

Abstract
Efferocytosis, the clearance of apoptotic cells (ACs) by macrophages, is critical for tissue resolution, with defects driving many diseases. Mechanisms of efferocytosis-mediated resolution are incompletely understood. Here, we show that AC-derived methionine regulates resolution through epigenetic repression of the ERK1/2 phosphatase Dusp4. We focus on two key efferocytosis-induced pro-resolving mediators, PGE2 and TGFβ1, and show that efferocytosis induces Ptgs2/COX2, leading to PGE2 synthesis and PGE2-mediated induction of TGFβ1. ERK1/2 phosphorylation/activation by AC-activated CD36 is necessary for Ptgs2 induction, but this is insufficient owing to an ERK-DUSP4 negative-feedback pathway that lowers phospho-ERK. However, subsequent AC engulfment and phagolysosomal degradation repress Dusp4, enabling enhanced phospho-ERK and induction of the Ptgs2-PGE2-TGFβ1 pathway. Mechanistically, AC-derived methionine is converted to S-adenosylmethionine (SAM), which is used by DNA-methyltransferase-3A (DNMT3A) to methylate Dusp4. Bone-marrow DNMT3A deletion in mice blocks COX2/PGE2, TGFβ1, and resolution in sterile-peritonitis, apoptosis-induced thymus injury, and atherosclerosis. Knowledge of how macrophages use AC-cargo and epigenetics to induce resolution provides mechanistic insight and therapeutic options for diseases driven by impaired resolution.
Macrophages play critical roles during the resolution of inflammation by clearing apoptotic cells (ACs), termed efferocytosis, and secreting pro-resolving factors that mediate tissue repair after injury1. Defects in efferocytosis and resolving mediator synthesis lead to cell and tissue necrosis, unresolved inflammation, and tissue damage, and impaired efferocytosis has been implicated in several chronic inflammatory diseases, including autoimmune diseases, atherosclerosis, chronic lung diseases, and neurodegenerative diseases2,3. For example, efferocytosis is impaired in advanced atherosclerotic lesions, promoting clinically unstable plaques characterized by necrosis, inflammation, and fibrous cap thinning2,4,5. Resolving mediators produced during efferocytosis not only promote resolution but also enhance efferocytosis itself, forming a highly beneficial positive-feedback cycle1,2. Accordingly, when efferocytosis becomes defective in diseases, impaired resolution ensues as part of a negative feedback cycle2,3.
Efferocytosis-induced macrophage resolution pathways are triggered by activation of AC receptors or molecules resulting from the phagolysosomal degradation of engulfed ACs2,3. For example, AC-derived arginine enhances the engulfment of subsequent ACs via an ornithine-putrescine-dependent metabolic pathway6; fatty acids resulting from efferocytosis promote macrophage synthesis of IL-107; and AC-derived cholesterol activates liver-X-receptor mediated resolution pathways8.
An important efferocytosis-mediated resolution program involves the synthesis and secretion of PGE2 and TGFβ19. PGE2 synthesis involves the conversion of arachidonic acid to PGH2 by cyclooxygenase 2 (COX2; encoded by Ptgs2), followed by conversion of PGH2 to PGE2 by PGES. Although COX2 and PGE2 have been implicated in inflammation and thrombosis10,11, their effects are context- and PGE2 receptor-dependent, and PGE2 has pro-resolving roles in certain settings, including efferocytosis, atherosclerosis, and cardiac ischemia/reperfusion9,12,13,14,15,16,17,18. Similarly, while TGFβ can promote inflammation in certain settings, it has potent pro-repair activities19, including in the settings of efferocytosis and atherosclerosis9,20.
A key gap in this area of research is how efferocytosis stimulates the production of PGE2 and TGFβ. In this report, we show that this process occurs through coordinated signaling involving AC-receptor binding and a DNA methylation pathway triggered by AC-derived methionine. Knowledge of this pathway may suggest new therapeutic strategies for the many diseases in which macrophage-mediated resolution is impaired.
Results
Efferocytosis-induced TGFβ1 requires AC degradation and PGE2.
We began by investigating whether the key process of efferocytosis-induced PGE2 and TGFβ19 might be linked to the phagolysosomal degradation of ACs. First, we showed that incubation of bone marrow-derived macrophages (BMDMs) with ACs causes increases in Ptgs2 mRNA, its product cyclooxygenase 2 (COX2), media PGE2, and media TGFβ1 (Extended Data Fig. 1a–c). The ACs were human Jurkat cells and the primers were based on mouse mRNA sequences, and thus the Ptgs2 and Tgfb1 mRNA was from the macrophages, not the engulfed ACs (Extended Data Fig. 1d). To test the importance of AC phagolysosomal degradation, we silenced Rubicon, a protein required for LC3-associated phagocytosis (LAP), which mediates lysosome-phagosome fusion and phagolysosomal AC degradation during efferocytosis21. We found that Rubicon silencing suppressed AC-induced increases in both Ptgs2 and Tgfb1 (Fig. 1a,b and Extended Data Fig. 1e). Similar results were found with macrophages pre-treated with bafilomycin A1, a lysosomal vacuolar ATPase inhibitor that blocks phagolysosomal AC degradation during efferocytosis6 (Fig. 1c,d), and with human monocyte-derived macrophages (HMDMs) (Fig. 1e,f). Note that neither siRbcn nor bafilomycin treatment blocked initial AC engulfment (Extended Data Fig. 1f). These data suggest that one or more AC-derived metabolites are involved in the post-efferocytic induction of Ptgs2/Tgfb1.
Fig. 1. Efferocytosis-induced TGFβ1 production requires phagolysosomal AC degradation and Ptgs2/COX2 induction.
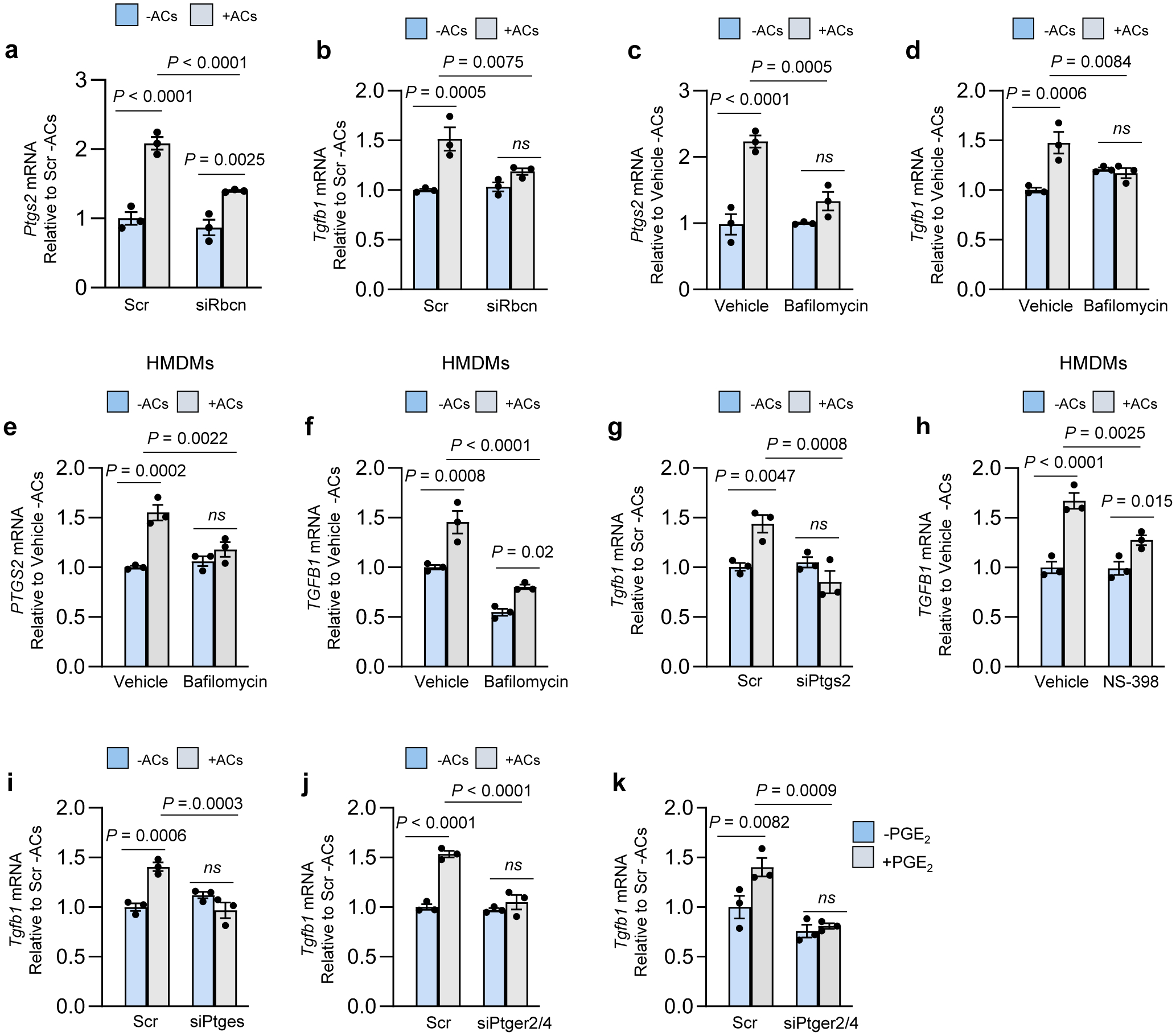
a-h, Bone marrow-derived macrophages (BMDMs) or human monocyte-derived macrophages (HMDMs) were pretreated with siRNAs for 72 h or inhibitors for 2 h, as indicated below. The cells were then incubated ± ACs for 45 min, after which noninternalized ACs were removed by rinsing. After an additional 1 or 6 h of incubation, the cells were assayed for Ptgs2 or Tgfb1 mRNA, respectively. a-b, BMDMs were transfected with scrambled RNA (Scr) or siRbcn. c-f, BMDMs or HMDMs were pretreated with vehicle or bafilomycin A1. g, BMDMs were transfected with Scr or siPtgs2. h, HMDMs were pretreated with vehicle or the COX2-specific inhibitor (NS-398). i, BMDMs were transfected with Scr or siPtges. j, BMDMs were transfected with Scr or siPtger2 and siPtger4. k, BMDMs were transfected with Scr or siPtger2 and siPtger4. After 72 h, the cells were incubated with vehicle or 10 uM PGE2 for 2 h and then assayed for Tgfb1 mRNA. The mRNA data are expressed relative to the first control group. Values are means ± SEM; ns, not significant (P > 0.05); n = 3 biological replicates. Two-sided P values were determined by one-way ANOVA with Fisher’s LSD posthoc analysis.
Based on previous work22, we hypothesized that AC-induced PGE2 might induce TGFβ1 in an autocrine/paracrine manner. Consistent with this idea, the AC-induced increase in Tgfb1 mRNA was prevented by Ptgs2 silencing in mouse macrophages and by the COX2-specific inhibitor NS-39823 in HMDMs (Fig. 1g,h and Extended Data Fig. 1g). Incubation of macrophages with ACs also increased in Ptges (PGE synthase), and silencing Ptges blocked AC-induced Tgfb1 mRNA (Fig 1h,i and Extended Data Fig. 1i). Furthermore, silencing two PGE2 receptors, EP2 and EP4, decreased AC-induced Tgfb1, and exogenous PGE2 increased Tgfb1, which was also blocked by silencing EP2/4 (Fig. 1j,k and Extended Data Fig. 1j,k). In contrast, silencing Tgfb1 did not affect AC-induced Ptsg2 (Extended Data Fig. 1l,m). These data suggest that phagolysosomal degradation of ACs triggers Ptgs2 induction, leading to COX2-mediated PGE2 synthesis/secretion and then PGE2-mediated induction of Tgfb1.
The pathway uses S-adenosyl methionine from AC-methionine.
Based on our previous work linking macrophage metabolism of AC-derived amino acids to gene regulation6, we became interested in methionine, as methionine-derived S-adenosylmethionine (SAM) can be used by histone and DNA methyltransferases to alter gene transcription24. Moreover, methionine was among the top 10 amino acids showing an increase in macrophages following efferocytosis6. SAM is formed by the addition of methionine to ATP, which is catalyzed by the enzyme methionine adenosyltransferase 2A (MAT2A)25. We observed that treating BMDMs with the MAT2A inhibitor PF-936626 or siMat2a blocked AC-induced increases in Ptgs2 and Tgfb1 mRNA and TGFβ1 secretion (Fig. 2a,b) without affecting initial AC uptake (Fig. 2a,b and Extended Data Fig. 2a–d). MAT2A inhibition also blocked AC-induced increases in PTGS2 and TGFB1 in HMDMs (Fig. 2c, d). Further, exogenously added SAM was sufficient to induce Ptgs2 and Tgfb1, and, as predicted, this was not significantly reduced by PF-9366 (Fig. 2e,f and Extended Data Fig. 2e). Note that the modest trend toward reduction by PF-9366 might be related to a small contribution by the methionine cycle, where SAM is converted to methionine, which can then be converted back to SAM via MAT2A27.
Fig. 2. The AC-induced Ptgs2-TGFβ1 pathway requires SAM formation from AC-derived methionine.
