

Abstract
Expression levels of long non-coding RNA (lncRNA) have been shown to associate with clinical outcome of patients with cytogenetically normal acute myeloid leukemia (CN-AML). However, the frequency and clinical significance of genetic variants in the nucleotide sequences of lncRNA in AML patients is unknown. Herein, we analyzed total RNA sequencing data of 377 younger adults (aged <60 years) with CN-AML, who were comprehensively characterized with regard to clinical outcome. We used available genomic databases and stringent filters to annotate genetic variants unequivocally located in the non-coding transcriptome of AML patients. We detected 981 variants, which are recurrently present in lncRNA that are expressed in leukemic blasts. Among these variants, we identified a cytosine-to-thymidine variant in the lncRNA RP5-1074L1.4 and a cytosine-to-thymidine variant in the lncRNA SNHG15, which independently associated with longer survival of CN-AML patients. The presence of the SNHG15 cytosine-to-thymidine variant was also found to associate with better outcome in an independent dataset of CN-AML patients, despite differences in treatment protocols and RNA sequencing techniques. In order to gain biological insights, we cloned and overexpressed both wild-type and variant versions of the SNHG15 lncRNA. In keeping with its negative prognostic impact, overexpression of the wild-type SNHG15 associated with higher proliferation rate of leukemic blasts when compared with the cytosine-to-thymidine variant. We conclude that recurrent genetic variants of lncRNA that are expressed in the leukemic blasts of CN-AML patients have prognostic and potential biological significance.
Introduction
Acute myeloid leukemia (AML) is heterogeneous with regard to the patients’ clinical course and the underlying molecular lesions that drive the disease.1-2 Research efforts of the past four decades have identified a growing list of genetic alterations associated with clinical outcome that could be used as biomarkers for the risk stratification of the patients’ treatment. These alterations include chromosomal abnormalities,3-5 gene mutations,6-11 and aberrant expression of RNA transcripts.12-16 The advent of next-generation sequencing has revealed that AML displays notable heterogeneity at the level of isolated cases; leukemic blasts of individual AML patients represent, in many instances, the sum of distinct clonal subpopulations, within which mutations in different genes co-exist and co-operate.17,18 While such sequencing efforts continue to expand our understanding of AML pathogenesis, the majority of them are focused on the protein-coding fraction of the genome, which is, comparatively, its smallest part.19
The non-protein-coding part of the genome, a large fraction of which is actively transcribed into non-coding RNA, is gaining gradual recognition for its important regulatory role.20,21 Long non-coding RNA (lncRNA), which are transcripts longer than 200 nucleotides and, per definition, lack protein-coding potential, regulate many key cellular functions in health and disease.22-24 Deregulated expression of individual lncRNA has been demonstrated to significantly affect the cancer phenotype and patients’ clinical outcome. 25-29 We and others have previously shown that aberrant expression of small subsets of lncRNA independently associate with the clinical outcome of patients with cytogenetically normal AML (CN-AML).30-33 With regard to variations in the nucleotide sequences of lncRNA, it has previously been reported that disease-associated single nucleotide polymorphisms (SNP) are enriched in the genetic loci encoding these transcripts.34-36 In addition, acquired mutations of lncRNA that are recurrently detectable in the leukemic blasts have previously been identified.37 However, to our knowledge, their prognostic and biologic significance in AML have not yet been studied.
Herein, we analyzed whole transcriptome sequencing data of younger adults with the goal to evaluate the clinical and biological relevance of lncRNA variants in CN-AML. We used a panel of currently available databases of human genomic polymorphisms to annotate recurrent genetic variants located within expressed lncRNA. We show that a subset of these variants independently associates with clinical outcome of CN-AML patients.
Methods
Patients and treatment
Exploratory analysis was conducted in pretreatment bone marrow (BM) or blood samples from 377 younger adults (aged <60 years; range, 18-59 years) with de novo CN-AML. Patients were treated with intensive, first-line chemotherapy on Cancer and Leukemia Group B (CALGB)/Alliance for Clinical Trials in Oncology (Alliance) trials. Confirmatory analyses were conducted in a set of 135 CN-AML patients (75 of whom were younger than 60 years and 60 were older) enrolled on clinical trials of the German AML Cooperative Group (AMLCG).38,39
All patients provided written informed consent regarding the research use of their specimens. All study protocols were in accordance with the Declaration of Helsinki and approved by Institutional Review Boards at each center.
Cytogenetic and molecular analyses
Cytogenetic analyses of CALGB/Alliance patients were performed in CALGB/Alliance-approved institutional laboratories and results were confirmed by central karyotype review.40 The diagnosis of normal karyotype was based on analysis of ≥20 metaphases obtained from BM specimens subjected to short-term (24- or 48-hour) unstimulated cultures.40
Mutational analyses of patient samples were conducted with Sanger sequencing (for the CEBPA gene), fragment analysis (for detection of FLT3-internal tandem duplications [FLT3-ITD]) and targeted amplicon sequencing (for all other prognostic gene mutations), as reported previously.31,41-43 Molecular and cytogenetic profiling of the AMLCG cohort were obtained as described previously. 38,39
Transcriptome analyses
RNA samples of the patients treated on CALGB/Alliance protocols were analyzed with total RNA sequencing (RNA Seq) after depletion of ribosomal and mitochondrial RNA using the Illumina HiSeq 2500 platform. The results of the RNA Seq analysis have been deposited in the functional genomics data repository GEO and are publicly available under the accession number GSE137851. Patients in the AMLCG cohort were analyzed with RNA Seq following selection for poly-adenylated transcripts (poly-A RNA Seq) as previously described.16
For exploratory analyses, after quality control, adaptor-trimmed 50 base-pair-long paired-end reads were mapped to the human reference genome and variant calling was performed following the Genome Analysis Toolkit best practice recommendations for RNA Seq datasets.44 A two-pass variant calling approach was applied to ensure variant detection and depth of coverage (Figure 1). Unique variant positions were identified on non-coding transcripts that do not overlap with coding exons and are located in low-complexity regions of the genome (i.e., excluding repeat masked regions and segmental duplications). These variants were further evaluated for associations with clinical outcome and the expression levels of other RNA transcripts.
Statistical analyses
Clinical endpoint definitions are provided in the Online Supplementary Appendix. For each examined lncRNA variant, only patients with detectable expression of the lncRNA and adequate coverage of the variant position (i.e., depth of coverage >8) were analyzed. The estimated probabilities of disease-free (DFS), overall (OS) and event-free (EFS) survival were calculated using the Kaplan–Meier method, and the log-rank test evaluated differences between survival distributions. Cox proportional hazard models were used to calculate hazard ratios for DFS, OS and EFS.45 Multivariable proportional hazards models were constructed using a backward selection procedure.45 All statistical analyses using CALGB/Alliance data were performed by the Alliance Statistics and Data Center.
In vitro experiments
LncRNA wild-type and variant transcripts were amplified with Phusion high fidelity polymerase by polymerase chain reaction (PCR). Amplicons were cloned into pcDNA using the Gibson technique, according to standard protocols. For primer sequences and further experimental details please see the Online Supplementary Appendix. K-562 and THP-1 cells were transfected with vectors containing either a cytosine (C)-to-thymidine (T) variant in the lncRNA SNHG15 (SNHG15varT) or wild-type lncRNA SNHG15 (SNHG15wt); cells were also transfected with empty pcDNA3.1 and were used as controls. Cell viability and apoptosis were assessed with annexin V staining. The colorimetric MTT assay was used to assess the proliferative capacity of the transfected blasts.
Results
Detection of genetic variants in the non-coding transcriptome of younger adults with cytogenetically normal acute myeloid leukemia
In order to examine whether recurrent genetic variants are present in the non-coding transcriptomes of CN-AML patients, we first analyzed total RNA sequencing data of 377 younger adults with CN-AML. In order to identify unequivocally non-coding genetic variants and to avoid ambiguity in their genomic location, we excluded from further analyses all variants, which overlapped with exons of protein-coding genes and those that mapped to segmental duplications or other repeat regions of the genome.
In order to evaluate the clinical and functional relevance of the lncRNA variants, additional filters were applied. Specifically, we focused on the variants that displayed: i) adequate expression and coverage in at least 100 samples (approximately 25% of a total number of samples), ii) detection of wild-type genotype in at least 5% of the samples, and iii) detection of variant allele frequency above 0.4 in at least 5% of the samples (Figure 1). Based on these criteria, 981 variants were selected for further analyses (Online Supplementary Table S1).
Detection of long non-coding RNA variants in the Cancer Genome Atlas (TCGA) dataset
In order to examine the validity and reproducibility of our experimental pipeline and results, we queried the publicly available TCGA total RNA Seq dataset.8 It is noteworthy that the TCGA dataset was generated with a different RNA Seq technique (i.e., poly-A RNA Seq), which is less suitable for the interrogation of the non-coding fraction of the transcriptome. 46 In addition, a relatively small number of patients in the TCGA cohort represent CN-AML cases (i.e., 44 of the 196 available cases).8 Despite these limitations, 277 out of the 981 variants that we tested were detectable in the transcriptomes of the CN-AML cases included in the TCGA study (Online Supplementary Table S2).
For a subset of TCGA cases, DNA sequencing data from both leukemic blasts and germline material are available in addition to the transcriptome sequencing data. We therefore sought: i) to validate whether the presence of the detected variants in the transcriptome is also detectable at the DNA level and ii) to examine whether these variants are bona fide acquired genetic events or are present in the germline configuration of the AML patient genomes. As the sequencing technique that was used to analyze the TCGA dataset (i.e., exome sequencing) preferentially captures and interrogates the coding fraction of the genome, only 20 variant positions were available for analyses at the DNA level. Overall, there was a complete concordance between the detection of a variant in the transcriptome and its detection in the genome. Furthermore, 11 of these variants were detected in both leukemic blasts and non-leukemic tissues and thus could be considered as germline genetic variants, whereas nine variants were only detectable in leukemic samples and could therefore represent acquired mutations (Online Supplementary Table S2).
Figure 1.

Outline of the two-pass experimental approach for the identification of recurrent genetic variants located within long non-coding RNA in younger adult patients with cytogenetically normal acute myeloid leukemia. In the first pass, variant calling was performed on alignment results (i.e., BAM files) following the Genome Analysis Toolkit (GATK) best practice recommendations for RNA sequencing datasets. Variants of non-coding transcripts that do not overlap with coding exons and are not located in low-complexity regions of the genome were selected. In the second pass, Samtools pile-up programs were used to identify sequencing depth, quality and alternative allele counts on selected unique variant positions. Resulting visual component framework (VCF) files were consolidated with annotation and the final variant call matrix was generated.
Figure 2.

Outcome of younger adult patients with cytogenetically normal acute myeloid leukemia who harbored the C-to-T variant of the RP5-1074L1.4 long non-coding RNA (IncRNA) (RP5-1074L1.4varT) and of those who had the wild-type lncRNA (RP5-1074L1.4wt). (A) Disease-free survival, (B) overall survival and (C) event-free survival.
Prognostic significance of recurrent long non-coding RNA variants in acute myeloid leukemia
We proceeded to examine the associations of the lncRNA variants with the clinical outcome of younger patients with CN-AML. Of the variants tested, 41 associated with more than one outcome endpoint (DFS, OS and/or EFS; Online Supplementary Table S3) above a threshold level of significance (P<0.05). There was no association between the presence of lncRNA variants and complete remission rates in our cohort.
Among the variants that showed significant association with clinical outcome in our dataset were a C-to-T variant in the lncRNA RP5-1074L1.4 (RP5-1074L1.4varT), and SNHG15varT, a C-to-T variant in the lncRNA SNHG15.
In patients with detectable RP5-1074L1.4 expression (n=243), the presence of the RP5-1074L1.4varT was found in 156 (64%) of them. Patients with the RP5-1074L1.4varT had longer DFS (P<0.001) than patients with wild-type RP5-1074L1.4 (RP5-1074L1.4wt); 5 years after diagnosis 44% of the RP5-1074L1.4varT patients were alive and leukemia-free in contrast to only 26% of those with RP5-1074L1.4wt. RP5-1074L1.4varT was also associated with longer EFS (P<0.001; 5-year rates: 38% vs. 21%) and showed a trend for longer OS (P=0.09; 5-year rates: 43% vs. 34%; Figures 2 A to C; Online Supplementary Table S4).
Among patients who expressed the SNHG15 lncRNA (n=306), SNHG15varT was detected in 78% of them (n=239). Patients who expressed the SNHG15varT had longer DFS (P=0.04; 5-year rates: 37% vs. 22%) than patients who expressed the SNHG15wt. SNHG15varT expressers also had longer EFS (P=0.04; 5-year rates: 31% vs. 19%) and a trend for longer OS (P=0.07; 5-year rates: 41% vs. 32%) compared with SNHG15wt expressers (Figures 3A to C; Online Supplementary Table S5).
Finally, we examined whether genetic variants were detectable in the set of 24 lncRNA, whose expression levels were previously shown to associate with the clinical outcome of younger adults with CN-AML.30 We found seven such variants in three of the prognostic lncRNA (annotated in bold lettering in the Online Supplementary Table S1). A guanosine (G)-to-C variant in the lncRNA AL122127.25 (AL122127.25varC) was the only one that associated with patient outcome. AL122127.25varC was detected in 72 of the 257 patients who expressed the AL122127.25 lncRNA (i.e., 28% of the AL122127.25 expressers). The presence of AL122127.25varC significantly associated with shorter DFS (P=0.01; 5-year rates: 17% vs. 35%), OS (P=0.01; 5-year rates: 22% vs. 40%) and EFS (P=0.002; 5-year rates: 12% vs. 30%; Online Supplementary Figures S1A to C; Online Supplementary Table S6) in younger adult CN-AML patients. Notably, the presence of AL122127.25varC had no impact on the expression levels of the AL122127.25 transcript, when compared to the AL122127.25wt (Online Supplementary Figure S2).
Associations of long non-coding RNA variants with pretreatment clinical and molecular characteristics
Next, we examined potential associations of RP5-1074L1.4varT, SNHG15varT and AL122127.25varC with pretreatment clinical characteristics and prognostic molecular features of younger adult CN-AML patients. Overall, there were only minor differences in these features between variant and wild-type lncRNA expressers. Patients expressing the RP5-1074L1.4varT were more likely to harbor tyrosine kinase domain mutations of the FLT3 gene (FLT3-TKD) than patients who expressed the RP5-1074L1.4wt (P=0.03; 12% vs. 4%; Online Supplementary Table S7). Patients expressing the SNHG15varT were older (P=0.03), more likely to be Caucasian (P=0.02) and more likely to have low expression of the MN1 gene (P=0.05; Online Supplementary Table S8) than SNHG15wt expressers. With regard to the lncRNA AL122127.25, there were no significant differences in the clinical features or frequencies of prognostic gene mutations or gene expression between AL122127.25varC and AL122127.25wt expressers (Online Supplementary Table S9).
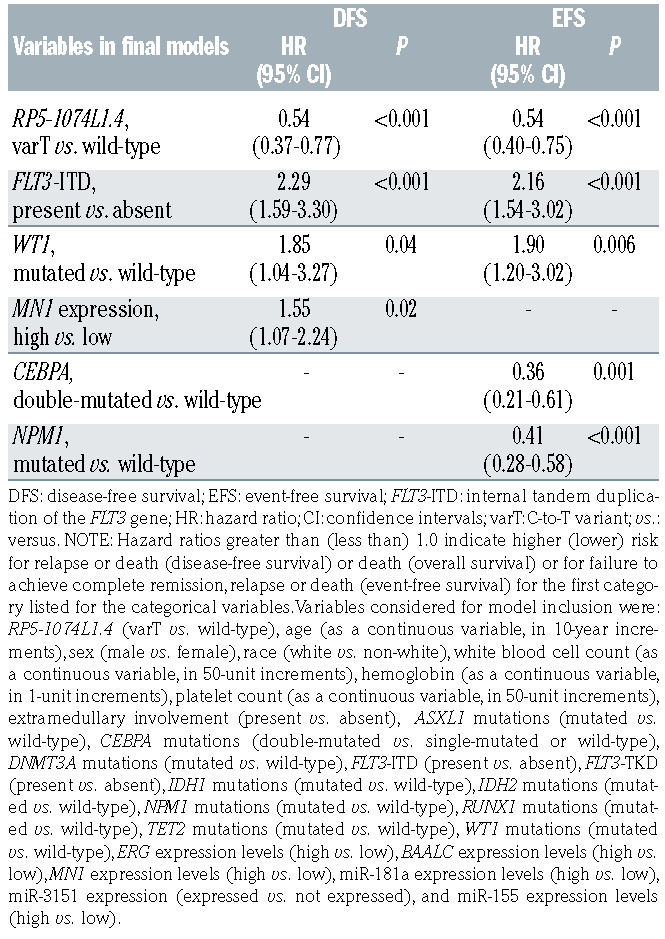
Table 1.
Multivariable analyses of outcome in younger adult patients with cytogenetically normal acute myeloid leukemia by expression of the C-to-T variant of the RP5-1074L1.4 long non-coding RNA (IncRNA) (RP5-1074L1.4varT) versus the wild-type lncRNA (RP5-1074L1.4wt).
Multivariable analyses
In order to examine prognostic significance of the detected lncRNA variants in the context of other established clinical and molecular prognostic markers, we constructed multivariable models.
Expression of RP5-1074L1.4varT was a significant marker for longer DFS (hazard ratio [HR]: 0.54; P<0.001) and longer EFS (HR: 0.54; P<0.001) after adjusting for other covariates (Table 1). Expression of SNHG15varT significantly associated with longer DFS (HR: 0.63; P=0.02), longer OS (HR: 0.63; P=0.008) and longer EFS (HR: 0.68; P=0.02) after adjusting for other variables for each outcome endpoint (Table 2). Finally, expression of the AL122127.25varC was an independent marker of shorter OS (HR: 1.57; P=0.009) and shorter EFS (HR: 1.59; P=0.004) after adjusting for other covariates (Online Supplementary Table S10).
Table 2.
Multivariable analyses of outcome in younger adult patients with cytogenetically normal acute myeloid leukemia by expression of the Cto- T variant of the SNHG15 long non-coding RNA (lncRNA) (SNHG15varT) versus the wild-type lncRNA (SNHG15wt).

RP5-1074L1.4 is an intronic lncRNA, which is embedded in intron 7 of the protein-coding SLC16A4 transcript. In order to ensure that the prognostic effect of RP5-1074L1.4varT was not due to perturbation of the expression levels of SLC16A4, we compared the SLC16A4 transcript abundance between 87 patients who expressed the RP5-1074L1.4wt and 156 patients who expressed the RP5-1074L1.4varT. We found no significant difference in expression levels of SLC16A4 between these two patient groups (Online Supplementary Figure S3).
Evaluation of the prognostic significance of long non-coding RNA variants in an independent cohort of cytogenetically normal acute myeloid leukemia patients
In order to examine whether our findings were reproducible in an independent cohort of CN-AML patients, we examined patients treated on AMLCG protocols,38,39 who had available clinical outcome data, and were analyzed with poly-A selected RNA Seq (n=135). As was the case with the TCGA dataset,8 the use of an alternative RNA Seq technique limited the number of lncRNA variants that could be detected and analyzed. The lncRNA transcripts, whose variants showed the strongest association with prognosis in our initial cohort were not captured by the poly-A RNA Seq and could not, therefore, be analyzed for associations with clinical outcome. Despite this limitation, there was concordance in the findings between the two cohorts. SNHG15varT was the one detectable lncRNA variant, which associated with multiple outcome endpoints in the AMLCG dataset. Specifically, the SNHG15varT was detected in 103 of the 120 AMLCG CN-AML patients who expressed the SNHG15 lncRNA. In agreement with our findings in the CALGB/Alliance dataset, the presence of SNHG15varT associated with longer DFS (P=0.04; Figure 3D) and EFS (P=0.007, Figure 3E), but not OS (P=0.17), in the AMLCG cohort.
Expression levels of SNHG15 in normal hematopoiesis
In order to further examine the functional significance of the lncRNA, which harbor prognostic variants we sought to determine their expression patterns during normal hematopoiesis. To this end, we used publicly available datasets of normal hematopoietic cells analyzed with microarrays or RNA Seq and deposited in the BloodSpot portal (www.bloodspot.eu). Of the lncRNA with prognostic genetic variants, only SNHG15 was annotated in the database and could be further analyzed. We found that SNHG15 was most abundantly expressed in common myeloid progenitors, granulocyte monocyte progenitors and megakaryocyte-erythroid progenitors. It was also highly expressed in hematopoietic stem cells and lymphoid cell populations. Among mature cell populations, SNHG15 was overexpressed in monocytes, whereas its expression levels were the lowest in polymorphonuclear leucocytes of the bone marrow and peripheral blood (Online Supplementary Figure S4).
Figure 3.

Prognostic significance of long non-coding RNA variants across different cytogenetically normal acute myeloid leukemia cohorts. (A) Diseasefree survival, (B) overall survival and (C) event-free survival of younger adult cytogeneti-cally normal acute myeloid leukemia (CN-AML) patients in the CALGB/Alliance cohort who had the C-to-T variant of the SNHG15 long non-coding RNA (SNHG15varT) and of patients with the wild-type SNHG15 lncRNA (SNHG15wt). (D) Disease-free survival and (E) eventfree survival of CN-AML patients in the AMLCG cohort with the C-to-T variant of the SNHG15 lncRNA (SNHG15varT) and of those the wild-type SNHG15 lncRNA (SNHG15wt).
Functional relevance of prognostic long non-coding RNA variants
We hypothesized that in addition to associations with outcome, the presence of genetic variants in lncRNA could have a functional impact and affect AML blast viability and proliferation. We focused on the lncRNA SNHG15varT, whose prognostic significance was validated in an independent cohort of AML patients. We isolated total RNA from AML cell lines and amplified both the SNHG15wt and SNHG15varT transcripts. We cloned the amplicons into pcDNA3.1 expression vectors and transfected two AML cell lines (i.e., K-562 and THP-1 cells). Expression levels of the SNHG15 lncRNA were similar in cells transfected with the SNHG15wt and those transfected with SNHG15varT-containing vectors, when compared with the empty vector controls (Figures 4A and B). Regarding cell viability, ectopic overexpression of the SNHG15wt and the SNHG15varT led to a discreet but consistent decrease in cell viability across cell lines, which did not reach statistical significance (Figures 4C and D).
In order to further evaluate the effect of SNHG15 on the growth kinetics of the AML cells, we performed colorimetric MTT assays. Forced overexpression of the SNHG15varT had no significant effect on blast growth when compared with controls. In contrast, overexpression of the SNHG15wt led to increased proliferative capacity of the leukemic cells when compared with both controls and cells overexpressing the SNHG15varT (Figures 4E and F). These findings are in line with our prognostic observations and the inferior outcome of CN-AML patients who express SNHG15wt, compared with those who express SNHG15varT.
Discussion
lncRNA are gaining recognition as important molecular mediators and regulators of key cellular functions in health and disease.22-29 In AML, lncRNA have been shown to associate with the clinical outcome of both younger and older patients.30-33 However, these previous studies have focused on the expression levels of the lncRNA transcripts; the effect of genetic variants within lncRNA has not been extensively studied. Accumulating evidence suggests that such genetic variants could impact on the function of the lncRNA and be relevant for disease pathogenesis.47 In support of this view, disease-associated SNP are more frequently found in regions of the genome that encode for non-coding RNA transcripts in several types of solid tumors34,35 and in AML.34,36 These findings suggest that the presence of a variant in the non-coding transcriptome could be the functional link that explains how genetic variants that do not alter the structure of protein molecules associate with malignant phenotypes. Recently, Klco et al.37 have reported the acquisition of somatic mutations in lncRNA transcripts of AML patients, as demonstrated by analyses of the leukemic blasts in parallel with germline material. However, the prognostic value of these mutations could not be tested due to sample size limitations. Herein, we analyzed total RNA Seq data of younger adult patients with CN-AML with the goal to detect recurrent lncRNA variants and evaluate their prognostic and biologic significance.
We used a stringent approach for detecting and filtering sequence variations of lncRNA. We generated a list of 981 recurrent variants, which are located within lncRNA in younger adult patients constituting the CALGB/Alliance cohort, and which had adequate sample sizes for meaningful survival analyses. In order to study potential associations of the detected variants with clinical outcome, we individualized analysis for each variant and limited comparisons to the group of patients that were expressers of the corresponding lncRNA transcript (i.e., we compared the expressers of a variant to the expressers of the wild-type lncRNA). Of the 981 candidate variants, a subset of 41 significantly associated with at least two clinical outcome endpoints of younger adults with CN-AML. LncRNA genetic variants RP5-1074L1.4varT and SNHG15varT were significantly associated with prognosis. RP5-1074L1.4 has not been previously associated with cancer pathogenesis or clinical outcome of cancer patients. In contrast, SNHG15, a MYC-regulated lncRNA, which harbors a small nucleolar RNA in one of its introns, has been implicated in the pathogenesis of multiple solid malignancies. SNHG15 has been shown to interact with the protein AIF and to associate with a clinically aggressive and prognostically unfavorable subset of colorectal carcinomas.48 SNHG15 has also been associated with aggressive phenotypes of hepatocellular and breast carcinomas via sponging and inhibiting the function of microRNAs 141-3p and 211-3p.49,50
Patients who expressed the RP5-1074L1.4varT had more favorable outcome (i.e., longer DFS, EFS and a trend for longer OS) than RP5-1074L1.4wt expressers. SNHG15varT also associated with better prognosis and SNHG15varT expressers had longer DFS and EFS compared with patients who expressed the SNHG15wt lncRNA.
Further, we examined whether recurrent genetic variants could be detected in lncRNA previously reported to be prognostic in younger adults with CN-AML.30 A G-to-C variant in the prognostic lncRNA AL122127.25 (AL122127.25varC) significantly associated with clinical outcome of CN-AML patients. Specifically, expression of the AL122127.25varC associated with shorter DFS, OS and EFS. Notably, the expression levels of the AL122127.25 lncRNA were not significantly affected by the presence of the AL122127.25varC. Thus, both the abundance and the nucleotide sequence of the AL122127.25 lncRNA associate with the clinical outcome of CN-AML patients via potentially independent mechanisms.
Currently, there is limited availability of AML datasets analyzed with RNA Seq techniques (i.e., with total RNA Seq) that are suitable for in-depth analyses of the non-coding transcriptome. In order to examine whether our observations could be reproduced in other datasets, we examined the presence of our curated variant list in an independent cohort of CN-AML patients who were treated on AMLCG protocols. As expected, the use of a different RNA Seq technique significantly limited the number of lncRNA transcripts that we could interrogate. Nevertheless, SNHG15varT was detectable and associated with longer DFS and EFS of the AMLCG patients, similarly to its prognostic effect in the CALGB/Alliance dataset.
Given the constantly increasing number of prognostic molecular alterations in AML, it is important to examine the prognostic value of novel markers in the context of other established clinical and molecular prognosticators. It is thus noteworthy that the lncRNA variants that we examined (RP5-1074L1.4varT, SNHG15varT, and AL122127.25varC) did not show associations with gene mutations that are currently used for the risk stratification of the treatment of AML patients.11 Mutations in the CEBPA, RUNX1, ASXL1 and NPM1 genes as well as the presence of the FLT3-ITD were similarly distributed between expressers of the lncRNA variants and expressers of the wild-type transcripts. In addition, in formal multivariable analyses that included prognostic clinical and molecular parameters, RP5-1074L1.4varT, SNHG15varT, and AL122127.25varC retained their prognostic significance after adjusting for other covariates. Consequently, detection of these variants could provide additional prognostic information and further refine risk-stratification of CN-AML patients.
In addition to testing their prognostic significance, we sought to evaluate whether the presence of recurrent variants in lncRNA sequences has functional implications. We focused on SNHG15 lncRNA and performed overexpression experiments with SNHG15wt- and SNHG15varT-containing vectors in the K-562 and THP-1 cell lines. Overexpression of the SNHG15varT had no significant effect on cell viability and no evident impact on cell proliferation, compared with controls. In contrast, overexpression of the SNHG15wt led to a significant increase in the proliferative capacity of the leukemic blasts in both cell lines that were tested. Despite the limitations of in vitro assays, these results indicate that expression of SNHG15wt associates with a more aggressive disease phenotype, when compared with SNHG15varT, and are in line with the inferior clinical outcome of SNHG15wt-expressers.
In summary, we have performed comprehensive characterization of genetic variants within lncRNA in younger adults with CN-AML. We present analyses that support the prognostic and potential biologic significance of lncRNA variants in CN-AML. We believe that our work will serve as a useful starting point for further studies on the role of genetic variants in the non-coding transcriptome of cancer patients.
Figure 4.

Biologic significance of recurrent long non-coding RNA variants in younger adult cytogenetically normal acute myeloid leukemia patients. Fold changes of SNHG15 long non-coding RNA (lncRNA) expression levels (A and B), percent of viable cells (C and D) and proliferation (E and F) of acute myeloid leukemia cell lines transfected with empty pcDNA3, SNHG15wt-containing or SNHG15varT-containing vectors. Results for K-562 (A, C and E) and THP-1 (B, D and F) cells are depicted. Proliferation is assessed with the MTT colorimetric assay, by light absorbance. *P<0.05; **P<0.01; ***P<0.001; N.S: not significant.
Supplementary Material
Acknowlegments
The authors would like to thank: the Alliance Hematology Malignancy Biorepository supported by Washington University subcontract WU-15-398/WU-16-51 and the Alliance NCTN Biorepository and Biospecimen Resource (U24CA196171) for sample processing and storage services, and Lisa J. Sterling (The Ohio State University, Comprehensive Cancer Center, Columbus, OH 43221) for data management. Celebrating the life and accomplishments of Dr. Clara D. Bloomfield (1942-2020).
Funding Statement
Funding: Research reported in this publication was supported in part by the National Cancer Institute of the National Institutes of Health under award numbers U10CA180821 and U10CA180882 (to the Alliance for clinical trials in oncology), U10CA077658, U10CA180850, U10CA180861, CA140158, CA16058, UG1CA233338 and R35CA197734. This work was also supported in part by the Leukemia Clinical Research Foundation, D. Warren Brown Foundation and the Pelotonia Fellowship Program. TH was supported by a Physician Scientists grant (G-509200-004) from the Helmholtz Zentrum München. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1.Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373(12):1136-1152. [DOI] [PubMed] [Google Scholar]
- 2.Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424-447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Byrd JC, Mrózek K, Dodge RK, et al. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461). Blood. 2002;100(13):4325-4336. [DOI] [PubMed] [Google Scholar]
- 4.Grimwade D, Hills RK, Moorman AV, et al. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood. 2010;116(3):354-365. [DOI] [PubMed] [Google Scholar]
- 5.Mrózek K, Heerema NA, Bloomfield CD. Cytogenetics in acute leukemia. Blood Rev. 2004;18(2):115-136. [DOI] [PubMed] [Google Scholar]
- 6.Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209-2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patel JP, Gönen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366(12):1079-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cancer Genome Atlas Research Network Ley TJ, Miller C, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059-2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Metzeler KH, Herold T, Rothenberg-Thurley M, et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood. 2016;128(5):686-698. [DOI] [PubMed] [Google Scholar]
- 10.Eisfeld A-K, Kohlschmidt J, Mrózek K, et al. Mutation patterns identify adult patients with de novo acute myeloid leukemia aged 60 years or older who respond favorably to standard chemotherapy: an analysis of Alliance studies. Leukemia. 2018;32(6):1338-1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mrózek K, Marcucci G, Paschka P, Whitman SP, Bloomfield CD. Clinical relevance of mutations and gene-expression changes in adult acute myeloid leukemia with normal cytogenetics: are we ready for a prognostically prioritized molecular classification? Blood. 2007;109(2):431-448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Valk PJM, Verhaak RGW, Beijen MA, et al. Prognostically useful gene-expression profiles in acute myeloid leukemia. N Engl J Med. 2004;350(16):1617-1628. [DOI] [PubMed] [Google Scholar]
- 13.Metzeler KH, Hummel M, Bloomfield CD, et al. An 86-probe-set gene-expression signature predicts survival in cytogenetically normal acute myeloid leukemia. Blood. 2008;112(10):4193-4201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Z, Herold T, He C, et al. Identification of a 24-gene prognostic signature that improves the European LeukemiaNet risk classification of acute myeloid leukemia: an international collaborative study. J Clin Oncol. 2013;31(9):1172-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marcucci G, Yan P, Maharry K, et al. Epigenetics meets genetics in acute myeloid leukemia: clinical impact of a novel sevengene score. J Clin Oncol. 2014;32(6):548-556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herold T, Jurinovic V, Batcha AMN, et al. A 29-gene and cytogenetic score for the prediction of resistance to induction treatment in acute myeloid leukemia. Haematologica. 2018;103(3):456-465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Welch JS, Ley TJ, Link DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150(2):264-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klco JM, Spencer DH, Miller CA, et al. Functional heterogeneity of genetically defined subclones in acute myeloid leukemia. Cancer Cell. 2014;25(3):379-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860-921. [DOI] [PubMed] [Google Scholar]
- 20.Kellis M, Wold B, Snyder MP, et al. Defining functional DNA elements in the human genome. Proc Natl Acad Sci U S A. 2014;111(17):6131-6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taylor J. Clues to function in gene deserts. Trends Biotechnol. 2005;23(6):269-271. [DOI] [PubMed] [Google Scholar]
- 22.Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guttman M, Rinn JL. Modular regulatory principles of large non-coding RNAs. Nature. 2012;482(7385):339-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43(6):904-914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gupta RA, Shah N, Wang KC, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464(7291):1071-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leucci E, Vendramin R, Spinazzi M, et al. Melanoma addiction to the long non-coding RNA SAMMSON. Nature. 2016;531(7595): 518-522. [DOI] [PubMed] [Google Scholar]
- 27.Trimarchi T, Bilal E, Ntziachristos P, et al. Genome-wide mapping and characterization of Notch-regulated long noncoding RNAs in acute leukemia. Cell. 2014;158(3): 593-606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Papaioannou D, Petri A, Dovey OM, et al. The long non-coding RNA HOXB-AS3 regulates ribosomal RNA transcription in NPM1-mutated acute myeloid leukemia. Nat Commun. 2019;10(1):5351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ji P, Diederichs S, Wang W, et al. MALAT-1, a novel noncoding RNA, and thymosin β4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene. 2003;22(39):8031-8041. [DOI] [PubMed] [Google Scholar]
- 30.Papaioannou D, Nicolet D, Volinia S, et al. Prognostic and biologic significance of long non-coding RNA profiling in younger adults with cytogenetically normal acute myeloid leukemia. Haematologica. 2017;102(8): 1391-1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garzon R, Volinia S, Papaioannou D, et al. Expression and prognostic impact of lncRNAs in acute myeloid leukemia. Proc Natl Acad Sci U S A. 2014;111(52):18679-18684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beck D, Thoms JAI, Palu C, et al. A fourgene LincRNA expression signature predicts risk in multiple cohorts of acute myeloid leukemia patients. Leukemia. 2018;32(2): 263-272. [DOI] [PubMed] [Google Scholar]
- 33.Mer AS, Lindberg J, Nilsson C, et al. Expression levels of long non-coding RNAs are prognostic for AML outcome. J Hematol Oncol. 2018;11(1):52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yan X, Hu Z, Feng Y, et al. Comprehensive genomic characterization of long non-coding RNAs across human cancers. Cancer Cell. 2015;28(4):529-540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iyer MK, Niknafs YS, Malik R, et al. The landscape of long noncoding RNAs in the human transcriptome. Nat Genet. 2015;47(3):199-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schwarzer A, Emmrich S, Schmidt F, et al. The non-coding RNA landscape of human hematopoiesis and leukemia. Nat Commun. 2017;8(1):218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klco JM, Miller CA, Griffith M, et al. Association between mutation clearance after induction therapy and outcomes in acute myeloid leukemia. JAMA. 2015;314(8):811-822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Büchner T, Berdel WE, Schoch C, et al. Double induction containing either two courses or one course of high-dose cytarabine plus mitoxantrone and postremission therapy by either autologous stem-cell transplantation or by prolonged maintenance for acute myeloid leukemia. J Clin Oncol. 2006;24(16):2480-2489. [DOI] [PubMed] [Google Scholar]
- 39.Braes J, Amler S, Kreuzer K-A, et al. Sequential high-dose cytarabine and mitoxantrone (S-HAM) versus standard double induction in acute myeloid leukemia―a phase 3 study. Leukemia. 2018;32(12):2558-2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mrózek K, Carroll AJ, Maharry K, et al. Central review of cytogenetics is necessary for cooperative group correlative and clinical studies of adult acute leukemia: the Cancer and Leukemia Group B experience. Int J Oncol. 2008;33(2):239-244. [PMC free article] [PubMed] [Google Scholar]
- 41.Eisfeld A-K, Mrózek K, Kohlschmidt J, et al. The mutational oncoprint of recurrent cytogenetic abnormalities in adult patients with de novo acute myeloid leukemia. Leukemia. 2017;31(10):2211-2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marcucci G, Maharry K, Radmacher MD, et al. Prognostic significance of, and gene and microRNA expression signatures associated with, CEBPA mutations in cytogenetically normal acute myeloid leukemia with highrisk molecular features: a Cancer and Leukemia Group B study. J Clin Oncol. 2008;26(31):5078-5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Whitman SP, Archer KJ, Feng L, et al. Absence of the wild-type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytogenetics and the internal tandem duplication of FLT3: a Cancer and Leukemia Group B study. Cancer Res. 2001;61(19):7233-7239. [PubMed] [Google Scholar]
- 44.Nekrutenko A, Taylor J. Next-generation sequencing data interpretation: enhancing reproducibility and accessibility. Nat Rev Genet. 2012;13(9):667-672. [DOI] [PubMed] [Google Scholar]
- 45.Vittinghoff E, Glidden DV, Shiboski SC, McCulloch CE. Regression methods in biostatistics: linear, logistic, survival and repeated measures models. New York, NY: Springer; 2005. [Google Scholar]
- 46.Zhao W, He X, Hoadley KA, Parker JS, Hayes DN, Perou CM. Comparison of RNASeq by poly (A) capture, ribosomal RNA depletion, and DNA microarray for expression profiling. BMC Genomics. 2014;15(1):419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Khurana E, Fu Y, Chakravarty D, Demichelis F, Rubin MA, Gerstein M. Role of non-coding sequence variants in cancer. Nat Rev Genet. 2016;17(2):93-108. [DOI] [PubMed] [Google Scholar]
- 48.Saeinasab M, Bahrami AR, González J, et al. SNHG15 is a bifunctional MYC-regulated noncoding locus encoding a lncRNA that promotes cell proliferation, invasion and drug resistance in colorectal cancer by interacting with AIF. J Exp Clin Cancer Res. 2019;38(1):172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ye J, Tan L, Fu Y, et al. LncRNA SNHG15 promotes hepatocellular carcinoma progression by sponging miR-141-3p. J Cell Biochem. 2019;120(12):19775-19783. [DOI] [PubMed] [Google Scholar]
- 50.Kong Q, Qiu M. Long noncoding RNA SNHG15 promotes human breast cancer proliferation, migration and invasion by sponging miR-211-3p. Biochem Biophys Res Commun. 2018;495(2):1594-1600. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.