

Abstract
The TDP-43 type C pathological form of frontotemporal lobar degeneration is characterized by the presence of immunoreactive TDP-43 short and long dystrophic neurites, neuronal cytoplasmic inclusions, neuronal loss and gliosis and the absence of neuronal intranuclear inclusions. Frontotemporal lobar degeneration-TDP-type C cases are commonly associated with the semantic variant of primary progressive aphasia or behavioural variant frontotemporal dementia. Here, we provide detailed characterization of regional distributions of pathological TDP-43 and neuronal loss and gliosis in cortical and subcortical regions in 10 TDP-type C cases and investigate the relationship between inclusions and neuronal loss and gliosis. Specimens were obtained from the first 10 TDP-type C cases accessioned from the Northwestern Alzheimer’s Disease Research Center (semantic variant of primary progressive aphasia, n = 7; behavioural variant frontotemporal dementia, n = 3). A total of 42 cortical (majority bilateral) and subcortical regions were immunostained with a phosphorylated TDP-43 antibody and/or stained with haematoxylin–eosin. Regions were evaluated for atrophy, and for long dystrophic neurites, short dystrophic neurites, neuronal cytoplasmic inclusions, and neuronal loss and gliosis using a semiquantitative 5-point scale. We calculated a ‘neuron-to-inclusion’ score (TDP-type C mean score – neuronal loss and gliosis mean score) for each region per case to assess the relationship between TDP-type C inclusions and neuronal loss and gliosis. Primary progressive aphasia cases demonstrated leftward asymmetry of cortical atrophy consistent with the aphasic phenotype. We also observed abundant inclusions and neurodegeneration in both cortical and subcortical regions, with certain subcortical regions emerging as particularly vulnerable to dystrophic neurites (e.g. amygdala, caudate and putamen). Interestingly, linear mixed models showed that regions with lowest TDP-type C pathology had high neuronal dropout, and conversely, regions with abundant pathology displayed relatively preserved neuronal densities (P < 0.05). This inverse relationship between the extent of TDP-positive inclusions and neuronal loss may reflect a process whereby inclusions disappear as their associated neurons are lost. Together, these findings offer insight into the putative substrates of neurodegeneration in unique dementia syndromes.
Keywords: frontotemporal lobar degeneration, primary progressive aphasia, behavioural variant frontotemporal dementia, TDP-43
Kawles et al. present detailed pathological findings from patients with FTLD-TDP-type C, and show that TDP-43 inclusions ‘disappear’ as their associated neurons are lost in both cortical and subcortical regions. These results provide insights into the link between cell death and neuropathology in dementia syndromes.
Introduction
Neurodegenerative diseases that cause dementia syndromes selectively ‘target’ anatomic regions responsible for cognition or behaviour. This selectivity leads to focal brain atrophy and to progressive clinical syndromes (i.e. dementias). In frontotemporal lobar degeneration (FTLD), there is progressive degeneration of the frontal and temporal cortices, which is almost exclusively associated with non-amnestic dementia clinical syndromes that affect personality, behaviour and/or language.1 FTLD gives rise to a number of clinical dementia presentations. These include primary progressive aphasia (PPA), a dementia syndrome characterized by selective language disruption, such as word-finding difficulties and impaired single word comprehension, and behavioural variant frontotemporal dementia (bvFTD), characterized by abnormalities in comportment, such as disinhibition, apathy and executive functioning.2,3 The one common denominator of nearly all cases of PPA is the leftward asymmetry of the atrophy in language-related regions that fits with the salience of the aphasic phenotype.4-6 In bvFTD, bifrontal or right-sided atrophy has been similarly considered a core biological feature of the syndrome.7–9
In the past, FTLD cases without tau accumulation were designated FTLD-U because the only abnormal protein precipitate detected was ubiquitin.10 We now know that the vast majority of these cases contain abnormal precipitates of a phosphorylated and truncated form of TDP-43, designated FTLD-TDP.10,11 Inclusions containing hyperphosphorylated transactive response DNA-binding protein 43 (TDP-43) are typically associated with frontotemporal dementia (FTD) and amyotrophic lateral sclerosis, and more recently have been found but in a much more restricted limbic distribution in pathologically confirmed autopsies with Alzheimer’s disease.2,12,13 Characteristic hallmarks of TDP-43 include neuronal intranuclear inclusions (NII), neuronal cytoplasmic inclusions (NCI), or dystrophic neurites (DNs). FTLD-TDP presents with mislocalization of TDP-43 from the nucleus to the cytoplasm,15 an important step in TDP-43 aggregation and potential toxicity. FTLD-TDP is classified into five distinct subtypes based on the presence, predominance, and distribution of NIIs, NCIs, and DNs in neocortical areas: A, B, C, D and E.12 Based on the Mackenzie et al.16 harmonized classification scheme, FTLD-TDP-type A is classified by moderate to numerous NCIs and short DNs in the upper cortical layers, and type B by predominance of NCIs in all cortical layers. Moderate NIIs are only observed in type D and occasionally type B.12,16 FTLD-TDP-type C (TDP-type C) is more easily distinguished from types A and B by abundance of long DNs in the superficial neocortex with rare NCIs.17 The significance of long DNs in particular remains unclear and intriguing, posited to be remnants of damaged axons or dendrites.