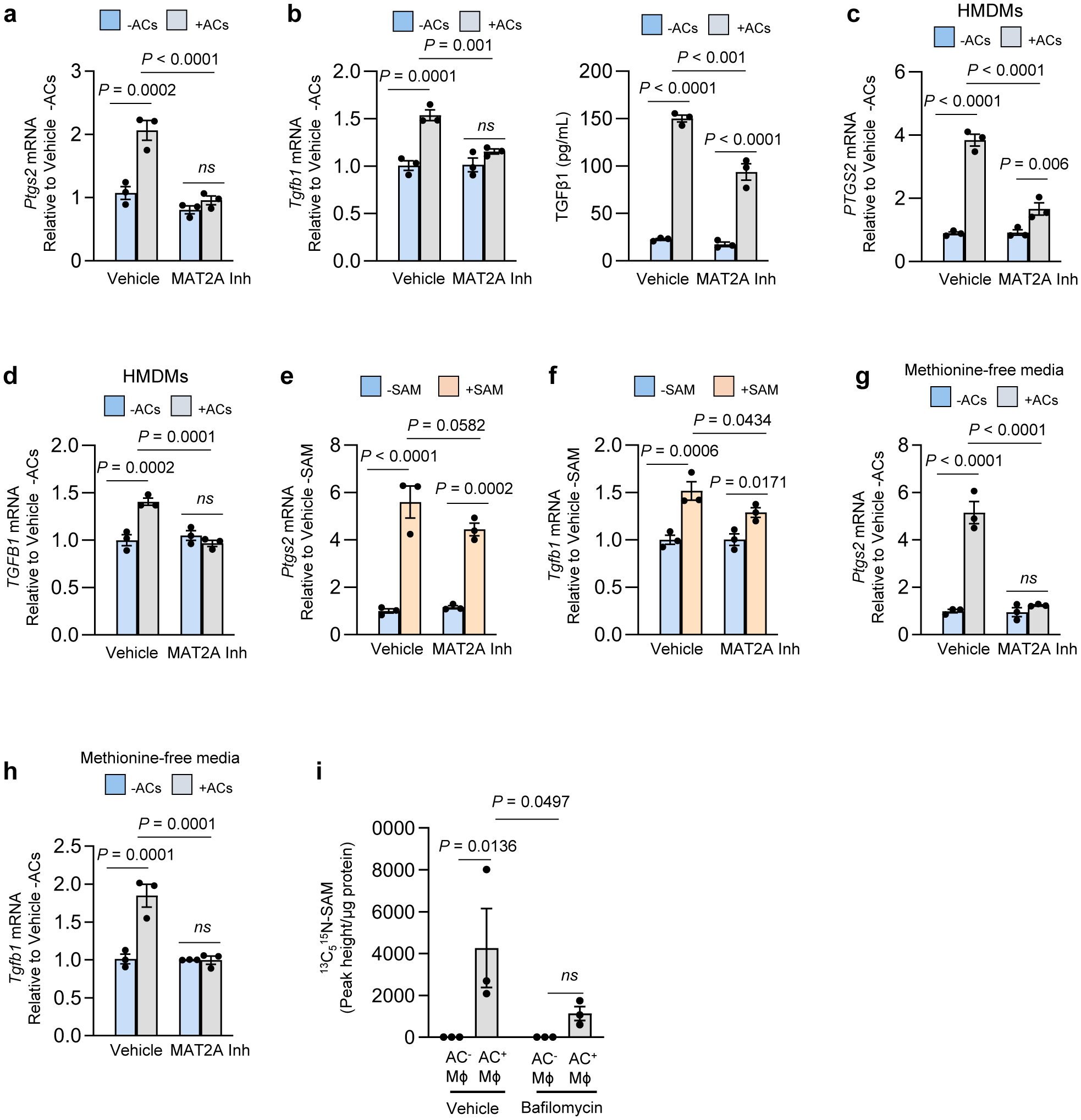
a-d, BMDMs (a-b) or human monocyte-derived macrophages (HMDMs) (c-d) were pretreated with vehicle or the 10 μM MAT2A inhibitor PF-9366 for 2 h. The cells were then incubated ± ACs for 45 min, after which noninternalized ACs were removed by rinsing. After an additional 1 or 6 h of incubation, the cells were assayed for Ptgs2/PTGS2 orTgfb1/TGFB1 mRNA, respectively. For media TGFβ1 in b, macrophages were pretreated for 45 min with vehicle or MAT2A inhibitor before the addition of ACs. The macrophages were rinsed to remove non-engulfed ACs and then incubated for an additional 24 h. The cell media were collected and assayed for TGFβ1. e-f, BMDMs were incubated with vehicle or 20 μM SAM and then assayed for Ptgs2 orTgfb1 mRNA after 1 h or 6 h, respectively. g-h, BMDMs were cultured in methionine-free DMEM supplemented with dialyzed FBS (D-FBS). Before the addition of ACs, cells were pretreated with vehicle or 10 μM MAT2A inhibitor. The cells were then incubated ± ACs for 45 min, after which noninternalized ACs were removed by rinsing. After an additional 1 or 6 h of incubation, the cells were assayed for Ptgs2 orTgfb1 mRNA, respectively. i, BMDMs were cultured in methionine-free media supplemented with D-FBS. The cells were then pretreated for 2 h with vehicle or bafilomycin A1 (Baf), followed by incubation for 45 mins with PKH26-labeled ACs whose proteins had been labeled with 13C515N-methionine before apoptosis induction. After non-engulfed ACs were removed by rinsing and then an additional 1 h of incubation, macrophage extracts were assayed for 13C515N-SAM by LC-MS/MS, with the data expressed as peak height per μg of cell protein. The mRNA data are expressed relative to the first control group. Values are means ± SEM; n.s., not significant (P > 0.05); n = 3 biological replicates. Two-sided P values were determined by a one-way ANOVA with Fisher’s LSD posthoc analysis.
We further explored the source of methionine in this pathway. First, AC-induced increases in Ptgs2 and Tgfb1 were not dependent on methionine in the media (Fig. 2g,h). Next, we sought to trace methionine from AC-proteins into macrophage SAM during efferocytosis. Jurkat cells were incubated with 13C515N-methionine, labeled with fluorescent PKH26 dye, and then rendered apoptotic. To prevent 13C515N-SAM formation in the Jurkat cells, labeling was done in the presence of the MAT2A inhibitor, and we showed by LC-MS/MS that the Jurkat cells had 13C515N-methionine but not 13C515N-SAM (Extended Data Fig. 2f). Macrophages were incubated with these ACs for 45 minutes, followed by the removal of non-internalized ACs by rinsing and further incubation in medium without ACs for 1 hour. The macrophages were then sorted into PKH26+ (AC+) and PKH26+ (AC−) cells and analyzed by LC-MS/MS for 13C515N-SAM. 13C515N-SAM was found only in AC+ macrophages and, importantly, this was markedly inhibited when phagolysosomal hydrolysis was blocked by bafilomycin (Fig. 2i and Extended Data Fig. 2g, h). These combined data provide evidence that methionine originating from phagolysosomal AC degradation during efferocytosis is converted into SAM, which leads to the induction of Ptgs2 and then PGE2-mediated induction of Tgfb1.
The DNMT3A-PGE2-TGFβ1 pathway requires DNMT3A.
SAM is used by DNA methyltransferases to methylate gene regulatory regions, and thus we investigated whether this process might somehow be involved in the PGE2-TGFβ1 pathway. We focused on the DNA methyltransferase DNMT3A, which becomes functional in late embryos and after birth28. We found that the AC-induced increases in Ptgs2, COX2, and Tgfb1 were blocked in BMDMs from Dnmt3afl/fl Vav1Cre+/− mice (hematopoietic (H)-DNMT3A KO mice) versus control Vav1Cre+/− mice, despite equal uptake of ACs (Fig. 3a–c, Extended Data Fig. 2i,j). Similarly, DNMT3A silencing blocked AC-induced PTGS2 and TGFβ1 in HMDMs (Fig. 3d,e, Extended Data Fig. 2k), and the BMDM data were similar when apoptotic macrophages were the source of ACs (Fig. 3f,g). To test another source of cellular methionine cargo, we incubated control and DNMT3A-KO macrophages with IgG-coated RBCs. By flow cytometry, RBC+ macrophages had a marked increase in COX2 compared with RBC− macrophages, and this increase was decreased by ~50% in DNMT3A-KO macrophages (Extended Fig. 2l). In contrast to these findings with engulfed cells, control and DNMT3A-KO macrophages, or macrophages treated with scrambled RNA or siDnmt3a, responded similarly to LPS (Il6 and Ptgs2), IFNγ + LPS (COX2 and Nos2), and IL4 (Arg1) (Extended Data Fig. 2m–p). Further, basal and AC-induced SAM levels were similar between control and H-DNMT3A KO macrophages (Extended Data Fig. 2q). Finally, the increases in Ptgs2 and Tgfb1 induced by exogenous SAM were also prevented in macrophages lacking DNMT3A (Fig. 3h,i).
Fig. 3. The AC-induced COX2-TGFβ1 pathway requires DNMT3A.
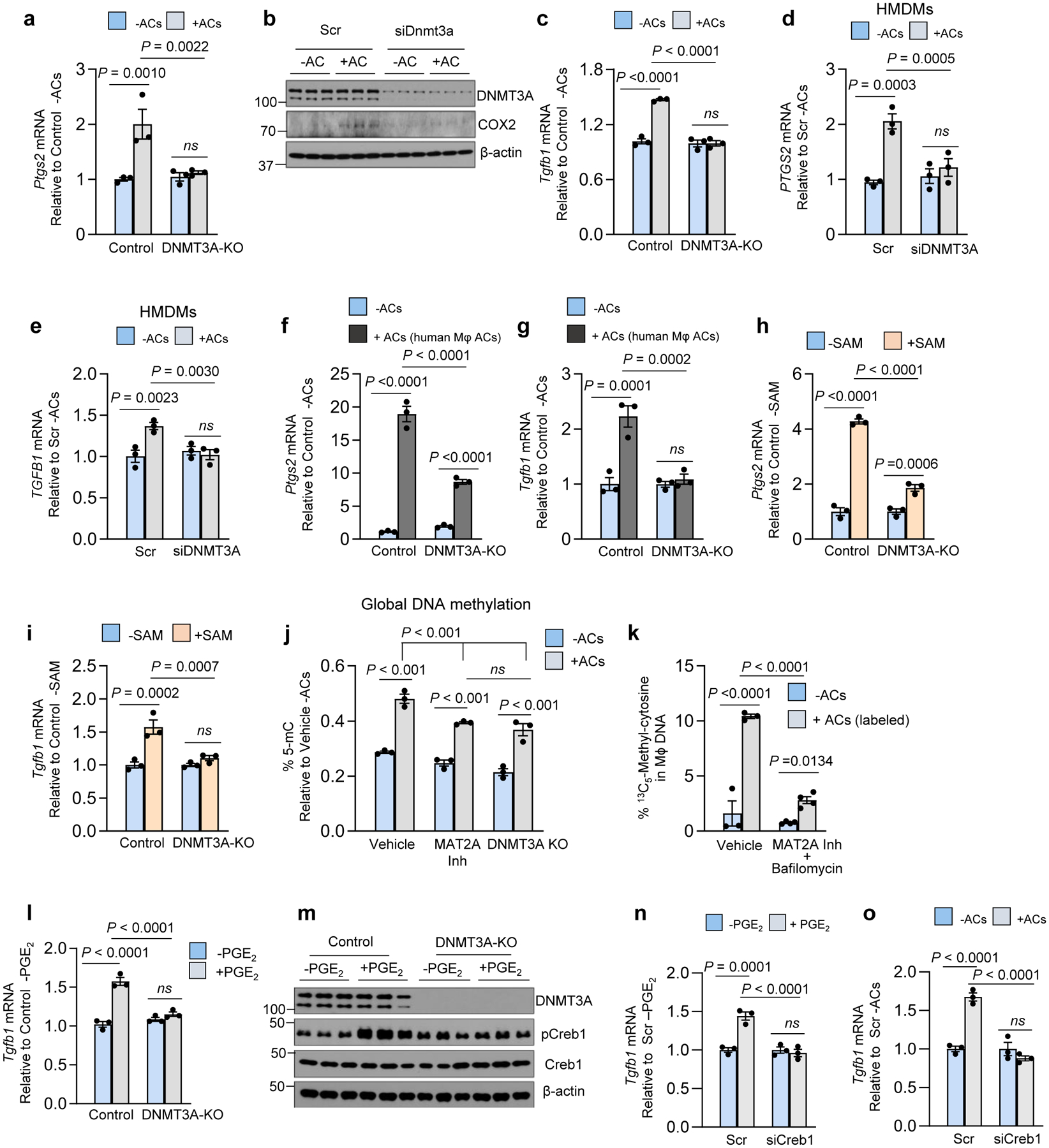
All incubations ± ACs were 45 min, followed by rinsing and then a 1-h or 6-h chase period for Ptgs2 or Tgfb1, respectively. a, BMDMs from Vav1Cre+/− (Control) or Dnmt3afl/fl Vav1Cre+/− (H-DNMT3A-KO) mice were incubated ± ACs and then assayed for Ptgs2. b, BMDMs treated with scrambled RNA (Scr) or siDnmt3a were incubated ± ACs and then immunoblotted for DNMT3A, COX2, and β-actin after 3 h. c, Control or DNMT3A-KO BMDMs were incubated ± ACs and then assayed for Tgfb1. d-e, HMDMs treated with Scr or siDNMT3A were incubated ± ACs and then assayed for PTGS2 or TGFB1. f-g, Control or DNMT3A-KO BMDMs were incubated ± apoptotic human macrophages and then assayed for Ptsg2 or Tgfb1. h-i, Control or DNMT3A-KO BMDMs were incubated with vehicle or SAM and then assayed for Ptgs2 or Tgfb1 mRNA after 1 h or 6 h, respectively. j, Control macrophages pretreated with vehicle or MAT2A inhibitor (PF9366) and DNMT3A-KO macrophages were incubated ± ACs and then assayed for global DNA methylation levels (% 5-mC). k, BMDMs pretreated for 1 h with vehicle or bafilomycin A1 (Baf) and the MAT2A inhibitor were incubated for 45 min ± ACs whose proteins were labeled with 13C515N-methionine. After rinsing and an additional 1 h of incubation, macrophage DNA was assayed for 13C5-methylcytosine. l-m, Control or DNMT3A-KO BMDMs were incubated with vehicle or PGE2 for 2 h and then assayed for Tgfb1 or immunoblotted for DNMT3A, pCreb1, Creb1 and β-actin (1 h). n-o, BMDMs were pre-treated with Scr or siCreb1 and then incubated ± PGE2 for 2 h (n) or ± ACs for 45 mins followed by a 6-h chase (o). The cells were then assayed for Tgfb1. The mRNA data in this figure are expressed relative to the first control group. Values are means ± SEM. n.s., not significant; n = 3 biological replicates. Two-sided P values were determined by a one-way ANOVA with Fisher’s LSD posthoc analysis.
The involvement of SAM and DNMT3A prompted us to look at global DNA methylation in efferocytosing macrophages. We found that ACs increased the percent of 5-methylcytosine in macrophage DNA, which was partially dampened by MAT2A inhibition or DNMT3A absence (Fig. 3j). We then incubated macrophages with 13C515N-methionine-labeled ACs (above) and found that 13CH3-DNA, assayed by LC-MS/MS, was detected in macrophages incubated with 13C515N-methionine-labeled ACs versus unlabeled ACs, and this was partially blocked by bafilomycin and markedly blocked by bafilomycin plus the MAT2A inhibitor (PF9366) (Extended Data Fig. 2r and Fig. 3k).
As a separate finding, the addition of exogenous PGE2 did not rescue the defect in Tgfb1 mRNA in DNMT3A KO macrophages (Fig. 3l). We then asked whether this finding might be linked to the fact that PGE2 receptors signal through p-CREB129. Indeed, PGE2-induced p-CREB1 was blocked in DNMT3A-KO macrophages (Fig. 3m). Most importantly, silencing CREB1 blocked the increase in Tgfb1 in macrophages treated with exogenous PGE2 or incubated with ACs (Fig. 3n,o, Extended Data Fig. 2s). These combined data suggest at least two roles for DNMT3A in efferocytosis-induced pro-resolving mediator production. First, efferocytosis/SAM-mediated Ptgs2 induction requires DNMT3A, and the finding that methyl groups from AC-derived methionine can be traced into macrophage DNA suggests a possible role for DNA methylation in this pathway (below). Second, the ability of PGE2 to induce Tgfb1 also involves DNMT3A via a pathway involving p-CREB.
The pathway requires DNMT3A-mediated Dusp4 repression.
As DNA methylation often suppresses gene transcription, we reasoned that suppression of one or more genes by DNA methylation might secondarily increase Ptgs2, e.g., by removing an inhibitory signal from a Ptgs2-inducing pathway. We began with the finding that ERK activation has been implicated in COX2 induction and PGE2 secretion in other cell types and settings30. In addition, we and others have shown previously that TGFβ1 production by macrophages requires ERK1/2 activation31,32. Consistent with these previous reports, incubation of macrophages with ACs increased the active form of ERK, phospho-ERK1/2, which was dampened by bafilomycin (Extended Data Fig. 3a,b). Most importantly, inhibition of ERK1/2 by U0126, or silencing of Makp1 and Mapk3, prevented AC-induced increases in Ptgs2 and Tgfb1 and decreased TGFβ1 secretion by mouse and human macrophages (Fig. 4a–d and Extended Data Fig. 3c–d).
Fig. 4. The role of ERK, CD36, and DUSP4 in the efferocytosis-Ptgs2/COX2-TGFβ1 pathway.
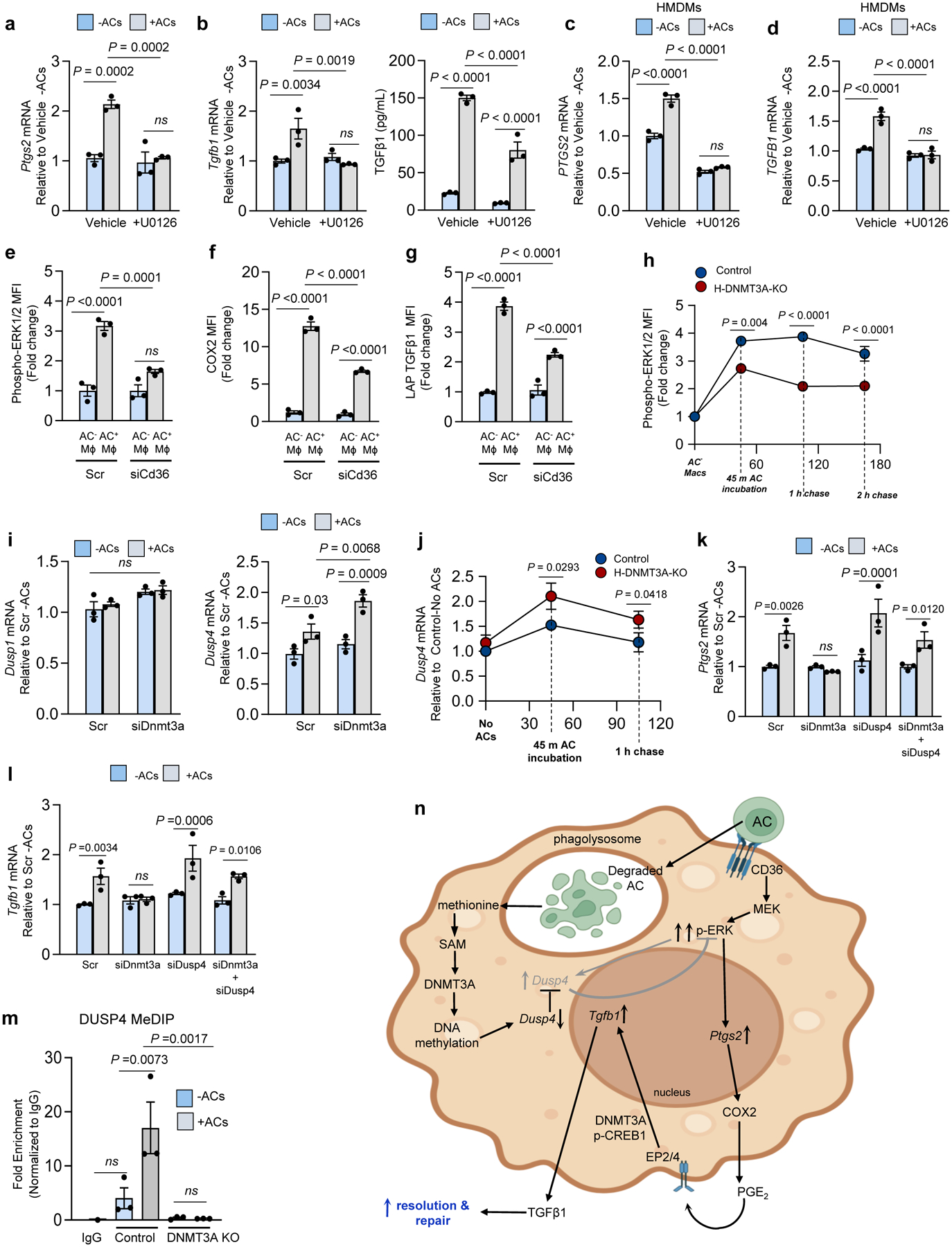
All incubations ± ACs were 45 min, followed by rinsing and then a 1-h or 6-h chase period for Ptgs2 or Tgfb1, respectively. a-d, BMDMs or HMDMs pretreated for 2 h with the MEK/ERK inhibitor U0126 were incubated ± ACs for 45 min and then assayed of Ptgs2/PTGS2 or Tgfb1/TGFB1. Media TGFβ1 was assayed 24 h after AC removal. e-g, BMDMs treated with scrambled RNA (Scr) or siCd36 were incubated with PKH26-labeled ACs for 45 min and then assayed by flow cytometry for p-ERK1/2 mean fluorescence intensity (MFI), or, after an additional 3 h or 18 h of incubation, for COX2 or TGFβ1 MFI, respectively, gating on PKH26+ (AC+) and PKH26− (AC−) macrophages. h, Control and DNMT3A-KO BMDMs were incubated with PKH26-labeled ACs for 45 min and then assayed by flow cytometry for p-ERK1/2 at the indicated time points after AC-removal. i, BMDMs transfected with Scr or siDnmt3a were incubated ± ACs for 45 mins, chased for 1h, and then assayed of Dusp1 or Dusp4. j, Control or DNMT3A-KO BMDMs were incubated ± ACs and then assayed for Dusp4 mRNA after 0 or 1-h chase. k-l, BMDMs treated with Scr, siDnmt3a, siDusp4, or siDnmt3a + siDusp4 were incubated ± ACs and then assayed for Ptgs2 or Tgfb1 mRNA. m, Control or DNMT3A KO BMDMs were incubated ± ACs and then after 1 h subjected to MeDIP to immunoprecipitate fragments of methylated DNA. PCR targeting the CpG-rich region of the Dusp4 promoter was used to determine the fold-enrichment of methylated DNA relative to IgG control. n, Pathway scheme, showing AC-CD36-mediated ERK activation, which is limited by Dusp4, and the subsequent AC-degradation pathway, which represses Dusp4 via DNMT3A-mediated DNA methylation to sustain p-ERK. The pathway leads to PGE2, which acts via EP2/4 and p-CREB to induce Tgfb1. The p-CREB pathway also involves DNMT3A. The data in a-l are expressed relative to the first control group. Values are means ± SEM; n.s., not significant; n = 3 biological replicates. Two-sided P values were determined by one-way ANOVA with Fisher’s LSD posthoc analysis.
Apoptotic cells are known to activate cell surface receptors that can activate ERK, notably MerTK and CD3631,33,34. We therefore targeted these receptors in macrophages, incubated them with PKH-26 or pHrodo-labeled ACs, and conducted flow cytometry assays, gating on labeled-AC+ and AC− macrophages to correct for the lower number of efferocytosing macrophages in receptor-silenced cells. In comparison with AC− WT macrophages, AC+ MerTK-KO macrophages showed a modest decrease in phospho-ERK and COX2 MFI (Extended Data Fig. 3e), while siCD36-treated versus scrambled RNA-treated AC+ macrophages showed a more robust reduction in phospho-ERK, COX2, and TGFβ1 MFI (Fig. 4e–g, Extended Data Fig. 3f,g). Thus, activation of CD36 by ACs is an important activator of ERK in this pathway.
We next questioned whether ERK activated by the early event of AC binding might be enhanced by DNMT3A after AC engulfment and degradation. Consistent with this idea, control macrophages showed an initial increase in p-ERK1/2 45 min after exposure to ACs, which was maintained after the cells were chased in the absence of ACs for 1 or 2 h (Fig. 4h, blue circles). In contrast, DNMT3A-KO macrophages showed a smaller increase in p-ERK1/2 at 45 min, and the difference between KO and control macrophages persisted for the following 2-h chase period (Fig. 4h, red circles). Based on these data, we hypothesized that DNMT3A might repress a gene that functions to lower p-ERK, e.g., an ERK phosphatase. ERK phosphorylation in cells is often transient owing to negative feedback by certain dual-specificity phosphatases (DUSPs), notably DUSP1 and DUSP435. We first found that when macrophages were exposed to ACs, there was an increase in Dusp4 but not Dusp1 mRNA in siDnmt3a-treated macrophages (Fig. 4i). Similar results with Dusp4 were found in a time-course experiment comparing control and DNMT3A-KO macrophages (Fig. 4j). In addition, AC-induced Dusp4 was blocked by the ERK inhibitor U0126, consistent with an ERK-DUSP4 negative-feedback pathway in these macrophages (Extended Data Fig. 3h). Most importantly, the suppression of Ptgs2, COX2, and Tgfb1 by siDnmt3a or DNMT3A-KO in efferocytosing macrophages was abrogated when Dusp4 was also silenced but not when Dusp1 was silenced (Fig. 4k,l and Extended Data Fig. 3i–m). Moreover, the reduction in Tgfb1 expression in siMat2a-treated macrophages was rescued by also silencing Dusp4 (Extended Data Fig. 3n,o). Finally, Dusp4 silencing could not rescue the defect in Tgfb1 expression in siMapk1/3-treated macrophages (Extended Data Fig. 3p,q), which is consistent with DUSP4 functioning in the pathway by increasing ERK1/2 activation. Finally, using methylated DNA immunoprecipitation (MeDIP), we found that the CpG-rich promoter region of the Dusp4 gene was methylated in response to ACs in wild-type but not DNMT3A-deficient macrophages (Fig. 4m). When considered together, the data suggest the following model (Fig. 4n): the initial binding of an AC to an efferocytosis receptor such as CD36 triggers an ERK pathway that induces Ptgs2, leading to the downstream COX2-PGE2-Tgfb1 pathway. The pathway requires more than transient ERK activation, and thus escape from DUSP4-mediated negative-feedback is required. This escape occurs during the next stage in efferocytosis, i.e., the phagolysosomal degradation of the engulfed AC, and involves a pathway that relies on AC-methionine, SAM, and DNMT3A-mediated suppression of Dusp4. Finally, the subsequent ability of PGE2 to induce Tgfb1 also involves DNMT3A, which functions through yet-to-be-defined pathway involving p-CREB.
Evidence for the DNMT3A-COX2-TGFβ1 pathway in vivo.
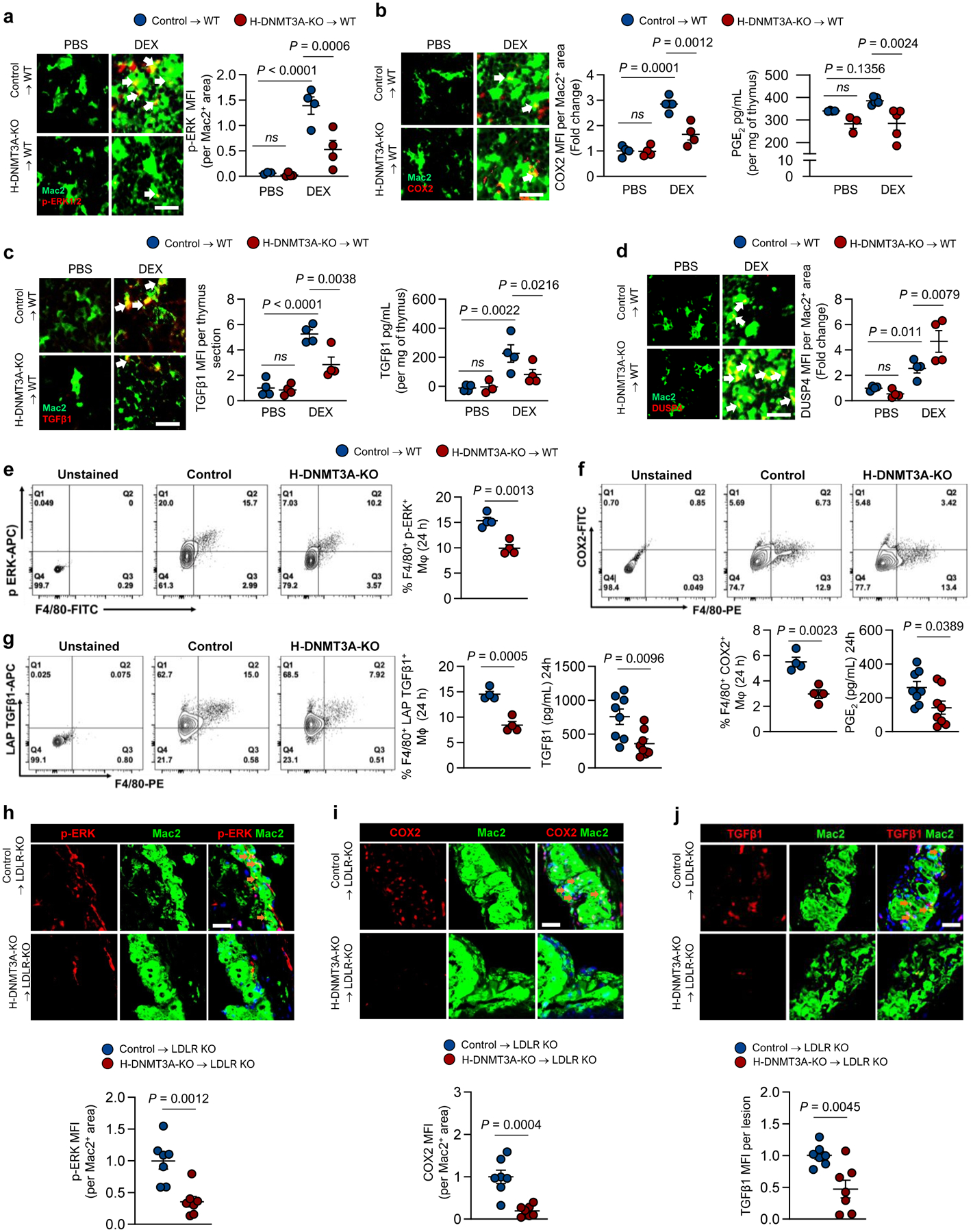
To test for the presence of the DNMT3A-COX2-TGFβ1 pathway in vivo, we compared control and H-DNMT3A KO bone marrow-transplanted mice (above) mice using three well-characterized models influenced by macrophage-mediated efferocytosis and resolution: dexamethasone-induced thymocyte apoptosis, Zymosan A1-induced peritonitis, and atherosclerosis. For the dexamethasone-thymus model, mice were injected with dexamethasone (dex) to induce thymocyte apoptosis or phosphate-buffered saline (PBS) as the vehicle control. In this model, apoptotic thymocytes are continually cleared by thymic macrophages, which is necessary to prevent dead thymocyte accumulation, tissue necrosis, and inflammation6. We first showed that H-DNMT3A-KO did not affect the initial apoptosis phase, as the number of annexin V+ (apoptotic) cells in the thymus 4 hours after dex injection was similar in the 2 groups of mice (Extended Data Fig. 4a). For pathway identification, we examined the thymi from control and H-DNMT3A-KO mice 18 h after the dex or PBS injection, a timepoint crucial for identifying macrophage-mediated resolution effects6. As designed, DNMT3A was absent in thymic macrophages of the H-DNMT3A-KO mice (Extended Data Fig. 4b, left). We observed that dex increased thymic macrophage p-ERK, COX2, PGE2, and TGFβ1 in the control mice, whereas these responses were suppressed in the H-DNMT3A-KO mice (Fig. 5a–c, Extended Data Fig. 4b, right). Importantly, in the dex-treated cohorts, the thymi of H-DNMT3A-KO mice had higher DUSP4 expression than control mice thymi (Fig. 5d).
Fig. 5. Evidence for the DNMT3A-COX2-TGFβ1 pathway in vivo.
a-d, Wildtype (WT) C57BL/6J mice were transplanted with bone marrow (BMT) from Vav1Cre+/− (Control) or Dnmt3afl/fl Vav1Cre+/− (H-DNMT3A-KO) mice and injected 4 weeks later with PBS or dexamethasone (DEX). After 18 h, the thymi were harvested and immunostained for Mac2 (macrophages) and the following proteins: (a) p-ERK1/2, n=4; (b) COX2, n=4; (c) TGFβ1, n=4; and (d) DUSP4, n=4. The data are quantified as MFI per Mac2+ area, expressed relative to the first control group. Scale bars, 50 μm. Panels b and c show the content of PGE2 and TGFβ1, respectively, in thymic extracts as assayed by ELISA (n=3 & 4 for PBS and DEX groups respectively). e-g, Control or H-DNMT3A-KO BMT mice were injected i.p. with 1 mg/mL Zymosan A1. After 24 h, peritoneal exudate cells were analyzed by flow cytometry for the following proteins, gating on F4/80+ cells (macrophages): (e) p-ERK, n=4; (f) COX2, n=4; (g) LAP-TGFβ1, n=4. Panels f and g also show the content of PGE2 (n=8) and TGFβ1 (n=8), respectively, in peritoneal exudates as assayed by ELISA. h-j, Ldlr−/− (LDLR-KO) mice were transplanted with bone marrow from Vav1Cre+/− (Control) or Dnmt3afl/fl Vav1Cre+/− (H-DNMT3A-KO) and, after 4 weeks, fed a western-type diet (WD) for 12 weeks. Aortic root sections were immunostained for Mac2 and the following proteins: (h) p-ERK1/2, (n=7); (i) COX2, (n=7); and (j) TGFβ1, (n=7). The data are quantified as MFI per Mac2+ area expressed relative to the control group. Scale bars, 50 μm. Values are means ± SEM, n.s., not significant (P > 0.05). Two-sided P values were determined by the Student’s t-test for 2 groups or one-way ANOVA with Fisher’s LSD posthoc analysis for 4 groups.
