does not appear to be above background in any population. It has been possible that the specific combination of mutations from BA.1 and BA.2 make it less fit than either of those variants [69].
Convalescent Patient and Polyclonal Therapeutic Approaches
As noted in OSM Section S4, convalescent patients recovering from infection with SARS-CoV-2 can mount a neutralizing antibody response to the virus. The use of convalescent plasma takes advantage of that response in an effort to try to help newly diagnosed patients sick with COVID-19. In the early days of the COVID-19 pandemic, there were no proven treatments and no available vaccines for protection against SARS-CoV-2 or the ramifications of the immune and physiological response to the virus. Thus, doctors turned to some of the oldest forms of immunoglobulin-based treatment available, such as plasma from convalescent patients [119], purified F(Ab′)2 fractions of sera from immunized horses [120–123], general (non-immune) [124, 125] and specific (hyperimmune) [125, 126] IVIg approaches, therapeutic plasma exchange [127], as well as a variety of other polyclonal approaches (Table 1). These treatments helped to bridge the gap until more directed and more potent therapies became available. Additionally, in countries or geographic areas in which advanced therapies are still not available, these polyclonal approaches continue to be used in efforts to save lives.
Table 1.
Clinical studies on pooled antibody-directed therapies against SARS-CoV-2
| Format (and example candidates) | Antibody source | Sponsor(s) | ROA | Number of clinical studiesa | Most advanced stage | Example NCTsb |
|---|---|---|---|---|---|---|
| Convalescent plasma | Convalescent patients | Multiple hospitals and medical facilities | IV | 192a | Phase III; US-FDA EUA 8/23/20 | NCT04361253 and many others |
| Specific (hyperimmune) IVIg | Convalescent patients | Green Cross Corp., Emergent Biosolutions, others | IV, SC | 21 | Phase III | NCT04555148 |
| Normal IVIg (e.g., Gamunex-C) | Pooled human donors | Grifols Therapeutics, Emergent BioSolutions, others | IV, SC, IM | 11 | Phase III | NCT04480424, NCT04561115 |
| Hyperimmune equine serac (e.g., INM005) | Immunized horses | Immunova SA; Hospital San Jose Tec de Monterrey; Bharat Serums and Vaccines Ltd; others | IV | 9 | Phase II/III | NCT04514302, NCT04494984, NCT04834908 |
| Non-specific plasma exchange | Pooled non-specific human donors | Multiple hospitals | IV | 6 | NA | NCT04751643 |
| Recombinant purified human IgGs from immunized cows (SAB-185) | Immunized tg cows | SAB Biotherapeutics | IV | 3 | Phase II/III | NCT04468958. NCT04469179, NCT04518410 |
| Bovine IgG (EnteraGam®) | Cows | Entera Health, Inc | Oral | 2 | NA | NCT04682041 |
| Glycoengineered hyperimmune porcine antibodies (XAV-19) | Immunized pigs | Xenothera SAS | IV | 2 | Phase II/III | NCT04453384, NCT04928430 |
| Polyclonal antibody preparation containing IgG (56%), IgA (21%), and IgM (23%) (Trimodulin; BT-588) | Purified from pooled plasma obtained from donors | Biotest AG | IV | 1 | Phase II | NCT04576728 |
| Polyclonal recombinant hyperimmune IgG (GIGA-2050) | Pooled recombinant IgGs | Gigagen | IV | 1 | Phase I | NCT04883138 |
| Polyclonal hyperimmune IgY | Immunized chickens | Stanford University | IN | 1 | Phase I | NCT04567810 |
| Hyperimmune antibodies | Colostrum from immunized cows | Icosagen Cell Factory | IN | 1 | Phase I | NCT04916574 |
EUA emergency use authorization (USA), Ig immunoglobulin, IM intramuscular, IN intranasal, IV intravenous (administration), IVIg intravenous immunoglobulin, NA not applicable (approved for other indication), NCT National Clinical Trial, ROA route of administration, SC subcutaneous (administration), tg transgenic, US-FDA United States Food and Drug Administration
aAt least this number of separate studies registered with NCT; studies registered with EudraCT (European Union Drug Regulating Authorities Clinical Trials Database; ANZCTR (Australian New Zealand Clinical Trials Registry), ChiCTR (Chinese Clinical Trial Registry), or other registries not included. Based on public data available by 9/1/21
bNCT registries can be found using reference [7]
cEither Fab or F(Ab′)2 fragments isolated from the serum of immunized horses
Convalescent Plasma Therapy
Convalescent plasma therapy (CPT) has been used for over a century as a therapeutic tool to treat patients infected with various viruses. While the origins of CPT have been disputed, the current thinking is that Cenci, during the 1901 measles outbreak in Italy, was the first to practice it [128]. Cenci used the blood of a patient who had recovered from measles to successfully protect four children from measles, even as their uninoculated cohabitating siblings became ill [128]. Since then, CPT has been used countless times as a first line of therapy against epidemic and pandemic virus outbreaks including, notably, the Spanish influenza epidemic of 1916-1918 [129], SARS in 2004 [130, 131], influenza A H1N1 pandemic of 2009 [132], Ebola in 2014 [133, 134], MERS in 2015 [135] and, most recently, COVID-19 [136–145].
CPT involves the extraction of plasma containing anti-viral antibodies from patients who have recovered (i.e., convalescent patients), followed by transfusion of the collected plasma into new patients suffering with the same disease. While blood typing to decrease the incidence of mismatched plasma was not practiced in the earliest examples of CPT, it has now long been the practice to match ABO blood types to ensure compatibility with the donated plasma [146].
The apparent first documented use of convalescent plasma therapy to treat COVID-19 was in China as early as February 2020 [147]. With the many precedents for using convalescent plasma to treat viral infectious diseases, the use of plasma from COVID-19-surviving convalescent patients to treat severe disease was quickly tested in clinical trials registered with Clinicaltrials.gov. Early on in the pandemic, the use of CPT was strongly encouraged for both prophylaxis from and treatment of COVID-19 [148], in part because no other good options were available at the time. By 20 July 2021, about 190 different clinical trials using convalescent plasma therapy have been registered with Clinicaltrials.gov. Based on the “totality of evidence” in early clinical trials, the FDA issued an EUA for the use of convalescent plasma for treatment of COVID-19 on 23 August 2020 [149]. According to the FDA, the use of convalescent plasma on patients with COVID-19 decreased the mortality rate in hospitalized patients by 37% (p = 0.03) [150]. By that time, approximately 8 months into the pandemic in the USA, more than 70,000 Americans had been treated with convalescent therapy. The World Health Organization followed shortly thereafter, on 25 August 2020, with their version of an EUA for the use of convalescent plasma therapy to treat COVID-19. The current guidelines are that COVID-19 patients who are not hospitalized may be considered for plasma therapy whereas those who are hospitalized should not receive it.
Since those EUAs were issued, several studies have been carried out at a wide variety of different clinical sites to confirm whether or not CPT would prove beneficial to the patients receiving it. Unfortunately, while certain, typically smaller studies showed at least some benefit to the use of CPT for treatment of COVID-19 [137, 139, 140], other more extensive studies often demonstrated no clear clinical benefit of COVID-19 treatment with CPT [138, 141, 142]. One such study was the Phase 3 Inpatient Treatment with Anti-Coronavirus Immunoglobulin (ITAC) clinical trial from the CoVIg-19 Plasma Alliance, formed in April 2020, by Takeda to help treat hospitalized patients who had very little other options at the time [145]. The trial ended up being halted early due to futility [151].
Successful CPT treatment of COVID-19 patients requires multiple factors to be in place, including adequate plasma titer, treatment timing, patient status, and desired endpoints [145]. One of the issues with CPT is that there is less control over the level and quality of the antibodies in serum therapy than with purified antibodies. It has been documented that high titers of IgGs in convalescent plasma used for CPT, such as 1:640 or higher, are required to see clinical benefit as compared with lower titers [137], especially if the patients have additional comorbidities or are immunocompromised [152]. In one study, high serum levels of IgGs (over 18.45) in patients treated with CPT correlated with improved clinical outcomes, including lower numbers of deaths in the study [140]. On the other hand, a recent study showed that CPT with donor plasma titers averaging 1:641 was no better than placebo in preventing patients from progressing to more severe disease or preventing the need for hospitalization [144]. To help standardize the plasma as part of the EUA covering use of CPT for COVID-19, the FDA has provided guidelines as to what constitutes high titer plasma, i.e., “neutralizing antibody titer of ≥ 250 in the Broad Institute’s neutralizing antibody assay” or cutoffs in other similar assays, and has provided guidance for testing [145, 153].
The second major factor is timing [137, 145]. In early CPT trials, there was a general lack of understanding about how critical timing and dose of CPT administration was to the success of the therapeutic approach, so too little focus was placed on administration of high titer CPT as soon as possible after infection. While this, in principle, sounds like a reasonable concept, a well-controlled, multicenter randomized trial that enrolled over 500 patients very recently demonstrated no clear benefit to early administration of CPT [142]. Similarly, a retrospective analysis of over a dozen randomized SARS-CoV-2 CPT trials have demonstrated little to no benefit for patients with mild to severe disease [145]. With all that said, a separate retrospective analysis determined that the mortality rates due to COVID-19 in hospitals that used convalescent plasma therapy to treat COVID-19 were significantly lower than in those who did not use that approach [143]. The authors went on to suggest that more aggressive use of CPT to treat COVID-19 could have saved as many as 29,000 lives in the USA [143].
As it became more obvious over the course of the pandemic that high titer and early administration were key factors for success, more trials resulted in statistically significant benefits to patients. In a recent example published in December 2021, a double-blinded randomized trial of 1225 outpatient subjects (NCT04373460), early administration of high titer (> 1:320) CPT showed a clear and statistically significant 54% risk reduction benefit over placebo [154]. In another study, a randomized control trial of sero-negative but hospitalized patients demonstrated a significant benefit to 28-day mortality [155]. Other similar trials often did not meet clinical endpoints, but at least in some cases did provide benefit in terms of limiting progression to ventilation and death [156], or overall survival [157, 158].
The quality and quantity of these factors other than IgG neutralizing titers are not typically used to qualify potential convalescent plasma. This could significantly underplay the potential of CPT, as it has been demonstrated, for example that both neutralizing IgM [159] and IgA [160] titers in convalescent plasma were correlated with better outcomes in COVID-19 patients treated with CPT. CPT has the potential benefits of not only direct neutralization of virus binding to receptor, but also immunological activity of the various antibody isotypes, including ADCC (primarily IgG1 and IgG3 isotypes), antibody dependent cellular phagocytosis (ADCP; all IgG isotypes); complement mediated cytotoxicity (CDC; IgG1, IgG2, IgG3, and IgM isotypes) [161]. In another study, Bégin et al. [141] demonstrated that the level of ADCC induced by IgGs in plasma was correlated directly with outcomes. Other potential factors with the quality of convalescent plasma used to treat COVID-19 that have not been fully analyzed are titers of neutralizing IgM [159] and IgA [160] isotypes, and levels in the convalescent plasma of other potentially protective factors, such as IL-1β, IL-2, IL-6, IL-8, IL-17, CCL2, and TNF-α [162].
While there appear to be potential benefits of using CPT in certain settings, for example when other more specific therapeutics are not readily available or for immunocompromised patients, there are potential downsides and limitations to its use. One of the significant limitations of CPT is the source of the convalescent plasma versus the SARS-CoV-2 variant infecting the patient to be treated. Unfortunately, SARS-CoV-2 has mutated significantly as hundreds of variants have been discovered and sequenced, not even to mention the variants that likely exist that have not yet been analyzed. As noted above, some of those variants (Fig. 2) may be very significant with respect to resisting treatments. It has been demonstrated already, for example, that convalescent plasma from wild-type infections is significantly less effective against variants possessing the D614G mutation [163]. Moreover, variants carrying the now ubiquitous E484K mutation (including all Beta, Gamma, Eta, P.2, P.3, Mu, and C.1.2 variants, as well as some Alpha and Iota variants; see Fig. 2) have significantly increased resistance (typically three- to fivefold, but not entirely resistant) to convalescent plasma derived from patients harboring SARS-CoV-2 lacking the E484K mutation [42, 164–168]. Additional mutations such as N440K, V483A, F490S, Q493R/K, and N501T also have been shown to contribute to immune evasion of CPT [169]. With these factors in mind, note that the quality of convalescent plasma with respect to new variants will change with the infected source, i.e., convalescent patients. For example, convalescent plasma from patients recovered from SARS-CoV-2 Beta variant infections have been demonstrated to protect against a broader set of variants (e.g., Delta, Omicron) [170] than plasma from patients recovered from infection with the ancestral strain. Also, as mentioned above, CPT has not worked well with the Omicron variants due to their inherent ability to evade antibodies in plasma derived from patients infected with previous variants [93–96].
An additional significant factor in the variants is the antigenic hotspot “supersite” in the NTD that is mutated away (deleted and/or mutated) in many of the VOCs and VOIs [171, 172]. Many of the SARS-CoV-2 neutralizing antibodies generated by patients during infection are focused on the supersite; the various deletions found in the NTDs of VOCs and VOIs can severely dampen the effects of those neutralizing antibodies [153, 154]. For example, deletions in the NTD such as ΔHV69-70 (Alpha, Eta, Omicron variants), ΔLGVY141-144 (Eta, Theta, Omicron variants) and ΔAL243-244 (Beta variant) (see Fig. 2), have contributed to immune evasion of CPT [169]. Perhaps even more insidious is that fact that it appears as if CPT can actually induce the NTD supersite escape mutations [164, 173].
In terms of other potential limitations and risks for use of CPT, in at least a few rare cases, the use of COVID-19 convalescent plasma was linked to a diagnosis of transfusion-related acute lung injury (TRALI) [174]. Finally, there is always the risk of transfusion-related infection [174]. Thus, while CPT was a great “band-aid” early on in the pandemic that clearly contributed to saved lives [143], its use in Western countries now is largely eclipsed by vaccine prophylaxis and therapeutic monoclonal antibodies (mAbs), both of which, with the exception of bamlanivimab used as a single agent, have fared reasonably well against the wide variety of variants that have sprung up since the pandemic started. The use of convalescent plasma therapy, however, continues to be of great value in regions and countries in which therapeutic mAbs are not widely available [166], or for elderly or immunocompromised patients still early in disease where other treatments are not readily available [175].
General and Specific Intravenous Immunoglobulin (IVIg)
Intravenous immunoglobulin (IVIg) comes in two flavors, general (i.e., non-specific) and specific [176]. General IVIg is typically a preparation purified from pooled serum from as many as 40 or more individuals who have not necessarily been vaccinated against a particular antigen of interest. General IVIg in various formats and preparations has been approved for over a dozen indications, mostly in the form of anti-inflammatory therapy [176]. As of 20 July 2021, there were approximately ten clinical trials registered with Clinicaltrials.gov for normal IVIg therapy of COVID-19. The use of normal IVIg therapy is intended as a broader anti-inflammatory treatment than the use of specific IVIg, and would fall under the general category of palliative therapy, similar to the use of specific mAbs against pro-inflammatory cytokines such as interleukin-6 and its receptor (IL-6, IL-6R) and/or granulocyte macrophage colony-stimulating factor (GM-CSF) (see Sect. 9).
So far, the results for use of general IVIg as an adjunct therapy for COVID-19 have been mixed, with some studies showing at least some clinical benefit [9, 177], particularly in decreasing the rate of patients progressing to mechanical ventilation [178], while others demonstrated no additional clinical benefit of using non-specific IVIg over standard of care [179]. Moreover, a very recent meta-study analyzing over 2400 patients in ten studies (for randomized, controlled; six non randomized) showed no statistically significant advantage of high dose IVIg in COVID-19 patients [180]. Trimodulin (BT-588), a polyclonal antibody from non-hyperimmune donors, is a preparation containing IgG (~ 56%), IgA (~ 21%), and IgM (~ 23%) that also is being tested as a treatment for COVI-19, although no results from this trial (NCT04576728) are yet available (Table 1). One aspect of normal IVIg therapy that will be constantly changing is the quality of the plasma donated; as more donors are vaccinated or have been exposed to SARS-CoV-2 and its variants, even normal IVIg will contain anti-SARS-CoV-2 IgGs. Currently, non-specific IVIg is not recommended for use as adjunctive therapy for COVID-19.
Specific IVIg, sometimes referred to as “hyperimmune” IVIg, is immunoglobulin purified from vaccinated subjects, or from convalescent patients, to provide protection against a specific pathogen or disease-causing antigen [10, 11, 176, 181–183]. As of 20 July 2021, there were at least 21 clinical trials registered with Clinicaltrials.gov using specific (“hyperimmune”) IVIg sourced from convalescent patients. This approach is similar to convalescent plasma therapy as noted above, with the exception that the immunoglobulin fraction has been purified away from other plasma proteins and concentrated [182]. While it is still too early to make critical assessments of the success, or lack thereof, for the use of hyperimmune IVIg to treat COVID-19 patients, there have been small studies showing positive results such as improved chest X-rays, significant improvement in lung function, and earlier discharge from hospital, and above standard of care [164]. Mechanistically, hyperimmune globulin has been demonstrated not only to block virus binding to ACE2, but also to kill SARS-CoV-2 by both ADCC and antibody-dependent cellular phagocytosis (ADCP) [180]. Nevertheless, similar to CPT, hyperimmune IVIg has limitations for use with the SARS-CoV-2 variants [183]. It was demonstrated that hyperimmune IVIg bound well to K417N mutant virus, moderately to N501Y mutant virus, but poorly to E484K mutant virus, the latter similar to CPT [167, 183]. Also, just like CPT, the source(s) of the IVIg, whether it be from vaccinated individuals or convalescing patients, would have an impact on the ability to bind and neutralize variants, especially when new variants such as Omicron come along that are highly resistant to most antibodies from vaccinees or convalescent patients, as noted previously. A recent study showed that with proper screening for relevant donors and using only high titer (i.e., > 1:320) preparations, that hyperimmune IVIg could be beneficial for use in the pre-exposure prophylaxis and treatment of post-exposure/seronegative patient groups, even in areas in which Delta or Omicron (BA.1) variants are prevalent [184].
Polyclonal IgG Approaches
Besides normal and specific human IVIg approaches, several other polyclonal approaches to therapy for COVID-19 have been attempted, including pooled equine antibodies from immunized horses, polyclonal IgY antibodies from immunized chickens [185, 186], glyco-engineered polyclonal antibodies from immunized swine [187–189], orally-administered hyper-immune bovine IgG (NCT04682041 [7]) [190], pooled human IgG from immunized transgenic cows [191], pooled polyvalent mixture containing IgG, IgA, and IgM [192, 193], and pooled recombinant human IgGs [194, 195] (Table 1).
One of the oldest polyclonal approaches dating back to the late nineteenth century, known as hyperimmune equine serum therapy, has also been used in efforts to treat COVID-19. For this approach, IgG from horses immunized with SARS-CoV-2 spike protein were collected, proteolytically cleaved to F(Ab′)2 fragments to reduce immunogenicity as well as to minimize potential adverse effects (such as antibody dependent enhancement, or ADE [196] (see also Sect. 4.3.2), and used to treat COVID-19 patients [5–7]. There are currently at least nine clinical trials testing hyperimmune equine F(Ab′)2 or FAb fragments, some of which are at the Phase II/III stage (Table 1). In one set of studies, hyperimmune equine sera targeting the spike protein were compared to hyperimmune sera immunized against a mixture of nuclear (N), envelope (E), and membrane (M) proteins (NCT04494984 [7]). Results from that study indicated that anti-spike equine hyperimmune sera were superior to the combined NEM sera, leading to a Phase II/III trial (NCT04838821 [7]) specifically to evaluate hyperimmune equine sera for treatment of severe COVID-19 disease. In a separate study, a preliminary readout of Phase II/III clinical data (NCT04494984 [7]) indicated that there was a beneficial effect based on the use of RBD-specific equine polyclonal F(Ab′)2 fragments, including an overall reduction in mortality to 6.9% (treated) from 11.4% (placebo) [122]. Additionally, these constructs appear to be safe in humans [122].
Another polyclonal anti-SARS-CoV-2 product of interest is XAV-19 (Table 1), from Xenothera, which is a heterologous glyco-humanized, polyclonal antibody from cytidine monophosphate-N-acetylneuraminic acid hydroxylase (CMAH) and α1,3-galactosyl-transferase (GGTA1)-double knockout swine immunized with SARS-CoV-2 spike protein [189, 190]. The glyco-humanization is required since swine produce proteins containing the N-glycolyl form of the neuraminic acid (Neu5Gc) and α-1,3-galactose, which typically trigger xenogeneic antibody responses in humans [178]. These polyclonal antibodies, which were found to be effective against the Alpha (B.1.1.7, UK) and Beta (B.1.351, South Africa) variants even though the swine were immunized with the “wildtype” Wuhan-D614G spike protein [190], are currently being evaluated in the POLYCOR Phase II clinical trial [191]. Recently, it was demonstrated that XAV-19 preparations were able to neutralize the Omicron BA.1 variant, potentially making this approach more attractive [197].
Perhaps the most advanced of the animal-derived anti-SARS-CoV-2 IgG pools is the product called SAB-185 (Table 1), from SAb Biotherapeutics, a polyclonal mixture of human antibodies to SARS-CoV-2 administered IV which is currently being tested clinically in the ACTIV-2 Phase II/III clinical trials (NCT04518410 [7]) along with several other anti-SARS-CoV-2 antibody product candidates. SAB-185 is purified human IgG mixture from transgenic (tg) cows [198, 199] immunized with plasmid DNA encoding SARS-CoV-2 spike protein, followed by booster immunizations with spike protein generated by insect cells. Prior to the COVID-19 pandemic, SAb Biotherapeutics had also evaluated SAB-301, a polyclonal mixture of human IgGs targeting middle east respiratory (MERS) virus, in Phase I clinical trials (NCT02788188 [7]) [193]. The potential upside of tg cattle-produced human IgGs is supply, consistency across lots, and the ability to vaccinate the cows with antigens not available for human vaccination due to regulatory and safety considerations. Recently, it was demonstrated that SAB-185 preparations were able to neutralize the Omicron BA.1 variant, also potentially making this approach more attractive [200].
The various polyclonal approaches described above, including human specific IVIg, IgGs, or IgG fragments from immunized cows, horses, and pigs, and other “natural” sourcing of hyperimmune antibodies come with some level of batch-to-batch variation and the potential for supply issues. A relatively new biotech company, GigaGen (recently acquired by Grifols), has generated a process for sorting and capturing high-value antibodies from B cells utilizing microfluidics and molecular genomics [194, 195]. Their anti-SARS-CoV-2 antibody product, called GIGA-2050, contains about 12,000 unique recombinant antibodies from 16 convalescent donors, selected from literally millions of antibodies sequences, that strongly and specifically bind SARS-CoV-2 spike protein. These antibodies have been site-specifically introduced into, and are produced by, Chinese Hamster Ovary (CHO) cell lines. GIGA-2050 is currently in Phase I clinical trials (NCT04883138 [7]) for treatment of COVID-19.
Anti-SARS-CoV-2 IgG Antibodies
Types of Antibodies
The full array of antibody and antibody-like structures has been employed by various groups in the efforts to develop anti-SARS-CoV-2 therapeutics, including single natural IgG isotypes, Fc-engineered IgGs both for increased Fc activity and decreased Fc activity, cocktails of multiple IgGs, IgMs, single and multiple domain antibodies, domain antibody-Fc fusions of various types, bispecific and multi-specific antibodies, and ACE2-Fc fusions (Table 2, Fig. 3). These different antibody formats each have their own strengths and weaknesses, and each offers a unique approach to neutralizing SARS-CoV-2, as will be discussed in the following sections. Several antibody-like formats have been tested as to neutralize SARS-CoV-2, the bulk of which are shown in Fig. 3. These include: (i) IgGs with intact Fc function or modified, enhanced Fc-γ receptor binding function; (ii) IgGs in which Fc function has been muted or eliminated; (iii) IgGs with Fc modifications to extend half-life and increase area under the curve (AUC); (iv) VHH or single domain antibodies, the smallest antibody formats used to date (one binding site, MW 12-15 kDa), and homologous concatemers of those; (v) single or multiple VHH molecules fused to an Fc for half-life extension; (vi) bispecific antibodies comprised of two different, non-overlapping anti-RBD domains; (vii) IgMs, the largest natural antibody structures (ten binding sites, MW 900 kDa); and (viii) ACE2-Fc fusions, either using native ACE2 or ACE2 which has been modified to improve its binding to SARS-CoV-2 RBD. Each of these molecules offers potential advantages and disadvantages in addressing SARS-CoV-2, as discussed in the following sections.
Table 2.
SARS-CoV-2 neutralizing antibodies in development
| Molecule | Sponsor | Development stage (example NCT)a | Source | Format | Dosing and comments | References |
|---|---|---|---|---|---|---|
|
Bamlanivimab (LY3819253; LY-CoV555) |
Eli Lilly/Abcellera |
US-FDA EUA 11/9/20; revoked 4/16/21 |
BC/CPs | Human IgG1κ | IV dosing; no longer in development as single agent; still used in combination with etesevimab | [203] |
|
Etesevimab (LY3832479; LY-CoV016; JS016; CB6-LALA) |
Eli Lilly/Shanghai Junshi Bioscience |
Phase I/II (studied as single agent) |
BC/CPs | Human IgG1κ | IV dosing | [204] |
|
Bamlanivimab plus Etesevimab |
Eli Lilly/Shanghai Junshi Bioscience |
US-FDA EUA 2/9/21; EMA EUA 5/3/21 |
BC/CPs | See above for each antibody | IV combination cocktail of etesevimab (1400 mg) and bamlanivimab (700 mg) | [203, 204] |
|
REGEN-COV™ (US); Ronapreve™ (UK and EU) (casirivimab plus imdevimab) |
Regeneron |
Approved by Japan 7/22/21, UK 8/26/21, EMA, 11/11/21; FDA EUA 11/20/20; NCT04452318 ANR |
CAS: immunized VLI tg mice; IMD: BC/CPs |
Casirivimab, Human IgG1κ; imdevimab, human IgG1λ | IV or SC dosing; combination cocktail; casirivimab (1200 mg) + imdevimab (1200 mg) and other doses; testing both therapeutic and prophylactic use | [205] |
|
Regkirona™ (regdanvimab) |
Celltrion |
Approved by Korea MFDS, 9/17/21 and EMA, 11/11/21; |
BC/CPs | Humanized IgG1κ | IV dose; recommended dose 40 mg/kg (2.8 g for 70 kg patient) | [206, 207] |
|
Xevudy™ (UK); sotrovimab (GSK4182136; VIR-7831) |
GSK/Vir Biologics |
Approved by UK MHRA 12/2/21; US-FDA EUA 5/26/21; EMA EUA 5/21/21 NCT04545060 ANR |
BC/CPs | Human IgG1κ LS HLE | IV and IM dosing being compared (NCT04913675); Also binds SARS-CoV-1 RBD; Derived from S309, which was isolated from SARS-CoV-1 patient B cells; neutralizes Omicron BA.1 | [208, 209] |
| EvuSheld™ (AZD7442; Tixagevimab [AZD8895, COV2-2196] plus cilgavimab [AZD1061, COV2-2130]) | AstraZeneca/Vanderbilt |
US-FDA EUA 12/8/21 Phase III R |
Both BC/CPs | Both human IgG1κ FE/YTE/S HLE and FCM | IV and IM dosing being compared; Fixed dose combination cocktail of tixagevimab (150 mg) + cilgavimab (150 mg); EUA for pre-exposure prophylaxis in immunocompromised subjects; two IM injections provide protection for up to 6 months; neutralizes Omicron BA.1 | [210, 211] |
| Amubarvimab (BRII-196, P2C-1F11) plus romlusevimab (BRII-198, P2B-1G5) | Brii Biosciences | Approved by China NMPA 12/8/21; Application for US-FDA EUA submitted; Phase III R NCT04518410 | Both BC/CPs | Human IgG1 YTE HLE, minor FCM | IV dose; Fixed dose combination cocktail of BRII-196 (1000 mg) and BRII-198 (1000 mg); testing for ambulatory patients; met Phase III clinical endpoints; EUA application submitted to US-FDA; neutralizes Omicron BA.1 | [212, 213] |
|
Bebtelovimab LY-3853113 (LY-CoV1404) |
Eli Lilly |
US-FDA EUA 2/11/22 Phase II C NCT04634409 |
BC/CPs | Human IgG1 | IV dosing; Newer antibody to address VOI, and VOC variants; neutralizes Omicron BA.1 | [214] |
| Ensovibep (MP0420) | Molecular Partners |
EUA application submitted to US-FDA (2/10/22) Phase II/III R NCT04870164 |
DARPin library | Trimeric DARPins | IV delivery; non-antibody protein-binding scaffold | [215] |
| TY027 | Tychan Pte., Ltd (Singapore) |
Phase III R |
In vitro designed and engineered | Human IgG | IV dose at 1500 mg; Reportedly effective against all VOCs, including Delta | [216] |
|
BMS-986414 (C135-LS) plus BMS-986413 (C144-LS) |
BMS/Rockefeller |
Phase II/III R |
Both BC/CPs | Both human IgG1 LS HLE | SC dosing; two injections of C135-LS 200 mg and two injections of C144-LS 200 mg for each dose | [217, 218] |
| ADG-20 (ADG-2 HLE) | Adagio Therapeutics |
Phase II/III R |
BC/CPs - YDAF | Human IgG; reported HLE | IV dosing; Affinity-matured in vitro; Testing therapeutic vs prophylactic (isolated from SARS-CoV-1 patient); Fc activities ADCP, ADCC present | [219, 220] |
| MAD0004J08 | Toscana Life Sciences Sviluppo |
Phase II/III R |
BC/CPs | Human IgG1κ LALAPG/LS FCM, HLE | IV dosing; Human IgG Fc modified to reduce effector function | [221] |
|
Meplazumab (Ketantin®) |
Jiangsu Pacific Meinuoke Bio- pharmaceutical |
Phase III (NYR) |
NA | Humanized IgG2 | Anti-malaria antibody clinical candidate that targets CD147 (EMMPRIN), a proposed alternative receptor for SARS-CoV-2 entry | [222] |
|
(COV2-2130-YTE-LALA and COV2-2381-YTE-LALA) |
Ology Bioservices |
Phase II/III NYR |
NA | Cocktail of 2 human IgGs with FCM, HLE | IM dosing; 1:1 mixture of two human IgG1-YTE-LALA non-competitive binding antibodies targeting SARS-CoV-2 | [223] |
|
ABP-300 (MW05 LALA, MW33) |
Abpro Biotech/Mabwell Biosciences |
Phase II R |
BC/CPs | Human IgG1κ LALA FCM | IV dosing; LALA mutation inserted to eliminate ADE activity observed in preclinical studies | [224, 225] |
| STI-2020 (COVI-AMGTM) | Sorrento |
Phase II R |
GMNDAL | Human IgG | IV dosing; affinity-matured version of STI-1499 | NP |
| STI-2099 (COVI-DROPS) | Sorrento |
Phase II NYR |
GMNDAL | Human IgG | IN delivered; affinity-matured version of STI-1499 | [226] |
|
BI 767551 (DZIF-10c) |
Boehringer Ingelheim |
Phase II/III W,T |
BC/CPs | Human IgG1 | Inhaled vs IV dosing (tested IN preclinically, as well); tested therapeutic vs prophylactic; Development recently discontinued (7/26/21) | [227] |
| VIR-7832 | GSK/Vir Biologics |
Phase I/II R |
BC/CPs | IgG1κ M428L, N434S HLE and GAALIE FCIN | IV dosing; FCIN version of VIR-7831, which itself was derived from anti-SARS-CoV-1 mAb S309. | [209] |
| REGN14256 | Regeneron |
Phase I/II (R) |
ND | ND (presumed human IgG) | SC dosing; alternative partner for imdevimab combination | NP |
| BGB-DXP593 (BD-368-2) | BeiGene/Singlomics |
Phase II C |
BC/CPs | Human IgG | IV dosing | [228] |
| BGB-DXP604 (BD-604) | BeiGene/Singlomics |
Phase I C |
BC/CPs | Human IgG | IV dosing; neutralizes Omicron BA.1 | [228] |
|
BGB-DXP604 plus BGB-DXP593 |
BeiGene/Singlomics |
Phase I C |
See above | Cocktail of two Human IgGs | IV dosing; neutralizes Omicron BA.1 | [228] |
| JS026 | Shanghai Junshi Bioscience |
Phase I R |
BC/CPs | Human IgG | IV dosing; alternative partner for JS016 combination | [229] |
| SI-F019 | Sichuan Baili Pharma/SystImmune |
Phase I R |
Recombinant FcFP | ACE2-Fc fusion | IV dosing; recombinant human bivalent ACE2-Fc fusion protein injection | NP |
|
LYCovMab BA4101 (CA521 FALA) |
Boan Biotech/Luye Pharma |
Phase I ANR |
Immunized Tg mice followed by phage library |
Human IgG4κ S228P/F234A/L235A | IV dosing; Reduced Fc effector function for reduced ADE | [230] |
|
ABBV-47D11 (HBM9022) |
Abbvie/Harbour Biomed |
Phase I C |
Immunized Tg mice |
Human IgG1κ | IV dosing; Harbour tg mice | [231, 232] |
| ABBV-2B04 | Wash U/Abbvie |
Phase I C |
Immunized C57BL/6 mice |
IgG1 | IV dosing; either chimeric or humanized | [233, 234] |
|
ABBV-47D11 plus ABBV-2B04 |
Abbvie |
Phase I C |
See above | IgG1 Cocktail | IV dosing; Fixed dose combination | NP |
|
HFB30132A (P4A1-2A) |
HiFiBiO/Shanghai Jiaotong Univ. |
Phase I ANR |
BC/CPs | Human IgG4κ-L234F, L235E, M252Y, S254T, T256E, P331S HLE and FCM | IV dosing; Human IgG Fc modified to reduce effector function | [235] |
| COR-101 (STE90-C11) | Corat Therapeutics |
Phase I R |
PDHAL-CP | Human IgG1κ PVALδGQS FCM | IV dosing; Library constructed from convalescent patients; Human IgG Fc modified to reduce effector function | [236] |
|
JMB2002 (Ab2001.08 N297A) |
Jemincare Group |
Phase I CTR2100042150 |
PDNHAL; yeast display selections | Human IgG1 N297A FCM | Human IgG1 Fc non-glycosylated at residue 297 to reduce effector function | [237] |
| XVR011 (humVHH_S56A/LALA-Fc/Gen2) | ExeVir Bio BV |
Phase I R |
Immunized llamas |
Humanized VHH-IgG1 LALA Fc fusion FCM | IV dosing; Unique Llama-derived VHH72-Fc antibody (XVR011) affinity optimized S56A | [238, 239] |
|
HMBD-115 (AOD01, SC31) |
Hummingbird Biosciences (Singapore) |
Phase I (Singapore) (No NCT) |
BC/CPs | Human IgG1 | IV dosing; Intact Fc function required for maximal activity (not engineered) | [240] |
| CT-P63 | Celltrion |
Phase III NYR |
NA | Human IgG | To be added to CT-P59 (regdanvimab) to make cocktail for a nebulized formulation for inhalation; neutralizes Omicron BA.1 | [241] |
| HLX71 | Hengenix Biotech Inc |
Phase I NYR |
Recombinant FcFP | ACE2-Fc fusion | IV dosing; Recombinant Human Angiotensin-Converting Enzyme 2-Fc fusion Protein | [242] |
| HLX70 (P17) |
Henlius Biotech / Hengenix Biotech |
Phase I NYR |
ST-ST-HuNAL | Human IgG1κ | IV dosing; Research papers suggest combining HLX70 mAb and HLX71 ACE2-Fc fusion protein | [242, 243] |
|
IGM-6268 (COV2-14) |
IGM Biosciences |
Phase I R |
PDNHAL | Human IgM | IN dosing; IgM format; neutralizes Omicron BA.1 | [244–246] |
| IBI314 | Innovent Biologics (Suzhou) |
Phase I/II R |
UNK | Cocktail of 2 human IgG1 antibodies | IBI-314A IgG plus IBI-314B IgG in 1:1 fixed dose ratio; ambulatory patients | NP |
| COVAB 36 | Memo Therapeutics AG |
Preclinical (No NCT yet) |
BC/CPs MD | Human IgG1 | Retains the ability to mediate ADCC, ADCP and CDC; being developed for inhalation delivery | [247, 248] |
| DIOS-202 and DIOS-203 | DiosCURE |
Preclinical (No NCT yet) |
Immunized llama | Humanized VHH-VHH heterodimers | Small (ca. 25 kDa) VHH dimers, possibly VHH-VE and VHH-EV (not confirmed) | [249] |
| TATX-03 | ImmunoPrecise |
Preclinical (No NCT yet) |
PDNHAL | Cocktail of 4 human IgGs | TATX-03 is a “Polytope” cocktail of four proprietary monoclonal antibodies (mAbs) directed against distinct regions of the SARS-CoV-2 spike protein | [250] |
|
AR712 (AR-711 (1212C2) + AR-720) |
Aridis Pharma-ceuticals |
Preclinical (No NCT yet) |
BC/CPs | Cocktail of 2 human IgG1 antibodies with HLE | Formulated for inhaled delivery | [251] |
| PiN-21 (Nb21) | University of Pittsburgh |
Preclinical (No NCT yet) |
Immunized llama | Homo-trimeric VHH | IN delivery; Homo-trimerized VHH; IN delivery at 0.2-0.5 mg/kg protects animals | [252, 253] |
| ZRC-3308 | Zydus Cadila |
Preclinical (No NCT yet) |
BC/CPs | Cocktail of 2 human IgGs with FCM, HLE | Combination of two antibodies binding different epitopes | NP |
| IMM-BCP-01 | Immunome |
Preclinical (IND filed; CRL) (No NCT yet) |
BC/CPs | Cocktail of 3 human IgG antibodies | Antibody cocktail neutralizes VOCs and VOIs, including Delta and Omicron, in preclinical studies | NP |
| RB-100 | RenBio/Columbia University |
Preclinical (No NCT yet) |
UNK; BC/CPs likely | Bispecific, bivalent IgG | DNA delivery; bispecific antibody targets RBD and NTD of the SARS-CoV-2 spike protein | NP |
| Centi-B9 | Centivax, Inc |
Preclinical (No NCT yet) |
PDNHAL most likely |
Likely human IgG | Dosing either SC or IM, not IV | NP |
| STI-9167 and STI-9199 | Sorrento Therapeutics, Inc/Mount Sinai |
Preclinical (No NCT yet) |
Immunized Tg mice |
Human IgG1-LALA (FCM) | STI-9167 (COVI-SHIELD) is likely IV dosed; STI-9199 is IN formulation of STI-9167; both are active against the Omicron variant | [254] |
| IDB003 | IDBiologics/Vanderbilt Univ |
Preclinical (No NCT yet) |
BC/CPs likely | Likely human IgG | Identified in Dr. James Crowe’s lab at the Vanderbilt Vaccine Center; Presumed IgG | NP |
ACE2 angiotensin converting enzyme-2, ADCC antibody-dependent cellular cytotoxicity, ADCP antibody-dependent cellular phagocytosis, ADE antibody-dependent enhancement, ANR active not recruiting (clinical trial), BC/CPs B-cells from convalescent patients, BMS Bristol Myers Squibb, C clinical trial completed, CRL complete response letter, DARPin designed ankyrin repeat protein, EMA European Medicines Agency, EMMPRIN extracellular matrix metalloproteinase inducer, EU European Union, EUA emergency use authorization, Fc fragment crystallizable, FcFP Fc fusion protein, FCIN Fc increased activity with Fc receptors, FCM Fc muting (silencing activity on Fc receptors), FE/YTE/S L234F L235E M252Y S254T T256E P331S mutations of hinge/Fc, GAALIE G236A/A330L/I332E Fc mutations to increase activity with Fc receptors, GMNDAL Sorrento’s G Mab Naive donor antibody library, GSK GlaxoSmithKline, HLE half-life extension, IM intramuscular, IN intranasal, IND investigational new drug (application), IV intravenous (administration), LALA L234A L235A mutations of IgG1 hinge to dampen Fc activity with Fc receptors, LALAPG/LS L234A/L235A/P329G/M428L/N434S hinge/Fc mutations, LS M428L/N434S mutations for increasing half-life, MD mammalian display, MHRA Medicines and Healthcare products Regulatory Agency (UK), NA not available, NCT National Clinical Trial (registry number prefix), ND no data available, NMPA National Medical Products Administration, NP no publication (press release only), NTD N-terminal domain, NYR not yet recruiting (clinical trial), PDHAL-CP phage displayed human antibody library constructed from convalescent patients, PDNHAL phage displayed naive human antibody library, PVALδGQS E233P/L234V/L235A/G236D/D265G/A327Q/A330S mutations to dampen Fc and complement activity, R recruiting (clinical trial), RBD receptor binding domain, SARS-CoV-1 severe acute respiratory syndrome coronavirus-1, SC subcutaneous (dosing), ST-ST-HuNAL name of Henlius phage displayed naïve human antibody library, T clinical trial terminated, Tg transgenic (mice producing human antibodies), UK United Kingdom, UNK unknown to authors, US-FDA United States Food and Drug Administration, VHH single domain antibodies (regardless of origin), VLI VelocImmune mice (producing human antibodies), VOCs variants of concern, W clinical trial withdrawn, YDAF yeast display affinity maturation, YTE M252Y/S254T/T256E mutations for increasing half-life
aNCT registries can be found using reference [7]
Fig. 3.

Antibody formats used to neutralize SARS-CoV-2 described here. A IgG, typical IgG1 isotype; B Fc-modified IgGs (the red parts in the structure shown represent mutations made in the hinge and/or Fc); C Cocktails of IgGs. IgG antibodies in A–C reconstructed using PDB ID 1IGT; D IgM antibody (J-chain in green), reconstructed using PDB IDs 6KXS and 1IGT; E, F three domain- and one domain-nanobodies fused to each arm of an IgG-Fc, respectively. The IgG-Fc is derived from PDB ID 1IGT; G domain/nanobody antibodies of about 12 kDa. All domain antibodies in (E–G) constructed using PDB ID 6ZXN of nanobody Ty1; H bivalent bispecific antibody targeting two distinct epitopes; and I human ACE2-IgG-Fc fusion protein, reconstructed using human Fc (PDB ID 1IGT) and human ACE2 (PDB ID 6M17). For all drawings, the PDB program [201, 202] was used to generate the structures
Sources of Antibodies Targeting SARS-CoV-2
With today’s antibody discovery technologies, there are multiple approaches to obtain neutralizing antibodies against important viral antigens quickly, including immunizing mice or transgenic mice producing human antibodies and then recovering the antibody genes via hybridoma (traditional and slower), via single B cell technology (faster, more efficient and now widely used), via next-generation sequencing and analysis, or by generating immune phage, yeast or mammalian libraries to select the antigen-binding antibodies. Alternatively, naïve libraries of human antibodies, generated from pools of human B cells or made synthetically, can be panned in any one of several display formats. Finally, and in the case of antibodies to SARS-CoV-2, the most widely used approach, antibody genes from B cells of infected patients can be isolated, expressed and selected either directly or via immune library approaches as noted above. In a few cases, antibodies to SARS-CoV-2 have even been derived from B cells taken from patients infected with SARS-CoV-1 [208, 255].
In theory, any of the surface proteins of SARS-CoV-2 could be used as potential antigens, including the envelope (E) protein, the M glycoprotein, or the trimeric spike protein (S). The spike protein, however, is required for both the targeting to ACE2 and mechanism for cell entry, so virtually all efforts have targeted various aspects of the spike protein, with most of those efforts focused on the receptor binding domain (RBD).
By far, the most frequently used platform for isolation of SARS-CoV-2 neutralizing antibodies (nAbs) is from the memory B cells of convalescent COVID-19 patients. Of the 36 identified clinical stage antibodies and 11 identified preclinical stage antibodies preparing for clinical development, at least 27/47 were isolated from human B cells (Table 2). Moreover, of the most advanced 12 nAbs that have been tested in Phase III clinical trials, ten nAbs were isolated using this platform (Table 2). With the advance of single B-cell cloning and advanced microfluidics technologies in the last decade, as well as more recently developed single B-cell RNA-sequencing technology, the genes encoding potent nAbs can be isolated in as little as 2 weeks [210, 256, 257].
After SARS-CoV-2 infection, the B cell response continues to evolve in patients. Therefore, time of sampling post infection affects quality of the isolated nAbs, such as potency and resistance to viral mutations [258]. By selecting patients who had pre-existing immune responses to seasonal endemic coronaviruses, SARS-CoV-2 nAbs with broader coverage of several members of sarbecoviruses (lineage B), and even relatively distant lineages A and C betacoronaviruses (β-CoV), also have been isolated [259–262]. However, these broadly active nAbs are rare and usually less potent than antibodies directed specifically towards SARS-CoV-2.
The second most likely source of antibodies to SARS-CoV-2 is transgenic, engineered mice that produce human antibodies [263, 264]. There are now several human antibody-producing transgenic mouse platforms, including, for examples, the Medarex HuMAb/UltiMAb mouse, Kirin TC mouse, Abgenix Xenomouse, KymAb mouse, Regeneron VelocImmune mouse, Harbour H2L2 mouse, Trianni Mouse, Alloy GX mouse, Ablexis AlivaMAb mouse, and Ligand OmniMouse. One limitation to this approach is that these engineered mice in some cases are company owned and not available for out-licensing (e.g., Abgenix mouse, Medarex mouse, VelocImmune mouse), or on the other hand, can be only accessed via licenses (e.g., AlivaMouse, OmniMouse) [205, 231].
By immunizing these transgenic mice with the spike protein of SARS-CoV-2, parts of the spike protein (e.g., RBD or RBM), or other antigens, fully human antibodies specific for those targeted antigens can be isolated. Interestingly, the predominant antibody genes used by these immunized mice to make anti-SARS-CoV-2 spike antibodies are different from those isolated from human B cells derived from convalescent patients [76]. By combining the two platforms, Hansen et al. [205] generated a collection of diverse nAbs that ultimately resulted in the identification of one convalescent patient human B-cell-derived antibody and one immunized VelocImmune mouse-derived antibody to generate a fixed-dose combination antibody cocktail called REGEN-COV™: REGN10987 was isolated from a VelocImmune mouse immunized with SARS-CoV-2 RBD protein and REGN10933 was isolated from a COVID-19 convalescent patient [205, 265]. ABBV-47D11, currently in Phase I clinical trials (Table 2), was derived from the Harbour H2L2 transgenic mouse [231, 232].
Human antibody libraries, including phage-, yeast-, or mammalian-displayed antibody libraries, are also platforms and sources of SARS-CoV-2 nAbs. There are essentially three sources of human antibody libraries, including: (i) libraries from B cells derived from vaccinated, infected, or diseased subjects in which the desired antigen or epitope is relevant to the infection or disease [230]. These are often termed “immune libraries”; (ii) libraries constructed from B cells derived from “naïve” donors, i.e., subjects who have not been specifically vaccinated, infected, or diseased in a manner that would skew the antibody repertoire. In this case, the retrieved antibodies are usually relatively low affinity and often need to be affinity matured to improve their chances of being therapeutically relevant; and (iii) synthetic human antibody libraries [266–268], which are made to resemble natural antibodies by sequence and/or structure, usually by modelling hundreds of antibodies for which sequence and X-ray crystallographic structure information is available [268]. In this third case, synthetic libraries can also be made to mimic unusual antibodies such as the pool of anti-viral VH1-69 germline antibodies that rely on CDR-H2 contacts as part of their binding capacity [270–272], or anti-viral antibodies with long CDR-H3s [262], another “phenotype” of antibody associated with some anti-viral neutralizing antibodies [274–276]. Recently, it was clearly demonstrated that library-derived antibodies were equal in quality to animal-derived antibodies for anti-SARS-CoV-2 activity [269].
Regdanvimab (CT-P59), which has been approved by Korea and EU-EMA under the trade name of Regkirona™ (Table 2), was a SARS-CoV-2 nAb isolated from a phage-displayed single-chain variable fragment (scFv) library that was constructed from the B-cell antibody genes of a convalescent COVID-19 patient in Korea. Regdanvimab potently neutralizes SARS-CoV-2 infection of host cells with a titer of 8.4 ng/mL and exhibits therapeutic efficacy in ferret, hamster, and rhesus monkey models of SARS-CoV-2 infection [206]. Additionally, Corat COR-101 was isolated from a phage library built by recovering antibody genes from B cells of COVID-19 convalescent patients [236]. Finally, LYCovMab BA4101 (aka CAS521 FALA) was generated by immunizing transgenic mice capable of producing human antibodies with SARS-CoV-2 spike protein, followed by harvesting the B cells and generating a phage displayed library, from which the lead antibody was selected in vitro [230].
Naïve phage- or yeast-displayed antibody libraries based on antibody genes from healthy donors can also be panned to isolating potent nAbs and cross-neutralizing nAbs [244, 267, 278–280]. One advantage of panning naïve libraries is that antibody selection can be initiated without recruitment of COVID-19 patients. In addition, the phage- or yeast-display platforms are powerful tools for antibody engineering to enhance antibody potency, which may be required because the antibody genes in these naïve libraries are of germline sequences or have minimal somatic mutations, so nAbs from this source are relatively less potent than those from COVID-19 patients or immunized mice. A SARS-CoV-2 nAb (ADG-2) was successfully engineered to enhance not only the neutralizing potency but also the neutralizing breadth using a yeast-display strategy [219]. Other SARS-CoV-2 antibodies in development that were derived from naïve libraries include Sorrento STI-2020 and STI-2099, Jemincare JMB2002, Immunoprecise TATX-03, and IGM Biosciences IGM-6268 (Table 2).
The final source of anti-SARS-CoV-2 nAbs is the B cells of convalescent patients who had previously been infected with SARS-CoV-1. SARS-CoV-1 and SARS-CoV-2 share significant sequence homology in some parts of the spike protein [270], and early on, antibodies such as CR3022 [255, 282] and S309 [208], both originally isolated years ago from SARS-CoV-1 patients. Antibody S309, which binds a cryptic proteoglycan site on the RBD distal from the ACE2 recognition site [277], is the preclinical precursor to sotrovimab (GSK4182136, VIR-7831) which has been approved in the UK under the trade name Xevudy™, granted a US-EUA (26 May 2021), and is in Phase III clinical trials targeting full approval (Table 2).
IgG isotypes
Human IgGs come in four natural isotypes, IgG1, IgG2, IgG3, and IgG4. Of these IgG3 is rarely used as a template to make therapeutic antibodies, but the other three isotypes have all been used to generate approved therapeutics [176, 283]. We and several others have reviewed the activities of each isotype in details elsewhere so this will not be repeated here. It is important to note, however, that with the ability to engineer the Fc and hinge regions of human IgGs, as well as incorporating different isotypes, the antibodies can be tuned to possess or delete desired functionalities including ADCC, ADCP, CDC, and ability to crosslink.
Of the antibodies listed in Table 2, at least 22 have normal human IgG1-based Fc functionality, one (VIR-7832) is engineered to have increased Fc function, and ten (etesevimab [JS-016], ABP-300 [MW05], tixagevimab [COV2-2196], cilgavimab [COV2-2130], CA521, COR-101, JMB2002, HFB30132A [P4A1-2A], MAD0004J08, STI-9167/9199) are engineered to reduce or eliminate Fc function for safety purposes.
There is an ongoing discussion concerning the importance and the potential risk of having active Fc activity in antibodies targeting SARS-CoV-2. On one hand, Fc effector functions have been reported to be essential for optimal therapeutic protection against SARS-CoV-2 [218, 240, 284–286]; on the other hand, at least in some cases, significant protection was achieved in animal models independent of Fc functionality, suggesting that antibody Fab-dependent neutralization in absence of Fc function was sufficient to eliminate the virus [287]. Additionally, Fc engagement of FcγRIIIa has been correlated with disease severity in COVID-19 patients [288, 289] and Fc engagement of FcγRIIa/b is a potential risk of increasing viral infection via an ADE mechanism [221, 288]. A final consideration is the engagement of the complement pathway, the results of which are still not fully understood. Thus, the benefits versus the risks of Fc-engineered antibody therapies for COVID-19 are still not fully understood, but the current wisdom indicates that good Fc activity provides a significantly better chance at protecting against SARS-CoV-2.
Standard Human IgG1 Fc-Related Activities
As noted above, most of the antibodies to SARS-CoV-2 in clinical trials or those known by the authors in late-stage preclinical development are human IgG1 isotype antibodies with intact or enhanced Fc function. These antibodies typically bind to RBD and obstruct the ability of RBD to bind to its target, ACE2. They also have the ability to engage immune cells such as macrophages, dendritic cells, natural killer (NK) cells, and neutrophils via their Fc functionality [176]. One of the perhaps less appreciated advantages to possessing Fc function is the ability of these antibodies to opsonize and form cross-linked immune complexes on the surface of SARS-CoV-2, which can improve both the blocking function and the ability of the antibodies to clear the virus via FcγR-mediated activities. As noted in Sect. 7.4, cross-linking spikes is one of the mechanisms that antibodies utilize to neutralize SARS-CoV-2 [290].