TDP-type C is commonly associated with severe anterior temporal atrophy, invariably resulting in poor word comprehension as the principal feature in the semantic variant of the PPA syndrome (PPA-S). Previous autopsy data show that nearly 90% of TDP-type C cases present with an antemortem clinical diagnosis of PPA-S, with the remaining 10% demonstrating a bvFTD syndrome in life.14,18 This clinicopathological correspondence provides an unparalleled model for studying selective vulnerability and putative correlates of neurodegeneration in clinical dementia syndromes. The goal of this study was to (i) characterize the distribution of TDP-type C pathological inclusions, with a focus on long DNs, in cases with PPA-S and bvFTD at a cortical and subcortical level; and (ii) identify the relationships between markers of pathological burden and patterns of neurodegeneration [neuronal loss and gliosis (NL/G)]. Through a granular focus on TDP-type C, our findings provide broader insights into the putative correlates of neurodegeneration in unique clinical dementia syndromes.
Materials and methods
Case characteristics
Ten FTLD-TDP-type C cases were consecutively accessioned from the brain holdings of the NIA-funded Alzheimer’s Disease Research Center at the Mesulam Center for Cognitive Neurology and Alzheimer’s Disease at Northwestern University’s Feinberg School of Medicine. All cases with a clinical diagnosis of PPA were co-enrolled in the PPA programme at the Mesulam Center for Cognitive Neurology and Alzheimer’s Disease. Written informed consent was obtained from all participants who committed to brain donation. Of the 10 cases, three were male and seven were female. All were right-handed. Age at death ranged from 55 to 82 (mean = 68.9 years) and age of onset ranged from 50 to 69 (mean = 55.9 years). Disease duration ranged from 8 to 20 years. The mean post-mortem interval was 17.44 h, and the mean brain weight was 1050.9 g. Seven of 10 cases carried a clinical diagnosis of PPA and three cases carried a clinical diagnosis of bvFTD. One case (Case 6) had additional evidence of motor neuron disease. The apolipoprotein E (ApoE) ε3,3 genotype was present in seven of nine cases with ApoE available. No cases had a family history of dementia or a known genetic mutation associated with FTLD-TDP. The diagnosis of PPA was based on the criteria of Mesulam19 and required a clinical history of progressive language impairment unaccompanied by consequential decline in other cognitive domains within the initial stages of the disease.20,21 All seven PPA cases displayed the semantic-variant subtype. A diagnosis of bvFTD was based on the 2011 work of the International Behavioural Variant FTD Consortium, which generated a set of sensitive and specific clinical diagnostic criteria.3 See Table 1 for demographic and participant information.
Table 1.
Case characteristics
| Case | Age of death (years) | Age at onset (years) | Disease duration (years) | Gender | PMI (h) | Brain weight (g) | Clinical diagnosis | MND | ApoE | Comorbid pathological diagnoses |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 74 | 62 | 12 | M | 26 | 1125 | PPA | No | 3,3 | ADNC A3, B1, C3; moderate vascular disease |
| 2 | 82 | 69 | 13 | F | 31 | 1250 | PPA | No | UNK | ADNC A1, B0, C2 |
| 3 | 71 | 55 | 15 | F | 19 | 910 | PPA | No | 3,3 | None |
| 4 | 55 | 45 | 10 | M | 18 | 1430 | PPA | No | 3,3 | Pigment spheroid degeneration, basal ganglia and substantia nigra |
| 5 | 65 | 52 | 13 | F | 5 | 810a | bvFTD | No | 2,3 | None |
| 6 | 71 | 63 | 8 | F | 28 | 970 | bvFTD | Yes | 3,4 | Severe vascular disease |
| 7 | 73 | 60 | 13 | M | 6 | 1370 | PPA | No | 3,3 | ADNC A1, B0, C0; moderate vascular disease |
| 8 | 61 | 50 | 11 | F | UNK | 840 | PPA | No | 3,3 | ADNC A1, B0, C0; hippocampal sclerosis |
| 9 | 73 | 53 | 20 | F | 12 | 904 | PPA | No | 3,3 | ADNC A1, B1, C0; UMN degeneration |
| 10 | 64 | 50 | 14 | F | 12 | 900 | bvFTD | No | 3,3 | ADNC, A1, B0, C0; PLS-like pathology |
ADNC = Alzheimer’s disease neuropathological change; F = female; M = male; MND = motor neuron disease; PMI = post-mortem interval; UNK = unknown.
Fixed brain weight.
Neuropathological evaluation and histological preparation
Following autopsy, the cerebral hemispheres were separated in the midsagittal plane, cut into 3- to 4-cm coronal slabs, fixed in 10% formalin for 2 weeks or 4% paraformaldehyde for 36 h, taken through sucrose gradients (10%–40%) for cryoprotection and stored in 40% sucrose at 4°C. Gross examination after autopsy showed generally severe atrophy in frontotemporal regions with hemispheric differences noted particularly in PPA cases (see below, Table 2). The pathological diagnosis of FTLD and specification of its variants was rendered by neuropathologists (E.B. and Q.M.) using the published consensus criteria of the Consortium for FTLD.1 Blocks containing regions of interest were embedded in paraffin and sections were cut to a thickness of 5 μm for immunohistochemical analysis using an antibody against human phosphorylated TDP-43 (pSer409/410, Cosmo Bio; polyclonal) as described previously,22 to visualize TDP-43-positive pathology. NL/G was assessed using haematoxylin and eosin stained sections. A total of 42 unique cortical and subcortical regions were analysed. Regions used for analysis are listed in Supplementary Table 1.
Table 2.
Gross atrophy in 10 FTLD-TDP-type C cases
| Case | Hippocampus | Frontal | Temporal | Parietal | Occipital | Caudate | Brainstem | Cerebellum |
|---|---|---|---|---|---|---|---|---|
| 1 | 0 | B ++ | B +++ | L ++, R+ | 0 | 0 | 0 | 0 |
| 2 | B +++ (L > R) | L ++, R + | L ++, R + | L +, R 0 | 0 | L +++, R+ | 0 | 0 |
| 3 | A +++, P ++ | B ++ (L > R) | L +++, R ++ | B ++ (L > R) | 0 | L +++, R+ | 0 | 0 |
| 4 | B + | B ++ | L +++, R ++ | B ++ | 0 | 0 | 0 | 0 |
| 5 | B + | B +++ | B +++ | B +++ | B + | +++ | MB +++ | 0 |
| 6 | B + | B +++ | B +++ | B ++ | B + | +++ | + | 0 |
| 7 | 0 | B ++ (L > R) | B +++ (L > R) | B ++ | 0 | 0 | 0 | 0 |
| 8 | L +++, R ++ | L +++, R ++ | L +++, R ++ | L ++, R+ | B + | ++ | 0 | 0 |
| 9 | B ++ | B +++ (R > L) | B +++ (R > L) | B ++ | B + | 0 | 0 | 0 |
| 10 | B + | B +++ | B +++ (L > R) | B ++ | B + | ++ | + | 0 |
With the exception of one case (Case 9), all PPA cases showed asymmetric atrophy of cortex consistent with the aphasic phenotype. Cases diagnosed with bvFTD (Cases 5, 6 and 10) showed relatively symmetric patterns of cortical atrophy at gross examination. Semiquantitative grading: 0 absent; + mild, ++ moderate; +++ severe/numerous. A = anterior; P = posterior; L = left; R = right; B = bilateral; MB = midbrain.
Semiquantitative analysis
The extent of TDP-type C pathology and NL/G were assessed using a semiquantitative 5-point scale of 0 to 4. TDP-positive inclusions were scored as follows: 0, absent; 1, rare (i.e. one inclusion in several 200× fields); 2, mild (maximum of one inclusion per 200× field); 3, moderate (maximum of two to three inclusions per 200× field); and 4, severe (more than three inclusions in most 200× field); see Bigio et al.23 for examples of cortical NL/G at varying degrees. TDP-type C pathology included NCIs, short DNs and long DNs. Consistent with prior reports,12,16 NIIs were virtually absent and therefore not included in our analysis in the majority of cases. TDP-43 positive lesions were assessed in both the superficial and deep cortical layers, corresponding to lamina II and IV–VI, respectively. In rare cases where additional sections were required, assessment was carried out by neuropathologists Q.M. and A.F. We calculated a ‘neuron-to-inclusion’ score for each region per case to assess the relationship between the extent of TDP-type C pathology versus NL/G; the ‘neuron-to-inclusion’ score was calculated by subtracting the mean NL/G score from the mean TDP-type C score for each TDP-type C pathological specie (NCIs, short DNs and long DNs). Two subgroups of regions were chosen to further examine this relationship: the ‘high neuronal dropout’ subgroup included three regions with the lowest neuron-to-inclusion score, indicative of greater NL/G than inclusion density (amygdala, temporal poles, transentorhinal and entorhinal cortices) and the ‘low neuronal dropout’ subgroup included three regions with the highest neuron-to-inclusion score, indicative of lower NL/G when compared with density of inclusions (right and left inferior frontal gyri, occipital cortex and somatosensory cortex).24
Statistical analysis
Spearman correlation was used to examine the relationship between TDP-type C pathology and NL/G across all cortical and subcortical regions and between the high and low neuron-to-inclusion subgroups. Differences in neuron-to-inclusion score between the high and low neuronal dropout regions were assessed with two different methods, each performed separately for the three different pathology types: (i) For each region comprising the high and low groups, the median neuron-to-inclusion score across all subjects was computed. Normality was determined with the Shapiro–Wilk test and a t-test was then used to compare whether medians differed between the high and low neuronal dropout groups; and (ii) A linear mixed-effects model with neuron-to-inclusion score as the response, high or low group as a binary covariate and a random intercept for subject was fit. The coefficient of group was used to determine if the neuron-to-inclusion score differed between the high and low groups. Significance level was set at P < 0.05 (two-sided).
Data availability
The data that support the findings of this study are available from the corresponding author, upon reasonable request.
Results
Gross patterns of atrophy in FTLD-TDP-type C cases
Regions including frontal, temporal, parietal and occipital cortices, hippocampus, caudate, brainstem and cerebellum were examined grossly for atrophy and patterns of hemispheric asymmetry. Of the seven PPA cases, six showed clear leftward gross asymmetry in at least one cortical region and one case showed slight rightward asymmetry in frontotemporal regions. All three bvFTD cases showed bilateral symmetry in frontotemporal cortical regions; Case 10 showed slight predilection towards leftward asymmetry in temporal regions. Atrophy patterns appreciated at gross examination are detailed in Table 2.
Anatomic characterization of FTLD-TDP-type C inclusions and neuronal loss/gliosis in cortical and subcortical regions at the group level
TDP-type C pathology distributions differed according to inclusion morphology and type (Fig. 1). Semiquantitative scoring data were consolidated to form heat maps of long DN, short DN, NCI and NL/G distribution (Fig. 2). Pathological inclusions were generally most abundant in frontal and temporal cortical regions, including the temporal poles and insula, and subcortical regions including caudate, putamen, amygdala and claustrum. Bilateral temporal poles, insula and transentorhinal and entorhinal cortices showed highest levels of NL/G among cortical regions, and the amygdala and caudate were the most affected subcortical regions. We observed a leftward asymmetry in the cortical regions in both TDP-type C pathology and NL/G, particularly in PPA cases. Interestingly, the thalamus, subthalamus and globus pallidus were either only mildly affected or entirely spared from pathological burden; NL/G followed the same pattern, except for the globus pallidus, which showed mild to moderate loss. The majority of brainstem regions and nuclei were spared from NL/G and TDP-type C inclusions, with a number of exceptions. The red nucleus of the tegmentum showed moderate short and long DN distribution. The substantia nigra showed mild NCIs and short DNs and mild NL/G, and the periaqueductal grey showed mild short DN distribution. The locus coeruleus showed mild NL/G only. These subcortical findings were consistent across PPA and bvFTD cases, further highlighting that the differences in pathological burden between clinical phenotype appear primarily in cortical regions.
