

Abstract
The enzyme phenylethanolamine N-methyltransferase (PNMT, EC 2.1.1.28) catalyzes the final step in the biosynthesis of epinephrine and is a potential drug target, primarily for the control of hypertension. Unfortunately, many potent PNMT inhibitors also possess significant affinity for the a2-adrenoceptor, which complicates interpretation of their pharmacology. A bisubstrate analogue approach offers the potential for development of highly selective inhibitors of PNMT. This paper documents the design, synthesis, and evaluation of such analogues, several of which were found to possess hPNMT inhibitory potency < 5 nM versus AdoMet. Site-directed mutagenesis studies were consistent with bisubstrate binding. Two of these compounds (19 and 29) were co-crystallized with hPNMT and the resulting structures revealed both compounds bound as predicted, simultaneously occupying both substrate binding domains. This bisubstrate inhibitor approach has resulted in one of the most potent (20) and selective (versus the a2-adrenoceptor) inhibitors of hPNMT yet reported.
Graphical Abstract
INTRODUCTION
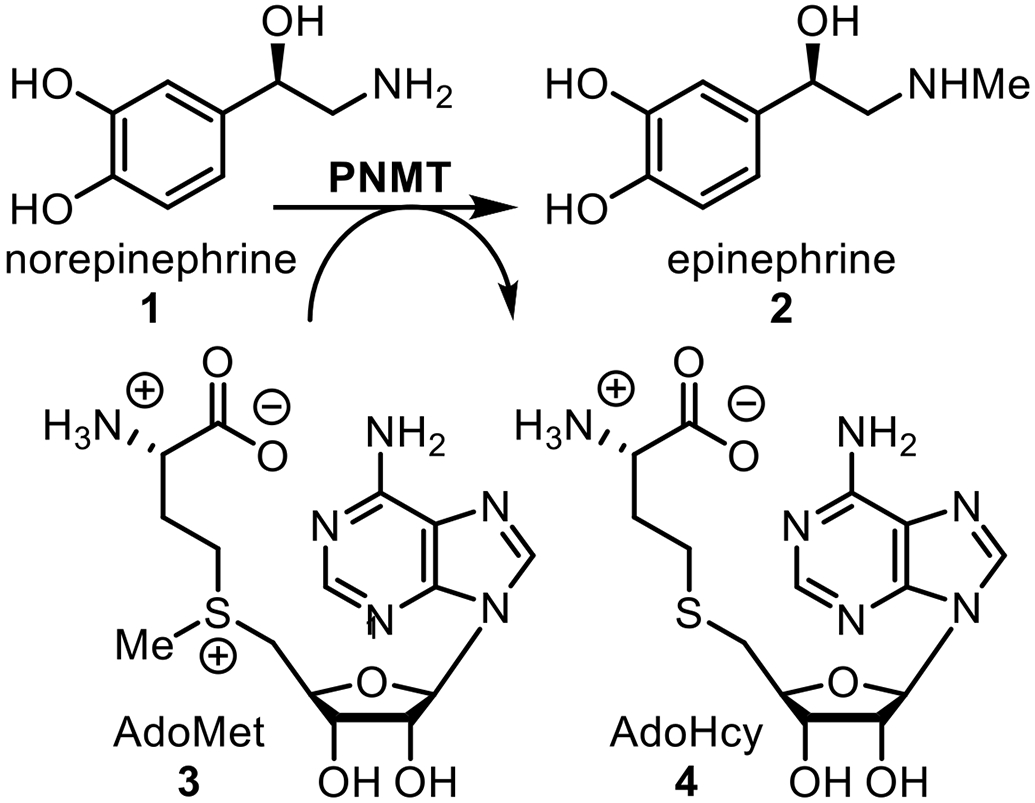
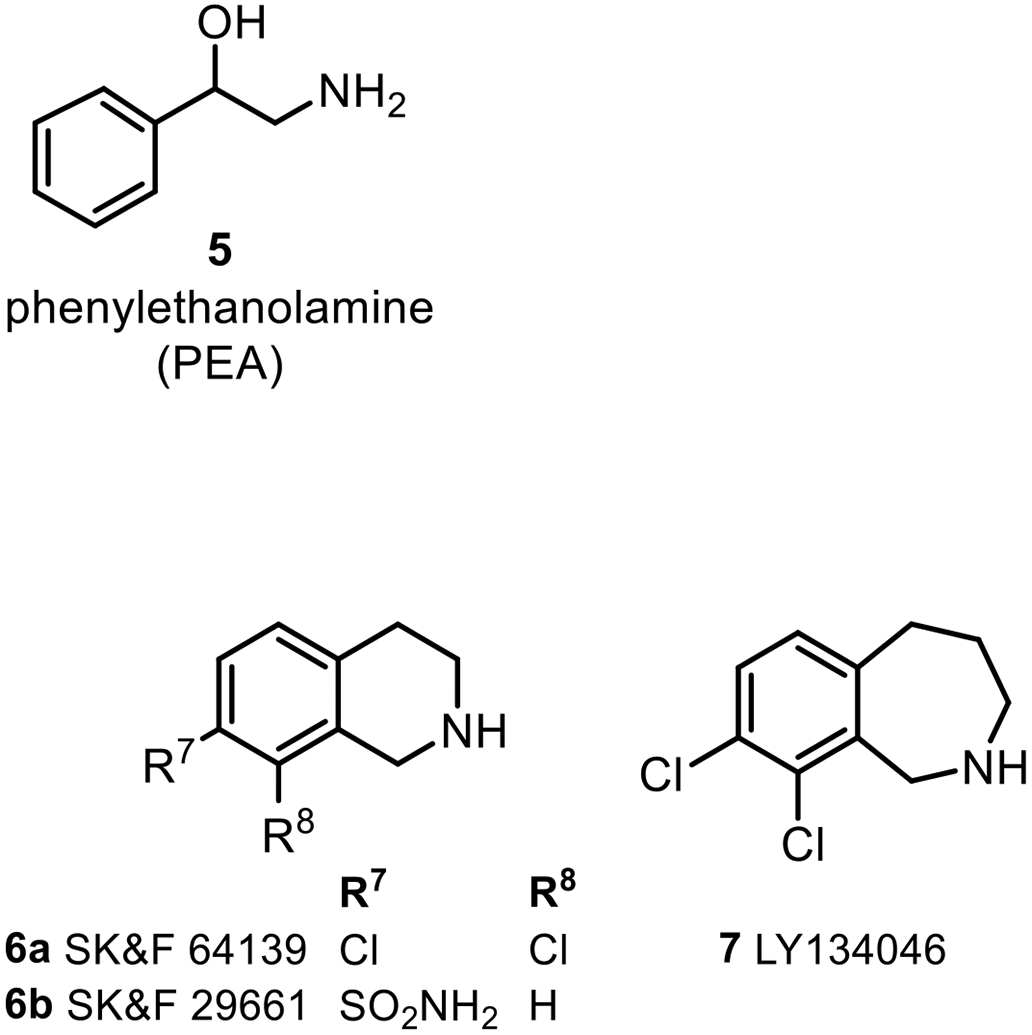
The enzyme phenylethanolamine N-methyltransferase (PNMT, EC 2.1.1.28) is found primarily in the adrenal glands and is also located in the brain, heart, lung, and retina.1-3 Therein, it catalyzes the biosynthetic transformation (Figure 1) of norepinephrine (1) to epinephrine (2).4 The enzyme is not selective for 1 as its methyl-acceptor substrate and Axelrod5 identified a variety of phenylethanolamines, such as 5 (phenylethanolamine, PEA), that could also function as methyl-acceptor substrates and, as a result, the enzyme was named phenylethanolamine N-methyltransferase.
Figure 1.
Biosynthesis of Epinephrine
As a bisubstrate enzyme, PNMT catalyzes the transfer of a methyl group from S-adenosyl-l-methionine (AdoMet, 3) to the amino group of 1. Inhibitors of this reaction fall into two general classes. One class consists of compounds that compete with the binding of the methyl-acceptor substrate (such as 1) and a number of these have been investigated, including hydrophobic ethanolamine alternate substrate inhibitors5-7 along with benzylamines and phenethylamines.8-10 Compounds based on structural modification of conformationally-constrained benzylamines, such as in the tetrahydroisoquinoline11-15 (e.g., 6a, 6b) or tetrahydrobenzazepine16,17 (e.g., 7) ring systems (Figure 2) have been extensively studied for their ability to inhibit PNMT in the central nervous system as a new approach for the control of hypertension.18-21 However, most of these compounds show significant affinity for the α2-adrenoceptor, which complicates the interpretation of their pharmacology.22
Figure 2.
Some Inhibitors of PNMT.
We have investigated numerous compounds in this class in order to improve inhibitory potency at the PNMT active site,23,24 CNS penetration,25,26 and selectivity versus the α2-adrenoceptor.27
A second class of inhibitors of PNMT consists of compounds that compete with the binding of the methyl-donor (3), which include the co-substrate product (4) and its analogues.28-32 However, inhibitors of this class show significantly diminished inhibitory potency at PNMT, compared to those that compete with the binding of 1 and also show poor selectivity for PNMT, as they inhibit other methyltransferases that utilize 3 as a co-substrate.33
A bisubstrate inhibitor, based on a combination of elements of these two inhibitor classes, could be used to achieve selectivity over competing binding domains. Previous studies indicated that the transfer of the methyl group from 3 to 1 occurs via a SN2 process, suggesting that the methyl transfer reaction proceeds through a transition state that includes both substrates bound in the active site of the enzyme.34-36 This suggested that it would be possible to develop a bisubstrate analogue inhibitor of PNMT containing structural features that could simultaneously inhibit the binding of both substrates. A bisubstrate inhibitor should possess high binding affinity for the enzyme due to an entropic advantage of reduced molecularity and an additive binding contribution from each of the molecules it mimics.37 Also, the combination of the analogues of both substrates in the same molecule should make it less likely to be recognized by other pharmacologically relevant sites and, for example, provide selectivity for PNMT versus the α2-adrenoceptor. Thus, the goal of this project is to design, synthesize, and evaluate potent and selective bisubstrate inhibitors of PNMT to explore the potential of the bisubstrate inhibitor approach for future inhibitor design.
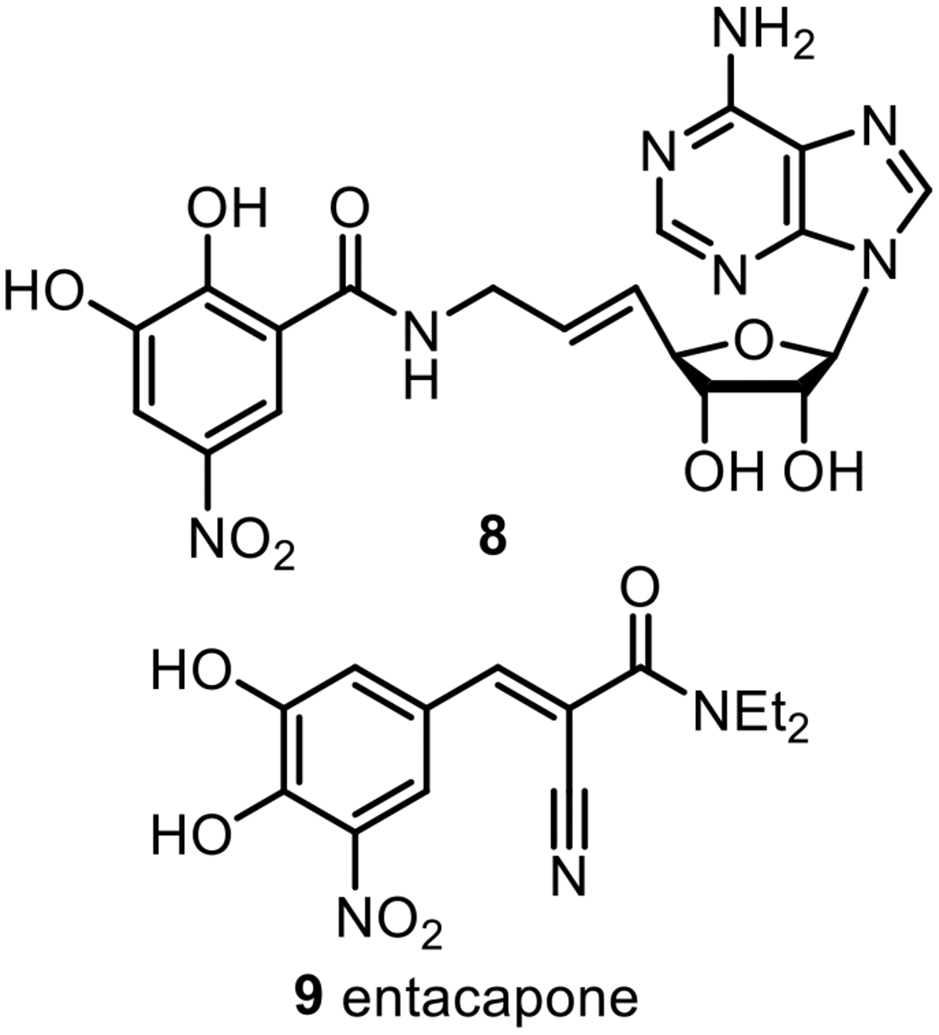
The bisubstrate analogue approach has been applied to develop inhibitors for other AdoMet-dependent methyltransferases.38-47 Most relevant among these is the 2.6 Å resolution crystal structure of a bisubstrate inhibitor (8, Figure 3) complexed with catechol O-methyltransferase (COMT) that showed good inhibitory potency (IC50 = 9 nM),48 as compared to entacapone (9), a clinically used inhibitor of COMT (IC50 = 14 nM).49
Figure 3.
Inhibitors of COMT.
Bisubstrate Inhibitor Design.
From our earlier work with the Martin group at the University of Queensland, we reported the X-ray crystal structures of a number of ligands bound to the active site of the enzyme,50-55 including the hPNMT-3+11 ternary complex.56 (See Figure 4 for THIQ structures.)
Figure 4.
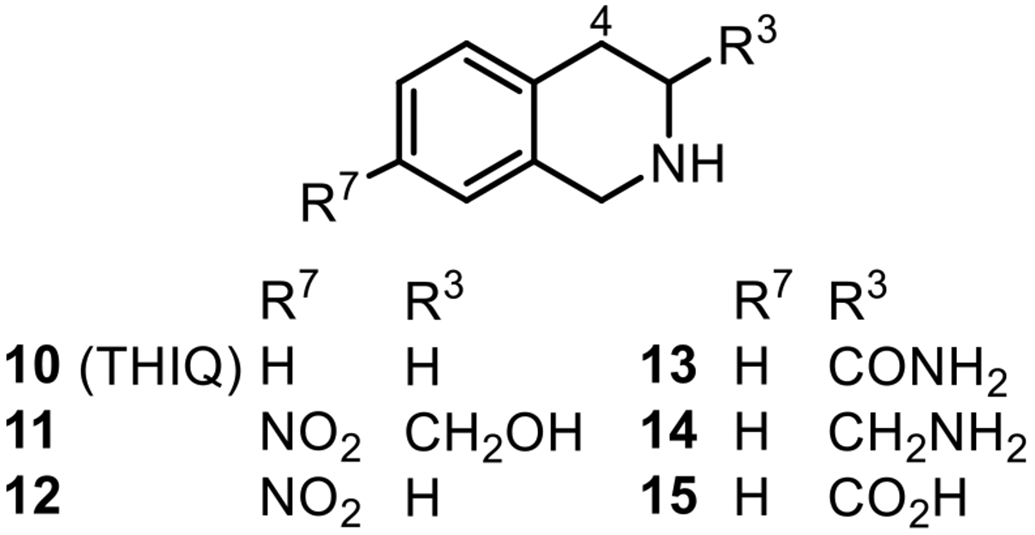
1,2,3,4-Tetrahydroisoquinolines (THIQs) used in the bisubstrate inhibitor design.
Examination of the active site in this crystal structure showed that the two substrate binding domains, designated the AdoMet domain and the PEA domain, are connected by an unobstructed channel (Figure 5). This strongly supports the idea that a bisubstrate inhibitor of hPNMT could be created by connecting a ligand binding in the PEA domain, such as 11,16 with an analogue of 3 using a suitable linker moiety.
Figure 5:

Active site cavityi of the hPNMT-3+11 complex57 (gray) [PDB: 2G70, Chain B] showing co-substrate 3 and inhibitor 11. Labeled amino acids have been shown by site directed mutagenesis to interact with the substrate or co-substrate.51,54 Carbon is shown in white, nitrogen in blue, oxygen in red and sulfur in yellow. Hydrogens are not shown for clarity.
i PyMOL Molecular Graphics System, Version 2.1c, Schrödinger, LLC.
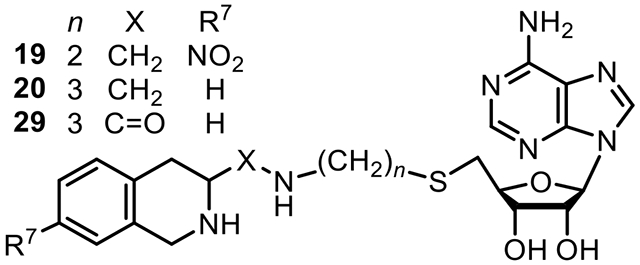
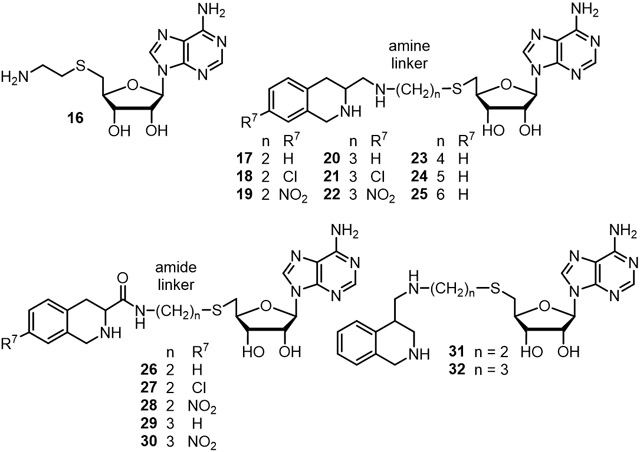
In the hPNMT-3+11 complex, the C-3 atom of 11 is approximately 5.8 Å from the sulfur atom of 3 and is very close to the channel that connects the two binding domains in the enzyme active site. Modeling studies showed that a four-atom linker could meet the distance requirement between the C-3 atom of 11 and the sulfur atom of 3. To allow for synthetic diversity, two linker types were considered. A four-atom amine linker, (e.g., 17, n = 2, Table 1), consisted of an ethylaminomethyl moiety [−(CH2)2NHCH2−], which could be synthesized via reductive amination using an amine and an aldehyde (Scheme 3). The corresponding amide linker, (e.g., 26, n = 2, Table 1), consisted of an N-ethylaminocarbonyl moiety [−(CH2)2NHC(=O)−], which could be synthesized via the coupling of an amine and an acid (Scheme 6).
Table 1.
In vitro activities of bisubstrate analogue inhibitors of hPNMT.
| |||||||
|---|---|---|---|---|---|---|---|
| Cmpd | n | R | PNMT KiAdoMet (nM)a,b |
PNMT KisPEA (nM)a,c |
PNMT KiiPEA (nM)a,c |
α2 Ki (nM)d | Selectivitye |
| 4 | – | – | 14,000 ± 1,000 | f | f | f | f |
| 11 g | – | – | f | 47 ± 5h | f | 19,000 ± 1,000 | f |
| 13 | – | – | f | 31,000 ± 3,000g,i | f | 250,000 ± 20,000j | f |
| 14 | – | – | f | 1,400 ± 200i | f | 1,500 ± 100 | f |
| 16 | 2 | – | 28,000 ± 2,000 | 58,000 ± 14,000 | 37,000 ± 12,000 | 1,100,000 ± 100,000 | 39 |
| C-3 Attachment Amine Linker Compounds | |||||||
| 17 | 2 | H | 38 ± 2 | 115 ± 18 | 220 ± 52 | 15,000 ± 2,000 | 390 |
| 18 | 2 | Cl | 3.2 ± 0.5k | 5.9 ± 0.7k | 8.8 ± 2.5k | 15,000 ± 1,000 | 4,700 |
| 19 | 2 | NO2 | 3.8 ± 0.2k | 7.8 ± 1.8k | 9.4 ± 4.0k | 64,000 ± 6,000 | 17,000 |
| 20 | 3 | H | 2.1 ± 0.2k | 10 ± 3k | 6.0 ± 3.0k | 11,000 ± 1,000 | 5,200 |
| 21 | 3 | Cl | 3.3 ± 0.3k | 15 ± 4k | 12 ± 6k | 13,000 ± 1,000 | 3,900 |
| 22 | 3 | NO2 | 3.5 ± 0.5k | 6.0 ± 0.9k | 13 ± 4k | 33,000 ± 3,000 | 9,400 |
| 23 | 4 | H | 3.9 ± 0.3k | 15 ± 4k | 42 ± 18k | 7,100 ± 500 | 1,800 |
| 24 | 5 | H | 57 ± 6 | 100 ± 12 | 120 ± 33 | 4,700 ± 400 | 82 |
| 25 | 6 | H | 18 ± 1 | 66 ± 10 | 145 ± 67 | 5,000 ± 500 | 280 |
| C-3 Attachment Amide Linker Compounds | |||||||
| 26 | 2 | H | 810 ± 50 | 1,300 ± 20 | 3,500 ± 1560 | 9%l | f |
| 27 | 2 | Cl | 71 ± 4 | 94 ± 11 | 520 ± 290 | 5%l | f |
| 28 | 2 | NO2 | 86 ± 3 | 160 ± 20 | 270 ± 39 | 8%l | f |
| 29 | 3 | H | 46 ± 2 | 81 ± 7 | 180 ± 40 | 130,000 ± 10,000 | 2,800 |
| 30 | 3 | NO2 | 70 ± 4 | 220 ± 30 | 174 ± 16 | 7%l | f |
| C-4 Attachment Amine Linker Compounds | |||||||
| 31 | 2 | H | 5,100 ± 200 | 12,600 ± 2,100 | 17,800 ± 3,600 | 52,000 ± 5,000 | 10 |
| 32 | 3 | H | 4,300 ± 200 | 8,400 ± 890 | 21,000 ± 5,250 | 15,000 ± 1,000 | 3.5 |
Recombinant human enzyme was used, unless otherwise noted.
Data were obtained by varying concentrations the inhibitor and 3 at a fixed concentration (100 μM) of 5. KiAdoMet values were obtained by fitting initial rate data to a competitive inhibition model.
Data were obtained by varying concentrations the inhibitor and 5 at a fixed concentration (5 μM) of 3. The slope and intercept inhibition constants, KisPEA and KiiPEA respectively, were obtained by fitting initial rate data to a mixed (non-competitive) inhibition model.
In vitro activities for the inhibition of [3H]clonidine binding to the α2-adrenoceptor.
α2 Ki/KiAdoMet.
Data have not been determined.
Reference 16.
Previously reported data was for bovine PNMT.
Data is for bovine enzyme.
Reference 24.
Calculated using the Tight Binding routine in SigmaPlot (Reference 83).
Percent inhibition of [3H]-clonidine binding at 100 μM. Ki value was not calculated due to compound showing poor solubility under assay conditions at concentrations ≥ 1,000 μM.
Scheme 3.
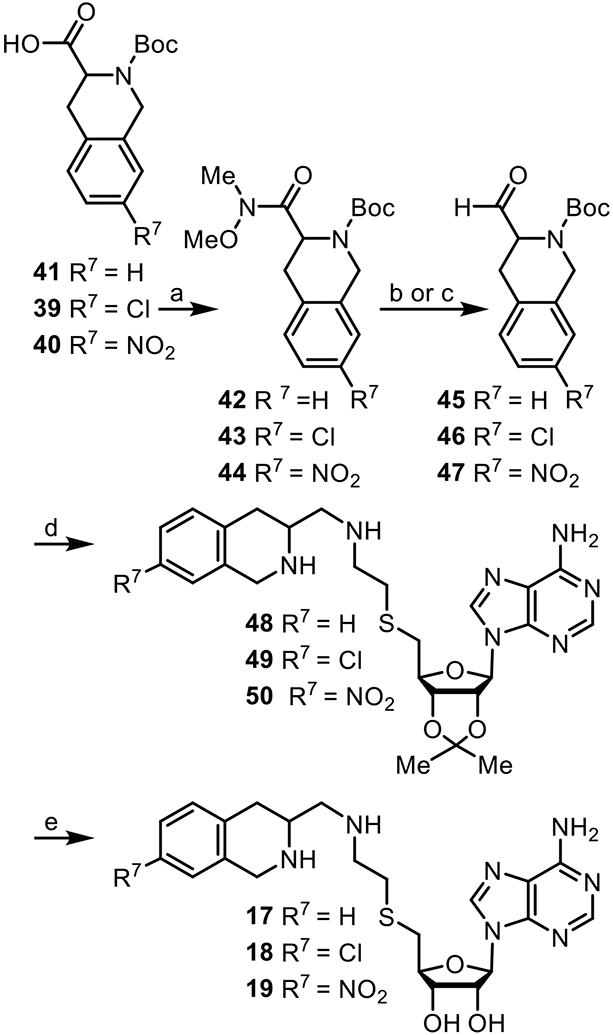
Reagents and conditions: (a) N, O-dimethylhydroxylamine HCl salt, BOP, Et3N (62%–83%); (b) for 42 or 43: LiAlH4 (77%–88%); (c) for 44: Cp2Zr(H)Cl (56%); (d) 34, NaBH4 (48%–57%); (e) TFA/H2O (9:1) (59%–65%).
Scheme 6.
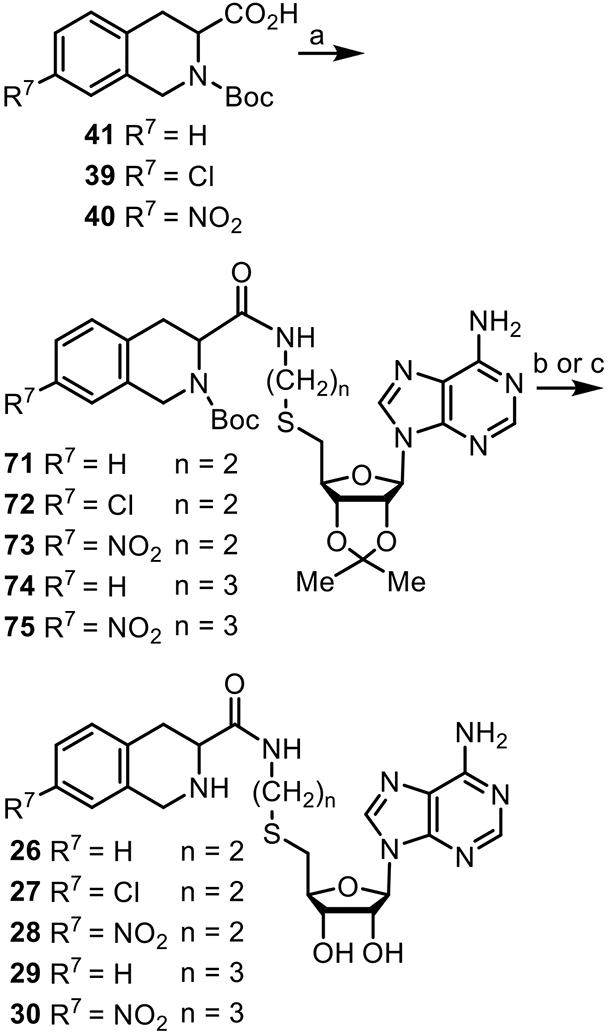
Reagents and conditions: (a) 34 or 61, EDCI, HOBT, N-methylmorpholine (68%– 98%); (b) HCO2H/H2O (4:1) for 71, (c) TFA/H2O (9:1) for 72–75 (40%–66%).
Docking studies were conducted on proposed compounds 17 and 26, based on the hPNMT active site from the crystal structure of the hPNMT-3+11 complex, using AutoDock57 and Sybyl.58 As expected, these studies showed that molecules of this type could fit the active site, with their THIQ and adenosyl moieties mimicking the binding of the corresponding inhibitor and co-substrate analogues. For example, the N-6 atom of the adenine ring in the AutoDock model of 17 is 3.5 Å from the carboxylate of D158, which suggested the possibility of a weak hydrogen bond interaction (3.7 Å in the crystal structure of hPNMT-3+11). The aliphatic ring nitrogen of the THIQ moiety in the model of 26 was placed within 3.2 Å of the E219 carboxylate, which is in range of a hydrogen bond interaction59 (2.9 Å in hPNMT-3+11). While these molecular modeling studies suggested that a linker with a length of four atoms would be a good fit for the hPNMT active site, we also proposed amine linker compounds 20 and 23–25 (5–8 atom length linkers, respectively, Table 1) and five-atom amide linker 29, to explore the effects of an extension of the linker.
Previous structure-activity relationship (SAR) studies revealed that electron-withdrawing groups on the 7-position of the THIQ ring (e.g., Cl and NO2) could increase the affinity of these compounds for hPNMT.15,60 The chlorine and nitro groups were chosen to represent an electron-withdrawing lipophilic or hydrophilic substituent, respectively. Thus, compounds 18, 19, 21, 22, 27, 28, and 30 (Table 1) were proposed for synthesis.
The crystal structure of the hPNMT-3+11 complex also showed that the C-4 atom of 11 is approximately 5.3 Å from the sulfur atom of 3 and close to the channel between the two ligands, although it is not as in line with the channel as is C-3. Amine linker compounds 31 (four-atom linker) and 32 (five-atom) were proposed to investigate the effect a change in the orientation and directionality of the THIQ (10) moiety would have on inhibitor potency.
Examination of the X-ray crystal structures of hPNMT with AdoMet (3) bound57,61 compared to structures with AdoHcy (4) bound62,63 suggested that the positively charged sulfonium species in 3 plays a minimal, if any, role in its binding. Thus, due to the potential instability of the sulfonium center,64-66 only a neutral sulfur atom was included in the design of these inhibitors. For the syntheses of the target compounds in this study, the proposed linker moiety would replace the amino acid portion of 4 in order to retain a neutral sulfur atom in the molecule. A comparison of the inhibitory potencies of 16 and 4 would provide direct evidence as to whether this structural simplification was justified.
hPNMT uses an ordered binding mechanism,36 with 3 binding first, and it possesses an enclosed active site.51 It has been hypothesized that PNMT must undergo a conformational change before a substrate can bind in the PEA domain,51 which may be triggered by the binding of 3 in a similar fashion as for the mechanism proposed for COMT.67 In PNMT, this change is hypothesized to open a cover on the active site that exposes the active site to the PEA domain ligand.36 Since, by this mechanism, 3 must bind first, it is not known if this conformational change will occur when using the bisubstrate compounds.
Most of the compounds in this study conform to Lipinski’s rules.68 For example, 20 has a ClogP of 0.78, nine rotatable bonds, five hydrogen bond donors, ten hydrogen bond acceptors, and a molecular weight of 485, with a total polar surface area69 of 140.70
Chemistry
Because the objective of this study was to explore the optimal nature and length of the linkage for bisubstrate inhibition, compounds were prepared as diastereomeric mixtures based on the unresolved nature of the C-3 or C-4 attachment points on the THIQ moiety. Key intermediate 34 was synthesized by reacting commercially available adenosine analogue 33 with 2-aminoethylthiol hydrochloride (Scheme 1).71,72 Deprotection of 34 with formic acid/water (4:1) yielded 16.
Scheme 1.
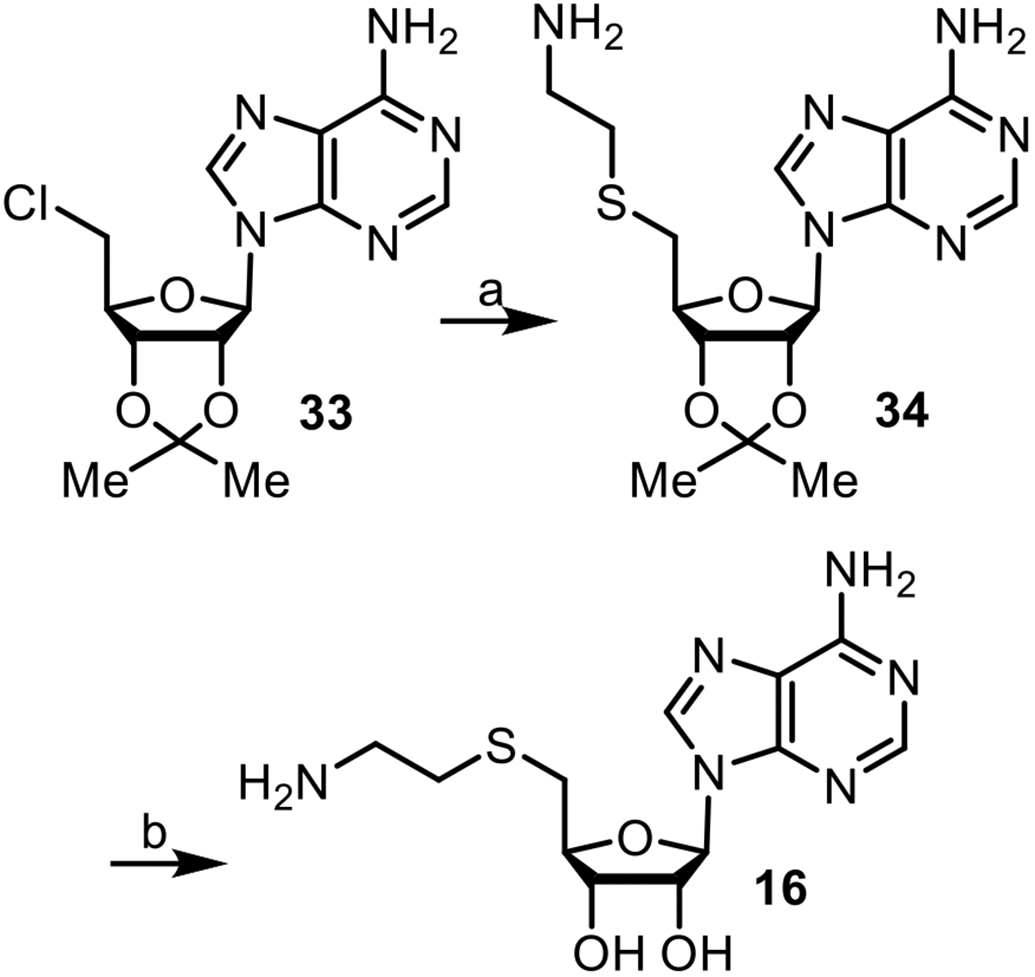
Reagents and conditions: (a) H2NCH2CH2SH•HCl, NaOEt (98%); (b) HCO2H/H2O (4:1) (90%).
For the synthesis of inhibitors 17–32, N-Boc-protected THIQ-3-carboxylic acids 39, 40, or 41 were used. Compound 39 was synthesized as shown (Scheme 2), 40 was synthesized by modification of the literature procedure,73 and 41 was commercially available.
Scheme 2.
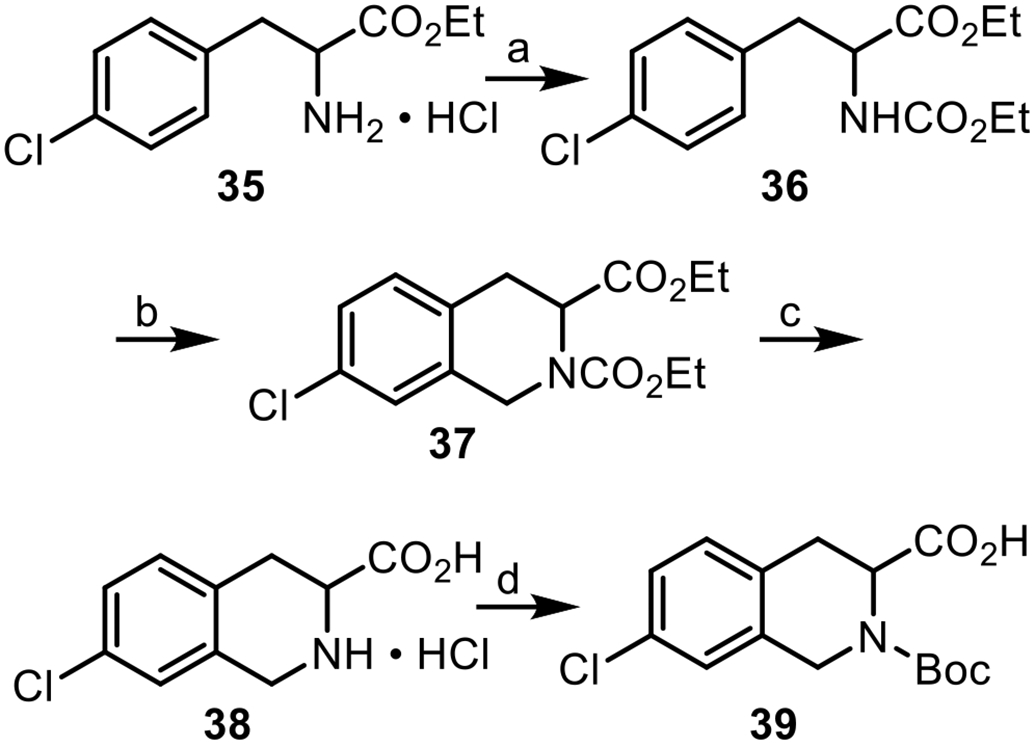
Reagents and conditions: (a) ClCO2Et, pyridine (99%); (b) (CH2O)n, HOAc/H2SO4 (69%); (c) 6 N HCl (48%); (d) (Boc)2O, Et3N (56%).
The synthesis of 39 began by reacting commercially available ester 35 with ethyl chloroformate to form carbamate 36, followed by a Pictet-Spengler reaction to yield 37.74 Treatment of 37 with HCl (6 N) at reflux resulted in the formation of acid hydrochloride 38,75 which was then treated with di-tert-butyl dicarbonate [(Boc)2O] to yield 39.
Inhibitors 17–19 were synthesized as shown (Scheme 3). Weinreb amides 42–44 were formed by treatment of 41, 39, or 40, respectively, with dimethylhydroxylamine hydrochloride and benzotriazol-1-yloxytris(dimethylamino)phosphonium hexafluorophospate (BOP). Reduction of 42 or 43 with LiAlH4 in anhydrous diethyl ether at room temperature afforded aldehydes 45 or 46,76 while reduction of 44 with Cp2Zr(H)Cl (Schwartz reagent) gave aldehyde 47.77 Reductive aminations of 45–47 with amine 34 and NaBH4 yielded 48–50, followed by deprotection of the THIQ amine and the 1,2-dihydroxyl groups to yield final products 17–19.
The syntheses of amines 61–64 are shown (Scheme 4). Subjection of commercially available adenosine analogue 51 to a Mitsunobu reaction with thioacetic acid provided thioester 52,78 followed by treatment with bromides 53–56 and sodium methoxide afforded 57–60,80 which were then hydrolyzed with hydrazine to form amines 61–64.
Scheme 4.
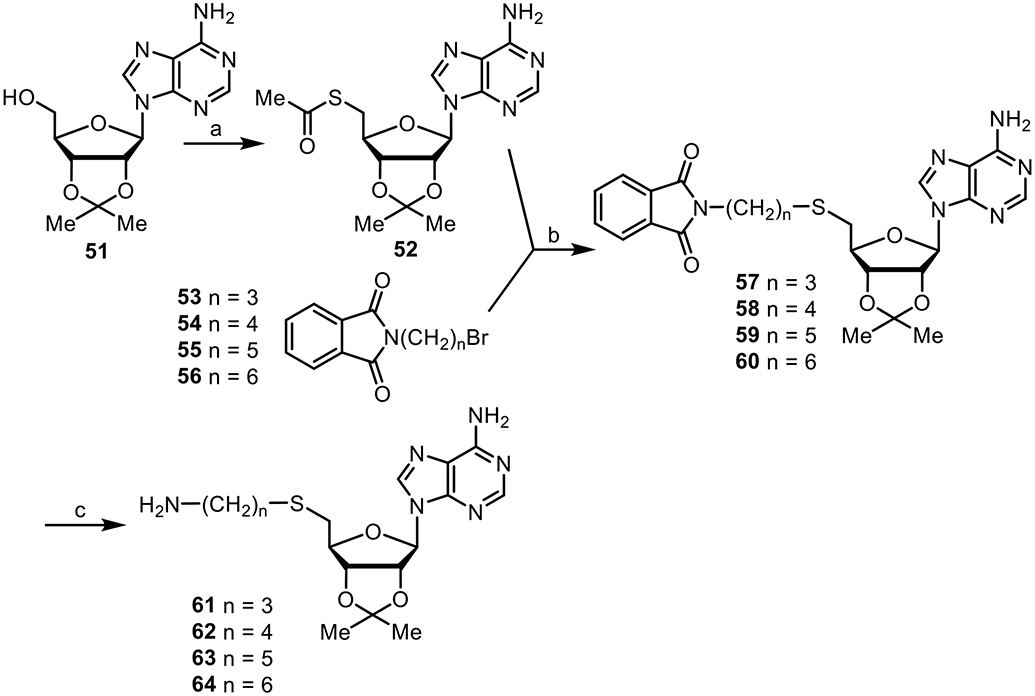
Reagents and conditions: (a) Thioacetic acid, DIAD, PPh3 (100%); (b) NaOMe (64%–85%); (c) NH2NH2 (81%–94%)
Reductive aminations of aldehydes 45–47 and the appropriate amine 61–64 with NaBH4, yielding 65–70, followed by deprotection with TFA/H2O (9:1) afforded inhibitors 20–25 (Scheme 5).
Scheme 5.
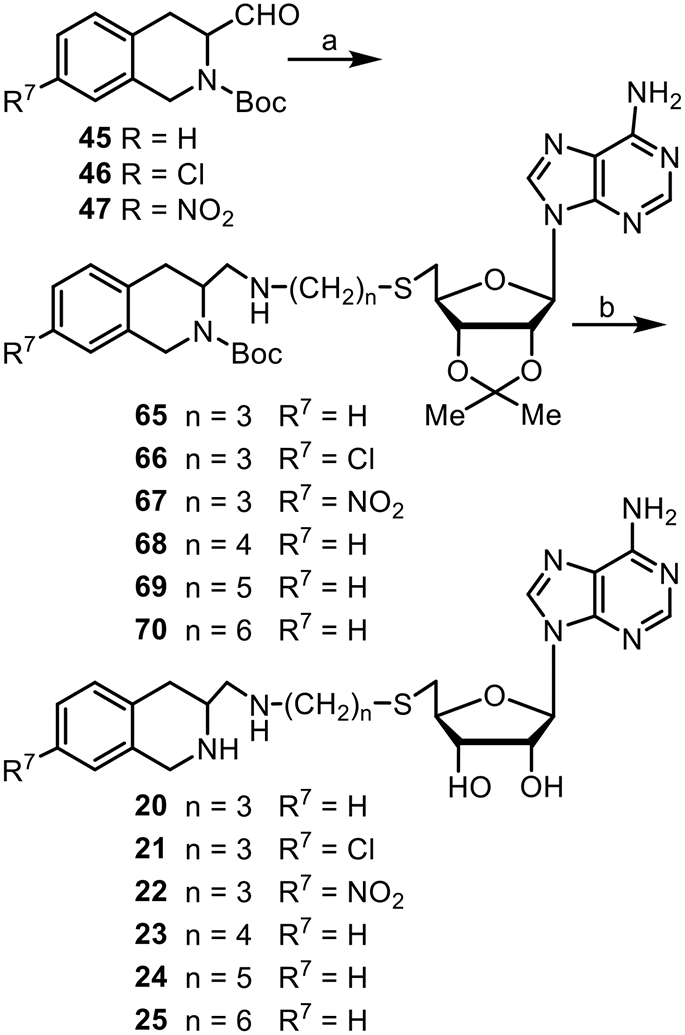
Reagents and conditions: (a) 61, 62, 63, or 64, NaBH4 (36%–68%); (b) TFA/H2O (9:1) (42%–75%).
Compounds 26–30 were synthesized (Scheme 6) by coupling 41, 39 or 40 with 34 or 61 to yield compounds 71–75.79 Treatment of 71 with formic acid/water (4:1) yielded 26. Deprotection of 72–75 with trifluoroacetic acid [TFA/H2O (9:1)] yielded final products 27–30.
Reduction of commercially available 76 with LiAlH4 gave amino alcohol 77 (Scheme 7). Selective protection of the amino group with di-tert-butyl dicarbonate, followed by Swern oxidation afforded aldehyde 79. Reductive amination with amines 34 or 61 formed 80 or 81, respectively, which were deprotected with TFA/H2O (9:1) to afford target products 31 or 32.
Scheme 7.
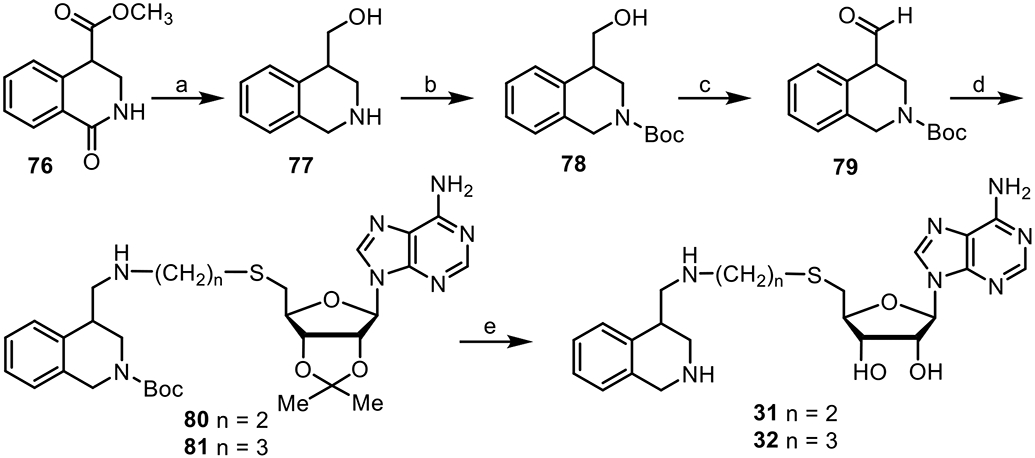
Reagents and conditions: (a) LiAlH4 (88%); (b) NaHCO3, (Boc)2O (88%); (c) oxalyl dichloride, DMSO, DIEA (71%); (d) 34 or 61, NaBH4 (54%–60%); (e) TFA/H2O (9:1) (74%–86%).
Biochemistry
Human PNMT (hPNMT) and its variants, all with a C-terminal hexahistidine tag, were expressed in E. coli and purified as described previously.26 Enzyme activity was monitored using a radiochemical assay,26,52 modified when appropriate to account for the high binding affinity of some inhibitors.80 Assays were carried out using four concentrations of 3 or 5 as the variable substrate, and three concentrations of inhibitor. Kinetic constants were obtained by fitting the initial rate data using the Enzyme Kinetics Module in SigmaPlot.81 At a minimum, assays were run in duplicate and the average result presented.
α2-Adrenergic receptor binding assays were performed using cortex obtained from male Sprague Dawley rats.82 [3H]Clonidine was used as the radioligand to define the specific binding and phentolamine was used to define the nonspecific binding. Clonidine was used as the ligand to define α2-adrenergic binding affinity for a direct comparison with previous studies. At a minimum, assays were run in duplicate and the average result presented.
RESULTS AND DISCUSSION
Results of the kinetic studies are shown in Table 1. hPNMT has been shown to function via an ordered, sequential mechanism wherein 3 binds first and facilitates the binding of the methyl-acceptor substrate.36 Initial rate data for each inhibitor were fit to equations for competitive, non-competitive and mixed type inhibition using the enzyme kinetics module in SigmaPlot.83 The bisubstrate inhibitors all displayed competitive inhibition versus AdoMet and non-competitive inhibition versus PEA (Table 1). This pattern is consistent with a steady-state ordered, mechanism, in which AdoMet binds first.83 In light of that observation, the data in Table 1 for the bisubstrate inhibitors were calculated using a competitive binding model versus 3 (KisAdoMet) and a mixed (non-competitive) model versus 5 (KisPEA and KiiPEA).84 It should be noted that the inhibition patterns were determined by measuring initial rates at varying concentrations of one reactant and a fixed concentration of the second reactant. Accordingly, the values in Table 1 should be treated as apparent Ki values. For AdoMet, appKis = Ki.86 For PEA, appKis = Ki (1 + [AdoMet]/KiaAdoMet) and appKii = Ki (1 + [AdoMet]/KmAdoMet).86 For 3 the fixed concentration was 5 μM, while for ±5, the concentration was 100 μM. Using ITC, KiaAdoMet was found to be 4.6 μM,36 thus appKisPEA should be ~ × KisAdoMet (i.e., 2 × Ki). Given the constraints of the assay it was gratifying to see that, in general, the Ki values determined from the competitive pattern versus AdoMet were broadly similar to those obtained from the mixed (noncompetitive) pattern versus PEA (Table 1).
Comparison of the inhibitory potency of 4 (AdoHcy) with that of 16 (the adenosyl fragment used in these bisubstrate inhibitors) shows only a two-fold increase in potency with the inclusion of the amino acid side chain, indicating that the presence of the homocysteine side chain is not required for binding.
All of the compounds with an amine linker were more potent as inhibitors of hPNMT than their corresponding amide analogues. For example, the best compound in each of the two linker types were 20 (KiAdoMet = 2.1 nM) for an amine linker and 29 (KiAdoMet = 46 nM) for an amide linker. Each of these compounds possess a five-atom length linker (Table 1, n = 3) with a hydrogen as the 7-substituent on the THIQ moiety. The potency difference between the corresponding compounds (comparing amine vs. amide linker compounds: 17 vs. 26, 18 vs. 27, 19 vs. 28, 20 vs. 29, and 22 vs. 30) was approximately 20-fold (Table 1) in all cases. These results are consistent with 3-substituted-THIQ 14 (aminomethyl) showing over 20-fold greater potency (Table 1) as an inhibitor of hPNMT than 13 (aminocarbonyl).
As expected, the introduction of an electron-withdrawing group to the 7-position of the THIQ fragment increased the hPNMT inhibitory potency of compounds possessing a four-atom (n = 2) length linker. Comparison of both amine linker compounds 18 (Cl) and 19 (NO2) compared to 17 (H) and amide linker compounds 27 (Cl) and 28 (NO2) compared to 26 (H) showed an approximately 10-fold increase in potency. However, this did not occur with compounds possessing a five-atom length linker, with both amine linker compounds 21 (Cl) or 22 (NO2) compared to 20 (H) and amide linker 30 (NO2) compared to 29 (H), showing at least a 40% reduction in hPNMT inhibitory potency (Table 1). This may be due to the increased length of the linker, such that the THIQ portion, for compounds possessing a five-atom length linker, would sit further into the hydrophobic pocket formed by V53, M258, V269, and V272 in the hPNMT active site,52 resulting in the five-atom length amine linker 20 and its amide linker analogue 29, which possess a hydrogen as the 7-substituent on the THIQ fragment, being the most potent compounds of their respective series. Because of this possible positioning, adding a 7-substituent to the aromatic ring of the THIQ moiety of 20 or 29 could result in unfavorable steric interactions that would offset the positive contribution of the electron-withdrawing THIQ 7-substituent.
Compounds possessing a six- (23), seven- (24) or eight-atom (25) amine linker were prepared to explore the effect of an extension of the linker. Compared to its five-atom linker analogue (20, KiAdoMet = 2.1 nM), the six-atom linker (23) was slightly less potent (KiAdoMet = 3.9 nM) and similar in potency to compounds 21 or 22 wherein substitutions at the 7-position of the THIQ moiety were incorporated (KiAdoMet = 3.3 or 3.5 nM, respectively). When the linker was increased to seven atoms, potency was further reduced as demonstrated for 24 (KiAdoMet = 57 nM), potentially due to negative steric interactions or increasing entropic demand. In contrast, the eight-atom linker analogue (25) showed a slight increase in potency (KiAdoMet = 18 nM) compared to 24, possibly resulting from conformational adjustments by the enzyme. Similar conformational adjustment had previously been shown to reveal a cryptic binding site in hPNMT.55
Compounds 31 and 32 (four- and five-atom length amine linkers, respectively) were synthesized to explore the effect of a different orientation of the THIQ moiety, with the linker being attached at the C-4 of THIQ (10). Both of these compounds showed reduced hPNMT inhibitory potency of ca. 4–5 μM compared to the equivalent C-3 attachment analogues and thus this series was not investigated further.
As mentioned previously, a bisubstrate inhibitor should show at least an additive increase in inhibitory potency over its component fragments38 and this is indeed the case for all of the bisubstrate compounds in this study. For example, the increase in potency for 20 compared to 16 is greater than ~13,000-fold and ~4,800-fold more potent when compared to 10 (KiPEA = 10 μM).85
Mutagenesis studies.
Since a number of the compounds were very effective inhibitors of hPNMT and docking suggested binding would be compatible with simultaneous occupancy of both the AdoMet and PEA binding domains, site-directed mutagenesis was used to probe whether 19, one of the most potent inhibitors in this study, was binding at both hPNMT substrate domains. The amino acid residues mutated in these studies were those with established interactions with either inhibitor 11 or co-substrate 3 based on the hPNMT-3+11 complex structure (Figure 5).54 Both residues E219 and K57 were mutated to alanine (E219A and K57A, respectively). As mentioned earlier, E219 forms a hydrogen bond with the aliphatic isoquinoline amine of tetrahydroisoquinoline-type inhibitors in the PEA binding domain. This structure also showed that K57 is another important amino acid residue in the PEA binding domain that forms a hydrogen bond with the oxygen of the nitro moiety in 11.53 Because of their considerable distances from the AdoMet domain, E219 and K57 were expected to have little effect on inhibitors binding only in the AdoMet domain. In contrast, residues D158 and C183 lie in the AdoMet domain and mutations of these residues were expected to have little effect on the binding of ligands binding in the PEA domain. D158 forms a hydrogen bond with the N-6 atom of the adenine ring of 3,51 while C183 also forms part of the binding domain for 3,51 being located under the plane of the adenine ring system. Both were mutated to alanine (D158A and C183A, respectively).
The effect of inhibitors 12 (THIQ fragment binding only in the PEA domain), 16 (adenosyl fragment binding only in the AdoMet domain), and 19 (bisubstrate inhibitor) on the inhibition of mutant hPNMTs were compared to the wild-type enzyme as shown in Table 2. As expected, both the K57A or E219A mutants had a larger impact on the binding of THIQ-type inhibitor 12, which binds only in the PEA domain, than did the D158A or C183A mutants, while the D158A and C183A mutants showed a much larger impact on the hPNMT inhibitory potency of 16, an inhibitor binding only in the AdoMet domain, than did the K57A or E219A mutants.
Table 2.
Relative activities of inhibitors 12, 16 and 19, at wild-type (WT) and mutant hPNMTs.
| Relative Potencya | |||
|---|---|---|---|
| hPNMT | 12 | 16 | 19 |
| Wild-type | 1.0 | 1.0 | 1.0 |
| K57A | 18 | 0.46 | 10 |
| E219A | 43 | 1.8 | 69 |
| D158A | 8.1 | 35 | 15 |
| C183A | 4.9 | 7.3 | 22 |
Inhibitory potency relative to wild-type (WT) hPNMT for 12 and 19: Ki/Ki (WT); for 16: IC50/IC50 (WT). Compound 5 was used as the methyl-acceptor substrate.
Notably, all four mutants showed a strong effect on the binding of inhibitor 19, which suggests that 19 interacts with all four residues and thus occupies both the AdoMet and PEA binding domains.
X-ray Structures of Bisubstrate Inhibitors in the hPNMT Active Site.
To confirm the general binding mode of these bisubstrate inhibitors and to elucidate the details of their interactions with hPNMT, crystals of 19 or 29 co-crystallized with hPNMT were obtained. The structures of these complexes were determined at 1.95 Å resolution for hPNMT-1986 (Figure 6a) and at 2.20 Å for hPNMT-2987 (Figure 6b). Consistent with previous studies,51-53 the asymmetric unit for both complexes contained two hPNMT molecules which showed root mean square deviations (rmsd)58 of 0.195 Å and 0.241 Å over all atoms of the hPNMT-19 and hPNMT-29 complexes, respectively. By comparison, the two molecules in the hPNMT-3+11 asymmetric unit57 showed an rmsd value of 0.307 Å. It was immediately apparent that the presence of the bisubstrate inhibitor had little effect on the overall conformation of hPNMT. This was confirmed by superposition of the monomers of the bisubstrate complexes on those of the hPNMT-3+11 complex, which provided average rmsd values (over all atoms) of 0.329 Å for hPNMT-19 (Figure 7a) and 0.347 Å for hPNMT-29.
Figure 6.

Active site cavity58 (gray) from the hPNMT-3+1157 crystal structure [PDB: 2G70]. Amino acids used in the site-directed mutagenesis investigations are labeled. A molecule of Tris was identified in both hPNMT:bisubstrate inhibitor complexes. Atom colors are described in Figure 5. The Figures show (a) the potent bisubstrate inhibitor 19 that contains a four-atom amine linker [PDB: 4MIK, Chain A] and (b) the bisubstrate inhibitor 29 [PDB: 4MQ4, Chain A] that contains a five-atom length amide linker.
Figure 7.

Active site cavity58 (shown in gray) based on the hPNMT-3+1157 crystal structure [PDB: 2G70]. Amino acids used in the site-directed mutagenesis investigations are labeled. The molecule of Tris in the active site is shown for completeness. Atom colors are described in Figure 5. The Figures show the (a) superimposition58 of the X-ray structures of (3+11) (red) and 19 (blue) [PDB: 2G70, Chain B and 4MIK, Chain A, respectively] and (b) superimposition58 of the X-ray structures of 19 (blue) and 29 (green) [PDB: 4MIK, Chain A and 4MQ4, Chain A, respectively].
In both structures, the electron density for some of the linker atoms was weak, implying flexibility, but clearly the main portions of these bisubstrate inhibitors occupy both substrate binding domains as predicted. The electron density was weaker and the B-factors higher for the linker atoms of 19, which incorporates a flexible amine linker, than for the corresponding atoms of 29 and its more rigid amide linker (Table 3).
Table 3.
X-ray data collection, refinement, and validation statistics
| hPNMT-19 | hPNMT-29 | |
|---|---|---|
| Data Collection | ||
| Space group | P43212 | P43212 |
| Cell dimensions: a, b, c (Å) | 94.66, 94.66, 188.46 | 94.15, 94.15, 188.16 |
| Resolution (Å)* | 38.62–1.95 (2.06-1.95) | 38.43–2.2 (2.32-2.2) |
| Total Reflections* | 1,613,327 (222,007) | 277,262 (40,988) |
| Unique Reflections* | 63,094 (8,974) | 43,470 (6,217) |
| Redundancy* | 25.6 (24.7) | 6.4 (6.6) |
| Rpim* | 0.015 (0.426) | 0.023 (0.371) |
| Mn(I/sd)* | 31.1 (1.7) | 20.5 (2.0) |
| Completeness* (%) | 99.8 (98.7) | 99.8 (99.8) |
| Refinement | ||
| Resolution (Å) | 38.62-1.95 | 38.43-2.2 |
| No. reflections | 62,657 | 43,250 |
| Rwork /R free | 0.1824 / 0.2295 | 0.1826 / 0.2207 |
| Residues modeled for molecules | A: 26-280 B: 16-280 |
A: 17-280 B: 26-280 |
| Wilson B factor | 40.22 | 47.57 |
| Number of atoms / B factor | ||
| Protein | 8,135 / 50.60 | 8,115 / 60.67 |
| Ligand (Tris) | 40 / 54.25 | 40 / 65.85 |
| Ligand (Inhibitor) | 128 / 82.59 | 128 / 63.68 |
| Water | 314 / 51 | 181 / 56.18 |
| RMS deviations | ||
| Bond lengths (Å) | 0.013 | 0.013 |
| Bond angles (°) | 1.493 | 1.443 |
| Structure Analysis | ||
| Ramachandran (%) | ||
| Most favored | 98.85 | 98.07 |
| Allowed | 1.15 | 1.93 |
| Disallowed | 0 | 0 |
highest resolution shell in parenthese
Compound 19, which possesses a four-atom length (n = 2) linker, is one of the most potent bisubstrate inhibitors in this study (Figure 6a).
Compound 29 (Figure 6b) is the most potent amide linker compound in this study and shares the five-atom linker length (n = 3) with the most potent amine linker compound 20 (KiAdoMet = 2.1 nM), which could provide insights into the reasons why 20 was such a potent compound. However, 29 was less potent as an inhibitor of hPNMT (KiAdoMet = 46 nM) than 19 (KiAdoMet = 3.8 nM).
Comparison of the crystal structure of hPNMT-19 with hPNMT-3+11 showed considerable similarity between how these compounds fit in the hPNMT active site (Figure 7a). In the AdoMet binding domain, the closest distance between D158 and the N-6 of the adenine ring is 2.9 Å in the hPNMT-19 structure, which is shorter than the corresponding distance in the hPNMT-3+11 structure (3.7 Å). This results from a slight torsion in the terminal atoms of the D158 side chain, rather than a change in the position of the adenine ring, and results in a stronger hydrogen bond to 19 as compared to 3. The position of C183, also in the AdoMet binding domain, is unchanged. The sugar ring is positioned similarly in both structures, conserving interactions between its hydroxyls and D101. Although the sugar C-5 is positioned slightly differently in the two structures, the position of the sulfur atom is highly conserved. In the PEA binding domain, the THIQ ring of 19 is shifted slightly toward E219 compared to the equivalent position of 11 in the hPNMT-3+11 structure. As a result, the closest distance between an E219 carbonyl oxygen and the ligand amine nitrogen was 2.6 Å, in the hPNMT-19 structure, while the equivalent distance in hPNMT-3+11 was 2.9 Å. Similarly, the distance between the THIQ nitro oxygen and the K57 amine was shorter for hPNMT-19 (2.7 Å), compared to the equivalent interaction in hPNMT-3+11 (3.4 Å). Again, these shorter interaction distances for both K57 and E219 may indicate an increase in the strength of the binding interactions of 19 compared to 11.
Despite the fact that the linker in 29 is one atom longer than for 19, an overlay of the hPNMT-19 and hPNMT-29 structures (Figure 7b) shows that they both bind in a similar fashion, with a root mean square deviation of 0.225 Å for the alignment of all atoms of their respective hPNMT A chains.
In the AdoMet binding domain, the adenosyl moieties of the two bisubstrate inhibitors superimpose well, with only 0.2 Å differences between their respective adenosine N-6s, ribose ring oxygens, and sulfurs, and no significant differences in the positions of D158 or C183. While the five-atom amide linker passes through the channel between the AdoMet and PEA binding domains, its longer length and the different positions of these atoms bury the THIQ moiety deeper into the PEA binding domain. There are two possible orientations of the THIQ moiety in the hPNMT-29 structure, with the plane of the THIQ being flipped by 180°. The electron density does not distinguish between them and, as a result, they were each modeled with 50% occupancy. Regardless, both orientations, the carbonyl oxygen of the linker amide appeared to be in contact with the E219 side chain carboxylate of PNMT at distances of 2.7 and 3.3 Å. These observations indicate no significant preference for binding of either of the C-3 stereoisomers. In contrast, the cocrystal structures of both 19 and 11 revealed that the THIQ nitrogen served as the hydrogen donor contact to E219. The cocrystal structure with 19 showed only the C-3 (S)-stereoisomer, which is consistent with the importance of the THIQ nitrogen-E219 contact (figure 5a).
The aromatic rings of the THIQ moieties in 19 and 29 lie in the same plane, being sandwiched between the sidechains of F182 and N39, as observed previously.51 Because the longer linker of 29 seats the THIQ moiety deeper into the PEA binding domain, the aromatic ring of the THIQ ring system occupies approximately the same area of space as that occupied by the nitro substituent of 19 (Figure 7b).
Unlike 19, which possesses a nitro group, 29 does not have a substituent on its THIQ ring that can interact with K57 and the C-7 of its THIQ ring is only 3.5 Å from the isopropyl carbons of V53 (Figure 6b), which, along with M258, V269, and V272, lines the end of the PEA binding domain to form a hydrophobic surface.52 The THIQ moiety of the five-atom linker compounds is seated deeper into this hydrophobic surface (figure 6b), which may be responsible for the increased inhibitory potency of 20 and 29 as compared to their four-atom linker analogues (20: KiAdoMet = 2.1 nM vs. 17: KiAdoMet = 38 nM and 29: KiAdoMet = 46 nM vs. 26: KiAdoMet = 810 nM, respectively). Addition of a chloro- or nitro-substituent to the C-7 of the THIQ moiety in the five-atom linker compounds was expected to grant a similar increase in inhibitory potency as it did in the case of the four-atom linker compounds, but unfavorable steric interactions with V53 may be responsible for the observed reduction in inhibitory potency of these compounds [21 (Cl, KiAdoMet = 3.3 nM) and 22 (NO2 , KiAdoMet = 3.5 nM)], compared to the equivalent hydrogen-substituted compound (20, KiAdoMet = 2.1 nM) or 30 (NO2 , KiAdoMet = 70 nM)], compared to its hydrogen-substituted analogue (29, KiAdoMet = 46 nM). Decreasing the distance between the THIQ moiety and V53 by increasing the length of the linker to six or seven atoms, as in 23 (KiAdoMet = 3.9 nM) or 24 (KiAdoMet = 57 nM), respectively, could be responsible for their decreased inhibitory potency by increasing the possibility of steric clash with V53.
It is notable that both the hPNMT-19 and hPNMT-29 structures contained electron density for another molecule in the portion of the AdoMet binding domain that would normally bind the homocysteine sidechain of AdoMet. A survey of all components in the protein and crystallization buffers revealed that this density was best fit as a molecule of the buffer Tris (tris(hydroxymethyl)amino methane). As shown (Figure 7a), two of the Tris oxygens are positioned in exactly the same locations as the oxygens in the homocysteine carboxylate, while the third hydroxyl group is directed into approximately the same space as the homocysteine amine group and the Tris amine is directed into the space normally occupied by the homocysteine sidechain. The density was strong in both structures. This is consistent with Tris inhibition being observed in PNMT assays, which prompted a switch to phosphate buffer, and may suggest that hPNMT strongly prefers a compatible molecule in the homocysteine-binding portion of the AdoMet binding domain. This adventitious occupancy of the homocysteine-binding area of the AdoMet binding domain by a molecule of Tris suggests that further improvements in the potency of these bisubstrate inhibitors may be achievable.
CONCLUSIONS
A series of compounds was developed in this study as potential bisubstrate inhibitors of hPNMT, based on information from the X-ray crystal structure of the hPNMT-3+11 ternary complex, molecular modeling studies, and previous SAR studies. Kinetic studies showed that many of these new analogues were not only low nanomolar inhibitors of hPNMT, but also displayed only micromolar affinity for the α2-adrenoceptor. This additional selectivity would be highly advantageous for delineating the separate pharmacological effects of PNMT inhibition and α2-adrenoceptor affinity. For example, 19 showed a six-fold increase in hPNMT inhibitory potency and a four-fold decrease in α2-adrenoceptor affinity as compared to 11, its mono-substrate analogue, making 19 one of the most potent (KiAdoMet = 3.8 nM) and selective (versus the α2-adrenoceptor, Table 1) inhibitors of hPNMT yet reported.
While docking studies predicted the bisubstrate binding nature of these new inhibitors and the site-directed mutagenesis results were consistent with these compounds occupying both binding domains, the binding mode of these inhibitors was confirmed by the X-ray crystal structures of hPNMT-19 and hPNMT-29.
The object of these studies was to determine whether co-occupancy of both the PEA and AdoMet binding sites might lead to a further enhancement of hPNMT inhibitory potency and selectivity. These compounds were successful at both of these objectives. While the most potent compound (20) obeys Lipinski’s rules, it is likely that further optimization of drug-like properties will be required before it could be considered as a clinical candidate.
EXPERIMENTAL
All of the reagents and solvents used were reagent grade or were purified by standard methods before use. Melting points were determined in open capillary tubes on a Thomas-Hoover melting point apparatus calibrated with known compounds but are otherwise uncorrected. All proton (1H NMR) and carbon (13C NMR) nuclear magnetic resonance spectra were taken on a Bruker DRX-400 or a Bruker AM-500 spectrometer. Multiplicity abbreviations are as follows: s, singlet; d, doublet; t, triplet; m, multiplet; br, broad; ex, exchangeable. Electron-impact mass spectra (MS (EI)), chemical–ionization mass spectra (MS (CI)) and high resolution mass spectra (HRMS) were obtained on a Varian Atlas CH-5, a Ribermag R 10-10, a VG ZAB, or an LCT Premier high resolution instrument. The intensity of each peak in the mass spectrum relative to the base peak is reported in parentheses. The purity of all biologically tested compounds was determined to be ≥95% by combustion analysis, as performed by Quantitative Technologies, Inc. (Whitehouse, New Jersey). Flash chromatography was performed using silica gel 60 (230–400 mesh) supplied by Universal Adsorbents, Atlanta, Georgia. Methanol (MeOH) and ethanol (EtOH) were anhydrous unless stated otherwise and were prepared by distillation over magnesium. Anhydrous tetrahydrofuran (THF) and diethyl ether (Et2O) were distilled from sodium-benzophenone ketyl. Hexanes refer to the mixture of hexane isomers (bp 40–70 °C). All reactions that required anhydrous conditions were performed under dried argon, and all glassware was either oven-dried or flame-dried before use. Chiral high-performance liquid chromatography (HPLC) was performed on a Shimadzu LC 6A system or an IPro500-Lambda-I system (IRIS Technologies). The Chiralcel OJ analytic column (column size: 250 × 4.6 mm, particle diameter: 10 μm) and semi-preparative column (column size: 250 × 20 mm, particle diameter: 10 μm) were purchased from Chiral Technologies, Inc., West Chester, PA.
The purity of some final products was determined using a Vydac 218TP54 column (C18 reversed-phase, column size: 250 × 4.6 mm, particle diameter: 5 μm, Grace Vydac, Hesperia, CA.) on a HPLC system consisted of a Schimadzu SLC-10A VP system controller, two LC-6AD pumps and a SPD-M10A VP diode array detector and monitored at wavelengths specified for each compound. System A parameters consisted of the following: a linear gradient solvent system of H2O (0.1% TFA)/CH3CN from 99/1 to 1/99 in 40 min, with a flow rate of 1 mL/min. System B parameters consisted of the following: a linear gradient solvent system of H2O (0.1%TFA)/MeOH from 99/1 to 1/99 in 40 min, with a flow rate of 1 mL/min.
AdoMet was obtained from Sigma–Aldrich (St. Louis, MO). [methyl-3H]AdoMet and [3H]clonidine were obtained from PerkinElmer (Boston, MA). Stock inhibitor solutions (10 mM) were made by dissolving the compound in H2O (for salts) or in 0.01 N HClO4 (for free amines).
Protein Expression and Purification.
Expression vectors containing genes for wt, K57A and E219A PNMT were available from previous studies.26,54 The D158A and C183A variants were obtained by mutagenesis of pET17PNMT26 using the QuikChange site-directed mutagenesis kit (Agilent, Santa Clara, CA).
Hexahistidine-tagged hPNMT was expressed and purified as described previously.26,54
Radiochemical Assay for hPNMT Inhibitory Potency.
The radiochemical assay used to measure the inhibitory potency of these compounds against hPNMT was performed as described previously.82
Kinetic data were fit to equations for competitive (Eq 1) or non-competitive (mixed) type (Eq 2) inhibition using SigmaPlot.83
| Eq. 1 |
| Eq. 2 |
Where appropriate, data were fit using the tight-binding competitive and mixed inhibition routines in the Enzyme Kinetics module of SigmaPlot.
α2-Adrenoceptor Radioligand Binding Assay.
Tissue samples were prepared and the radioligand receptor binding assay was performed according to the method of U’Prichard et al.84 All animal use was conducted in accordance with institutional guidelines.
Molecular Modeling.
Initial molecular modeling studies were performed on a Silicon Graphics Octane workstation using Sybyl.60 Docking of inhibitors into the PNMT active site was performed using AutoDock 3.0 (default settings).59 The initial ligand was overlayed with the cocrystallized ligand and minimized with the Tripos force field before it was docked into the active site of PNMT. Graphics for Figures 5-7 were generated on an iMac i7 using PyMOL.58
Protein Crystallization, Data Collection, and Structure Determination.
Purified hPNMT was mixed with 19 (final concentration of 240 μM) and concentrated to 35 mg/mL final protein concentration. DTT was added to 10 μM. Crystals were grown from 1 μL of the 35 mg/mL hPNMT-19 protein complex mixed with 1 μL well solution (0.1 M Na cacodylate pH 5.6 (Hampton Research, Aliso Viejo, CA), 0.17 M LiCl, and 9% (w/v) PEG 6000 (Hampton Research)) equilibrated against 750 μL of this same solution by the hanging drop vapor diffusion method. Tetragonal rods appeared after 10 days of incubation at 17 °C, growing from a background of heavy precipitation. Crystals were immersed in cryoprotectant (well solution with 25% ethylene glycol), and flash cooled in liquid nitrogen for storage until data collection.
Crystals of hPNMT-29 were grown in a similar manner with slight modifications. hPNMT was concentrated to 40 mg/mL and a 200 mM stock solution of 29 dissolved in 100% DMSO was added directly to the protein sample to a final concentration of 800 μM. Crystals were grown with the same crystallization reagents and conditions used for hPNMT-19. Crystals with tetragonal morphology grew after 4 days of incubation at 17 °C. Crystal harvesting and storage was performed as described previously.
Data sets were collected from single crystals at the Stanford Synchrotron Radiation Lightsource (Stanford, CA) Beamline 12-2 at a wavelength of 1.03 Å. Data integration and scaling were carried out employing the programs XDS88 and SCALA.89 The structures were solved by molecular replacement using Phaser90 with the hPNMT-3-fluoromethyl-7-thiomorpholinosulfonamide-THIQ/AdoMet structure as a search model55 (Protein Data Bank code 2G72, molecule A) using the entire resolution range to 2.0 Å. The solution for both hPNMT inhibitor complexes (P432121 space group) had an asymmetric unit consisting of two hPNMT molecules. Log likelihood gain values were 6058 for hPNMT-19 and 8361 for hPNMT-29 respectively. Iterative model building and refinement were carried out using COOT 91 and PHENIX.92 The statistics for data collection, refinement, and validation are shown in Table 3. Structures were deposited to the Protein Data Bank under accession codes 4MIK (hPNMT-19) and 4MQ4 (hPNMT-29).
5′-(2-Aminoethyl)thio-5′-deoxy-adenosine (16).
Compound 34 (200 mg, 0.546 mmol) was dissolved in HCOOH/H2O (4:1, 10 mL). The solution was stirred at room temperature for 8 h and Na2CO3 was added until the mixture was basic. The solvents were removed under reduced pressure and the residue was purified by flash chromatography (silica gel) with CHCl3/CH3OH/NH3·H2O (10:1:0.1) as eluent to yield 16 as a white solid foam (160 mg, 90%): 1H NMR (400 MHz, CD3OD) δ 8.29 (s, 1H), 8.18 (s, 1H), 5.98 (d, J = 5.0 Hz, 1H), 4.77 (t, J = 5.2 Hz, 1H), 4.32 (t, J = 5.0 Hz, 1H), 4.17 (quartet, J = 4.6 Hz, 1H), 3.29 (m, 1H), 2.91 (m, 2H), 2.72 (m, 2H), 2.63 (m, 2H); 13C NMR (100 MHz, CD3OD) δ 156.3, 152.9, 149.6, 140.4, 119.5, 89.1, 84.7, 73.8, 73.0, 40.7, 35.6, 33.9; MS (CI) m/z (relative intensity) 327 (MH+, 25), 199 (75), 136 (90); HRMS (ESI+) m/z calcd for C12H19N6O3S (MH+) 327.1239, obsd 327.1211; Anal. Calcd for C12H18N6O3S·1/3(CHCl3·H2O): C, 39.80; H, 5.15; N, 22.58. Found: C, 39.90; H, 5.00, N, 22.44.
5′-Deoxy-5′-{2-[(1,2,3,4-tetrahydroisoquinolin-3-yl)methyl]amino-ethyl}thio-adenosine (17).
A solution of 48 (500 mg, 0.820 mmol) in TFA/H2O (9:1, 9 mL) was stirred at room temperature for 1 h, and then the solution was evaporated under vacuum. The residue was dissolved in MeOH (5 mL), Na2CO3 solution (sat, 5 mL) was added to the solution and the mixture was stirred for 10 min. The solvent was evaporated under reduced pressure. The resulting brown residue was purified by flash column chromatography (silica gel) eluting with CHCl3/MeOH/NH3OH/EtOAc/hexane (10:1:0.1:0.1:0.1) to yield 17 (300 mg, 78%) as a white foam: 1H NMR (500 MHz, CDCl3/CD3OD, 10:1) δ 8.30 (d, J = 3.3 Hz, 1H), 8.21 (s, 1H), 7.10–7.02 (m, 4H), 6.02 (d, J = 4.7 Hz, 1H), 4.79 (t, J = 5.0 Hz, 1H), 4.36 (t, J = 5.0 Hz, 1H), 4.24 (d, J = 5.2 Hz, 1H), 3.98 (br s, 2H), 3.01 (m, 3H), 2.80–2.51 (m, 8H); 13C NMR (125.7 MHz, CDCl3/CD3OD, 10:1) δ 155.9, 152.5, 149.2, 139.9, 134.4, 133.6, 128.7, 125.9, 125.7, 125.5, 119.1, 88.7, 84.3, 73.4, 72.6, 53.8, 52.9, 48.3, 48.2, 33.7, 32.4, 32.0; MS (CI) m/z (relative intensity) 472 (MH+, 25), 284 (40), 189 (50), 79 (80); HRMS (FAB+) m/z calcd for C22H30N7O3S (MH+) 472.2131, obsd 472.2152; Anal. Calcd for C22H29N7O3S·1/2CHCl3: C, 50.87; H, 5.60; N, 18.46. Found: C, 50.93; H, 5.51, N, 18.14.
5′-{2-[(7-Chloro-1,2,3,4-tetrahydroisoquinolin-3-yl)methyl]amino-ethyl}thio-5′-deoxy-adenosine (18).
This compound was prepared in a similar manner as 17 but using 49 (1.30 g, 2.01 mmol) as the starting material to afford 18 (370 mg, 36%) as a white foam: 1H NMR (500 MHz, CD3OD) δ 8.32 (d, J = 0.8 Hz, 1H), 8.22 (s, 1H), 7.10–7.02 (m, 3H), 6.03 (d, J = 4.7 Hz, 1H), 4.80 (t, J = 5.0 Hz, 1H), 4.37 (t, J = 5.0 Hz, 1H), 4.24 (d, J = 5.3 Hz, 1H), 3.93 (m, 2H), 2.98 (m, 2H), 2.86 (m, 1H), 2.84–2.66 (m, 6H), 2.43 (m, 2H); 13C NMR (125.7 MHz, CD3OD) δ 155.8, 152.5, 149.2, 139.9, 136.8, 132.6, 130.9, 130.2, 125.9, 125.5, 119.1, 88.7, 84.3, 73.4, 72.6, 53.3, 52.7, 48.4, 48.3, 33.7, 32.0, 31.8; MS (CI) m/z (relative intensity) 506 (MH+, 50), 257 (10), 166 (50), 136 (70); HRMS (FAB+) m/z calcd for C22H29ClN7O3S (MH+) 506.1741, obsd 506.1735; Anal. Calcd for C22H28ClN7O3S·2/3H2O: C, 51.01; H, 5.71; N, 18.93. Found: C, 51.09; H, 5.46, N, 18.61.
5′-Deoxy-5′-{2-[(7-nitro-1,2,3,4-tetrahydroisoquinolin-3-yl)methyl]aminoethyl}thio-adenosine (19).
This compound was prepared in a similar manner as 17 but using 50 (150 mg, 0.23 mmol) as the starting material to afford 19 (70 mg, 59%) as a slightly yellow foam: 1H NMR (500 MHz, CDCl3/CD3OD, 10:1) δ 8.33 (s, 1H), 8.24 (s, 1H), 8.01 (m, 2H), 7.34 (d, J = 8.2 Hz, 1H), 6.03 (d, J = 4.6 Hz, 1H), 4.79 (t, J = 5.0 Hz, 1H), 4.39 (t, J = 5.0 Hz, 1H), 4.26 (d, J = 5.2 Hz, 1H), 4.11 (m, 2H), 3.06–3.01 (m, 3H), 2.90–2.76 (m, 6H), 2.64 (m, 2H); 13C NMR (125.7 MHz, CDCl3/CD3OD, 10:1) δ 155.8, 152.5, 149.1, 146.1, 142.0, 139.9, 136.6, 129.9, 120.8, 120.7, 119.1, 88.8, 84.2, 73.4, 72.5, 52.9, 52.1, 48.1, 47.0, 33.7, 32.4, 31.8; MS (CI) m/z (relative intensity) 517 (MH+, 40), 327 (20), 290 (25), 136 (80); HRMS (ESI+) m/z calcd for C22H29N8O5S (MH+) 517.1982, obsd 517.1961; Anal. Calcd for C22H28N8O5S·1/2CF3COOH: C, 48.16; H, 5.01; N, 19.54. Found: C, 48.19; H, 5.21, N, 19.31.
5′-Deoxy-5′-{3-[(1,2,3,4-tetrahydroisoquinolin-3-yl)methyl]amino-propyl}thio-adenosine (20).
This compound was prepared in a similar manner as 17 using the procedure outlined previously, but using 65 (1.00 g, 1.60 mmol) as the starting material to afford 20 (420 mg, 54%) as a white foam: IR (KBr) 3424, 3181, 2925, 2827, 1650, 1593, 1470, 1327, 1127, 748 cm−1; 1H NMR (500 MHz, CD3OD) δ 8.26 (s, 1H), 8.16 (s, 1H), 7.12–7.03 (m, 4H), 5.99 (d, J = 4.9 Hz, 1H), 4.76 (t, J = 5.0 Hz, 1H), 4.32 (t, J = 4.9 Hz, 1H), 4.19 (m, 1H), 4.09 (s, 2H), 3.27 (m, 1H), 3.08–2.95 (m, 2H), 2.92–2.61 (m, 8H), 1.86 (quintet, J = 7.0 Hz, 2H); 13C NMR (125.7 MHz, CD3OD) δ 154.2, 150.9, 147.6, 138.3, 130.5, 130.4, 127.1, 125.0, 124.6, 124.1, 117.5, 87.0, 82.6, 71.8, 71.0, 49.8, 49.1, 46.8, 44.1, 32.2, 28.8, 27.7, 25.1; MS (CI) m/z (relative intensity) 486 (MH+, 45), 284 (20), 250 (40), 132 (80); HRMS (ES+) m/z calcd for C23H32N7O3S (MH+) 486.2287, obsd 486.2265; Anal. Calcd for C23H31N7O3S·1/2 H2O: C, 55.85; H, 6.52; N, 19.82. Found: C, 55.72; H, 6.45, N, 19.39.
5′-Deoxy-5′-{3-[(7-chloro-1,2,3,4-tetrahydroisoquinolin-3-yl)methyl]amino-propyl}thio-adenosine (21).
This compound was prepared in a similar manner as 17 using the procedure outlined previously, but using 66 (270 mg, 0.410 mmol) as the starting material to afford 21 (130 mg, 61.0%) as a white foam: 1H NMR (400 MHz, CD3OD) δ 8.32 (s, 1H), 8.21 (s, 1H), 7.10 (m, 3H), 5.99 (d, J = 5.0 Hz, 1H), 4.78 (t, J = 5.0 Hz, 1H), 4.33 (t, J = 5.0 Hz, 1H), 4.23 (m, 1H), 3.97 (s, 2H), 2.95 (m, 3H), 2.79–2.45 (m, 8H), 1.79 (quintet, J = 7.1 Hz, 2H); 13C NMR (125.7 MHz, CD3OD) δ 155.9, 152.4, 149.2, 139.9, 136.8, 132.6, 131.0, 130.2, 125.9, 125.5, 119.1, 88.7, 84.3, 73.4, 72.5, 53.8, 52.6, 48.4, 48.1, 33.8, 31.9, 30.1, 28.8; MS (CI) m/z (relative intensity) 520 (MH+, 45), 271 (20), 239 (15), 136 (80); HRMS (ESI+) m/z calcd for C23H31ClN7O3S (MH+) 520.1898, obsd 520.1892; Anal. Calcd for C23H30ClN7O3S·MeOH: C, 52.21; H, 6.21; N, 17.76. Found: C, 52.36; H, 5.91, N, 17.55.
5′-Deoxy-5′-{3-[(7-nitro-1,2,3,4-tetrahydroisoquinolin-3-yl)methyl]amino-propyl}thio-adenosine (22).
This compound was prepared in a similar manner as 17 using the procedure outlined previously, but using 67 (400 mg, 0.600 mmol) as the starting material to afford 22 (210 mg, 66%) as a slightly yellow foam: 1H NMR (400 MHz, CDCl3/CD3OD, 10:1) δ 8.30 (s, 1H), 8.23 (s, 1H), 7.99 (m, 2H), 7.32 (d, J = 8.3 Hz, 1H), 6.02 (d, J = 4.6 Hz, 1H), 4.76 (t, J = 4.6 Hz, 1H), 4.36 (t, J = 5.1 Hz, 1H), 4.27 (m, 1H), 4.12 (m, 2H), 2.98 (m, 4H), 2.71 (m, 7H), 1.82 (quintet, J = 7.1 Hz, 2H); 13C NMR (100 MHz, CDCl3/CD3OD, 10:1) δ 156.2, 152.9, 149.6, 146.5, 142.6, 140.2, 137.1, 130.4, 121.4, 121.3, 119.6, 89.3, 84.4, 74.1, 72.9, 54.2, 52.7, 34.4, 33.2, 30.7, 29.3; MS (CI) m/z (relative intensity) 531 (MH+, 40), 145 (20), 79 (70); HRMS (ESI+) m/z calcd for C23H31N8O5S (MH+) 531.2138, obsd 531.2132; Anal. Calcd for C23H30N8O5S·1/2CHCl3: C, 47.82; H, 5.21; N, 18.98. Found: C, 47.85; H, 5.17, N, 18.82.
5′-Deoxy-5′-{4-[(1,2,3,4-tetrahydroisoquinolin-3-yl)methyl]amino-butyl}thio-adenosine (23).
This compound was prepared in a similar manner as 17 using the procedure outlined previously, but using 68 (450 mg, 0.700 mmol) as the starting material to afford 23 (240 mg, 68%) as a white foam: 1H NMR (400 MHz, CDCl3/CD3OD, 10:1) δ 8.29 (s, 1H), 8.24 (s, 1H), 7.11 (m, 3H), 7.04 (m, 1H), 6.02 (s, 1H), 4.76 (m, 1H), 4.36 (m, 1H), 4.25 (m, 1H), 4.02 (s, 2H), 3.02 (m, 2H), 2.99 (m, 1H), 2.77 (m, 2H), 2.61 (m, 6H), 1.61 (m, 4H); 13C NMR (100 MHz, CDCl3/CD3OD, 10:1) δ 156.3, 152.9, 149.6, 140.2, 134.9, 134.1, 129.2, 126.5, 126.3, 126.1, 119.6, 89.2, 84.6, 74.1, 72.9, 54.6, 53.4, 49.5, 48.8, 34.3, 33.0, 32.9, 28.7, 27.5; MS (CI) m/z (relative intensity) 500 (MH+, 50), 251 (20), 164 (25), 136 (70); HRMS (ESI+) m/z calcd for C24H34N7O3S (MH+) 500.2444, obsd 500.2439; Anal. Calcd for C24H33N7O3S·1/3(CHCl3·H2O): C, 53.58; H, 6.28; N, 17.98. Found: C, 53.75; H, 6.26, N, 17.71.
5′-Deoxy-5′-{5-[(1,2,3,4-tetrahydroisoquinolin-3-yl)methyl]amino-pentyl}thio-adenosine (24).
This compound was prepared in a similar manner as 17 using the procedure outlined previously, but using 69 (0.75 g, 1.2 mmol) as the starting material to afford 24 (0.44 g, 75%) as a slightly yellow foam: 1H NMR (500 MHz, CDCl3) δ 8.20 (s, 1H), 8.08 (s, 1H), 7.08 (br s, 2H, NH2), 7.00 (m, 4H), 6.02 (s, 1H), 4.72 (m, 1H), 4.34 (m, 1H), 4.29 (m, 1H), 4.00 (s, 2H), 3.02 (m, 1H), 2.87–2.48 (m, 10H), 1.47–1.28 (m, 6H); 13C NMR (125.7 MHz, CDCl3) δ 155.9, 152.9, 149.4, 139.6, 135.3, 134.1, 129.4, 126.5, 126.3, 126.1, 119.8, 89.4, 84.4, 74.4, 73.0, 54.6, 53.2, 49.8, 48.0, 34.8, 33.2, 29.5, 29.2, 26.4; MS (ESI+) m/z (relative intensity) 514 (MH+, 30); HRMS (ESI+) m/z calcd for C25H36N7O3S (MH+) 514.2600, obsd 514.2614; HPLC analysis of the diastereomeric product mixture observed a single peak at 11.2 min (System A, 250 nm, purity 96%), and at 14.2 min (System B, 240 nm, purity 96%).
5′-Deoxy-5′-{6-[(1,2,3,4-tetrahydroisoquinolin-3-yl)methyl]amino-hexyl}thio-adenosine (25).
This compound was prepared in a similar manner as 17 using the procedure outlined previously, but using 70 (0.70 g, 1.0 mmol) as the starting material to afford 25 (0.24 g, 43%) as a slightly yellow foam: 1H NMR (400 MHz, CDCl3) δ 8.17 (s, 1H), 8.03 (s, 1H), 7.05 (br ex s, 2H, NH2), 7.00 (m, 4H), 6.02 (s, 1H), 4.69 (m, 1H), 4.27 (m, 2H), 3.97 (m, 2H), 2.97 (m, 1H), 2.86–2.45 (m, 10H), 1.40 (m, 4H), 1.20 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 156.0, 153.0, 149.4, 139.5, 135.5, 134.2, 129.4, 126.4, 126.3, 126.1, 119.8, 89.3, 84.2, 74.4, 73.0, 54.9, 53.4, 50.1, 48.1, 34.7, 33.2, 31.2, 29.8, 29.6, 28.7, 26.9; MS (ESI+) m/z (relative intensity) 528 (MH+, 20), 405 (50); HRMS (ESI+) m/z calcd for C26H38N7O3S (MH+) 528.2757, obsd 528.2759; HPLC analysis of the diastereomeric product mixture observed a single peak at 15.6 min (System A, 255 nm, purity 97%), and at 15.7 min (System B, 250 nm, purity 99%).
5′-Deoxy-5′-[2-(1,2,3,4-tetrahydroisoquinoline-3-carboxamido)ethyl]thio-adenosine (26).
Compound 71 (4.40 g, 7.04 mmol) was dissolved in H2O/HCOOH (1:4, 20 mL). The solution was stirred at the room temperature for 40 h. The H2O and HCOOH were removed under reduced pressure, the residue was mixed with CH3OH (10 mL), and NH3·H2O was added until pH = 12. The solvents were removed under reduced pressure and the residue was purified by flash chromatography (silica gel) with CHCl3/MeOH (10:1) as eluent to yield 26 as a white foam (2.60 g, 76%): IR (KBr) 3421, 2914, 1650, 1598, 1475, 1424, 1327, 1250, 1132, 748 cm−1; 1H NMR (400 MHz, CD3OD) δ 8.32 (s, 1H), 8.21 (s, 1H), 7.13–7.02 (m, 4H), 6.03 (d, J = 5.0 Hz, 1H), 4.80 (t, J = 5.1 Hz, 1H), 4.35 (t, J = 4.9 Hz, 1H), 4.25–4.21 (m, 1H), 4.04–3.94 (m, 2H), 3.53 (dd, J = 4.7, 10.5 Hz, 1H), 3.42 (m, 2H), 3.02–2.86 (m, 4H), 2.74–2.70 (m, 2H); 13C NMR (100 MHz, CD3OD) δ 174.4, 156.3, 152.9, 149.7, 140.4, 135.2, 133.8, 129.0, 126.5, 126.2, 125.9, 119.6, 89.1, 84.9, 73.9, 73.0, 56.7, 46.8, 38.9, 34.1, 32.1, 31.5; MS (CI) m/z (relative intensity) 486 (MH+, 60), 237 (15), 164 (80), 132 (80); HRMS (FAB+) m/z calcd for C22H28N7O4S (MH+) 486.1923, obsd 486.1926; Anal. Calcd for C22H27N7O4S·1/2H2O: C, 53.43; H, 5.71; N, 19.82. Found: C, 53.19; H, 5.51; N, 19.52.
5′-[2-(7-Chloro-1,2,3,4-tetrahydroisoquinoline-3-carboxamido)ethyl]thio-5′-deoxy-adenosine (27).
A solution of 72 (200 mg, 0.300 mmol) in TFA/H2O (9:1, 3 mL) was stirred at room temperature for 2 h, and the solvents were evaporated under the vacuum. The residue was dissolved in MeOH (5 mL), and NaOH (10%) was added until pH = 12. The solvents were removed under reduced pressure. The resulting brown residue was purified by flash column chromatography (silica gel) eluting with CHCl3/MeOH (20:1) to yield 27 (80 mg, 51%) as a white foam: 1H NMR (500 MHz, CDCl3/CD3OD, 10:1) δ 8.45 (s, 1H), 8.43 (s, 1H), 7.31 (m, 1H), 7.26 (m, 2H), 6.21 (d, J = 4.6 Hz, 1H), 4.94 (t, J = 5.1 Hz, 1H), 4.54 (t, J = 4.9 Hz, 1H), 4.45 (quartet, J = 5.4 Hz, 1H), 4.18–4.13 (m, 2H), 3.72 (dd, J = 5.4, 10.2 Hz, 1H), 3.64 (t, J = 6.6 Hz, 2H), 3.23–3.18 (m, 2H), 3.16–3.00 (m, 2H), 2.94 (m, 2H); 13C NMR (125.7 MHz, CDCl3/CD3OD, 10:1) δ 173.8, 155.8, 152.5, 149.1, 139.7, 137.0, 132.2, 131.4, 130.0, 126.2, 125.4, 119.3, 88.9, 84.0, 73.7, 72.4, 56.0, 46.2, 38.4, 33.8, 31.9, 30.5; MS (FAB+) m/z (relative intensity) 520 (MH+, 29), 329 (80), 176 (100), 154 (100); HRMS (FAB+) m/z calcd for C22H27ClN7O4S (MH+) 520.1534, obsd 520.1531; Anal. Calcd for C22H26ClN7O4S·2/3CHCl3: C, 45.40; H, 4.48; N, 16.35. Found: C, 45.62; H, 4.53; N, 16.36.
5′-Deoxy-5′-[2-(7-nitro-1,2,3,4-tetrahydroisoquinoline-3-carboxamido)ethyl]thio-adenosine (28).
This compound was prepared in a similar manner as 26 but using 73 (0.70 g, 1.0 mmol) as the starting material to afford 28 (220 mg, 40%) as a yellow foam: 1H NMR (400 MHz, DMSO-d6) δ 8.37 (s, 1H), 8.15 (s, 1H), 8.10 (ex m, 1H), 7.97 (m, 2H), 7.39 (d, J = 8.8 Hz, 1H), 7.31 (ex s, 1H), 5.89 (d, J = 5.8 Hz, 1H), 5.53–5.35 (ex m, 1H), 4.75 (t, J = 5.0 Hz, 1H), 4.14 (m, 1H), 4.05–3.91 (m, 2H), 4.01 (m, 1H), 3.46 (quartet, J = 4.7 Hz, 1H), 3.34 (br ex m, 2H), 3.26 (m, 2H), 3.06–2.83 (m, 4H), 2.61 (t, J = 6.6 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 172.9, 156.9, 153.5, 150.3, 146.3, 144.1, 140.7, 139.0, 131.1, 121.5, 121.4, 120.0, 88.1, 84.6, 73.4, 73.3, 55.9, 47.1, 39.1, 34.1, 32.1, 31.7; MS (ESI+) m/z (relative intensity) 531 (MH+, 100), 396 (15), 266 (10); HRMS (ESI+) m/z calcd for C22H27N8O6S (MH+) 531.1774, obsd 531.1782; HPLC analysis of the diastereomeric product mixture observed a single peak at 11.1 min (System A, 289 nm, purity 95%), and at 13.1 min (System B, 300 nm, purity 96%).
5′-Deoxy-5′-[3-(1,2,3,4-tetrahydroisoquinoline-3-carboxamido)propyl]thio-adenosine (29).
This compound was prepared in a similar manner as 26 using the procedures outlined previously, but using 74 (0.70 g, 1.1 mmol) as the starting material to afford 29 (300 mg, 55%) as a white foam: 1H NMR (500 MHz, CD3OD) δ 8.26 (s, 1H), 8.15 (s, 1H), 7.05 (m, 3H), 6.97 (t, J = 4.2 Hz, 1H), 5.96 (d, J = 4.9 Hz, 1H), 4.73 (m, 1H), 4.30 (t, J = 5.0 Hz, 1H), 4.17 (q, J = 5.0 Hz, 1H), 3.92 (q, J = 10.3 Hz, 2H), 3.44 (dd, J = 4.7, 10.5 Hz, 1H), 3.25 (m, 2H), 2.90 (m, 4H), 2.54 (t, J = 7.1 Hz, 2H), 1.73 (quintet, J = 7.1 Hz, 2H); 13C NMR (125.7 MHz, CD3OD) δ 174.0, 155.8, 152.4, 149.2, 139.9, 134.7, 133.4, 128.5, 125.9, 125.8, 125.4, 119.1, 88.6, 84.2, 73.5, 72.5, 56.3, 46.4, 37.7, 33.8, 31.2, 29.6, 28.9; MS (CI) m/z (relative intensity) 500 (MH+, 50), 219 (10), 136 (70); HRMS (ESI+) m/z calcd for C23H30N7O4S (MH+) 500.2080, obsd 500.2082; Anal. Calcd for C23H29N7O4S·H2O: C, 53.37; H, 6.04; N, 18.94. Found: C, 53.29; H, 5.68, N, 18.66.
5′-Deoxy-5′-[3-(7-nitro-1,2,3,4-tetrahydroisoquinoline-3-carboxamido)propyl]thio-adenosine (30).
This compound was prepared in a similar manner as 26 using the procedure outlined previously, but using 75 (0.90 g, 1.3 mmol) as the starting material to afford 30 (470 mg, 66%) as a white foam: 1H NMR (400 MHz, CDCl3/CD3OD, 10:1) δ 8.28 (s, 1H), 8.17 (s, 1H), 7.96 (d, J = 8.3 Hz, 1H), 7.94 (s, 1H), 7.29 (d, J = 8.3 Hz, 1H), 5.98 (d, J = 4.8 Hz, 1H), 4.73 (t, J = 5.0 Hz, 1H), 4.32 (t, J = 5.0 Hz, 1H), 4.21 (quartet, J = 5.1 Hz, 1H), 4.08 (m, 2H), 3.53 (m, 1H), 3.29 (m, 2H), 3.06–2.87 (m, 4H), 2.59 (t, J = 7.2 Hz, 2H), 1.77 (quintet, J = 7.0 Hz, 2H); 13C NMR (100 MHz, CDCl3/CD3OD, 10:1) δ 173.8, 156.3, 152.9, 149.6, 146.7, 142.2, 140.2, 137.3, 130.2, 121.2, 121.0, 119.6, 89.1, 84.6, 74.0, 72.9, 56.0, 46.7, 38.3, 34.3, 31.6, 30.1, 29.3; (FAB+) m/z (relative intensity) 545 (MH+, 45), 460 (15), 307 (55), 242 (20), 154 (100), 136 (85); HRMS (FAB+) m/z calcd for C23H29N8O6S (MH+) 545.1931, obsd 545.1912; HPLC analysis of the diastereomeric product mixture observed a single peak at 11.1 min (System A, 280 nm, purity 96%), and at 14.2 min (System B, 280 nm, purity 98%).
5′-Deoxy-5′-{2-[(1,2,3,4-tetrahydroisoquinolin-4-yl)methyl]amino-ethyl}thio-adenosine (31).
A solution of 80 (0.930 g, 1.49 mmol) in TFA/H2O (9:1, 18 mL) was stirred at room temperature for 1 h, and then the solution was evaporated under vacuum. The residue was dissolved in MeOH (20 mL). Na2CO3 solution (sat, 7 mL) was added to the solution and the mixture was stirred for 10 min. The solvent was evaporated under reduced pressure. The resulting brown residue was purified by flash column chromatography (silica gel) eluting with CHCl3/MeOH/NH4OH/EtOAc/hexanes (10:1:0.1:0.1:0.1) to yield 31 (0.60 g, 86%) as a white foam: 1H NMR (500 MHz, CD3OD) δ 8.27 (s, 1H), 8.20 (s, 1H), 7.64–7.36 (m, 1H), 7.31 (m, 2H), 7.15 (dd, J = 7.6, 25.4 Hz, 1H), 5.98 (m, 1H), 4.79 (m, 1H), 4.31 (m, 1H), 4.20 (m, 2H), 4.14 (m, 1H), 3.57 (m, 1H), 3.34–3.19 (m, 2H), 3.14 (m, 2H), 2.93–2.58 (m, 6H); 13C NMR (125.7 MHz, CD3OD) δ 155.9, 152.5, 149.1, 140.0, 137.1, 130.9, 128.4, 128.2, 127.9, 125.9, 119.1, 88.9, 84.3, 73.1, 72.7, 66.6, 58.4, 54.0, 52.1, 45.6, 33.8, 30.7; MS (CI) m/z (relative intensity) 472 (MH+, 60), 339 (25), 204 (30), 144 (80), 115 (40); HRMS (FAB+) m/z calcd for C22H30N7O3S (MH+) 472.2112, obsd 472.2112; HPLC analysis of the diastereomeric product mixture observed two partially resolved peaks at 9.3 and 9.7 min for System A (251 nm, combined purity 95%), and at 10.7 and 10.9 min for System B (250 nm, combined purity 99%).
5′-Deoxy-5′-{3-[(1,2,3,4-tetrahydroisoquinolin-4-yl)methyl]amino-propyl}thio-adenosine (32).
This compound was prepared in a similar manner as 31 using the procedures outlined previously but using 81 (1.10 g, 1.76 mmol) as the starting material to afford 32 (0.65 g, 74%) as a white foam: 1H NMR (500 MHz, CD3OD) δ 8.30 (s, 1H), 8.19 (s, 1H), 7.45–7.15 (m, 1H), 7.17 (m, 2H), 7.00 (m, 1H), 5.99 (d, J = 5.0 Hz, 1H), 4.78 (t, J = 5.0 Hz, 1H), 4.33 (t, J = 5.0 Hz, 1H), 4.20 (q, J = 5.0 Hz, 1H), 3.90 (d, J = 10.0 Hz, 2H), 3.15–3.02 (m, 2H), 2.96–2.79 (m, 5H), 2.63–2.54 (m, 4H), 1.76–1.67 (m, 2H); 13C NMR (125.7 MHz, CD3OD) δ 157.4, 154.0, 150.8, 141.5, 137.6, 136.8, 129.9, 127.6, 127.4, 127.3, 120.6, 90.2, 85.8, 74.9, 74.0, 70.1, 59.7, 55.3, 47.2, 37.9, 35.4, 31.8, 30.3; MS (CI) m/z (relative intensity) 486 (MH+, 60), 237 (20), 136 (80); HRMS (ESI+) m/z calcd for C23H32N7O3S (MH+) 486.2287, obsd 486.2292; Anal. Calcd for C23H31N7O3S·1/2CHCl3: C, 51.76; H, 5.82; N, 17.98. Found: C, 52.08; H, 5.92, N, 17.75.
5′-(2-Aminoethyl)thio-5′-deoxy-2′,3′-O-(1-methylethylidene)-adenosine (34).
Sodium (2.80 g, 123 mmol) was dissolved in EtOH (200 mL) at 0 °C, and the mixture was stirred for 0.5 h. 2-Aminoethylthiol·HCl salt (7.00 g, 61.6 mmol) was added slowly into the sodium ethoxide solution at 0°C and the mixture was allowed to warm up to room temperature, and then stirred for 20 min. Compound 33 (10.0 g, 30.6 mmol) was added, and the mixture was refluxed for 8 h. The solvent was removed under reduced pressure, and the residue was purified by flash column chromatography (silica gel) eluting with CHCl3/MeOH/NH4OH (10:1:0.1) to yield 34 as a yellowish foam (11.0 g, 98%): 1H NMR (400 MHz, CD3OD) δ 8.29 (s, 1H), 8.24 (s, 1H), 6.20 (d, J = 2.3 Hz, 1H), 5.53 (dd, J = 2.4, 6.4 Hz, 1H), 5.08 (dd, J = 3.0, 6.4 Hz, 1H), 4.34 (m, 1H), 2.81 (m, 2H), 2.70 (m, 2H), 2.60 (m, 2H), 1.60 (s, 3H), 1.21 (s, 3H); 13C NMR (100 MHz, CD3OD) δ 156.4, 153.0, 149.2, 140.9, 119.6, 114.5, 90.7, 87.2, 84.2, 84.1, 40.7, 35.4, 33.9, 26.4, 24.5; MS (CI) m/z (relative intensity) 367 (MH+, 70), 232 (35), 164 (75), 136 (80), 44 (45); HRMS (FAB+) m/z calcd for C15H23N6O3S (MH+) 367.1552, obsd 367.1571.
(±)-4-Chloro-N-(ethoxycarbonyl)phenylalanine, ethyl ester (36).93
4-Chlorophenylalanine ethyl ester hydrochloride 35 (2.00 g, 7.57 mmol) was mixed with pyridine (2.27 g, 28.8 mmol) and CHCl3 (100 mL) at 0 °C. Ethyl chloroformate (0.990 g, 9.08 mmol) was added dropwise and the suspension was warmed to room temperature. The mixture was stirred for 8 h. Ice water (80 mL) was added, and the mixture was stirred for 10 min. The phases were separated, and the aqueous phase was extracted with CHCl3 (2 × 40 mL). The organic phases were combined, washed with HCl (3 N, 3 × 30 mL), brine (2 × 30 mL), and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure to afford 36 as a colorless oil (2.25 g, 99%): 1H NMR (400 MHz, CDCl3) δ 7.26 (d, J = 8.0 Hz, 2H), 7.06 (d, J = 8.0 Hz, 2H), 5.32 (br s, 1H), 4.58 (m, 1H), 4.17–4.05 (m, 4H), 3.18–2.99 (m, 2H), 1.23 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 171.8, 156.3, 134.9, 133.3, 131.1, 128.9, 61.9, 61.5, 55.0, 38.0, 14.9, 14.6; MS (FAB+) m/z (relative intensity) 300 (MH+, 75), 226 (100), 154 (30); HRMS (ESI+) m/z calcd for C14H19ClNO4 (MH+) 300.1003, obsd 300.1003.
(±)-Diethyl 7-chloro-3,4-dihydro-2,3(1H)-isoquinolinedicarboxylate (37).
To a solution of carbamate 36 (2.30 g, 7.69 mmol) in AcOH (9 mL)/H2SO4 (3 mL) was added paraformaldehyde (240 mg). After stirring overnight, the mixture was poured into ice water (50 mL). The resulting solution was extracted with EtOAc (3 × 20 mL). The combined organic extracts were washed with sodium bicarbonate (sat, 30 mL), brine (30 mL), and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure to afford 37 as a colorless oil (1.66 g, 69%): 1H NMR (500 MHz, CDCl3) δ 7.02–6.95 (m, 3H), 5.05–4.83 (m, 1H), 4.63–4.42 (m, 2H), 4.14–3.92 (m, 4H), 3.14–2.96 (m, 2H), 1.24–1.13 (m, 3H), 1.01 (t, J = 7.1 Hz, 3H); 13C NMR (125.7 MHz, CDCl3) δ (170.8, 170.6), (155.9, 155.3), (134.9, 134.3), (132.2, 132.1), (130.2, 130.1), (129.3, 128.6), (126.7, 126.6), (126.0, 125.9), (62.1, 61.8), 61.0, (53.3, 52.6), (43.9, 43.8), (30.8, 30.5), (14.5, 14.4), 13.9; MS (EI) m/z (relative intensity) 311 (M+, 60), 238 (100), 164 (40), 103 (25); HRMS (ESI+) m/z calcd for C15H18ClNO4 (M+) 311.0924, obsd 311.0918.
(±)-7-Chloro-1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid Hydrochloride (38•HCl).
A solution of 37 (500 mg, 1.61 mmol) in HCl (6 N, 20 mL) was heated at reflux for 40 h. The reaction mixture was cooled to room temperature, Et2O (20 mL) was added, and the mixture was filtered to afford 38•HCl (490 mg, 48%) as a white solid: mp ≥ 285 °C; 1H NMR (500 MHz, DMSO-d6) δ 7.41 (s, 1H), 7.35–7.30 (m, 2H), 4.40 (dd, J = 4.9, 11.0 Hz, 1H), 4.33 (m, 2H), 3.34–3.10 (m, 2H); 13C NMR (125.7 MHz, DMSO-d6) δ 170.0, 131.6, 131.2, 131.0, 130.3, 127.8, 126.6, 53.2, 43.7, 27.9; MS (CI) m/z (relative intensity) 212 (MH+, 90), 166 (75); HRMS (FAB+) m/z calcd for C10H11ClNO2 (MH+) 212.0478, obsd 212.0464.
(±)-7-Chloro-3,4-dihydro-2(1H)-(1,1-dimethylethoxy)carbonyl-isoquinoline-3-carboxylic acid (39).
A mixture of 38•HCl (200 mg, 0.810 mmol) and CH2Cl2 (5 mL) was cooled to 0 °C and Et3N (326 mg, 3.20 mmol) was added slowly. A solution of (Boc)2O (352 mg, 1.61 mmol) in CH2Cl2 (1 mL) was then added over 10 min. The mixture was stirred at room temperature overnight. Aqueous citric acid (sat, 6 mL) was added and the mixture was stirred for 5 min. The organic phase was washed with brine (2 × 5 mL), and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the residue was purified by flash column chromatography (silica gel) eluting with CHCl3/MeOH (20:1) to yield 39 as a white solid (140 mg, 56%): mp 103–105 °C; 1H NMR (400 MHz, CDCl3) δ 7.14–7.02 (m, 3H), 5.15–4.78 (m, 1H), 4.67–4.41 (m, 2H), 3.24–3.06 (m, 2H), 1.51, 1.42 (two peaks, 9H); 13C NMR (100 MHz, CDCl3) δ (177.4, 176.9), (155.9, 155.2), (135.8, 134.8), (132.9, 132.8), (130.7, 130.5), (130.3, 129.7), (127.5, 127.3), (126.8, 126.6), 81.7, (54.2, 52.5), (44.6, 44.0), (31.2, 30.8), 28.8, 28.6, 28.4; MS (CI) m/z (relative intensity) 312 (MH+, 60), 212 (80), 166 (80), 84 (25); HRMS (FAB+) m/z calcd for C15H18ClNO4 (M+) 311.0924, obsd 311.0888.
(±)-3,4-Dihydro-2(1H)-(1,1-dimethylethoxy)carbonyl-7-nitro-isoquinoline-3-carboxylic acid (40).
THIQ-3-carboxylic acid hydrochloride (15•HCl, 2.00 g, 9.36 mmol) was dissolved in concentrated H2SO4 (8 mL) at 0 °C. KNO3 (1.04 g, 10.3 mmol) was added in small portions over 30 min. The reaction mixture was stirred at room temperature for 8 h and then was poured onto ice (200 g). NH3·H2O was added to the mixture until pH = 12. The water was removed under reduced pressure to yield a yellow residue. The residue was mixed with CH2Cl2 (100 mL) and the mixture was cooled to 0 °C. Et3N (1.40 g, 13.8 mmol) was added slowly. A solution of (Boc)2O (3.06 g, 14.0 mmol) in CH2Cl2 (10 mL) was then added over 10 min. The mixture was warmed to room temperature and stirred overnight. Aqueous citric acid (sat, 20 mL) was added and the mixture was stirred for 5 min. The organic phase was washed with brine (2 × 40 mL), and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the residue was purified by chromatography on a silica gel column eluting with CHCl3/MeOH (30:1) to yield a yellow foam (1.70 g), which was purified by the reported procedures to afford regiopure 40.75
(±)-1,1-Dimethylethyl 3,4-dihydro-3-(N-methoxy-N-methylcarbamoyl)-isoquinoline-2(1H)-carboxylate (42).78
Weinreb amide 42 was prepared by following the literature procedure.16 BOP (3.67 g, 8.30 mmol) was added to a stirred solution of 41 (2.30 g, 8.30 mmol) and Et3N (1.16 mL, 8.30 mmol) in CH2Cl2 (70 mL). After five minutes, N,N-dimethylhydroxylamine hydrochloride (972 mg, 9.96 mmol) and Et3N (1.34 mL, 9.96 mmol) were added to the solution. The reaction was stirred for 24 h at room temperature. The mixture was diluted with CH2Cl2 (70 mL), and washed with HCl (3 N, 2 × 50 mL), aqueous NaHCO3 (sat, 50 mL), brine (50 mL), and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and the resulting crude product was purified by chromatography on a silica gel column eluting with EtOAc/hexanes (1:2) to yield 42 (2.21 g, 83%) as colorless crystals: mp 128–130 °C; 1H NMR (400 MHz, CDCl3) δ 7.20–7.14 (m, 4H), 5.24–4.47 (m, 3H), 3.85, 3.78 (two peaks, 3H), 3.19, 3.16 (two peaks, 3H), 3.08–2.97 (m, 2H), 1.52, 1.46 (two peaks, 9H); 13C NMR (100 MHz, CDCl3) δ (173.4, 172.9), (155.7, 155.1), (135.9, 134.9), (133.4, 132.5), (128.3, 127.7), (127.4, 127.2), (127.1, 127.0), (126.4, 126.2), (80.8, 80.6), (61.9, 61.7), (53.2, 51.1), (45.8, 44.9), (32.8, 32.5), (31.6, 31.4), 28.9.
(±)-1,1-Dimethylethyl 7-chloro-3,4-dihydro-3-(N-methoxy-N-methylcarbamoyl)-isoquinoline-2(1H)-carboxylate (43).
This compound was synthesized using the same procedure as 42 except 39 (1.90 g, 6.11 mmol) was used as the starting material to afford 43 (1.77 g, 82%) as a white solid: mp 175–177 °C; IR (KBr) 3011, 2975, 2929, 1690, 1670, 1486, 1393, 1168, 1004, 861, 763 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.15–7.05 (m, 3H), 5.28–4.47 (m, 3H), 3.84–3.68 (m, 3H), 3.15–2.97 (m, 5H), 1.51, 1.39 (two peaks, 9H); 13C NMR (125.7 MHz, CDCl3) δ (172.5, 172.1), (155.1, 154.4), (136.8, 135.9), (132.3, 132.1), (131.0, 130.3), (129.3, 128.7), (126.9, 126.7), (126.0, 125.8), (80.6, 80.4), (61.4, 61.2), (52.2, 50.1), (45.0, 44.2), (32.3, 32.0), (30.6, 30.4), 28.4; MS (FAB+) m/z (relative intensity) 355 (MH+, 50), 255 (100), 154 (75); HRMS (FAB+) m/z calcd for C17H24ClN2O4 (MH+) 355.1425, obsd 355.1321.
(±)-1,1-Dimethylethyl 3,4-dihydro-3-(N-methoxy-N-methylcarbamoyl)-7-nitro-isoquinoline-2(1H)-carboxylate (44).
BOP (2.33 g, 5.28 mmol) was added to a stirred solution of crude 40 (1.70 g, 5.28 mmol) and Et3N (0.74 mL, 5.3 mmol) in CH2Cl2 (60 mL). After five minutes, N-dimethylhydroxylamine hydrochloride (0.46 g, 6.3 mmol) and Et3N (0.88 mL, 6.3 mmol) were added to the solution. The mixture was stirred for 24 h at room temperature. The mixture was then diluted with CH2Cl2 (20 mL), and washed with HCl (3 N, 2 × 20 mL), aqueous NaHCO3 (sat, 30 mL), and brine (30 mL), and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and the resulting crude product was purified by chromatography on a silica gel column eluting with EtOAc/hexanes (1:5) to yield amide 44 (1.20 g, 35% from 15) as slightly yellow crystals: mp 174–175 °C; 1H NMR (400 MHz, CDCl3) δ 8.04 (br s, 2H), 7.30 (m, 1H), 5.42–5.07 (m, 1H), 4.85 (m, 2H), 3.86, 3.81 (two peaks, 3H), 3.30–3.10 (m, 5H), 1.57, 1.49 (two peaks, 9H); 13C NMR (100 MHz, CDCl3) δ (172.4, 172.1), (155.3, 154.7), 147.1, (140.5, 139.9), (136.8, 136.0), (129.6, 129.0), (122.4, 122.1), (121.7, 121.6), (81.4, 81.3), (62.0, 61.8), (51.9, 49.9), (45.6, 44.7), (32.7, 32.4), 31.6, 28.8; MS (ESI+) m/z (relative intensity) 366 (MH+, 62), 266 (60), 177 (65); HRMS (ESI+) m/z calcd for C17H24N3O6 (MH+) 366.1665, obsd 366.1662.
(±)-1,1-Dimethylethyl 3,4-dihydro-3-formyl-isoquinoline-2(1H)-carboxylate (45).
To a stirred solution of 42 (160 mg, 0.500 mmol) in Et2O (6 mL), LiAlH4 (24 mg, 0.63 mmol) was added at 0 °C. The reaction was stirred for 1 h at room temperature and the reaction mixture was then hydrolyzed with a solution of KHSO4 (120 mg, 0.880 mmol) in water (2.5 mL). The aqueous phase was separated and extracted with Et2O (3 × 5 mL). The organic phases were combined, washed with HCl (3 N, 10 mL), NaHCO3 (sat, 10 mL), brine (10 mL) and then dried over anhydrous Na2SO4. After solvent evaporation, aldehyde 45 (100 mg, 77%) was obtained in pure form as an oil: 1H NMR (500 MHz, CDCl3) δ 9.52, 9.48 (two peaks, 1H), 7.21–7.11 (m, 4H), 4.74–4.57 (m, 3H), 3.12–3.10 (m, 2H), 1.55–1.47 (m, 9H).78,94
(±)-1,1-Dimethylethyl 7-chloro-3,4-dihydro-3-formyl-isoquinoline-2(1H)-carboxylate (46).
This compound was prepared in a similar manner as 45 but using 43 (1.70 g, 4.79 mmol) as the starting material to afford 46 (1.25 g, 88%) as a colorless oil: IR (KBr) 3436, 3058, 2971, 2925, 2802, 2704, 2249, 1736, 1690, 1598, 1490, 1388, 1250, 1158, 1024, 866, 814, 732 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.53, 9.49 (two peaks, 1H), 7.18–7.12 (m, 3H), 4.87–4.49 (m, 3H), 3.22–3.04 (m, 2H), 1.55, 1.48 (two peaks, 9H); 13C NMR (100 MHz, CDCl3) δ (201.1, 200.5), 155.0, (136.1, 135.1), (133.2, 133.0), (131.1, 130.7), (130.3, 129.6), (127.9, 127.6), (126.9, 126.7), (81.9, 81.7), (60.6, 59.3), (45.0, 44.4), (29.0, 28.0), (28.8, 28.7); MS (ESI+) m/z (relative intensity) 296 (MH+, 100), 240 (35), 196 (30); HRMS (ESI+) m/z calcd for C15H19ClNO3 (MH+) 296.1053, obsd 296.1039.
(±)-1,1-Dimethylethyl 3,4-dihydro-3-formyl-7-nitro-isoquinoline-2(1H)-carboxylate (47).
Weinreb amide 44 (300 mg, 0.820 mmol) was mixed with Cp2Zr(H)Cl (Schwartz reagent) (0.630 g, 2.47 mmol) and THF (15 mL). The mixture was stirred at room temperature for 1 h. Water (0.1 mL) was added to the reaction and the solvent was removed under reduced pressure. The residue was purified by chromatography on a silica gel column eluting with hexanes/EtOAc (4:1) to yield aldehyde 47 (140 mg, 56%) as a slightly yellow oil: IR (KBr) 3012, 2976, 2919, 2858, 1737, 1701, 1521, 1393, 1352, 1163, 892, 856, 738 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.55 (d, J = 12.0 Hz, 1H), 8.07 (m, 2H), 7.38 (d, J = 8.5 Hz, 1H), 4.99–4.61 (m, 3H), 3.38–3.16 (m, 2H), 1.56, 1.50 (two peaks, 9H); 1H NMR (500 MHz, CDCl3) δ (199.7, 199.3), (155.1, 154.7), (146.9, 146.8), (139.7, 139.5), 134.3, (129.6, 129.0), (122.3, 122.0), 121.5, (81.8, 81.7), (59.6, 58.2), (44.7, 44.0), (28.8, 27.9), 28.2; MS (ESI+) m/z (% relative intensity) 307 (MH+, 95), 294 (50), 265 (80), 189 (45); HRMS (ESI+) m/z calcd for C15H19N2O5 (MH+) 307.1294, obsd 307.1288.
5′-Deoxy-5′-{2-{[3,4-dihydro-2(1H)-(1,1-dimethylethoxy)carbonyl-isoquinolin-3-yl]methyl}amino-ethyl}thio-2′,3′-O-(1-methylethylidene)-adenosine (48).
A mixture of aldehyde 45 (1.20 g, 4.56 mmol), amine 34 (1.90 g, 5.19 mmol), molecular sieves (4Å, 2.0 g) in THF (60 mL) was stirred at room temperature for 3 h. The mixture was filtered and the solvent was removed under reduced pressure. The residue was dissolved in ethanol (60 mL), and sodium borohydride (240 mg, 6.30 mmol) was added to the solution over 1.5 h. The mixture was stirred for 12 h at room temperature, and water (0.3 mL) was added. The solvent was removed under reduced pressure and the residue was purified by chromatography on a silica gel column eluting with CHCl3/MeOH (30:1) to yield 48 (1.34 g, 48%) as a white foam: 1H NMR (500 MHz, CDCl3) δ 8.34 (s, 1H), 7.92 (s, 1H), 7.18–7.10 (m, 4H), 6.08 (s, 1H), 5.96 (br s, 2H), 5.51 (dd, J = 1.8, 6.4 Hz, 1H), 5.05 (dd, J = 2.6, 6.2 Hz, 1H), 4.73 (m, 1H), 4.59–4.36 (m, 2H), 4.23 (m, 1H), 3.06–2.82 (m, 2H), 2.79–2.43 (m, 8H), 1.62 (s, 3H), 1.49 (s, 9H), 1.40 (s, 3H); 13C NMR (125.7 MHz, CDCl3) δ 155.6, 155.3, 153.1, 149.1, 140.0, 132.9, 129.1, 126.5, 126.1, 120.2, 114.4, 90.8, 86.8, 83.9, 83.7, 79.9, 50.2, (49.9, 48.4), 48.2, (43.3, 42.5), 34.1, 32.8, 30.9, 28.5, 27.0, 25.3; MS (CI) m/z (relative intensity) 612 (MH+, 45), 512 (50), 379 (40), 132 (70); HRMS (FAB+) m/z calcd for C30H42N7O5S (MH+) 612.2968, obsd 612.2944.
5′-{2-{[7-Chloro-3,4-dihydro-2(1H)-(1,1-dimethylethoxy)carbonyl-isoquinolin-3-yl]methyl}amino-ethyl}thio-5′-deoxy-2′,3′-O-(1-methylethylidene)-adenosine (49).
This compound was prepared in a similar manner as 48 but using aldehyde 46 (1.20 g, 4.05 mmol) as the starting material to afford 49 (1.50 g, 57%) as a white foam: 1H NMR (500 MHz, CDCl3) δ 8.27 (s, 1H), 7.89 (s, 1H), 7.06–6.97 (m, 3H), 6.70 (br s, 2H), 6.05 (s, 1H), 5.48 (d, J = 6.4 Hz, 1H), 5.02 (dd, J = 2.5, 5.8 Hz, 1H), 4.67 (m, 1H), 4.53–4.32 (m, 2H), 4.11 (m, 1H), 2.93–2.77 (m, 2H), 2.72–2.34 (m, 8H), 1.56 (s, 3H), 1.43 (s, 9H), 1.35 (s, 3H); 13C NMR (125.7 MHz, CDCl3) δ 156.0, 155.0, 153.0, 149.0, 139.8, 134.7, 131.6, 131.3, 130.4, 126.6, 126.0, 120.1, 114.2, 90.7, 86.8, 83.9, 83.7, 80.0, 49.9, 49.5, 48.1, (42.8, 42.1), 34.0, 32.8, 30.3, 28.5, 27.0, 25.2; MS (CI) m/z (relative intensity) 646 (MH+, 50), 379 (50), 244 (25), 164 (40), 136 (60); HRMS (FAB+) m/z (MH+) calcd for C30H41ClN7O5S (MH+) 646.2578, obsd 646.2572.
5′-Deoxy-5′-{2-{[3,4-dihydro-2(1H)-(1,1-dimethylethoxy)carbonyl-7-nitro-isoquinolin-3-yl]methyl}amino-ethyl}thio-2′,3′-O-(1-methylethylidene)-adenosine (50).
This compound was prepared in a similar manner as 48 but using aldehyde 47 (140 mg, 0.460 mmol) as the starting material to afford 50 (150 mg, 50%) as a slightly yellow foam: 1H NMR (500 MHz, CDCl3) δ 8.35 (s, 1H), 8.04 (d, J = 8.3 Hz, 1H), 8.02 (s, 1H), 7.94 (s, 1H), 7.31 (d, J = 8.3 Hz, 1H), 6.09 (s, 1H), 5.73 (br s, 2H, NH2), 5.53 (dd, J = 2.0, 6.4 Hz, 1H), 5.07 (t, J = 2.9 Hz, 1H), 5.03–4.20 (m, 4H), 3.08–2.94 (m, 2H), 2.87–2.44 (m, 8H), 1.62 (s, 3H), 1.51 (s, 9H), 1.41 (s, 3H); 13C NMR (125.7 MHz, CDCl3) δ 155.7, 155.1, 153.1, 149.2, 146.5, 141.0, 140.1, 134.6, 130.2, 121.7, 121.4, 120.2, 114.5, 90.9, 86.9, 84.0, 83.8, 80.6, 53.5, 50.1, 48.1, (42.8, 42.1), 34.1, 32.8, 31.3, 28.5, 27.1, 25.3; MS (ESI+) m/z (% relative intensity) 657 (MH+, 100), 557 (60), 422 (40), 301 (55), 191 (60); HRMS (ESI+) m/z calcd for C30H41N8O7S (MH+) 657.2819, obsd 657.2802.
Compounds 52 and 57 were synthesized by following the literature procedures.80
5′-Deoxy-5′-[4-(1,3-dioxoisoindolin-2-yl)butyl]thio-2′,3′-O-(1-methylethylidene)-adenosine (58).
Protected thionucleoside 52 (3.60 g, 10.0 mmol) and N-(4-bromobutyl)phthalimide (54) (4.23 g, 15.0 mmol) were added to oxygen-free absolute MeOH (150 mL) under argon. The suspension was cooled to −20 °C. NaOMe (1.20 g, 22.2 mmol, in 10 mL MeOH) solution was freshly prepared and slowly added into the suspension. The mixture was then allowed to warm up to room temperature and was stirred for 12 h. The solvent was removed under reduced pressure and the residue was mixed with H2O/CHCl3 (1:1, 100 mL). The water layer was extracted with CHCl3 (3 × 30 mL). The chloroform phases were combined together and dried with anhydrous Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by flash column chromatography (silica gel) eluting with CH2Cl2/MeOH (10:1) to yield 58 (4.4 g, 82%) as a white solid: mp 174–176 °C; 1H NMR (500 MHz, CDCl3) δ 8.35 (s, 1H), 7.95 (s, 1H), 7.83 (m, 2H), 7.70 (m, 2H), 6.20 (br s, 2H, NH2), 6.09 (d, J = 2.1 Hz, 1H), 5.52 (dd, J = 2.1, 6.4 Hz, 1H), 5.07 (m, 1H), 4.38 (m, 1H), 3.65 (t, J = 7.0 Hz, 2H), 2.83–2.77 (m, 2H), 2.54 (t, J = 7.0 Hz, 2H), 1.73 (m, 2H), 1.61 (s, 3H), 1.58 (m, 2H), 1.40 (s, 3H); 13C NMR (125.7 MHz, CDCl3) δ 168.3, 155.6, 153.0, 149.1, 139.9, 133.8, 132.0, 123.1, 120.2, 114.3, 90.8, 86.8, 83.9, 83.7, 37.3, 34.2, 31.9, 27.5, 27.0, 26.7, 25.3; MS (CI) m/z (relative intensity) 525 (MH+, 80), 234 (50), 202 (20), 164 (60); HRMS (FAB+) m/z calcd for C25H29N6O5S (MH+) 525.1920, obsd 525.1929.
5′-Deoxy-5′-[5-(1,3-dioxoisoindolin-2-yl)pentyl]thio-2′,3′-O-(1-methylethylidene)-adenosine (59).
This compound was prepared in a similar manner as 58 using the procedure outlined previously, but using phthalimide 55 (3.6 g, 10 mmol) as the starting materials to afford 59 (3.41 g, 64%) as a white foam: 1H NMR (500 MHz, CDCl3) δ 8.35 (s, 1H), 7.93 (s, 1H), 7.83 (m, 2H), 7.70 (m, 2H), 6.09 (d, J = 2.0 Hz, 1H), 5.99 (br s, 2H, NH2), 5.54 (dd, J = 2.0, 6.3 Hz, 1H), 5.06 (dd, J = 3.0, 6.3 Hz, 1H), 4.39 (m, 1H), 3.65 (t, J = 7.0 Hz, 2H), 2.83–2.77 (m, 2H), 2.46 (t, J = 7.0 Hz, 2H), 1.64 (m, 2H), 1.61 (s, 3H), 1.52 (m, 2H), 1.40 (s, 3H), 1.36 (m, 2H); 13C NMR (125.7 MHz, CDCl3) δ 168.3, 155.6, 153.1, 149.2, 140.0, 133.8, 132.1, 123.1, 120.3, 114.3, 90.8, 87.0, 83.9, 83.8, 37.7, 34.2, 32.3, 29.0, 28.0, 27.0, 25.9, 25.3; MS (FAB+) m/z (relative intensity) 539 (MH+, 70), 307 (60), 154 (100); HRMS (FAB+) m/z calcd for C26H31N6O5S (MH+) 539.2077, obsd 539.2077.
5′-Deoxy-5′-[6-(1,3-dioxoisoindolin-2-yl)hexyl]thio-2′,3′-O-(1-methylethylidene)-adenosine (60).
This compound was prepared in a similar manner as 58 using the procedure outlined previously, but using phthalimide 56 (5.00 g, 16.1 mmol) as the starting material to afford 60 (5.40 g, 71%) as a white solid: mp 85–87 °C; 1H NMR (400 MHz, CDCl3) δ 8.33 (s, 1H), 7.96 (s, 1H), 7.83 (m, 2H), 7.70 (m, 2H), 6.48 (br s, 2H, NH2), 6.11 (d, J = 2.1 Hz, 1H), 5.54 (dd, J = 2.1, 6.4 Hz, 1H), 5.07 (dd, J = 3.1, 6.4 Hz, 1H), 4.40 (m, 1H), 3.66 (t, J = 7.3 Hz, 2H), 2.80 (m, 2H), 2.46 (t, J = 7.4 Hz, 2H), 1.65 (m, 2H), 1.62 (s, 3H), 1.47 (m, 2H), 1.41 (s, 3H), 1.30 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 168.9, 156.3, 153.5, 149.5, 140.4, 134.2, 132.5, 123.6, 120.6, 114.7, 91.3, 87.5, 84.4, 84.3, 38.3, 34.6, 32.8, 29.8, 28.8, 28.6, 27.5, 26.8, 25.7; MS (ESI+) m/z (relative intensity) 553 (MH+, 80), 418 (30); HRMS (ESI+) m/z calcd for C27H33N6O5S (MH+) 553.2233, obsd 553.2228.
5′-(3-Aminopropyl)thio-5′-deoxy-2′,3′-O-(1-methylethylidene)-adenosine (61).29,73,95
Phthalimide 57 (3.20 g, 6.27 mmol) was mixed with ethanol (60 mL), and was heated to 50 °C until 57 was completely dissolved. Hydrazine (610 mg, 18.8 mmol) was added dropwise and the reaction mixture was stirred at 80 °C for 3.5 h. The solid was filtered off and washed with MeOH (20 mL). The filtrates were combined together. The solvent was removed under reduced pressure, and the residue was purified by chromatography on a silica gel column with CHCl3/MeOH/NH4OH (10:1:0.1) to yield 61 (2.08 g, 87%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 8.33 (s, 1H), 8.02 (s, 1H), 7.51 (br s, 2H, NH2), 6.17 (s, 1H), 5.58 (d, J = 6.4 Hz, 1H), 5.08 (m, 1H), 4.41 (m, 1H), 2.81(m, 2H), 2.70 (t, J = 6.8 Hz, 2H), 2.65 (m, 2H), 1.65 (br s, 2H), .161 (s, 3H), 1.46 (m, 2H), 1.40 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 156.7, 153.2, 149.2, 140.1, 120.3, 114.4, 91.0, 87.1, 84.2, 84.1, 40.9, 34.5, 33.3, 30.1, 27.2, 25.6; MS (CI) m/z (relative intensity) 381 (MH+, 50), 3222 (50), 290 (25), 186 (80), 164 (25), 136 (50); HRMS (FAB+) m/z calcd for C16H25N6O3S (MH+) 381.1709, obsd 381.1713.
5′-(4-Aminobutyl)thio-5′-dexoy-2′,3′-O-(1-methylethylidene)-adenosine (62).
This compound was prepared in a similar manner as 61 using the procedure outlined previously, but using phthalimide 58 (1.50 g, 2.86 mmol) as the starting material to afford 62 (1.10 g, 98%) as a colorless oil: 1H NMR (400 MHz, CD3OD) δ 8.26 (s, 1H), 8.21 (s, 1H), 6.17 (d, J = 2.2 Hz, 1H), 5.54 (dd, J = 2.2, 6.3 Hz, 1H), 5.05 (dd, J = 2.9, 6.3 Hz, 1H), 4.33 (m, 1H), 2.76 (d, J = 6.9 Hz, 2H), 2.54 (t, J = 6.9 Hz, 2H), 2.45 (t, J = 6.4 Hz, 2H), 1.57 (s, 3H), 1.44 (m, 4H), 1.37 (s, 3H); 13C NMR (100 MHz, CD3OD) δ 156.4, 153.0, 149.2, 140.9, 119.6, 114.3, 90.8, 87.5, 84.2, 84.1, 41.0, 34.1, 32.1, 31.8, 27.0, 26.4, 24.5; MS (CI) m/z (relative intensity) 395 (MH+, 30), 203 (10), 96 (60); HRMS (FAB+) m/z calcd for C17H27N6O3S (MH+) 395.1865, obsd 395.1877.
5′-(5-Aminopentyl)thio-5′-dexoy-2′,3′-O-(1-methylethylidene)-adenosine (63).
This compound was prepared in a similar manner as 61 using the procedure outlined previously, but using phthalimide 59 (3.2 g, 5.9 mmol) as the starting material to afford 63 (2.3 g, 94%) as a colorless oil: 1H NMR (500 MHz, CDCl3) δ 8.35 (s, 1H), 7.98 (s, 1H), 6.30 (br s, 2H, NH2), 6.09 (d, J = 1.7 Hz, 1H), 5.55 (dd, J = 1.7, 6.3 Hz, 1H), 5.06 (dd, J = 2.9, 6.3 Hz, 1H), 4.40 (m, 1H), 3.15 (br s, 2H, NH2), 2.79 (m, 2H), 2.71 (t, J = 7.1 Hz, 2H), 2.44 (t, J = 7.4 Hz, 2H), 1.61 (s, 3H), 1.45 (m, 4H), 1.40 (s, 3H), 1.32 (m, 2H); 13C NMR (125.7 MHz, CDCl3) δ 155.7, 153.0, 149.1, 140.1, 120.2, 114.3, 90.9, 87.2, 83.9, 83.8, 41.4, 34.2, 32.4, 32.0, 29.2, 27.0, 25.8, 25.3; MS (FAB+) m/z (relative intensity) 409 (MH+, 100), 274 (55), 215 (65), 185 (95), 136 (78); HRMS (FAB+) m/z calcd for C18H29N6O3S (MH+) 409.2022, obsd 409.1997.
5′-(6-Aminohexyl)thio-5′-dexoy-2′,3′-O-(1-methylethylidene)-adenosine (64).
This compound was prepared in a similar manner as 61 using the procedure outlined previously, but using phthalimide 60 (5.4 g, 9.8 mmol) as the starting material to afford 64 (4.1 g, 99%) as a colorless oil: 1H NMR (400 MHz, CDCl3/CD3OD 10:1) δ 8.33 (s, 1H), 7.97 (s, 1H), 6.82 (br ex s, 2H, NH2), 6.11 (s, 1H), 5.53 (d, J = 6.3 Hz, 1H), 5.08 (m, 1H), 4.40 (m, 1H), 2.84 (m, 2H), 2.66 (t, J = 7.0 Hz, 2H), 2.47 (t, J = 7.3 Hz, 2H), 2.37 (br ex s, 2H, NH2), 1.62 (s, 3H), 1.50 (m, 2H), 1.41 (s, 3H), 1.39 (m, 2H), 1.27 (m, 4H); 13C NMR (100 MHz, CDCl3/CD3OD 10:1) δ 156.5, 153.1, 149.0, 140.1, 120.2, 114.2, 90.8, 87.2, 84.0, 83.9, 41.9, 34.3, 33.4, 32.5, 29.6, 28.6, 27.1, 26.5, 25.4; MS (ESI+) m/z (relative intensity) 423 (MH+, 50), 288 (100), 230 (50); HRMS (ESI+) m/z calcd for C19H31N6O3S (MH+) 423.2178, obsd 423.2153.
5′-Deoxy-5′-{3-{[3,4-dihydro-2(1H)-(1,1-dimethylethoxy)carbonyl-isoquinolin-3-yl]methyl}amino-propyl}thio-2′,3′-O-(1-methylethylidene)-adenosine (65).
This compound was prepared in a similar manner as 48 using the procedure outlined previously, but using aldehyde 45 (1.53 g, 5.85 mmol) and amine 61 (2.00 g, 5.26 mmol) to afford 65 (1.30 g, 36%) as a white foam: 1H NMR (400 MHz, CDCl3) δ 8.25 (s, 1H), 7.90 (s, 1H), 7.06 (m, 4H), 6.97 (br s, 2H, NH2), 6.05 (d, J = 1.6 Hz, 1H), 5.48 (dd, J = 1.6, 6.3 Hz, 1H), 5.01 (m, 1H), 4.74–4.14 (m, 4H), 2.98–2.73 (m, 2H), 2.70 (m, 2H), 2.57–2.46 (m, 4H), 2.39–2.34 (m, 2H), 1.58 (m, 2H), 1.54 (s, 3H), 1.42 (s, 9H), 1.33 (s, 3H); 13C NMR (125.7 MHz, CDCl3) δ 156.1, 155.2, 153.0, 148.9, 139.7, 132.8, 129.0, 128.8, 126.4, 126.0, 120.1, 114.1, 90.7, 86.7, 83.9, 83.7, 79.7, 50.4, (49.6, 48.4), 48.0, (43.1, 42.4), 34.2, 30.9, 30.1, 29.8, 28.4, 27.0, 25.2; MS (ESI+) m/z (relative intensity) 626 (MH+, 100), 526 (75), 391 (20), 313 (50); HRMS (ESI+) m/z calcd for C31H44N7O5S (MH+) 626.3125, obsd 626.3122.
5′-Deoxy-5′-{3-{[7-chloro-3,4-dihydro-2(1H)-(1,1-dimethylethoxy)carbonyl-isoquinolin-3-yl]methyl}amino-propyl}thio-2′,3′-O-(1-methylethylidene)-adenosine (66).
This compound was prepared in a similar manner as 48 using the procedure outlined previously, but using aldehyde 46 (450 mg, 1.52 mmol) and amine 61 (600 mg, 1.58 mmol) as the starting materials to afford 66 (0.68 g, 68%) as a white foam: 1H NMR (500 MHz, CDCl3) δ 8.28 (s, 1H), 7.91 (s, 1H), 7.06–6.97 (m, 3H), 6.63 (br s, 2H, NH2), 6.06 (d, J = 1.6 Hz, 1H), 5.50 (d, J = 6.2 Hz, 1H), 5.02 (dd, J = 2.9, 6.2 Hz, 1H), 4.72 (m, 1H), 4.54–4.13 (m, 2H), 4.34 (m, 1H), 2.93–2.76 (m, 2H), 2.70 (m, 2H), 2.57–2.36 (m, 6H), 1.56 (m, 5H), 1.44 (s, 9H), 1.35 (s, 3H); 13C NMR (125.7 MHz, CDCl3) δ 155.9, 155.1, 153.0, 149.0, 139.8, 134.7, 131.6, 131.3, 130.3, 126.6, 125.9, 120.1, 114.2, 90.8, 86.7, 83.9, 83.7, 80.0, 50.3, 49.5, 48.1, (42.8, 42.1), 34.3, 30.4, 30.2, 29.8, 28.4, 27.0, 25.2; MS (FAB−) m/z (relative intensity) 660 (MH+, 68), 560 (20), 307 (90), 289 (55), 154 (100), 136 (70); HRMS (ESI+) m/z calcd for C31H43ClN7O5S (MH+) 660.2735, obsd 660.2759.
5′-Deoxy-5′-{3-{[3,4-dihydro-2(1H)-(1,1-dimethylethoxy)carbonyl-7-nitro-isoquinolin-3-yl]methyl}amino-propyl}thio-2′,3′-O-(1-methylethylidene)-adenosine (67).
This compound was prepared in a similar manner as 48 using the procedure outlined previously, but using aldehyde 47 (500 mg, 1.63 mmol) and amine 61 (500 mg, 1.32 mmol) as the starting materials to afford 67 (420 mg, 48%) as a yellow foam: 1H NMR (500 MHz, CDCl3) δ 8.36 (s, 1H), 8.03 (m, 2H), 7.94 (s, 1H), 7.30 (d, J = 8.3 Hz, 1H), 6.09 (d, J = 1.4 Hz, 1H), 5.72 (br s, 2H, NH2), 5.55 (dd, J = 1.7, 6.3 Hz, 1H), 5.06 (dd, J = 3.0, 6.3 Hz, 1H), 4.88 (m, 1H), 4.56–4.28 (m, 2H), 4.39 (m, 1H), 3.07–2.96 (m, 2H), 2.77 (m, 2H), 2.62–2.44 (m, 6H), 1.64 (m, 2H), 1.63 (s, 3H), 1.51 (s, 9H), 1.41 (s, 3H); 13C NMR (125.7 MHz, CDCl3) δ 155.5, 155.0, 153.0, 149.2, 146.5, 140.9, 140.0, 134.7, 130.0, 121.6, 121.3, 120.3, 114.3, 90.9, 86.9, 83.9, 83.8, 80.4, 50.5, 49.4, 48.1, 42.5, 34.4, 31.2, 30.3, 29.8, 28.4, 27.0, 25.3; MS (CI) m/z (relative intensity) 671 (MH+, 75), 571 (30), 240 (75); HRMS (ESI+) m/z calcd for C31H43N8O7S (MH+) 671.2975, obsd 671.2987.
5′-Deoxy-5′-{4-{[3,4-dihydro-2(1H)-(1,1-dimethylethoxy)carbonyl-isoquinolin-3-yl]methyl}amino-butyl}thio-2′,3′-O-(1-methylethylidene)-adenosine (68).
This compound was prepared in a similar manner as 48 using the procedure outlined previously, but using aldehyde 45 (440 mg, 1.69 mmol) and amine 62 (0.670 g, 1.69 mmol) as the starting materials to afford 68 (0.52 g, 48%) as a white foam: 1H NMR (400 MHz, CDCl3) δ 8.35 (s, 1H), 7.94 (s, 1H), 7.13 (m, 4H), 6.09 (d, J = 2.0 Hz, 1H), 5.96 (br s, 2H, NH2), 5.53 (dd, J = 2.0, 6.3 Hz, 1H), 5.06 (dd, J = 3.0, 6.3 Hz, 1H), 4.82 (m, 1H), 4.71–4.23 (m, 2H), 4.40 (m, 1H), 3.07–2.83 (m, 2H), 2.77 (m, 2H), 2.57 (m, 6H), 1.62 (s, 3H), 1.50 (s, 9H), 1.45 (m, 4H), 1.41 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 155.7, 155.4, 153.0, 149.1, 139.9, 132.9, 130.8, 129.0, 128.8, 126.5, 126.1, 120.2, 114.3, 90.8, 86.8, 83.9, 83.8, 79.9, 50.6, (49.6, 48.5), 48.9, (43.3, 42.4), 34.2, 32.4, 31.1, 29.0, 28.4, 27.1, 27.0, 25.3; MS (CI) m/z (relative intensity) 640 (MH+, 30), 407 (20), 292 (15), 136 (60); HRMS (ESI+) m/z calcd for C32H46N7O5S (MH+) 640.3281, obsd 640.3292.
5′-Deoxy-5′-{5-{[3,4-dihydro-2(1H)-(1,1-dimethylethoxy)carbonyl-isoquinolin-3-yl]methyl}amino-pentyl}thio-2′,3′-O-(1-methylethylidene)-adenosine (69).
This compound was prepared in a similar manner as 48 using the procedure outlined previously, but using aldehyde 45 (0.760 g, 2.91 mmol) and amine 63 (1.18 g, 2.91 mmol) as the starting materials to afford 69 (0.81 g, 43%) as a white foam: 1H NMR (500 MHz, CDCl3) δ 8.33 (s, 1H), 7.94 (s, 1H), 7.16 (m, 4H), 6.13 (br s, 2H, NH2), 6.09 (s, 1H), 5.54 (m, 1H), 5.06 (dd, J = 3.0, 6.3 Hz, 1H), 4.82 (m, 1H), 4.70–4.24 (m, 2H), 4.40 (m, 1H), 3.07–2.83 (m, 2H), 2.77 (m, 2H), 2.59–2.49 (m, 6H), 1.62 (s, 3H), 1.49 (s, 9H), 1.45 (m, 4H), 1.40 (s, 3H), 1.36 (m, 2H); 13C NMR (125.7 MHz, CDCl3) δ 155.7, 155.5, 153.2, 149.2, 140.0, 132.9, 129.2, 129.1, 126.6, 126.2, 126.1, 120.2, 114.3, 90.9, 87.1, 84.0, 83.9, 80.0, 50.8, (49.6, 48.6), 49.4, (43.4, 42.5), 34.3, 32.5, 31.2, 29.6, 29.4, 28.5, 27.1, 26.4, 25.3; MS (ESI+) m/z (relative intensity) 654 (MH+, 45), 554 (20), 327 (100); HRMS (ESI+) m/z calcd for C33H48N7O5S (MH+) 654.3438, obsd 654.3446.
5′-Deoxy-5′-{6-{[3,4-dihydro-2(1H)-(1,1-dimethylethoxy)carbonyl-isoquinolin-3-yl]methyl}amino-hexyl}thio-2′,3′-O-(1-methylethylidene)-adenosine (70).
This compound was prepared in a similar manner as 48 using the procedures outlined previously, but using aldehyde 45 (0.60 g, 2.3 mmol) and amine 64 (0.97 g, 2.3 mmol) as the starting materials to afford 70 (0.90 g, 59%) as a white foam: 1H NMR (400 MHz, CDCl3) δ 8.34 (s, 1H), 7.94 (s, 1H), 7.14 (m, 4H), 6.35 (br s, 2H, NH2), 6.09 (d, J = 2.0 Hz, 1H), 5.54 (dd, J = 2.0, 6.3 Hz, 1H), 5.07 (dd, J = 3.0, 6.3 Hz, 1H), 4.90–4.45 (m, 2H), 4.40 (m, 1H), 4.23 (m, 1H), 3.10–2.85 (m, 2H), 2.79 (m, 2H), 2.64–2.44 (m, 6H), 1.62 (s, 3H), 1.50 (s, 9H), 1.45 (m, 2H), 1.40 (s, 3H), 1.34 (m, 2H), 1.25 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 156.3, 155.7, 153.5, 149.6, 140.4, 133.4, 133.3, 129.6, 129.5, 127.0, 126.5, 120.7, 114.8, 91.3, 87.4, 84.4, 84.3, 80.3, 51.1, 49.9, 48.6, (43.7, 42.9), 34.7, 33.0, 31.6, 30.4, 29.9, 29.1, 28.9, 27.5, 27.2, 25.7; MS (ESI+) m/z (relative intensity) 668 (MH+, 100), 568 (30); HRMS (ESI+) m/z calcd for C34H50N7O5S (MH+) 668.3594, obsd 668.3593.
5′-Deoxy-5′-{2-[3,4-dihydro-2(1H)-(1,1-dimethylethoxy)carbonyl-isoquinoline-3-carboxamido]ethyl}thio-2′,3′-O-(1-methylethylidene)-adenosine (71).
To a stirred mixture of acid 41 (2.50 g, 9.02 mmol), amine 34 (3.30 g, 9.02 mmol) and HOBt·H2O (1.22 g, 9.02 mmol) in DMF (25 mL), were added EDCI•HCl (1.73 g, 9.02 mmol) and then N-methylmorpholine (1.0 mL, 9.0 mmol) under argon at 0 °C. The reaction mixture was warmed to room temperature and stirred for 12 h. The solvent was removed under reduced pressure and the residue was purified by flash column chromatography (silica gel) eluting with CHCl3/MeOH (40:1) to yield 71 (5.50 g, 98%) as a white foam: 1H NMR (500 MHz, CD3OD) δ 8.26 (s, 1H), 8.23 (s, 1H), 7.15 (m, 4H), 6.17 (s, 1H), 5.49 (m, 1H), 5.03 (br s, 1H), 4.60–4.46 (m, 3H), 4.30 (br s, 1H), 3.21–3.08 (m, 4H), 2.76 (m, 2H), 2.46–2.40 (m, 2H), 1.58 (s, 3H), 1.51, 1.42 (two peaks, 9H), 1.38 (s, 3H); 13C NMR (100 MHz, CD3OD) δ (173.8, 173.1), 156.3, (156.1, 156.4), 152.9, 149.2, 140.9, (134.9, 134.2), (133.9, 133.5), (128.0, 127.5), (127.4, 127.2), 126.9, 126.2, 119.6, 114.5, 90.6, 86.9, 84.2, 84.1, 81.1, (56.9, 55.3), (45.4, 44.5), (38.9, 38.8), 33.9, (32.5, 31.9), 31.5, 27.8, 26.5, 24.7; MS (FAB+) m/z (relative intensity) 626 (MH+, 50), 524 (15), 136 (100); HRMS (FAB+) m/z calcd for C30H40N7O6S (MH+) 626.2761, obsd 626.2763.
5′-{2-[7-Chloro-3,4-dihydro-2(1H)-(1,1-dimethylethoxy)carbonyl-isoquinoline-3-carboxamido]ethyl}thio-5′-deoxy-2′,3′-O-(1-methylethylidene)-adenosine (72).
This compound was prepared in a similar manner as 71 using the procedures outlined previously but using 39 (170 mg, 0.550 mmol) as the starting material to afford 72 (300 mg, 83%) as a white foam: IR (KBr) 3334, 3191, 2981, 2930, 1660, 1593, 1486, 1383, 1209, 1163, 1096, 871, 738 cm−1; 1H NMR (500 MHz, CD3OD) δ 8.26 (s, 1H), 8.23 (s, 1H), 7.16–7.09 (m, 3H), 6.17 (s, 1H), 5.49 (m, 1H), 5.03 (br s, 1H), 4.58–4.46 (m, 3H), 4.30 (br s, 1H), 3.21–3.05 (m, 4H), 2.77 (m, 2H), 2.49 (m, 2H), 1.58 (s, 3H), 1.50, 1.42 (two peaks, 9H), 1.37 (s, 3H); 13C NMR (125.7 MHz, CD3OD) δ (172.9, 172.3), 155.9, (155.4, 155.0), 152.6, 148.7, 140.3, (136.4, 135.7), (132.2, 131.9), (129.1, 128.6), (128.5, 127.8), (126.9, 126.7), 125.7, 119.1, 114.0, 90.2, 86.5, 83.7, 83.6, 80.7, (56.0, 54.4), (44.6, 43.7), (38.4, 38.3), 33.4, (31.4, 30.9), 31.1, 27.3, 26.0, 24.2; MS (CI) m/z (relative intensity) 660 (M+, 25), 560 (20), 324 (25), 292 (60), 136 (80), 120 (90); HRMS (FAB+) m/z calcd for C30H39ClN7O6S (MH+) 660.2371, obsd 660.2366.
5′-Deoxy-5′-{2-[3,4-dihydro-2(1H)-(1,1-dimethylethoxy)carbonyl-7-nitro-isoquinoline-3-carboxamido]ethyl}thio-2′,3′-O-(1-methylethylidene)-adenosine (73).
This compound was prepared in a similar manner as 71 using the procedures outlined previously, but using 40 (500 mg, 1.55 mmol) as the starting material to afford 73 (0.86 g, 83%) as a white foam: 1H NMR (500 MHz, CDCl3) δ 8.30 (s, 1H), 8.00 (m, 2H), 7.95 (s, 1H), 7.32 (m, 1H), 6.74–6.39 (br, 2H), 6.07 (br s, 1H), 5.49 (br s, 1H), 5.01 (br s, 1H), 4.76–4.48 (m, 3H), 4.30 (br s, 1H), 3.29 (m, 2H), 3.05 (m, 2H), 2.72 (m, 2H), 2.49 (m, 2H), 1.59 (s, 3H), 1.56 (s, 9H), 1.39 (s, 3H); 13C NMR (125.7 MHz, CDCl3) δ 170.9, 155.6, 155.0, 152.9, 149.0, 146.9, 141.3, 140.1, 134.4, 129.4, 122.1, 121.2, 120.1, 114.4, 90.7, 86.7, 83.9, 83.6, 81.8, 52.8, 44.5, 38.0, 33.7, 32.1, 30.7, 28.2, 26.9, 25.2; MS (ESI+) m/z (% relative intensity) 671 (MH+, 100), 571 (10), 480 (5), 436 (10), 292 (10); HRMS (ESI+) m/z calcd for C30H39N8O8S (MH+) 671.2612, obsd 671.2614.
5′-Deoxy-5′-{3-[3,4-dihydro-2(1H)-(1,1-dimethylethoxy)carbonyl-isoquinoline-3-carboxamido]propyl}thio-2′,3′-O-(1-methylethylidene)- adenosine (74).
To a stirred mixture of acid 41 (0.50 g, 1.8 mmol), amine 61 (0.69 g, 1.81 mmol) and HOBt (244 mg, 1.81 mmol) in DMF (12 mL) were added EDCI•HCl (347 mg, 1.81 mmol) followed by N-methylmorpholine (183 mg, 1.81 mmol) under argon at 0 °C. The reaction mixture was warmed to room temperature and stirred for 12 h. The solvent was removed under reduced pressure and the residue was purified by flash column chromatography (silica gel) eluting with CHCl3/MeOH (40:1) to yield 74 (0.78 g, 68%) as a white foam: 1H NMR (400 MHz, CDCl3) δ 8.25 (s, 1H), 7.89 (s, 1H), 7.11 (m, 4H), 6.73 (br s, 2H, NH2), 6.04 (s, 1H), 6.03 (br s, 1H), 5.48 (m, 1H), 4.98 (m, 1H), 4.77 (m, 1H), 4.59–4.40 (m, 2H), 4.28 (m, 1H), 3.13 (m, 2H), 3.05 (m, 2H), 2.68 (m, 2H), 2.13 (m, 2H), 1.54 (s, 3H), 1.49 (m, 2H), 1.39 (s, 9H), 1.34 (s, 3H); 13C NMR (100 MHz, CDCl3) δ (172.4, 171.7), 156.4, (156.1, 155.6), 153.4, 149.4, 140.3, 134.3, 133.6, 128.6, 128.0, 127.0, 126.6, 120.5, 114.7, 91.3, 87.1, 84.4, 84.2, 81.5, (57.1, 54.9), (45.6, 45.0), 38.4, 34.6, (32.6, 31.2), 29.8, 29.3, 28.8, 27.5, 25.7; MS (CI) m/z (relative intensity) 640 (MH+, 75), 292 (20), 219 (30), 132 (75); HRMS (ESI+) m/z calcd for C31H42N7O6S (MH+) 640.2917, obsd 640.2934.
5′-Deoxy-5′-{3-[3,4-dihydro-2(1H)-(1,1-dimethylethoxy)carbonyl-7-nitro-isoquinoline-3-carboxamido]propyl}thio-2′,3′-O-(1-methylethylidene)-adenosine (75).
This compound was prepared in a similar manner as 74 using the procedure outlined previously for 74, but using acid 40 (500 mg, 1.55 mmol) as the starting material to afford 75 (1.0 g, 94%) as a white foam: 1H NMR (400 MHz, CDCl3) δ 8.22 (s, 1H), 7.93 (m, 2H), 7.91 (s, 1H), 7.27 (d, J = 6.7 Hz, 1H), 6.80 (br s, 2H, NH2), 6.68 (br s, 1H), 6.04 (s, 1H), 5.46 (dd, J = 1.4, 6.1 Hz, 1H), 4.96 (dd, J = 2.6, 6.1 Hz, 1H), 4.66 (m, 2H), 4.42 (m, 1H), 4.26 (m, 1H), 3.34–3.09 (m, 2H), 3.08 (m, 2H), 2.65 (m, 2H), 2.28 (m, 2H), 1.52 (s, 3H), 1.46 (m, 2H), 1.42 (s, 9H), 1.32 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 171.0, 156.4, (156.0, 155.2), 153.4, 149.4, 146.8, 141.9, 140.4, (135.7, 134.7), 129.7, 122.9, 121.5, 120.5, 114.7, 91.2, 87.1, 84.3, 84.2, 82.1, (56.0, 53.5), (45.0, 44.5), 38.7, 34.6, (32.2, 30.9), 30.0, 29.3, 28.7, 27.4, 25.7; (ESI+) m/z (relative intensity) 685 (MH+, 100), 306 (15), 262 (25); HRMS (ESI+) m/z calcd for C31H41N8O8S (MH+) 685.2768, obsd 685.2760.
(±)-4-Hydroxymethyl-1,2,3,4-tetrahydroisoquinoline (77).
Lactam 76 (200 mg, 0.980 mmol) was dissolved in THF (20 mL). LiAlH4 (111 mg, 2.93 mmol) was added slowly into the solution at 0 °C. The reaction mixture was warmed to room temperature, after which it was heated at reflux for 8 h. The reaction mixture was cooled to 0 °C. H2O (0.1 mL), 15% NaOH (0.1 mL) and then H2O (3.3 mL) were added slowly to the mixture. The mixture was stirred at room temperature for 30 min and then filtered through a Celite pad. The filtrate was concentrated and the resulting residue was purified by flash column chromatography (silica gel) eluting with CHCl3/MeOH/NH4OH (5:1:0.1) to yield 77 (140 mg, 88%) as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 7.19 (m, 3H), 7.04 (d, J = 7.2 Hz, 1H), 4.05 (ex s, 2H), 3.98 (br s, 2H), 3.96–3.82 (m, 2H), 3.42–3.09 (m, 2H), 2.80 (m, 1H); 13C NMR (125.7 MHz, CDCl3) δ 135.5, 135.3, 128.9, 126.5, 126.3, 126.1, 68.0, 47.9, 47.0, 38.6; MS (CI) m/z (relative intensity) 163 (M+, 20), 162 (50), 130 (75), 105 (98), 91 (45); HRMS (FAB+) m/z calcd for C10H14NO (MH+) 164.1075, obsd 164.1057.
(±)-1,1-Dimethylethyl 3,4-dihydro-4-hydroxymethyl-isoquinoline-2(1H)-carboxylate (78).
To a stirred solution of 77 (70 mg, 0.43 mmol) in MeOH (6.6 mL) was added NaHCO3 (253 mg, 3.01 mmol). (Boc)2O (281 mg, 1.29 mmol) was then added and the mixture was stirred at room temperature overnight. The solvent was removed under reduced pressure, and the residue was mixed with water (10 mL) and then extracted with CH2Cl2 (3 × 10 mL). The organic layers were combined together, washed with brine (20 mL) and dried with anhydrous Na2SO4. The solvent was removed and the residue was purified by column chromatography (silica gel) eluting with hexanes/EtOAc (4:1) to provide 78 (100 mg, 88%) as a colorless oil: 1H NMR (500 MHz, CDCl3) δ 4.92 (d, J = 17.0 Hz, 1H), 4.51–4.34 (m, 2H), 3.65 (m, 2H), 3.40 (br s, 1H), 3.10–3.00 (m, 2H), 1.51 (s, 9H); 13C NMR (125.7 MHz, CDCl3) δ 156.2, 135.0, 133.3, 129.4, 126.8, 126.4, 126.0, 80.5, 64.1, (46.6, 45.4), (42.2, 41.5), 41.0, 28.3; MS (CI) m/z (relative intensity) 264 (MH+, 50), 233 (18), 208 (40), 162 (90), 130 (40); HRMS (FAB+) m/z calcd for C15H22NO3 (MH+) 264.1600, obsd 264.1592.
(±)-1,1-Dimethylethyl 3,4-dihydro-4-formyl-isoquinoline-2(1H)-carboxylate (79).
To a solution of oxalyl chloride (135 mg, 1.06 mmol) in CH2Cl2 (3 mL) was added dimethyl sulfoxide (0.14 mL, 1.9 mmol) at −78 °C. The mixture was then stirred for 30 min. A solution of alcohol 78 (100 mg, 0.380 mmol) in CH2Cl2 (4 mL) was added dropwise to the reaction at −78 °C and the reaction mixture was then stirred for another 1.5 h. N,N-Diisopropylethylamine (0.60 mL) was added slowly to the reaction and the mixture was warmed up to room temperature. The solution was poured into 1M HCl solution (20 mL). The organic layer was washed with 1M HCl (10 mL), NaHCO3 (sat, 10 mL), brine (10 mL) and then dried with anhydrous Na2SO4. The solvent was removed under reduced pressure and the residue was purified by column chromatography (hexanes/EtOAc 6:1) to provide aldehyde 79 (70 mg, 71%) as a colorless oil: 1H NMR (500 MHz, CDCl3) δ 9.73 (s, 1H), 7.30–7.20 (m, 4H), 4.90–4.50 (m, 2H), 4.34 (m, 1H), 3.60–3.30 (m, 2H), 1.50 (s, 9H); 13C NMR (125.7 MHz, CDCl3) δ 199.6, 154.4, 134.1, 129.7, 128.9, 127.8, 127.0, 126.8, 80.5, 51.6, (45.8, 45.0), (41.4, 40.2), 28.1; MS (ESI+) m/z (relative intensity) 262 (MH+, 100), 206 (88), 188 (40), 145 (40), 117 (32); HRMS (FAB+) m/z calcd for C15H20NO3 (MH+) 262.1443, obsd 262.1421.
5′-Deoxy-5′-{2-{[3,4-dihydro-2(1H)-(1,1-dimethylethoxy)carbonyl-isoquinolin-4-yl]methyl}amino-ethyl}thio-2′,3′-O-(1-methylethylidene)-adenosine (80).
A mixture of aldehyde 79 (70 mg, 0.27 mmol), amine 34 (110 mg, 0.300 mmol), molecular sieves (4A, 110 mg), and THF (6 mL) was stirred at room temperature for 3 h. The mixture was filtered and the solvent was removed under reduced pressure. The residue was combined with ethanol (6 mL), and sodium borohydride (15 mg, 0.40 mmol) was added over 1.5 h. The mixture was stirred for 12 h at room temperature, and water (0.1 mL) was added. The solvent was removed under reduced pressure and the residue was purified by chromatography on a silica gel column in CHCl3/MeOH (50:1) to yield 80 (90 mg, 54%) as a white foam: 1H NMR (400 MHz, CDCl3) δ 8.32 (s, 1H), 7.95 (br s, 1H), 7.60–7.23 (m, 1H), 7.16 (m, 2H), 7.08 (m, 1H), 6.56 (ex m, 2H), 6.09 (m, 1H), 5.51 (m, 1H), 5.08 (m, 1H), 4.82 (m, 1H), 4.42 (m, 1H), 4.29 (m, 2H), 3.15 (m, 1H), 2.89–2.66 (m, 10H), 1.54 (s, 3H), 1.48 (s, 9H), 1.40 (s, 3H); 13C NMR (125.7 MHz, CDCl3) δ 155.6, 155.0, 153.0, 149.1, 139.9, (136.8, 136.2), 133.0, 128.9, 126.8, 126.3, 126.0, 120.2, 114.3, 90.8, 86.8, 84.0, 83.7, 79.8, 52.5, 48.7, (46.1, 45.2), (43.0, 41.8), (39.1, 38.7), 34.2, 32.8, 28.4, 27.0, 25.3; MS (ESI+) m/z (relative intensity) 612 (MH+, 100), 512 (40), 377 (10); HRMS (ESI+) m/z calcd for C30H42N7O5S (MH+) 612.2968, obsd 612.2957.
5′-Deoxy-5′-{3-{[3,4-dihydro-2(1H)-(1,1-dimethylethoxy)carbonyl-isoquinolin-4-yl]methyl}amino-propyl}thio-2′,3′-O-(1-methylethylidene)-adenosine (81).
This compound was prepared in a similar manner as 80 using the procedures outlined previously but using aldehyde 79 (1.30 g, 5.00 mmol) and amine 61 (2.40 g, 6.30 mmol) as the starting materials to afford 81 (1.87 g, 60%) as a white foam: 1H NMR (500 MHz, CDCl3) δ 8.36 (s, 1H), 7.95 (s, 1H), 7.18 (m, 3H), 7.10 (m, 1H), 6.09 (s, 1H), 5.82 (br s, 2H, NH2), 5.53 (d, J = 6.1 Hz, 1H), 5.07 (m, 1H), 4.85 (m, 1H), 4.42 (m, 1H), 4.32 (m, 2H), 3.20–2.90 (m, 2H), 2.84 (m, 2H), 2.80 (m, 1H), 2.70 (m, 4H), 2.60 (m, 2H), 1.73 (t, J = 6.6 Hz, 2H), 1.62 (s, 3H), 1.49 (s, 9H), 1.42 (s, 3H); 13C NMR (125.7 MHz, CDCl3) δ 155.6, 155.0, 153.1, 149.2, 139.9, 136.0, 133.2, 128.8, 126.7, 126.5, 126.3, 120.2, 114.3, 90.9, 86.8, 84.0, 83.8, 79.8, 52.8, (48.8, 48.2), (46.1, 45.3), (43.0, 41.7), (39.0, 38.6), 34.4, 30.3, (29.9, 29.7), 28.4, 27.0, 25.3; MS (CI) m/z (relative intensity) 626 (MH+, 75), 393 (20), 102 (50); (ESI+) m/z calcd for C31H44N7O5S (MH+) 626.3125, obsd 626.3108.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Michael F. Rafferty for helpful and detailed comments on this manuscript. GLG thanks all of the research students he has had the pleasure of working with over the past five decades. In particular the work (and friendship) of his first five students (Bharat V. Kamdar, Ann Marie Warner, Winton Dennis Jones, Jr., Dale B. Mirth, and Allan H. Mizoguchi) were especially important to a young assistant professor beginning a research career and their work made that of later students much easier. Molecular modeling studies by a later student (James A. Monn), conducted long before anything about the structure of hPNMT was known, led to the undertaking of the project described in this manuscript. Early work on much simpler approaches by postdoctoral research associates John R. Ross, Akmal Bhatti and Meri Slavica were instrumental in the ultimate success described in this paper.
This work was supported by NIH grant HL34193 “Synthesis of Inhibitors of Epinephrine Biosynthesis”. Structural work by A. G. B. was partially supported by a K-INBRE Undergraduate Scholarship funded by NIH P20 GM103418 and startup funds from the University of Kansas (to E. E. S). We thank David VanderVelde and Sarah Neuenswander of the University of Kansas Nuclear Magnetic Resonance Laboratory for their assistance. The 500 MHz NMR spectrometer was partially funded by the National Science Foundation Grant CHE-9977422. We also thank Gerald Lushington of the University of Kansas Molecular Graphics and Modeling Laboratory and Todd Williams of the University of Kansas Mass Spectrometry Laboratory for their assistance. Crystallization employed facilities of the University of Kansas Protein Structure Laboratory (PSL). The PSL was partially supported by NIH through the KU COBRE Center for Protein Structure and Function (NIH RR17708). X-ray diffraction data were collected at the Stanford Synchrotron Radiation Lightsource. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences.
Abbreviations Used
- AdoHcy
S-adenosyl-homocysteine
- AdoMet
S-adenosyl-l-methionine
- hPNMT
human phenylethanolamine N-methyltransferase
- PNMT
phenylethanolamine N-methyltransferase
- THIQ
1,2,3,4-tetrahydroisoquinoline
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01475.
HPLC tracings for 24, 25, 28, 30, and 31 (CSV)
Molecular formula strings for all new compounds (PDF)
The authors declare no competing financial interest.
REFERENCES
- 1.Kitahama K; Denoroy L; Goldstein M; Jouvet M; Pearson J Immunohistochemistry of tyrosine hydroxylase and phenylethanolamine N-methyltransferase in the human brain stem: description of adrenergic perikarya and characterization of longitudinal catecholaminergic pathways. Neurosci. 1988, 25, 97–111. [DOI] [PubMed] [Google Scholar]
- 2.Kennedy B; Bigby TD; Ziegler MG Nonadrenal epinephrine-forming enzymes in humans. Characteristics, distribution, regulation, and relationship to epinephrine levels. J. Clin. Invest 1995, 95, 2896–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ziegler MG; Bao X; Kennedy BP; Joyner A; Enns R; Location, development, control, and function of extraadrenal phenylethanolamine N-methyltransferase. Ann. N. Y. Acad. Sci 2002, 971, 76–82. [DOI] [PubMed] [Google Scholar]
- 4.Axelrod J Purification and properties of phenylethanolamine-N-methyl transferase. J. Biol. Chem 1962, 237, 1657–1660. [PubMed] [Google Scholar]
- 5.Grunewald GL; Grindel JM; Vincek WC; Borchardt RT Importance of the aromatic ring in adrenergic amines. Nonaromatic analogs of phenylethanolamine as substrates for phenylethanolamine N-methyltransferase. Mol. Pharmacol 1975, 11, 694–699 [PubMed] [Google Scholar]
- 6.Grunewald GL; Vincek WC; Davis DP; Borchardt RT; Some new inhibitors of epinephrine biosynthesis. Importance of the aromatic ring in adrenergic amines. 4. Catecholamines: Basic Clin. Front., Proc. Int. Catecholamine Symp. 4th. 1979, 1, 189–191. [Google Scholar]
- 7.Rafferty MF; Wilson DS; Monn JA; Krass P; Borchardt RT; Grunewald GL Comparison of the stereoselectivity of norepinephrine N-methyltransferase for aromatic versus non-aromatic substrates and Inhibitors. The importance of the aromatic ring in adrenergic amines. 7. J. Med. Chem 1982, 25, 1198–1204. [DOI] [PubMed] [Google Scholar]
- 8.Grunewald GL; Rafferty MF Stereochemical aspects of binding of aromatic and non-aromatic substrates and inhibitors to phenylethanolamine N-methyltransferase. In Function and Regulation of Monoamine Enzymes: Basic and Clinical Aspects. Usdin E, Weiner N, Youdim MBH, Eds. (Macmillan and Company, New York: ), 1981, pp. 691–699. [Google Scholar]
- 9.Grunewald GL; Borchardt RT; Rafferty MF Krass P Conformational preferences of amphetamine analogues for inhibition of phenylethanolamine N-methyltransferase. Conformationally defined adrenergic agents. 5. Mol. Pharmacol, 1981, 20, 377–381. [PubMed] [Google Scholar]
- 10.Rafferty MF; Wilson DS; Monn JA; Krass P; Borchardt RT; Grunewald GL Comparison of the stereoselectivity of norepinephrine N-methyltransferase for aromatic versus non-aromatic substrates and inhibitors. The importance of the aromatic ring in adrenergic amines. 7. J. Med. Chem 1982, 25, 1198–1204. [DOI] [PubMed] [Google Scholar]
- 11.Bondinell WE; Chapin FW; Girard GR; Kaiser C; Krog AJ; Pavloff AM; Schwartz MS; Silvestri JS; Vaidya PD; Lam BL; Wellman GR; Pendleton RG Inhibitors of phenylethanolamine N-methyltransferase and epinephrine biosynthesis. 1. Chloro-substituted 1,2,3,4-tetrahydroisoquinolines. J. Med. Chem 1980, 23, 506–11. [DOI] [PubMed] [Google Scholar]
- 12.Blank B; Krog AJ; Weiner G; Pendleton RG Inhibitors of phenylethanolamine N-methyltransferase and epinephrine biosynthesis. 2. 1,2,3,4-Tetrahydroisoquinoline-7-sulfonanilides. J. Med. Chem 1980, 23, 837–40. [DOI] [PubMed] [Google Scholar]
- 13.Sall DJ; Grunewald GL Inhibition of phenylethanolamine N-methyltransferase (PNMT) by aromatic hydroxy-substituted 1,2,3,4-tetrahydroisoquinolines: further studies on the hydrophilic pocket of the aromatic ring binding region of the active site. J. Med. Chem, 1987, 30, 2208–2216. [DOI] [PubMed] [Google Scholar]
- 14.Grunewald GL; Dahanukar VH; Jalluri RK Criscione KR Synthesis, biochemical evaluation, and classical and three-dimensional quantitative structure-activity relationship studies of 7-substituted-1,2,3,4-tetrahydroisoquinolines and their relative affinities toward phenylethanolamine N-methyltransferase and the α2-adrenoceptor. J. Med. Chem 1999, 42, 118–134. [DOI] [PubMed] [Google Scholar]
- 15.Grunewald GL; Sall DJ; Monn JA Synthesis and evaluation of 3-substituted analogues of 1,2,3,4-tetrahydroisoquinoline as inhibitors of phenylethanolamine N-methyltransferase (PNMT). J. Med. Chem 1988, 31, 824–830. [DOI] [PubMed] [Google Scholar]
- 16.Fuller RW; Molloy BB; Hemrick SK Inhibition in vitro of rabbit adrenal norepinephrine N-methyltransferase by 2,3,4,5-tetrahydro-1H-2-benzazepines. Biochem. Pharmacol 1979, 28, 528–530. [DOI] [PubMed] [Google Scholar]
- 17.Grunewald GL; Dahanukar VH; Criscione KR Effects of a 3-alkyl-, 4-hydroxy- and/or 8-aromatic-substituent on the phenylethanolamine N-methyltransferase inhibitor potency and α2-adrenoceptor affinity of 2,3,4,5-tetrahydro-1H-2-benzazepines. Bioorg. Med. Chem. Lett 2001, 9, 1957–1965. [DOI] [PubMed] [Google Scholar]
- 18.Liang NY; Chandra A; Tessel RE; Grunewald GL; Borchardt RT Inhibitors of phenylethanolamine-N-methyltransferase: effects on brain catecholamine content and blood pressure in DOCA-salt hypertensive rats. Res. Commun. Chem. Path 1984, 46, 319–29. [PubMed] [Google Scholar]
- 19.Epinephrine in the Central Nervous System. Stolk JM; U'Pritchard DC; Fuxe K; Eds.; Oxford Univ. Press: New York, N. Y. 1987. [Google Scholar]
- 20.Chatelain RE; Manniello MJ; Dardik BN; Rizzo M; Brosnihan KB Antihypertensive effects of CGS 19281A, an inhibitor of phenylethanolamine-N-methyltransferase. J. Pharmacol. Exp. Ther 1990, 252, 117–125. [PubMed] [Google Scholar]
- 21.Kennedy B; Elayan H; Ziegler MG Glucocorticoid hypertension and nonadrenal phenylethanolamine N-methyltransferase. Hypertension. 1993, 21, 415–19. [DOI] [PubMed] [Google Scholar]
- 22.Toomey RE; Horng JS; Hemrick-Luecke SK; Fuller RW Alpha-2 Adrenoreceptor affinity of some inhibitors of norepinephrine N-methyltransferase. Life Sci. 1981, 29, 2467–2472. [DOI] [PubMed] [Google Scholar]
- 23.Grunewald GL; Dahanukar VH; Teoh B Criscione KR 3,7-Disubstituted-1,2,3,4-tetrahydroisoquinolines display remarkable potency and selectivity as inhibitors of phenylethanolamine N-methyltransferase versus the α2-adrenoceptor. J. Med. Chem 1999, 42, 1982–1990. [DOI] [PubMed] [Google Scholar]
- 24.Grunewald GL; Caldwell TM; Li Q Criscione KR Synthesis and evaluation of 3-trifluoromethyl-7-substituted-1,2,3,4-tetrahydroisoquinolines as selective inhibitors of phenylethanolamine N-methyltransferase versus the α2-adrenoceptor. J. Med. Chem 1999, 42, 3315–3323. [DOI] [PubMed] [Google Scholar]
- 25.Romero FA; Vodonick SM; Criscione KR; McLeish MJ; Grunewald GL Inhibitors of phenylethanolamine N-methyltransferase that are predicted to penetrate the blood-brain barrier: design, synthesis and evaluation of 3-fluoromethyl-7-(n-substituted aminosulfonyl)-1,2,3,4-tetrahydroisoquinolines that possess low affinity toward the α2-adrenoceptor. J. Med. Chem 2004, 47, 4483–4493. [DOI] [PubMed] [Google Scholar]
- 26.Grunewald GL; Caldwell TM; Li Q; Slavica M; Borchardt RT; Wang W Criscione KR Synthesis and biochemical evaluation of 3-fluoromethyl-1,2,3,4-tetrahydroisoquinolines as selective inhibitors of phenylethanolamine N-methyltransferase versus the α2-adrenoceptor. J. Med. Chem 1999, 42, 3588–3601. [DOI] [PubMed] [Google Scholar]
- 27.Grunewald GL; Romero FA; Criscione KR Nanomolar inhibitors of CNS epinephrine biosynthesis: (R)-(+)-3-fluoromethyl-7-(N-substituted aminosulfonyl)-1,2,3,4-tetrahydroisoquinolines as potent and highly selective inhibitors of phenylethanolamine N-methyltransferase. J. Med. Chem 2005, 48, 1806–1812. [DOI] [PubMed] [Google Scholar]
- 28.Borchardt RT; Wu YS; Potential inhibitors of S-adenosylmethionine-dependent methyltransferases. 1. Modification of the amino acid portion of S-adenosylhomocysteine. J. Med. Chem 1974, 17, 862–868. [DOI] [PubMed] [Google Scholar]
- 29.Borchardt RT; Huber JA; Wu YS Potential inhibitors of S-adenosylmethionine-dependent methyltransferases. 2. Modification of the base portion of S-adenosylhomocysteine. J. Med. Chem 1974, 17, 868–873. [DOI] [PubMed] [Google Scholar]
- 30.Borchardt RT; Wu YS Potential inhibitors of S-adenosylmethionine-dependent methyltransferases. 3. Modifications of the sugar portion of S-adenosylhomocysteine. J. Med. Chem 1975, 18, 300–304. [DOI] [PubMed] [Google Scholar]
- 31.Borchardt RT; Huber JA; Wu YS Potential inhibitors of S-adenosylmethionine-dependent methyltransferases. 4. Further modifications of the amino acid and base portions of S-adenosyl-l-homocysteine. J. Med. Chem 1976, 19, 1094–1099. [DOI] [PubMed] [Google Scholar]
- 32.Borchardt RT; Wu Y-S S-Aristeromycinyl-l-homocysteine, a potent inhibitor of S-adenosylmethionine-dependent transmethylations. J. Med. Chem 1976, 19, 197–198. [DOI] [PubMed] [Google Scholar]
- 33.Borchardt RT Synthesis and biological activity of analogs of adenosylhomocysteine as inhibitors of methyltransferases. Biochem. Adenosylmethionine, Proc. Int. Symp.; Salvatore F; Borek E; Zappia V, Eds.; Columbia Univ. Press: Irvington-on-Hudson, N. Y, 1977; pp 151–171. [Google Scholar]
- 34.Georgieva P; Wu Q; McLeish MJ; Himo F The reaction mechanism of phenylethanolamine N-methyltransferase: A density functional theory study. Biochim. Biophys. Acta 2009, 1794, 1831–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu Q; McLeish MJ Kinetic and pH studies on human phenylethanolamine N-methyltransferase. Arch. Biochem. Biophys 2013, 539, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stratton CF; Poulin MB; Du Q; Schramm VL Kinetic isotope effects and transition state structure for human phenylethanolamine N-methyltransferase. ACS Chem. Biol 2017, 12, 342–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jencks WP Binding energy, specificity, and enzymic catalysis: the Circe effect. Adv. Enzymol. Relat. Areas Mol. Biol 1975, 43, 219–410. [DOI] [PubMed] [Google Scholar]
- 38.Anderson GL; Bussolotti DL; Coward JK Synthesis and evaluation of some stable multisubstrate adducts as inhibitors of catechol O-methyltransferase. J. Med. Chem 1981, 24, 1271–1277. [DOI] [PubMed] [Google Scholar]
- 39.Benghiat E; Crooks PA Multisubstrate adducts as potential inhibitors of S-adenosylmethionine dependent methylases: inhibition of indole N-methyltransferase by (5H-deoxyadenosyl)[3-(3-indolyl)propyl-1-yl]methyl-sulfonium and (5'-deoxyadenosyl)[4-(3-indolyl)but-1-yl]methylsulfonium salts. J. Med. Chem 1983, 26, 1470–1477. [DOI] [PubMed] [Google Scholar]
- 40.Benghiat E; Crooks PA; Goodwin R; Rottman F Inhibition of vaccinia RNA guanine 7-methyltransferase by compounds designed as multisubstrate adducts. J. Pharm. Sci 1986, 75, 142–145. [DOI] [PubMed] [Google Scholar]
- 41.Crooks PA; Hassan SF; Benghiat E; Hemrick-Luecke SK; Fuller RW 5’-Thioadenosine derivatives as potent and selective inhibitors of histamine N-methyltransferase. Drug Metab. Drug Interactions 1989, 7, 111–141. [DOI] [PubMed] [Google Scholar]
- 42.Babault N; Allali-Hassani A; Li F; Fan J; Yue A; Ju K; Liu F; Vedadi M; Liu J; Jin J; Discovery of bisubstrate inhibitors of nicotinamide N-methyltransferase (NNMT). J. Med. Chem 2018, 61, 1541–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gao Y; van Haren MJ; Moret EE; Rood JJM; Sartini D; Salvucci A; Emanuelli M; Craveur P; Babault N; Jin J; Martin N Bisubstrate inhibitors of nicotinamide N-methyltransferase (NNMT) with enhanced activity. J. Med. Chem 2019, 62, 6597–6614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Policarpo RL; Decultot L; May E; Kuzmič P; Carlson S; Huang D; Chu V; Wright BA; Dhakshinamoorthy S; Kannt A; Rani S; Dittakavi S; Panarese JD; Gaudet R; Shair MD High-affinity alkynyl bisubstrate inhibitors of nicotinamide N-methyltransferase (NNMT). J. Med. Chem 2019, 62, 9837–9873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen D; Li L; Diaz K Iyamu ZD; Yadav R; Noinaj N; Huang R Novel propargyl-linked bisubstrate analogues as tight-binding inhibitors for nicotinamide N-methyltransferase. J. Med. Chem 2019, 62, 10783–10797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen D; Dong C; Dong G; Srinivasan K; Min J; Noinaj N; Huang R Probing the plasticity in the active site of protein N-terminal methyltransferase 1 using bisubstrate analogues. J. Med. Chem 2020, 63, 8419–8431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hassan SF The synthesis and inhibitory activities of compounds designed as mechanism-based inhibitors of phenylethanolamine-N-methyltransferase. Ph.D. Thesis, University of Kentucky, Lexington, KY, 1987. ProQuest Dissertation Express #8802015. [Google Scholar]
- 48.Lerner C; Ruf A; Gramlich V; Masjost B; Zürcher G; Jakob-Roetne R; Borroni E; Diederich F X-ray Crystal structure of a bisubstrate inhibitor bound to the enzyme catechol-O-methyltransferase: A dramatic effect of inhibitor preorganization on binding affinity. Angew. Chem. Int. Ed 2001, 40, 4040–4042. [DOI] [PubMed] [Google Scholar]
- 49.Forsberg M; Lehtonen M; Heikkinen M; Savolainen J; Järvinen T; Männistö PT Pharmacokinetics and pharmacodynamics of entacapone and tolcapone after acute and repeated administration: a comparative study in the rat. J. Pharmacol. Exp. Ther 2003, 304, 498–506. [DOI] [PubMed] [Google Scholar]
- 50.Martin JL; Begun J; McLeish MJ; Caine JM; Grunewald GL Getting the adrenaline going. Crystal structure of the adrenaline-synthesizing enzyme PNMT. Structure 2001, 9, 977–985. [DOI] [PubMed] [Google Scholar]
- 51.McMillan FM; Archbold J; McLeish MJ; Caine JM; Criscione KR; Grunewald GL; Martin JL Molecular recognition of sub-micromolar inhibitors by the epinephrine-synthesizing enzyme phenylethanolamine N-methyltransferase. J. Med. Chem 2004, 47, 37–44. [DOI] [PubMed] [Google Scholar]
- 52.Gee CL; Tyndall JDA; Grunewald GL; Wu Q; McLeish MJ; Martin JL Mode of binding of methyl acceptor substrates to the adrenaline-synthesizing enzyme phenylethanolamine N-methyltransferase: Implications for catalysis. Biochemistry 2005, 44, 16875–16885. [DOI] [PubMed] [Google Scholar]
- 53.Wu Q; Gee CL; Lin F; Tyndall JD; Martin JL; Grunewald GL; McLeish MJ Structural, mutagenic, and kinetic analysis of the binding of substrates and inhibitors of human phenylethanolamine N-methyltransferase. J. Med. Chem 2005, 48, 7243–7252. [DOI] [PubMed] [Google Scholar]
- 54.Gee CL; Drinkwater N; Tyndall JD; Grunewald GL; Wu Q; McLeish MJ; Martin JL Enzyme adaptation to inhibitor binding: a cryptic binding site in phenylethanolamine N-methyltransferase. J. Med. Chem 2007, 50, 4845–4853. [DOI] [PubMed] [Google Scholar]
- 55.Drinkwater N; Vu H; Lovell KM; Criscione KR; Collins BM; Prisinzano TE; Poulsen S-A; McLeish MJ; Grunewald GL; Martin JL Fragment-based screening by X-ray crystallography, MS and isothermal titration calorimetry to identify PNMT (phenylethanolamine N-methyltransferase) inhibitors. Biochem. J 2010, 431, 51–61. [DOI] [PubMed] [Google Scholar]
- 56.PDB ID: 2G70 Gee CL; Drinkwater N; Tyndall JD; Grunewald GL; Wu Q; McLeish MJ; Martin JL Enzyme adaptation to inhibitor binding: a cryptic binding site in phenylethanolamine N-methyltransferase. J. Med. Chem 2007, 50, 4845–4853. [DOI] [PubMed] [Google Scholar]
- 57.Morris GM; Goodsell DS; Halliday RS; Huey R; Hart WE; Belew RK; Olson AJ Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem 1998, 19, 1639–1662. [Google Scholar]
- 58.Tripos SYBYL, version 6.9; Tripos Inc. 1699 South Hanley Rd., St. Louis, MO, 63144, USA.: 2001. [Google Scholar]
- 59.Jeffrey GA; Editor, An Introduction to Hydrogen Bonding. Oxford University Press, Inc.: New York, 1997. [Google Scholar]
- 60.Bondinell WE; Chapin FW; Girard GR; Kaiser C; Krog AJ; Pavloff AM; Schwartz MS; Silvestri JS; Vaidya PD; Lam BL; Wellman GR; Pendleton RG Inhibitors of phenylethanolamine N-methyltransferase and epinephrine biosynthesis. 1. Chloro-substituted 1,2,3,4-tetrahydroisoquinolines. J. Med. Chem 1980, 23, 506–511. [DOI] [PubMed] [Google Scholar]
- 61.PDB ID: 2G72 Gee CL; Drinkwater N; Tyndall JD; Grunewald GL; Wu Q; McLeish MJ; Martin JL Enzyme adaptation to inhibitor binding: a cryptic binding site in phenylethanolamine N-methyltransferase. J. Med. Chem 2007, 50, 4845–4853. [DOI] [PubMed] [Google Scholar]
- 62.PDB ID: 1HNN Martin JL; Begun J; McLeish MJ; Caine JM; Grunewald GL Getting the adrenaline going. Crystal structure of the adrenaline-synthesizing enzyme PNMT. Structure 2001, 9, 977–985. [DOI] [PubMed] [Google Scholar]
- 63.PDB ID: 3HCD Drinkwater N; Gee CL; Puri M; Criscione KR; McLeish MJ; Grunewald GL; Martin JL Molecular recognition of physiological substrate noradrenaline by the adrenaline-synthesizing enzyme PNMT and factors influencing its methyltransferase activity. 2009, Biochem.J 422, 463–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Borchardt RT Mechanism of alkaline hydrolysis of S-adenosyl-l-methionine and related sulfonium nucleosides. J. Am. Chem. Soc 1979, 101, 458–463. [Google Scholar]
- 65.Wu SE; Huskey WP; Borchardt RT; Schowen RL Chiral instability at sulfur of S-adenosylmethionine. Biochemistry 1983, 22, 2828–2832. [DOI] [PubMed] [Google Scholar]
- 66.Hoffman JL Chromatographic analysis of the chiral and covalent instability of S-adenosyl-l-methionine. Biochemistry 1986, 25, 4444–4449. [DOI] [PubMed] [Google Scholar]
- 67.Masjost B; Ballmer P; Borroni E; Zurcher G; Winkler FK; Jakob-Roetne R; Diederich F Structure-based design, synthesis, and in vitro evaluation of bi-substrate inhibitors for catechol O-methyltransferase (COMT). Chem. Eur. J 2000, 6, 971–982. [DOI] [PubMed] [Google Scholar]
- 68.Lipinski CA; Lombardo F; Dominy BW; Feeney PJ Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliver. Rev 1997, 23, 3–25. [DOI] [PubMed] [Google Scholar]
- 69.Veber DF; Johnson SR; Cheng H-Y; Smith BR; Ward KW; Kopple KD Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem 2002, 45, 2615–2623. [DOI] [PubMed] [Google Scholar]
- 70.ClogP and tPSA were calculated using ChemDraw 18, copyright 2018. by PerkinElmer Informatics. [Google Scholar]
- 71.Jamieson GA The synthesis of 5'-deoxy-5'-S-(3-methylthiopropylamine)sulfoniumadenosine ("decarboxylated S-adenosylmethionine"). J. Org. Chem 1963, 28, 2397–2400. [Google Scholar]
- 72.Anglin JL; Deng L; Yao Y; Cai G; Liu Z; Hong J; Cheng G; Chen P; Dong S; Song Y Synthesis and structure-activity relationship investigation of adenosine-containing inhibitors of histone methyltransferase DOT1L J. Med. Chem 2012, 55, 8066–8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Matter H; Schudok M; Schwab W; Thorwart W; Barbier D; Billen G; Haase B; Neises B; Weithmann K-U; Wollmann T Tetrahydroisoquinoline-3-carboxylate based matrix-metalloproteinase inhibitors: design, synthesis and structure-activity relationship. Bioorg. Med. Chem 2002, 10, 3529–3544. [DOI] [PubMed] [Google Scholar]
- 74.Murakami S; Tomozane H; Togo Y; Morimoto Y Preparation of N-(isoquinolinylmethyl)benzamide analogs for treatment of schizophrenia. 9310089, 19930527, 1993. [Google Scholar]
- 75.Ortwine DF; Malone TC; Bigge CF; Drummond JT; Humblet C; Johnson G; Pinter GW Generation of N-methyl-D-aspartate agonist and competitive antagonist pharmacophore models. Design and synthesis of phosphonoalkyl-substituted tetrahydroisoquinolines as novel antagonists. J. Med. Chem 1992, 35, 1345–1370. [DOI] [PubMed] [Google Scholar]
- 76.Schiller P Preparation of opioid peptide analogs as opioid receptor antagonists. 9535316, 19951228, 1995. [Google Scholar]
- 77.White JM; Tunoori AR; Georg GI A novel and expeditious reduction of tertiary amides to aldehydes using Cp2Zr(H)Cl. J. Am. Chem. Soc 2000, 122, 11995–11996. [Google Scholar]
- 78.Pignot M; Pljevaljcic G; Weinhold E Efficient synthesis of S-adenosyl-l-homocysteine natural product analogues and their use to elucidate the structural determinant for cofactor binding of the DNA methyltransferase M.HhaI. Eur. J. Org. Chem 2000, 3, 549–555. [Google Scholar]
- 79.Ohmoto K; Yamamoto T; Okuma M; Horiuchi T; Imanishi H; Odagaki Y; Kawabata K; Sekioka T; Hirota Y; Matsuoka S; Nakai H; Toda M; Cheronis JC; Spruce LW; Gyorkos A; Wieczorek M Development of orally active nonpeptidic inhibitors of human neutrophil elastase. J. Med. Chem 2001, 44, 1268–1285. [DOI] [PubMed] [Google Scholar]
- 80.Wu Q; Criscione KR; Grunewald GL; McLeish MJ Phenylethanolamine N-methyltransferase inhibition: re-evaluation of kinetic data. Bioorg. Med. Chem. Lett 2004, 14, 4217–4220. [DOI] [PubMed] [Google Scholar]
- 81.SigmaPlot, version 12.5; Systat Software, San Jose, CA: 2012. [Google Scholar]
- 82.U'Prichard DC; Greenberg DA; Snyder SH Binding characteristics of a radiolabeled agonist and antagonist at central nervous system alpha noradrenergic receptors. Mol. Pharmacol 1977, 13, 454–473. [PubMed] [Google Scholar]
- 83.McLeish MJ; Kenyon GL Approaches to the Rational Design of Enzyme Inhibitors, In Burger’s Medicinal Chemistry and Drug Discovery, Volume 1, Sixth Edition, Abraham DJ, Ed.; John Wiley & Sons, Inc.:Hoboken, NJ, 2003; 1, p. 715–779. [Google Scholar]
- 84.Yu M, Magalhaes ML Cook PF and Blanchard JS Bisubstrate inhibition: Theory and application to N-acetyltransferases. Biochemistry 2006, 45, 14788–14794. [DOI] [PubMed] [Google Scholar]
- 85.Grunewald GL; Dahanukar VH; Ching P; Criscione KR Effect of ring size or an additional heteroatom on the potency and selectivity of bicyclic benzylamine-type inhibitors of phenylethanolamine N-methyltransferase. J. Med. Chem 1996, 39, 3539–3546. [DOI] [PubMed] [Google Scholar]
- 86.PDB ID: 4MIK Bart AG; Scott EE Crystal Structure of hPNMT in Complex with bisubstrate inhibitor (2R,3R,4S,5S)-2-(6-amino-9H-purin-9-yl)-5-(((2-(((7-nitro-1,2,3,4-tetrahydroisoquinolin-3-yl)methyl)amino)ethyl)thio)methyl)tetrahydrofuran-3,4-diol. (2013) [Google Scholar]
- 87.PDB ID: 4MQ4 Bart AG; Scott EE Crystal Structure of hPNMT in Complex with bisubstrate inhibitor N-(3-((((2S,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)thio)propyl)-1,2,3,4-tetrahydroisoquinoline-3-carboxamide. (2013) [Google Scholar]
- 88.Kabsch W XDS. Acta Crystallogr. D 2010, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Collaborative Computational Project, N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D 1994, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- 90.McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phaser crystallographic software. J. Appl. Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Emsley P; Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr. D 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- 92.Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tada M; Shijima H; Nakamura M Smiles-type free radical rearrangement of aromatic sulfonates and sulfonamides: syntheses of arylethanols and arylethylamines. Org. Biomol. Chem 2003, 1, 2499–2505 [DOI] [PubMed] [Google Scholar]
- 94.Wright DL; McMills MC Intramolecular aziridination: decomposition of diazoamides with tethered imino bonds. Org. Lett 1999, 1, 667–670. [Google Scholar]
- 95.Orr GR; Danz DW; Pontoni G; Prabhakaran PC; Gould SJ; Coward JK Synthesis of chirally deuteriated (S-adenosyl-S-methylsulfonio)propylamines and spermidines. Elucidation of the stereochemical course of putrescine aminopropyltransferase (spermidine synthase). J. Am. Chem. Soc 1988, 110, 5791–5799. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.