

Abstract
Introduction:
Spinocerebellar ataxias (SCA) are a group of rare neurodegenerative diseases that dramatically affect the lives of affected individuals and their families. Despite having a clear understanding of SCA’s etiology, there are no current symptomatic or neuroprotective treatments approved by the FDA.
Areas covered:
Research efforts have greatly expanded the possibilities for potential treatments, including both pharmacological and non-pharmacological interventions. Great attention is also being given to novel therapeutics based in gene therapy, neurostimulation, and molecular targeting. This review article will address the current advances in the treatment of SCA and what potential interventions are on the horizon.
Expert Opinion:
SCA is a highly complex and multifaceted disease family with the majority of research emphasizing symptomatic pharmacologic therapies. As pre-clinical trials for SCA and clinical trials for other neurodegenerative conditions illuminate the efficacy of disease modifying therapies such as AAV-mediated gene therapy and ASOs, the potential for addressing SCA at the pre-symptomatic stage is increasingly promising.
Keywords: Spinocerebellar ataxia, treatment, neurodegenerative diseases, gene therapy, neurostimulation
1. Introduction
Spinocerebellar ataxias (SCAs) are autosomal dominantly inherited, progressive neurodegenerative disorders marked by cerebellar degeneration[1]. SCAs are numbered in the order in which they were chronologically identified, with over 40 of them being genetically and phenotypically characterized (Table 1) [2]. Although SCAs are symptomatically heterogeneous disorders, they share ataxia as a core symptom. Other symptoms may include extrapyramidal and pyramidal signs, although at least one SCA, SCA6, solely involves the cerebellum[1]. Cognitive impairment can also be observed among patients with SCA, such as executive dysfunction, depression, verbal fluency, and memory [3,4]. Symptom onset in SCA patients usually occurs in early mid-life, although it may also manifest in childhood or older age [5]. In neuropathology analysis, neurodegeneration can be observed in the cerebellum but the interconnected nervous system, such as the brainstem, spinal cord, peripheral nerves, basal ganglia, and autonomic nerves, may also be affected [1,6].
Table 1.
| SCA Type | Locus | Gene/protein | Genetic etiology | Clinical Manifestations (all present with ataxia) |
|---|---|---|---|---|
| SCA 1 | 6p24-p23 | ATXN1/ Ataxin 1 | CAG triplet repeat expansion (36–81 repeats); mutant protein forms nuclear aggregates | dysarthria, bulbar dysfunction, increased tone, hyperreflexia; some patients present with generalized fasciculations and wasting of extremities |
| SCA 2 | 12q24.1 | ATXN2/ Ataxin 2 | CAG triplet repeat expansion (35–64 repeats); mutant protein forms cytosplasmic aggregates | slow saccadic eye movements, hypotonia, hyporeflexia, dysarthria, nystagmus |
| SCA 3 | 14q32 | ATXN3/ Ataxin 3 | CAG triplet repeat expansion(40–200 repeats); mutant protein forms nuclear aggregates | most common; slow saccades, upper and lower motor neuropathy symptoms–abnormalities in tone and reflexes – rigidity, dystonia, muscle cramps, sleep disorders |
| SCA 4 | 16q22.1 | Unknown | Unknown | Rare; Associated with exaggerated sensoral neuropathy and extensor plantar reflexes; auditory impairments, diplopia, dysarthroa and dysphagia |
| SCA 5 | 11q13.2 | SPTBN2/ beta-III spectrin | Heterozygous mutation; mutant Beta-III spectrin affects glutamate transport in Purkinje cells | slowly progressive form of ataxia; almost pure cerebellar syndrome; dysarthria, downbeat nystagmus, impaired vibration detection, impaired lateral gaze |
| SCA 6 | 19p13.13 | CACNA1A/ αlA (P/Qtype, α12.1) | CAG triplet repeat expansion (21–33 repeats); mutant αlA (P/Qtype, α12.1) subunit of voltage-gated calcium channel causes neuronal degeneration | Pure cerebellar ataxia; dysarthria, dysphagia, diplopia, horizontal and vertical nystagmus, impaired vestibuloocular reflex |
| SCA 7 | 3p14.1 | ATXN7/Ataxin-7 | CAG triplet repeat expansion(37–200 repeats) | Progressive macular degeneration specifically affecting color vision and central visual acuity-visual abnormalities can present before ataxia; slow saccades, sensory neuropathy, anticipation, seizures |
| SCA 8 | 13q21 | ATXN8 and ATXN8OS | CTG expansion in 3′untranslated region and CAG′ repeat expansion in open reading frame (80–250 repeats); toxic gain-of-function mRNA and protein | cerebellar ataxia that affects gait, speech, swallowing, eye movements, and limb function; manifests in saccades, hyperreflexia, nystagmus, and extensor plantar responses |
| SCA 10 | 22q13 | ATXN10/ Ataxin 10 | heterozygous ATTCT pentanucleotide repeat expansion (800–4500 repeats) | oculomotor impairments, scanning dysarthria, seizures, cognitive deficits, and peripheral neuropathy |
| SCA 11 | 15q14-q21.3 | TTBK2/tau-tubulin kinase-2 | heterozygous mutation resulting in tau protein deposition | mild, pure cerebellar ataxia; horizontal and vertical nystagmus, jerky pursuits |
| SCA 12 | 5q31–5q32 | PPP2R2B/ PPP2R2B | CAG repeat expansion (66–78 repeats) at 5′ UTR of PPP2R2B-encodes regulatory subunit of brain-specific protein phosphatase | pronounced action tremor of limbs and head, bradykinesia, hyperreflexia, sensorimotor neuropathy |
| SCA 13 | 19q13.13 | KCNC3/KCNC3 | heterozygous mutation (missense mutation) | intellectual disability, motor deficits, occasional seizures, hyperreflexia, pyramidal signs |
| SCA 14 | 19q13.42 | PRKCG /PKC-gamma | heterozygous mutation(missense mutation, deletion)- most like a gain of function mutation | very rare; dysarthria, episodic axial and multifocal myoclonus, parkinsonism, myokymia, sensory loss, and cognitive deficits |
| SCA 15/16 | 3p26.1 | ITPR1 gene/ inositol 1,4,5triphophsate receptor 1 | heterozygous mutations (missense mutation, deletion) | pure gait ataxia, dysphagia, dysarthria, kinetic and postural tremors in upper extremities, gaze-evoked nystagmus, impaired vestibulo ocular reflex gain, mild executive dysfunction |
| SCA 17 | 6q27 | TBP | CAG/CAA repeat expansion (37–200 repeats) in TBP, which encodes a transcription initiation factor | dementia, dysmetira, bradykinesia, hyperreflexia, dysdiadochokinesis, chorea, intellectual impairment, psychiatric symptoms, and parkinsonism |
| SCA 18 | 7q31-q32 | unknown (IFRD1 potential candidate) | unknown | sensorimotor neuropathy with ataxia; dysmetira, muscle atrophy, hyporeflexia, and pyramidal tract signs |
| SCA 19/22 | 1p21-q21 | KCND3 | heterozygous mutation(missense mutation, deletion) in KCND3- important in cerebellar development | dysphagia and dysarthria, gaze-evoked nystagmus, hyporeflexia, tremor, intellectual impairments, and myoclonus |
| SCA 20 | 11q12 | SCA-20 | unknown | dysarthria, gait/upper limb ataxia, dysphoniam palatal tremor, hypermetric horizontal saccades, and postural tremor of arms |
| SCA 21 | 7p15.1-p21.3 | TMEM240 | heterozygous mutations (missense and nonsense) | postural and resting tremor, dysarthria, dysphagia, hyporeflexia, delayed psychomotor development, intellectual impairment, and behavioral abnormalities(apathy, aggression, impulsivity) |
| SCA 23 | 20p13 | PDYN/prodynorphin | heterozygous mutations | gait and limb ataxia, sensory neuropathy, upper extremities and head tremor, slow saccades, ocular dysmetria, and dysarthria |
| SCA 25 | 2p15-p21 | SCA-25 | unknown | sensory neuropathy, facial tics and myokymia, decreased visual acuity, urinary problems, and gastrointestinal symptoms |
| SCA 26 | 19p13.3 | EEF2 | heterozygous mutation | gait and limb ataxia, irregular visual pursuit, and dysarthria |
| SCA 27 | 13q33.1 | FGF14 | heterozygous mutation results in altered protein stability | childhood onset low amplitude, high-frequency hand tremors, unsteady gait, disrupted visual pursuit, gaze-evoked nystagmus, cognitive deficits, behavioral problems |
| SCA 28 | 18p11.21 | AFG3L2/ ATP-dependent metalloprotease | heterozygous mutations (missense mutations, small insertions or deletions) | oculomotor impairments, pyramidal tract signs,slow saccades, dysarthria, hyperrflexia of lower extremities, ptosis, mitigated vibration detection at ankles |
| SCA 29 | 3p26.1 | ITPR1 | unknown | infantile onset of impaired motor development, learning disability, dysarthria, tremor, and nystagmus |
| SCA 30 | 4q34.3-q35.1 | Unknown/ ODZ3 potential candidate | unknown | dysarthria, hypermetric saccades, lower-limb hyperreflexia, and minor pyramidal signs |
| SCA 31 | 16q21 | BEAN1/ BEAN 1 | non-coding pentanucleotide repeat insertion at Alu sequence | dyarthria, horizontal gaze nystagmus, decreased musce tone, and hearing loss |
| SCA 32 | 7q32-q33 | SCA-32 | unknown | impaired intellect, azoospermia in males |
| SCA 34 | 6q14.1 | ELOVL4/very-long-chain fatty ac | heterozygous missense mutations- loss of function imutation | neurocutaneous syndromic ataxia with childhood onset- erythrokeratodermia; at later age, severe gait ataxia, nystagmus dysarthria, mitigated tendon reflexes |
| SCA 35 | 20p13 | TGM6/ transglutaminase 6 | heterozygous mutations | gait and limb ataxia, intention tremor, dysarthria, ocular dysmetria, saccadic eye movements, hyperreflexia, torticollis, and some intellectual deficits |
| SCA 36 | 20p13 | NOP56 | pathogenic heterozygous GGCCTG repeat expansion in intron 1 (650 to 2500 repeats) | adult onset gait ataxia, tongue atrophy and fasciculations, dysarthria, oculomotor apraxia, hearing loss, motor neuron degeneration, and nystagmus |
| SCA 37 | 1p32.2 | DAB1 | pathogenic pentanucleotide ATTTC insertion within the 1p32 5′ non-coding regulatory region | early onset of dysmetric vertical saccades and irregular vertical pursuit, dysarthria, and some nystagmus |
| SCA 38 | 6p12.1 | ELOVL5 | heterozygous missense mutation | nystagmus, slow saccades, dysarthria, and distal sensory impairment |
| SCA 40 | 14q32 | CCDC88C | heterozygous missense mutation (R464H) in the coding region of the coiled-coil domain containing 88C (CCDC88C) gene | unsteady gait, dysarthria, intention tremor, ocular dysmetria, dysdiadochokinesis, hyperreflexia, and spastic paraparesis |
| SCA 41 | 4q27 | TRPC3 | heterozygous missense mutation | progressive imbalance and gait ataxia |
| SCA 42 | 17q21.33 | CACNA1G | heterozygous missense mutation | dysarthria, diplopia, saccadic persuits, decreased vibration detection, mild cognitive deficits, nystagmus, resting tremor, and mild pyramidal signs |
| SCA 43 | 3q25.2 | MME | heterozygous missense mutation | gait and limb ataxia, sensorimotor axonal polyneuropathy, pectus carinatum, pes cavus, hypometric saccades, and parkinsonism |
| SCA 44 | 6q24.3 | GRM1 | heterozygous missense mutation | childhood onset ataxia, cognitive impairments, spasticity |
| SCA 45 | 5q33.1 | FAT2 | heterozygous missense mutation | limb and gait ataxia, nystagmus, and dysarthria |
| SCA 46 | 19q13.2 | PLD3 | heterozygous missense mutation | adult onset oculomotor abnormalities and sensory impairment/ ataxia neuropathy |
| SCA 47 | 1p35.2 | PUM1 | heterozygous missense mutation | gait ataxia, dysarthria, dysmetria, some diplopia |
| SCA 48 | 16p13.3 | STUB1 | heterozygous mutation (missense and frameshift mutation) | dysarthria, dysphagia, executive function impairments, behavioral abnormalities, parkinsonism, chorea, dystonia, tremor |
The most common SCAs are caused by polyglutamine (poly Q)-encoding CAG repeat expansions in respective genes, which cause dysfunctional conformation of protein structure and subsequent aggregation and intranuclear inclusions [10]. The length of the repeats in these polyglutamine SCAs correlates with aggregation tendency and inversely with age of onset; increasing repeat length is linked with earlier age of onset and increased aggregation tendency [1,6]. The exact role of inclusions is unclear, but the dysfunctional confirmation of the polyglutamine disease protein causes neuronal stress and disruption of cellular homeostasis [6,10]. SCAs with polyglutamine repeat expansions (SCA 1, 2, 3, 6, 7, and 17) tend to exhibit ‘anticipation,’ such that ensuing generations suffer earlier onset and more severe symptoms [1]. While SCA12 is also caused by pathological expanded CAG repeats, these CAG repeats are located in the untranslated region; therefore, SCA12 is not considered a polyglutamine SCA [11]. The non-polyglutamine SCA diseases may also cause altered protein function via point mutations and repeat expansions in noncoding regions, as proteotoxicity appears to play a role in disease pathogenesis [6]. Mitochondrial dysfunction and voltage-dependent ion channel dysfunction of cerebellar neurons also contribute to SCA pathophysiology [12,13].