While it has been demonstrated that antibodies to SARS-CoV-2 can neutralize the virus in the absence of Fc functionality [287], it has become clear that Fc activity enhances the ability of IgGs to neutralize SARS-CoV-2. Several groups have recently demonstrated using in vitro and/or in vivo experiments that an intact Fc, which interacts with immune cells such as NK cells to promote ADCC and phagocytes to promote antibody dependent cellular phagocytosis (ADCP), is required for optimal anti-SARS-CoV-2 antiviral activity [218, 284, 291], just as it is with HIV [292, 293]. The fact that monocytes, neutrophils and NK cells all contribute to this activity points to the importance of both ADCC and ADCP activities [291]. Additionally, it has been demonstrated that antibodies induced by vaccines utilize both their Fab function (binding to the virus) and Fc function (ADCC, ADCP) to neutralize SARS-CoV-2 [294]. This Fc-related activity clearly is related to Fc-γ receptor engagement, but complement-dependent cytotoxicity (CDC) does not appear to be critical to clearing SARS-CoV-2 virus [286], similar to what was previously found with HIV, where FcγR function was critical in helping to clear the virus, whereas CDC activity was found to be dispensable [293]. On the other hand, several different viruses employ complement-neutralizing factors [295], so the innate complement pathways must have some effect on certain viruses.
The anti-SARS-CoV-2 antibody, S309, an RBD-5B epitope IgG1 antibody that does not block RBD binding to ACE2, can neutralize SARS-CoV-2, at least in part, utilizing its strong ADCC, ADCP, and CDC activities [197]. Importantly, however, not all anti-SARS-CoV-2 IgG1 antibodies with normal Fc sequence have identical Fc functionality. Certain anti-SARS-CoV-2 antibodies, such as S2H13 [296], S309 [208], ADG20 [220], MTX-COVAB [247], S2P6 [297], Ab1 [298], and S2M11 [299], have been shown to be strong inducers of ADCC (NK cell, FcγRIIIa driven) activity [296]. Similarly, antibodies to SARS-CoV-2 such as S309 [208], MTX-COVAB [247], S2P6 [297], and ADG20 [220] have been demonstrated to induce ADCP (macrophage, FcγRIIa-driven) activity [296]. On the other hand, other antibodies such as S2A4, S2H14, and S304 were shown either not to induce these Fc-mediated activities (e.g., ADCC) or induce very modest activities (e.g., ADCP) on SARS-CoV-2 infected cells [296]. This suggests that epitope, geometry, affinity, and access of FcγRs to the antibody Fc may play important roles in determining which antibodies engage FcγRs and complement factors in vivo, which may contribute to the overall potency of certain antibodies over others [296]. In an interesting twist, Winkler et al. [284] demonstrated that Fc activity is required for optimal neutralization and killing of SARS-CoV-2 in a therapeutic setting, but was not necessary for pre-exposure prophylaxis. This suggests that neutralization alone may be enough to protect from virus, but not enough to eliminate virus once it sets up an infection [284].
Other than ADCC and ADCP, the other major pathway for elimination of foreign antigens and cells is the CDC pathway. S2H13 and S309 were shown to induce complement-dependent cytotoxicity (CDC) whereas several other antibodies, such as S2A4, S2H14, and S304, did not [296]. A recent study showed a correlation between antibody-dependent complement deposition (ADCD), a marker for CDC, and the severity of COVID-19 [300]. ADCD also was correlated with the overall inflammation state [300], while increased ADCP was actually correlated with reduced inflammation. Unfortunately, it appears that complement activation plays a potentially harmful role in COVID-19 [301–304]. While it is still under investigation, complement activation and dysfunction during COVID-19 have been suggested to be one of the key drivers of severe COVID-19 disease, and have been linked to ARDS, pro-coagulation and micro-thrombosis, systemic inflammation, and kidney failure [301–304].
Consideration for Antibody-Dependent Enhancement (ADE) of Infection
It has been known for nearly 40 years that viruses opsonized with IgGs generated as a result of previous infection, or alternatively vaccination, can bind to Fc-gamma receptors (FcγRs) and/or complement receptors, and function to cross-link the virus and receptor-positive immune cells (e.g., macrophages, monocytes, NK cells, B cells). This can result in viral-receptor-independent, increased viral entry into the cells, a mechanism dubbed “antibody-dependent enhancement” (ADE) [305, 306]. Thus, Fc-mediated ADE can enhance viral infection rather than clearing it [307]. ADE has been demonstrated for several viruses, including HIV-1, dengue virus, Ross River virus, and Epstein-Barr virus (EBV) [307, 308]. For vaccines, poor or waning titers can result in ADE, but also non-neutralizing antibodies to “non-required” epitopes may be a cause, so vaccine designers typically focus the anti-viral immune response as much as possible on epitopes that will induce neutralizing antibodies.
While ADE is usually associated with non-neutralizing antibodies, neutralizing antibodies also can be involved. Recently, Wan et al. [309] demonstrated that a specific neutralizing, anti-MERS-CoV spike antibody could mediate ADE. Additionally, ADE has been demonstrated for antibody pools generated from vaccines using SARS-CoV-1 spike protein [310–312]. In one case, the vaccine-induced antibodies were protective, even in the presence of in vitro-demonstrated ADE via FcγRII into B cells [310].
Thus far, there is no concrete clinical evidence supporting the hypothesis that antibodies induced by vaccines against SARS-CoV-2 spike protein or therapeutic antibodies targeting the RBD of SARS-CoV-2 can lead to ADE [313, 314]. As noted in the introduction to this paper, however, there are several coronaviruses that can infect humans, including the endemic strains known as NL63 and 229E. It has been demonstrated that previous infection with endemic CoVs, NL63 and/or 229E, followed later by infection with SARS-CoV-2 resulted in worse COVID-19 clinical outcome [315]. This was traced to non-SARS-CoV-2-neutralizing antibodies targeting nucleocapsid protein (NP), which are cross-reactive between the various coronavirus strains, resulting in ADE upon SARS-CoV-2 infection [315]. Thus, at least in this case driven by the presence of preexisting, non-neutralizing antibodies to NP, SARS-CoV-2 mediated ADE may occur.
ADE activity has been discovered in preclinical studies of certain potential anti-SARS-CoV-2 clinical candidates. Scientists at Abpro found that the anti-SARS-CoV-2 antibody MW05 (being developed as ABP-300) caused ADE via interaction with FcγRIIb, whereas MW07 [214] and MW06 [305], both of which bind different epitopes, did not. This suggests that the ability of a particular SARS-CoV-2 targeting antibody to cause ADE may not only be Fc-activity specific, but also epitope-specific [214, 305]. It is also the reason that ADP-300 (MW05) has been reconfigured into an Fc-partially muted IgG1 LALA (IgG1 with L234A, L235A modifications in the lower hinge) antibody [225]. Jemincare Group’s JMB2002 had a result similar to MW05 in that the wild-type IgG1 demonstrated ADE in preclinical studies, so it also was reconfigured into an Fc-muted format, this time by engineering it to be an aglycosylated IgG1 (Ab2001.08-N297A) by removing the N297 glycosylation site [237]. As noted above, besides these two Fc-muted antibodies to SARS-CoV-2, seven others have entered clinical trials with Fc functionality reduced or eliminated out of potential concern for ADE (Table 2). Nevertheless, the over-riding opinion two years into the COVID-19 epidemic is that ADE is not a serious issue for anti-SARS-CoV-2 therapeutic antibodies [314, 317].
Improved Fc Functionality
Recently Yamin et al. [286] examined the potential impact of improving FcγR activity on the ability of an antibody to neutralize SARS-CoV-2. They generated antibodies with reduced activity or improved activity to test in comparison to standard human IgG1. Their Fc-improved version, IgG1-GAALIE (G236A/A330L/I332E Fc mutations to increase activity with Fc receptors), was by far the most active at neutralizing SARS-CoV-2 in vitro and in vivo, suggesting that instead of reducing Fc activity for fear of ADE, increasing Fc activity might be a better strategy. VIR-7832, which is currently in Phase I clinical trials, incorporates the GAALIE mutation (Table 2).
This approach, however, may run some additional risk, since recent data suggest that antibodies that have the ability to engage FcγRIIIa may be correlated with disease severity. Recently, this mechanism of action was described for dengue infections, in which the degree of IgG1 N297 glycan afucosylation, which increases the binding of IgG1 to FcγRIIIa resulting in higher levels of ADCC [318], in non-neutralizing anti-dengue antibodies was directly correlated with disease severity of a second dengue infection [319]. This same phenomenon has now been observed with SARS-CoV-2 infection, in which higher levels of afucosylated antibodies generated by patients in response to infection are correlated with more severe disease, including increased risk of developing ARDS [320]. Thus, there are still some questions as to the role of increased Fc activity in protection from SARS-CoV-2 on one hand, and potential risk of greater immunopathology and more severe disease, on the other hand.
Mixtures or Cocktails of Specific IgGs
Although hundreds of potent nAbs have been successfully isolated (cf., [290, 321]), studies on antibody resistance have demonstrated that rapid viral escape arises with any monotherapy regardless of antibody neutralizing activity and epitope conservation [322–324]. Thus, many researchers and companies have turned to a rational combination of at least two neutralizing antibodies that possess different, non-overlapping epitopes together as a combination therapeutic to provide broader epitope coverage, and hopefully, greater resistance against variants that may arise over time [211, 245, 261, 325].
There are currently eight clinical stage anti-SARS-CoV-2 antibody cocktails, including: Eli Lilly/Shanghai Junshi Bioscience’s bamlanivimab (LY3819253; LY-CoV555) and etesevimab (LY3832479; LY-CoV016; JS016; CB6-LALA), Regeneron and Roche’s (REGEN-COV™ (USA); Ronapreve™ (UK) (casirivimab [CAS] and imdevimab [IMD]), AstraZeneca’s EvuSheld™ (AZD7442; tixagevimab [AZD8895, COV2-2196] and cilgavimab [AZD1061, COV2-2130]), Brii Biosciences amubarvimab (BRII-196, P2C-1F11) plus romlusevimab (BRII-198, P2B-1G5), Bristol Myers-Squibb’s BMS-986414 (C135-LS) and BMS-986413 (C144-LS), Beigene/Singlomics’ BGB-DXP604 and BGB-DXP593, Ology Bioservices ADM03820 (COV2-2130-YTE-LALA and COV2-2381-YTE-LALA), and AbbVie’s ABBV-47D11 and ABBV-2B04 (Table 2).
Of these antibody cocktails, three have received EUAs (REGEN-COV™, bamlanivimab/etesevimab, and EvuSheld™), one (BRII-196/198) has been fully approved by the China National Medical Products Association (NMPA) but not yet awarded EUAs in the west, and the other (BMS-986413/’414) is in late-stage clinical trials (Table 2). Note that REGEN-COV™ has also been granted full approval in Japan and the UK over the last few months (Table 2). Additionally, Celltrion has recently placed CT-P63 into clinical trials with the intent to add it to regdanvimab to make a cocktail for those antibodies as well [241].
Beyond the current clinical candidates, there are several preclinical candidates that show promise against variants in both the VOI and VOC categories. These include AR712 (AR-711 [antibody 1212C2] plus AR-720), both engineered with half-life extension technology, from Aridis Pharmaceuticals, TATX-03 from ImmunoPrecise, a combination of four neutralizing antibodies, each recognizing a distinct epitope, ZRC-3308, a combination of two nAbs from Zydus Cadila, and IMM-BCP-01, and an antibody cocktail of 3 nAbs from Immunome that has been demonstrated to neutralize VOCs and VOIs, including Delta and Omicron, in preclinical studies (Table 2).
As will be described in greater detail in Sect. 5, the results of using antibody mixtures can nearly be predicted entirely on the additive ability of each component antibody to resistant mutants or variants. For example, with Regeneron’s REGEN-COV™, imdevimab covers for the inability of casirivimab to neutralize the Beta variant [211, 325]. Similarly, the combination of bamlanivimab and etesevimab were effective against Kappa, Epsilon, and Iota even though bamlanivimab alone failed to neutralize those variants [326]. These and other studies indicate that not all antibody combinations are equally effective at reducing resistance. By comparing different antibody combinations, it has been proposed that nAbs targeting non-overlapping epitopes are more effective than those targeting overlapping epitopes [290, 325]. This may explain why the bamlanivimab plus etesevimab cocktail is not as effective against certain variants as the other cocktails. As a result of bamlanivimab plus etesevimab having overlapping epitopes (see Sect. 5), several single mutations, including I472D, G485P and Q493K/R, simultaneously affect the neutralization abilities of both antibodies [326].
Bispecific IgG-Based Antibodies
The cocktail approach requires combination of two antibodies, which can potentially complicate the development process and increase manufacturing cost. To overcome these issues, a bispecific antibody, which combines two nAbs into one molecule, is an alternative approach [327, 328]. De Gasparo et al. [328] used the “CrossMAb” platform and engineered a bispecific IgG1-like molecule (CoV-X2) based on two nAbs C121 and C135, which binds to non-overlapping epitopes of RBD. CoV-X2 enhances binding to RBD compared to the parental nAbs via a mechanism utilizing the avidity effect. Importantly, CoV-X2 neutralizes the escape mutants generated by the individual parental nAbs, although its neutralizing activities against these mutants are relatively lower than that against the wild type virus [328]. Lacking in this study, however, was a direct comparison of CoV-X2 with the cocktail of C121 and C135, which would have provided further insights on the development of bispecific and cocktail- based therapeutics. It is also valuable to investigate whether different formats of bispecific antibodies will impact efficacy. Cho et al. [327] used an alternative approach for engineering bispecific antibodies based on six NTD-targeting and three RBD-targeting nAbs to generate a series of bispecific antibody candidates. Several candidates with ultra-potent neutralizing activity (< 1 ng/mL) and good coverage of VOCs were identified. Interestingly, three of their bispecific antibodies exhibited a novel model of action that is beyond that observed for the individual parental nAbs [327].
Another interesting bispecific antibody construct was recently reported by Sanyou Biopharmaceuticals in China. They described SYZ001, which is a human IgG targeting one epitope on SARS-CoV-2 RBD fused with the domain antibody, P14-F8, targeting a second RBD domain epitope, resulting in a tetravalent, bispecific antibody which they reported had stronger activity against Delta and Epsilon than against the wild-type SARS-CoV-2 virus [329].
Finally, new bispecific antibodies were recently reported that neutralized all variants of SARS-CoV-2, including Omicron BA.1 [330, 331]. One of these was a biparatopic nanobody comprised of Nb1 and Nb2 fused to an Fc that broadly neutralized all variants tested, including Omicron [330]. In the second study, a bispecific, bivalent CrossMAb design combining CoV-14, the antibody used to make the potent and broadly neutralizing IgM antibody, IGM-6268 [245, 246], and CoV-06 (Table 2) [244, 331] was compared with a tetravalent IgG-(scFv)2 like construct. Both of these bispecific antibodies exhibited higher therapeutic efficacy than the equivalent cocktail of CoV-14 and CoV-06 antibodies [244], indicating the potential power of combining antibodies into a single molecule over using cocktails of multiple antibodies [331]. As might be expected from previous work [321], the tetravalent IgG-(scFv)2 bispecific antibody comprised of CoV-14 and CoV-06 was superior to the CrossMAb format as well as the two-antibody cocktail in SARS-CoV-2 neutralization activity in vitro, including broad neutralization of escape variants, and enhanced protective efficacy in vivo [331]. This is one example of the principle, described throughout this work, that an increase in antibody valency correlates with an increased ability to neutralize SARS-CoV-2 [332, 333].
IgA Isotype
Secretory IgA (sIgA) is the predominant Ig isotype in airway secretions and plays a crucial role in protecting mucosal surfaces against pathogens by neutralizing respiratory viruses or impeding their attachment to epithelial cells [334]. Moreover, sIgA is a dimer of two IgA monomers and thus has four antigen binding sites versus two in an IgG. Several groups have therefore investigated the functionality and potency of IgA antibodies directed against the SARS-CoV-2 RBD, both as monomers (IgA) and as dimers (dIgA) [193].
Ejemel et al. [335] described the evaluation of a cross-reactive human IgA antibody, MAb362, that binds to both SARS-CoV-1 and SARS-CoV-2. To better understand potency, these authors converted the antibody into other formats, including IgG, monomeric IgA, dimeric IgA (dIgA) and secretory IgA (sIgA), which is the dimeric dIgA form co-expressed with the secretory component. Of particular interest is the fact that the IC50 for these different forms against a SARS-CoV-2 pseudovirus varied considerably, with IC50 potency following the pattern of IgG (two binding sites; 58.7 μg/mL) < IgA (two binding sites; 1.26 μg/mL) < dIgA (four binding sites; 30 ng/mL) < sIgA (four binding sites; 10 ng/mL). When tested against authentic SARS-CoV-2 virus, the most potent form of MAb362 (sIgA) exhibited a relatively poor potency with an IC50 of 9.54 μg/mL [335].
Wang et al. [336] characterized the natural IgA response to SARS-CoV-2 in a cohort of 149 convalescent individuals diagnosed with COVID-19. In this study, plasma IgA monomers specific to SARS-CoV-2 proteins were found to be twofold less potent than the IgG equivalents. However, IgA dimers, which are the primary form of antibody in the nasopharynx, were typically 15-fold more potent than the IgA monomers against the same target [336]. This is consistent with the notion of increased antibody valency correlating well with an increased ability to neutralize SARS-CoV-2 [333].
IgM Isotype
IgM isotype antibodies are the largest natural form of antibodies generated, with J-chain-expressed pentamers of approximately 970 kDa possessing ten antigen-binding sites [193, 326]. As noted above for bispecific antibody and IgA constructs, valency appears to play a major role in determining the potency and breath of neutralization for antibodies and antibody-like constructs, with more binding sites typically leading to greater potency. Along these same lines, we recently described the production and evaluation of six antibodies to SARS-CoV-2 that were recombinantly class switched from IgG to dimeric IgA (dIgA, four antigen binding sites) and pentameric IgM (ten antigen binding sites) antibodies [245]. In all cases, the IgM versions were consistently more potent at neutralizing authentic SARS-CoV-2 than the IgG versions, and the IgA versions were positioned in between the two isotypes, IgG and IgM. For one of the antibodies, the IgM version (IGM-6268, IgM-14 in Ku et al. [245]) was 230-fold more potent at neutralizing authentic virus than was the IgG and exhibited an IC50 of 10 ng/mL (~ 10 pM). Importantly, the IgM version also retained high potency against authentic SARS-CoV-2 virus expressing a mutation at position E484, where it was > 2,000-fold more potent than the parental IgG. Similarly, the IgM version was also much more potent in vitro against authentic virus expressing the Alpha, Beta and Gamma VOC spike proteins. When tested in vivo, IGM-6268 was highly potent in both prophylactic and therapeutic settings at doses as low as 0.04 mg/kg and 0.4 mg/kg, respectively. These data further demonstrate the relative benefits of multivalency and avidity to improve potency and function [332, 333], most likely due to significant decrease in the binding off-rates and overcoming steric hindrance.
Single-Domain Antibodies (VHH)
Single-domain antibodies (sometimes referred to as “VHH”) are antibodies that utilize only a single scaffold, essentially half of a normal Fv, for binding to the antigen. These antibodies can come from a variety of sources, including the camelid family, which includes camels, llamas and alpacas, that produce a subclass of antibodies that possess an unpaired heavy-chain variable domain [338]. These camelid heavy chain variable domains, can be expressed as a single domain, typically referred to as a “nanobody,” with a molecular weight of about 12 kDa, roughly 1/12th that of the full-length IgG, and 1/80th the size of the IgM isotype described above. Other typical sources of single domain antibodies are camelid antibody libraries [339] and libraries of engineered human single domain antibodies [340].
One potential shortcoming of the VHH structure is their small size, which typically leads to rapid clearance following intravenous administration. Two approaches that potentially overcome this limitation include (i) fusing the VHH to the Fc region of an IgG, which not only improves clearance but also typically improves potency as a dimeric structure, and (ii) targeting delivery approaches that are less susceptible to clearance issues. The first approach has been used quite broadly for many single domain antibodies, and tetrameric versions have also been evaluated (see Table 3). The second approach, utilizing delivery approaches other than IV infusion such as inhalation, is discussed below and in Section 8. Hundreds of nanobodies have been identified that are capable of neutralizing SARS-CoV-2, as well as several of its variants [341]. There are now several examples of high potency single domain (VHH) antibodies neutralizing SARS-CoV-2 that have reached at least the preclinical development stage, three (VHH72/XVR011, Nb21/PiN-21, DIOS-202/DIOS-203) of which are described below.
Table 3.
Examples of VHH formats for targeting SARS-CoV-2
| Name | Format | Development stage | Affinity to RBD (KD) | ACE2 block | IC50 pseudovirus | IC50 live virus | Epitope | Notes | Reference |
|---|---|---|---|---|---|---|---|---|---|
| VHH72 (original) | VHH monomer | See below | 39 nM | No | 200 ng/mL (~ 12 nM) | ND | Class 4 (Margulies et al.); RBD-6 | Binds MERS, SARS1 and SARS2—neutralizes SARS1 but not MERS | [238, 239] |
| XVR011 (from VHH72) | Fc-VHH72 fusion | Phase I/II (NCT04884295 [R]) | 8 nM; 47 pM a | No | 840 ng/mL (~ 12 nM) | 130 ng/mL | Class 4 (Margulies et al.); RBD-6 | Humanized and optimized VHH72; muted Fc; (HumVHH72_S56A/LALA-Fc/Gen2) | [239] |
| Nb21 | VHH monomer | Preclinical | ND | Yes | ND | ND | Class 1 (Margulies et al.); RBD-2 | Precursor to PiN-21, which is in preclinical development | [252, 253, 342] |
| VHH-EV | VHH-VHH dimer-Fc | Preclinical | ND | Yes | 2.9 nM | 0.7 nM | RBD-2/RBD-6 | Fused to Fc; May be DIOS-202 or DIOS-203 | [249] |
| VHH-VE | VHH-VHH dimer-Fc | Preclinical | ND | Yes | 4.1 nM | 1.32 nM | RBD-6/RBD-2 | Fused to Fc; May be DIOS-202 or DIOS-203 | [249] |
| VHH-E | VHH monomer | Preclinical | 2 nM | Yes | 60 nM | 48 nM | Class 1 (Margulies et al. [345]); RBD-2 | Blocks ACE2 | [249] |
| VHH-EE | VHH-VHH dimer | Research | ND | Yes | 0.93 nM | 0.18 nM | Class 1 (Margulies et al. [345]); RBD-2 | Blocks ACE2 | [249] |
| VHH-EEE | VHH-VHH-VHH trimer | Research | ND | Yes | 0.52 nM | 0.17 nM | Class 1 (Margulies et al. [345]); RBD-2 | Blocks ACE2 | [249] |
| VHH-V | VHH monomer | Research | 9 nM | No | 198 nM | 142 nM | Class 4 (Margulies et al. [345]); RBD-6 | ND | [249] |
| Re5D06 | VHH monomer | Research | 2 pM | Yes | ND | ND | RBD-2 | Blocks ACE2 | [343] |
| Ty1 | VHH monomer | Research | ND | Yes | 0.77 μg/ml (54 nM) | 12 ng/mL as Fc fusion | RBD-2 | Blocks ACE2 | [344] |
ACE2 angiotensin-converting enzyme-2, KD affinity (M), MERS Middle East Respiratory Syndrome, ND no data, R recruiting, RBD receptor binding domain, SARS Severe Acute Respiratory Syndrome, VHH single domain antibody (ca. 12 kDa in size)
a8 nM monomeric; 47 pM as dimeric Fc fusion so avidity also is in play
In one of the first reports on VHH antibodies targeting SARS-CoV-2, Wrapp et al. [238], immunized llamas with pre-fusion-stabilized forms of the coronavirus spike protein. In an effort to get cross-reactive nanobodies, the llama was immunized with S protein from SARS-CoV-1, then MERS-CoV, SARS-CoV-1, S and again with both SARS-CoV-1 and MERS-CoV S proteins. Several of the VHHs so obtained were able to neutralize pseudovirus displaying the SARS-CoV-1 and MERS-CoV S proteins, and one (VHH-72) was able to weakly neutralize SARS-CoV-1 and SARS-CoV-2 pseudoviruses. However, when displayed as a dimeric Fc fusion (VHH-72-Fc), the antibody construct exhibited a more potent neutralization of SARS-CoV-2 pseudovirus with an IC50 of 200 ng/mL [239]. This molecule was affinity matured (modification of residue S56A), and fused to an Fc carrying the LALA mutation to silence FcγR-binding activity [239]. This construct (VHH72 HumVHH_S56A/LALA-Fc/Gen2) was renamed XVR011 by the startup company, ExeVir Bio BV, who have advanced it to a Phase I/II trial (Tables 2 and 3).
For a second example, Sun et al. [342] categorized three different classes of VHH antibodies based on their epitopes. Class I VHH antibodies, characterized by Nb21, targeted both open and closed RBDs and were described as ultrapotent (Table 3). Nb21, which Margulies et al. classify as binding a “Class 1” epitope [345], blocks ACE2 binding to RBD. Nb21 is highly sensitive to the E484K mutation, so while it neutralizes Alpha mutants lacking E484K, Nb21 is not active against Beta [342]. Nb21 is being developed as an intranasally delivered VHH antibodies to combat SARS-CoV-2 infections. Several reports have appeared suggesting that VHH antibodies may be ideal candidates for aerosol delivery to the sinus and lungs of infected individuals, given their small size and exceptional biophysical stability to withstand aerosolization [252, 346, 347]. The Fc-fused, sequence optimized Nb21, known as PiN-21 (Table 3), has been delivered by intranasal delivery [253]. PiN-21 at 0.6 mg/kg protected infected animals from weight loss and substantially reduced viral load in both lower and upper airways. Furthermore, aerosol delivery of PiN-21 facilitated deposition throughout the respiratory tract and allowed the reduction of a protective dose to 0.2 mg/kg [253]. PiN-21 is currently in preclinical studies.
A third example of a preclinical stage VHH antibody is a multivalent VHH antibody, based on two or more single VHH antibodies targeting the same or different RBD epitopes [249, 252, 346, 348, 349]. Some of these engineered VHH antibodies show remarkable neutralizing activity (< 0.1 ng/mL) and may perform better to reduce virus escape mutations [249, 252, 346]. Because these engineered multivalent nAbs are stable, they may be developed as aerosol-delivered therapeutics for treatment of COVID-19 [252, 346].
To generate multi-specific VHH antibodies, Koenig et al. [249] isolated 23 llama-derived nanobodies, four of which bound SARS-CoV-2 RBD with relatively high affinity and potently neutralized viral entry. They [249] tested the ability of monomers, E, which bound one epitope, and U, V, and W, which bound a separate non-overlapping epitope, to form homodimers (e.g., E–E), homotrimers (e.g., E–E–E), and heterodimers (e.g., E–V, V–E, E–W) of the nanobodies to neutralize SARS-CoV-2. VHH E alone was capable of locking spike into a fusogenic 3-RBD-up position in the absence of ACE2, which neutralizes the virus due to premature activation of the fusion mechanism, a novel MOA. The trimeric VHH EEE demonstrated the same activity with 100-fold enhanced potency, but was highly sensitive to the single site mutation S494P. Similarly, VHH V, another potent neutralizer, but was sensitive to mutations S371P and K378Q [249]. The heterodimeric VHHs, EV and VE were both potent and more resistant to mutations than monomers or homomeric oligomers [249]. The biotech company DiosCURE Therapeutics is taking two of these heterodimeric VHHs (DIOS-202, DIOS-203) into development (Tables 2 and 3) with the expectation of initiating clinical trials soon.
Several other VHH antibodies also have been isolated with potent neutralizing activity against SARS-CoV-2, a few of which will be mentioned briefly here. Synthetic “camelid-inspired” yeast-displayed libraries have been used to isolate several VHH antibodies, including Nb6 [346], which competes with ACE2 and displays a KD on RBD of 41 nM. That antibody bound spike in a 3-RBD-down conformation and locked the spike in the fully inactive conformation inaccessible down state incapable of binding ACE2. Other VHH antibodies from “camelid-inspired” libraries include Sb14, Sb16, and Sb45 [345], all of which competed with ACE2, and Sb68, which bound more on the periphery of the ACE2 binding site. Based on structural data, they mapped the epitopes to the four antibody classes described by Barnes et al. [276].
Güttler et al. [343] isolated two VHH antibodies from phage displayed SARS-CoV-2 spike-immunized llama libraries, Re5D06 and Re9B09, to generate tandem, multispecific VHH constructs. A heterodimeric bivalent bispecific VHH dimer of Re5D06 and Re9B09 exhibited sub-30 pM KD binding to the Alpha, Beta, Gamma and Epsilon variants [343]. In another example, a trimer of Re6D06, a sub-pM RBD binder, had a minimum neutralizing concentration of only 17 pM [344]. Interestingly, though, the trimer with the greatest potency, neutralizing down to a concentration of only 1.7 pM, was derived from a monomer that neutralized rather poorly [343].
Ty1 is a nanobody described by Hanke et al. [344] that binds to RBD in both the up and down conformations and competes with ACE2 binding (Table 3). Based on its epitope, which buries 860Å2, it appears to belong to the RBD-2 epitope group (Table 3). As a stand-alone VHH, Ty1 was not particularly potent (770 ng/mL (54 nM) IC50 in pseudovirus assay), but when fused to an Fc to make a bivalent VHH-Fc fusion protein, its potency was increased to an IC50 of 12 ng/mL [344]. Hanke et al. [344] also reported the identification of a monomeric VHH, Fu2, that interacts with RBDs on two different spike trimers and neutralizes pseudovirus with an IC50 of 106 ng/mL. Cryo-EM studies demonstrated that the bound structure was a dimer of SARS-CoV-2 spike timers containing six Fu2 VHH molecules [344].
Xu et al. [350] isolated anti-SARS-CoV-2 VHH antibodies from llamas and from transgenic mice engineered to produce llama antibodies. Their VHH antibodies were low pM binders, but had modest potency on wild-type SARS-CoV-2 as monomers (320–7,145 pM IC50s) and for the most part did not neutralize E484K or N501Y mutants well. However, when the VHHs were constructed into multivalent homotrimers, the IC50s on wild-type virus ranged from 12 to 91 pM and four out of the five tested retained high neutralizing activity (mostly sub-100 pM IC50) on all the mutants tested (R683G, K417N, E484K, N501Y) [350]. This is another excellent example, similar to what was observed with IGM-6268 [245], of higher avidity constructs helping to overcome mutants [333].
Finally, Bracken et al. [348] reported the isolation of 85 VH binders from a synthetic human VHH library that recognized two non-overlapping sites within the ACE2 binding site on the SARS-CoV-2 RBD. These VHH constructs were subsequently linked into multimeric and biparatopic formats that showed considerable improvements in potency (up to 1400-fold) when tested against SARS-CoV-2 pseudovirus. When tested against authentic SARS-CoV-2, the most potent construct, a trivalent VH, exhibited an IC50 of 180 ng/mL [348]. None of these constructs have yet advanced to human clinical trials. Combined, these data all indicate that small, single-domain VHH constructs can be generated that potently block the interaction between the SARS-CoV-2 RBD and ACE2, and that multi-specificity and multivalency can increase potency significantly, as noted previously [333].
Multibodies
Taking multivalency one step further, Rujas et al. [333] used the self-assembly of human apoferritin to generate large structures that express 24 identical polypeptides. Each apoferritin polypeptide was fused to an anti-SARS-CoV-2 VHH (e.g., VHH72) or a single chain Fab plus single chain Fc, generating “multibodies” that have 24 binding sites for the SARS-CoV-2 RBD and, in the case of the Fab-Fc version, Fc fragments capable of interacting with the FcRn recycling receptor and thereby improving the in vivo half-life. When tested in vitro against SARS-CoV-2 pseudovirus, both multibody constructs were considerably more potent in vitro than the corresponding parental constructs—10,000-fold for the VHH multibody and ~ 2000-fold for one of the Fab-Fc multibodies [333]. When combined with the multi-VHH-Fc-fusion and IgM results noted previously, it is apparent that increased valency may be an important approach to improving the potency of anti-SARS-CoV-2 antiviral antibodies.
DARPins
Non-antibody, protein-binding scaffolds are also being used to build monovalent and multivalent agents capable of neutralizing SARS-CoV-2. Similar to VHH antibodies, DARPins (Designed Ankyrin Repeat Proteins) are roughly one-tenth the size of a typical IgG and are antibody mimetics that typically exhibit highly specific binding to target proteins. By fusing several DARPins in a row, a range of molecular functions can be built into a single molecule. Walser et al. [351] recently described the development of five-domain, tri-specific DARPins that binds to multiple regions of the SARS-CoV-2 spike protein (virus neutralization) and to human serum albumin (HSA; to improve pharmacokinetics). Some of these constructs exhibited IC50 values against SARS-CoV-2 virus of 1 ng/mL (~ 12 pM), and one such construct (MP0420) is being tested in a Phase II clinical trial (Table 2).
ACE2-Fc Fusion Approaches
Human ACE2 exodomain spans residues 18–740 (1–17 are signal sequence, and > 740 are membrane spanning and intracellular domains). The enzymatic peptidase domain, responsible for cleavage of the vaso-constrictive peptide, angiotensin II, to the vasodilator, angiotensin, is found in residues 18–615, while the Collectrin-like domain (CLD), which is involved in amino acid transport, is encoded by residues 616–740. Various studies have demonstrated that soluble ACE2 protein can be truncated at residue 615, resulting in a soluble, well-behaved, biologically active N-terminal enzymatic domain that binds well to RBDs of SARS-CoV-1 and SARS-CoV-2 [352, 353]. These soluble ACE2 fragments, or receptor traps, have also been shown to interfere with SARS-CoV-2 binding to cellular ACE2, thereby neutralizing viral infectivity [354].
The binding of SARS-CoV-1 and CoV-2 spike proteins to their cognate receptor, ACE2, depend on a series of specific interactions, including at least 17 residues on SARS-CoV-2 RBM known to make contact with the N-terminal domain of ACE2 (see OSM Fig. S2). Similarly, from the co-crystal structure of SARS-CoV-2 RBD-HuACE2, the key specific ACE2 residues responsible for binding to SARS-CoV-2 RBM are Q24, D30, H34, Y41, Q42, M82, K353, R357 [352, 353]. Suryamohan et al. [355] analyzed polymorphisms of ACE2 across hundreds of individuals and found that the mutations S19P, I21V, E23K, K26R, T27A, N64K, T92I, Q102P and H378R were likely to increase susceptibility to SARS-CoV-2, suggesting that they may increase the affinity of the human ACE2-SARS-CoV-2 interaction. Of these, K26R and T92I mutants were confirmed in vitro to increase the affinity of the interaction [355]. Predictably, a mutant ACE2 K26R and T92I protein was more effective at interfering with entry of pseudotype virus than wild-type ACE2 [355]. These studies clearly demonstrated the potential for modifying human ACE2 for fusion with Fc to make an infection blocker, or trap, with activity akin to antibodies targeting SARS-CoV-2.
As discussed above, antibody domains that bind RBD have been fused to the Fc portion of IgG antibodies to increase valency and improve pharmacokinetics, and this approach has also been applied to ACE2. Typically, ACE2-Fc fusion proteins are constructed with amino acids 18–614 (start of the mature protein through the end of the protease domain), while some constructs have also included residues 615–740 to make a larger fusion protein [356]. Additionally, efforts have been made to increase the potency of ACE2-Fc fusion proteins via mutation of the RBM-binding motif of ACE2 to increase the binding affinity to SARS-CoV-2 RBM [356]. A combination of four mutations of ACE2 (K31F, N33D, H34S, E35Q) increased the affinity of mutant ACE2-Fc, CVD313, to SARS-CoV-2 RBM by about 40-fold (20.4 nM to 0.52 nM KD). Moreover, the IC50 value of the CVD313 mutant, using a pseudovirus assay, was improved more than tenfold over wildtype ACE2(18-614)-Fc, from 0.43 to 0.028 µg/mL. An additional mutation, H345L, was also included to remove ACE2 enzymatic activity to decrease the possibility of adverse effects during therapeutic use [356].
Chan et al. [357] also generated high affinity, mutant ACE2-Fc fusion proteins by modifying the residues T27Y, L79T, and N330Y, which resulted in an increase in avidity for ACE2-Fc constructs from 22 nM (wild type ACE2-Fc) to 0.6 nM (mutant V2.4) [357]. This construct, engineered by scientists at Orthogonal Biologics, appears to be in preclinical development at this time. Similarly, Svilenov et al. [358] generated modified ACE2-IgG4-Fc fusion proteins that were highly potent against wild-type SARS-CoV-2 virus, as well as the Alpha, Beta, and Delta variants with sub-nM IC50s. Based on one of these constructs [347], Formycon AG currently has FYB207, which retains ACE2 enzymatic activity that may potentially help protect against acute respiratory distress syndrome (ARDS), in preclinical studies [359].
In an effort to make a more potent, high avidity Fc fusion of ACE2, Miller et al. [360] constructed a tetrameric ACE2-Fc fusion by fusing two Fcs together using the tetramerization domain of P53 as the linking domain. By tetramerizing the Fc (four ACE2 domains vs. two of a normal Fc), they increased the avidity from 22 nM to 3.9 nM KD, and the IC50 for pseudovirus neutralization about 14-fold from 456 to 33 ng/mL [360]. Similarly, in live virus assays, the tetrameric Fc decreased viral load about a log more than the dimeric Fc fusion protein [360].
Again, using an avidity-based approach, Guo et al. [361] constructed a trimeric ACE2-three helix bundle (3HB) fusion protein with very high avidity towards trimeric spike protein in the sub-1 pM KD range, without the benefit of mutations to modify binding of ACE2 to RBM. This trimeric ACE2-THB fusion protein was demonstrated by cryo-electron microscopy (cryo-EM) to force all three RBDs of a spike protein into the up position in vitro [361]. Given that VOIs and VOCs are typically evolved to bind human ACE2 with higher affinity, it is expected that this trimeric ACE2-3HB fusion would bind and neutralize all high affinity VOI/VOC variants. While not an antibody-like Fc fusion protein, these types of constructs are very instructive for potential design of future ACE2-Fc fusions for clinical use.
As shown in OSM Table S1, the earliest potential ACE2-Fc fusion protein candidate for use as a COVID-19 therapeutic was Sorrento’s STI-4398 (COVIDTRAP™), but even after > 18 months, that asset still appears to be in preclinical studies. There are currently two ACE2-Fc fusion proteins registered for Phase I clinical trials, HLX71 from Hengenix Biotech (Henlius), and SI-F019, from Suchuan Baili Pharmaceutical Co, however, only the latter appears to be recruiting patients (Table 2). Additionally, according to the Hengenix/Henlius Biotech website [362], their ACE2-Fc fusion protein candidate, HLX71, has just achieved first dosed patient (NCT04583228 [7]). Very little is known about their structure other than they are described as “human ACE2-Fc fusion proteins”. Additionally, Apeiron Biologics has run Phase II clinical trials (NCT04335136 [7]) on APN01, a soluble recombinant human ACE2 (residues 1–740) [354], but this protein lacks an Fc and would be expected to have a relatively short half-life.
White et al. [363] described the construction of bifunctional ACE2-Fc fusion proteins that also targeted the pIg receptor (pIgR). pIgR is responsible for transcytosis of J-chain-containing IgAs and IgMs from circulation to the mucosa [363]. These constructs were designed to distribute the SARS-CoV-2 neutralizing fusion proteins to the environment in which the majority of SARS-CoV-2 exists, especially early on in infection. Their constructs, some of which demonstrated significant transcytosis activity, did not result in Fc-mediated activity against pIgR-expressing cells [363]. It will be interesting to see the results of future studies on the efficacy of antibodies that distribute into mucosal surfaces as compared with those that mainly stay in circulation.
Descriptions of Key Antibodies Targeting SARS-CoV-2
Introduction
Since the beginning of the pandemic a little over 2 years ago, several hundred antibodies have been isolated that bind and neutralize SARS-CoV-2. Many of these have been advanced to clinical trials, and a select few have received either EUAs or full approvals in countries across the world. This section focuses on those antibodies that have been placed into clinical development or that appear to be poised to enter clinical development. These antibodies virtually all target the receptor binding domain (RBD) of SARS-CoV-2, and are identified not only by their neutralizing activities but also by the epitopes to which they bind. In this section, the epitopes are identified in general and can be mapped to the epitope groups identified in Figure 4, which shows the location of the receptor binding motif (RBM; Fig. 4A), the residues to which ACE2 binds (Fig. 4B), and the general epitope groupings as described by Barnes et al. (Classes 1–4 in Fig. 4B) [276], Yuan et al. (RBS-A/B, -C, -D, and CR3022 site in Fig. 4B) [277] and Hastie et al. (Fig. 4C) [290]. In Sect. 6, the specific epitopes to which key IgGs, IgM, and VHH antibodies bind will be described in significantly greater detail.
Fig. 4.

A, B Structure of the SARS-CoV-2 RBD (from PDB ID 7CH5) showing the receptor binding motif (RBM; in yellow, residues 437–507 as per OSM Fig. S2) and the core (non-RBM sequences of RBD in blue). The landscape of the RBM, including the peak (top as shown here; characterized by residues S477, E484 and F486), valley (the “indentation motif”; characterized by residues K417 and Y453), and the mesa (large hump; characterized by residues Q498 and N501), are shown as described by Hastie et al. [290]. The escarpment is the area occupying the base under the steep slope, as depicted by residues N343, V367, Y369, and S375. The inner face, which is buried when the RBD is in the closed (or down) position, is on the left. The outer face, which is exposed with the RBD in both the closed (down) and open (up) positions, is on the right of the RBD as depicted here. B The 17 residues that specifically contact human ACE2 receptor (see OSM Fig. S2) are shown in white, and several residues throughout the RBD are labeled for reference. As depicted here, the Class I antibodies described by Barnes et al. [276], as well as the RBS-A and B antibodies of Yuan et al. [277] tend to bind in the area of the peak toward the inner face; Class 2 antibodies [276] and RBS-C epitope antibodies [277] tend to bind on the outer face of peak area, Class 3 [276]/RBS-D [277] antibodies bind at the lower outer face, also known as the S309 site or the proteoglycan epitope site [277] and typically interact with the glycan attached to residue N343 [277], and Class 4 antibodies [276] such as CR3022 bind on the inner side of the lower part in the area of residues Y369 and S383. C The RBD with the overlapping epitope groups described by Hastie et al. [290], RBD-4, RBD-5, and RBD-7 are shown in red, orange, green, light blue and pink, respectively. These seven epitope groups described by Hastie et al. [290] and adopted herein are shown in more detail in Table 5. The PDB program [201, 202] was used to generate and annotate the structures. ACE2 angiotensin-converting enzyme-2, PDB Protein Data Bank, RBD receptor binding domain, RBM receptor binding motif, RBS receptor binding site
Antibodies to SARS-CoV-2 That Have Received Emergency Use Authorizations (EUAs) or Full Approvals
Table 4 shows the antibodies that have received EUAs as well as a few others who have completed, or nearly completed, trials to support such authorizations. As noted previously, the EUA for use of convalescent plasma was approved in August 2020, based on data suggesting that CPT provided a 37% reduction in mortality of hospitalized patients over seven days [150]. It would seem that this set a relatively low bar for mAb therapy to pass in order to achieve potential EUAs.
Table 4.
SARS-CoV-2 neutralizing antibodies receiving Emergency Use Authorizations (EUAs) or are likely close to receiving EUAs
| Antibody | Sponsor | Significant development date(s) a | Clinical Trial(s) b | Dose | Clinical sample size | Efficacy (as compared with placebo) | Grant population and notes | Referencesc |
|---|---|---|---|---|---|---|---|---|
| Convalescent plasma | Multiple |
US EUA 8/23/20; WHO EUA 8/25/20 |
> 100 trials | NA | NA | 37% reduction in mortality over 7 days | Hospitalized patients with COVID-19 | [150] |
| Bamlanivimab | Eli Lilly/Abcellera |
US EUA 11/9/20; US EUA revoked 4/16/21; EMA EUA 5/3/21 |
Phase II BLAZE-1 trial (NCT04427501) 6/17/20–9/20/20 |
700 mg (2.8 g and 7.0 g also tested) | 777 patients | 70% reduction over placebo in ERV or hospitalizations | Non-hospitalized patients with moderate COVID-19 disease; revoked due to inactivity on variants | [370, 373] |
|
REGEN-COV™ (US), Ronapreve™ (UK) (casirivimab plus imdevimab) |
Regeneron |
Approved by UK 8/26/21; EMA, 11/11/21; US EUA 11/20/20; EMA EUA 2/26/21; India EUA 5/5/21d |
Phase III (NCT04425629, NCT04452318) 6/16/20–9/20/20 and continuing |
700 mg and up | 5531 patients | 70% reduction in COVID-19 related hospitalization and death | Non-hospitalized patients with moderate COVID-19 disease; EUA approved for new dose and subcutaneous formulation; EUA expanded in August 2021 to include post-exposure prevention | [265, 366, 369, 374] |
|
Regkirona™ Regdanvimab (CT-P59) |
Celltrion |
Approved by Korea MFDS, 9/17/21 and EMA, 11/11/21; Korea EUA 2/5/21; EMA EUA 3/26/21 |
Phase II/III (NCT04602000; EudraCT: 2020-003369-20) |
40 mg/kg | 327 patients | Reduced risk of hospitalization or death in high-risk patients by 72%; 70% for all patients | Non-hospitalized patients with moderate COVID-19 disease | [367, 371, 375] |
|
Bamlanivimab (LY3819253) plus Etesevimab (LY3832479) |
Eli Lilly (Abcellera)/Shanghai Junshi Bioscience |
Approved in UK 8/26/21; US EUA 2/9/21; EMA EUA 5/3/21 |
Phase III BLAZE-1 trial |
700 mg bamlanivimab plus 1400 mg etesevimab | 769 high-risk patients | 70% reduction (high dose) and 87% reduction (low dose) in risk of hospitalization or death | Non-hospitalized, ambulatory patients; use halted nationwide in US June, 2021 due to concern about inactivity on variants; then reinstated to combat Delta | [368, 376, 377] |
|
Xevudy™ (UK); Sotrovimab (GSK4182136; VIR-7831) |
GSK/Vir Biologics |
US EUA 5/26/21; EMA EUA 5/21/21 |
Phase III COMET-ICE trial (NCT04545060) | Single dose IV 500 mg | 868 patients | 85% reduction in hospitalization for more than 24 h or death | Treatment of mild-to-moderate non-hospitalized COVID-19 adult patients and pediatric patients (12 years of age and older weighing at least 40 kg) | [365, 371, 378] |
|
Evusheld™ (US); AZD7442 (Tixagevimab plus cilgavimab) |
AstraZeneca/Vanderbilt | US EUA 12/8/21 |
Phase III PROVENT trial (NCT04625725); Phase II/III ACTIV-2 trial (NCT04518410) |
Tixagevimab (300 mg) + cilgavimab (300 mg) | 5197 subjects | Reduced risk of developing symptomatic COVID-19 by 77% over a period of 183 days | Prophylactic; Risk of developing symptomatic COVID-19, even in subjects with significant comorbidities | [322]e |
|
Bebtelovimab (LY-CoV1404; LY3853113) |
Eli Lilly | US EUA 2/11/22 | Phase II BLAZE-4 trial (NCT04634409) | Bebtelovimab 175 mg IV single dose | > 500 patients | NMSDf ; Potently neutralizes all SARS-CoV-2 variants including Omicron | Treatment of mild-to-moderate COVID-19 adult patients and pediatric patients (12 years of age and older weighing at least 40 kg) | [214, 380, 381]e |
| Amubarvimab (BRII-196, P2C-1F11) plus romlusevimab (BRII-198, P2B-1G5) | Brii Biosciences | Approved in China 12/8/21; EUA application submitted to US-FDA 10/11/21 |
Phase II/III ACTIV-2 trial (NCT04518410) |
1000 mg Amubarvimab and 1000 mg romlusevimab | 837 non-hospitalized patients | Cut hospitalization and deaths 78%; reduction in hospitalization of 80% | Mild-to-moderate outpatients | [365, 382, 383]e |
ACTIV-2 Accelerating COVID-19 therapeutic interventions and vaccines, BMS Bristol Myers Squibb, EMA European Medicines Agency, EUA Emergency Use Authorization, ERV emergency room visits, GSK GlaxoSmithKline, IV intravenous, NA not applicable, NMSD no meaningful statistical data
aUS-FDA reference for EUAs [384]
bNCT registries can be found using reference [7]
cThe listed references are examples, and do not necessarily include all of these data supporting EUAs, as much of that is still unpublished. Also, refer to Kreuzberger et al. [372] for additional details
dAlso, EUAs approved in over 100 other countries
eNon-clinical reference to candidates
fWhile several hundred patients were treated with bebtelovimab, the EUA-supporting clinical trials were not powered for statistical significance [381]
On the other hand, in trials in which hospitalized patients were included, it appeared that none of the antibody therapies were significantly better at reducing advancement to next disease stages than placebo [364]. These findings were expanded recently in an ACTIV-3 randomized, controlled clinical study comparing the use of BRII-196/BRII-198 versus sotrovimab for adults hospitalized with COVID-19; neither antibody treatment provided improved clinical outcomes as compared with placebo [365]. Regeneron recently reported that use of their antibody cocktail by hospitalized patients resulted in a 36% reduction in risk of death over a 29-day period. Regeneron has submitted their data to the FDA for possible inclusion in their EUA. All of the other EUAs and most of the trials for anti-viral MAbs or combination MAb therapy for patients with COVID-19 focus on the non-hospitalized (i.e., ambulatory) patient population (Table 4) [265, 366–371]. As shown in Table 4, all of the antibodies that have been approved, granted EUAs, or are positioned for near-future EUAs have demonstrated at least 70% reduction over placebo controls in rates of hospitalization, emergency room visits, admission to ICUs, and/or death. Recently, the EUAs for both Regeneron’s REGEN-COV™ and Lilly’s bamlanivimab/etesevimab cocktail have been expanded to include prevention of COVID-19 disease in post-exposure subjects. Kreuzberger et al. [372] have provided an up-to-date, in-depth analysis of the clinical trials for antibodies targeting SARS-CoV-2. A few of these approved and emergency use antibodies are described in the following sections.
Descriptions of the Most Advanced Antibodies
Bamlanivimab LY3819253 (LY-CoV555)
Bamlanivimab (aka LY3819253, LY-CoV555, Ab169) is a human IgG1κ antibody, derived from VH1-69 and Vk1-39 germline families, isolated at Abcellera from B cells obtained from a convalescent patient and licensed to Eli Lilly for development [203]. Bamlanivimab IgG has an affinity to SARS-CoV-2 RBD of 3.5 nM, a potency in pseudovirus assays of about 6-12 ng/mL, and potency against authentic virus of about 36 ng/mL [203] (Table 5). Bamlanivimab, which binds key residues G482, E484, G485, F490, L492, N493, and S494, belongs to the RBD-2 epitope group (Table 5) and has been shown to bind both the open (up) and closed (down) conformations of RBD [203]. As would be expected from its key binding residues, bamlanivimab is sensitive to mutations in residues V483, E484, F490, and S494, which either decrease or eliminate binding and neutralization activity [326]. Conversely, mutations in residues V367, K417, S477, or N501 have no effect on binding or neutralization [326].
Table 5.