Figure 1.
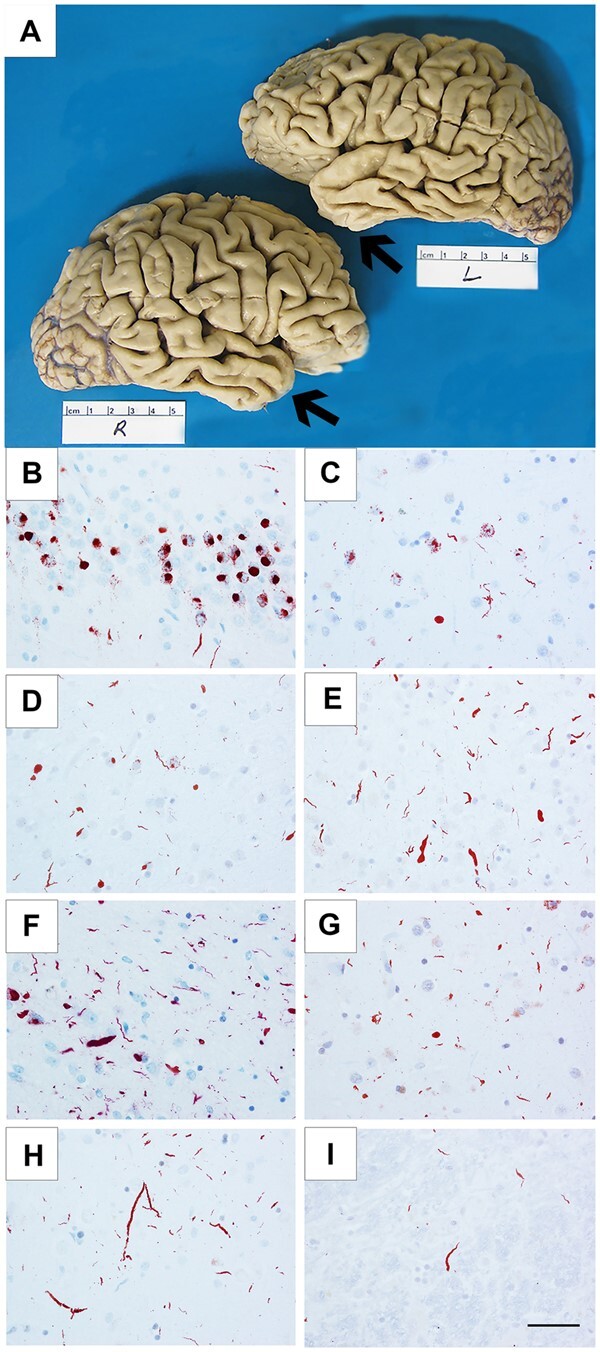
Cortical atrophy and TDP-43 immunoreactive pathology in FTLD-TDP-type C cases. Cortical atrophy of FTLD-TDP-type C is distinct in its selective and severe targeting of the temporal poles (arrows), seen in Case 3, a 71-year-old female with a 15-year duration of the semantic form of primary progressive aphasia (PPA-S), more severe on the left (A). Distribution of TDP-43-positive inclusions varied according to inclusion morphology and type (NCIs, short DNs, long DNs), suggesting that certain neuronal populations may be differentially vulnerable in TDP-type C. TDP-43-positive NCIs were severe in the dentate granular cells of hippocampus (B) and in the striatum (C), and mild in the middle frontal cortex (D). Short DNs were severe in the superior temporal cortex (E) and amygdala (F), and mild in the striatum (G). Long DNs were severe in the superior temporal cortex (H) and mild in the red nucleus (I). (B and H) Case 8; (C) Case 7; (D, E and I) Case 10; (F) Case 1; (G) Case 6. Scale bar = 50 μm in I, and also applies to B–H.
Figure 2.

Heat maps of TDP-type C inclusions (NCIs, short DNs, and long DNs) and neuronal loss and gliosis in 10 cases. Each colour block indicates the severity of the disease burden according to the semiquantitative rating scale; 0 = absent, 1 = rare, 2 = mild, 3 = moderate, and 4 = severe; grey shading indicates that the region was not available for pathological inspection. Heat maps were arranged by the highest to lowest average score across all 10 cases for each inclusion type or NL/G. At the group level, we observed an overall positive correlation (Spearman) across all regions between NL/G and NCIs (r = 0.654, P < 0.0001), short DNs (r = 0.662, P < 0.0001) and long DNs (r = 0.537, P < 0.001).
At the group level (n = 10), we observed an overall positive correlation across all regions between NL/G and NCIs (r = 0.654, P < 0.0001), short DNs (r = 0.662, P < 0.0001) and long DNs (r = 0.537, P < 0.001), suggesting that, in general, pathological inclusions accompany neuronal loss and gliosis. We noticed regional differences between the densities of NCIs and DNs; for example, while the granule cell layer of the dentate gyrus was selectively vulnerable to NCIs, it was relatively devoid of DNs (Fig. 2). At the neuronal level, granule cells were relatively intact despite harbouring NCIs; this was not the case in regions containing DNs, in which neurons were lost.