In the absence of a cure for SCAs, symptoms of the disease should be treated if they interfere with a patient’s quality of life. In addition to ataxia, other symptoms that can be mitigated with treatment include spasticity, tremor, dystonia, pain, bladder dysfunction, mood disorders such as depression and anxiety, and sleep dysfunction. Potential therapies such as medications, botulinum toxin, physical and occupational therapy may be considered. Occupational and physical therapy are of utmost importance in SCA patients to treat debilitating symptoms and increase quality of life; speech therapy is also critical, as dysphagia may lead to aspiration. Furthermore, diet and nutrition may play a critical role in overall wellness and quality of life for SCA patients.
There are several rating scales that are used to evaluate symptomatic severity as well as overall disease progression for SCAs [14]. These scales often function as measures of treatment efficacy in clinical trials. One of the most commonly used scales is the Scale for the Assessment and Rating of Ataxia (SARA) [14]. The SARA has eight parts that measure gait, sitting, stance, speech, finger chase, nose-finger test, fast alternative hand movements, and heel shin slide [14]. This is the most widely used scale in assessment of SCA due to the high reliability – both inter-rater and test-retest [14]. The other popular scale for measuring ataxia severity is the International Cooperative Ataxia Rating Scale (ICARS). The ICARS has 19 parts with 4 subgroups of posture and gait disturbance, dysarthria, limb ataxia, and oculomotor disorders [14]. In order to evaluate additional movement disturbances that are not ataxia related, the Neurological Examination Score for the Assessment of Spinocerebellar Ataxia (NESSCA) is utilized [14]. This scale includes assessment of visual disturbances, such as optic atrophy and eyelid retraction, parkinsonism – bradykinesia and rigidity–, fasciculations, sensory loss, and vertigo [14]. In addition to the aforementioned scales, there are two common performance assessments used to evaluate SCA: the Composite Cerebellar Functional Severity Score (CCFS) and the SCA Functional Index (SCAFI) [14]. These scales provide more information about SCA patients’ functional performance and coordination [14]. Both are composed of varying activities, including the 9-hole peg test and the click test; the 8-m walk is conducted as part of the SCAFI but not the CCFS [14].
Currently, there are no symptomatic or neuroprotective treatments approved by the United States (US) Food and Drug Administration (FDA) for SCAs, although research has dramatically expanded in the past decade (Table 2). Novel future treatments of SCAs may include gene therapy, clustered regularly interspaced short palindromic repeats (CRISPR) gene editing, stem cell therapy, antisense oligonucleotides (ASOs), and pharmacologic agents. Several ‘off-label’ medications have shown some promise in a few randomized, double-blind, placebo-controlled studies, although their efficacy has not been firmly established. The goal of this review is to evaluate multiple avenues of therapeutic intervention for SCA and the potential efficacy of such treatments.
Table 2.
Therapeutic interventions for treatment of spinocerebellar ataxias and notable clinical trials.
| Name | Notable Clinical Trials | SCA Types | Intervention | Outcome |
|---|---|---|---|---|
| SYMPTOMATIC INTERVENTION | ||||
| Pharmacologics | ||||
| Riluzole | Ristori et al [15] | 1, 2, 28 | 100 mg daily | The proportion of patients who experienced a 5-point decrease in ICARS score was significantly higher in patients taking riluzole compared to the placebo group after four weeks (9/19 vs. 1/19; odds ratio [OR] 16.2; 95% confidence interval [CI] 1.8–147.1) and eight weeks (13/19 vs. 1/19; OR 39.0; 95% CI 4.2–364.2)[16]. There was also a significant mean change in the total ICARS in the riluzole group compared to placebo (7.05 [4.96] vs. 0.16 [2.65] (p < 0.001)) |
| Romani et al [16] | NR(38 subjects with SCA) | 50 mg twice daily | The study found that 50% (14/28) of patients in the riluzole group had a one point decrease in SARA score compared to 11% (3/27) of patients in the placebo group (OR 8.00, 95% CI 1.95–32.83; p = 0.002) | |
| Thyrotropin Releasing Hormone | Sobue et al [35] | NR(254 patients with SCD) | 2 mg or .5 mg daily | Overall, patients randomized to receive TRH 2 mg had better standing, gait, speech, and writing when compared to the placebo group. However, about 50% of patients taking 2 mg TRH had adverse effects, including headache and nausea |
| Rotaviterelin | Nishizawa et al (2 trials) [36] | 6, 31 | 1.6 mg or 2.4 mg daily | in the pooled analysis of data from the two studies, patients presented a more significant reduction in SARA total score when taking rovatirelin compared with placebo (1.64 vs. 1.03; 95% CI- 1.16 to 0.06; p = 0.029) |
| Varenicline | Zesiewicz et al [41] | 3 | 1 mg twice daily | Patients taking varenicline experienced improvements in the SARA subsections for gait (p = 0.04), stance (p = 0.03), rapid alternating movements (p = 0.003), timed 25-foot walk (p = 0.05) and Beck Depression Inventory scores (p = 0.03) compared to patients taking placebo |
| Buspirone | Assadi et al [47] | 1,2,3,6,17 | 30 mg twice daily | Buspirone was not found to be superior to placebo: ICARS scores (baseline = 42.63 ± 18.89; placebo = 46 ± 18.98 buspirone = 44.26 ± 19.64, p =0.24) |
| Valproic Acid | Lei et al [48] | 3 | 800 mg or 1200 mg daily | The mean change in the total SARA total score was significantly greater in the 1200-mg/d group(−2.05) versus the 800-mg/d (−1.58) and the placebo (−0.75) groups (analysis of variance p = 0.021) |
| Lithium | Saute et al [52] | 3 | 300 mg daily | While lithium was determined to be safe and tolerable, it had no statistically significant effect on overall NESSCA scores (p =0.222). The lithium treatment group presented statistically significant progression in secondary outcome assessments, including the nondominant Click Test (p =0.023), the PATA rate test (p =0.002), the SCAFI (p=0.003), and the CCFS (p =0.029) |
| Sacca et al [53] | 2 | 150 mg twice daily(up to 1500 mg) | Lithium was found to be safe but did not demonstrate significant differences in SARA scores. BDI-II score significantly decreased in lithium group (p < 0.05). | |
| Amantadine | Botez et al [55] | None (FRDA and MSA-C | 200 mg daily | Significant improvement was noted in MSA-C but not in FRDA |
| Acetazolamide | Yabe et al [62] | 6 | 500 mg daily | Cerebral ataxia was shown to be reduced with acetazolamide treatment, as evidenced by a statistically significant decrease in total ataxia rating scores, and posture and kinetics subscores (p < 0.05) |
| Trehalose | Noorsaykin et al [66] | 3 | 100 mg daily | Results demonstrated statistically significant improvements in SARA scores (p = 0.05) and 8-minute walking test scores(p = 0.007) |
| Non-Pharmacologic Rehabilitative Therapy | Miyai et al [67] | 6,31 | PT and OT, 2 hours on weekdays and 1 hour on weekends | The immediate treatment group demonstrated significantly greater improvements in SARA score in the short-term, especially in truncal ataxia, compared with the delayed-entry treatment group over the 4 weeks (p < 0.001). In addition, at the 24-week follow-up, more than 50% of all individuals maintained these improvements. |
| Bastian et al [68] | 3,5,6,8,17 | Home balance exercises | Neurological and functional assessments done 5 times over the course of the exercise program revealed significant improvements in walking speed, dynamic gait index, stride length, and percent double limb support time (p < 0.05). However, no change was seen in ICARS scores or ABC scores | |
| Non-Invasive Neurostimulation | Shiga et al [73] | 1,3,6 | rTMS | In comparing the active TMS and sham groups, improvement of the TMS group was found to be statistically significant for the timed 10-meter walk, 10-meter step assessment, and standing capacity (p < 0.05). Among the active-TMS group, blood flow to the pons and cerebellum was significantly increased (p < 0.005) but no significant increase was noted in the cerebral cortices. |
| Manor et al [74] | 1,2,3,6,8,13 | rTMS | Subjects receiving rTMS demonstrated a significant improvement in the SARA sub-score for “stance” (p = 0.002) from baseline to 1-month follow-up when compared to the sham group. No significant changes were observed in the 9-hole peg test or TUG assessment | |
| Franca et al [75] | 3 | rTMS | The rTMS group was shown to have statistically significant improvement in SARA scores (median 10.2 for rTMS vs. median 12.8 for sham (p = 0.002)). Improvements in ICARS scores were also noted to be significant between rTMS and sham groups (median 29.0 for rTMS and 32.8 for sham (p = 0.005) | |
| Benussi et al 2015 [78] | 1,2,38 | tDCS | In comparing the sham and tDCS group, tDCS subjects demonstrated a statistically significant improvement in scores measured pre and post stimulation for all assessments (p < 0.001 for SARA, ICARS, and 8-meter walk; p = 0.012 for 9-hole-peg test) | |
| Benussi et al 2018 [79] | 6,14,38 | tDCS | Statistically significant improvements in functional performance assessments of SARA and ICARS(p < 0.001) | |
| BCAAs | Mori et al [85] | 6,7 | 1.5,3.0, or 6.0 mg | When comparing placebo and treatment groups, the ICARS scores significantly improved (p < 0.01) from pre- to post-treatment |
2. Symptomatic therapies
2.1. Pharmacologics
2.1.1. Riluzole
Riluzole, a drug used to treat amyotrophic lateral sclerosis (ALS), improved cerebellar symptoms in patients with various types of degenerative ataxia in two small clinical trials [15,16]. The proposed mechanism of action for riluzole is to open calcium-activated potassium channels that regulate the firing of deep cerebellar neurons and/or Purkinje cells, thus decreasing neuronal hyperexcitability. It also appears to interrupt glutamatergic transmission, thus offering potential protection against excitotoxic neurodegeneration [17].