In the peritonitis model, Zymosan A1 elicits a neutrophil-mediated sterile inflammatory response, which is followed by a gradual accumulation of efferocytosing macrophages that clear neutrophils as they become apoptotic36,37. To test for the presence of the DNMT3A-COX2-TGFβ1 pathway in this model, control and H-DNMT3A-KO mice were injected intraperitoneally with Zymosan A1. After 24 hours, but not 12 hours, exudate macrophages from the H-DNMT3A-KO mice had decreases in phospho-ERK, COX2, and LAP-TGFβ1 and decreases in PGE2 and TGFβ1 (Fig. 5e–g, Extended Data Fig. 4c–d).
To look for the pathway in atherosclerotic lesions, control and H-DNMT3A-KO mice were fed a western-type diet for 12 weeks. As designed, the atherosclerotic lesions of the chimeric knockout mice had undetectable macrophage DNMT3A (Extended Data Fig. 4e). Blood glucose, body weight, total cholesterol, and systemic blood parameters were similar in both groups after harvesting (Extended Data Fig. 4f–m). Most importantly, lesional macrophages of H-DNMT3A-KO mice had decreases in phospho-ERK, COX2, and TGFβ1 (Fig. 5h–j). In summary, DNMT3A-dependent phospho-ERK, COX2, and TGFβ1 are evident in the macrophages of three different models where macrophage-mediated efferocytosis and resolution are known to play important roles.
The pathway is functional in resolution in vitro and in vivo.
PGE2 and TGFβ1 promote tissue resolution, including efferocytosis, which is an important effector arm of the resolution response9,12,13,14,19. In this context, we reasoned that activation of the DNMT3A pathway by an initial efferocytic event might further promote efferocytosis via autocrine/paracrine effects of TGFβ1. To begin, we confirmed that incubation of macrophages with TGFβ1 in vitro enhances efferocytosis38, which was blocked by inhibiting TGFβRI-mediated SMAD phosphorylation with LY3200882 (Extended Data Fig. 5a). More importantly, incubation of macrophages with conditioned media (CM) from AC-exposed control macrophages, but not AC-exposed DNMT3A-KO macrophages, increased efferocytosis, and the increase seen with control-macrophage CM was blocked by an anti-TGFβ1 antibody (Fig. 6a). These data are consistent with the idea that efferocytosis-induced TGFβ1 secretion further increases efferocytosis by a paracrine mechanism.
Fig. 6. Loss of hematopoietic cell DNMT3A impairs efferocytosis in vitro and resolution in vivo.
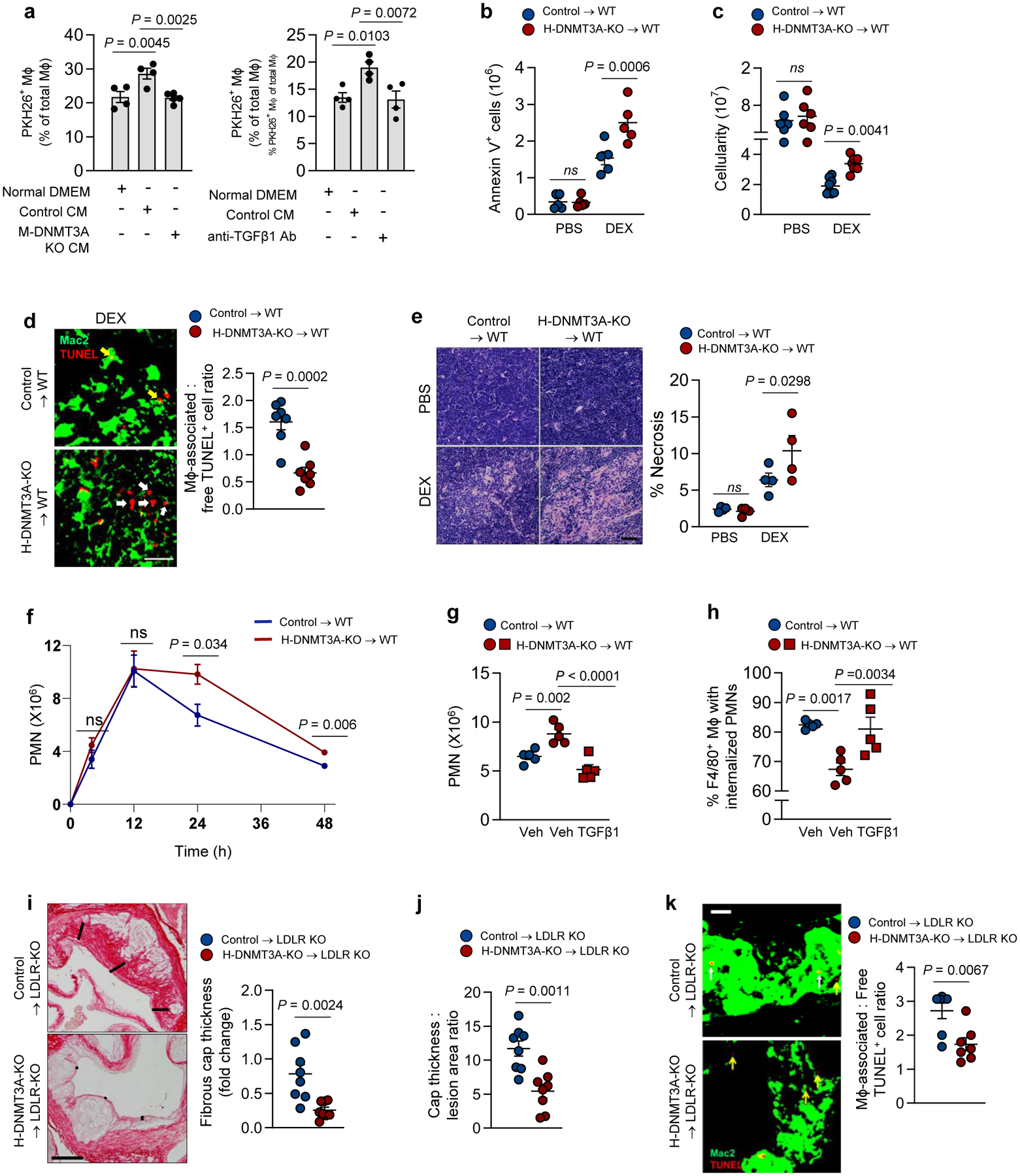
a, 18-h conditioned media (CM) from control or DNMT3A-KO BMDMs incubated ± AC, or DMEM control, were added to recipient BMDMs (left). In a parallel experiment (right), CM from control BMDMs were treated ± anti-TGFβ1 antibody and then added to BMDMs. After 1 h, the BMDMs were incubated ± PKH26 labeled-ACs and then assayed for percent PKH26-AC+ of total macrophages (n=4 biological replicates). b-e, The thymi of PBS and DEX-treated control and H-DNMT3A-KO BMT mice similar to those in Fig. 5a–d were assayed for annexin V+ cells, n=5 mice/group (b); cellularity, n=6 (PBS) and 8 (DEX) mice (c); efferocytosis, n=7 mice/group; yellow and white arrows indicate macrophage-associated or free TUNEL+ cells, respectively (d); and percent necrosis, n=4 mice/group (e). Scale bars, 50 μm (d) & 200 μm (e). f, The peritoneal exudates of Zymosan A1-injected control and H-DNMT3A-KO BMT mice similar to those in Fig. 5 e–g were assayed for Ly6G+ polymorphonuclear cells (PMN) as a function of time after Zymosan A1 injection, n=5 mice/group. g-h, As in panel f, but a cohort of KO mice received 200 ng/mL TGFβ1 i.p., while cohorts of control and KO mice received vehicle (Veh). After 24 h, the exudates were assayed for the number of PMNs, n=5 mice/group, and for the percent of F4/80+ macrophages that were Gr1+ by flow cytometry (efferocytosis of PMNs), n=5 mice/group. i-k, Aortic root lesions of 12-week WD-fed control and H-DNMT3A-KO BMT Ldlr−/− mice similar to those in Fig. 5h–j were assayed for fibrous cap thickness, expressed relative to the control cohort, n= 8 mice/group (i); fibrous cap thickness as a ratio of the lesion area, n=8 mice/group (j); and efferocytosis, n=7 mice/group (k). Scale bar, 200 μm (i) and 50 μm (k). For k, yellow and white arrows indicate macrophage-associated (top) or free TUNEL+ (bottom) cells, respectively. Values are means ± SEM, n.s., not significant. Two-sided P values were determined by the Student’s t-test for 2 groups or one-way ANOVA with Fisher’s LSD posthoc analysis for 4 groups.
To test the functionality of this pathway in vivo, we turned to the dex-thymus model and looked for signs of impaired efferocytosis and resolution in the H-DNMT3A-KO cohort. As alluded to above, within 4 h of dex treatment, there is marked thymocyte apoptosis, but after 18 h the number of apoptotic thymocytes is modest owing to their clearance by thymic macrophages6. Accordingly, although there was a modest increase in ACs in the thymi of the control mice 18 h after dex, thymus weight and cellularity were decreased owing to macrophage-mediated clearance of apoptotic thymocytes (Fig. 6b, c, Extended Data Fig. 5b, groups 1 and 3). Most importantly, the thymi of the H-DNMT3A-KO cohort showed a greater increase in ACs and cellularity and a smaller decrease in thymus weight after dex, indicative of impaired continual efferocytosis (Fig. 6b,c and Extended Data Fig. 5b, groups 2 and 4). To assess efferocytosis directly, we stained thymic sections in the dex-treated cohorts for macrophages using anti-Mac2 antibody and TUNEL to mark dead thymocytes and then quantified the ratio of macrophage-associated TUNEL+ cells: free TUNEL+ cells as a measure of efferocytosis6. Note that “macrophage-associated TUNEL+ cells” refers to macrophages with cytoplasmic-TUNEL+ living macrophages, not nuclear-TUNEL+ dead macrophages, which are extremely rare in this model (Extended Data Fig. 5c). We observed that the ratio of macrophage-associated: free TUNEL+ thymocytes was lower in the thymi of dex-treated H-DNMT3A-KO versus dex-treated control mice (Fig. 6d), with similar levels of thymic macrophages (Extended Data Fig. 5d), further supporting a defect in efferocytosis.
Apoptotic cells become necrotic when they remain uncleared. As such, we examined sections from PBS and dex-treated control and H-DNMT3A-KO mice for evidence of necrosis in H&E-stained sections. As expected, there was no observable difference between the control and KO groups after PBS treatment, but 18 h after dex, the KO cohort showed a greater increase in percent necrosis than the control cohort (Fig. 6e). Further, the thymi of dex-treated H-DNMT3A-KO mice had a higher level of immunoreactive tumor necrosis factor-alpha (TNF⍺) and a trend toward higher interleukin-6 (IL6) compared with the thymi of dex-treated control mice (Extended Data Fig. 5e, f). Thus, in the dex-thymus model, hematopoietic DNMT3A deletion impairs efferocytosis and resolution, which, when considered together with the decreases in COX2 and TGFβ1 in this model shown in the previous section, provide evidence for the functionality of the DNMT3A-COX2- TGFβ1 pathway in vivo.
We next examined the Zymosan sterile peritonitis model. Peritoneal PMN counts were similar between control and H-DNMT3A-KO mice during the inflammatory stage at 12 h, but the KO cohort showed a relatively impaired decrease in PMNs compared with the control cohort at both 24 and 48 h (Fig. 6f). We also showed in a separate experiment that the exudate PMN numbers were also different between the two cohorts at 18 h after Zymosan (Extended Data Fig. 5g). Based on previous studies using this model36,37, this finding suggests that loss of DNMT3A in macrophages impairs resolution in sterile peritonitis and likely compromises the efferocytic clearance of apoptotic PMNs by exudate macrophages. Following this observation, we determined whether intraperitoneal injection of TGFβ1, whose production is impaired in DNMT3A-deficient mice after injection of Zymosan (above), would restore efferocytosis and resolution. As an initial test, we first showed that injection of WT mice with 200 ng TGFβ1 i.p. 15 and 20 hours after Zymosan lowered exudate PMN number 4 hours later (Extended Data Fig. 5h), which is consistent with the above in-vitro data showing that TGFβ1 enhances efferocytosis. Most importantly, we found that treatment of H-DNMT3A-KO mice with TGFβ1 15 and 20 hours after Zymosan lowered the number of PMNs to the level seen in the control cohort (Fig. 6g). We then immunostained cells from the peritoneal exudate for the neutrophil marker Gr1 and the macrophage marker F4/80 and used flow cytometric analysis to evaluate the percentage of macrophages (F4/80+ cells) with internalized PMNs (F4/80+Gr1+ cells), which is an indicator of in vivo efferocytosis33. TGFβ1 treatment increased the percentage of efferocytosing macrophages (F4/80+Gr1+) to the level seen in the control chimeric cohort (Fig. 6h). Thus, as in the dex-thymus model, H-DNMT3A-KO impairs resolution and efferocytosis, with direct evidence for the role of TGFβ1 in this model.
Lastly, we examined the 12-week atherosclerosis model, focusing on the TGFβ1-mediated endpoints of fibrous cap thickness and efferocytosis, both of which are features of plaque resolution and stability2,4,5,20,39. H-DNMT3A-KO resulted in marked decreases in cap thickness when compared with lesions from control mice (Fig. 6i). Importantly, this finding was not simply a reflection of smaller lesion size (Fig. 6j), as lesion size was not affected by H-DNMT3A-KO (Extended Data Fig. 5i). Moreover, lesional efferocytosis was impaired in the lesions of H-DNMT3A-KO versus control mice (Fig. 6k). Thus, in this complex, disease-relevant model, there is evidence that the DNMT3A pathway plays a role in processes linked to resolution and efferocytosis.
Discussion
A fascinating concept that has emerged in the area of inflammation resolution relates to the ability of macrophages to use molecules derived from the phagolysosomal hydrolysis of ACs to drive resolution and continual efferocytosis6,7,8. Thus far, the mechanisms have focused on AC metabolite-induced pathways that result in specific transcription factor activation, mRNA stabilization, or cellular energy metabolism alteration6,7,8. Here we report on a new mechanism that involves epigenetic gene regulation. In this case, AC-derived methionine is converted to SAM, which provides the substrate for DNMT3A-mediated DNA methylation. The result is methylation-induced suppression of Dusp4, which enables prolonged ERK phosphorylation to induce Ptgs2 and subsequent activation of the PGE2-TGFβ1 resolution pathway (Fig. 4n). This pathway is supported by mechanistic data using BMDMs and HMDMs; by the presence of the unique biochemical signature of the pathway in resolution settings in vivo; and, most importantly, by DNMT3A-causation experiments in vivo in settings where macrophage-mediated efferocytosis and resolution are important, notably, atherosclerosis. We showed DNMT3A KO or silencing in efferocytosing macrophages suppresses the incorporation of methyl groups from AC-methionine into SAM and DNA and decreases Dusp4 promoter methylation. However, future studies will be needed to show that AC-derived methionine per se is the source of methyl groups on the Dusp4 promoter and that methylation of this promoter is directly responsible for AC-induced repression of Dusp4 expression.
The major premise for the study was that efferocytosis promotes resolution by stimulating the synthesis and secretion of PGE2 and TGFβ19. Moreover, another study showed that a CD36-activating antibody led to the induction of TGFβ1 in a leukemia-derived macrophage cell line31. Our studies in BMDMs and HMDMs indicate that AC-induced activation of CD36-ERK1/2 is limited by a DUSP4-mediated negative-feedback pathway and that PGE2 and TGFβ1 induction requires Dusp4 repression via the subsequent step of AC engulfment and phagolysosomal degradation. Other efferocytosis receptor-ERK pathways may have a more modest role than CD36, as demonstrated by our data with MerTK-KO macrophages.
Our data suggest that methionine/SAM is rate-limiting for at least certain DNA methylation events in macrophages and that AC-derived methionine, or exogenous SAM, provides the necessary substrate for DNA methylation. Other DNA and histone methylation reactions that are known to be active in differentiating macrophages40 may deplete local stores of endogenous SAM, which could contribute to the rate-limiting property of AC-derived methionine/SAM. Previous examples suggesting that methionine may be rate-limiting in DNA methylation-dependent processes include the use of exogenous methionine by T cells to expand the Th17 lineage41 and by macrophages to inhibit LPS-induced inflammation42. In theory, it is also possible that efferocytosis increases the enzymatic activity of DNMT3A methyltransferase, e.g., by suppressing negative-regulatory processes that limit DNMT3A activity43,44.
Our findings raise the possibility that enhancers or promoters in addition to the Dusp4 promoter are methylated in efferocytosing macrophages, leading to changes in the expression of other genes. Global methylation analysis coupled with ATAC-Seq and RNA-Seq in control and DNMT3A-knockout macrophages incubated with or without ACs will help shed light on this issue. In a more general sense, the actions of other DNA methyltransferases and/or histone methyltransferase45 may also be stimulated by post-efferocytotic SAM formation, leading to yet additional changes in gene expression following the uptake and degradation of ACs. Finally, prolonged ERK activation in efferocytosing macrophages caused by Dusp4 repression is likely to have effects beyond Ptgs2 induction, and elucidation of these may reveal additional insight into post-efferocytosis resolution signaling.
Advanced atherosclerosis was one of the models we used to show that the DNMT3A pathway is present and functional in vivo. Human and experimental advanced atherosclerotic lesions are associated with impaired resolution, and molecular-genetic and pharmacological studies in mice have shown that impaired resolution contributes to plaque progression, while pharmacological restoration of resolution suppresses plaque progression2,4,5,20,39,46,47. We showed here that homozygous deletion of hematopoietic DNMT3A led to fibrous cap thinning and impaired efferocytosis, two features of unstable plaques in humans that we predicted would occur via the suppression of TGFβ1 synthesis9,19,20. Moreover, the lower expression level of COX2/PGE2 in hematopoietic DNMT3A-deficient mice may contribute to the overall defect in resolution, as COX2/PGE2 has been implicated in several anti-inflammatory responses9,12,13,14,48. Furthermore, experimental atherosclerotic mouse models have revealed a direct role of COX2 in protecting against lesion development and suppressing pro-inflammatory IL-6 and TNF-⍺ production15,16,17,18. Age-related loss-of-function somatic mutations in DNMT3A in humans (“clonal hematopoiesis of indeterminate potential” [CHIP]) are associated with increased incidence of coronary artery disease (CAD)49, with the mechanism remaining unknown. Thus, a topic for future study will be to determine if the degree of DNMT3A lowering in CHIP is enough to compromise the DNMT3A-PGE2-TGFβ1 pathway in atherosclerotic lesional macrophages, thus providing at least one mechanism linking DNMT3A CHIP to CAD. This mechanism may be relevant to a common dominant-negative DNMT3A CHIP mutation associated with CAD, DNMT3AR882H. In addition, the findings from this study might be particularly applicable to atherosclerosis regression, where efferocytosis and resolution play important roles in plaque stabilization6,50. Finally, conceiving ways to therapeutically enhance the DNMT3A-PGE2-TGFβ1 pathway may provide a novel way to boost resolution in atherosclerosis and other key diseases in which resolution is impaired. In this context, our findings may provide a mechanistic basis to an emerging area in resolution therapeutics in which infusion of apoptotic cells, including apoptotic mesenchymal stems cells, is used to trigger efferocytosis-mediated resolution pathways51. This type of therapy is effective in experimental models of autoimmune disease and has been shown in a small clinical trial to decrease the incidence of graft-versus-host disease in subjects receiving hematopoietic stem cell transplantation for hematologic malignancies51,52. Several studies have suggested that stimulation of PGE2 secretion by efferocytosing macrophages may be an important pro-resolving mechanism of this therapeutic strategy53,54 and, based upon the findings here, this process may be linked to TGFβ1 secretion as well. To the extent that infusion of apoptotic cells may cause adverse effects, i.e., if the infused cells become necrotic51, knowledge of the DNMT3A-PGE2-TGFβ1 pathway may suggest alternative, safer strategies to enhance resolution in disease settings.
Methods
Experimental Animals.
Animal protocols used for experiments were approved by Columbia University’s Institutional Animal Care and Use Committee and were cared for according to NIH guidelines for the care and use of laboratory animals under the protocol number AAAY9450. The mice were socially housed in standard cages at 22°C and under a 12–12 h light-dark cycle in a barrier facility with ad libitum access to water and food. Eight-week-old male C57BL/6J wild-type mice (Jackson Laboratory #000664) were used as bone marrow-transplantation (BMT) recipient mice for the dexamethasone-thymus and Zymosan A1 experiments, with n numbers indicated in the figure legends. Eight-week-old male LDLR-KO mice on the C57BL/6J background (B6.129S7-Ldlrtm1Her/J; Jackson Laboratory #002207) were used as BMT recipient mice for the atherosclerosis experiments, with n numbers indicated in the figure legends. All purchased mice were allowed to adapt to housing in the animal facility for 1 week before the commencement of experiments. For BMT donor mice, we used 8-wk/old Dnmt3afl/flVav1Cre+/− and Vav1Cre+/− control male mice, both on the C57BL/6J background, which were a generous gift from Dr. Siddhartha Mukherjee and Alan Burke. The ratio of donor:recipient mice for BMT was 10:1. Mertkfl/fl Lyz2cre male mice on the C57BL/6J background were generated as previously described55 and used at 8 wks of age. Investigators were blinded for atherosclerosis experiments but were not blinded for experiments from Dexamethasone-induced thymocyte apoptosis model and Zymosan-induced peritonitis model.
Generation of bone marrow-derived macrophages (BMDMs).
Bone marrow cells from either male or female 8–12-week-old mice were cultured in DMEM supplemented with 10 % heat-inactivated (HI) FBS, 100 mg/mL streptomycin,10 U/mL penicillin, and 20 % L-929 fibroblast-conditioned media for 6–8 days, after which are ready to use for experiments.
Generation of human monocyte-derived macrophages (HMDMs).
Peripheral human blood leukocytes were isolated from buffy coats of anonymous, de-identified healthy adult volunteers, with informed consent (New York Blood Center). The leukocytes were purified in a discontinuous gradient of Histopaque solution and added to 24-well tissue culture plates in a humidified CO2 incubator at 37°C. After 4 h to allow for adhesion, the monolayers were rinsed to remove unattached cells, and the medium was changed to RPMI-1640 (GIBCO) containing 10% HI-FBS, 100 mg/mL streptomycin, 10 U/mL penicillin, and 20 ng/mL GM-CSF (Peprotech). After 10 days, the HMDMs were used for experiments.
Cell lines.
Jurkat human T lymphocytes (TIB-152), and L-929 mouse fibroblasts (CCL-1) cells were obtained from ATCC and cultured in DMEM (GIBCO) supplemented with 10% (vol/vol) heat-inactivated fetal bovine serum (HI-FBS; GIBCO), and 10 U/mL penicillin and 100 mg/mL streptomycin (Corning). Cells were cultured in a humidified CO2 incubator at 37°C.
Induction of apoptosis and labeling of Jurkat cells.
Jurkat cells were irradiated under a 254-nm UV lamp for 15 min, followed by incubation in 1X PBS for 2–3 h, and then used for experiments. For some experiments, the apoptotic cells (ACs) were rinsed once with serum-free DMEM, resuspended at a concentration of 2 × 107 cells/mL in Diluent C (Sigma-Aldrich), and incubated for 5 min with 1 mL of Diluent C containing concentrated PKH26 dye (Sigma-Aldrich). The reaction was stopped with 2 mL FBS, and the ACs were then rinsed twice with DMEM containing 10% heat-inactivated FBS and used for experiments. For other experiments, ACs were stained with pHrodo dye (ThermoFisher) as described by the manufacturer's protocol. For experiments using apoptotic human macrophages, HMDMs were made apoptotic by treatment with 1 μM staurosporine in DMEM media for 48 h.
In vitro macrophage efferocytosis assay.
BMDMs were plated in 24-well tissue culture plates and incubated with PHK26- or pHrodo-labeled Jurkat cells rendered apoptotic by 254 nM UV at a ratio of 5:1 (ACs: Mϕ). After 45 min, unbound apoptotic cells were vigorously washed off and fluorescence and bright field images were taken using the Leica epifluorescence microscope (DMI6000B) to identify the uptake of labeled cells. Images were quantified using Image J software. For some experiments, Mϕ were incubated with ACs for 3 h, whilst other experiments involved incubating Mϕ for an additional 1 h or 6 h in normal cell culture media following the removal of unbound ACs.
Immunoblotting.
After experiments, cells were lysed in 2X Laemmli lysis buffer (Bio-Rad) containing 50 mM DTT. Lysates were heated at 95°C for 5 min, separated on 4%–20% SDS-PAGE gradient gels (Invitrogen) at 120V for 2 h, and electro-transferred to 0.45-mm PVDF membranes at 100V for 1.5 h. The membranes were incubated at 4°C with primary antibodies in PBS containing 5% BSA overnight and detected using HRP-conjugated secondary antibodies (Pierce) the following day. Densitometric analysis was performed using ImageJ software.
Enzyme-linked immunosorbent assay.
TGFβ1 or PGE2 were measured in cell culture media or peritoneal exudates using kits purchased from Biolegend and Cayman Chemicals, respectively, and conducted as described by the manufacturer's protocol.
Methylated DNA capture (MeDIP).
Control and H-DNMT3A KO macrophages were incubated with ACs for 45 mins. Non-internalized ACs were removed by media removal and monolayer rinsing, and the macrophages were incubated in fresh media for an additional hour. The macrophages were then harvested, and DNA was isolated using the PureLink™ Genomic DNA Kit (ThermoFisher). Next, using 1 μg of DNA from each sample, methylated DNA was captured and detected by antibodies using the Methylamp Methylated DNA Capture Kit from EPIGENTEK, following the manufacturer’s protocol. PCR was conducted using primers targeting the CpG-rich region in the promoter of Dusp4, and the data were quantified as fold-enrichment of methylated DNA in the Dusp4 promoter relative to control IgG antibodies that do not recognize methylated DNA.
siRNA-mediated gene silencing.
50 nM of scrambled siRNA control or siRNA targeting specific gene were transfected into macrophages using Lipofectamine RNAiMAX (Life Technologies) following the manufacturer’s protocol. Briefly, cells seeded in 450 uL BMDM media were incubated with 50 uL of OptiMEM containing 1.5 uL iMAX and 50 nM siRNA. After 48 h or 72 h of transfection, experiments were conducted.
Quantitative real-time PCR.
RNA was extracted from samples using the PureLink™ RNA Mini Kit (Life Technologies) following the manufacturer’s protocol. The purity and concentration of RNA were determined using a NanoDrop spectrophotometer (ThermoScientific). Complementary DNA was synthesized from 200 ng of RNA from samples with a 260/280 ratio greater than 1.8 using oligo (dT) and Superscript II (Applied Biosystems). Quantitative RT-PCR was performed using a 7500 Real-Time PCR system (Applied Biosystems) and SYBR Green Master Mix reagents (Applied Biosystems). All sequences for primers and RNAi have been included as a supplementary table.
Zymosan-induced peritonitis and uptake of apoptotic neutrophils.
8– 10-week-old male mice were injected i.p. with 500 uL of 1 mg/mL Zymosan A (Sigma-Aldrich) to induce sterile peritonitis. For some experiments, mice received 200 ng/mL recombinant TGFβ1 at 15 h and 20 h after Zymosan A injection. Exudate fluid was collected and assayed for TGFβ1 and PGE2 by ELISA, and cells in the exudate were stained with mouse anti-Gr1 (neutrophils; Biolegend) and assayed by flow cytometry to quantify neutrophil numbers. To assay apoptotic neutrophil uptake by flow cytometry, exudate cells were stained with anti-F4/80 antibody (macrophages; Biolegend), permeabilized (below), and stained with anti-Gr1. Macrophages that had engulfed apoptotic neutrophils were identified as F4/80+Gr1+ cells.
Flow cytometry.
Cells were fixed in 4% PFA (ThermoScientific), suspended in FACS staining buffer (PBS containing 2% FBS and 1 mM EDTA), and incubated with TruStain FcX™ PLUS (anti-mouse CD16/32) Fc-blocking antibody from Biolegend. Cell surface receptor staining was carried out by incubating with fluorescent antibodies for 60 min at 4°C. For intracellular staining, cells were permeabilized with BD Perm/Wash (BD Biosciences), followed by fluorescent antibody staining at 4°C for 1 h. Cells were washed in FACS buffer twice and then resuspended for analysis on a BD FACS Canto II flow cytometer. For detection of apoptosis, cells were washed twice with cold FACS buffer, resuspended in annexin V-binding buffer at a concentration of 1×106 cells/mL, and incubated with FITC-conjugated Annexin V antibody for 15 min at room temperature. Samples were analyzed by BD FACS Canto II flow cytometer. Data analysis was carried out using the FlowJo Software, version 10.
Dexamethasone thymus experiment.
8–10 weeks old male mice were injected i.p. with 250 uL PBS containing 250 mg dexamethasone (Sigma-Aldrich) dissolved in DMSO. Eighteen hours after injection, the mice were euthanized, and thymi were harvested and weighed. One lobe of the thymus was mechanically disaggregated, and cells number was determined using Countess II (Invitrogen). 1×106 cells were stained with either FITC-conjugated Annexin V antibody (Biolegend) or anti-F4/80 antibody (Biolegend) to determine apoptotic cells and macrophage number, respectively. The other thymus lobe was formalin-fixed, paraffin-embedded, and sectioned, followed by immunostaining the sections with TUNEL reagent (Roche), anti-TGFβ1 (Abcam), anti-phospho-ERK1/2 (Cell Signaling), anti-ERK1/2 (Cell Signaling), anti-DUSP4 (Abcam), and/or anti-Mac2 antibodies (Cedarlane). In situ efferocytosis was quantified by counting TUNEL+ cells that were associated with Mac2+ cells versus macrophage-free TUNEL+ cells. Macrophage-associated apoptotic cells followed the criteria of TUNEL+ nuclei surrounded by or in contact with neighboring Mac2+ macrophages. Free apoptotic cells exhibited nuclear condensation and were not in contact with neighboring macrophages.
Tissue collection and lesion analysis.
Male Ldlr−/− mice (8–10 weeks/old) were prepared one week before irradiation by giving them acidic water containing 100 mg/L neomycin and 10 g/L polymyxin. The mice were given 1,000 rads using a 137 Cesium GammaCell source. Four-6 hours later, the mice were injected i.v. with 2.5 × 106 bone marrow (BM) cells from 8–10-week-old male control Vav1Cre+/− mice or Dnmt3afl/fl Vav1Cre+/− mice56. To isloated the BM cells, femurs from the donor mice were flushed with RPMI-1640 containing 10 units/mL of heparin, 10 U/mL penicillin, and 100 mg/mL streptomycin to collect the BM cells, which were then passed through a 40-mm cell strainer, collected by centrifugation at 500 × g for 5 min, and washed twice before loading into syringe for injection. Six weeks after BM cell injection, the mice were fed a Western-type diet (Envigo, TD 88137) for 12 weeks. The work-up of the mice and all atherosclerosis measurements were conducted as previously described6,33. In brief, the night before sacrifice, food was withdrawn, and the next morning blood glucose was assayed using a OneTouch Ultra device; plasma cholesterol was measured using a kit from WAKO Diagnostics; and complete blood cell counts and leukocyte differential were quantified using a FORCYTE Hematology Analyzer (Oxford Science). The mice were then euthanized, followed by PBS perfusion through the left ventricle. Aortic roots were formalin-fixed, paraffin-embedded, sectioned, and stained with hematoxylin and eosin6,33. For each mouse, we quantified 6 sections that were separated by 30 mm, starting at the base of the aortic root. The area from the internal elastic lamina to the lumen (total lesion area) was quantified using image analysis software. The lesions were also stained for collagen using picrosirius red (Polysciences, catalog 24901A) to quantify fibrous cap thickness at the cap midpoint and both shoulder regions. These measurements were averaged and expressed as the ratio of collagen cap thickness to lesion area.
Tissue immunohistochemistry and immunofluorescence microscopy.
6-mm paraffin-embedded sections were deparaffinized with xylene and rehydrated in decreasing concentrations of ethanol. Sections were incubated with TUNEL staining reagents at 37°C for 60 min and then washed three times with PBS. Alternatively, rehydrated sections were blocked for 60 min with 2 %. BSA in 1X PBS, and immunostained with anti-Mac2, anti-COX2, anti-TGFβ1 or anti-DUSP4 antibodies overnight at 4°C. Sections were stained with fluorescently labeled secondary antibodies and then counterstained with DAPI. Images were captured using a Leica epifluorescence microscope (DMI6000B).
Detection of 13C515N-labeled SAM in BMDMs incubated with ACs labeled with 13C515N-methionine.
Jurkat cells incubated with media containing 13C515N-methionine were labeled with fluorescent PKH26 dye and then rendered apoptotic. To prevent 13C515N-SAM formation in the Jurkat cells themselves, the cells were pre-incubated with the MAT2A inhibitor PF9366. Next, macrophages were pre-incubated with or without bafilomycin and then incubated with the labeled ACs for 45 minutes, followed by the removal of unbound ACs by rinsing and further incubation in medium without ACs for 1 hour. The macrophages were then sorted into PKH26+ (AC+) and PKH26− (AC−) macrophages and analyzed by LC-MS/MS for 13C515N-SAM. Briefly, 500 μL of pre-chilled cold methanol was added to each cell pellet, followed by vortexing and incubation at −80°C for 20 minutes. Samples were centrifuged at 14,000 × g for 5 minutes, and the metabolite extract was collected in a clean microcentrifuge tube. The pellet was re-extracted as described above, and the metabolite extracts were pooled, dried in a vacuum centrifuge (Savant SC210A SpeedVac Concentrator, ThermoFisher Scientific), and re-suspended in liquid chromatography loading solvent. For isotope tracer analysis, an ultra-high performance liquid chromatograph (Vanquish, ThermoFisher Scientific, San Jose, CA) interfaced with an electrospray Q Exactive Focus mass spectrometer (ThermoFisher Scientific, San Jose, CA) was used in full scan mode. The following solvent systems were used: solvent A was 100% LC-MS grade water (Burdick & Jackson, Honeywell, Muskegon, MI) containing 0.1% formic acid (28905, ThermoFisher); and solvent B was aqueous 100% LC-MS grade acetonitrile (34967, Burdick & Jackson, Honeywell, Muskegon, MI). For each sample, a 5 μl aliquot of the metabolite mixture was loaded onto an ACE C18-PFP column, (2.1 mm ID × 100 mm length with 2 μm particle size). The column was equilibrated at 0% B with a flow rate of 0.350 mL/min for 3 minutes. A gradient from 0% B to 80% B was applied over 10 min with a flow rate of 0.350 mL/min, held at 80% B for 3 minutes, followed by re-equilibration over 5 minutes, for a total of 21 minutes for the LC experiment. Mass spectrometry instrument parameters for full Scan MS on the Q Exactive Focus included the following: resolution 70,000; AGC target 3e6; and maximum IT 100 ms. Peak heights for unlabeled methionine and SAM and 13C515N-labeled methionine and 13C515N-labeled SAM were extracted from the chromatograms using Xcalibur Quan Browser (version 4.2.47, ThermoFisher Scientific, San Jose, CA).
Detection and quantification of 13C-CH3 in genomic DNA.
Macrophages pre-treated with or without bafilomycin and/or PF9366 were incubated for 45 minutes with or without 13C515N-methionine-labeled Jurkat cells (above). Unengulfed ACs were removed by rinsing and then incubated for an additional 1 hour. DNA was extracted from the cells and digested into single nucleosides by incubation with the Nucleoside Digestion Mix (New England BioLabs) at 37 °C for 2 hours. 13C-DNA methylation was identified and quantified by nano-liquid chromatography (nLC) coupled online with mass spectrometry (nLC-MS). nLC was configured with a two-column system consisting in a 300-μm ID × 0.5 cm C18 trap column (Dionex) and a 75-μm ID × 25 cm Reprosil-Pur C18-AQ (3 μm; Dr. Maisch GmbH, Germany) analytical nano-column packed in-house using a Dionex RSLC Ultimate 3000 (Thermo Scientific, San Jose, CA, USA). nLC was coupled to an Orbitrap Fusion Lumos mass spectrometer (Thermo Scientific). The voltage of the spray was set to 2.3 kV, and the temperature of the heated capillary was set to 275 °C. A full scan range of 110−600 m/z was acquired in the Orbitrap at a resolution of 120,000. The source fragmentation energy was set at 30 V, and the RF lens % was set at 50. The instrument was optimized to fragment the protonated nucleosides deoxy-methylcytidine (dmC) and labeled 13C-dmC into protonated nucleobases methylcytosine (mC) and 13C-mC with m/z at 126.0662 and 127.0695, respectively. The percentage of 13C-labeled methylation was calculated using the intensity of mC over mC + 13C-mC.
Statistical analysis.
GraphPad Prism software (version 9.2.0) was used for all statistical analyses. We first subjected data to the D’Agostino-Pearson or Shapiro-Wilk test and found that all data were normally distributed. For comparison of 2 groups, we used the Studen’s t-test to calculate P values, and for more than 2 groups we used one-way ANOVA with Fisher’s Least Significant Difference (LSD) posthoc analysis. Data are reported as means ± S.E.M, and differences were considered statistically significant at P < 0.05. Using power calculations based on our previous studies in mice using the dexamethasone-thymus, Zymosan-peritonitis, and mouse atherosclerosis models, the numbers of mice chosen for each cohort was sufficient to enable the testing of our hypotheses based on an expected 20%–30% coefficient of variations and an 80% chance of detecting a 33% difference in key plaque parameters (lesion size and fibrous cap thickness). Before the start of any analyses, mice were excluded if they died, suffered an injury requiring, or loss > 15% of their body weight.
Reporting summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1. Additional experiments documenting the efferocytosis-Ptgs2/COX2-TGFβ1 pathway in macrophages. Related to Fig. 1.a–d.
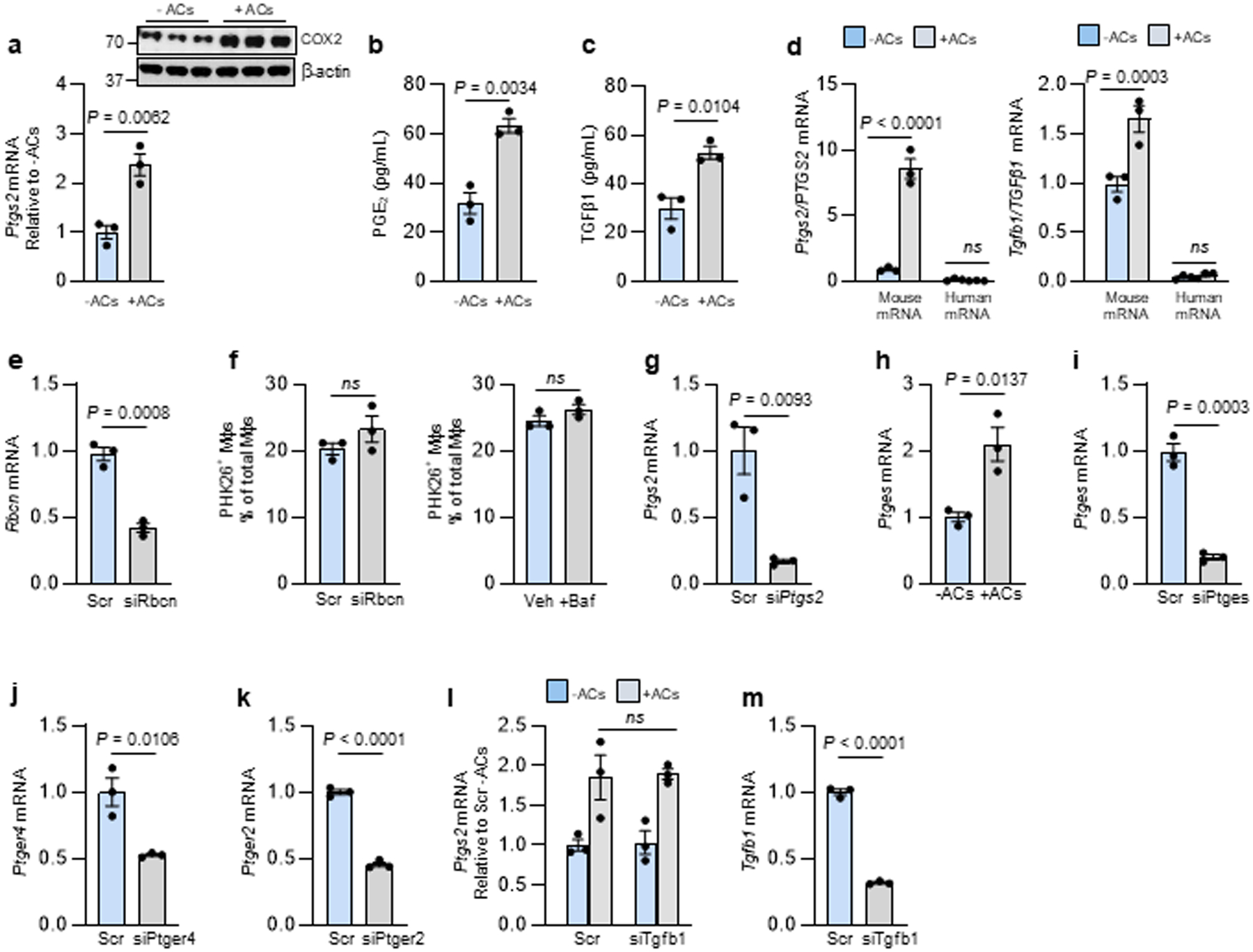
BMDMs were incubated ± ACs, after which noninternalized ACs were removed by rinsing. The cells were assayed for Ptgs2 mRNA after an additional 1 h incubation and COX2 protein after 3 h (a); PGE2 in the media after 3 h (b); and TGFβ1 in the media after 18 h (c). For (d), experiments similar to those in panel a were analyzed for mouse and human Ptgs2/PTGS2 or Tgfb1/TGFβ1 mRNA to prove that mRNA being measured is not residual human mRNA derived from human apoptotic Jurkat cells. e, BMDMs were transfected with scrambled RNA (Scr) or siRbcn and then, after 72 h, assayed for Rbcn mRNA. f, BMDMs were transfected with Scr or siRbcn or treated with vehicle or bafilomycin A1 and then incubated with PKH26-labeled ACs for 45 min, followed by rinsing and quantification of percent PKH26+ macrophages of total macrophages. g, BMDMs were transfected with Scr or siPtgs2 and then, after 72 h, assayed for Ptgs2 mRNA. h, BMDMs were incubated ± ACs for 45 min, after which noninternalized ACs were removed by rinsing. The cells were assayed for Ptges mRNA after an additional 1 h incubation (left). i, BMDMs were transfected with scrambled RNA (Scr) or siPtges and then, after 72 h, assayed for Ptges mRNA. j-k, BMDMs were transfected with Scr, siPtger4, or siPtger2 and then, after 72 h, assayed for Ptger4 or Ptger2 mRNA, respectively. l, BMDMs were transfected with Scr or siTgfb1. After 72 h, the cells were incubated ± ACs for 45 min, after which noninternalized ACs were removed by rinsing. After an additional 1 h of incubation, the cells were assayed for Ptgs2 mRNA. m, BMDMs were transfected with Scr or siTgfb1 and then, after 72 h, assayed for Tgfb1 mRNA. All mRNA data are expressed relative to the first control group. Values are means ± SEM. ns, not significant (P > 0.05); n = 3 biological replicates. Two-sided P values were determined by a Student’s t-test for two groups or one-way ANOVA with Fisher’s LSD posthoc analysis for three or more groups.
Extended Data Fig. 2. Additional experiments documenting the role of SAM in the efferocytosis-Ptgs2/COX2-TGFβ1 pathway. Related to Fig. 2.
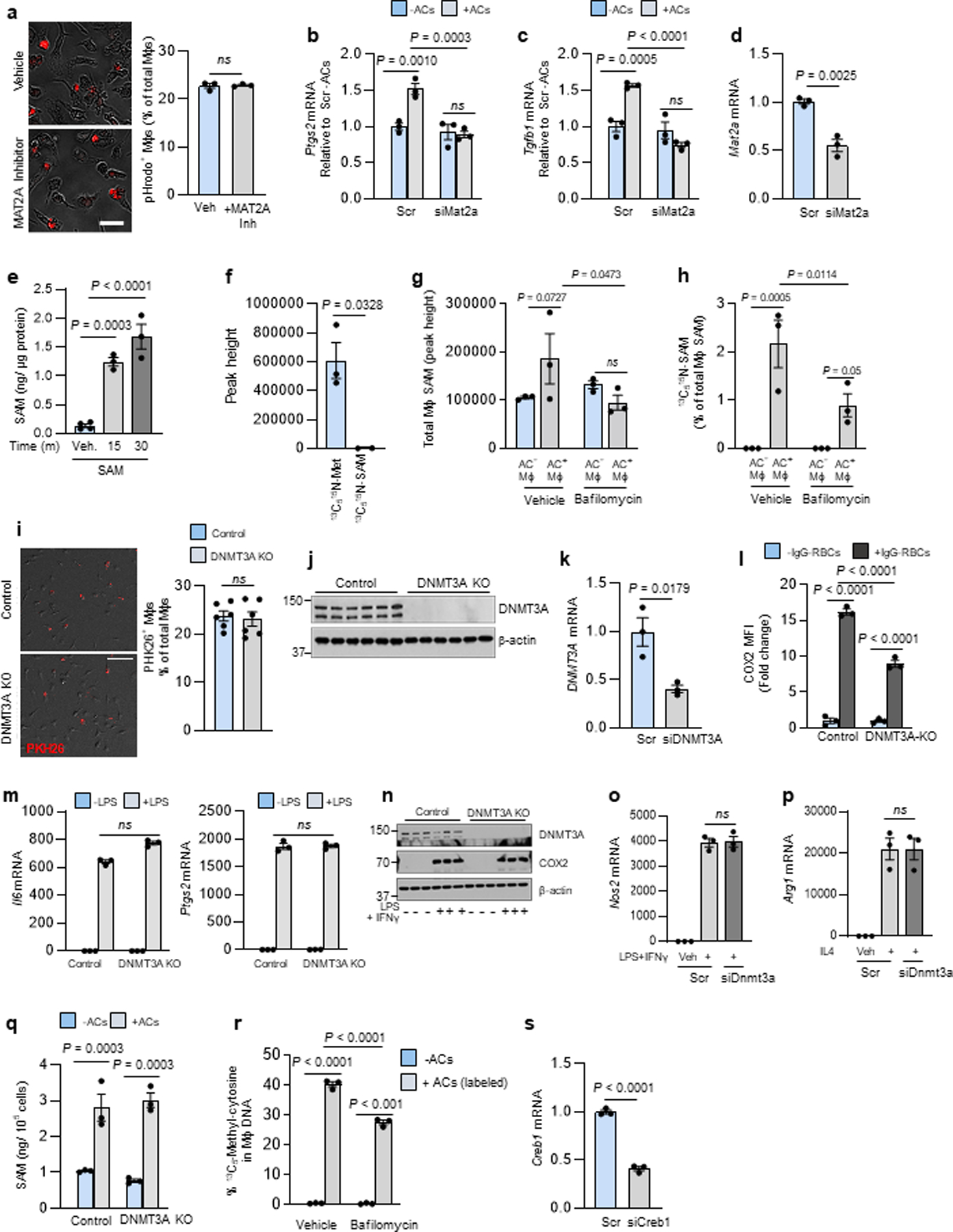
All AC incubations were 45 min, and Ptgs2 or Tgfb1 were assayed 1 or 6 h after AC removal, respectively. a, BMDMs pretreated 2 h with vehicle or the MAT2A inhibitor PF9366 were incubated with pHrodo-labeled ACs and quantified for the percent pHrodo-AC+ macrophages. Scale bar, 50 μm. b-c, BMDMs treated with scrambled RNA (Scr) or siMat2a were incubated ± ACs and then assayed for Ptgs2 orTgfb1. d, BMDMs treated with Scr or siMat2a were assayed for Mat2a. e, BMDMs treated with vehicle or SAM were assayed for SAM content. f, BMDMs cultured in methionine-free media with D-FBS and pretreated for 2 h with vehicle or bafilomycin A1 (Baf) were incubated 1 h with PKH26-labeled ACs whose proteins were labeled with 13C515N-methionine. AC+ and AC− macrophages were sorted and assayed for SAM content (g) and percent 13C515N-SAM of total SAM (h). i, Control or DNMT3A-KO BMDMs were incubated with PKH26-labeled ACs and then quantification for the percent PKH26-AC+ macrophages. Scale bar, 50 μm. j, Control and DNMT3A-KO BMDMs were immunoblotted for DNMT3A and β-actin. k, HMDMs treated with Scr or siDNMT3A were assayed for DNMT3A. l, BMDMs were incubated with IgG-coated RBCs for 45 min and then assayed 3 h later for COX2 MFI by flow cytometry. m-n, Control and DNMT3A-KO BMDMs treated with LPS or LPS + IFNγ for 4 h were assayed for Il6, Ptgs2, or COX2 (n). o-p, BMDMs treated with Scr or siDnmt3a were incubated for 4 h with vehicle and LPS + IFNγ or IL4 and assayed for Nos2 or Arg1. q, Control and DNMT3A-KO BMDMs were incubated ± ACs for 45 min and then assayed for SAM. r, BMDMs pretreated for 2 h with vehicle or bafilomycin A1 were incubated ± ACs whose proteins were labeled with 13C515N-methionine and then assayed 1 h after AC removal for 13C5-methylcytosine in DNA. s, BMDMs treated with Scr or siCreb1 were assayed for Creb1. All mRNA data are expressed relative to the first control group. Values are means ± SEM. n.s., not significant (P > 0.05); n = 3 biological replicates for all bar graphs except i (n = 6). Two-sided P values were determined by the Student’s t-test for two groups or one-way ANOVA with Fisher’s LSD posthoc analysis for three or more groups.
Extended Data Fig. 3. Experiments documenting the role of ERK, CD36, and DUSP4 in the efferocytosis-Ptgs2/COX2-TGFβ1 pathway. Related to Fig. 4.
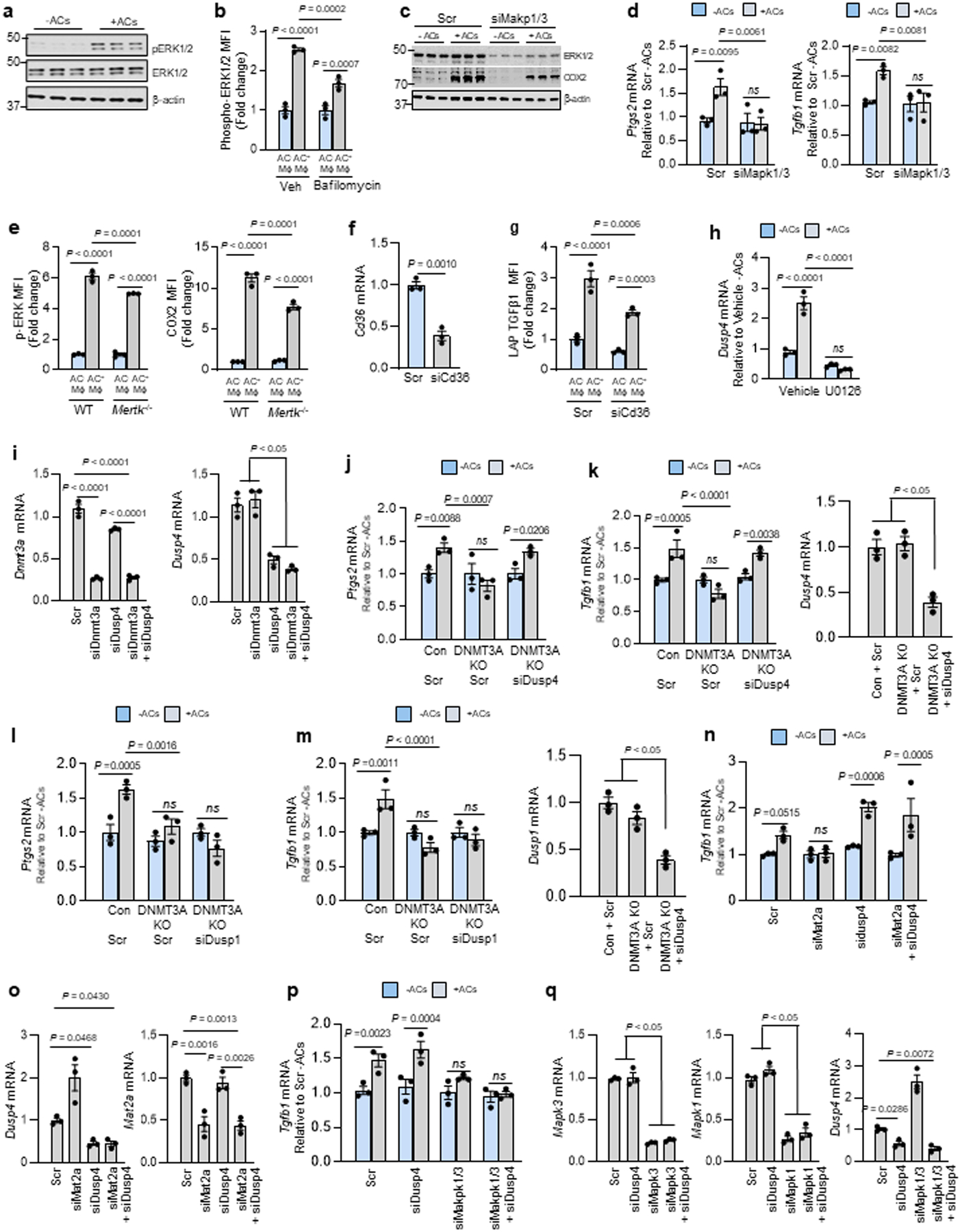
AC incubations were 45 min, and Ptgs2 or Tgfb1 were assayed 1 or 6 h after AC removal, respectively. a, BMDMs incubated ± ACs were immunoblotted for p-ERK1/2, ERK1/2, and β-actin. b, BMDMs pretreated with vehicle or bafilomycin A1 were incubated ± PKH26-labeled ACs for 45 min and assayed by flow cytometry for p-ERK1/2 in PKH26+ (AC+) and PKH26− (AC−) macrophages. c-d, BMDMs transfected with scrambled RNA (Scr) or siMapk1 and siMapk3 were incubated ± ACs for 45 min and immunoblotted for ERK1/2, COX2, and β-actin or assayed for Ptgs2 or Tgfb1. e, WT or MerTK-KO BMDMs were incubated with PKH26-labeled ACs and assayed by flow cytometry for p-ERK1/2 or, after a 3-h chase, COX2. (f) BMDMs treated with Scr or siCd36 were assayed for Cd36. (g) BMDMs treated with Scr or siCd36 were incubated with pHrodo-labeled ACs and, after an 18-h chase, assayed by flow cytometry for TGFβ1 in pHrodo+ (AC+) and pHrodo− (AC−) macrophages. h, BMDMs pretreated for 2 h with vehicle or U0126 (MEK inhibitor) were incubated ± ACs and assayed for Dusp4. i, BMDMs treated with Scr, siDnmt3a, siDusp4, or siDnmt3a + siDusp4 were assayed for Dnmt3a or Dusp4. j-k, Control or DNMT3A-KO BMDMs transfected with Scr or siDusp4 as indicated were incubated ± ACs and assayed for Ptgs2 or Tgfb1. l-m, Control or DNMT3A-KO BMDMs treated with Scr or siDusp1 as indicated were incubated ± ACs and assayed for Ptgs2 or Tgfb. n-o, BMDMs treated with Scr, siMat2a, siDusp4, or siMat2a + siDusp4 were incubated ± ACs and assayed for Tgfb1. p-q, BMDMs treated with Scr, siMapk1/3, siDusp4, or siMapk1/3 + siDusp4 were incubated ± ACs and assayed for Tgfb1, Mapk3, Mapk1, and Dusp4 after a 6-h chase. Data are expressed relative to the first control group. Values are means ± SEM. n.s., not significant (P > 0.05); n = 3 biological replicates. Two-sided P values were determined by the Student’s t-test for two groups or one-way ANOVA with Fisher’s LSD posthoc analysis for three or more groups.
Extended Data Fig. 4. In vivo evidence of AC-induced COX2-TGFβ1 pathway. Related to Fig. 5. a–b.
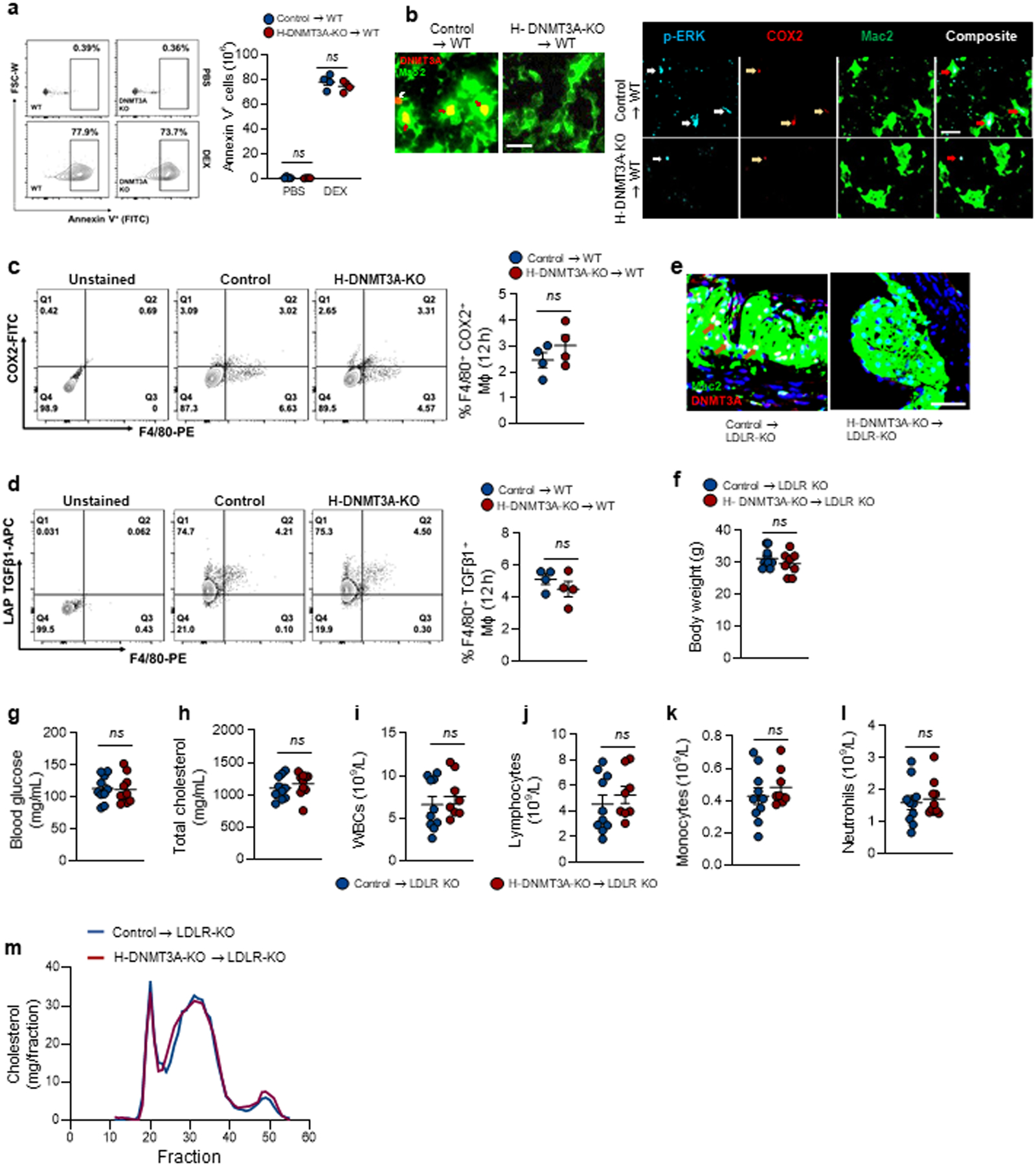
Wild-type (WT) C57BL/6J mice were transplanted with bone marrow from Vav1Cre+/− (Control) or Dnmt3afl/fl Vav1Cre+/− (H-DNMT3A-KO) mice and, after 4 weeks, injected with PBS or dexamethasone (DEX). After 4h, the thymus was harvested and immunostained with anti-annexin V to document the initial increase in apoptosis after PBS or DEX injection, n=4 mice per group (a). For b (left), thymi were immunostained with Mac2 (green) and DNMT3A (Red). White arrows indicate non-macrophage DNMT3A, and brown arrows indicate macrophage DNMT3A. For b (right), documentation of co-localized p-ERK1/2, COX2, and Mac2. Representative image from n=4 mice per group. Scale bar, 50 μm. c-d, Wildtype (WT) C57BL/6J mice transplanted with bone marrow from control or H-DNMT3A-KO mice were injected i.p. with 1 mg/mL Zymosan A1. After 12 h, peritoneal exudate cells were analyzed by flow cytometry for F4/80+ and COX2+ cells (n=4 mice/group) LAP-TGFβ1+ cells (n= 4 mice/group). e-m, Ldlr−/− (LDLR-KO) mice were transplanted with bone marrow from control or H-DNMT3A-KO mice and, after 4 weeks, fed a western-type diet (WD) for 12 weeks. Aortic root sections were immunostained for Mac2 and DNMT3A. Brown arrows indicate macrophage DNMT3A. Representative image from n=8 mice per group (e); body weight, n=10 mice/group (f); and the plasma or blood was assayed for the indicated metabolic and immune cell parameters, n=10 mice/group (g-l) and the lipoprotein-cholesterol profile by FPLC (m). Scale bar, 50 μm. Values are means ± SEM; n.s., not significant (P > 0.05). Two-sided P values were determined by the Student’s t-test for two groups or one-way ANOVA with Fisher’s LSD posthoc analysis for three or more groups.
Extended Data Fig. 5. DNMT3A mediates efferocytosis and resolution in vivo. Related to Fig. 6. a.
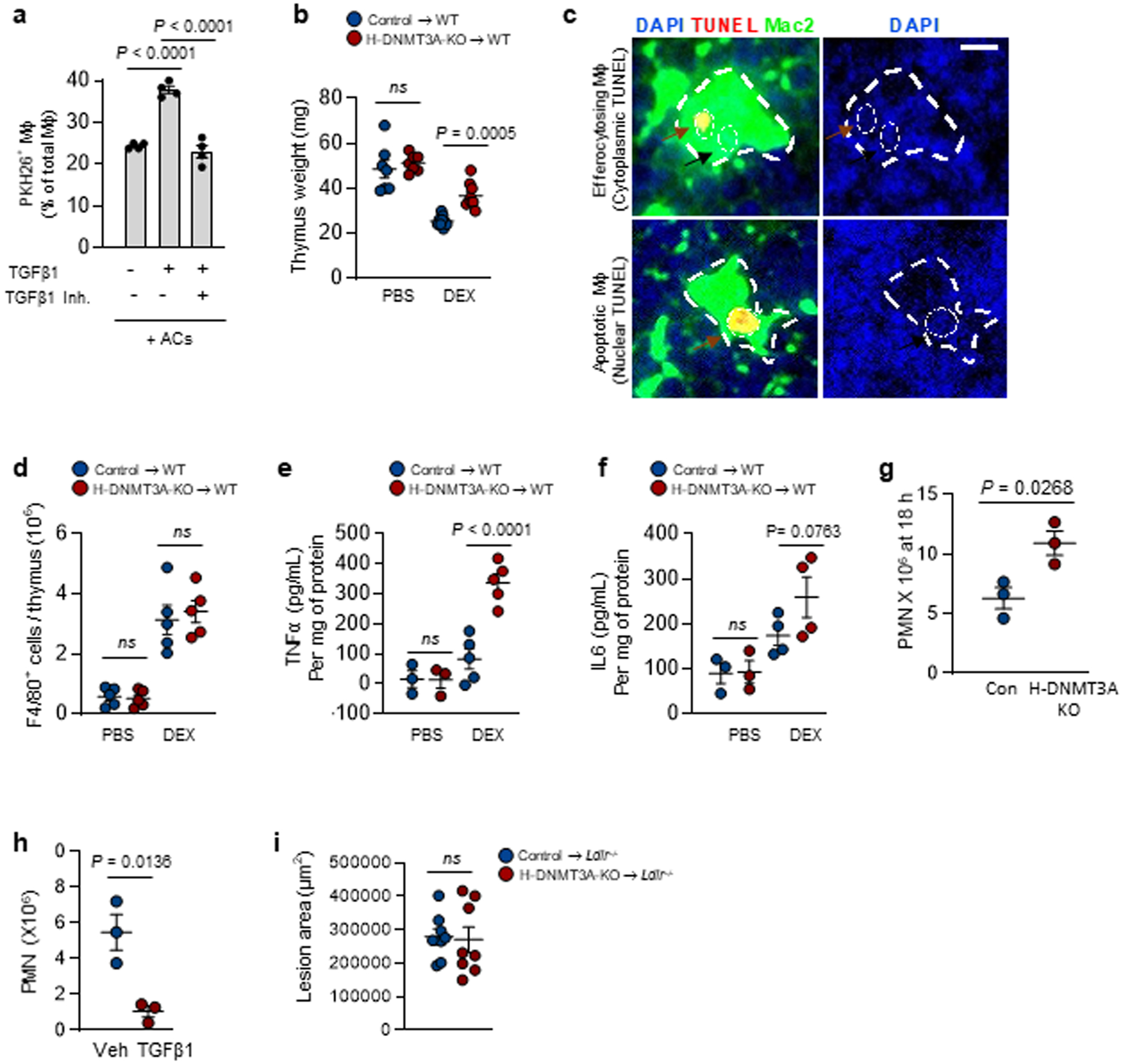
BMDMs were pretreated with vehicle or a TGFβ1R inhibitor for 2 h and with vehicle or recombinant TGFβ1 for 1 h, as indicated. The macrophages were then incubated with PKH26-labeled ACs for 45 mins, followed by rinsing and quantification of percent PKH26-AC+ macrophages of total macrophages, n=4 biological replicates. b-f, Wildtype (WT) C57BL/6J mice were transplanted with bone marrow from control or H-DNMT3A-KO mice and, after 4 weeks, injected with PBS or dexamethasone (DEX). After 18 h, the thymi were weighed, n= 7 and 9 mice for PBS and DEX groups respectively (b); immunostained for DAPI, TUNEL, and Mac2 (c); assayed for F4/80+ cells, n=5 mice per group (d); and assayed for TNF-a, n=3 and 5 mice for PBS and DEX groups respectively and IL-6 by ELISA, n=3 and 4 mice for PBS and DEX groups respectively (e-f). The image in b illustrates thymic macrophages with cytoplasmic TUNEL as an example of efferocytosing thymic macrophages. Scale bar, 100 μm. g, The peritoneal exudates were assayed for Ly6G+ polymorphonuclear cells (PMN) 18 hours after Zymosan A1 injection, n=3 mice per group. h, Wild-type mice received 200 ng/mL recombinant TGFβ1 i.p. or vehicle control 15 and 20 hours after Zymosan injection and then assayed for the number of PMNs 4 hours later, n=3 mice/group. i, Ldlr−/− (LDLR-KO) mice were transplanted with bone marrow from control or H-DNMT3A-KO mice and, after 4 weeks, fed a western-type diet (WD) for 12 weeks. Aortic root sections were quantified for lesional area, n=8 mice/group. Values are means ± SEM; n.s., not significant (P > 0.05). Two-sided P values were determined by the Student’s t-test for two groups or one-way ANOVA with Fisher’s LSD posthoc analysis for three or more groups.
Supplementary Material
Acknowledgments
We thank Dr. Siddhartha Mukherjee and Alan Burke (Columbia University) for the Dnmt3afl/fl Vav1Cre+/− mice; Drs. Robert Bowman and Ross Levine (Memorial Sloan Kettering Cancer Center) for initial discussions about the experimental approach using Dnmt3a-targeted mice; and Drs. Jianlong Wang and Xin Huang (Columbia) for initial discussions on DNA methylation assays. We acknowledge Dr. Caisheng Lu of the Columbia Center for Translational Immunology Core Facility for assisting in the flow cytometry and immunofluorescent imaging experiments, which were conducted in the Columbia Center for Translational Immunology Core Facility, funded by NIH grants P30CA013696 and S10OD020056 and S10RR027050. This study was funded by a Transatlantic Network of Excellence (TNE-18CVD04) grant from the Leducq Foundation (to A.R.T. and I.T.) and by the following NIH grants: R00 DK115778 (to B.C.); T32 5T32HL007343-42 (to B.D.G.); K99 HL145131 (to A.Y.); R01 HL127464 (to I.T.); and R01 HL087123, and R35 HL145228 (to I.T.). B.C. received funding support from R00DK115778. S.S. and Y.S. acknowledge the Leukemia Research Foundation (Hollis Brownstein New Investigator Research Grant); AFAR (Sagol Network GerOmic Award); the Einstein Nathan Shock Center for the Biology of Aging, Deerfield (Xseed award); NIH P30 grant CA01333047; shared instrument grant NIH 1 S10 OD030286-01; and NIGMS grant 5 R01GM129350-04 (PI: Brenowitz). This work has also been supported in part by the Proteomics & Metabolomics Core Facility at the H. Lee Moffitt Cancer Center & Research Institute, an NCI designated Comprehensive Cancer Center (P30-CA076292).
Footnotes
Competing interests
The authors declare no competing interests.
Data availability
This study did not generate any unique datasets or codes. All other data can be made available from the authors on reasonable request. Additional data associated with the paper can be found in the Supplementary information.
References
- 1.Dalli J & Serhan CN Pro-resolving mediators in regulating and conferring macrophage function. Front Immunol 8, 1400 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doran AC, Yurdagul A Jr. & Tabas I Efferocytosis in health and disease. Nat Rev Immunol 20, 254–267 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morioka S, Maueroder C & Ravichandran KS Living on the edge: Efferocytosis at the interface of homeostasis and pathology. Immunity 50, 1149–1162 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Linton MF et al. Macrophage apoptosis and efferocytosis in the pathogenesis of atherosclerosis. Circ J 80, 2259–2268 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kojima Y, Weissman IL & Leeper NJ The role of efferocytosis in atherosclerosis. Circulation 135, 476–489 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yurdagul A Jr. et al. Macrophage metabolism of apoptotic cell-derived arginine promotes continual efferocytosis and resolution of injury. Cell Metab 31, 518–533 e510 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang S et al. Efferocytosis fuels requirements of fatty acid oxidation and the electron transport chain to polarize macrophages for tissue repair. Cell Metab 29, 443–456 e445 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.A-Gonzalez et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity 31, 245–258 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fadok VA et al. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest 101, 890–898 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakanishi M & Rosenberg DW Multifaceted roles of PGE2 in inflammation and cancer. Semin Immunopathol 35, 123–137 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Funk CD & FitzGerald GA COX-2 inhibitors and cardiovascular risk. J Cardiovasc Pharmacol 50, 470–479 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Cheng H, Huang H, Guo Z, Chang Y & Li Z Role of prostaglandin E2 in tissue repair and regeneration. Theranostics 11, 8836–8854 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang EH, Libby P, Vanhoutte PM & Xu A Anti-inflammation therapy by activation of prostaglandin EP4 receptor in cardiovascular and other inflammatory diseases. J Cardiovasc Pharmacol 59, 116–123 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takayama K et al. Prostaglandin E2 suppresses chemokine production in human macrophages through the EP4 receptor. J Biol Chem 277, 44147–44154 (2002). [DOI] [PubMed] [Google Scholar]
- 15.Gitlin JM & Loftin CD Cyclooxygenase-2 inhibition increases lipopolysaccharide-induced atherosclerosis in mice. Cardiovasc Res 81, 400–407 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kirkby NS et al. COX-2 protects against atherosclerosis independently of local vascular prostacyclin: identification of COX-2 associated pathways implicate Rgl1 and lymphocyte networks. PLoS One 9, e98165 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu Z et al. Disruption of the 5-lipoxygenase pathway attenuates atherogenesis consequent to COX-2 deletion in mice. Proc Natl Acad Sci U S A 109, 6727–6732 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Narasimha A et al. A novel anti-atherogenic role for COX-2--potential mechanism for the cardiovascular side effects of COX-2 inhibitors. Prostaglandins Other Lipid Mediat 84, 24–33 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoshimura A, Wakabayashi Y & Mori T Cellular and molecular basis for the regulation of inflammation by TGF-beta. J Biochem 147, 781–792 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mallat Z et al. Inhibition of transforming growth factor-beta signaling accelerates atherosclerosis and induces an unstable plaque phenotype in mice. Circ Res 89, 930–934 (2001). [DOI] [PubMed] [Google Scholar]
- 21.Martinez J et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A 108, 17396–17401 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Mao Y et al. Inhibition of tumor-derived prostaglandin-e2 blocks the induction of myeloid-derived suppressor cells and recovers natural killer cell activity. Clin Cancer Res 20, 4096–4106 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Sun WH et al. Induction of cyclooxygenase-2 in rat gastric mucosa by rebamipide, a mucoprotective agent. J Pharmacol Exp Ther 295, 447–452 (2000). [PubMed] [Google Scholar]
- 24.Mato JM, Martinez-Chantar ML & Lu SC S-adenosylmethionine metabolism and liver disease. Ann Hepatol 12, 183–189 (2013). [PMC free article] [PubMed] [Google Scholar]
- 25.Parkhitko AA, Jouandin P, Mohr SE & Perrimon N Methionine metabolism and methyltransferases in the regulation of aging and lifespan extension across species. Aging Cell 18, e13034 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quinlan CL et al. Targeting S-adenosylmethionine biosynthesis with a novel allosteric inhibitor of Mat2A. Nat Chem Biol 13, 785–792 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Finkelstein JD Pathways and regulation of homocysteine metabolism in mammals. Semin Thromb Hemost 26, 219–225 (2000). [DOI] [PubMed] [Google Scholar]
- 28.Okano M, Bell DW, Haber DA & Li E DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257 (1999). [DOI] [PubMed] [Google Scholar]
- 29.Cho KN et al. Prostaglandin E2 induces MUC8 gene expression via a mechanism involving ERK MAPK/RSK1/cAMP response element binding protein activation in human airway epithelial cells. J Biol Chem 280, 6676–6681 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Xia Q et al. Induction of COX-2-PGE2 synthesis by activation of the MAPK/ERK pathway contributes to neuronal death triggered by TDP-43-depleted microglia. Cell Death Dis 6, e1702 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiong W, Frasch SC, Thomas SM, Bratton DL & Henson PM Induction of TGF-beta1 synthesis by macrophages in response to apoptotic cells requires activation of the scavenger receptor CD36. PLoS One 8, e72772 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cai B et al. Macrophage MerTK promotes liver fibrosis in nonalcoholic steatohepatitis. Cell Metab 31, 406–421 e407 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gerlach BD et al. Efferocytosis induces macrophage proliferation to help resolve tissue injury. Cell Metab 33, 2445–2463 e2448 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nishi C, Yanagihashi Y, Segawa K & Nagata S MERTK tyrosine kinase receptor together with TIM4 phosphatidylserine receptor mediates distinct signal transduction pathways for efferocytosis and cell proliferation. J Biol Chem 294, 7221–7230 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lang R, Hammer M & Mages J DUSP meet immunology: dual specificity MAPK phosphatases in control of the inflammatory response. J Immunol 177, 7497–7504 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Newson J et al. Inflammatory resolution triggers a prolonged phase of immune suppression through COX-1/mPGES-1-derived Prostaglandin E2. Cell Rep 20, 3162–3175 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cai B et al. MerTK cleavage limits proresolving mediator biosynthesis and exacerbates tissue inflammation. Proc Natl Acad Sci U S A 113, 6526–6531 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ren Y & Savill J Proinflammatory cytokines potentiate thrombospondin-mediated phagocytosis of neutrophils undergoing apoptosis. J Immunol 154, 2366–2374 (1995). [PubMed] [Google Scholar]
- 39.Tse K & Ley K Transforming growth factor-beta: transforming plaque to stability. Eur Heart J 34, 3684–3686 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takeuch O & Akira S Epigenetic control of macrophage polarization. Eur J Immunol 41, 2490–2493 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Roy DG et al. Methionine metabolism shapes t helper cell responses through regulation of epigenetic reprogramming. Cell Metab 31, 250–266.e259 (2020). [DOI] [PubMed] [Google Scholar]
- 42.Ji J et al. Methionine attenuates lipopolysaccharide-induced inflammatory responses via DNA methylation in macrophages. ACS Omega 4, 2331–2336 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shikauchi Y et al. SALL3 interacts with DNMT3A and shows the ability to inhibit CpG island methylation in hepatocellular carcinoma. Mol Cell Biol 29, 1944–1958 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guo X et al. Structural insight into autoinhibition and histone H3-induced activation of DNMT3A. Nature 517, 640–644 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Dai Z, Mentch SJ, Gao X, Nichenametla SN & Locasale JW Methionine metabolism influences genomic architecture and gene expression through H3K4me3 peak width. Nat Commun 9, 1955 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fredman G et al. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat Commun 7, 12859 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Merched AJ, Ko K, Gotlinger KH, Serhan CN & Chan L Atherosclerosis: evidence for impairment of resolution of vascular inflammation governed by specific lipid mediators. FASEB J 22, 3595–3606 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takayama K, Sukhova GK, Chin MT & Libby P A novel prostaglandin E receptor 4-associated protein participates in antiinflammatory signaling. Circ Res 98, 499–504 (2006). [DOI] [PubMed] [Google Scholar]
- 49.Jaiswal S et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med 377, 111–121 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sharma M et al. Regulatory T cells license macrophage pro-resolving functions during atherosclerosis regression. Circ Res 127, 335–353 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Toussirot E, Bonnefoy F, Vauchy C, Perruche S & Saas P Mini-Review: The administration of apoptotic cells for treating rheumatoid arthritis: current knowledge and clinical perspectives. Front Immunol 12, 630170 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mevorach D et al. Single infusion of donor mononuclear early apoptotic cells as prophylaxis for graft-versus-host disease in myeloablative HLA-matched allogeneic bone marrow transplantation: a phase I/IIa clinical trial. Biol Blood Marrow Transplant 20, 58–65 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Zhang Z et al. Clearance of apoptotic cells by mesenchymal stem cells contributes to immunosuppression via PGE2. EBioMedicine 45, 341–350 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pujol-Autonell I et al. Efferocytosis promotes suppressive effects on dendritic cells through prostaglandin E2 production in the context of autoimmunity. PLoS One 8, e63296 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fourgeaud L et al. TAM receptors regulate multiple features of microglial physiology. Nature 532, 240–244 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kasikara C et al. Deficiency of macrophage PHACTR1 impairs efferocytosis and promotes atherosclerotic plaque necrosis. J Clin Invest 131 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or codes. All other data can be made available from the authors on reasonable request. Additional data associated with the paper can be found in the Supplementary information.