

Summary
A complete understanding of synaptic-vesicle recycling requires the use of multiple microscopy methods to obtain complementary information. However, many currently available probes are limited to a specific microscopy modality, which necessitates the use of multiple probes and labeling paradigms. Given the complexity of vesicle populations and recycling pathways, having new single-vesicle probes that could be used for multiple microscopy techniques would complement existing sets of tools for studying vesicle function. Here, we present a probe based on the membrane-binding C2 domain of cytosolic phospholipase A2 (cPLA2) that fulfills this need. By conjugating the C2 domain with different detectable tags, we demonstrate that a single, modular probe can allow synaptic vesicles to be imaged at multiple levels of spatial and temporal resolution. Moreover, as a general endocytic marker, the C2 domain may also be used to study membrane recycling in many cell types.
Keywords: synaptic vesicles, synapse, super-resolution, STED, PALM, TIRF, C2 domain, electron microscopy
Graphical abstract
Highlights
-
•
Purified cPLA2 C2 domain labels recycling membranes by binding to the cell surface
-
•
Ca2+ dependence of membrane binding enables efficient removal of uninternalized probe
-
•
C2 domain can be conjugated to a variety of useful dyes and reporter proteins
-
•
Synaptic vesicles can be imaged by disparate microscopy methods using the C2 domain
Motivation
Currently, imaging probes for synaptic vesicles have unique strengths and weaknesses, which not only limit their utility but also confine their application to certain types of imaging approaches. We therefore developed a method of labeling synaptic vesicles that has the flexibility to be applied across many microscopy modalities. Our probe is based on the cytosolic phospholipase A2 (cPLA2) C2 domain, which reversibly binds membranes in a Ca2+-dependent manner. Through conjugation to different dyes, enzymes, and fluorescent proteins, the purified cPLA2 C2 domain can serve as a versatile probe for synaptic vesicles and other recycling membranes.
An et al. demonstrate that the purified cPLA2 C2 domain can label recycling membranes in neurons and non-neuronal cells. By conjugating the C2 domain to various tags, the authors visualize synaptic vesicles by disparate microscopy methods, including electron microscopy and super-resolution techniques, such as STED microscopy and FPALM.
Introduction
Much has been learned about the synaptic-vesicle cycle by using fluorescent probes, which label vesicle membranes (Kavalali and Jorgensen, 2014). Broadly, vesicle probes can be divided into ones that are taken up by endocytosis and recycled into synaptic vesicles and those that are fusion proteins concatenating synaptic-vesicle proteins to fluorescent proteins. The two most widely used vesicle probes are the FM family (named after the dye developer, Fei Mao) of lipophilic fluorescent dyes (Betz and Bewick, 1992), which can label vesicles that recycle during neuronal activity, and synaptic-vesicle membrane proteins genetically fused to pHluorin, a highly pH-sensitive variant of GFP (Miesenbock et al., 1998), which can report pH changes that occur during vesicle recycling when targeted to the acidic vesicle lumen (Sankaranarayanan and Ryan, 2000). Despite their immense usefulness, FM dyes and pHluorin-based reporters suffer from unique drawbacks (for review, see Kavalali and Jorgensen, 2014). Compared with some commonly available organic dyes, FM dyes are not very bright or photostable, which makes sensitive, low-light-level applications such as single-vesicle imaging challenging (Ryan et al., 1997; Murthy and Stevens, 1998; Zenisek et al., 2000; Aravanis et al., 2003; Joselevitch and Zenisek, 2009, 2020). FM dyes also possess very broad excitation and emission spectra, which are unfavorable for multicolor experiments. From a practical standpoint, FM dyes require extensive washout times (>15 min) for labeled synaptic vesicles to be detected sufficiently above the background fluorescence emanating from surface-bound dye. A long washing step not only allows unwanted loss of labeled vesicles through spontaneous exocytosis but also prevents the observation of early stages in vesicle recycling.
pHluorin-based reporters provide an easier readout of vesicle recycling because they do not require loading and washing steps like FM dyes. Moreover, their excellent dynamic range and signal to noise enable the detection of single-vesicle recycling events (Gandhi and Stevens, 2003; Balaji and Ryan, 2007; Gramlich and Klyachko, 2019; Maschi and Klyachko, 2020). However, because pHluorin is useful insofar as it reports pH changes, it can reveal information about vesicles only just during and right after fusion. For instance, it is not feasible to use a pHluorin-based reporter to track the movement of vesicles prior to exocytosis or to observe endocytosed vesicles after they have reacidified. These two examples bring up a second fundamental limitation of pHluorin and other genetically encoded reporters (Martineau et al., 2017), which is that any genetically encoded reporter will produce a density of labeled vesicles that is far too great to allow individual vesicles to be tracked. This is because synapse maturation requires weeks for cultured neurons, which is sufficient time for most if not all vesicles to acquire a copy of the exogenously expressed reporter. This limitation can be overcome only if individual vesicles will contain far fewer than one fluorescent molecule per vesicle either by photoconversion or low expression (Vaithianathan et al., 2016, 2019). While this has been achieved in some approaches, the signal is limited to single molecules per vesicle under these conditions.
Lastly, a great limitation shared by both FM dyes and pHluorin-based reporters is that they are not well suited for super-resolution imaging. Techniques such as stimulated emission depletion (STED) microscopy and (fluorescence) photoactivation localization microscopy/stochastic optical reconstruction microscopy ([F]PALM/STORM) have emerged as indispensable tools for neurobiological imaging applications (Westphal et al., 2008; Dani et al., 2010; Berning et al., 2012). However, the successful application of any of these techniques requires probes with superior photophysical properties (Gould et al., 2012). For STED microscopy, it is advantageous to use fluorophores with outstanding photostability (Donnert et al., 2006). Currently, to label synaptic vesicles for STED microscopy, a traditional immunolabeling approach is required, which can entail a complicated labeling regimen and also labels surface proteins not associated with vesicles (Willig et al., 2006; Westphal et al., 2008). On the other hand, single-molecule approaches such as (F)PALM/STORM and others rely on switching single fluorophores stochastically between a detectable and non-detectable state (Gould et al., 2012). This modality therefore requires probes that can be switched efficiently between these two states and that are sufficiently bright to be detected at the single-molecule level. Neither pHluorin nor FM dyes have been demonstrated to work under any of these imaging conditions. These incompatibilities are particularly unfortunate since synaptic vesicles are much smaller (∼40-nm diameter) than the diffraction limit of conventional light microscopy (∼200 nm).
More recently, labeling approaches based on antibodies or nanobodies to lumenal domains of synaptic-vesicle proteins (Hua et al., 2011; Joensuu et al., 2016; Martineau et al., 2017; Seitz and Rizzoli, 2019; Park et al., 2021) or probes possessing a membrane anchor (Revelo et al., 2014) have been used to label synaptic vesicles with a selection of fluorophores conducive to super-resolution microscopy. While both approaches have proven useful for imaging vesicle turnover in several synaptic preparations, both approaches are limited due to their essentially irreversible binding to their targets requiring extracellular quenching to see intracellular organelles.
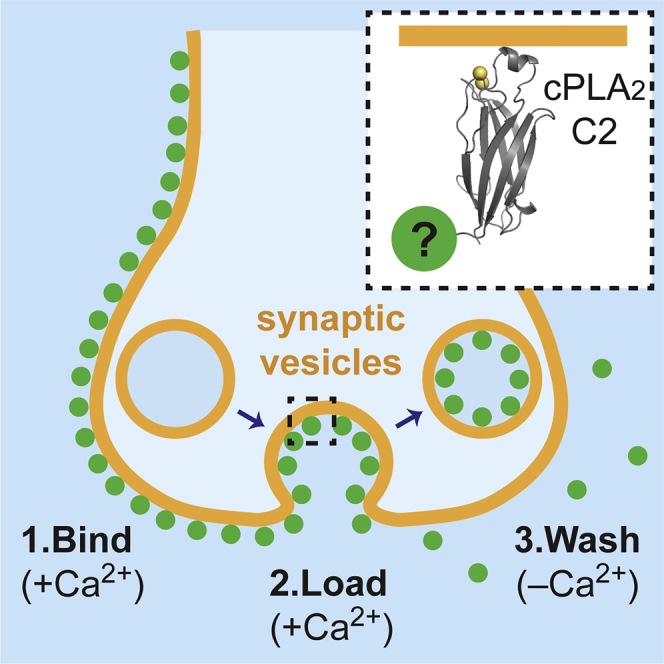
Here, we sought to add to the existing tool set for imaging vesicles. Our goal was to develop a membrane tracer that can be used like FM dyes but has the ability to be washed off of membranes efficiently and conjugated with any type of detectable tag, fluorescent or non-fluorescent (e.g., for electron microscopy). We based our probe on the cytosolic phospholipase A2 (cPLA2) C2 domain, which is a small domain that binds to phosphatidylcholine (PC) in a Ca2+-dependent manner (Nalefski et al., 1998). Since PC, unlike most phospholipids, resides in both leaflets of the plasma membrane, we anticipated that the C2 domain would even bind to the outer surface of cells. As expected, we found that the fluorescently tagged C2 domain can become internalized within recycling vesicles when briefly applied to neurons. By conjugating the C2 domain with different types of tags, we demonstrate that a single, modular probe can allow synaptic vesicles to be imaged not only by conventional fluorescence microscopy but also by STED microscopy, (F)PALM/STORM, total internal reflection fluorescence microscopy (TIRFM), and electron microscopy.