Anatomic heat maps were derived from data displayed in Fig. 2 to visualize regional distributions and relationships between NL/G and long DNs (Fig. 3). First, we were able to appreciate a distinct leftward asymmetric pattern of long DNs and NL/G in cortical regions. At the group level, a positive relationship between long DNs and NL/G was generally appreciated (Fig. 3), whereby higher pathological burden was accompanied by greater NL/G. However, we were struck by what appeared to be areas that did not follow this trend. Right and left temporal poles, areas shown to undergo atrophy in the initial course of disease propagation,25 showed severe neuronal loss, but only mild-to-moderate long DN inclusion burden. The same inverse trend was observed in the amygdala and transentorhinal cortex, areas also involved early in disease course. By contrast, at the group level, distribution heat maps showed that long DNs were most abundant in the middle frontal, left superior temporal and inferior parietal gyri, and inferior frontal gyri bilaterally, but showed mild-to-moderate NL/G. We concluded that areas at the highest and lowest ratings of NL/G display a reverse pattern of inclusion density (Supplementary Table 2).
Figure 3.

Mean cortical distribution (lateral view) and mean subcortical/cortical distribution (coronal view) of long DNs and NL/G in TDP-type C. Cortical and subcortical distribution maps depict the relationship between TDP-type C long dystrophic neurites (long DNs) and neuronal loss and gliosis at the group level (n = 10). Semiquantitative ratings for (A and B) long DNs and (C and D) NL/G in cortical regions were averaged across all 10 cases. Scale bar on the bottom shows the colour associated with mean scores. Through the anatomic depiction of long DN and NL/G distribution, we found areas that showed an inverse relationship between long DNs and NL/G that was not appreciated through standard heat maps. These areas included the right and left temporal poles, the entorhinal and transentorhinal cortices and the amygdala, which are three regions that are first affected in FTLD-TDP-type C disease progression. (A and C) CBL = cerebellum; IFG = inferior frontal gyrus; IPL = inferior parietal lobule; LH = left hemisphere; MFG = middle frontal gyrus; OCC = occipital; RH = right hemisphere; STG = superior temporal gyrus; TP = temporal pole. (B and D) cd = caudate; cg = cingulate gyrus; cls = claustrum; dg = dentate gyrus; erc = entorhinal cortex; gp = globus pallidus; ic = internal capsule; ins = insula; itg = inferior temporal gyrus; LH = left hemisphere; mc = motor cortex; mtg = middle temporal gyrus; pon = pons; pre-sub = pre-subiculum; pt = putamen; RH = hemisphere; rn = red nucleus; sc = somatosensory cortex; sn = substantia nigra; stg = superior temporal gyrus; stn = subthalamic nucleus; sub = subiculum; th = thalamus; trs erc = transentorhinal cortex. *Refer to Fig. 2 to view NL/G and long DN distribution within the amygdala. Created with BioRender.com.
Inverse relationship between TDP-type C inclusions and neuronal loss/gliosis at the individual level
We used the neuron-to-inclusion score to investigate the complex association between the extent of TDP-type C pathology versus NL/G. We identified the three regions with the highest levels of neurodegeneration (i.e. high neuronal loss and gliosis): amygdala, right and left temporal poles and transentorhinal/entorhinal cortices. We found that these three regions had the lowest (most negative) neuron-to-inclusion score, indicating high neuronal ‘dropout’ with low densities of inclusions at autopsy. Conversely, we identified three regions that did not show significant neurodegeneration (i.e. low NL/G), which interestingly were accompanied by high density of inclusions. These three regions included right and left inferior frontal gyri, the occipital cortex and the somatosensory cortex, and were found to have the highest (most positive) neuron-to-inclusion score. We designated these subgroups the ‘high neuronal dropout’ and the ‘low neuronal dropout’, respectively. The ‘high neuronal dropout’ group showed low pathology and high neuronal loss, while the ‘low neuronal dropout’ group showed abundant pathology and relatively preserved neuronal densities. A linear mixed-effects model showed that, at the case-by-case level, the ‘neuron-to-inclusion’ score of the ‘low neuronal dropout’ group was significantly greater than that of the ‘high neuronal dropout’ group across NCIs (P < 0.0001), short DNs (P < 0.0001) and long DNs (P < 0.0001; Fig. 4). This difference was also evident when the neuron-to-inclusion scores were averaged across subjects: separate t-tests for each pathology type showed significant differences in median score between the high and low group for NCIs (P < 0.05), short DNs (P < 0.01) and long DNs (P < 0.001), with the low group always larger than the high group (Fig. 5). Results suggest that pathological burden and NL/G in TDP-type C cases are positively and significantly correlated; however, this relationship is reversed in areas with severe neurodegeneration (e.g. temporal poles). Regions with intact neurons harbour high densities of pathological inclusions, giving the impression that there is abundant pathology in absolute terms (Fig. 6). These relationships were observed regardless of inclusion subtype (NCI, short DNs or long DNs). At both the group and individual levels, our findings reflect a remarkable process in which TDP-43 inclusions disappear with the degeneration of accompanying neurons in regions extremely afflicted by disease (Supplementary Table 3).
Figure 4.

Relationship between TDP-type C inclusions and NL/G at the individual case level. Neuron-to-inclusion scores were calculated for (A) NCIs, (B) short DNs and (C) long DNs in the selected regions in the high and low neuronal dropout groups, and scores from all 10 cases were plotted. When scores were available from homotypic regions in both hemispheres, such as inferior frontal gyrus (IFG) and temporal poles, scores were averaged for the individual inclusion and NL/G in the right and left hemispheres and used in the analysis and are thus notated as bilateral temporal poles and bilateral IFG. Entorhinal and transentorhinal cortices also had similar neuron-to-inclusion scores and were therefore also averaged, notated as Trans/ERC. Neuron-to-inclusion scores differed significantly between high and low neuronal dropout regions at an individual level across NCIs (P < 0.0001), short DNs (P < 0.0001) and long DNs (P < 0.0001).
Figure 5.

Relationship between TDP-type C long DNs, short DNs and NCI in high versus low neuronal dropout at the group level. Neuron-to-inclusion scores were averaged across all 10 cases for long DNs, short DNs and NCIs in the regions designated as part of the high neuronal dropout group and low neuronal dropout group. We found that the median neuron-to-inclusion scores differed significantly (P < 0.05) between these two groups.
Figure 6.

Inverse relationship between TDP-type C DNs and NL/G in temporal pole (TP) and middle frontal gyrus (MFG). (B and C) Images were obtained from Case 4, a 55-year-old male with a 10-year duration of the semantic form of primary progressive aphasia (PPA-S). (A) Image was obtained from Case 1, a 74-year-old female with a 12-year duration of PPA-S. (D) Image was obtained from Case 8, a 61-year-old female with an 11-year duration of PPA-S. In the left temporal pole, severe neuronal loss and gliosis are observed (score = 4) (A), with mild-to-moderate TDP-type C pathology (score = 2.5) (C), suggesting that inclusions may wane as neuronal dropout occurs. This is juxtaposed with B and D, which highlight the reverse relationships; in the left MFG, NL/G (B) was mild (score = 1), while the presence of TDP-type C inclusions (D) were severe (score = 4). Scale bar = 50 µm in D, and also applies to A–C.
Discussion
In the post-mortem neuropathological evaluation, distribution and morphological features of TDP-43-positive inclusions are used to distinguish between FTLD-TDP subtypes.1 FTLD-TDP-type C in particular is diagnosed by the presence of thick, long DNs in the upper neocortical layers and the lower densities of neuronal cytoplasmic inclusions. A staging scheme utilizing volumetric MRI scans has been offered for TDP-type C, which affirms the amygdala, temporal poles and the medial temporal lobe as regions first affected by the disease.10,24 The present study aimed to characterize with granularity the distribution of TDP-type C pathological inclusions, with a focus on long DNs, in cortical and subcortical regions from 10 autopsy-confirmed cases, who presented during life with PPA or bvFTD. The goal was to ascertain the unique fingerprint of FTLD-TDP-type C, and through this characterization, glean relationships between inclusions and patterns of neurodegeneration (neuronal loss and gliosis).
There were three main findings. First, dedicated heat maps based on quantitative scoring showed extensive cortical and subcortical TDP-type C inclusions and neuronal loss. Prior studies have examined inclusions and neuronal loss in TDP-type C but not as extensively in bilateral cortex and subcortical regions.26,27 While the amygdala is known to be severely affected in TDP-type C,10,24 subcortical regions caudate, putamen and claustrum were also devastated. The amygdala in particular has been considered a ‘locus of pathological protein misfolding’ across nearly all neurodegenerative diseases.28 Reports by Mackenzie and Neumann in 2020 showed that cases with types A and B display more NCIs and short DNs than type C in the substantia nigra, which, consistent with our findings, showed mild DN pathology.26 The locus coeruleus and substantia nigra, nuclear components of the reticular activating system that are commonly targeted in both Alzheimer’s and alpha-synucleinopathies, showed mild neuronal loss and gliosis.
Preceding the harmonized TDP-43 classification scheme,16 a number of studies described the patterns of TDP-43 immunoreactive NCIs and DNs in neocortex, subcortical regions and hippocampus.29-31 In 2009, Josephs and colleagues31 extended their survey to include the amygdala, putamen, thalamus and brainstem, in addition to cortex and hippocampal subregions. Over the last decade our understanding of clinical correlates of TDP-type C pathology has informed diagnosis, uniform criteria and an appreciation of brain–behaviour relationships. The current examination that includes 42 cortical and subcortical regions, many bilateral, is likely to offer a more comprehensive view of the anatomic vulnerability profile of TDP-type C and provide insight into cell loss and disease spread. In some neuropathological entities, such as Alzheimer’s disease, neurodegeneration seems to spread along projection pathways, usually in a corticofugal direction.32–34 It is interesting that the globus pallidus, with no direct connections with the cerebral cortex, is spared in TDP-type C. However, the striatum, which receives extensive cortical input, was heavily burdened by NCIs and short DNs, but not long DNs. The origins of DNs in TDP-43 are not currently known. In Alzheimer’s disease, DNs are processes that form around neuritic plaques and are microscopically noticeable because of abnormal sprouting, expansion and aberrant collection of cytoskeletal proteins35; it is likely that they reflect both axonal and dendritic damage. In Alzheimer’s disease, DNs also have been shown to correlate with synaptic impairments,36 suggesting that they may hamper neurocircuitry during disease spread. A closer study of network-level progression trajectories of TDP-C inclusions may help elucidate the biology of TDP spread, anatomic vulnerabilities and resultant functional impairments.
We have shown that dementia syndromes are associated with sites of peak atrophy within components of the relevant neurocognitive network, and that dysfunction is associated with the distribution of underlying pathology.37–39 In the PPA cases in this study, both cortical TDP-type C inclusions and NL/G followed the well-characterized leftward asymmetry concordant with the laterality of the language network in this right-handed sample.4,5,40,41 At group level, neuronal loss and gliosis were positively correlated with the quantity of TDP-43 inclusions. However, we also observed inverse relationships in regions with predilection for severe atrophy. For example, the anterior temporal lobe displayed profound neuronal loss but sparse TDP inclusions. In fact, the density of anterior temporal lobe inclusions was similar to that of somatosensory cortex, a region with negligible neuronal loss. It seemed that the pattern of severe neuronal loss combined with relatively sparse TDP-43 abnormality marked regions of early involvement whereas lesser cell loss in the presence of severe TDP-43 abnormality marked areas that were affected later. Somatosensory cortex, with little neuronal loss or TDP-43 abnormality, reflected a third pattern of relative sparing. Consistent with previous reports,42,43 our findings suggest that with increasing neurodegeneration, the density of TDP-43 aggregates may be reduced, perhaps through microglial absorption. A different pattern is seen in Alzheimer’s Disease, where the neurofibrillary tangles linger after neurons die, forming ‘ghost tangles’ which represent the transformation of intracellular tau deposits into insoluble extracellular tangles.44–46 No such tombstones exist in FTLD-TDP type C. In the future, stereological quantification in large groups of participants with PPA and bvFTD will enable a more precise view of regional differences across TDP-C cases, and the extent to which inclusions associate with activated glia, synaptic loss, dendritic abnormalities, and cognitive function.
Supplementary Material
Acknowledgements
We are grateful to our research participants for their invaluable contributions to scientific discovery.
Funding
This study was supported by grants from the National Institute on Aging (P30 AG013854, R01 AG056258, R01 AG062566, K08 AG065463), National Institute of Neurological Disorders and Stroke (R01 NS085770, R01 NS075075, T32 NS047987), the National Institute on Deafness and Other Communication Disorders (R01 DC008552) and a National Alzheimer’s Coordinating Center New Investigator Award (U01 AG016976).
Competing interests
The authors report no competing interests. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Supplementary material
Supplementary material is available at Brain online.
Abbreviations
- bvFTD
-
behavioural variant frontotemporal dementia
DNs = dystrophic neurites
- FTLD
-
frontotemporal lobar degeneration
NCI = neuronal cytoplasmic inclusions
NL/G = neuronal loss and gliosis
- PPA
primary progressive aphasia
References
- 1. Cairns NJ, Bigio EH, Mackenzie IR, et al. ; Consortium for Frontotemporal Lobar Degeneration. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: Consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007;114(1):5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mesulam MM. Primary progressive aphasia—a language-based dementia. N Engl J Med. 2003;349(16):1535–1542. [DOI] [PubMed] [Google Scholar]
- 3. Rascovsky K, Hodges JR, Knopman D, et al. . Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134(Pt 9):2456–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rogalski E, Cobia D, Martersteck A, et al. . Asymmetry of cortical decline in subtypes of primary progressive aphasia. Neurology. 2014;83(13):1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mesulam MM. Primary progressive aphasia and the left hemisphere language network. Dement Neurocogn Disord. 2016;15(4):93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mesulam M, Weintraub S, Parrish T, Gitelman D. Primary progressive aphasia: Reversed asymmetry of atrophy and right hemisphere language dominance. Case Reports Research Support, N.I.H., Extramural Research Support, U.S. Gov't, P.H.S. Neurology. 2005;64(3):556–557. [DOI] [PubMed] [Google Scholar]
- 7. Bertoux M, O'Callaghan C, Flanagan E, Hodges JR, Hornberger M. Fronto-striatal atrophy in behavioral variant frontotemporal dementia and Alzheimer's disease. Front Neurol. 2015;6:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lu PH, Mendez MF, Lee GJ, et al. . Patterns of brain atrophy in clinical variants of frontotemporal lobar degeneration. Dement Geriatr Cogn Disord. 2013;35(1-2):34–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Seeley WW, Crawford R, Rascovsky K, et al. . Frontal paralimbic network atrophy in very mild behavioral variant frontotemporal dementia. Arch Neurol. 2008;65(2):249–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rohrer JD, Geser F, Zhou J, et al. . TDP-43 subtypes are associated with distinct atrophy patterns in frontotemporal dementia. Neurology. 2010;75(24):2204–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Forman MS, Trojanowski JQ, Lee VM. TDP-43: A novel neurodegenerative proteinopathy. Curr Opin Neurobiol. 2007;17(5):548–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee EB, Porta S, Michael Baer G, et al. . Expansion of the classification of FTLD-TDP: Distinct pathology associated with rapidly progressive frontotemporal degeneration. Acta Neuropathol. 2017;134(1):65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Irwin DJ, McMillan CT, Xie SX, et al. . Asymmetry of post-mortem neuropathology in behavioural-variant frontotemporal dementia. Brain. 2018;141(1):288–301. 10.1093/brain/awx319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Landin-Romero R, Tan R, Hodges JR, Kumfor F. An update on semantic dementia: Genetics, imaging, and pathology. Alzheimers Res Ther. 2016;8(1):52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Grossman M, Wood EM, Moore P, et al. . TDP-43 pathologic lesions and clinical phenotype in frontotemporal lobar degeneration with ubiquitin-positive inclusions. Arch Neurol. 2007;64(10):1449–1454. [DOI] [PubMed] [Google Scholar]
- 16. Mackenzie IR, Neumann M, Baborie A, et al. . A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 2011;122(1):111–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mackenzie IR, Neumann M, Bigio EH, et al. . Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: An update. Acta Neuropathol. 2010;119(1):1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gefen T, Ahmadian SS, Mao Q, et al. . Combined pathologies in FTLD-TDP types A and C. J Neuropathol Exp Neurol. 2018;77(5):405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mesulam MM. Primary progressive aphasia. Ann Neurol. 2001;49(4):425–432. [PubMed] [Google Scholar]
- 20. Mesulam M. Primary progressive aphasia: A dementia of the language network. Dement Neuropsychol. 2013;7(1):2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mesulam MM. Slowly progressive aphasia without generalized dementia. Ann Neurol. 1982;11(6):592–598. [DOI] [PubMed] [Google Scholar]
- 22. Mao Q, Wang D, Li Y, et al. . Disease and region specificity of granulin immunopositivities in Alzheimer disease and frontotemporal lobar degeneration. J Neuropathol Exp Neurol. 2017;76(11):957–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bigio EH, Weintraub S, Rademakers R, et al. . Frontotemporal lobar degeneration with TDP-43 proteinopathy and chromosome 9p repeat expansion in C9ORF72: Clinicopathologic correlation. Neuropathology. 2013;33(2):122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bocchetta M, Iglesias Espinosa MDM, Lashley T, Warren JD, Rohrer JD. In vivo staging of frontotemporal lobar degeneration TDP-43 type C pathology. Alzheimers Res Ther. 2020;12(1):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Borghesani V, Battistella G, Mandelli ML, et al. . Regional and hemispheric susceptibility of the temporal lobe to FTLD-TDP type C pathology. Neuroimage Clin. 2020;28:102369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mackenzie IR, Neumann M. Subcortical TDP-43 pathology patterns validate cortical FTLD-TDP subtypes and demonstrate unique aspects of C9orf72 mutation cases. Acta Neuropathol. 2020;139(1):83–98. [DOI] [PubMed] [Google Scholar]
- 27. Brandmeir NJ, Geser F, Kwong LK, et al. . Severe subcortical TDP-43 pathology in sporadic frontotemporal lobar degeneration with motor neuron disease. Acta Neuropathol. 2008;115(1):123–131. [DOI] [PubMed] [Google Scholar]
- 28. Nelson PT, Abner EL, Patel E, et al. . The amygdala as a locus of pathologic misfolding in neurodegenerative diseases. J Neuropathol Exp Neurol. 2018;77(1):2–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mackenzie IR, Baborie A, Pickering-Brown S, et al. . Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: Classification and relation to clinical phenotype. Acta Neuropathol. 2006;112(5):539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sampathu DM, Neumann M, Kwong LK, et al. . Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am J Pathol. 2006;169(4):1343–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Josephs KA, Stroh A, Dugger B, Dickson DW. Evaluation of subcortical pathology and clinical correlations in FTLD-U subtypes. Acta Neuropathol. 2009;118(3):349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Braak H, Brettschneider J, Ludolph AC, Lee VM, Trojanowski JQ, Del Tredici K. Amyotrophic lateral sclerosis—A model of corticofugal axonal spread. Nat Rev Neurol. 2013;9(12):708–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim EJ, Hwang JL, Gaus SE, et al. . Evidence of corticofugal tau spreading in patients with frontotemporal dementia. Acta Neuropathol. 2020;139(1):27–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Braak H, Braak E. Staging of Alzheimer's disease-related neurofibrillary changes. Neurobiol Aging. 1995;16(3):271–278. Discussion 278–84. [DOI] [PubMed] [Google Scholar]
- 35. Yan XX. Formation of dystrophic neurites in Alzheimer's disease. FASEB J. 2017;31:91.3. [Google Scholar]
- 36. Sharoar MG, Hu X, Ma XM, Zhu X, Yan R. Sequential formation of different layers of dystrophic neurites in Alzheimer's brains. Mol Psychiatry. 2019;24(9):1369–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gefen T, Gasho K, Rademaker A, et al. . Clinically concordant variations of Alzheimer pathology in aphasic versus amnestic dementia. Brain. 2012;135(Pt 5):1554–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Weintraub S, Mesulam M. With or without FUS, it is the anatomy that dictates the dementia phenotype. Brain. 2009;132(Pt 11):2906–2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mesulam MM, Weintraub S, Rogalski EJ, Wieneke C, Geula C, Bigio EH. Asymmetry and heterogeneity of Alzheimer's and frontotemporal pathology in primary progressive aphasia. Brain. 2014;137(Pt 4):1176–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim G, Bolbolan K, Gefen T, et al. . Atrophy and microglial distribution in primary progressive aphasia with transactive response DNA-binding protein-43 kDa. Ann Neurol. 2018;83(6):1096–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kim G, Vahedi S, Gefen T, et al. . Asymmetric TDP pathology in primary progressive aphasia with right hemisphere language dominance. Neurology. 2018;90(5):e396–e403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yousef A, Robinson JL, Irwin DJ, et al. . Neuron loss and degeneration in the progression of TDP-43 in frontotemporal lobar degeneration. Acta Neuropathol Commun. 2017;5(1):68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nishihira Y, Tan CF, Hoshi Y, et al. . Sporadic amyotrophic lateral sclerosis of long duration is associated with relatively mild TDP-43 pathology. Acta Neuropathol. 2009;117(1):45–53. [DOI] [PubMed] [Google Scholar]
- 44. Bancher C, Brunner C, Lassmann H, et al. . Tau and ubiquitin immunoreactivity at different stages of formation of Alzheimer neurofibrillary tangles. Prog Clin Biol Res. 1989;317:837–848. [PubMed] [Google Scholar]
- 45. Ikeda K, Haga C, Oyanagi S, Iritani S, Kosaka K. Ultrastructural and immunohistochemical study of degenerate neurite-bearing ghost tangles. J Neurol. 1992;239(4):191–194. [DOI] [PubMed] [Google Scholar]
- 46. Endoh R, Ogawara M, Iwatsubo T, Nakano I, Mori H. Lack of the carboxyl terminal sequence of tau in ghost tangles of Alzheimer's disease. Brain Res. 1993;601(1-2):164–172. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, upon reasonable request.