Affinity, neutralization, and biochemical data for IgG and IgM antibodies in development and comparators
| Antibody or binding protein | Source | VH/VL; isotype | Epitope classa | ACE2 block | Binds SARS-CoV1 |
Bind RBDb | Cause S1 shed | Buried Surface area Å2 | KD (nM) to CoV-2 RBD c |
IC50 (ng/mL) | PDB IDd | Key contact residues | References | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pseudo virus | Live virus | |||||||||||||
| Human ACE2 | SARS-CoV-2 | NA | NA | NA | NA | Open only | NA | 863 | 23 nM | NA | NA | 6M0J |
17 residues (See Fig. 4) |
[388] |
|
Etesevimab (LY-CoV016, CB6) |
BC/CP | VH3-66/VK1-39; HuIgG1κ LALA (FCM) |
B1, RBS-A; RBD-1 |
Yes | No | Open only | ND | 1,049 | 2.5 nM | 36 ng/mL | 380 ng/mL | 7C01 | G416, K417, Y473, A475, G476, F486, N487 | [204, 276, 323] |
|
Amubarvimab (BRII-196, P2C-1F11) |
BC/CPs |
VH3-66/VK3-20; HuIgG1κ (YTE HLE) |
RBD-1 | Yes | No | Open only | Yes | 955 | 1.7 nM | 30 ng/mL | 30 ng/mL | 7CDI | L455, K458, S459, A475, E476, F486, N487 | [212, 213] |
| C102 | BC/CPs | VH3-53/VK3-20 |
B1; RBD-1 |
Yes | No | Open only | ND | 1,022 | 27 nM | ND | 34 ng/mL | 7K8M | R457, K458, Y473, A475, S477, F486, N501, G502, Y505 | [276] |
|
HFB30132A (P4A1-2A) |
BC/CPs |
VH3-53/VK1-12 HuIgG4κ FE/YTE/S; (HLE, FCM) |
RBD-1 | Yes | No | Open only | ND | 1196 | 0.1 nM | 300 ng/mL | Ca 750 ng/mL | 7CJF | Y421, R457, K458, Y473, A475, G476, S477, Y489, N501, G502 | [235] |
|
COR-101 (STE90-C11) |
PDHAL-ND/CP |
VH3-66/VK1-9 HuIgG1κ PVALδGQS (FCM) |
RBD-1 | Yes | No | Open only | ND | 1133 | Ca. 8 nM | ND | Ca 80 ng/mL | 7B3O | N409, K416, F417, Q474, A475, S477, N487, V503, G504, Y505 | [236, 389] |
| BD-629 | BC/CPs |
VH3-53/VK3-6 HuIgG |
RBS-A; RBD-1 | Yes | No | Open only | ND | ND | 0.78 nM | 6 ng/mL | ND | 7CH5 | K417, L455, F456, A475-478, F486, N487, Y489 | [228, 277] |
| CC12.3 | BC/CPs | VH3-53/VK3-20 HuIgG1λ |
B1, RBS-A; RBD-1 |
Yes | No | Open only | ND | 863 | 8.6 nM | 18 ng/mL | 26 ng/mL | 6XC4 | K417, A419, D420, Y421, K458, S459, Y473, Q474, N487 | [257, 277] |
| S2E12 | BC/CPs | VH1-58/VK3-20; HuIgG1 | RBD-2 | Yes | No | Open only | Yes | ND | 1.7 nM | 2 ng/mL | 4.5 ng/mL | 7K45 | Y473, A475, S477, F486, N487 | [299, 390] |
| Casirivimab (REGN10933) | ITGM-VLI | VH3-11/VK1-33 HuIgG1λ |
B1; RBD-2 |
Yes | No | ND | ND | 935 | 3.4 nM | 43 ng/mL | 37 ng/mL | 6XDG | Y453, E484, G485, F486, C488, Y489 | [205, 323] |
|
Tixagevimab (AZD8895; COV2-2196) |
BC/CPs |
VH1-58/VK3-20 HuIgG1λ FE/YTE/S (HLE, FCM) |
RBD-2 | Yes | No | Open only | ND | 650 | ND |
0.7 ng/mL |
15 ng/mL | 7L7E | A475, G485, F486, N487, C488 | [210, 211, 390] |
| B1-182.1 | BC/CP | VH1-58/VK3-20; HuIgG1λ | RBD-2 | Yes | No | Open only | ND | ND | 2.6 nM | 3.4 ng/mL | 2.4 ng/mL | 7MLZ | Y473, G476, S477, N487 | [391] |
| C144 (BMS) | BC/CPs |
VH3-53/VL2-14; HuIgG1λ LS (HLE) |
B2; RBD-2 |
Yes | No | Open & closed | ND | 689 | 18 nM | 4 ng/mL | 2.6 ng/mL | 7K90 | Y449, Y473, A475, F486, Y489, F490, P491 | [217, 276] |
| HLX70 (P17) | ST-ST-HuNAL |
VH3-30/VK1-39 HuIgG1λ |
RBD-2 | Yes | No | Open & closed | ND | ND | 0.1 nM | 22-30 ng/mL | 150 ng/mL | 7CWL | N481, R484, F486, Y489 | [242, 243] |
|
Bamlanivimab (LY3819253; LY-CoV555) |
BC/CPs |
VH1-69/Vk1-39 HuIgG1λ |
RBD-2 | Yes | No | Open & closed | ND | ND | 3.5 nM | 12 ng/mL | 36 ng/mL | 7KMG | G482, E484, G485, F490, L492, N493, S494 | [203] |
|
Regkirona (Regdanvimab; CT-P59) |
BC/CPs-PL | VH2-70/VL1-51 HuIgG1λ | RBD-2 | Yes | No | Open only | ND | 938 | 0.25 nM | 10 ng/mL | 8 ng/mL | 7CM4 | Y449, N450, G485, F486, S494, Q495 - TURNED | [206] |
|
LYCovMab BA4101 (CA521 FALA) |
ITGM-PL |
VH4-34/VK3-6 HuIgG4λ-PAA (FCM) |
RBD-2 | Yes | No | Open & closed | ND | ND | 0.7 nM | 15–18 ng/mL | Ca. 105 ng/mL | 7E23 | G485, F486, Y489 | [230] |
| COVA2-39 | BC/CPs |
VH3-53/VL2-23 HuIgG1 |
B2, RBS-B; RBD-2 |
Yes | No | Open only | ND | 744 | 21 nM | 36 ng/mL | 54 ng/mL | 7JMP | E484, F486, N487, C488, Y489, F490 | [277, 392–394] |
| CV07-250 | BC/CPs |
VH1-18/VL2-8 IgG1κ |
RBS-B; RBD-2 | Yes | No | Open only | ND | 958 | 56 pM | 3.5 ng/mL | ND | ND | G446, Y449, Y453, A475, S477, T478, F486, N487, Y489, Q493, S494 | [277, 394, 395] |
| Ab2-4 | BC/CP |
VH1-2/VL2-8 IgG1κ |
RBS-B; RBD-2 | Yes | No | Open & closed | ND | ND | ND | 394 ng/mL | ND | 6XEY | F486, Y489, L492, S494 | [277, 396] |
|
IGM-6268 IgM (COV2-14) |
PDNHAL |
VH6-1/VL2-14 HuIgM |
RBD-2 | Yes | No | Mostly open | ND | ND |
100 pMe,f; 4.3 nM g |
12 ng/mL | 11 ng/mL | ND | F456, A475, E484, F486, Y489 | [244, 245] |
| P2B-2F6 | BC/CP | VH4-38/VL2-8; HuIgG1λ | RBD-4 | Yes | No | Open & closed | No | 626 | 5.1 nM | 50 ng/mL | 410 ng/mL | 7BWJ | G447, N448, Y449, N450, L452, E484, F490 | [212, 213] |
| CV07-270 | BC/CPs |
VH3-11/VL2-14 HuIgG1λ |
RBS-C; RBD-4 | Weak to no | No | Open only | ND | 825 | ND | 82 ng/mL | ND | 6XKP | G447, Y449, N450, E484, N498 | [277, 394, 395] |
| ABP-300 (MW05, MW33) | BC/CPs |
VH1-69/VK3-2 HuIgG1λ LALA (FCM) |
RBD-4 | Yes | No | ND | ND | ND | 0.4 nM | 30 ng/mL | 1000 ng/mL | 7DK0 | Y449, N450, E484, G485, Y489, F490, L492, Q493, S494, Q498, | [224, 316] |
| BD-368-2 (BGB-DXP593) | BC/CPs |
VH3-53/VK3-6 HuIgG1λ |
RBD-4 | Yes | No | Open & closed | ND | ND | 0.54 nM | 1.2 ng/mL | 15 ng/mL | 7CHC | Y449, N450, C480, N481, E484 | [397] |
|
Cilgavimab AZD1061 – COV2-2130 |
BC/CPs |
VH3-15/VK4-1 HuIgG1λ- FE/YTE/S (HLE, FCM) |
RBD-4 | Yes | No | Open & closed | ND | 740 | ND |
1.6 ng/mL |
107 ng/mL | 7L7E | K417, K444, V445, G485, F486, N487, C488 | [211, 390, 398] |
| BG10-19 | BC/CPs |
VH5-51/VL1-13-7 HuIgG1λ |
RBD-4 | No | Yes | Open & closed | ND | 1090 | ND | 3 ng/mL | 4 ng/mL | 7M6E | D420, N343, Y449 | [399] |
| CoV-2 06 | PDNHAL |
VH4-4/VL2-14 HuIgG |
Likely RBD-4 |
Yes | No | Open & closed | ND | ND | 20.8 nM | 150 ng/mL | ND | ND | T345, R346, K444, G446, G447, N448, Y449, N450 | [244, 245] |
| Imdevimab (REGN10987) | BC/CPs |
VH3-30/VL2-14 HuIgG1λ |
B3; RBD-5A |
Yes | No | Open & closed | ND | 607 | 45 nM | 41 ng/mL | 42 ng/mL | 6XDG | V445, N498 | [205, 323, 400] |
|
Bebtelovimab LY-3853113 (LY-CoV1404) |
BC/CPs |
VH2-5/VL2-14 HuIgG1λ |
RBD-5A | Yes | No | Open & closed | ND | 584 | 1.6 nM | 1-3 ng/mL | 9-22 ng/mL | 7MMO | N439, N440, V445, G447, N448, Q498, T500 | [214] |
| C110 | BC/CPs |
VH5-51/VK1-5 HuIgG1λ |
B3; RBD-5A | Yes | No | Open & closed | ND | ND | ND | 18.4 ng/mL | ND | 7K8V | Y449, N450, F490 | [276] |
| ADG-20 | BC/CPs (SARS-1 patient)-YDAF | HuIgG (HLE) | Likely RBD-5A | Yes | Yes | Open | ND | ND | 0.26 nM | 1 ng/mL | 1 ng/mL | ND | Structure shows epitope on mesa | [219, 220] |
| S309 (sotrovimab precursor) | BC/CPs (SARS-1 patient) |
VH1-18/VK3-20; HuIgG1λ |
B3; Site IV; RBD-5B | No | Yes | Open & closed | No | 1150 | 0.001 nM | 120-180 ng/mL |
69 ng/mL Pinto |
6WPS | P337, E340, N343, N343-glycan, T345, R346 | [208, 209, 276, 277, 390] |
|
Xevudy™ Sotrovimab (GSK4182136, VIR-7831) |
BC/CPs |
VH1-18/VK3-20; HuIgG1λ LS (HLE) |
RBD-5B | No | Yes | Open & closed | No | 1150 | 0.21 nM | 100 ng/mL | 100 ng/mL | 6WPS | P337, E340, N343, N343GLCN, T345, R346 | [209] |
| C135 (BMS) | BC/CPs |
VH3-30/VK1-5; HuIgG1λ LS (HLE) |
B3; RBD-5B |
No | No | Open & closed | ND | 700 | 6 nM | 17 ng/mL | 3 ng/mL | 7K8Z | N343, N343-glycan, N439, N440, L441 | [217, 276] |
|
ABBV-47D11 (HBM9022) |
ITGM | HuIgG1λ | RBD-5B | Yes | Yes | Closed only | ND | 800 | 11 nM | 61 ng/mL | ND | 7AKD | L335, P337, G339 | [231, 232] |
| COVA1-16 | BC/CPs |
VH1-46/VK1-33 HuIgG1λ |
RBD-6 | Yes | Yes | Open | ND | 844 | 46 nM | 20 | 750 | 7JMW | T500, V503 | [277, 393] |
| MW06 | BC/CPs | HuIgG1 | RBD-6 | No | Yes | 3/3 RBDs open | ND | 1555 | 5.5 nM | 252-338 ng/mL | 119-214 ng/mL | 7DPM | Y369, F374, S375, T376, F377, C379, Y508 | [316] |
| H014 | M-PL | Mouse IgG | RBD-6 | Yes | Yes | Open only | ND | 1000 | 0.1 nM | 400 ng/mL | 38,100 | 7CAH | S375, T376, F377, K378, C379, S383, R408, G413 | [277, 401, 402] |
| S2X259 | BC/CP |
VH1-69/VL1-40 HuIgG1 |
RBD-6 | No | Yes | 2/3 RBDs open | Yes | 950 | 0.5 nM | 213 ng/mL | 144 ng/mL | 7RAL | N370, T376, F377, C379, R408, V503, G504 | [262, 390] |
| CR3022 | BC/CPs SARS-CoV-1 |
VH5-51/VK4-1 HuIgG1 |
B4; RBD-7 |
No | Yes | 2/3 RBDs open | ND | 991 | Ca. 20 nM | nd | > 400 ng/mL | 6XC3, 6XC7 | F377, K378, C379, G381, G431, V433 | [255, 282] |
| EY6A | BC/CPs |
VH3-30/VK1-2 HuIgG1 |
B4; RBD-7 |
No | Yes | Open only | ND | 895 | 2 nM | ND | 70 ng/mL | 6ZER | C379, Y380, G381, V382, S383, T385 | [403, 404] |
| S2H97 | BC/CP | VH5-51/VL2-14; HuIgG1 | Cryptic siteh | No | Yes | Open only | Yes | ND | 0.04 nM | 338 ng/mL | 749 ng/mL | 7M7W | K424, D427, K462, H519 | [261] |
| S2X58 | BC/CP | VH1-46/VK1-33; HuIgG1 | ND | Yes | No | Open only | Yes | ND | 0.6 nM | 2 ng/mL | 4 ng/mL | ND | ND | [323] |
|
BI 767551 DZIF-10c i |
BC/CPs | HuIgG1 | ND | No | ND | At least one RBD open | ND | ND | 1.1 nM | 7 ng/mL | 10 ng/mL | ND | ND | [227] |
| MAD0004J08 | BC/CPs |
HuIgG1λ LALAPG/LS (FCM, HLE) |
ND | Yes | No | Open only | ND | ND | 0.02 nM | ND | 3.9 ng/mL | ND | ND | [221] |
|
JMB2002 (Ab2001.08) |
PDNHAL; YDS | HuIgG1 N297A (FCM) | ND | Yes | ND | ND | ND | ND | 3.3–5.2 nM | 4.25 ng/mL | – | ND | ND | [237] |
ACE2 angiotensin converting enzyme-2, BC/CPs B-cells from convalescent patients, BMS Bristol Myers-Squibb, Ca. approximately, Fc fragment crystallizable, FCIN Fc increased activity with Fc receptors, FCM Fc muting (silencing activity on Fc receptors), FE/YTE/S L234F L235E M252Y S254T T256E P331S mutations of hinge/Fc, GAALIE G236A/A330L/I332E Fc mutations to increase activity with Fc receptors, GMNDAL Sorrento’s G Mab Naive donor antibody library, GSK GlaxoSmithKline, HLE half-life extension, HuIgG1 human immunoglobulin G-1, HuIgM human immunoglobulin M, ITGM-PL Immunized Tg mice—phage library, LALA L234A L235A mutations of IgG1 hinge to dampen Fc activity with Fc receptors, LALAPG/LS L234A/L235A/P329G/M428L/N434S hinge/Fc mutations to dampen Fc activity with Fc receptors and increase half-life, LS M428L/N434S mutations for increasing half-life, MD mammalian display, M-PL mouse antibodies panned by phage display library, NA not available, ND no data, NR no published reference, NTD N-terminal domain, NYR not yet recruiting (clinical trial), PDB Protein Data Bank, PDHAL-CP phage displayed human antibody library constructed from convalescent patients, PDNHAL phage displayed naive human antibody library, PL phage library, PVALδGQS E233P/L234V/L235A/G236D/D265G/A327Q/A330S mutations to dampen Fc and complement activity, RBD receptor binding domain, SARS-CoV-1 severe acute respiratory syndrome coronavirus-1, ST-ST-HuNAL name of Henlius phage displayed naïve human antibody library, Tg transgenic (mice producing human antibodies), Unk unknown to authors, VK variable kappa (light chain), VL variable lambda (light chain), VLI VelocImmune mice (producing human antibodies), YDAF yeast display affinity maturation, YDS yeast display selection, YTE M252Y/S254T/T256E IgG Fc mutations for increasing half-life
aEpitope classes, “B-1” through “B-4” represent the classes described by Barnes et al. [276]; RBS-A through “RBS-C” represent the epitope groups described by Yuan et al., [277]
bAbility of antibody to bind RBD in open (up) and/or closed (down) conformation
cRounded to two significant digits
dPDB entries reference [201]
eNT100 (100% neutralization)
fData are presented based on IgM format, which adds significant avidity effects [245, 246]
gData based on IgG format
hOn backside of RBD (see Fig. 5)
iDevelopment discontinued (7/26/21)
Bamlanivimab was the first antibody to be granted an EUA by the FDA for treatment of COVID-19 (EUA granted 11/9/2020) (Tables 2 and 3), but it suffered from losing potency against several key variants, particularly Alpha, Beta, and Gamma, all three of which possess the E484K mutation (Fig. 2), and Delta [385–387] (Table 6). This ultimately led the FDA to revoke EUA on 16 April 2021 for use of bamlanivimab as a single agent, which resulted in bamlanivimab no longer being developed as a single agent for treatment of COVID-19.
Table 6.
Neutralization of SARS-CoV-2 wild-type virus and variants by selected monoclonal antibodiesa
| Antibody | Epitope group | Neutralization (IC50 in ng/mL in pseudovirus assays) against SARS-CoV-2 variants | Referencesb | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Wild-type | WT-D614G | Alpha (B.1.1.7) |
Beta (B.1.351) |
Gamma (P.1, B.1.1.28) |
Delta b (B.1.617.2) |
Epsilon (B.1.427/B.1.429) |
Omicron (B.1.1.529.1; BA.1) b |
|||
| Bamlanivimab (LY3819253; LY-CoV555) | RBD-2 | 6–12 | 3–5 | 4–9 | > 10,000c | > 10,000 | 8311–> 10,000 | > 10,000 | > 10,000 | [80, 386, 391, 405] |
| Etesevimab (LY-CoV016, CB6) | RBD-1 | 26–35 | 31–52 | 22–3225 | > 10,000 | > 10,000 | 12–15 | 23–54 | 7600 – > 10,000 | [386, 391, 405] |
| Bamlanivimab plus Etesevimab | RBD-2/RBD-1 | ND | 9 | 11 | > 10,000 | > 10,000 | ND | 63 | > 10,000 | [391] |
| Casirivimab (REGN10933) | RBD-2 | 4–43 | 5–7 | 7–13 | 3284–> 10,000 | 1046–6177 | 3–7 | 4–9 | > 10,000 | [80, 386, 391, 405] |
| Imdevimab (REGN10987) | RBD-5A | 32–71 | 12–20 | 7–28 | 4–24 | 4–13 | 17–455 | 75–114 | > 10,000 | [80, 386, 391, 405] |
| REGEN-COV™ (Casirivimab plus Imdevimab) | RBD-2/RBD-5A | ND | 5 | 3 | 8 | 7 | 5 | 4 | > 10,000 | [80, 386, 391] |
| Cilgavimab (AZD1061; COV2-2130) | RBD-4 | 2–13 | 2–26 | 1–12 | 3–19 | 2–14 | 4–80 | 55 | 2178–5850 | [80, 386, 391, 405–407] |
|
Tixagevimab (AZD8895; COV2-2196) |
RBD-2 | 1–5 | 1–4 | 2–11 | 4–46 | 2–46 | 2–3 | 11 | 270–1150 | [80, 386, 391, 405–407] |
| Cilgavimab plus Tixagevimab | RBD-4/RBD-2 | 9 | 4 | 4–7 | 11–12 | 6–7 | ND | 5 | 51–418 | [80, 386, 405] |
|
Bebtelovimab (LY-CoV1404) |
RBD-5A | 3 | 3 | 2–31 | 2–4 | 1–12 | ND | 1–4 | 5 | [214, 405] |
| Amubarvimab (BRII-196) | RBD-1 | 30 | 53 | 31 | 30 | 41 | 42 | ND | 7258 | [407] |
| Romlusevimab (BRII-198) | RBD-5B | ND | ~ 180 | ~ 45–70 | ~ 580 | ~ 320 | ~ 1600 | ~ 430 | 82 | [382, 408] |
| ABBV-2B04 | UNK | ND | 1 | 1 | > 10,000 | > 10,000 | ND | 3 | ND | [386] |
| ABBV-47D11 | RBD-5B | ND | 319 | 305 | 240 | 277 | ND | 456 | ND | [386] |
| ABBV-2B04 plus 47D11 |
UNK/ RBD-5B |
ND | 3 | 2 | 431 | 384 | ND | 4 | ND | [386] |
| BMS C144 | RBD-2 | ND | 4–5 | 6 | > 10,000 | > 10,000 | 5 | ND | > 10,000 | [405] |
| BMS C135 | RBD-5B | ND | 11–17 | 14 | 34 | 15 | 25 | ND | 5850 | [405] |
| SARS2-38 | Likely RBD-5A | 2 | 2 | 3 | 4 | 2 | 4 | 1 | ND | [409] |
|
Sotrovimab (GSK4182136, VIR-7831) |
RBD-5B | 100 | 58 | 80 | 50 | 66 | 42 | ND | 180–340 | [80, 407] |
| S309 (precursor to Sotrovimab) | RBD-5B | 40 | 156 | 78–209 | 82–98 | 76 | 113 | 20 | 256–281 | [80, 386, 405] |
| B1-182.1 | RBD-2 | 3 | 1 | <1 | 1 | <1 | 2 | 2 | 281 | [391, 405] |
| S2E12 | RBD-2 | 2 | 5 | 21 | 10 | 2 | 2 | 2 | 38 | [386, 405] |
| S309/S2E12 combined | RBD-5B/RBD-2 | –– | 7 | 8 | 15 | 3 | ND | 5 | ND | [386] |
| IGM-6268 (as an IgM) | RBD-2 | 6 | ND | 31 | 31 | 23 | ND | ND | 230 | [245, 410] |
| Regkirona (Regdanvimab; CT-P59) | RBD-2 | <1–10 | 2–10 | 2–6 | 66–330 | 13–40 | 15–1,237 | 366 | > 10,000 | [405, 406, 411, 412] |
| ADG20 | RBD-5Ad | 1–4 | 5 | 2–6 | 5–16 | 3–9 | 1–8 | 1 | 1100–2037 | [80, 220, 405] |
| MW05 | RBD-4 | 30 | ND | Ca. 100 | > 10,000 | > 10,000 | ND | ND | ND | [316] |
| MW06 | RBD-4 | 250–340 | ND | Ca. 300 | Ca. 500 | Ca. 500 | ND | ND | ND | [316] |
| S2X259 | RBD-6 | 74–213 | ND | 205 | 358 | 458 | 101 | ~ 220 | 588 | [262, 406] |
| DZIF-10ce | UNK | 10 (LV) | ND | 14 (LV) | 170 (LV) | ND | ND | ND | ND | [227] |
| MAD0004J08 | UNK | 4 | 4 | 4 | ND | ND | ND | ND | ND | [221] |
| CAB-B37 | RBD-1 | ND | 63 | 46 | 25 | 34 | 82 | ND | 20 | [413] |
ND no data, RBD receptor-binding domain, UNK unknown, LV authentic live virus assay instead of pseudovirus assay
aAll numbers converted to nearest integer, and where multiple data were available, ranges spanning those data are given. These numbers are from many different experiments, protocols, laboratories, and using different wild-type comparators (different wild-type viruses and sometimes D614G was used as control/comparator), so the specific numbers may not be exactly comparable. Nevertheless, they represent the overall trends of potency which are critical to neutralization of the variants
bSome of the data on Delta neutralization are from Mlcochova et al. [58]; Data for Omicron (BA.1 only) are from: Dejnirattisai et al. [80]; Iketani et al. [109] (ng/mL values extrapolated from fold increase/decrease based on D614G data provided by other authors, Zhou et al. [405] and Cameroni [406]; Brii-198 actual neutralization figures in ng/mL have not been given, but figures of neutralization curves were provided, so the data presented here were extrapolated from figures shown by Wang et al. [382] and Liu et al. [408]
cPartial loss of activity (ca. 500–8000 ng/mL) in italics; essentially complete loss is noted by > 10,000 in bold; d Presumed based on description and figure of epitope [220], but not confirmed due to absence of detailed epitope mapping data
eDiscontinued from development 7/25/21
Etesevimab LY3832479 (JS016, LY-CoV016, CB6)
Etesevimab was isolated from B cells from a convalescing patient by scientists at the Chinese Academy of Sciences as antibody CB6, which was selected from two key leads and taken into development by Shanghai Junshi Biosciences, Ltd as JS016 [204]. After Junshi agreed to a licensing deal with Eli Lilly, JS016 was further developed and given the USAN name of etesevimab. Etesevimab is a human IgG1κ antibody, derived from VH3-66/VK1-39 germline families, and modified in the lower hinge by L235A, L236A which significantly mutes Fc activity to reduce any chance of ADE [204] (Table 5). The affinity of etesevimab for SARS-CoV-2 RBD is 2.49 nM and binding to SARS-CoV-2 RBD buried a surface of 1,088 Å2 [204]. The key residues bound by etesevimab include G416, K417, Y473, A475, G476, F486, and N487, and it belongs to epitope group RBD-1 (Table 5). The potency of etesevimab in pseudovirus assays is about 35 ng/mL, and potency against authentic virus is about 380 ng/mL. Etesevimab is most sensitive to potential escape mutations at positions K417, N460, Y473, A475, E484, N487, Y489, and Q493 [169, 322, 323].
While etesevimab has been developed in some clinical trials as a stand-alone antibody (Table 2), its significance is as a partner antibody in a bamlanivimab-etesevimab cocktail, which was granted an EUA by the FDA and the EU on 2/9/21 and 5/3/21, respectively. This cocktail went out of style when Alpha and Beta were dominant mutants, but it came back into significant usage once Delta became the dominant variant, mostly due to the strength of etesevimab against certain SARS-CoV-2 variants, especially Delta (Table 6). The combination of bamlanivimab/etesevimab are still sensitive to mutations E484D, Q493R/K, and S494P [169]. Because bamlanivimab contributes so little efficacy over and above etesevimab in this cocktail, its inclusion has been questioned recently [414]. The combination of bamlanivimab-etesevimab is ineffective against Omicron BA.1 [391] and BA.2 [109], which resulted on 24 January 2022 in a modification of the FDA EUA resulting in the significant reduction in use of bamlanivimab-etesevimab to treat COVID-19 [415].
REGEN-COV™
REGEN-COV™ (known as Ronapreve™ in the UK and EU) is a combination of casirivimab (REGN10933) and imdevimab (REGN10987) for treatment of ambulatory patients infected with SARS-CoV-2, as well as prophylaxis for subjects post-exposure to COVID-19. Casirivimab (REGN10933) is a human IgG1κ antibody derived from immunized VelocImmune transgenic mice [205]. It is a VH3-11/VK1-33 germline derived antibody that has an affinity for SARS-CoV-2 RBD of 3.4 nM, burying a surface of 935 Å2 on RBD [345]. The potency of casirivimab in pseudovirus assays and authentic virus assays is 43 ng/mL and 37 ng/mL, respectively (Table 5). The key residues bound by casirivimab include E484, G485, F486, C488, and Y489 (Table 5), placing it into epitope group RBD-2 (Table 5). Casirivimab is most sensitive to potential escape mutations K417E, Y453F, L455F, F486V, and Q493K [325].
Imdevimab (REGN10987) is a human IgG1λ antibody derived from B cells from a convalescing patient [205]. It is a VH3-30/VL2-14 germline-derived antibody that has an affinity for SARS-CoV-2 RBD of 45 nM, burying a surface of 607 Å2 on RBD [345]. The potency of casirivimab in pseudovirus assays and authentic virus assays is 41 ng/mL and 42 ng/mL, respectively (Table 5). The key residues bound by casirivimab include V445 and N498 (Table 5), placing it into epitope group RBD-5A (Table 5), a non-overlapping epitope with casimirimab (Protein Data Bank [PDB] ID 6XKG; [205]), which allows the two antibodies to bind SARS-CoV-2 RBD simultaneously. Casirivimab is most sensitive to potential escape mutations N440D, K444Q and V445A [323, 325]. As noted above, casirivimab and imdevimab are both IgG1 isotype antibodies. Both antibodies induce ADCC and ADCP, but have not exhibited ADE of viral activity [205], a theoretical concern that some antibody developers have taken seriously, as discussed in Section 4.3.2.
Casirivimab and imdevimab have been developed from the beginning as a cocktail of two antibodies to treat SARS-CoV-2 infections [205] (Tables 2 and 3). REGEN-COV™ is indicated for non-hospitalized patients with moderate COVID-19 disease, and more recently received expanded EUAs that include post-exposure prevention (Table 4). New studies have demonstrated that seronegative hospitalized patients also received significant benefit from treatment with REGEN-COV for reduction in viral load, risk of death, and risk of mechanical ventilation [416]. This antibody combination, called REGEN-COV™ in the United States and Ronapreve™ in the UK where it recently (26 August 21) gained full approval and EU, the most widely used therapeutic and prophylactic antibody (either alone or in combination) in the world.
When combined, the only mutation that was thought to be able to escape the two antibodies was a non-contact residue, E406W [169, 416], which is not found in any of the major variants thus far (Fig. 2). This, however, was pre-Omicron. Unfortunately, even the combination of these two antibodies is ineffective against Omicron BA.1 [80, 386, 391] and BA.2 [109], which resulted on 24 January 2022 in a modification of the FDA EUA resulting in the significant reduction in use of REGEN-COV™ to treat COVID-19 [415].
Regkirona™ (Regdanvimab; CT-P59)
Regdanvimab (VT-P59) is a human IgG1λ antibody isolated from a phage library constructed of genes from peripheral B cells from a COVID-19 convalescent patient [206]. It is derived from VH2-70/VL1-51 germlines and has a high affinity for SARS-CoV-2 RBD of 27 pM, burying a surface of 938 Å2 on RBD [206]. The potency of regdanvimab in pseudovirus assays and authentic virus assays is 10 ng/mL and 8 ng/mL, respectively (Table 5). The key residues bound by casirivimab include Y449, N450, G485, F486, S494, Q495 (Table 5), placing it into epitope group RBD-2 (Table 5). Regdanvimab is resistant against key RBD mutations K417N, E484K, and N501Y, as well as other mutations A435S, W436R, K458R, and V483A [206, 412]. Additionally, regdanvimab has demonstrated in vitro and in vivo resistance to SARS-CoV-2 variants Gamma [411], Delta [411], and Alpha [412], while showing some reduction of neutralization activity against Beta [412] (Table 6). Regdanvimab is most sensitive (approximately 35-fold loss in activity) to the potential escape mutation L452R, a mutation found in the Delta, Epsilon, and Kappa variants [411]. Nevertheless, regdanvimab protected animals infected with the Delta variant, suggesting that it retained sufficient potency in spite of the loss of neutralizing activity [411]. Despite the high potency of regdanvimab and its overall lack of sensitivity to key RBD mutations, Celltrion has recently forwarded a second antibody, CT-P63, into clinical trials (Table 2) with the intention of combining the two antibodies in a cocktail for potential parenteral as well as nebulized formulations and delivery [241].
Sotrovimab GSK4182136 (VIR-7831) and VIR-7832
Sotrovimab (GSK4182136, VIR-7831, CB6), now approved in the UK as Xevudy™, is an IgG1κ antibody that binds SARS-CoV-2 RBD away from the RBM. Sotrovimab (VIR-7831) and VIR-7832 are both derived from antibody S309, which was isolated from B cells from a 2003 SARS-CoV-1 convalescent patient [208, 209]. Thus, both antibodies bind an epitope shared by both SARS-CoV-1 and -CoV-2. Both antibodies have been modified with the Xencor Xtend® M428L/N434S (“LS” mutant) half-life extension mutations [417] and both have active Fc functionality with full capability to interact with FcγRs on immune cells [209].
Sotrovimab binds to an epitope in the spike RBD that is highly conserved in the Sarbecovirus subgenus which does not compete with ACE2 binding [208]. This epitope, which we are labeling as RBD-5B based on descriptions by Hastie et al. [290], mostly focuses around residues N334-to-R346, also contains a glycan attached to N343, does not overlap with any of the mutations associated with VOCs prior to Omicron [209]. Omicron BA.1.1 has the R346K mutation which, based on epitopes, should cause significant alteration in binding to antibodies of the RBD-4, RBD-5A, or RBD-5B groups (Fig. 6A).
Fig. 6.


A, B The sequence of SARS-CoV-2 RBD from residues 317 to 424 (A) and residues 425 to 532 (B), annotated with sequence of the RBM (underlined in B) and the 17 residues to which human ACE2 bind (blue shaded residues and asterisks). Additionally, mutations found in each of the major variants (yellow shaded residues; gray shaded for those mutations only found in some of the isolates of that variant), and epitopes for 40 antibodies known to target SARS-CoV-2 RBD (red letters/green shade) are provided. Purple shaded residues are those epitopes to which those particular antibodies bind to a second RBD at the distal end of the RBD (intraspike cross-linkers). The epitopes were retrieved from several references, including Barnes et al. [276], Yuan et al. [277], Niu et al. [435], Deshpande et al. [436], Dumet et al. [400], and Wang et al. [391]. Additional epitopes not adequately described in the literature were determined or corrected using the Protein Data Bank entries for the RBD-antibody co-crystal structures, with identification of the epitopes based on residues within 5 Å of the antibody CDR loops. The PDB ID entries used are provided in Table 5. Epitopes for IGM-6268 (COV2-14) and COV2-06 [244, 245] were determined based on mutational analysis, so are likely incomplete. At the right are notations for Classes into which Barnes et al. [276] placed these antibody epitopes (Classes 1-4), notations for how Yuan et al. [277] categorized the RBD-binding antibodies (RBS A-through-C and “S309-like” and “CR3022-like”), and the epitope groups into which we are currently placing these antibodies (RBD1-7, based on the epitope groupings described by Hastie et al. [290]. In this Figure, we are equating the epitope of sotrovimab with the antibody from which it was derived, S309. ACE2 angiotensin-converting enzyme-2, CDR complementarity determining region, PDB Protein Data Bank, RBD receptor binding domain, RBM receptor binding motif, RBS receptor binding site, SARS-CoV-2 severe acute respiratory syndrome coronavirus-2
Due to its epitope on the outer face of the RBD, sotrovimab can bind RBDs in both the open (up) and closed (down) conformations. RBD binding ELISA EC50 values for VIR-7831 and VIR-7832 were 20.40 ng/mL and 14.9 ng/mL, respectively, the affinity (KD) measured by SPR for sotrovimab was 210 pM. S309, the precursor to both VIR-7831 and VIR-7832, buries a surface area on RBD of 1150 Å2 (Table 5; [208]). In a cell-based assay, VIR-7831 and VIR-7832 demonstrate viral neutralization with IC50 values of 100.1 and 78.3 ng/mL, respectively, and IC90 values of 186.3 and 253.1 ng/mL, respectively [209]. Sotrovimab also neutralized live virus with an IC50 of about 100 ng/mL (Table 5). Key residues for binding of S309, the precursor to sotrovimab, to RBD include P337, E340, N343, N343GLCN, T345, R346 (Table 5). Based on those data, it is not surprising that the neutralizing activity of sotrovimab (VIR-7831) was abolished by mutations P337L/R or E340A/K [209]. Due to the very different epitope from many of the antibodies toSARS-CoV-2 (Table 5), sotrovimab was found to be resistant to all of the RBM-based mutations found in the VOCs and VOIs [209]. Additionally, both sotrovimab (VIR-7831) and VIR-7832 are resistant to VOCs Alpha (B.1.1.7, UK variant), Beta (B.1.351, South African variant), and Epsilon (P.1, Brazilian variant) [209], but lose approximately two- to threefold neutralization activity against Omicron [405] (Table 6).
Sotrovimab, which is currently being studied in Phase II/III clinical trials, received an EUA from the FDA on 26 May 2021, shortly after receiving an EUA from the EU on 21 May 2021. The EUAs are based on the Phase II/III COMET-ICE clinical trials , in which it demonstrated an efficacy of 85% [155] (Table 3). Currently, the EUAs are limited to 500 mg IV infusions, but sotrovimab is also being studied for possible IM injections (Table 2). Sotrovimab recently (2 December 2021) received full approval in the UK (Table 2). Due to its partial loss of activity against Omicron BA.1 and significant loss of activity against BA.2 [109, 407], the FDA limited use of sotrovimab in certain georgraphical areas in which sotrovimab-resistant Omicron variants were prevelant [418].
VIR-7832, which is currently in Phase I/II clinical trials (NCT04746183) also has been modified in its Fc with the “GAALIE” (G236A, A330L, I332E) mutations in the Fc domain which have been associated with activation of CD8+ T cells in other respiratory viral infections [419]. The GAALIE mutations in VIR-7832 were shown to enhance binding to FcγRIIa and FcγRIIIa without a concomitant increase in binding to FcγRIIb, which reportedly activates CD8+ T cells to respond to respiratory viral infections [209]. The increased activating receptor Fc activity has not been associated with a concomitant increase in ADE [209].
AZD7442
AZD7442 is a fixed-dose combination of two antibodies, cilgavimab (aka AZD1061, COV2-2130) and tixagevimab (aka AZD8895, COV2-2196) (Tables 2 and 3), both of which were isolated from the B cells of convalescent patients at Vanderbilt University [210]. Both tixagevimab (AZD8895) and cilgavimab (AZD1061) are modified IgG1κ isotypes that bind non-overlapping epitopes on the RBD of SARS-CoV-2 [210, 211]. Both antibodies possess the “TM” triple mutations L234F/L235E/P331S described by Oganesyan et al. [420] to decrease their binding to human FcγRI, FcγRIIa, FcγRIIIa, and C1q, resulting in reduced ADCC, ADCP, and complement-dependent cytotoxicity (CDC). These mutations also reduce the theoretical risk of ADE [196]. Additionally, both antibodies have the M252Y/S254T/T256E “YTE” modification [421] in their Fc to extend their half-life in circulation (called “LAAB, long-acting antibody”). It was recently demonstrated that a 300 mg intramuscular dose of AZD7442 resulted in serum titers tenfold and threefold above those of convalescent plasma at 3 and 9 months, respectively, indicating that one dose of AZD7442 could potentially provide protection for up to a full year [422].
Cilgavimab (AZD1061, COV2-2130) is a human IgG1κ antibody derived from B cells from a convalescing patient. It is a VH3-15/VK4-1 germline derived antibody that buries a surface of 740 Å2 on RBD [211]. The potency of cilgavimab in pseudovirus assays and authentic virus assays is 1.6 ng/mL and 107 ng/mL, respectively (Table 5). The key residues bound by cilgavimab include K417, K444, V445, G485, F486, N487, C488 (Table 5), placing it into epitope group RBD-4 (Table 5). This is a non-overlapping epitope with tixagevimab (PDB ID 7L7E), which allows the two antibodies to bind SARS-CoV-2 RBD simultaneously [211]. Cilgavimab is most sensitive to potential escape mutations N444R/E [211].
Tixagevimab (aka AZD8895, COV2-2196) is a human IgG1κ antibody derived from B cells from a convalescing patient. It is a VH1-58/VK3-20 germline derived antibody, similar to S2E12 [211, 299], and has significant sequence similarity and nearly identical RBD-binding interface as S2E12, the prototypical antibody of the RBD-2 epitope group. Tixagevimab displays an interesting binding motif, forming an “aromatic cage” at the VH/VL interface using essentially germline-encoded residues from CDR-L1, CDR-L3, CDR-H2, and CDR-H3 [211]. This similar binding motif has apparently been found in several antibodies isolated from convalescent patients [211]. Tixagevimab also has a disulfide bond “staple” in CDR-H3 that is required for optimal activity; again, this feature has been observed in several clonotypes from convalescent patients, suggesting that these features together are common in the B cell response to SARS-CoV-2 [211].
Tixagevimab buries a surface of 650 Å2 on RBD [82], and is very potent, with neutralization in pseudovirus and authentic virus assays of 0.7 ng/mL and 15 ng/mL, respectively (Table 5). The key residues bound by tixagevimab include A475, G485, F486, N487, and C488 (Table 5), placing it into epitope group RBD-2 (Table 5). Tixagevimab is most sensitive to potential escape mutations at residues G476, F486, and N487 [323, 371]. Together, cilgavimab (AZD1061, COV2-2130) and tixagevimab (AZD8895, COV2-2196) are very potent against both specific mutations [211] and current VOCs/VOIs [386, 423], including retention of activity against Omicron BA.1 (Table 6).
To date, AZD7442 is being developed as a prophylactic antibody combination rather than as a therapeutic combination as many of the other lead antibodies such as REGEN-COV™. Additionally, AZD7442 is being developed as both an IV infusion as well as an intramuscular (IM) injection. In June 2021, readouts from the Phase III STORM CHASER trial (NCT04625972 [7]) on the safety and efficacy of AZD7442 for the prevention of symptomatic COVID-19 in participants recently exposed to SARS-CoV-2 did not meet primary endpoints. While AstraZeneca continues to mine the data from that trial and run additional trials on prophylaxis for subsets of subjects such as those who have not been vaccinated but remain PCR-negative, development will now be more difficult and restrictive. On the other hand, in the PROVENT trial (NCT04625725 [7]), AZD7442 was demonstrated to reduce development of symptomatic COVID-19 by 77% for up to 183 days (~ 6 months), demonstrating a path forward for long term prevention [379], which led the FDA to issue an EUA on December 8 2021 for AZD7442 (tradename Evusheld™) for long-term (up to 6 months) pre-exposure prophylaxis in immunocompromised subjects, and individuals who cannot be vaccinated against SARS-CoV-2 for medical reasons [424].
Amubarvimab (BRII-196, P2C-1F11) and Romlusevimab (BRII-198, P2B-1G5)
Amubarvimab (BRII-196, P2C-1F11) and romlusevimab (BRII-198, P2B-1G5) are two human IgG1 antibodies derived from B cells from convalescent patients, isolated as part of a huge antibody isolation and analysis program in China [212]. In that screen, P2C-1F11, P2B-2F6, and P2C-1A3 were the most potent, with IC50 values in pseudovirus assays of 30, 50 and 620 ng/mL, and 30, 410 and 280 ng/mL in SARS-CoV-2 live virus assays, respectively [212, 213]. From these data, P2C-1F11 was chosen to be one of two antibodies in a cocktail. Eventually, the non-overlapping antibody, P2B-1G5 (BRII-198), was chosen as the second of the pair, although little information has been published on that candidate.
Amubarvimab (BRII-196, P2C-1F11) is a human IgG1κ antibody derived from B cells from a convalescing patient. It is a VH3-66/VK3-20 germline derived antibody that binds RBD with an affinity of 1.7 nM and buries a surface on RBD of 955 Å2 [212]. BRII-196 has a neutralization potency of 30 ng/mL in both pseudovirus and authentic virus assays (Table 5). BRII-196 belongs to the RBD-1 epitope group (Table 5) and the key residues it binds include L455, K458, S459, A475, E476, F486, N487 (Table 5). A key feature of BRII-196 is that it causes rapid shedding of the S1 subunit, inhibiting its ability to enter cells [213]. This additional mechanism of action is shared by some (e.g., S2E12 and S2X259 induce S1 shedding as well; Table 5), but not all (i.e., P2B-2F6 was shown not to induce S1 shedding), RBD binding antibodies. BRII-196 is most sensitive to potential escape mutations Y421A, L455A, F456A, R457A, Y473A, N487A, and Y489A [213]. The amubarvimab-romlusevimab combination product received full approved by the China National Medical Products Administration (NMPA) on 8 December 2021, and is currently in late-stage clinical trials for broader geographical registration [212] (Table 2). Brii Biosciences Ltd has recently (10/8/21) submitted an EUA application to the FDA for use of amubarvimab plus romlusevimab for treatment of COVID-19.
BMS-986413 (C144-LS) and BMS-986414 (C135-LS)
BMS-986413 (C144-LS) is a human IgG1λ antibody derived from B cells from a convalescing patient. It is a VH3-53/VL2-14 germline derived antibody that competes with ACE2 binding [276], has an affinity for SARS-CoV-2 RBD of 18 nM [217], and buries a surface area of 689 Å2 on RBD [345]. The potency of C144 in pseudovirus assays and authentic virus assays is 4 ng/mL and 2.6 ng/mL, respectively (Table 5). The key residues bound by C144 include Y449, Y473, A475, F486, Y489, F490, P491 (Table 5), consistent with its addition to the RBD-2 epitope group (Table 5). A characteristic of C144, shared by certain other RBD-binding antibodies such as BG10-19, is the ability to bind two RBDs simultaneously, one with primary binding activity that competes with ACE2 binding, while binding a second RBD at a distal site [276]. This cross-linking locks the RBDs in a closed conformation adding to the overall potency of the response. C144 is most sensitive to potential escape mutations E484K and Q493R [277, 425].
BMS-986414 (C135-LS) is a human IgG1κ antibody derived from B cells from a convalescing patient. It is a VH3-30/VK1-5 germline derived antibody that has an affinity for SARS-CoV-2 RBD of 6 nM, burying a surface of 700 Å2 on RBD [276]. The potency of C135 in pseudovirus assays and authentic virus assays is 17 ng/mL and 3 ng/mL, respectively (Table 5). The key residues bound by C135 include N343, the N343 glycan, N439, N440, and L441, similar to S309 (sotrovimab) (Table 5). Thus, it was placed into epitope group RBD-5B (Table 5). The epitope for C135 does not overlap with the primary epitope of C144, but interestingly, the C135 epitope overlaps significantly with the C144 distal binding site (its binding site on the “second” RBD). As an RBD-5B epitope mAb, C135 is essentially resistant to all mutations in the RBMs exhibited by VOCs and VOIs. C135 is most sensitive to the potential escape mutations R346S and N440K, with partial sensitivity to mutation N439K [276].
Both C135 and C144 have been modified by insertion of the M428L/N434S (“LS“) mutations which increase the circulating half-life of the antibodies by modifying the interaction with the recycle receptor, FcRn [417]. The C135/C144 cocktail is currently in Phase II/III clinical trials, including participation in the large ACTIV-2 trial of ambulatory patients (Table 2).
LY-CoV1404
Eli Lilly placed a back-up antibody, LY-CoV1404, into clinical trials, where it is currently in Phase II (Table 2). LY-CoV1404 belongs to the RBD-5A epitope group, so it interacts with RBD residues mostly at the periphery or outside the ACE2 binding site, with an epitope that significantly overlaps that of imdevimab [214]. This antibody, however, is both more potent than imdevimab, with IC50s for pseudovirus and authentic virus neutralization assays of 1–3 ng/mL and 9–22 ng/mL, respectively [214], and more broadly acting than imdevimab. Even though LY-CoV1404 binds N439 and N501, both residues of which are found in certain variants (Fig. S2), it retains complete neutralization capability on variants possessing those mutations. As a result, LY-CoV1404 has sub-10 ng/mL neutralization potency against all variants tested (Table 6), making it a very interesting candidate of the future, against a known and proven epitope. In the face of the new Omicron mutants BA.1 and BA.2, both of which are potently neutralized by bebtelovimab (Tables 6, 7), the FDA granted bebtelovimab an EUA on 11 February 2022 [380], even though clinical data had yet to reach statistical significance [381].
Table 7.
Examples of antibodies effective against Omicron BA.1 and/or BA.2 variantsa
| Antibodies | Company/institution | Most advanced stage |
Epitope group | Description | Neutralization IC50 (ng/mL) against Omicron variant | Reference | ||
|---|---|---|---|---|---|---|---|---|
| BA.1 | BA.1.1 | BA.2 | ||||||
| Bebtelovimab (LY-CoV1404) | Lilly | EUA | RBD-5A | Human IgG1 | 2–5 | 2 | 2 | [109, 214] |
| Tixagevimab (COV2-2196) | AstraZeneca | EUA | RBD-2 | Human IgG1 (HLE, FCM) | 270–1150 | 470 | ND | [109, 214, 254, 405] |
| Cilgavimab (COV2-2130) | AstraZeneca | EUA | RBD-2 | Human IgG1 (HLE, FCM) | 2178-5850 | > 10,000 | ~ 30-50 b | [109, 254] |
| EvuSheld™ (AZD7442; tixagevimab + cilgavimab) | AstraZeneca | EUA | RBD-2/RBD-4 | Human IgG1 (HLE, FCM) | 51–418 | ND | 20 | [406, 407, 409] |
| Sotrovimab/S309 VIR-7831 | Vir Biotechnology | EUA | RBD-5B | Human IgG1 (HLE, FCM) | 181–356 | 314 | 944-2,200 b | [109, 214, 405, 407] |
| Romlusevimab (BRII-198) | Brii Biosciences | Approved in China | RBD-5 | IgG | ~ 82 b | ~ 5760 b | ND | [109, 382, 408] |
| VIR-7832 | Vir Biotechnology | Phase I | RBD-5B | Human IgG1 (HLE, FCE) | 165 | ND | ND | [406] |
| IGM-6268 | IGM Biosciences | Phase I | RBD-2 | IgM; IN | 230 | ND | ND | [410] |
| DXP-604 | Beigene | Phase I | RBD-1 | IgG | 287 | ND | ND | [407] |
| A23.58.1 | NIH | PC | RBD-2 | IgG | 231 | ND | ND | [405] |
| A19-46.1 | NIH | PC | RBD-2 | IgG | 223 | ND | ND | [405] |
| B1.182.1 | NIH/Academic | PC | RBD-2 | IgG | 281 | ND | ND | [405] |
| S2E12 | Vir Biotechnology | PC | RBD-2 | Human IgG1 | 38 | ND | ND | [405] |
| CAB-B37 | Academic | PC | RBD-1 | Affinity-matured IgG | 20 | ND | ND | [413] |
| Nb1-Nb2-Fc | Academic | PC | ND | Biparatopic, bispecific nanobody-Fc construct | ~ 0.2 | ND | ND | [330] |
| STI-9167/STI-9199 c | Sorrento | PC | ND | Human IgG1 FCM; IN | 15 | 24 | SNd | [254] |
| ZCB11 | Academic | PC | ND | Human IgG1 | 37 | 12 | ND | [426] |
| Bn03 | Academic | PC | RBD-4 | Bispecific domain antibodies fused; INH | ~ 100 to 300 | ND | ND | [427] |
| mAbs 58, 222, β29, β40, β47, β54 | Academic | PC | ND | Various IgGs | 12–261 | ND | ND | [80] |
| S2N12, S2N28, S2K146, S2X324 | Vir Biotechnology | PC | RBM | Compete with ACE2 | 3–17e | ND | ND | [406] |
| BD55-5840, BD55-3546, BD55-5549 | Academic | PC | RBD-5 (“E1”) | Do not compete with ACE2 | 4-27e | 4–14 | 16-58 | [407] |
| BD55-3372, BD55-5514, BD55-5483, BD55-5558 | Academic | PC | RBD-7 (“F3”) | Do not compete with ACE2 | 2–20 e | 3–14 | 19-105 | [407] |
| Hu33 | Academic | PC | RBD-5B | ND | 154 | ND | ND | [428] |
| NA8 | NIH | PC | RBD-4 | IgG IN | 4 | ND | ND | [429] |
| 3B6 | NIH | PC | ND | ND | 58 | ND | ND | [429] |
| 3B8 | Academic | PC | ND | ND | < 20 | ND | ND | [430] |
ACE2 angiotensin converting enzyme-2, HLE half-life extended, EUA Emergency Use Authorization received, FCE Fc enhanced for superior Fc function, FCM Fc muted to reduce Fc activity, IN intranasal delivery, INH inhaled delivery, ND no data, NIH US National Institutes of Health, PC preclinical, RBD receptor binding domain, RBM receptor binding motif (would compete with ACE2 binding, SN strong neutralizer
aCut-off for inclusion ca. 500 ng/mL against at least one Omicron variant
bData extrapolated from neutralization baseline information on wild-type virus and fold-change reported, so should be construed as an estimate
cSTI-9199 is the intranasal formulated version of STI-9167
dWhile no numerical data have been presented, Sorrento reported that neutralization of BA.2 was on par with that of LY-CoV1404, which would make it a strong neutralizer
eSince multiple antibody candidates from the same epitope grouping were reported, these are reported together as a group, with range of results reported
New, Highly Potent Antibodies
The most pressing issues with antibodies to SARS-CoV-2 are the ability to neutralize at high potency, and the ability to potently neutralize variants that arise over time. As shown in OSM Fig. S3, even within a single immunocompromised patient, multiple mutations arose in less than 6 months. Some of the early antibodies, such as bamlanivimab, were able to potently neutralize wild-type SARS-CoV-2 (e.g., Wuhan B.1), but were susceptible to many of the mutations that quickly arose in the VOCs (Fig. 2). In an analysis of polyclonal sera, it was demonstrated that the three most sensitive positions in the RBD to potential escape mutations were found to be F456, E484, and F486 [322]. Similarly, in a separate study, mutations in residues K417, N439, L452, E484, and N501 were found to have the greatest negative effect on both antibodies and vaccine-induce antibody responses [431]. These residues are all part of the RBD and all but K417 are part of the RBM linear sequence to which ACE2 binds (OSM Fig. S2). Additionally, many of these residues are mutated in the current VOIs and VOCs (Fig. 2). Thus, these is a constant search for new antibodies that bind SARS-CoV-2 RBD with high affinity and neutralize both wild-type and all known variants with IC50s <100 ng/mL. Some new candidate antibodies that fit this profile are noted below.
ADG-20
ADG-20, which is a half-life extended version of ADG-2, a human antibody isolated from SARS-CoV-1 convalescent patients and affinity matured using yeast display [219], also appears to fall into the RBD-5A epitope group based on a figure of it binding to RBD [220], but this will need to be confirmed once structural data are published (Table 5). ADG-2 is highly potent, with an affinity of 260 pM for RBD and neutralization IC50s against both SARS-CoV-1 and SARS-CoV-2 of 1 ng/mL [219]. One of the key features to ADG-2 is that it recognizes all Clade I Sarbecoviruses, indicating that it has enormous breadth of coverage [219], which could be important both for future variants and pandemics. While data for ADG-2/ADG-20 against important variants are not yet published in peer-reviewed journal, data from Adagio’s S1 filing indicate that ADG-20 has 1–5 ng/mL IC50 neutralization activity against all variants tested, including all current VOCs [220], except Omicron, where it loses significant (approximately 250- to 500-fold) activity against BA.1 (Table 6) and essentially all neutralization activity against BA.2 [109, 407]. ADG-20 is currently in Phase II/III clinical trials (Table 2) and is being developed as an intranasally delivered antibody [220].
IGM-6268
IGM-6268 is a potent IgM that is derived from CoV2-14, an antibody isolated from a naïve human antibody library [244–246]. This antibody appears to belong to the RBD-2 epitope class and as an IgG has an affinity for SARS-CoV-2 RBD of 4.3 nM (Table 5). Conversion to an IgM isotype, however, improved the binding avidity to RBD by about 14-fold through the high avidity of IgM [245]. Similarly, the improvement in neutralization activity by converting an IgG to an IgM is about 230-fold against wild-type virus [245]. Interestingly, with mutant viruses such as Gamma and Beta, the IgG format exhibited approximately a 100-fold reduction in potency, whereas the IgM format retained very high potency [245]. IGM-6268 has demonstrated significant neutralization potency against those variants thus far tested including Omicron BA.1 [410] (Tables 6 and 7). This antibody, formatted as an IgM, is being developed by IGM Biosciences as an intranasally delivered antibody and is currently in Phase I clinical trials [410] (Table 2).
SARS2-38
A very recent report described a new anti-SARS-CoV-2 antibody, SARS2-38, that also fits into the epitope group as imdevimab and LY-CoV1404 [409]. This murine-derived antibody had good affinity for RBD at 6.5 nM and was highly potent, with IC50s for neutralization around 2 ng/mL. Importantly, SARS2-38 was able to neutralize all the VOCs tested, including Delta, at IC50s below 7 ng/mL [409] (Table 6). Additionally, SARS2-38 provided full protection of mice against both Beta and Kappa variants of SARS-CoV-2 [409]. With the expectation that this antibody retains its fully functionality upon humanization, it could be another exciting, highly potent and variant-resistant candidate from the RBD-5A epitope group.
B.1-182.1 and S2E12
S2E12 [299, 390, 404] and B.1-182.1 [391] are highly similar antibodies that share the same germlines, similar CDRs [391], similar epitopes (both belong to the RBD-2 epitope group; Table 5), and potency against wild-type SARS-CoV-2 (IC50s of 2 ng/mL). Importantly, both antibodies retain virtually all of their potency against wild-type SARS-CoV-2 and its variants, including Delta (Table 6). While neither of these antibodies currently appears to be in development, both of them have the characteristics that make them attractive as next-generation antibodies to SARS-CoV-2. Both antibodies have some activity against Omicron BA.1, with S2E12 having about fourfold better potency in pseudovirus assays (Table 7).
6D6, 7D6, and S2H97
Two recently described broadly-CoV neutralizing murine antibodies, 6D6 and 7D6, are unique amongst those described in this review, for several reasons. First, they bind RBD with very high affinity (low pM KD) to a cryptic epitope region (SARS-CoV-2 RBM residues 346–355, 466–471) that is on the opposite side of the RBD from the RBM [432]. The epitope for these antibodies is not available in either the open or closed RBD state, but appears to be exposed transiently during inter-domain movements within the spike protein. While these antibodies were not overly potent in viral infectivity assays (low μg/mL IC50 values), likely due to the limited access to the cryptic epitope site, they are resistant to all of the currently known VOCs up to Omicron, which has not been tested with these antibodies to our knowledge. As noted in Section 7, the proposed MOA for these antibodies is the destabilization of the spike and induction of S1 shedding to reduce infectivity [432].
S2H97, described in a separate study [261], shares some characteristics with 6D6 and 7D6. It also binds a cryptic epitope opposite of the SARS-CoV-2 RBM with high affinity, with about a 25% overlap of the 6D6/7D6 epitope (Fig. 5). The epitope for S2H97, which skews slightly more towards the RBD-7 epitope shown in Figure 3, is present across all clades of Sarbecovirus. Additionally, S2H97 appeared to be highly resistant to mutations, but results against VOCs were not reported [261]. S2H97 also induces S1 shedding and inhibits conversion of S to the post-fusion state, blocking cell entry [261]. Interestingly, considering its highly conserved epitope, S2H97 neutralizes Omicron BA.1 only at about an IC50 of 1.3 μg/mL, about four- to fivefold poorer than its neutralization of wild-type virus [406].
Fig. 5.
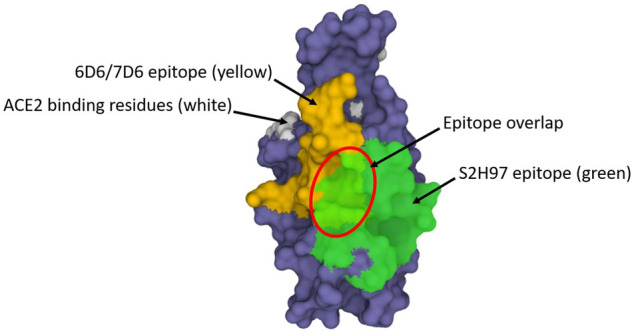
A filled structure (PDB ID 7CH5) showing the “backside” of SARS-CoV-2 RBD (rotated 180° from Fig. 4) and the epitopes for antibodies S2H97 (green; [261]) and 6D6/7D6 (yellow; [432]). A small portion of the ACE2 binding site (white) can be seen peaking around the edge. The S2H97 and 7D6 epitopes are based on the structures shown in PDB IDs 7M7W and 7EAM, respectively. The area denoted by the red circle indicates the area in which the epitopes overlap. The PDB program [201, 202] was used to generate and annotate the structures. ACE2 angiotensin-converting enzyme-2, PDB Protein Data Bank
Antibodies Against Omicron Variants
The emergence of Omicron BA.1, BA.1.1, and BA.2 since November 2021 has resulted in a significant shift in the use and discovery of new antibodies targeting SARS-CoV-2. Currently in the US, BA.1/BA1.1 comprise about 96.2% of all COVID-19 infections (CDC), with BA.1.1 (BA.1 + R346K mutation) making up three-quarters of that total [76]. BA.2, which has lagged behind BA.1 in infections, currently comprises about 3.8% of all COVID-19 infection in the US [76]. As shown in Table 6, several of the key antibodies that have been approved via the EUA process, including REGEN-COV™, bamlanivimab plus etesevimab, and Regkirona™, do not neutralize Omicron BA.1. Similarly, as compared with wild-type SARS-CoV-2 (e.g., WA-1), Sotrovimab lost two- to threefold activity against BA.1 and BA.1.1 but 10- to 20-fold against BA.2 (Table 7). Additionally, the vast majority of other antibodies, especially those that compete with ACE2 binding, tested against BA.1 were inactive or marginally active against that variant [109, 405–408]. As pointed out by others [109, 406–408, 428, 433], antibodies that compete with ACE2 (largely RBD-1 epitope antibodies; Figs. 6A, B, 7A) are usually ineffective against Omicron BA.1. Interestingly, the S371F/L mutation present in both BA.1/BA.1.1 and BA.2 appears to play a significant role in resistance of the Omicron variants to several antibodies [109, 433], even though that particular residue is part of the epitope only of a few antibodies, particularly those of the RBD-6/RBD-7 epitope groups (Figs. 6A, B and 7B) and C144 and BG10-19, which bind a second RBD using that residue as part of their epitope (Fig. 6A).
Fig. 7.

A, B Structures of SARS-CoV-2 RBD and anti-RBD antibodies from co-crystal structures split into six epitope groups, RBD1-2 (A) and RBD4-7 (B). For each antibody, two structures are shown: (i) a ribbon diagram based on published co-crystal structure showing attachment site on RBD and angle of attachment. For these structures, the RBD structure (in purple) is held constant so the different attachment angles for the antibodies can be compared. The PDB ID entry used to generate top is given below for each [202]; and (ii) a filled structure of RBD (PDB ID 7CH5) showing the ACE2 binding site (white; refer to Fig. 4 for baseline information) and the epitope on the RBD to which the antibody binds (green). The PDB program [201, 202] was used to generate and annotate the structures. A Structures B-T for antibodies of RBD-1 and RBD-2 epitope groups and B Structures U-LL for antibodies in RBD-4, 5, 6, and 7 epitope groups. Epitope group RBD-3, as described by Hastie et al. [290], is not represented in this group of antibodies. Figure 5A, Structure A. SARS-CoV-2 RBD (purple), with human ACE2 binding site identified (white) (PDB ID 7CH5); Epitope group RBD-1 antibodies: B. Etesevimab (LY-CoV016, CB6) Fab [204, 276], PDB ID 7C01; C. Amubarvimab (BRII-196, P2C-1F11) Fab [211, 212], PDB ID 7CDI; D. C102 Fab [276], PDB ID 7K8M; E. HFB30132A (P4A1-2A) Fab [235], PDB ID 7CJF; F. COR-101 Fab [236, 389], PDB ID 73BO; G. BD-629 Fab [228, 277], PDB ID 7CH5; H. CC12.3 Fab [257, 277], PDB ID 6XC4; Epitope group RBD-2 antibodies: I. S2E12 Fv only [299, 390], PDB ID 7K45; J. Casarivimab (REGN10933) Fab [205, 323], PDB ID 6XDG; K. Tixagevimab (AZD8895; COV2-2196) Fab [210, 211, 390], PDB ID 7L7E; L. B1.182.1 Fab [391], PDB ID 7MLZ; M. C144 Fv only [217, 276]; a second Fv is shown bound to the distal portion of the RBD as described in the text; PDB ID 7K90; N. HLX70 (P17) Fv only [242, 243], PDB ID 7CWL; O. Bamlanivimab (LY3819253; LY-CoV555) Fab [203, 326], 7KMG; P. Regdanvimab (CT-P59) Fab [206], PDB ID 7CM4; Q. LYCovMab BA4101 (CA521) Fv only [230], PDB ID 7E23; R. COVA2-39 Fab [277, 393, 394], PDB ID 7JMP; S. CV07-250 Fv only [277, 395], PDB ID 6XKQ; T. Fab2-4 Fv only [277, 396], PDB ID 6XEY. Figure 5B, Epitope group RBD-4 antibodies: U. P2B-2F6 Fab [212, 213, 277], PDB ID 7BWJ; V. CV07-270 Fab [277, 395], PDB ID 6XKP; W. MW05 Fab [224, 316], PDB ID 7DK0; X. BD-368-2 Fab [397], PDB ID 7CHC; Y. AZD1061 Fab [211, 390, 398], PDB ID 7L7E; Z. BG10-19 Fv only [399], a second Fv is shown bound to the distal portion of the RBD as described in the text, PDB ID 7M6E; Epitope group RBD-5A antibodies: AA. Imdevimab (REGN10987) Fab, [205, 277, 323, 400] PDB ID 6XDG; BB. LY-CoV1404 Fab [214], PDB ID 7MMO; CC. C110 Fv only [276], PDB ID 7K8V; Epitope group RBD-5B antibodies: DD. S309 Fv only [208, 276, 277, 390], PDB ID 6WPS; EE. C135 Fv only [217, 276] (some missing sequence from structure in database), PDB ID 7K8Z; FF. ABV-47D11 Fv only [231, 232], PDB ID 7AKD; Epitope group RBD-6 antibodies: GG. COVA-1-16 Fv only [277, 392–394] PDB ID 7JMW; HH. MW06 Fv only [316], PDB ID 7DPM; II. H014 Fv only (some missing sequence from structure in database) [277, 401], PDB ID 7CAH; JJ. S2X259 Fv only (some missing sequence from structure in database) [262, 390], PDB ID 7RAL; KK. CR3022 Fab [255, 276, 277, 282], PDB ID 6XC3; LL. EY6A Fab [276, 403, 404], PDB ID 6ZFO. ACE2 angiotensin-converting enzyme-2, Fab fragment antigen binding, Fv fragment-variable, PDB ID Protein Data Bank Identifier, RBD receptor binding domain, SARS-CoV-2 severe acute respiratory syndrome coronavirus-2
Thus far, LY-CoV1404, an RBD-5A epitope group antibody, appears to have the greatest activity against all three Omicron variants (Table 7). Interestingly, BRII-198 (P2B-1G5) has good activity against BA.1, but loses activity against BA.1.1, which has only the additional E346K mutation [109, 408]. Since the epitope of BRII-198 has not been made public yet, the structural basis for this difference is not apparent. Additionally, several antibodies can neutralize BA.1 but lose significant activity against BA.2, which is significantly different from BA.1 in its mutational profile (see Fig. 6A, B). These include sotrovimab (RBD-5B), DH1047 (RBD-6), and S2X259 (RBD-6) [109]. Conversely, cilgavimab has no activity against BA.1 or BA.1.1, but is quite potent against BA.2 [109] (Table 7).
These data have translated into regulatory activity in the USA, including limiting EUA-authorized use of REGEN-COV™ and bamlanivimab plus etesevimab in the USA while Omicron is still dominant [415], limiting the use of sotrovimab to geographical areas apparently not impacted by Omicron BA.2 [418], granting of an EUA for EvuSheld™ (AZD7442; tixagevimab + cilgavimab) for pre-exposure prophylaxis in high risk populations [424], and granting of an EUA for bebtelovimab, for treatment of COVID-19 for patients with any variant [380]. The EUA for EvuSheld™ in the face of Omicron BA.1 depends on the combined activity of the combined antibodies, neither of which alone is very potent (Tables 6 and 7); it is known that cilgavimab has excellent neutralization activity against BA.2, which is critical moving forward. Similarly, the EUA granted for the use of bebtelovimab is interesting because it was granted based on clinical safety data and preclinical data showing incredible potency against Omicron BA.1, BA1.1, and BA.2, but in the complete absence of statistically relevant clinical data [381].
Table 7 shows several examples of existing and new antibodies with activity against Omicron variants. Unfortunately, as of the final writing of this paper, most have been tested against just BA.1 and/or BA1.1. Of those antibodies tested against all three important Omicron variants, several very recently described antibodies that bind away from the RBM (BD55-5840, BD55-3546, BD55-5549, BD55-3372, BD55-5514, BD55-5483, BD55-5558) have excellent activity against all three variants [407] (Table 7).
It is noteworthy that at least three of the antibodies targeting Omicron variants are intended to be dosed locally into the respiratory tract via either inhalation or intranasal dosing. CT-P63, which has been reported to neutralize Omicron, is combined with regdanvimab (CT-P59) and is preparing to enter Phase III clinical testing as an inhaled biologic [241]. IGM-6268, which neutralizes BA.1 with an IC50 of 230 ng/mL is in Phase I clinical trials for intranasal dosing [410]. STI-9199, which is the intranasal formulated version of STI-9167, has potent neutralizing activity against both BA.1, BA.1.1, and BA.2 and is being readied for an IND in early 2022 (Table 7).
Finally, Sheward et al. [413] demonstrated that RBD-1 group antibodies of the IGV3-53 germline, i.e., ACE2-competing antibodies that would normally not be expected to bind or neutralize the Omicron variants well, could be affinity matured to be potent Omicron neutralizers. One of their affinity-matured antibodies, CAB-B37, was a potent neutralizer of every variant tested, including Omicron BA.1 (Tables 6 and 7). In a separate high affinity approach, two nanobodies, Nb1 and Nb2, were fused to an IgG1-Fc to make a biparatopic, bispecific antibody-like construct that neutralized Omicron BA.1 with an IC50 reported to be 1.7 pM, which would translate to approximately 0.2 ng/mL [330]. It will be interesting to see if these ultra-high potency constructs translate into development candidates at some point.
Beyond these individual antibodies targeting Omicron, antibody mixtures such as Immunome IMM20253 [434], SAB-185 (Table 1), the fully human IgG antibody hyperimmune plasma from transgenic cows [200], and the glyco-humanized polyclonal preparation from swine, XAV-19 (Table 1) [197], all have been demonstrated to neutralize Omicron BA.1. Their activity against BA.2 is as yet unknown publicly.
Epitope Classes of SARS-CoV-2 Antibodies
Overview of SARS-CoV-2 Epitopes
Essentially all antibodies studied for the ability to neutralize SARS-CoV-2 target the spike protein (OSM Fig. S1), i.e., the viral protein mechanistically responsible for both tropism and target cell entry. Of these, most are focused on three different types of epitopes in the RBD. The first of these is the RBM sub-domain, which is responsible for recognizing and binding the SARS-CoV-2 receptor, ACE2 (Fig. 4A). The second and third epitope regions in the RBD core are cryptic sites on the inner and outer sides of trimeric interface (Fig. 4A), which serve as a scaffold to maintain the RBD structure [388]. All three of these epitope regions contain susceptible epitopes for nAbs [276, 277, 290] (Figs. 6A, B). Two other major regions in the spike protein that provide interesting targets for nAbs are the NTD and the S2 domain, which will be discussed later in this section.
Several different research groups have categorized RBD-binding antibodies according to the epitopes to which they bind, as well as characteristics associated with those epitopes. The most widely recognized epitope classification of anti-RBD antibodies has been generated by Barnes et al. [276], who subdivided RBD-binding antibodies into four classes. Since that classification, Niu et al. [435], Deshpande et al. [436], Wang et al. [382], and others have further refined the four classes proposed by Barnes et al. [276] and included additional members for each class. Moreover, Yuan et al. [277], who analyzed the anti-SARS-CoV-2 RBD epitopes on the basis of buried surface area (BSA) instead of linear sequence, arranged groups of anti-SARS-CoV-2 anti-RBD antibodies into three groups RBS-A, RBS-B, and RBS-C, the A group of which largely overlaps Class 1 of Barnes et al. [276]. Similarly, Piccoli et al. [296] studied several anti-SARS-CoV-2 RBD antibodies recovered from patients and categorized the epitopes of six of them into various groups, including site 1a, which appears to belong the Barnes epitope group 1, and 1b, which appears close to Barnes et al. group 2 [277]. Dumet et al. [400] analyzed all of the available structures and epitope sequences of antibodies to SARS-CoV-2 and used a modeling program to place them into seven separate epitope bins. Liu et al. [396] also described seven epitope groups, A-H, of which groups A, B, and C bound to epitopes in the NTD, group D was an outlier antibody, and groups E-H all bound epitopes on the RBD. Importantly, they described their epitope grouping as a Venn diagram, with many significant overlaps between the groups. Most recently, a consortium of researchers funded by the Gates Foundation analyzed 186 different anti-SARS-CoV-2 RBD antibodies in detail [290] and separated them into seven major epitope groups, a few of which were further subdivided based on competitive binding experiments (Fig. 4, Table 8). The strength of their analysis, which also resulted in a Venn diagram of epitopes [290], included both the sheer numbers of antibodies analyzed and the depth of the analysis. This is offset, however, by the fact that they code-blinded the data to protect individual intellectual property, so very few antibodies known in the literature or which are in development were identified with particular epitope classes, making the broader translation of their work more difficult. In this work, we are attempting to bring together these various epitope groupings, as well as adding in our own analysis (Figs. 6A, 6B, 7A, and B), to align antibodies currently in clinical or late preclinical development, as well as widely studied antibodies as comparators, by epitope groups.
Table 8.
Seven major epitope groups of antibodies targeting SARS-CoV-2 RBD#
| Class# | Other epitope grouping a |
Block ACE2 | Binding location | RBD confor-mation | Key binding residues b,c | Mutations that affect binding | Mutations or variants mostly resisted | Other comments | Antibodies in groupd |
|---|---|---|---|---|---|---|---|---|---|
| RBD-1 |
B-1, Y-RBS-A |
Yes | Largely overlap RBM | Open only | R403, K417, D420, Y421, Y453, L455, A475, N501 |
Quite variable; Beta, Gamma; K417N/T e |
Alpha, Epsilon, Delta | Amongst most potent but also most susceptible to mutations; Fully occupy all three RBDs per spike; often cross-link spike trimers | CB6, CC12.3, C102, BD-629, P4A1, etc |
| RBD-2 |
B-2, Y-RBS-B |
Yes | Shifted toward the peak of RBM | Open only | (K417), L455, F456, E484, G485, F486, N487, Q493 | Beta, Gamma; combination of Y453F, E484K/Q, F486L, N501T | Alpha, Epsilon, Delta | Largest group of mabs; amongst most potent but also most susceptible to mutations; tend to bind bivalently within one spike | S2E12, REGN10933, AZD8895, B1-182.1, C144, HLX70, LY-CoV555, CT-P59, CA521, COVA2-39, CV07-250, Fab2-04 |
| RBD-3 | NA | Yes | RBM; Bind center of ACE2 binding site near “mesa” | Open only | ND | N501T/Y, E484K | K417N | Competes with both ACE2 and CR3022; IgGs can cross-link spikes; some IgGs will bind bivalently intraspike | ADI-56046 [437] |
| RBD-4 |
B-2, Y-RBS-C |
Yes (4A)/no (4B) | Outer face of RBD; bind toward the outer edge of the RBM | Open or closed | R346, K444, G446, N450, E484, Y489, Q493 |
E484K and/or L452R; B.1.429 (epsilon) |
L455, F456, E484, F490, Q493 |
Some can crosslink spike trimers in solution |
P2B-2F6, CV07-270, MW05, BD-368-2, AZD1061, BG10-19 |
| RBD-5 |
B-3, Y-RBS-D |
No | Outer face of RBD; toward S309 site | Open or closed |
T345, R346; N343 (5B); L452R |
Few residues | Broad resistance against variants | Some can cross-link spikes, which leads to potent neutralizing activity | REGN10987, LY-1404, C110, S309, C135, 47D11 |
| RBD-6/7 |
B-4, Y-CR3022 site |
Yes (6, 7A); no (7B, 7C) | Bind inner face of RBD; access cryptic epitope | Two RBDs must be open | Y369, T376, S383, T385, (D427, D428) | ND | ND | Generally less potent; tendency to cross-link spike trimers; RBD-6 epitope extends closer to RBM | COVA1-16, MW06, H014, S2X259, CR3022, EY6A |
B-1, etc., Barnes et al., Class 1 antibodies, etc. [276], NA not applicable, ND no data, RBD receptor binding domain, RBM receptor binding motif, Y-RBS-A, etc., Yuan et al., RBS epitope grouping A, etc. [277]
aAs guided by the epitope groupings by Hastie et al. [290]
bGroupings by Barnes et al. [276] (B) and Yuan et al. [277] (Y)
cResidues included in the epitopes of the majority of group members. Those residues in parentheses are more variably included in the epitope
dSee Figs. 6A, B, 7A, and B for more information. Note that many other antibodies not included in this paper also fall into these epitope groups
e reference [322]
SARS-CoV-2 RBD Antibody Epitopes
With respect to SARS-CoV-2 neutralizing antibodies and the epitopes to which they bind, there are three characteristics that define antibody activity: (i) neutralization potency; (ii) breadth of neutralization; and (iii) resistance to mutations. In most cases, high potency also comes with relatively narrow breadth and increased risk of activity loss due to mutations, as will be described in the following sections. On the other hand, antibodies with broad neutralization capabilities often are more resistant to mutations. These three properties largely depend on two factors, binding (e.g., affinity/avidity) and epitope.
In general, RBD-targeting nAbs, especially those that interfere directly with ACE2 binding, are generally more potent than those targeting other domains of the spike protein [396]. The neutralizing titers for many of the potent RBD-targeting nAbs can be as low as 1 ng/mL [395]. The RBM epitopes, such as the epitope for VH3-53 nAbs described below, are dominant in the B cell response after infection. Therefore, these epitopes may be under stronger immune pressure and more likely to generate escape mutations during repeated worldwide viral transmissions. The RBD-targeting nAbs have been extensively reviewed elsewhere [276, 277], so for this review, we are focused primarily on the nAbs in development, along with a few well characterized antibodies used as comparators (Table 5).
Among the 15 nAbs that have entered Phase III clinical trials (Table 2), 12 nAbs target epitopes associated with the RBM [203–205, 212, 213], two nAbs (REGN10987 [205] and ADG-20 [219]) target epitopes associated with the outer face of the mesa (Fig. 4), and one nAb (sotrovimab, derived from S309) targets an epitope on the RBD core region [208, 209, 276]. The epitope information for the other three nAbs has not been fully disclosed yet (Table 5).
We have categorized the epitopes for 40 different nAbs that bind SARS-CoV-2 RBD and have placed them into seven epitope groups, RBD-1, RBD-2, RBD-4, RBD-5A, RBD-5B, RBD-6, and RBD-7, along the lines described by Hastie et al. [290], and one outlier, S2H97, a pan-CoV antibody that targets a novel cryptic epitope on the “backside” of the RBD [261] (Table 5, Fig. 5). None of the antibodies in our analysis appears to bind to the RBD-3 epitope described by Hastie et al. [290].
Epitope Group RBD-1
Epitope group RBD-1 (Table 5) is essentially identical to Barnes et al. Class 1 [276], Yuan et al. RBS-A [277], and Hastie et al. [290] RBD-1 epitope group and contains seven antibodies described here (Figs. 6A, B, 7A, B). Additionally, several other antibodies that fit cleanly into Barnes et al. Class 1 [276] also belong to this group. Class 1 antibodies directly compete with ACE2 for binding and, as such, they only bind RBDs that are in the “up” or “open” conformation, similar to ACE2 [276] (Tables 5 and 8), and they tend to fully occupy all three RBDs per spike [290]. Most of these antibodies are derived from the VH3-53 and VH3-66 germline families (Table 5), and they tend to bind to RBD at similar angles to block ACE2 binding [228, 276, 277, 392, 397, 438] (Fig. 6A). These Class 1 RBM-directed nAbs function through steric clash with ACE2 (Fig. 7A) and they typically have higher potency than core region-directed nAbs [290], but also suffer from lack of breadth (i.e., they typically target only SARS-CoV-2), and are prone to loss of activity by mutations in the RBM [322, 425].
Examples of Class 1 antibodies from the original designation [276] include C102, C105, B38, CC12.3, etesevimab (LY-CoV016; CB6), casirivimab (REGN10933), and COVA2-4. Similarly, Yuan et al. [277] described CB6 (etesevimab), CC12.3, BD-629, and similar antibodies in their “group A.” In our analysis, we also find most of those antibodies to be grouped together in what we are calling, based on epitope groupings by Hastie et al., [290], group RBD-1 (Tables 5 and 8; Figs. 6A, B, and 7A). Figures 6A, B, and 7A show that additional antibodies such as amubarvimab (BRII-196, P2C-1F11), HiFiBio HFB30132A (P4A1-2A), and Corat Therapeutics COR-101 (STE90-C11), also belong to this group (Tables 5 and 8; Figs. 6A, B and 7A). These antibodies are all encoded by either a VH3-53 or VH3-66 heavy chain (Table 5). Note that casirivimab (REGN10933), which was categorized as Class 1 by Barnes et al. [276], has been re-categorized as belonging with the RBD-2 group due to its specific epitope (Fig. 6A, B) and to the angle at which the Fab binds RBD [277] (Fig. 7A). Many of these antibodies share significant attributes, so it is expected that the various epitope groupings would have at least partial overlaps, as exemplified by Fig. 4 and described in more detail by Hastie et al. [290].
Four of the six described RBD-1 antibodies are currently in clinical trials, including Eli Lilly’s etesevimab ((LY3832479; LY-CoV016; JS016; CB6-LALA), Brii Biosciences amubarvimab (BRII-196, P2C-1F11), HiFiBio’s HFB30132A (P4A1-2A), and Corat Therapeutics’ COR-101 (STE90-C11)). In addition to the antibodies listed above, dozens of other antibodies likely belong to the RBD-1 epitope group including, for example, CC12.1, COVA2-04, B38, CV30, C105, and BD-604, all of which are encoded by antibody germline genes VH3-53 or VH3-66 [277]. All of these RBD-1 epitope group antibodies are most sensitive to a potential escape mutation at position A475 [276] and K417 [277], but also have sensitivities at positions L455, F456, N460, and Y473 [322] and potentially also at N501, based on the linear epitope shown in Fig. 6B. Finally, it was recently demonstrated that Alpha, Beta, and Gamma variants are either partially or completely resistant to most (but not all) VH3-53 antibodies [439].
Epitope Group RBD-2
The RBD-2 epitope group largely overlaps the epitope of RBD-1 antibodies, but is shifted more toward the “peak” of the RBD (see Fig. 4), overlapping only about half of the ACE2 binding residues (Fig. 6A). Hastie et al. [290] noted that RBD-2 is the largest epitope group of anti-SARS-CoV-2 RBD antibodies, and accordingly, they subdivided RBD-2 into several subgroups based on competition experiments. For example, of the antibodies described here, Hastie et al. [290] suggested that REGN10933 overlapped with their RBD-2A group, that COVA2-39 clustered with their RBD-2b.1 group, that C144 bound more toward the RBD outer face similar to their RBD-2b.2 group, and that S2E12 bound more toward the peak like their RBD-2b.3 group. Since we do not have enough competitive binding information for the entire group, we will group them all together in RBD-2 here. Nevertheless, as can be seen in Figs. 6A, B, 7A, and B, there is certainly some heterogeneity in this group which could allow for potential sub-grouping when more information becomes available. Key residues to which most RBD-2 antibodies bind include L455, F456, E484, G485, F486, N487, and Q493 (Table 8, Fig. 6A, B). As compared with members of RBD-1, most RBD-2 antibodies do not bind R403, D420, Y421, N501, G502, or Y505 (Fig. 6A, B). These antibodies all compete with ACE2, they tend to bind bivalently within a single spike [290], and about half of them bind both the open (up) and closed conformations of RBD in the spike protein (Table 5). This last characteristic may help to subdivide this epitope group further. As would be expected from the list of key residues, Barnes Class 2 antibodies are most sensitive to potential escape mutations at positions L455, F456, E484, F490 and Q493 [322], with E484K being their Achilles heel [425].
The RBD-2 epitope group described here includes 13 antibodies, five of which had been previously described as “Class 2” antibodies [276] or RBS-B group antibodies [277], or both. This group also includes one antibody, casirivimab (REGN10933) that was included in the Class 1 epitope group [276], and three antibodies, COVA2-39, CV07-250, and Ab2-4, that Yuan et al. [277] categorized together as RBS-B. Also included in this epitope group are S2E12, tixagevimab (AZD8895), B1.182.1, C144, HLX70 (P17), bamlanivimab (LY3819253; LY-CoV555), regdanvimab (CT-P59), LYCovMab BA4101 (CA521), and IGM-6268 (Figs. 6A, 6B, and 7A; Table 5). It should be noted that B1.182.1 is highly similar to S2E12, including sharing the same germline families and a high degree of sequence identity [391]. A very newly described antibody, P5C3, also likely belongs to the RBD-2 epitope group [441]. Similar to S2E12, B.1-182.1, and tixagevimab (AZD8895, COV2-2196), P5C3 is fully active against all VOCs prior to Omicron, including variant Beta [441], to which the first generation RBD-2 epitope antibodies, casirivimab (REGN10933) and bamlanivimab (LY-CoV555), are inactive. Thus, it is becoming clear that a subset of RBD-2 antibodies is fully capable of neutralizing VOCs Alpha, Beta, Gamma, and Delta.
One key feature of this group of antibodies is that they interact with F486 (Fig. 8), which is inserted into the groove between the antibody heavy and light chains as they straddle the tip of the RBS knob [277]. These antibodies can bind at different angles and even rotate around the key F486 residue at the tip of the RBS “knob” [277]. Due to their attachment angles, COVA2-39 and CV07-250 can only bind RBD in the up position, whereas Fab2-4 can bind to both open and closed conformations of RBD. As expected, these antibodies are sensitive to mutations in F486, as shown in Table 8.
Fig. 8.

Structure of the interface of the RBM with example antibodies in the RBD-2 epitope group showing how RBM residue F486 fits into the groove between the VH and VL chains of each of these antibodies as previously described by Yuan et al. [277] for antibodies in their RBS-B epitope group. RBD is in cyan, VH is in purple, VL is in blue, residue F486 Is in red. A Fab2-4, from PDB ID 6XEY; B COVA2-39, from PDB ID 7JMP; C. REGN10933, from PDB ID 6XDG; and D. CV07-250, from PDB ID 6XKQ. The PDB program [201, 202] was used to generate and annotate the structures. Fab fragment antigen binding, PDB ID Protein Data Bank Identifier, RBD receptor binding domain, RBM receptor binding motif, RBS receptor binding site, VH variable heavy (chain), VL variable light (chain)
Eight of these RBD-2 epitope group antibodies are currently being studied in clinical trials for treatment of COVID-19. These include Regeneron’s casirivimab (REGN10933), AstraZeneca’s tixagevimab (AZD8895), Bristol-Myers Squibb’s BMS-986413 (C144-LS), Hengenix Biotech’s HLX70 (P17), Eli Lilly’s bamlamivimab (LY-CoV555), Celltrion’s CT-P59, Boan Biotech’s LYCovMab BA4101 (CA521-FALA), and IGM Biosciences IGM-6268. Many RBD-2 antibodies are generally ineffective against Beta and Gamma variants due to the E484K mutation found in those variants [290]. This is not the case for the IgM antibody IGM-6268, however, which appears to overcome many individual escape mutations likely because of its higher avidity [245].
Epitope Group RBD-4
The RBD-4 epitope group was described by Hastie et al. [290] as targeting the outer face of the RBD, binding at the outer edge of the RBM (see Figs. 4 and 7B). Most of the antibodies in this epitope group bind RBD in both its closed and open conformation, they weakly block ACE2 binding to RBD, and they bind on the outer face of the RBD with a partial overlap of the ACE2 binding site (Tables 5 and 8, Figs. 6A, B, 7A, and B). These antibodies would likely compete with epitope group RBD-5A antibodies (Figs. 6B and 7B). Two antibodies in epitope group RBD-4, P2B-4F6, and CV07-270 were described by Yuan et al. [277] as belonging to their RBS-C epitope group. P2B-2F6 also was described by Barnes et al. [276] as part of their Class 2 epitope group. The Class 2 Barnes RBD antibodies, included in RBD-4, were described to compete with ACE2 binding, but due to their epitope angle of attachment, are able to bind RBDs in both the “up” (open) and “down” (closed) conformations [276]. Key residues for the RBD-4 epitope group include R346, K444, G446, N450, E484, Y489, and Q493 (Table 8).
We list seven antibodies that cluster together in epitope group RDB-4, three (Abpro Biotech’ ABP-300, Beigene/Singlomics’ BGB-DXP593, AstraZeneca’s cilgavimab) of which are currently in clinical trials for treatment of COVID-19 (Table 2).
Epitope Group RBD-5A,B
Antibodies in epitope group RBD-5 bind in the escarpment outer face region of the SARS-CoV-2 RBD (Fig. 4), they do not compete with ACE2 for binding (Fig. 6B), and they can access RBDs in both the “up” (open) and “down” (closed) conformations [323]. This group is essentially defined by antibody S309, an antibody described as binding the “proteoglycan” site due to its interaction with the glycan on N343 [277]. S309, the precursor to sotrovimab (VIR-7831), was first isolated in 2003 from the B cells of a SARS patient [209]. The sequences of the core region across different SARS-like Sarbecovirus (lineage B) are more conserved than that of the RBM, which results in core region-directed nAbs have greater breadth of binding and neutralization compared with RBM-directed nAbs [261, 262]. Thus, after the COVID-19 pandemic broke out, S309 was tested against SARS-CoV-2 and was found to be a high affinity binder to the RBDs of both SARS-CoV-1 and SARS-CoV-2 [208].
The original list of Class 3 antibodies [276] includes the prototype antibody, S309, as well as antibodies C135, C110, and REGN10987. Key residues for the RBD-5 antibodies combined include T345 and R346. Of the six RBD-5 antibodies, three (REGN10987, LY-1404, C110) have epitopes that reach toward the outer face of the mesa, where they can potentially compete with RBD-4 epitope group antibodies. In keeping with the definition proposed by Hastie et al. [290], we are calling these BRD-5A epitope antibodies. None of these three antibodies appears to interact with residue N343 (Fig. 6A). Additionally, a very newly described, highly potent antibody, 54042-4, which is effective against all VOCs prior to Omicron [442], appears to belong to epitope group RBD-5A. The other three antibodies in the RBD-5 epitope group, including S309, C135, and 47D11, all interact with N343 and its glycan (Fig. 6A) and have a more focused epitope on the lower outer face of the escarpment (Fig. 7B). Yuan et al. [425] made this same sub-group distinction, separating out C110, C119, and REGN10987 into their RBS-D group, while placing S309, C135, and CV38-142 into their “S309 site” epitope group. We are placing these three antibodies, S309 (and thus, also sotrovimab), C135, and 47D11, into epitope group RBD-5B.
These core region-targeting nAbs are good candidates for further development as antibody therapeutics with broader protection against emerging coronaviruses in the future. Unlike RBM-directed nAbs, core region-directed nAbs, such as S309, neutralize SARS-CoV-2 through ACE2-noncompeting mechanisms. The exact modes of action for neutralization for core region targeting nAbs are yet to be defined, although RBD-5 antibodies capable of inter-spike cross-linking activity had significantly more potent neutralization activity, so it may be possible that the inter-spike cross-linking may lead to a blockage of spike maturation or internalization process [290]. Moreover, RBD-5 antibodies are active against all current VOC and VOI variants [290].
Of the six antibodies in the RBD-5 epitope group described here, five of them are currently in clinical trials; the only antibody in this group not currently in the clinic is C110 [276]. The other five are important clinical candidates, including Regeneron’s imdevimab (REGN10987) and the GSK/Vir antibody, sotrovimab, which is derived from the prototypical S309, both of which have received EUAs, as well as Eli Lilly’s second-generation antibody bebtelovimab (LY-CoV1404), Bristol Myers Squibb BMS-986414 (C135-LS), and AbbVie and Harbour Biomed’s ABBV-47D11 (aka HBM9022) (Table 2). Sotrovimab was very recently (2 December 2021) approved by the UK Medicines and Healthcare products Regulatory Agency (MHRA) under the trade name of Xevudy™, and bebtelovimab recently received an EUA from the FDA [380].
Epitope Groups RBD-6 and 7
Antibodies in the RBD-6 and 7 epitope groups bind to cryptic epitopes on the inner face of the escarpment (Figs. 4 and 7B) which, in the “down” or “closed” position, is normally buried in the trimeric interface. Similar to S309 described above, the prototypical member of this group, CR3022, was originally isolated from B cells from a patient infected with SARS-CoV-1 [282], and later found to bind and neutralize SARS-CoV-2 [255], although CR3022 neutralization of SARS-CoV-2 is rather weak (Table 5). Based on the epitope of CR3022, the RBD-7 antibodies have been previously described as Class 4 antibodies [276] or “CR3022-like” [277]. The original group of Class 4 antibodies, which were defined as binding the CR3022 epitope region, not competing with ACE2 (Figs. 6B and 7B), and ability to bind RBDs only in the “up” or “open” conformation [276], included the prototype, CR3022, as well as COV1-16, EY6A, S304, and S2A4 [276]. Nevertheless, this is a heterogeneous group. CR3022 and EY6A both have a more compact epitope confined to the inner face of the escarpment than COVA1-16, MW06, H014, and S2X59 (Fig. 7B), and the two groups differ in binding to residues F392, R408, T430, and F515. Thus, we are defining the RBD-7 epitope antibodies to include only CR3022 and EY6A. The other antibodies in this group, including COVA1-16, MW06, H014, and the new broad CoV-neutralizing antibody, S2X259 [262], are being placed into RBD-6. These antibodies span a broader epitope than CR3022 and EY6A, and even encroach on the binding site for ACE2 with the possibility to block ACE2 binding, making them much closer to the description of the RBD-6 epitope group by Hastie et al. [290]. Indeed, COVA1-16 and H014 do block ACE2 binding, whereas MW06 and S2X259 appear not to block ACE2 binding. Nevertheless, with the expanded epitopes for all of them, we are placing these four antibodies into RBD-6 (Figs. 6A, B, and 7B and Table 8).
Epitopes for Bispecific Antibodies or Combinations
As noted previously, one of the potential approaches to increase both potency and breadth in neutralization of SARS-CoV-2 variants is to generate bispecific antibodies utilizing antibodies with non-overlapping epitopes [193, 331]. It already has been demonstrated that a bispecific antibody of Ab-06 and Ab-14 was significantly more potent at neutralizing SARS-CoV-2 than the two IgG antibodies together [331]. This same paradigm also was true for the nanobodies Nb1 and Nb2 which, when fused into a bispecific antibody format with an IgG1 Fc, resulted in a significantly more potent bispecific antibody against SARS-CoV-2 than if Nb1 and Nb2 were used as a mixture [330].
In the huge study carried out by Hastie et al. [290], several combinations based on the most likely non-overlapping RBD group (in their case, analyzed by competition experiments) were suggested. Based on the data shown in Figs. 6A, B, 7A and B, one could imagine that RBD-1 epitope group nAbs should work well in a bispecific antibody format with antibodies from epitope groups of RBD-5A, RBD-5B, or RBD-6, but not likely from RBD-2 or RBD-4. Newer antibodies like S2X259, which in its own right is effective against all of the current variants [262] (Table 6), should work very well in a bispecific antibody format with antibodies from epitope groups RBD-1, RBD-2, or RBD-4. Several of these combinations are sure to be made in the coming years in efforts to find highly potent, pan-Sarbecovirus antibodies.
RBD Epitopes of Key VHH Antibodies and Bispecific VHH Antibodies
Single domain, or VHH, antibodies (including the camelid nanobodies) have been isolated against a wide variety of RBD epitopes. Interestingly, though, a large proportion of neutralizing VVH antibody epitopes have focused on two epitope groups (Fig. 9). Koenig et al. [249] reported isolating four nanobodies that bound SARS-CoV-2 RBD with relatively high affinity and potently neutralized viral entry. One of the nanobodies, named E, bound one epitope (Fig. 9), whereas the three other nanobodies, U, V, and W, bound a separate non-overlapping epitope, which we interpret as the RBD-6 epitope group (Fig. 9 [for V]). VHH E was described as having an epitope similar to the IgG CC12.3 [276], which would put it into the RBD-1 epitope group, but Koenig et al. [249] reported that it bound in a different orientation (attachment angle) from the other neutralizing VHHs, and it does not fulfill the criteria for an RBD-1 epitope antibody, so we have placed it and similar nanobodies into the RBD-2 epitope group (Fig. 9). Nanobodies E (RBD-2)/V (RBD-6) are the likely components of the preclinical candidates DIOS-202 and DIOS-203 [249]. Similarly, Nb21, the precursor to the potential development candidate PiN-21 [253], also binds to RBD-2, as does Re5D06 [343]. Finally, VHH-72, the precursor to the Phase I/II clinical candidate, XVR011, is also an RBD-2 epitope antibody [239]. In an interesting twist to VHH antibody epitopes, the single VHH antibody Fu2 binds two distinct epitopes on RBD, i.e., the RBD-2 and RBD-6 epitopes (Fig. 9), which resulted in spike cross-linking and potential neutralization [344], as further described in Section 7.5.
Fig. 9.

A The sequence of SARS-CoV-2 RBD from residue 317 to residue 532, annotated with sequence of the RBM (thick underline) and the 17 residues to which human ACE2 bind (blue shaded). Additionally, mutations found in each of the major variants (yellow shaded residues; gray shaded for those mutations only found in some of the isolates of that variant), and epitopes for ten single-domain nanobodies known to target SARS-CoV-2 RBD (shaded green) are provided. The epitopes were retrieved from several references, including Wrapp et al. [238], Koenig et al. [249], Margulies et al. [345], Sun et al. [342], Gűttler et al. [343], Schoof et al. [346], and Hanke et al. [344]. Additional epitopes not adequately described in the literature were determined or corrected using the Protein Data Bank entries for the RBD-antibody co-crystal structures, with identification of the epitopes based on residues within 5Å of antibody loops. B Structures of SARS-CoV-2 RBD, the ACE2 binding site, and the epitopes for anti-RBD nanobodies from co-crystal structures. The PDB ID entry used to generate top is given below for each [202]; and (ii) a filled structure of RBD (PDB ID 7CH5 [351]) showing the ACE2 binding site (white; refer to Fig. 4 for baseline information) and the epitope on RBD to which the antibody binds (green). B1. RBD showing ACE2 binding site (white); B2. Epitope for Nb6, overlapping ACE2 binding site (PDB ID 7KKK [255]); B3. Epitope for Sb14, overlapping ACE2 binding site (PDB ID 7MFU [240]); B4. Epitope for Re5D06, overlapping ACE2 binding site (PDB ID 7OLZ [253]); B5. Epitope for VHH-E, overlapping ACE2 binding site (PDB ID 7KSG [115]); B6. Epitope for Nb21 (precursor to PiN21), overlapping ACE2 binding site (PDB ID 7N9B [252]); B7. Epitope for VHH-72, which does not overlap ACE2 binding site (PDB ID 6WAQ [105]); B8. Epitope for VHH-V, which does not overlap ACE2 binding site (PDB ID 7B11 [249]). Note that VHH antibodies Nb6, Sb14, Re5D06, VHH-E, and Nb21 fall into our RBD-2 epitope class (see Figs. 6 and 7) and VHH-72 and VHH-V fall into our RBD-6 epitope class. Nanobody Fu2 [344] binds to both the RBD-2 and the RBD-6 epitopes. The PDB program [201, 202] was used to generate and annotate the structures. ACE2 angiotensin-converting enzyme-2, PDB ID Protein Data Bank Identifier, RBD receptor binding domain, RBM receptor binding motif, SARS-CoV-2 severe acute respiratory syndrome coronavirus-2, VHH single domain antibody
N-Terminal Domain (NTD)-Targeting Antibodies
Multiple research groups have demonstrated the presence of an antigenic hotspot “supersite” on the spike protein to which a strong natural antibody response is often mounted. For example, in an analysis of 121 SARS-CoV-2 spike-binding antibodies isolated from B cells of five convalescent patients by Liu et al. [396], the non-RBD binders, many of which were NTD binders, outnumbered the RBD binders by more than two-to-one. NTD-targeting antibodies include both antagonist antibodies [396, 443, 444] and antibodies possessing infectivity enhancing activity [445], each group of which have specific epitopes (Fig. 10).
Fig. 10.

The sequence of the NTD of SARS-CoV-1 (blue letters) and SARS-CoV-2 (black letters) spike from residue 1 to 293 and 306, respectively. Mutations found in each of the major variants (yellow shaded residues), and epitopes for seven antibodies known to target SARS-CoV-2 NTD are provided. Epitopes for antagonist and agonist antibodies are shaded green and blue, respectively. The epitopes were retrieved from several references, including Suryadevara et al. [444], Cerutti et al. [172], and Liu et al. [445]. Comb common combined epitope, NTD N-terminal domain, WT wild-type (WA-1)
The core region of this conformational antigenic site, also called site “i" [282] and the “Site 1 antigenic supersite” [171], is formed by a β-hairpin comprised of residues 141–156 and a loop comprised of residues 246-260 in the NTD [172, 447] (Fig. 10). Additionally, the N-terminal residues 14–20 and residues 67–79 are adjacent and possibly interactive with the supersite [172]. While the antigenic supersite is surrounded by glycans, the supersite itself is largely glycan-free (in fact, it is the largest glycan-free area in the spike) and is highly electropositive, providing a focused, highly antigenic epitope that is easily mutated, allowing for escape from a natural antibody response [172, 448].
The neutralizing mechanisms for NTD-targeting nAbs are only partly understood. These nAbs do not compete with ACE2 binding to the S protein [396], but they appear to neutralize SARS-CoV-2 by blocking entry of the virus into cells [171, 172], at titers ranging from very potent (1 ng/mL) to poorly potent (1000 ng/mL) [172, 396, 443]. It has been proposed that these nAbs may prevent the conformational changes in the S protein to indirectly inhibit membrane fusion between virus and host cells [172], likely at the post-virus/cell attachment stage [444]. The amino acid sequence homology between SARS-CoV-2 and SARS-CoV-1 NTDs is low (51%) [449], so NTD-targeting nAbs are typically not cross-reactive with other SARS-like coronaviruses. There are no NTD-targeting nAbs that have advanced to clinical trials.
OSM Fig. S2A shows the general location in the S1 region of the spike protein to which NTD antagonist antibodies bind. Cerutti et al. [172] characterized the binding of eight NTD-binding antagonist antibodies, and found a significant overlap in their epitopes. The epitopes bound by various NTD-targeting antagonist antibodies, as defined by mutagenesis studies [444], are shown in Fig. 10. As can be seen, these epitopes are in a region of the spike protein that is specific to SARS-CoV-2 around the S1 supersite β-hairpin, with two epitope residues in the hairpin itself (Fig. 10). Unfortunately, of the five residues identified as key residues for the NTD-binding antagonist antibody, COV2-2489, three coincide with mutations found in COVs Alpha and Delta, suggesting that those variants could potentially be resistant to these particular NTD binding antibodies. Similarly, the NTD-targeting nAb, 4-8, a representative example of a family of NTD-binding antagonist antibodies (including 4A8, 1-87, 2-17, 2-51, 4-18, 5-24, FC05, S2L28, S2M28, S2X333, and DH1050.1) targeting the supersite, loses virtually all of its activity against variants Alpha (B.1.1.7), Beta (B.1.351), and Epsilon (B.1.429) [172]. Similarly, 4A8, the epitope for which is shown in Fig. 10, is completely inactive against Alpha, Beta, and any variants with deletions at Δ144 or Δ242-244 [396]. Additionally, based on the epitopes and sequence shown in Fig. 8, it is expected that 4A8 also would be inactive against Lambda, due to the large deletion at Δ247-253. Recently, Hastie et al. [290] defined three different epitopes within the NTD for antagonist antibodies. Their NTD-1 epitope group, which binds from the top of the NTD, overlaps the epitope of antibody 4A8 [290], shown in Fig. 8. Their NTD-2 epitope group coincides with what had previously been called the “antigenic site V” [171], encompassing residues H69, V70, Y144, W152, and G261 [290]. Finally, NTD-3 appears to represent a novel epitope focused around residue W152 [290].
Importantly, several of the VOC and VOI variants of SARS-CoV-2, including VOC Alpha (Δ69-70 [ΔHV], Δ144-145 [ΔYY]), VOC Beta (Δ241-243 [ΔLLA], R246I), VOI Eta (Δ69-70 [ΔHV], Δ144 [ΔY]), VOI Theta (Δ141-143 [ΔLGV], Δ243–244 [ΔAL]), and VOI Lambda (Δ247-253 [ΔSYLTPGD]), have mutations and/or deletions in this NTD supersite region [282, 447] (Figs. 2B and 10). As noted above, mutations in this NTD supersite have been shown to help enable escape of the virus from convalescent plasma [339]. It has been postulated that there is significant selective pressure for the virus to modify this supersite to escape neutralizing antibodies directed at the NTD [171, 172].
One unique NTD-targeting antibody, called 5–7, has an epitope that is significantly different from other NTD-targeting nAbs such as 4-8, 4A8, and COV2-2489 (Fig. 8) [449]. This antibody, which buries an area of 1223.6 Å2, binds to a hydrophobic pocket outside the supersite. This highly novel epitope would be the backside of the NTD in OSM Fig. S1, opposite the shown NTD antibody binding site, and does not correspond to any of the three NTD antibody epitopes described by Hastie et al. [290]. One key feature of antibody 5–7 is a long, 24-amino acid residue CDR-H3 that contributes most of the binding energy [449]. Importantly, NTD-binding nAb 5–7 has at least partial efficacy against Alpha (B.1.1.7), Beta (B.1.351), and Iota (B.1.526) variants [449]. Taking advantage of these properties, RenBio Therapeutics has generated a bispecific antibody in which one arm targets an epitope on the RBD and the other arm targets an epitope in the NTD. While it is not certain from publicly available information that nAb 5–7 is part of this bispecific antibody, its properties certainly would be appropriate for such an antibody. This bispecific antibody is being developed to be delivered as plasmid DNA to muscle, where the muscle itself becomes the protein manufacturer. This promising approach of genetic delivery of the antibody has been described elsewhere [447, 450], and already is being used to deliver clinical candidate antibodies, such as INO-A002, a DNA-delivered antibody targeting dengue virus (NCT03831503 [7], recruiting), mRNA-1944, an mRNA-encoded antibody targeting chikungunya virus (NCT03829384 [7], now completed), BNT141, an mRNA-encoded antibody targeting claudin 18.2 for solid tumors (NCT04683939 [7], not yet recruiting) and others in which antibodies are delivered using viral vectors such as adeno-associated virus (AAV) or oncolytic viruses.
Not all NTD-targeting antibodies are antagonists and/or nAbs. Several NTD-binding antibodies that stimulate spike function and increase infectivity of SARS-CoV-2 also have been isolated and characterized [445]. The SARS-CoV-2 epitopes for two of these, COV2-2490 and COV2-8D2, have been mapped to three distinct loops around S1 residues 70–76, 183–187, and 211–215 (Fig. 10). Several other NTD infectivity enhancing antibodies (e.g., COV2-2660, COV2-2210, COV2-2582, and COV2-2369) competed for binding to S1 with COV2-2490 and COV2-8D2, and while certain differences were observed in responses to specifically mutated residues, they appear to bind in the same manner [445], suggesting there is something specific to the binding site that allows for infectivity enhancing activity. The infectivity enhancing activity of these antibodies was different from classical antibody-dependent enhancement (ADE) because it is Fc-independent and did not require FcγR or complement receptor for inducing cell entry. Additionally, only cells with ACE2 were affected by this activity. The mechanism of action (MOA), however, did require the presence of both Fab arms; i.e., monomeric or single Fab arm-antibodies did not enhance infectivity. Thus, Liu et al. [445] hypothesized that the MOA for these infection enhancing antibodies appears to be the induction of RBD open conformations due to the coupling of NTD domains of two adjacent spikes. They also suggested that the levels of infectivity enhancing antibodies in plasma could potentially be a factor in both disease severity and in antibody treatment efficacy.
S2-Targeting Antibodies
The S2 subunit is more conserved than the S1 subunit between SARS-CoV-2 and SARS-CoV-1 (90% vs. 64%), and across different lineages of beta-coronavirus (β-CoV) [451]. Therefore, S2-targeting nAbs may have the advantage for developing broad-protective or even pan-coronavirus therapies. Unlike the success of isolating RBD- and NTD-targeting potent nAbs with recombinant domain proteins as baits, selecting S2-targeting antibodies with recombinant S2 protein as a bait has typically yielded binders with almost no neutralizing activity [437]. One important reason is that recombinant S2 proteins might adopt a post-fusion conformation without stabilization by the S1 subunit [452]. To overcome this challenge, a stabilized S2 of the MERS-CoV was designed by introducing the S-2P (V1060P and L1061P) mutations to retain the prefusion conformation [453] and adding a C-terminal T4 phage fibritin (foldon) domain to facilitate trimer formation. Using this optimized hybrid S2 to immunize humanized mice, four MERS-CoV S2-targeting antibodies (3A3, 4A5, 4H2 and 3E11) were isolated [454]. These antibodies have cross-reactivity to SARS-CoV-2, SARS-CoV-1 and HKU1 to varying degrees. Antibody 3A3 shows neutralizing activity against SARS-CoV-2 pseudovirus with titers of 25.4 µg/mL against the ancestral spike and 1.6 µg/mL against the D614G spike [454]. The 3A3 epitope is located at residues 980-1006 (see “X” area in Fig. 2A) in the flexible hinge region at the S2 apex. A recently described camelid nanobody, 7A3, also binds this same region with key epitope residues of D985, P987, and E988 [455]. Interestingly, 7A3 binds deep into a cross-CoV, highly conserved pocket in the spike protein likely unavailable to a normal IgG [455].
Very recently, another S2-targeting nAb (CC40.8) was reported to be isolated from a COVID-19 patient whose serum exhibits broad reactivity with human β-CoV [456]. Antibody CC40.8 neutralizes SARS-CoV-2 and SARS-CoV-1 with potencies of 11.5 µg/mL and 14.8 µg/ml, respectively. It exhibits protective efficacy against SARS-CoV-2 challenge in a hamster infection model. The CC40.8 epitope locates at residues 1140–1164 (see “Y” area in Fig. 2A) in the stem-helix region [457]. Using an alternative approach, a nAb (28D9) that also targets the stem-helix region (residues 1229–1243; see “Z” area in Fig. 2A) was isolated from humanized mice that were sequentially immunized with the spikes of HCoV-OC43, SARS-CoV-1 and MERS-CoV [458]. Antibody 28D9 has strong and comparable binding activity to spikes of five coronaviruses of lineage A (Embecovirus), lineage B (Sarbecovirus) and lineage C (Merbecovirus) of β-CoV. Antibody 28D9 potently neutralize MERS-CoV (IC50 = 0.13 µg/mL) but very weakly neutralizes SARS-CoV-2 (IC50 = 45.3 µg/mL), SARS-CoV-1 (IC50 = 60.5 µg/mL) and HCoV-OC43 (IC50 = 64.9 µg/mL) [458].
The S2-targeting nAbs with broad breadth against pathogenic coronaviruses are of great value for engineering pan-coronavirus therapies to confront next and future waves of coronavirus-related diseases. It is critical to continue to isolate more promising S2-targeting nAbs and to identify new susceptible epitopes in the S2 subunit. Engineering of existing S2-targeting nAbs could also help to enhance neutralizing potency and breadth. Such strategies have shown promising results for several RBD-targeting nAbs [219, 459].
Other Potential SARS-CoV-2 Receptors and Antibodies Against Them
All of the antibodies described above are focused on the interaction of SARS-CoV-2 spike protein with the cell surface receptor, ACE2, and the ability of antibodies to inhibit that interaction. Nevertheless, key tissues such as nasal epithelial cells [460], lung epithelial cells [461], and vascular endothelial cells [462] have relatively low levels of ACE2, the primary receptor for SARS-CoV-2. This suggests that alternative receptors may be involved in SARS-CoV-2 cell entry. Based on relatively minor evidence, it appears that several other receptors, including CD147 [463, 464], neuropilin-1 [460, 465], CD209 [466, 467], CD209L [467], and heparin sulfate [468], may be involved in either implementing or facilitating SARS-CoV-2 cellular entry, although the exact binding sequences and mechanisms of action for these potential alternative cell entry receptors is still lacking. Finally, a receptome profiling study recently identified additional potential receptors for SARS-CoV-2 spike protein, including asialoglycoprotein receptor-1 (ASGR1) and kringle containing transmembrane protein 1 (KREMEN1), that may play a role in SARS-CoV-2 cell entry either independently, or by ACE2/ASGR1/KREMEN1 (ASK) receptor combinations [469]. If any of these alternative pathways for cell entry are eventually found to be physiologically and/or pharmacologically relevant, they represent additional challenges, as well as potential therapeutic targets, for treatment of COVID-19. A few of these potential alternative receptors are described in more detail as follows.
CD147
CD147, also called basigin and EMMPRIN (extracellular matrix metalloproteinase inducer), has been proposed as an alternative receptor for the SARS-CoV-2 spike protein [463, 470]. This is somewhat controversial, as it comes from a single group [463], and has only been verified by one other independent research group [464]. The potential significance of this is that CD147 is a receptor on T-cells, which have low expression levels of ACE2, and there are reports that SARS-CoV-2 infection can also kill T-cells [470]. The validity of the data supporting CD147 as an alternative SARS-CoV-2 receptor, however, has been challenged by Shilts et al. [471], who provided evidence against CD147 being a second receptor for SARS-CoV-2 spike protein. Nevertheless, Geng et al. [472] generated humanized transgenic mice expressing human CD147, and found that meplazumab could protect those mice from SARS-CoV-2 caused pneumonia. Meplazumab (HP6H8) is a humanized anti-human CD147 IgG2 antibody that has been studied in clinical trials for treatment of malaria, based on the finding that it inhibits the cell entry of the malaria parasite, Plasmodium falciparum [473]. The apparent IC50 for the ability of meplazumab to suppress SARS-CoV-2 viral titer is 15 µg/mL [474], which is quite high and would suggest that huge doses would be required for functional activity in vivo. Nevertheless, a Phase I/II dose escalation clinical trial with doses ranging from 0.05 to 0.56 mg/kg was run with a total of 59 patients to test the ability of meplazumab to treat SARS-CoV-2 infected patients [474]. Despite the fact that blood concentrations of meplazumab topped out at 1–2 µg/mL, about tenfold below the IC50 for viral suppression [463], the clinical researchers reported statistically significant differences in time to hospital discharge, disease severity scores, and time to virus-negative status between patients treated with meplazumab and control patients [474]. Since this does not make pharmacological sense at this point, it remains to be seen whether anti-CD147 antibodies will provide meaningful benefit for COVID-19 patients. CD147 is a receptor on several tissues, including T lymphocytes, and has been implicated in other viral diseases [464]. Thus, if these results are confirmed in larger clinical trials and supported by further preclinical studies by a broader group of scientists, this could be a significant finding.
Neuropilin-1
Neuropilin-1 (NRP1; also known as CD304) was shown to bind to the C-terminus of cleaved S1 during the proteolytic processing step of SARS-CoV-2 cell entry, resulting in NRP1 receptor mediated viral entry [465]. This process was inhibited down-modulation of the receptor via RNA interference [465], and by modifying the furin cleavage site in SARS-CoV-2 S protein [460], demonstrating specificity for both the NRP1-mediated viral entry and the substrate for that entry [465]. Moreover, autopsies of COVID-19 victims revealed that NRP1-positive, ACE2-low to negative, epithelial cells in the nasal cavity were infected with SARS-CoV-2 [460], suggesting that NRP1 may play a significant role in SARS-CoV-2 infection in the nasal passages. An anti-NRP1 antibody, ASP1948 (also called PTZ-329) is currently in Phase I clinical trials for inhibition of T-regulatory cells in a cancer setting (NCT03565445 [7]). Thus far, this antibody has not been tested against SARS-CoV-2.
CD209 and CD209-L
The C-type lectin receptor, CD209, also known as DC-SIGN (Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin), and its ligand, CD209-L (L-SIGN; Ligand for Specific Intercellular adhesion molecule-3-Grabbing Non-integrin), have been shown previously to act as alternative receptors for SARS-CoV-1 [475–477]. Along a similar vein, it has recently been reported that the C-type lectin receptor, CD209, also known as DC-SIGN (Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin), can bind B8-dIgA1- and B8-dIgA2-bound SARS-CoV-2 RBD, thereby acting as an alternative receptor for SARS-CoV-2 in mucosal passages [466]. A separate study has also shown in vitro evidence suggesting that SARS-CoV-2 RBD also can bind, and be internalized by, CD209 in the absence of bound IgA [467]. Additionally, SARS-CoV-2 can bind, and be internalized by, CD209 ligand (L-SIGN; Ligand for Specific Intercellular adhesion molecule-3-Grabbing Non-integrin), which was enhanced with non-glycosylated CD209-L [467]. CD209-L in particular is interesting due to its significant expression in lung and kidney epithelial and endothelial cells [467]. Currently there are no antibodies in development for either CD209 or CD209-L.
Mechanisms of Action
While direct competition with ACE2 binding is the primary mechanism of action (MOA) for many antibodies targeting SARS-CoV-2, it is not the only MOA used by antibodies to neutralize the virus. Recent publications have highlighted at least five other distinct mechanisms of action, including FcγR-dependent ADCC and/or ADCP, lock-down of RBDs within a spike to prevent interaction with ACE2, degradation of spike protein, blocking internalization post-ACE2-binding, and cross-linking spikes, which may also function to block internalization. It is clear that many antibodies possess multiple MOAs, which should add to their potency and potentially breadth of activity. These MOAs, which may turn out to be as, or more, important across the board than interfering with ACE2 binding, will be covered in more detail below.
Fc Functional Activity
As discussed in detail in Sect. 4.3, Fc activity is now considered as a potentially critical function for many SARS-CoV-2 antibodies, including ADCC and ADCP [197, 205, 220, 247, 296–299], while CDC activities have been more associated with immune reaction-based side effects [300]. Additionally, engineering the human IgG Fc to extend the circulating half-life, which has been incorporated into several SARS-CoV-2 antibodies [417, 421], increases the AUC which can improve functionality over time. Since these topics were covered earlier, they will not be covered further here.
Locking down RBDs
As mentioned previously, C144 has the ability to not only bind its primary RBD epitope, but also to bind a second RBM at a distal site, resulting in the ability to lock the RBDs into a closed conformational state that prevents interaction with the SARS-CoV-2 receptor, ACE2 [276]. C144 accomplishes this by inserting its long CDR-H3 into a hydrophobic patch in the neighboring RBD (see Fig. 7A, M (“second” epitope on RBD marked in yellow)). BG10-9 has the same type of binding (Fig. 7B, Z); it binds to a novel epitope on one RBD, either in the open or closed position, while it also binds a neighboring RBD, locking the spike trimer in a closed conformation, neutralizing SARS-CoV-2 [399]. Because this epitope is also relatively conserved, BG10-9 also cross-reacts and neutralizes SARS-CoV1 and WIV1-CoV.
Other antibodies, such as S2M11 [299], Fab2-4 [396], Ab1-57 [478], XG014 [479] and COVOX-316 [480], are also known to possess this same mechanism, i.e., locking down RBDs in a closed conformation, as a mechanism for neutralization of SARS-CoV-2 [276]. One significant subtlety within this group of antibodies is that C144 [276], S2M11 [299], BG10-9 [399], and XG014 [479] all bind two RBDs simultaneously to lock down all RBDs, whereas COVOX16 [480], Fab2-4 [173], and Ab1-57 [478] are capable to lock down the RBDs even though they only bind one RBD at a time [479].
Blocking Internalization Post ACE2-Binding
As noted in Section 6.5, NTD-targeting nAbs do not compete with ACE2 binding to RBD [396], so their ability to neutralize relies instead on blocking entry of the virus into cells after attachment of the viral spike protein to its receptor, ACE2 [171, 444, 449]. It has been suggested that anti-NTD antibodies may force conformational changes in the S protein to indirectly inhibit membrane fusion between virus and host cells [449]. Additionally, antibodies that bind the outer face cryptic site (e.g., RBD-5 epitope group including S309, C135, 47D11, VHH antibody n3113) block membrane-membrane fusion by a still undefined mechanism, thereby interrupting internalization [481]. Neither of these mechanisms inhibits ACE2 binding to RBD.
Cross-Linking Spike Proteins
Tan et al. [482] demonstrated that the dimensions of a typical IgG are approximately 8.5 nm (height) × 14.5 nm (Fab arm to Fab arm width) × ca. 4 nm depth. They also calculated the average distance between antigen combining sites to be 13.7 nm. A caveat to these dimensions is the well-known ability of IgG Fab arms to rotate and flex on their axis, as well as the substantial differences in Fab arm angles exhibited by IgG1, IgG2, and IgG4 isotypes [483]. SARS-CoV-2 virions have been measured to be approximately 90 nm in diameter, with each virion containing approximately 100 spike trimers, yielding an average distance between spike trimers of about 20 nm [361]. Given that the trimer heads are about 15 nm across (Fig. 11), this means that there is ample opportunity for IgGs to engage in inter-spike cross-linking. The optimal distance between combining sites of a human IgG for greatest avidity was found to be approximately 13 nm, approximately the average distance as noted above, provided the greatest avidity effect based on both Fab arms [484]. Based on the theoretical measurements (Fig. 11) and data from Hastie et al. [290], it appears that both intraspike and inter-spike binding could be achieved with maximal avidity.
Fig. 11.

A model of the surface of a SARS-CoV-2 virion showing sizes and distances of spike and antibodies. Top: A The size and shape of a human IgG1 [480]. B The size, shape, and dimensions of SARS-CoV-2 spike proteins [361], informed also by SARS-CoV-1 spike protein anatomy [485, 486]. There are approximately 100 spikes per 90 nm diameter virion [361]. C A model showing how the sizes and spacings allow for potential inter-spike cross-linking by both IgGs, as recently observed by Hastie et al. [290], as well as potential for crosslinking by triple-VHH-Fc fusions. This model also shows the approximate coverage of an IgM, which has a diameter of about 30 nm [337]. Bottom: Model of SARS-CoV-2 targeted by antibodies. SARS-CoV-2 spike proteins with one open RBD or all closed RBDs based on PDB ID 6VYB and 6VXX, respectively. A IgGs (PDB ID 1IGT) crosslinking two spikes; B an IgM (based on PDB ID 2RCJ) binding to multiple spikes similar to a hand palming a basketball; C a trimeric VHH-Fc fusion protein (based on PDB IDs 6ZXN and 1IGT as per Fig. 2) cross-linking spikes; and D individual VHHs (PDB ID 6ZXN) binding to RBDs with no cross-linking ability. For the Bottom drawing, the PDB program [201, 202] was used to generate the structures. Approx approximate, dia diameter, IgG immunoglobulin G, IgM immunoglobulin M, nm nanometers, PDB ID Protein Data Bank Identifier, RBD receptor binding domain, SARS-CoV-2 severe acute respiratory syndrome coronavirus-2, VHH single domain antibody, VHH-Fc single domain antibody fused to an IgG-Fc domain
Hastie et al. [290] demonstrated clearly how some antibodies targeting SARS-CoV-2, based on epitope classes, are capable of inter-spike cross-linking, whereas others either bind a spike monovalently or intraspike, bivalently. While this may be a characteristic of a particular epitope [290], in natural responses to infection, this also could be a reflection of the IgG isotype, as the different IgG isotypes exhibit markedly different flexibilities and hinge length and Fab arm angles. The epitope classes noted by Hastie et al. [290] to engage in inter-spike cross-linking include antibodies in epitope groups RBD-1, RBD-5, RBD-6 and RBD-7. On the other hand, antibodies in epitope groups RBD-2, RBD-3, RBD-4 do not appear to be able to engage in inter-spike cross-linking [290].
Besides epitope and perhaps isotype influences on inter-spike crosslinking, there are at least two other mechanisms by which antibodies to SARS-CoV-2 may crosslink spikes. The first is via the natural interaction of antibody Fc with immune cells such as macrophages, monocytes, NK cells and neutrophils. While these binding activities may lead to neutralization via ADCC or ADCP, they also cluster antibodies together, resulting in the ability to cross-link spikes by cluster effect. Those antibodies lacking Fc activity, as noted above, would lack this effect and likely lose some in vivo potency as a result of that, on top of the loss of potency due to the absence of ADCC and ADCP MOAs.
Similar to Fc-muted IgGs, domain or VHH antibodies bind RBD and block its ability to bind ACE2 effectively neutralizing the virus, but they do not bring immune cell activity to the fight. One of the greatest perceived advantages of VHH antibodies is their ability to be dosed via the intranasal or inhaled route of administration with the potential for deep access in the lungs [252, 253, 486]. One type of VHH construct is a homotrimeric VHH in which three anti-RBD VHH domain antibodies are linked together via standard GS type linkers, allowing for targeting of more than one RBD at a time [252, 253, 346]. These trimers may engage in inter-spike crosslinking as well, based on the distances.
The anti-SARS-CoV-2 IgM antibody, IGM-6268, is currently in Phase I clinical trials. As can be seen in Fig. 11, IgM, with its ten antigen-binding Fab domains, can theoretically cover the area occupied by about three and possibly, with expected virus membrane fluidity, four spike trimers. In light of the observation by Hastie et al. [290] that the potency of RBD-5 epitope group antibodies correlates with their ability to cross-link spike proteins, an IgM format may be preferred over IgG to neutralize SARS-CoVs. IGM-6268 (CoV-14) had excellent activity on SARS-CoV-2 Beta and Gamma variants despite its’ IgG version exhibiting considerable loss of activity on those variants [245], likely due to the difference in avidity.
Induction of Spike Disorder and S1 Shedding
Antibodies that bind the inner face cryptic sites (e.g., the RBD-6 and -7 epitope groups, including CR3022, EY6A, etc.) are known to disrupt the structural integrity of the spike protein [255, 403]. While this activity effectively impedes cell entry, CR3022 is poorly neutralizing, whereas EY6A, which possesses a very similar epitope (Fig. 6A, B), is a strongly neutralizing antibody.
As shown in Table 5, several anti-SARS-CoV-2 RBD antibodies (e.g., amubarvimab, S2X259, S2X58, S2H97), amongst others, have the ability to induce shedding of S1 upon binding RBD [213, 261, 487]. Additionally, two new antibodies, 7D6 and 6D6, also have shown the ability to promote S1 shedding, leading Li et al. [432] to propose that the primary MOAs for neutralization of SARS-CoV-2 by those antibodies is via induction of spike disorder and, ultimately, shedding of the S1 subunit [432]. In the context that the D614G mutation helps to stabilize the spike protein and reduce S1 shedding, which results in greater infectivity [186], an antibody MOA which induces S1 shedding to reduce viral infectivity makes functional sense. In a similar vein, while CR3022 neutralizes SARS-CoV-2 via disruption of the conformation of the prefusion spike protein, it has not been shown whether that antibody causes S1 shedding [255].
In a new report demonstrating an interesting twist on inter-spike crosslinking, Hanke et al. [344] demonstrated that a novel nanobody, Fu2, was able to cross-link spike proteins head-to-head rather than side-by-side. Fu2 binds two distinct overlapping epitopes on RBD, i.e., the RBD-2 area and the RBD-6 area (Fig. 9), which resulted in two significant MOAs. First, all three RBDs were locked in the up position blocking ACE2 binding, and second, the nanobody cross-linked the spike with another spike in a head-to-head conformation to generate “spike trimer dimers.” Since these would necessarily be inter-virion cross-links, Fu2 caused virion aggregation and potent neutralization [344]. A unique aspect of this MOA is that the nanobody functioned to cross-link the spike trimers as a monomeric unit [344].
Alternative Modes of Delivery
The requirement for cell surface-based infection of a cell by SARS-CoV-2 are the presence of the viral receptor, ACE2, and the protease, TMPRSS2, that assists in processing the spike protein during cell entry [488–490]. These two proteins are most highly co-expressed in the lung, as described earlier [82, 86–88], but also have a wide distribution throughout the respiratory system and other organs [490]. ACE2 and TMPRSS2 are expressed on nasal epithelial cells in the nasal passages [491] and it has been demonstrated that the sinus and nasopharynx are the initial sites of SARS-CoV-2 replication upon infection [488]. Moreover, high nasopharyngeal viral loads of SARS-CoV-2 have been shown to directly correlate with several critical parameters in COVID-19 patients, including hypoxemia and disease outcomes [492], increased organ damage and disease severity, and risk of intubation and in-hospital mortality [440]. This is now even more amplified as noted earlier, since the Delta variant rapidly replicates in the upper respiratory system to produce more than 1000-fold higher titer in nasopharyngeal swabs compared to the initial SARS-CoV-2 [493]. As noted previously, the Omicron variant, which relies more heavily on cathepsin-dependent endosomal entry, also replicates to high viral loads in the upper respiratory system [82, 86]. Finally, it was recently discovered that the oral cavity also is a major site of primary infection and a carrier of high viral load [494]. Thus, getting therapeutic levels of antibody to the nasopharynx/oral cavity and the luminal surface of the lung, the initial and secondary sites of SARS-CoV-2 replication, respectively [446], is critical to blocking infection and treating the disease.
The first round of antibodies to SARS-CoV-2, including the four that have been granted EUA, have all been developed as IV infusions. This approach has been shown to require very high doses, in some cases as high as 4-8 grams (approximately 60–120 mg/kg for a 70 kg patient), in order to achieve efficacy. A potential reason for the need for such large doses is the poor bioavailability of IV-dosed antibodies to the lung, which has been estimated to be in the range of 1% [495]. While such doses have been effective in treating patients with mild to moderate disease and have resulted in EUA approvals (Table 3), considerable effort and resources are needed to produce and distribute such large quantities of recombinant proteins. Second, IV administration requires an infusion that can only be performed in hospital or outpatient settings where the necessary equipment and trained personnel are available. This can be a complication since clustering COVID-positive patients in hospital suites or infusion centers poses a potential risk to patients and staff. And third, not all patients have ready access to infusion centers, resulting in large numbers of patients who cannot get treatment.
One approach to partially mitigate the issues associated with IV administration of anti-SARS-CoV-2 antibodies is to use subcutaneous (SC) or intramuscular (IM) delivery (Table 9). However, delivery volume considerations may limit the antibody doses that can be delivered IM, whereas SC delivery has greater flexibility. Indeed, Regeneron has tested SC dosing of their REGEN-COV™ antibody combination product and has recently received an EUA for SC delivery, and several other antibody therapeutics are currently being tested for IM delivery (see Table 9). One of these antibodies, ADG20, has reported good tolerability of IM doses up to 600 mg [496] and is currently in studies for post-exposure and pre-exposure prophylaxis. But as with IV administration, doses of 300 to 1,200 mg are still being used and, again due to volume constraints, multiple SC and IM injections may be required. Evusheld™ (AZD7442), which received an EUA from the FDA on 12/8/21 for pre-exposure long term prophylaxis with high patients, is dosed in two IM injections of 300 mg, one for each antibody (tixagevimab and cilgavimab) in the preparation [497]. Still, this approach represents an important improvement in being able to rapidly treat patients, as infusion centers are not required, and more patients can be more easily treated. Beyond IV, SC and IM, several other approaches are also being explored for the delivery of SARS-CoV-2 neutralizing antibodies to COVID-positive patients and to subjects at high risk of infection.
Table 9.
Alternative modes of delivery for antibodies targeting SARS-CoV-2
| Candidate | Sponsor | SOD | Format | Dose (mg) | ROA | MOA | Strategy | Clinical trial # | References |
|---|---|---|---|---|---|---|---|---|---|
| Evusheld™ | AstraZeneca | EUA | 2x IgGs mixture | 125 mg x two injections | IM | INJ | Px (long term) | NCT04625725 (ANR) | [497] |
| REGEN-COV™ | Regeneron | EUA; Phase III | 2x IgGs mixture | 1200 mga | SC | INJ | Rx | NCT04452318 (C) | [498] |
| CT-P63 and CT-P59 | Celltrion | Phase III | 2x IgGs mixture | ND | INH | NEB | Rx | NCT05224856 (NYR) | [499] |
|
Xevudy™ (UK); Sotrovimab (VIR-7831) |
Vir |
EUA; Phase II/III |
IgG | 250 mga, 500 mga | IM | INJ | Rx | NCT04913675 (ANR) | [209] |
| AZD7442 | AstraZeneca | Phase III | 2x IgGs mixture | 300 mg a | IM | INJ | Rx | NCT04625972 (ANR) | [211] |
| MAD0004108 | Toscana Life Sciences Sviluppo | Phase II/III | IgG | 100 mga, 400 mga | IM | INJ | Rx | NCT04952805 (R) | [221] |
| ADG20 | Adagio | Phase II/III | IgG | 400 mga | IM | INJ | Px/Rx | NCT04859517 (R), NCT04805671 (R) | [219] |
|
BI 767551 (DZIF-10c) c |
Boehringer Ingelheim | Phase II/III | IgG | ND | INH | NEB | Rx | NCT04894474 (W) | [227] |
|
STI-2099 (COVI-DROPS) |
Sorrento | Phase II | IgG | 10-40 mga | IN | Drops | Rx | NCT04906694 (NYR), NCT04900428 (R) | [226] |
| IGM-6268 (CoV-14) | IGM Biosciences | Phase I | IgM | 7.5 mg dose/day a | IN | Drops | Px/Rx |
NCT05160402 (R), NCT05184218 (R) |
[245, 246, 410] |
| IgY-110 | Stanford | Phase I | IgY | 2-24 mga | IN | Drops | Rx | NCT04567810 (C) | [500] |
| EU126-M2 | Eureka | PC | IgG | 1.25–10 mg/kgb | IN | Drops | Px | NA | [501, 502] |
| Nb15-NbH-Nb15 | Abrev Biotech-nology Co., Ltd. | PC | Bispecific nanobody | 10 mg/kgb | IN | Drops | Px/Rx | NA | [503] |
| PiN21 | University of Pittsburg | PC | Nanobody | 0.2 mg/kgb | IN | NEB | Px | NA | [253] |
| 1212C2 d | Aridis | PC | IgG | 0.6 mg/kgb | INH | NEB | Px/Rx | NA | [251] |
| AUG-3387 | TFF Pharma-ceuticals/Augmenta Bioworks | PC | IgG | 0.3-1.0 mg/kgb | INH | Dry powder | Px/Rx | NA | [504] |
|
STI-9199 e (COVISHIELD™ IN) |
Sorrento Therapeutics, Inc/Mount Sinai | PC | Human IgG1-LALA | 0.5 mg/kgb | IN | Drops | Rx | NA | [254] |
ANR active not recruiting, C completed, EUA emergency use authorization granted, IM intramuscular, IN intranasal, INH inhalation, INJ injection, MOA mode of administration, NA not applicable, NCT National Clinical Trial (registry), ND not disclosed, NEB nebulizer, NYR not yet recruiting, PC preclinical, Px prophylaxis, R recruiting, ROA route of administration, Rx therapeutic treatment, SC subcutaneous, SOD stage of development, W withdrawn (from clinical development)
aClinical dose in mg (total)
bEfficacious preclinical dose in mg/kg
cDiscontinued from development 7/26/21
dPreclinical study was with antibody 1212C2, but plans are to move forward with AR-712, a cocktail consisting of two anti-SARS-CoV-2 IgGs [505]
eSTI-9199 is IN formulation of STI-9167, which has been shown to have strong neutralization activity against Omicron
Based on the data presented above, multiple intranasal (IN) approaches are also being explored for the delivery of neutralizing antibodies, in an effort to prevent or control infections by SARS-CoV-2 (see Table 9). The first antibodies targeting SARS-CoV-2 to enter clinical trials in this category were nose drops containing IgY-110, an IgY antibody isolated from immunized chickens [500], and STI-2099, an IN formulation of the STI-9167 human anti-SARS-CoV-2 IgG antibody, has been tested in healthy adults in Phase I studies and has announced the initiation of a Phase II study [276] (NCT04900428 [7]). In an interesting variation on this approach, the appropriate genes are being incorporated into adenovirus vectors for direct infection of the sinus, which are rich in sialic acid glycans, the receptors for many adenoviruses, to achieve local production of the antibodies [506] or ACE2 decoys [507].
At the preclinical stage, significant protection has also been reported in animal models using IN administration of antibodies to SARS-CoV-2 (see Table 9). Included in this group are antibody-based products such as EU126-M2 [501, 502] and the Nb15-NbH-Nb15 bispecific nanobody derived from llamas [503], each of which has reported significant protection in mouse models of SARS-CoV-2 infection. Similarly, a trimeric nanobody has been reported that can potently neutralize SARS-CoV-2, and that may be useful for intranasal delivery [346].
In addition to IgGs and nanobodies, other Ig forms may also have particular relevance for modes of delivery besides the typical IV route. We recently described the development of a highly potent human IgM antibody, IGM-6268, that can neutralize authentic SARS-CoV-2 virus at low pM concentrations [245]. Moreover, when administered intranasally, IGM-6268 provided significant in vivo protection in mice infected with Alpha, Beta, and Gamma SARS-CoV-2 VOCs at therapeutic and prophylactic doses as low as 0.4 and 0.044 mg/kg, respectively [245]. IGM-6268, which has significant neutralization activity against Delta and Omicron BA.1 [410], is currently in dose escalation Phase I clinical trials, dosing up to 7.5 mg/day via the intranasal route (NCT05160402, NCT05184218; [7]) (Table 9).
Lastly, inhalation is also being explored as an alternative route for the delivery of antibody therapeutics for COVID-19. Following initial replication of SARS-CoV-2 in the sinus and nasopharynx, the virus spreads to the lungs where it creates a strong pulmonary infection [488]. Inhalation of neutralizing antibodies may therefore be able to interfere with infection in the lung and possibly prevent pulmonary damage. There is precedence for delivery of antibodies and other biologics to the lung via inhalation; recently, Liang et al. [508] described at least 18 biologics that have been under clinical investigation for inhalation delivery. The most advanced SARS-CoV-2 neutralizing antibodies are the IgGs, CT-P59 (regdanvimab) plus CT-P63 [241], co-formulated for inhalation delivery. This combination inhaled product is currently preparing for a Phase III clinical trial (NCT05224856; [7]) [499]. BI 767551 (DZIF-10c) was formulated and under development for inhaled delivery [227], but has since been dropped after reaching at least Phase II clinical trials. Aridis 1212C2 [251] and TFF Pharmaceuticals AUG-3387 [504], both of which are currently in preclinical studies, are being formulated for inhalation delivery (Table 9).
Antibodies for Palliative Therapy
Progression of COVID-19 after infection is quite varied from one individual to the next, but in broad terms the infection course tends to follow a given pattern, even if quantity and identity of cells, cytokines, and other response elements may be varied. COVID-19 typically progresses through a viral infection stage, in which the virus rapidly infects alveolar cells, followed by replication, release, and further expansion of the infection phase to cells outside the respiratory tract (Fig. 1). This first stage of infection is often accompanied by a dry cough, fever, and fatigue as the body begins to respond to the infection. This phase, which occurs within the first 24–48 h after exposure, is the period in which anti-SARS-CoV-2 antibody therapy has the greatest chance to succeed. Overlapping the viral infection stage is the initiation of an immune response stage, which can be from asymptomatic to very severe. This stage is typically characterized by lymphocytopenia, likely due to T cells migrating into tissues at sites of infection [509], and increased expression of type I interferons to counteract the viral invasion [510]. This, in turn, drives the overexpression of proinflammatory cytokines such as IL-6 [511], and chemokines such as IL-8 (aka CXCL8 [C-X-C motif chemokine ligand 8]) [512, 513]. The lymphopenia usually appears within the first week of infection, whereas the cytokine storm, typically associated with macrophage activation, typically occurs later as the disease progresses [514]. Nevertheless, there may be a link between these two processes, as it has been demonstrated that neutralization of IL-6 helped to restore circulatory T-cell counts [515]. The proinflammatory signals can ultimately lead to vascular leakage, alveolar edema and hypoxia (ARDS), and ultimately, multi-organ failure [516]. These signals begin during the second stage of COVID-19 and, in some patients, can become overwhelming in the third stage of COVID-19 (Fig. 1).
Zhang et al. [509] categorized COVID-19 disease into four reasonably well-defined categories: (i) mostly asymptomatic, PCR-positive patients with no fever, no respiratory issues, and no lung damage as determined by X-ray tests. These patents, who make up about 30% of all COVID-19 cases, are known as “asymptomatic carriers”; (ii) mild cases in which patients had fever, and showed signs of pulmonary inflammation, indicating pneumonia, in X-ray scans; (iii) patients with severe disease who experienced difficulty breathing and possessed lung damage visualized by X-ray as “ground-glass opacities”; and (iv) critically ill patients who developed ARDS, typically requiring invasive mechanical ventilation (IMV) to support breathing. These patients, who make up some 20% of all cases, typically had mortality rates of 45–60% in the earlier days of the epidemic [517], although these rates have come down as COVID-19-specific critical care has improved. The US NIH has added a fifth category of “moderate illness” in patients who are demonstrated to have respiratory damage, but able to maintain blood oxygen levels above 94% [518].
United States National Institutes of Health (US NIH) guidelines for treatment of severe and critical COVID-19 include the use of several drugs as palliative or supportive care in treatment of immune dysregulation associated with response to infection. Table 9 provides a listing of antibodies in clinical trials that have been, or are being, tested as potential palliative care drugs for COVID-19. As of November 2021, the only antibodies on the NIH recommendation list for use in the most severe cases are the anti-IL-6R mAbs, tocilizumab (Actemra®) and sarilumab (Kevzara®; recommended only if Actemra® is not available), which have a BIIa rating (B, moderate; IIa, based on randomized trials with sub-group analyses) [519].
Beyond the NIH recommendations, several other antibodies have been studied in clinical trials for potential use to treat various aspects of the immune response to SARS-CoV-2 infection, including macrophage activation and trafficking, cytokine storm, complement issues, immune cell over-activation, and T-cell trafficking. Table 10 shows 38 antibodies that have been, or are being, studied in clinical trials for palliative treatment of COVID-19. Of these, only the anti-IL-6R mAbs, tocilizumab (Actemra®; USA, UK, WHO) and sarilumab (Kevzara®; UK, WHO), have received EUAs in the western world. Additionally, the anti-CD6 mAb, itolizumab (Alzumab®), has been granted EUA in India for treatment of COVID-19 (Table 10). Of the remaining candidates, five target the GM-CSF pathway to block macrophage activation and trafficking [522], several block inflammatory cytokines such as IL-1β, IL-6, TNF-α, and IFN-γ, and others block various steps in either immune activation or immune cell migration [30].
Table 10.
Clinical-stage antibodies and Fc-fusion proteins repurposed as palliative treatment for COVID-19
| Drug or candidate | Sponsor | COVID-19 most advanced status | COVID-19 clinical trial(s)a | Molecular target | Indication (non-COVID-19) |
Development Stage (US-FDA) | Format/Description |
|---|---|---|---|---|---|---|---|
| Alzumab (itolizumab) | Biocon (India) | EUA granted in India July 2020b | NA | CD6 | PsO | Approved in India, Jan 2013 | Humanized IgG1κ |
|
Actemra®; (RoACTEMRA® in EU) (Tocilizumab) |
Roche/Chugai/Genentech |
UK EUA 1/8/21; US-FDA EUA 6/24/21; WHO EUA 7/7/21; EC EUA 12/7/21 |
IL-6R (CD126) | CaD, RA |
US-FDA approved 1/09/10 |
Humanized IgG1κ | |
| Kevzara® (Sarilumab) | Sanofi/Regeneron |
UK EUA 1/8/21; WHO EUA 7/7/21 |
NCT04386239 | IL-6R (CD126) | RA |
US-FDA approved 5/22/17 |
Human IgG1κ |
|
Lenzilumab (KB003) |
Humanigen, Inc. | US-FDA EUA rejectedc; UK granted speedy review | NCT04583969 | GM-CSF | CMML, JMML | Phase III | Humanized IgG1κ |
| Ilaris® Canakinumab | Novartis |
Phase III (R) d Phase II (C) |
IL-1β | CAPS |
FDA Approved 06/19/09 |
Human IgG1κ | |
|
Ultomiris® (Ravulizumab-cwvz) |
Alexion Pharma | Phase III (R) | Complement C5 | PNH, CMD | FDA approved 12/21/18 | Humanized IgG2/4κ hybrid, modified Fc; Xencor Xtend HLE | |
|
Takhzyro® (Lanadelumab-flyo) |
Shire/Dyax | Phase III (ANR) | NCT04590586 | pKal | HAE | FDA approved 8/23/18 | Human IgG1κ |
| Avastin® (Bevacizumab) | Genentech | Phase III (NYR) | VEGF | CRC | FDA approved 02/26/04 | Humanized IgG1κ, | |
| Remicade® (Infliximab) | Johnson & Johnson | Phase II/III (R) | TNF-α | CrD, RA, 14 other indications | FDA approved 8/24/98 | Chimeric IgG1κ | |
|
Mavrilimumab (KPL-301) |
Kiniksa Phar-maceuticals | Phase II/III (R) | NCT04447469 | GM-CSF-Rα | GCA | Phase IIb/III | Human IgG4κ |
| TJM2 (aka TJ003234) | I-Mab Biopharma | Phase II/III (R) | NCT04341116 | GM-CSF | RA | Phase II/III | Humanized IgG1 |
|
Gimsilumab (KIN-1901) |
Kinevant Sciences GmbH | Phase II (C) | NCT04351243 | GM-CSF | RA | Phase II | Human IgG1κ |
|
Otilimab (GSK3196165) |
GSK/MorphoSys | Phase II (ANR) (D) | NCT04376684 | GM-CSF | RA, MS, OA | Phase III | Human IgG1κ |
| CERC-002 (AEVI-002, MDGN-002) f | Cerecor, Inc | Phase II (C, FTD) | NCT04412057 | LIGHT (TNFSF14) | CrD | Phase I | Fully human IgG4 |
|
Garadacimab (CSL312) |
CSL Behring | Phase II (C) | NCT04409509 | Factor XIIa antagonist | HAE, PICC-ADVT | Phase III | Human IgG4κ |
| Soliris® (Eculizumab) | Alexion Pharma | Phase II (R) | Complement C5 | PNH, aHUS | FDA approved 03/16/07 | Humanized IgG2/4κ hybrid, modified Fc | |
|
Avdoralimab (IPH5401) |
Innate Pharma/Novo Nordisk | Phase II (R) | C5a receptor (C5aR1) | INFL | Phase II | Human IgG1 | |
|
Narsoplimab (OMS721) |
Omeros | Phase II (R) | NCT04488081 | MASP-2 | HSCT-TMA, AHUS |
Phase III – BLA submitted |
Human IgG4κ |
|
Dupixent® (dupilumab) |
Regeneron/Sanofi | Phase II (R) | NCT04920916 | IL-4Rα | AD, asthma |
FDA Approved 3/28/17 |
Human IgG4κ-S/P; hinge modified |
|
Sylvant® (siltuximab) |
Fundacion Clinic per a la Recerca Biomédica | Phase II (R) | NCT04329650 | IL-6 | MCD |
FDA approved 4/23/14 |
Chimeric IgG1κ |
|
Clazakizumab (ALD518) |
CSL Behring | Phase II (R) |
NCT04363502 |
IL-6 | KTR | Phase III |
Humanized IgG1κ, non-glycosylated |
|
Olokizumab (CDP-6038) |
RPharm | Unstated (C) | NCT05196477 | IL-6 | RA | Phase III | Humanized IgG4κ |
|
BMS-986253 HuMax® IL-8 (MDX-018) |
BMS /Genmab/Cormorant Pharmaceuticals | Phase II (R) | NCT04347226 | IL-8 | Cancer | Phase II | Human IgG |
| hzVSF-v13 | ImmuneMed, Inc. | Phase II (R) | Vimentin ectodomain | Viral diseases | Phase II | Humanized IgG4 | |
|
Efineptakin alfa (Hyleukin-7; GX-17; NT-I7; rhIL-7-hyFc) |
NeoImmune-Tech/I-Mab | Phase II (R) | NCT04810637 | IL-7 receptor | HPV, GB, PML, MMel | Phase I/II | Fc fusion protein; Human IL-7 fused to a hybrid Fc (hyFc). |
|
Monalizumab (IPH2201) |
MedImmune/ Innate Pharma/Novo Nordisk |
Phase II (R) | NCT04333914 | NKG2A | INFL | Phase III | Humanized IgG4 |
| IC14 | Implicit Bioscience | Phase II (R) | CD14 | ALS, MND | Phase I | Chimeric IgG | |
|
Leronlimabe (PRO140) |
CytoDyn |
Phase III (NYR) Phase II (R) |
CCR5 | HIV, TNBC, NASH |
Phase II/III BLA submitted |
Humanized IgG4κ | |
| Opdivo® (nivolumab) | BMS | Phase II (NYR) | NCT04413838 | PD-1 | MMel | FDA approved 12/22/14 | Human IgG4κ |
| Simulect® (basiliximab) | Fort Worth Clinical Sciences Working Group | Phase II (NYR) | NCT05013034 | IL-2 | AOR | FDA approved 12/5/98 | Chimeric IgG1κ |
| Foralumab | Tiziana Life Sciences, PLC | Phase II (NYR) | NCT04983446 | CD3ε | CD, MS | Phase II | Human IgG1κ |
|
Adakveo® (Crizanlizumab-tmca) |
Novartis | Phase II (C, NKFD) | NCT04435184 | PSGL-1 | SCDA-VOC | US-FDA approved 11/15/19 | Humanized IgG2κ |
|
Astegolimab (RG6149; AMG 282; MSTT1041A) |
Genentech | Phase II (C, D) | NCT04386616 | ST2 (IL-33R) | Atopic asthma | Phase II | Human IgG2κ |
|
Efmarodocokin alfa UTTR1147A |
Genentech | Phase II (C, D) | NCT04386616 | IL-22 | GVHD, NIH | Phase II | IL-22-IgG4-Fc fusion protein |
|
CD24Fc g (MK-7110) |
Merck (OncoImmune) | Phase III (C, D) | NCT04317040 | P-selectin | GVHD, ALL, AML, MDS | Phase II/III | CD24-Fc fusion protein |
|
Gamifant® (Emapalumab-lzsg) |
NovImmune/SOBI | Phase II/III (T, D) | NCT04324021 | IFN-γ | HLH | US-FDA approved 11/20/18 | Fully Human IgG1λ |
|
Pamrevlumab (FG-3019) |
Fibrogen | Phase II (T, NKFD) | NCT04432298 | CTGF | DMD, IPF | Phase III | Human IgG1κ |
| CPI-006 | Corvus |
Phase III (SUS) Phase I (C) |
CD73 | Solid tumors | Phase III | Humanized IgG1 |
AD Atopic dermatitis, aHUS atypical hemolytic uremic syndrome, ALS amyotrophic lateral sclerosis, AML adult acute myeloid leukemia, ANR clinical trial status as active but not recruiting, AS Ankylosing spondylitis, BI Boehringer Ingelheim, BMS Bristol-Myers Squibb, C completed, CaD Castleman’s disease, CAPS Cryopyrin-associated periodic syndrome, CCR5 C-C chemokine receptor type 5, CD cluster of differentiation, CrD Crohn’s disease, CMD Complement-mediated diseases, CMML chronic myelomonocytic leukemia, CRC colorectal cancer, CTGF connective tissue growth factor, D discontinued (typically based on press release from sponsor), DMD Duchenne Muscular Dystrophy, EC European Commission, EUA emergency use authorization, Fc IgG fragment (crystallizable), FTD fast track designation (by US-FDA), GB glioblastoma, GCA giant cell arteritis, GM-CSF granulocyte-macrophage colony-stimulating factor, GVHD graft-versus-host disease, HAE hereditary angioedema, HIV human immunodeficiency virus, HLE half-life extension, HLH hemophagocytic lympho-histiocytosis, HPV human papillomavirus, HSCT-TMA hematopoietic stem cell transplant-associated thrombotic microangiopathy, IFN interferon, IL interleukin, INFL inflammation/inflammatory disease, IPF idiopathic pulmonary fibrosis, JMML juvenile myelomonocytic leukemia, KTR kidney transplant rejection, LIGHT homologous to lymphotoxin exhibits inducible expression and competes with HSV glycoprotein D for binding to herpesvirus entry mediator a receptor expressed on T lymphocytes, MASP-2 Mannan-binding lectin-associated serine protease-2, MCD Multicentric Castleman’s disease, MMel metastatic melanoma, MND motor neuron diseases, NA not applicable, NASH nonalcoholic steatohepatitis, NKFD no known future development plans, NKG2ACD94/NK (natural killer) group 2 member A, PML progressive multifocal leukoencephalopathy, PICC-ADVT peripherally inserted central venous catheter (PICC)-associated deep vein thrombosis (DVT), pKal plasma kallikrein, PNH paroxysmal nocturnal hemoglobinuria, PSGL-1 P-selectin glycoprotein ligand-1 (aka CD162), PsA psoriatic arthritis, PsO Psoriasis, R recruiting, RA rheumatoid arthritis, rh recombinant human, RYR clinical trial registered but not yet recruiting, SCDA-VOC sickle cell disease associated vaso-occlusive crises, SUS clinical trial suspended (sponsor decision), T terminated clinical trial, TNBC triple-negative breast cancer, TNF tumor necrosis factor, UK United Kingdom, US-FDA United States Food and Drug Administration, VEGF vascular endothelial growth factor, WHO World Health Organization
aNot all COVID-19-related trials are included here
bAtal et al. [520]
c[521]
dIn diabetic patients who contract COVID-19, after not meeting endpoints for treatment of COVID-19 directly
eUS-FDA stated on 5/17/21 that the data did not support clinical benefit for use of leronlimab to treat COVID-19
fGranted fast track designation by US-FDA on 11 May 2021 based on Phase 2 data, so further development expected
gIn Merck 10-k filing on 2/25/21, Merck stated that FDA requested additional studies to support an EUA filing, ultimately resulting in Merck discontinuing on development of MK-7110 for COVID-19
Perhaps the most significant cytokine driving COVID-19 disease severity is IL-6 [511]. Currently, four antibodies are in clinical trials targeting the IL-6 pathway, including two targeting IL-6 cytokine (siltuximab [Sylvant®], clazakizumab) and two targeting the IL-6 receptor (tocilizumab [Actemra®], sarilumab [Kevzara®]). The most advanced of these, as noted above, is tocilizumab, which has been granted multiple EUAs. The data supporting tocilizumab EUAs, however, are mixed, with some studies demonstrating significant improvements in patient outcomes such as time to hospital discharge or survival [523–525], whereas in other studies, no significant differences were observed [526]. Recent studies have suggested that focusing on patients with high circulatory IL-6 concentrations [527] or timing in administration of tocilizumab [528] were potential key factors in the success of tocilizumab in improving outcomes. One of the potential problems in these studies is that most of the data supporting or not supporting tocilizumab come from retrospective multi-study analyses. Nevertheless, a recent analysis has shown that the use of tocilizumab in supportive care for COVID-19 is cost-effective in reducing mortality as measured in QALYs (quality-adjusted life years) [529]. One of the keys for successful use of tocilizumab, just the same as for anti-SARS-CoV-2 approaches, is early administration [530, 531]. Interestingly, the use of tocilizumab for COVID-19 has been wide enough to cause a world-wide temporary shortage of the drug, as announced by Genentech in August 2021 [532]. Thus, it seems likely that tocilizumab has been used more widely in COVID-19 palliative treatment regimens than as recommended by the NIH.
The EUA in India for the use of itolizumab in treatment of COVID-19 was based on the results of a small clinical trial that demonstrated potential [520]. Itolizumab targets CD6, which is involved in continuation of T-cell activation responses. It has been demonstrated that blocking CD6 can reduce production of proinflammatory cytokines interferon-γ (IFN-γ), interleukin (IL)-6, and tumor necrosis factor-α (TNF-α) and adhesion molecules that eventually leads to reduced T-cell numbers at inflammatory sites [520].
GM-CSF is a cytokine that activates myeloid cells and stimulates their proliferation and migration to inflammatory sites [513, 533]. As such, GM-CSF, due to its potential role in myeloid dysregulation, is an important target for inflammatory diseases that have cellular components, including rheumatoid arthritis and possibly also multiple sclerosis. Thus, anti-GM-CSF antibodies have been studied for the past several years for their potential use in a variety of inflammatory diseases. While no anti-GM-CSF antibodies have yet been approved by major regulatory agencies for any indication, there are multiple candidates in mid-to-late-stage clinical trials, including lenzilumab, otilimab, mavrilimumab, and plonmarlimab, all of which have advanced to at least in Phase II clinical trials. These anti-GM-CSF antibodies offer a potentially unique advantage for use in COVID-19 treatment due to the likely central role played by activated macrophages in disease progression [534]. Currently, no anti-GM-CSF antibodies have been approved or granted EUAs for treatment of COVID-19, although Humanigen filed an EUA application with the FDA in June 2021 for treatment of COVID-19 with lenzilumab. A Phase III trial supporting the EUA filing indicated that treatment of severely diseased COVID-19 patients improved survival without ventilation in hospitalized, hypoxic patients from approximately 50% to 200%, depending on the specific subpopulation of patients included in the analysis [522].
Dysregulation of the complement pathway has been shown to play a role in sepsis and may play a role in increasing the severity of COVID-19 [301–304, 320]. Production of C5a anaphylatoxin from C5 can have multiple detrimental effects, including overproduction of cytokines, activation of macrophages, induction of tissue factor expression that can result in disseminated intravascular coagulopathy (DIC), and development of ARDS [510]. Similarly, inhibition of C5a receptor-1 in a mouse influenza model was shown to relieve symptoms associated with ARDS [535]. Additionally, evidence suggests that the alternative complement pathway plays a role in COVID-19 disease severity, mediated through the inhibition of Factor H function by SARS-CoV-2 viral proteins [304]. While it is still early, four complement pathway inhibition antibodies, three of which target the C5 step to eliminate production of anaphylatoxin C5a, are being tested clinically (see Table 9) for their potential to reduce the effects of complement-mediated exacerbation of COVID-19 disease.
Other antibodies, such as the anti-IL-1βmAb, canakinumab (Ilaris®), have been tested for the treatment of CRS associated with COVID-19, but have not demonstrated significant clinical efficacy (patient survival) over SOC. Based on clinical data, the US-CDC recommends against the use of canakinumab for treatment of COVID-19 [519]. Nevertheless, similar to tocilizumab and antibodies targeting SARS-CoV-2, early use of canakinumab was shown to provide superior efficacy over standard of care [536]. Similarly, clinical studies on antibodies targeting IL-33R, IL-22, P-selectin, IFN-γ, CTGF, and CD73 to improve COVID-19 outcomes all have been discontinued due to lack of clinical efficacy (Table 10).
It is clear from the broad and deeps efforts made by several companies and research institutions that there is significant difficulty in applying existing drugs to improve COVID-19 outcomes. The best hopes still remaining for tamping down the out-of-control immune system, without undermining the ability of the immune system to help clear the virus, are the use of anti-IL-6R inhibitors, GM-CSF inhibitors, and potentially complement pathway inhibitors.
Access and Costs of COVID-19 Antibodies
Access to the antibodies for treatment of COVID-19 that have been given EUAs is quite varied. Several governments have bought up stockpiles of antibodies for distribution to their citizens, while in other cases, insurance and/or government programs (e.g., Medicare in USA [537]) either provide or help with costs associated with the drugs. The publicly reported price-points are: REGEN-COV™, US$2,100 per dose; bamlanivimab/etesevimab, US$2,100 per dose; sotrovimab, US$2,202 per dose; Regkirona™, US$3,650 per dose. It is noteworthy that of these available anti-SARS-CoV-2 antibody therapeutics, REGEN-COV™ has led the way over the other available antibodies up to December 2021, when Omicron became the dominant variant; they reported sales of U$2.59B of REGEN-COV™ in 1Q21, largely due to government stockpiling, but nevertheless approximately 2.5-fold more than the US$959M Eli Lilly brought in for both 1Q and 2Q201 for their antibodies to SARS-CoV-2. Tocilizumab (Actemra®), which has been approved since 2009 (EU)-2010 (USA) for treatment of rheumatoid arthritis, has been reported to cost US$5,304 for the single 800 mg dose used to treat COVID-19 [532]. The most recent US government purchase of antibody was 600,000 doses of bebtelovimab to fight Omicron BA.1 and BA.2 at US$1,200/dose [538].
Note in Table 3 the doses for Regkirona™ (40 mg/kg) and the amubarvimab plus romlusevimab (BRII-196/BRII-198) antibody cocktail (40 mg/kg plus 80 mg/kg). For your typical 70 kg patient, these doses would amount to totals of 2.8 g for Regkirona™ and 2.8 g-plus-5.6 g (total, 8.4 g) for the Brii cocktail. At a nominal cost for active pharmaceutical ingredient (API) of approximately US$175/g (average costs for midsized biotech company using early-stage fed-batch manufacturing process; [539]), the cost of goods (COGs) on a per gram basis for API for a single dose would likely exceed US$490 and US$1,470, respectively, for these antibodies. Certainly, if the API costs are lowered due to efficiencies in manufacturing, these costs can be reduced significantly. Nevertheless, as compared with the price per dose above for Regkirona™, a theoretical COG of $490 for API alone takes up about 13% of the price. Thus, there will be significant pressure to find lower, but still efficacious doses. An example of this is REGEN-COV™. The first doses applied, including the dose given to President Donald Trump, was 8.0 g, which using the math above would have incurred at COG of at least $1,400 per dose. Regeneron eventually found that 1,200 mg casirivimab/1,200 mg imdevimab was efficacious, and this dose was approved in the original EUA. At the US$175/g API mark used as a model here, the COG for the original REGEN-COV™ dose would have been US$420, essentially 20% of the price per dose. Regeneron continued to investigate the efficacy of lower doses successfully, and in June 2021, the FDA amended the EUA for yet a lower dose of 600 mg casirivimab and 600 mg imdevimab for REGEN-COV™, cutting in half the API COGs/dose, again showing the importance on the economics of the drug to reduce the dose as much as possible. Note that none of these COG projections include the cost of research and development, packaging, distribution, or storage, so actual total costs to the manufacturers is actually significantly higher.
Summary
It has now been a little over 2 years since the beginning of the world-wide COVID-19 pandemic. Starting from the earliest days of the COVID-19 pandemic, enormous effort by legions of researchers, biopharmaceutical companies, and research institutes has been put into developing antibodies to either block SARS-CoV-2 infection directly or to modulate the dysfunctional immune response mounted against the virus, especially in severe cases.
Antibodies for therapeutic, prophylactic and palliative purposes have played a large role in saving lives, reducing hospitalization and lowering the risk of mechanical ventilation [540], as well as possibly limiting the number of “long COVID-19” cases, i.e., those suffering long-term physical and psychological effects of the disease [541]. A recent analysis of all potential therapeutics for treatment of COVID-19 highlighted the few drugs that have shown enough efficacy to warrant continued use in COVID-19 treatment [542]. These included the antibodies to SARS-CoV-2 for which EUAs have been approved and convalescent plasma, if administered in the early stages of the disease, and glucocorticoids administered with tocilizumab in later stages of the disease [541]. While it is clear that antibody therapy is no substitute for vaccination [543], these therapies have undoubtably saved thousands of lives.
This pandemic and the incredible response to it has provided us with a basket full of lessons learned. First and foremost, antibodies can make a huge difference in the course of the pandemic and save lives that might be lost otherwise. The second and perhaps most significant lesson is that the earlier the diagnosis and treatment, the much greater the possibility to intervene successfully. While this general principle has played out for all modes of therapeutic treatment, it is especially important for antibody treatment, whether it be via convalescent plasma or manufactured recombinant antibodies. Finally, this pandemic has taught us that the first successful drugs may not be the drugs that ultimately save lives because of the speed of antigenic drift. Multiple times in this pandemic we have witnessed the emergence of new variants that took over and dominated within 2 months of first detection, an incredible view into the power of the virus to adapt and change.
Additional important learnings that are still playing out but could be key approaches for future pandemics and anti-viral therapeutics are that multi-valency and biparatopic/bispecific approaches can significantly improve potency of the antibody constructs. Added to that was the strong data supporting the use of antibodies with full or even improved Fc functionality, both for FcγR interactions (e.g., ADCC, ADCP, ability to cross-link) and FcRn interactions (i.e., longer circulating half-life). Thus, the full activity of natural antibodies plays out as critical for recombinant therapeutic antibodies as well. Finally, it is clear that direct competition with the receptor, i.e., competition-based neutralization is not the only MOA of importance. Several of the antibodies described herein do not block SARS-CoV-2 from binding to human ACE2 but are highly potent neutralizers in any case utilizing very different MOAs than just blocking. These “novel” MOAs include intra- and inter-spike crosslinking, destabilization of the spike complex, locking RBDs in conformations that do not allow ACE2 binding, and so forth. This is critical for future anti-COV antibody design because many of the most highly conserved sequences, which might be employed to derived pan-COV antibody therapeutics, are away from the RBM, or ACE2 binding site. These could be important future approaches to account for both novel COVs different from SARS-CoV-2 as well as for continued antigenic drift of SARS-CoV-2 giving rise to future variants.
The COVID-19 pandemic has galvanized many aspects of antibody discovery, leading the way to faster and more efficient antibody discovery and development processes. Human B cells have been used for several years to derive antibodies against infectious disease agents [176], and microfluidics approaches coupled with advanced RNA recovery technology [203, 210, 256, 544–546] have been increasingly used in antibody discovery over the past several years. Nevertheless, the pandemic brought about a forced evolution in antibody discovery, demonstrating how the use of captured antibody genes from B cells, automation, parallel and overlapping processes, and focus could cut years off the process of going from antigen to the clinic [203, 214]. Additionally, with so many efforts focused on essentially a single antigen, i.e., SARS-CoV-2 spike protein, this has resulted in a greater appreciation for the nuances of specific epitopes, antibody binding angles, mixed mechanisms of action, and avidity effects, and their combined roles in producing highly potent antibodies. Hopefully, there will not be another pandemic to match COVID-19, but if there is, the antibody industry will be more prepared to take a leading role in treating whatever comes next.
We have endeavored to take most of our information directly from primary literature wherever possible, so very few SARS-CoV-2 antibody reviews are cited thus far. There are, however, dozens of excellent reviews that have taken different approaches than taken here, as well as some highly informative websites, a few of which are cited here for reference [547–554].
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
We thank Betsy Haines and Jessica Jarecki for reading through and editing the manuscript. Both have provided written approval to be named in this manuscript.
Declarations
Funding Support
This work was supported in part by a Welch Foundation grant AU-0042-20030616 and Cancer Prevention and Research Institute of Texas (CPRIT) Grants RP150551 and RP190561 (ZA).
Potential Conflicts of Interest
The University of Texas System has filed a patent on the SARS-CoV-2 antibodies and the reverse genetic system and reporter SARS-CoV-2. The University of Texas System and IGM Biosciences Inc. have filed a joint patent on the SARS-CoV-2 IgM antibodies. Z.A. and Z.K. are employed by The University of Texas System. The antibody, IGM-6268 is under development by IGM Biosciences Inc. for prophylactic and therapeutic treatment of COVID-19. B.A.K. and S.F.C. are employees of IGM Biosciences, Inc. W.R.S. is a member of the Board of Directors, IGM Biosciences, Inc., and is a member of the Scientific Advisory Board for Immunome, two companies mentioned in this paper. Other authors declare no competing interests.
Ethics approval
Not applicable.
Consent to participate/publish
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable; all codes mentioned herein are public.
Author contributions
All authors contributed to concepts used to build the manuscript, data analysis, writing, and editing the manuscript. LMS and WRS carried out structural analyses from public databases and made all of the figures.
Contributor Information
William R. Strohl, Email: wrstrohl@gmail.com
Zhiqiang Ku, Email: Zhiqiang.Ku@uth.tmc.edu.
Zhiqiang An, Email: Zhiqiang.An@uth.tmc.edu.
Stephen F. Carroll, Email: steve@igmbio.com
Bruce A. Keyt, Email: bruce@IGMbio.com
Lila M. Strohl, Email: strohl2b@gmail.com
References
- 1.Von Behring E, Kitasato S. Ueber das Zustandekommen der Diphtherie-Immunitat and der Tetanus-Immunitat bei Thieren. Dtsch Med Wochenschr. 1890;16:1113–1114. doi: 10.1055/s-0029-1207589. [DOI] [Google Scholar]
- 2.Casadevall A, Scharff MD. Return to the past: the case for antibody-based therapies in infectious diseases. Clin Infect Dis. 1995;21(1):150–161. doi: 10.1093/clinids/21.1.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berry JD, Gaudet RG. Antibodies in infectious diseases: polyclonals, monoclonals and niche biotechnology. N Biotechnol. 2011;28(5):489–501. doi: 10.1016/j.nbt.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Salazar G, Zhang N, Fu TM, An Z. Antibody therapies for the prevention and treatment of viral infections. NPJ Vaccines. 2017;2:19. doi: 10.1038/s41541-017-0019-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumar D, Gauthami S, Bayry J, Kaveri SV, Hegde NR. Antibody therapy: From diphtheria to cancer, COVID-19, and beyond. Monoclon Antib Immunodiagn Immunother. 2021;40(2):36–49. doi: 10.1089/mab.2021.0004. [DOI] [PubMed] [Google Scholar]
- 6.Krilov LR. Palivizumab in the prevention of respiratory syncytial virus disease. Expert Opin Biol Ther. 2002;2(7):763–769. doi: 10.1517/14712598.2.7.763. [DOI] [PubMed] [Google Scholar]
- 7.ClinicalTrials.gov. https://clinicaltrials.gov/. Accessed 18 Feb2022.
- 8.Bergeron HC, Tripp RA. Breakthrough therapy designation of nirsevimab for the prevention of lower respiratory tract illness caused by respiratory syncytial virus infections (RSV) Expert Opin Investig Drugs. 2021 doi: 10.1080/13543784.2022.2020248. [DOI] [PubMed] [Google Scholar]
- 9.Mahomed S, Garrett N, Baxter C, Abdool Karim Q, Abdool Karim SS. Clinical trials of broadly neutralizing monoclonal antibodies for human immunodeficiency virus prevention: A review. J Infect Dis. 2021;223(3):370–380. doi: 10.1093/infdis/jiaa377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sedeyn K, Saelens X. New antibody-based prevention and treatment options for influenza. Antivir Res. 2019;170:104562. doi: 10.1016/j.antiviral.2019.104562. [DOI] [PubMed] [Google Scholar]
- 11.Xu J, Jia W, Wang P, Zhang S, Shi X, Wang X, et al. Antibodies and vaccines against Middle East respiratory syndrome coronavirus. Emerg Microbes Infect. 2019;8(1):841–856. doi: 10.1080/22221751.2019.1624482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saxena D, Kaul G, Dasgupta A, Chopra S. Atoltivimab/maftivimab/odesivimab (Inmazeb) combination to treat infection caused by Zaire ebolavirus. Drugs Today (Barc) 2021;57(8):483–490. doi: 10.1358/dot.2021.57.8.3280599. [DOI] [PubMed] [Google Scholar]
- 13.Gorshkov K, Shiryaev SA, Fertel S, Lin YW, Huang CT, Pinto A, Farhy C, Strongin AY, Zheng W, Terskikh AV. Zika virus: origins, pathological action, and treatment strategies. Front Microbiol. 2019;9:3252. doi: 10.3389/fmicb.2018.03252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.August A, Attarwala HZ, Himansu S, Kalidindi S, Lu S, Pajon R, et al. A phase 1 trial of lipid-encapsulated mRNA encoding a monoclonal antibody with neutralizing activity against Chikungunya virus. Nat Med. 2021;27(12):2224–2233. doi: 10.1038/s41591-021-01573-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Corti D, Benigni F, Shouval D. Viral envelope-specific antibodies in chronic hepatitis B virus infection. Curr Opin Virol. 2018;30:48–57. doi: 10.1016/j.coviro.2018.04.002. [DOI] [PubMed] [Google Scholar]
- 16.McClain JB, Chuang A, Reid C, Moore SM, Tsao E. Rabies virus neutralizing activity, pharmacokinetics, and safety of the monoclonal antibody mixture SYN023 in combination with rabies vaccination: results of a phase 2, randomized, blinded, controlled trial. Vaccine. 2021;39(40):5822–5830. doi: 10.1016/j.vaccine.2021.08.066. [DOI] [PubMed] [Google Scholar]
- 17.Borucki MJ, Spritzler J, Asmuth DM, Gnann J, Hirsch MS, Nokta M, et al. A phase II, double-masked, randomized, placebo-controlled evaluation of a human monoclonal anti-Cytomegalovirus antibody (MSL-109) in combination with standard therapy versus standard therapy alone in the treatment of AIDS patients with Cytomegalovirus retinitis. Antivir Res. 2004;64(2):103–111. doi: 10.1016/j.antiviral.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 18.Mujib S, Liu J, Rahman AKMN, Schwartz JA, Bonner P, Yue FY, Ostrowski MA. Comprehensive cross-clade characterization of antibody-mediated recognition, complement-mediated lysis, and cell-mediated cytotoxicity of HIV-1 envelope-specific antibodies toward eradication of the HIV-1 reservoir. J Virol. 2017;91(16):e00634–e717. doi: 10.1128/JVI.00634-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Erp EA, Luytjes W, Ferwerda G, van Kasteren PB. Fc-mediated antibody effector functions during respiratory syncytial virus infection and disease. Front Immunol. 2019;10:548. doi: 10.3389/fimmu.2019.00548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lewis GK, Ackerman ME, Scarlatti G, Moog C, Robert-Guroff M, Kent SJ, et al. Knowns and unknowns of assaying antibody-dependent cell-mediated cytotoxicity against HIV-1. Front Immunol. 2019;10:1025. doi: 10.3389/fimmu.2019.01025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao R, Sheng Z, Sreenivasan CC, Wang D, Li F. Influenza A virus antibodies with antibody-dependent cellular cytotoxicity function. Viruses. 2020;12(3):276. doi: 10.3390/v12030276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nelson CS, Huffman T, Jenks JA, Cisneros de la Rosa E, Xie G, Vandergrift N, et al. HCMV glycoprotein B subunit vaccine efficacy mediated by nonneutralizing antibody effector functions. Proc Natl Acad Sci USA. 2018;115:6267–6272. doi: 10.1073/pnas.1800177115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richardson SI, Ayres F, Manamela NP, Oosthuysen B, Makhado Z, Lambson BE, et al. HIV broadly neutralizing antibodies expressed as IgG3 preserve neutralization potency and show improved Fc effector function. Front Immunol. 2021;12:733958. doi: 10.3389/fimmu.2021.733958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kugelman JR, Kugelman-Tonos J, Ladner JT, Pettit J, Keeton CM, Nagle ER, et al. Emergence of Ebola virus escape variants in infected nonhuman primates treated with the MB-003 antibody cocktail. Cell Rep. 2015;12(12):2111–2120. doi: 10.1016/j.celrep.2015.08.038. [DOI] [PubMed] [Google Scholar]
- 25.Both L, Banyard AC, van Dolleweerd C, Wright E, Ma JK, Fooks AR. Monoclonal antibodies for prophylactic and therapeutic use against viral infections. Vaccine. 2013;31(12):1553–1559. doi: 10.1016/j.vaccine.2013.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Antibody treatment guidelines for COVID-19, US National Institutes of Health. https://www.covid19treatmentguidelines.nih.gov/therapies/anti-sars-cov-2-antibody-products/anti-sars-cov-2-monoclonal-antibodies/ Accessed 27 Jan 2022.
- 27.COVID-19 worldwide morbidity and mortality count. https://www.worldometers.info/coronavirus/. Accessed 27 Feb 2022.
- 28.Tumpey TM, Basler CF, Aguilar PV, Zeng H, Solórzano A, Swayne DE, et al. Characterization of the reconstructed 1918 Spanish influenza pandemic virus. Science. 2005;310(5745):77–80. doi: 10.1126/science.1119392. [DOI] [PubMed] [Google Scholar]
- 29.Short KR, Kedzierska K, van de Sandt CE. Back to the future: lessons learned from the 1918 influenza pandemic. Front Cell Infect Microbiol. 2018;8:343. doi: 10.3389/fcimb.2018.00343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patel S, Saxena B, Mehta P. Recent updates in the clinical trials of therapeutic monoclonal antibodies targeting cytokine storm for the management of COVID-19. Heliyon. 2021;7(2):e06158. doi: 10.1016/j.heliyon.2021.e06158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.World Health Organization. Tracking SARS-CoV-2 variants. https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/. Accessed 17 Feb 2022. [PubMed]
- 32.CDC COVID-19. https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-info.html. Accessed 17 Feb 2022.
- 33.CDC report on omicron. https://www.cdc.gov/coronavirus/2019-ncov/science/science-briefs/scientific-brief-omicron-variant.html. Accessed 17 Feb 2022.
- 34.Liu Y, Liu J, Johnson BA, Xia H, Ku Z, Schindewolf C, et al. Delta spike P681R mutation enhances SARS-CoV-2 fitness over Alpha variant. bioRxiv [preprint]. 2021 Sep 5:2021.08.12.456173. doi: 10.1101/2021.08.12.456173. [DOI] [PMC free article] [PubMed]
- 35.Kraemer MUG, Hill V, Ruis C, Dellicour S, Bajaj S, McCrone JT, et al. Spatiotemporal invasion dynamics of SARS-CoV-2 lineage B.1.1.7 emergence. Science. 2021;373(6557):889–895. doi: 10.1126/science.abj0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Galloway SE, Paul P, MacCannell DR, Johansson MA, Brooks JT, MacNeil A, et al. Emergence of SARS-CoV-2 B.1.1.7 Lineage—United States, December 29 2020-January 12 2021. MMWR Morb Mortal Wkly Rep. 2021;70(3):95–99. doi: 10.15585/mmwr.mm7003e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ostrov DA. Structural consequences of variation in SARS-CoV-2 B.1.1.7. J Cell Immunol. 2021;3(2):103–108. doi: 10.33696/immunology.3.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grint DJ, Wing K, Williamson E, McDonald HI, Bhaskaran K, Evans D, et al. Case fatality risk of the SARS-CoV-2 variant of concern B.1.1.7 in England, 16 November to 5 February. Euro Surveill. 2021;26(11):2100256. doi: 10.2807/1560-7917.ES.2021.26.11.2100256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meng B, Kemp SA, Papa G, Datir R, Ferreira IATM, Marelli S, et al. Recurrent emergence of SARS-CoV-2 spike deletion H69/V70 and its role in the Alpha variant B.1.1.7. Cell Rep. 2021;35(13):109292. doi: 10.1016/j.celrep.2021.109292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barton MI, MacGowan SA, Kutuzov MA, Dushek O, Barton GJ, van der Merwe PA. Effects of common mutations in the SARS-CoV-2 Spike RBD and its ligand the human ACE2 receptor on binding affinity and kinetics. Elife. 2021;10:e70658. doi: 10.7554/eLife.70658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cai Y, Zhang J, Xiao T, Lavine CL, Rawson S, Peng H, et al. Structural basis for enhanced infectivity and immune evasion of SARS-CoV-2 variants. Science. 2021;373(6555):642–648. doi: 10.1126/science.abi9745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wibmer CK, Ayres F, Hermanus T, Madzivhandila M, Kgagudi P, Oosthuysen B, et al. SARS-CoV-2 501Y.V2 escapes neutralization by South African COVID-19 donor plasma. Nat Med. 2021;27(4):622–625. doi: 10.1101/2021.01.18.427166. [DOI] [PubMed] [Google Scholar]
- 43.Liu H, Zhang J, Cai J, Deng X, Peng C, Chen X, et al. Herd immunity induced by COVID-19 vaccination programs and suppression of epidemics caused by the SARS-CoV-2 Delta variant in China. medRxiv [Preprint]. 2021:2021.07.23.21261013. doi: 10.1101/2021.07.23.21261013.
- 44.Wang P, Casner RG, Nair MS, Wang M, Yu J, Cerutti G, et al. Increased resistance of SARS-CoV-2 variant P.1 to antibody neutralization. Cell Host Microbe. 2021;29(5):747–7514. doi: 10.1101/2021.03.01.433466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cherian S, Potdar V, Jadhav S, Yadav P, Gupta N, Das M, et al. SARS-CoV-2 spike mutations, L452R, T478K, E484Q and P681R, in the second wave of COVID-19 in Maharashtra, India. Microorganisms. 2021;9(7):1542. doi: 10.3390/microorganisms9071542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tchesnokova V, Kulasekara H, Larson L, Bowers V, Rechkina E, Kisiela D, et al. Acquisition of the L452R mutation in the ACE2-binding interface of Spike protein triggers recent massive expansion of SARS-CoV-2 variants. J Clin Microbiol. 2021 doi: 10.1128/JCM.00921-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Augusto G, Mohsen MO, Zinkhan S, Liu X, Vogel M, Bachmann MF. In vitro data suggest that Indian delta variant B.1.617 of SARS-CoV-2 escapes neutralization by both receptor affinity and immune evasion. Allergy. 2022;77(1):111–117. doi: 10.1111/all.15065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Neerukonda SN, Vassell R, Lusvarghi S, Wang R, Echegaray F, Bentley L, et al. SARS-COV-2 Delta variant displays moderate resistance to neutralizing antibodies and spike protein properties of higher soluble ACE2 sensitivity, enhanced cleavage and fusogenic activity. Viruses. 2021;13(12):2485. doi: 10.3390/v13122485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peacock TP, Sheppard CM, Brown JC, Goonawardane N, Zhou J, Whiteley M, et al. The SARS-CoV-2 variants associated with infections in India, B.1.617, show enhanced spike cleavage by furin. bioRxiv [preprint] 2021.05.28.446163; 10.1101/2021.05.28.446163
- 50.CDC report on SARS-CoV-2 and Delta Variant July 29 2021. https://context-cdn.washingtonpost.com/notes/prod/default/documents/8a726408-07bd-46bd-a945-3af0ae2f3c37/note/57c98604-3b54-44f0-8b44-b148d8f75165.#page=1. Accessed 24 Oct 2021.
- 51.Li B, Deng A, Li K, Hu Y, Li Z, Xiong Q, et al. Viral infection and transmission in a large, well-traced outbreak caused by the SARS-CoV-2 Delta variant. Viral infection and transmission in a large, well-traced outbreak caused by the SARS-CoV-2 Delta variant. Nat Commun. 2022;13(1):460. doi: 10.1038/s41467-022-28089-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xin H, Wong JY, Murphy C, Yeung A, Ali ST, Wu P, Cowling BJ, The incubation period distribution of coronavirus disease, (COVID-19): a systematic review and meta-analysis. Clin Infect Dis. 2019 doi: 10.1093/cid/ciab501. [DOI] [PubMed] [Google Scholar]
- 53.Kang M, Xin H, Yuan J, Ali ST, Liang Z, Zhang J, et al. Transmission dynamics and epidemiological characteristics of Delta variant infections in China. medRxiv [preprint] 2021.08.12.21261991; 10.1101/2021.08.12.21261991.
- 54.Brown CM, Vostok J, Johnson H, Burns M, Gharpure R, Sami S, et al. Outbreak of SARS-CoV-2 infections, including COVID-19 vaccine breakthrough infections, associated with large public gatherings - Barnstable County, Massachusetts, July 2021. MMWR Morb Mortal Wkly Rep. 2021;70(31):1059–1062. doi: 10.15585/mmwr.mm7031e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Riemersma KK, Grogan BE, Kita-Yarbro A, Halfmann P, Kocharian A, Florek KR, et al. Shedding of infectious SARS-CoV-2 despite vaccination when the delta variant is prevalent - Wisconsin, July 2021. medRxiv [preprint] 2021.07.31.21261387; 10.1101/2021.07.31.21261387.
- 56.Yadav PD, Mohandas S, Shete AM, Nyayanit DA, Gupta N, Patil DY, et al. SARS CoV-2 variant B.1.617.1 is highly pathogenic in hamsters than B.1 variant. bioRxiv [preprint] 2021.05.05.442760; 10.1101/2021.05.05.442760.
- 57.Public Health England. https://www.gov.uk/government/news/vaccines-highly-effective-against-b-1-617-2-variant-after-2-doses. Accessed 3 Oct 2021
- 58.Mlcochova P, Kemp SA, Dhar MS, Papa G, Meng B, Ferreira IATM, et al. SARS-CoV-2 B.1.617.2 Delta variant replication and immune evasion. Nature. 2021;599(7883):114–119. doi: 10.1038/s41586-021-03944-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brown E. AY.4.2 detected in 32 states as U.K. declares variant under investigation. https://www.newsweek.com/ay-4-2-delta-variant-cases-detected-us-states-uk-declares-investigation-ay42-1642091. Accessed 24 Oct 2021.
- 60.Aljindan RY, Al-Subaie AM, Al-Ohali AI, Kumar DT, Doss CGP, Kamaraj B. Investigation of nonsynonymous mutations in the spike protein of SARS-CoV-2 and its interaction with the ACE2 receptor by molecular docking and MM/GBSA approach. Comput Biol Med. 2021;135:104654. doi: 10.1016/j.compbiomed.2021.104654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Viana R, Moyo S, Amoako DG, Tegally H, Scheepers C, Althaus CL, et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Afriapproximately. Nature. 2022 doi: 10.1038/s41586-022-04411-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kupferschmidt K. Where did ‘weird’ Omicron come from? https://www.science.org/content/article/where-did-weird-omicron-come. doi: 10.1126/science.acx9754. Accessed 7 Dec 2021. [DOI] [PubMed]
- 63.Kannan SR, Spratt AN, Sharma K, Chand HS, Byrareddy SN, Singh K. Omicron SARS-CoV-2 variant: Unique features and their impact on pre-existing antibodies. J Autoimmun. 2022;126:102779. doi: 10.1016/j.jaut.2021.102779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Planas D, Saunders N, Maes P, Guivel-Benhassine F, Planchais C, Buchrieser J, et al. Considerable escape of SARS-CoV-2 Omicron to antibody neutralization. Nature. 2022;602(7898):671–675. doi: 10.1038/s41586-021-04389-z. [DOI] [PubMed] [Google Scholar]
- 65.Enhancing Readiness for Omicron (B.1.1.529): Technical Brief and Priority Actions for Member States. https://www.who.int/docs/default-source/coronaviruse/20211217-global-technical-brief-and-priority-action-on-omicron_latest-2.pdf. Accessed 14 Feb 2022.
- 66.Kumar S, Karuppanan K, Subramaniam G. Omicron (BA.1) and sub-Variants (BA.1, BA.2 and BA.3) of SARS-CoV-2 spike infectivity and pathogenicity: A comparative sequence and structural-based computational assessment. bioRxiv [preprint] 2022.02.11.480029; 10.1101/2022.02.11.480029. [DOI] [PMC free article] [PubMed]
- 67.Haseltine W. Whither the Omicron Family: BA.1, BA.1.1, BA.2, BA.2.H78Y, BA.3? February 23 2022. https://www.forbes.com/sites/williamhaseltine/2022/02/23/whither-the-omicron-family-ba1-ba11-ba2-ba2h78y-ba3/?sh=3af9b7dd862e. Accessed 27 Feb 2022.
- 68.Mykytyn AZ, Rissmann M, Kok A, Rosu ME, Schipper D, Breugem TI, et al. Omicron BA.1 and BA.2 are antigenically distinct SARS-CoV-2 variants. bioRxiv [preprint] 2022.02.23.481644; doi: 10.1101/2022.02.23.481644. [DOI] [PMC free article] [PubMed]
- 69.Desingu PA, Nagarajan K, Dhama K. Emergence of Omicron third lineage BA.3 and its importance. J Med Virol. 2022 doi: 10.1002/jmv.27601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ito K, Piantham C, Nishiura H. Relative instantaneous reproduction number of Omicron SARS-CoV-2 variant with respect to the Delta variant in Denmark. J Med Virol. 2021 doi: 10.1002/jmv.27560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nishiura H, Ito K, Anzai A, Kobayashi T, Piantham C, Rodríguez-Morales AJ. Relative reproduction number of SARS-CoV-2 Omicron (B.1.1.529) compared with Delta variant in South Afriapproximately. J Clin Med. 2021;11(1):30. doi: 10.3390/jcm11010030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Omicron information. https://www.nebraskamed.com/COVID/how-quickly-do-diseases-spread. Accessed 16 Feb 2022.
- 73.Syal A. Do vaccines work against omicron? Lab studies are coming, but won't tell whole story. https://www.nbcnews.com/health/health-news/omicron-lab-studies-learn-wont-tell-us-rcna7536. Accessed 6 Dec 2021.
- 74.Chen J, Wang R, Gilby NB, Wei GW. Omicron Variant (B.1.1.529): Infectivity, vaccine breakthrough, and antibody resistance. J Chem Inf Model. 2022;62(2):412–422. doi: 10.1021/acs.jcim.1c01451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Network for Genomic Surveillance in South Afriapproximately https://www.nicd.ac.za/wp-content/uploads/2021/12/Update-of-SA-sequencing-data-from-GISAID-3-Dec-21-Final.pdf. Accessed 15 Dec 2021.
- 76.United States Centers for Disease Control COVID data. https://covid.cdc.gov/covid-data-tracker/#variant-proportions. Accessed 24 Feb 2022.
- 77.Omicron mutations. https://covdb.stanford.edu/page/mutation-viewer/#omicron. Accessed 10 Feb 2022.
- 78.Zahradník J, Marciano S, Shemesh M, Zoler E, Harari D, Chiaravalli J, et al. SARS-CoV-2 variant prediction and antiviral drug design are enabled by RBD in vitro evolution. Nat Microbiol. 2021;6(9):1188–1198. doi: 10.1038/s41564-021-00954-4. [DOI] [PubMed] [Google Scholar]
- 79.Han P, Li L, Liu S, Wang Q, Zhang D, Xu Z, et al. Receptor binding and complex structures of human ACE2 to spike RBD from omicron and delta SARS-CoV-2. Cell. 2022;185(4):630–640.e10. doi: 10.1016/j.cell.2022.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dejnirattisai W, Huo J, Zhou D, Zahradník J, Supasa P, Liu C, et al. SARS-CoV-2 Omicron-B.1.1.529 leads to widespread escape from neutralizing antibody responses. Cell. 2022 doi: 10.1016/j.cell.2021.12.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wu L, Zhou L, Mo M, Liu T, Wu C, Gong C, et al. SARS-CoV-2 Omicron RBD shows weaker binding affinity than the currently dominant Delta variant to human ACE2. Signal Transduct Target Ther. 2022;7(1):8. doi: 10.1038/s41392-021-00863-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Peacock TP, Brown JC, Zhou J, Thakur N, Newman J, Kugathasan R et al. The SARS-CoV-2 variant, Omicron, shows rapid replication in human primary nasal epithelial cultures and efficiently uses the endosomal route of entry. bioRxiv preprint] 2021.12.31.474653; 10.1101/2021.12.31.474653.
- 83.Cerutti G, Guo Y, Liu L, Liu L, Zhang Z, Luo Y, et al. Cryo-EM structure of the SARS-CoV-2 Omicron spike. Cell Rep. 2022 doi: 10.1016/j.celrep.2022.110428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McCallum M, Czudnochowski N, Rosen LE, Zepeda SK, Bowen JE, Walls AC, et al. Structural basis of SARS-CoV-2 Omicron immune evasion and receptor engagement. Science. 2022 doi: 10.1126/science.abn8652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cui Z, Liu P, Wang N, Wang L, Fan K, Zhu Q, et al. Structural and functional characterizations of infectivity and immune evasion of SARS-CoV-2 Omicron. Cell. 2022;S0092–8674(22):00077. doi: 10.1016/j.cell.2022.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Meng B, Abdullahi A, Ferreira IATM, Goonawardane N, Saito A, Kimura I, et al. Altered TMPRSS2 usage by SARS-CoV-2 Omicron impacts tropism and fusogenicity. Nature. 2022 doi: 10.1038/s41586-022-04474-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lamers MM, Mykytyn AZ, Breugem TI, Groen N, Knoops K, Schipper D, et al. SARS-CoV-2 Omicron efficiently infects human airway, but not alveolar epithelium. bioRxiv 2022.01.19.476898; 10.1101/2022.01.19.476898
- 88.Shuai H, Chan JF, Hu B, Chai Y, Yuen TT, Yin F, et al. Attenuated replication and pathogenicity of SARS-CoV-2 B.1.1.529 Omicron. Nature. 2022 doi: 10.1038/s41586-022-04442-5. [DOI] [PubMed] [Google Scholar]
- 89.Zhao H, Lu L, Peng Z, Chen LL, Meng X, Zhang C, et al. SARS-CoV-2 Omicron variant shows less efficient replication and fusion activity when compared with Delta variant in TMPRSS2-expressed cells. Emerg Microbes Infect. 2022;11(1):277–283. doi: 10.1080/22221751.2021.2023329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hui KPY, Ho JCW, Cheung MC, Ng KC, Ching RHH, Lai KL, et al. SARS-CoV-2 Omicron variant replication in human bronchus and lung ex vivo. Nature. 2022 doi: 10.1038/s41586-022-04479-6. [DOI] [PubMed] [Google Scholar]
- 91.Fantini J, Yahi N, Colson P, Chahinian H, La Scola B, Raoult D. The puzzling mutational landscape of the SARS-2-variant Omicron. J Med Virol. 2022 doi: 10.1002/jmv.27577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zeng C, Evans JP, King T, Zheng YM, Oltz EM, Whelan SPJ, et al. SARS-CoV-2 spreads through cell-to-cell transmission. Proc Natl Acad Sci U S A. 2022;119(1):e2111400119. doi: 10.1073/pnas.2111400119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Carreño JM, Alshammary H, Tcheou J, Singh G, Raskin AJ, Kawabata H, et al. Activity of convalescent and vaccine serum against SARS-CoV-2 Omicron. Nature. 2022;602(7898):682–688. doi: 10.1038/s41586-022-04399-5. [DOI] [PubMed] [Google Scholar]
- 94.Banerjee A, Lew J, Kroeker A, Baid K, Aftanas P, Nirmalarajah K, et al. Immunogenicity of convalescent and vaccinated sera against clinical isolates of ancestral SARS-CoV-2, beta, delta, and omicron variants. bioRxiv [preprint] 2022.01.13.475409; 10.1101/2022.01.13.475409 [DOI] [PMC free article] [PubMed]
- 95.Cele S, Jackson L, Khoury DS, Khan K, Moyo-Gwete T, Tegally H, et al. Omicron extensively but incompletely escapes Pfizer BNT162b2 neutralization. Nature. 2022;602(7898):654–656. doi: 10.1038/s41586-021-04387-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hoffmann M, Krüger N, Schulz S, Cossmann A, Rocha C, Kempf A, et al. The Omicron variant is highly resistant against antibody-mediated neutralization: Implications for control of the COVID-19 pandemic. Cell. 2021;S0092–8674(21):01495–1501. doi: 10.1016/j.cell.2021.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Muik A, Lui BG, Wallisch AK, Bacher M, Mühl J, Reinholz J, et al. Neutralization of SARS-CoV-2 Omicron by BNT162b2 mRNA vaccine-elicited human sera. Science. 2022;375(6581):678–680. doi: 10.1126/science.abn7591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lusvarghi S, Pollett SD, Nath Neerukonda S, Wang W, Wang R, Vassell R, et al. SARS-CoV-2 Omicron neutralization by therapeutic antibodies, convalescent sera, and post-mRNA vaccine booster. bioRxiv 2021.12.22.473880; 10.1101/2021.12.22.473880
- 99.Wesemann DR. Omicron's message on vaccines: Boosting begets breadth. Cell. 2022;185(3):411–413. doi: 10.1016/j.cell.2022.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nakagawa M, Greenfield W, Moerman-Herzog A, Coleman HN. Cross-reactivity, epitope spreading, and de novo immune stimulation are possible mechanisms of cross-protection of nonvaccine human papillomavirus (HPV) types in recipients of HPV therapeutic vaccines. Clin Vaccine Immunol. 2015;22(7):679–687. doi: 10.1128/CVI.00149-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Keeton R, Richardson SI, Moyo-Gwete T, Hermanus T, Tincho MB, Benede N, et al. Prior infection with SARS-CoV-2 boosts and broadens Ad26.COV2.S immunogenicity in a variant-dependent manner. Cell Host Microbe. 2021;29(11):1611–1619.e5. doi: 10.1016/j.chom.2021.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yu J, Collier A-rY, Rowe M, Mardas F, Ventura JD, Wan H, et al. Comparable neutralization of the SARS-CoV-2 Omicron BA.1 and BA.2 variants. medRxiv [preprint] 2022.02.06.22270533; doi: 10.1101/2022.02.06.22270533 [DOI] [PMC free article] [PubMed]
- 103.Diamond M, Halfmann P, Maemura T, Iwatsuki-Horimoto K, Iida S, Kiso M, et al. The SARS-CoV-2 B.1.1.529 Omicron virus causes attenuated infection and disease in mice and hamsters. Res Sq [Preprint]. 2021 doi: 10.21203/rs.3.rs-1211792/v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Halfmann PJ, Iida S, Iwatsuki-Horimoto K, Maemura T, Kiso M, Scheaffer SM, et al. SARS-CoV-2 Omicron virus causes attenuated disease in mice and hamsters. Nature. 2022 doi: 10.1038/s41586-022-04441-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Iuliano AD, Brunkard JM, Boehmer TK, Peterson E, Adjei S, Binder AM, et al. Trends in disease severity and health care utilization during the early Omicron variant period compared with previous SARS-CoV-2 high transmission periods—United States, December 2020-January 2022. MMWR Morb Mortal Wkly Rep. 2022;71(4):146–152. doi: 10.15585/mmwr.mm7104e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Abdullah F, Myers J, Basu D, Tintinger G, Ueckermann V, Mathebula M, et al. Decreased severity of disease during the first global Omicron variant covid-19 outbreak in a large hospital in Tshwane, South Afriapproximately. Int J Infect Dis. 2021;116:38–42. doi: 10.1016/j.ijid.2021.12.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Suzuki R, Yamasoba D, Kimura I, Wang L, Kishimoto M, Ito J, et al. Attenuated fusogenicity and pathogenicity of SARS-CoV-2 Omicron variant. Nature. 2022 doi: 10.1038/s41586-022-04462-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bhattacharyya RP, Hanage WP. Challenges in inferring intrinsic severity of the SARS-CoV-2 Omicron variant. N Engl J Med. 2022;386(7):e14. doi: 10.1056/NEJMp2119682. [DOI] [PubMed] [Google Scholar]
- 109.Iketani S, Liu L, Guo Y, Liu L, Huang Y, Wang M, et al. Antibody evasion properties of SARS-CoV-2 Omicron sublineages. bioRxiv [preprint] 2022.02.07.479306; 10.1101/2022.02.07.479306 [DOI] [PMC free article] [PubMed]
- 110.Immune status against Omicron. https://www.usnews.com/news/health-news/articles/2022-02-17/estimated-73-of-us-now-immune-to-omicron-is-that-enough. Accessed 19 February 2022.
- 111.Desingu PA, Nagarajan K. Omicron BA.2 lineage spreads in clusters and is concentrated in Denmark. J Med Virol. 2022 doi: 10.1002/jmv.27659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Metzger CMJA, Lienhard R, Seth-Smith HMB, Roloff T, Wegner F, Sieber J, et al. PCR performance in the SARS-CoV-2 Omicron variant of concern? Swiss Med Wkly. 2021;151:w30120. doi: 10.4414/smw.2021.w30120. [DOI] [PubMed] [Google Scholar]
- 113.Yamasoba D, Kimura I, Nasser H, Morioka Y, Nao M, Ito J, et al. Virological characteristics of SARS-CoV-2 BA.2 variant. bioRxiv [preprint] 2022.02.14.480335; 10.1101/2022.02.14.480335
- 114.Lyngse FP, Kirkeby CT, Denwood M, Engbo Christiansen L, Mølbak K, Holten Møller C, et al. Transmission of SARS-CoV-2 Omicron VOC subvariants BA.1 and BA.2: Evidence from Danish Households medRxiv [preprint] 2022.01.28.22270044; 10.1101/2022.01.28.22270044
- 115.Surveillance report UK, Week 4. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1050721/Vaccine-surveillance-report-week-4.pdf. Accessed 5 Feb 2022.
- 116.Chen J, Wei GW. Omicron BA.2 (B.1.1.529.2): high potential to becoming the next dominating variant. ArXiv [preprint]. 2022 Feb 10: arXiv:2202.05031v1.
- 117.Tapp T. Omicron BA.2 spreading. February 16 2022. https://deadline.com/2022/02/omicron-ba-2-spreading-covid-cases-1234935129/. Accessed 25 Feb 2022.
- 118.Callaway E. Omicron sub-variant: What scientists know so far. Nature. 2022;602:556–557. doi: 10.1038/d41586-022-00471-2. [DOI] [PubMed] [Google Scholar]
- 119.Chen B, Xia R. Early experience with convalescent plasma as immunotherapy for COVID-19 in China: Knowns and unknowns. Vox Sang. 2020;115(6):507–514. doi: 10.1111/vox.12968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Pan X, Zhou P, Fan T, Wu Y, Zhang J, Shi X, et al. Immunoglobulin fragment F(ab')2 against RBD potently neutralizes SARS-CoV-2 in vitro. Antiviral Res. 2020;182:104868. doi: 10.1016/j.antiviral.2020.104868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zylberman V, Sanguineti S, Pontoriero AV, Higa SV, Cerutti ML, Morrone Seijo SM, et al. Development of a hyperimmune equine serum therapy for COVID-19 in Argentina. Medicina (B Aires). 2020;80(Suppl 3):1–6. [PubMed] [Google Scholar]
- 122.Lopardo G, Belloso WH, Nannini E, Colonna M, Sanguineti S, Zylberman V, et al. RBD-specific polyclonal F(ab´)2 fragments of equine antibodies in patients with moderate to severe COVID-19 disease: A randomized, multicenter, double-blind, placebo-controlled, adaptive phase 2/3 clinical trial. EClinicalMedicine. 2021;34:100843. doi: 10.1016/j.eclinm.2021.100843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.León G, Herrera M, Vargas M, Arguedas M, Sánchez A, Segura Á, et al. Development and characterization of two equine formulations towards SARS-CoV-2 proteins for the potential treatment of COVID-19. Sci Rep. 2021;11(1):9825. doi: 10.1038/s41598-021-89242-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gharebaghi N, Nejadrahim R, Mousavi SJ, Sadat-Ebrahimi SR, Hajizadeh R. The use of intravenous immunoglobulin gamma for the treatment of severe coronavirus disease 2019: a randomized placebo-controlled double-blind clinical trial. BMC Infect Dis. 2020;20(1):786. doi: 10.1186/s12879-020-05507-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Nguyen AA, Habiballah SB, Platt CD, Geha RS, Chou JS, McDonald DR. Immunoglobulins in the treatment of COVID-19 infection: Proceed with caution! Clin Immunol. 2020;216:108459. doi: 10.1016/j.clim.2020.108459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tharmalingam T, Han X, Wozniak A, Saward L. Polyclonal hyper immunoglobulin: a proven treatment and prophylaxis platform for passive immunization to address existing and emerging diseases. Hum Vaccin Immunother. 2021;19:1–20. doi: 10.1080/21645515.2021.1886560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ranganathan C, Fusinski SD, Obeid IM, Ismail KM, Ferguson DT, Raminick MF, et al. Therapeutic plasma exchange for persistent encephalopathy associated with Covid-19. eNeurologicalSci. 2021;22:100327. doi: 10.1016/j.ensci.2021.100327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Marson P, Cozza A, De Silvestro G. The true historical origin of convalescent plasma therapy. Transfus Apher Sci. 2020;59(5):102847. doi: 10.1016/j.transci.2020.102847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Luke TC, Kilbane EM, Jackson JL, Hoffman SL. Meta-analysis: convalescent blood products for Spanish influenza pneumonia: a future H5N1 treatment? Ann Intern Med. 2006;145(8):599–609. doi: 10.7326/0003-4819-145-8-200610170-00139. [DOI] [PubMed] [Google Scholar]
- 130.Yeh KM, Chiueh TS, Siu LK, Lin JC, Chan PK, Peng MY, et al. Experience of using convalescent plasma for severe acute respiratory syndrome among healthcare workers in a Taiwan hospital. J Antimicrob Chemother. 2005;56(5):919–922. doi: 10.1093/jac/dki346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cheng Y, Wong R, Soo YO, Wong WS, Lee CK, Ng MH, et al. Use of convalescent plasma therapy in SARS patients in Hong Kong. Eur J Clin Microbiol Infect Dis. 2005;24(1):44–46. doi: 10.1007/s10096-004-1271-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hung IF, To KK, Lee CK, Lee KL, Chan K, Yan WW, et al. Convalescent plasma treatment reduced mortality in patients with severe pandemic influenza A (H1N1) 2009 virus infection. Clin Infect Dis. 2011;52(4):447–456. doi: 10.1093/cid/ciq106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Florescu DF, Kalil AC, Hewlett AL, Schuh AJ, Stroher U, Uyeki TM, et al. Administration of brincidofovir and convalescent plasma in a patient with Ebola Virus disease. Clin Infect Dis. 2015;61(6):969–973. doi: 10.1093/cid/civ395. [DOI] [PubMed] [Google Scholar]
- 134.van Griensven J, Edwards T, Baize S, Ebola-Tx Consortium Efficacy of convalescent plasma in relation to dose of Ebola Virus antibodies. N Engl J Med. 2016;375(23):2307–2309. doi: 10.1056/NEJMc1609116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Arabi YM, Hajeer AH, Luke T, Raviprakash K, Balkhy H, Johani S, et al. Feasibility of using convalescent plasma immunotherapy for MERS-CoV infection Saudi Arabia. Emerg Infect Dis. 2016;22(9):1554–1561. doi: 10.3201/eid2209.151164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Shen C, Wang Z, Zhao F, Yang Y, Li J, Yuan J, et al. Treatment of 5 critically ill patients with COVID-19 with convalescent plasma. JAMA. 2020;323(16):1582–1589. doi: 10.1001/jama.2020.4783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Duan K, Liu B, Li C, Zhang H, Yu T, Qu J, et al. Effectiveness of convalescent plasma therapy in severe COVID-19 patients. Proc Natl Acad Sci U S A. 2020;117(17):9490–9496. doi: 10.1073/pnas.2004168117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Agarwal A, Mukherjee A, Kumar G, Chatterjee P, Bhatnagar T, Malhotra P. Convalescent plasma in the management of moderate covid-19 in adults in India: open label phase II multicentre randomised controlled trial (PLACID Trial) BMJ. 2020;371:m3939. doi: 10.1136/bmj.m3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Salazar MR, González SE, Regairaz L, Ferrando NS, González Martínez VV, Carrera Ramos PM, et al. Risk factors for COVID-19 mortality: The effect of convalescent plasma administration. PLoS ONE. 2021;16(4):e0250386. doi: 10.1371/journal.pone.0250386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Joyner MJ, Carter RE, Senefeld JW, Klassen SA, Mills JR, Johnson PW, et al. Convalescent plasma antibody levels and the risk of death from Covid-19. N Engl J Med. 2021;384(11):1015–1027. doi: 10.1056/NEJMoa2031893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bégin P, Callum J, Jamula E, Cook R, Heddle NM, Tinmouth A, et al. Convalescent plasma for hospitalized patients with COVID-19: an open-label, randomized controlled trial. Nat Med. 2021 doi: 10.1038/s41591-021-01488-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Korley FK, Durkalski-Mauldin V, Yeatts SD, Schulman K, Davenport RD, Dumont LJ, et al. Early convalescent plasma for high-risk outpatients with Covid-19. N Engl J Med. 2021 doi: 10.1056/NEJMoa2103784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Casadevall A, Dragotakes Q, Johnson PW, Senefeld JW, Klassen SA, Wright RS, et al. Convalescent plasma use in the USA was inversely correlated with COVID-19 mortality. Elife. 2021;10:e69866. doi: 10.7554/eLife.69866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Korley FK, Durkalski-Mauldin V, Yeatts SD, Schulman K, Davenport RD, Dumont LJ, et al. Early convalescent plasma for high-risk outpatients with Covid-19. N Engl J Med. 2021;385(21):1951–1960. doi: 10.1056/NEJMoa2103784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tobian A, Cohn CS, Shaz B. COVID-19 convalescent plasma. Blood. 2021 doi: 10.1182/blood.2021012248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Focosi D, Anderson AO, Tang JW, Tuccori M. Convalescent plasma therapy for COVID-19: State of the art. Clin Microbiol Rev. 2020;33(4):e00072–e120. doi: 10.1128/CMR.00072-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Chen L, Xiong J, Bao L, Shi Y. Convalescent plasma as a potential therapy for COVID-19. Lancet Infect Dis. 2020;20(4):398–400. doi: 10.1016/S1473-3099(20)30141-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Casadevall A, Pirofski LA. The convalescent sera option for containing COVID-19. J Clin Invest. 2020;130(4):1545–1548. doi: 10.1172/JCI138003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Katz LM. (A Little) clarity on convalescent plasma for Covid-19. N Engl J Med. 2021;384(7):666–668. doi: 10.1056/NEJMe2035678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.FDA Issues Emergency Use Authorization for Convalescent Plasma as Potential Promising COVID–19 Treatment, Another Achievement in Administration’s Fight Against Pandemic. Aug 23 2020. https://www.fda.gov/news-events/press-announcements/fda-issues-emergency-use-authorization-convalescent-plasma-potential-promising-covid-19-treatment. Accessed 25 Oct 2021
- 151.CoVIg-19 Plasma Alliance. https://www.takeda.com/newsroom/newsreleases/2021/covig-19-plasma-alliance-announces-topline-results-from-nih-sponsored-clinical-trial-of-investigational-covid-19-hyperimmune-globulin-medicine/. Accessed 15 Feb 2022.
- 152.Focosi D, Franchini M. Potential use of convalescent plasma for SARS-CoV-2 prophylaxis and treatment in immunocompromised and vulnerable populations. Expert Rev Vaccines. 2021;27:1–8. doi: 10.1080/14760584.2021.1932475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.FDA guidelines on plasma. https://www.fda.gov/media/141477/download. Accessed 30 Jan 2022.
- 154.Sullivan DJ, Gebo KA, Shoham S, Bloch EM, Lau B, Shenoy AG, et al. Randomized controlled trial of early outpatient COVID-19 treatment with high-titer convalescent plasma. medRxiv [Preprint]. 2021 Dec 21:2021.12.10.21267485.
- 155.Bar KJ, Shaw PA, Choi GH, Aqui N, Fesnak A, Yang JB, et al. A randomized controlled study of convalescent plasma for individuals hospitalized with COVID-19 pneumonia. J Clin Invest. 2021;131(24):e155114. doi: 10.1172/JCI155114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Avendaño-Solá C, Ramos-Martínez A, Muñez-Rubio E, Ruiz-Antorán B, Malo de Molina R, Torres F, et al. A multicenter randomized open-label clinical trial for convalescent plasma in patients hospitalized with COVID-19 pneumonia. J Clin Invest. 2021;131(20):e152740. doi: 10.1172/JCI152740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.O'Donnell MR, Grinsztejn B, Cummings MJ, Justman JE, Lamb MR, Eckhardt CM, et al. A randomized double-blind controlled trial of convalescent plasma in adults with severe COVID-19. J Clin Invest. 2021;131(13):e150646. doi: 10.1172/JCI150646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ortigoza MB, Yoon H, Goldfeld KS, Troxel AB, Daily JP, Wu Y, et al. Efficacy and safety of COVID-19 convalescent plasma in hospitalized patients: a randomized clinical trial. JAMA Intern Med. 2022;182(2):115–126. doi: 10.1001/jamainternmed.2021.6850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Gasser R, Cloutier M, Prévost J, Fink C, Ducas É, Ding S, et al. Major role of IgM in the neutralizing activity of convalescent plasma against SARS-CoV-2. Cell Rep. 2021;34(9):108790. doi: 10.1016/j.celrep.2021.108790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Verkerke H, Saeedi BJ, Boyer D, Allen JW, Owens J, Shin S, et al. Are we forgetting about IgA? A re-examination of coronavirus disease 2019 convalescent plasma. Transfusion. 2021;61(6):1740–1748. doi: 10.1111/trf.16435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Natarajan H, Crowley AR, Butler SE, Xu S, Weiner JA, Bloch EM, et al. Markers of polyfunctional SARS-CoV-2 antibodies in convalescent plasma. MBio. 2021;12(2):e00765-21. doi: 10.1128/mBio.00765-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Moubarak M, Kasozi KI, Hetta HF, Shaheen HM, Rauf A, Al-Kuraishy HM, et al. The rise of SARS-CoV-2 variants and the role of convalescent plasma therapy for management of infections. Life (Basel). 2021;11(8):734. doi: 10.3390/life11080734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hu J, He C-L, Gao Q-Z, Zhang G-J, Cao X-X, Long Q-X, et al. D614G mutation of SARS-CoV-2 spike protein enhances viral infectivity. bioRxiv [preprint] 2020.06.20.161323; 10.1101/2020.06.20.161323
- 164.Andreano E, Piccini G, Licastro D, Casalino L, Johnson NV, Paciello I, et al. SARS-CoV-2 escape from a highly neutralizing COVID-19 convalescent plasma. Proc Natl Acad Sci U S A. 2021;118(36):e2103154118. doi: 10.1073/pnas.2103154118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Cele S, Gazy I, Jackson L, Hwa SH, Tegally H, Lustig G, et al. Escape of SARS-CoV-2 501Y.V2 from neutralization by convalescent plasma. Nature. 2021;593(7857):142–146. doi: 10.1038/s41586-021-03471-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Jangra S, Ye C, Rathnasinghe R, Stadlbauer D, Krammer F, Simon V, et al. The E484K mutation in the SARS-CoV-2 spike protein reduces but does not abolish neutralizing activity of human convalescent and post-vaccination sera. medRxiv [preprint]. 2021 Jan 29:2021.01.26.21250543. 10.1101/2021.01.26.21250543.
- 167.Annavajhala MK, Mohri H, Wang P, Nair M, Zucker JE, Sheng Z, et al. Emergence and expansion of SARS-CoV-2 B.1.526 after identification in New York. Nature. 2021;597(7878):703–708. doi: 10.1038/s41586-021-03908-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ho D, Wang P, Liu L, Iketani S, Luo Y, Guo Y, et al. Increased resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7 to antibody neutralization. Res Sq [preprint]. 2021 doi: 10.21203/rs.3.rs-155394/v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Focosi D, Maggi F, Franchini M, McConnell S, Casadevall A. Analysis of immune escape variants from antibody-based therapeutics against COVID-19: a systematic review. Int J Mol Sci. 2021;23(1):29. doi: 10.3390/ijms23010029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Reincke SM, Yuan M, Kornau HC, Corman VM, van Hoof S, Sánchez-Sendin E, et al. SARS-CoV-2 Beta variant infection elicits potent lineage-specific and cross-reactive antibodies. Science. 2022;375(6582):782–787. doi: 10.1126/science.abm5835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.McCallum M, De Marco A, Lempp FA, Tortorici MA, Pinto D, Walls AC, et al. N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2. Cell. 2021;184(9):2332–2347.e16. doi: 10.1016/j.cell.2021.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Cerutti G, Guo Y, Zhou T, Gorman J, Lee M, Rapp M, et al. Potent SARS-CoV-2 neutralizing antibodies directed against spike N-terminal domain target a single supersite. Cell Host Microbe. 2021;29(5):819–833.e7. doi: 10.1016/j.chom.2021.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kemp SA, Collier DA, Datir RP, Ferreira IATM, Gayed S, Jahun A, et al. SARS-CoV-2 evolution during treatment of chronic infection. Nature. 2021;592(7853):277–282. doi: 10.1038/s41586-021-03291-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wang KY, Shah P, Pierce M. Convalescent plasma for COVID-19 complicated by ARDS due to TRALI. BMJ Case Rep. 2021;14(1):e239762. doi: 10.1136/bcr-2020-239762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Focosi D, Maggi F. Neutralising antibody escape of SARS-CoV-2 spike protein: Risk assessment for antibody-based Covid-19 therapeutics and vaccines. Rev Med Virol. 2021;31(6):e2231. doi: 10.1002/rmv.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Strohl WR, Strohl LM. 2012. Therapeutic Antibody Engineering: Current and Future Advances Driving the Strongest Growth Area in the Pharma Industry. Woodhead Publishing Series in Biomedicine No. 11 (Cambridge), 656 pp. ISBN 9781907568374. Published October 22, 2012.
- 177.Tzilas V, Manali E, Papiris S, Bouros D. Intravenous immunoglobulin for the treatment of COVID-19: A promising tool. Respiration. 2020;99(12):1087–1089. doi: 10.1159/000512727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Sakoulas G, Geriak M, Kullar R, Greenwood K, Habib M, Vyas A, et al. Use of intravenous immunoglobulin therapy reduces progression to mechanical ventilation in COVID-19 patients with moderate to severe hypoxia. Poster 1746, ASH Annual Meeting and Exposition. Dec 6 2020. https://ash.confex.com/ash/2020/webprogram/Paper141003.html. Accessed 25 Oct 2021.
- 179.Hou X, Tian L, Zhou L, Jia X, Kong L, Xue Y, et al. Intravenous immunoglobulin-based adjuvant therapy for severe COVID-19: a single-center retrospective cohort study. Virol J. 2021;18(1):101. doi: 10.1186/s12985-021-01575-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Focosi D, Franchini M, Tuccori M, Cruciani M. Efficacy of high-dose polyclonal intravenous immunoglobulin in COVID-19: A systematic review. Vaccines (Basel). 2022;10(1):94. doi: 10.3390/vaccines10010094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Díez JM, Romero C, Cruz M, Vandeberg P, Merritt WK, Pradenas E, et al. Anti-SARS-CoV-2 hyperimmune globulin demonstrates potent neutralization and antibody-dependent cellular cytotoxicity and phagocytosis through N and S proteins. J Infect Dis. 2021 doi: 10.1093/infdis/jiab540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Ali S, Uddin SM, Ali A, Anjum F, Ali R, Shalim E, et al. Production of hyperimmune anti-SARS-CoV-2 intravenous immunoglobulin from pooled COVID-19 convalescent plasma. Immunotherapy. 2021;13(5):397–407. doi: 10.2217/imt-2020-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Tang J, Lee Y, Ravichandran S, Grubbs G, Huang C, Stauft CB, et al. Epitope diversity of SARS-CoV-2 hyperimmune intravenous human immunoglobulins and neutralization of variants of concern. iScience. 2021;24(9):103006. doi: 10.1016/j.isci.2021.103006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Zahra FT, Bellusci L, Grubbs G, Golding H, Khurana S. Neutralisation of circulating SARS-CoV-2 delta and omicron variants by convalescent plasma and SARS-CoV-2 hyperimmune intravenous human immunoglobulins for treatment of COVID-19. Ann Rheum Dis. 2022 doi: 10.1136/annrheumdis-2022-222115. [DOI] [PubMed] [Google Scholar]
- 185.Lu Y, Wang Y, Zhang Z, Huang J, Yao M, Huang G, et al. Generation of chicken IgY against SARS-COV-2 spike protein and epitope mapping. J Immunol Res. 2020;2020:9465398. doi: 10.1155/2020/9465398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Somasundaram R, Choraria A, Antonysamy M. An approach towards development of monoclonal IgY antibodies against SARS CoV-2 spike protein (S) using phage display method: a review. Int Immunopharmacol. 2020;85:106654. doi: 10.1016/j.intimp.2020.106654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Vanhove B, Duvaux O, Rousse J, Royer PJ, Evanno G, Ciron C, et al. High neutralizing potency of swine glyco-humanized polyclonal antibodies against SARS-CoV-2. Eur J Immunol. 2021;51(6):1412–1422. doi: 10.1002/eji.202049072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Vanhove B, Marot S, Gaborit B, Evanno G, Malet I, Ciron C, et al. XAV-19, a novel swine glyco-humanized polyclonal antibody against SARS-CoV-2 spike, efficiently neutralizes B.1.1.7 British and B.1.351 South-African variants. bioRxiv [preprint] 2021.04.02.437747; 10.1101/2021.04.02.437747.
- 189.Gaborit B, Vanhove B, Vibet MA, Le Thuaut A, Lacombe K, Dubee V, et al. Evaluation of the safety and efficacy of XAV-19 in patients with COVID-19-induced moderate pneumonia: study protocol for a randomized, double-blinded, placebo-controlled phase 2 (2a and 2b) trial. Trials. 2021;22(1):199. doi: 10.1186/s13063-021-05132-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Entera Health Announces Approval of COVID-19 Clinical Trial using EnteraGam in Spain. July 2 2020. https://www.biospace.com/article/releases/entera-health-announces-approval-of-covid-19-clinical-trial-using-enteragam-in-spain/. Accessed 26 Oct 2021.
- 191.Luke T, Wu H, Egland KA, Sullivan EJ, Bausch CL. Fully human antibody immunoglobulin from transchromosomic bovines is potent against SARS-CoV-2 variant pseudoviruses. bioRxiv [preprint] 2021.08.09.454215; 10.1101/2021.08.09.454215
- 192.Welte T, Dellinger RP, Ebelt H, Ferrer M, Opal SM, Singer M, et al. Efficacy and safety of trimodulin, a novel polyclonal antibody preparation, in patients with severe community-acquired pneumonia: a randomized, placebo-controlled, double-blind, multicenter, phase II trial (CIGMA study) Intensive Care Med. 2018;44(4):438–448. doi: 10.1007/s00134-018-5143-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Zhang J, Zhang H, Sun L. Therapeutic antibodies for COVID-19: is a new age of IgM, IgA and bispecific antibodies coming? MAbs. 2022;14:1. doi: 10.1080/19420862.2022.2031483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Keating SM, Mizrahi RA, Adams MS, Asensio MA, Benzie E, Carter KP, et al. Capturing and recreating diverse antibody repertoires as multivalent recombinant polyclonal antibody drugs.bioRxiv [preprint] 2020.08.05.232975; 10.1101/2020.08.05.232975.
- 195.Keating SM, Mizrahi RA, Adams MS, Asensio MA, Benzie E, Carter KP, et al. Generation of recombinant hyperimmune globulins from diverse B-cell repertoires. Nat Biotechnol. 2021;39(8):989–999. doi: 10.1038/s41587-021-00894-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Farshadpour F, Taherkhani R. Antibody-dependent enhancement and the critical pattern of COVID-19: possibilities and considerations. Med Princ Pract. 2021;9:212–219. doi: 10.1159/000516693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Vanhove B, Marot S, Evanno G, Malet I, Rouvray G, Shneiker F, et al. Anti-SARS-CoV-2 swine glyco-humanized polyclonal antibody XAV-19 retains neutralizing activity against SARS-CoV-2 B.1.1.529 (Omicron). bioRxiv [preprint] 2022.01.26.477856; 10.1101/2022.01.26.477856.
- 198.Matsushita H, Sano A, Wu H, Jiao JA, Kasinathan P, Sullivan EJ, et al. Triple immunoglobulin gene knockout transchromosomic cattle: bovine lambda cluster deletion and its effect on fully human polyclonal antibody production. PLoS ONE. 2014;9(3):e90383. doi: 10.1371/journal.pone.0090383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Matsushita H, Sano A, Wu H, Wang Z, Jiao JA, Kasinathan P, et al. Species-specific chromosome engineering greatly improves fully human polyclonal antibody production profile in cattle. PLoS ONE. 2015;10(6):e0130699. doi: 10.1371/journal.pone.0130699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.SAB-185 neutralizes Omicron press release. https://www.biospace.com/article/releases/sab-biotherapeutics-announces-sab-185-retains-neutralization-against-omicron-sars-cov-2-variant-in-vitro/. Accessed 17 Feb 2022.
- 201.Protein Data Bank. https://www.rcsb.org/. Accessed on 26 Feb 2022.
- 202.Sehnal D, Bittrich S, Deshpande M, Svobodová R, Berka K, Bazgier V, et al. Mol* Viewer: modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acids Res. 2021;49(W1):W431–W437. doi: 10.1093/nar/gkab314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Jones BE, Brown-Augsburger PL, Corbett KS, Westendorf K, Davies J, Cujec TP, et al. The neutralizing antibody, LY-CoV555, protects against SARS-CoV-2 infection in nonhuman primates. Sci Transl Med. 2021;13(593):eabf1906. doi: 10.1126/scitranslmed.abf1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Shi R, Shan C, Duan X, Chen Z, Liu P, Song J, et al. A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature. 2020;584(7819):120–124. doi: 10.1038/s41586-020-2381-y. [DOI] [PubMed] [Google Scholar]
- 205.Hansen J, Baum A, Pascal KE, Russo V, Giordano S, Wloga E, et al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science. 2020;369(6506):1010–1014. doi: 10.1126/science.abd0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Kim C, Ryu DK, Lee J, Kim YI, Seo JM, Kim YG, et al. A therapeutic neutralizing antibody targeting receptor binding domain of SARS-CoV-2 spike protein. Nat Commun. 2021;12(1):288. doi: 10.1038/s41467-020-20602-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Syed YY. Regdanvimab: first approval. Drugs. 2021;81(18):2133–2137. doi: 10.1007/s40265-021-01626-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Pinto D, Park YJ, Beltramello M, Walls AC, Tortorici MA, Bianchi S, et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature. 2020;583(7815):290–295. doi: 10.1038/s41586-020-2349-y. [DOI] [PubMed] [Google Scholar]
- 209.Cathcart AL, Havenar-Daughton C, Lempp FA, Ma D, Schmid MA, Agostini ML, et al. The dual function monoclonal antibodies VIR-7831 and VIR-7832 demonstrate potent in vitro and in vivo activity against SARS-CoV-2. bioRxiv [preprint] 2021.03.09.434607; 10.1101/2021.03.09.434607.
- 210.Zost SJ, Gilchuk P, Chen RE, Case JB, Reidy JX, Trivette A, et al. Rapid isolation and profiling of a diverse panel of human monoclonal antibodies targeting the SARS-CoV-2 spike protein. Nat Med. 2020;26(9):1422–1427. doi: 10.1038/s41591-020-0998-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Dong J, Zost S, Greaney A, Starr TN, Dingens AS, Chen EC, et al. Genetic and structural basis for recognition of SARS-CoV-2 spike protein by a two-antibody cocktail. bioRxiv [preprint]. 2021 doi: 10.1101/2021.01.27.428529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Ju B, Zhang Q, Ge J, Wang R, Sun J, Ge X, et al. Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature. 2020;584(7819):115–119. doi: 10.1038/s41586-020-2380-z. [DOI] [PubMed] [Google Scholar]
- 213.Ge J, Wang R, Ju B, Zhang Q, Sun J, Chen P, et al. Antibody neutralization of SARS-CoV-2 through ACE2 receptor mimicry. Nat Commun. 2021;12(1):250. doi: 10.1038/s41467-020-20501-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Westendorf K, Wang L, Žentelis S, Foster D, Vaillancourt P, Wiggin M, et al. LY-CoV1404 (bebtelovimab) potently neutralizes SARS-CoV-2 variants. bioRxiv [preprint]. 2022 doi: 10.1101/2021.04.30.442182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Novartis press release on ensovibep (MP0420). https://www.novartis.com/news/media-releases/novartis-and-molecular-partners-report-positive-topline-data-from-phase-2-study-ensovibep-mp0420-darpin-antiviral-therapeutic-covid-19. Accessed 19 Feb 2022.
- 216.Lee DCP, Raman R, Ghafar NA, Budigi Y. An antibody engineering platform using amino acid networks: a case study in development of antiviral therapeutics. Antiviral Res. 2021;192:105105. doi: 10.1016/j.antiviral.2021.105105. [DOI] [PubMed] [Google Scholar]
- 217.Robbiani DF, Gaebler C, Muecksch F, Lorenzi JCC, Wang Z, Cho A, et al. Convergent antibody responses to SARS-CoV-2 in convalescent individuals. Nature. 2020;584(7821):437–442. doi: 10.1038/s41586-020-2456-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Schäfer A, Muecksch F, Lorenzi JCC, Leist SR, Cipolla M, Bournazos S, et al. Antibody potency, effector function, and combinations in protection and therapy for SARS-CoV-2 infection in vivo. J Exp Med. 2021;218(3):e20201993. doi: 10.1084/jem.20201993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Rappazzo CG, Tse LV, Kaku CI, Wrapp D, Sakharkar M, Huang D, et al. Broad and potent activity against SARS-like viruses by an engineered human monoclonal antibody. Science. 2021;371(6531):823–829. doi: 10.1126/science.abf4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Adagio Therapeutics S-1. July 16 2021. https://www.sec.gov/Archives/edgar/data/1832038/000119312521217506/d549501ds1.htm. Accessed 25 Oct 2021.
- 221.Andreano E, Nicastri E, Paciello I, Pileri P, Manganaro N, Piccini G, et al. Extremely potent human monoclonal antibodies from COVID-19 convalescent patients. Cell. 2021;184(7):1821–1835.e16. doi: 10.1016/j.cell.2021.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Bian H, Zheng ZH, Wei D, Wen A, Zhang Z, Lian JQ, et al. Safety and efficacy of meplazumab in healthy volunteers and COVID-19 patients: a randomized phase 1 and an exploratory phase 2 trial. Signal Transduct Target Ther. 2021;6(1):194. doi: 10.1038/s41392-021-00603-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Cobb RR, Nkolola J, Gilchuk P, Chandrashekar A, Yu J, House RV, et al. A combination of two human neutralizing antibodies prevents SARS-CoV-2 infection in cynomolgus macaques. Med (N Y). 2022 doi: 10.1016/j.medj.2022.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Wang S, Peng Y, Wang R, Jiao S, Wang M, Huang W, et al. Characterization of neutralizing antibody with prophylactic and therapeutic efficacy against SARS-CoV-2 in rhesus monkeys. Nat Commun. 2020;11(1):5752. doi: 10.1038/s41467-020-19568-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Meng X, Wang P, Xiong Y, Wu Y, Lin X, Lu S, et al. Safety, tolerability, pharmacokinetic characteristics, and immunogenicity of MW33: a Phase 1 clinical study of the SARS-CoV-2 RBD-targeting monoclonal antibody. Emerg Microbes Infect. 2021;10(1):1638–1648. doi: 10.1080/22221751.2021.1960900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Sorrento news release. July 21 2021. https://investors.sorrentotherapeutics.com/news-releases/news-release-details/sorrento-announces-dosing-covid-19-patients-phase-2-clinical. Accessed 1 Nov 2021.
- 227.Halwe S, Kupke A, Vanshylla K, Liberta F, Gruell H, Zehner M, et al. Intranasal administration of a monoclonal neutralizing antibody protects mice against SARS-CoV-2 infection. Viruses. 2021;13(8):1498. doi: 10.3390/v13081498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Du S, Cao Y, Zhu Q, Yu P, Qi F, Wang G, et al. Structurally resolved SARS-CoV-2 antibody shows high efficacy in severely infected hamsters and provides a potent cocktail pairing strategy. Cell. 2020;183(4):1013–1023.e13. doi: 10.1016/j.cell.2020.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Wang F, Li L, Dou Y, Shi R, Duan X, Liu H, et al. Etesevimab in combination with JS026 neutralizing SARS-CoV-2 and its variants. Emerg Microbes Infect. 2022;11(1):548–551. doi: 10.1080/22221751.2022.2032374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Song D, Wang W, Dong C, Ning Z, Liu X, Liu C, et al. Structure and function analysis of a potent human neutralizing antibody CA521FALA against SARS-CoV-2. Commun Biol. 2021;4(1):500. doi: 10.1038/s42003-021-02029-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Wang C, Li W, Drabek D, Okba NMA, van Haperen R, Osterhaus ADME, et al. A human monoclonal antibody blocking SARS-CoV-2 infection. Nat Commun. 2020;11(1):2251. doi: 10.1038/s41467-020-16256-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Fedry J, Hurdiss DL, Wang C, Li W, Obal G, Drulyte I, et al. Structural insights into the cross-neutralization of SARS-CoV and SARS-CoV-2 by the human monoclonal antibody 47D11. Sci Adv. 2021;7(23):eabf5632. doi: 10.1126/sciadv.abf5632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Alsoussi WB, Turner JS, Case JB, Zhao H, Schmitz AJ, Zhou JQ, et al. A potently neutralizing antibody protects mice against SARS-CoV-2 infection. J Immunol. 2020;205(4):915–922. doi: 10.4049/jimmunol.2000583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Liu Z, VanBlargan LA, Bloyet LM, Rothlauf PW, Chen RE, Stumpf S, et al. Landscape analysis of escape variants identifies SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe. 2021;29(3):477–488.e4. doi: 10.1101/2020.11.06.372037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Guo Y, Huang L, Zhang G, Yao Y, Zhou H, Shen S, et al. A SARS-CoV-2 neutralizing antibody with extensive spike binding coverage and modified for optimal therapeutic outcomes. Nat Commun. 2021;12(1):2623. doi: 10.1038/s41467-021-22926-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Bertoglio F, Fühner V, Ruschig M, Heine PA, Abassi L, Klünemann T, et al. A SARS-CoV-2 neutralizing antibody selected from COVID-19 patients binds to the ACE2-RBD interface and is tolerant to most known RBD mutations. Cell Rep. 2021;36(4):109433. doi: 10.1016/j.celrep.2021.109433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Gu C, Cao X, Wang Z, Hu X, Yao Y, Zhou Y, et al. A human antibody with blocking activity to RBD proteins of multiple SARS-CoV-2 variants including B.1.351 showed potent prophylactic and therapeutic efficacy against SARS-CoV-2 in rhesus macaques. bioRxiv [preprint] 2021.02.07.429299; doi: 10.1101/2021.02.07.429299.
- 238.Wrapp D, De Vlieger D, Corbett KS, Torres GM, Wang N, Van Breedam W, et al. Structural basis for potent neutralization of betacoronaviruses by single-domain camelid antibodies. Cell. 2020;181(6):1436–1441. doi: 10.1016/j.cell.2020.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Schepens B, van Schie L, Nerinckx W, Roose K, Van Breedam W, Fijalkowska D, et al. Drug development of an affinity enhanced, broadly neutralizing heavy chain-only antibody that restricts SARS-CoV-2 in rodents. bioRxiv [preprint] 2021.03.08.433449; 10.1101/2021.03.08.433449. [DOI] [PMC free article] [PubMed]
- 240.Chan CEZ, Seah SGK, Chye H, Massey S, Torres M, Lim APC, et al. The Fc-mediated effector functions of a potent SARS-CoV-2 neutralizing antibody, SC31, isolated from an early convalescent COVID-19 patient, are essential for the optimal therapeutic efficacy of the antibody. PLoS ONE. 2021;16(6):e0253487. doi: 10.1371/journal.pone.0253487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Celltrion press release on development of CT-P63 and nebulized formulation. https://www.celltrionhealthcare.com/en-us/board/newsdetail?modify_key=543&pagenumber=1&keyword=&keyword_type. Accessed on 4 Dec 2021.
- 242.Liu J, Chen Q, Yang S, Li Y, Dou Y, Deng YQ, et al. hACE2 Fc-neutralization antibody cocktail provides synergistic protection against SARS-CoV-2 and its spike RBD variants. Cell Discov. 2021;7(1):54. doi: 10.1038/s41421-021-00293-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Yao H, Sun Y, Deng YQ, Wang N, Tan Y, Zhang NN, et al. Rational development of a human antibody cocktail that deploys multiple functions to confer pan-SARS-CoVs protection. Cell Res. 2021;31(1):25–36. doi: 10.1038/s41422-020-00444-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Ku Z, Xie X, Davidson E, Ye X, Su H, Menachery VD, et al. Molecular determinants and mechanism for antibody cocktail preventing SARS-CoV-2 escape. Nat Commun. 2021;12(1):469. doi: 10.1038/s41467-020-20789-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Ku Z, Xie X, Hinton PR, Liu X, Ye X, Muruato AE, et al. Nasal delivery of an IgM offers broad protection from SARS-CoV-2 variants. Nature. 2021;595(7869):718–723. doi: 10.1038/s41586-021-03673-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Carroll S. An IgM antibody platform to tackle SARS-CoV-2 and its variants. https://www.drugtargetreview.com/article/97106/an-igm-antibody-platform-to-tackle-sars-cov-2-and-its-variants/2/. Accessed 5 Dec 2021.
- 247.Schmitt S, Weber M, Hillenbrand M, Seidenberg J, Zingg A, Townsend C, et al. MTX-COVAB, a human-derived antibody with potent neutralizing activity against SARS-CoV-2 infection in vitro and in a hamster model of COVID-19. bioRxiv [preprint] 2020.12.01.406934; 10.1101/2020.12.01.406934.
- 248.New inhaled SARS-CoV-2 antibody. https://www.biopharma-reporter.com/Article/2021/12/13/Memo-Therapeutics-AG-to-clinically-develop-SARS-CoV-2-antibody. Accessed 19 Feb 2022.
- 249.Koenig PA, Das H, Liu H, Kümmerer BM, Gohr FN, Jenster LM, et al. Structure-guided multivalent nanobodies block SARS-CoV-2 infection and suppress mutational escape. Science. 2021;371(6530):eabe6230. doi: 10.1126/science.abe6230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Roodink I, van Erp M, Li A, Potter S, van Duijnhoven SMJ, Kuipers AJ, et al. Cornering an ever-evolving coronavirus: TATX-03, a fully human synergistic multi-antibody cocktail targeting the SARS-CoV-2 spike protein with in vivo efficacy. bioRxiv [preprint] 2021.07.20.452858; 10.1101/2021.07.20.452858.
- 251.Piepenbrink MS, Park JG, Oladunni FS, Deshpande A, Basu M, Sarkar S, et al. Therapeutic activity of an inhaled potent SARS-CoV-2 neutralizing human monoclonal antibody in hamsters. Cell Rep Med. 2021;2(3):100218. doi: 10.1016/j.xcrm.2021.100218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Xiang Y, Nambulli S, Xiao Z, Liu H, Sang Z, Duprex WP, et al. Versatile and multivalent nanobodies efficiently neutralize SARS-CoV-2. Science. 2020;370(6523):1479–1484. doi: 10.1126/science.abe4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Nambulli S, Xiang Y, Tilston-Lunel NL, Rennick LJ, Sang Z, Klimstra WB, et al. Inhalable nanobody (PiN-21) prevents and treats SARS-CoV-2 infections in Syrian hamsters at ultra-low doses. Sci Adv. 2021;7(22):eabh0319. doi: 10.1126/sciadv.abh0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Duty JA , Kraus T, Zhou H, Zhang Y, Shaabani N, Yildiz S, et al. Discovery of a SARS-CoV-2 broadly-acting neutralizing antibody with activity against Omicron and Omicron + R346K variants. 10.1101/2022.01.19.476998. [DOI] [PMC free article] [PubMed]
- 255.Huo J, Zhao Y, Ren J, Zhou D, Duyvesteyn HME, Ginn HM, et al. Neutralization of SARS-CoV-2 by destruction of the prefusion spike. Cell Host Microbe. 2020;28(3):497. doi: 10.1016/j.chom.2020.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Xiao X, Chen Y, Varkey R, Kallewaard N, Koksal AC, Zhu Q, et al. A novel antibody discovery platform identifies anti-influenza A broadly neutralizing antibodies from human memory B cells. MAbs. 2016;8(5):916–927. doi: 10.1080/19420862.2016.1170263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Rogers TF, Zhao F, Huang D, Beutler N, Burns A, He WT, et al. Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model. Science. 2020;369(6506):956–963. doi: 10.1126/science.abc7520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Gaebler C, Wang Z, Lorenzi JCC, Muecksch F, Finkin S, Tokuyama M, et al. Evolution of antibody immunity to SARS-CoV-2. Nature. 2021;591(7851):639–644. doi: 10.1038/s41586-021-03207-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Jette CA, Cohen AA, Gnanapragasam PNP, Muecksch F, Lee YE, Huey-Tubman KE, et al. Broad cross-reactivity across sarbecoviruses exhibited by a subset of COVID-19 donor-derived neutralizing antibodies. Cell Rep. 2021 doi: 10.1101/2021.04.23.441195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Martinez DR, Schaefer A, Gobeil S, Li D, De la Cruz G, Parks R, et al. A broadly neutralizing antibody protects against SARS-CoV, pre-emergent bat CoVs, and SARS-CoV-2 variants in mice. bioRxiv [preprint]. 2021 doi: 10.1101/2021.04.27.441655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Starr TN, Czudnochowski N, Liu Z, Zatta F, Park YJ, Addetia A, et al. SARS-CoV-2 RBD antibodies that maximize breadth and resistance to escape. Nature. 2021;597(7874):97–102. doi: 10.1038/s41586-021-03807-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Tortorici MA, Czudnochowski N, Starr TN, Marzi R, Walls AC, Zatta F, et al. Broad sarbecovirus neutralization by a human monoclonal antibody. Nature. 2021;597(7874):103–108. doi: 10.1038/s41586-021-03817-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Green LL. Transgenic mouse strains as platforms for the successful discovery and development of human therapeutic monoclonal antibodies. Curr Drug Discov Technol. 2014;11(1):74–84. doi: 10.2174/15701638113109990038. [DOI] [PubMed] [Google Scholar]
- 264.Brüggemann M, Osborn MJ, Ma B, Hayre J, Avis S, Lundstrom B, et al. Human antibody production in transgenic animals. Arch Immunol Ther Exp (Warsz) 2015;63(2):101–108. doi: 10.1007/s00005-014-0322-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Weinreich DM, Sivapalasingam S, Norton T, Ali S, Gao H, Bhore R, et al. REGN-COV2, a neutralizing antibody cocktail, in outpatients with Covid-19. N Engl J Med. 2021;384(3):238–251. doi: 10.1056/NEJMoa2035002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Kim YJ, Lee MH, Lee SR, Chung HY, Kim K, Lee TG, et al. Neutralizing human antibodies against severe acute respiratory syndrome coronavirus 2 isolated from a human synthetic Fab phage display library. Int J Mol Sci. 2021;22(4):1913. doi: 10.3390/ijms22041913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Bertoglio F, Meier D, Langreder N, Steinke S, Rand U, Simonelli L, et al. SARS-CoV-2 neutralizing human recombinant antibodies selected from pre-pandemic healthy donors binding at RBD-ACE2 interface. Nat Commun. 2021;12(1):1577. doi: 10.1038/s41467-021-21609-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Valadon P, Pérez-Tapia SM, Nelson RS, Guzmán-Bringas OU, Arrieta-Oliva HI, Gómez-Castellano KM, et al. ALTHEA Gold Libraries™: antibody libraries for therapeutic antibody discovery. MAbs. 2019;11(3):516–531. doi: 10.1080/19420862.2019.1571879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Ferrara F, Erasmus MF, D'Angelo S, Leal-Lopes C, Teixeira AA, Choudhary A, et al. A pandemic-enabled comparison of discovery platforms demonstrates a naïve antibody library can match the best immune-sourced antibodies. Nat Commun. 2022;13(1):462. doi: 10.1038/s41467-021-27799-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Avnir Y, Tallarico AS, Zhu Q, Bennett AS, Connelly G, Sheehan J, et al. Molecular signatures of hemagglutinin stem-directed heterosubtypic human neutralizing antibodies against influenza A viruses. PLoS Pathog. 2014;10(5):e1004103. doi: 10.1371/journal.ppat.1004103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Wu NC, Wilson IA. Influenza hemagglutinin structures and antibody recognition. Cold Spring Harb Perspect Med. 2020;10(8):a038778. doi: 10.1101/cshperspect.a038778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Chen F, Tzarum N, Wilson IA, Law M. VH1-69 antiviral broadly neutralizing antibodies: genetics, structures, and relevance to rational vaccine design. Curr Opin Virol. 2019;34:149–159. doi: 10.1016/j.coviro.2019.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Bonvin P, Venet S, Kosco-Vilbois M, Fisher N. Purpose-oriented antibody libraries incorporating tailored CDR3 sequences. Antibodies. 2015;4:103–122. doi: 10.3390/antib4020103. [DOI] [Google Scholar]
- 274.Larimore K, McCormick MW, Robins HS, Greenberg PD. Shaping of human germline IgH repertoires revealed by deep sequencing. J Immunol. 2012;189(6):3221–3230. doi: 10.4049/jimmunol.1201303. [DOI] [PubMed] [Google Scholar]
- 275.Briney BS, Willis JR, Crowe JE., Jr Human peripheral blood antibodies with long HCDR3s are established primarily at original recombination using a limited subset of germline genes. PLoS ONE. 2012;7(5):e36750. doi: 10.1371/journal.pone.0036750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Barnes CO, Jette CA, Abernathy ME, Dam KA, Esswein SR, Gristick HB, et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature. 2020;588(7839):682–687. doi: 10.1038/s41586-020-2852-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Yuan M, Liu H, Wu NC, Wilson IA. Recognition of the SARS-CoV-2 receptor binding domain by neutralizing antibodies. Biochem Biophys Res Commun. 2021;538:192–203. doi: 10.1016/j.bbrc.2020.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Li W, Schäfer A, Kulkarni SS, Liu X, Martinez DR, Chen C, et al. High potency of a bivalent human VH domain in SARS-CoV-2 animal models. Cell. 2020;183(2):429–441.e16. doi: 10.1016/j.cell.2020.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Fagre AC, Manhard J, Adams R, Eckley M, Zhan S, Lewis J, et al. A potent SARS-CoV-2 neutralizing human monoclonal antibody that reduces viral burden and disease severity in Syrian hamsters. Front Immunol. 2020;11:614256. doi: 10.3389/fimmu.2020.614256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Bell BN, Powell AE, Rodriguez C, Cochran JR, Kim PS. Neutralizing antibodies targeting the SARS-CoV-2 receptor binding domain isolated from a naïve human antibody library. Protein Sci. 2021;30(4):716–727. doi: 10.1002/pro.4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Baruah V, Bose S. Immunoinformatics-aided identification of T cell and B cell epitopes in the surface glycoprotein of 2019-nCoV. J Med Virol. 2020;92(5):495–500. doi: 10.1002/jmv.25698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.ter Meulen J, van den Brink EN, Poon LL, Marissen WE, Leung CS, Cox F, et al. Human monoclonal antibody combination against SARS coronavirus: synergy and coverage of escape mutants. PLoS Med. 2006;3(7):e237. doi: 10.1371/journal.pmed.0030237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Strohl WR. Current progress in innovative engineered antibodies. Protein Cell. 2018;9(1):86–120. doi: 10.1007/s13238-017-0457-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Winkler ES, Gilchuk P, Yu J, Bailey AL, Chen RE, Chong Z, et al. Human neutralizing antibodies against SARS-CoV-2 require intact Fc effector functions for optimal therapeutic protection. Cell. 2021;184(7):1804–1820.e16. doi: 10.1016/j.cell.2021.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Keeler SP, Fox JM. Requirement of Fc-Fc gamma receptor interaction for antibody-based protection against emerging virus infections. Viruses. 2021;13(6):1037. doi: 10.3390/v13061037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Yamin R, Jones AT, Hoffmann HH, Schäfer A, Kao KS, Francis RL, et al. Fc-engineered antibody therapeutics with improved anti-SARS-CoV-2 efficacy. Nature. 2021;599(7885):465–470. doi: 10.1038/s41586-021-04017-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Noy-Porat T, Edri A, Alcalay R, Makdasi E, Gur D, Aftalion M, et al. Fc-Independent protection from SARS-CoV-2 infection by recombinant human monoclonal antibodies. Antibodies (Basel). 2021;10(4):45. doi: 10.3390/antib10040045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Chakraborty S, Gonzalez J, Edwards K, Mallajosyula V, Buzzanco AS, Sherwood R, et al. Proinflammatory IgG Fc structures in patients with severe COVID-19. Nat Immunol. 2021;22(1):67–73. doi: 10.1038/s41590-020-00828-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Ricke DO. Two different antibody-dependent enhancement (ADE) risks for SARS-CoV-2 antibodies. Front Immunol. 2021;12:640093. doi: 10.3389/fimmu.2021.640093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Hastie KM, Li H, Bedinger D, Schendel SL, Dennison SM, Li K, et al. Defining variant-resistant epitopes targeted by SARS-CoV-2 antibodies: a global consortium study. Science. 2021;374(6566):472–478. doi: 10.1126/science.abh2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Ullah I, Prévost J, Ladinsky MS, Stone H, Lu M, Anand SP, et al. Live imaging of SARS-CoV-2 infection in mice reveals that neutralizing antibodies require Fc function for optimal efficacy. Immunity. 2021;54(9):2143–2158.e15. doi: 10.1016/j.immuni.2021.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Asokan M, Dias J, Liu C, Maximova A, Ernste K, Pegu A, et al. Fc-mediated effector function contributes to the in vivo antiviral effect of an HIV neutralizing antibody. Proc Natl Acad Sci U S A. 2020;117(31):18754–18763. doi: 10.1073/pnas.2008236117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Hessell AJ, Hangartner L, Hunter M, Havenith CE, Beurskens FJ, Bakker JM, et al. Fc receptor but not complement binding is important in antibody protection against HIV. Nature. 2007;449(7158):101–104. doi: 10.1038/nature06106. [DOI] [PubMed] [Google Scholar]
- 294.Gorman MJ, Patel N, Guebre-Xabier M, Zhu AL, Atyeo C, Pullen KM, et al. Fab and Fc contribute to maximal protection against SARS-CoV-2 following NVX-CoV2373 subunit vaccine with Matrix-M vaccination. Cell Rep Med. 2021;2(9):100405. doi: 10.1016/j.xcrm.2021.100405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Ng N, Powell CA. Targeting the complement cascade in the pathophysiology of COVID-19 disease. J Clin Med. 2021;10(10):2188. doi: 10.3390/jcm10102188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Piccoli L, Park YJ, Tortorici MA, Czudnochowski N, Walls AC, Beltramello M, et al. Mapping neutralizing and immunodominant sites on the SARS-CoV-2 spike receptor-binding domain by structure-guided high-resolution serology. Cell. 2020;183(4):1024–1042.e21. doi: 10.1016/j.cell.2020.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Pinto D, Sauer MM, Czudnochowski N, Low JS, Tortorici MA, Housley MP, et al. Broad betacoronavirus neutralization by a stem helix-specific human antibody. Science. 2021;373(6559):1109–1116. doi: 10.1126/science.abj3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Li W, Chen C, Drelich A, Martinez DR, Gralinski LE, Sun Z, et al. Rapid identification of a human antibody with high prophylactic and therapeutic efficacy in three animal models of SARS-CoV-2 infection. Proc Natl Acad Sci U S A. 2020;117(47):29832–29838. doi: 10.1073/pnas.2010197117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Tortorici MA, Beltramello M, Lempp FA, Pinto D, Dang HV, Rosen LE, et al. Ultrapotent human antibodies protect against SARS-CoV-2 challenge via multiple mechanisms. Science. 2020;370(6519):950–957. doi: 10.1126/science.abe3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Adeniji OS, Giron LB, Purwar M, Zilberstein NF, Kulkarni AJ, Shaikh MW, et al. COVID-19 severity is associated with differential antibody Fc-mediated innate immune functions. MBio. 2021;12(2):e00281-21. doi: 10.1128/mBio.00281-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.de Nooijer AH, Grondman I, Janssen NAF, Netea MG, Willems L, van de Veerdonk FL, et al. Complement activation in the disease course of coronavirus disease 2019 and its effects on clinical outcomes. J Infect Dis. 2021;223(2):214–224. doi: 10.1093/infdis/jiaa646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Li Q, Chen Z. An update: the emerging evidence of complement involvement in COVID-19. Med Microbiol Immunol. 2021;210:101–109. doi: 10.1007/s00430-021-00704-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Bonaventura A, Vecchié A, Dagna L, Martinod K, Dixon DL, Van Tassell BW, et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat Rev Immunol. 2021;21(5):319–329. doi: 10.1038/s41577-021-00536-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Yu J, Gerber GF, Chen H, Yuan X, Chaturvedi S, Braunstein EM, et al. Complement dysregulation is associated with severe COVID-19 illness. Haematologiapproximately. 2021 doi: 10.3324/haematol.2021.279155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Iwasaki A, Yang Y. The potential danger of suboptimal antibody responses in COVID-19. Nat Rev Immunol. 2020;20(6):339–341. doi: 10.1038/s41577-020-0321-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Ubol S, Halstead SB. How innate immune mechanisms contribute to antibody-enhanced viral infections. Clin Vaccine Immunol. 2010;17(12):1829–1835. doi: 10.1128/CVI.00316-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Taylor A, Foo SS, Bruzzone R, Dinh LV, King NJ, Mahalingam S. Fc receptors in antibody-dependent enhancement of viral infections. Immunol Rev. 2015;268(1):340–364. doi: 10.1111/imr.12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Chan KR, Ong EZ, Mok DZ, Ooi EE. Fc receptors and their influence on efficacy of therapeutic antibodies for treatment of viral diseases. Expert Rev Anti Infect Ther. 2015;13(11):1351–1360. doi: 10.1586/14787210.2015.1079127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Wan Y, Shang J, Sun S, Tai W, Chen J, Geng Q, et al. Molecular mechanism for antibody-dependent enhancement of coronavirus entry. J Virol. 2020;94(5):e02015–e2019. doi: 10.1128/JVI.02015-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Kam YW, Kien F, Roberts A, Cheung YC, Lamirande EW, Vogel L, et al. Antibodies against trimeric S glycoprotein protect hamsters against SARS-CoV challenge despite their capacity to mediate FcgammaRII-dependent entry into B cells in vitro. Vaccine. 2007;25(4):729–740. doi: 10.1016/j.vaccine.2006.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Wang SF, Tseng SP, Yen CH, Yang JY, Tsao CH, Shen CW, et al. Antibody-dependent SARS coronavirus infection is mediated by antibodies against spike proteins. Biochem Biophys Res Commun. 2014;451(2):208–214. doi: 10.1016/j.bbrc.2014.07.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Jaume M, Yip MS, Kam YW, Cheung CY, Kien F, Roberts A, et al. SARS CoV subunit vaccine: antibody-mediated neutralisation and enhancement. Hong Kong Med J. 2012;18(Suppl 2):31–36. [PubMed] [Google Scholar]
- 313.Lee WS, Wheatley AK, Kent SJ, DeKosky BJ. Antibody-dependent enhancement and SARS-CoV-2 vaccines and therapies. Nat Microbiol. 2020;5(10):1185–1191. doi: 10.1038/s41564-020-00789-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.García-Nicolás O, V’kovski P, Zettl F, Zimmer G, Thiel V, Summerfield A. No evidence for human monocyte-derived macrophage infection and antibody-mediated enhancement of SARS-CoV-2 infection. Front Cell Infect Microbiol. 2021;11:644574. doi: 10.3389/fcimb.2021.644574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Focosi D, Genoni A, Lucenteforte E, Tillati S, Tamborini A, Spezia PG, et al. Previous humoral immunity to the endemic seasonal alpha coronaviruses NL63 and 229E is associated with worse clinical outcome in COVID-19 and suggests original antigenic sin. Life (Basel). 2021;11(4):298. doi: 10.3390/life11040298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Jiang W, Wang J, Jiao S, Gu C, Xu W, Chen B, et al. Characterization of MW06, a human monoclonal antibody with cross-neutralization activity against both SARS-CoV-2 and SARS-CoV. MAbs. 2021;13(1):1953683. doi: 10.1080/19420862.2021.1953683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Li D, Edwards RJ, Manne K, Martinez DR, Schäfer A, Alam SM, et al. In vitro and in vivo functions of SARS-CoV-2 infection-enhancing and neutralizing antibodies. Cell. 2021;184(16):4203–4219.e32. doi: 10.1016/j.cell.2021.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Pereira NA, Chan KF, Lin PC, Song Z. The, "less-is-more" in therapeutic antibodies: afucosylated anti-cancer antibodies with enhanced antibody-dependent cellular cytotoxicity. MAbs. 2018;10(5):693–711. doi: 10.1080/19420862.2018.1466767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Bournazos S, Vo HTM, Duong V, Auerswald H, Ly S, Sakuntabhai A, et al. Antibody fucosylation predicts disease severity in secondary dengue infection. Science. 2021;372(6546):1102–1105. doi: 10.1126/science.abc7303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Larsen MD, de Graaf EL, Sonneveld ME, Plomp HR, Nouta J, Hoepel W, et al. Afucosylated IgG characterizes enveloped viral responses and correlates with COVID-19 severity. Science. 2021;371(6532):eabc8378. doi: 10.1126/science.abc8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Kombe Kombe AJ, Zahid A, Mohammed A, Shi R, Jin T. Potent molecular feature-based neutralizing monoclonal antibodies as promising therapeutics against SARS-CoV-2 infection. Front Mol Biosci. 2021;8:670815. doi: 10.3389/fmolb.2021.670815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Greaney AJ, Starr TN, Barnes CO, Weisblum Y, Schmidt F, Caskey M, et al. Mapping mutations to the SARS-CoV-2 RBD that escape binding by different classes of antibodies. Nat Commun. 2021;12(1):4196. doi: 10.1038/s41467-021-24435-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Starr TN, Greaney AJ, Addetia A, Hannon WW, Choudhary MC, Dingens AS, et al. Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. Science. 2021;371(6531):850–854. doi: 10.1126/science.abf9302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Baum A, Kyratsous CA. SARS-CoV-2 spike therapeutic antibodies in the age of variants. J Exp Med. 2021;218(5):e20210198. doi: 10.1084/jem.20210198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Baum A, Fulton BO, Wloga E, Copin R, Pascal KE, Russo V, et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science. 2020;369(6506):1014–1018. doi: 10.1126/science.abd0831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Starr TN, Greaney AJ, Dingens AS, Bloom JD. Complete map of SARS-CoV-2 RBD mutations that escape the monoclonal antibody LY-CoV555 and its cocktail with LY-CoV016. Cell Rep Med. 2021;2(4):100255. doi: 10.1016/j.xcrm.2021.100255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Cho H, Gonzales-Wartz KK, Huang D, Yuan M, Peterson M, Liang J, et al. Ultrapotent bispecific antibodies neutralize emerging SARS-CoV-2 variants. bioRxiv [preprint] 2021.04.01.437942, 10.1101/2021.04.01.437942.
- 328.De Gasparo R, Pedotti M, Simonelli L, Nickl P, Muecksch F, Cassaniti I, et al. Bispecific IgG neutralizes SARS-CoV-2 variants and prevents escape in mice. Nature. 2021;593(7859):424–428. doi: 10.1038/s41586-021-03461-y. [DOI] [PubMed] [Google Scholar]
- 329.Sanyou Biopharmaceuticals news release. https://www.prnewswire.com/news-releases/bispecific-antibody-drug-against-covid-19-jointly-developed-by-shanghai-zj-bio-tech-and-sanyou-biopharmaceuticals-appearing-at-china-international-import-expo-ciie-301428939.html. Accessed on 4 Dec 2021.
- 330.Chi X, Zhang X, Pan S, Yu Y, Lin T, Duan H, et al. An ultrapotent RBD-targeted biparatopic nanobody neutralizes broad SARS-CoV-2 variants. bioRxiv 2021.12.25.474052; 10.1101/2021.12.25.474052 [DOI] [PMC free article] [PubMed]
- 331.Ku Z, Xie X, Lin J, Gao P, El Sahili A, Su H, et al. Engineering SARS-CoV-2 cocktail antibodies into a bispecific format improves neutralizing potency and breadth. bioRxiv 2022.02.01.478504; 10.1101/2022.02.01.478504 [DOI] [PMC free article] [PubMed]
- 332.Miersch S, Li Z, Saberianfar R, Ustav M, Brett Case J, Blazer L, et al. Tetravalent SARS-CoV-2 neutralizing antibodies show enhanced potency and resistance to escape mutations. J Mol Biol. 2021;433(19):167177. doi: 10.1016/j.jmb.2021.167177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Rujas E, Kucharska I, Tan YZ, Benlekbir S, Cui H, Zhao T, et al. Multivalency transforms SARS-CoV-2 antibodies into ultrapotent neutralizers. Nat Commun. 2021;12(1):3661. doi: 10.1038/s41467-021-23825-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Mantis NJ, Forbes SJ. Secretory IgA: arresting microbial pathogens at epithelial borders. Immunol Invest. 2010;39(4–5):383–406. doi: 10.3109/08820131003622635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Ejemel M, Li Q, Hou S, Schiller ZA, Tree JA, Wallace A, et al. A cross-reactive human IgA monoclonal antibody blocks SARS-CoV-2 spike-ACE2 interaction. Nat Commun. 2020;11(1):4198. doi: 10.1038/s41467-020-18058-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Wang Z, Lorenzi JCC, Muecksch F, Finkin S, Viant C, Gaebler C, et al. Enhanced SARS-CoV-2 neutralization by dimeric IgA. Sci Transl Med. 2021;13(577):eabf1555. doi: 10.1126/scitranslmed.abf1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Keyt BA, Baliga R, Sinclair AM, Carroll SF, Peterson MS. Structure, function, and therapeutic use of IgM antibodies. Antibodies (Basel). 2020;9(4):53. doi: 10.3390/antib9040053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Muyldermans S. Nanobodies: natural single-domain antibodies. Annu Rev Biochem. 2013;82:775–797. doi: 10.1146/annurev-biochem-063011-092449. [DOI] [PubMed] [Google Scholar]
- 339.Zupancic JM, Desai AA, Tessier PM. Facile isolation of high-affinity nanobodies from synthetic libraries using CDR-swapping mutagenesis. STAR Protoc. 2022;3(1):101101. doi: 10.1016/j.xpro.2021.101101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Yuan TZ, Garg P, Wang L, Willis JR, Kwan E, Hernandez AGL, et al. Rapid discovery of diverse neutralizing SARS-CoV-2 antibodies from large-scale synthetic phage libraries. MAbs. 2022;14(1):2002236. doi: 10.1080/19420862.2021.2002236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Bessalah S, Jebahi S, Mejri N, Salhi I, Khorchani T, Hammadi M. Perspective on therapeutic and diagnostic potential of camel nanobodies for coronavirus disease-19 (COVID-19) 3 Biotech. 2021;11(2):89. doi: 10.1007/s13205-021-02647-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Sun D, Sang Z, Kim YJ, Xiang Y, Cohen T, Belford AK, et al. Potent neutralizing nanobodies resist convergent circulating variants of SARS-CoV-2 by targeting diverse and conserved epitopes. Nat Commun. 2021;12(1):4676. doi: 10.1038/s41467-021-24963-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Güttler T, Aksu M, Dickmanns A, Stegmann KM, Gregor K, Rees R, et al. Neutralization of SARS-CoV-2 by highly potent, hyperthermostable, and mutation-tolerant nanobodies. EMBO J. 2021;40(19):e107985. doi: 10.15252/embj.2021107985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Hanke L, Vidakovics Perez L, Sheward DJ, Das H, Schulte T, Moliner-Morro A, et al. An alpaca nanobody neutralizes SARS-CoV-2 by blocking receptor interaction. Nat Commun. 2020;11(1):4420. doi: 10.1038/s41467-020-18174-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Margulies D, Ahmad J, Jiang J, Boyd L, Zeher A, Huang R, et al. Structures of synthetic nanobody-SARS-CoV-2-RBD complexes reveal distinct sites of interaction and recognition of variants. J Biol Chem. 2021;297(4):101202. doi: 10.1016/j.jbc.2021.101202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Schoof M, Faust B, Saunders RA, Sangwan S, Rezelj V, Hoppe N, et al. An ultrapotent synthetic nanobody neutralizes SARS-CoV-2 by stabilizing inactive Spike. Science. 2020;370(6523):1473–1479. doi: 10.1126/science.abe3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Esparza TJ, Martin NP, Anderson GP, Goldman ER, Brody DL. High affinity nanobodies block SARS-CoV-2 spike receptor binding domain interaction with human angiotensin converting enzyme. Sci Rep. 2020;10(1):22370. doi: 10.1038/s41598-020-79036-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Bracken CJ, Lim SA, Solomon P, Rettko NJ, Nguyen DP, Zha BS, et al. Bi-paratopic and multivalent VH domains block ACE2 binding and neutralize SARS-CoV-2. Nat Chem Biol. 2021;17(1):113–121. doi: 10.1038/s41589-020-00679-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Hunt AC, Case JB, Park Y-J, Cao L, Wu W, Walls AC, et al. Multivalent designed proteins protect against SARS-CoV-2 variants of concern. bioRxiv [preprint] 2021.07.07.451375; 10.1101/2021.07.07.451375.
- 350.Xu J, Xu K, Jung S, Conte A, Lieberman J, Muecksch F, et al. Nanobodies from camelid mice and llamas neutralize SARS-CoV-2 variants. Nature. 2021;595(7866):278–282. doi: 10.1038/s41586-021-03676-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Walser M, Rothenberger S, Hurdiss DL, Schlegel A, Calabro V, Fontaine S, et al. Highly potent anti-SARS-CoV-2 multivalent DARPin therapeutic candidates. bioRxiv [preprint] 2020.08.25.256339; 10.1101/2020.08.25.256339.
- 352.Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581(7807):215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 353.Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367(6485):1444–1448. doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Monteil V, Dyczynski M, Lauschke VM, Kwon H, Wirnsberger G, Youhanna S, et al. Human soluble ACE2 improves the effect of remdesivir in SARS-CoV-2 infection. EMBO Mol Med. 2021;13(1):e13426. doi: 10.15252/emmm.202013426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Suryamohan K, Diwanji D, Stawiski EW, Gupta R, Miersch S, Liu J, et al. Human ACE2 receptor polymorphisms and altered susceptibility to SARS-CoV-2. Commun Biol. 2021;4(1):475. doi: 10.1038/s42003-021-02030-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Glasgow A, Glasgow J, Limonta D, Solomon P, Lui I, Zhang Y, et al. Engineered ACE2 receptor traps potently neutralize SARS-CoV-2. Proc Natl Acad Sci U S A. 2020;117(45):28046–28055. doi: 10.1073/pnas.2016093117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Chan KK, Dorosky D, Sharma P, Abbasi SA, Dye JM, Kranz DM, et al. Engineering human ACE2 to optimize binding to the spike protein of SARS coronavirus 2. Science. 2020;369(6508):1261–1265. doi: 10.1126/science.abc0870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Svilenov HL, Sacherl J, Reiter A, Wolff LS, Cheng CC, Stern M, et al. Picomolar inhibition of SARS-CoV-2 variants of concern by an engineered ACE2-IgG4-Fc fusion protein. Antivir Res. 2021;196:105197. doi: 10.1016/j.antiviral.2021.105197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Formycon AG web site. https://www.formycon.com/en/pipeline/fyb207/. Accessed 2 Dec 2021.
- 360.Miller A, Leach A, Thomas J, McAndrew C, Bentley E, Mattiuzzo G, et al. A super-potent tetramerized ACE2 protein displays enhanced neutralization of SARS-CoV-2 virus infection. Sci Rep. 2021;11(1):10617. doi: 10.1038/s41598-021-89957-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Guo L, Bi W, Wang X, Xu W, Yan R, Zhang Y, et al. Engineered trimeric ACE2 binds viral spike protein and locks it in "three-up" conformation to potently inhibit SARS-CoV-2 infection. Cell Res. 2021;31(1):98–100. doi: 10.1038/s41422-020-00438-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Hengenix Biotech (Henlius) website. https://hengenix.com/products/. Accessed 30 Oct 2021.
- 363.White I, Tamot N, Doddareddy R, Ho J, Jiao Q, Harvilla PB, et al. Bifunctional molecules targeting SARS-CoV-2 spike and the polymeric Ig receptor display neutralization activity and mucosal enrichment. MAbs. 2021;13(1):1987180. doi: 10.1080/19420862.2021.1987180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Lundgren JD, Grund B, Barkauskas CE, Holland TL, Gottlieb RL, Sandkovsky SM, et al. A neutralizing monoclonal antibody for hospitalised patients with COVID-19. N Engl J Med. 2021;384:905–914. doi: 10.1056/NEJMoa2033130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.ACTIV-3/Therapeutics for Inpatients with COVID-19 (TICO) Study Group Efficacy and safety of two neutralising monoclonal antibody therapies, sotrovimab and BRII-196 plus BRII-198, for adults hospitalised with COVID-19 (TICO): a randomised controlled trial. Lancet Infect Dis. 2021 doi: 10.1016/S1473-3099(21)00751-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Horby PW, Mafham M, Peto L, Campbell M, Pessoa-Amorim G, Spata E, et al. Casirivimab and imdevimab in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. medRxiv [preprint]. 2021 doi: 10.1101/2021.06.15.21258542. [DOI] [Google Scholar]
- 367.Eom JS, Ison M, Streinu-Cercel A, Sandulescu O, Preotescu LL, Kim YS, et al. Efficacy and safety of CT-P59 plus standard of care: a phase 2/3 randomized, double-blind, placebo-controlled trial in outpatients with mild-to-moderate SARS-CoV-2 infection. Research Square [preprint] researchsquare.com/article/rs-296518/v1. 10.21203/rs.3.rs-296518/v1.
- 368.Dougan M, Nirula A, Azizad M, Mocherla B, Gottlieb RL, Chen P, et al. Bamlanivimab plus etesevimab in mild or moderate Covid-19. N Engl J Med. 2021;385(15):1382–1392. doi: 10.1056/NEJMoa2102685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Razonable RR, Pawlowski C, O'Horo JC, Arndt LL, Arndt R, Bierle DM, et al. Casirivimab-Imdevimab treatment is associated with reduced rates of hospitalization among high-risk patients with mild to moderate coronavirus disease-19. EClinicalMedicine. 2021;40:101102. doi: 10.1016/j.eclinm.2021.101102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Ganesh R, Pawlowski CF, O'Horo JC, Arndt LL, Arndt RF, Bell SJ, et al. Intravenous bamlanivimab use associates with reduced hospitalization in high-risk patients with mild to moderate COVID-19. J Clin Invest. 2021;131(19):e151697. doi: 10.1172/JCI151697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Gupta A, Gonzalez-Rojas Y, Juarez E, Crespo Casal M, Moya J, Falci DR, et al. Early treatment for Covid-19 with SARS-CoV-2 neutralizing antibody sotrovimab. N Engl J Med. 2021;385(21):1941–1950. doi: 10.1056/NEJMoa2107934. [DOI] [PubMed] [Google Scholar]
- 372.Kreuzberger N, Hirsch C, Chai KL, Tomlinson E, Khosravi Z, Popp M, et al. SARS-CoV-2-neutralising monoclonal antibodies for treatment of COVID-19. Cochrane Database Syst Rev. 2021;9(9):CD013825. doi: 10.1002/14651858.CD013825.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Chen P, Nirula A, Heller B, Gottlieb RL, Boscia J, Morris J, et al. SARS-CoV-2 neutralizing antibody LY-CoV555 in outpatients with Covid-19. N Engl J Med. 2021;384(3):229–237. doi: 10.1056/NEJMoa2029849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Fact sheet for health care providers. Emergency use authorization (EUA) of REGEN-COV™ (casirivimab and imdevimab). https://www.fda.gov/media/145611/download. Accessed 23 Oct 2021.
- 375.ANNEX I. Conditions of use, conditions for distribution and patients targeted and conditions for safety monitoring addressed to member states. https://www.ema.europa.eu/en/documents/referral/celltrion-use-regdanvimab-treatment-covid-19-article-53-procedure-conditions-use-conditions_en.pdf. Accessed 23 Oct 2021.
- 376.Gottlieb RL, Nirula A, Chen P, Boscia J, Heller B, Morris J, et al. Effect of bamlanivimab as monotherapy or in combination with etesevimab on viral load in patients with mild to moderate COVID-19: a randomized clinical trial. JAMA. 2021;325(7):632–644. doi: 10.1001/jama.2021.0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Fact sheet for health care providers. Emergency use authorization (EUA) of bamlanivimab and etesevimab. http://pi.lilly.com/eua/bam-and-ete-eua-factsheet-hcp.pdf. Accessed 23 Oct 2021.
- 378.Fact sheet for health care providers. Emergency use authorization (EUA) of sotrovimab. https://www.fda.gov/media/149534/download. Accessed 23 Oct 2021.
- 379.Ward A. PROVENT Trial: AZD7442 as long-acting pre-exposure prophylaxis protects against symptomatic COVID-19. Sept 30 2021. https://www.contagionlive.com/view/provent-trial-azd7442-pre-exposure-prophylaxis-symptomatic-covid-19. Accessed 23 Oct 2021.
- 380.FDA Letter dated 11 February 2022 granting EUA for bebtelovimab. https://www.fda.gov/media/156151/download. Accessed 21 Feb 2022.
- 381.Factsheet on bebtelovimab. https://www.fda.gov/media/156152/download. Accessed 21 Feb 2022.
- 382.Wang R, Zhang Q, Ge J, Ren W, Zhang R, Lan J, et al. Analysis of SARS-CoV-2 variant mutations reveals neutralization escape mechanisms and the ability to use ACE2 receptors from additional species. Immunity. 2021;54(7):1611–1621.e5. doi: 10.1016/j.immuni.2021.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Singh M, de Wit E. Antiviral agents for the treatment of COVID-19: progress and challenges. Cell Rep Med. 2022 doi: 10.1016/j.xcrm.2022.100549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.United States Food and Drug Administration information on COVID-19 EUAs. https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization#coviddrugs. Accessed 22 Feb 2022.
- 385.Hoffmann M, Arora P, Groß R, Seidel A, Hörnich BF, Hahn AS, et al. SARS-CoV-2 variants B.1.351 and P.1 escape from neutralizing antibodies. Cell. 2021;184(9):2384–2393.e12. doi: 10.1016/j.cell.2021.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Chen RE, Winkler ES, Case JB, Aziati ID, Bricker TL, Joshi A, et al. In vivo monoclonal antibody efficacy against SARS-CoV-2 variant strains. Nature. 2021;596(7870):103–108. doi: 10.1038/s41586-021-03720-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Tao K, Tzou PL, Nouhin J, Gupta RK, de Oliveira T, Kosakovsky Pond SL, et al. The biological and clinical significance of emerging SARS-CoV-2 variants. Nat Rev Genet. 2021 doi: 10.1038/s41576-021-00408-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Shang J, Ye G, Shi K, Wan Y, Luo C, Aihara H, et al. Structural basis of receptor recognition by SARS-CoV-2. Nature. 2020 doi: 10.1038/s41586-020-2179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Dübel S, Herrmann A, Schirrmann T, Frenzel A, Hust M. COR-101, ein menschlicher Antikörper gegen COVID-19. Biospektrum (Heidelb). 2021;27(1):46–48. doi: 10.1007/s12268-021-1512-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Chen RE, Zhang X, Case JB, Winkler ES, Liu Y, VanBlargan LA, et al. Resistance of SARS-CoV-2 variants to neutralization by monoclonal and serum-derived polyclonal antibodies. Nat Med. 2021;27(4):717–726. doi: 10.1038/s41591-021-01294-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Wang L, Zhou T, Zhang Y, Yang ES, Schramm CA, Shi W, et al. Ultrapotent antibodies against diverse and highly transmissible SARS-CoV-2 variants. Science. 2021;373(6556):eabh1766. doi: 10.1126/science.abh1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Wu NC, Yuan M, Liu H, Lee CD, Zhu X, Bangaru S, et al. An alternative binding mode of IGHV3-53 antibodies to the SARS-CoV-2 receptor binding domain. Cell Rep. 2020;33(3):108274. doi: 10.1016/j.celrep.2020.108274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Brouwer PJM, Caniels TG, van der Straten K, Snitselaar JL, Aldon Y, Bangaru S, et al. Potent neutralizing antibodies from COVID-19 patients define multiple targets of vulnerability. Science. 2020;369(6504):643–650. doi: 10.1126/science.abc5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Mejdani M, Haddadi K, Pham C, Mahadevan R. SARS-CoV-2 receptor-binding mutations and antibody contact sites. Antibod Therap. 2021;4(3):149–158. doi: 10.1093/abt/tbab015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Kreye J, Reincke SM, Kornau HC, Sánchez-Sendin E, Corman VM, Liu H, et al. A therapeutic non-self-reactive SARS-CoV-2 antibody protects from lung pathology in a COVID-19 hamster model. Cell. 2020;183(4):1058–1069.e19. doi: 10.1016/j.cell.2020.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Liu L, Wang P, Nair MS, Yu J, Rapp M, Wang Q, et al. Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike. Nature. 2020;584(7821):450–456. doi: 10.1038/s41586-020-2571-7. [DOI] [PubMed] [Google Scholar]
- 397.Cao Y, Su B, Guo X, Sun W, Deng Y, Bao L, et al. Potent neutralizing antibodies against SARS-CoV-2 identified by high-throughput single-cell sequencing of convalescent patients' B cells. Cell. 2020;182(1):73–84.e16. doi: 10.1016/j.cell.2020.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Zost SJ, Gilchuk P, Case JB, Binshtein E, Chen RE, Nkolola JP, et al. Potently neutralizing and protective human antibodies against SARS-CoV-2. Nature. 2020;584(7821):443–449. doi: 10.1038/s41586-020-2548-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Scheid JF, Barnes CO, Eraslan B, Hudak A, Keeffe JR, Cosimi LA, et al. B cell genomics behind cross-neutralization of SARS-CoV-2 variants and SARS-CoV. Cell. 2021;184(12):3205–3221.e24. doi: 10.1016/j.cell.2021.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Dumet C, Jullian Y, Musnier A, Rivière Ph, Poirier N, Watier H, et al. Exploring epitope and functional diversity of anti-SARS-CoV2 antibodies using AI-based methods bioRxiv [preprint]. 2020.12.23.424199; 10.1101/2020.12.23.424199.
- 401.Lv Z, Deng YQ, Ye Q, Cao L, Sun CY, Fan C, et al. Structural basis for neutralization of SARS-CoV-2 and SARS-CoV by a potent therapeutic antibody. Science. 2020;369(6510):1505–1509. doi: 10.1126/science.abc5881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Verkhivker GM, Di Paola L. Integrated biophysical modeling of the SARS-CoV-2 spike protein binding and allosteric interactions with antibodies. J Phys Chem B. 2021;125(18):4596–4619. doi: 10.1021/acs.jpcb.1c00395. [DOI] [PubMed] [Google Scholar]
- 403.Zhou D, Duyvesteyn HME, Chen CP, Huang CG, Chen TH, Shih SR, et al. Structural basis for the neutralization of SARS-CoV-2 by an antibody from a convalescent patient. Nat Struct Mol Biol. 2020;27(10):950–958. doi: 10.1038/s41594-020-0480-y. [DOI] [PubMed] [Google Scholar]
- 404.Sartor ITS, Varela FH, Meireles MR, Kern LB, Azevedo TR, Giannini GLT, et al. Y380Q novel mutation in receptor-binding domain of SARS-CoV-2 spike protein together with C379W interfere in the neutralizing antibodies interaction. Diagn Microbiol Infect Dis. 2022 doi: 10.1016/j.diagmicrobio.2022.115636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Zhou T, Wang L, Misasi J, Pegu A, Zhang Y, Harris DR, et al. Structural basis for potent antibody neutralization of SARS-CoV-2 variants including B.1.1.529. bioRxiv [Preprint] 2021.12.27.474307; 10.1101/2021.12.27.474307 [DOI] [PMC free article] [PubMed]
- 406.Cameroni E, Bowen JE, Rosen LE, Saliba C, Zepeda SK, Culap K, et al. Broadly neutralizing antibodies overcome SARS-CoV-2 Omicron antigenic shift. Nature. 2022;602(7898):664–670. doi: 10.1038/s41586-021-04386-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Cao Y, Yisimayi A, Jian F, Xiao T, Song W, Wang J, et al. Comprehensive epitope mapping of broad Sarbecovirus neutralizing antibodies. bioRxiv 2022.02.07.479349; 10.1101/2022.02.07.479349
- 408.Liu L, Iketani S, Guo Y, Chan JF, Wang M, Liu L, et al. Striking antibody evasion manifested by the Omicron variant of SARS-CoV-2. Nature. 2022;602(7898):676–681. doi: 10.1038/s41586-021-04388-0. [DOI] [PubMed] [Google Scholar]
- 409.VanBlargan LA, Adams LJ, Liu Z, Chen RE, Gilchuk P, Raju S, et al. A potently neutralizing SARS-CoV-2 antibody inhibits variants of concern by utilizing unique binding residues in a highly conserved epitope. Immunity. 2021 doi: 10.1016/j.immuni.2021.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.IGM Biosciences press release on Omicron neutralization. February 9 2022. https://www.biospace.com/article/releases/igm-biosciences-advances-novel-antibody-igm-6268-into-clinical-trials-for-the-treatment-and-prevention-of-covid-19-/. Accessed 28 Febr 2022.
- 411.Ryu DK, Kang B, Noh H, Woo SJ, Lee MH, Nuijten PM, et al. The in vitro and in vivo efficacy of CT-P59 against Gamma, Delta and its associated variants of SARS-CoV-2. Biochem Biophys Res Commun. 2021;578:91–96. doi: 10.1016/j.bbrc.2021.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Ryu DK, Song R, Kim M, Kim YI, Kim C, Kim JI, et al. Therapeutic effect of CT-P59 against SARS-CoV-2 South African variant. Biochem Biophys Res Commun. 2021;566:135–140. doi: 10.1016/j.bbrc.2021.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Sheward DJ, Pushparaj P, Das H, Kim C, Kim S, Hanke L, et al. Structural basis of Omicron neutralization by affinity-matured public antibodies. bioRxiv [preprint] 2022.01.03.474825; 10.1101/2022.01.03.474825.
- 414.Doggrell SA. Do we need bamlanivimab? Is etesevimab a key to treating Covid-19? Expert Opin Biol Ther. 2021;21(11):1359–1362. doi: 10.1080/14712598.2021.1985458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.FDA Statement on Omicron. https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-limits-use-certain-monoclonal-antibodies-treat-covid-19-due-omicron. Accessed 25 Feb 2022.
- 416.Somersan-Karakaya S, Mylonakis E, Menon VP, Wells JC, Ali S, Sivapalasingam S, et al. COVID-19 Phase 2/3 Hospitalized Trial Team. Casirivimab and imdevimab for the treatment of hospitalized patients with COVID-19. medRxiv [preprint] 21.11.05.21265656; 10.1101/2021.11.05.21265656
- 417.Zalevsky J, Chamberlain AK, Horton HM, Karki S, Leung IW, Sproule TJ, et al. Enhanced antibody half-life improves in vivo activity. Nat Biotechnol. 2010;28(2):157–159. doi: 10.1038/nbt.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Dunleavy K. FDA limits use of Sotrovimab in some areas due to Omicron BA.2. https://www.fiercepharma.com/pharma/fda-will-limit-use-glaxosmithkline-and-vir-s-covid-19-antibody-treatment-some-geographic. Accessed 25 Feb 2022.
- 419.Bournazos S, Corti D, Virgin HW, Ravetch JV. Fc-optimized antibodies elicit CD8 immunity to viral respiratory infection. Nature. 2020;588(7838):485–490. doi: 10.1038/s41586-020-2838-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Oganesyan V, Gao C, Shirinian L, Wu H, Dall'Acqua WF. Structural characterization of a human Fc fragment engineered for lack of effector functions. Acta Crystallogr D Biol Crystallogr. 2008;64(Pt 6):700–704. doi: 10.1107/S0907444908007877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Dall'Acqua WF, Kiener PA, Wu H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn) J Biol Chem. 2006;281(33):23514–23524. doi: 10.1074/jbc.M604292200. [DOI] [PubMed] [Google Scholar]
- 422.Loo YM, McTamney PM, Arends RH, Abram ME, Aksyuk AA, Diallo S, et al. The SARS-CoV-2 monoclonal antibody combination, AZD7442, is protective in non-human primates and has an extended half-life in humans. Sci Transl Med. 2022 doi: 10.1126/scitranslmed.abl8124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Wang P, Nair MS, Liu L, Iketani S, Luo Y, Guo Y, et al. Antibody resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7. Nature. 2021;593(7857):130–135. doi: 10.1038/s41586-021-03398-2. [DOI] [PubMed] [Google Scholar]
- 424.FDA EUA for AZD7442. https://www.fda.gov/media/154704/download. Accessed 26 Feb 2022.
- 425.Yuan M, Huang D, Lee CD, Wu NC, Jackson AM, Zhu X, et al. Structural and functional ramifications of antigenic drift in recent SARS-CoV-2 variants. Science. 2021;373(6556):818–823. doi: 10.1126/science.abh1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Zhou B, Zhou R, Chan JF-W, Luo M, Peng Q, Yuan S, et al. An elite broadly neutralizing antibody protects SARS-CoV-2 Omicron variant challenge. bioRxiv [Preprint] 2022.01.05.475037; 10.1101/2022.01.05.475037.
- 427.Li C, Zhan W, Yang Z, Tu C, Zhu Y, Song W, et al. Broad neutralization of SARS-CoV-2 variants by an inhalable bispecific single-domain antibody. bioRxiv [prepint] 2021.12.30.474535; 10.1101/2021.12.30.474535 [DOI] [PMC free article] [PubMed]
- 428.Duan X, Shi R, Liu P, Huang Q, Wang F, Chen X, et al. A non-ACE2-blocking neutralizing antibody against Omicron-included SARS-CoV-2 variants. Signal Transduct Target Ther. 2022;7(1):23. doi: 10.1038/s41392-022-00879-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Chen Z, Zhang P, Matsuoka Y, Tsybovsky Y, West K, Santos C, et al. Extremely potent monoclonal antibodies neutralize Omicron and other SARS-CoV-2 variants. medRxiv [preprint]. 2022 doi: 10.1101/2022.01.12.22269023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Imbrechts M, Kerstens W, Rasulova M, Vercruysse T, Maes W, Ampofo L, et al. Anti-SARS-CoV-2 human antibodies retaining neutralizing activity against SARS-CoV-2 B.1.1.529 (omicron). bioRxiv [preprint] 2021.12.21.473706; 10.1101/2021.12.21.473706.
- 431.Thomson EC, Rosen LE, Shepherd JG, Spreafico R, da Silva FA, Wojcechowskyj JA, et al. Circulating SARS-CoV-2 spike N439K variants maintain fitness while evading antibody-mediated immunity. Cell. 2021;184(5):1171–1187.e20. doi: 10.1016/j.cell.2021.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Li T, Xue W, Zheng Q, Song S, Yang C, Xiong H, et al. Cross-neutralizing antibodies bind a SARS-CoV-2 cryptic site and resist circulating variants. Nat Commun. 2021;12(1):5652. doi: 10.1038/s41467-021-25997-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Li M, Lou F, Fan H. SARS-CoV-2 variant Omicron: currently the most complete "escapee" from neutralization by antibodies and vaccines. Signal Transduct Target Ther. 2022;7(1):28. doi: 10.1038/s41392-022-00880-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Immunome antibody cocktail effective against Omicron. https://www.businesswire.com/news/home/20220208005608/en/Immunome%E2%80%99s-Antibody-Cocktail-Effective-Against-SARS-CoV-2-Omicron-Variant-in-In-Vitro-Live-Virus-Testing. Accessed 27 Feb 2022.
- 435.Niu L, Wittrock KN, Clabaugh GC, Srivastava V, Cho MW. A structural landscape of neutralizing antibodies against SARS-CoV-2 receptor binding domain. Front Immunol. 2021;12:647934. doi: 10.3389/fimmu.2021.647934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Deshpande A, Harris BD, Martinez-Sobrido L, Kobie JJ, Walter MR. Epitope classification and RBD binding properties of neutralizing antibodies against SARS-CoV-2 variants of concern. Front Immunol. 2021;12:691715. doi: 10.3389/fimmu.2021.691715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Wec AZ, Wrapp D, Herbert AS, Maurer DP, Haslwanter D, Sakharkar M, et al. Broad neutralization of SARS-related viruses by human monoclonal antibodies. Science. 2020;369(6504):731–736. doi: 10.1126/science.abc7424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Fagiani F, Catanzaro M, Lanni C. Molecular features of IGHV3-53-encoded antibodies elicited by SARS-CoV-2. Signal Transduct Target Ther. 2020;5(1):170. doi: 10.1038/s41392-020-00287-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Dejnirattisai W, Zhou D, Supasa P, Liu C, Mentzer AJ, Ginn HM, et al. Antibody evasion by the P.1 strain of SARS-CoV-2. Cell. 2021;184(11):2939–2954.e9. doi: 10.1016/j.cell.2021.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Magleby R, Westblade LF, Trzebucki A, Simon MS, Rajan M, Park J, et al. Impact of SARS-CoV-2 viral load on risk of intubation and mortality among hospitalized patients with coronavirus disease 2019. Clin Infect Dis. 2020 doi: 10.1093/cid/ciaa851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Fenwick C, Turelli P, Perez L, Pellaton C, Esteves-Leuenberger L, Farina A, et al. A highly potent antibody effective against SARS-CoV-2 variants of concern. Cell Rep. 2021;21:109814. doi: 10.1016/j.celrep.2021.109814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Kramer KJ, Johnson NV, Shiakolas AR, Suryadevara N, Periasamy S, Raju N, et al. Potent neutralization of SARS-CoV-2 variants of concern by an antibody with an uncommon genetic signature and structural mode of spike recognition. Cell Rep. 2021;37(1):109784. doi: 10.1016/j.celrep.2021.109784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Chi X, Yan R, Zhang J, Zhang G, Zhang Y, Hao M, et al. A neutralizing human antibody binds to the N-terminal domain of the spike protein of SARS-CoV-2. Science. 2020;369(6504):650–655. doi: 10.1126/science.abc6952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Suryadevara N, Shrihari S, Gilchuk P, VanBlargan LA, Binshtein E, Zost SJ, et al. Neutralizing and protective human monoclonal antibodies recognizing the N-terminal domain of the SARS-CoV-2 spike protein. Cell. 2021;184(9):2316–2331.e15. doi: 10.1016/j.cell.2021.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Liu Y, Soh WT, Kishikawa JI, Hirose M, Nakayama EE, Li S, et al. An infectivity-enhancing site on the SARS-CoV-2 spike protein targeted by antibodies. Cell. 2021;184(13):3452–3466.e18. doi: 10.1016/j.cell.2021.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Wölfel R, Corman VM, Guggemos W, Seilmaier M, Zange S, Müller MA, et al. Virological assessment of hospitalized patients with COVID-2019. Nature. 2020;581(7809):465–469. doi: 10.1038/s41586-020-2196-x. [DOI] [PubMed] [Google Scholar]
- 447.Zankharia US, Kudchodkar S, Khoshnejad M, Perales-Puchalt A, Choi H, Ho M, et al. Neutralization of hepatitis B virus by a novel DNA-encoded monoclonal antibody. Hum Vaccin Immunother. 2020;16(9):2156–2164. doi: 10.1080/21645515.2020.1763686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Weisblum Y, Schmidt F, Zhang F, DaSilva J, Poston D, Lorenzi JC, et al. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. Elife. 2020;9:e61312. doi: 10.7554/eLife.61312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Cerutti G, Guo Y, Wang P, Nair MS, Huang Y, Yu J, et al. Neutralizing antibody 5–7 defines a distinct site of vulnerability in SARS-CoV-2 spike N-terminal domain. bioRxiv [preprint]. 2021 doi: 10.1101/2021.06.29.450397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Patel A, Bah MA, Weiner DB. In vivo delivery of nucleic acid-encoded monoclonal antibodies. BioDrugs. 2020;34(3):273–293. doi: 10.1007/s40259-020-00412-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451.Jaimes JA, André NM, Chappie JS, Millet JK, Whittaker GR. Phylogenetic analysis and structural modeling of SARS-CoV-2 spike protein reveals an evolutionary distinct and proteolytically sensitive activation loop. J Mol Biol. 2020;432(10):3309–3325. doi: 10.1016/j.jmb.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Cai Y, Zhang J, Xiao T, Peng H, Sterling SM, Walsh RM, Jr, et al. Distinct conformational states of SARS-CoV-2 spike protein. Science. 2020;369(6511):1586–1592. doi: 10.1126/science.abd4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Pallesen J, Wang N, Corbett KS, Wrapp D, Kirchdoerfer RN, Turner HL, et al. Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen. Proc Natl Acad Sci U S A. 2017;114(35):E7348–E7357. doi: 10.1073/pnas.1707304114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Huang Y, Nguyen AW, Hsieh C-L, Silva R, Olaluwoye OS, Wilen RE, et al. Identification of a conserved neutralizing epitope present on spike proteins from all highly pathogenic coronaviruses. bioRxiv [preprint] 2021.01.31.428824; 10.1101/2021.01.31.428824. [DOI] [PMC free article] [PubMed]
- 455.Hong J, Kwon HJ, Cachau R, Chen CZ, Butay KJ, Duan Z, et al. Camel nanobodies broadly neutralize SARS-CoV-2 variants. bioRxiv [Preprint]. 2021 doi: 10.1101/2021.10.27.465996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Song G, He WT, Callaghan S, Anzanello F, Huang D, Ricketts J, et al. Cross-reactive serum and memory B-cell responses to spike protein in SARS-CoV-2 and endemic coronavirus infection. Nat Commun. 2021;12(1):2938. doi: 10.1038/s41467-021-23074-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Zhou P, Yuan M, Song G, Beutler N, Shaabani N, Huang D, et al. A protective broadly cross-reactive human antibody defines a conserved site of vulnerability on beta-coronavirus spikes. bioRxiv [preprint]. 2021 doi: 10.1101/2021.03.30.437769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Wang C, van Haperen R, Gutiérrez-Álvarez J, Li W, Okba NMA, Albulescu I, et al. A conserved immunogenic and vulnerable site on the coronavirus spike protein delineated by cross-reactive monoclonal antibodies. Nat Commun. 2021;12(1):1715. doi: 10.1038/s41467-021-21968-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Jiang S, Zhang X, Yang Y, Hotez PJ, Du L. Neutralizing antibodies for the treatment of COVID-19. Nat Biomed Eng. 2020;4(12):1134–1139. doi: 10.1038/s41551-020-00660-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Cantuti-Castelvetri L, Ojha R, Pedro LD, Djannatian M, Franz J, Kuivanen S, et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science. 2020;370(6518):856–860. doi: 10.1126/science.abd2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Hikmet F, Méar L, Edvinsson Å, Micke P, Uhlén M, Lindskog C. The protein expression profile of ACE2 in human tissues. Mol Syst Biol. 2020;16(7):e9610. doi: 10.15252/msb.20209610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Ganier C, Du-Harpur X, Harun N, Wan B, Arthurs C, Luscombe NM, et al. CD147 (BSG) but not ACE2 expression is detectable in vascular endothelial cells within single cell RNA sequencing datasets derived from multiple tissues in healthy individuals. bioRxiv [preprint] 2020.05.29.123513; 10.1101/2020.05.29.123513.
- 463.Wang K, Chen W, Zhang Z, Deng Y, Lian JQ, Du P, et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct Target Ther. 2020;5(1):283. doi: 10.1038/s41392-020-00426-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Fenizia C, Galbiati S, Vanetti C, Vago R, Clerici M, Tacchetti C, et al. SARS-CoV-2 entry: at the crossroads of CD147 and ACE2. Cells. 2021;10(6):1434. doi: 10.3390/cells10061434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Daly JL, Simonetti B, Klein K, Chen KE, Williamson MK, Antón-Plágaro C, et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science. 2020;370(6518):861–865. doi: 10.1126/science.abd3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Zhou B, Zhou R, Chan JFW, Zeng J, Zhang Q, Yuan D, et al. SARS-CoV-2 hijacks neutralizing dimeric IgA for enhanced nasal infection and injury. bioRxiv [preprint] 2021.10.05.463282; 10.1101/2021.10.05.463282.
- 467.Amraei R, Yin W, Napoleon MA, Suder EL, Berrigan J, Zhao Q, et al. CD209L/L-SIGN and CD209/DC-SIGN act as receptors for SARS-CoV-2. ACS Cent Sci. 2021;7(7):1156–1165. doi: 10.1021/acscentsci.0c01537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Clausen TM, Sandoval DR, Spliid CB, Pihl J, Perrett HR, Painter CD, et al. SARS-CoV-2 infection depends on cellular heparan sulfate and ACE2. Cell. 2020;183(4):1043–1057.e15. doi: 10.1016/j.cell.2020.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Gu Y, Cao J, Zhang X, Gao H, Wang Y, Wang J, et al. Receptome profiling identifies KREMEN1 and ASGR1 as alternative functional receptors of SARS-CoV-2. Cell Res. 2022;32(1):24–37. doi: 10.1038/s41422-021-00595-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Behl T, Kaur I, Aleya L, Sehgal A, Singh S, Sharma N, et al. CD147-spike protein interaction in COVID-19: get the ball rolling with a novel receptor and therapeutic target. Sci Total Environ. 2022;808:152072. doi: 10.1016/j.scitotenv.2021.152072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Shilts J, Crozier TWM, Greenwood EJD, Lehner PJ, Wright GJ. No evidence for basigin/CD147 as a direct SARS-CoV-2 spike binding receptor. Sci Rep. 2021;11(1):413. doi: 10.1038/s41598-020-80464-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Geng J, Chen L, Yuan Y, Wang K, Wang Y, Qin C, et al. CD147 antibody specifically and effectively inhibits infection and cytokine storm of SARS-CoV-2 and its variants delta, alpha, beta, and gamma. Signal Transduct Target Ther. 2021;6(1):347. doi: 10.1038/s41392-021-00760-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.Zhang K, Zhao Y, Zhang Z, Zhang M, Wu X, Bian H, et al. Nonclinical safety, tolerance and pharmacodynamics evaluation for meplazumab treating chloroquine-resistant Plasmodium falciparum. Acta Pharm Sin B. 2020;10(9):1680–1693. doi: 10.1016/j.apsb.2020.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Bian H, Zheng Z-H, Wei D, Zhang Z, Kang W-Z, Hao C-Q, et al. Meplazumab treats COVID-19 pneumonia: an open-labelled, concurrent controlled add-on clinical trial. medRxiv [preprint] 2020.03.21.20040691; 10.1101/2020.03.21.20040691.
- 475.Jeffers SA, Tusell SM, Gillim-Ross L, Hemmila EM, Achenbach JE, Babcock GJ, et al. CD209L (L-SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proc Natl Acad Sci U S A. 2004;101(44):15748–15753. doi: 10.1073/pnas.0403812101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Marzi A, Gramberg T, Simmons G, Möller P, Rennekamp AJ, Krumbiegel M, et al. DC-SIGN and DC-SIGNR interact with the glycoprotein of Marburg virus and the S protein of severe acute respiratory syndrome coronavirus. J Virol. 2004;78(21):12090–12095. doi: 10.1128/JVI.78.21.12090-12095.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Yang ZY, Huang Y, Ganesh L, Leung K, Kong WP, Schwartz O, et al. pH-dependent entry of severe acute respiratory syndrome coronavirus is mediated by the spike glycoprotein and enhanced by dendritic cell transfer through DC-SIGN. J Virol. 2004;78(11):5642–5650. doi: 10.1128/JVI.78.11.5642-5650.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478.Cerutti G, Rapp M, Guo Y, Bahna F, Bimela J, Reddem ER, et al. Structural basis for accommodation of emerging B.1.351 and B.1.1.7 variants by two potent SARS-CoV-2 neutralizing antibodies. Structure. 2021;29(7):655–663.e4. doi: 10.1016/j.str.2021.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479.Liu Z, Xu W, Chen Z, Fu W, Zhan W, Gao Y, et al. An ultrapotent pan-β-coronavirus lineage B (β-CoV-B) neutralizing antibody locks the receptor-binding domain in closed conformation by targeting its conserved epitope. Protein Cell. 2021 doi: 10.1007/s13238-021-00871-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Dejnirattisai W, Zhou D, Ginn HM, Duyvesteyn HME, Supasa P, Case JB, et al. The antigenic anatomy of SARS-CoV-2 receptor binding domain. Cell. 2021;184(8):2183–2200.e22. doi: 10.1016/j.cell.2021.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.Yang Z, Wang Y, Jin Y, Zhu Y, Wu Y, Li C, et al. A non-ACE2 competing human single-domain antibody confers broad neutralization against SARS-CoV-2 and circulating variants. Signal Transduct Target Ther. 2021;6(1):378. doi: 10.1038/s41392-021-00810-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482.Tan YH, Liu M, Nolting B, Go JG, Gervay-Hague J, Liu GY. A nanoengineering approach for investigation and regulation of protein immobilization. ACS Nano. 2008;2(11):2374–2384. doi: 10.1021/nn800508f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Roux KH, Strelets L, Michaelsen TE. Flexibility of human IgG subclasses. J Immunol. 1997;159(7):3372–3382. [PubMed] [Google Scholar]
- 484.Jendroszek A, Kjaergaard M. Nanoscale spatial dependence of avidity in an IgG1 antibody. Sci Rep. 2021;11(1):12663. doi: 10.1038/s41598-021-92280-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.Lin Y, Yan X, Cao W, Wang C, Feng J, Duan J, et al. Probing the structure of the SARS coronavirus using scanning electron microscopy. Antivir Ther. 2004;9(2):287–289. doi: 10.1177/135965350400900204. [DOI] [PubMed] [Google Scholar]
- 486.Neuman BW, Adair BD, Yoshioka C, Quispe JD, Orca G, Kuhn P, et al. Supramolecular architecture of severe acute respiratory syndrome coronavirus revealed by electron cryomicroscopy. J Virol. 2006;80(16):7918–7928. doi: 10.1128/JVI.00645-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Li W, Schäfer A, Kulkarni SS, Liu X, Martinez DR, Chen C, et al. High potency of a bivalent human VH domain in SARS-CoV-2 animal mModels. Cell. 2020;183(2):429–441.e16. doi: 10.1016/j.cell.2020.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Hou YJ, Okuda K, Edwards CE, Martinez DR, Asakura T, Dinnon KH, 3rd, et al. SARS-CoV-2 reverse genetics reveals a variable infection gradient in the respiratory tract. Cell. 2020;182(2):429–446.e14. doi: 10.1016/j.cell.2020.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Sungnak W, Huang N, Bécavin C, Berg M, Queen R, Litvinukova M, et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat Med. 2020;26(5):681–687. doi: 10.1038/s41591-020-0868-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Muus C, Luecken MD, Eraslan G, Sikkema L, Waghray A, Heimberg G, et al. Single-cell meta-analysis of SARS-CoV-2 entry genes across tissues and demographics. Nat Med. 2021;27(3):546–559. doi: 10.1038/s41591-020-01227-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491.Klingenstein M, Klingenstein S, Neckel PH, Mack AF, Wagner AP, Kleger A, et al. Evidence of SARS-CoV2 entry protein ACE2 in the human nose and olfactory bulb. Cells Tissues Organs. 2020;209(4–6):155–164. doi: 10.1159/000513040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.Shlomai A, Ben-Zvi H, Glusman Bendersky A, Shafran N, Goldberg E, Sklan EH. Nasopharyngeal viral load predicts hypoxemia and disease outcome in admitted COVID-19 patients. Crit Care. 2020;24(1):539. doi: 10.1186/s13054-020-03244-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493.Li B, Deng A, Li K, Hu Y, Li Z, Xiong Q, et al. Viral infection and transmission in a large, well-traced outbreak caused by the SARS-CoV-2 Delta variant. Nat Commun. 2022;13(1):460. doi: 10.1038/s41467-022-28089-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494.Huang N, Pérez P, Kato T, Mikami Y, Okuda K, Gilmore RC, et al. SARS-CoV-2 infection of the oral cavity and saliva. Nat Med. 2021;27(5):892–903. doi: 10.1038/s41591-021-01296-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 495.Guilleminault L, Azzopardi N, Arnoult C, Sobilo J, Hervé V, Montharu J, et al. Fate of inhaled monoclonal antibodies after the deposition of aerosolized particles in the respiratory system. J Control Release. 2014;196:344–354. doi: 10.1016/j.jconrel.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 496.Adagio press release. May 5 2021. https://adagiotx.com/wp-content/uploads/2021/05/Adagio-EVADE-Ph1-HV-Data-PR-210505-FINAL.pdf. Accessed 1 Nov 2021.
- 497.Evusheld™ Fact Sheet for Healthcare Providers. https://www.fda.gov/media/154701/download. Accessed 28 Feb 2022.
- 498.Emergency Use Authorization 091. Sept 9 2021. https://www.fda.gov/media/145610/download. Accessed 1 Nov 2021.
- 499.Celltrion News Release. https://www.biospace.com/article/releases/celltrion-submits-investigational-new-drug-ind-application-to-initiate-a-global-phase-iii-clinical-trial-evaluating-an-inhaled-covid-19-antibody-cocktail-therapy/. Accessed 28 Feb 2022.
- 500.Lee L, Samardzic K, Wallach M, Frumkin LR, Mochly-Rosen D. Immunoglobulin Y for potential diagnostic and therapeutic applications in infectious diseases. Front Immunol. 2021;12:696003. doi: 10.3389/fimmu.2021.696003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 501.Pilicheva B, Boyuklieva R. Can the nasal cavity help tackle COVID-19? Pharmaceutics. 2021;13(10):1612. doi: 10.3390/pharmaceutics13101612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Zhang H, Yang Z, Xiang J, Cui Z, Liu J, Liu C. Intranasal administration of SARS-CoV-2 neutralizing human antibody prevents infection in mice. bioRxiv [preprint] 2020.12.08.416677; 10.1101/2020.12.08.416677.
- 503.Wu X, Cheng L, Fu M, Huang B, Zhu L, Xu S, et al. A potent bispecific nanobody protects hACE2 mice against SARS-CoV-2 infection via intranasal administration. Cell Rep. 2021;37(3):109869. doi: 10.1016/j.celrep.2021.109869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Emig CJ, Mena MA, Henry SJ, Vitug A, Ventura CJ, Fox D, et al. AUG-3387, a human-derived monoclonal antibody neutralizes SARS-CoV-2 variants and reduces viral load from therapeutic treatment of hamsters in vivo. bioRxiv [preprint] 2021.10.12.464150; 10.1101/2021.10.12.464150.
- 505.Aridis news release. July 12 2021. https://investors.aridispharma.com/2021-07-12-Aridis-Pharmaceuticals-COVID-mAb-AR-712-Neutralizes-SARS-CoV-2-Delta-Variant. Accessed 1 Nov 2021.
- 506.University of Pennsylvania news release. Nov 30 2020. https://www.pennmedicine.org/news/news-releases/2020/november/penn-medicine-collaborates-with-regeneron-to-investigate-delivery-of-covid-antibody. Accessed 1 Nov 2021.
- 507.Sims JJ, Greig JA, Michalson KT, Lian S, Martino RA, Meggersee R, et al. Intranasal gene therapy to prevent infection by SARS-CoV-2 variants. PLoS Pathog. 2021;17(7):e1009544. doi: 10.1371/journal.ppat.1009544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508.Liang W, Pan HW, Vllasaliu D, Lam JKW. Pulmonary delivery of biological drugs. Pharmaceutics. 2020;12(11):1025. doi: 10.3390/pharmaceutics12111025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509.Zhang X, Tan Y, Ling Y, Lu G, Liu F, Yi Z, et al. Viral and host factors related to the clinical outcome of COVID-19. Nature. 2020;583(7816):437–440. doi: 10.1038/s41586-020-2355-0. [DOI] [PubMed] [Google Scholar]
- 510.Fodil S, Annane D. Complement inhibition and COVID-19: the story so far. Immunotargets Ther. 2021;10:273–284. doi: 10.2147/ITT.S284830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 511.Awasthi S, Wagner T, Venkatakrishnan AJ, Puranik A, Hurchik M, Agarwal V, et al. Plasma IL-6 levels following corticosteroid therapy as an indicator of ICU length of stay in critically ill COVID-19 patients. Cell Death Discov. 2021;7(1):55. doi: 10.1038/s41420-021-00429-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 512.Cabaro S, D'Esposito V, Di Matola T, Sale S, Cennamo M, Terracciano D, et al. Cytokine signature and COVID-19 prediction models in the two waves of pandemics. Sci Rep. 2021;11(1):20793. doi: 10.1038/s41598-021-00190-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 513.Gu R, Mao T, Lu Q, Tianjiao SuT, Wang J. Myeloid dysregulation and therapeutic intervention in COVID-19. Semin Immunol. 2021 doi: 10.1016/j.smim.2021.101524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 514.Oliynyk O, Barg W, Slifirczyk A, Oliynyk Y, Gurianov V, Rorat M. Efficacy of tocilizumab therapy in different subtypes of COVID-19 cytokine storm syndrome. Viruses. 2021;13(6):1067. doi: 10.3390/v13061067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515.Giamarellos-Bourboulis EJ, Netea MG, Rovina N, Akinosoglou K, Antoniadou A, Antonakos N, et al. Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe. 2020;27(6):992–1000.e3. doi: 10.1016/j.chom.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516.Tang D, Comish P, Kang R. The hallmarks of COVID-19 disease. PLoS Pathog. 2020;16(5):e1008536. doi: 10.1371/journal.ppat.1008536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517.Tzotzos SJ, Fischer B, Fischer H, Zeitlinger M. Incidence of ARDS and outcomes in hospitalized patients with COVID-19: a global literature survey. Crit Care. 2020;24(1):516. doi: 10.1186/s13054-020-03240-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518.Clinical Spectrum of SARS-CoV-2 infection. Updated Oct 19 2021. https://www.covid19treatmentguidelines.nih.gov/overview/clinical-spectrum/. Accessed 1 Nov 2021.
- 519.Immunomodulators under evaluation for the treatment of COVID-19. Updated Oct 19 2021. https://www.covid19treatmentguidelines.nih.gov/therapies/immunomodulators/summary-recommendations/. Accessed 30 Nov 2021.
- 520.Atal S, Fatima Z, Balakrishnan S. Approval of itolizumab for COVID-19: a premature decision or need of the hour? BioDrugs. 2020;34(6):705–711. doi: 10.1007/s40259-020-00448-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521.Philippidis A. FDA rejects EUA for Humanigen’s lenzilumab. Sept 13 2021. https://www.genengnews.com/news/fda-rejects-eua-for-humanigens-lenzilumab/. Accessed 1 Nov 2021.
- 522.Temesgen Z, Burger CD, Baker J, Polk C, Libertin C, Kelley C, et al. Lenzilizumab efficacy and safety in newly hospitalized COVID-19 subjects: Results from the live-air Phase 3 randomized double-blind placebo-controlled trial. medRxiv [preprint]. 2021 doi: 10.1101/2021.05.01.21256470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 523.Wei Q, Lin H, Wei RG, Chen N, He F, Zou DH, et al. Tocilizumab treatment for COVID-19 patients: a systematic review and meta-analysis. Infect Dis Poverty. 2021;10(1):71. doi: 10.1186/s40249-021-00857-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524.Khan FA, Stewart I, Fabbri L, Moss S, Robinson K, Smyth AR, et al. Systematic review and meta-analysis of anakinra, sarilumab, siltuximab and tocilizumab for COVID-19. Thorax. 2021;76(9):907–919. doi: 10.1136/thoraxjnl-2020-215266. [DOI] [PubMed] [Google Scholar]
- 525.Mariette X, Hermine O, Tharaux PL, Resche-Rigon M, Steg PG, Porcher R, et al. Effectiveness of tocilizumab in patients hospitalized with COVID-19: a follow-up of the CORIMUNO-TOCI-1 randomized clinical trial. JAMA Intern Med. 2021;181(9):1241–1243. doi: 10.1001/jamainternmed.2021.2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Burlacu R, London J, Fleury A, Sené T, Diallo A, Meyssonnier V, et al. No evidence of tocilizumab treatment efficacy for severe to critical SARS-CoV2 infected patients: results from a retrospective controlled multicenter study. Medicine (Baltimore) 2021;100(21):e26023. doi: 10.1097/MD.0000000000026023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527.Flisiak R, Jaroszewicz J, Rogalska M, Łapiński T, Berkan-Kawińska A, Bolewska B, et al. Tocilizumab improves the prognosis of COVID-19 in patients with high IL-6. J Clin Med. 2021;10(8):1583. doi: 10.3390/jcm10081583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 528.Milic J, Banchelli F, Meschiari M, Franceschini E, Ciusa G, Gozzi L, et al. The impact of tocilizumab on respiratory support states transition and clinical outcomes in COVID-19 patients A Markov model multi-state study. PLoS ONE. 2021;16(8):e0251378. doi: 10.1371/journal.pone.0251378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 529.Sinha P, Linas BP. Combination therapy with tocilizumab and dexamethasone cost-effectively reduces Coronavirus disease 2019 mortality. Clin Infect Dis. 2021 doi: 10.1093/cid/ciab409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 530.Kaya S, Kavak S. Efficacy of tocilizumab in COVID-19: single-center experience. Biomed Res Int. 2021;2021:1934685. doi: 10.1155/2021/1934685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531.Durán-Méndez A, Aguilar-Arroyo AD, Vivanco-Gómez E, Nieto-Ortega E, Pérez-Ortega D, Jiménez-Pérez C, et al. Tocilizumab reduces COVID-19 mortality and pathology in a dose and timing-dependent fashion: a multi-centric study. Sci Rep. 2021;11(1):19728. doi: 10.1038/s41598-021-99291-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532.Genentech press release. Update on Actemra® (tocilizumab) supply in the U.S. September 3 2021. https://www.gene.com/media/statements/ps_081621. Accessed 1 Nov 2021.
- 533.Lotfi N, Thome R, Rezaei N, Zhang GX, Rezaei A, Rostami A, et al. Roles of GM-CSF in the pathogenesis of autoimmune diseases: an update. Front Immunol. 2019;10:1265. doi: 10.3389/fimmu.2019.01265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534.Lee JS, Koh JY, Yi K, Kim YI, Park SJ, Kim EH, et al. Single-cell transcriptome of bronchoalveolar lavage fluid reveals sequential change of macrophages during SARS-CoV-2 infection in ferrets. Nat Commun. 2021;12(1):4567. doi: 10.1038/s41467-021-24807-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.Song N, Li P, Jiang Y, Sun H, Cui J, Zhao G, et al. C5a receptor1 inhibition alleviates influenza virus-induced acute lung injury. Int Immunopharmacol. 2018;59:12–20. doi: 10.1016/j.intimp.2018.03.029. [DOI] [PubMed] [Google Scholar]
- 536.Mastroianni A, Greco S, Chidichimo L, Urso F, Greco F, Mauro MV, et al. Early use of canakinumab to prevent mechanical ventilation in select COVID-19 patients: A retrospective, observational analysis. Int J Immunopathol Pharmacol. 2021;35:20587384211059675. doi: 10.1177/20587384211059675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 537.Centers for Medicare & Medicaid Services. Monoclonal Antibody COVID-19 Infusion. https://www.cms.gov/medicare/COVID-19/monoclonal-antibody-COVID-19-infusion. Accessed 1 Nov 2021.
- 538.US Government purchase of bebtelovimab. https://news.bloomberglaw.com/health-law-and-business/lilly-to-supply-u-s-up-to-600k-bebtelovimab-doses-for-720m. Accessed 28 Feb 2022.
- 539.Mahal H, Branton H, Farid SS. End-to-end continuous bioprocessing: Impact on facility design, cost of goods, and cost of development for monoclonal antibodies. Biotechnol Bioeng. 2021;118(9):3468–3485. doi: 10.1002/bit.27774. [DOI] [PubMed] [Google Scholar]
- 540.Wynia MK, Beaty LE, Bennett TD, Carlson NE, Davis CB, Kwan BM, et al. Real world evidence of neutralizing monoclonal antibodies for preventing hospitalization and mortality in COVID-19 outpatients. medRxiv [preprint]. 2022 doi: 10.1101/2022.01.09.22268963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541.Baig AM. Deleterious outcomes in long-hauler COVID-19: The effects of SARS-CoV-2 on the CNS in chronic COVID syndrome. ACS Chem Neurosci. 2020;11(24):4017–4020. doi: 10.1021/acschemneuro.0c00725. [DOI] [PubMed] [Google Scholar]
- 542.Alunno A, Najm A, Machado PM, Bertheussen H, Burmester GR, Carubbi F, et al. Update of the EULAR points to consider on the use of immunomodulatory therapies in COVID-19. Ann Rheum Dis. 2021 doi: 10.1136/annrheumdis-2021-221366. [DOI] [PubMed] [Google Scholar]
- 543.Reardon S. Do monoclonal antibodies help COVID patients? Scientific American, Sept 27 2021. https://www.scientificamerican.com/article/do-monoclonal-antibodies-help-covid-patients/. Accessed 1 Nov 2021.
- 544.Van Lent J, Breukers J, Ven K, Ampofo L, Horta S, Pollet F, Imbrechts M, et al. Miniaturized single-cell technologies for monoclonal antibody discovery. Lab Chip. 2021;21(19):3627–3654. doi: 10.1039/d1lc00243k. [DOI] [PubMed] [Google Scholar]
- 545.Rajan S, Dall'Acqua WF. Emerging strategies for therapeutic antibody discovery from human B cells. Adv Exp Med Biol. 2020;1255:221–230. doi: 10.1007/978-981-15-4494-1_18. [DOI] [PubMed] [Google Scholar]
- 546.Gaa R, Menang-Ndi E, Pratapa S, Nguyen C, Kumar S, Doerner A. Versatile and rapid microfluidics-assisted antibody discovery. MAbs. 2021;13(1):1978130. doi: 10.1080/19420862.2021.1978130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 547.Corti D, Purcell LA, Snell G, Veesler D. Tackling COVID-19 with neutralizing monoclonal antibodies. Cell. 2021;184(12):3086–3108. doi: 10.1016/j.cell.2021.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 548.Valdez-Cruz NA, García-Hernández E, Espitia C, Cobos-Marín L, Altamirano C, Bando-Campos CG, et al. Integrative overview of antibodies against SARS-CoV-2 and their possible applications in COVID-19 prophylaxis and treatment. Microb Cell Fact. 2021;20(1):88. doi: 10.1186/s12934-021-01576-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549.Renn A, Fu Y, Hu X, Hall MD, Simeonov A. Fruitful neutralizing antibody pipeline brings hope to defeat SARS-Cov-2. Trends Pharmacol Sci. 2020;41(11):815–829. doi: 10.1016/j.tips.2020.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 550.Xiaojie S, Yu L, Lei Y, Guang Y, Min Q. Neutralizing antibodies targeting SARS-CoV-2 spike protein. Stem Cell Res. 2020;50:102125. doi: 10.1016/j.scr.2020.102125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 551.Sun Y, Ho M. Emerging antibody-based therapeutics against SARS-CoV-2 during the global pandemic. Antib Ther. 2020;3(4):246–256. doi: 10.1093/abt/tbaa025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 552.Yin R, Guest JD, Taherzadeh G, Gowthaman R, Mittra I, Quackenbush J, et al. Structural and energetic profiling of SARS-CoV-2 receptor binding domain antibody recognition and the impact of circulating variants. PLoS Comput Biol. 2021;17(9):e1009380. doi: 10.1371/journal.pcbi.1009380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 553.Antibody Society COVID-19 Biologics. https://www.antibodysociety.org/covid-19/. Accessed 23 Feb 2022.
- 554.Stanford SARS-CoV-2 data site. https://covdb.stanford.edu/page/susceptibility-data/. Accessed 24 Feb 2022.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.