Results
Cell-surface binding by the C2 domain
The C2 domain of cPLA2 forms an antiparallel β sandwich that binds two Ca2+ ions cooperatively through three interstrand loops at one end of the domain (Figure 1A; Perisic et al., 1998). Ca2+ in the low micromolar range induces an over 10-fold enhancement in the affinity of the C2 domain for PC but not for anionic lipids such as phosphatidylserine (Nalefski et al., 1998). Removal of free Ca2+ triggers membrane undocking on the seconds timescale (Nalefski et al., 1997). We reasoned that this reversible membrane interaction could be exploited to label recycling synaptic vesicles (Figure 1A).
Figure 1.

The C2 domain binds to the cell surface in a Ca2+-dependent manner
(A) Rationale behind using the C2 domain (gray) to label recycling vesicles (amber) and the crystal structure of the cPLA2 C2 domain bound to Ca2+ (yellow spheres) (PDB: IRLW; Perisic et al., 1998).
(B) Adherent PC12 cells were incubated with the indicated concentrations of purified C2 domain for 5 min on ice, and the amount of C2 domain bound to cells after washing was detected by immunoblotting (black trace; n = 6 dishes). To examine the lipid interactions involved in membrane binding, cells were pre-treated with either phosphatidylcholine-specific phospholipase D (red trace; n = 5 dishes) or sphingomyelinase (blue trace; n = 5 dishes) for 1 h. Cell integrity was assayed by checking for β-actin leakage under all conditions (data not shown).
(C) Divalent cation specificity of membrane binding. Cells were incubated with 1 μM C2 domain for 5 min in the indicated free Ca2+ and Mg2+ concentrations (n = 3).
(D) Reversibility of membrane binding. Cells were incubated with 1 μM C2 domain (no wash) or further incubated without the C2 domain for 5 min in normal extracellular solution (Ca2+ wash), in a pH 5.5 solution containing 2 mM Ca2+ (pH 5.5 wash), or in 1 mM EGTA solution containing 1 mM Mg2+ (EGTA wash) (n = 4). Error bars, SEM.
When added to PC12 cells that were chilled on ice (to block endocytosis), the purified C2 domain bound saturably to membranes (Figure 1B). Despite the fact that PC is the physiological substrate for the catalytic domain of cPLA2, the C2 domain has been reported to bind to other lipids with non-anionic headgroups (Nalefski et al., 1998) including sphingomyelin, which is also found on the extracellular leaflet of the plasma membrane. Accordingly, we tested the effects of pre-treating PC12 cells with either phospholipase D or sphingomyelinase to reduce PC or sphingomyelin levels, respectively. Both treatments shifted the binding curve to the right, but phospholipase D had a much greater effect. This suggested that the C2 domain can bind to lipids other than PC on the cell surface but that PC is bound with higher affinity.
We next tested the divalent cation selectivity for cell-surface binding. We found that Ca2+ promoted C2-domain binding to PC12 cell membranes by 22-fold, with a half maximal effective concentration (EC50) value of 1 μM Ca2+ (Figure 1C). In contrast, Mg2+ had a negligible effect up to 3 mM, while 10 mM Mg2+ only had 2.5-fold greater binding—an order of magnitude less than 1 mM Ca2+. To test the reversibility of membrane binding, we washed cells pre-labeled with the C2 domain with extracellular solution containing the Ca2+ chelator EGTA (1 mM) for 5 min, again on ice. Only 3.4% ± 2.8% of the C2 domain was left bound to membranes after the washout (Figure 1D). When cells were washed with solution containing Ca2+ instead, 68% ± 5.9% of the C2 domain was still bound after 5 min, indicating that dissociation from the cell surface requires minutes when Ca2+ is continuously present. Since our goal was to label recycling vesicles, it was of interest to determine how the acidic environment of the vesicle (pH 5.5) might affect membrane dissociation. Similar to the washout with Ca2+ at pH 7.4, 65% ± 11.1% of the C2 domain was still bound in the presence of Ca2+ at pH 5.5.
Confocal microscopy of C2-domain-loaded synaptic terminals
We first tested the C2 domain as a vesicle probe in cultured hippocampal neurons. To label synaptic vesicles, we bathed VGlut1-pHluorin-expressing neurons with the C2-domain protein conjugated to Texas red (C2-TR) for 5 min and stimulated vesicle recycling by raising the extracellular K+ concentration (40 mM) for 90 s. After a 20-min recovery period to allow vesicle reformation from endocytic intermediates, we removed uninternalized C2-TR by washing the neurons with EGTA solution twice for 1 min each (Figure 2A). This staining protocol yielded numerous C2-TR-labeled puncta that clearly colocalized with VGlut1-pHluorin-identified terminals after vesicles were alkalinized with ammonium chloride (NH4Cl) (Figure 2B). When neurons labeled this way were stimulated with high K+ (70 mM), VGlut1-pHluorin fluorescence first increased, as pHluorin was unquenched by exocytosis, and then decreased, as pHluorin was endocytosed within reacidifying vesicles (Figures 2C [green] and S1). At the same terminals, C2-TR fluorescence dropped sharply over the first ∼15 s and then continued to decrease slowly, reaching ∼45% of its initial value after 90 s (Figures 2C [red] and S1), reminiscent of the way FM1-43 is released with high K+ stimulation (Ryan et al., 1993).
Figure 2.

Spinning-disk confocal microscopy of synaptic vesicles labeled with C2-TR
(A) Experimental protocol for stimulation-dependent loading of synaptic terminals.
(B) Spinning-disk confocal images of hippocampal neurons expressing vGLUT-pHluorin labeled with 1 μM C2-TR. Acid quenching of vGLUT-pHluorin was relieved by addition of 50 mM NH4Cl to better identify terminals (bottom images).
(C) Time course of average fluorescence intensity (F) normalized to initial fluorescence (Fo) of terminals colabeled with vGLUT-pHluorin and C2-TR during continuous high K+ stimulation, starting at 0 s (n = 193 terminals, 10 experiments).
(D) Average normalized vGLUT-pHluorin responses with (red circles) or without (black circles; n = 207 terminals, 4 experiments) C2-TR loading. Inset, higher temporal resolution of responses.
(E) NH4Cl-induced brightening of vGLUT-pHluorin terminals as a function of C2-TR loading. Each data point is a bin of 150 terminals (n = 5 experiments).
(F) Average normalized C2-TR responses to hypertonic-sucrose stimulation. The fluorescence loss from readily releasable vesicles was related to the fluorescence of total recycling vesicles by completely destaining terminals with multiple rounds of high K+ stimulation (n = 38 terminals, 2 experiments). Error bars, SEM.
Stimulation-dependent VGlut1-pHluorin signals in terminals with and without C2-TR showed a high degree of similarity (Figure 2D). However, VGlut1-pHluorin fluorescence rose slightly less rapidly if terminals also released C2-TR (Figure 2D [inset]). One explanation for this is that C2-TR quenched VGlut1-pHluorin through fluorescence resonance energy transfer (FRET) before it diffused away from the terminal. Since FRET quenching requires closeness, based on the proximity of the excited donor fluorophore and the acceptor fluorophore, the brightening of VGlut1-pHluorin (donor) that occurs when synaptic vesicles are alkalinized, either by exocytosis or NH4Cl application, should diminish as the intravesicular concentration of C2-TR (acceptor) increases. We tested this by loading over 1,600 terminals with varying amounts of C2-TR (Figure 2E). We found that as terminals contained more C2-TR, the NH4Cl-induced brightening of VGlut1-pHluorin became smaller. This apparent quenching was half maximal at a relatively low C2-TR load (∼500 AU), and even at the highest C2-TR load (∼5,800 AU), the brightening was reduced by only ∼57%. These results suggest that synaptic vesicles could be saturably labeled by C2-TR but that either the FRET efficiency is less than 100% or only a fraction of all vesicles take up the probe. The latter possibility is supported by the existence of a reserve pool of vesicles that does not undergo recycling and is thus inaccessible to FM dyes (Murthy and Stevens, 1999).
To investigate the possibility that the C2 domain had somehow functionally compromised synaptic vesicles that are immediately capable of release, which might also explain the slower initial rise of VGlut1-pHluorin fluorescence, we exposed C2-TR-loaded neurons to a hypertonic sucrose solution (800 mOsM) that is known to selectively trigger the readily releasable pool of vesicles (Rosenmund and Stevens, 1996). We further subjected neurons to three successive rounds of high K+ stimulation to relate the size of the sucrose-responsive pool to that of the total recycling pool. We found that ∼30% of the total pool fluorescence could be lost with the hypertonic challenge (Figure 2F), which is similar to the case when terminals are loaded with FM dye (Pyle et al., 2000).
Electron microscopy of C2-domain-labeled vesicles
The above experiments provided strong indications that the C2 domain had labeled synaptic vesicles. However, to demonstrate this unequivocally, it was necessary to examine terminals loaded with the C2 domain by electron microscopy. To this end, we conjugated the C2 domain with horseradish peroxidase (HRP), which converts 3,3′-diaminobenzidine (DAB) into an electron-dense precipitate that can fill the interior of organelles. Since HRP is bigger than the C2 domain (∼40 versus ∼14 kDa), we inserted a flexible, extended linker (XL) between the C2 domain and the conjugatable cysteine residue to reduce potential clashing between HRP and the membrane. We bathed neurons with C2-XL-HRP for 5 min, then stimulated recycling with high K+ (90 mM) application for 90 s, and finally allowed them to recover for 20 min, always in the presence of the probe. In this manner, C2-XL-HRP labeled the plasma membrane, the synaptic vesicles (Figure 3A), and also the transient endosome-like vacuoles (Figure 3A [arrows]). As expected, when we included a short (1 min) wash step with EGTA solution before the recovery period (10 min), the intense staining of the plasma membrane did not occur (Figure 3B). The loading protocols with and without the EGTA wash step gave synaptic-vesicle-labeling efficiencies of 21.7% ± 1.9% (±SEM) and 29.4% ± 1.4%, respectively, which are similar to the labeling efficiency (∼18%) that can be achieved using the technique of FM1-43 photoconversion (Harata et al., 2001).
