

Abstract
Solution-phase hydrogen/deuterium exchange (HDX) coupled to mass spectrometry (MS) is a widespread tool for structural analysis across academia and the biopharmaceutical industry. By monitoring the exchangeability of backbone amide protons, HDX-MS can reveal information about higher-order structure and dynamics throughout a protein, can track protein folding pathways, map interaction sites, and assess conformational states of protein samples. The combination of the versatility of the hydrogen/deuterium exchange reaction with the sensitivity of mass spectrometry has enabled the study of extremely challenging protein systems, some of which cannot be suitably studied using other techniques. Improvements over the past three decades have continually increased throughput, robustness, and expanded the limits of what is feasible for HDX-MS investigations. To provide an overview for researchers seeking to utilize and derive the most from HDX-MS for protein structural analysis, we summarize the fundamental principles, basic methodology, strengths and weaknesses, and the established applications of HDX-MS while highlighting new developments and applications.
1. Introduction
1.1. General Application of H/D Exchange
Hydrogen/deuterium exchange (HDX) exchange as a general approach exploits the natural exchange of hydrogens that occurs at backbone amides within proteins.1−3 In a folded protein, the exchange kinetics for individual amides are strongly influenced by the local electronic and solvent environment, as well as the higher-order structure. The incorporated deuterium acts as a label along the backbone that can reveal information about each amide’s relative accessibility in the native protein. The tools for measuring amide hydrogen exchange evolved considerably over the years.4,5 The pioneering experiments that provided the foundational theory of H/D exchange in proteins were accomplished using sensitive densitometry measurements of intact proteins in solution.1 Using tritium instead of deuterium to do hydrogen/tritium exchange later became a more sensitive and insightful tool for measuring amide hydrogen exchange and also allowed for proteolysis to localize exchange to a peptide level.6−8 The foundational work and principles established from these early studies allowed HDX to be effectively coupled to mass spectrometry (MS) and offered advantages over previous methods of detection.9−13
The approach for most HDX-MS studies remains relatively simple. A protein, typically under native conditions, is transferred from an aqueous buffer to a deuterium-rich buffer and allowed to undergo exchange. The exchange is then slowed and the levels of deuterium incorporation are readily measured with MS, thanks to the approximately one Dalton mass difference between the mass of a protium (1H) and a deuterium (2H). While the experimental concept is simple, there are many considerations necessary for obtaining and interpreting HDX-MS data sets. One of the reasons HDX-MS has been so successful is its inherent versatility; it can be applied to nearly any protein system, many of which are too large, flexible, heterogeneous, or sample limited to be analyzed by other existing structural tools. Here we detail the chemistry, basic principles, and methodologies of HDX-MS and cover the many applications for which it is used. We also refer the reader to several recent reviews on different aspects of HDX-MS.14−19
1.2. Structural Factors Determining Amide Exchange Kinetics
The simple model that still serves as a foundational principle for amide HDX was proposed by Linderstrom-Lang.1 A given amide within a protein can exist in a closed or open state, depending on whether the amide is protected (Figure 1). In the closed state, the amide is not accessible for deprotonation. When local conformational fluctuations or global unfolding render the amide accessible, the hydrogen can undergo exchange with solvent at a specific rate, termed the chemical exchange rate (kch). After deuterium exchange, the protein may then revert back to a closed state. We note that in the schema there is a single arrow for the kch in cases with D2O in high excess; at this step, amides equilibrate to the final percentage of deuterium in the surrounding solution. It is generally accepted that hydrogen bonding is a primary determinant of amide protection4,20,21 but that solvent occlusion also plays a role in determining protection from exchange.22−24 While protection is strongly correlated to the degree of hydrogen bonding, there remain gaps in our full understanding on the combination of all the molecular determinants that govern amide HDX kinetics,25 as highlighted by recent studies where the observed protection cannot be fully rationalized.26
Figure 1.
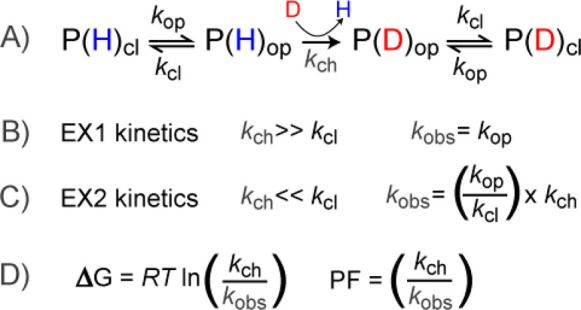
(A) Overview of the basic principles of amide exchange in proteins. The protein samples a closed (cl) and open (op) forms, of which only the open form is accessible for deuterium exchange. The relative rates of opening (kop), closing (kcl), and the chemical exchange rate of the accessible amide (kch) govern whether the experimental observed rates will fall in the realm of (B) EX1 kinetics or (C) EX2 kinetics. (D) For EX2 kinetics, the free energy of the local stability can be calculated using the ratio of the kch and the observed rate kobs, a ratio referred to as the protection factor (PF) associated with the local structure.
The rates of the closing/opening motions relative to the kch determine what the HDX-MS data will look like and the type of information that can be obtained. If the closing rate (kcl) of an amide is much slower (more than 10-fold slower) than the kch, then the amide will be deuterated before it can close and the observed rate kobs will be governed by the opening rate (kop).27 This kinetic regime is referred to as EX1 kinetics. On the other hand, if kcl is much faster than kch, the amide will have to undergo many opening motions before it becomes deuterated (EX2 kinetics). The observed rate will therefore reflect the combination of kch and the equilibrium of the opening/closing motions within that amide. With EX2 kinetics, the free energy associated with the structural stabilization can be calculated from the ratio of kch to kobs, which is also referred to as the local protection factor (PF) (Figure 1D). Depending on the local stability of the protein, PFs can vary from 1 (kobs = kch) up to >109 for very highly protected amides.3,20
It is also important to appreciate how the solution conditions that determine kch can impact the exchange regime for HDX. Changing the conditions, for example, by raising the solution pH, will alter the kch and can potentially shift exchange from EX2 kinetics to EX1 (Figure 2). Manipulation of kch can therefore be used to differentiate EX1/EX2 mechanisms and extract more information from HDX analysis. However, this approach assumes that the protein dynamics will be invariant at drastically different solution conditions, which is often not the case.
Figure 2.

(A) Deuterium uptake at pulse times in D2O is plotted across a range of pH. (B) Exchange rates as a function of pH show the transition point from the EX2 to and EX1 exchange regime. Above pH 11, the exchange reaches the EX1 limit where kch ≫ kop and the rate is entirely governed by the opening rate of the amide. Reproduced with permission from ref (4). Copyright 2006 American Chemical Society.
Through decades of research, it has been observed that under physiological conditions the vast majority of proteins undergo fast local structural fluctuations resulting in EX2 kinetics. Most proteins exist in populations in solution where the natively folded closed state is preferred. For peptide level analysis, this results in a gradual mass shift over the course of deuteration, with the labeling following a binomial distribution that is convoluted with the natural isotope distribution of the peptide (Figure 3A).10 The width of the isotopic envelope is dependent on the number of amides in the peptide that are exchanging at that time point.28−31 EX1 kinetics, while rare, continue to be observed across a wide range of protein systems,32−34 and the peptide mass spectra manifest as a bimodal distribution (Figure 3B). The undeuterated species reflects the population that has not yet undergone the correlated opening motion, whereas the highly deuterated species is the population that has gone through an opening motion and a large portion of the amides have fully exchanged. Over a deuterium exchange time course, the relative EX1 populations will convert from the undeuterated to the highly deuterated species as governed by the local kop.3,28,35,36 Distinct conformers of a protein that coexist in solution can also cause the appearance of multiple species within HDX-MS spectra.37 This case is distinct from EX1, as the two conformers of the protein do not interconvert, and the spectra will therefore show two features whose relative intensities do not change over the time course (Figure 3C). We note that in Figure 3C, the faster exchanging species is fully deuterated by the earliest time point, but this may not always be the case as it will depend on the accessibility of amides in this less protected conformer. Because proteins often undergo many local and global conformational changes on various time scales, it is also possible for a region of the protein to fall into both EX1 and EX2 regimes. Spectra from such regions will show both shifting population intensities and a gradual deuterium uptake for the slow-exchanging state (Figure 3D). This scenario can be distinguished from a mixture of noninterconverting conformations by the change in the relative peak intensities across all time points. This ability to see minor subpopulations of alternate conformers is one of the unique strengths of HDX-MS. Tracking protein subpopulations has given insight into several biological systems, including quasi-equivalent subunits in viral particles,38 heterogeneity in capsid assembly39 and prion fibrils,40 the presence of degraded or misfolded forms of a protein,41,42 and asymmetry in protein assemblies.43,44 However, it is important to be aware of artifactual bimodal profiles that can arise from several sources, for example, sample carry over (Figure 3E; see section 2.3.3)
Figure 3.

Examples of mass spectra at various time points showing the isotopic distributions resulting from pure EX2 (A), pure EX1 (B), a noninterconverting mixture of conformers (C), mixed EX1/EX2 (D), and artifactual bimodal spectra attributed to sample carryover (E). The minor subpopulation in (C) is denoted with a *. The arrow in (E) reflects the contribution to the signal from a previous injection. Reproduced with permission from ref (665). Copyright 2016 Elsevier.
1.3. Intrinsic Hydrogen Exchange within Proteins
The chemical process of backbone amide exchange is broadly described in solution using proton transfer theory.45 This process is highly sensitive to exchange reaction conditions and the local electronic environment.46,47 The complex relationships between reaction parameters and amide exchange kinetics have been extensively studied by numerous investigators.1,3,8,10,47−62 Here we will attempt to distill the core concepts underlying these relationships and illuminate some of the more subtle aspects of kch for backbone amides. While not covered in detail here, it is also important to be aware of the hydrogen exchange processes in other positions, such as at side chain positions, which have recently been reviewed in detail.63
Amide exchange in an aqueous medium proceeds through three mechanisms: acid, base, and water catalysis.51 Acid catalysis proceeds through two distinct mechanisms shown in Schemes 1 and 2. Scheme 1 describes O-protonation (acid–imidic exchange), which is understood to be the predominant pathway of acid catalysis for backbone amides.54,56,63,64 This process begins with the transfer of a deuteron from the solvent to the peptide carbonyl oxygen. The imidic nitrogen is then deprotonated, which is followed by transfer of a deuteron from the solvent back to the nitrogen. Scheme 2 displays the pathway for N-protonation or the direct deuteration of the amide. This pathway begins with the transfer of a deuteron to the neutral amide, followed by removal of the hydrogen, leaving behind a deuterated amide. It should be noted that the N-protonation pathway is not the primary exchange pathway for backbone amides, with the possible exception being the N-terminal amide.54
Scheme 1.

Scheme 2.

The process of base catalyzed amide exchange, on the other hand, is understood to proceed through a single pathway (Scheme 3). Base catalysis begins with the direct removal of a hydrogen from the amide, and the resulting highly basic nitrogen then picks up a deuteron from the solvent. Deuterium oxide in its un-ionized form is also capable of initiating the protonation/deprotonation steps in these three mechanisms to mediate hydrogen/deuterium exchange, albeit at a much slower rate. The second-order rates for acid (kD+), base (kOD–), and water (kD2O) catalyzed exchange can be calculated using proton transfer theory, provided the reaction conditions are well characterized. These values are summed using eq 1 to yield the observed rate of exchange for a particular amide (kch). For solution HDX-MS, the rate of water catalyzed exchange (kD2O) is considered negligible and can be ignored which simplifies eq 1 to eq 2. Furthermore, because of the tendency of amide exchange to proceed primarily through base catalysis above pH 2.3–2.6, the contribution of acid catalysis (kD+) to the observed rate amide exchange (kch) is considered minimal. This allows for the use of eq 3 to describe kch near physiological pH (pH 5–10).65 Accurate estimation of kch is central to informative measurements from HDX-MS experiments.49,66 Therefore, much effort has been devoted to understand how structural influences and solution conditions combine to impact amide exchange rates.1,3,8,10,47−62,64,67
| 1 |
| 2 |
| 3 |
Scheme 3.

The value of kch is unique for each type of amino acid and varies for each amide within a sequence depending on its position within the sequence and neighboring residues. This is because the side chain chemistry of a particular amino acid influences the buildup of charge on the amide during the proton transfer process.47,48,59 The term “side chain effects” is often used to describe the structural influences responsible for the distinct exchange behavior of amino acids. These structural nuances have more far-reaching effects within a polypeptide sequence. Often referred to as “nearest neighbor effects” or “sequence effects”, the arrangement of amino acids relative to one another has a significant impact on kch.3,47−49,51 Additionally, the proximity of a particular residue to the N- or C-terminus also greatly impacts the exchange behavior. Although N- and C-terminal effects have little influence on intrinsic exchange behavior in full proteins, these effects become pronounced in small unstructured peptides.47,48,51,68 The positive charge at the N-terminus accelerates kOD- of the neighboring amino acid,47 sometimes to the point where the second amino acid (first backbone amide) exchanges so quickly that it cannot be probed by HDX-MS.49 As a result, N- and C-terminal effects are frequently considered when gathering higher resolution HDX data.63 The Englander group has compiled a set of spreadsheets for calculating kch for amides in the context of their sequence and buffer conditions, which are available at: http://hx2.med.upenn.edu/download.html. Alternatively, the online tool, Server Program for Hydrogen Exchange Rate Estimation (Sphere) can also calculate predicted kch for a given peptide sequence and condition: https://protocol.fccc.edu/research/labs/roder/sphere/sphere.html.
1.3.1. Factors that Govern kch: pH
Controlling solution pH is fundamental to the HDX-MS experiment. In fact, the sensitivity of amide exchange rates to pH is what enables the retention of deuterium for analysis by MS. Proteins are typically labeled around neutral pH or under near-physiological conditions (pH 5–10). Within this pH range kch is relatively fast, enabling the probing of diverse protein motions on a reasonable time scale.31,65,69 To actually observe the localization of deuterium and gain any kind of structural insight from the HDX-MS experiment, it is necessary to slow the exchange process or “quench” the reaction, otherwise the presence of H2O in subsequent sample handling steps will result in the loss of the deuterium label. Although the quench step serves additional purposes, the slowing of amide exchange is accomplished here by acidifying the exchange reaction buffer to a range where the rates of acid and base catalysis are minimized.3,8,50,56,70,71 The “V-shaped” plot in Figure 4 is a useful approximation of the pH dependency of amide exchange. The negative sloping region on the left side of the plot represents the region where amide exchange proceeds primarily through acid catalysis, and the corresponding positive sloping region on the right represents the region where amide exchange proceeds primarily through base catalysis. The minimum in the center represents the region where both acid and base catalyzed exchange rates are at their lowest values, referred to as the pHmin. Although pHmin varies for individual amino acids, it is generally approximated at pH 2.5 for proteins and is what most HDX-MS researchers target for their quench pH.8,71
Figure 4.
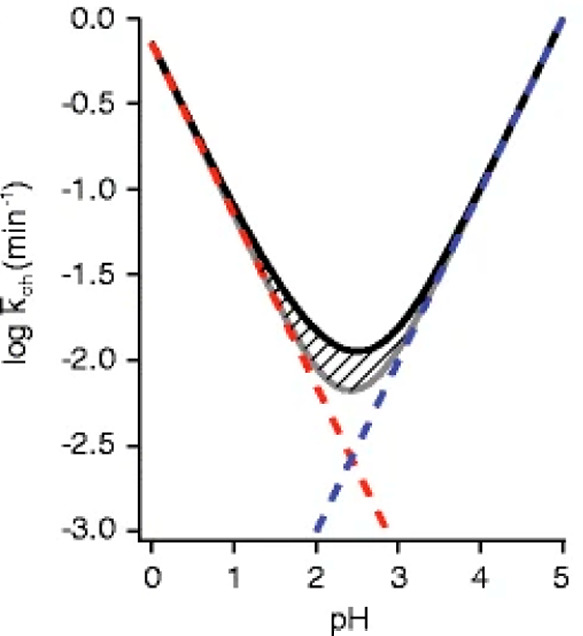
Rate of exchange of an unstructured amide is shown as a function of pH. The gray line is the summed rate from the acid and base catalyzed exchange contributions, which are individually depicted with the red and blue dashed lines, respectively. The black line above the hatch marks is the net rate accounting for the contribution of water catalysis. Reproduced with permission from ref (71). Copyright 2012 American Chemical Society.
Amide exchange in the deuterium rich labeling buffer is primarily catalyzed by the acidic (D3O+) and basic (OD–) species in solution. Equation 2 provides a way to calculate the impact that the relative quantities of these two species have on the observed rate of amide exchange in the HDX-MS experiment. Although the second-order rate constants for acid and base catalysis vary with some solution parameters, the effect on kch is usually minimal.3,51 Consequently, special care should be given to controlling labeling reaction and quench buffer pH values (or more appropriately, the pD values when working in high concentrations of D2O), as the concentration of D3O+ and OD– has a direct impact on exchange kinetics. In the physiologically relevant pH range, an increase of 1 pH unit will result in a 10-fold increase in kch. The use of glass electrodes to measure the pD of labeling and quench buffers is ubiquitous, however, it is necessary to correct the pH measured in a D2O rich solution (pHread or “pH*”) to account for the variation in ionic activities of H+ and D+.72 There are several methods for calculating pD from pH*, each of which features different assumptions regarding the variations on the pH scale.63,73−75 It should also be noted that pH* varies with the concentration of deuterium in solution (%D).73 As a result, the use of a different approach to convert pH* can result in a slightly different calculated pD. For this reason, it is recommended that investigators simply report pH* associated with HDX-MS experiments to avoid confusion.76
1.3.2. Factors that Govern kch: Temperature
Temperature is another major factor that affects kch and should be controlled throughout the HDX-MS experiment. We note that temperature will likely also affect the solution structure and dynamics of a protein, but here we focus only on the effects related to kch. Solution temperature is directly tied to the ionization constant of deuterium oxide (KD2O) in a given buffer system, which therefore effects the concentration of D3O+ and OD– in solution.3,72,77,78 A theoretical value for kch at a specific temperature can be computed using a modified Arrhenius equation (eq 4).3,46,54 In this equation, T refers to the experimental temperature in kelvin, kch(293) is the reference rate for the target at 293 K, Ea refers to the activation energy for the target, and R refers to the appropriate molar gas constant.
| 4 |
Accordingly, kch increases 10-fold with every 22 °C increase in temperature (Figure 5). This relationship appears to be maintained even below freezing, as observed from exchange studies of ultralow temperature HDX or the observed deuterium loss in solid frozen samples.79,80 While this relationship between temperature and kch is adequate for planning and interpreting most solution HDX-MS experiments, there are several assumptions associated with this treatment that should be noted, particularly if experiments are to be compared across different temperatures. The activation energies for acid, base, and water catalyzed amide exchange are often reported as 14, 17, and 19 kcal/mol, respectively.48,49,63 Although these fixed values are appropriate for most amide rate predictions, the actual acid, base, and water catalyzed activation energies can be moderately offset by solution parameters, like dissolved salts.52,57,59,81,82 Furthermore, temperature dependent changes to KD2O are not uniform for all buffer systems.82−86 This inconsistency can result in a temperature dependent change in pH, which is unique to a particular buffer system. For example, phosphate shows minimal variation while TRIS, ACES, acetate, and citrate show more significant variation in pH with temperature and should be pH adjusted at their intended temperatures.
Figure 5.
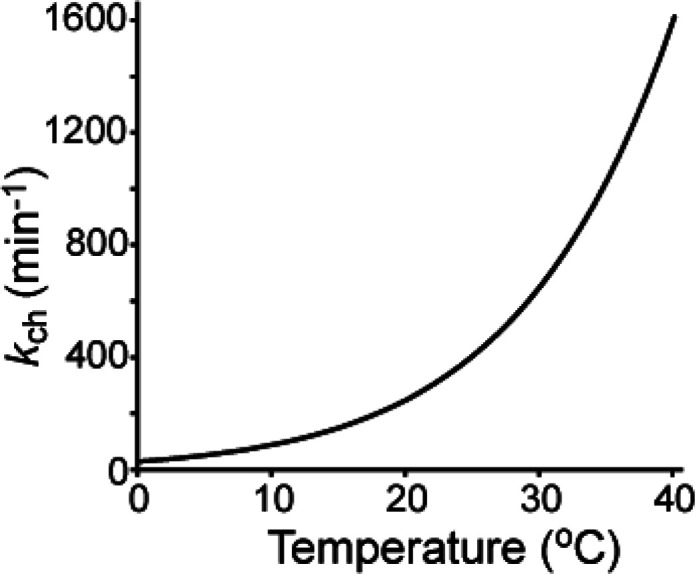
kch of polyalanine is plotted as a function of temperature based on calculations from Bai et al.41
1.3.3. Factors that Govern kch: Pressure
Solution HDX-MS experiments are typically conducted at atmospheric pressure, however, elevated pressures are common during downstream processes like digestion and chromatographic separation. Like temperature, system pressure exerts an influence on KD2O, and the resulting change in pH impacts amide intrinsic exchange.46,87 Under certain conditions, it is possible to use established empirical relationships to model the effects of pressure, but in many cases, this approach is not adequate for describing the full impact of pressure on kch.3,60,88,89 In general, the ionization of weak electrolytes, like phosphoric acid, increases with pressure.46,87 As a result, the effects of pressure on the rate of amide intrinsic exchange in some common buffer systems can be generalized.46,90 It is possible to move beyond this generalization to provide a semiquantitative estimate for the pressure dependence of pH for specific buffered systems using Planck’s equation.87,90,91 This approach brings to light the more complex relationship between buffer pH and pressure. For example, phosphate buffer is understood to drop by almost half a pH unit per 100 MPa, while the pH of a MOPS buffer increases by approximately the same amount (Figure 6).88,90,92,93 Although these effects are minor compared to pH and temperature, they may need to be considered when estimating kch at elevated pressures.
Figure 6.

Relationship between solution pressure and pH for phosphate (white) and MOPS (black) buffer are shown. Circles, squares, and diamonds reflect a starting solution pH of 7.0, 7.5, and 8.0, respectively. Reproduced with permission from ref (92). Copyright 2005 Elsevier.
1.3.4. Factors that Govern kch: Ionic Strength
The identities and concentrations of salts added to the labeling buffer are understood to effect deuterium uptake, and this phenomena has been extensively investigated.2,3,47,48,50−52,59 These experiments have clearly demonstrated that ions in solution exert a direct influence on kch through altering the local electronic environment of the amide. These data demonstrate that charged residues exhibit a greater response to KCl than neutral residues and that the response of a negatively charged residue differs from that of a positively charged residue. This selective and directional modulation of kch is often attributed to the exclusion of deuterium oxide and its ionization products by salt ions interacting preferentially with charged regions of the protein sequence.46,52,94 While there is strong empirical evidence to support this model, it is also clear that dissolved salts like NaCl and KCl alter the activities of other charged species in the solvent, which manifests as a change in buffer pH. Therefore the effect of salt content on buffer pH should be considered for studies conducted at high ionic strength.
On a related note, glass electrode pH probes are unrelible for the interpretation of H+ activity at higher ionic strength.95 As a result, many investigators use empirically determined activity values for water to improve estimates of pH in the bulk solution at elevated ionic strength.52,59,78,95,96 Although this is commmon practice, it should be noted that the effect of salts like NaCl and KCl on activity is not uniform for all buffer systems.82,85,86,97 Therefore, the use of empirically determined activity values from noncomparable buffer systems may misrepresent the actual pH of the bulk solution. Figure 7 shows the effect of salt concentration for several different chloride salts on the pH of citric acid and triethanolamine/trienthanolammonium chloride (TEA) buffers. Note that the addition of NaCl (square) results in a decrease in pH in the citric acid buffered system, while the addition of the same salt to the TEA buffer results in an increase in pH. Although there is little evidence to suggest that such a discrepancy could lead to significant misinterpretation of kch, performing HDX-MS experiments involving high salt concentrations in well-studied systems where appropriate empirically determined activty values are available will allow for a more accurate estimate of pH in the bulk solution, thereby enabling a more informed interpretation of salt effects on deuterium uptake. Although not necessarily related to ionic strength, it has also been noted that the solution viscosity can affect kch in ways that may need to be considered for certain studies.98,99
Figure 7.

Relationship between the salt concentration and pH for triethanolamine (TEA) buffer and citrate buffer (CA) are shown. The salts used in the study were tetramethylammonium chloride (▷), choline chloride (◇), cesium chloride (○), potassium chloride (▽) sodium chloride (□), and lithium chloride (△). Solid lines are predictions based upon extended Debye–Huckel equation using ionic size parameter 4 × 10–10 m. Reproduced with permission from ref (85). Copyright 2006 American Chemical Society.
1.3.5. Factors that Govern kch: Organic Solvent
Organic solvents are often involved in HDX-MS during sample processing and are also occasionally necessary in the deuterium labeling step to facilitate probing of ligand interactions. It is common for studies of protein–ligand interactions to include low amounts of organic solvent (e.g., DMSO) to aid in the solubilization of hydrophobic small molecules. Despite the extensive use of organic solvents in HDX-MS, there are few studies of the effects of organic cosolvents on kch.3,8,100
It is widely accepted that organic cosolvents influence kch indirectly through several mechanisms: (1) the addition of the organic component reduces the concentration of water, thereby reducing the number of interactions between the analyte and the aqueous components that catalyze exchange, (2) miscible organic solvents decrease KD2O in certain systems, resulting in fewer ionization products and a lower kch, (3) the organic component reduces the dielectric constant of the solution, which shifts the equilibrium to favor neutral products reducing the availability of catalytic ions to suppress kch.3,46,47,63,100 Using these assumptions, the depression of amide exchange in the presence of organic cosolvents has been predicted for a variety of solvent systems (Figure 8). However, there have been examples suggesting that an organic solvent may accelerate kch.98,101−104 Interestingly, there is a considerable body of literature to suggest that the acceleration of amide HDX in the presence of an organic cosolvent is expected for certain systems due to an increase in pH or change in buffer capacity.105−112 These discrepancies may arise from the inability to uniformly relate proton activity measured in an aqueous reference to that of a nonaqueous or mixed (hydroorganic) solvent using a universal pH scale.106 This is because pH scale length depends on an activity coefficient, which is a system-specific parameter that depends not only upon the particular organic modifier and the pH of the aqueous component but also upon the nature of the buffering system, i.e., the concentration and identities of the buffering agents.105,113 At the very least, these findings indicate that organic cosolvents can influence kch in different ways, and additional care should be taken to ensure the utilization of organic cosolvents is consistent, especially in comparative HDX-MS studies.
Figure 8.
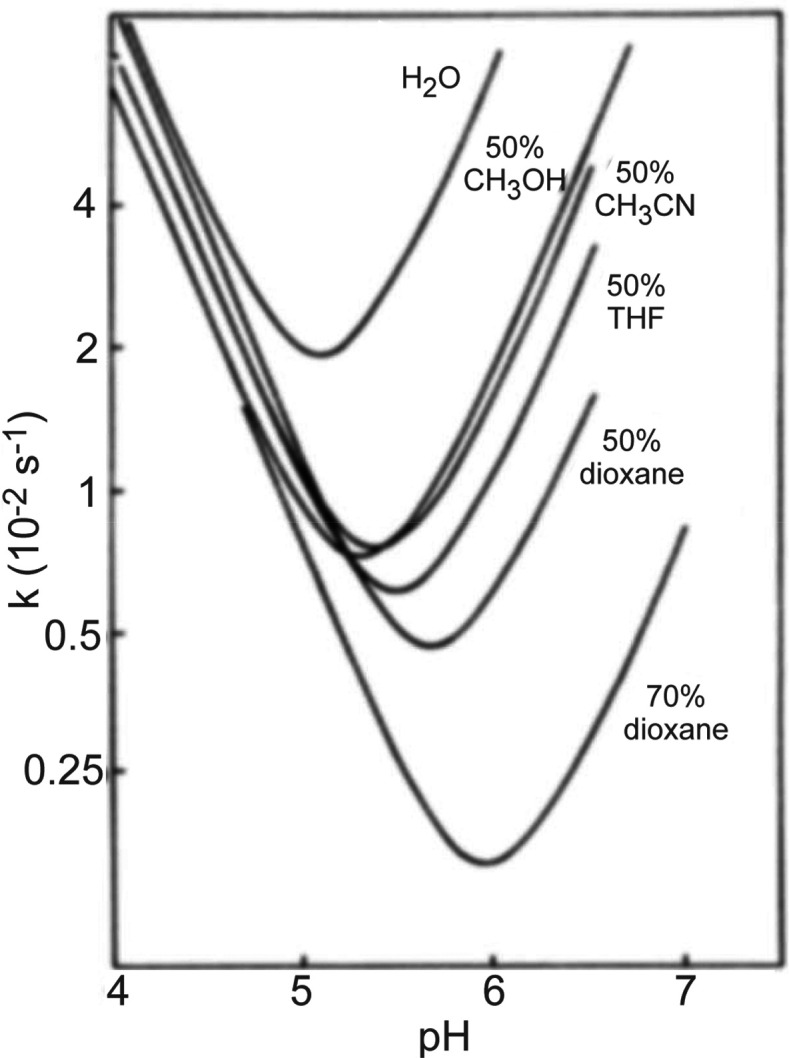
Predicted relationship between kch and pH at different levels of organic solvent. The general shift in the position of the “V” shaped curves results from offsets to solution conditions and the lower concentration of water available for catalyzing amide exchange. Reproduced with permission from ref (8). Copyright 1985 Elsevier.
1.3.6. Factors that Govern kch: Isotope Effects
The hydrogen isotope effect only has a small effect on kch and is rarely considered in the HDX-MS experiment. For base-catalyzed exchange, the slight isotope effect is primarily attributed to the rate limiting step which is breaking of the N–H or N–D bond to form the imidate ion (Scheme 3). This is corroborated by the rate of hydrogen exchange being higher than that of deuterium and tritium exchange, which is consistent with the primary kinetic isotope effect.55 For the acid catalyzed reaction, there is an associated slight inverse isotope effect that is attributed to the slightly higher acidity of the D3O+ ion compared to the H3O+ ion.77 Although isotopic effects have relatively little impact on kch, it has been noted that the stabilities, activities, and dynamics within native proteins can be offset by changing from H2O to D2O.114 This can at least be partially explained by the isotope effect of deuterium leading to slightly weaker hydrogen bonding.115,116
2. Measuring Hydrogen Exchange
2.1. Sample Considerations for HDX-MS
Two primary considerations for setting up informative HDX-MS experiments are the time scale and the reaction conditions. Unstructured amides will exchange at rates approaching the chemical exchange rate (kch), which under standard conditions will have a half-life on the order of hundreds of milliseconds.3,48 On the other hand, the most stable protein folds have immense protection factors and may take years under standard conditions to fully exchange.21 Because amides can exchange with kinetics over many orders of magnitude, capturing as much of the kinetic range of amides as possible requires sampling a wide temporal range. Sampling a limited temporal range risks information loss and perhaps even concluding that two protein samples are identical because of insufficient temporal measurement (Figure 9). To enable collection of wide time scales, it is sometimes necessary to offset conditions, thereby adjusting the kch to achieve different effective temporal ranges without requiring prohibitively long incubations. This is most commonly achieved by sampling exchange at different pHs, where the low pH data will be able to capture fast dynamics and the high pH data will be able to probe the exchange of the very slow exchanging regions.31,65,69 It is important to point out that these pH and temperature adjustments make the assumption that the pH will offset kch but not impact the structural dynamics of the protein, which may not be valid for many proteins.65 Obtaining meaningful results through offsetting the pH to expand the time window for HDX measurements may thus be inaccurate for probing the physiological conformation, the effects of binding under physiological conditions, or the actions of molecular machines under physiological conditions.
Figure 9.
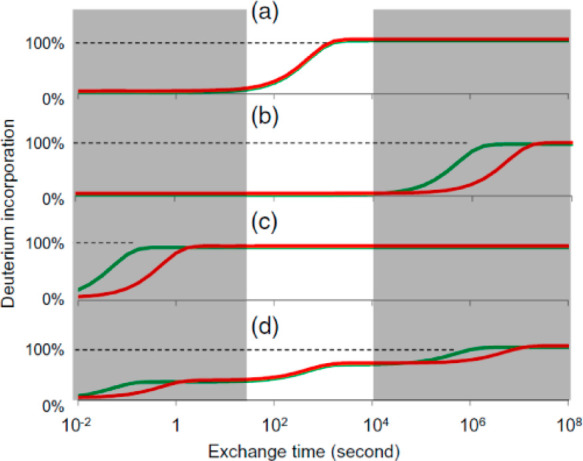
Amide exchange kinetics in proteins vary over 8 orders of magnitude. Comparative studies that sample only a limited temporal range can lead to missed information. Plots a–d show various kinetics for two states of a protein (red and green). Only in plot a is the comparison truly the same for all exchange times. Plots b–d have actual difference in exchange rates, but they are invisible due to the limited temporal sampling (highlighted region of the plot). Reproduced with permission from ref (24). Copyright 2017 American Chemical Society.
The kinetics of amide exchange are typically measured using an “on-exchange” approach, where a protiated protein (all 1H) is incubated in D2O for a set amount of time. However, off-exchange experiments have also been utilized, particularly in the case of folding studies, where proteins are first fully unfolded in deuterated buffer and then diluted into protiated buffer to initiate exchange of amides.37,117−121 In general, the higher the D2O content in the deuterium labeling step, the larger the observed mass shift, and thereby the increased accuracy of the deuterium measurement. However, in some cases, it might be advantageous to use lower %D for the labeling step to restrict the broadening of the mass isotopic envelope and obtain better signal-to-noise and less spectral overlap among many peptides in the mass spectrum.122
When performing exchange reactions, it is important to note that although only the amide hydrogens are monitored, all of the fast-exchanging hydrogens may need to be taken into account as well.63 Most modern HDX-MS platforms include some form of a desalting (trapping) step,10 which removes all of the deuterium at the fast-exchanging sites including side chains and at the peptide termini. However, some approaches may still have residual levels of deuterium as samples are analyzed by mass spectrometry, which necessitates accounting for all deuterium associated at the fast-exchanging sites.9,119,121
Effective HDX-MS studies also necessitate monitoring a large portion, ideally all, backbone amides in the protein of interest. With the common “bottom-up” approach, the protein is digested in small peptides which are used to obtain information across the protein sequence. Most people strive for the highest number of peptides which will maximize the sequence coverage (the portion of the protein sequence that HDX-MS is monitoring) and redundancy (how many unique peptides cover each amide position in a protein). While sequence coverage is important for monitoring all portions of the protein, redundancy is important for adding rigor to the method and providing a way of obtaining higher sequence resolution by utilizing information from all overlapping peptides.123
2.2. Controls for HDX-MS Experiments
An undeuterated sample is a necessary starting point for HDX-MS studies as it is used to establish sample handling conditions, collect MS/MS data to identify the peptides, and serves as an undeuterated reference for calculation of deuterium content in the deuterated samples. This sample is prepared identically to all deuterated samples, with replacement of D2O for H2O in the labeling step. While the undeuterated control is essential for all HDX-MS studies, there are several other useful controls which may be critical depending on the questions being sought to address. For comparative HDX-MS studies that seek to identify changes within two states of a protein, for example a side-by-side comparison of two protein samples or to map changes associated with binding, elaborate controls may not be critical.124 On the other hand, HDX-MS studies for fine structural studies or quantitative dynamics measurements require proper controls to account for the minimal and maximal extent of possible deuterium labeling.
One of the most useful controls to include for HDX-MS is a maximally deuterated standard.10,125,126 This control serves as the most accurate way of measuring levels of back-exchange during analysis, an important metric for evaluating the platform suitability for reliable HDX measurements.76 Using a maximally deuterated control is also the most accurate approach for determining the maximal deuteration point in calculating extents of labeling and exchange kinetics (Figure 10). The importance of this control can be illustrated using the highlighted range of the data in Figure 9D. Without the 100% reference standard, we might (incorrectly) infer that the peptide has fully exchanged by the 104 second time point, as its plateaued final observed exchange is close to what we would expect based on the deuterium incorporation for a certain number of amides. Having the 100% reference here is critical for properly interpreting that there are actually a few amides within the peptide that are highly protected even at very long time points. With the maximally deuterated control, it is also possible to calculate percent deuterium for each time points using eq 5, with m being the average isotopic mass for a given time point and m0 and m100 reflecting the masses of the undeuterated and maximally deuterated standards, respectively.10
| 5 |
Figure 10.
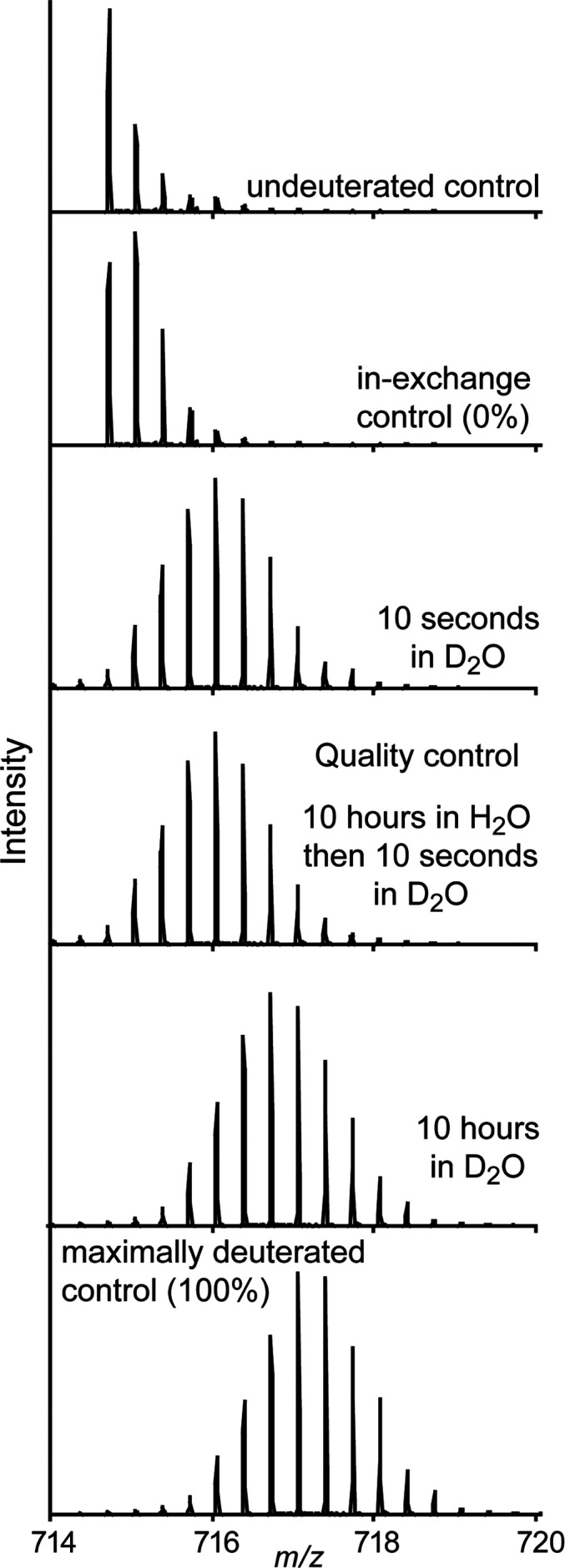
Example peptide spectra as undeuterated, 0% (in-exchange control), after 10 s labeling in D2O, after 10 h of labeling in D2O, and 100% exchanged control (top to bottom). The quality control in the middle is used to verify that the protein has not been perturbed during the 10 h incubation by verifying that the spectra looks identical to the first 10 s time point.
In practice, the preparation of an appropriate maximally deuterated standard can be challenging. Historically, preparation of a maximally deuterated standard was accomplished by heating the protein (e.g., 75 °C for several hours) in the presence of D2O to equilibrate all amides with deuterium.10 However, some proteins are prone to aggregation or may be too stable to maximally exchange even under such extreme conditions.124 Furthermore, high temperatures can lead to exchange of the C2 proton on histidine side chains (see section 3.7), which will inflate the levels of observed deuterium uptake observed.127 Additionally, there are reports that under exhaustive deuterium exchange other carbon–hydrogens can start to exchange, which may further distort the level of deuterium in a harshly treated deuterated sample.128,129 A safer approach is to incubate the protein for a long period of time (12–24 h) at a low pH (between 2.5 and 4) and at room temperature in the presence of denaturants (guanidine or urea).76,130 When preparing a maximally deuterated control, it is important to match the final percentage of deuterium to the other samples in the data set. High urea/guanidine content adds a high concentration of fast-exchanging hydrogens to the solution, which can offset the final percentage of deuterium in the resulting mixture.131 We note that the low pH can help destabilize proteins to facilitate complete exchange while also preventing chemical modifications (for example, carbamylation of amines can occur in the presence of urea at high pH132). In the coming years, with more study of a variety of proteins, a more generally reliable protocol for preparation of maximally deuterated controls may emerge.
Without additional techniques, there is always a level of uncertainty with regard to whether a maximally deuterated control is in fact actually completely deuterated at all possible positions (hence the preference for calling such a control “maximally” deuterated as opposed to “totally” or “fully” deuterated). An alternative approach for obtaining a fully deuterated control is to prepare a predigested sample that is dried, resuspended in deuterium, and then quenched to mimic all samples.125 Although the back-exchange rates might vary slightly for a quenched free peptide vs the same peptide in the context of a full protein,48,63 this approach provides quite a reliable method for generating a fully deuterated standard, at least in the proteins for which it has been tested.125,126,133
Another consideration with a bottom-up HDX-MS workflow is the time samples spend in the quench buffer. During the digestion step, peptides will continue to equilibrate to the final percentage of deuterium in solution. In some quenched samples, the solution deuterium level can be significantly higher than the deuterium level of the peptides. For example, if the labeling reaction was 90% deuterium, and quenching was accomplished with a 1:1 mixture of all-H2O quench buffer at 0 °C, then the peptides will experience 45% deuterium upon the quench. They can equilibrate to a maximum of 45% deuterium until the 45% deuterium level is reduced by the aqueous (100% H2O) solution used for trapping, desalting, and chromatography. This effect can cause peptides that were natively completely undeuterated during the actual deuterium labeling step to take on considerable amounts of deuterium even at 0 °C in the few minutes a quench step might last, leading to artifactual “in exchange” (Figure 11).125 An easy control to account for this exchange during the quench step is to prepare a sample where the protein is diluted into a premixed solution of quench and D2O labeling solution. This sample, often referred to as a “quench exchange” control, serves as a better reference standard for 0% exchange, to more accurately determine if regions are completely unexchanged vs slightly exchanged.125 An alternative approach is to quench with a higher than 1:1 ratio of quench buffer to dilute down the deuterium and/or very quickly get the protein loaded for online desalting.
Figure 11.
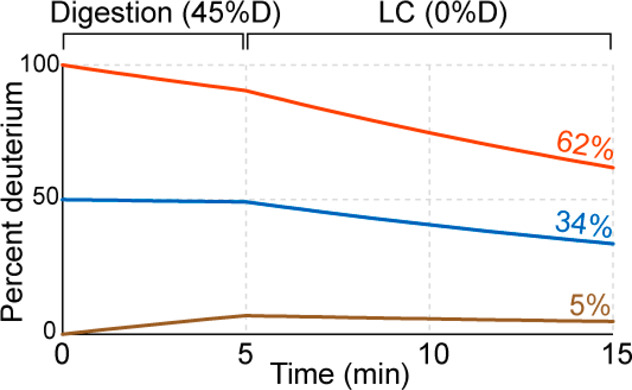
Deuteration levels are shown throughout a 5 min digestion step and a 10 min LC step for peptides starting with 0% (tan), 50% (blue), and 100% (red) deuterium labeling. Final values recorded by the experiments are shown to the far right with the same coloring. Rates were estimated from Bai et al.48
Over long deuterium exchange time courses, it is possible for proteins to undergo chemical modifications, degradation, oligomerization, aggregation, unfolding, etc., all of which will lead to confounding HDX-MS results in the long time points. An additional HDX control that has been increasingly utilized to detect such long-term modifications during labeling is a “deuterium pulse quality control”.76 The protein is incubated under the identical conditions as found in long exchange times (pH, buffer, temperature, etc.) but in an all-H2O solution. At a time matching the longest time point, the protein is labeled with a short pulse of deuterium (say 10 s of labeling). Comparison of this sample to the equivalent short deuterium incubation time can reveal whether the protein has undergone any structural changes during the long deuterium exchange incubation.
2.3. Bottom-up LC-MS
The classic and still most widely adopted approach for analyzing HDX of proteins is a bottom-up approach based on the pre-MS protocol from Rosa and Richards,7 where proteins are digested with an acid-active protease after the quench step and the mixture of peptides is analyzed by mass spectrometry.10 Over the past two decades, there have been many developments and advances in approaches for protein denaturation, digestion, and tools for resolving and detecting the maximum number of observable peptides for analyses utilizing continued improvements in LC-MS technology. In each step, there are inherent considerations that often lead to a balancing act with regard to data quality, processing time, and back-exchange that often needs to be tailored to a given protein analyte which we outline here.
2.3.1. Denaturation and Digestion
Theoretically, the conditions used to quench deuterium labeling should denature the protein to allow it to be easily digested and favor high activity for the protease utilized to effectively cleave the protein, all while keeping the sample as cold as possible and keeping sample processing times as short as possible to minimize back-exchange. Practically, there is some leeway in quench conditions and parameters for denaturation, protease activity, time, temperature, and buffer compositions that are often adjusted, in most cases established on an empirical basis, to obtain the best coverage for a given protein analyte.
Many proteins are inherently destabilized by the low pH used for the quench step and may not require additional denaturants for effective digestion under the quench condition constraints of time and temperature. However, the addition of chaotropic agents such as urea and guanidinium are often required for effective unfolding and subsequent digestion of the target protein.134−136 Use of high concentrations of chaotropic agents does not compromise back-exchange, but it is important to consider that high levels may also inhibit the proteases used for the digestion. For example, pepsin, the most widely employed protease for HDX-MS, retains activity up to 4 M urea but loses nearly all activity above 3 M guanidine.137 For some recalcitrant proteins that require higher concentrations of denaturant, studies have opted for a two-step quench approach in which the deuterium labeled protein is first added to a very high concentration of denaturant to induce unfolding, and the denaturant is subsequently diluted just prior to the digestion step.
Denaturants may also be required, not just to help a protein unfold but to minimize the possibilities of acid-induced protein aggregation, which has been observed for some systems.138 Proteins in complex with nucleic acids also can precipitate in quench conditions, for which specialized quench buffers have been established.139 Detergents have also been explored as additional additives to aid in protein denaturation and solubilization.135 Recent studies have illustrated the potential of low concentrations of organic solvents to aid with denaturation and digestion of recalcitrant proteins.140 Specifically, dimethylformamide, acetonitrile, and methanol were found to empirically achieve better denaturation and digestion of proteins, with the caveat that the solution had to be diluted prior to digestion, as pepsin cannot tolerate more than 5% organic solvents. Ultimately, the selection of buffer conditions for optimal denaturation and digestion remains an empirical task for each protein system and is one of the first steps to applying HDX-MS to study a protein system.135
The need to maintain low pH during digestion imposes a major limit on the number of available proteases for HDX-MS. The typical proteases used for proteomics (e.g., trypsin) cleave with very high specificity but are not active at the low pH necessary for minimizing back-exchange.141,142 All available proteases active at low pH and reliable for HDX-MS cleave at a wide range of amino acid sequences. This is both a strength and a weakness. The lack of specific sequences for cleavage complicates identification and effective prediction of peptides generated from the protein analyte. At the same time, the broad cleavage specificity enables the proteases to cleave the proteins in different combinations of sites to generate many overlapping peptides which provide information across the protein sequence.7,123,141,143
Porcine pepsin is the original and by far the most common protease utilized for HDX-MS studies. It has reliably high activity under HDX quench conditions, is readily available in highly purified forms, and has relatively broad cleavage preference.10 In general, pepsin prefers to cleave adjacent to hydrophobic residues but is able to cleave C-terminal to all amino acids except histidine, lysine, arginine, and proline.144−146 While pepsin generally produces a wide array of peptides covering the analyte protein sequence, there has been a continued desire for more peptides to increase coverage, redundancy, and spatial resolution.7 To this end, several additional proteases with different cleavage preferences have been explored and some implemented into HDX-MS workflows to provide complementary coverage to pepsin. Protease type XIII from Aspergillus saitoi (also known as aspergillopepsin or Fungal XIII) and Protease type XVIII (Rhizopuspepsin) were found to be effective proteases for HDX-MS with different cleavage preferences compared to pepsin.141,143 Other acid-active enzymes have been explored, including rice field eel pepsin,147 pepsin from Antarctic rock cod,148 and cathepsin-L.34 An aspartic protease from Nepenthes carnivorous plants was shown to have strong protease activity under quench conditions with cleavage preference that was broader than pepsin, namely it is able to efficiently cleave after basic residues.149,150 A homologue, nepenthesin II (“NepII”) has similar protease activity and exhibits much greater resistance to denaturants, on par with pepsin137,146 (Figure 12). More recently, a prolyl endopeptidase (An-PEP) was found suitable for HDX-MS151 and has some similarities to pepsin. Importantly, it cleaves after proline residues, which most available acid active proteases cannot do,145 thereby adding another useful option for expanding sequence coverage for HDX-MS studies. Beyond having a range of protease options for HDX-MS, several groups have combined multiple proteases to greatly improve coverage and spatial resolution.141 In fact, the practice of utilizing multiple proteases simultaneously is now common practice for many bottom-up HDX-MS approaches.123,152−154
Figure 12.