In one randomized, double-blind, placebo-controlled pilot trial, 40 cerebellar ataxia patients were randomized to receive either riluzole (100 mg/day) or a placebo for 8 weeks [15]; 8 of these patients had diagnoses of SCAs (2 SCA1, 4 SCA2, 2 SCA28), and the rest presented with Friedreich’s ataxia (FRDA), sporadic ataxia, multiple-system atrophy type C (MSA-C), and ataxias of unknown origin [15]. The study outcome measures included the difference between placebo and riluzole groups in the proportion of patients who experienced a decrease of at least 5 points in the ICARS total score after 4 and 8 weeks relative to baseline, the difference in mean ICARS score changes after 8 weeks, and the difference in tolerability and safety [15]. The proportion of patients who experienced a 5-point decrease in ICARS score was significantly higher in patients taking riluzole compared to the placebo group after 4 weeks (9/19 vs. 1/19; odds ratio [OR] 16.2; 95% confidence interval [CI] 1.8–147.1) and 8 weeks (13/19 vs. 1/19; OR 39.0; 95% CI 4.2–364.2) [15]. There was also a significant mean change in the total ICARS in the riluzole group compared to placebo (7.05 [4.96] vs. 0.16 [2.65] (p < 0.001)). Adverse events were mild, although riluzole use requires regular liver function monitoring [15,18].
The second study was a 12-month, double-blind, placebo-controlled trial (NCT01104649) [16] that randomized 55 patients, 38 with genetically confirmed SCA and 17 with FRDA, to either the riluzole (50 mg orally, twice daily) or placebo group. The primary endpoint of the study was the proportion of patients with a decrease of at least one point in their SARA score [16]. The study found that 50% (14/28) of patients in the riluzole group met this endpoint compared to 11% (3/27) of patients in the placebo group (OR 8.00, 95% CI 1.95–32.83; p = 0.002) [16]. Riluzole was well tolerated with no serious adverse events [16]. Currently, a prodrug of riluzole, troriluzole, is being tested in SCA patients in a study sponsored by Biohaven Pharmaceuticals, Inc [19].
Several SCAs – SCA15, SCA19/22, SCA13 – have been directly associated with mutations in ion channels, such as SK and BK ion channels, which are both calcium activated potassium channels [13]. Other SCAs, such as SCA1, SCA2, and SCA3, have demonstrated ion channel dysfunction that is secondary to the disease-causing polyglutamine repeats, but still plays a role in disease manifestation [13]. Modulation of these ion channels has demonstrated positive results in SCA1 [20], SCA2 [21,22], and SCA3 mice models [23], including improvements in performance in rotarod and balance beam walk tests, increased neuronal excitability, and decreased Purkinje cell death. In addition, the issue of deranged calcium signaling has been highlighted as a potential pathogenic mechanism for SCA, which may also be helped by ion channel modulators [24–31]. In this same vein, a recent study demonstrating that riluzole molecules can bind to an intracellular allosteric site of the SK2 ion channel has revealed critical information about riluzole’s mechanism of action [32]. The combination of chlorzoxazone and baclofen has also been shown to be helpful in treating ion channel dysfunction linked to misfiring of Purkinje cells in animal models [33]. Considering the common theme of ion channel dysfunction, modulators like riluzole should be given increased attention as potential therapeutic modalities [13].
2.1.2. Thyrotropin-releasing hormone (TRH)
TRH promotes thyroid-stimulating hormone in the pituitary and promotes prolactin release [34]. Several case reports in the 1980s found anecdotal improvement of ataxia with TRH use [35]. One study published in 1983 evaluated the efficacy of TRH in 254 patients with ‘spinocerebellar degeneration’ (SCD) in a two-week double-blind, placebo-controlled trial [35]. Patients were randomized to receive either TRH intramuscularly (2 mg or 0.5 mg) or a placebo daily [35]. The primary endpoints included a ‘global improvement rating’ and an ‘ataxia improvement rating,’ which both showed significant improvements in patients taking TRH 2 mg or 0.5 mg compared to placebo [35].
Overall, patients randomized to receive TRH 2 mg had better standing, gait, speech, and writing when compared to the placebo group [35]. However, about 50% of patients taking 2 mg TRH had adverse effects, including headache and nausea [35]. While the results were favorable, the study pre-dated genetic testing, and many patients in the study were thought to have ‘olivopontocerebellar atrophy’ [35,36]. The study also did not utilize a currently validated clinical rating scale and the follow-up time for the study endpoint was a mere 2 weeks [35,36]. Nevertheless, an analog of TRH was approved in Japan in 1985 to treat ataxia associated with SCD [36]. An American Academy of Neurology consensus guideline noted that TRH ‘possibly improves some signs of ataxia over 10–14 days’[37].
Another analog of TRH, rovatirelin, improved ataxia in a rolling Nagoya mice model carrying the mutation in the CACNA1A gene [38]. Two randomized, double-blind clinical trials (KPS1301 and KPS1305) investigated the safety and efficacy of rovatirelin as a treatment of cerebellar ataxia [36]. In KPS1301, 165 patients had diagnoses of SCA6 and 72 had SCA31 diagnoses; in KPS1304, 83 and 57 patients had diagnoses of SCA6 and SCA31, respectively [36]. Patients with cerebellar ataxia, including SCA6, SCA31, or cortical cerebellar atrophy, were enrolled in the studies and randomized to rovatirelin 1.6 mg, 2.4 mg, or placebo in KPS1301, and 2.4 mg or placebo in KPS1305 [36]. The primary endpoint for both studies was the change in the total SARA score in the rovatirelin group relative to placebo [36]. There were no significant changes in the SARA between rovatirelin or placebo in either study [36]. However, in the pooled analysis of data from the two studies, patients presented a more significant reduction in SARA total score when taking rovatirelin compared with placebo (1.64 vs. 1.03; 95% CI- 1.16 to 0.06; p = 0.029) [36]. The mechanism of action behind TRH therapy is unclear. One study found that TRH was associated with increased cerebellar regional cerebral blood flow (rCBF) [39].
2.1.3. Varenicline
Varenicline (Chantix; Pfizer, New York, NY) is used as a smoking cessation drug that acts as a partial agonist at α4β2 nicotinic acetylcholine receptors [40]. Case reports have noted that varenicline improved cerebellar symptoms in various types of ataxia [41,42]. In a double-blind, placebo controlled study, 20 SCA3 patients (mean age = 51 ± 10.98 years; mean disease duration = 14 ± 9.82 years; mean SARA score = 16.13 ± 4.67) were randomized to either varenicline (4 weeks for titration and 4 weeks at a dose of 1 mg twice daily) or placebo [41]. The primary outcome measure was the change in the SARA scale at the study endpoint (8 weeks) compared to baseline [41]. Patients taking varenicline experienced improvements in the SARA subsections for gait (p = 0.04), stance (p = 0.03), rapid alternating movements (p = 0.003), timed 25-foot walk (p = 0.05) and Beck Depression Inventory scores (p = 0.03) compared to patients taking placebo [41]. However, there was a 40% dropout rate in the placebo group, and 20% of subjects in the varenicline group discontinued the study; nausea was the most common side effect [41]. The American Academy of Neurology guidelines in cerebellar dysfunction reported Class II evidence that varenicline improved the axial functions of gait, stance, and timed 25-foot walk-in adult patients with genetically confirmed SCA3; however, the evidence was determined insufficient to conclude efficacy of varenicline in treatment of SCA 3 [37].
The effect of nicotinic agonists in treating some of the behavioral deficits in olivocerebellar ataxia was evaluated in one preclinical study of rats that underwent destruction of their olivocerebellar pathways with 3-AP [43]. Nicotine administered daily (0.33 mg free base/kg) improved rotarod performance by 50% following the first week after 3-AP administration [43]. Varenicline in doses of 1.0 and 3.0 mg free base/kg daily also improved rotarod performance by about 50% after the first week of administration [43]. Additional research is needed to confirm these findings.
2.1.4. Buspirone
Buspirone is a 5-HT1A and dopamine D2 agonist anxiolytic that has been evaluated as treatment for ataxia with mixed results [44]. The use of buspirone in ataxia was based on evidence of extensive cerebellar serotonergic innervation [45,46]. Several early case studies found buspirone to mitigate gait and leg ataxia in patients with various types of cerebellar degeneration [44–46]. However, one double-blind, placebo-controlled trial in 20 subjects – 10 with genetically confirmed SCAs (1 SCA1, 5 SCA2, 2 SCA3, 1 SCA6, 1 SCA17) – which randomized patients to either buspirone 30 mg twice daily or placebo for 3 months – did not find buspirone superior to placebo [47].
2.1.5. Valproic acid (VPA)
VPA is an anticonvulsant and histone deacetylase (HDAC) inhibitor used to treat both seizures and bipolar disorder, that demonstrated positive results in a randomized, double-blind, placebo-controlled study in 12 SCA3 patients [48]. Patients were randomized to low dose VPA (800 mg/day), high-dose VPA (1200 mg/day), or placebo for 12 weeks [48]. The mean change in the total SARA total score was significantly greater in the 1200-mg/d group (−2.05) versus the 800-mg/d (−1.58) and the placebo (−0.75) groups (analysis of variance p = 0.021) [48]. Mild-to-moderate side effects included dizziness and gastrointestinal issues. VPA has also been found to mitigate locomotor deficits, in a Drosophila model of SCA3 [48].
The mechanism of action is unclear but may involve VPA’s action as an HDAC inhibitor. However, a bothersome side effect of valproic acid is tremor, which may already be a symptom in SCA patients. Prevalence of tremor among SCA patients is highest in those with SCA2, SCA3, and SCA6 and correlated with more severe ataxia [49,50]. A literature review of randomized controlled trials utilizing valproic acid as an intervention found an overall tremor incidence of 14%, correlating with higher dose and treatment duration [51]. While subjects in the clinical trial conducted by Lei et al. 2016 did not present with tremor as side effect [48], further investigation must be conducted to determine potential tremor manifestations resulting from VPA and consequences for SCA patients.
2.1.6. Lithium
While there has been interest regarding lithium as treatment for ataxia, at least one double-blind, randomized, placebo-controlled, study in 62 patients with genetically confirmed SCA3 failed to note improvement in cerebellar function [52]. The study took place over 48 weeks with subjects randomized into either lithium group (300 mg tablets) or placebo group [52]. The primary endpoint of the study was the difference in mean scores on the NESSCA from baseline to 48 weeks between lithium and placebo groups and the safety of the intervention – the mean progression in NESSCA scores was not statistically significant (p = 0.222) [52]. The lithium treatment group presented statistically significant progression in secondary outcome assessments [52], including the nondominant Click Test (p = 0.023), the PATA rate test (p = 0.002), the SCAFI (p=0.003), and the CCFS (p = 0.029) [52]. While lithium was determined to be safe and tolerable – as measured by comparing the number of adverse events – it had no statistically significant effect on overall NESSCA scores [52]. Another 48-week double-blind, randomized, placebo-controlled trial assessing lithium for treatment of SCA2 in 20 subjects found the intervention safe but did not demonstrate significant differences in SARA scores [53]. Furthermore, acute lithium intoxication may be associated with neurological symptoms including tremor, ataxia, dysarthria, seizures; treatment of SCA with lithium could potentially exacerbate the condition, even at acceptable levels [54]. Due to lithium’s narrow therapeutic window and different reactions between individuals, it is difficult to predict whether accepted doses will cause toxicity [54]. Ultimately, future studies must focus on determining efficacy of lithium for treatment of SCA and establishing safe dosage.
2.1.7. Amantadine
Amantadine is a noncompetitive N-methyl-D-aspartate agonist that has been shown to be beneficial in treatment of parkinsonian features and degenerative ataxias [55]. In a randomized double-blind placebo-controlled trial of 57 patients with FRDA and MSA-C, efficacy of amantadine was assessed over the course of 4 months [56]. Patients randomized into the amantadine treatment group were dosed with 200 mg/day and both groups were assessed on reaction time and movement time, along with neurological exams [56]. Subjects with MSA-C showed significant improvement with amantadine treatment, whereas FRDA patients did not demonstrate this level of improvement [56]. Another trial conducted as a non-randomized open-label study (NCT00950196) evaluated amantadine in treatment of ataxia telangiectasia [57]. 17 children were treated with amantadine for an 8-week period and assessed using the ICARS, the Unified Parkinson’s Disease Rating Scale, and the Abnormal Involuntary Movement Scale [57]. Functional improvements were significant across all three assessments (25.3%, 29.5%, 32.5%, respectively; p <0.001) [57]. While these studies were not specifically addressing SCA, a recent publication details the positive effects of amantadine on a patient with SCA7 [58]. This patient experienced a 16% improvement in his SARA score after 4 weeks of amantadine 100 mg; this score decrease was further corroborated by positive anecdotal evidence from the subject’s family [58]. While this study cannot confirm the benefits of amantadine, it does indicate the potential for a new treatment for SCA [58].
2.1.8. Acetazolamide
Acetazolamide is a carbonic anhydrase inhibitor that is used in the treatment of epilepsy, congestive heart failure, and glaucoma [59]. Acetazolamide was first demonstrated to be useful in the treatment of ataxia by Griggs et al. (1978); this study revealed that acetazolamide was effective in mitigating and preventing episodic ataxia symptoms [60]. A subsequent case study showed acetazolamide to be useful in treatment of episodic ataxia and positional vertigo with central positional nystagmus seen in SCA6, but ineffective for chronic ataxia [61]. The potential of acetazolamide as a treatment for SCA6 was further evaluated in an open clinical trial of 9 SCA6 patients treated with 500 mg acetazolamide for 88 weeks [62]. Cerebral ataxia was shown to be reduced with acetazolamide treatment, as evidenced by a statistically significant decrease in total ataxia rating scores, and postural and kinetic subscores (p < 0.05) [62]. To properly assess the use of acetazolamide as a treatment for SCA6, additional placebo-controlled clinical trials must be conducted.
2.1.9. Trehalose
Trehalose is a disaccharide that has shown promising effects in stabilizing the progression of SCA17 in mice models [63]. One open-label trial of 14 SCA3 patients showed trehalose administration to be safe and tolerable, as well as effective in stabilizing SARA scores [64,65]. A more recent clinical trial of 13 SCA3 subjects taking 100 mg trehalose demonstrated statistically significant improvements in SARA scores(p = 0.05) and 8-minute walking test scores (p = 0.007) [66]. Due to the lack of placebo-controlled and randomized trials for trehalose, the potential benefits of this drug must be better assessed to determine its efficacy and safety.
2.2. Non-Pharmacologics
2.2.1. Rehabilitative therapy
Physical and occupational therapy has long been considered a critical component in treatment of movement disorders, and this is not an exception when caring for patients with SCAs. Since there are no studies specifically evaluating the effect of rehabilitation therapies on SCA, we will review those in patients with general cerebellar ataxias, which often include subjects with SCA. One randomized controlled trial evaluated both short and long-term effects of rehabilitation therapy on gait, ataxia, and activities of daily living [67]. The physical therapy protocol included muscle strengthening, general conditioning, and improving mobility. The occupational therapy protocol focused on activities of daily living, such as hygiene, writing, eating, and bathing [67]. 42 subjects presenting with pure cerebellar degeneration – including 20 with SCA6 and 6 with SCA31–were randomized into an immediate or delayed-entry treatment group [67]. The immediate treatment group began inpatient occupational and physical therapy for 2 hours on weekdays and 1 hour on weekends over the course of 4 weeks [67]. The delayed-entry treatment group underwent the same treatment, but after a waiting period of 4 weeks – in the short term, this delayed-entry treatment group functioned as the control group [67]. Subjects were assessed using various outcome measures, including the SARA, the Functional Independence Measure, and number of falls [67]. The immediate treatment group demonstrated significantly greater improvements in SARA score in the short-term, especially in truncal ataxia, compared with the delayed-entry treatment group over the 4 weeks [67]. In addition, at the 24-week follow-up, more than 50% of all individuals maintained these improvements [67].
Another investigation looked at the potential benefit of a 6-week individualized and home-based balance exercise program to treat 14 patients with cerebellar ataxia; 6 individuals had genetically confirmed diagnoses of SCA (2 SCA6, 1 SCA8, 1 SCA3, 1 SCA5, 1 SCA17) [68]. The physical therapy protocol included balance exercises from various positions and surfaces [68]. Neurological and functional assessments done 5 times over the course of the exercise program revealed significant improvements in walking speed, dynamic gait index, stride length, and percent double limb support time [68]. However, no change was seen in ICARS scores or Activity-specific Balance Confidence (ABC) Scale [69] scores. It is important to note that this study emphasized the importance of individualizing the exercise program based on the patient’s functional status and needs. Improvements were noted specifically in those subjects whose programs really challenged their balance [68]. Another topic of interest is the use of whole-body-controlled videogames or ‘exergames’ to assist in coordinative training for SCA patients and others with degenerative ataxia [70]. Just as was noted in studies on physical and occupational therapy, the efficacy of these treatments is highly dependent on consistency and frequency [68] – two factors that can be highly influenced by a patient’s functional status and external uncontrolled factors, such as accessibility. Ultimately, rehabilitative therapy is a beneficial addition to pharmacological treatment of cerebellar ataxia but must be further studied in placebo-controlled trials to better understand the efficacy specifically for SCA.
2.2.2. Noninvasive neurostimulation
Increasing attention has been directed at non-pharmacological and noninvasive methods for treatment of cerebellar disease or dysfunction. Interventions such as transcranial magnetic stimulation (TMS) and anodal or cathodal transcranial direct current stimulation (tDCS) have both been shown to positively affect cerebellar functions – motor, affective, and cognitive – that are disrupted in SCAs [71]; TMS and tDCS help to modulate cerebellar excitability via regulation of the association between the primary motor cortex and cerebellum [71].
TMS was first introduced as a potential therapeutic agent for treatment of major depressive disorder following FDA approval in 2008 [72]. It was later approved for use in treatment of migraines in 2013 and for obsessive compulsive disorder (OCD) in 2018 [72]. Several studies have demonstrated the positive effect of TMS in the treatment of hereditary cerebellar ataxias, like SCAs. One double-blind sham-controlled trial found significant improvement in truncal ataxia with active TMS treatment [73]. 74 patients presenting with cerebellar ataxia – including patients with SCA6, SCA1, and SCA3 – were randomized to receive active repetitive TMS (rTMS) or sham intervention every day over a 21-day period [73]. Truncal ataxia was evaluated through the timed 10-m walk, 10-m step assessment, and standing capacities; regional blood flow was also measured utilizing single-photon emission computed tomography (SPECT) [73]. In comparison to the active TMS and sham groups, improvement of the TMS group was found to be statistically significant for the timed 10-m walk, 10-m step assessment, and standing capacity (p < 0.05) [73]. Among the active-TMS group, blood flow to the pons and cerebellum was significantly increased (p < 0.005) but no significant increase was noted in the cerebral cortices [73].
A more recent randomized, double-blind sham-controlled trial evaluated the effect of rTMS (20 sessions over 4 weeks) on clinical impression, gait and posture among 20 subjects with genetically confirmed SCAs – 13 SCA3s, 3 SCA6s, and 1 patient each with SCA1, SCA2, SCA8, SCA13 [74]. The primary outcome of this study was the total SARA score and the secondary outcome included functional tests like the 9-hole peg test and the timed up-and-go test (TUG) [74]. Subjects receiving rTMS demonstrated a significant improvement in the SARA sub-score for ‘stance’ (p = 0.002) from baseline to 1-month follow-up when compared to the sham group [74]. No significant changes were observed in the 9-hole peg test or TUG assessment [74]. Another recent clinical trial (NCT03213106) on the effect of rTMS on patients with SCA3, MSA-C, and post-lesion ataxia yielded more promising results [75]. The study included 24 subjects – including 9 SCA3 patients – and measured improvements using the SARA and ICARS [75]. When comparing baseline to post-treatment scores of the treatment and sham groups, the rTMS group was shown to have statistically significant improvement in SARA scores (median 10.2 for rTMS vs. median 12.8 for sham (p = 0.002)) [75]. Improvements in ICARS scores were also noted to be significant between rTMS and sham groups (median 29.0 for rTMS and 32.8 for sham (p = 0.005) [75]. While these results are promising, further investigations must be conducted to confidently conclude the potential benefits of TMS treatment for SCAs [76].
Although tDCS is a new treatment option, studies have revealed potential for therapeutic benefit in the treatment of SCAs and other hereditary ataxias. Initial studies implementing tDCS therapy demonstrated improvements in SARA scores, specifically in stance, gait, and sitting sub-scores in cerebellar ataxia patients [71,77]. Following these preliminary studies, one randomized, double-blind, sham-controlled study conducted by Benussi et al. assessed the effect of tDCS on 19 subjects with ataxia of varying origins, including 5 SCA2, 1 SCA1, 2 SCA38, 6 MSA-C and 1 FRDA [78]. For subjects in the tDCS group, current was applied for 20 minutes at 2 mA; the cathode was placed on the right deltoid and the anode on the scalp area above the cerebellum [78]. All subjects were evaluated pre and post stimulation utilizing the SARA, ICARS, 9-hole-peg test, and timed 8-meter walk [78]. In comparing the sham and tDCS group, tDCS subjects demonstrated a statistically significant improvement in scores measured pre and post stimulation for all assessments (p < 0.001 for SARA, ICARS, and 8-meter walk; p = 0.012 for 9-hole-peg test) [78]. A subsequent trial conducted by Benussi et al. attempted to increase the aforementioned improvements with tDCS intervention for ataxia by focusing on spinal cord stimulation [79]. This randomized, double-blind, sham-controlled trial of 21 ataxia patients (6 SCA6s, 1 SCA38, and 1 SCA14 patient) used tDCS treatment with the anode on the cerebellar area of the scalp and the cathode over the spinal lumbar enlargement [79]. This spino-cerebellar focused stimulation also presented statistically significant improvements in functional performance assessments [79,80].
Conversely, two clinical trials did not demonstrate significant improvements in ataxia symptoms with tDCS intervention among similar patient types, including those with SCAs [81,82]. In both studies, patients underwent 22 minutes of tDCS with the cathode on the right buccinator muscle and the anode over the right cerebellar hemisphere [81,82]. Discrepancies in findings are further indication that more intensive investigation must be conducted on tDCS treatment. One current clinical trial being conducted in the Netherlands is focusing on tDCS treatment for SCA3 (NTR7537) [83].
2.2.3. Branched-chain amino acids (BCAAs)
Branched-chain amino acids (BCAAs) are the essential amino acids valine, leucine, and isoleucine [84]. BCAAs have been shown to be most effective in stimulating glutamate metabolism, which in turn improves transmission between cerebellar neurons [84]. A double-blind crossover study of BCAA intervention was conducted with 16 SCD patients – 8 of these patients had genetically confirmed diagnoses of SCA6 and 1 with SCA7 [85]. Individuals were randomized to receive 1.5, 3.0, or 6.0 mg BCAAs, or placebo over a 4-week period [85]. When comparing placebo and treatment groups, the ICARS scores significantly improved (p < 0.01) from pre- to post-treatment [85]. Additional double-blind, placebo-controlled studies with larger sample sizes should be conducted to confirm the positive findings of this study.
2.2.4. Coenzyme Q10 & vitamin E
Coenzyme Q10 and vitamin E have been evaluated for potential use as therapeutic agents in the treatment of ataxia. Both have been shown to be promising in treatment of Friedreich’s ataxia [86]; however, conclusive evidence has yet to be produced to confirm efficacy for SCA. One longitudinal study looked at natural history data to evaluate the effect of Coenzyme Q10, vitamin E, and other pharmacological agents on SARA, Unified Huntington’s Disease Rating Scale IV, and PHQ-9 scores in a large cohort of SCA1, 2, 3, and 6 patients [87]. Cross-sectional analysis showed that SCA1 and SCA3 patients demonstrated lower SARA and UHDRS-IV scores with coenzyme Q10 exposure [87]. However, neither coenzyme Q10 nor vitamin E were correlated with the improvement of SARA or UHDRS-IV scores [87]. While these results are not conclusive, the potential for positive effects warrants further investigation [87].
3. Disease modifying therapies
3.1. CRISPR/Cas9
Advances in gene editing technology-like CRISPR/Cas9 (Figure 1) have opened a new avenue of research into alternative options for treatment of genetic conditions, like SCA. The precision editing of the CRISPR/Cas9 technology has critical application for SCA, especially the types that are caused by a CAG triplet expansion [88]. In fact, CRISPR has been used to successfully delete CAG repeats in induced pluripotent stem cells from a SCA3 individual [89]. Another study by Marthaler et al. was able to produce three CRISPR-edited SCA2 cellular models [90–92]. While these findings are exciting, there was no investigation into the physiological effects of these deletions. In addition, CRISPR/Cas9 has been mostly implemented in cellular models and this technology will need to be further tested in relevant animal models to understand the effects at the brain circuitry and behavioral levels. Another concern for CRISPR/Cas9 is the off-target effect, which may cause unintended genetic alterations outside of SCA genes. In summary, studies focused on CRISPR editing and subsequent evaluation of potential physiological repercussions are critical to establish the viability of CRISPR therapy for SCAs [93].
Figure 1.
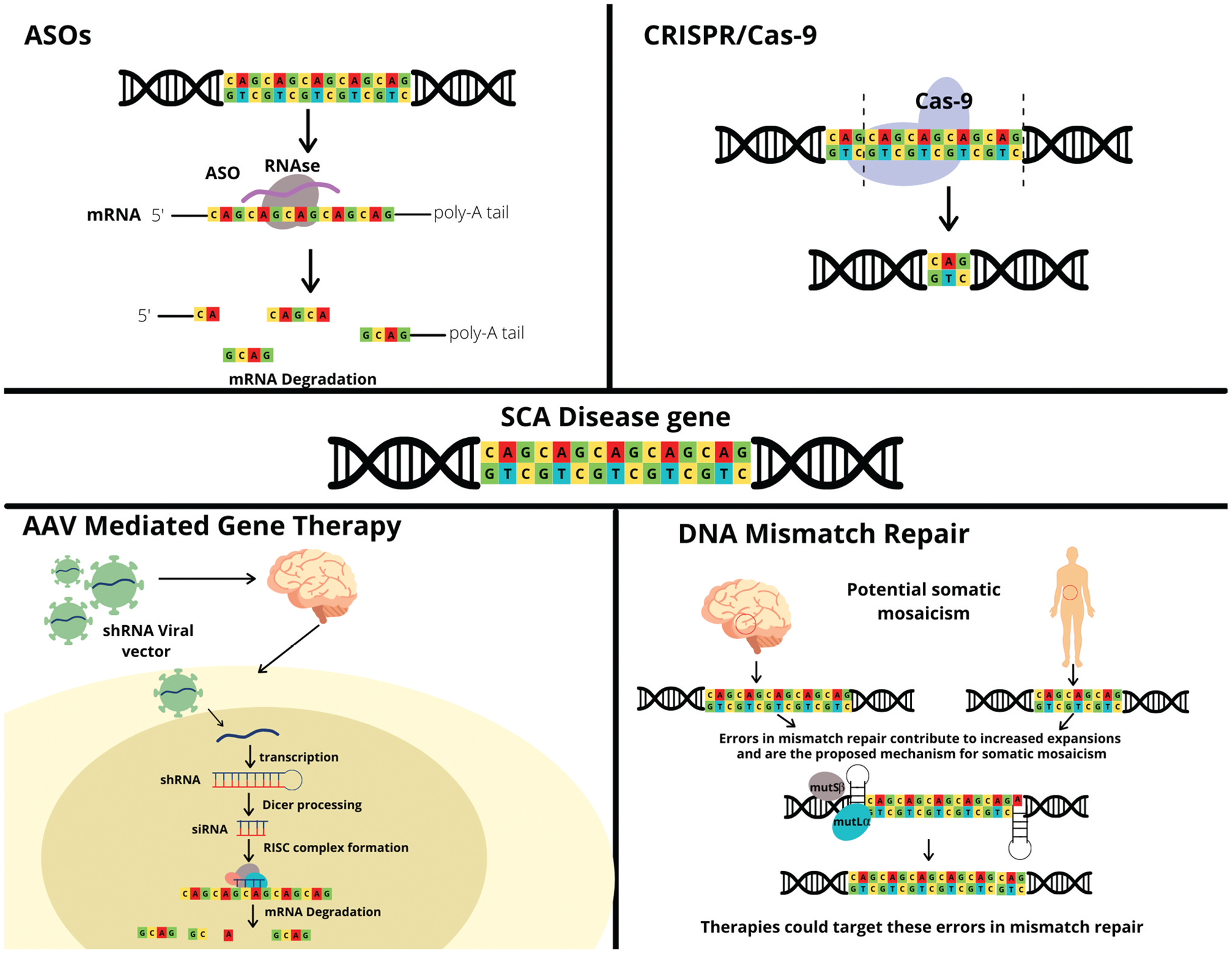
Gene therapy for SCA.
3.2. Antisense oligonucleotides (ASOs)
Antisense oligonucleotides (ASOs) are short, synthetic oligonucleotides that are gaining increasing popularity in treatment of neurodegenerative conditions [88,94]. ASOs can be designed to target specific RNA molecules to modulate their function via degradation or modification of translation [94]. For example, ASOs can target RNA molecules responsible for production of a toxic protein in order to decrease adverse effects of the protein’s activity [94]. ASO therapy has been successfully implemented in treatment of spinal muscular atrophy (SMA) and has potential for treatment of Huntington’s disease (HD) [88,94,95]. Studies on the potential use of ASOs for treatment of SCAs have yet to reach clinical trials, although preclinical studies have yielded promising results. One study evaluated the efficacy of ASO therapy in SCA2 mouse models [96]. ASOs were designed to target the ATXN2 RNA and were found to significantly down-regulate ATXN2 expression in the cerebellum and improve motor function in SCA2 mouse models when compared to placebo treatment [96]; similar promising preclinical studies have been conducted in SCA1, SCA3, and SCA7 [97–99]. Currently, there is an ongoing Phase 1 clinical trial investigating the treatment of ALS or poly-CAG ALS with ASOs targeting the ATXN2 gene to determine safety and tolerability [100]. Six experimental cohorts will receive 3 intrathecal administrations of ASOs over the course of 3 days and maintenance doses at 2 later days [100]. Considering the results demonstrating the efficacy of ASOs in preclinical trials and the establishment of clinical trials treating ALS with ASOs, further pre-clinical studies should be conducted to usher this treatment into the clinical phase for SCA [100].
3.3. Adeno-Associated Virus (AAV)-mediated gene therapy
In recent years, AAV-mediated gene therapy has risen in interest for treatment of neurodegenerative conditions like FRDA, SMA, and HD [101]. In fact, AAV mediated gene therapy was the first of its kind to be approved for treatment of SMA in 2019; the approved treatment delivers the SMN1 gene using an AAV9 vector that can cross the blood–brain barrier [102–104]. There is concern over the strong host-mediated immune response that has been noted with AAV-mediated gene therapy [101]. Other considerations include serotypes of the AAV, which may have differential tropism in the cerebellum and related brainstem areas, and the mode of delivery to ensure that enough cells are being adequately treated. Furthermore, other gene therapy methods such as targeting heat shock proteins to affect protein folding or lysosomal proteins to enhance protein degradation may be considered for further research – these methods allow for increased precision to treat such rare diseases like SCAs [105].
3.4. DNA mismatch repair
Recent genetic evidence has revealed that DNA repair mechanisms, such as mismatch repair (MMR), may be responsible for the repeat expansions seen in CAG-related ataxia [106]. One proposed mechanism is that of a toxic cycle linking DNA damage and repair to expansion of CAG repeats [106]. While DNA repair mechanisms are critical for correcting DNA damage such as CAG repeats, these same mechanisms can sometimes facilitate expansion of these repeats. Once the disease threshold for repeat number has been reached, these repeats are more likely to continue expanding among germ-line and somatic cells [106]. Since these expansions in CAG repeats can occur in genes encoding DNA repair proteins, the repeats themselves can impair the DNA damage repair process, furthering DNA damage [106].
Biochemical and genetic evidence, as well as mouse models, have demonstrated the critical role that the mismatch-repair mechanism plays in development of CAG-repeat diseases like HD and SCA [106]. Genome-wide association studies (GWAS) have identified several MMR genes linked to HD, such as MSH3, MLH1, and PMS1; these MMR genes have been shown to affect HD progression, age of onset, and manifestation [107]. In addition, GWAS revealed that many of the genetic variants associated with age of onset and disease severity were found in or close to DNA repair genes, and especially MMR genes [106]–these variants could be an important target for therapeutic intervention. It is also interesting to consider the presence of somatic mosaicism in HD and SCA, which is observed prominently in the central nervous system and is linked to DNA repair mechanisms [108,109]. Because the DNA repair mechanism has been thoroughly studied and the molecular and biochemical level, therapeutic interventions will focus on targeting proteins and RNA-based gene activation to mediate disease development and progression [107]. While CAG repeat diseases like HD and SCA may have differing phenotypes, the potential for a common pathogenic mechanism could allow for disease modifying therapies for multiple debilitating neurodegenerative conditions [106].
4. Conclusion
While SCAs are groups of devastating neurological disorders without effective treatments, therapeutic developments have been rapidly emerging; several trials are on the horizon. SCAs are unique, as many patients are at a younger age, and therefore less susceptible to aging associated cardiovascular comorbidities that can be more vulnerable to side effects. In addition, SCAs are monogenetic disorders with very high disease penetrance; thus, SCAs are ideal to test targeted treatments such as gene therapies or ASOs. Finally, SCAs have long-standing natural history studies in North America and Europe, using almost the same outcome measurements, which further promotes international collaboration on these rare diseases.
Despite these promising prospects, there are challenges to be overcome to increase the chance of success for clinical trials. Specifically, there is insufficient validated biomarkers for SCAs. Biomarkers are critical for target engagement and to track disease progression. Current biomarkers include molecules involved in autophagy, growth factors, enzymes, inflammatory and oxidative stress response, and chaperones [64]. Recently, the measurement of mutant ataxin-3 levels has been validated in patient spinal fluid as a target engagement biomarker for gene therapies or ASOs to reduce mutant ataxin-3 protein levels [110]. In addition, neurofilament light chains in the serum and magnetic resonance spectroscopy may be useful as a biomarker for disease progression [111]. Furthermore, recent investigation into volumetric biomarkers has revealed potential utility, specifically of fixel-based analysis [112]. One study found that volumetric changes in the brain, specifically in the brain stem and striatum, among SCA1 individuals were reliably differentiated from unaffected controls and were detected under a year [113]. All these developments can help to further accelerate therapeutic development. One major gap for biomarker development is a physiological biomarker, which can track the function of the cerebellum and can potentially provide more direct measurements of responses to therapies. The discovery of a physiological biomarker would be a huge step in treatment of SCA as it would allow for objective evaluation of therapies and direct evaluation of treatments’ effects on brain structure and function. Other challenges include a lack of patient reported outcomes and a lack of symptomatic therapies to treat ataxia.
Hopefully, in the next five to 10 years, SCAs will have a major breakthrough in therapy development that will serve as a model for treatment advancement for rare neurological disorders.
5. Expert opinion
The lack of FDA-approved treatments for SCA is a testament to the complexity and multifaceted nature of this disease family. Several clinical trials of a small sample size have reported promising results with pharmacologic intervention; however, more evidence in support of these drugs’ effects must be provided to warrant official approval. Establishing statistical significance at a higher confidence level requires larger sample sizes, which is difficult considering the fact that SCA is a rare group of diseases. Furthermore, patients may be unwilling to enter clinical trials, with a chance of receiving a placebo. That being said, current clinical trials provide an exciting look into a potential future in which SCA disease progression may be slowed to maintain quality of life for SCA patients.
The majority of clinical trials thus far have emphasized treating the symptoms of SCA with pharmacologics; while these drugs are shown to have potential, they are not specifically targeting the mutant genes and their toxic products. Methods such as AAV-mediated gene therapy, ASOs, and CRISPR open a world of possibilities for treating SCA at the source to alter the disease trajectory. Furthermore, considering the long presymptomatic period of SCA, efforts should be made to recruit preataxic individuals in an attempt to slow or prevent disease progression prior to the disease onset. This is of course will require earlier diagnosis which in itself poses unique challenges. Ultimately, early diagnosis combined with early therapeutic intervention could be critical for curing SCA.
Article highlights.
Spinocerebellar ataxias (SCAs) are autosomal dominantly inherited, progressive disorders marked by cerebellar degeneration
Currently, there are no symptomatic or neuroprotective treatments approved by the United States (US) Food and Drug Administration (FDA) for SCAs
Research efforts have focused on symptomatic and pharmacologic treatments for SCA, with the majority of clinical trials involving oral pharmaceutical agents
Emerging disease modifying therapies are being developed, such as AAV-mediated gene therapy and ASOs to address SCA at its source
Biomarker identification can facilitate therapy development and thus must continue to take high priority in SCA research
Declaration of interests
SH Kuo received research funding from National Institutes of Health and National Ataxia Foundation. SH Kuo also serves as a consultant for Praxis, Neurocrine, uniQure, and Sage Therapeutics. TA Zesiewicz has received personal compensation for serving on the advisory boards of Boston Scientific; Reata Pharmaceuticals, Inc; and Steminent Biotherapeutics. TA Zesiewicz has received personal compensation as senior editor for Neurodegenerative Disease Management and as a consultant for Steminent Biotherapeutics. Dr Zesiewicz has received royalty payments as co-inventor of varenicline for treating imbalance (patent number 9,463,190) and non-ataxic imbalance (patent number 9,782,404). TA Zesiewicz has received research/grant support as principal investigator/investigator for studies from AbbVie Inc; Biogen; Biohaven Pharmaceutics; Boston Scientific; Bukwang Pharmaceuticals Co, Ltd; Cala Health, Inc; Cavion; Friedreich’s Ataxia Research Alliance; Houston Methodist Research Institute; National Institutes of Health (READISCA U01); Retrotope Inc; and Takeda Development Center Americas, Inc. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or conflict with the subject matter or materials discussed in this manuscript apart from those disclosed.
Footnotes
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Paulson HL. The spinocerebellar ataxias. J Neuro-Ophthalmol Off J North Am Neuro-Ophthalmol Soc. 2009;29(3):227–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ashizawa T, Ö G, Paulson HL. Spinocerebellar ataxias: prospects and challenges for therapy development. Nat Rev Neurol. 2018;14 (10):590–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klinke I, Minnerop M, Schmitz-Hübsch T, et al. Neuropsychological features of patients with spinocerebellar ataxia (SCA) types 1, 2, 3, and 6. Cerebellum Lond Engl. 2010;9(3):433–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Almeida-Silva UC, Hallak JEC, Júnior WM, et al. Association between spinocerebellar ataxias caused by glutamine expansion and psychiatric and neuropsychological signals - a literature review. Am J Neurodegener Dis. 2013;2(2):57–69. [cited 2021 Dec 2]. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3703120/ [PMC free article] [PubMed] [Google Scholar]
- 5.Bird TD. Hereditary Ataxia Overview. In: Adam MP, Ardinger HH, and Pagon RA, et al. , editors. GeneReviews®. eattle: University of Washington; 1993. 1–17. [cited 2021 Aug 17]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1138/ [Google Scholar]
- 6.Klockgether T, Mariotti C, Paulson HL. Spinocerebellar ataxia. Nat Rev Dis Primer. 2019;5(1):1–21. [DOI] [PubMed] [Google Scholar]
- 7.Soong BW, and Morrison PJ. Chapter 10 - Spinocerebellar ataxias. In: Manto M, and Huisman TAGM, editors. Handbook of Clinical Neurology. Vol. 155. The Cerebellum: Disorders and Treatment, Cambridge, MA: Elsevier; 2018. p. 143–174. [DOI] [PubMed] [Google Scholar]
- 8.Opal P, and Zoghbi H. The spinocerebellar ataxias. In: Eichler AF, editor. UpToDate. Waltham, MA: UpToDate; 2021. https://www.uptodate.com/contents/the-spinocerebellar-ataxias Accessed 17 Aug 2021 [Google Scholar]
- 9.Duenas AM. Molecular pathogenesis of spinocerebellar ataxias. Brain. 2006;129(6):1357–1370. [DOI] [PubMed] [Google Scholar]
- 10.Buijsen RAM, Toonen LJA, Gardiner SL, et al. Genetics, mechanisms, and therapeutic progress in polyglutamine spinocerebellar ataxias. Neurotherapeutics. 2019;16(2):263–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Honti V, Vécsei L. Genetic and molecular aspects of spinocerebellar ataxias. Neuropsychiatr Dis Treat. 2005;1(2):125–133. [cited 2021 Dec 14]. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2413192/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sullivan R, Yau WY, O’Connor E, et al. Spinocerebellar ataxia: an update. J Neurol. 2019;266(2):533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bushart DD, Shakkottai VG. Ion channel dysfunction in cerebellar ataxia. Neurosci Lett. 2019;688:41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen ML, Lin CC, Rosenthal LS, et al. Rating scales and biomarkers for CAG-repeat spinocerebellar ataxias: implications for therapy development. J Neurol Sci. 2021;424:117417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ristori G, Romano S, Visconti A, et al. Riluzole in cerebellar ataxia: a randomized, double-blind, placebo-controlled pilot trial. Neurology. 2010;74(10):839–845. [DOI] [PubMed] [Google Scholar]; • This is the first randomized placebo-controlled trial to show significant improvements in functional measures of SCA in patients taking riluzole.
- 16.Romano S, Coarelli G, Marcotulli C, et al. Riluzole in patients with hereditary cerebellar ataxia: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2015;14(10):985–991. [DOI] [PubMed] [Google Scholar]
- 17.Shakkottai VG, Hua CC, Oddo S, et al. Ovid: enhanced neuronal excitability in the absence of neurodegeneration induces cerebellar ataxia. J Clin Invest. 2004;113(4):582–590. [cited 2021 Aug 8]. Available from: https://ovidsp-dc2-ovid-com.ezproxy.lib.usf.edu/ovid-a/ovidweb.cgi?Link+Set+Ref=00006114-201003090-00011|00004686_2004_113_582_shakkottai_neurodegeneration_%7c00006114-201003090-00011%23xpointer%28id%28R11-11%29%29%7c10%7c%7covftdb%7c00004686-200402040-00015&P=63&S=AHLJFPAPAJEBPOFFIPOJLGEHEAPBAA00&WebLinkReturn=Full+Text%3dL%7cS.sh.22.23%7c0%7c00006114-201003090-00011&Counter5=FTV_bibliographic%7c00004686-200402040-00015%7covft%7covftdb%7covftg [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Riluzole. In: LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. National Institute of Diabetes and Digestive and Kidney Diseases; 2012. [cited 2021 Aug 17]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK548919/ [PubMed] [Google Scholar]
- 19.Biohaven Pharmaceuticals, Inc. A Phase III, Long-Term, Randomized, Double-Blind, Placebo-Controlled Trial of Troriluzole in Adult Subjects With Spinocerebellar Ataxia. clinicaltrials.gov; 2021. [cited 2021 Aug 5]. Available from: https://clinicaltrials.gov/ct2/show/NCT03701399
- 20.Bushart DD, Chopra R, Singh V, et al. Targeting potassium channels to treat cerebellar ataxia. Ann Clin Transl Neurol. 2018;5(3):297–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kasumu AW, Hougaard C, Rode F, et al. Selective positive modulator of calcium-activated potassium channels exerts beneficial effects in a mouse model of spinocerebellar ataxia type 2. Chem Biol. 2012;19(10):1340–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Egorova PA, Zakharova OA, Vlasova OL, et al. In vivo analysis of cerebellar purkinje cell activity in SCA2 transgenic mouse model. J Neurophysiol. 2016;115(6):2840–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shakkottai VG, Do Carmo Costa M, Dell’Orco JM, et al. Early changes in cerebellar physiology accompany motor dysfunction in the polyglutamine disease spinocerebellar ataxia type 3. J Neurosci. 2011;31(36):13002–13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kasumu A, Bezprozvanny I. Deranged calcium signaling in purkinje cells and pathogenesis in spinocerebellar ataxia 2 (SCA2) and other ataxias. Cerebellum Lond Engl. 2012;11(3):630–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meera P, Pulst SM, Otis TS. Cellular and circuit mechanisms under-lying spinocerebellar ataxias. J Physiol. 2016;594(16):4653–4660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mark MD, Schwitalla JC, Groemmke M, et al. Keeping our calcium in balance to maintain our balance. Biochem Biophys Res Commun. 2017;483(4):1040–1050. [DOI] [PubMed] [Google Scholar]
- 27.Egorova PA, Bezprozvanny IB. Inositol 1,4,5-trisphosphate receptors and neurodegenerative disorders. FEBS J. 2018;285(19):3547–3565. [DOI] [PubMed] [Google Scholar]
- 28.Hisatsune C, Hamada K, Mikoshiba K. Ca2+ signaling and spinocerebellar ataxia. Biochim Biophys Acta Mol Cell Res. 2018;1865(11 Pt B):1733–1744. [DOI] [PubMed] [Google Scholar]
- 29.Shimobayashi E, Kapfhammer JP. Calcium signaling, PKC gamma, IP3R1 and CAR8 link spinocerebellar ataxias and purkinje cell dendritic development. Curr Neuropharmacol. 2018;16(2):151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Egorova PA, Bezprozvanny IB. Molecular mechanisms and therapeutics for spinocerebellar ataxia type 2. Neurother J Am Soc Exp Neurother. 2019;16(4):1050–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prestori F, Moccia F, D’Angelo E. Disrupted calcium signaling in animal models of human spinocerebellar ataxia (SCA). Int J Mol Sci. 2019;21(1):216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cho LTY, Alexandrou AJ, Torella R, et al. An intracellular allosteric modulator binding pocket in SK2 ion channels is shared by multiple chemotypes. Structure. 2018;26(4):533–544.e3. [DOI] [PubMed] [Google Scholar]; • The recent discovery of Riluzole’s ability to allosterically bind to SK2 channels has provided a better understanding of the drug’s mechanism action.
- 33.Bushart DD, Huang H, Man LJ, et al. A chlorzoxazone-baclofen combination improves cerebellar impairment in spinocerebellar ataxia type 1. Mov Disord. 2021;36(3):622–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fröhlich E, Wahl R. The forgotten effects of thyrotropin-releasing hormone: metabolic functions and medical applications. Front Neuroendocrinol. 2019;52:29–43. [DOI] [PubMed] [Google Scholar]
- 35.Sobue I, Takayanagi T, Nakanishi T, et al. Controlled trial of thyrotropin releasing hormone tartrate in ataxia of spinocerebellar degenerations. J Neurol Sci. 1983;61(2):235–248. [DOI] [PubMed] [Google Scholar]
- 36.Nishizawa M, Onodera O, Hirakawa A, et al. Effect of rovatirelin in patients with cerebellar ataxia: two randomised double-blind placebo-controlled phase 3 trials. J Neurol Neurosurg Psychiatry. 2020;91(3):254–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zesiewicz TA, Wilmot G, Kuo SH, et al. Comprehensive systematic review summary: treatment of cerebellar motor dysfunction and ataxia. Neurology. 2018;90(10):464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ijiro T, Yaguchi A, Yokoyama A, et al. Ameliorating effect of rovatirelin on the ataxia in rolling mouse Nagoya. Eur J Pharmacol. 2020;882:173271. [DOI] [PubMed] [Google Scholar]
- 39.Kimura N, Kumamoto T, Masuda T, et al. Evaluation of the effect of thyrotropin releasing hormone (TRH) on regional cerebral blood flow in spinocerebellar degeneration using 3DSRT. J Neurol Sci. 2009;281(1):93–98. [DOI] [PubMed] [Google Scholar]
- 40.Singh D, and Saadabadi A. Varenicline. In: StatPearls. Treasure Island, FL: StatPearls Publishing; 2021;1–7. [cited 2021 Aug 8]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK534846/ [Google Scholar]
- 41.Zesiewicz TA, Greenstein PE, Sullivan KL, et al. A randomized trial of varenicline (Chantix) for the treatment of spinocerebellar ataxia type 3. Neurology. 2012;78(8):545–550. [DOI] [PubMed] [Google Scholar]; • This is one of the first studies to demonstrate potential efficacy of Varenicline in the treatment of SCA.
- 42.Ilg W, Bastian AJ, Boesch S, et al. Consensus paper: management of degenerative cerebellar disorders. The Cerebellum. 2014;13(2):248–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wecker L, Engberg M, Philpot R, et al. Neuronal nicotinic receptor agonists improve gait and balance in olivocerebellar ataxia. Neuropharmacology. 2013;73. DOI: 10.1016/j.neuropharm.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Loane C, Politis M. Buspirone: what is it all about? Brain Res. 2012;1461:111–118. [DOI] [PubMed] [Google Scholar]
- 45.Trouillas P, Xie J, Adeleine P. Treatment of cerebellar ataxia with buspirone: a double-blind study. Lancet. 1996;348 (9029):759. [DOI] [PubMed] [Google Scholar]
- 46.Lou JS, Goldfarb L, McShane L, et al. Use of buspirone for treatment of cerebellar ataxia. An open-label study. Arch Neurol. 1995;52 (10):982–988. [DOI] [PubMed] [Google Scholar]
- 47.Assadi M, Campellone JV, Janson CG, et al. Treatment of spinocerebellar ataxia with buspirone. J Neurol Sci. 2007;260(1–2):143–146. [DOI] [PubMed] [Google Scholar]
- 48.Lei LF, Yang GP, Wang JL, et al. Safety and efficacy of valproic acid treatment in SCA3/MJD patients. Parkinsonism Relat Disord. 2016;26:55–61. [DOI] [PubMed] [Google Scholar]
- 49.Gan SR, Wang J, Figueroa KP, et al. Postural tremor and ataxia progression in spinocerebellar ataxias. Tremor Hyperkinetic Mov N Y N. 2017;7:492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lai RY, Tomishon D, Figueroa KP, et al. Tremor in the degenerative cerebellum: towards the understanding of brain circuitry for tremor. Cerebellum Lond Engl. 2019;18(3):519–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qi ZC, Ming HB, Ling HM, et al. Risk of valproic acid-related tremor: a systematic review and meta-analysis. Front Neurol. 2020;11:576579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saute JAM, de Castilhos RM, Monte TL, et al. A randomized, phase 2 clinical trial of lithium carbonate in Machado-Joseph disease. Mov Disord Off J Mov Disord Soc. 2014;29(4):568–573. [DOI] [PubMed] [Google Scholar]
- 53.Saccà F, Puorro G, Brunetti A, et al. A randomized controlled pilot trial of lithium in spinocerebellar ataxia type 2. J Neurol. 2015;262 (1):149–153. [DOI] [PubMed] [Google Scholar]
- 54.Schneider MA, Smith SS. Lithium-Induced neurotoxicity: a case study. J Neurosci Nurs J Am Assoc Neurosci Nurses. 2019;51 (6):283–286. [DOI] [PubMed] [Google Scholar]
- 55.van de W BPC, van G J, Boesch S, et al. EFNS/ENS Consensus on the diagnosis and management of chronic ataxias in adulthood. Eur J Neurol. 2014;21(4):552–562. [DOI] [PubMed] [Google Scholar]
- 56.Botez MI, Botez-Marquard T, Elie R, et al. Amantadine hydrochloride treatment in heredodegenerative ataxias: a double blind study. J Neurol Neurosurg Psychiatry. 1996;61(3):259–264. [cited 2021 Aug 15]. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC486548/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nissenkorn A, Hassin-Baer S, Lerman SF, et al. Movement disorder in ataxia-telangiectasia: treatment with amantadine sulfate. J Child Neurol. 2013;28(2):155–160. [DOI] [PubMed] [Google Scholar]
- 58.Pacheco LP, Thakur N Amantadine therapy for ataxia management in patients with spinocerebellar ataxia type 7. MDS Abstracts. [cited 2021 Aug 18]. Available from: https://www.mdsabstracts.org/abstract/amantadine-therapy-for-ataxia-management-in-patients-with-spinocerebellar-ataxia-type-7/
- 59.Farzam K, and Abdullah M. Acetazolamide. In: StatPearls. Treasure Island, FL: StatPearls Publishing; 2021;1–5. [cited 2021 Nov 7]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK532282/ [Google Scholar]
- 60.Griggs RC, Moxley RT, Lafrance RA, et al. Hereditary paroxysmal ataxia: response to Acetazolamide. Neurology. 1978;28(12):1259–1264. [DOI] [PubMed] [Google Scholar]
- 61.Jen JC, Yue Q, Karrim J, et al. Spinocerebellar ataxia type 6 with positional vertigo and Acetazolamide responsive episodic ataxia. J Neurol Neurosurg Psychiatry. 1998;65(4):565–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yabe I, Sasaki H, Yamashita I, et al. Clinical trial of acetazolamide in SCA6, with assessment using the ataxia rating scale and body stabilometry: clinical trial of acetazolamide for SCA6. Acta Neurol Scand. 2001;104(1):44–47. [DOI] [PubMed] [Google Scholar]
- 63.Chen ZZ, Wang CM, Lee GC, et al. Trehalose attenuates the gait ataxia and gliosis of spinocerebellar ataxia type 17 mice. Neurochem Res. 2015;40(4):800–810. [DOI] [PubMed] [Google Scholar]
- 64.Chen YS, Hong ZX, Lin SZ, et al. Identifying therapeutic targets for spinocerebellar ataxia type 3/Machado–Joseph disease through integration of pathological biomarkers and therapeutic strategies. Int J Mol Sci. 2020;21(9):3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ltd BP. Bioblast announces phase 2a results of trehalose in patients with spinocerebellar ataxia type 3 (SCA3). GlobeNewswire News Room. Published January 18, 2017. [cited 2021 Nov 8]. Available from: https://www.globenewswire.com/news-release/2017/01/18/906630/32455/en/Bioblast-Announces-Phase-2a-Results-of-Trehalose-in-Patients-with-Spinocerebellar-Ataxia-Type-3-SCA3.html
- 66.Noorasyikin MA, Azizan EA, Teh PC, et al. Oral trehalose maybe helpful for patients with spinocerebellar ataxia 3 and should be better evaluated. Parkinsonism Relat Disord. 2020;70:42–44. [DOI] [PubMed] [Google Scholar]
- 67.Miyai I, Ito M, Hattori N, et al. Cerebellar ataxia rehabilitation trial in degenerative cerebellar diseases. Neurorehabil Neural Repair. 2012;26(5):515–522. [DOI] [PubMed] [Google Scholar]
- 68.Bastian A, Keller JL. A home balance exercise program improves walking in people with cerebellar ataxia. Neurorehabil Neural Repair. 2014;28(8):770–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Winser S, Smith C, Hale L, et al. Balance outcome measures in cerebellar ataxia: a Delphi survey. Disabil Rehabil. 2014;37. DOI: 10.3109/09638288.2014.913709 [DOI] [PubMed] [Google Scholar]
- 70.Synofzik M, Ilg W. Motor training in degenerative spinocerebellar disease: ataxia-specific improvements by intensive physiotherapy and exergames. BioMed Res Int. 2014;2014:e583507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Grimaldi G, Argyropoulos GP, Boehringer A, et al. Non-invasive cerebellar stimulation—a consensus paper. Cerebellum. 2014;13 (1):121–138. [DOI] [PubMed] [Google Scholar]
- 72.Commissioner O of the. FDA permits marketing of transcranial magnetic stimulation for treatment of obsessive compulsive disorder. FDA. Published March 24, 2020. [cited 2021 Aug 15]. Available from: https://www.fda.gov/news-events/press-announcements/fda-permits-marketing-transcranial-magnetic-stimulation-treatment-obsessive-compulsive-disorder
- 73.Shiga Y, Tsuda T, Itoyama Y, et al. Transcranial magnetic stimulation alleviates truncal ataxia in spinocerebellar degeneration. J Neurol Neurosurg Psychiatry. 2002;72(1):124–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Manor B, Greenstein PE, Davila-Perez P, et al. Repetitive transcranial magnetic stimulation in spinocerebellar ataxia: a pilot randomized controlled trial. Front Neurol. 2019;10:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.França C, de Andrade DC, Silva V, et al. Effects of cerebellar transcranial magnetic stimulation on ataxias: a randomized trial. Parkinsonism Relat Disord. 2020;80:1–6. [DOI] [PubMed] [Google Scholar]
- 76.Chen TX, Yang CY, Willson G, et al. The efficacy and safety of transcranial direct current stimulation for cerebellar ataxia: a systematic review and meta-analysis. Cerebellum Lond Engl. 2021;20(1):124–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pozzi NG, Minafra B, Zangaglia R, et al. Transcranial direct current stimulation (tDCS) of the cortical motor areas in three cases of cerebellar ataxia. Cerebellum. 2014;13(1):109–112. [DOI] [PubMed] [Google Scholar]
- 78.Benussi A, Koch G, Cotelli M, et al. Cerebellar transcranial direct current stimulation in patients with ataxia: a double-blind, randomized, sham-controlled study. Mov Disord Off J Mov Disord Soc. 2015;30(12):1701–1705. [DOI] [PubMed] [Google Scholar]
- 79.Benussi A, Dell’Era V, Cantoni V, et al. Cerebello-spinal tDCS in ataxia: a randomized, double-blind, sham-controlled, crossover trial. Neurology. 2018;91(12):e1090–e1101. [DOI] [PubMed] [Google Scholar]
- 80.Benussi A, Pascual-Leone A, Borroni B. Non-Invasive cerebellar stimulation in neurodegenerative ataxia: a literature review. Int J Mol Sci. 2020;21(6):1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hulst T, John L, Küper M, et al. Cerebellar patients do not benefit from cerebellar or M1 transcranial direct current stimulation during force-field reaching adaptation. J Neurophysiol. 2017;118(2):732–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.John L, Küper M, Hulst T, et al. Effects of transcranial direct current stimulation on grip force control in patients with cerebellar degeneration. Cerebellum Ataxias. 2017;4(1):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Maas RPPWM, Toni I, Doorduin J, et al. Cerebellar transcranial direct current stimulation in spinocerebellar ataxia type 3 (SCA3-tDCS): rationale and protocol of a randomized, double-blind, sham-controlled study. BMC Neurol. 2019;19:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Holeček M Branched-chain amino acids in health and disease: metabolism, alterations in blood plasma, and as supplements. Nutr Metab. 2018;15(1):33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mori M, Adachi Y, Mori N, et al. Double-blind crossover study of branched-chain amino acid therapy in patients with spinocerebellar degeneration. J Neurol Sci. 2002;195(2):149–152. [DOI] [PubMed] [Google Scholar]
- 86.Cooper JM, Korlipara LVP, Hart PE, et al. Coenzyme Q10 and vitamin E deficiency in Friedreich’s ataxia: predictor of efficacy of vitamin E and coenzyme Q10 therapy. Eur J Neurol. 2008;15 (12):1371–1379. [DOI] [PubMed] [Google Scholar]
- 87.Lo RY, Figueroa KP, Pulst SM, et al. Coenzyme Q10 and spinocerebellar ataxias. Mov Disord Off J Mov Disord Soc. 2015;30(2):214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lin CC, Ashizawa T, Kuo SH. Collaborative efforts for spinocerebellar ataxia research in the United States: CRC-SCA and READISCA. Front Neurol. 2020;11:902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ouyang S, Xie Y, Xiong Z, et al. CRISPR/Cas9-Targeted deletion of polyglutamine in spinocerebellar ataxia type 3-derived induced pluripotent stem cells. Stem Cells Dev. 2018;27(11):756–770. [DOI] [PubMed] [Google Scholar]
- 90.Marthaler AG, Schmid B, Tubsuwan A, et al. Generation of an isogenic, gene-corrected control cell line of the spinocerebellar ataxia type 2 patient-derived iPSC line H196. Stem Cell Res. 2016;16(1):162–165. [DOI] [PubMed] [Google Scholar]
- 91.Marthaler AG, Schmid B, Tubsuwan A, et al. Generation of an isogenic, gene-corrected control cell line of the spinocerebellar ataxia type 2 patient-derived iPSC line H271. Stem Cell Res. 2016;16(1):180–183. [DOI] [PubMed] [Google Scholar]
- 92.Marthaler AG, Tubsuwan A, Schmid B, et al. Generation of an isogenic, gene-corrected control cell line of the spinocerebellar ataxia type 2 patient-derived iPSC line H266. Stem Cell Res. 2016;16(1):202–205. [DOI] [PubMed] [Google Scholar]
- 93.Babačić H, Mehta A, Merkel O, et al. CRISPR-cas gene-editing as plausible treatment of neuromuscular and nucleotide-repeat-expansion diseases: a systematic review. PLoS ONE. 2019;14(2): e0212198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bennett CF, Krainer AR, Cleveland DW. Antisense oligonucleotide therapies for neurodegenerative diseases. Annu Rev Neurosci. 2019;42:385–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tabrizi SJ, Leavitt BR, Landwehrmeyer GB, et al. Targeting huntingtin expression in patients with huntington’s disease. N Engl J Med. 2019. May 6; Published online. DOI: 10.1056/NEJMoa1900907. [DOI] [PubMed] [Google Scholar]
- 96.Scoles DR, Meera P, Schneider M, et al. Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature. 2017;544 (7650):362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.O’Callaghan B, Hofstra B, Handler HP, et al. Antisense oligonucleotide therapeutic approach for suppression of ataxin-1 expression: a safety assessment. Mol Ther Nucleic Acids. 2020;21:1006–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Moore L, Rajpal G, Dillingham I, et al. Evaluation of ASOs targeting ATXN3 in SCA3 mouse models. Mol Ther Nucleic Acids. 2017;7. DOI: 10.1016/j.omtn.2017.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Niu C, Prakash TP, Kim A, et al. Antisense oligonucleotides targeting mutant Ataxin-7 restore visual function in a mouse model of spinocerebellar ataxia type 7. Sci Transl Med. 2018;10(465): eaap8677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Biogen. A Phase 1 Multiple-Ascending-Dose Study to Assess the Safety, Tolerability, and Pharmacokinetics of BIIB105 Administered Intrathecally to Adults With Amyotrophic Lateral Sclerosis With or Without Poly-CAG Expansion in the Ataxin-2 Gene. clinicaltrials.gov; 2021. [cited 2021 Nov 7]. Available from: https://clinicaltrials.gov/ct2/show/NCT04494256.; *This is the first clinical trial to employ ASOs in the treatment of a neurodegenerative disease.
- 101.Perez BA, Shutterly A, Chan YK, et al. Management of neuroinflammatory responses to AAV-Mediated gene therapies for neurodegenerative diseases. Brain Sci. 2020;10(2):119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Al-Zaidy SA, Kolb SJ, Lowes L, et al. AVXS-101 (Onasemnogene Abeparvovec) for SMA1: comparative study with a prospective natural history cohort. J Neuromuscul Dis. 2019;6(3):307–317. [DOI] [PubMed] [Google Scholar]
- 103.Schwartz M, Likhite S, Meyer K. Onasemnogene abeparvovec-xioi: a gene replacement strategy for the treatment of infants diagnosed with spinal muscular atrophy. Drugs Today Barc Spain 1998. 2021;57(6):387–399. [DOI] [PubMed] [Google Scholar]
- 104.Mendell JR, Al-Zaidy S, Shell R, et al. Single-Dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377 (18):1713–1722. [DOI] [PubMed] [Google Scholar]
- 105.Bushart DD, Murphy GG, Shakkottai VG. Precision medicine in spinocerebellar ataxias: treatment based on common mechanisms of disease. Ann Transl Med. 2016;4(2):2–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Massey TH, Jones L. The central role of DNA damage and repair in CAG repeat diseases. Dis Model Mech. 2018;11(1):dmm031930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Iyer RR, Pluciennik A. DNA mismatch repair and its role in huntington’s disease. J Huntingt Dis. 2021;10(1):75–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kacher R, Lejeune FX, and Noël S, et al. In: Orr HT, Zoghbi HY, and Wheeler VC, editors. eLife. Vol. 10; 2021. p. e64674.33983118 [Google Scholar]
- 109.Cancel G, Gourfinkel-An I, Stevanin G, et al. Somatic mosaicism of the CAG repeat expansion in spinocerebellar ataxia type 3/Machado-Joseph disease. Hum Mutat. 1998;11(1):23–27. [DOI] [PubMed] [Google Scholar]
- 110.Hübener-Schmid J, Kuhlbrodt K, Peladan J, et al. Polyglutamine-Expanded ataxin-3: a target engagement marker for spinocerebellar ataxia type 3 in peripheral blood. Mov Disord Off J Mov Disord Soc. 2021. Aug 16;36(11):2675–2681. Published online. [DOI] [PubMed] [Google Scholar]
- 111.Wilke C, Haas E, Reetz K, et al. Neurofilaments in spinocerebellar ataxia type 3: blood biomarkers at the preataxic and ataxic stage in humans and mice. EMBO Mol Med. 2020;12(7):e11803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Adanyeguh IM, Perlbarg V, Henry PG, et al. Autosomal dominant cerebellar ataxias: imaging biomarkers with high effect sizes. NeuroImage Clin. 2018;19:858–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Koscik TR, Sloat L, van der Plas E, et al. Brainstem and striatal volume changes are detectable in under 1 year and predict motor decline in spinocerebellar ataxia type 1. Brain Commun. 2020;2(2):fcaa184. [DOI] [PMC free article] [PubMed] [Google Scholar]