Figure 3.

Electron microscopy of synaptic vesicles labeled with C2-XL-HRP
(A and B) Representative electron micrographs of terminals loaded with 4 μM C2-XL-HRP by high K+ stimulation followed with (B) or without (A) a 1-min EGTA wash step. Post-stimulation recovery time was 10–20 min. Arrows indicate endosome-like vacuoles.
(C) Distributions of vesicle diameters in terminals loaded with C2-XL-HRP by high K+ stimulation. Black bars, C2-positive vesicles (n = 359); white bars, C2-negative vesicles (n = 783).
(D) Distributions of vesicle diameters in terminals loaded by spontaneous activity (overnight incubation with C2-XL-HRP). Black bars, C2-positive vesicles (n = 199); white bars, C2-negative vesicles (n = 492).
High K+ stimulation produced labeled vesicles that were slightly bigger than unlabeled vesicles at the same terminal (52.98 ± 0.41 versus 49.14 ± 0.21 nm; Figure 3C). This effect was not specific to C2-XL-HRP, since it was also seen when cholera toxin-HRP, a GM1 ganglioside-binding marker, was used to label vesicles (data not shown). Interestingly, when C2-XL-HRP was internalized through spontaneous recycling, labeled and unlabeled vesicles were similar in size (50.86 ± 0.24 versus 50.98 ± 0.46 nm; Figure 3D), which argued against the C2 domain causing vesicles to be larger. Instead, these results suggest that the size of labeled synaptic vesicles may reflect the existence of different endocytic mechanisms, given that vesicle recycling occurs at least in part through endosome-like intermediates during strong stimulatory conditions, like high K+ depolarization, while it is thought to occur primarily via clathrin-mediated budding during spontaneous neuronal activity (Hayashi et al., 2008).
STED microscopy of C2-domain-labeled vesicles
To visualize synaptic vesicles with super resolution in living neurons, we conjugated the C2 domain with ATTO647N next, a highly photostable dye that is useful for STED microscopy (Westphal et al., 2008). We applied C2-ATTO647N to neurons for 2 min at 37°C to label spontaneously recycling vesicles. After a 3-min wash with EGTA solution, neurons were imaged on a commercial STED microscope (TCS STED, Leica Microsystems) featuring a 640-nm pulsed excitation laser synchronized to the output of a Ti:Sapphire laser tuned to 770 nm for depletion of ATTO647N. Large diffraction-limited spots that were seen with confocal imaging revealed themselves to be comprised of smaller spots when viewed in STED mode (Figures 4A and 4B). The average background noise in a STED image was 0.4 counts/pixel, corresponding to a peak signal-to-background ratio of 65. The fluorescence profile, or the full width at half maximum (FWHM), of the STED-resolved spots approached the resolution limit (∼60 nm) of the microscope, confirming that the labeled objects were not larger than the expected size of synaptic vesicles. Comparing the FWHM of the same stationary spot in confocal and STED images showed that the spatial resolution was increased by ∼3.5-fold, or by ∼13-fold when considering the focal area of the spot (Figure 4C). A similar improvement in spatial resolution has been reported for STED imaging of synaptic vesicles labeled with an antibody directed against the lumenal domain of synaptotagmin (Abbott et al., 2018).
Figure 4.

STED microscopy of synaptic vesicles labeled with C2-ATTO647N
(A) Raw confocal and STED images of a small field of neurons briefly exposed to 4 μM C2-ATTO647N at 37°C. Although dim, STED-resolved spots can be clearly distinguished from random noise since they appeared in consecutive frames (middle and right column panels).
(B) Smoothing the above images with a 2D Gaussian filter with an FWHM of 80 nm better differentiates individual labeled objects in STED mode, but not in confocal mode.
(C) Intensity profiles of a stationary vesicle (measured across arrowheads in A) were fitted to either a Gaussian (confocal) or a Lorentzian function (STED) to give the indicated FWHM values.
(D) Image sequence (smoothed) from an STED movie recorded at 4.9 Hz. The white line in the last frame depicts the trajectory of the vesicle.
(E) Examples of different types of vesicle mobility in one movie. Vesicles show either directed (red) or diffusive motion (green) or remain stationary (black) throughout the whole recording. Insets show expanded views.
Some spots observed by STED microscopy were not stationary but highly mobile, sometimes exhibiting directed motion over long distances, as if movement was confined within axonal processes (Figure 4D). A previous super-resolution study of vesicle mobility demonstrated that vesicles can move either diffusively or directionally, in a motor-driven manner, or become stuck at “hot spots” (Balaji and Ryan, 2007). We recapitulated these findings by tracking the trajectory of multiple labeled objects in a single field of view. In the traces shown in Figure 4E, one object (red trace) traveled 3.09 μm, stopped for 3.65 s, and then continued to move along a linear path for another 4.47 μm that coincided with the position of a second object (black trace) that was stationary throughout the recording. At a different location, another object (green trace) showed diffusive motion that was limited to a region compatible in size with a synaptic terminal. Taken together with our functional (Figure 2) and ultrastructural-labeling data (Figure 3), these results strongly suggest that C2-ATTO647N-labeled objects represented synaptic vesicles.
FPALM of C2-domain-labeled vesicles
To image synaptic vesicles by FPALM using the C2 domain, we turned to monomeric Eos2 (mEos2), a green-to-red photoconvertible fluorescent protein that is useful for localization-based super-resolution techniques due to its high contrast ratio and brightness (Hayashi et al., 2008). We fused mEos2 to the N terminus of the C2 domain via a flexible linker region and purified the probe after ∼48 h of bacterial expression to ensure chromophore maturation. As expected, terminals loaded with mEos2-C2 initially showed green fluorescence but photoswitched to red emission when briefly illuminated with a 405-nm light (Figures 5A and 5B). When subjected to high K+ stimulation, loaded terminals released mEos2-C2 robustly (Figures 5C and 5D). The time dependence and extent of fluorescence loss were similar to those of C2-TR destaining experiments (Figure 2C), which indicated that the C2 domain could functionally label synaptic vesicles whether it was coupled to a small dye (∼700 Da) or to a much larger tag like a fluorescent protein (∼25 kDa).
Figure 5.

FPALM of synaptic vesicles labeled with mEos2-C2
(A and B) Spinning-disk confocal images of a field of neurons incubated overnight with 1 μM mEos2-C2. Neurons were washed with EGTA solution for 3 min prior to imaging. Region of interest (dashed polygon) was illuminated with 405-nm light to photoswitch mEos2-C2.
(C) Image sequence of mEos2-C2-loaded terminals subjected to high K+ stimulation.
(D) Average normalized mEos2-C2 response to high K+ stimulation (n = 16); Error bars represent SEM.
(E–H) FPALM experiments. Prior to fixation, neurons were exposed to 4 μM mEos2-C2 for 2 min at 37°C to label vesicles and then with 4 μM C2-Alexa405 for 5 min on ice to label the cell surface. (E) Widefield image of C2-Alexa405 labeling. (F) FPALM image of mEos2-C2 labeling. (G) Expanded view of the boxed regions, merged. (H) Normalize radial distribution of molecules within the particles.
Since acquiring FPALM images would require that neurons are fixed, it was desirable to obtain a corresponding image of total membrane staining so that vesicles could be distinguished from background localization events. For this purpose, after loading terminals with mEos2-C2 for 2 min at 37°C, we bathed neurons with C2 domain conjugated with Alexa 405 (C2-Alexa 405) for 5 min on ice before fixation. C2-Alexa 405 was excited with low levels of a 405-nm light to capture the widefield image of neurons (Figure 5E) prior to mEos2 measurements on a custom-built biplane FPALM microscope (Juette et al., 2008). In each FPALM image (Figure 5F), localized molecules were plotted in x-y and strongly smoothed with a Gaussian. The peaks were identified in each smoothed image and taken to be the center position of each particle. All molecules within a 200-nm radius of each peak were identified and then further filtered so that particles containing 5–15 molecules within a 75-nm radius were accepted as viable particles. We identified nearly 11,000 particles in 19 datasets and plotted them in a histogram (Figure 5H) based on molecule distance from the center of the peak and normalized by radial distance from center. The mean particle diameter was ∼50 nm. Given that our FPALM setup can achieve a resolution of ∼30 nm, these particles likely represented synaptic vesicles.
In addition to FPALM, we note that mEos2-C2 allows for the second main application of photoswitchable proteins, i.e., highlighting and tracking the movements of proteins or, in this case, synaptic vesicles. Selective photoswitching of green mEos2-C2-labeled vesicles (Figures 5A and 5B) would allow their tracking in neurons without having to employ fluorescence recovery after photobleaching (Darcy et al., 2006) or methods of sparsely labeling vesicles (Westphal et al., 2008; Gramlich et al., 2017; Gramlich and Klyachko, 2017).
TIRFM of C2-domain-labeled vesicles
We next tested whether the C2 domain could be used to image single vesicles using TIRFM. To do so, we used type Mb goldfish retinal bipolar cells, an established preparation for live imaging of single synaptic vesicles labeled with FM1-43 (e.g., Zenisek et al., 2000). These neurons are well suited for TIRFM because they possess large bulbous pre-synaptic terminals (∼10-μm diameter) that can be plated onto a glass coverslip, allowing vesicles to be investigated within the evanescent field. As such, for a membrane tracer to be useful for TIRFM, its ability to be washed efficiently from the membrane-glass interface is critical. Bipolar cells were loaded with a brief (10–15 s) application of C2-TR (1 μM) in a 25 mM K+ solution, followed by a 1- to 5-min EGTA wash, which was in turn followed by a 5- to 10-min recovery period in low Ca2+ to reduce exocytosis until the cell was stimulated. After recovery, bipolar cell terminals imaged using TIRFM exhibited discrete spots that resembled FM1-43-labeled synaptic vesicles in terms of their diffraction-limited size and mobility (Figures 6A–6C; Zenisek et al., 2000; Holt et al., 2004; Midorikawa et al., 2007). As previously described for FM1-43-labeled cells, a small subset of spots (∼3%) exhibited high fluorescence intensities (>3 SD above the mean; Figure 6B), which may represent endosomal labeling (Coggins et al., 2007), but most spots showed relatively uniform fluorescence intensities, consistent with synaptic-vesicle labeling.
Figure 6.

TIRFM of synaptic vesicles labeled with C2-TR
(A) Single frame (left) and average of 600 frames (4 recordings, right). Boxed region shows expanded views in (C).
(B) Histogram of spot intensities. Intensities (129 spots in 3 cells) were normalized to mean intensity of each cell and then pooled.
(C) Expanded region from (A). Red circles indicate spots that appear or disappear during one recording. Green arrows point to sites of exocytosis. Images were low-pass filtered.
(D) Image sequence of a single fusion event. Time is relative to the moment of fusion. Dotted concentric circles in the rightmost panel depict center and annulus regions.
(E) Fluorescence changes in center (filled) and annulus (open) of the fusion event in (D). Arrow indicates moment of fusion.
(F–I) Fusion events versus undocking events. (F) Image sequence of averaged fusion events (n = 10 events in 3 cells). (G) Image sequence of averaged undocking events (n = 10 events in 3 cells). (H) Center-minus-annulus trace for fusion events in (F). (I) Center-minus-annulus trace for undocking events in (G). Error bars, SEM.
Previous TIRFM studies of bipolar cells have revealed that docked synaptic vesicles and fusion events are concentrated in discrete active zones demarcated by synaptic ribbons (Zenisek et al., 2000; Midorikawa et al., 2007). Bipolar cell terminals labeled with C2-TR exhibited similar active zones, which can be seen as bright spots in time-averaged images (Figure 6A [right]) due to vesicles occupying preferred sites. Figure 6C shows a sequence of images from a terminal region that demonstrates the mobility of individual vesicles in and out of the evanescent field (outlined by red circles). Notably, in this region, two of three sites where vesicles underwent exocytosis (see below) coincided with locations where vesicles repeatedly appeared (green arrows).
To image exocytosis of individual vesicles, bipolar cells were super fused with a solution containing 25 mM K+ to stimulate neurotransmitter release. Upon stimulation, immobilized vesicles were occasionally seen to brighten transiently and then disappear with a spreading cloud of fluorescence (Figure 6D), as expected for the exocytic release of a probe and similar to what is observed for fusion events using FM1-43 (Zenisek et al., 2000). The release of C2-TR during fusion is plotted in Figure 6E, which shows the fluorescence within a circle encompassing a fusing vesicle (filled circles) and an annular region outside the vesicle (open circles), as indicated in the rightmost panel of Figure 6D. On average, the fluorescence of fusing vesicles (n = 10) peaked within a single 30-ms frame before it decayed over several frames (Figures 6F and 6H). These fusion events were readily distinguishable from occurrences where vesicles abruptly disappeared without brightening or producing a visible cloud (Figures 6G and 6I), which we interpret to be undocking events (Zenisek et al., 2000).
The C2 domain as an endocytic marker in non-neuronal cells
Finally, since the C2 domain of cPLA2 binds PC, which is a phospholipid present in all membranes, we tested the utility of the C2 domain as a general endocytic marker in non-neuronal cells. Using structured-illumination microscopy, we compared the post-endocytic trafficking of C2-Alexa568 in HeLa cells with that of two membrane-receptor ligands, transferrin-Alexa488 (Figure S2), or EGF-Alexa488 (Figure S3). As expected for a non-specific membrane marker, C2-Alexa568 showed partial colocalization with each ligand. Interestingly, within some intracellular structures, transferrin-Alexa488 was spatially segregated from C2-Alexa568, which was more homogenously distributed (Figure S2B [arrow]). These results demonstrate that the C2 domain could be used like FM dyes (Hao and Maxfield, 2000) to analyze the recycling of membranes in non-neuronal cells.
Discussion
Our C2-based probe complements existing strategies for imaging synaptic vesicles and provides added value for several experimental purposes. Specifically, antibody-based approaches are currently limited to species and cell types where antibodies to the lumenal domains of relevant proteins have been created. While this hurdle has been overcome for mouse neurons (Willig et al., 2006; Westphal et al., 2008), similar antibodies are not commercially available for numerous other preparations, such as goldfish retinal bipolar cells. Moreover, the two approaches—antibody- and C2-based—can be used for complementary purposes since one tracks a specific protein and the other tracks the internalized membrane generally. Styryl-dye-based approaches also have their limitations. For example, to visualize single vesicles in the goldfish bipolar cell preparation, high concentrations of FM1-43 permeabilize the cells (data not shown), and one needs to take care to minimize the time and concentration of the dye exposure to the cells to ensure good recordings. Such toxicity is not observed with our C2-based approach. Additionally, styryl dyes have been found to permeate certain channel types, such as mechanotransduction channels of hair cells (Nishikawa and Sasaki, 1996; Gale et al., 2001), rendering them unusable in some cases.
Beyond complementing existing methods for imaging synaptic vesicles, our C2-domain-based probe offers several key advantages. First, due to its modular functionality, the C2 domain can be applied across multiple microscopy techniques and bridge the gap between fluorescence microscopy and electron microscopy (EM) using the same tagging strategy (and thus the same class of vesicles). Although it is currently possible to visualize vesicles by methods other than conventional fluorescence microscopy, to do so usually requires the use of different single-purpose probes, each with a distinct mode of membrane binding. This necessitates multiple paradigms for loading neurons, which may affect what is being labeled in each type of experiment. Moreover, given that the molecular composition of vesicles has been reported to be heterogeneous (Hua et al., 2011; Ramirez et al., 2012), it is possible that different vesicle probes may favor the labeling of different vesicles, depending on whether the probe intercalates into the membrane (FM dyes), recognizes a specific vesicle protein (synaptotagmin antibody), or binds to a specific lipid (cholera toxin-HRP). For these reasons, we believe that using a single type of probe for all microscopy techniques would allow for a more reliable synthesis of different imaging data.
Second, due to its controllable membrane binding, the C2 domain can be washed off the cell surface quickly and efficiently. This is an advantage over FM dyes, which stick to the membrane non-specifically and thus can be slow to wash out, and antibodies or probes that irreversibly bind to the plasma membrane. This improves routine experimentation by not only reducing the time it takes to record from loaded neurons but also, importantly, by increasing the signal of labeled synaptic vesicles over background fluorescence. The fast washout time of the C2 domain should enable the design of pulse-chase studies with improved temporal resolution. Combined with the use of spectrally distinct dyes on the C2 domain, such experiments represent a potentially powerful way of dissecting vesicle-recycling pathways.
Third, the ability to conjugate the C2 domain with virtually any fluorophore opens up numerous experimental possibilities. In principle, pH-sensitive dyes that are acid quenched, like fluorescein, should allow the C2 domain to report exocytosis with a greater dynamic range. Conversely, alkaline-quenched dyes, like cypher5E (Hua et al., 2011), may permit a reacidification-based readout of endocytosis. In addition to being useful for FPALM/STORM, mEos2 could be used to study the trafficking of vesicles photoswitched at specific regions within neurons. Ni-nitrilotriacetic acid-linked quantum dots, by binding the polyhistidine tag engineered onto the C2 domain, should produce very bright and photostable labeling of synaptic vesicles (Zhang et al., 2009). Enzymatic or bioluminescent tags, through signal amplification, could make possible high-throughput assays of pre-synaptic function. As the C2 domain is intrinsically adaptable as a vesicle probe, we anticipate that its usefulness will grow as tags with novel functionalities are continually developed.
In conclusion, we have demonstrated that the purified C2 domain can be used to label synaptic vesicles for disparate microscopy purposes. Since synaptic vesicles are the smallest organelles, visualizing other types of larger secretory vesicles should be feasible, even by technically demanding microscopy techniques. Recently, STED microscopy has been applied to study fusion-pore dynamics of large dense-core vesicles in neuroendocrine cells (Zhao et al., 2016; Shin et al., 2018). Our C2-based probe may enrich the tools of study for these vesicular membranes as well, to allow visualization of fusion-pore dynamics and single-vesicle dynamics before and after fusion.
Limitations of the study
Currently, a limitation of the cPLA2 C2 domain is that membrane binding occurs at very low Ca2+ concentrations (∼1 μM). For some purposes, it may be desirable to maintain extracellular Ca2+ at millimolar levels at all times, which would preclude washing out the uninternalized probe as we have described here. However, Ca2+-binding residues in the C2 domain can be mutated to shift the Ca2+ dependence of membrane binding by several orders of magnitude (up to an EC50 of 3.5 mM; Bittova et al., 1999). Thus, it should be possible to use low-Ca2+-affinity versions of the C2 domain to perform the wash step at higher Ca2+ concentrations.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-His tag | GenScript | Cat# A00186-100; RRID:AB_914704 |
| Mouse monoclonal anti-beta-actin | Abcam | Cat# ab8226 RRID:AB_306371 |
| Bacterial and virus strains | ||
| BL21 Gold competent cells | Agilent | Cat# 230130 |
| Chemicals, peptides, and recombinant proteins | ||
| Texas Red C2 Maleimide | ThermoFisher Scientific | Cat# T6008 |
| EZ-Link Maleimide Activated Horseradish Peroxidase | ThermoFisher Scientific | Cat# 31485 |
| ATTO 647N maleimide | ATTO TEC GmbH | Cat# AD 647N-41 |
| Alexa Fluor 568 C5 Maleimide | ThermoFisher Scientific | Cat# A20341 |
| Transferrin-Alexa488 | ThermoFisher Scientific | Cat# T13342 |
| EGF-Alexa488 | ThermoFisher Scientific | Cat# E13345 |
| Phospholipase D | Enzo | Cat# BML-SE301-0025 |
| Sphingomyelinase | Enzo | Cat# BML-SE108-0010 |
| PreScission protease | Cytiva | Cat# 27084301 |
| Ampicillin | Sigma-Aldrich | Cat# A0166 |
| IPTG | Sigma-Aldrich | Cat# I5502 |
| Lysozyme | Sigma-Aldrich | Cat# L6876 |
| Protease inhibitor cocktail | Roche | Cat# 4693159001 |
| DTT | Sigma-Aldrich | Cat# D0632 |
| Imidazole | Sigma-Aldrich | Cat# I2399 |
| Poly-L-lysine solution | Sigma-Aldrich | Cat# P4707 |
| Triton X-100 | Sigma-Aldrich | Cat# 93443 |
| N-Lauroylsarcosine sodium salt | Sigma-Aldrich | Cat# 61745 |
| Matrigel matrix | BD Biosciences | Cat# 356234 |
| Fibronectin | Sigma-Aldrich | Cat# F0895 |
| BSA | Sigma-Aldrich | Cat# A9418 |
| Versene | Gibco | Cat# 15040066 |
| Critical commercial assays | ||
| QIAprep Spin Miniprep Kit | Qiagen | Cat# 27104 |
| EndoFree Plasmid Maxi Kit | Qiagen | Cat# 12362 |
| Glutathione sepharose 4B resin | Cytiva | Cat# 17075601 |
| ProBond Nickel-Chelating Resin | Invitrogen | Cat# R80101 |
| Experimental models: Cell lines | ||
| PC12-GR5 | R. Nishi, University of. Vermont, Burlington | N/A |
| HeLa | ATCC | CCL-2 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6 | The Jackson Laboratory | Stock # 000664 |
| Goldfish | Slither and Swim | 5”-6” comets |
| Oligonucleotides | ||
| Primer: C2 Forward: GGCGGCGAATT CGGTGGTTCTGGTGGTTCTTCCCAT AAGTTTACAGTAGTGGTTCTACGTGC |
Sigma-Aldrich | N/A |
| Primer: C2 Reverse: GGCGGCCTCGAGTTAACAGTG ATGGTGATGGTGGTGGCCCTG GAAGTACAGGTTCTCAGAACC ACCAGAACCACCGCTTGAGG CAACTTCAAGAGA CATTTCC |
Sigma-Aldrich | N/A |
| Primer: C2-XL Forward: GGCGGC GAATTCGGTGGTTCTGGTGGTTC TGGTGGTTCTGGTGGTTCTGGTG GTTCTGGTGGTTCTGGTGGTTCTT CCCATAAGTTTACAGTAGTGGTTC TACGTGC |
Sigma-Aldrich | N/A |
| Primer: C2-XL Reverse: GGCGGCCTCGAGTTAACAGT GATGGTGATGGTGGTGGCCC TGGAAGTACAGGTTCTCAGA ACCACCAGAACCACCAGAAC CACCAGAACCACCAGA ACCA CCGCTTGAGGCAACTTCAAGA GACATTTCC |
Sigma-Aldrich | N/A |
| Primer: mEso2-C2 Forward: GGCGGC GAATTCGGTGGTTCTGGTGGTTCTGG TGGTTCTGGTGGTTCTGGTGGTTCTG GTGGTTCTATGAGTGCGATTAAGCCA GACATGAAG |
Sigma-Aldrich | N/A |
| Primer: mEso2-C2 Reverse: GGCGGC CTCGAGTTAACAGTGATGGTGATGG TGGTGGCCCTGGAAGTACAGGTTCT CAGAACCACCAGAACCACCGCTT GAGGCAACTTCAAGAGA CATTTCC |
Sigma-Aldrich | N/A |
| Recombinant DNA | ||
| Pla2g4a (NM_133551) cDNA clone | OriGene | Cat# RN204286 |
| pGEX-6P-1 | Cytiva | Cat# 28-9546-48 |
| pGEX-6P-C2 | This paper | N/A |
| pGEX-6P-C2-XL | This paper | N/A |
| pGEX-6P-mEos2-C2 | This paper | N/A |
| vGLUT1-pHluorin | Voglmaier et al., 2006 | N/A |
| Other | ||
| LB broth | Sigma-Aldrich | Cat# L3522 |
| DMEM, high glucose | Gibco | Cat# 11965092 |
| New calf serum | Sigma-Aldrich | Cat# 12023C |
| Horse serum | Gibco | Cat# 26050070 |
| Neurobasal-A Medium | Gibco | Cat# 10888022 |
| B-27 Supplement | Gibco | Cat# 17504044 |
| Fetal calf serum | PAN-Biotech | Cat# P30-1401 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, David Zenisek (david.zenisek@yale.edu).
Materials availability
Unique reagents generated in this study are available from the lead contact with a Materials Transfer Agreement.
Experimental model and subject details
PC12 cells
PC12-GR5 cell stocks were maintained in T80 flasks (Nalgene/Nunc) at 37°C and 10% CO2 in DMEM high glucose (Invitrogen) supplemented with 5% new calf serum (vol/vol) and 5% horse serum (vol/vol). Cells were passaged by dissociation using Versene (EDTA) solution (Gibco). For experiments, cells were replated to 50% confluence on 6-well multiwell plates (BD Biosciences) pretreated with poly-L-lysine (Sigma-Aldrich). After 24 h, cells were bathed in extracellular solution (130 mM NaCl, 2.8 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES and 10 mM glucose; pH 7.4) and prechilled on ice for 5 min to block endocytosis.
HeLa cells
HeLa cells (CCL-2, ATCC) were maintained in T-75 flasks (Falcon) at 37°C and 5% CO2 in DMEM (Gibco) supplemented with 4.5 g/L glucose, 1 mM sodium pyruvate, 1× non-essential amino acids (Gibco), 10% (vol/vol) FBS (Sigma) and 100 U/mL penicillin-streptomycin mix (Gibco). 100,000 cells were plated on glass-bottom dishes coated with 10 μg/mL fibronectin (MatTek Corporation) in complete DMEM media. The next day, cells were incubated for 2 h in serum-free and phenol-red free DMEM media containing 0.5% BSA. After starvation, cells were incubated with 1 μM C2-568 and 10 μg/mL transferrin-Alexa488 or 100 ng/mL EGF-Alexa488 for 30 min in starvation media to allow the labeled molecules to get internalized by the cells. Labeled cells were washed twice with PBS and fixed with 4% PFA for 10 min. Fixed cells were then washed with PBS before imaging.
Mouse hippocampal culture
All procedures with goldfish were in accordance with the Yale Institutional Animal Care and Use Committee (IACUC) (protocol 2019-11170). Dissociated mouse neurons were prepared from hippocampi of C57BL/6 mice embryos (E17) of both male and females as described previously (Stagi et al., 2010). Briefly, hippocampi were isolated, dispersed mechanically and seeded in 8-well chamber culture dishes. The dishes were pretreated with 0.1 mg/mL poly-L-lysine (Sigma-Aldrich) or alternatively on Matrigel (BD Biosciences). The cells were cultured in neurobasal A medium (Life Technologies) supplemented with 2% B-27 supplement (Life Technologies) and 0.5% fetal calf serum (PAN-Biotech). Cells were cultured for 20–30 days to obtain morphologically mature neurons with formed synapses. For transfections, the plasmid vGLUT1-pHluorin was purified using the EndoFree Maxi Kit (Qiagen). Transfection was performed using electroporation at the time of dissociation using an Amaxa Nucleofector system (Lonza), following the manufacturer’s instructions (program O-005). The average transfection efficiency was 30–60%, and the majority of transfected neurons showed plasmid expression from 3–5 days in vitro (DIV) until 30 DIV.
Goldfish bipolar neurons
All procedures with goldfish were in accordance with the Yale Instutional Animal Care and Use Committee (IACUC) (protocol 2020-10598). Bipolar neurons from adult male and female goldfish were enzymatically and mechanically isolated from goldfish retina as previously described (Coggins et al., 2007). Briefly, goldfish were decapitated, pithed and enucleated. The sclera and lenses were removed from the dissected eyes. The eyecups were then placed in an oxygenated solution containing 120 mM NaCl, 0.5 mM CaCl2, 2.5 mM KCl, 1 mM Mg Cl2, 10 mM glucose, 10 mM HEPES (pH 7.4 with NaOH) and 1100 u/mL type V Hyaluronidase (from Sheep Testes; Sigma) for 20 minutes. Following incubation, each retina was removed from the eyecup, cut into 6-8 pieces and placed in a solution containing: 120 mM NaCl, 0.5 mM CaCl2, 2.5 mM KCl, 1 mM Mg Cl2, 10 mM glucose, 10 mM HEPES (pH 7.4 with NaOH), 0.3 mg/mL D/L cysteine (Sigma) and 35 u/mL Papain (from Carica Papaya; Sigma) for 30-35 minutes. After digestion in Papain solution, retina pieces were rinsed three times in oxygenated solution (120 mM NaCl, 0.5 mM CaCl2, 2.5 mM KCl, 1 mM Mg Cl2, 10 mM glucose, 10 mM HEPES (pH 7.4 with NaOH) and placed in tissue culture dish for up to 6 hours. To isolate cells, individual pieces were placed in a microcentrifuge tube and mechanically triturated using a fire-polished Pasteur pipette.
For imaging, cells were plated in a solution containing 120 mM NaCl, 2.5 mM CaCl2, 2.5 mM KCl, 1.0 mM MgCl2, 10 mM glucose and 10 mM HEPES (pH 7.4 with NaOH) on special coverslips made from high refractive index glass (n488 = 1.80; Plan Optik, Germany). Bipolar neurons were recognized by their large synaptic terminal and unique cell body morphology. A selected neuron was locally superfused for 10–15 s with a labeling solution containing 1 μM C2-TR, 2.5 mM CaCl2, 25 mM KCl, 95 mM NaCl, 1.0 mM MgCl2, 10 mM glucose and 10 mM HEPES (pH 7.4 with NaOH). Next, the neuron was washed by local superfusion for 1–5 min with a low calcium solution designed to remove extracellular C2-TR. It contained 120 mM NaCl, 2.5 mM KCl, 1.0 mM MgCl2, 10 mM glucose, 10 mM HEPES and 1.0 mM EGTA (pH 7.4 with NaOH). After this brief washing period, the cells were superfused with a solution containing 120 mM NaCl, 0.5 mM CaCl2, 2.5 mM KCl, 1.0 mM MgCl2, 10 mM glucose, 10 mM HEPES and 1.25 mM EGTA (pH 7.4 with NaOH). To stimulate exocytosis, bipolar cells were superfused with a solution containing 2.5 mM CaCl2, 25 mM KCl, 95 mM NaCl, 1.0 mM MgCl2, 10 mM glucose and 10 mM HEPES (pH 7.4 with NaOH).
Method details
Plasmid preparation
The cDNA of rat cPLA2 C2 domain (residues 17-141; GenBank: NM_133551), with engineered Cys139Ala and Cys141Ser substitutions, was subcloned into the pGEX-6P-1 bacterial expression vector (Cytiva) using EcoRI and XhoI restriction sites. A polyhistidine tag was appended to the C-terminus of the C2 domain, followed by a unique cysteine for conjugation with thiol-reactive dyes or enzymes. This yielded an N-terminal glutathione S-transferase (GST) tag that could be cleaved by PreScission protease (Cytiva) and a C-terminal polyhistidine tag for sequential affinity chromatography. A second version of the probe, C2-XL, was made by inserting a (GlyGlySer)7 repeat between the polyhistidine tag and the unique cysteine. To generate mEos2-C2, the ORF of mEos2 was subcloned into pGEX-6P-1 using BamHI and EcoRI sites, flanked by N-terminal (GlyGlySer)6 and C-terminal (GlyGlySer)6 repeats. Subsequently, residues 17-141 of the C2 domain were subcloned into pGEX-6P-1-mEos2 using EcoRI and XhoI sites.
Protein purification and labeling
Single colonies of BL21 Gold competent cells (Agilent Technologies) transformed with the pGEX-6P-1-C2, pGEX-6P-1-C2-XL or pGEX-6P-1-mEos2-C2 plasmids were cultured in 5 mL of LB medium containing 50 μg/mL ampicillin at 37°C for 16 h and diluted into 500 mL of LB medium containing ampicillin for 4 h. Protein expression was induced by addition of 0.1 mM isopropyl-β-D-thiogalactopyranoside for up to 16 h at 25°C for C2 and C2-XL, or for up to 48 h at 25°C for mEos2-C2. Cells were harvested by centrifugation at 7,000g and then resuspended in lysis buffer (10 mM Tris, pH 8.0, 150 mM NaCl, 1 mM EDTA), followed by incubation with 100 μg/mL lysozyme (chicken egg white; Sigma) at 4°C for 15 min. Protease inhibitor cocktail (Roche Applied Science), 5 mM dithiothreitol and 1.5% sarkosyl (w/v) were then added, and the cells were vortexed for 30 s before lysis by sonication. After centrifugation at 10,000g, the supernatant was collected and 2% Triton X-100 (w/v) was added. The mixed detergent extract was vortexed for 30 s and then incubated with glutathione sepharose 4B beads (Cytiva) at 4°C for 1 h. After washing with lysis buffer extensively (6–8x) to remove detergent, to label the C2 domain with dyes, the beads were incubated with 0.1 mg/mL of either Texas Red C2-maleimide (ThermoFisher Scientific), Alexa Flour 568 C5-maleimide (ThermoFisher Scientific) or ATTO647N maleimide (ATTO-TEC) at 4°C for up to 16 h. After extensive washing with lysis buffer, the beads were incubated with 20 U PreScission protease at 4°C for 16 h to liberate dye-labeled C2 domain, which was then mixed with ProBond nickel-chelating beads at 4°C for 1 h and eluted off the resin using phosphate-buffered solution (pH 7.4) containing 160–400 mM imidazole. Eluted dye-labeled C2 domain was finally dialyzed in HEPES-buffered solution (HBS) (20 mM HEPES, pH 7.4, 150 mM NaCl). To label C2-XL with HRP, after the GST-affinity chromatography step, C2-XL bound to ProBond beads was incubated with 8 mg/mL EZ-link maleimide activated HRP (Thermo Fisher Scientific) at 4°C for 16 h and further prepared as described above.
Spinning-disk confocal microscopy
Neurons (25–30 DIV) were imaged at room temperature in modified Tyrode solution (pH 7.4) on a Perkin-Elmer UltraView VoX Spinning Disk microscope with an autofocus system, a motorized stage, 488 nm/50 mW and 561 nm/50 mW diode lasers and a Hamamatsu C9100-50 camera. This system included a PhotoKinesis feature for fluorescence recovery after photobleaching that was used in the case of photoswitching experiments. All experiments were performed using a 60X/1.4 numerical aperture (NA) oil immersion objective lens (CFI Plan Apo VC, Nikon), and images were acquired at 4 frames/s. One dendritic and one axonal arborization were randomly selected to perform each experiment.
Electron microscopy
Neurons were fixed with 1.2% glutaraldehyde in 0.1 M sodium cacodylate buffer, postfixed in 1% OsO4, 1.5% K4Fe(CN)6, 0.1M sodium cacodylate, en bloc stained with 0.5% uranyl magnesium acetate, dehydrated, and embedded in Embed 812. Where indicated, HRP reactions were developed with diaminobenzidene and H2O2 after the glutaraldehyde fixation step. Electron microscopy reagents were purchased from Electron Microscopy Sciences. Ultrathin sections were observed in a Philips CM10 microscope at 80 kV and images were taken with a Morada 1kx1k CCD camera (Olympus). For quantification, 20–30 pictures from each sample were used for calculating the C2-XL-HRP-labeled synaptic vesicles. 200–500 vesicles were measured for synaptic vesicle diameter analysis.
STED microscopy
STED microscopy was performed on a commercial setup (TCS STED, Leica Microsystems) equipped with a 640 nm pulsed excitation laser (LDH-P-C-640B, PicoQuant Photonics) synchronized to the output of a Ti:Sapphire laser (Mai Tai, Spectra Physics) tuned to 770 nm for depletion. Laser intensities (measured at the objective back aperture) of ∼80–150 μW and ∼180 mW were used for excitation and depletion, respectively. Images of a 7.75 × 7.75 μm2 area were acquired at ∼5 frames/s (256x256 pixel format scanned at 1400 lines/s). Fluorescence from the sample was collected by a 100X/1.4 NA oil immersion objective lens (HCX PL APO STED, Leica Microsystems), bandpass filtered (665–705 nm) and imaged through a one Airy unit pinhole onto a single photon counting avalanche photodiode. Vesicle trajectories were identified using custom software written in Matlab (Mathworks). Raw STED images were first smoothed with a Gaussian filter with a FWHM of 80 nm, and intensity peaks were then fitted to a two-dimensional Gaussian to determine the x- and y-positions of each vesicle.
FPALM
Measurements were performed with a custom built Biplane FPALM setup based on a Zeiss microscope stand (Axio Observer D1, Carl Zeiss MicroImaging) and an Andor iXon EM-CCD camera (DU-897 DCS-BV, Andor Technology). A multi-edge dichroic (Di01-R405/488/561/635, Semrock) reflected all laser lines to the sample through a 63X/1.2 NA water immersion objective (Plan-Apo 63X/1.2w, Carl Zeiss MicroImaging). Fluorescence was collected by the objective and then passed through the dichroic and two multiline bandpass filters (FF01 446/523/600/677-25 and FF01-594/730, Semrock). mEos2 was activated with a 405-nm laser (Crystalaser) and its fluorescence was excited by a 568-nm laser (Evergreen Laser Corporation). Approximately 10,000 frames at 50 ms/frame were recorded for each FPALM measurement. Localization was performed with the Vutara SRX software package (Vutara Inc.) as described previously (Mlodzianoski et al., 2011). Vesicle sizes were measured using custom written scripts in Matlab (Mathworks). In each FPALM image, localized molecules were plotted in x-y and strongly smoothed with a Gaussian. The peaks were identified in each smoothed image and taken to be the center position of each particle. All molecules within a 200-nm radius of each peak were identified and then further filtered so that particles containing between 5 and 15 particles within a 75-nm radius were accepted as viable particles.
TIRFM
Bipolar neurons were imaged as previously described (Coggins et al., 2007). Briefly, cells were observed through an inverted microscope (Olympus IX-70) modified for objective-type evanescent field illumination (Stout and Axelrod, 1989; Axelrod, 2001) using a 1.65 NA objective and a 561-nm solid state laser (Melles Griot). Fluorescence images were captured using an EM-CCD camera (Cascade 512B, Roper Scientific). Vesicle image analysis was performed using a custom-written image analysis program written in Matlab (Mathworks) (Zenisek et al., 2002)
Structure-illuminated microscopy
Images were acquired using a U-PLANAPO 60X/1.42 PSF, oil immersion objective lens (Olympus, Center Valley, PA) and CoolSNAP HQ2 CCD cameras with a pixel size of 0.080μm (Photometrics, Tucson, AZ) on the OMX version 3 system (Applied Precision) equipped with 488-, 561-, and 642-nm solid-state lasers (Coherent and MPB communications). Samples were illuminated by a coherent scrambled laser light source that had passed through a diffraction grating to generate the structured illumination by interference of light orders in the image plane to create a 3D sinusoidal pattern, with lateral stripes approximately 0.270 nm apart. The pattern was shifted laterally through five phases and through three angular rotations of 60° for each Z-section, separated by 0.125 nm. Exposure times were typically between 200 and 500 ms, and the power of each laser was adjusted to achieve optimal intensities of between 2,000 and 4,000 counts in a raw image of 16-bit dynamic range, at the lowest possible laser power to minimize photo bleaching. Raw images were processed and reconstructed to reveal structures with 100–125 nm resolution (Gustafsson et al., 2008). The channels were then aligned in x, y, and rotationally using predetermined shifts as measured using a target lens and the Softworx alignment tool (Applied Precision).
Quantification and statistical analysis
Statistical analysis
Data analysis was performed using Excel (Microsoft) and Origin (Microcal); all data are presented in plots as mean ± s.e.m. The n’s for each experiment and what they represent can be found in the figure legends. No statistical tests were used. Image analysis procedures are described in the sections highlighting each imaging modality in the STAR Methods sections above.
Acknowledgments
This work was supported by R01DC019057, R01EY021195, and R01EY032396 (to D.Z.), R01AG034924 and R01NS074319 (to S.M.S.), NIH NS36251 (to P.D.C.), RC1 GM091791 (to J.B.), the Yale Core Grant for Vision Research (P30 EY026878), and a James Hudson Brown–Alexander Brown Coxe Postdoctoral Fellowship (to T.J.G.).
Author contributions
S.J.A. conceived the study, designed constructs, and purified proteins. M.S. prepared neuronal cultures and performed spinning-disk confocal microscopy. T.J.G. performed STED microscopy experiments and analyzed data. Y.W. prepared neuronal cultures, performed EM, and analyzed data. M.M. performed FPALM experiments and analyzed data. F.R.-M. performed structure-illumination microscopy with S.J.A. D.Z. performed TIRFM experiments and analyzed data with S.J.A. All authors discussed the results and wrote the manuscript.
Declaration of interests
J.B. declares competing financial interests.
Published: April 25, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2022.100199.
Contributor Information
Seong J. An, Email: seong.an@yale.edu.
David Zenisek, Email: david.zenisek@yale.edu.
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Abbott B.P., Abbott R., Abbott T.D., Abraham S., Acernese F., Ackley K., Adams C., Adya V.B., Affeldt C., Agathos M. Prospects for observing and localizing gravitational-wave transients with Advanced LIGO, Advanced Virgo and KAGRA. Living Rev. Relat. 2018;21:3. doi: 10.1007/s41114-018-0012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravanis A.M., Pyle J.L., Tsien R.W. Single synaptic vesicles fusing transiently and successively without loss of identity. Nature. 2003;423:643–647. doi: 10.1038/nature01686. [DOI] [PubMed] [Google Scholar]
- Axelrod D. Total internal reflection fluorescence microscopy in cell biology. Traffic. 2001;2:764–774. doi: 10.1034/j.1600-0854.2001.21104.x. [DOI] [PubMed] [Google Scholar]
- Balaji J., Ryan T.A. Single-vesicle imaging reveals that synaptic vesicle exocytosis and endocytosis are coupled by a single stochastic mode. Proc. Natl. Acad. Sci. U S A. 2007;104:20576–20581. doi: 10.1073/pnas.0707574105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berning S., Willig K.I., Steffens H., Dibaj P., Hell S.W. Nanoscopy in a living mouse brain. Science. 2012;335:551. doi: 10.1126/science.1215369. [DOI] [PubMed] [Google Scholar]
- Betz W.J., Bewick G.S. Optical analysis of synaptic vesicle recycling at the frog neuromuscular junction. Science. 1992;255:200–203. doi: 10.1126/science.1553547. [DOI] [PubMed] [Google Scholar]
- Bittova L., Sumandea M., Cho W. A structure-function study of the C2 domain of cytosolic phospholipase A2. Identification of essential calcium ligands and hydrophobic membrane binding residues. J. Biol. Chem. 1999;274:9665–9672. doi: 10.1074/jbc.274.14.9665. [DOI] [PubMed] [Google Scholar]
- Coggins M.R., Grabner C.P., Almers W., Zenisek D. Stimulated exocytosis of endosomes in goldfish retinal bipolar neurons. J. Physiol. 2007;584:853–865. doi: 10.1113/jphysiol.2007.140848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani A., Huang B., Bergan J., Dulac C., Zhuang X. Superresolution imaging of chemical synapses in the brain. Neuron. 2010;68:843–856. doi: 10.1016/j.neuron.2010.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darcy K.J., Staras K., Collinson L.M., Goda Y. Constitutive sharing of recycling synaptic vesicles between presynaptic boutons. Nat. Neurosci. 2006;9:315–321. doi: 10.1038/nn1640. [DOI] [PubMed] [Google Scholar]
- Donnert G., Keller J., Medda R., Andrei M.A., Rizzoli S.O., Luhrmann R., Jahn R., Eggeling C., Hell S.W. Macromolecular-scale resolution in biological fluorescence microscopy. Proc. Natl. Acad. Sci. U S A. 2006;103:11440–11445. doi: 10.1073/pnas.0604965103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale J.E., Marcotti W., Kennedy H.J., Kros C.J., Richardson G.P. FM1-43 dye behaves as a permeant blocker of the hair-cell mechanotransducer channel. J. Neurosci. 2001;21:7013–7025. doi: 10.1523/JNEUROSCI.21-18-07013.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi S.P., Stevens C.F. Three modes of synaptic vesicular recycling revealed by single-vesicle imaging. Nature. 2003;423:607–613. doi: 10.1038/nature01677. [DOI] [PubMed] [Google Scholar]
- Gould T.J., Hess S.T., Bewersdorf J. Optical nanoscopy: from acquisition to analysis. Annu. Rev. Biomed. Eng. 2012;14:231–254. doi: 10.1146/annurev-bioeng-071811-150025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gramlich M.W., Klyachko V.A. Actin/Myosin-V- and activity-dependent inter-synaptic vesicle exchange in central neurons. Cell Rep. 2017;18:2096–2104. doi: 10.1016/j.celrep.2017.02.010. [DOI] [PubMed] [Google Scholar]
- Gramlich M.W., Klyachko V.A. Nanoscale organization of vesicle release at central synapses. Trends Neurosci. 2019;42:425–437. doi: 10.1016/j.tins.2019.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gramlich M.W., Conway L., Liang W.H., Labastide J.A., King S.J., Xu J., Ross J.L. Single molecule investigation of kinesin-1 motility using engineered microtubule defects. Sci. Rep. 2017;7:44290. doi: 10.1038/srep44290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson M.G., Shao L., Carlton P.M., Wang C.J., Golubovskaya I.N., Cande W.Z., Agard D.A., Sedat J.W. Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophys. J. 2008;94:4957–4970. doi: 10.1529/biophysj.107.120345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao M., Maxfield F.R. Characterization of rapid membrane internalization and recycling. J. Biol. Chem. 2000;275:15279–15286. doi: 10.1074/jbc.275.20.15279. [DOI] [PubMed] [Google Scholar]
- Harata N., Ryan T.A., Smith S.J., Buchanan J., Tsien R.W. Visualizing recycling synaptic vesicles in hippocampal neurons by FM 1-43 photoconversion. Proc. Natl. Acad. Sci. U S A. 2001;98:12748–12753. doi: 10.1073/pnas.171442798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M., Raimondi A., O'Toole E., Paradise S., Collesi C., Cremona O., Ferguson S.M., De Camilli P. Cell- and stimulus-dependent heterogeneity of synaptic vesicle endocytic recycling mechanisms revealed by studies of dynamin 1-null neurons. Proc. Natl. Acad. Sci. U S A. 2008;105:2175–2180. doi: 10.1073/pnas.0712171105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt M., Cooke A., Neef A., Lagnado L. High mobility of vesicles supports continuous exocytosis at a ribbon synapse. Curr. Biol. 2004;14:173–183. doi: 10.1016/j.cub.2003.12.053. [DOI] [PubMed] [Google Scholar]
- Hua Y., Sinha R., Thiel C.S., Schmidt R., Huve J., Martens H., Hell S.W., Egner A., Klingauf J. A readily retrievable pool of synaptic vesicles. Nat. Neurosci. 2011;14:833–839. doi: 10.1038/nn.2838. [DOI] [PubMed] [Google Scholar]
- Joensuu M., Padmanabhan P., Durisic N., Bademosi A.T., Cooper-Williams E., Morrow I.C., Harper C.B., Jung W., Parton R.G., Goodhill G.J., et al. Subdiffractional tracking of internalized molecules reveals heterogeneous motion states of synaptic vesicles. J. Cell Biol. 2016;215:277–292. doi: 10.1083/jcb.201604001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joselevitch C., Zenisek D. Imaging exocytosis in retinal bipolar cells with TIRF microscopy. J. Vis. Exp. 2009:1305. doi: 10.3791/1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joselevitch C., Zenisek D. Direct observation of vesicle transport on the synaptic ribbon provides evidence that vesicles are mobilized and prepared rapidly for release. J. Neurosci. 2020;40:7390–7404. doi: 10.1523/JNEUROSCI.0605-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juette M.F., Gould T.J., Lessard M.D., Mlodzianoski M.J., Nagpure B.S., Bennett B.T., Hess S.T., Bewersdorf J. Three-dimensional sub-100 nm resolution fluorescence microscopy of thick samples. Nat. Methods. 2008;5:527–529. doi: 10.1038/nmeth.1211. [DOI] [PubMed] [Google Scholar]
- Kavalali E.T., Jorgensen E.M. Visualizing presynaptic function. Nat. Neurosci. 2014;17:10–16. doi: 10.1038/nn.3578. [DOI] [PubMed] [Google Scholar]
- Martineau M., Somasundaram A., Grimm J.B., Gruber T.D., Choquet D., Taraska J.W., Lavis L.D., Perrais D. Semisynthetic fluorescent pH sensors for imaging exocytosis and endocytosis. Nat. Commun. 2017;8:1412. doi: 10.1038/s41467-017-01752-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maschi D., Klyachko V.A. Spatiotemporal dynamics of multi-vesicular release is determined by heterogeneity of release sites within central synapses. eLife. 2020;9:e55210. doi: 10.7554/eLife.55210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midorikawa M., Tsukamoto Y., Berglund K., Ishii M., Tachibana M. Different roles of ribbon-associated and ribbon-free active zones in retinal bipolar cells. Nat. Neurosci. 2007;10:1268–1276. doi: 10.1038/nn1963. [DOI] [PubMed] [Google Scholar]
- Miesenbock G., De Angelis D.A., Rothman J.E. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- Mlodzianoski M.J., Schreiner J.M., Callahan S.P., Smolkova K., Dlaskova A., Santorova J., Jezek P., Bewersdorf J. Sample drift correction in 3D fluorescence photoactivation localization microscopy. Opt. express. 2011;19:15009–15019. doi: 10.1364/OE.19.015009. [DOI] [PubMed] [Google Scholar]
- Murthy V.N., Stevens C.F. Synaptic vesicles retain their identity through the endocytic cycle. Nature. 1998;392:497–501. doi: 10.1038/33152. [DOI] [PubMed] [Google Scholar]
- Murthy V.N., Stevens C.F. Reversal of synaptic vesicle docking at central synapses. Nat. Neurosci. 1999;2:503–507. doi: 10.1038/9149. [DOI] [PubMed] [Google Scholar]
- Nalefski E.A., Slazas M.M., Falke J.J. Ca2+-signaling cycle of a membrane-docking C2 domain. Biochemistry. 1997;36:12011–12018. doi: 10.1021/bi9717340. [DOI] [PubMed] [Google Scholar]
- Nalefski E.A., McDonagh T., Somers W., Seehra J., Falke J.J., Clark J.D. Independent folding and ligand specificity of the C2 calcium-dependent lipid binding domain of cytosolic phospholipase A2. J. Biol. Chem. 1998;273:1365–1372. doi: 10.1074/jbc.273.3.1365. [DOI] [PubMed] [Google Scholar]
- Nishikawa S., Sasaki F. Internalization of styryl dye FM1-43 in the hair cells of lateral line organs in Xenopus larvae. J. Histochem. Cytochem. 1996;44:733–741. doi: 10.1177/44.7.8675994. [DOI] [PubMed] [Google Scholar]
- Park C., Chen X., Tian C.L., Park G.N., Chenouard N., Lee H., Yeo X.Y., Jung S., Tsien R.W., Bi G.Q., Park H. Unique dynamics and exocytosis properties of GABAergic synaptic vesicles revealed by three-dimensional single vesicle tracking. Proc. Natl. Acad. Sci. U S A. 2021;118 doi: 10.1073/pnas.2022133118. e2022133118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perisic O., Fong S., Lynch D.E., Bycroft M., Williams R.L. Crystal structure of a calcium-phospholipid binding domain from cytosolic phospholipase A2. J. Biol. Chem. 1998;273:1596–1604. doi: 10.1074/jbc.273.3.1596. [DOI] [PubMed] [Google Scholar]
- Pyle J.L., Kavalali E.T., Piedras-Renteria E.S., Tsien R.W. Rapid reuse of readily releasable pool vesicles at hippocampal synapses. Neuron. 2000;28:221–231. doi: 10.1016/s0896-6273(00)00098-2. [DOI] [PubMed] [Google Scholar]
- Ramirez D.M., Khvotchev M., Trauterman B., Kavalali E.T. Vti1a identifies a vesicle pool that preferentially recycles at rest and maintains spontaneous neurotransmission. Neuron. 2012;73:121–134. doi: 10.1016/j.neuron.2011.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revelo N.H., Kamin D., Truckenbrodt S., Wong A.B., Reuter-Jessen K., Reisinger E., Moser T., Rizzoli S.O. A new probe for super-resolution imaging of membranes elucidates trafficking pathways. J. Cell Biol. 2014;205:591–606. doi: 10.1083/jcb.201402066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C., Stevens C.F. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- Ryan T.A., Reuter H., Smith S.J. Optical detection of a quantal presynaptic membrane turnover. Nature. 1997;388:478–482. doi: 10.1038/41335. [DOI] [PubMed] [Google Scholar]
- Ryan T.A., Reuter H., Wendland B., Schweizer F.E., Tsien R.W., Smith S.J. The kinetics of synaptic vesicle recycling measured at single presynaptic boutons. Neuron. 1993;11:713–724. doi: 10.1016/0896-6273(93)90081-2. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S., Ryan T.A. Real-time measurements of vesicle-SNARE recycling in synapses of the central nervous system. Nat. Cell Biol. 2000;2:197–204. doi: 10.1038/35008615. [DOI] [PubMed] [Google Scholar]
- Seitz K.J., Rizzoli S.O. GFP nanobodies reveal recently-exocytosed pHluorin molecules. Sci. Rep. 2019;9:7773. doi: 10.1038/s41598-019-44262-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin W., Ge L., Arpino G., Villarreal S.A., Hamid E., Liu H., Zhao W.D., Wen P.J., Chiang H.C., Wu L.G. Visualization of membrane pore in live cells reveals a dynamic-pore theory governing fusion and endocytosis. Cell. 2018;173:934–945.e12. doi: 10.1016/j.cell.2018.02.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagi M., Fogel A.I., Biederer T. SynCAM 1 participates in axo-dendritic contact assembly and shapes neuronal growth cones. Proc. Natl. Acad. Sci. USA. 2010;107:7568–7573. doi: 10.1073/pnas.0911798107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stout A.L., Axelrod D. Evanescent field excitation of fluorescence by epi-illumination microscopy. Appl. Opt. 1989;28:5237–5242. doi: 10.1364/AO.28.005237. [DOI] [PubMed] [Google Scholar]
- Vaithianathan T., Henry D., Akmentin W., Matthews G. Nanoscale dynamics of synaptic vesicle trafficking and fusion at the presynaptic active zone. eLife. 2016;5:e13245. doi: 10.7554/eLife.13245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaithianathan T., Wollmuth L.P., Henry D., Zenisek D., Matthews G. Tracking newly released synaptic vesicle proteins at ribbon active zones. iScience. 2019;17:10–23. doi: 10.1016/j.isci.2019.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voglmaier S.M., Kam K., Yang H., Fortin D.L., Hua Z., Nicoll R.A., Edwards R.H. Distinct endocytic pathways control the rate and extent of synaptic vesicle protein recycling. Neuron. 2006;51:71–84. doi: 10.1016/j.neuron.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Westphal V., Rizzoli S.O., Lauterbach M.A., Kamin D., Jahn R., Hell S.W. Video-rate far-field optical nanoscopy dissects synaptic vesicle movement. Science. 2008;320:246–249. doi: 10.1126/science.1154228. [DOI] [PubMed] [Google Scholar]
- Willig K.I., Rizzoli S.O., Westphal V., Jahn R., Hell S.W. STED microscopy reveals that synaptotagmin remains clustered after synaptic vesicle exocytosis. Nature. 2006;440:935–939. doi: 10.1038/nature04592. [DOI] [PubMed] [Google Scholar]
- Zenisek D., Steyer J.A., Almers W. Transport, capture and exocytosis of single synaptic vesicles at active zones. Nature. 2000;406:849–854. doi: 10.1038/35022500. [DOI] [PubMed] [Google Scholar]
- Zenisek D., Steyer J.A., Feldman M.E., Almers W. A membrane marker leaves synaptic vesicles in milliseconds after exocytosis in retinal bipolar cells. Neuron. 2002;35:1085–1097. doi: 10.1016/s0896-6273(02)00896-6. [DOI] [PubMed] [Google Scholar]
- Zhang Q., Li Y., Tsien R.W. The dynamic control of kiss-and-run and vesicular reuse probed with single nanoparticles. Science. 2009;323:1448–1453. doi: 10.1126/science.1167373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W.D., Hamid E., Shin W., Wen P.J., Krystofiak E.S., Villarreal S.A., Chiang H.C., Kachar B., Wu L.G. Hemi-fused structure mediates and controls fusion and fission in live cells. Nature. 2016;534:548–552. doi: 10.1038/nature18598. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.