Comparison of peptides for aprataxin and PNKP-like factor (APLF). Each bar under the primary sequence shows a peptide from either pepsin (red) or NepII (blue) digest. Sites indicated with triangles show sites that are uniquely cleaved by pepsin (red) or NepII (blue). Reproduced with permission from ref (137). Copyright 2015 American Chemical Society.
The development of immobilized proteases in a column format155,156 offers several advantages over the traditional in-solution protease digest. The major advantage is the high enzyme:substrate ratio that can be achieved during the relatively short contact time. Pepsin immobilized onto bead support was found to be highly efficient and allowed for short digestion times (under 1 min), online sample processing, and minimal interference from the peptides generated from pepsin autolysis, which was common with solution pepsin digestion. Optimized protocols have since been generated for fabrication of pepsin columns and their tolerance to various conditions (including reducing agents and denaturants) has been well-established.157 Several of the other proteases utilized for HDX-MS also work effectively in a column format137,158 and, in some cases, in a mixed bed of different proteases within a single column.152,153 Because of the relatively short digestion times needed with protease columns, it is possible to use elevated temperatures to enhance proteolysis without a large impact on levels of back-exchange.147 For example, increasing the protease column temperature from 0 to 10 °C significantly improved digestion efficiency and without increasing back-exchange.159
High pressures for enhanced protease digestions have emerged as a tool for streamlining proteomics applications.160 Increased digestion efficiency with pressure has been known since at least 1995,161,162 and it was shown that incorporation of high pressures into an HDX-MS experiment also enhanced pepsin digestion efficiency under quench conditions138,163 (Figure 13). Conventional pepsin digestion of the HIV-1 NBSA protein showed poor coverage with only 10 observable peptic peptides. High pressure (>9000 psi) digestion yielded more than triple the number of peptides and doubled the sequence coverage. The increased pressure utilized for the digestion had no apparent influence on the levels of observed back-exchange, which is not surprising as it is known that high-pressure mainly affects the folded forms of proteins and improves digestion efficiency through pressure-induced partial denaturation.159 High pressure digestions have also been demonstrated in a protease column format using a combination of a high strength silica matrix that can tolerate pressures in excess of 10 000 psi and a flow restrictor to regulate the pressure within the column.159 This column that can tolerate high-pressure is now commercially available and shows improvement in digestion efficiency and the number of peptides as compared to low pressure columns, where enzyme is immobilized on particles that routinely operate at around 1000–1500 psi.
Figure 13.
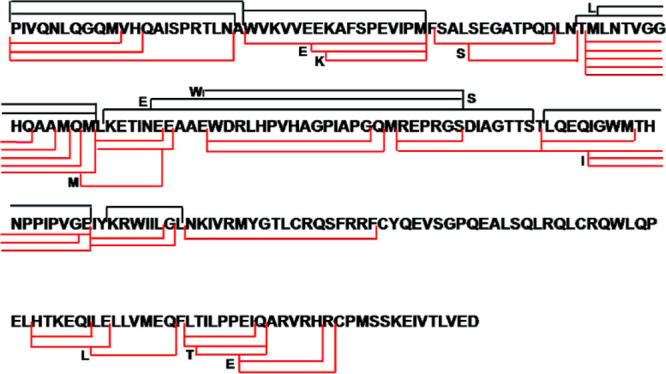
Comparison of peptides obtained from pepsin digest of HIV-1 capsid mutant protein at either ambient pressure (black) or >9000 psi (red). Lines represent start and end positions for observable peptides. Reproduced with permission from ref (138). Copyright 2010 American Chemical Society.
2.3.2. Reduction of Disulfide Bonds
Disulfide bonds are an abundant post-translational modification in proteins, and effective HDX-MS analysis of such proteins requires reduction of disulfide bonds for efficient denaturation and proteolysis under quench conditions. As traditional thiol-based reducing agents (e.g., dithiothreitol) rely on a deprotonated thiol to serve as a nucleophile, they exhibit negligible activity at the low pH needed for HDX quench conditions. Instead, HDX-MS analysis have relied on phosphine-based reducing agents such as tris-carboxyethyl phosphine (TCEP),164 which retains moderate activity under HDX quench conditions165 (Figure 14). The less optimal reducing activity at low pH is often offset by using high concentrations of TCEP in the quench step (up to 1 M). Modifications to TCEP, such as esterification, have been shown to improve reactivity, especially at lower pH,166 but at the cost of decreased water solubility, which has limited their utility for HDX-MS studies. While TCEP has been shown to induce unwanted side reactions in cysteines such as desulfurization,167 to our knowledge no such activity has been observed using TCEP for HDX-MS workflows under quench conditions.
Figure 14.

Chemistry or disulfide bond reduction using TCEP.
The amount of TCEP used in the quench step must be optimized empirically for each protein system and can range from μM concentrations up to >1 M. It is generally desirable to keep TCEP concentrations no higher than needed, as the TCEP has an affinity for PEEK tubing and is often challenging to completely wash away during the desalting step, which can cause residual signal in the MS that interferes with detection of peptides.135,168 Furthermore, some enzymes used for HDX-MS sample processing also cannot tolerate high concentrations of TCEP (e.g., An-PEP and PNGaseA).151,169
Electrochemical reduction cells (ERCs) have recently been introduced as a means for tightly controlled online protein reduction.170 Protein or peptide samples are flown through a pair of electrodes with oscillating potentials resulting in a reduction of disulfide bonds in the sample. This approach has been shown effective for processing samples under quench conditions (0 °C, pH 2.5, and can handle up to 3600 psi) and generally amenable to maintaining low levels of back-exchange.171 The benefits of electrochemical reduction were illustrated through a study of nerve grown factor β, a highly stable protein which contains three disulfide bonds. Even high levels of TCEP and denaturant were unable to provide efficient reduction, resulting in poor sequence coverage of regions spanning the disulfide bonded cysteines.172 In contrast, the electrochemical reduction method was able to efficiently reduce the sample to regain coverage of all cysteine containing peptides (Figure 15). A few caveats were noted on the use of ECR, including its low tolerance to salt (inability to use most denaturants or salts), frequent cleaning of the electrode surface, and side reactions including methionine oxidation were also commonly observed. Recently, an improved platform was developed that shows a more robust performance while minimizing oxidative side reactions,173 and this technology will likely continue to make more proteins amenable to detailed HDX-MS analysis.
Figure 15.
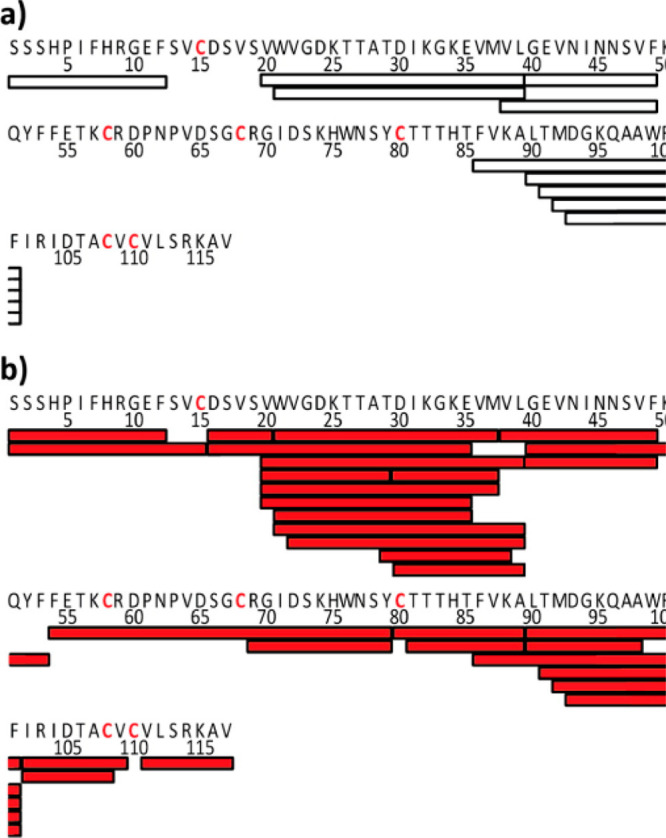
Peptic coverage maps are shown for nerve grown factor β using either (a) TCEP for reduction or (b) electrochemical reduction prior to pepsin digestion. Bars under the primary sequence show each unique peptide observed. Cysteine residues involved in disulfide bonds are shown in red. Figure adapted from Trabjerg et al.433
2.3.3. Liquid Chromatography
Once the protein is quenched and digested, the next task is desalting and separating the peptides for analysis. A major analytical challenge in HDX-MS is the analysis of deuterium incorporation without unacceptable loss of the deuterium label during sample handling. With nearly all approaches, quenched samples must be desalted prior to electrospray mass spectrometry to remove nonvolatile salts and other buffer components that would suppress the MS peptide signal. Incorporation of a liquid chromatography (LC) step conveniently provides a way of desalting the sample and simultaneously provides an opportunity to wash away all deuterium that was incorporated into side chain positions, leaving the deuterium label at just the backbone amides, which vastly simplifies data interpretation.10
In modern LC systems for HDX-MS, there is a trapping step utilizing a short column to first capture and desalt peptides, which are then eluted and resolved over a longer analytical column. The trapping step allows for easy integration with online protease columns155,156,174 and rapid cleaning of the trapping stage between samples.175 The vast majority of HDX-MS protocols use an analytical column packed with reverse phase hydrophobic stationary phases, most commonly C18; however, shorter hydrocarbon chain length chemistries have also been used, for example, when resolving very hydrophobic peptides derived from membrane proteins.136 Other studies have demonstrated the potential for alternative chromatographic separations such as hydrophilic interaction chromatography (HILIC)176 and supercritical-fluid chromatography.177
Maintaining a quench pH of 2–3 during the LC stage is relatively straightforward, as the most common mobile phases for LC-MS, such as water:acetonitrile with formic acid or trifluoroacetic acid, are perfectly suited for this pH range. The need to keep the sample cold introduces a larger technical hurdle, although as was shown very early in the development of HDX-MS that this can be easily accomplished by keeping the injection loop, LC lines, and HPLC column in an ice bath.10 More modern systems have placed the entire LC component in a refrigerator178,179 (see also ref (180)) or incorporated Peltier cooling devices181,182 to maintain stable temperatures, minimize condensation, and accommodate a digestion chamber with independent temperature control for which there are now several commercial options (Figure 16).
Figure 16.

Schematics of the plumbing system for analyzing HDX samples based on Wang, Pan, and Smith.147 Samples are injected onto a loading loop through the injection valve (left). The loading pump flows digestion buffer to push the sample over the pepsin column onto a peptide trap column. The analytical valve (right) is then toggled so the gradient elutes peptides from the trapping column through the analytical column and out.
Another major analytical challenge in the LC separation of HDX-MS samples is the need to keep separation times as short as possible to retain deuterium labels. Even at 0 °C, amides will exchange with a half-life in the range of 30–120 min.48 This means that peptides should enter the gas phase of the mass spectrometer in <10 min to keep back-exchange levels low.176 For smaller or simple protein systems, the rapid gradient needed to accomplish such a separation is usually sufficient for providing adequate separation of all peptides. For larger systems, coelution of peptides quickly limits the ability to resolve and detect all the peptides of interest.5 This problem is further exacerbated by the need to keep the analytical column cold, which is detrimental to peak shape.181 The general need for higher resolution in HPLC applications, not just in HDX-MS, led to the development of ultrahigh pressure LC (UHPLC/UPLC) systems. The increased pressure limits (>15 kpsi) enabled the use of columns with smaller particles sizes (2 μm or less) greatly improving separations [reviewed in ref (183)]. The initial application of UPLC to HDX-MS showed that the resolving power of UPLC at 0 °C was vastly superior to HPLC.181,184,185 Shorter run times and better separation at low temperature afforded by utilizing UPLC were extremely useful for HDX applications for resolving even complex peptide mixtures with short gradients.181,184,185 The increased pressure during chromatography is not detrimental to back-exchange levels and resulted in sharp LC peaks even at 0 °C.
The ability of the chromatographic system to resolve the hundreds to thousands of peptides generated from a digest (termed “peak capacity”) is vital for effectively monitoring deuterium uptake across all observable peptides.19 Often, the separation power of the LC step is one inherent limitation to the complexity of the systems amenable to study by HDX-MS, and there have been efforts to tune sample-handling conditions to achieve longer LC separations while minimizing back-exchange.71 While nearly all forms of chromatography utilize a gradient from water to acetonitrile for peptide elution, some studies have explored the incorporation of other aprotic solvents to help reduce back exchange by reducing the exchangeable proton content of the LC buffers. Valeja et al. found that incorporation of cosolvents such as dimethylformamide into the aqueous buffer could moderately reduce back-exchange during chromatography but at the cost of decreased LC resolution.186
An emerging effort in the HDX-MS field has been to perform LC separations at subzero temperatures.79,119,187−190 An early version of such a platform was developed by Venable et al. capable of LC separations at −30 °C.79 The study found that −20 °C was sufficiently cold to extend LC gradients out to 120 min without any effect on the observed back-exchange. One major consideration for subzero LC was the solvent additives, typically ethylene glycol or organic cosolvents like methanol, required in the aqueous buffer to prevent the solvent from freezing. Antifreeze additives are sometimes detrimental to chromatographic resolution, may prevent retention of the more hydrophilic peptides, or may be detrimental to peptide detection by MS. Wales et al. tested the incorporation of a subzero LC compartment into an established HDX-MS platform capable of stable temperatures down to −20 °C.190 The system was modified to introduce methanol into the loading buffer after the online digestion to prevent the sample from freezing as it enters the subzero chamber. The expected reduction of back-exchange at lower temperatures was achieved while the chromatographic profiles were not compromised (14.7 vs 15.1 s for the median LC peak width fwhm at 0 °C and −20 °C, respectively) (Figure 17). More recent studies have demonstrated effective LC separation of an entire deuterated Echerichia coli lysate digest separated at −10 °C using a 90 min gradient with only moderate back exchange.191
Figure 17.

(A) Comparison of UPLC separations at either 0 °C, −10 °C, or −20 °C showing the total ion chromatogram. (B) Deuterium levels across different peptides are shown for each temperature (top) and the difference between predicted levels of deuterium vs measured (below). Reproduced with permission from ref (190). Copyright 2017 Elsevier.
There has also been a common need to increase the sensitivity of HDX-MS to enable studies of highly material limited samples. Nanospray LC-MS has been a cornerstone of modern proteomics techniques thanks to its inherent low sample requirements and flow rates down to the scale of nL/min, which greatly improves ionization resulting in huge gains in sensitivity.192 Similarly, for HDX-MS, the incorporation of nanospray has long been known to reduce sample requirements nearly 100-fold.193 The inherent challenge with nanospray has been the general lack of robustness and run-to-run chromatographic precision, which dissuaded users and led to most researchers continuing to use larger bore columns with flow rates in the μL/min range.194 Thanks to developments by the proteomics community, the prospect of robust nanospray for HDX-MS has been revisited. Sheff et al. reported an integrated nanospray ion source developed specifically for HDX-MS that has demonstrated chromatographic precision and robustness for routine HDX-MS analyses.195 The system was capable of chromatographic peak widths under 6 s (fwhm) and while keeping back-exchange levels on par with conventional LC-MS.
Another approach that has promise for HDX-MS is capillary electrophoresis coupled to MS (CE-MS) thanks to its inherently low flow rates, fast separations, and most critically, very low sample requirements [reviewed in ref (196)]. Black et al. demonstrated the potential of CE-MS for bottom-up HDX-MS using a microchip format.197 Peptides were well-resolved with just a quarter of the time required for LC-MS analysis, demonstrating the superior peak capacity of CE-MS (Figure 18). The short run times also led to similar levels of back-exchange with LC-MS at 0 °C, despite the CE chip being at room temperature. A major hurdle that prevents CE-MS from taking over LC-based HDX-MS has been the effective desalting and loading of the sample onto the CE device, although this could be incorporated into a chip format. With the need for more sensitive separation techniques for challenging protein targets, less sample demanding options such as nanospray and CE-MS will likely continue to gain ground in the HDX-MS field in the coming years.
Figure 18.
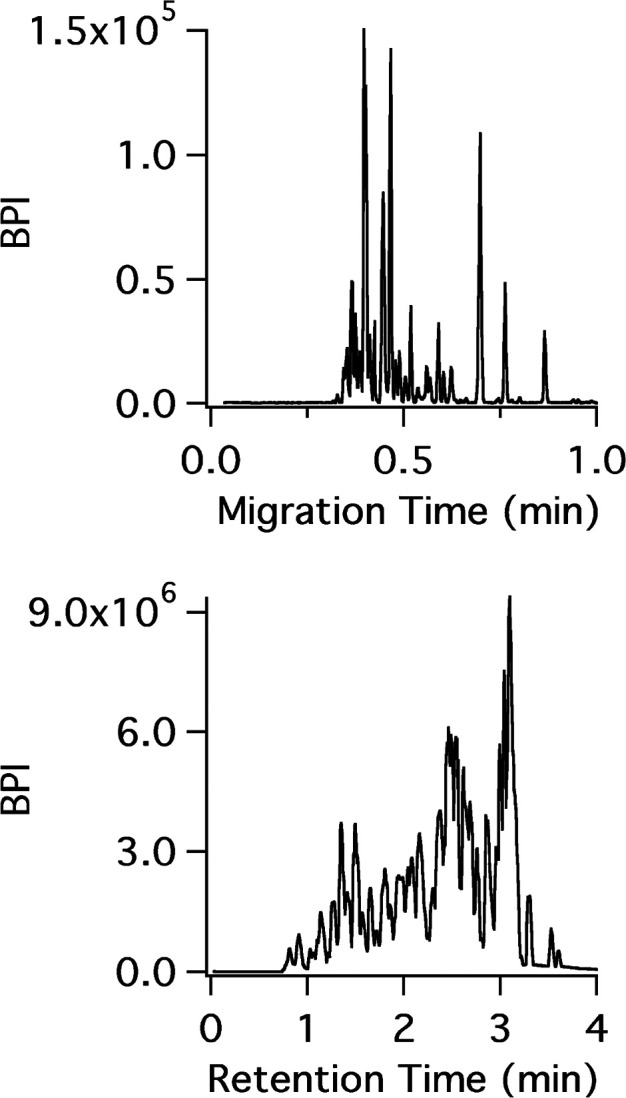
CE-MS (top) and LC-MS (bottom) separation of a bovine hemoglobin pepsin digest. Reproduced with permission from (197). Copyright 2015 American Chemical Society.
As with any separation involving chromatography, a consideration for the LC-MS stage of HDX-MS analysis is sample carryover between runs.175 Any portion of a sample not completely washed from the chromatographic system can be detected in the subsequent injection. For deuterated samples, this can be highly problematic as the previous sample will likely have lost deuterium by the time it elutes in the next run and may therefore appear as a low-deuterated subpopulation. Certain “sticky” peptides may elute in several subsequent injections, therefore, simply running a single inter-run blank may not be enough. Carryover will at best complicate, and at worst completely confound, the measure of deuterium uptake. Washing protocols have been developed to address carryover in HDX-MS analysis. There are two main sources of carryover: the protease column and the trapping/analytical columns. For the protease column, stringent washes, and sometimes back-flushing are often necessary.135,198 The trapping and analytical columns can also be sources of carryover, for which cleaning steps have been described.175 Some studies of membrane proteins require extensive rounds of stringent cleaning using chlorinated solvents to mitigate issues with sample carryover.199 Proteins prone to formation of aggregates are also problematic as denatured “globs” of protein can entrench themselves on the head of the digestion column. Frequent checking for levels of carryover using an injection of a true blank (a sample containing all components except the protein) is very highly recommended to ensure high quality HDX-MS data sets.
2.3.4. Automation of HDX-MS
The complicated combination of obtaining a broad range of time points together with maintaining specific conditions to minimize back-exchange in sample handling has impeded the automation of HDX-MS. Preparation of deuterium labeled samples by hand remains common in HDX-MS. Quenched samples are either analyzed immediately or flash frozen and kept at −80 °C for downstream thawing and MS analysis. While automation for HDX-MS has many challenges, the ability to enable high throughput analyses, particularly for routine analysis of the same protein, has driven the development and incorporation of automation into HDX-MS.
One approach to reduce manual sample handling has been to focus on automation solely of the LC-MS analysis. For this, a set of exchanged samples can be prepared, stored at −80 °C, and then batch analyzed by LC-MS in a queue. While this still requires exchange samples to be prepared by hand, it reduces variability in the thawing and injection process and automates the LC-MS analysis stage. An early solution for HDX LC-MS automation was to use a −80 °C sample holder coupled to an autosampler for controlled thawing and injection of samples over a custom HDX-LC system with inline proteolysis.178,179 Quenched samples included 10% glycerol to help with reliable thawing of exchanged samples. Since then, more sophisticated cooling systems for automated thawing and LC-MS analysis of HDX-MS have been developed to provide the capacity and flexibility to accommodate large batches of samples.200 Another approach was recently introduced that uses a dry ice–ethanol bath to hold quenched samples below −60 °C, which was found to be suitable for storing samples without any detectable back-exchange for at least 20 h.80 This partial automation greatly facilitates the LC-MS portion of the analysis by alleviating the cumbersome task of extensive manual injections, while allowing complete flexibility for sampling a wide range of time points, exchange conditions, and performing complicated postquench sample manipulation that might be required with some protein systems.
In parallel, there has been development in automation approaches that cover both sample preparation and HDX-MS analysis steps. An early implementation of automated sample preparation used a sample handling robot to perform the deuterium labeling and quenching steps, followed by online pepsin digestion coupled to LC-MS.174 As input, the setup required a stock protein solution, deuteration buffer, and quench buffer and was programmed to prepare and collect MS data for a series of samples with time points ranging from 30 s to >24 h. The throughput afforded with this type of automation was used for HDX-MS studies that screened a wide array of ligands for protein binding.201 A limit of the early sample handling systems was the restriction in the time range it could sample due to the physical movements necessary for transferring and mixing solutions. The use of an air gap approach for deuterium labeling widens the range of deuteration times, mainly by expanding the range on the lower (shorter) end of the time scale. The deuterated labeling solution and the quench are aspirated into the same syringe with a gap in between. The syringe dispenses these two volumes into a tube at different flow rates to achieve deuteration times down to 130 ms.202 This ability is now incorporated into commercial options for integrated sample HDX-MS handling systems.
Other methods to increase sample throughput include the use of a dual column LC-systems for HDX-MS. While column one is performing a gradient elution, the second is washed, equilibrated, and loaded for seamless LC-MS data collection. This approach provides a 2-fold improvement in throughput, while also maximizing the use of the mass spectrometer time, as it is essentially collecting useful data the entire time.203 Some of the ongoing limitations of automated systems are being addressed, for example, solid-phase cartridges for automated lipid removal were recently introduced.204 Future advances will likely continue to expand the abilities of automated systems to conduct more challenging HDX-MS sample preparation and make a broader set of protein systems amenable to automation.
2.4. Mass Spectrometry
Starting with the first studies of HDX by mass spectrometry,9 electrospray ionization (ESI) continues to be the most prevalent method for monitoring proteins and peptides to measure deuterium uptake. This is largely attributed to the ease of direct online integration with LC systems, which were already incorporated into the first bottom-up HDX-MS studies.10 One caveat associated with the ESI process is that effective desolvation of the analyte typically requires elevated temperatures. Even though the spray process is expected to last only a few milliseconds, this time frame is still enough for complete back-exchange of all fast-exchanging side chains and at higher temperatures can start to induce back-exchange of backbone amides as well.9 Optimal source conditions have been established for nearly all instruments commonly used for HDX-MS, often finding that considerably lower than ideal source temperatures are necessary for minimizing amide back-exchange during ionization.71,205,206 The electrical potentials in the first regions of the mass spectrometer where there is still ample solvent could also influence deuterium loss.207 We note that it is also advisible to tune the source conditions to minimize the risk of in-source fragmentation of peptides. This is not always possible, as higher potentials needed for optimal signal are often tied to inherently higher risk of generating in-source fragmentation.208 This fragmentation can be especially problematic for HDX-MS studies which employ nonspecific proteases, and some of the in-source fragments can be misidentified as intact peptides.
An alternative approach to electrospray ionization in HDX-MS has been matrix assisted laser desorption ionization (MALDI) mass spectrometry. Samples are mixed and crystallized with a small organic acid on a sample stage, and a laser is used to generate ions from the cocrystallized sample. The feasibility of MALDI for HDX-MS was first demonstrated by Mandell et al. using a precooled stage, rapid vacuum drying of the sample, and immediate analysis by MALDI-TOF.117 This approach was unique as it could be performed directly from quenched samples with acceptable levels of back-exchange, thus negating the need for an LC step. Later studies utilizing HDX-MS by MALDI achieved lower levels of back-exchange by rapid processing of quenched samples cooled to −20 °C209 or incorporation of organic cosolvents into the MALDI quench step.101 While not a major platform for HDX-MS, due to the inherently limited number of peptides it can resolve without an LC separation, MALDI has some notable strengths, including the ability to analyze many samples in parallel101,210 and the ability to probe the HDX of proteins within tissue samples in situ.211
Although not a strict requirement, a mass resolution sufficient for resolving isotopic peaks within peptide isotopic distributions is desirable for tracking levels of deuterium uptake. Nowadays most HDX-MS studies utilize either quadrupole time-of-flight (Q-TOF) mass analyzers or Fourier transform (FT)-based analyzers (Orbitrap or ion cyclotron resonance), which are both able to provide the resolution needed for resolving isotopes in peptides and small proteins (mass resolution >10 000).212 It has been observed that minor offsets to the measured deuterium content may arise on different mass analyzer types. Burns et al. noted that Orbitrap instruments can sometimes underreport deuterium uptake due to isotopic interference effects in the FT-MS measurement at certain resolution settings.213 On the other hand, Q-TOF instruments have observed detector saturation effects that can distort measured deuterium levels especially when combined with ion mobility, as it leads to temporally compressed ion packets hitting the detector simultaneously.214,215 While it is important to be aware of caveats associated with mass spectrometers for HDX-MS, we note that these effects have generally only influenced a minor fraction of peptides from HDX-MS data sets.
As outlined in the section 2.3.3, the major size limit of what HDX-MS can handle boils down to the analytical platform’s ability to resolve and detect all of the peptides generated. To this end, higher mass resolution has some impact, as it can help resolve coeluting peptides that may be very close in m/z but not directly overlapping216 (Figure 19). The increase in resolving power and sensitivity of mass spectrometers has aided the ability of HDX-MS studies to resolve more complex samples while also detecting more low-abundance peptides for increased sequence coverage and redundancy.
Figure 19.

Example illustrating the benefits of high mass resolution. The overlapped spectra contains two deuterated peptides (blue, top; red, bottom) and some of the isotopic peaks closely overlap in m/z (inset). Without the high mass resolution, these isotopic peaks would confound accurate measure of deuterium uptake for both peptides. Reproduced with permission from ref (216). Copyright 2010 American Chemical Society.
In the past decade, the mass spectrometry field has seen a large boom in the development and commercialization of ion mobility (IM) separation.217,218 IM separation offers a way to resolve peptides by their mass, charge, and shape due to differences in their interactions with a neutral drift gas. Importantly, IM separations can be accomplished on a millisecond time scale, making it amenable for adding a third dimension of separation to LC-MS without requiring any additional time for data collection. Early IM-enabled instruments were used to show the potential of IM for HDX-MS through resolving species that overlapped in the LC dimension but were resolved in the IM dimension.219 IM technology has since achieved higher resolution and has become increasingly incorporated for HDX-MS, notably with the Waters Synapt platform.215 A minor technical drawback of utilizing ion mobility is the increased risk of detector saturation due to the high flux of compressed ions from the IM stage simultaneously hitting the detector (Figure 20), which can be largely alleviated by acquisition methods that account for this effect. Currently emerging implementations in IM capable of even higher resolving power (in the mobility dimension) on several MS platforms will undoubtably continue to expand the limits of complexity that are amenable to HDX-MS.
Figure 20.
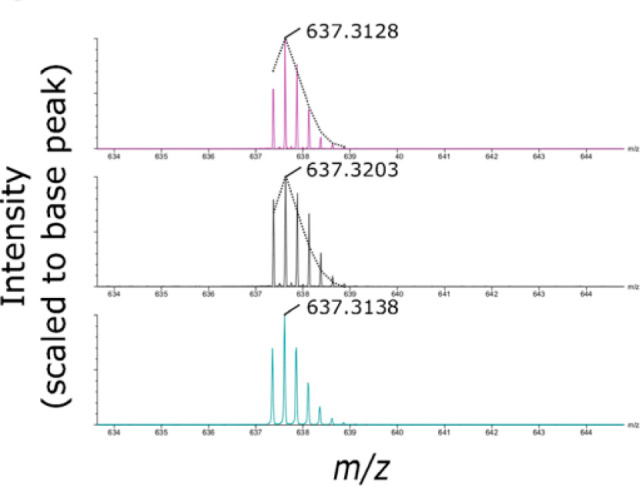
Detector saturation can lead to distorted isotopic envelopes that offset deuterium measurements. An undeuterated peptide was analyzed by TOF (cyan, bottom), showing the expected isotopic distribution. The same peptide analyzed using an additional ion mobilty separation leads to detector saturation distorting the isotopic profile (gray, middle). The dashed lines indicate the expected isotopic profile. Using a dynamic range extension feature to account for the large ion flux largely mitigates the distortions attributed to detector saturation (magenta, top). Reproduced with permission from ref (215). Copyright 2017 American Chemical Society.
2.5. Peptide Identification
One of the first steps in HDX-MS analysis of a target protein is mapping the peptides produced during protease digestion. This is accomplished by obtaining MS/MS spectra of all observable peptides from undeuterated samples. MS/MS fragmentation by collision induced dissociation (CID) is now an integrated feature on nearly all MS platforms, and most researchers use a single platform for both MS/MS and HDX-MS measurements. The traditional approach for obtaining MS/MS spectra is LC-MS using data-dependent analysis (DDA), where the abundant precursor ions are immediately recognized, mass isolated, and fragmented to obtain clean MS/MS spectra for all prevalent ions in the LC chromatogram. While DDA analysis can be performed on nearly all modern MS instruments used for HDX-MS, it has been shown to be most effective on ion-trap instruments which are capable of very rapid MS/MS scans.213 To gain more identifications, alternative strategies have started using data-independent acquisition (DIA) where MS/MS spectra are collected on as many observable peptides as possible.220 With DIA, many peptides are fragmented simultaneously, and their MS/MS spectra and precursor ions are assigned through alignment in their retention times. The ability to perform ion mobility separations with DIA adds an important dimension that greatly increases the ability to resolve and characterize low-abundance peptides.221,222
The MS/MS spectra are then used to identify peptide sequences. The fragment ion spectra along with the exact mass of the precursor ions are compared to those predicted from a in silico digestion of a protein sequence database to match and identify MS features to peptides.124 Because proteases compatible with HDX-MS are relatively nonspecific, cleavage after all residues is considered when performing these peptide searches. However, knowledge of protease specificity can help identify in-source fragmentation products or potential degradation products within the protein sample. For example, Hamuro et al. showed that the presence of peptides generated by cleavage after proline or basic residues, where pepsin is known not to cleave,145 suggests the presence of degraded forms of the protein, which can complicate deuterium analysis.135
As HDX-MS samples are most often performed on purified proteins, the sequence libraries used for searches often only include the target protein, any proteases used for sample processing, and any known contaminants. A common addition for searches is to include the reverse sequences for assessing the levels of false positives in peptide identifications.223 While the use of limited sequence libraries reduces the search space and computational requirements, there is a risk of false positive identifications due to unknown contaminants not included in the sequence library. Dobbs et al. recently mapped out the prevalent protein contaminants derived from the most common expression and purification systems used for protein structural studies.224 Inclusion of these known potential contaminants helps minimize the risk of false positives and improve statistics associated with peptide identification.
As some proteins may bear post-translation modifications (PTMs), and in some cases multiple unique PTMs, the masses of several peptides may be altered by the presence of a modification. The signal intensity of modified peptides may be different from the unmodified forms, and modification may not be complete for all protein molecules. To achieve peptide identification in these cases, the PTMs must be accounted for. The extensive possibilities for modifications greatly increase the search space when identifying peptides, especially for HDX-MS, where proteases are relatively nonspecific. Such large search spaces can also increase the rates of false positives. Liu et al. proposed a workflow where proteins are first analyzed by a conventional proteomics methods to map out all PTMs, which are next incorporated into the search for the HDX-MS peptides.225 An example of this is shown in Figure 21, where knowledge of the phosphorylation states of vasopressin type 2 receptor (V2R) was used to assign pepsin fragments from this region and fill in what would have been missing sequence coverage.
Figure 21.
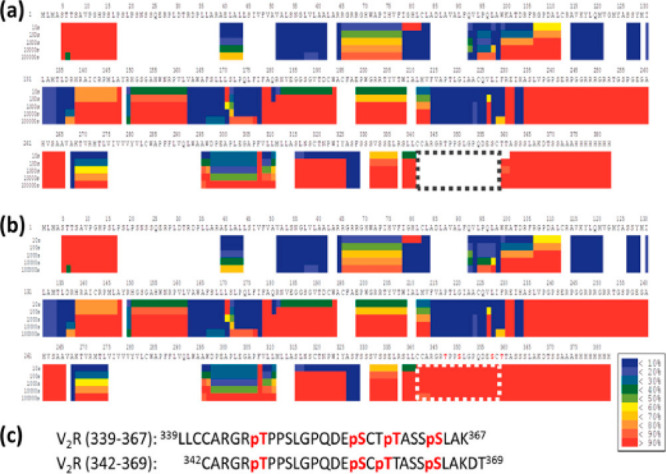
Comparison of the sequence coverage for V2R either without (a) or with (b) parallel proteomic analysis to map out PTMs. The dashed segment contains phosphorylation sites, which were identified from trypsin digested samples and subsequently used to identify two pepsin peptides from this region (c). Reproduced with permission from ref (225). Copyright 2019 American Chemical Society.
2.6. Fragmentation Approaches for Higher Spatial Resolution
An inherent limitation with bottom-up HDX-MS studies has been peptide-level spatial resolution, where the measured deuterium level reflects the sum total of all exchange at each backbone amide in a peptide (average length is 12–13 residues). While the analysis of overlapping peptides can provide increased spatial resolution (section 5.4), many studies have also explored using MS/MS to measure deuterium levels in fragment ions and thereby localize deuterium to much smaller sequences, perhaps even single residues. Early reports indicated that CID-based fragmentation could reliably localize deuterium, as judged by consistency with known NMR HDX of well-studied proteins.226 The deuterium incorporation from b and y peptide fragment ion series could be tracked to calculate exchange rates for nearly all amides within peptides. However, studies investigating the fundamental chemistry of peptide fragmentation appreciated that the energetic barrier of proton migration is far lower than the energy required to induce fragmentation of a peptide bond.227 With partially deuterated peptides, migration would cause both hydrogen and deuterium to be redistributed across all exchangeable sites on a peptide, an effect termed “scrambling”. The fragment ions would therefore have lost all relevant information on their deuteration levels and simply represent an average for the entire precursor peptide.
The issue of scrambling in HDX-MS/MS experiments has been heavily studied.228−231 More investigations into the fundamental process of scrambling have revealed that scrambling can produce a nonuniform distribution of deuterium across a peptide, which may explain some of the earlier conflicting findings.232 It is now generally agreed that CID-based fragmentation, especially for b/y ions of protonated peptides, is not a reliable approach for achieving accurate localization of deuterium in peptides deuterated at backbone amide positions.233,234 Further studies showed that deprotonated peptides (anions) are also susceptible to scrambling.235,236 A few notable exceptions have been studies using very fast activation times (10 ns or less), such as in-source decay with MALDI mass spectrometry, to induce fragmentation before scrambling can take place.237 Despite the inability to report site-specific deuterium incorporation, HDX-MS with CID MS/MS has still proven useful. Percy et al. showed that one way to expand peak capacity for HDX-MS is to perform CID fragmentation and measure deuterium levels of fragment ions. The deuterium levels of the fragment ions are assumed to be completely scrambled so they can be used as surrogates to measure deuterium uptake of precursor ions, which may be too overlapped in the spectra to measure their deuterium levels directly.238 Their approach worked best when combined with a DIA approach, and lower levels of deuterium in the labeling step to maintain narrow isotopic distributions to further minimize spectral overlap among peptides.
Alternative peptide fragmentation methods such as electron capture dissociation (ECD) or electron transfer dissociation (ETD) have become increasingly widespread due to their complementarity to CID and ability to generate peptide fragments without loss of labile post-translational modifications.239 These electron-driven processes generate c and z type fragments through a radical mechanism that can be accomplished with very little vibrational excitation of the peptide ions.240 The potential of ETD/ECD to fragment deuterium labeled peptides without inducing scrambling was soon appreciated241 (Figure 22). To methodically study all instrument parameters relevant to scrambling, a 12 amino acid peptide probe was developed with the sequence HHHHHHIIKIIK.242 In deuterated solution, all the amides will take up deuterium rapidly as the peptide is largely unstructured. At the quench step the 6 N-terminal amides (sequence HHHHHH) will all lose deuterium very rapidly due to side chain and neighboring effects.48 Therefore, when the partially deuterated peptide enters the mass spectrometer, it is selectively deuterated only on the 6 C-terminal amides (sequence IIKIIK). The deuterium levels of the various c/z fragment ions generated can therefore be used to detect the level of scrambling that has occurred in the course of ionization and fragmentation, as there should only be deuterium in ions derived from the C-terminal residues. By fine-tuning fragmentation and ionization conditions, it was possible to keep the general levels of scrambling low (less than 10%). Optimal parameters for minimizing scrambling have since been established for nearly all MS platforms capable of electron-based fragmentation.243−246 Thanks to this foundational work, ETD is increasingly being incorporated to bottom-up HDX-MS workflows to gain higher spatial resolution and more insight into many protein systems154,247−252
Figure 22.
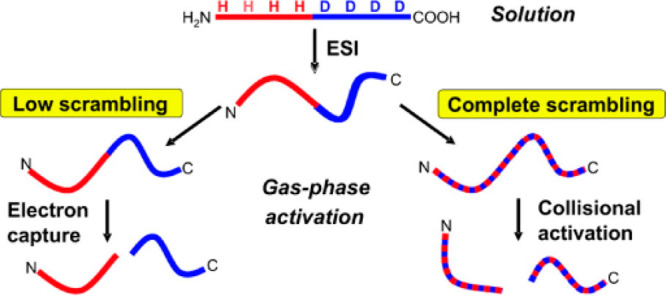
Depiction of deuterium scrambling in a partially deuterated peptide. The deuterium is localized to only the c-terminal half, but upon collision activation, the protons and deuterium migrate along the exchangeable sites on the peptide, leading to loss of the deuterium localization (“scrambling”). With electron capture dissociation, there is very little ion excitation, which leaves the deuterium in place to provide informative fragment ions. Reproduced with permission from ref (241). Copyright 2008 American Chemical Society.
The ECD/ETD studies have also discovered that different peptide sequence and charge states have inherently different susceptibilities to scrambling.242 A robust approach for directly measuring the level of scrambling within a specific peptide was demonstrated through measurement of the deuterium level of the ammonia-loss product, commonly generated during ETD.253 In a quenched HDX-MS sample, the amine at the N-terminus on peptides should be fully back-exchanged. By measuring the deuterium content attributed to the N-terminal amine, it is possible to empirically measure the level or scrambling that has occurred within each peptide. More recent studies have revealed additional details about how the sequence of peptides and the presence of different exchangeable sites influences scrambling.254 Through a targeted study of cytochrome c, a small protein with well-established amide exchange kinetics from NMR measurements, Hamuro et al. found that only a portion of the ETD fragment ions were accurate for measuring site-specific deuterium uptake kinetics255 (Figure 23). These studies described some extra considerations that should be taken when interpreting site-specific HDX-MS data obtained from ETD fragments.
Figure 23.

Site-specific determination of amide exchange kinetics using ETD. Amide exchange within peptide 67–82 of cytochrome c were analyzed from the available c (left) and z (right) ions, indicated by ×s. As a reference, the predicted deuterium levels in the absence of scrambling (blue) and complete scrambling (red) are shown. Reproduced with permission from ref (254). Copyright 2017 American Chemical Society.
Another emerging method for gas-phase fragmentation has been photon-based ion excitation, including ultraviolet photodissociation (UVPD). The ability to generate different cleavage pathways in peptides and other biological macromolecules has led to UVPD being more broadly applied for biological mass spectrometry.256 UVPD has been shown effective for fragmentation without inducing scrambling,257,258 presenting a new tool for achieving site-specific information from HDX-MS studies (Figure 24). An additional strength of UVPD is that it can be applied for fragmentation of singly charged peptides, which cannot be examined by ETD/ECD techniques. Systematic examination of UVPD parameters revealed long UV irradiation can induce high levels of scrambling, even before fragmentation occurs.259 Only below 25 ms UV irradiation was there a low level of scrambling, which is consistent with previous uses of UVPD for HDX-MS.251 The study highlights the need to keep the irradiation times short to obtain fragments for accurately measuring deuterium content. The increased availability of instrumentation capable of electron- or photon-based dissociation, and likely additional novel approaches for peptide fragmentation in the future, is expected to propel the use of site-specific HDX-MS studies in the coming years.
Figure 24.
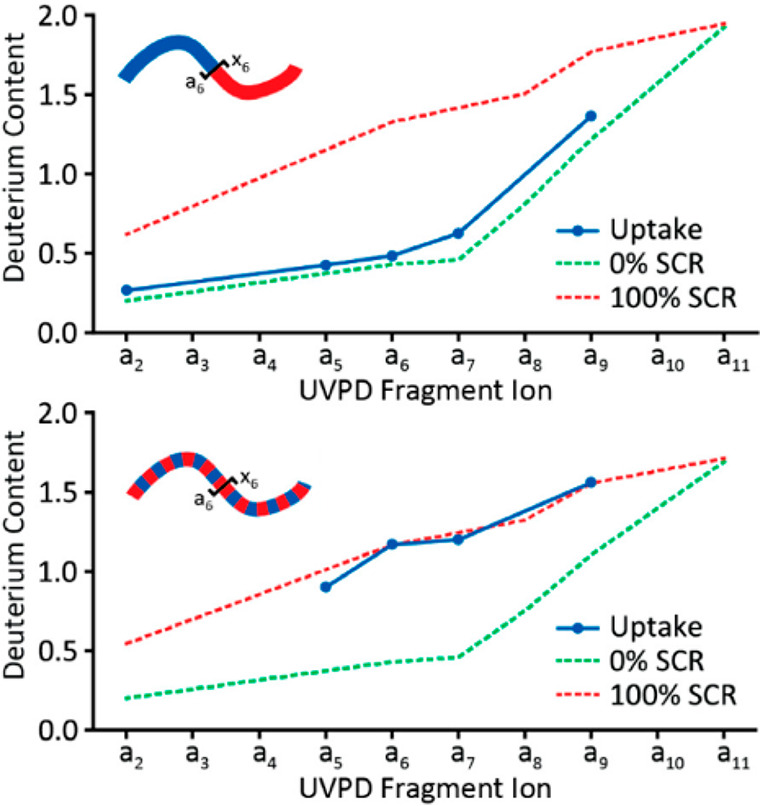
UVPD of deuterated peptides can occur without scrambling. Deuterium content of various a ions are shown at conditions that prevent (top) or induce scrambling (bottom). The uptake measured is plotted in blue. Theoretical positions of the deuterium levels assuming no (green) or full (red) scrambling are indicated in dashed lines. Reproduced with permission from ref (258). Copyright 2018 American Chemical Society.
2.7. Global and Top-down HDX-MS
From the early years of HDX-MS, deuterium exchange kinetics have also been monitored on an intact protein level.9,37,260 While this does not provide local information throughout the protein sequence, it does offer a way to monitor the sum-total exchange of all amides on a global level. This approach has proven useful for a variety of applications over the years, including detecting conformational changes in different protein states, quantifying ligand-binding affinities, and enabling high throughput ligand binding screening for target proteins.261−266 Global HDX-MS also has an inherent advantage of being well-suited for detecting subpopulations of conformers and tracking slow correlated protein conformational changes (EX1 kinetics) [reviewed in ref (267)]. This later approach was used recently to track how detergents and ligand binding influence the global conformational profiles of membrane proteins.199
An emerging approach in proteomics has been the application of a top-down strategy for characterizing proteins.268 The intact proteins are ionized and activated to generate a large series of fragment ions to track the sequence and map post-translational modifications throughout the sequence. An inherent advantage of examining the intact protein directly is the ability to resolve various populations resulting from different combinations of PTMs across the protein (“proteoforms”) that would otherwise all be pooled together with a typical bottom-up approach (see Figure 26 for an example). The advent of fragmentation techniques like ECD and ETD that have circumvented the issues of deuterium scrambling have paved the way for a top-down approach to enable high spatial resolution HDX-MS analysis from an intact protein analyte.118,269 The mass shifts of all the c and z ions in the MS/MS spectra are used to calculate deuterium incorporation throughout the sequence (Figure 25).
Figure 26.

Top-down analysis of the various phosphorylated states of calmodulin. The top spectrum shows the unphosphorylated calmodulin (CaMp0) and four different phosphorylated proteoforms. The bottom spectrum is after 20 s of deuterium exchange. Each proteoform could be mass-isolated and the amide exchange characterized with high spatial resolution using ETD. Reproduced with permission from ref (271). Copyright 2016 from Elsevier.
Figure 25.
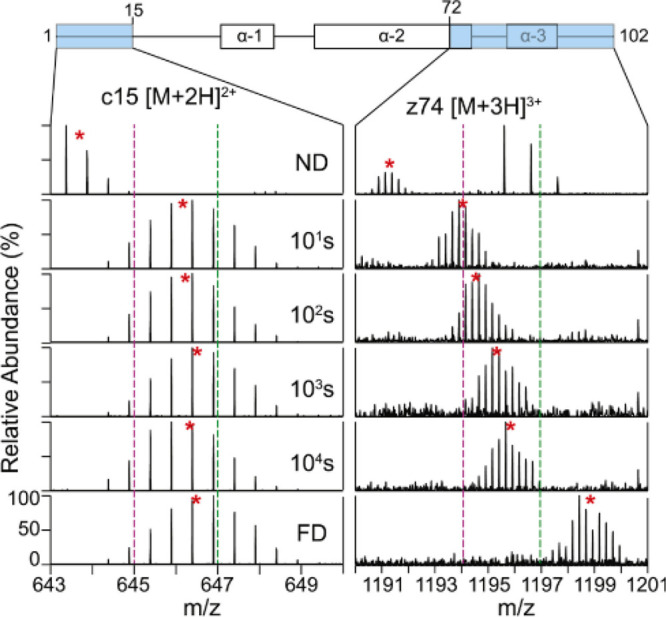
Example of top-down HDX-MS to study the dynamics of histone tails. The deuterium uptake for the c15 ion (left) and z74 ion (right) generated by ETD are shown for each deuterium exchange time point and the fully deuterated (FD) control. Reproduced with permission from ref (275). Copyright 2018 Elsevier.
A major advantage of top-down analysis is the ability is to mass-select different conformers within a sample of a protein based on global deuterium uptake. The mass-resolved conformers can then be individually characterized by their deuterium exchange profiles from all the fragment ions.121,270 A similar mass-resolved top-down approach was used to study how different phosphorylation states influenced the conformational dynamics of calmodulin.271 With four known phosphorylation sites, there are 16 possible phosphorylated calmodulin proteoforms. Using top-down fragmentation, it was found that only six of these potential species exist in vitro and that phosphorylation of these sites occurs in a sequential manner. Using a top-down HDX-MS approach, they discovered that structural differences only arise in the tetra-phosphorylated calmodulin, directly identifying how the degree and sites of phosphorylation affect conformational dynamics, which would not have been possible to resolve with a bottom-up approach (Figure 26). Integration of capillary electrophoresis has proven a highly complementary tool when combined with HDX-MS.42 CE under native conditions is used to resolve different conformers, which are then structurally characterized by top-down HDX-MS using ETD. The ability to both resolve and characterize conformationally distinct subpopulations through multiple means is a powerful increase for the comprehensive characterization of biopharmaceutical proteins.272
The frequent challenge with top-down studies is low sensitivity, which often necessitates collecting and averaging spectra over longer periods, which can be up to several minutes depending on the signal quality. To enable long collection times, several approaches have opted to use volatile buffers and directly analyze the quenched sample by mass spectrometry, thereby alleviating the need for a desalting or LC step. However, this imposes a restriction on which buffers can be incorporated for the protein labeling step, as most nonvolatile salts at even low mM levels can be problematic for MS analysis. Another complication with bypassing a desalting step is the inability to use high concentrations of reducing agents to reduce of disulfide bonds, which present a barrier for effective ECD/ETD fragmentation. To this end, Wang et al. demonstrated using β2-microglobulin that even low concentrations of TCEP (5 mM) could be included in the infused protein sample to effectively reduce disulfide bonds while still being able to observe protein signal in the MS.273 Another downside to not using a desalting step is that deuterium incorporated into side chain positions will not be washed away, meaning there can be variable numbers of deuterium on different residues according to the number of exchangeable positions at the side chains. Such data can be complex to analyze, whereas a short desalting step washes away deuterium in the side chains, leaving only exchanged deuterium at backbone amide positions, thereby simplifying data interpretation.
Whether or not a desalting step is used, minimizing back-exchange is critical for any top-down HDX-MS study. Deuterium loss during the MS acquisition will result in a mass shift that, when averaged, can artificially broaden the isotopic distribution and greatly complicate deuterium uptake calculations. To this end, several groups have adopted subzero cooling systems for top-down HDX-MS analysis that are capable of sufficiently reducing back-exchange to allow for collection of data for several minutes with stable deuterium levels.119,187
The spatial resolution obtainable with top-down analysis is determined by the number of fragment ions that are observed in the MS/MS spectra. Efficient fragmentation becomes increasingly challenging for larger proteins. To expand both sequence coverage and spatial resolution, bottom-up and top-down HDX-MS analyses can be combined.274,275 A “middle-down” approach has also been used in proteomics to obtain better sequence coverage. Unlike a bottom-up approach, very highly specific proteases are used to predominantly generate large peptides, which are then typically analyzed by ETD/ECD.276 For HDX-MS, all available proteases are relatively nonspecific, making it nearly impossible to achieve efficient proteolysis at only a limited set of sites on the protein. Pan et al. developed a middle-down HDX-MS approach to maximize coverage for Herceptin.277 Limited pepsin digestion was achieved by not reducing disulfide bonds until after the digestion step to minimize the number of accessible cleavage sites. Pepsin was inhibited in the subsequent disulfide reduction step by the addition of pepstatin and produced three large fragments that could be well-resolved and produce a rich set of fragment ions (Figure 27). The combination of top-down and middle-down analysis yielded a coverage for the heavy chain of 95%, where with top-down alone it was only 50%.187 Other proteases with more limited substrate specificity may offer another way to obtain limited fragmentation for middle-down HDX-MS analysis34 (see section 2.3.1).
Figure 27.
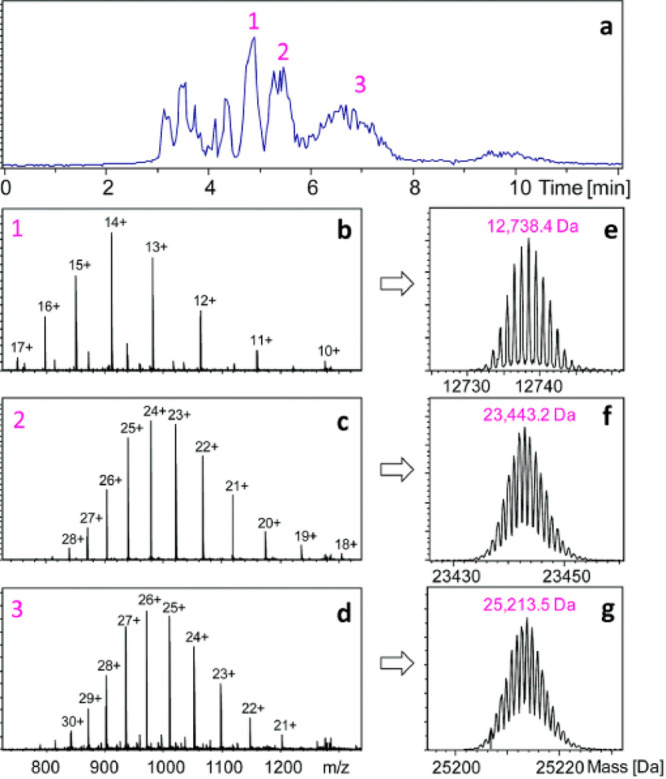
Middle-down HDX-MS was used to study herceptin. Limited pepsin digestion yielded three large fragments (1–3) that were resolved and independently analyzed by ETD to increase the sequence coverage that was obtained from direct top-down ETD analysis. Figure adapted from ref (277).
Supercharging has been popular in the proteomics field to enable higher charge states that are more suitable for top-down analysis. Cosolvents such as m-nitrobenzyl alcohol or sulfolane added to the infusion solution can result in a much higher charge state distribution for proteins. Supercharging has been demonstrated as an effective means to increase the sensitivity and spatial resolution for top-down HDX-MS studies without influencing scrambling or back-exchange levels.278 A more recent approach of electrothermal supercharging, using higher source potentials to unfold proteins during the ionization process, was even more effective for improving spatial resolution without inducing scrambling.279 An alternative approach for expanding spatial resolution has been to incorporate additional fragmentation methods such as UVPD.257 Importantly, UVPD generated a different set of fragments compared to ETD, thereby providing a new and highly complementary approach for expanding the spatial resolution of top-down HDX-MS studies (Figure 28).
Figure 28.
Comparison of ETD (top) and UVPD (bottom) fragmentation used for high resolution top-down HDX-MS. Each notch in the sequence represents a fragment ion that was observable for HDX-MS. Reproduced with permission from ref (257). Copyright 2018 American Chemical Society.
3. Current Uses of HDX-MS
3.1. Characterizing Protein–Ligand and Protein–Protein Interactions
3.1.1. Mapping Interactions
The interaction of proteins with other proteins and ligands is central to nearly all biological processes, and the study of these processes is vital for the development of new therapeutics. HDX-MS is particularly powerful for investigating protein binding interactions, and this has been one of the most prevalent applications of HDX-MS.280 By comparing the exchange kinetics between a free (unbound) vs a bound protein, one can localize and track the structural changes associated with the binding event. In most cases, ligand binding leads to stabilization of the local structure at the interface, leading to slowed deuterium exchange,280,281 and provides an efficient way to map interaction surfaces.282−284
To best resolve differences upon binding, it is important to make the comparison between a fully free population and an as-fully bound-as-possible population. With weak binding interactions it is often difficult to achieve saturation, as it may require very high concentrations of ligand.285,286 Often even a several-fold molar excess of ligand is not sufficient for achieving >99% of the protein bound in solution. Furthermore, a complex should be formed prior to deuterium exchange to ensure the fully bound state is being probed. For ligands with fast dissociation kinetics (>1 x 105 s–1), it is important to consider that the protein will spend time in the unbound state during deuterium exchange,287,288 which in some cases leads to the appearance of two species in the deuteration profiles.251,289,290 Although data like this can be confounding, such experiments actually offer a way to gain more insight into an interaction. For example, Zhang and Vachet were able to use this effect to measure dissociation kinetics for the β-lactoglobulin dimer.291
Another consideration with free vs bound comparative HDX-MS is that small ligands can act to globally stabilize a protein. The measured changes in the deuterium exchange profiles will reflect both global domain stabilization and local contacts with the ligand, which can sometimes be resolved by varying the concentrations of the ligand.292 Although generally ligand binding increases protection of amides, thereby slowing deuterium exchange (“type 1” binding), it is also possible for ligand binding to actually lead to increased deuterium exchange at the binding site (“type 2” binding).130,293 It is also possible for an interaction to not perturb the backbone amide hydrogen environment, thereby producing no observable HDX changes at all.294
With antibodies, comparative HDX-MS is now well-defined. Most antibodies generally have strong affinities (KD ∼ low nM) with sufficiently slow dissociation kinetics and recognize a very specific surface patch on proteins. As such, HDX-MS has been particularly useful for mapping the protein surfaces on antigens that are recognized by monoclonal antibodies (mAbs) (often termed “epitope mapping”) (Figure 29). Epitopes can be linear, where the binding site is a single stretch of amino acids on the antigen protein, or conformational, which are made up of discrete segments of the protein sequence that are close together in three-dimensional structure. HDX-MS has proven particularly useful for mapping conformational epitopes for which there are fewer techniques to characterize. In fact, epitope mapping is now one of the primary uses of HDX-MS in the biopharmaceutical industry.295
Figure 29.
Epitope mapping of three monoclonal antibodies to Staphylococcus aureus manganese transporter protein (MntC). Regions in yellow exhibited slowed exchange when bound to the antibody. Figure adapted from ref (284).
3.1.2. Monitoring Allosteric Effects
While HDX-MS is effective in characterizing protein interfaces and ligand binding events, in many instances, there are protein–ligand interactions that require other considerations and caution must be exercised as data interpretation depends on the protein system. Allostery, global protein stabilization, and lack of influence on backbone amide hydrogens as a result of various binding modes (hydrophobic, electrostatic, etc.) can present challenges in some systems. Even with antibodies, allosteric effects can occur at distal sites on the protein(s) that are not always structurally foreseeable.296,297 Depending on the system being examined, the intrinsic ability to see all changes across a protein upon binding can be either tremendously insightful or confounding.
For systems where the interaction interface has been established and especially when high resolution structures of complexes are available, detection of allosteric effects presents a powerful way to detect indirect effects of binding.15,298,299 A very clear demonstration of allosteric changes were observed in Nipah Virus G ectodomain upon binding its receptor, epherinB2.300 Comparative HDX-MS revealed the surface of the ectodomain directly interacting with epherinB2 becoming more protected, while several sites on the opposite side of the protein show increased exchange (Figure 30). Interestingly, the allosteric effects are relayed all the way to the stalk helices, implicating this binding event in the mechanism of activation leading to host/virus membrane fusion.
Figure 30.
Allosteric changes observed in the Nipah virus G ectodomain upon binding ephrinB2. Regions of the G ectodomain that do not change (white) become more protected (blue) or less protected (red) are indicated on the structure. The N-terminal stalk helices that also become less protected are shown in the bottom with their predicted positions. The position of EprhinB2 is shown in yellow. Figure adapted from ref (300).
Pinpointing allosteric effects has also been tremendously insightful in recent years for understanding how antibodies are able to neutralize their targets. For example, Dagbay et al.301 found that binding of mAb SRK-015 elicits extensive changes on its target protein myostatin (Figure 31). In addition to increased protection at the epitope at the end of the arms, there was a large degree of protection at the center of the protein, particularly around the furin cleavage site. This long-range allosteric effect provides a rationale for how SRK-015 inhibits the activation of myostatin, inhibiting access to the furin cleavage site. Similar insights into how antibodies are able to protect from various pathogens have been elucidated by HDX-MS including rhinovirus particles,302 diptheria toxin,303 Japanese encephalovirus envelope protein,304Neisseria Meningitis adhesin A,305,306 factor H binding protein,307 heparin binding antigen,308Staphylococcus aureus manganese transporter protein,284 HIV envelope glycoprotein,293,309 influenza hemagglutinin,310,311 Malaria Plasmodium vivax Duffy binding protein,312,313 and Dengue virus.314 A recent nanoparticle vaccine against SARS-CoV-2 utilized HDX-MS to elucidate binding epitopes of known neutralizing mAbs.315 The ability to detail the mechanisms of neutralization, in part from HDX-MS input, has paved the way for advanced therapeutic and vaccination approaches for a multitude of diseases.
Figure 31.
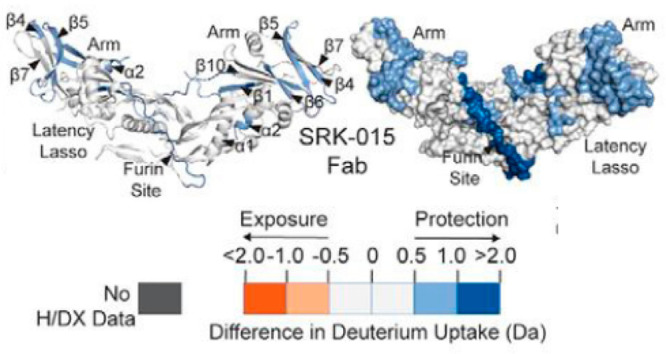
Mechanism of action of Mab SRK-015 on the inhibition of myostatin. Antibody binding to the distal arm of myostatin elicits allosteric effects throughout the protein including protection around the furin cleavage site to limit its accessibility for proteolytic cleavage. Figure adapted from ref (301).
When HDX-MS is used to characterize a protein–protein interface with no prior structural information, allosteric effects can become a complicating factor because the allosteric changes can be misinterpreted as part of a binding interface.316,317 A common strategy is to use HDX-MS to identify regions of change in a protein, then use targeted site-directed mutagenesis to probe the involvement of the regions shown by HDX-MS. If mutation prevents binding, as compared to decoupling allosteric effects, the nonbinding mutants can be identified in binding assays (usually not HDX-MS). HDX-MS is also often combined with other approaches such as chemical cross-linking to help better define interaction surfaces.250 It has been observed that shorter deuterium labeling times (<5 min) are more likely to reveal changes associated solvent accessibility and protein interaction surfaces, whereas longer time points can reveal allosteric changes at structured regions and other global structural effects.15,22,318−320 A “kinetic” millisecond HDX workflow was recently proposed as an alternative approach to differentiate direct binding from allosteric changes.321 Instead of preincubating the protein with the antibody prior to exchange, the deuterium labeling step is combined with the antibody binding step. Using a time-resolved ESI system on a chip, rapid in-line exchange and quenching on short time scales (as low as 200 ms) were achieved.322 Only the changes at the direct binding interface were observed by the kinetic HDX-MS approach as compared to a preincubated (“equilibrium”) complex. The study suggests that HDX-MS experiments at very short time scales during the binding event may help specifically identify surfaces directly involved in the binding interface.
3.1.3. Mapping Complex Interactions
HDX-MS also serves as a useful tool for assessing interactions with polyclonal antibodies. Mapping polyclonal antibody response provides critical insight into the immune response, but they are much more challenging to characterize than mAbs as the mixtures of antibodies are present at different concentrations, bind at different rates, and may be cross-reactive.323 To address these challenges, Zhang et al. digested and purified Fab fragments from a polyclonal mixture before analyzing the sample using HDX.324 The experiments revealed and localized four epitopes relevant to the measured immune response to nut allergen Ana o 2. In other work, HDX-MS was used to track epitope specificity and diversity in polyclonal serum following immunizations.325 To get around the issue of matrix affects associated with the complexity of serum, an affinity purification step was incorporated into the workflow to fractionate only the antigen-reactive antibody population. A similar study compared the binding of various mAbs to human cystatin C with that obtained from polyclonal serum and used a similar enrichment procedure to fractionate only the antigen-reactive antibody population.326 These few examples already show the potential of HDX-MS to aid in mapping epitopes of complex polyclonal responses.
Beyond epitope mapping, HDX-MS continues to be utilized for studying protein–protein interactions in complex protein systems and revealing mechanisms of activation in a range of systems including: clotting factors,320,327−329 metabolic enzymes,67,298,330−338 heat shock proteins317,339,340 and other chaperones,341−348 signaling complexes,349−360 and complexes exclusively residing in membranes.361 HDX-MS has also been extensively used to study interactions between proteins with nucleic acids, including activation of transcription factors.362−371 Other notable applications are the characterization of ligands with weak affinity such as metals299,371−376 or carbohydrates.285,377 For proteins with multiple distinct ligand binding sites, it has been shown that by performing a concentration series it is possible to characterize individual interactions.264,378 The strength of HDX-MS to provide detailed insight into such a wide range of interactions continues to drive its use to study increasingly complex biological systems.
3.2. Pulsed Labeling HDX-MS and Protein Folding Studies
The HDX-MS described so far has been primarily continuous-labeling experiments in which the protein is exposed to D2O while structural fluctuations occur. Deuteration levels in continuously labeled proteins integrate the number of molecules that behave dynamically during the labeling period, which can range from milliseconds to days. In contrast, in pulsed-labeling experiments, the duration of D2O exposure is fixed and short compared to the to the time scale of structural changes. Short pulses of labeling can deuterate primarily unfolded regions, and this strategy has been shown to be applicable to all manner of unfolded proteins, including polypeptides that are in the process of conversion to/from the native structure through protein folding/unfolding as well as intrinsically disordered proteins (IDPs) (discussed in the next section). Pulsed-labeling provides structural snapshots that can reveal the fraction of each region of the protein that is folded at different stages in the folding process and reveal intermediate folding states (Figure 32).37,341,379−383 In a recent review on the folding of apomyoglobin, Nishimura highlights the power of HDX in combination with NMR to probe these intermediate folding states.384 Recent folding studies have detected a foldon pathway in cytochrome c consisting of units that exhibit the same energetic steps in both the forward and reverse reactions.385
Figure 32.

Pulse labeling HDX-MS was used to study the folding kinetics of cytochrome c. Fully deuterated unfolded protein was mixed with D2O from 5 to 506 ms to initiate folding. The samples were then immediately pulse labeled with H2O at pH 10.1 for 11 ms to rapidly label unstructured amides, followed by immediate quenching of the sample. The bimodal profiles during the folding process can be used to track the folding kinetics. The top and bottom panels are the controls for pulse labeling of the fully unfolded and folded states, respectively. Reproduced with permission from ref (380). Copyright 1997 American Chemical Society.
Other groups have employed pulsed HDX experiments to identify interaction sites between protein subunits or binding partners. Many of the original intact-level protein folding studies were performed with pulsed labeling.37,260,379 Short labeling times were shown to be more appropriate for revealing changes associated with protein interfaces.318,319 In more recent years, pulsed HDX methods have been used to study the interfaces within potassium channels386 and the interactions with lipoprotein lipase.387,388 Dornan et al.389 combine pulsed and continuous HDX experiments with electron microscopy to characterize the heterotrimeric PIKIIIα/TTC7B/FAM126A complex. By pulsed HDX, the authors determine the extent of secondary structure throughout the protein complex, finding that regions unobserved in the EM structure appear dynamic by HDX.389
Benhaim et al.390 used a pulsed HDX approach to monitor complex structural changes during the fusion activation of influenza hemagglutinin (HA) on the surface of intact infectious virions. Sequential local conformational changes in HA were tracked after activation to reveal two on-pathway intermediates during HA activation. The previous understanding of activation had been based on studies of the isolated soluble ectodomain of HA, and the study revealed how activation of the soluble HA ectodomain was distinct from that of the intact protein on the surface of the virion.
Pulsed HDX can provide critical structural information on the process of protein aggregation. Wang et al.391 monitored the oligomerization and accompanying structural changes in CsgE, which is thought to act as a chaperone-like subunit active during amyloid formation of the curli protein. They found evidence of at least three intermediate oligomerization states and observed a large structural rearrangement upon CsgE oligomerization. In a study on CsgA, the major component of curli protein, Wang et al. deciphered the effects of deamidation of CsgA on aggregation.392 Sabareesan and Udgaonkar393 tracked the aggregation of the wild-type prion protein (PrP) to the N-terminal region of the protein, consistent with positions of mutations known to increase aggregation. Finally, Renawala et al.394 studied calcitonin, an aggregation-prone therapeutic peptide hormone, to determine the effects of disulfide reduction on its aggregation kinetics. Interestingly, they found that reduced calcitonin aggregation involves different residues than wild-type (Figure 33), demonstrating the utility of pulsed HDX approaches to therapeutic protein development.
Figure 33.

Pulse labeling HDX-MS used to track the aggregation of calcitonin. Each spectrum shows the deuteration profile (after a 2 min pulse of D2O) starting from initial conditions (0 min) to the final time point at 1440 min. Populations I and II are clearly resolved and shift as the aggregates form over time. Reproduced with permission from ref (394). Copyright 2021 Elsevier.
Beyond providing structural and kinetic insight, pulse labeling has also been useful for characterization of protein ligand binding properties. By varying ligand concentration, it is possible to extract binding affinities395,396 and assess the effects of numerous ligands on protein stability.210,397 These approaches have paved the way to high throughput studies to identify inhibitors against therapeutic protein targets.249
Several methodological and data processing advances in pulsed HDX have been introduced in recent years. Pansca et al. mined existing pulsed HDX data from rapid folding experiments and found a correlation between the backbone rigidity of a sequence and early protection from HDX during folding.398 Raimondi et al., introduced EFoldMine, an early folding prediction tool built on similar pulsed HDX-NMR data of early folding states.399 The authors validated the EFoldMine predictions with pulsed HDX-MS experiments. On the experimental side, Makepeace et al. demonstrate that a 2.5 s deuterium pulse in the ligand bound and unbound states followed by intact protein MS can be used to estimate the number of exchange-competent amides in each state.266 Finally, Tsirigotaki et al. described a pulsed HDX approach to study the non-native and disordered translocation-competent states of secretory proteins to identify short, structured regions.400
3.3. HDX to Monitor Intrinsic Disorder
Intrinsically disordered proteins (IDPs) and regions (IDRs) are estimated to make up 30–50% of the human proteome.401 IDP conformations span a range of disorder from fully extended chains to mostly folded proteins that have persistent disordered loops (Figure 34).401−403 As the level of disorder increases and compaction decreases, the structural fluctuations exhibited by IDPs increase. This results in IDPs populating conformational ensembles instead of exhibiting a single well-defined tertiary structure. Because IDPs demonstrate extensive conformational plasticity, they frequently play roles in biology that demand structural rearrangements that folded proteins cannot undergo. As such, IDPs aid in cell signaling, cytoskeletal architecture, and other cellular physiology roles.401−403
Figure 34.
Schematic representation of the continuum model of protein structure. The color gradient represents a continuum of conformational states ranging from highly dynamic, expanded conformational ensembles (red) to compact, dynamically restricted, fully folded globular states (blue). Dynamically disordered states are represented by heavy lines, stably folded structures as cartoons. A characteristic of IDPs is that they rapidly interconvert between multiple states in the dynamic conformational ensemble. In the continuum model, the proteome would populate the entire spectrum of dynamics, disorder, and folded structure depicted. Reproduced with permission from ref (403). Copyright 2014 American Chemical Society.
Interest in IDPs has risen sharply in recent decades, particularly in the areas of pathological aggregation and liquid–liquid phase separation.401,402,404 Even with this added interest, the structural study of disordered regions and proteins has remained a challenge as techniques to resolve and model disordered regions have fallen behind advances in structure-resolving techniques such as cryoEM.405,406 Of the available structural characterization techniques, HDX-MS is particularly well-suited to the study of IDPs because it can detect and resolve changes in protection resulting from weak or transient hydrogen bonding without perturbing the IPD structural ensemble.407
3.3.1. Theoretical Considerations
Although HDX-MS experiments can offer valuable insight into IDPs, the detection of transient hydrogen bonding remains challenging. Very short time points (milliseconds) are often required to access relevant amide exchange kinetics for IDPs, a significant departure from the seconds–hours long time points used for typical HDX-MS samples. Furthermore, quantifying the level of transient hydrogen bonding in IDPs and IDRs requires an accurate intrinsic exchange rate (kch) for the fully unstructured state for comparison. In the case of IDPs, it is challenging to accurately create and measure the exchange of a truly unstructured reference.407 The kch estimates are commonly calculated from values obtained from extensive NMR studies of unprotected amides. In this method, the intrinsic exchange rate for a particular amide is predicted by accounting for its position in the sequence and all relevant solution conditions.47,48 Despite the sophisticated calculations established for accurately predicting kch, some studies have noted instances where the measured exchange rate of IDPs and IDRs is faster than the prediction66,408−411 (Figure 35A,B). These findings have led to updates in the tools used for predictions of kch for proteins as outlined recently by Nguyen et al.49 Walters introduced a similar empirical approach for accurately calculating local energetic differences in comparative HDX-MS studies without the need for estimated exchange rates.412
Figure 35.

HDX-MS to measure residual structure in peptides. The exchange of two peptides compared to the predicted exchange rate is shown in (A) and (B). The predicted rates from kch are shown in dashed lines. (C,D) Exchange rates for predigested peptides (open circles) and peptide from intact protein (triangles) are compared directly to assess transient structure in the protein. Reproduced with permission from ref (66). Copyright 2017 American Chemical Society.
In an effort to alleviate possible mispredictions in kch, Al-Naqshabandi and Weis66 developed a different approach to quantify the level of transient structure in IDPs. The authors introduce a strategy for empirically determining intrinsic exchange rates of IDPs using predigested peptides from the proteins of interest as references. By measuring protection in both the full protein and the peptides from the predigested protein, it is possible to directly calculate the protection ratio within each region independent of theoretical predictions. Although this value resembles a protection factor, the authors caution that thermodynamic interpretations applicable to protection factors may not hold in the case of protection ratios. The authors demonstrate the efficacy of this approach on the fully disordered ACTR and the molten globule CBP, finding that some predigested peptides exchange on a faster time scale than the experimental peptides (Figure 35C,D).
3.3.2. Methods for IDPs
With IDRs and IDPs, at physiological conditions, amide exchange happens on the millisecond time scale.48,69,408,413,414 Perturbing the experimental conditions by lowering the pH or temperature to slow the kch offers a way to probe the fast kinetics in both structured and unstructured proteins69 but will likely reduce the physiological relevance of the study. The millisecond exchange time scale of IDPs and the desire to remain near physiologically relevant conditions has prompted the use of millisecond HDX. Traditional quench-flow systems are well-suited for tracking fast dynamics in proteins and have been employed for probing fast time scales for HDX-MS with high precision.319,380,381,383,415 Simpler rapid mixing systems have also been described which are capable of probing time scales relevant to many disordered regions.408 Deuterium exposure time in quench-flow or rapid mixing devices is varied by changing either the flow rate or the length of the deuteration loop between the D2O/protein mixing stage and the introduction of quench in a second mixing chamber. Offline quench-flow HDX-MS sample preparation does not directly interface with the mass spectrometer; samples may be prepared and stored at −80 °C for later analysis. Keppel et al.416 made use of such an apparatus in their work on EGFR and HER3 to access time points from 108 ms to 2.033 s. The authors also performed manual HDX-MS to extend their observations from 5 s to 2 h, showing the utility of this method for bridging time scales relevant to IDP exchange kinetics. More recently, chip-based rapid mixing devices have been fabricated using thiol–ene photochemistry that offer a convenient approach for rapid mixing and quenching with a time scale of 140 ms to 1.1 s.417 Related to rapid mixing devices, automated sample handing systems have employed mixing strategies to also sample down the time ranges approaching 100 ms, adding another approach for probing fast time scales202 (see automation section).
A strategy that uses nested capillaries was also shown to be effective for monitoring exchange on very short labeling times. Time-resolved electrospray ionization (TRESI)-HDX, developed by Wilson and Konnerman in 2003,418 consists of a nested capillary mixing system connected to a variable volume reaction chamber. The effluent from the reaction chamber is quenched and sprays directly into the mass spectrometer ESI source (i.e., an online method). This setup allows investigation of time scales ranging from 42 ms to 8 s, making it a powerful tool to resolve fast kinetics. More recent implementations have incorporated a pepsin digestion chamber to enable bottom-up analyses.322 The main downside to spraying the quenched sample directly into the mass spectrometer is the potential for nonvolatile buffer components to interfere with MS analysis (see section 2.7). TRESI-HDX has recently been utilized to resolve fast kinetics on native and phospho-tau419 and investigate the amyloidogenic shift attributed to tau phosphorylation.420 These studies were able to detect increased exposure of the hexapeptide two region in phospho-tau, a key region for pathological tau aggregation.
3.3.3. Localizing Disorder in Proteins
One of the most straightforward application of HDX-MS is the ability to readily identify disordered regions within proteins. This approach continues to be utilized to pinpoint disordered regions in isolated proteins416,421−424 and viral accessory proteins.425,426 HDX-MS was also able to identify Ana2 as an IDP427 and to confirm contraction is disordered in solution and thereby resolve a long-standing debate in the field.428 Killoran et al.429 used HDX-MS to assess the structure and dynamics of Cby, a wnt-signaling antagonist that had evaded previous structural characterization due to line broadening observed in NMR studies. In this case, the results allowed the authors to locate the boundaries of the Cby coiled-coil domain and to elucidate the role of the C-terminal disordered region in Cby solubility. HDX has also been effective for probing dynamics in large macromolecular complexes.389,430−433 In many cases, HDX-MS was used in combination with other biophysical and biochemical characterization techniques to determine complex stoichiometry and the overall molecular architecture. HDX has also become an important tool for the structural characterization of aggregation prone IDPs such as α-synuclein434 and antithrombin,435 to investigate the structural effects of PTMs known to increase aggregation,419,436 and to probe the influence of other protein domains on the aggregation properties.437
Identification of specific disordered segments has greatly aided efforts for full structural determination of proteins. Pantazatos et al.438 demonstrated the application of HDX-MS to localize disordered regions of proteins to guide re-engineered protein constructs to enhance crystallography. This proof-of-concept work on 24 proteins from Thermotoga maritima demonstrated that some disorder-depleted proteins can maintain the integrity of their folded domains and thereby be amenable for crystallography studies. While this strategy does not work for all proteins, Fowler et al. recently showed the applicability of this technique in the identification and deletion of disordered regions of P14KIIIβ to create a construct amenable for crystallography.439 They further demonstrated the role of HDX-MS in crystallography by comparing the dynamics of the wild-type P14KIIIβ and its disorder-depleted form, showing that the deuterium uptake for the remaining regions is unaffected, implying that the dynamics of the entire protein were not disturbed by the deletions.
HDX-MS has also been useful for resolving how residual structure of IDPs and IDRs are integral to their function and probe the influence of disordered regions on the other folded domains of the protein. Trabjerg et al.440 investigated the opposing biological activity of dimerized proNGF and dimerized NGF by HDX-MS and found that the pro- region of proNGF is unstructured and, interestingly, protects mature (folded) regions of proNGF from exchange. In a similar investigation of the impact an unstructured region can exert on a structured region, Clouser et al. found that the N-terminal disordered region of HSPB1 dimerizes and binds to a single groove in multiple orientation-specific conformations.340 Mutations in noncontiguous regions of the disordered domain influence the deuterium uptake observed elsewhere on HSPB1. Mysling et al.387 observed that the N-terminal disordered region of GPIHBP1 functions to promote an encounter complex that results in tighter binding between the folded domain of GPIHBP1 and the LPL homodimer. On a similar theme, Saikusa et al.441 used HDX-MS to study how histones H2A and H2B exhibit differences in dynamics between their wild type and disorder-depleted forms. The authors caution that deletion mutants should be characterized by HDX-MS to determine if deletion influences global protein structure or dynamics.
An array of recent studies has also shown that IDPs can be highly perturbed by mutations442,443 and post-translational modifications, particularly phosphorylation.420,444−446 IDPs generally exhibit disorder-to-order transitions upon binding,403 however, Kacirova et al. noted that phosducin actually did not show ordering upon binding its regulatory partner 14–3–3 protein.447 Papanastasiou et al. used similar methods to observe the difference between complement protein iC3b and its mature form C3b, finding that removing the immature portion of iC3b leads to a disorder-to-order transition, resulting in a structured CUB domain only observed in the mature C3b protein.448 Other HDX-MS studies of IDPs have disorder-to-order transitions upon binding ligands such a metals,376 small molecules,266,449 and lipid membranes.450
Hamdi et al. exemplified the investigation of disorder-to-order transitions in IDPs in their study of ripening proteins HvASR1 (barley) and TtASR1 (wheat).451 With a combination of Stoke’s radius, SAXS, CD, and HDX-MS, the authors found that the addition of glycerol, zinc, and TFE can increase order within the proteins. By HDX, the authors determined that the addition of TFE alone does not affect HDX, but either zinc alone or TFE and zinc together induce global reductions in HDX due to increased structure formation (Figure 36). The authors localized the area affected by the addition of TFE and zinc as residues 105–115 in HvASR1 and 104–113 in TtASR1, identifying these regions as bona fide molecular recognition elements and highlighting the applicability of HDX-MS for resolving factors that affect protein folding.
Figure 36.
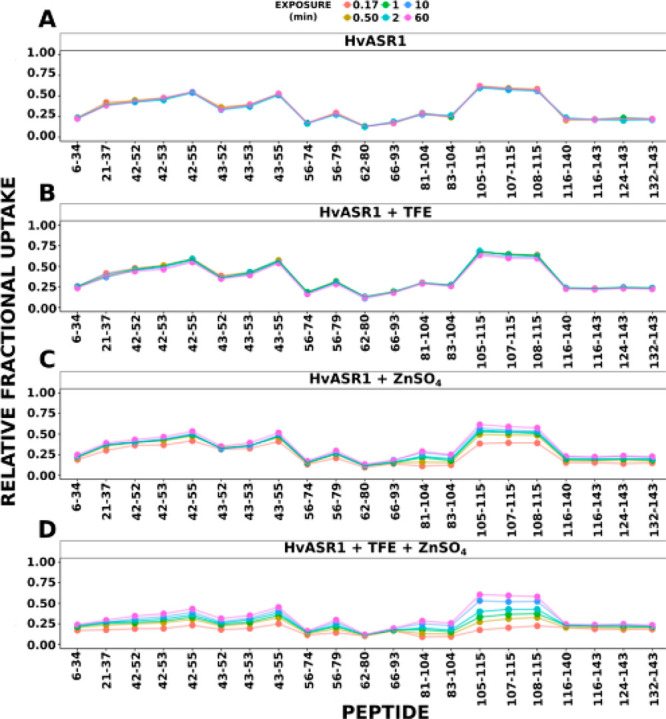
Deuterium uptake plots are shown for each peptide of unliganded HvASR1 (A), or bound to TFE (B), ZnSO4 (C), or both TFE and ZnSO4 (D). Points represented different time points of exchange denoted in the top of the figure. Figure adapted from ref (451).
HDX-MS is well-suited for tracking ordering associated with complex formation. Ramirez et al. characterized a molten globule unfolding intermediate of the obligate homodimer phosphofructokinase-2, which exhibits strong coupling between dissociation and unfolding.15 In a similar study, Dembinski et al. monitored the folding state of IκBα in a ternary complex with NFκB and found that IκBα begins to fold during the release of DNA. The authors resolved that two regions of IκBα remain partially unfolded in the ternary complex, which helps to explain line broadening observed in NMR experiments of the ternary complex.452 De Vera et al. examined another DNA binding complex composed of SRC-2 and the PPARγ/RXRα heterodimer and concluded that ligand and DNA cooperatively recruit the SRC-2 interaction domain to the heterodimer.453 Other groups have employed HDX to monitor domain-swapping of proteins such as FoxP, which functions as a domain-swapped dimer. Medina et al. found that there are several intermediate domain-swapped states of FoxP that vary in the degree of disorder they exhibit.367
IDPs behave distinctly from structured proteins, and understanding their response to the cellular environment (e.g., crowding) is necessary to understand their biochemistry. Rusinga and Weis characterized the effects of molecular crowders on the IDP ATCR to interpret structural changes, which could previously not be sufficiently explained by volume exclusion theory.454 Using predeuterated Ficoll as a crowding agent, they found that IDPs display complex behaviors in crowded environments that depend on the structure and concentration of the crowder. The concentration-dependent effects on secondary structure by crowded environments may play a functional role in modulating IDP activity.
3.4. Application to Membrane Proteins
Membrane proteins have historically been a particularly problematic class of proteins for structural studies. The native state of the protein requires stabilization of the membrane interacting portion, which introduces major challenges. Over the past decade, there have been several advances and workflows developed for HDX-MS that have enabled detailed studies of peripheral and integral membrane proteins as discussed in several recent reviews.17,455−457 Several systems have been analyzed in their active states through detergent solubilization, but for many systems it is generally preferable to incorporate the proteins into a more native-like membrane environment. In fact, several studies have observed differences in detergent solubilized vs membrane-incorporated integral membrane proteins.458−460 To this end, studies have been performed using artificial lipid membranes including liposomes,44,309,424,461,462 bicelles,463 Langmuir monolayers,464 lipid nanodiscs,17,465−467 and natural bacterial membranes.38 Beyond the effects of membrane presentation, recent studies have also revealed how just the lipid composition can have significant effects on membrane protein structure and dynamics (Figure 37).468,469
Figure 37.
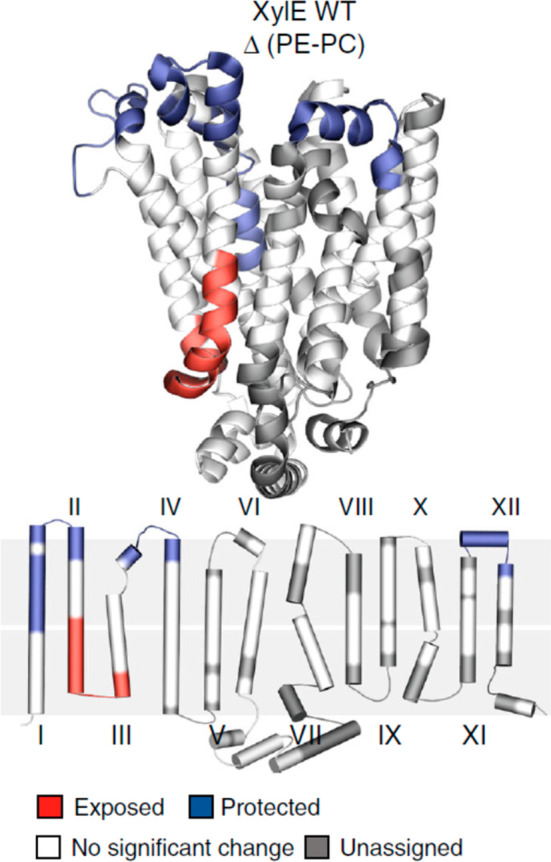
HDX-MS comparison of xylose transporter in nanodiscs with different lipid compositions. The lower depiction shows the transmembrane helices in relation to the two membrane leaflets (gray). Differences are color coded onto the crystal structure. Figure adapted from ref (468).
A major hurdle with any membrane protein study is the need for detergents and lipid components, which can both greatly complicate the HDX-MS analysis. While some detergents (e.g., Tween, Triton, CHAPS) are very problematic because they produce strong and overshadowing signals in MS that swamp the signal from peptides, less problematic detergents such as DDM, DM, or LMNG can be used at low concentrations without being detrimental to the LC-MS analysis.135,199,463,470−472 For the more problematic detergents, techniques have been established for postquench removal prior to LC-MS.473 Luckily, phospholipids do not present nearly the challenge that some detergents do and can mostly be removed prior to MS or sprayed directly into the instrument without much decrease in performance.474−476 An additional challenge for membrane proteins is the inability to achieve full sequence coverage, particularly for the transmembrane regions. Combinations of proteases and the use of stationary phases with shorter alkyl chains for chromatography have been reported to help maximize coverage for more elaborate studies of membrane proteins.136
Lipid nanodiscs have offered a way to prepare membrane integral proteins using a membrane scaffold protein that encapsulates a lipid bilayer and have proven to be highly useful for HDX-MS analysis of proteins in their native membrane environment.465,466,477 An additional advantage of the nanodisc platform is that it enables the customization of the lipid content. After deuterium labeling, removal of the phospholipid components can be accomplished through zirconium oxide depletion,465 which can be performed in a column format.204 The presence of the scaffold protein presents a challenge as the digested fragments contribute to spectral complexity, although control experiments of empty nanodiscs can reduce complexity by providing a library of scaffold protein signals that can be used to filter experiments on loaded nanodiscs.474 Another approach is to use biotinylated scaffold protein, which is then depleted during the postquench sample workup.461 A major downside to lipid nanodiscs is that the lipid–protein ratio is fixed; should a membrane protein need to change conformation (and therefore the amount of space it occupies in the disc of lipids) or insert new or more regions into the lipids, this is generally inhibited by the scaffold protein. For this reason, liposomes are preferred wherein there is no fixed lipid:protein ratio and proteins are free to change shape and size as they would normally do in a native membrane environment.
Thanks to all the developments in methodology, HDX-MS has facilitated more meaningful investigations into many membrane spanning systems including, to name a few, transporters,133,459−461,478−482 ion channels,133,483 apoptotic protein assembly,484 viral glycoproteins309,314,390,485,486 and matrix proteins,487,488 pore forming toxins,489 ATP synthase,44 receptor complexes,463,471,490,491 and enzymes.466 HDX-MS has also been used to probe peripheral membrane proteins in the context of their native environments such as drug metabolizing enzymes at the lipid membrane surface492−494 and the interactions and structural transitions mediating lipoprotein assembly.474,476,495,496 One interesting finding from the slew of HDX-MS studies is that EX1 kinetics, while rare in most globular proteins, have been encountered quite frequently with membrane proteins. EX1 has been observed and characterized in several lipoproteins, interestingly even in the lipid-free state.474,476,497,498 Correlated motions occurring near or in the membrane have been noted for several systems, and by tracking the EX1 profiles the kinetics of the conformational transitions could be characterized.445,459−461,499,500 Further advances in HDX-MS may enable the study of membrane proteins in their fully native membrane environment in situ. This has been demonstrated in the study of the ADP/ATP carrier (bAnc1p) on intact mitochondria.501 However, the inherent complexity of most native membrane environments will likely require continued advances in HDX-MS techniques.
3.5. Application to Glycoproteins
3.5.1. Challenges Brought on with Glycosylation
Glycosylation is arguably one of the most complex types of post-translational modification that occurs on mammalian proteins. Proteins can be glycosylated at either asparagine side chains (N-linked) or serine/threonine side chains (O-linked) and can occur co- and post-translationally.502 Complexity stems from not just the potentially large carbohydrate structures attached at each site but also from inherent heterogeneity, both in terms of glycan occupancy at a specific site, as well as many diverse types of glycan structures being present within the same site.503,504 The interest in glycoprotein structural studies has risen in parallel with the emergence of biotherapeutics, the majority of which are based on the IgG framework that includes a conserved N-linked glycan that is critical for the structure and effector function of most therapeutics.505 The caveats attributed to the intrinsic heterogeneity and high degree of flexibility of the glycans has historically limited the structural analysis of glycoproteins. Despite many additional challenges with respect to sample preparation and data analysis,506 HDX-MS has provided an invaluable tool for analyzing the structure and interactions of complex glycoproteins.
A common theme among many HDX-MS studies of glycoproteins to date is suboptimal sequence coverage, which can result from poor digestion, poor detection, poor identification, or a combination of these factors. One of the core functions of glycosylation is to stabilize protein structure and shield a protein from proteolysis. Accordingly, glycoproteins are inherently more challenging to efficiently digest in bottom-up LC-MS approaches.507 This is especially true for proteins that contain dense clusters of glycosylation.508 Even when glycopeptides of suitable size for peptide-based LC-MS are generated, these peptides may not be readily detected during LC-MS. The glycan moieties are often very hydrophilic, and the glycopeptides may not be sufficiently hydrophobic to be effectively retained by the typical reverse phase columns used for LC-MS.509 Glycopeptides are also inherently difficult to detect because the intrinsic heterogeneity of the glycans splits the MS signal for a unique peptide among many “glycoforms” (same peptide with different glycan structures).169,504 Next, the identification of glycopeptides during the peptide mapping stage presents an additional hurdle for glycoprotein analysis by HDX-MS. When performing conventional proteomics with specific proteases (e.g., trypsin), a few b and y ions generated can be sufficient for peptide identification, but this is not always sufficient for confident assignment with the nonspecific proteases used for HDX-MS. In conventional CID MS/MS, the fragmentation is dominated by glycan neutral losses510 and fragmentation of the peptide backbone is modest. While the use of elevated collision energies with different stages of activation has been shown to be fruitful for obtaining peptide fragmentation useful for sequence identification,511,512 effective glycan assignments are best accomplished with softer fragmentation techniques. ETD and ECD allow for directed fragmentation at the peptide backbone while leaving the glycan intact, thus making the peptide ID straightforward.504 The widespread availability of electron-based fragmentation approaches to the general HDX-MS community has not only enabled single-residue analysis (section 2.6) but also facilitated studies that include glycopeptides in their analysis. Finally, it is also highly beneficial to have knowledge of the glycan structures associated with a glycoprotein before assigning peptides from HDX-MS digests. Parallel analysis with conventional proteomics approach to map PTMs in this case is highly informative for aiding in the assignment of glycopeptides.225 As a result of all these additional challenges, many studies have excluded glycosylated peptides in the HDX-MS analysis. Modern improvements have begun to reverse this general trend, but more work is clearly needed.
Another effect that must be considered in HDX-MS analysis of glycoproteins is the exchange at the glycan moieties themselves. While the many hydroxyl groups in a glycan exchange on a time frame that is much faster than what is monitored by HDX-MS, namely backbone amide hydrogen exchange,63 the acetamido groups on N-acetylhexosamines and sialic acids exchange more slowly513 and can retain deuterium under HDX-MS quench conditions.514 ETD analysis of deuterated glycopeptides confirmed that glycan chains retain significant levels of deuterium, enough to offset the deuterium content measured in glycopeptides.515 Interpretation of deuterium uptake of intact glycopeptides therefore needs to account for deuteration at both the peptide backbone amides and glycan acetamido groups. This caveat needs to be considered for comparative studies utilizing glycopeptides to make comparisons among different glycoforms of the protein.
3.5.2. Strategies to Make Glycoproteins More Amenable to HDX-MS
Despite the caveats of glycosylation, a number of studies have succeeded in obtaining meaningful HDX-MS via several strategies. Because glycoproteins can have variable degrees of glycan occupancy, some studies have relied on HDX-MS data from a nonglycosylated subpopulation. In such cases, the nonglycosylated peptides analyzed may only reflect a small fraction of the total population of the protein. In another strategy, some studies have opted to enzymatically trim the glycans prior to deuterium labeling168,516,517 or to use constructs that have site-specific mutants that knock out glycosylation to simplify the analysis.459,482 However, these removal strategies can be problematic, as many glycoproteins need to be studied in their fully glycosylated native state to understand their biologically relevant structure. One strategy that has garnered considerable attention is development of methods to perform exchange with the glycans intact but then remove them just before LC-MS analysis. This way the glycoprotein is labeled in its fully native glycosylated state and the problematic glycans do not interfere with the analytical stages of the analysis (Figure 38). To accomplish this, enzymatic removal of the glycans under quench conditions is required.
Figure 38.

HDX-MS of glycoproteins employing a postquench deglycosylation step. Glycoproteins are deuterium labeled in their fully native glycosylated state and after the quench step the glycopeptides generated through proteolysis are further digested using an acid-active endoglycosidase to remove the glycan and simply the LC-MS analyses of the deglycosylated peptides. Reproduced with permission from ref (520). Copyright 2020 American Chemical Society.
Nearly all commonly used endoglycosidases for glycoproteomics, such as peptide N-glycanase F (PNGaseF), do not retain activity at the low pH of HDX-MS quench conditions.518 However, PNGaseA, an endoglycosidase with similar activity, was shown to retain sufficient levels of activity at quench conditions to achieve effective deglycosylation of a deuterium labeled and pepsin digested glycoprotein.169,519 The ability to remove the glycan not only aids with identification and detection of glycosylated segments of the protein sequence but also resolves deuterium content associated with the glycan from the peptide backbone of the glycosylated regions. It is important to note that the action of PNGaseA not only removes the glycan but also converts the asparagine side chain to an aspartic acid. In cases of partial glycosylation occupancy, the resulting peptides for a glycosylated segment will be a mixture of both nonglycosylated (containing the native asparagine residue) and deglycosylated (resulting in glycosylated asparagine being converted to aspartic acid).169 Deamidation often alters the peptide’s hydrophobicity and often the nonglycosylated and deglycosylated species can be resolved chromatographically, thereby largely alleviating the caveat of the two species coeluting and overlapping in spectra to complicate interpretation of the deuterium content.520 One practical drawback to PNGaseA as an enzyme for postquench deglycosylation is the enzyme’s poor tolerance of denaturants and reducing agents (TCEP), which are both commonly employed in quench conditions to aid in unfolding and proteolytic digestion.
PNGaseH+, a more recently isolated endoglycosidase, was found to have deglycosidase activity similar to PNGaseA and with an optimal pH of around 2.6.521 When tested for HDX-MS applications, PNGaseH+ was found to be more robust than PNGaseA and was also able to tolerate moderate concentrations of TCEP (0.25M).520 Incorporation of PNGaseH+ into sample processing improved the sequence coverage of α-antichymotrypsin, namely by providing peptides spanning all 5 N-linked glycosylation sites. Additional homologues of PNGaseH+ have now been identified and characterized, several of which have desirable activities including broad glycan specificity. More importantly some of the newly identified variants were found to be more suitable for expression, purification, and immobilization onto a stationary support for inline deglycosylation.522 These reagents are expected to greatly simplify glycoprotein structural studies. Furthermore, by comparing the exchange of the glycosylated vs postquench deglycosylated peptides, it is possible to directly probe deuterium exchange attributed solely to the glycan. This may prove to be useful for probing the conformational dynamics of the glycan chains, for which very few approaches exist.
3.5.3. How Glycosylation Affects IgG Structure and Dynamics
Immunoglobulin g (IgG) is the primary platform for the majority of modern biopharmaceutics. It should therefore come as no surprise that it is by far the most studied glycoprotein by HDX-MS. The IgG is a 150 kDa dimeric molecule that has a conserved N-linked glycan within the Fc portion that is critical for the interactions with the immune system and the overall function of the antibody molecule.523 The first application of HDX-MS to examine IgG identified several areas in the vicinity of the conserved N-linked glycan in the CH2 domain whose dynamics are directly influenced by the presence of the glycan chain.524 The changes in dynamics were further investigated by looking at systematic modifications to the glycan structure that revealed the effects of galactosylation on the dynamics of the Fc region.525 More recent studies have examined changes in Fc dynamics attributed to mutations and post-translational modifications, including glycosylation, for a number of monoclonal IgG constructs.277,526−533 HDX-MS comparisons of the IgG glycoforms led to the consensus that larger glycans within the Fc leads to the increased conformational dynamics within the CH2 domain. An example of the profound effects attributed to different glycosylation across the entire IgG is shown in Figure 39.532 More targeted studies of glycoengineered forms of IgG showed that it is specifically the galactosylation of the 6′ mannose arm that influences both CH2 dynamics and function.534 This finding was remarkably consistent with a parallel study that noted the effect specifically of a α(2–3) linked sialic acid on the 6′ mannose arm of the IgG glycan in destabilizing the CH2 domain.530 A similar approach of comparing glycoforms was also used to understand the mechanism of action of the Fc-specific endoglycosidase EndoS2.535
Figure 39.
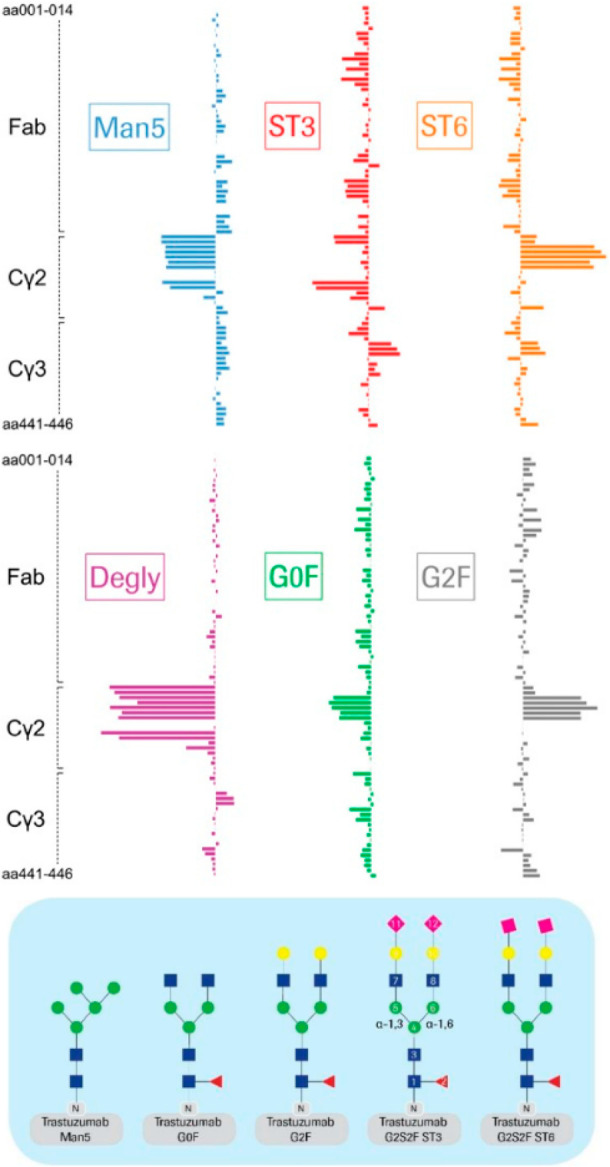
Difference in deuterium uptake for various glycoforms of trastuzumab (IgG) relative to reference material that is predominantly G0F with or without a single galactose (yellow circle, “G1F”). The structural effect of the glycosylation most readily evident in the Cγ2 domain. Figure adapted from ref (532).
Beyond the effects of mutations and modifications of the IgG itself, HDX-MS has been utilized to map interactions and track structural changes within IgG upon engagement of its many receptors, including FcγRIIIa,525 FcRn,536,537 and FcγIII.538 These studies have elucidated regions directly involved in the receptor interfaces, while also revealing some unexpected changes within the Fab region, that are suggestive of long-range allosteric interactions between the IgG Fc and Fab domains. Interestingly, a subset of the studies comparing glycoforms of IgG have noted similar long-range effects within the Fabs.525 The strongest evidence of this Fab–Fc interaction was from the direct observation of distal effects within the CH2 and CH3 regions in a human IgG1 upon antigen binding at the Fab.539 The allosteric link between Fab and Fc was also recently tested directly by looking at the influence of protein A binding to the Fc, which noted no significant alterations in the Fab regions, at least within the context of an antibody-protein A interaction.540 Overall, the HDX-MS studies to date on IgG provide strong evidence for a functional allosteric link between the Fc and Fab domains, which are likely context dependent. We expect that future HDX-MS studies looking at the interaction between antibodies, antigens, and immune complexes will elucidate such allosteric networks and examine their potential role in regulating the immune response.
3.5.4. Analysis of Highly Glycosylated Protein Systems
In addition to the analysis of IgG as just described, over the past decade HDX-MS has been applied to examine many other classes of glycoproteins including receptor complexes and serum glycoprotein. As we have noted, due to many of the challenges associated with glycopeptides, only a subset of the studies included analysis of the deuterium exchange at the glycosylated regions of the protein. Even with limited sequence coverage, HDX-MS has been a reliable tool for mapping binding interfaces and tracking structural changes upon protein–protein and protein–ligand interactions involving serum glycoproteins375,443,541−546 and receptors.432,466,517,547 Several studies have also used HDX-MS to assess the influence of glycosylation itself on the overall structure of serum glycoproteins with some interesting variability that reveals a complex relationship between a protein’s glycosylation state and its conformational dynamics. In some systems, HDX-MS showed mixed effects with some areas becoming stabilized and other regions showing increased accessibility upon glycan removal.548−551 In other glycoproteins, the only observed changes were increased dynamics through the protein upon glycan removal.541,552 Some studies observed only very minor changes in HDX kinetics despite known effects on overall thermal stability upon glycan removal.553 The available studies have thus far revealed a complex and very context-dependent relationship between glycosylation and structural dynamics.
Structural studies of viral surface glycoproteins have been a particularly widespread application where HDX-MS has shined in the past decade. Comparative HDX-MS studies of viral glycoproteins in different conformational states have been a highly informative approach for understanding their fundamental structure and function.300,390,485,486,516,554−559 As noted for many systems, the ability to detect long-range effects in proteins has also been observed in key viral glycoproteins.556,558 Most recently this was observed in the complex interaction between the ACE2 receptor and the SARS-CoV2 spike glycoprotein (Figure 40).558 The binding interaction between the receptor binding domain (RBD) of the spike glycoprotein not only elicits the expected changes directly at the binding site but also induces long-range effects all the way to the furin cleavage site. This link reveals how receptor binding primes the rest of the spike glycoprotein for further activation.
Figure 40.

Changes observed in the SARS-CoV-2 spike glycoprotein upon receptor binding. Beyond the stabilization seen at the receptor binding domain, there are also large changes observed within the furin cleavage site (672–690) and the stalk region (1175–1188). The long-range changes implicate the spike shifting to a primed state upon receptor binding, which leads to the activation of the protein to initiate membrane fusion. Figure adapted from ref (558).
The structural characterization of viral glycoproteins as they exist in the native form on a virion surface is of profound importance to the development of modern therapeutics and vaccine design strategies. To this end, HDX-MS has been an incredibly useful tool for assessing the conformational authenticity of engineered proteins designed to mimic the antigen on the surface of the virus and to be used for vaccine development.315,560−572 These studies have also shed critical insight on the most effective antibody epitopes and, more importantly, on the mechanisms of how neutralizing antibodies or targeted molecules are able to bind and neutralize viral surface glycoproteins.293,311,314,573−575
While many studies have focused on soluble versions of the viral glycoproteins, the general feasibility of using HDX-MS to examine the structures of viral glycoproteins within intact infectious virions has also been established.390,485,555,576 The ability to study these proteins within their native environments on the viral surface is a unique strength of structural mass spectrometry approaches. Future advances in methodology and instrumentation are expected to open more complex viral surface protein systems as well as proteins on other pathogens for in situ HDX-MS analysis.
3.6. Protein Therapeutic Development
Over the past two decades, HDX-MS has played an increasing role in the development of biotherapeutic proteins.249,272,295,524,577−582 Routine applications include epitope mapping (see section 3.1) and assessing how conformational states are impacted by mutations533,583 or chemical modifications such as oxidation, deamidation, glycation, or cross-linking,525,584−588 to name a few. Interestingly, it was recently shown that HDX-MS is highly predictive of the rates of deamidation occurring in proteins through long-term storage.589 Thanks to its versatility, HDX-MS has also been utilized for several other applications for therapeutic development. HDX-MS is an excellent tool to confirm the conformational authenticity and heterogeneity of designed protein therapeutics590 and antibody–drug conjugates.274,591 The technique has also proven effective for monitoring the structural effects of hydrocarbon stapling of therapeutic peptides592,593 or the addition of engineered disulfide bonds.247
Techniques capable of assaying the conformation of therapeutic proteins in their formulation buffers are highly valuable because they are often prepared at very high concentrations of protein (>100 mg/mL) and with multiple excipients. To enable HDX-MS to screen the effects of a wide range of excipients used to stabilize proteins, Nazari et al. developed a rapid postquench desalting step that removes components that would otherwise be problematic for LC-MS analyses.98 To address the challenges associated with analysis of highly concentration samples, Houde et al.594 utilized a dialysis coupled HDX platform. As an alternatively approach, Tian et al.595 were able to take advantage of the phase separation that occurs with highly concentrated samples to facilitate deuteration and isolation of the desired species. Another critical aspect of biotherapeutics is understanding their potential to form oligomers and aggregates. To this end, HDX-MS has been applied to monitor self-association of antibodies and reveal hot spots of aggregation.580,596−598
3.7. Other Applications of HDX-MS
The versatility of HDX-MS has also enabled detailed studies of conformational changes due to light. Light sensing proteins in their resting and activated forms have been studied to provide revolutionary insights into their mechanisms of photoactivation.599−608 For example, Etzl et al.605 were able to discern the changes in the structure of phytochrome-activated guanylate cyclase upon illumination with red light (Figure 41). This application is another illustration of the capability of HDX-MS to provide detailed insight into a system that cannot be analyzed by other means.
Figure 41.
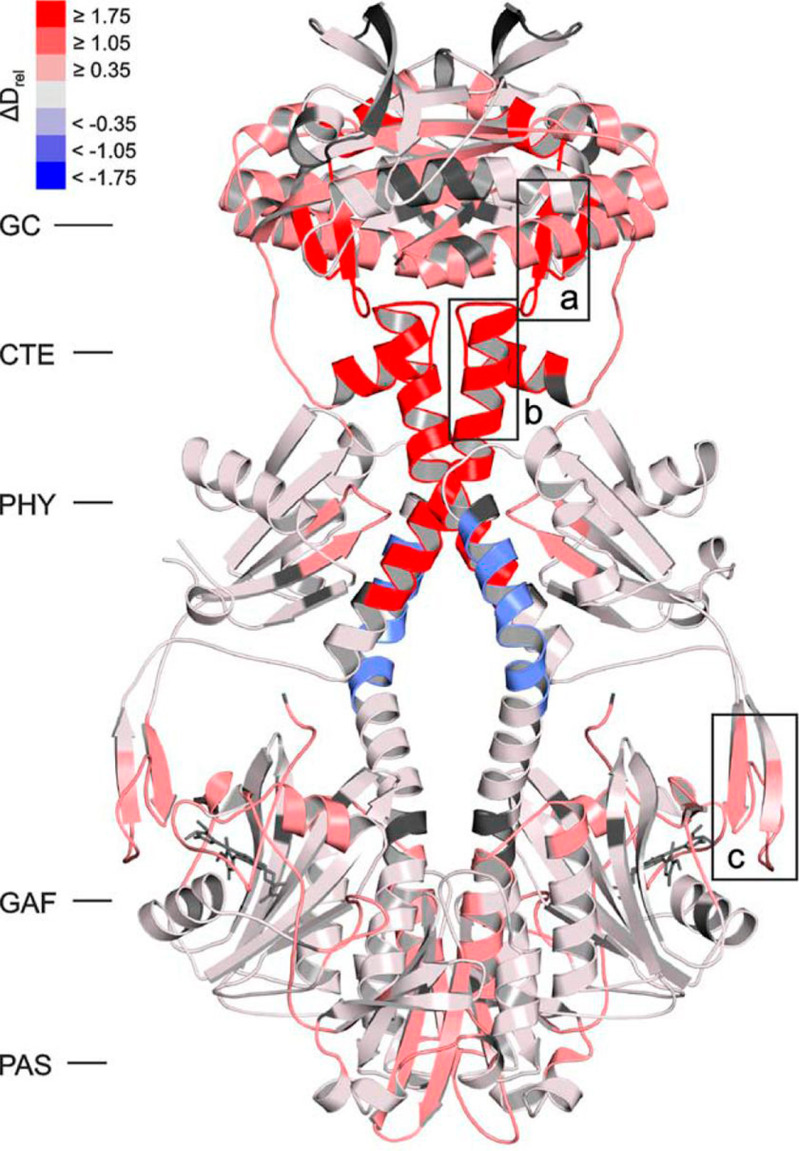
Structural changes in phytochrome-activated guanylate cyclase upon red light illumination detected by HDX-MS. Regions becoming more accessible or more protected relative to the inactive (nonilluminated state) are shown in red and blue notes, respectively. Figure adapted ref (605).
While most of the focus is aimed at the structure and dynamics of proteins in their solution state, there is a growing need for techniques capable of characterizing the conformations and dispositions of proteins in their solid or dried state. Li et al. originally demonstrated that incubation of lyophilized protein samples with D2O vapors can enable exchange of amides.609,610 The exchanged solids can then be resolubilized in quench buffer and processed by standard HDX-MS procedures to provide a readout of the conformational states of the protein. This solid-state HDX (ssHDX) (Figure 42) has since been used to track how various excipients,611−613 humidity during exchange,614 or different protein drying methods615 alter the exchangeability of proteins in an amorphous solid. The exchange profiles measured by ssHDX correlate with long-term stability measurements of the lyophilized protein,616,617 making it an excellent tool for assessing the stability of lyophilized formulations of biotherapeutics.618−620 An outstanding caveat of ssHDX is the limited understanding of the mechanism of exchange in the solid phase. It is known from early work that the exchange is not adequately described by the Linderstrom–Lang model, and the deuterium uptake profiles appear complex and multimodal.612 Recent efforts have used model systems to elucidate the factors that govern ssHDX,621−623 revealing that both secondary structure and matrix interactions in the amorphous solids contribute to the observed exchange behavior.
Figure 42.
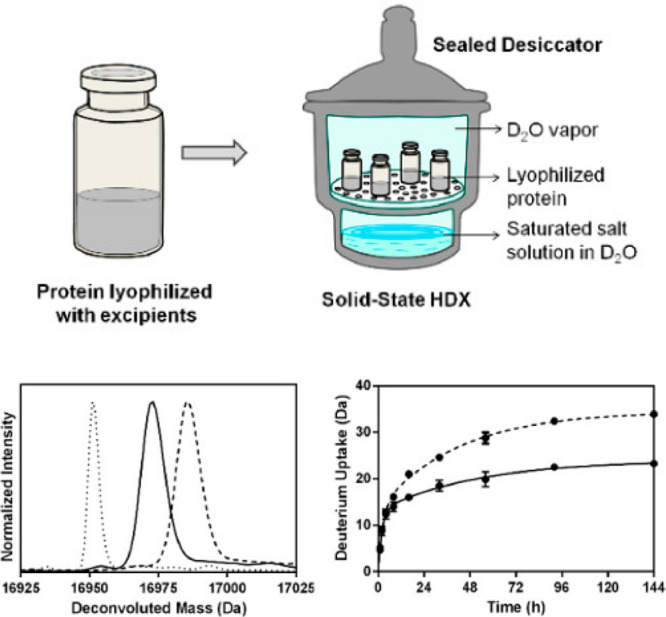
Overview of solid-state HDX-MS. Lyophilized proteins are incubated with D2O vapors to initiate exchange (top). Samples are then resuspended in quench buffer and processed by conventional HDX-MS approaches to measure deuterium uptake kinetics. Deconvoluted mass spectra for myoglobin are shown for the undeuterated (dotted line), dried with trehalose (solid line), and dried with sorbitol (dashed line) and the corresponding kinetic uptake curves (bottom). Figure is adapted from with permission from ref (697). Copyright 2015 MyJove.
While the vast majority of HDX-MS focuses purely on backbone amides, another realm of HDX-MS utilizes the exchangeability of the C2 proton on the histidine imidazole side chain (Figure 43).127 Miyagi et al. were able to utilize the measured C2 exchange rates across different pH conditions to accurately measure the pKa of each histidine in ribonuclease. By analyzing both the exchange rates and measured pKa of histidines, this approach was found to be useful for probing the local environment of histidines to provide structural insight into the protein.624 The main caveat of measuring C2 exchange is the slow exchange rates, which can require deuteration of the protein for several days at 37 °C. However, this slowed exchange also has some advantages. Back-exchange is not a complicating factor, which greatly simplifies sample handling. Furthermore, the deuterium label in the C2 position does not undergo scrambling during CID, making it easy to localize by conventional MS/MS methods.625 C2 exchange has since been used for mechanistic studies, elucidating the involvement of specific histidines in proteases,128,626 and was found to be an effective general approach for assessing protein folding stability and changes in stability upon ligand binding.627
Figure 43.

Overview of histidine C2 HDX-MS. The C2 proton can undergo exchange through the formation of a ylide intermediate. Reproduced with permission from ref (127). Copyright 2008 American Chemical Society.
HDX-MS has been useful for other macromolecules besides proteins and peptides. Solution HDX-MS with fast time scales has recently628 been capable of probing the accessibility of groups on nucleic acids. This pioneering study was able to examine the exchangeability of protons within nucleosides, whereas the phosphate backbone protons were exchanged away during the quench step. This study establishes the use of HDX-MS as a general technique for studying nucleic acid structure and dynamics. Taraban et al.629 showed the utility of solution HDX-MS for studying the solution conformation of dendrimers. By monitoring the amide groups within the dendrimers, they were able to resolve distinct conformers in solution using their amide groups as reporters. As noted in section 3.5.1, n-acetyl groups within glycans can also be directly probed by HDX-MS.514,515,553 These emerging applications illustrate the potential of HDX-MS as a tool for structural studies of biological macromolecules beyond proteins.
4. Precision and Reproducibility of HDX-MS Measurements
In response to the widespread use and growing interest for regulatory filings, numerous studies have focused on the analytical performance of HDX-MS. Particular attention has been directed toward the assessment of the repeatability and reproducibility of HDX-MS studies. HDX-MS experiments conducted within a single lab on the same day often report very high precision, as little as 1% variation in deuterium uptake; as one would expect, when measurements are made over an extended period of time or carried out in different laboratories, variability increases,213,630,631 as it would for any analytical technique. Automated sample handling systems offer one way to reduce some of the variables and have been implemented in efforts to increase reproducibility of HDX-MS.409,630 Cummins et al. demonstrated the potential of improving reproducibility by reporting impressively comparable HDX-MS data collected in two different laboratories.203 In this particular study, the reagents and analyses were rigorously harmonized, and identical automated systems were used to eliminate platform dependency and provide better control over each step in the HDX-MS experiment. Despite the success of this example, the disparate systems used across many laboratories performing HDX-MS renders this kind of harmonized approach untenable. As a result, numerous strategies have been developed to improve the reproducibility of HDX-MS measurements collected using distinct platforms and assess the level of variation one should expect.
A recent study carried out by the National Institute of Standards and Technology (NIST) compared HDX-MS data collected on a wide variety of platforms by a large group of unique investigators.631 Participants in this study were provided with a kit containing buffer components, a fragment of a standardized monoclonal antibody designed and produced by NIST, and explicit instructions. By providing participants with identical kits, NIST was able to assess the interlab variability of HDX-MS independent of buffer and sample conditions. The measurement of deuterium incorporation varied by as much as 9% between laboratories despite the use of numerous controls, including a system-specific back-exchange correction (Figure 44). As the downstream processes such as digestion and liquid chromatography were not standardized between laboratories in this study, much of the reported variability was attributed to those parameters. Robust comparative studies require careful matching of solution exchange conditions to minimize interlab variability.102,131,409,632
Figure 44.
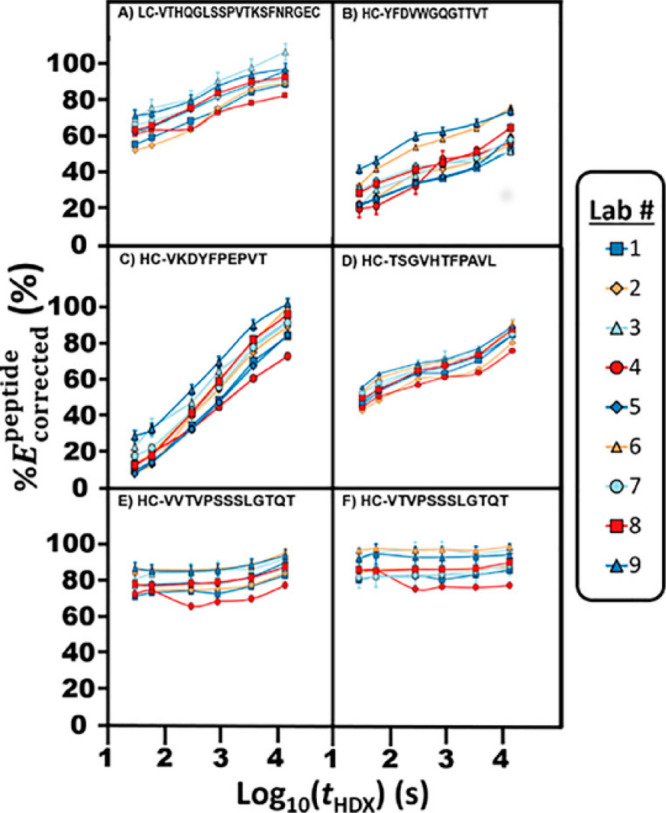
Various peptides from an interlab comparative HDX-MS study that controlled sample and labeling/quench buffers. Results for the same peptide from nine different laboratories are plotted to illustrate the interlab variability in the study. Reproduced with permission from ref (631). Copyright 2019 American Chemical Society.
There are many parameters that can potentially create variability within the final measurement of deuteration in HDX-MS.5,633 These variables arise from both the samples themselves (proteins, protein systems, inherent chemical exchange rates3) as well as the analytical conditions used for the measurements. Measurement variability can be attributed to a number of variables ranging from altered solution conditions, different sample handling, inherent differences between analytical platforms, and data processing approaches.5,76,213,219,409,632,633 Even small changes in these variables can affect the reproducibility of an HDX-MS experiment. In the next sections, we look more broadly into some sources of analytical variability in HDX-MS data and discuss efforts directed towards improving reproducibility for general HDX-MS measurements.
4.1. Changes in Digestion Profiles
Variation in the proteolytic digestion of proteins can be a problematic step in bottom-up HDX-MS affecting method reproducibility. While digestion variability does not alter the measured deuterium levels, it is a critical factor in comparative HDX-MS studies: if a peptide is present in one digestion but absent in another, it will hinder quantitative sequence-level comparisons. The efficiency of proteolytic digestion can be impacted by numerous parameters including pH, ionic strength, temperature, solvent composition, pressure, flow rate, digestion mode (online or offline), and of course the protease itself (e.g., refs (76, 138, 163, and 631)). Digestion conditions are often empirically optimized to improve sequence coverage for a particular system or even a particular protein (see section 2.3.1). While much attention has been directed toward improving digestion efficiency to maximize spatial resolution, considerably less attention has been directed toward understanding the reproducibility of digestion. Several studies145−147,160 have shown that intralaboratory reproducibility of digestion, where conditions are well controlled, often as a result of the comparisons being done on an identical instrumental setup, is high but not identical. It generally only takes a small number of replicates to identify all peptides that will be observed reproducibly between replicate digestions. One study634 showed that five replicate digestion and mapping experiments were sufficient to identify all peptides that were observed for a given digest condition.
In contrast, interlaboratory digestion reproducibility where only the identity of the enzyme is conserved does not fare as well as intralaboratory comparisons. The recent NIST interlaboratory comparison reported a surprisingly large disparity in the sets of peptides observed by each participating lab in that the number of reproducible peptides (those seen in each digestion) was low, even when the same protease was utilized.631 In fact, only two peptides could be compared across all 15 of the participating laboratories. These observations point out that standardization of conditions and reagents is critical for generating consistent digests so that peptides can be compared directly between sample sets. Using a standard peptide or well-established protein to quantitatively assess proteolytic efficiency is an effective way to achieve more reproducible digestion profiles among data sets and across different laboratories.635
4.2. Internal Standards and Ways to Improve Reproducibility
Analytical conditions may vary during HDX-MS measurements. As part of the recent growth of HDX-MS as an analytical tool in the biopharmaceutical industry, Houde et al.577 demonstrated how HDX-MS could be a valuable tool for establishing comparability in biopharmaceuticals, showing how HDX-MS was able to readily discern several modifications made to a reference protein. In that study, a major goal was to understand the variability of the method itself so that differences in products produced over extended periods of time or due to changes to manufacturing processes could be elucidated. The authors demonstrated more variability in data collected on nonconsecutive days than in data collected on the same day. Not surprisingly, other investigators have noted higher precision within data generated in a single day than in data generated over several days.185,203,630,636 Burkitt et al. attributed day-to-day variation to instrument conditions, inconsistent chromatography, and sample concentration.630 Other studies implicated ambient lab temperature and the shift in the pH of buffers over time as possible contributors to this kind of variability.203 Moreover, the bias inherent to different ionization methods and mass analyzers, often referred to as platform dependency, can further complicate the comparison of HDX-MS data.213 Day-to-day variability in HDX-MS must be known both to establish the range of expected variation in measurements and to facilitate efforts to minimize the variation. Despite the many compounding factors that make HDX-MS data challenging to reproduce over extended periods of time or in different laboratories, numerous controls have been developed to help mitigate these effects, including measurements of back-exchange during analysis and forward-exchange conditions during deuterium labeling.
4.3. Measuring Back-exchange Variation
An inherent variable common to any HDX-MS analysis is the extent of back-exchange that occurs throughout all downstream processes after the labeling step. Consistent conditions that yield reproducible back-exchange are crucial to the comparability of HDX-MS data. With bottom-up HDX-MS, reducing the back-exchange itself can limit variability. Many factors have been optimized to minimize back-exchange during digestion and separation71,176,182,184,186,637 and within the mass spectrometer (e.g., ion source temperature71,205). However, reducing overall back-exchange only limits this inherent variation, it does not alleviate the problem. The use of a well-matched fully deuterated protein sample as a means of more accurately accounting and correcting for back-exchange has been effective.28,76,125,638 By referencing the exchange of all time points to the fully deuterated standard, it is possible to dampen the effects of day-to-day variation and increase the precision of HDX-MS measurements.409 However, it was noted from well-harmonized comparisons that correction using a fully deuterated standard actually did not help improve interlab reproducibility,203 likely due to the larger number of variables one must account for between laboratories. Ultimately, for the most robust comparative studies, all samples should be prepared and analyzed with the same set of reagents and experiments should be performed as close in time as possible. To account for back-exchange variation, researchers have started to include standards capable of reporting on exact levels of back-exchange,409 which is now recognized as a metric that should be reported for HDX-MS studies.76
4.4. Variation in Percent Deuterium
The deuterium incorporation of peptides is directly influenced by the deuterium content in the surrounding solution. Variations in the percent deuterium (%D) of the labeling solution/buffer can occur from sample to sample either through dispensing error or by changes in the quality of the deuterium buffer (e.g., deuterium solutions open to air will exchange with atmospheric moisture and slowly lose %D over time). In an effort to reduce variation attributed to %D, Sheff et al. developed an isotopic approach for assessing and correcting for variations in solution dispensing.632 This approach works by adding known quantities of light and heavy isotopically labeled caffeine to the protein solution and deuterium labeling solution. When the two solutions are mixed for the exchange reaction, the observed ratio of light and heavy caffeine can be used to estimate the relative proportions of D2O and H2O in each sample (Figure 45). In this study, the incorporation of the caffeine dispensing standards was able to improve the precision of deuterium measurements across multiple replicates. While there is clearly utility in a dispensing standard, more attention has been directed toward the development of exchangeable standards capable of directly reporting on solution conditions.
Figure 45.

Light and heavy (13C) caffeine standards used to measure variations in percent deuterium. The ratio of the heavy and light caffeine that were spiked into the protein solution and deuterium labeling solution accurately report the percentage of deuterium during the labeling step and account for solution dispending variability. Reproduced with permission from ref (632). Copyright 2014 American Chemical Society.
4.5. Measuring Forward-exchange Variation
Variation in the conditions during deuterium labeling can dramatically affect the reproducibility of the HDX-MS experiment.5,71,76,632 Focusing on the solution reactions, changes in solution conditions (i.e., pH, temperature, concentrations of salts or organic solvent) will affect kch while simultaneously potentially perturbing protein structure. In the past decade, there have been several efforts to account for variations in solution conditions during the deuterium labeling step by incorporating an internal exchange reporter (IER). The IER is designed to exchange alongside the protein analyte and respond predictably to changes in solution parameters like pH, ionic strength, and temperature. Several groups have proposed the use of small unstructured peptides as IERs.98,131,409 The lack of higher-order structure for these peptides means the exchange behavior of these compounds is solely governed by kch (not by secondary, tertiary, or quaternary structure), thus directly reflecting solution conditions. Small peptide IERs only have a single slowly exchanging C-terminal amide that exchanges in a time regime relevant to most HDX-MS studies and retains deuterium during downstream processes. Furthermore, these IERs are efficiently ionized and sufficiently retained under most relevant chromatographic conditions, making them relatively simple to incorporate and detect using a wide variety of HDX-MS systems. Incorporation of these peptide IERs has been shown to enable more robust comparisons where solution conditions are not matched, for example, looking at the effects of high concentrations of chaotropes or buffer additives on the structural dynamics of proteins98,131,409 (Figure 46). One notable limitation of using the C-terminal amide as a reporter is that their exchange rates are not linear with respect to pH, rendering them unsuitable for accurately correcting offsets in solution pH.639 An alternative chemistry proposed as an exchange reporter has been the imidazolium scaffold. Murphree et al. described a series of benzimidazolium compounds that exchange solely at a single acidic carbon on a wide time scale that is relevant for HDX-MS studies.102 The exchange of the imidazolium group is purely base-catalyzed, so the exchange rate is linearly correlated with solution pH. While further developments are necessary to establish more reliable and versatile IERs, such standards may be valuable for addressing interday and even interlab variability in HDX-MS studies by providing users with a more robust method of detecting and correcting for variation in solution conditions during the deuterium labeling step.
Figure 46.

Deuterium uptake kinetics for the tripeptide YPI are shown in the presence of various buffer additives. The offset in the exchange profiles can be used to correct for the offset to the kch in the different solution, thereby enabling robust comparisons for proteins in the various solutions. Reproduced with permission from ref (131). Copyright 2017 American Chemical Society.
5. Analysis of HDX-MS Data
5.1. Software for HDX-MS Analysis
The data analysis stage has historically been a bottleneck for HDX-MS studies. The combination of peak integration, isotopic peak-picking, and deuterium incorporation analysis for several time points, replicates, and multiple conditions is time-consuming even for a single peptide. When considered for dozens to hundreds of peptides, the magnitude of data analysis can be daunting. For many years, the inability to cope with larger and larger data sets was one of the main factors limiting the size of proteins amenable to HDX-MS. A variety of software options have emerged and have been continually refined to accommodate the need for the analysis of larger HDX-MS data sets.640 Modern algorithms generally offer sophisticated peak picking, isotopic distribution fitting, and quality assessment of data, limiting the required manual analysis to predominantly curation and validation. Today there are several established HDX-MS software options, both academic and commercial. These software options have some subtle differences regarding their included features or their approach for picking species of interest to calculate deuterium incorporation from the isotopic profiles.
A summary of currently available software for various levels of analyzing and visualizing HDX-MS data is shown Table 1. Several of these programs include tools for extraction and integration of the data from raw MS acquisition files, while others rely on external programs for data extraction from the acquisition files and are therefore used primarily for deuterium calculation or visualization of the results. Some software only supports specific data file formats, limiting their use to certain instrument types and vendors. On this note, ion mobility-enabled LC-MS is increasingly being used to resolve overlapping peptides,215,641 but only a subset of the available software supports ion mobility-enabled data. Here we describe different algorithms and features of programs that have been developed to accelerate and enhance HDX-MS analysis.
Table 1.
| software | ref | data extraction | notes |
|---|---|---|---|
| deMix | Na et al. 201941 | yes | bimodal analysis |
| ExMS2 | Kan et al. 2019644 | yes | residue-level analysis from overlapping peptides, bimodal analysis |
| HDX-Analyzer | Liu et al. 2012689 | yes | statistical tools for HDX comparisons |
| HDXFinder | Miller et al. 2012654 | yes | web-based application for analysis and visuzliation |
| HeXicon2 | Lindner et al. 2014650 | yes | deconvolution and bimodal analysis possible |
| Mass spec Studio | Raval et al. 2021651 | yes | integrated structural mass spectromtery tools, statistics and visualization options |
| MassAnalyzer | Zhang et al. 2012409 | yes | extensive rate fitting for calculation of protection factors. |
| HX-Express2 | Guttman et al. 2013648 | fitting overlapped spectra, bimodal analysis | |
| HDX Match | Petrotchenko et al. 2015660 | top-down HDX-MS analysis | |
| QUDeX-MS | Salisbury et al. 2014655 | fitting isotopomers for analyzing ultrahigh mass resolution data. | |
| Commercial Options | |||
| Dynamx | Waters | yes | supports Waters data, includes several visualization tools |
| HDExaminer | Sierra Analytics | yes | supports multiple instrument vendors, various statistical and visualization tools |
| HDX Workbench | Omics Informatics, Pascal et al. 2012643 | yes | supports Thermo and Waters data, various statistical and visualization tools |
| HDX Workflow | Protein Metrics | yes | supports all instrument vendors, various visualization and statistical tools |
| Postprocessing Software | |||
| Deuteros 2.0 | Lau et al. 2020680 | visualization and statistical tools | |
| MEMHDX | Hourdel et al. 2016678 | visualization and statistical tools; web interface | |
| HaDeX | Puchla et al. 2020 | visualization and statistical tools; web interface or standalone | |
| HR-HDXMS | Gessner et al. 2017668 | improves resolution of HDX data | |
| HDX Modeler | Salmas et al. 2021670 | web-based tools to analyze overlapping peptides | |
| HDX Viewer | Bouyssie et al. 2019679 | structural visualization tools | |
| MS Tools | Kavan and Man et al. 2011672 | primarily HDX-MS visualization tools | |
| DECA | Lumpkin et al. 2019681 | statistical tools, back-exchange correction | |
| HD-eXplosion | Zhang et al. 2020683 | web-based, visualization tools | |
5.2. Feature Extraction and Fitting
Regardless of how a specific piece software works, there are a number of considerations that all analyses must take, whether done manually or by software.124 The first stage of data analysis is the identification of the observable peptides and the generation of a list of peptides to be analyzed. This is most often performed independently of the software used for integrating peaks and calculating deuterium uptake. Whatever the method for assigning peptides from tandem MS, it is important to have some form of quality control for the identification of the peptides, often by the scores and statistics generated from the peptide search.642 Once the peptide list is established, the next stage is integration of the isotopic peak profiles from all the deuterated LC-MS data. This is most commonly performed by using each peptide’s retention time and mass to extract specific portions of the LC-MS data.41,643,644 Although chromatographic reproducibility is highly sought after for HDX-MS data sets to minimize variability in back-exchange, there can still be minor variation in retention times across different LC-MS runs. Small differences in retention time can be accounted for by a restricted search to find the optimal integration ranges to use within each individual LC-MS run. Alternatively, retention time alignment algorithms based on spectral features have been developed and proven useful for aligning HDX-MS runs to achieve more accurate extraction of deuterium uptake.645
Of all the identified peptides in an HDX-MS data set, generally only a portion will provide clean, resolved peaks that are suitable for deuterium uptake analysis. The loss of some peptides from the identified list can arise from a variety of factors, including low signal-to-noise or spectral overlap once species are deuterated. While there have been efforts to measure accurate deuterium uptake information for complicated peptide signals,216,646−649 overlapping isotopic distributions are generally filtered off the peptide list through manual curation of the data. This manual assessment of what peaks are sufficiently “clean” introduces some level of bias that can contribute to slightly different sets of peptides obtained from different researchers. New software tools have begun to address this inherent source of bias by providing metrics on the suitability of a given peptide to be used for deuterium analysis and by making the process more automated. As an example, HeXicon uses a random-forest classifier using abundance, m/z values, retention time, and D-distribution to quantitatively select superior peptides.650 HX-PIPE (a recent tool in MassSpec Studio) combines analysis of resolved features within the MS1 spectra together with identifications from MS/MS data for an initial working set of peptides.651 The full scans are then further scrutinized to identify and omit any peptides that have potential spectral contamination from nearby overlapping features, thereby offering a rapid tool for curation of a peptide list to only the “clean” peptides (Figure 47).
Figure 47.

HX-PIPE combines both identification of clean features from the MS1 scans and peptide assignment from MS/MS data to accomplish peptide curation with less need for manual intervention. Reproduced with permission from ref (651). Copyright 2021 American Chemical Society.
A more rigorous approach to extraction of data from HDX-MS data sets is to identify all features within the MS1 spectra and track their deuterium uptake. Once the integrations are complete, the features are matched to a list of peptides to assign a peptide sequence to each integrated feature.409,650,652−654 The generation of an “overcomplete” set of candidate isotope patterns from spectral features is a notable advantage, given it essentially looks at everything and can help identify additional peptides that were absent from the curated list. Several programs can also match in silico predicted peptides to the observed features in the MS1 data.650,655,656 It is important that the user confirm any matches to predicted peptides. In silico calculation of peptide mass by the user without consulting tandem MS spectra can lead to erroneous peak assignment due to the potentially identical masses of different peptic fragments.657 The ability to output the unmatched features is highly valuable as it can be used for additional targeted MS/MS data to fill in the assignments, and thereby maximize the amount of information derived from HDX-MS data sets. One current limitation with peptide assignment is that most peptide identification tools have been established for general proteomics and are less well-suited for identification of peptides derived from nonspecific proteases. Therefore, there can potentially be highly useful peptides with clean signal in an HDX-MS data set that simply do not fragment and generate the type and quality MS/MS spectra required by the existing proteomics tools for confident identification.651
5.3. Deuterium Uptake Calculation
Once all mass spectra for a series of deuterium exchange time points are integrated, the next step is to quantify the deuterium uptake from the isotopic ratios. The mass envelopes will encompass the isotopic distribution that arises from natural abundance isotopes (e.g., 2H, 13C, 15N, 18O, and 34S), merged together with the extent of deuterium uptake. With most modern high-resolution mass spectrometers, the isotopic peaks can be well-resolved on a peptide or a small protein level, whereas isotopomers (e.g., the isotopic peak for a peptide with a single 13C vs a single 2H) are generally not mass-resolved (Figure 48). With ultrahigh resolution (>200 000), it becomes possible to separate isotopomers,649 for which new software tools have been established.655 Classically, the deuterium uptake is calculated from the geometric mean of the deuterated mass envelope (“centroid”) relative to the undeuterated spectra. This is a very simple and rapid way to quantify the deuterium content as a simple numerical value and is often sufficient for assessing protein structure and comparative studies to localize changes within proteins.
Figure 48.
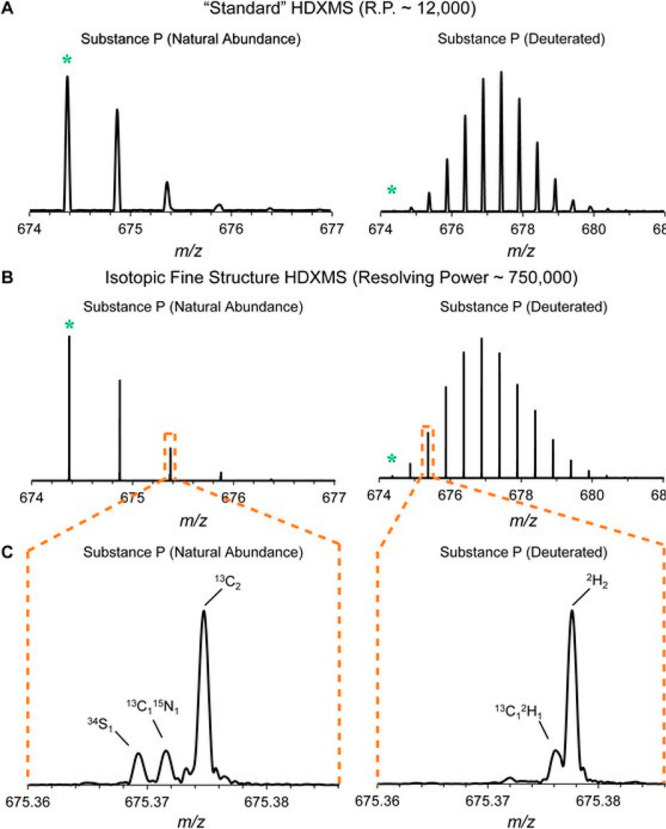
Spectra are shown for an undeuterated (left) and deuterium labeled (right) peptide at either a mass resolution of 12 000 (a) or 750 000 (b). At ultrahigh resolution, the isotopomers within each apparent isotopic peak become resolved (c). Figure adapted from ref (655).
A weakness of the centroid approach is that it sacrifices additional information that might be present within the shape and width of the deuterated mass envelope.658 Weis et al. demonstrated that including the measured distribution width of the isotopic envelopes over the time course could be used to detect the presence of multiple exchange profiles.28 Fitting the isotopic distribution to a theoretical binomial expansion provides a robust approach for calculating deuterium levels29 that is less prone to being offset by weak signal, spectral artifacts, and overlap of unrelated peptides.648,654,659 Even for a very clean spectrum of a peptide that is undergoing only EX2 kinetics, the isotopic distributions can reveal information about the fraction of amides in the peptide that have yet to exchange, which can aid in the analysis of overlapping peptides for improving spatial resolution.30 For these reasons, a theoretical fitting approach has been increasingly incorporated within many HDX-MS software algorithms.41,643,644,650−652,655,660,661
Fitting the deuterated isotopic profiles to theoretical distributions also offers a robust approach for analyzing and resolving when multiple conformers (species) are present. Using ideas developed very early in HDX-MS (e.g., refs (662−664)), including some primarily used for pulsed-labeling experiments, fitting a wide isotopic mass envelope to two distributions makes it possible to deconvolute the individual components within the spectra (Figure 49). To prevent overfitting, most algorithms first attempt to fit the deuterated mass envelope using a single distribution, and when that fails, they implement multiple distributions. For example, deMix uses a complex fitting algorithm if it suspects the data is inconsistent with a single isotopic distribution.41 Bimodal isotopic profiles can arise from EX1 kinetics, a mixture of EX1 and EX2 kinetics, or from a conformationally heterogeneous sample (outlined in section 1.2).665 By extracting information on the deuterium levels and relative intensities of both populations, it is possible to characterize the biological origin of the bimodal spectrum as well as any associated kinetic parameters. As noted earlier, it is also important to stress that bimodal spectra may arise from experimental artifacts, most notably from sample carryover,135,175,198 which can result in variable proportions of a low deuterated species. Therefore, appropriate controls should be included in data sets where bimodal analysis is to be implemented and interpreted.
Figure 49.
Deconvolution of multiple isotopic distributions using deMix. Isotopic distributions for three deuterium exchange time points are shown. The distributions could not be fit to a single distribution and thus were fit using two species (red and blue dashed lines). The w term in the inset shows the relative intensities of each species used in the fits. Figure adapted from ref (41).
5.4. Improving Spatial Resolution
Because the proteases for HDX-MS are relatively nonspecific, it is very common to obtain overlapping peptides that cover similar regions. Using several proteases in addition to or in combination with pepsin allows for even larger sets of peptides that not only improves coverage and redundancy but can also be used to calculate higher spatial resolution. Through a subtractive approach, higher resolution data can be gleaned from the combination of deuterium uptake across different sets of overlapping peptides. In the simplest case, if two peptides share a common starting or ending residue, the difference in deuteration level used to resolve the exchange at the overlapping region.646,666
More elaborate algorithms have since been developed that segmentally analyze portions of any overlapped peptides to calculate spatially resolved deuterium uptake.31,61,152,409,667 Bayesian models can account for uncertainties generated from overlapping peptides and experimental error by using residues-resolved exchange rates to simultaneously fit data from multiple samples in the HDX experiment.656 Another program, HR-HDXMS generates a “stability fingerprint” at the single amide level using thermodynamics and information from overlapping peptides to improve resolution668 (Figure 50). Skinner et al. introduced an approach to prediction and clustering of the most likely pattern of protection factors derived from all overlapping peptides.669 A recently established online Web server, HDXmodeller, uses a similar approach to improve HDX data spatial resolution from peptide-level deuterium uptake data.670
Figure 50.
HR-HDXMS uses a combination of overlapping peptides broken down into subfragments to estimate the thermodynamic properties with high spatial resolution, revealing a predicted stability fingerprint across the protein sequence. Figure adapted from ref (668).
With all approaches, the fitting of each amide’s exchange rate can be uncertain as the data is often insufficient to effectively restrict the fitting to a single confident solution. Both broad temporal sampling of the exchange kinetics and a large number of peptides with diverse overall coverage are needed to constrain the number of potential solutions and provide single residue or near-single residue exchange information. It is not just peptide redundancy but the overall peptide distribution throughout the sequence that has a big impact on constraining the fitting to a unique solution that is capable of accurately reporting amide-level exchange.670 For this reason, several groups utilize a combination of proteases to generate more diverse peptides for calculating residue-level HDX rates.123,667,668 Many of the algorithms necessitate critical assumptions; for example, most assume that all peptide exchange will follow pure EX2 kinetics. Some assume that the first two amino acids in a given peptide will not retain deuterium for HDX-MS, which is not always the case.63 Incorporation of the shape of the isotopic distribution across all time points can further help refine spatial resolution and alleviates the need to assume all exchange data follows strict EX2 kinetics.644,661 One remaining caveat to these subtractive analyses is that the levels of back-exchange can vary drastically and unpredictably, even for closely related peptides due to residual secondary structure and column interactions during LC-MS, which can offset the accuracy of subtractive analysis.637 The recent advances in subzero temperature LC-MS, which achieve very low levels of back-exchange, could drastically mitigate this source of uncertainty for subtractive analyses (see section 2.3.3).
Another approach for increased spatial resolution has been the incorporation of tandem mass spectrometry without inducing deuterium scrambling (see section 2.6). Analysis of ETD or ECD coupled HDX-MS/MS data is now supported on some software platforms including EXMS2,644 HDX-Workbench,643 HD Examiner (Sierra Analytics), and DynamX (Waters). Software tools have also been developed to aid with top-down HDX-MS analysis such as HDX Match.660 The ability of software to analyze deuteration in all resolved c/z ions greatly reduces the time required for a thorough analysis of MS/MS-enabled HDX-MS data.
5.5. Data Interpretation and Visualization
With the inherent need to rigorously apply statistical analyses to HDX-MS data sets, several tools have been developed that provide postprocessing options. The direct output from a bottom-up HDX-MS experiment is a deuterium uptake plot for each peptide analyzed. Because data sets can encompass hundreds of peptides, researchers have employed new visualization options for illustrating elaborate HDX-MS data sets. One of the earliest tools was to summarize the deuterium uptake in the form of a heat map.671 The exchange levels for various peptides are plotted using a color gradient across the sequence of the protein to comprehensively show data across all time points and reveal which regions of the sequence are protected672 (Figure 51).
Figure 51.
Heat maps show deuterium uptake represented by different colors for each peptide across the primary sequence of the protein. Each bar shows the data for different time points, and the upper and lower set of bars represent two conditions of the protein. Reproduced with permission from ref (672). Copyright 2011 Elsevier.
Another illustration to view comparative HDX-MS data sets is the use of a butterfly plot (or mirror plot as it is sometimes called), which shows deuterium uptake of all features simultaneously for a comparative HDX-MS data set (Figure 52A).577 Multiple time points can be plotted on the same graph, which allows for the tracking of changes that occur on the entire time scale of the experiment. Each side of the “mirror” will depict the experimental time course of a given protein sample. While subtle changes might be tough to detect on a traditional butterfly plot, plotting the differences (“difference plot”) allows for a more direct visualization of the areas and time points of difference on a given protein (Figure 52B). Butterfly plots are an easy way to compress all the exchange data (all time points) for two conditions into a small figure that nicely illustrates where any changes occur. A notable drawback is that a butterfly plot does not include information about the peptide’s location in the protein sequence, and it is often hard to see where coverage is lacking.
Figure 52.
Butterfly plots are used to compare two HDX experimental data sets. (A) The fraction deuterium uptake is plotted for all time points for all observable peptides for one protein sample (positive y-axis) and mirrored with another (negative y-axis). (B) The corresponding difference plot shows the subtracted result of the two data sets across all peptides and time points, revealing where changes are occurring. Reproduced with permission from ref (577). Copyright 2011 Elsevier.
Woods plots offer another option for depiction of large comparative data set. Peptides are depicted as horizontal bars showing their positions and lengths across the sequence. The positions on the y-axis reveal the difference in deuterium uptake between the two HDX-MS data sets.178 The Woods plot is notably strong at revealing information on coverage, level of deuteration, and peptide redundancy (Figure 53). If statistically relevant cutoffs are known, they can be depicted on the plot to make the visualization more robust. The Woods plots are similar to the difference plots used for butterfly plots but also include information about peptide length and position. As a drawback, information on uptake is generally limited to a single time point, so full data sets may require several plots to encompass all of the data.
Figure 53.
HDX-MS studies of the transporter XylE. Effects of mutation on the exchange of XylE are plotted on the crystal structure (left). Woods plots show differences in the exchange between WT and mutant for all peptides (right). Regions becoming destabilized (red) and stabilized (blue) in the mutant are plotted according to their difference in deuterium uptake. Figure adapted from ref (478).
Often HDX-MS analyses will make elaborate sets of comparisons between more than two states of a proteins (e.g., mutations, different ligands). Chiclet plots have been introduced to comprehensively compare HDX-MS data across a protein among more than two different protein states.450,673−675 Colors, usually in gradients, are used to show ranges of difference between two states for each peptide at each time point (Figure 54). One major advantage of chiclets is the ability to easily and quickly see differences and make comparisons across very large data sets (e.g.,674,676,677). A drawback of chiclet plots is the inability to see the actual extent of exchange for each peptide.
Figure 54.

HDX-MS comparisons of Escherichia coli DinB polymerase represented by Chiclet plots. Differences are shown between free polymerase (E), the pol-DNA complex (ED), and the ternary pol-DNA-dNTP complex (EDN) with either the correct or incorrect dNTP. Comparisons for each peptide at each time point across different comparative sets are shown in the plots with coloring from more exposed (red) to no difference (gray) to more protected (blue). Areas with missing data are shown in white. Figure adapted from Nevin et al.674
If a high-resolution structure is available, then a common approach for illustration and interpretation of the data is to show the changes in HDX-MS patterns across the structures (example shown in Figure 53). There are now several software tools for depiction of HDX-MS results on 3D protein structures, including web-based applications such as HX-Tools,672 MEMHDX,678 and most recently HDX-Viewer,679 which accept direct outputs from commercial HDX-MS software. With the need for more rigorous analysis, several groups have developed postprocessing software to provide visualization and statistical analysis that general workflows might not offer. For example, Deuteros 2.0, DECA, HaDeX, and HD-eXplosion are designed to function primarily with Waters’ DynamX data format680−683 but offer some flexibility. With these tools, data have already been extracted, fitted, and the resulting calculated deuterium outputs used as input for the programs to do statistical analyses or create visualization in various formats.
As one of the main applications of HDX-MS is comparative analysis, it is important to establish statistical metrics for assigning when differences are real or where two samples can be confidently deemed to be equivalent. Individual and global confidence intervals have been deployed to test significance between HDX-MS data sets (e.g., refs (185,577,597, and 684)). However, the error intervals estimated for these approaches do not account for type I or II error when evaluating significance. A hybrid approach introduced by Hageman et al. relies on replicate error to test reliability while also considering the magnitude of the difference in deuterium uptake to evaluate plausibility.685 The resulting “volcano plots” are 2-D scatterplots, with each point representing a time point of a particular peptide. The change in deuterium uptake is plotted against a p-value (y-axis) measured from replicates, with dashed boxes revealing significance cutoffs (Figure 55). By including a second criteria based on the difference in deuterium uptake on the x-axis, the relative magnitude of the change in uptake is considered for plausibility to determine potential significance. While the relative location on the protein sequence of the peptide is not shown, volcano plots offer a rapid means of detecting significant changes in deuterium uptake across all the peptides in the data set. Weis recently described the appropriate approaches for error propagation when processing HDX-MS data sets to evaluate statistically relevant differences for comparative data sets.686
Figure 55.
Examples of a volcano plots generated using Deuteros 2.0. The difference between two states of a protein are shown for all peptides and each time point. The x and y axis show the differences in deuterium uptake (ΔDU) and the p value, respectively. Quadrants that reflect statistically significant differences in exchange are shown in blue and red. Figure adapted from ref (680).
In terms of structural interpretation, there remains a limitation to deriving concrete 3-D structural information from HDX-MS data (i.e., exact details about atomic positions akin to NMR, crystallography, or cryoEM). While the predominant determinants of amide exchange are well-established, there is still an incomplete understanding of all the factors that contribute to observed amide exchange kinetics.26,687,688 Using HDX-MS in conjunction with molecular dynamics (MD) simulations has been a promising approach for acquiring more structural insight from HDX-MS. Vast ensembles of predicted structures or models can be validated by comparison to experimental HDX-MS profiles.689−691 Over the years, several tools have been developed for predicting HDX-MS properties from structures25,692 to expand the information content of HDX-MS studies for structural modeling,434 more accurate modeling of interaction surfaces,428,693 or understanding allosteric networks,694 even for complex systems such as membrane proteins.695,696 From these recent trends, we expect that MD and other structural computational approaches will play a more prevalent role in both the analysis and structural interpretation of HDX-MS data.697
6. Conclusions
What was once a highly specialized tool for studying simple proteins is firmly entrenched some 30 years after it was initially described. HDX-MS is now a widespread and commercialized tool that is routinely used to analyze some of the most complex protein systems, including membrane proteins, glycoproteins, and intrinsically disordered proteins. Over the past three decades, HDX-MS has revealed critical insight into protein dynamics, folding pathways, and ligand interactions, with new uses continually emerging that have expanded the utility of HDX-MS into applications even beyond proteins. Advances in sampling technology and software have streamlined HDX data analysis, making the study of giant molecular complexes feasible. At the same time, the increased accessibility of high-resolution mass spectrometers and commercial options for HDX-MS have lowered the barrier to entry for new users to apply HDX-MS to more diverse systems. With the increasing accessibility to electron-based fragmentation approaches and continued software advances, single residue resolution HDX-MS may become routine. Continued improvements in separation technologies and mass spectrometry sensitivity will undoubtably propel HDX-MS to address even more sophisticated and exciting biological questions to the point where studying protein dynamics and interactions within living cells might be possible by HDX-MS.
Acknowledgments
This work was supported in part by grants T32GM007750 (E.I.J.), R01-CA233978 (J.R.E.), R01-AI153191 (M.G.), and R01-GM127579 (M.G.) from the National Institutes of Health. We also thank David D. Weis, Lars Konermann, Elizabeth A. Komives, Thomas E. Wales, Thomas J. Jorgensen, Kasper D. Rand, John E. Burke, Jeffrey W. Hudgens, Ben T. Walters, and many other colleagues over the years for insightful discussions.
Glossary
Abbreviations
- CD
circular dichroism
- CE
capillary electrophoresis
- CID
collision induced dissociation
- DDA
data-dependent acquisition
- DDM
dodecyl β-d-maltopyranoside
- DIA
data-independent acquisition
- DM
decyl β-d-maltopyranoside
- DMSO
dimethyl sulfoxide
- ECD
electron capture dissociation
- ECR
electrochemical reduction cell
- EM
electron microscopy
- ESI
electrospray ionization
- ETD
electron transfer dissociation
- FT
Fourier transform
- HDX
hydrogen/deuterium exchange
- HILIC
hydrophilic interaction chromatography
- IDP
intrinsically disordered protein
- IDR
intrinsically disordered region
- IER
internal exchange reporter
- IM
ion mobility
- IgG
immunoglobulin gamma
- LC
liquid chromatography
- LMNG
lauryl maltose neopentyl glycol
- mAb
monoclonal antibody
- MALDI
matrix assisted laser desorption ionization
- MS
mass spectrometry
- NepII
nepenthesin II protease
- NMR
nuclear magnetic resonance
- PEEK
polyether ether ketone
- PF
protection factor
- PNGase
peptide N-glycanase
- PTM
post-translational modification
- Q-TOF
quadrupole time-of-flight
- SAXS
small-angle X-ray scattering
- ssHDX
solid-state HDX
- TCEP
tris-carboxyethyl phosphine
- TEA
triethanolamine
- TFE
trifluoroethanol
- TRESI
time-resolved electrospray ionization
- UPLC
ultrahigh pressure liquid chromatography
- UVPD
ultraviolet photodissociation
Biographies
Ellie I. James received her B.S. in Biochemistry from Western Washington University in 2018, where she was recognized as the Outstanding Graduate of the Biochemistry Department. She then moved to Seattle to attend the University of Washington in pursuit of a Ph.D. in Molecular Engineering. Her work in the laboratories of Dr. Abhinav Nath and Dr. Miklos Guttman (UW Medicinal Chemistry) aims to characterize the effect of modulators on IDP structural ensembles, particularly those relevant to pathological protein aggregation.
Taylor A. Murphree received his B.S in Chemistry from Western Carolina University in 2015. After graduating, he began working in Dr. Aaron Wright’s lab at Pacific Northwest National Laboratory (PNNL). While at PNNL, he focused on facilitating chemical proteomics through the design and synthesis of chemical probes capable of covalently modifying targets. He is currently working towards his Ph.D. under the guidance of Dr. Miklos Guttman at the University of Washington in Seattle. His research is focused on the development of internal standards for HDX-MS for more reliable comparative studies.
Clint Vorauer received B.A. degrees in Chemistry and German Studies from Whitman College. Prior to graduate school, Clint worked on the drug discovery team of the Seattle-based biopharmaceutical company, Omeros. He is currently working toward his Ph.D. at the University of Washington with Dr. Miklos Guttman to employ mass spectrometry-based approaches to map epitopes and study the higher-order structure of antibody–toxin interactions.
John R. Engen is the James L. Waters Chair in Analytical Chemistry and a Distinguished Professor of Chemistry and Chemical Biology at Northeastern University in Boston. He obtained his Ph.D. in Chemistry from the University of Nebraska—Lincoln in 1999 under the watchful eye of one of the pioneers of HDX MS, Prof. David L. Smith. His laboratory (neu.hxms.com) is focused on HDX MS and how it can be used to understand proteins of relevance to human disease; he has studied hundreds of proteins with HDX MS. Current areas of research focus are proteins related to apoptosis, kinases, large proteins and protein complexes, improving the analytical aspects of the technology including resolving power, and commercial availability.
Miklos Guttman is an Assistant Professor in the Department of Medicinal Chemistry at the University of Washington, Seattle (UW). He earned a B.S. in Chemistry from the University of California, Irvine, and completed his Ph.D. in Biochemistry at the University of California, San Diego, under the direction of Dr. Elizabeth Komives. He performed postdoctoral research at the University of Washington with Dr. Kelly Lee, focusing on structural mass spectrometry studies of viral surface glycoproteins. His group focusses on structural analysis of glycoprotein complexes using HDX-MS and development of novel approaches for analyzing protein glycosylation.
Author Contributions
§ E.I.J., T.M.M., and C.V. contributed equally.
The authors declare no competing financial interest.
References
- Hvidt A.; Linderstrøm-Lang K. The Kinetics of the Deuterium Exchange of Insulin with D2o. An Amendment. Biochim. Biophys. Acta 1955, 16, 168–169. 10.1016/0006-3002(55)90200-6. [DOI] [PubMed] [Google Scholar]
- Hvidt A.; Nielsen S. O.. Hydrogen Exchange in Proteins. In Advances in Protein Chemistry; Anfinsen C. B., Anson M. L., Edsall J. T., Richards F. M., Eds.; Academic Press, 1966; Vol. 21, pp 287–386. [DOI] [PubMed] [Google Scholar]
- Englander S. W.; Kallenbach N. R. Hydrogen Exchange and Structural Dynamics of Proteins and Nucleic Acids. Q. Rev. Biophys. 1983, 16, 521–655. 10.1017/S0033583500005217. [DOI] [PubMed] [Google Scholar]
- Englander S. W. Hydrogen Exchange and Mass Spectrometry: A Historical Perspective. J. Am. Soc. Mass Spectrom. 2006, 17, 1481–1489. 10.1016/j.jasms.2006.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engen J. R.; Wales T. E. Analytical Aspects of Hydrogen Exchange Mass Spectrometry. Annu. Rev. Anal. Chem. 2015, 8, 127–148. 10.1146/annurev-anchem-062011-143113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englander S. W. A Hydrogen Exchange Method Using Tritium and Sephadex: Its Application to Ribonuclease. Biochemistry 1963, 2, 798–807. 10.1021/bi00904a030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa J. J.; Richards F. M. An Experimental Procedure for Increasing the Structural Resolution of Chemical Hydrogen-Exchange Measurements on Proteins: Application to Ribonuclease S Peptide. J. Mol. Biol. 1979, 133, 399–416. 10.1016/0022-2836(79)90400-5. [DOI] [PubMed] [Google Scholar]
- Englander J. J.; Rogero J. R.; Englander S. W. Protein Hydrogen Exchange Studied by the Fragment Separation Method. Anal. Biochem. 1985, 147, 234–244. 10.1016/0003-2697(85)90033-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katta V.; Chait B. T. Hydrogen/Deuterium Exchange Electrospray Ionization Mass Spectrometry: A Method for Probing Protein Conformational Changes in Solution. J. Am. Chem. Soc. 1993, 115, 6317–6321. 10.1021/ja00067a054. [DOI] [Google Scholar]
- Zhang Z.; Smith D. L. Determination of Amide Hydrogen Exchange by Mass Spectrometry: A New Tool for Protein Structure Elucidation. Protein Sci. 1993, 2, 522–531. 10.1002/pro.5560020404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englander S. W.; Sosnick T. R.; Englander J. J.; Mayne L. Mechanisms and Uses of Hydrogen Exchange. Curr. Opin. Struct. Biol. 1996, 6, 18–23. 10.1016/S0959-440X(96)80090-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chance M. R.Mass Spectrometry Analysis for Protein-Protein Interactions and Dynamics; Wiley, 2008. [Google Scholar]
- Weis D. D.Hydrogen Exchange Mass Spectrometry of Proteins: Fundamentals, Methods, and Applications; Wiley, 2016. [Google Scholar]
- Trabjerg E.; Nazari Z. E.; Rand K. D. Conformational Analysis of Complex Protein States by Hydrogen/Deuterium Exchange Mass Spectrometry (Hdx-Ms): Challenges and Emerging Solutions. TrAC, Trends Anal. Chem. 2018, 106, 125–138. 10.1016/j.trac.2018.06.008. [DOI] [Google Scholar]
- Ramirez-Sarmiento C. A.; Komives E. A. Hydrogen-Deuterium Exchange Mass Spectrometry Reveals Folding and Allostery in Protein-Protein Interactions. Methods 2018, 144, 43–52. 10.1016/j.ymeth.2018.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oganesyan I.; Lento C.; Wilson D. J. Contemporary Hydrogen Deuterium Exchange Mass Spectrometry. Methods 2018, 144, 27–42. 10.1016/j.ymeth.2018.04.023. [DOI] [PubMed] [Google Scholar]
- Martens C.; Politis A. A Glimpse into the Molecular Mechanism of Integral Membrane Proteins through Hydrogen-Deuterium Exchange Mass Spectrometry. Protein Sci. 2020, 29, 1285–1301. 10.1002/pro.3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X. R.; Zhang M. M.; Gross M. L. Mass Spectrometry-Based Protein Footprinting for Higher-Order Structure Analysis: Fundamentals and Applications. Chem. Rev. 2020, 120, 4355–4454. 10.1021/acs.chemrev.9b00815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engen J. R.; Komives E. A. Complementarity of Hydrogen/Deuterium Exchange Mass Spectrometry and Cryo-Electron Microscopy. Trends Biochem. Sci. 2020, 45, 906–918. 10.1016/j.tibs.2020.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne J. S.; Mayne L.; Roder H.; Wand A. J.; Englander S. W. Determinants of Protein Hydrogen Exchange Studied in Equine Cytochrome C. Protein Sci. 1998, 7, 739–45. 10.1002/pro.5560070323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner J. J.; Lim W. K.; Bédard S.; Black B. E.; Englander S. W. Protein Hydrogen Exchange: Testing Current Models. Protein Sci. 2012, 21, 987–995. 10.1002/pro.2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truhlar S. M. E.; Croy C. H.; Torpey J. W.; Koeppe J. R.; Komives E. A. Solvent Accessibility of Protein Surfaces by Amide H/2h Exchange Maldi-Tof Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2006, 17, 1490–1497. 10.1016/j.jasms.2006.07.023. [DOI] [PubMed] [Google Scholar]
- Sowole M. A.; Alexopoulos J. A.; Cheng Y. Q.; Ortega J.; Konermann L. Activation of Clpp Protease by Adep Antibiotics: Insights from Hydrogen Exchange Mass Spectrometry. J. Mol. Biol. 2013, 425, 4508–19. 10.1016/j.jmb.2013.08.005. [DOI] [PubMed] [Google Scholar]
- Mohammadiarani H.; Shaw V. S.; Neubig R. R.; Vashisth H. Interpreting Hydrogen–Deuterium Exchange Events in Proteins Using Atomistic Simulations: Case Studies on Regulators of G-Protein Signaling Proteins. J. Phys. Chem. B 2018, 122, 9314–9323. 10.1021/acs.jpcb.8b07494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claesen J.; Politis A. Poppet: A New Method to Predict the Protection Factor of Backbone Amide Hydrogens. J. Am. Soc. Mass Spectrom. 2019, 30, 67–76. 10.1007/s13361-018-2068-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister R. G.; Konermann L. Challenges in the Interpretation of Protein H/D Exchange Data: A Molecular Dynamics Simulation Perspective. Biochemistry 2015, 54, 2683–92. 10.1021/acs.biochem.5b00215. [DOI] [PubMed] [Google Scholar]
- Konermann L.; Tong X.; Pan Y. Protein Structure and Dynamics Studied by Mass Spectrometry: H/D Exchange, Hydroxyl Radical Labeling, and Related Approaches. J. Mass Spectrom. 2008, 43, 1021–36. 10.1002/jms.1435. [DOI] [PubMed] [Google Scholar]
- Weis D. D.; Wales T. E.; Engen J. R.; Hotchko M.; Ten Eyck L. F. Identification and Characterization of Ex1 Kinetics in H/D Exchange Mass Spectrometry by Peak Width Analysis. J. Am. Soc. Mass Spectrom. 2006, 17, 1498–1509. 10.1016/j.jasms.2006.05.014. [DOI] [PubMed] [Google Scholar]
- Chik J. K.; Vande Graaf J. L.; Schriemer D. C. Quantitating the Statistical Distribution of Deuterium Incorporation to Extend the Utility of H/D Exchange Ms Data. Anal. Chem. 2006, 78, 207–214. 10.1021/ac050988l. [DOI] [PubMed] [Google Scholar]
- Kan Z. Y.; Walters B. T.; Mayne L.; Englander S. W. Protein Hydrogen Exchange at Residue Resolution by Proteolytic Fragmentation Mass Spectrometry Analysis. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 16438–43. 10.1073/pnas.1315532110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamuro Y. Determination of Equine Cytochrome C Backbone Amide Hydrogen/Deuterium Exchange Rates by Mass Spectrometry Using a Wider Time Window and Isotope Envelope. J. Am. Soc. Mass Spectrom. 2017, 28, 486–497. 10.1007/s13361-016-1571-1. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Yang L.; DeColli A.; Freel Meyers C.; Nemeria N. S.; Jordan F. Conformational Dynamics of 1-Deoxy-Xylulose 5-Phosphate Synthase on Ligand Binding Revealed by H/D Exchange Ms. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 9355–9360. 10.1073/pnas.1619981114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deredge D.; Wintrode P. L.; Tulapurkar M. E.; Nagarsekar A.; Zhang Y.; Weber D. J.; Shapiro P.; Hasday J. D. A Temperature-Dependent Conformational Shift in P38α Mapk Substrate-Binding Region Associated with Changes in Substrate Phosphorylation Profile. J. Biol. Chem. 2019, 294, 12624–12637. 10.1074/jbc.RA119.007525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papanastasiou M.; Mullahoo J.; DeRuff K. C.; Bajrami B.; Karageorgos I.; Johnston S. E.; Peckner R.; Myers S. A.; Carr S. A.; Jaffe J. D. Chasing Tails: Cathepsin-L Improves Structural Analysis of Histones by Hx-Ms. Mol. Cell Proteomics 2019, 18, 2089–2098. 10.1074/mcp.RA119.001325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraro D. M.; Lazo N. D.; Robertson A. D. Ex1 Hydrogen Exchange and Protein Folding. Biochemistry 2004, 43, 587–594. 10.1021/bi035943y. [DOI] [PubMed] [Google Scholar]
- Sivaraman T.; Robertson A. D. Kinetics of Conformational Fluctuations by Ex1 Hydrogen Exchange in Native Proteins. Methods Mol. Biol. 2001, 168, 193–214. 10.1385/1-59259-193-0:193. [DOI] [PubMed] [Google Scholar]
- Miranker A.; Robinson C. V.; Radford S. E.; Aplin R. T.; Dobson C. M. Detection of Transient Protein Folding Populations by Mass Spectrometry. Science 1993, 262, 896–900. 10.1126/science.8235611. [DOI] [PubMed] [Google Scholar]
- Gertsman I.; Komives E. A.; Johnson J. E. Hk97 Maturation Studied by Crystallography and H/2h Exchange Reveals the Structural Basis for Exothermic Particle Transitions. J. Mol. Biol. 2010, 397, 560–574. 10.1016/j.jmb.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanman J.; Lam T. T.; Emmett M. R.; Marshall A. G.; Sakalian M.; Prevelige P. E. Jr. Key Interactions in Hiv-1 Maturation Identified by Hydrogen-Deuterium Exchange. Nat. Struct. Mol. Biol. 2004, 11, 676–7. 10.1038/nsmb790. [DOI] [PubMed] [Google Scholar]
- Smirnovas V.; Kim J. I.; Lu X.; Atarashi R.; Caughey B.; Surewicz W. K. Distinct Structures of Scrapie Prion Protein (Prpsc)-Seeded Versus Spontaneous Recombinant Prion Protein Fibrils Revealed by Hydrogen/Deuterium Exchange. J. Biol. Chem. 2009, 284, 24233–41. 10.1074/jbc.M109.036558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na S.; Lee J. J.; Joo J. W. J.; Lee K. J.; Paek E. deMix: Decoding Deuterated Distributions from Heterogeneous Protein States Via Hdx-Ms. Sci. Rep. 2019, 9, 3176. 10.1038/s41598-019-39512-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y.; Zhao X.; Wang G.; Chen D. D. Y. Differential Hydrogen/Deuterium Exchange During Proteoform Separation Enables Characterization of Conformational Differences between Coexisting Protein States. Anal. Chem. 2019, 91, 3805–3809. 10.1021/acs.analchem.9b00558. [DOI] [PubMed] [Google Scholar]
- Murcia Rios A.; Vahidi S.; Dunn S. D.; Konermann L. Evidence for a Partially Stalled Γ Rotor in F(1)-Atpase from Hydrogen-Deuterium Exchange Experiments and Molecular Dynamics Simulations. J. Am. Chem. Soc. 2018, 140, 14860–14869. 10.1021/jacs.8b08692. [DOI] [PubMed] [Google Scholar]
- Vahidi S.; Bi Y.; Dunn S. D.; Konermann L. Load-Dependent Destabilization of the Γ-Rotor Shaft in Fof1 Atp Synthase Revealed by Hydrogen/Deuterium-Exchange Mass Spectrometry. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 2412–2417. 10.1073/pnas.1520464113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eigen M. Proton Transfer, Acid-Base Catalysis, and Enzymatic Hydrolysis. Part I: Elementary Processes. Angew. Chem., Int. Ed. Engl. 1964, 3, 1–19. 10.1002/anie.196400011. [DOI] [Google Scholar]
- Jensen P. F.; Rand K. D. Hydrogen Exchange. In Hydrogen Exchange Mass Spectrometry of Proteins 2016, 1–17. 10.1002/9781118703748.ch1. [DOI] [Google Scholar]
- Molday R. S.; Englander S. W.; Kallen R. G. Primary Structure Effects on Peptide Group Hydrogen Exchange. Biochemistry 1972, 11, 150–8. 10.1021/bi00752a003. [DOI] [PubMed] [Google Scholar]
- Bai Y.; Milne J. S.; Mayne L.; Englander S. W. Primary Structure Effects on Peptide Group Hydrogen Exchange. Proteins: Struct., Funct., Genet. 1993, 17, 75–86. 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D.; Mayne L.; Phillips M. C.; Walter Englander S. Reference Parameters for Protein Hydrogen Exchange Rates. J. Am. Soc. Mass Spectrom. 2018, 29, 1936–1939. 10.1007/s13361-018-2021-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englander S. W.; Poulsen A. Hydrogen-Tritium Exchange of the Random Chain Polypeptide. Biopolymers 1969, 7, 379–393. 10.1002/bip.1969.360070309. [DOI] [Google Scholar]
- Molday R. S.; Kallen R. G. Substituent Effects on Amide Hydrogen Exchange Rates in Aqueous Solution. J. Am. Chem. Soc. 1972, 94, 6739–6745. 10.1021/ja00774a029. [DOI] [Google Scholar]
- Kim P. S.; Baldwin R. L. Influence of Charge on the Rate of Amide Proton Exchange. Biochemistry 1982, 21, 1–5. 10.1021/bi00530a001. [DOI] [PubMed] [Google Scholar]
- Tüchsen E.; Woodward C. Mechanism of Surface Peptide Proton Exchange in Bovine Pancreatic Trypsin Inhibitor Salt Effects and O-Protonation. J. Mol. Biol. 1985, 185, 421–430. 10.1016/0022-2836(85)90413-9. [DOI] [PubMed] [Google Scholar]
- Perrin C. L. Proton Exchange in Amides: Surprises from Simple Systems. Acc. Chem. Res. 1989, 22, 268–275. 10.1021/ar00164a002. [DOI] [Google Scholar]
- Connelly G. P.; Bai Y.; Jeng M.-F.; Englander S. W. Isotope Effects in Peptide Group Hydrogen Exchange. Proteins: Struct., Funct., Genet. 1993, 17, 87–92. 10.1002/prot.340170111. [DOI] [PubMed] [Google Scholar]
- Eriksson M. A.; Härd T.; Nilsson L. On the Ph Dependence of Amide Proton Exchange Rates in Proteins. Biophys. J. 1995, 69, 329–339. 10.1016/S0006-3495(95)79905-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoffersen M.; Bolvig S.; Tüchsen E. Salt Effects on the Amide Hydrogen Exchange of Bovine Pancreatic Trypsin Inhibitor. Biochemistry 1996, 35, 2309–2315. 10.1021/bi951711q. [DOI] [PubMed] [Google Scholar]
- Liepinsh E.; Otting G. Proton Exchange Rates from Amino Acid Side Chains— Implications for Image Contrast. Magn. Reson. Med. 1996, 35, 30–42. 10.1002/mrm.1910350106. [DOI] [PubMed] [Google Scholar]
- Fogolari F.; Esposito G.; Viglino P.; Briggs J. M.; McCammon J. A. Pkashift Effects on Backbone Amide Base-Catalyzed Hydrogen Exchange Rates in Peptides. J. Am. Chem. Soc. 1998, 120, 3735–3738. 10.1021/ja963133m. [DOI] [Google Scholar]
- Hitchens T. K.; Bryant R. G. Pressure Dependence of Amide Hydrogen-Deuterium Exchange Rates for Individual Sites in T4 Lysozyme†. Biochemistry 1998, 37, 5878–5887. 10.1021/bi972950b. [DOI] [PubMed] [Google Scholar]
- Althaus E.; Canzar S.; Ehrler C.; Emmett M. R.; Karrenbauer A.; Marshall A. G.; Meyer-Bäse A.; Tipton J. D.; Zhang H.-M. Computing H/D-Exchange Rates of Single Residues from Data of Proteolytic Fragments. BMC Bioinformatics 2010, 11, 424. 10.1186/1471-2105-11-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajoddin N. N.; Konermann L. Analysis of Temperature-Dependent H/D Exchange Mass Spectrometry Experiments. Anal. Chem. 2020, 92, 10058. 10.1021/acs.analchem.0c01828. [DOI] [PubMed] [Google Scholar]
- Hamuro Y. Tutorial: Chemistry of Hydrogen/Deuterium Exchange Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2021, 32, 133–151. 10.1021/jasms.0c00260. [DOI] [PubMed] [Google Scholar]
- Martin R. B. O-Protonation of Amides in Dilute Acids. J. Chem. Soc., Chem. Commun. 1972, 793–794. 10.1039/c39720000793. [DOI] [Google Scholar]
- Coales S. J.; E S. Y.; Lee J. E.; Ma A.; Morrow J. A.; Hamuro Y. Expansion of Time Window for Mass Spectrometric Measurement of Amide Hydrogen/Deuterium Exchange Reactions. Rapid Commun. Mass Spectrom. 2010, 24, 3585–3592. 10.1002/rcm.4814. [DOI] [PubMed] [Google Scholar]
- Al-Naqshabandi M. A.; Weis D. D. Quantifying Protection in Disordered Proteins Using Millisecond Hydrogen Exchange-Mass Spectrometry and Peptic Reference Peptides. Biochemistry 2017, 56, 4064–4072. 10.1021/acs.biochem.6b01312. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Balsbaugh J. L.; Gao S.; Ahn N. G.; Klinman J. P. Hydrogen Deuterium Exchange Defines Catalytically Linked Regions of Protein Flexibility in the Catechol O-Methyltransferase Reaction. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 10797–10805. 10.1073/pnas.1917219117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheff J. G.; Rey M.; Schriemer D. C. Peptide–Column Interactions and Their Influence on Back Exchange Rates in Hydrogen/Deuterium Exchange-Ms. J. Am. Soc. Mass Spectrom. 2013, 24, 1006–1015. 10.1007/s13361-013-0639-4. [DOI] [PubMed] [Google Scholar]
- Goswami D.; Devarakonda S.; Chalmers M. J.; Pascal B. D.; Spiegelman B. M.; Griffin P. R. Time Window Expansion for Hdx Analysis of an Intrinsically Disordered Protein. J. Am. Soc. Mass Spectrom. 2013, 24, 1584–1592. 10.1007/s13361-013-0669-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englander S. W.; Mayne L.; Bai Y.; Sosnick T. R. Hydrogen Exchange: The Modern Legacy of Linderstrøm-Lang. Protein Sci. 1997, 6, 1101–1109. 10.1002/pro.5560060517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters B. T.; Ricciuti A.; Mayne L.; Englander S. W. Minimizing Back Exchange in the Hydrogen Exchange-Mass Spectrometry Experiment. J. Am. Soc. Mass Spectrom. 2012, 23, 2132–2139. 10.1007/s13361-012-0476-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington A. K.; Robinson R. A.; Bates R. G. The Ionization Constant of Deuterium Oxide from 5 to 50°. J. Phys. Chem. 1966, 70, 3820–3824. 10.1021/j100884a011. [DOI] [Google Scholar]
- Krȩżel A.; Bal W. A Formula for Correlating Pka Values Determined in D2o and H2o. J. Inorg. Biochem. 2004, 98, 161–166. 10.1016/j.jinorgbio.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Glasoe P. K.; Long F. A. Use of Glass Electrodes to Measure Acidities in Deuterium Oxide1,2. J. Phys. Chem. 1960, 64, 188–190. 10.1021/j100830a521. [DOI] [Google Scholar]
- Covington A. K.; Paabo M.; Robinson R. A.; Bates R. G. Use of the Glass Electrode in Deuterium Oxide and the Relation between the Standardized Pd (Pad) Scale and the Operational Ph in Heavy Water. Anal. Chem. 1968, 40, 700–706. 10.1021/ac60260a013. [DOI] [Google Scholar]
- Masson G. R.; Burke J. E.; Ahn N. G.; Anand G. S.; Borchers C.; Brier S.; Bou-Assaf G. M.; Engen J. R.; Englander S. W.; Faber J.; Garlish R.; Griffin P. R.; Gross M. L.; Guttman M.; Hamuro Y.; Heck A. J. R.; Houde D.; Iacob R. E.; Jorgensen T. J. D.; Kaltashov I. A.; Klinman J. P.; Konermann L.; Man P.; Mayne L.; Pascal B. D.; Reichmann D.; Skehel M.; Snijder J.; Strutzenberg T. S.; Underbakke E. S.; Wagner C.; Wales T. E.; Walters B. T.; Weis D. D.; Wilson D. J.; Wintrode P. L.; Zhang Z.; Zheng J.; Schriemer D. C.; Rand K. D. Recommendations for Performing, Interpreting and Reporting Hydrogen Deuterium Exchange Mass Spectrometry (Hdx-Ms) Experiments. Nat. Methods 2019, 16, 595–602. 10.1038/s41592-019-0459-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoesmith D. W.; Lee W. The Ionization Constant of Heavy Water (D2O) in the Temperature Range 298 to 523 K. Can. J. Chem. 1976, 54, 3553–3558. 10.1139/v76-511. [DOI] [Google Scholar]
- Lim W. K.; Rösgen J.; Englander S. W. Urea, but Not Guanidinium, Destabilizes Proteins by Forming Hydrogen Bonds to the Peptide Group. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 2595–2600. 10.1073/pnas.0812588106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venable J. D.; Okach L.; Agarwalla S.; Brock A. Subzero Temperature Chromatography for Reduced Back-Exchange and Improved Dynamic Range in Amide Hydrogen/Deuterium Exchange Mass Spectrometry. Anal. Chem. 2012, 84, 9601–9608. 10.1021/ac302488h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson M. J.; Harkewicz R.; Hodge E. A.; Vorauer C.; Palmer J.; Lee K. K.; Guttman M. Simple Platform for Automating Decoupled Lc–Ms Analysis of Hydrogen/Deuterium Exchange Samples. J. Am. Soc. Mass Spectrom. 2021, 32, 597–600. 10.1021/jasms.0c00341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe Y.; Ueda T.; Iwashita H.; Hashimoto Y.; Motoshima H.; Tanaka Y.; Imoto T. Effect of Salt Concentration on the pka of Acidic Residues in Lysozyme. J. Biochem. 1995, 118, 946–952. 10.1093/jb/118.5.946. [DOI] [PubMed] [Google Scholar]
- Critchfield F. E.; Johnson J. B. Effect of Neutral Salts on Ph of Acid Solutions. Anal. Chem. 1959, 31, 570–572. 10.1021/ac50164a034. [DOI] [Google Scholar]
- Reineke K.; Mathys A.; Knorr D. Shift of Ph-Value During Thermal Treatments in Buffer Solutions and Selected Foods. Int. J. Food Prop. 2011, 14, 870–881. 10.1080/10942910903456978. [DOI] [Google Scholar]
- Padró J. M.; Acquaviva A.; Tascon M.; Gagliardi L. G.; Castells C. B. Effect of Temperature and Solvent Composition on Acid Dissociation Equilibria, I: Sequenced Psska Determination of Compounds Commonly Used as Buffers in High Performance Liquid Chromatography Coupled to Mass Spectroscopy Detection. Anal. Chim. Acta 2012, 725, 87–94. 10.1016/j.aca.2012.03.015. [DOI] [PubMed] [Google Scholar]
- Voinescu A. E.; Bauduin P.; Pinna M. C.; Touraud D.; Ninham B. W.; Kunz W. Similarity of Salt Influences on the Ph of Buffers, Polyelectrolytes, and Proteins. J. Phys. Chem. B 2006, 110, 8870–8876. 10.1021/jp0600209. [DOI] [PubMed] [Google Scholar]
- Kennedy C. Ionic Strength and the Dissociation of Acids. Biochem. Educ. 1990, 18, 35–40. 10.1016/0307-4412(90)90017-I. [DOI] [Google Scholar]
- Lown D. A.; Thirsk H. R.; Wynne-Jones L. Effect of Pressure on Ionization Equilibria in Water at 25°C. Trans. Faraday Soc. 1968, 64, 2073–2080. 10.1039/TF9686402073. [DOI] [Google Scholar]
- Carter J. V.; Knox D. G.; Rosenberg A. Pressure Effects on Folded Proteins in Solution. Hydrogen Exchange at Elevated Pressures. J. Biol. Chem. 1978, 253, 1947–1953. 10.1016/S0021-9258(19)62340-9. [DOI] [PubMed] [Google Scholar]
- Hamann S. Chemical Equilibria in Condensed Systems High Pressure. High Pressure Phys. Chem. 1963, 2, 131. [Google Scholar]
- Gayan E.; Condon S.; Alvarez I.; Nabakabaya M.; Mackey B. Effect of Pressure-Induced Changes in the Ionization Equilibria of Buffers on Inactivation of Escherichia Coli and Staphylococcus Aureus by High Hydrostatic Pressure. Appl. Environ. Microbiol. 2013, 79, 4041–4047. 10.1128/AEM.00469-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacs N. S.Liquid Phase High Pressure Chemistry; John Wiley & Sons, 1981. [Google Scholar]
- Quinlan R. J.; Reinhart G. D. Baroresistant Buffer Mixtures for Biochemical Analyses. Anal. Biochem. 2005, 341, 69–76. 10.1016/j.ab.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Balasubramaniam V. M.; Ting E. Y.; Stewart C. M.; Robbins J. A. Recommended Laboratory Practices for Conducting High-Pressure Microbial Inactivation Experiments. Innovative Food Sci. Emerging Technol. 2004, 5, 299–306. 10.1016/j.ifset.2004.04.001. [DOI] [Google Scholar]
- Manning G. S. The Molecular Theory of Polyelectrolyte Solutions with Applications to the Electrostatic Properties of Polynucleotides. Q. Rev. Biophys. 1978, 11, 179–246. 10.1017/S0033583500002031. [DOI] [PubMed] [Google Scholar]
- Illingworth J. A. A Common Source of Error in Ph Measurements. Biochem. J. 1981, 195, 259–262. 10.1042/bj1950259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rösgen J.; Pettitt B. M.; Perkyns J.; Bolen D. W. Statistical Thermodynamic Approach to the Chemical Activities in Two-Component Solutions. J. Phys. Chem. B 2004, 108, 2048–2055. 10.1021/jp036325u. [DOI] [Google Scholar]
- Harned H. S.; Owen B. B.. The Physical Chemistry of Electrolytic Solutions. Reinhold Publishing Corporation: 1950. [Google Scholar]
- Nazari Z. E.; van de Weert M.; Bou-Assaf G.; Houde D.; Weiskopf A.; Rand K. D. Rapid Conformational Analysis of Protein Drugs in Formulation by Hydrogen/Deuterium Exchange Mass Spectrometry. J. Pharm. Sci. 2016, 105, 3269–3277. 10.1016/j.xphs.2016.07.006. [DOI] [PubMed] [Google Scholar]
- Rusinga F. I.; Weis D. D. Automated Strong Cation-Exchange Cleanup to Remove Macromolecular Crowding Agents for Protein Hydrogen Exchange Mass Spectrometry. Anal. Chem. 2017, 89, 1275–1282. 10.1021/acs.analchem.6b04057. [DOI] [PubMed] [Google Scholar]
- Klotz I. M.; Frank B. H. Deuterium-Hydrogen Exchange in Amide N—H Groups1. J. Am. Chem. Soc. 1965, 87, 2721–2728. 10.1021/ja01090a033. [DOI] [PubMed] [Google Scholar]
- Pirrone G. F.; Wang H.; Canfield N.; Chin A. S.; Rhodes T. A.; Makarov A. A. Use of Maldi-Ms Combined with Differential Hydrogen–Deuterium Exchange for Semiautomated Protein Global Conformational Screening. Anal. Chem. 2017, 89, 8351–8357. 10.1021/acs.analchem.7b01590. [DOI] [PubMed] [Google Scholar]
- Murphree T. A.; Vorauer C.; Brzoska M.; Guttman M. Imidazolium Compounds as Internal Exchange Reporters for Hydrogen/Deuterium Exchange by Mass Spectrometry. Anal. Chem. 2020, 92, 9830–9837. 10.1021/acs.analchem.0c01328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutcheon G. A.; Parker M. C.; Moore B. D. Measuring Enzyme Motility in Organic Media Using Novel H-D Exchange Methodology. Biotechnol. Bioeng. 2000, 70, 262–269. . [DOI] [PubMed] [Google Scholar]
- Fasoli E.; Ferrer A.; Barletta G. L. Hydrogen/Deuterium Exchange Study of Subtilisin Carlsberg During Prolonged Exposure to Organic Solvents. Biotechnol. Bioeng. 2009, 102, 1025–1032. 10.1002/bit.22147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen B. B. Direct Measurement of the Primary, Secondary and Total Medium Effects of Acetic Acid. J. Am. Chem. Soc. 1932, 54, 1758–1769. 10.1021/ja01344a006. [DOI] [Google Scholar]
- Subirats X.; Rosés M.; Bosch E. On the Effect of Organic Solvent Composition on the Ph of Buffered Hplc Mobile Phases and the Pk a of Analytes—a Review. Sep. Purif. Rev. 2007, 36, 231–255. 10.1080/15422110701539129. [DOI] [Google Scholar]
- Rosés M.; Canals I.; Allemann H.; Siigur K.; Bosch E. Retention of Ionizable Compounds on Hplc. 2. Effect of Ph, Ionic Strength, and Mobile Phase Composition on the Retention of Weak Acids. Anal. Chem. 1996, 68, 4094–4100. 10.1021/ac960105d. [DOI] [PubMed] [Google Scholar]
- Sarmini K.; Kenndler E. Ionization Constants of Weak Acids and Bases in Organic Solvents. J. Biochem. Biophys. Methods 1999, 38, 123–137. 10.1016/S0165-022X(98)00033-5. [DOI] [PubMed] [Google Scholar]
- Yang J.-Z.; Xu W.-G. Medium Effect of an Organic Solvent on the Activity Coefficients of Hcl Consistent with Pitzer?S Electrolyte Solution Theory. J. Solution Chem. 2005, 34, 71–76. 10.1007/s10953-005-2073-0. [DOI] [Google Scholar]
- Marcus Y. Effect of Ions on the Structure of Water: Structure Making and Breaking. Chem. Rev. 2009, 109, 1346–1370. 10.1021/cr8003828. [DOI] [PubMed] [Google Scholar]
- Espinosa S.; Bosch E.; Rosés M. Retention of Ionizable Compounds on Hplc. 12. The Properties of Liquid Chromatography Buffers in Acetonitrile-Water Mobile Phases That Influence Hplc Retention. Anal. Chem. 2002, 74, 3809–3818. 10.1021/ac020012y. [DOI] [PubMed] [Google Scholar]
- Barbosa J.; Bergés R.; Sanz-Nebot V.; Toro I. Chromatographic Behavior of Ionizable Compounds in Liquid Chromatography. Part 2. Standardization of Potentiometric Sensors and Effect of Ph and Ionic Strength on the Retention of Analytes Using Acetonitrile–Water Mobile Phases. Anal. Chim. Acta 1999, 389, 43–52. 10.1016/S0003-2670(99)00132-4. [DOI] [Google Scholar]
- Inczédy J.; Lengyel T.; Ure A. M.; Gelencsér A.; Pure I. U. o.; Chemistry A.. Compendium of Analytical Nomenclature: Definitive Rules 1997; Blackwell Science, 1998. [Google Scholar]
- Prasannan C. B.; Artigues A.; Fenton A. W. Monitoring Allostery in D2o: A Necessary Control in Studies Using Hydrogen/Deuterium Exchange to Characterize Allosteric Regulation. Anal. Bioanal. Chem. 2011, 401, 1083–1086. 10.1007/s00216-011-5133-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efimova Y. M.; Haemers S.; Wierczinski B.; Norde W.; van Well A. A. Stability of Globular Proteins in H2O and D2O. Biopolymers 2007, 85, 264–273. 10.1002/bip.20645. [DOI] [PubMed] [Google Scholar]
- Krantz B. A.; Moran L. B.; Kentsis A.; Sosnick T. R. D/H Amide Kinetic Isotope Effects Reveal When Hydrogen Bonds Form During Protein Folding. Nat. Struct. Biol. 2000, 7, 62–71. 10.1038/71265. [DOI] [PubMed] [Google Scholar]
- Mandell J. G.; Falick A. M.; Komives E. A. Measurement of Amide Hydrogen Exchange by Maldi-Tof Mass Spectrometry. Anal. Chem. 1998, 70, 3987–95. 10.1021/ac980553g. [DOI] [PubMed] [Google Scholar]
- Pan J.; Han J.; Borchers C. H.; Konermann L. Hydrogen/Deuterium Exchange Mass Spectrometry with Top-Down Electron Capture Dissociation for Characterizing Structural Transitions of a 17 Kda Protein. J. Am. Chem. Soc. 2009, 131, 12801–8. 10.1021/ja904379w. [DOI] [PubMed] [Google Scholar]
- Amon S.; Trelle M. B.; Jensen O. N.; Jorgensen T. J. Spatially Resolved Protein Hydrogen Exchange Measured by Subzero-Cooled Chip-Based Nanoelectrospray Ionization Tandem Mass Spectrometry. Anal. Chem. 2012, 84, 4467–4473. 10.1021/ac300268r. [DOI] [PubMed] [Google Scholar]
- Walters B. T.; Mayne L.; Hinshaw J. R.; Sosnick T. R.; Englander S. W. Folding of a Large Protein at High Structural Resolution. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 18898–18903. 10.1073/pnas.1319482110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G.; Abzalimov R. R.; Bobst C. E.; Kaltashov I. A. Conformer-Specific Characterization of Nonnative Protein States Using Hydrogen Exchange and Top-Down Mass Spectrometry. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 20087–92. 10.1073/pnas.1315029110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slysz G. W.; Percy A. J.; Schriemer D. C. Restraining Expansion of the Peak Envelope in H/D Exchange-Ms and Its Application in Detecting Perturbations of Protein Structure/Dynamics. Anal. Chem. 2008, 80, 7004–7011. 10.1021/ac800897q. [DOI] [PubMed] [Google Scholar]
- Mayne L.; Kan Z.-Y.; Sevugan Chetty P. S.; Ricciuti A.; Walters B. T.; Englander S. W. Many Overlapping Peptides for Protein Hydrogen Exchange Experiments by the Fragment Separation-Mass Spectrometry Method. J. Am. Soc. Mass Spectrom. 2011, 22, 1898–1905. 10.1007/s13361-011-0235-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wales T. E.; Eggertson M. J.; Engen J. R. Considerations in the Analysis of Hydrogen Exchange Mass Spectrometry Data. Methods Mol. Biol. (N. Y., NY, U. S.) 2013, 1007, 263–288. 10.1007/978-1-62703-392-3_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoofnagle A. N.; Resing K. A.; Ahn N. G. Protein Analysis by Hydrogen Exchange Mass Spectrometry. Annu. Rev. Biophys. Biomol. Struct. 2003, 32, 1–25. 10.1146/annurev.biophys.32.110601.142417. [DOI] [PubMed] [Google Scholar]
- Hoofnagle A. N.; Resing K. A.; Ahn N. G. Practical Methods for Deuterium Exchange/Mass Spectrometry. Methods Mol. Biol. 2004, 250, 283–298. 10.1385/1-59259-671-1:283. [DOI] [PubMed] [Google Scholar]
- Miyagi M.; Nakazawa T. Determination of Pka Values of Individual Histidine Residues in Proteins Using Mass Spectrometry. Anal. Chem. 2008, 80, 6481–7. 10.1021/ac8009643. [DOI] [PubMed] [Google Scholar]
- Elferich J.; Williamson D. M.; David L. L.; Shinde U. Determination of Histidine Pk a Values in the Propeptides of Furin and Proprotein Convertase 1/3 Using Histidine Hydrogen-Deuterium Exchange Mass Spectrometry. Anal. Chem. 2015, 87, 7909–7917. 10.1021/acs.analchem.5b01721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ba̧chor R.; Rudowska M.; Kluczyk A.; Stefanowicz P.; Szewczuk Z. Hydrogen-Deuterium Exchange of A-Carbon Protons and Fragmentation Pathways in N-Methylated Glycine and Alanine-Containing Peptides Derivatized by Quaternary Ammonium Salts. J. Mass Spectrom. 2014, 49, 529–536. 10.1002/jms.3371. [DOI] [PubMed] [Google Scholar]
- Sowole M. A.; Konermann L. Effects of Protein–Ligand Interactions on Hydrogen/Deuterium Exchange Kinetics: Canonical and Noncanonical Scenarios. Anal. Chem. 2014, 86, 6715–6722. 10.1021/ac501849n. [DOI] [PubMed] [Google Scholar]
- Toth R. T.; Mills B. J.; Joshi S. B.; Esfandiary R.; Bishop S. M.; Middaugh C. R.; Volkin D. B.; Weis D. D. Empirical Correction for Differences in Chemical Exchange Rates in Hydrogen Exchange-Mass Spectrometry Measurements. Anal. Chem. 2017, 89, 8931–8941. 10.1021/acs.analchem.7b01396. [DOI] [PubMed] [Google Scholar]
- Sun S.; Zhou J.-Y.; Yang W.; Zhang H. Inhibition of Protein Carbamylation in Urea Solution Using Ammonium-Containing Buffers. Anal. Biochem. 2014, 446, 76–81. 10.1016/j.ab.2013.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisinger M. L.; Dörrbaum A. R.; Michel H.; Padan E.; Langer J. D. Ligand-Induced Conformational Dynamics of the Escherichia Coli Na+/H+ Antiporter Nhaa Revealed by Hydrogen/Deuterium Exchange Mass Spectrometry. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 11691–11696. 10.1073/pnas.1703422114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brezski R. J.; Jordan R. E. Cleavage of Iggs by Proteases Associated with Invasive Diseases: An Evasion Tactic against Host Immunity?. mAbs 2010, 2, 212–220. 10.4161/mabs.2.3.11780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamuro Y.; Coales S. J. Optimization of Feasibility Stage for Hydrogen/Deuterium Exchange Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2018, 29, 623–629. 10.1007/s13361-017-1860-3. [DOI] [PubMed] [Google Scholar]
- Möller I. R.; Slivacka M.; Hausner J.; Nielsen A. K.; Pospíšilová E.; Merkle P. S.; Lišková R.; Polák M.; Loland C. J.; Kádek A.; Man P.; Rand K. D. Improving the Sequence Coverage of Integral Membrane Proteins During Hydrogen/Deuterium Exchange Mass Spectrometry Experiments. Anal. Chem. 2019, 91, 10970–10978. 10.1021/acs.analchem.9b00973. [DOI] [PubMed] [Google Scholar]
- Yang M.; Hoeppner M.; Rey M.; Kadek A.; Man P.; Schriemer D. C. Recombinant Nepenthesin Ii for Hydrogen/Deuterium Exchange Mass Spectrometry. Anal. Chem. 2015, 87, 6681–6687. 10.1021/acs.analchem.5b00831. [DOI] [PubMed] [Google Scholar]
- Jones L. M.; Zhang H.; Vidavsky I.; Gross M. L. Online, High-Pressure Digestion System for Protein Characterization by Hydrogen/Deuterium Exchange and Mass Spectrometry. Anal. Chem. 2010, 82, 1171–1174. 10.1021/ac902477u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliakov A.; Jardine P.; Prevelige P. E. Hydrogen/Deuterium Exchange on Protein Solutions Containing Nucleic Acids: Utility of Protamine Sulfate. Rapid Commun. Mass Spectrom. 2008, 22, 2423–8. 10.1002/rcm.3627. [DOI] [PubMed] [Google Scholar]
- Guo C.; Steinberg L. K.; Henderson J. P.; Gross M. L. Organic Solvents for Enhanced Proteolysis of Stable Proteins for Hydrogen–Deuterium Exchange Mass Spectrometry. Anal. Chem. 2020, 92, 11553–11557. 10.1021/acs.analchem.0c02194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cravello L.; Lascoux D.; Forest E. Use of Different Proteases Working in Acidic Conditions to Improve Sequence Coverage and Resolution in Hydrogen/Deuterium Exchange of Large Proteins. Rapid Commun. Mass Spectrom. 2003, 17, 2387–2393. 10.1002/rcm.1207. [DOI] [PubMed] [Google Scholar]
- Giansanti P.; Tsiatsiani L.; Low T. Y.; Heck A. J. R. Six Alternative Proteases for Mass Spectrometry–Based Proteomics Beyond Trypsin. Nat. Protoc. 2016, 11, 993–1006. 10.1038/nprot.2016.057. [DOI] [PubMed] [Google Scholar]
- Zhang H. M.; Kazazic S.; Schaub T. M.; Tipton J. D.; Emmett M. R.; Marshall A. G. Enhanced Digestion Efficiency, Peptide Ionization Efficiency, and Sequence Resolution for Protein Hydrogen/Deuterium Exchange Monitored by Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal. Chem. 2008, 80, 9034–41. 10.1021/ac801417d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers J. C.; Harley A. D.; Myers D. V. Subsite Specificity of Porcine Pepsin. Adv. Exp. Med. Biol. 1977, 95, 141–57. 10.1007/978-1-4757-0719-9_9. [DOI] [PubMed] [Google Scholar]
- Hamuro Y.; Coales S. J.; Molnar K. S.; Tuske S. J.; Morrow J. A. Specificity of Immobilized Porcine Pepsin in H/D Exchange Compatible Conditions. Rapid Commun. Mass Spectrom. 2008, 22, 1041–1046. 10.1002/rcm.3467. [DOI] [PubMed] [Google Scholar]
- Zheng J.; Strutzenberg T. S.; Reich A.; Dharmarajan V.; Pascal B. D.; Crynen G. C.; Novick S. J.; Garcia-Ordonez R. D.; Griffin P. R. Comparative Analysis of Cleavage Specificities of Immobilized Porcine Pepsin and Nepenthesin Ii under Hydrogen/Deuterium Exchange Conditions. Anal. Chem. 2020, 92, 11018–11028. 10.1021/acs.analchem.9b05694. [DOI] [PubMed] [Google Scholar]
- Ahn J.; Cao M.-J.; Yu Y. Q.; Engen J. R. Accessing the Reproducibility and Specificity of Pepsin and Other Aspartic Proteases. Biochim. Biophys. Acta, Proteins Proteomics 2013, 1834, 1222–1229. 10.1016/j.bbapap.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brier S.; Maria G.; Carginale V.; Capasso A.; Wu Y.; Taylor R. M.; Borotto N. B.; Capasso C.; Engen J. R. Purification and Characterization of Pepsins A1 and A2 from the Antarctic Rock Cod Trematomus Bernacchii. FEBS J. 2007, 274, 6152–6166. 10.1111/j.1742-4658.2007.06136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey M.; Yang M.; Burns K. M.; Yu Y.; Lees-Miller S. P.; Schriemer D. C. Nepenthesin from Monkey Cups for Hydrogen/Deuterium Exchange Mass Spectrometry. Molecular & cellular proteomics: MCP 2013, 12, 464–472. 10.1074/mcp.M112.025221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadek A.; Mrazek H.; Halada P.; Rey M.; Schriemer D. C.; Man P. Aspartic Protease Nepenthesin-1 as a Tool for Digestion in Hydrogen/Deuterium Exchange Mass Spectrometry. Anal. Chem. 2014, 86, 4287–94. 10.1021/ac404076j. [DOI] [PubMed] [Google Scholar]
- Tsiatsiani L.; Akeroyd M.; Olsthoorn M.; Heck A. J. R. Aspergillus Niger Prolyl Endoprotease for Hydrogen-Deuterium Exchange Mass Spectrometry and Protein Structural Studies. Anal. Chem. 2017, 89, 7966–7973. 10.1021/acs.analchem.7b01161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamuro Y.; Zhang T. High-Resolution Hdx-Ms of Cytochrome C Using Pepsin/Fungal Protease Type Xiii Mixed Bed Column. J. Am. Soc. Mass Spectrom. 2019, 30, 227–234. 10.1007/s13361-018-2087-7. [DOI] [PubMed] [Google Scholar]
- Nirudodhi S. N.; Sperry J. B.; Rouse J. C.; Carroll J. A. Application of Dual Protease Column for Hdx-Ms Analysis of Monoclonal Antibodies. J. Pharm. Sci. 2017, 106, 530–536. 10.1016/j.xphs.2016.10.023. [DOI] [PubMed] [Google Scholar]
- Mullahoo J.; Zhang T.; Clauser K.; Carr S. A.; Jaffe J. D.; Papanastasiou M. Dual Protease Type Xiii/Pepsin Digestion Offers Superior Resolution and Overlap for the Analysis of Histone Tails by Hx-Ms. Methods 2020, 184, 135–140. 10.1016/j.ymeth.2020.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehring H. Hydrogen Exchange/Electrospray Ionization Mass Spectrometry Studies of Structural Features of Proteins and Protein/Protein Interactions. Anal. Biochem. 1999, 267, 252–259. 10.1006/abio.1998.3000. [DOI] [PubMed] [Google Scholar]
- Wang L.; Pan H.; Smith D. L. Hydrogen Exchange-Mass Spectrometry: Optimization of Digestion Conditions. Mol. Cell Proteomics 2002, 1, 132–8. 10.1074/mcp.M100009-MCP200. [DOI] [PubMed] [Google Scholar]
- Busby S. A.; Chalmers M. J.; Griffin P. R. Improving Digestion Efficiency under H/D Exchange Conditions with Activated Pepsinogen Coupled Columns. Int. J. Mass Spectrom. 2007, 259, 130–139. 10.1016/j.ijms.2006.08.006. [DOI] [Google Scholar]
- Rey M.; Man P.; Brandolin G.; Forest E.; Pelosi L. Recombinant Immobilized Rhizopuspepsin as a New Tool for Protein Digestion in Hydrogen/Deuterium Exchange Mass Spectrometry. Rapid Commun. Mass Spectrom. 2009, 23, 3431–8. 10.1002/rcm.4260. [DOI] [PubMed] [Google Scholar]
- Ahn J.; Jung M. C.; Wyndham K.; Yu Y. Q.; Engen J. R. Pepsin Immobilized on High-Strength Hybrid Particles for Continuous Flow Online Digestion at 10,000 Psi. Anal. Chem. 2012, 84, 7256–7262. 10.1021/ac301749h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Ferrer D.; Petritis K.; Hixson K. K.; Heibeck T. H.; Moore R. J.; Belov M. E.; Camp D. G. 2nd; Smith R. D. Application of Pressurized Solvents for Ultrafast Trypsin Hydrolysis in Proteomics: Proteomics on the Fly. J. Proteome Res. 2008, 7, 3276–3281. 10.1021/pr7008077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour E.; Hervé G.; Haertle T. Hydrolysis of B-Lactoglobulin by Thermolysin and Pepsin under High Hydrostatic Pressure. Biopolymers 1995, 35, 475–483. 10.1002/bip.360350506. [DOI] [Google Scholar]
- Stapelfeldt H.; Petersen P. H.; Kristiansen K. R.; Qvist K. B.; Skibsted L. H. Effect of High Hydrostatic Pressure on the Enzymic Hydrolysis of Beta-Lactoglobulin B by Trypsin, Thermolysin and Pepsin. J. Dairy Res. 1996, 63, 111–8. 10.1017/S0022029900031587. [DOI] [PubMed] [Google Scholar]
- López-Ferrer D.; Petritis K.; Robinson E. W.; Hixson K. K.; Tian Z.; Lee J. H.; Lee S.-W.; Tolić N.; Weitz K. K.; Belov M. E.; Smith R. D.; Paša-Tolić L. Pressurized Pepsin Digestion in Proteomics: An Automatable Alternative to Trypsin for Integrated Top-Down Bottom-up Proteomics. Mol. Cell. Proteomics 2011, 10, S1–S11. 10.1074/mcp.M110.001479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns J. A.; Butler J. C.; Moran J.; Whitesides G. M. Selective Reduction of Disulfides by Tris(2-Carboxyethyl)Phosphine. J. Org. Chem. 1991, 56, 2648–2650. 10.1021/jo00008a014. [DOI] [Google Scholar]
- Zhang H.-M.; McLoughlin S. M.; Frausto S. D.; Tang H.; Emmett M. R.; Marshall A. G. Simultaneous Reduction and Digestion of Proteins with Disulfide Bonds for Hydrogen/Deuterium Exchange Monitored by Mass Spectrometry. Anal. Chem. 2010, 82, 1450–1454. 10.1021/ac902550n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline D. J.; Redding S. E.; Brohawn S. G.; Psathas J. N.; Schneider J. P.; Thorpe C. New Water-Soluble Phosphines as Reductants of Peptide and Protein Disulfide Bonds: Reactivity and Membrane Permeability. Biochemistry 2004, 43, 15195–15203. 10.1021/bi048329a. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Rejtar T.; Zhou Z. S.; Karger B. L. Desulfurization of Cysteine-Containing Peptides Resulting from Sample Preparation for Protein Characterization by Mass Spectrometry. Rapid Commun. Mass Spectrom. 2010, 24, 267–75. 10.1002/rcm.4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Chien E. Y. T.; Chalmers M. J.; Pascal B. D.; Gatchalian J.; Stevens R. C.; Griffin P. R. Dynamics of the Beta2-Adrenergic G-Protein Coupled Receptor Revealed by Hydrogen-Deuterium Exchange. Anal. Chem. 2010, 82, 1100–1108. 10.1021/ac902484p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen P. F.; Comamala G.; Trelle M. B.; Madsen J. B.; Jørgensen T. J.; Rand K. D. Removal of N-Linked Glycosylations at Acidic Ph by Pngase a Facilitates Hydrogen/Deuterium Exchange Mass Spectrometry Analysis of N-Linked Glycoproteins. Anal. Chem. 2016, 88, 12479–12488. 10.1021/acs.analchem.6b03951. [DOI] [PubMed] [Google Scholar]
- Kraj A.; Brouwer H. J.; Reinhoud N.; Chervet J. P. A Novel Electrochemical Method for Efficient Reduction of Disulfide Bonds in Peptides and Proteins Prior to Ms Detection. Anal. Bioanal. Chem. 2013, 405, 9311–20. 10.1007/s00216-013-7374-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysling S.; Salbo R.; Ploug M.; Jorgensen T. J. Electrochemical Reduction of Disulfide-Containing Proteins for Hydrogen/Deuterium Exchange Monitored by Mass Spectrometry. Anal. Chem. 2014, 86, 340–5. 10.1021/ac403269a. [DOI] [PubMed] [Google Scholar]
- Trabjerg E.; Jakobsen R. U.; Mysling S.; Christensen S.; Jorgensen T. J.; Rand K. D. Conformational Analysis of Large and Highly Disulfide-Stabilized Proteins by Integrating Online Electrochemical Reduction into an Optimized H/D Exchange Mass Spectrometry Workflow. Anal. Chem. 2015, 87, 8880–8. 10.1021/acs.analchem.5b01996. [DOI] [PubMed] [Google Scholar]
- Comamala G.; Wagner C.; de la Torre P. S.; Jakobsen R. U.; Hilger M.; Brouwer H. J.; Rand K. D. Hydrogen/Deuterium Exchange Mass Spectrometry with Improved Electrochemical Reduction Enables Comprehensive Epitope Mapping of a Therapeutic Antibody to the Cysteine-Knot Containing Vascular Endothelial Growth Factor. Anal. Chim. Acta 2020, 1115, 41–51. 10.1016/j.aca.2020.04.014. [DOI] [PubMed] [Google Scholar]
- Hamuro Y.; Coales S. J.; Southern M. R.; Nemeth-Cawley J. F.; Stranz D. D.; Griffin P. R. Rapid Analysis of Protein Structure and Dynamics by Hydrogen/Deuterium Exchange Mass Spectrometry. J. Biomol. Technol. 2003, 14, 171–182. [PMC free article] [PubMed] [Google Scholar]
- Fang J.; Rand K. D.; Beuning P. J.; Engen J. R. False Ex1 Signatures Caused by Sample Carryover During Hx Ms Analyses. Int. J. Mass Spectrom. 2011, 302, 19–25. 10.1016/j.ijms.2010.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.-M.; Bou-Assaf G. M.; Emmett M. R.; Marshall A. G. Fast Reversed-Phase Liquid Chromatography to Reduce Back Exchange and Increase Throughput in H/D Exchange Monitored by Ft-Icr Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2009, 20, 520–524. 10.1016/j.jasms.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmett M. R.; Kazazic S.; Marshall A. G.; Chen W.; Shi S. D.; Bolaños B.; Greig M. J. Supercritical Fluid Chromatography Reduction of Hydrogen/Deuterium Back Exchange in Solution-Phase Hydrogen/Deuterium Exchange with Mass Spectrometric Analysis. Anal. Chem. 2006, 78, 7058–7060. 10.1021/ac060693n. [DOI] [PubMed] [Google Scholar]
- Woods V. L. Jr.; Hamuro Y. High Resolution, High-Throughput Amide Deuterium Exchange-Mass Spectrometry (Dxms) Determination of Protein Binding Site Structure and Dynamics: Utility in Pharmaceutical Design. J. Cell. Biochem. 2001, 84, 89–98. 10.1002/jcb.10069. [DOI] [PubMed] [Google Scholar]
- Englander J. J.; Del Mar C.; Li W.; Englander S. W.; Kim J. S.; Stranz D. D.; Hamuro Y.; Woods V. L. Jr. Protein Structure Change Studied by Hydrogen-Deuterium Exchange, Functional Labeling, and Mass Spectrometry. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 7057–7062. 10.1073/pnas.1232301100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalmers M. J.; Busby S. A.; Pascal B. D.; West G. M.; Griffin P. R. Differential Hydrogen/Deuterium Exchange Mass Spectrometry Analysis of Protein-Ligand Interactions. Expert Rev. Proteomics 2011, 8, 43–59. 10.1586/epr.10.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wales T. E.; Fadgen K. E.; Gerhardt G. C.; Engen J. R. High-Speed and High-Resolution Uplc Separation at Zero Degrees Celsius. Anal. Chem. 2008, 80, 6815–6820. 10.1021/ac8008862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keppel T. R.; Jacques M. E.; Young R. W.; Ratzlaff K. L.; Weis D. D. An Efficient and Inexpensive Refrigerated Lc System for H/D Exchange Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2011, 22, 1472–6. 10.1007/s13361-011-0152-6. [DOI] [PubMed] [Google Scholar]
- De Vos J.; Broeckhoven K.; Eeltink S. Advances in Ultrahigh-Pressure Liquid Chromatography Technology and System Design. Anal. Chem. 2016, 88, 262–278. 10.1021/acs.analchem.5b04381. [DOI] [PubMed] [Google Scholar]
- Wu Y.; Engen J. R.; Hobbins W. B. Ultra Performance Liquid Chromatography (Uplc) Further Improves Hydrogen/Deuterium Exchange Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2006, 17, 163–7. 10.1016/j.jasms.2005.10.009. [DOI] [PubMed] [Google Scholar]
- Chalmers M. J.; Busby S. A.; Pascal B. D.; Southern M. R.; Griffin P. R. A Two-Stage Differential Hydrogen Deuterium Exchange Method for the Rapid Characterization of Protein/Ligand Interactions. J. Biomol. Technol. 2007, 18, 194–204. [PMC free article] [PubMed] [Google Scholar]
- Valeja S. G.; Emmett M. R.; Marshall A. G. Polar Aprotic Modifiers for Chromatographic Separation and Back-Exchange Reduction for Protein Hydrogen/Deuterium Exchange Monitored by Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2012, 23, 699–707. 10.1007/s13361-011-0329-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J.; Zhang S.; Parker C. E.; Borchers C. H. Subzero Temperature Chromatography and Top-Down Mass Spectrometry for Protein Higher-Order Structure Characterization: Method Validation and Application to Therapeutic Antibodies. J. Am. Chem. Soc. 2014, 136, 13065–71. 10.1021/ja507880w. [DOI] [PubMed] [Google Scholar]
- Pan J.; Zhang S.; Chou A.; Hardie D. B.; Borchers C. H. Fast Comparative Structural Characterization of Intact Therapeutic Antibodies Using Hydrogen-Deuterium Exchange and Electron Transfer Dissociation. Anal. Chem. 2015, 87, 5884–90. 10.1021/ac504809r. [DOI] [PubMed] [Google Scholar]
- Pan J.; Zhang S.; Borchers C. H. Comparative Higher-Order Structure Analysis of Antibody Biosimilars Using Combined Bottom-up and Top-Down Hydrogen-Deuterium Exchange Mass Spectrometry. Biochim. Biophys. Acta, Proteins Proteomics 2016, 1864, 1801–1808. 10.1016/j.bbapap.2016.08.013. [DOI] [PubMed] [Google Scholar]
- Wales T. E.; Fadgen K. E.; Eggertson M. J.; Engen J. R. Subzero Celsius Separations in Three-Zone Temperature Controlled Hydrogen Deuterium Exchange Mass Spectrometry. J. Chromatogr. A 2017, 1523, 275–282. 10.1016/j.chroma.2017.05.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang M.; Wang Z.; Cupp-Sutton K. A.; Welborn T.; Smith K.; Wu S. High-Throughput Hydrogen Deuterium Exchange Mass Spectrometry (Hdx-Ms) Coupled with Subzero-Temperature Ultrahigh Pressure Liquid Chromatography (Uplc) Separation for Complex Sample Analysis. Anal. Chim. Acta 2021, 1143, 65–72. 10.1016/j.aca.2020.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilm M.; Mann M. Analytical Properties of the Nanoelectrospray Ion Source. Anal. Chem. 1996, 68, 1–8. 10.1021/ac9509519. [DOI] [PubMed] [Google Scholar]
- Wang L.; Smith D. L. Downsizing Improves Sensitivity 100-Fold for Hydrogen Exchange-Mass Spectrometry. Anal. Biochem. 2003, 314, 46–53. 10.1016/S0003-2697(02)00620-6. [DOI] [PubMed] [Google Scholar]
- Bian Y.; Zheng R.; Bayer F. P.; Wong C.; Chang Y.-C.; Meng C.; Zolg D. P.; Reinecke M.; Zecha J.; Wiechmann S.; Heinzlmeir S.; Scherr J.; Hemmer B.; Baynham M.; Gingras A.-C.; Boychenko O.; Kuster B. Robust, Reproducible and Quantitative Analysis of Thousands of Proteomes by Micro-Flow Lc–Ms/Ms. Nat. Commun. 2020, 11, 157. 10.1038/s41467-019-13973-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheff J. G.; Hepburn M.; Yu Y.; Lees-Miller S. P.; Schriemer D. C. Nanospray Hx-Ms Configuration for Structural Interrogation of Large Protein Systems. Analyst 2017, 142, 904–910. 10.1039/C6AN02707E. [DOI] [PubMed] [Google Scholar]
- Wu H.; Tang K. Highly Sensitive and Robust Capillary Electrophoresis-Electrospray Ionization-Mass Spectrometry: Interfaces, Preconcentration Techniques and Applications. Rev. Anal. Chem. 2020, 39, 45–55. 10.1515/revac-2020-0112. [DOI] [Google Scholar]
- Black W. A.; Stocks B. B.; Mellors J. S.; Engen J. R.; Ramsey J. M. Utilizing Microchip Capillary Electrophoresis Electrospray Ionization for Hydrogen Exchange Mass Spectrometry. Anal. Chem. 2015, 87, 6280–6287. 10.1021/acs.analchem.5b01179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar R.; Manikwar P.; Hickey J. M.; Arora J.; Middaugh C. R.; Volkin D. B.; Weis D. D. Minimizing Carry-over in an Online Pepsin Digestion System Used for the H/D Exchange Mass Spectrometric Analysis of an Igg1Monoclonal Antibody. J. Am. Soc. Mass Spectrom. 2012, 23, 2140–2148. 10.1007/s13361-012-0485-9. [DOI] [PubMed] [Google Scholar]
- Möller I. R.; Merkle P. S.; Calugareanu D.; Comamala G.; Schmidt S. G.; Loland C. J.; Rand K. D. Probing the Conformational Impact of Detergents on the Integral Membrane Protein Leut by Global Hdx-Ms. J. Proteomics 2020, 225, 103845. 10.1016/j.jprot.2020.103845. [DOI] [PubMed] [Google Scholar]
- Espada A.; Haro R.; Castanon J.; Sayago C.; Perez-Cozar F.; Cano L.; Redero P.; Molina-Martin M.; Broughton H.; Stites R. E.; Pascal B. D.; Griffin P. R.; Dodge J. A.; Chalmers M. J. A Decoupled Automation Platform for Hydrogen/Deuterium Exchange Mass Spectrometry Experiments. J. Am. Soc. Mass Spectrom. 2019, 30, 2580–2583. 10.1007/s13361-019-02331-2. [DOI] [PubMed] [Google Scholar]
- Chalmers M. J.; Pascal B. D.; Willis S.; Zhang J.; Iturria S. J.; Dodge J. A.; Griffin P. R. Methods for the Analysis of High Precision Differential Hydrogen Deuterium Exchange Data. Int. J. Mass Spectrom. 2011, 302, 59–68. 10.1016/j.ijms.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalmers M. J.; Busby S. A.; Pascal B. D.; He Y.; Hendrickson C. L.; Marshall A. G.; Griffin P. R. Probing Protein Ligand Interactions by Automated Hydrogen/Deuterium Exchange Mass Spectrometry. Anal. Chem. 2006, 78, 1005–1014. 10.1021/ac051294f. [DOI] [PubMed] [Google Scholar]
- Cummins D. J.; Espada A.; Novick S. J.; Molina-Martin M.; Stites R. E.; Espinosa J. F.; Broughton H.; Goswami D.; Pascal B. D.; Dodge J. A.; Chalmers M. J.; Griffin P. R. Two-Site Evaluation of the Repeatability and Precision of an Automated Dual-Column Hydrogen/Deuterium Exchange Mass Spectrometry Platform. Anal. Chem. 2016, 88, 6607–6614. 10.1021/acs.analchem.6b01650. [DOI] [PubMed] [Google Scholar]
- Anderson K. W.; Gallagher E. S.; Hudgens J. W. Automated Removal of Phospholipids from Membrane Proteins for H/D Exchange Mass Spectrometry Workflows. Anal. Chem. 2018, 90, 6409–6412. 10.1021/acs.analchem.8b00429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coales S. J.; Tomasso J. C.; Hamuro Y. Effects of Electrospray Capillary Temperature on Amide Hydrogen Exchange. Rapid Commun. Mass Spectrom. 2008, 22, 1367–1371. 10.1002/rcm.3512. [DOI] [PubMed] [Google Scholar]
- Morgan C. R.; Engen J. R. Investigating Solution-Phase Protein Structure and Dynamics by Hydrogen Exchange Mass Spectrometry. Curr. Protoc. Protein Sci. 2009, 58, 1–17. Unit 17.6. 10.1002/0471140864.ps1706s58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M.; Wales T. E.; Whittington D.; Engen J. R.; Brown J. M.; Lee K. K. Tuning a High Transmission Ion Guide to Prevent Gas-Phase Proton Exchange During H/D Exchange Ms Analysis. J. Am. Soc. Mass Spectrom. 2016, 27, 662–668. 10.1007/s13361-015-1330-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.-S.; Monroe M. E.; Camp D. G. 2nd; Smith R. D.; Qian W.-J. In-Source Fragmentation and the Sources of Partially Tryptic Peptides in Shotgun Proteomics. J. Proteome Res. 2013, 12, 910–916. 10.1021/pr300955f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipping M.; Schierhorn A. Improving Hydrogen/Deuterium Exchange Mass Spectrometry by Reduction of the Back-Exchange Effect. J. Mass Spectrom. 2003, 38, 271–6. 10.1002/jms.437. [DOI] [PubMed] [Google Scholar]
- Ghaemmaghami S.; Fitzgerald M. C.; Oas T. G. A Quantitative, High-Throughput Screen for Protein Stability. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 8296–8301. 10.1073/pnas.140111397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quanico J.; Franck J.; Salzet M.; Fournier I. On-Tissue Direct Monitoring of Global Hydrogen/Deuterium Exchange by Maldi Mass Spectrometry: Tissue Deuterium Exchange Mass Spectrometry (Tdxms). Molecular & cellular proteomics: MCP 2016, 15, 3321–3330. 10.1074/mcp.O116.059832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross J. H., Instrumentation. In Mass Spectrometry: A Textbook; Gross J. H., Ed.; Springer International Publishing: Cham, 2017; pp 151–292. [Google Scholar]
- Burns K. M.; Rey M.; Baker C. A. H.; Schriemer D. C. Platform Dependencies in Bottom-up Hydrogen/Deuterium Exchange Mass Spectrometry. Mol. Cell. Proteomics 2013, 12, 539–548. 10.1074/mcp.M112.023770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shliaha P. V.; Bond N. J.; Gatto L.; Lilley K. S. Effects of Traveling Wave Ion Mobility Separation on Data Independent Acquisition in Proteomics Studies. J. Proteome Res. 2013, 12, 2323–2339. 10.1021/pr300775k. [DOI] [PubMed] [Google Scholar]
- Cryar A.; Groves K.; Quaglia M. Online Hydrogen-Deuterium Exchange Traveling Wave Ion Mobility Mass Spectrometry (Hdx-Im-Ms): A Systematic Evaluation. J. Am. Soc. Mass Spectrom. 2017, 28, 1192–1202. 10.1007/s13361-017-1633-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazazic S.; Zhang H. M.; Schaub T. M.; Emmett M. R.; Hendrickson C. L.; Blakney G. T.; Marshall A. G. Automated Data Reduction for Hydrogen/Deuterium Exchange Experiments, Enabled by High-Resolution Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2010, 21, 550–8. 10.1016/j.jasms.2009.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris C. B.; Poland J. C.; May J. C.; McLean J. A. Fundamentals of Ion Mobility-Mass Spectrometry for the Analysis of Biomolecules. Methods Mol. Biol. 2020, 2084, 1–31. 10.1007/978-1-0716-0030-6_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldrid C.; Thalassinos K. Developments in Tandem Ion Mobility Mass Spectrometry. Biochem. Soc. Trans. 2020, 48, 2457–2466. 10.1042/BST20190788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacob R. E.; Engen J. R. Hydrogen Exchange Mass Spectrometry: Are We out of the Quicksand?. J. Am. Soc. Mass Spectrom. 2012, 23, 1003–10. 10.1007/s13361-012-0377-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopmans F.; Ho J. T. C.; Smit A. B.; Li K. W. Comparative Analyses of Data Independent Acquisition Mass Spectrometric Approaches: Dia, Wisim-Dia, and Untargeted Dia. Proteomics 2018, 18, 1700304. 10.1002/pmic.201700304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond N. J.; Shliaha P. V.; Lilley K. S.; Gatto L. Improving Qualitative and Quantitative Performance for Ms(E)-Based Label-Free Proteomics. J. Proteome Res. 2013, 12, 2340–2353. 10.1021/pr300776t. [DOI] [PubMed] [Google Scholar]
- Hansen K.; Politis A. Improving Peptide Fragmentation for Hydrogen-Deuterium Exchange Mass Spectrometry Using a Time-Dependent Collision Energy Calculator. J. Am. Soc. Mass Spectrom. 2020, 31, 996–999. 10.1021/jasms.9b00133. [DOI] [PubMed] [Google Scholar]
- Moore R. E.; Young M. K.; Lee T. D. Qscore: An Algorithm for Evaluating Sequest Database Search Results. J. Am. Soc. Mass Spectrom. 2002, 13, 378–86. 10.1016/S1044-0305(02)00352-5. [DOI] [PubMed] [Google Scholar]
- Dobbs J. M.; Jenkins M. L.; Burke J. E. Escherichia Coli and Sf9 Contaminant Databases to Increase Efficiency of Tandem Mass Spectrometry Peptide Identification in Structural Mass Spectrometry Experiments. J. Am. Soc. Mass Spectrom. 2020, 31, 2202–2209. 10.1021/jasms.0c00283. [DOI] [PubMed] [Google Scholar]
- Liu H.; Wang D.; Zhang Q.; Zhao Y.; Mamonova T.; Wang L.; Zhang C.; Li S.; Friedman P. A.; Xiao K. Parallel Post-Translational Modification Scanning Enhancing Hydrogen-Deuterium Exchange-Mass Spectrometry Coverage of Key Structural Regions. Anal. Chem. 2019, 91, 6976–6980. 10.1021/acs.analchem.9b01410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y.; Pan H.; Smith D. L. Selective Isotope Labeling Demonstrates That Hydrogen Exchange at Individual Peptide Amide Linkages Can Be Determined by Collision-Induced Dissociation Mass Spectrometry. J. Am. Chem. Soc. 1999, 121, 1966–1967. 10.1021/ja982814+. [DOI] [Google Scholar]
- Wysocki V. H.; Tsaprailis G.; Smith L. L.; Breci L. A. Mobile and Localized Protons: A Framework for Understanding Peptide Dissociation. J. Mass Spectrom. 2000, 35, 1399–406. . [DOI] [PubMed] [Google Scholar]
- Hoerner J. K.; Xiao H.; Dobo A.; Kaltashov I. A. Is There Hydrogen Scrambling in the Gas Phase? Energetic and Structural Determinants of Proton Mobility within Protein Ions. J. Am. Chem. Soc. 2004, 126, 7709–7717. 10.1021/ja049513m. [DOI] [PubMed] [Google Scholar]
- Buijs J.; Hagman C.; Håkansson K.; Richter J. H.; Håkansson P.; Oscarsson S. Inter- and Intra-Molecular Migration of Peptide Amide Hydrogens During Electrospray Ionization. J. Am. Soc. Mass Spectrom. 2001, 12, 410–419. 10.1016/S1044-0305(01)00204-5. [DOI] [PubMed] [Google Scholar]
- Monson de Souza B.; Palma M. S. Monitoring the Positioning of Short Polycationic Peptides in Model Lipid Bilayers by Combining Hydrogen/Deuterium Exchange and Electrospray Ionization Mass Spectrometry. Biochim. Biophys. Acta, Biomembr. 2008, 1778, 2797–2805. 10.1016/j.bbamem.2008.09.005. [DOI] [PubMed] [Google Scholar]
- Demmers J. A.; Rijkers D. T.; Haverkamp J.; Killian J. A.; Heck A. J. Factors Affecting Gas-Phase Deuterium Scrambling in Peptide Ions and Their Implications for Protein Structure Determination. J. Am. Chem. Soc. 2002, 124, 11191–11198. 10.1021/ja0125927. [DOI] [PubMed] [Google Scholar]
- Ferguson P. L.; Pan J.; Wilson D. J.; Dempsey B.; Lajoie G.; Shilton B.; Konermann L. Hydrogen/Deuterium Scrambling During Quadrupole Time-of-Flight Ms/Ms Analysis of a Zinc-Binding Protein Domain. Anal. Chem. 2007, 79, 153–160. 10.1021/ac061261f. [DOI] [PubMed] [Google Scholar]
- Jorgensen T. J.; Gardsvoll H.; Ploug M.; Roepstorff P. Intramolecular Migration of Amide Hydrogens in Protonated Peptides Upon Collisional Activation. J. Am. Chem. Soc. 2005, 127, 2785–93. 10.1021/ja043789c. [DOI] [PubMed] [Google Scholar]
- Hamuro Y.; Tomasso J. C.; Coales S. J. A Simple Test to Detect Hydrogen/Deuterium Scrambling During Gas-Phase Peptide Fragmentation. Anal. Chem. 2008, 80, 6785–6790. 10.1021/ac800645f. [DOI] [PubMed] [Google Scholar]
- Bache N.; Rand K. D.; Roepstorff P.; Ploug M.; Jørgensen T. J. D. Hydrogen Atom Scrambling in Selectively Labeled Anionic Peptides Upon Collisional Activation by Maldi Tandem Time-of-Flight Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2008, 19, 1719–1725. 10.1016/j.jasms.2008.05.021. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Borotto N. B.; Håkansson K. Gas-Phase Hydrogen/Deuterium Scrambling in Negative-Ion Mode Tandem Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2019, 30, 855–863. 10.1007/s13361-019-02143-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bache N.; Rand K. D.; Roepstorff P.; Jørgensen T. J. Gas-Phase Fragmentation of Peptides by Maldi in-Source Decay with Limited Amide Hydrogen (1h/2h) Scrambling. Anal. Chem. 2008, 80, 6431–5. 10.1021/ac800902a. [DOI] [PubMed] [Google Scholar]
- Percy A. J.; Slysz G. W.; Schriemer D. C. Surrogate H/D Detection Strategy for Protein Conformational Analysis Using Ms/Ms Data. Anal. Chem. 2009, 81, 7900–7. 10.1021/ac901148u. [DOI] [PubMed] [Google Scholar]
- Zhurov K. O.; Fornelli L.; Wodrich M. D.; Laskay Ü. A.; Tsybin Y. O. Principles of Electron Capture and Transfer Dissociation Mass Spectrometry Applied to Peptide and Protein Structure Analysis. Chem. Soc. Rev. 2013, 42, 5014. 10.1039/c3cs35477f. [DOI] [PubMed] [Google Scholar]
- Syka J. E. P.; Coon J. J.; Schroeder M. J.; Shabanowitz J.; Hunt D. F. Peptide and Protein Sequence Analysis by Electron Transfer Dissociation Mass Spectrometry. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 9528–9533. 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand K. D.; Adams C. M.; Zubarev R. A.; Jorgensen T. J. Electron Capture Dissociation Proceeds with a Low Degree of Intramolecular Migration of Peptide Amide Hydrogens. J. Am. Chem. Soc. 2008, 130, 1341–9. 10.1021/ja076448i. [DOI] [PubMed] [Google Scholar]
- Rand K. D.; Jørgensen T. J. Development of a Peptide Probe for the Occurrence of Hydrogen (1h/2h) Scrambling Upon Gas-Phase Fragmentation. Anal. Chem. 2007, 79, 8686–93. 10.1021/ac0710782. [DOI] [PubMed] [Google Scholar]
- Rand K. D.; Lund F. W.; Amon S.; Jørgensen T. J. D. Investigation of Amide Hydrogen Back-Exchange in Asp and His Repeats Measured by Hydrogen (1h/2h) Exchange Mass Spectrometry. Int. J. Mass Spectrom. 2011, 302, 110–115. 10.1016/j.ijms.2010.08.027. [DOI] [Google Scholar]
- Rand K. D.; Pringle S. D.; Morris M.; Engen J. R.; Brown J. M. Etd in a Traveling Wave Ion Guide at Tuned Z-Spray Ion Source Conditions Allows for Site-Specific Hydrogen/Deuterium Exchange Measurements. J. Am. Soc. Mass Spectrom. 2011, 22, 1784. 10.1007/s13361-011-0196-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landgraf R. R.; Chalmers M. J.; Griffin P. R. Automated Hydrogen/Deuterium Exchange Electron Transfer Dissociation High Resolution Mass Spectrometry Measured at Single-Amide Resolution. J. Am. Soc. Mass Spectrom. 2012, 23, 301–309. 10.1007/s13361-011-0298-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollenberg D. T. W.; Pengelley S.; Mouritsen J. C.; Suckau D.; Jorgensen C. I.; Jorgensen T. J. D. Avoiding H/D Scrambling with Minimal Ion Transmission Loss for Hdx-Ms/Ms-Etd Analysis on a High-Resolution Q-Tof Mass Spectrometer. Anal. Chem. 2020, 92, 7453. 10.1021/acs.analchem.9b05208. [DOI] [PubMed] [Google Scholar]
- Seger S. T.; Breinholt J.; Faber J. H.; Andersen M. D.; Wiberg C.; Schjødt C. B.; Rand K. D. Probing the Conformational and Functional Consequences of Disulfide Bond Engineering in Growth Hormone by Hydrogen-Deuterium Exchange Mass Spectrometry Coupled to Electron Transfer Dissociation. Anal. Chem. 2015, 87, 5973–80. 10.1021/ac504782v. [DOI] [PubMed] [Google Scholar]
- Arndt J. R.; Brown R. J.; Burke K. A.; Legleiter J.; Valentine S. J. Lysine Residues in the N-Terminal Huntingtin Amphipathic A-Helix Play a Key Role in Peptide Aggregation. J. Mass Spectrom. 2015, 50, 117–126. 10.1002/jms.3504. [DOI] [PubMed] [Google Scholar]
- Masson G. R.; Maslen S. L.; Williams R. L. Analysis of Phosphoinositide 3-Kinase Inhibitors by Bottom-up Electron-Transfer Dissociation Hydrogen/Deuterium Exchange Mass Spectrometry. Biochem. J. 2017, 474, 1867–1877. 10.1042/BCJ20170127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M. M.; Huang R. Y.; Beno B. R.; Deyanova E. G.; Li J.; Chen G.; Gross M. L. Epitope and Paratope Mapping of Pd-1/Nivolumab by Mass Spectrometry-Based Hydrogen-Deuterium Exchange, Cross-Linking, and Molecular Docking. Anal. Chem. 2020, 92, 9086–9094. 10.1021/acs.analchem.0c01291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leurs U.; Lohse B.; Ming S.; Cole P. A.; Clausen R. P.; Kristensen J. L.; Rand K. D. Dissecting the Binding Mode of Low Affinity Phage Display Peptide Ligands to Protein Targets by Hydrogen/Deuterium Exchange Coupled to Mass Spectrometry. Anal. Chem. 2014, 86, 11734–11741. 10.1021/ac503137u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R. Y.; Kuhne M.; Deshpande S.; Rangan V.; Srinivasan M.; Wang Y.; Chen G. Mapping Binding Epitopes of Monoclonal Antibodies Targeting Major Histocompatibility Complex Class I Chain-Related a (Mica) with Hydrogen/Deuterium Exchange and Electron-Transfer Dissociation Mass Spectrometry. Anal. Bioanal. Chem. 2020, 412, 1693–1700. 10.1007/s00216-020-02409-x. [DOI] [PubMed] [Google Scholar]
- Rand K. D.; Zehl M.; Jensen O. N.; Jørgensen T. J. Loss of Ammonia During Electron-Transfer Dissociation of Deuterated Peptides as an Inherent Gauge of Gas-Phase Hydrogen Scrambling. Anal. Chem. 2010, 82, 9755–62. 10.1021/ac101889b. [DOI] [PubMed] [Google Scholar]
- Hamuro Y. Regio-Selective Intramolecular Hydrogen/Deuterium Exchange in Gas-Phase Electron Transfer Dissociation. J. Am. Soc. Mass Spectrom. 2017, 28, 971–977. 10.1007/s13361-017-1612-4. [DOI] [PubMed] [Google Scholar]
- Hamuro Y.; E S. Y. Determination of Backbone Amide Hydrogen Exchange Rates of Cytochrome C Using Partially Scrambled Electron Transfer Dissociation Data. J. Am. Soc. Mass Spectrom. 2018, 29, 989–1001. 10.1007/s13361-018-1892-3. [DOI] [PubMed] [Google Scholar]
- Brodbelt J. S.; Morrison L. J.; Santos I. Ultraviolet Photodissociation Mass Spectrometry for Analysis of Biological Molecules. Chem. Rev. 2020, 120, 3328–3380. 10.1021/acs.chemrev.9b00440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodie N. I.; Huguet R.; Zhang T.; Viner R.; Zabrouskov V.; Pan J.; Petrotchenko E. V.; Borchers C. H. Top-Down Hydrogen–Deuterium Exchange Analysis of Protein Structures Using Ultraviolet Photodissociation. Anal. Chem. 2018, 90, 3079–3082. 10.1021/acs.analchem.7b03655. [DOI] [PubMed] [Google Scholar]
- Mistarz U. H.; Bellina B.; Jensen P. F.; Brown J. M.; Barran P. E.; Rand K. D. Uv Photodissociation Mass Spectrometry Accurately Localize Sites of Backbone Deuteration in Peptides. Anal. Chem. 2018, 90, 1077–1080. 10.1021/acs.analchem.7b04683. [DOI] [PubMed] [Google Scholar]
- Modzel M.; Wollenberg D. T. W.; Trelle M. B.; Larsen M. R.; Jørgensen T. J. D. Ultraviolet Photodissociation of Protonated Peptides and Proteins Can Proceed with H/D Scrambling. Anal. Chem. 2021, 93, 691–696. 10.1021/acs.analchem.0c02957. [DOI] [PubMed] [Google Scholar]
- Robinson C. V.; Gross M.; Eyles S. J.; Ewbank J. J.; Mayhew M.; Hartl F. U.; Dobson C. M.; Radford S. E. Conformation of Groel-Bound Alpha-Lactalbumin Probed by Mass Spectrometry. Nature 1994, 372, 646–51. 10.1038/372646a0. [DOI] [PubMed] [Google Scholar]
- Sperry J. B.; Shi X.; Rempel D. L.; Nishimura Y.; Akashi S.; Gross M. L. A Mass Spectrometric Approach to the Study of DNA-Binding Proteins: Interaction of Human Trf2 with Telomeric DNA. Biochemistry 2008, 47, 1797–807. 10.1021/bi702037p. [DOI] [PubMed] [Google Scholar]
- Tong Y.; Wuebbens M. M.; Rajagopalan K. V.; Fitzgerald M. C. Thermodynamic Analysis of Subunit Interactions in Escherichia Coli Molybdopterin Synthase. Biochemistry 2005, 44, 2595–601. 10.1021/bi047762h. [DOI] [PubMed] [Google Scholar]
- Xiao H.; Kaltashov I. A.; Eyles S. J. Indirect Assessment of Small Hydrophobic Ligand Binding to a Model Protein Using a Combination of Esi Ms and Hdx/Esi Ms. J. Am. Soc. Mass Spectrom. 2003, 14, 506–15. 10.1016/S1044-0305(03)00135-1. [DOI] [PubMed] [Google Scholar]
- Zhu M. M.; Rempel D. L.; Zhao J.; Giblin D. E.; Gross M. L. Probing Ca2+-Induced Conformational Changes in Porcine Calmodulin by H/D Exchange and Esi-Ms: Effect of Cations and Ionic Strength. Biochemistry 2003, 42, 15388–97. 10.1021/bi035188o. [DOI] [PubMed] [Google Scholar]
- Kaltashov I. A.; Bobst C. E.; Abzalimov R. R.; Berkowitz S. A.; Houde D. Conformation and Dynamics of Biopharmaceuticals: Transition of Mass Spectrometry-Based Tools from Academe to Industry. J. Am. Soc. Mass Spectrom. 2010, 21, 323–337. 10.1016/j.jasms.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makepeace K. A. T.; Brodie N. I.; Popov K. I.; Gudavicius G.; Nelson C. J.; Petrotchenko E. V.; Dokholyan N. V.; Borchers C. H. Ligand-Induced Disorder-to-Order Transitions Characterized by Structural Proteomics and Molecular Dynamics Simulations Hhs Public Access. J. Proteomics 2020, 211, 103544–103544. 10.1016/j.jprot.2019.103544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltashov I. A.; Bobst C. E.; Abzalimov R. R. H/D Exchange and Mass Spectrometry in the Studies of Protein Conformation and Dynamics: Is There a Need for a Top-Down Approach?. Anal. Chem. 2009, 81, 7892–7899. 10.1021/ac901366n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrie S. M. Top-Down Mass Spectrometry: Proteomics to Proteoforms. Adv. Exp. Med. Biol. 2016, 919, 171–200. 10.1007/978-3-319-41448-5_8. [DOI] [PubMed] [Google Scholar]
- Abzalimov R. R.; Kaplan D. A.; Easterling M. L.; Kaltashov I. A. Protein Conformations Can Be Probed in Top-Down Hdx Ms Experiments Utilizing Electron Transfer Dissociation of Protein Ions without Hydrogen Scrambling. J. Am. Soc. Mass Spectrom. 2009, 20, 1514–1517. 10.1016/j.jasms.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J.; Han J.; Borchers C. H.; Konermann L. Structure and Dynamics of Small Soluble Aβ(1–40) Oligomers Studied by Top-Down Hydrogen Exchange Mass Spectrometry. Biochemistry 2012, 51, 3694–703. 10.1021/bi3002049. [DOI] [PubMed] [Google Scholar]
- Pan J.; Zhang S.; Borchers C. H. Protein Species-Specific Characterization of Conformational Change Induced by Multisite Phosphorylation. J. Proteomics 2016, 134, 138–143. 10.1016/j.jprot.2015.12.002. [DOI] [PubMed] [Google Scholar]
- Leurs U.; Mistarz U. H.; Rand K. D. Getting to the Core of Protein Pharmaceuticals--Comprehensive Structure Analysis by Mass Spectrometry. Eur. J. Pharm. Biopharm. 2015, 93, 95–109. 10.1016/j.ejpb.2015.03.012. [DOI] [PubMed] [Google Scholar]
- Wang G.; Kaltashov I. A. Approach to Characterization of the Higher Order Structure of Disulfide-Containing Proteins Using Hydrogen/Deuterium Exchange and Top-Down Mass Spectrometry. Anal. Chem. 2014, 86, 7293–7298. 10.1021/ac501789e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R. Y.; O’Neil S. R.; Lipovšek D.; Chen G. Conformational Assessment of Adnectin and Adnectin-Drug Conjugate by Hydrogen/Deuterium Exchange Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2018, 29, 1524–1531. 10.1007/s13361-018-1966-2. [DOI] [PubMed] [Google Scholar]
- Karch K. R.; Coradin M.; Zandarashvili L.; Kan Z. Y.; Gerace M.; Englander S. W.; Black B. E.; Garcia B. A. Hydrogen-Deuterium Exchange Coupled to Top- and Middle-Down Mass Spectrometry Reveals Histone Tail Dynamics before and after Nucleosome Assembly. Structure 2018, 26, 1651–1663. e3 10.1016/j.str.2018.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristobal A.; Marino F.; Post H.; van den Toorn H. W. P.; Mohammed S.; Heck A. J. R. Toward an Optimized Workflow for Middle-Down Proteomics. Anal. Chem. 2017, 89, 3318–3325. 10.1021/acs.analchem.6b03756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J.; Zhang S.; Chou A.; Borchers C. H. Higher-Order Structural Interrogation of Antibodies Using Middle-Down Hydrogen/Deuterium Exchange Mass Spectrometry. Chem. Sci. 2016, 7, 1480–1486. 10.1039/C5SC03420E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterling H. J.; Williams E. R. Real-Time Hydrogen/Deuterium Exchange Kinetics Via Supercharged Electrospray Ionization Tandem Mass Spectrometry. Anal. Chem. 2010, 82, 9050–9057. 10.1021/ac101957x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Going C. C.; Xia Z.; Williams E. R. Real-Time Hd Exchange Kinetics of Proteins from Buffered Aqueous Solution with Electrothermal Supercharging and Top-Down Tandem Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2016, 27, 1019–1027. 10.1007/s13361-016-1350-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H.; Ma L.; Wang L.; Xiao P.; Li H.; Zhou M.; Song D. Research Advances in Hydrogen-Deuterium Exchange Mass Spectrometry for Protein Epitope Mapping. Anal. Bioanal. Chem. 2021, 413, 2345–2359. 10.1007/s00216-020-03091-9. [DOI] [PubMed] [Google Scholar]
- Hager-Braun C.; Tomer K. B. Determination of Protein-Derived Epitopes by Mass Spectrometry. Expert Rev. Proteomics 2005, 2, 745–756. 10.1586/14789450.2.5.745. [DOI] [PubMed] [Google Scholar]
- Vance D. J.; Tremblay J. M.; Rong Y.; Angalakurthi S. K.; Volkin D. B.; Middaugh C. R.; Weis D. D.; Shoemaker C. B.; Mantis N. J. High-Resolution Epitope Positioning of a Large Collection of Neutralizing and Nonneutralizing Single-Domain Antibodies on the Enzymatic and Binding Subunits of Ricin Toxin. Clin. Vaccine Immunol. 2017, 24, e00236-17 10.1128/CVI.00236-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; Yang J.; Bautista J.; Badithe A.; Olson W.; Liu Y. Epitope Mapping by Hdx-Ms Elucidates the Surface Coverage of Antigens Associated with High Blocking Efficiency of Antibodies to Birch Pollen Allergen. Anal. Chem. 2018, 90, 11315–11323. 10.1021/acs.analchem.8b01864. [DOI] [PubMed] [Google Scholar]
- Gribenko A. V.; Parris K.; Mosyak L.; Li S.; Handke L.; Hawkins J. C.; Severina E.; Matsuka Y. V.; Anderson A. S. High Resolution Mapping of Bactericidal Monoclonal Antibody Binding Epitopes on Staphylococcus Aureus Antigen Mntc. PLoS Pathog. 2016, 12, e1005908 10.1371/journal.ppat.1005908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Kitova E. N.; Li J.; Eugenio L.; Ng K.; Klassen J. S. Localizing Carbohydrate Binding Sites in Proteins Using Hydrogen/Deuterium Exchange Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2016, 27, 83–90. 10.1007/s13361-015-1263-2. [DOI] [PubMed] [Google Scholar]
- Kochert B. A.; Iacob R. E.; Wales T. E.; Makriyannis A.; Engen J. R. Hydrogen-Deuterium Exchange Mass Spectrometry to Study Protein Complexes. Methods Mol. Biol. 2018, 1764, 153–171. 10.1007/978-1-4939-7759-8_10. [DOI] [PubMed] [Google Scholar]
- Mandell J. G.; Baerga-Ortiz A.; Akashi S.; Takio K.; Komives E. A. Solvent Accessibility of the Thrombin-Thrombomodulin Interface. J. Mol. Biol. 2001, 306, 575–89. 10.1006/jmbi.2000.4416. [DOI] [PubMed] [Google Scholar]
- Percy A. J.; Rey M.; Burns K. M.; Schriemer D. C. Probing Protein Interactions with Hydrogen/Deuterium Exchange and Mass Spectrometry-a Review. Anal. Chim. Acta 2012, 721, 7–21. 10.1016/j.aca.2012.01.037. [DOI] [PubMed] [Google Scholar]
- Jørgensen T. J. D.; Gårdsvoll H.; Danø K.; Roepstorff P.; Ploug M. Dynamics of Urokinase Receptor Interaction with Peptide Antagonists Studied by Amide Hydrogen Exchange and Mass Spectrometry. Biochemistry 2004, 43, 15044–15057. 10.1021/bi048706j. [DOI] [PubMed] [Google Scholar]
- Guttman M.; Kahn M.; Garcia N. K.; Hu S. L.; Lee K. K. Solution Structure, Conformational Dynamics, and Cd4-Induced Activation in Full-Length, Glycosylated, Monomeric Hiv Gp120. J. Virol. 2012, 86, 8750–8764. 10.1128/JVI.07224-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Vachet R. W. Kinetics of Protein Complex Dissociation Studied by Hydrogen/Deuterium Exchange and Mass Spectrometry. Anal. Chem. 2015, 87, 11777–11783. 10.1021/acs.analchem.5b03123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildes D.; Marqusee S. Hydrogen Exchange and Ligand Binding: Ligand-Dependent and Ligand-Independent Protection in the Src Sh3 Domain. Protein science: a publication of the Protein Society 2005, 14, 81–88. 10.1110/ps.04990205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M.; Cupo A.; Julien J. P.; Sanders R. W.; Wilson I. A.; Moore J. P.; Lee K. K. Antibody Potency Relates to the Ability to Recognize the Closed, Pre-Fusion Form of Hiv Env. Nat. Commun. 2015, 6, 6144. 10.1038/ncomms7144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engen J. R. Analysis of Protein Complexes with Hydrogen Exchange and Mass Spectrometry. Analyst 2003, 128, 623–628. 10.1039/b212800b. [DOI] [PubMed] [Google Scholar]
- Deng B.; Lento C.; Wilson D. J. Hydrogen Deuterium Exchange Mass Spectrometry in Biopharmaceutical Discovery and Development - a Review. Anal. Chim. Acta 2016, 940, 8–20. 10.1016/j.aca.2016.08.006. [DOI] [PubMed] [Google Scholar]
- Ahn J.; Engen J. R. The Use of Hydrogen/Deuterium Exchange Mass Spectrometry in Epitope Mapping. Chimica Oggi 2013, 31, 25–28. [Google Scholar]
- Konermann L. Heavy Lessons in Protein Allostery. Nat. Struct. Mol. Biol. 2016, 23, 511–2. 10.1038/nsmb.3234. [DOI] [PubMed] [Google Scholar]
- Sowole M. A.; Simpson S.; Skovpen Y. V.; Palmer D. R. J.; Konermann L. Evidence of Allosteric Enzyme Regulation Via Changes in Conformational Dynamics: A Hydrogen/Deuterium Exchange Investigation of Dihydrodipicolinate Synthase. Biochemistry 2016, 55, 5413–5422. 10.1021/acs.biochem.6b00764. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Li J.; Craig T. A.; Kumar R.; Gross M. L. Hydrogen–Deuterium Exchange Mass Spectrometry Reveals Calcium Binding Properties and Allosteric Regulation of Downstream Regulatory Element Antagonist Modulator (Dream). Biochemistry 2017, 56, 3523–3530. 10.1021/acs.biochem.7b00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong J. J. W.; Young T. A.; Zhang J.; Liu S.; Leser G. P.; Komives E. A.; Lamb R. A.; Zhou Z. H.; Salafsky J.; Jardetzky T. S. Monomeric Ephrinb2 Binding Induces Allosteric Changes in Nipah Virus G That Precede Its Full Activation. Nat. Commun. 2017, 8, 781. 10.1038/s41467-017-00863-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagbay K. B.; Treece E.; Streich F. C.; Jackson J. W.; Faucette R. R.; Nikiforov A.; Lin S. C.; Boston C. J.; Nicholls S. B.; Capili A. D.; Carven G. J. Structural Basis of Specific Inhibition of Extracellular Activation of Pro- or Latent Myostatin by the Monoclonal Antibody Srk-015. J. Biol. Chem. 2020, 295, 5404–5418. 10.1074/jbc.RA119.012293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng B.; Lan K.; Xie J.; Lerner R. A.; Wilson I. A.; Yang B. Inhibitory Antibodies Identify Unique Sites of Therapeutic Vulnerability in Rhinovirus and Other Enteroviruses. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 13499. 10.1073/pnas.1918844117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S.; Liuni P.; Ettorre L.; Chen T.; Szeto J.; Carpick B.; James D. A.; Wilson D. J. Hydrogen–Deuterium Exchange Epitope Mapping Reveals Distinct Neutralizing Mechanisms for Two Monoclonal Antibodies against Diphtheria Toxin. Biochemistry 2019, 58, 646–656. 10.1021/acs.biochem.8b01123. [DOI] [PubMed] [Google Scholar]
- Fernandez E.; Kose N.; Edeling M. A.; Adhikari J.; Sapparapu G.; Lazarte S. M.; Nelson C. A.; Govero J.; Gross M. L.; Fremont D. H.; Crowe J. E. Jr.; Diamond M. S. Mouse and Human Monoclonal Antibodies Protect against Infection by Multiple Genotypes of Japanese Encephalitis Virus. mBio 2018, 9, e00008-18 10.1128/mBio.00008-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malito E.; Biancucci M.; Faleri A.; Ferlenghi I.; Scarselli M.; Maruggi G.; Lo Surdo P.; Veggi D.; Liguori A.; Santini L.; Bertoldi I.; Petracca R.; Marchi S.; Romagnoli G.; Cartocci E.; Vercellino I.; Savino S.; Spraggon G.; Norais N.; Pizza M.; Rappuoli R.; Masignani V.; Bottomley M. J. Structure of the Meningococcal Vaccine Antigen Nada and Epitope Mapping of a Bactericidal Antibody. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 17128–33. 10.1073/pnas.1419686111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grauslund L. R.; Calvaresi V.; Pansegrau W.; Norais N.; Rand K. D. Epitope and Paratope Mapping by Hdx-Ms Combined with Spr Elucidates the Difference in Bactericidal Activity of Two Anti-Nada Monoclonal Antibodies. J. Am. Soc. Mass Spectrom. 2021, 32, 1575–1582. 10.1021/jasms.0c00431. [DOI] [PubMed] [Google Scholar]
- Malito E.; Faleri A.; Lo Surdo P.; Veggi D.; Maruggi G.; Grassi E.; Cartocci E.; Bertoldi I.; Genovese A.; Santini L.; Romagnoli G.; Borgogni E.; Brier S.; Lo Passo C.; Domina M.; Castellino F.; Felici F.; van der Veen S.; Johnson S.; Lea S. M.; Tang C. M.; Pizza M.; Savino S.; Norais N.; Rappuoli R.; Bottomley M. J.; Masignani V. Defining a Protective Epitope on Factor H Binding Protein, a Key Meningococcal Virulence Factor and Vaccine Antigen. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 3304–9. 10.1073/pnas.1222845110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domina M.; Lanza Cariccio V.; Benfatto S.; Venza M.; Venza I.; Donnarumma D.; Bartolini E.; Borgogni E.; Bruttini M.; Santini L.; Midiri A.; Galbo R.; Romeo L.; Patanè F.; Biondo C.; Norais N.; Masignani V.; Teti G.; Felici F.; Beninati C. Epitope Mapping of a Monoclonal Antibody Directed against Neisserial Heparin Binding Antigen Using Next Generation Sequencing of Antigen-Specific Libraries. PLoS One 2016, 11, e0160702 10.1371/journal.pone.0160702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M.; Sun Z. Y.; Rand K. D.; Shi X.; Song L.; Cheng Y.; Fahmy A. F.; Majumdar S.; Ofek G.; Yang Y.; Kwong P. D.; Wang J. H.; Engen J. R.; Wagner G.; Reinherz E. L. Antibody Mechanics on a Membrane-Bound Hiv Segment Essential for Gp41-Targeted Viral Neutralization. Nat. Struct. Mol. Biol. 2011, 18, 1235–43. 10.1038/nsmb.2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornburg N. J.; Zhang H.; Bangaru S.; Sapparapu G.; Kose N.; Lampley R. M.; Bombardi R. G.; Yu Y.; Graham S.; Branchizio A.; Yoder S. M.; Rock M. T.; Creech C. B.; Edwards K. M.; Lee D.; Li S.; Wilson I. A.; García-Sastre A.; Albrecht R. A.; Crowe J. E. Jr. H7n9 Influenza Virus Neutralizing Antibodies That Possess Few Somatic Mutations. J. Clin. Invest. 2016, 126, 1482–94. 10.1172/JCI85317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puchades C.; Kűkrer B.; Diefenbach O.; Sneekes-Vriese E.; Juraszek J.; Koudstaal W.; Apetri A. Epitope Mapping of Diverse Influenza Hemagglutinin Drug Candidates Using HDX-MS. Sci. Rep. 2019, 9, 4735. 10.1038/s41598-019-41179-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen E.; Salinas N. D.; Huang Y.; Ntumngia F.; Plasencia M. D.; Gross M. L.; Adams J. H.; Tolia N. H. Broadly Neutralizing Epitopes in the Plasmodium Vivax Vaccine Candidate Duffy Binding Protein. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 6277–6282. 10.1073/pnas.1600488113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urusova D.; Carias L.; Huang Y.; Nicolete V. C.; Popovici J.; Roesch C.; Salinas N. D.; Dechavanne S.; Witkowski B.; Ferreira M. U.; Adams J. H.; Gross M. L.; King C. L.; Tolia N. H. Structural Basis for Neutralization of Plasmodium Vivax by Naturally Acquired Human Antibodies That Target Dbp. Nat. Microbiol. 2019, 4, 1486–1496. 10.1038/s41564-019-0461-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim X. X.; Chandramohan A.; Lim X. E.; Crowe J. E. Jr.; Lok S. M.; Anand G. S. Epitope and Paratope Mapping Reveals Temperature-Dependent Alterations in the Dengue-Antibody Interface. Structure 2017, 25, 1391–1402. e3 10.1016/j.str.2017.07.007. [DOI] [PubMed] [Google Scholar]
- Walls A. C.; Fiala B.; Schäfer A.; Wrenn S.; Pham M. N.; Murphy M.; Tse L. V.; Shehata L.; O’Connor M. A.; Chen C.; Navarro M. J.; Miranda M. C.; Pettie D.; Ravichandran R.; Kraft J. C.; Ogohara C.; Palser A.; Chalk S.; Lee E. C.; Guerriero K.; Kepl E.; Chow C. M.; Sydeman C.; Hodge E. A.; Brown B.; Fuller J. T.; Dinnon K. H. 3rd; Gralinski L. E.; Leist S. R.; Gully K. L.; Lewis T. B.; Guttman M.; Chu H. Y.; Lee K. K.; Fuller D. H.; Baric R. S.; Kellam P.; Carter L.; Pepper M.; Sheahan T. P.; Veesler D.; King N. P. Elicitation of Potent Neutralizing Antibody Responses by Designed Protein Nanoparticle Vaccines for Sars-Cov-2. Cell 2020, 183, 1367–1382. e17 10.1016/j.cell.2020.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandit D.; Tuske S. J.; Coales S. J.; E S. Y.; Liu A.; Lee J. E.; Morrow J. A.; Nemeth J. F.; Hamuro Y. Mapping of Discontinuous Conformational Epitopes by Amide Hydrogen/Deuterium Exchange Mass Spectrometry and Computational Docking. J. Mol. Recognit. 2012, 25, 114–24. 10.1002/jmr.1169. [DOI] [PubMed] [Google Scholar]
- Chandramohan A.; Krishnamurthy S.; Larsson A.; Nordlund P.; Jansson A.; Anand G. S. Predicting Allosteric Effects from Orthosteric Binding in Hsp90-Ligand Interactions: Implications for Fragment-Based Drug Design. PLoS Comput. Biol. 2016, 12, e1004840 10.1371/journal.pcbi.1004840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell J. G.; Falick A. M.; Komives E. A. Identification of Protein-Protein Interfaces by Decreased Amide Proton Solvent Accessibility. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 14705–10. 10.1073/pnas.95.25.14705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharmasiri K.; Smith D. L. Mass Spectrometric Determination of Isotopic Exchange Rates of Amide Hydrogens Located on the Surfaces of Proteins. Anal. Chem. 1996, 68, 2340–2344. 10.1021/ac9601526. [DOI] [PubMed] [Google Scholar]
- Peacock R. B.; Davis J. R.; Markwick P. R. L.; Komives E. A. Dynamic Consequences of Mutation of Tryptophan 215 in Thrombin. Biochemistry 2018, 57, 2694–2703. 10.1021/acs.biochem.8b00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng B.; Zhu S.; Macklin A. M.; Xu J.; Lento C.; Sljoka A.; Wilson D. J. Suppressing Allostery in Epitope Mapping Experiments Using Millisecond Hydrogen/Deuterium Exchange Mass Spectrometry. mAbs 2017, 9, 1327–1336. 10.1080/19420862.2017.1379641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resetca D.; Wilson D. J. Characterizing Rapid, Activity-Linked Conformational Transitions in Proteins Via Sub-Second Hydrogen Deuterium Exchange Mass Spectrometry. FEBS J. 2013, 280, 5616–5625. 10.1111/febs.12332. [DOI] [PubMed] [Google Scholar]
- Donnarumma D.; Faleri A.; Costantino P.; Rappuoli R.; Norais N. The Role of Structural Proteomics in Vaccine Development: Recent Advances and Future Prospects. Expert Rev. Proteomics 2016, 13, 55–68. 10.1586/14789450.2016.1121113. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Noble K. A.; Mao Y.; Young N. L.; Sathe S. K.; Roux K. H.; Marshall A. G. Rapid Screening for Potential Epitopes Reactive with a Polycolonal Antibody by Solution-Phase H/D Exchange Monitored by Ft-Icr Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2013, 24, 1016–25. 10.1007/s13361-013-0644-7. [DOI] [PubMed] [Google Scholar]
- Yang D.; Frego L.; Lasaro M.; Truncali K.; Kroe-Barrett R.; Singh S. Efficient Qualitative and Quantitative Determination of Antigen-Induced Immune Responses. J. Biol. Chem. 2016, 291, 16361–74. 10.1074/jbc.M116.736660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pra̧dzińska M.; Behrendt I.; Astorga-Wells J.; Manoilov A.; Zubarev R. A.; Kołodziejczyk A. S.; Rodziewicz-Motowidło S.; Czaplewska P. Application of Amide Hydrogen/Deuterium Exchange Mass Spectrometry for Epitope Mapping in Human Cystatin C. Amino Acids 2016, 48, 2809–2820. 10.1007/s00726-016-2316-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar Barroeta A.; van Galen J.; Stroo I.; Marquart J. A.; Meijer A. B.; Meijers J. C. M. Hydrogen-Deuterium Exchange Mass Spectrometry Highlights Conformational Changes Induced by Factor Xi Activation and Binding of Factor Ix to Factor Xia. J. Thromb. Haemostasis 2019, 17, 2047–2055. 10.1111/jth.14632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeppe J. R.; Seitova A.; Mather T.; Komives E. A. Thrombomodulin Tightens the Thrombin Active Site Loops to Promote Protein C Activation. Biochemistry 2005, 44, 14784–91. 10.1021/bi0510577. [DOI] [PubMed] [Google Scholar]
- Handley L. D.; Treuheit N. A.; Venkatesh V. J.; Komives E. A. Thrombomodulin Binding Selects the Catalytically Active Form of Thrombin. Biochemistry 2015, 54, 6650–6658. 10.1021/acs.biochem.5b00825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karamitros C. S.; Murray K.; Sugiyama Y.; Kumada Y.; Johnson K. A.; Georgiou G.; D’Arcy S.; Stone E. M. Conformational Dynamics Contribute to Substrate Selectivity and Catalysis in Human Kynureninase. ACS Chem. Biol. 2020, 15, 3159–3166. 10.1021/acschembio.0c00676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson E. J.; Paul A.; Iavarone A. T.; Klinman J. P. Identification of Thermal Conduits That Link the Protein-Water Interface to the Active Site Loop and Catalytic Base in Enolase. J. Am. Chem. Soc. 2021, 143, 785–797. 10.1021/jacs.0c09423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S.; Thompson E. J.; Barrow S. L.; Zhang W.; Iavarone A. T.; Klinman J. P. Hydrogen-Deuterium Exchange within Adenosine Deaminase, a Tim Barrel Hydrolase, Identifies Networks for Thermal Activation of Catalysis. J. Am. Chem. Soc. 2020, 142, 19936–19949. 10.1021/jacs.0c07866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S.; Offenbacher A. R.; Thompson E. M.; Gee C. L.; Wilcoxen J.; Carr C. A. M.; Prigozhin D. M.; Yang V.; Alber T.; Britt R. D.; Fraser J. S.; Klinman J. P. Biophysical Characterization of a Disabled Double Mutant of Soybean Lipoxygenase: The “Undoing” of Precise Substrate Positioning Relative to Metal Cofactor and an Identified Dynamical Network. J. Am. Chem. Soc. 2019, 141, 1555–1567. 10.1021/jacs.8b10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offenbacher A. R.; Iavarone A. T.; Klinman J. P. Hydrogen-Deuterium Exchange Reveals Long-Range Dynamical Allostery in Soybean Lipoxygenase. J. Biol. Chem. 2018, 293, 1138–1148. 10.1074/jbc.M117.817197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyukhtenko S.; Ma X.; Rajarshi G.; Karageorgos I.; Anderson K. W.; Hudgens J. W.; Guo J. J.; Nasr M. L.; Zvonok N.; Vemuri K.; Wagner G.; Makriyannis A. Conformational Gating, Dynamics and Allostery in Human Monoacylglycerol Lipase. Sci. Rep. 2020, 10, 18531. 10.1038/s41598-020-75497-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corless E. I.; Saad Imran S. M.; Watkins M. B.; Bacik J. P.; Mattice J. R.; Patterson A.; Danyal K.; Soffe M.; Kitelinger R.; Seefeldt L. C.; Origanti S.; Bennett B.; Bothner B.; Ando N.; Antony E. The Flexible N-Terminus of Bchl Autoinhibits Activity through Interaction with Its [4fe-4s] Cluster and Released Upon Atp Binding. J. Biol. Chem. 2021, 296, 100107. 10.1074/jbc.RA120.016278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsirigotaki A.; Elzen R. V.; Veken P. V. D.; Lambeir A.-M.; Economou A. Dynamics and Ligand-Induced Conformational Changes in Human Prolyl Oligopeptidase Analyzed by Hydrogen/Deuterium Exchange Mass Spectrometry. Sci. Rep. 2017, 7, 2456. 10.1038/s41598-017-02550-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fast C. S.; Vahidi S.; Konermann L. Changes in Enzyme Structural Dynamics Studied by Hydrogen Exchange-Mass Spectrometry: Ligand Binding Effects or Catalytically Relevant Motions?. