

Abstract
Background:
Dual-specificity tyrosine phosphorylation-regulated kinase 1a (DYRK1a) contributes to the control of cycling cells, including cardiomyocytes. However, the effects of inhibition of DYRK1a on cardiac function and cycling cardiomyocytes after myocardial infarction (MI) remains unknown.
Methods:
We investigated the impacts of pharmacological inhibition and conditional genetic ablation of DYRK1a on endogenous cardiomyocyte cycling and left ventricular (LV) systolic function in ischemia-reperfusion (I/R) MI using αMHC-MerDreMer-Ki67p-RoxedCre::Rox-Lox-tdTomato-eGFP (RLTG) (denoted αDKRC::RLTG) and αMHC-Cre::Fucci2aR::DYRK1aflox/flox mice.
Results:
We observed that harmine, an inhibitor of DYRK1a, improved LV Ejection Fraction (LVEF) (39.5 ± 1.6% and 29.1 ± 1.6%, harmine versus placebo, respectively), two weeks after I/R MI. Harmine also increased cardiomyocyte cycling after I/R MI in αDKRC::RLTG mice, 10.8 ± 1.5 verses 24.3 ± 2.6 enhanced Green Fluorescent Protein (eGFP)+ cardiomyocytes, placebo verses harmine, respectively, p=1.0x10−3). The effects of harmine on LVEF were attenuated in αDKRC::DTA mice that expressed an inducible diphtheria toxin in adult cycling cardiomyocytes. The conditional cardiomyocyte-specific genetic ablation of DYRK1a in αMHC-Cre::Fucci2aR::DYRK1aflox/flox (denoted DYRK1a k/o) mice caused cardiomyocyte hyperplasia at baseline (210 ± 28 vs. 126 ± 5 cardiomyocytes per 40x field, DYRK1a k/o versus controls, respectively, p = 1.7x10−2) without changes in cardiac function compared to controls, or compensatory changes in the expression of other DYRK isoforms. After I/R MI, DYRK1a k/o mice had improved LV function (LVEF 41.8 ± 2.2% and 26.4 ± 0.8%, DYRK1a k/o versus control, respectively, p = 3.7x10−2). RNAseq of cardiomyocytes isolated from αMHC-Cre::Fucci2aR::DYRK1aflox/flox and αMHC-Cre::Fucci2aR mice after I/R MI or Sham surgeries identified enrichment in mitotic cell cycle genes in αMHC-Cre::Fucci2aR::DYRK1aflox/flox compared to αMHC-Cre::Fucci2aR.
Conclusions:
The pharmacological inhibition or cardiomyocyte-specific ablation of DYRK1a caused baseline hyperplasia and improved cardiac function after I/R MI, with an increase in cell cycle gene expression, suggesting the inhibition of DYRK1a may serve as a therapeutic target to treat MI.
Keywords: Animal Models of Human Disease, Basic Science Research, Genetically Altered and Transgenic Models, Myocardial Infarction, Myocardial Regeneration
Graphical Abstract
Introduction:
Rate-limiting pathways that potentially enhance cardiomyocyte cycling may serve as targets to treat cardiovascular disease. Endogenously cycling adult cardiomyocytes are scarce; however, our previous findings suggest that their ablation worsens myocardial function after infarction.1 Conversely, enhancing cardiomyocyte proliferation is associated with improved LV function after injury, suggesting signals that induce cycling are potential avenues to treat MI.2-5
Generally, cardiomyocyte cycling involves canonical cell cycle proteins and redundant brakes such that removing one or even several brakes will not be sufficient to generate enough cardiomyocytes to confer substantial overall functional benefit. However, upstream signaling nodes that induce global changes in cycling programs may circumvent several brakes simultaneously and increase endogenous cardiomyocyte cycling. Clearly, manipulating critical signaling nodes such as the Hippo/YAP axis induced proliferation and improvement in myocardial function after injury.2, 3, 5, 6 Thus, manipulating upstream nodal signals that control G1/S and G2/M transitions, rather than canonical cell cycle proteins, may also induce cardiomyocyte cycling and improve cardiac function.
Many pathways regulate the cell cycle. In some cells, the Myb-MuvB/Dimerization partner, RB-like, E2F, and multi-vulval class B (DREAM) multiprotein complex controls G1/S and G2/M checkpoints at the level of genes targeted by E2F and Myb.7-11 The MuvB/DREAM complex acts as a “brake,” preventing the expression of genes at the G1/S and G2/M transitions in several cell types.7, 10 Growth signals release the “brake” at G1/S and change the MuvB/DREAM complex into an “accelerator” at G2/M, enhancing the exit from quiescence to active cell cycling (Supplemental Figure 1). Cardiomyocyte cycling involves E2Fs12-17; however, the contribution of the MuvB/DREAM complex is unclear. The ablation of one or a few components of the MuvB/DREAM complex likely impacts cell growth.9, 11, 18 Therefore, we focused on upstream regulators that potentially regulate the expression of many genes controlled by the MuvB/DREAM complex. The dual-specificity tyrosine phosphorylation-regulated kinase 1a (DYRK1a) is a critical regulator of MuvB/DREAM and influences cycling in several cells.9 The inhibition of DYRK1a promotes cell cycling and proliferation in mouse and human pancreatic islet cells19-24, and DYRK1a has been implicated in cardiomyocyte growth through cyclin D125. Given the importance of E2F signals in cardiomyocyte growth and the potential of DYRK1a as one regulator of genes expressed at the G1/S and G2/M boundaries in cycling cells, we investigated the effects of pharmacologic inhibition and genetic ablation of DYRK1a on cardiomyocyte cycling and cardiac function after I/R MI.
Using our recently published reporter mouse, αDKRC::RLTG, that labels endogenously cycling adult cardiomyocytes1, we observed the DYRK1a inhibitor, harmine, enhanced cardiomyocyte cycling, and improved myocardial function after I/R MI. The expression of Diphtheria toxin to ablate cardiomyocytes that cycle attenuated the beneficial effects of harmine in LV systolic function after MI. Next, we generated αMHC-Cre::Fucci2aR::DYRK1aflox/flox mice to label cardiomyocyte nuclei and ablated DYRK1a. At baseline, αMHC-Cre::Fucci2aR::DYRK1aflox/flox mice had cardiomyocyte hyperplasia; however, cardiac function was similar to control mice. The cardiomyocyte-specific ablation of DYRK1a improved cardiac function after I/R MI. To understand the pathways involved in DYRK1a ablation, we performed RNAseq of cardiomyocytes isolated from αMHC-Cre::Fucci2aR::DYRK1aflox/flox after I/R MI or sham surgeries and identified differential enrichment in mitotic cell cycle genes, including pathways controlled by the MuvB/DREAM complex. Collectively, our findings suggest the inhibition of DYRK1a is a potential therapeutic target to enhance cardiomyocyte cycling and improve heart function after MI.
Methods:
Data Availability.
The data that support the findings of this study are available from the corresponding author upon reasonable request.
See Supplemental Materials for detailed methods.
Results:
DYRK1a is a potential regulator of postnatal cardiomyocyte growth.
Analyses of gene expression during postnatal heart growth is complex but can implicate candidate signals controlling the exit of cardiomyocytes from the cell cycle. Therefore, we analyzed three publically available datasets of gene expression of cardiomyocytes from the adult, neonatal, and embryonic mouse hearts to identify pathways potentially controlling postnatal cardiomyocyte growth that may serve as targets to induce adult cardiomyocyte cycling after injury. First, we examined RNA expression data obtained from FACS-purified cardiomyocytes of embryonic E11-12, neonatal P3-4, and adult (10 weeks old) C57BL/6 mice described by Walsh et al. (NCBI Gene Expression Omnibus (GEO Accession number GSE17020).26 Genes clustered into seven categories based on expression defined as (1) slow down-regulation, (2) rapid down-regulation, (3) moderately rapid down-regulation, (4) transiently up-regulated in neonatal, (5) slow up-regulation, (6) rapid up-regulation, (7) moderately rapid up-regulation (Supplemental Figure 2A and Supplemental Table 1). Clusters 5 to 7 corresponding to the genes upregulated in hearts of adults and neonates compared to embryos were enriched for pathways associated with extracellular matrix organization and Interleukin-10 signaling (Supplemental Figure 2B and C). To identify pathways that were down-regulated during the exit of cardiomyocytes from the cell cycle, we analyzed Clusters 1 to 3 using Signatures Database (MSigDB) gene set enrichment analysis27, 28 (Supplemental Figure 2B and C). Consistent with the findings reported by Walsh et al., we identified enrichment in genes associated with E2F targets, G2/M checkpoint gene sets, and E2F1 signals, known to participate in the control of cell cycle and DNA replication.
Second, we analyzed data from Talman et al.29, who produced a detailed molecular atlas of postnatal mouse heart development at ages P1, P4, P9, and P23 using RNAseq, proteomics, and metabolomics approaches. Recognizing the importance of the MuvB/DREAM complex in the regulation of G1/S and G2/M checkpoints at the level of genes targeted by E2F and Myb, we examined the temporal change in 214 genes predicted to be regulated by the MuvB/DREAM complex, based on the presence of proximal E2F or CHR sites (Supplemental Figure 2D).7, 8 Peak gene expression of the 214 genes occurred at P4 during the postnatal heart growth, corresponding to the window when cardiomyocytes exit the mitotic cell cycle.30 The findings suggested a correlation between the expression of genes regulated by the MuvB/DREAM complex and postnatal cardiomyocyte growth and cycling.
Third, we analyzed data from a multicellular transcriptional analysis of mammalian heart regeneration.31 Consistent with the observations of E2Fs in normal postnatal cardiomyocyte growth, several transcription factors regulated by the MuvB-DREAM complex, including E2F1, E2F2, FoxM1, Mybl2, and Myb, were differentially expressed in cardiomyocytes in P1 hearts. Based on our analyses of the three published investigations26, 29, 31, we hypothesized that a unifying theme was the MuvB-DREAM complex as a candidate regulator of cardiomyocyte cycling.
Next, we quantified the expression of seventeen genes encoding components of the MuvB-DREAM complex of hearts isolated from P1, P3, P5, P7, P10, P14, and P28 C57B6J mice (Supplemental Figure 2E). E2F1, E2F3, E2F4, and E2F5 had peak expression between P5 and P7, the postnatal period when cardiomyocytes exit the cell cycle. Rbl1 (p107) and Rbl2 (p130) had reciprocal changes, consistent with changes in expression of these genes in proliferating and quiescent cells.14, 15, 32, 33 Components of the DREAM complex, including Lin52, Lin9, Lin37, and Lin54, were differentially expressed in postnatal hearts.
Since the MuvB/DREAM complex is a multimeric protein structure, the ablation of one or a few complex proteins is likely detrimental to cell growth. Therefore, we focused our attention on upstream regulators of the MuvB/DREAM complex since this was predicted to influence the subsequent expression of a large number of cell cycle genes. Dual-specificity tyrosine phosphorylation-regulated kinase 1a (DYRK1a) is one regulator of MuvB/DREAM and implicated in cell cycling.9, 10 For example, the inhibition of DYRK1a promotes cell cycling and proliferation in mouse and human pancreatic islet cells19-24, and DYRK1a has been implicated in cardiomyocyte growth through cyclin D125. Given the importance of E2F signals in cardiomyocyte growth and the potential of DYRK1a as a regulator of genes expressed at the G1/S and G2/M boundaries in cycling cells, we investigated the effects of pharmacologic inhibition and genetic ablation of DYRK1a inhibition on cardiomyocyte cycling and cardiac function after I/R MI.
Harmine, an inhibitor of DYRK1a, improved left ventricular function and increased cardiomyocyte cycling after IR MI.
Recently, we described a new transgenic mouse, αMHC (alpha myosin heavy chain)-MerDreMer-Ki67p-RoxedCre::Rox-Lox-tdTomato-eGFP (denoted αDKRC::RLTG) that restricted Cre expression to cycling adult cardiomyocytes and uniquely integrated spatial and temporal adult cardiomyocyte cycling events.1 αDKRC::RLTG mice provide the unique opportunity to quantify the effects of inhibitors on endogenous cardiomyocyte cycling after MI, in vivo. Therefore, we investigated if harmine, an ATP-competitive inhibitor of DYRK1a shown to promote pancreatic islet cell proliferation23, 34, induced endogenous cardiomyocyte cycling and changes in myocardial function after I/R MI. Harmine was administered after I/R MI via osmotic minipump at a dose of 10 mg/kg per animal over two-week interval, comparable to the dose of harmine reported in in vivo investigations. Adult αDKRC::RLTG mice underwent pre-treatment with tamoxifen (1 mg/day intraperitoneal (IP) injection x 5 days for 2 cycles) to activate the αDKRC reporter. Two weeks after recovery from tamoxifen, we performed baseline echocardiography and 60 minutes of LAD ligation-mediated ischemia followed by reperfusion (Figure 1A). After reperfusion, but before the conclusion of surgeries, mice underwent the insertion of subcutaneous osmotic minipumps containing harmine (10 mg/kg/ml) or saline. Echocardiography was repeated two weeks after surgery, and eGFP+ cardiomyocytes and infarct sizes were quantified using protocols previously described. Harmine improved LVEF (Figure 1 B and C) and LV end-systolic volume (Figure 1 D and E), but not LV end-diastolic volume (Figure 1 F and G) compared to saline controls.
Figure 1. Harmine improved left ventricular function after ischemia-reperfusion myocardial infarction.
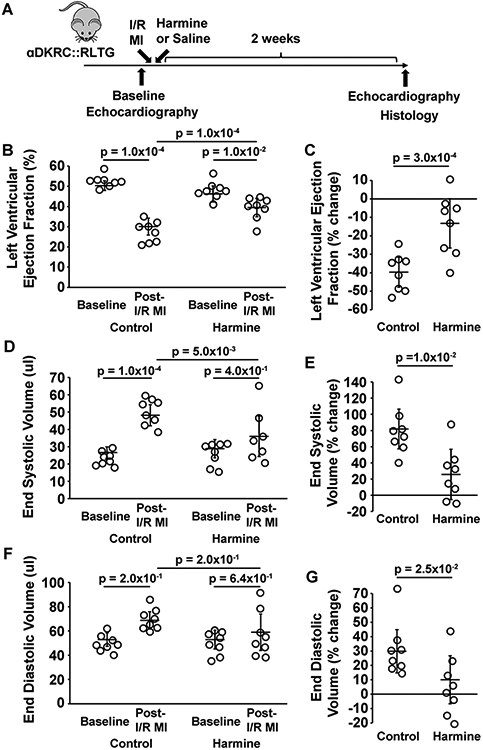
(A) Outline of the experimental protocol testing the effects of harmine and Saline (Control) in adult αDKRC::RLTG mice after 60 minutes of LAD ligation-mediated ischemia and reperfusion (I/R) myocardial infarction (MI). (B and C) The absolute values (%) (B) and percent changes (C) in left ventricular (LV) ejection fraction (EF) of mice treated with harmine minipumps (10 mg/kg/day) or saline after 60 minutes of I/R MI. Bars are the mean ± SD at baseline and two weeks after I/R MI in panel B. Open circles are measurements for individual animals. p = 1.0x10−2 for baseline vs. I/R MI in the harmine treatment group, p = 1.0x10−4 for baseline vs. I/R MI in the Saline treatment group, and p = 1.0x10−4 for harmine vs. saline treatment in the I/R MI groups. p-values were calculated by two-way ANOVA with Bonferroni’s multiple comparison test using six comparisons. Values are means ± SD. N = 8 animals per group. In panel C, p = 3.0x10−4 for the percent change in LVEF between harmine and saline treatments. p-values calculated by student’s t-test. (D and E) The absolute values (ul) (D) and percent changes (E) in LV end-systolic volume (ESV) of mice treated with harmine minipumps (10 mg/kg/day) or saline after 60 minutes of I/R MI. Bars are the mean ± SD at baseline and two weeks after I/R MI in panel D. Open circles are measurements for individual animals. p = 4.0x10−2 for LV ESV at baseline vs. I/R MI in the harmine treatment group, p = 1.0x10−4 for LV ESV baseline vs. I/R MI in the saline treatment group and p=5.0x10−3 for LV ESV harmine vs. saline treatment in the I/R MI groups. p-values were calculated by two-way ANOVA with Bonferroni’s multiple comparison test using six comparisons. Values are means ± SD. N = 8 animals per group. In panel E, p = 1.0x10−2 for the percent change in LV ESV between harmine and saline treatments. p-values calculated by student’s t-test. (F and G) The absolute values (%) (F) and percent changes (G) in LV end-diastolic volumes (EDV) of mice treated with Harmine minipumps (10 mg/kg/day) or saline after 60 minutes of I/R MI. Bars are the mean ± SD at baseline and two weeks after I/R MI in panel F. Open circles are measurements for individual animals. p = 1.4x10−1 for LV EDV at baseline vs. I/R MI in the Harmine treatment group, p=2.0x10−4 for LV EDV at baseline vs. I/R MI in the saline treatment group, and p=9.0x10−2 for LV EDV harmine vs. saline treatment in the I/R MI groups. p-values were calculated by two-way ANOVA with Bonferroni’s multiple comparison test using six comparisons. Values are means ± SD. N = 8 animals per group. In panel G, p = 2.5x10−2 for the percent change in LV ESV between harmine and saline treatments. P-values calculated by student’s t-test.
The infarcts of mice treated with harmine were similar to saline controls (Figure 2 A and B). Two weeks after I/R MI, eGFP+ cardiomyocytes representing adult cardiomyocytes that re-entered the cell cycle were readily identified in hearts of αDKRC::RLTG (Figure 2 C-E. Supplemental Figure 3). GFP+ cardiomyocytes increased in number after I/R MI in harmine-treated animals compared to saline controls (24.3 ± 2.6 versus 10.8 ± 1.5 cardiomyocytes per ten-micron section, harmine versus saline, p=1.0x10−3) with the majority of eGFP+ cardiomyocytes localized in the ischemic and border zones (Figure 2 F). Additionally, LVEF, EDD and ESD were linearly related to the number of eGFP+ cardiomyocytes (Figure 2 G-I), suggesting that an increase in cycling cardiomyocytes corresponded to improved LV function after MI despite similar infarct sizes (Figure 2 A). Thus, the pharmacological inhibition of DYRK1a increased the number of cycled cardiomyocytes and improved LV systolic function after I/R MI.
Figure 2. Harmine enhanced cardiomyocyte cycling after ischemia-reperfusion myocardial infraction.

(A) Infarct sizes for each group corresponding to Figure 1. Open circles are values for individual animals. Bars are group means ± SD. p = 4.5x10−1 by student t-test. (B) Representative Masson-Trichrome stained short-axis sections of hearts of mice treated with harmine or saline. Scale bars denote 1 mm. (C) Representative short-axis immunohistochemical (IHC) images of a heart from αDKRC::RLTG mouse treated with saline 2 weeks after I/R MI. White circles denote eGFP+ cardiomyocytes. Scale bar denotes 1 mm. (D) Representative short-axis IHC images of a heart from αDKRC::RLTG mouse treated with harmine 2 weeks after I/R MI. White circles denote eGFP+ cardiomyocytes. Scale bar denotes 1 mm. (E) Representative Immunohistochemical image of eGFP+ cardiomyocytes in the border zone of a harmine-treated animal. Arrows denote eGFP+ cardiomyocytes. Scale bar denotes 500 microns. (F) Quantification of eGFP+ cardiomyocytes per section from I/R MI hearts treated with Saline or Harmine and the infarct, border, and remote zones of I/R MI hearts. Bars are group means ± SD. Open circles are individual animals. N = 8 animals in the saline group and N = 7 animals in the harmine group. p = 1.0x10−3 for total eGFP+ cardiomyocytes of saline vs. harmine-treated animals after I/R MI. p = 6.0x10−2 for eGFP+ cardiomyocytes in the ischemia area of saline vs. harmine-treated animals after I/R MI. p = 2.0x10−2 for eGFP+ cardiomyocytes in the border area of saline vs. harmine-treated animals after I/R MI. p = 5.0x10−2 for eGFP+ cardiomyocytes in the remote area of saline vs. harmine-treated animals after I/R MI. p-values calculated by Mann-Whitney U test. (G-I) Relationship between echocardiographic parameters and total number of cycled cardiomyocytes per animal. Animals treated with saline (gray circles) and harmine (open circles) are shown for LVEF, r2 = 0.61 (G), EDV, r2 = 0.39 (H), and ESD, r2 = 0.52 (I). The Pearson coefficient (r2) calculated by linear regression.
The ablation of cycling cardiomyocytes attenuated the beneficial effects of Harmine on LV function after MI.
Next, we used αDKRC::RLTG/DTA mice to ablate cardiomyocytes that re-entered the cell cycle and investigated if the beneficial effects of Harmine on LV function after I/R MI were in part attributable to cardiomyocyte cycling. Male and female αDKRC::RLTG/DTA and littermate controls (+::RLTG/DTA) were pretreated with Tamoxifen and underwent I/R MI followed by the immediate implantation of an osmotic pump to deliver Harmine (10 mg/kg/ml) (Figure 3A). Serial echocardiography showed that αDKRC::RLTG/DTA treated with harmine had progressively decreased LVEF (Figure 3B and C) and increased end systolic volume (Figure 3D and E), with no differences in end diastolic volume (Figure 3F and G) compared to littermate controls (+::RLTG/DTA) treated with harmine. The changes in LVEF were similar to prior observations using αDKRC::DTA mice (Supplemental Figure 4).1 The changes in LV parameters suggested that the effects of Harmine on LV remodeling after I/R MI required cycling cardiomyocytes.
Figure 3. The ablation of cycling cardiomyocytes attenuated the beneficial effects of Harmine on LV systolic function after I/R MI.
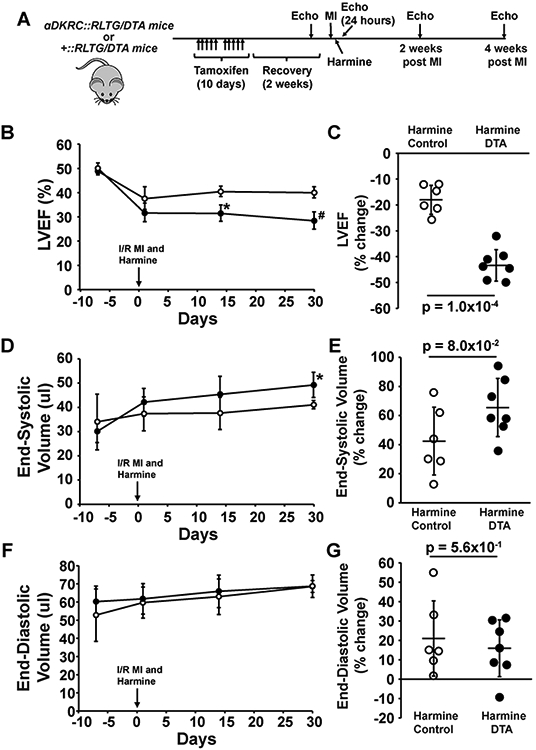
(A) Protocol using αDKRC (cardiomyocyte-specific αMHC-MerDreMer-Ki67p-RoxedCre)::RLTG (Rox-Lox-tdTomato-eGFP)/Rosa26-DTA (Diphtheria Toxin) (αDKRC::RLTG/DTA) and +::RLTG/DTA littermate controls treated with Tamoxifen (1 mg/kg IP [intraperitoneal] daily×5 d×2 cycles) followed by recovery period before undergoing 60 min of left anterior descending (LAD) artery-ligation ischemia and reperfusion (I/R myocardial infarction [MI]) and implantation of an osmotic minipump to deliver Harmine (10 mg/kg/day). Serial echocardiography was performed before and after MI. N=7 in the αDKRC::RLTG/DTA and N = 6 in the +::RLTG/DTA group. (B and C) The absolute values (%) (B) and percent changes (C) in left ventricular (LV) ejection fraction (EF) of αDKRC::RLTG/DTA and +::RLTG/DTA mice treated with harmine minipumps (10 mg/kg/day) after 60 minutes of I/R MI. Open and closed circles are means ± SDs of +::RLTG/DTA and αDKRC::RLTG/DTA mice, respectively. * denotes p = 1.0x10−4 and # p = 1.0x10−4 for the indicated time. p-values were calculated by student’s t-test. N=7 in the αDKRC::RLTG/DTA and N = 6 in the +::RLTG/DTA group. In panel C, p = 1.0x10−4 for the percent change in LVEF between +::RLTG/DTA and αDKRC::RLTG/DTA mice. p-values calculated by student’s t-test. (D and E) The absolute values (ul) (D) and percent changes (E) in LV end-systolic volume (ESV) of αDKRC::RLTG/DTA and +::RLTG/DTA mice treated with harmine minipumps (10 mg/kg/day) after 60 minutes of I/R MI. Open and closed circles are means ± SDs of +::RLTG/DTA and αDKRC::RLTG/DTA mice, respectively. * p = 4.0x10−3 for the indicated time. p-values were calculated by student’s t-test. N=7 in the αDKRC::RLTG/DTA and N = 6 in the +::RLTG/DTA group. In panel D, p = 8.0x10−2 for the percent change in LVEF between +::RLTG/DTA and αDKRC::RLTG/DTA mice. P-values calculated by student’s t-test. (F and G) The absolute values (%) (F) and percent changes (G) in LV end-diastolic volumes (EDV) of αDKRC::RLTG/DTA and +::RLTG/DTA mice treated with harmine minipumps (10 mg/kg/day) after 60 minutes of I/R MI. Open and closed circles are means ± SDs of +::RLTG/DTA and αDKRC::RLTG/DTA mice, respectively. N=7 in the αDKRC::RLTG/DTA and N = 6 in the +::RLTG/DTA group. In panel D, p = 5.6x10−1 for the percent change in LVEF between +::RLTG/DTA and αDKRC::RLTG/DTA mice. P-values calculated by student’s t-test.
The cardiomyocyte-specific ablation of DYRK1a caused baseline hyperplasia.
Pharmacologic studies are clinically more practical but inherently less specific in terms of the cell populations affected by drug treatment and potential off-target effects. Therefore, we investigated the effects of cardiomyocyte-specific genetic ablation of DYRK1a on cardiac function after I/R MI. We generated conditional knockouts, αMHC-Cre::Fucci2aR::DYRK1aflox/flox mice, using DYRK1aflox/flox mice created and validated by Thompson et al.35 in the context of a Cre-inducible second-generation Fluorescence Ubiquitin Cell Cycle Indicator, Fucci2aR, to label cardiomyocyte nuclei (Figure 4A)36. αMHC-Cre::Fucci2aR mice served as controls. Of note, the αMHC-Cre::Fucci2aR::DYRK1aflox/flox and αMHC-Cre::Fucci2aR mice were bred >9 generations into a C57BL/6J genetic background.
Figure 4. The cardiomyocyte-specific ablation of DYRK1a caused baseline hyperplasia.

(A) Overview of the αMHC-Cre::Fucci2aR (second-generation Fluorescence Ubiquitin Cell Cycle Indicator)::DYRK1aflox/flox (denoted DYRK1a k/o) mouse. (B) Quantitative polymerase chain reaction (qPCR) of DYRK1a, DYRK1b, and DYRK2 expression of total RNA extracted from cardiomyocytes isolated from adult αMHC-Cre::Fucci2aR::DYRK1aflox/flox (DYRK1a k/o) and αMHC-Cre::Fucci2aR (Control) mice. The probe set for DYRK1a corresponded to Exons 5 and 6, the exons flanked by LoxP sites (Panel A). p-values calculated by Mann-Whitney test. Values are mean ± SD. Open circles are values for individual animals. N = 3 for αMHC-Cre::Fucci2aR::DYRK1aflox/flox (DYRK1a k/o) and N = 5 for αMHC-Cre::Fucci2aR animals (Control). (C) Number of cardiomyocytes per 40 x field for αMHC-Cre::Fucci2aR::DYRK1aflox/flox and αMHC-Cre::Fucci2aR animals. Values are means ± SD. Open circles are individual animals. N = 5 animals per group, n = 8 fields per animal. p=1.7x10−2 for number of cardiomyocytes per 40 x field of αMHC-Cre::Fucci2aR::DYRK1aflox/flox and αMHC-Cre::Fucci2aR animals. p-value by student’s t-test. (D) Violin plots of the cross-sectional areas (CSAs) (um2) of cardiomyocytes of αMHC-Cre::Fucci2aR::DYRK1aflox/flox and αMHC-Cre::Fucci2aR animals. N = 3 animals per group, n = 317 cells for αMHC-Cre::Fucci2aR and n = 257 cells for αMHC-Cre::Fucci2aR::DYRK1aflox/flox. p=1.0x10−1 for cardiomyocyte CSA of αMHC-Cre::Fucci2aR::DYRK1aflox/flox and αMHC-Cre::Fucci2aR animals. P-value by Mann-Whitney test. The mean values for each animal was used to account for non-independence. (E and F) Representative immunofluorescence images of hearts of αMHC-Cre::Fucci2aR (E) and αMHC-Cre::Fucci2aR::DYRK1aflox/flox (F) mice. Red – mCherry labeling cardiomyocyte nuclei. Gray – wheat germ agglutinin. Scale bars = 25 microns. (G) LV end-diastolic volume (EDV) (ul) of αMHC-Cre::Fucci2aR::DYRK1aflox/flox and αMHC-Cre::Fucci2aR mice. Values are mean ± SD. Open circles are measurements for individual animals. N = 17 animals for αMHC-Cre::Fucci2aR and N = 13 animals for αMHC-Cre::Fucci2aR::DYRK1aflox/flox. p=8.0x10−2 for LV EDV of αMHC-Cre::Fucci2aR::DYRK1aflox/flox and αMHC-Cre::Fucci2aR animals. P-value by student’s t-test. (H) LV end-systolic volume (ESV) (ul) of αMHC-Cre::Fucci2aR::DYRK1aflox/flox and αMHC-Cre::Fucci2aR mice. Values are mean ± SD. Open circles are measurements for individual animals. N = 17 animals for αMHC-Cre::Fucci2aR and N = 13 animals for αMHC-Cre::Fucci2aR::DYRK1aflox/flox. p=1.1x10−1 for LV ESV of αMHC-Cre::Fucci2aR::DYRK1aflox/flox and αMHC-Cre::Fucci2aR animals. P-value by student’s t-test. (I) LV Ejection Fraction (EF) (%) of αMHC-Cre::Fucci2aR::DYRK1aflox/flox and αMHC-Cre::Fucci2aR mice. Values are mean ± SD. Open circles are measurements for individual animals. N = 17 animals for αMHC-Cre::Fucci2aR and N = 13 animals for αMHC-Cre::Fucci2aR::DYRK1aflox/flox. p=8.8x10−2 for LVEF of αMHC-Cre::Fucci2aR::DYRK1aflox/flox and αMHC-Cre::Fucci2aR animals. p-value by student’s t-test. (J and K) Gravimetric measurements of hearts from αMHC-Cre::Fucci2aR::DYRK1aflox/flox and αMHC-Cre::Fucci2aR mice expressed as heart weight (mg) to tibia length (mm) (J) and heart weight (mg) to body weight (g) (K). Values are mean ± SD. Open circles are measurements for individual animals. N = 8 animals per group. p-value by student’s t-test.
αMHC-Cre::Fucci2aR::DYRK1aflox/flox mice were viable and fertile. Adult cardiomyocytes isolated from αMHC-Cre::Fucci2aR::DYRK1aflox/flox mice had reduced expression of DYRK1a without compensatory changes in the expression of other DYRKs, compared to cardiomyocytes of αMHC-Cre::Fucci2aR controls (Figure 4B). At baseline, the hearts of αMHC-Cre::Fucci2aR::DYRK1aflox/flox mice had smaller cardiomyocyte cross-sectional areas and increased cardiomyocytes per area, consistent with hyperplasia (Figure 4C-F). Baseline left ventricular end-diastolic volume, end-systolic volume, and ejection fractions were also similar between αMHC-Cre::Fucci2aR::DYRK1aflox/flox and αMHC-Cre::Fucci2aR mice (Figure 4G-I). Heart weights were similar between αMHC-Cre::Fucci2aR::DYRK1aflox/flox and αMHC-Cre::Fucci2aR mice, but with a trend towards slightly increased heart size in the DYRK1a k/o mice (Figure 4 J and K). The results suggested that the ablation of DYRK1a caused cardiomyocyte hyperplasia.
Next, we performed 60-minute I/R MI and sham surgeries using αMHC-Cre::Fucci2aR::DYRK1aflox/flox and αMHC-Cre::Fucci2aR mice (Figure 5A). Four weeks after I/R MI, αMHC-Cre::Fucci2aR::DYRK1aflox/flox mice had improved LVEF (Figure 5 B and C) and LV end-systolic volumes (Figure 5 D and E) compared to αMHC-Cre::Fucci2aR. LV end-diastolic volumes were similar between αMHC-Cre::Fucci2aR::DYRK1aflox/flox and αMHC-Cre::Fucci2aR mice (Figure 5 F and G). Next, we analyzed the infarct characteristic of αMHC-Cre::Fucci2aR::DYRK1aflox/flox and αMHC-Cre::Fucci2aR mice (Figure 6 A-E). The infarct sizes were quantified as the percentage infarct area (PIA) (Figure 6A), the percent infarct circumference of the short-axis (PIC) (Figure 6B), the ratio of the PIA to PIC (Figure 6C) at the level of the mid-ventricle that corresponds to infarct thickness in the infarcted area, and the ratio of PIC to PIA (Figure 6D) that corresponds to thickness of non-infarcted myocardium in the infarcted area. Thus, αMHC-Cre::Fucci2aR::DYRK1aflox/flox has similar PIAs compared to control αMHC-Cre::Fucci2aR mice. However, αMHC-Cre::Fucci2aR::DYRK1aflox/flox had smaller PICs compared to control αMHC-Cre::Fucci2aR mice, suggesting that the cardiomyocyte-specific ablation of DYRK1a influenced post-I/R MI LV remodeling to produce thicker scars with smaller circumferences.
Figure 5. The cardiomyocyte-specific ablation of DYRK1a improved left ventricular function after I/R MI.
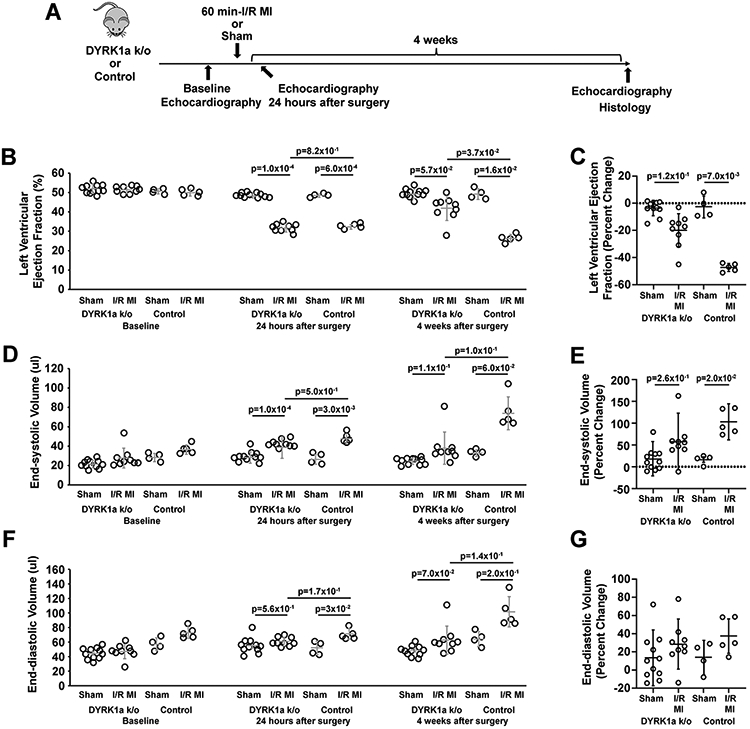
(A) Outline of the experimental protocol testing the effects of the cardiomyocyte-specific ablation of DYRK1a in αMHC-Cre::Fucci2aR::DYRK1aflox/flox (DYRK1a k/o) and αMHC-Cre::Fucci2aR (Control) mice after 60 minutes of LAD ligation-mediated ischemia and reperfusion (I/R) myocardial infarction (MI). (B and C) The absolute values (%) (B) and percent changes (C) in left ventricular (LV) ejection fractions (EFs) of αMHC-Cre::Fucci2aR::DYRK1aflox/flox and αMHC-Cre::Fucci2aR mice after 60 minutes of I/R MI. Bars are the means +/− SDs at baseline, 24 hours after surgery, and four weeks after I/R MI in panel B. Open circles are measurements for individual animals. p-values were calculated by a two-way ANOVA with Tukey’s multiple comparison test. In panel C, p=7.0x10-3 for the percent change in LVEF between sham and I/R MI at four weeks compared to baseline in the αMHC-Cre::Fucci2aR group. p=1.2x10-2 for the percent change in LVEF between sham and I/R MI in the αMHC-Cre::Fucci2aR::DYRK1aflox/flox group. p-values were calculated by a Kruskal-Wallis test with Dunn’s multiple comparison test. Values are means ± SD. N = 11 animals in the αMHC-Cre::Fucci2aR::DYRK1aflox/flox sham group, N = 9 animals in the αMHC-Cre::Fucci2aR::DYRK1aflox/flox I/R MI group, N = 4 animals in the αMHC-Cre::Fucci2aR sham group, N = 5 animals in the αMHC-Cre::Fucci2aR I/R MI group. (D and E) The absolute values (%) (D) and percent changes (E) in left ventricular (LV) end-systolic volume (ESD) (ul) of αMHC-Cre::Fucci2aR::DYRK1aflox/flox and αMHC-Cre::Fucci2aR mice after 60 minutes of I/R MI. Bars are the means +/− SDs at baseline, 24 hours after surgery, and four weeks after I/R MI in panel B. Open circles are measurements for individual animals. p-values were calculated by a two-way ANOVA with Tukey’s multiple comparison test. In panel E, the percent changes in LV ESD at four weeks compared to baseline are shown. Values are means ± SD. p=0.02 for the percent change in LV EDD at four weeks compared to baseline between sham and I/R MI in the αMHC-Cre::Fucci2aR group. p=2.6x10−1 for the percent change in LV EDD between sham and I/R MI in the αMHC-Cre::Fucci2aR::DYRK1aflox/flox group. p-values were calculated by a Kruskal-Wallis test with Dunn’s multiple comparison test. N = 11 animals in the αMHC-Cre::Fucci2aR::DYRK1aflox/flox sham group, N = 9 animals in the αMHC-Cre::Fucci2aR::DYRK1aflox/flox I/R MI group, N = 4 animals in the αMHC-Cre::Fucci2aR sham group, N = 5 animals in the αMHC-Cre::Fucci2aR I/R MI group. (F and G) The absolute values (%) (F) and percent changes (G) in left ventricular (LV) end-diastolic volume (EDD) (ul) of αMHC-Cre::Fucci2aR::DYRK1aflox/flox and αMHC-Cre::Fucci2aR mice after 60 minutes of I/R MI. Bars are the means +/− SDs at baseline, 24 hours after surgery, and four weeks after I/R MI in panel B. Open circles are measurements for individual animals. p-values were calculated by two-way ANOVA with Tukey’s multiple comparison test. N = 11 animals in the αMHC-Cre::Fucci2aR::DYRK1aflox/flox sham group, N = 9 animals in the αMHC-Cre::Fucci2aR::DYRK1aflox/flox I/R MI group, N = 4 animals in the αMHC-Cre::Fucci2aR sham group, N = 5 animals in the αMHC-Cre::Fucci2aR I/R MI group.
Figure 6. The cardiomyocyte-specific ablation of DYRK1a altered infarct parameters.

(A) Percent infarct area for αMHC-Cre::Fucci2aR (Control) and αMHC-Cre::Fucci2aR::DYRK1aflox/flox (DYRK1a k/o) animals corresponding to Figure 5. Open circles are values for individual animals. Bars are group means ± SD. p=3.9x10−1 for the percent infarct area between αMHC-Cre::Fucci2aR and αMHC-Cre::Fucci2aR::DYRK1aflox/flox animals. p-values were calculated by student’s t-test. N = 8 animals per group. (B) Percent circumference of the infarct for αMHC-Cre::Fucci2aR and αMHC-Cre::Fucci2aR::DYRK1aflox/flox animals corresponding to Figure 5. Open circles are values for individual animals. Bars are group means ± SD. p=5.2x10−2 for the percent circumference of the infarct between αMHC-Cre::Fucci2aR and αMHC-Cre::Fucci2aR::DYRK1aflox/flox animals. p-value were calculated by student’s t-test. N = 8 animals per group. (C) Ratio of the percent infarct area to percent circumference of the infarct for αMHC-Cre::Fucci2aR and αMHC-Cre::Fucci2aR::DYRK1aflox/flox animals corresponding to Figure 5. Open circles are values for individual animals. Bars are group means ± SD. P=4.6x10−2 for percent infarct area to percent circumference of the infarct between αMHC-Cre::Fucci2aR and αMHC-Cre::Fucci2aR::DYRK1aflox/flox animals. p-value were calculated by student’s t-test. N = 8 animals per group. (D) Ratio of the percent circumference of the infarct to percent infarct area for αMHC-Cre::Fucci2aR and αMHC-Cre::Fucci2aR::DYRK1aflox/flox animals corresponding to Figure 5. Open circles are values for individual animals. Bars are group means ± SD. P=4.6x10−2 for percent infarct area to percent circumference of the infarct between αMHC-Cre::Fucci2aR and αMHC-Cre::Fucci2aR::DYRK1aflox/flox animals. p-value were calculated by student’s t-test. N = 8 animals per group. (E) Representative short axis base to apex sections of hearts from αMHC-Cre::Fucci2aR and αMHC-Cre::Fucci2aR::DYRK1aflox/flox animals four weeks after I/R MI. Hearts were stained with Masson trichrome. Infarcted areas are shown. (F) Representations of the infarct area and circumference of αMHC-Cre::Fucci2aR and αMHC-Cre::Fucci2aR::DYRK1aflox/flox animals four weeks after I/R MI.
Since the knockout of DYRK1a could potentially alter the degree of injury occurring with ischemia, we performed (2,3,5-Triphenyltetrazolium chloride (TTC) staining (Supplemental Figure 5A-C). The Area-at-Risk (AAR) and Area-of-Necrosis (AON) 24 hours after MI were similar between αMHC-Cre::Fucci2aR::DYRK1aflox/flox and αMHC-Cre::Fucci2aR mice (Supplemental Figure 5B and C). The results suggested that the cardiomyocyte-specific genetic ablation of DYRK1a improved LV function after I/R MI without a change in the area at risk. In light of the hyperplasia observed in DYRK1a k/o mouse hearts, the changes in LV remodeling after I/R MI could be a result of a larger initial number of cardiac myocytes contributing to the remodeling response after injury. The change in infarct parameters may also contribute to improved LV function after I/R MI in DYRK1a k/o mouse hearts.
The ablation of DYRK1a promotes the expression of cell cycle genes after I/R MI.
Next, we investigated gene expression of cardiomyocytes to identify potential mechanisms responsible for the of DYRK1a ablation. Using Langendorff methods to retro-aortically perfuse collagenase ex vivo, we isolated cardiomyocytes from αMHC-Cre::Fucci2aR::DYRK1aflox/flox and control αMHC-Cre::Fucci2aR mice after I/R MI or Sham surgeries seven days after I/R MI, corresponding to a time when cardiomyocyte cycling occurs after injury, and performed RNAseq (Figure 7 A). Principal component analysis to reduce dimensionality of the datasets identified the clustering of samples into four distinct groups with 44% and 32% of the variance in gene expression attributed to MI compared to sham, and αMHC-Cre::Fucci2aR::DYRK1aflox/flox to αMHC-Cre::Fucci2aR, respectively (Figure 7 B). Volcano plot of differential gene expression of sham control and sham DYRK1a k/o hearts showed an increase in expression of DREAM target genes and cardiac transcription factors (Nkx 2.5, Txb5, Gata4, and Gata6) in DYRK1a k/o cardiomyocyte preparations (Figure 7C). Volcano plots of cardiomyocyte gene expression comparing Sham and I/R MI for αMHC-Cre::Fucci2aR (Figure 8 A and B) and αMHC-Cre::Fucci2aR::DYRK1aflox/flox identified that more genes had increased than decreased expression after I/R MI (Figure 8C). To refine the analyses of genes that had increased expression between αMHC-Cre::Fucci2aR and αMHC-Cre::Fucci2aR::DYRK1aflox/flox after MI, we generated a Venn diagram of genes that had an adjusted p-value of less than 0.05 and a four-fold or greater expression (Figure 6 E). One hundred and seventy-six genes and one hundred seventy-nine genes increased expression after I/R MI compared to Sham in cardiomyocytes isolated from αMHC-Cre::Fucci2aR and αMHC-Cre::Fucci2aR::DYRK1aflox/flox hearts, respectively (Supplemental Table 2). Eighty-nine genes were uniquely expressed in αMHC-Cre::Fucci2aR::DYRK1aflox/flox cardiomyocytes after I/R MI. Eighty-six genes were uniquely expressed in αMHC-Cre::Fucci2aR cardiomyocytes after I/R MI. Ninety genes had shared expression in αMHC-Cre::Fucci2aR::DYRK1aflox/flox and αMHC-Cre::Fucci2aR cardiomyocytes. Using the Reactome database, we identified enrichment of pathways associated with mitotic cell cycle and cell cycle checkpoints in genes uniquely expressed in αMHC-Cre::Fucci2aR::DYRK1aflox/flox cardiomyocytes after I/R MI (Figure 8D and Supplemental Figure 6). These genes included Anillin (Anln), Aurora Kinase B (Aurkb), Cyclin B1 (Ccnb1), Cyclin B2 (Ccnb2), Cyclin Dependent Kinase 1 (Cdk1), Cell Division Cycle 20 (Cdc20), Forkhead Box M1 (FOXM1), Kinetochore Localized Astrin (SPAG5) Binding Protein (KNSTRN), MYB Proto-Oncogene Like 2 (MYBL2), Polo Like Kinase 1 (PLK1), and Protein Regulator Of Cytokinesis 1 (PCR1), among others. Interleukin-10 and RUNX signaling were expressed in αMHC-Cre::Fucci2aR cardiomyocytes after I/R MI. Extracellular matrix formation genes has shared expression in αMHC-Cre::Fucci2aR::DYRK1aflox/flox and αMHC-Cre::Fucci2aR cardiomyocytes (Figure 8C). MsigDB gene set enrichment of the eighty-nine genes uniquely expressed in αMHC-Cre::Fucci2aR::DYRK1aflox/flox cardiomyocytes after I/R MI identified G2/M checkpoint and E2F targets as the top hits, similar to the findings identified in the initial analyses of Cluster 1 from the gene expression of embryonic and postnatal hearts (Supplemental figure 1B). Thirty-five of the eighty-nine genes (39%) were targets of the DREAM complex (Supplemental figure 7). Collectively, the results suggest that after I/R MI, the loss of DYRK1a in cardiomyocytes was associated with the expression of cell cycle genes.
Figure 7. The ablation of DYRK1a promotes the expression of DREAM target genes.

(A) Overview of the experimental protocol of I/R MI and sham surgeries in αMHC-Cre::Fucci2aR::DYRK1aflox/flox (DYRK1a k/o) and αMHC-Cre::Fucci2aR (Control) followed by cardiomyocyte isolation, and RNA and sequencing. (B) Principal components analysis (PCA) plot of the four groups: αMHC-Cre::Fucci2aR::DYRK1aflox/flox (DYRK1a k/o) Sham, αMHC-Cre::Fucci2aR::DYRK1aflox/flox (DYRK1a k/o) I/R MI, αMHC-Cre::Fucci2aR (Control) Sham, and αMHC-Cre::Fucci2aR (Control) I/R MI. (C) Volcano plot of gene expression of αMHC-Cre::Fucci2aR and αMHC-Cre::Fucci2aR::DYRK1aflox/flox cardiomyocytes after Sham surgeries. Green dots denote genes predicted to be targets of the DREAM complex, Red dots denote genes encoding sarcomere proteins, and Blue dots denote cardiac transcription factors.
Figure 8. The ablation of DYRK1a promotes the expression of cell cycle genes after I/R MI.

(A) Volcano plot of gene expression of αMHC-Cre::Fucci2aR cardiomyocytes after I/R MI or Sham surgeries. Red dots represent genes with a more than a 2-fold increase in expression and an adjusted p-value of a more than 0.05. Blue dots represent genes with less than a 2-fold increase in expression and an adjusted p-value of more than 0.05. Black dots represent genes with an adjusted p-value of a more than 0.05. Vertical dashed lines denote four-fold levels of gene expression. The horizontal dashed line denotes an adjusted P-value of 0.05. (B) Volcano plot of gene expression of αMHC-Cre::Fucci2aR::DYRK1aflox/flox cardiomyocytes after Sham or I/R MI. Red dots represent genes with more than 2-fold increase in expression and an adjusted p-value of more than 0.05. Blue dots represent genes with less than 2-fold increase in expression and an adjusted p-value of more than 0.05. Black dots represent genes with an adjusted p-value more than 0.05. Vertical dashed lines denote four-fold levels of gene expression. The horizontal dashed line denotes an adjusted P-value of 0.05. (C) Venn diagram of genes from panels C and D that had more than 2-fold increase in expression and an adjusted p-value of more than 0.05. Eighty-nine genes were uniquely expressed in αMHC-Cre::Fucci2aR::DYRK1aflox/flox cardiomyocytes after I/R MI. Eighty-six genes were uniquely expressed in αMHC-Cre::Fucci2aR cardiomyocytes after I/R MI. Ninety genes had shared expression in αMHC-Cre::Fucci2aR::DYRK1aflox/flox and αMHC-Cre::Fucci2aR cardiomyocytes. (D) Pathway enrichment or the genes in Panel E based on Reactome database. Genes unique to the αMHC-Cre::Fucci2aR::DYRK1aflox/flox cardiomyocytes after I/R MI were enriched in mitotic cell cycle and cell cycle checkpoints. Genes that were unique to the αMHC-Cre::Fucci2aR cardiomyocytes after I/R MI were enriched in Interleukin-10 and RUNX signaling. Genes in common between αMHC-Cre::Fucci2aR::DYRK1aflox/flox and αMHC-Cre::Fucci2aR cardiomyocytes after I/R MI were enriched in collagen formation and extracellular matrix organization. The tables list the Reactome pathway name, the number of genes in panel E found in the Reactome pathway, the number of total genes in the Reactome pathway, the ratio of genes found to total genes in the pathway, the p-value associated with over-represented genes within the Reactome pathway, and the false discovery rate (FDR).
Discussion:
Cardiomyocyte cycling, like other cell types, involves canonical signaling proteins and redundant brakes to prevent aberrant growth. The removal of one or even several brakes is likely insufficient to generate enough functional cardiomyocytes to confer substantial overall benefit to cardiac function after injury, assuming the benefits are restricted to contractile function. Therefore, we sought to identify candidate upstream signals that potentially regulate genes expressed at the G1/S and G2/M boundaries to induce adult cardiomyocyte cycling after myocardial injury. We postulated that investigating gene expression during the normal transition when cardiomyocytes exit the cell cycle during neonatal heart growth would identify potential nodal signals for manipulation in adult cardiomyocytes.
Publically available gene expression data of embryonic, neonatal, and adult cardiomyocytes and hearts identified enrichments in pathways of E2Fs and G2/M checkpoints, and temporal changes in the expression of genes containing proximal E2F or CHR DNA binding sites, known to be controlled by the DREAM complex. Additionally, genes encoding components of the DREAM complex and E2Fs had peak expression in postnatal hearts during the window when cardiomyocytes exit the cell cycle. These observations suggested that the MuvB-DREAM complex represented a potential mechanism to induce cardiomyocyte cycling, and the inhibition of DYRK1a by pharmacologic inhibition and genetic ablation may enhance cardiac function in part through enhanced cardiomyocyte cycling.
The administration of harmine at the time of reperfusion after MI improved systolic function and increased cardiomyocyte cycling in αDKRC::RLTG transgenic mice, without changes in infarct sizes. The harmine-induced cycled cardiomyocytes were predominantly in the ischemic and border zones. The observation that an increase in cycling cardiomyocytes corresponded to improved LV function after MI despite similar infarct sizes is particularly interesting in light of our recent findings that the ablation of cycling cardiomyocytes worsens cardiac function after MI. The number of cycled cells in response to harmine was sparse, and the fact that harmine did not induce uniform cardiomyocyte cycling suggests that harmine may activate cycling of the subpopulation of cardiomyocytes prone to reenter the cell cycle, particularly in the ischemic and border zones. Importantly, the I/R MI model did not produce a transmural infarct as seen with permanent LAD ligation MI models, and therefore cardiomyocytes were present in the infarcted area. Moreover, the I/R MI model more closely recapitulates the clinical scenario of patients presenting with ST-elevation MI who undergo percutaneous revascularization within 90 minutes, a performance quality metric in the treatment of MI.37 Although harmine was started after reperfusion and continued for two weeks after MI, shorter dosing intervals of DYRK1a inhibitors after MI may provide similar beneficial results without the potential untoward sequelae of prolonged administration of pro-proliferative compounds that can theoretically induce cancer.
Cycled cardiomyocytes and cardiac function (LVEF) after I/R MI were linearly related. Previously, we observed that the ablation of cycled cardiomyocytes worsened cardiac function in animals that had similar infarct sizes after I/R MI.1 Moreover, the ablation of cycling cardiomyocytes attenuated the beneficial effects of Harmine on LV function after I/R MI. One interpretation is that cycling cardiomyocytes express paracrine factors that contribute to myocardial function after injury, consistent with the observation that cycled cardiomyocytes are predominantly individual cells and not paired or clustered as would be expected of proliferation. Future investigations will characterize the gene expression profiles of cycling and non-cycling cardiomyocytes in our αDKRC::RLTG transgenic mice to address this possible mechanism.
Since harmine likely has effects on multiple cell types and organs, we used a transgenic approach to examine the effects of the cardiomyocyte-specific ablation of DYRK1a after MI. At baseline, the ablation of DYRK1a in αMHC-Cre::Fucci2aR::DYRK1aflox/flox caused cardiomyocyte hyperplasia; however, the systolic function was similar to controls. Similarly, the ablation of Salvador or activated YAP expression caused cardiomyocyte hypertrophy with normal baseline systolic function.2, 5, 6 The genetic ablation of DYRK1a improved LV systolic function after MI and reduced infarct sizes. αMHC-Cre::Fucci2aR::DYRK1aflox/flox mice had AAR and AON similar to αMHC-Cre::Fucci2aR controls, suggesting that DYRK1a ablation did not reduce the susceptibility to I/R injury. We then quantified infarct characteristics four weeks after I/R MI as the percentage infarct area (PIA), the percent infarct circumference of the short-axis (PIC), the ratio of the PIA to PIC at the level of the mid-ventricle that corresponds to infarct thickness in the infarcted area, and the ratio of PIC to PIA that corresponds to thickness of non-infarcted myocardium in the infarcted area. These measurements provide an estimation of scar thickness and size. The cardiomyocyte-specific knockout of DYRK1a produced thicker scars with smaller circumferences and these changes in remodeling may improve LV systolic function after injury.38 Future investigations using chronic ischemic cardiomyopathy models will potentially address the long-term impact of DYRK1a ablation in post-MI LV remodeling.
To explore potential mechanisms responsible for the beneficial effects of DYRK1a ablation in cardiomyocytes, we performed RNAseq on cardiomyocytes isolated from αMHC-Cre::Fucci2aR::DYRK1aflox/flox and αMHC-Cre::Fucci2aR mice seven days after I/R MI or sham surgeries. Interestingly, the loss of DYRK1a was associated with the upregulation of cell cycle genes and downregulation of genes encoding contractile proteins after MI, compared to controls. One interpretation of the results is that the loss of DYRK1a promotes genetic programs associated with dedifferentiation and proliferation, consistent with observations of proliferating caridomyocytes.39-43 The gene signatures upregulated in cardiomyocytes of αMHC-Cre::Fucci2aR::DYRK1aflox/flox after I/R MI were similar differentially expressed in embryonic and neonatal cardiomyocytes described by Walsh et al.26 (Clusters 1-2 in Supplemental Figure 2A and B, Supplemental Figure 8). Conversely, the gene signatures upregulated in cardiomyocytes of αMHC-Cre::Fucci2aR control mice after I/R MI were similar to those differentially expressed in adult cardiomyocytes compared to embryonic and neonates (Clusters 5-6 in Supplemental Figure 2A and C). Together, the results demonstrate the utility of gene expression data of developing hearts to identify candidate signaling nodes and potentially enhance adult cardiomyocyte cycling. DYRK1a inhibition represents one potential nodal regulator and the genetic ablation of DYRK1a in cardiomyocytes after myocardial injury recapitulates aspects of gene expression of developing myocardium.
Despite the unanticipated findings, there were limitations. First, the ablation of DYRK1a in αMHC-Cre::Fucci2aR::DYRK1aflox/flox mice occurred during cardiac development and, therefore, the adult phenotypes reflected the absence of DYRK1a during normal embryonic and post-natal heart growth. The hearts of αMHC-Cre::Fucci2aR::DYRK1aflox/flox had cardiomyocyte hyperplasia without changes in baseline echocardiographic parameters. Additionally, the Fucci2aR signal of αMHC-Cre::Fucci2aR::DYRK1aflox/flox was mCherry+/mVenus-, similar to αMHC-Cre::Fucci2aR control mice, indicative of non-cycling cardiomyocytes. Future investigations using tamoxifen-induced deletion of DYRK1a in cardiomyocytes harboring a mosaic analysis with double markers (MADM) reporter of cell proliferation44, 45 (cTNTMerCreMer::MADM::DYRK1afl/fl) will address the potential of post-developmental ablation of DYRK1a to enhance cardiomyocyte cycling in the context of a reporter of mitosis. Importantly, cells that reenter the cell cycle can proliferate to generate new daughter cells or undergo endoreplication to multinucleate and increase polyploidy, processes that have important implications.46 Future investigations will quantify polyploidy in the context of DYRK1a.
Second, Harmine, an ATP-competitive Inhibitor for DYRK1a, could have off-target effects.34, 47, 48 Additional kinases may contribute to the observed effects in vivo despite the observations that cardiomyocyte-specific ablation of DYRK1a recapitulates harmine’s effects after MI. Investigation of structurally-diverse DYRK1a inhibitors will be informative, particularly to the development of therapeutics to enhance cardiomyocyte cycling in the setting of myocardial infarction and percutaneous catheter-based reperfusion strategies.
Third, DYRK1a has many substrates in addition to the MuvB-DREAM complex that likely contribute to the overall effects observed with cell cycling.9-11, 49 For example, DYRKs phosphorylate NFATs50, Cyclins25, 35, p27/p2151, and Lin52 of the DREAM complex9. A genome-wide Drosophila RNAi screen identified DYRK-family kinases as regulators of NFAT.52 DYRK1a phosphorylates mammalian NFATc1, interfering with degradation and leading to NFAT1c stability.53 In cultured rat cardiomyocytes, adenovirus-mediated overexpression of DYRK1A antagonized calcineurin-mediated nuclear NFAT translocation and phenylephrine-induced hypertrophic growth response.50 However, a mouse model of post-developmental DYRK1a expression, myocyte diameter, heart weight/body weight ratio, and echocardiographic measurements showed that myocardial expression of DYRK1A failed to reduce hypertrophy induced after transverse aortic banding.50 The results suggested insufficient long-term inhibition of NFAT by DYRK1a in the mouse model or additional pathways influenced by DYRK1a activity in vivo.
DYRK1a regulates the cardiomyocyte cell cycle through Cyclin D-dependent Rb/E2f-signalling.25, 35 Transgenic mice with cardiac-specific overexpression of DYRK1a had increased phosphorylation of Ccnd2 with subsequent proteasomal degradation and hypo-phosphorylated Rb1, suppression of Rb/E2F-signalling, and reduced expression of E2F-target genes, which ultimately results in impaired cell cycle progression and lead to dilated cardiomyopathy.25
DYRK1a is located on chromosome 21 and has a naturally occurring gene dosage increase in trisomy 21.54 Dyrk1a mediates a dose-dependent increase in the duration and variability of the G1 phase and has been shown to shift cells within a p21-cyclin D1 signaling map to control the decision to enter the cell cycle.51 Additionally, DYRK1A is recruited preferentially to promoters of genes actively transcribed by RNA polymerase II, functionally associated with translation, RNA processing, and the cell cycle.55 Further investigations will address other DYRK1a-mediated signals in cycling cardiomyocytes.
Fourth, the RNAseq experiments have the limitation of contamination by non-cardiomyocytes. All samples were prepared similarly and gene expression attributed to non-cardiomyocytes should be filtered from the differential gene expression analyses. However, non-cardiomyocyte cell populations could differ between αMHC-Cre::Fucci2aR::DYRK1aflox/flox and αMHC-Cre::Fucci2aR hearts after MI. For example, genes associated with Interleukin-10 signals are enriched in αMHC-Cre::Fucci2aR hearts after MI. The improvement in cardiac function observed in αMHC-Cre::Fucci2aR::DYRK1aflox/flox could represent more favorable inflammatory responses mediated by changes in signals emanating from cardiomyocytes lacking DYRK1a.35 Future investigations of the temporal changes in inflammatory infiltrate after MI in hearts where cardiomyocytes lacking DYRK1a will be informative. Nonetheless, cardiomyocytes lacking DYRK1a the enrichment of genes associated with cycling after MI and recapitulated gene signatures associated with post-natal heart growth. Identifying the exact pathways mediating the effects of DYRK1a ablation on cycling programs in cardiomyocytes will improve our knowledge of targets to manipulate to improve myocardial function after MI.
Collectively, our findings suggest the inhibition of DYRK1a is a potential therapeutic target to enhance cardiomyocyte cycling and improve heart function after MI. Moreover, the MuvB-DREAM complex may serve as a regulator of cardiomyocyte cycling and merits further investigation.
Supplementary Material
Novelty and Significance.
What is Known?
Adult mammalian cardiomyocytes have limited cycling after an injury.
Signaling nodes active in developing hearts and downregulated in adult hearts have been implicated as potential pathways controlling cardiomyocyte cycling.
Dual-specificity Tyrosine Phosphorylation-regulated Kinase 1a (DYRK1a) regulates cell growth in several tissues.
What New Information Does This Article Contribute?
Our findings show that the pharmacological inhibition of DYRK1a after ischemia-reperfusion myocardial infarction enhances adult cardiomyocyte cycling and improves left ventricular function.
The cardiomyocyte-specific ablation of DYRK1a causes hyperplasia and improved cardiac function after myocardial infarction.
Cardiomyocytes that lack DYRK1a increase the expression of cell cycle-related genes after myocardial infarction.
Summary.
Enhancing the adult mammalian cardiomyocytes cycling after an injury is an attractive strategy to improve cardiac function after myocardial infarction. One approach to identifying pathways that enhance cardiomyocyte cycling is to compare gene expression during normal development when cells exit the cycle. Therefore, we analyzed gene expression of embryonic, neonatal, and adult hearts and hypothesized that DYRK1a might serve as one potential signal controlling cardiomyocyte cycling. Using our αDKRC [cardiomyocyte-specific αMHC-MerDreMer-Ki67p-RoxedCre] transgenic reporter mouse, we observed a chemical inhibitor of DYRK1a, Harmine, increased cardiomyocyte cycling, and improved ventricular function after ischemia-reperfusion myocardial infarction. The ablation of cycling cardiomyocytes attenuated the beneficial effects of harmine. We also observed that the cardiomyocyte-specific ablation of DYRK1a caused hyperplasia, improved cardiac function, and increased the expression of cell cycle-related genes after I/R MI. The results suggest that enhancing cardiomyocyte cycling, possibly through the inhibition of DYRK1a, may serve as a strategy to improve cardiac function after MI.
Sources of Funding:
Research reported in this publication was supported by the University of Virginia Center of Excellence in Cardiovascular Genetics and the National Heart, Lung, And Blood Institute of the National Institutes of Health under Award Number R01HL158718 (M.J.W.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.”
Nonstandard Abbreviations and Acronyms:
- DYRK1a
Dual-specificity Tyrosine Phosphorylation-regulated Kinase 1a
- EDV
End-diastolic Volume
- EF
Ejection fraction
- eGFP
enhanced Green Fluorescent Protein
- ESV
End-systolic Volume
- I/R
Ischemia-Reperfusion
- MI
Myocardial Infarction
Footnotes
Study Approval: All animal care and surgeries were in accordance with UVA ACUC Policy on Rodent Surgery and Perioperative Care under ACUC-approved animal protocol (UVA ACUC Wolf Laboratory protocol No. 4080).
Disclosures:
None.
References:
- 1.Bradley LA, Young A, Li H, Billcheck HO and Wolf MJ. Loss of Endogenously Cycling Adult Cardiomyocytes Worsens Myocardial Function. Circ Res. 2021;128:155–168. [DOI] [PubMed] [Google Scholar]
- 2.Heallen T, Zhang M, Wang J, Bonilla-Claudio M, Klysik E, Johnson RL and Martin JF. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science. 2011;332:458–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leach JP, Heallen T, Zhang M, Rahmani M, Morikawa Y, Hill MC, Segura A, Willerson JT and Martin JF. Hippo pathway deficiency reverses systolic heart failure after infarction. Nature. 2017;550:260–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mohamed TM, Stone NR, Berry EC, Radzinsky E, Huang Y, Pratt K, Ang YS, Yu P, Wang H, Tang S, Magnitsky S, Ding S, Ivey KN and Srivastava D. Chemical Enhancement of In Vitro and In Vivo Direct Cardiac Reprogramming. Circulation. 2017;135:978–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xin M, Kim Y, Sutherland LB, Murakami M, Qi X, McAnally J, Porrello ER, Mahmoud AI, Tan W, Shelton JM, Richardson JA, Sadek HA, Bassel-Duby R and Olson EN. Hippo pathway effector Yap promotes cardiac regeneration. Proc Natl Acad Sci U S A. 2013;110:13839–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.von Gise A, Lin Z, Schlegelmilch K, Honor LB, Pan GM, Buck JN, Ma Q, Ishiwata T, Zhou B, Camargo FD and Pu WT. YAP1, the nuclear target of Hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proc Natl Acad Sci U S A. 2012;109:2394–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engeland K Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 2018;25:114–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fischer M, Grossmann P, Padi M and DeCaprio JA. Integration of TP53, DREAM, MMB-FOXM1 and RB-E2F target gene analyses identifies cell cycle gene regulatory networks. Nucleic Acids Res. 2016;44:6070–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Litovchick L, Florens LA, Swanson SK, Washburn MP and DeCaprio JA. DYRK1A protein kinase promotes quiescence and senescence through DREAM complex assembly. Genes Dev. 2011;25:801–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sadasivam S and DeCaprio JA. The DREAM complex: master coordinator of cell cycle-dependent gene expression. Nat Rev Cancer. 2013;13:585–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sadasivam S, Duan S and DeCaprio JA. The MuvB complex sequentially recruits B-Myb and FoxM1 to promote mitotic gene expression. Genes Dev. 2012;26:474–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Angelis E, Zhao P, Zhang R, Goldhaber JI and Maclellan WR. The role of E2F-1 and downstream target genes in mediating ischemia/reperfusion injury in vivo. J Mol Cell Cardiol. 2011;51:919–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kirshenbaum LA, Abdellatif M, Chakraborty S and Schneider MD. Human E2F-1 reactivates cell cycle progression in ventricular myocytes and represses cardiac gene transcription. Dev Biol. 1996;179:402–11. [DOI] [PubMed] [Google Scholar]
- 14.MacLellan WR, Garcia A, Oh H, Frenkel P, Jordan MC, Roos KP and Schneider MD. Overlapping roles of pocket proteins in the myocardium are unmasked by germ line deletion of p130 plus heart-specific deletion of Rb. Mol Cell Biol. 2005;25:2486–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.MacLellan WR, Xiao G, Abdellatif M and Schneider MD. A novel Rb- and p300-binding protein inhibits transactivation by MyoD. Mol Cell Biol. 2000;20:8903–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park DS, Tompkins RO, Liu F, Zhang J, Phoon CK, Zavadil J and Fishman GI. Pocket proteins critically regulate cell cycle exit of the trabecular myocardium and the ventricular conduction system. Biol Open. 2013;2:968–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sdek P, Zhao P, Wang Y, Huang CJ, Ko CY, Butler PC, Weiss JN and Maclellan WR. Rb and p130 control cell cycle gene silencing to maintain the postmitotic phenotype in cardiac myocytes. J Cell Biol. 2011;194:407–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Litovchick L, Sadasivam S, Florens L, Zhu X, Swanson SK, Velmurugan S, Chen R, Washburn MP, Liu XS and DeCaprio JA. Evolutionarily conserved multisubunit RBL2/p130 and E2F4 protein complex represses human cell cycle-dependent genes in quiescence. Mol Cell. 2007;26:539–51. [DOI] [PubMed] [Google Scholar]
- 19.Ackeifi C, Swartz E, Kumar K, Liu H, Chalada S, Karakose E, Scott DK, Garcia-Ocana A, Sanchez R, DeVita RJ, Stewart AF and Wang P. Pharmacologic and genetic approaches define human pancreatic beta cell mitogenic targets of DYRK1A inhibitors. JCI Insight. 2020;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dirice E, Walpita D, Vetere A, Meier BC, Kahraman S, Hu J, Dancik V, Burns SM, Gilbert TJ, Olson DE, Clemons PA, Kulkarni RN and Wagner BK. Inhibition of DYRK1A Stimulates Human beta-Cell Proliferation. Diabetes. 2016;65:1660–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rachdi L, Kariyawasam D, Aiello V, Herault Y, Janel N, Delabar JM, Polak M and Scharfmann R. Dyrk1A induces pancreatic beta cell mass expansion and improves glucose tolerance. Cell Cycle. 2014;13:2221–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rachdi L, Kariyawasam D, Guez F, Aiello V, Arbones ML, Janel N, Delabar JM, Polak M and Scharfmann R. Dyrk1a haploinsufficiency induces diabetes in mice through decreased pancreatic beta cell mass. Diabetologia. 2014;57:960–9. [DOI] [PubMed] [Google Scholar]
- 23.Wang P, Alvarez-Perez JC, Felsenfeld DP, Liu H, Sivendran S, Bender A, Kumar A, Sanchez R, Scott DK, Garcia-Ocana A and Stewart AF. A high-throughput chemical screen reveals that harmine-mediated inhibition of DYRK1A increases human pancreatic beta cell replication. Nat Med. 2015;21:383–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang P, Karakose E, Liu H, Swartz E, Ackeifi C, Zlatanic V, Wilson J, Gonzalez BJ, Bender A, Takane KK, Ye L, Harb G, Pagliuca F, Homann D, Egli D, Argmann C, Scott DK, Garcia-Ocana A and Stewart AF. Combined Inhibition of DYRK1A, SMAD, and Trithorax Pathways Synergizes to Induce Robust Replication in Adult Human Beta Cells. Cell Metab. 2019;29:638–652 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hille S, Dierck F, Kuhl C, Sosna J, Adam-Klages S, Adam D, Lullmann-Rauch R, Frey N and Kuhn C. Dyrk1a regulates the cardiomyocyte cell cycle via D-cyclin-dependent Rb/E2f-signalling. Cardiovasc Res. 2016;110:381–94. [DOI] [PubMed] [Google Scholar]
- 26.Walsh S, Ponten A, Fleischmann BK and Jovinge S. Cardiomyocyte cell cycle control and growth estimation in vivo--an analysis based on cardiomyocyte nuclei. Cardiovasc Res. 2010;86:365–73. [DOI] [PubMed] [Google Scholar]
- 27.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP and Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES and Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Talman V, Teppo J, Poho P, Movahedi P, Vaikkinen A, Karhu ST, Trost K, Suvitaival T, Heikkonen J, Pahikkala T, Kotiaho T, Kostiainen R, Varjosalo M and Ruskoaho H. Molecular Atlas of Postnatal Mouse Heart Development. J Am Heart Assoc. 2018;7:e010378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alkass K, Panula J, Westman M, Wu TD, Guerquin-Kern JL and Bergmann O. No Evidence for Cardiomyocyte Number Expansion in Preadolescent Mice. Cell. 2015;163:1026–36. [DOI] [PubMed] [Google Scholar]
- 31.Quaife-Ryan GA, Sim CB, Ziemann M, Kaspi A, Rafehi H, Ramialison M, El-Osta A, Hudson JE and Porrello ER. Multicellular Transcriptional Analysis of Mammalian Heart Regeneration. Circulation. 2017;136:1123–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cui M, Wang Z, Bassel-Duby R and Olson EN. Genetic and epigenetic regulation of cardiomyocytes in development, regeneration and disease. Development. 2018;145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Henley SA and Dick FA. The retinoblastoma family of proteins and their regulatory functions in the mammalian cell division cycle. Cell Div. 2012;7:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumar K, Wang P, Sanchez R, Swartz EA, Stewart AF and DeVita RJ. Development of Kinase-Selective, Harmine-Based DYRK1A Inhibitors that Induce Pancreatic Human beta-Cell Proliferation. J Med Chem. 2018;61:7687–7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thompson BJ, Bhansali R, Diebold L, Cook DE, Stolzenburg L, Casagrande AS, Besson T, Leblond B, Desire L, Malinge S and Crispino JD. DYRK1A controls the transition from proliferation to quiescence during lymphoid development by destabilizing Cyclin D3. J Exp Med. 2015;212:953–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mort RL, Ford MJ, Sakaue-Sawano A, Lindstrom NO, Casadio A, Douglas AT, Keighren MA, Hohenstein P, Miyawaki A and Jackson IJ. Fucci2a: a bicistronic cell cycle reporter that allows Cre mediated tissue specific expression in mice. Cell Cycle. 2014;13:2681–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O'Gara PT, Kushner FG, Ascheim DD, Casey DE Jr., Chung MK, de Lemos JA, Ettinger SM, Fang JC, Fesmire FM, Franklin BA, Granger CB, Krumholz HM, Linderbaum JA, Morrow DA, Newby LK, Ornato JP, Ou N, Radford MJ, Tamis-Holland JE, Tommaso CL, Tracy CM, Woo YJ and Zhao DX. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2013;61:485–510. [DOI] [PubMed] [Google Scholar]
- 38.Richardson WJ, Clarke SA, Quinn TA and Holmes JW. Physiological Implications of Myocardial Scar Structure. Compr Physiol. 2015;5:1877–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.D'Uva G, Aharonov A, Lauriola M, Kain D, Yahalom-Ronen Y, Carvalho S, Weisinger K, Bassat E, Rajchman D, Yifa O, Lysenko M, Konfino T, Hegesh J, Brenner O, Neeman M, Yarden Y, Leor J, Sarig R, Harvey RP and Tzahor E. ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation. Nat Cell Biol. 2015;17:627–38. [DOI] [PubMed] [Google Scholar]
- 40.Jopling C, Sleep E, Raya M, Marti M, Raya A and Izpisua Belmonte JC. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature. 2010;464:606–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Senyo SE, Lee RT and Kuhn B. Cardiac regeneration based on mechanisms of cardiomyocyte proliferation and differentiation. Stem Cell Res. 2014;13:532–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang WE, Li L, Xia X, Fu W, Liao Q, Lan C, Yang D, Chen H, Yue R, Zeng C, Zhou L, Zhou B, Duan DD, Chen X, Houser SR and Zeng C. Dedifferentiation, Proliferation, and Redifferentiation of Adult Mammalian Cardiomyocytes After Ischemic Injury. Circulation. 2017;136:834–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y, Li TS, Lee ST, Wawrowsky KA, Cheng K, Galang G, Malliaras K, Abraham MR, Wang C and Marban E. Dedifferentiation and proliferation of mammalian cardiomyocytes. PLoS One. 2010;5:e12559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tasic B, Miyamichi K, Hippenmeyer S, Dani VS, Zeng H, Joo W, Zong H, Chen-Tsai Y and Luo L. Extensions of MADM (mosaic analysis with double markers) in mice. PLoS One. 2012;7:e33332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zong H, Espinosa JS, Su HH, Muzumdar MD and Luo L. Mosaic analysis with double markers in mice. Cell. 2005;121:479–92. [DOI] [PubMed] [Google Scholar]
- 46.Derks W and Bergmann O. Polyploidy in Cardiomyocytes: Roadblock to Heart Regeneration? Circ Res. 2020;126:552–565. [DOI] [PubMed] [Google Scholar]
- 47.Frost D, Meechoovet B, Wang T, Gately S, Giorgetti M, Shcherbakova I and Dunckley T. beta-carboline compounds, including harmine, inhibit DYRK1A and tau phosphorylation at multiple Alzheimer's disease-related sites. PLoS One. 2011;6:e19264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Patel K, Gadewar M, Tripathi R, Prasad SK and Patel DK. A review on medicinal importance, pharmacological activity and bioanalytical aspects of beta-carboline alkaloid "Harmine". Asian Pac J Trop Biomed. 2012;2:660–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boni J, Rubio-Perez C, Lopez-Bigas N, Fillat C and de la Luna S. The DYRK Family of Kinases in Cancer: Molecular Functions and Therapeutic Opportunities. Cancers (Basel). 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grebe C, Klingebiel TM, Grau SP, Toischer K, Didie M, Jacobshagen C, Dullin C, Hasenfuss G and Seidler T. Enhanced expression of DYRK1A in cardiomyocytes inhibits acute NFAT activation but does not prevent hypertrophy in vivo. Cardiovasc Res. 2011;90:521–8. [DOI] [PubMed] [Google Scholar]
- 51.Chen JY, Lin JR, Tsai FC and Meyer T. Dosage of Dyrk1a shifts cells within a p21-cyclin D1 signaling map to control the decision to enter the cell cycle. Mol Cell. 2013;52:87–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gwack Y, Sharma S, Nardone J, Tanasa B, Iuga A, Srikanth S, Okamura H, Bolton D, Feske S, Hogan PG and Rao A. A genome-wide Drosophila RNAi screen identifies DYRK-family kinases as regulators of NFAT. Nature. 2006;441:646–50. [DOI] [PubMed] [Google Scholar]
- 53.Liu H, Wang K, Chen S, Sun Q, Zhang Y, Chen L and Sun X. NFATc1 phosphorylation by DYRK1A increases its protein stability. PLoS One. 2017;12:e0172985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Duchon A and Herault Y. DYRK1A, a Dosage-Sensitive Gene Involved in Neurodevelopmental Disorders, Is a Target for Drug Development in Down Syndrome. Front Behav Neurosci. 2016;10:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Di Vona C, Bezdan D, Islam AB, Salichs E, Lopez-Bigas N, Ossowski S and de la Luna S. Chromatin-wide profiling of DYRK1A reveals a role as a gene-specific RNA polymerase II CTD kinase. Mol Cell. 2015;57:506–20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
See Supplemental Materials for detailed methods.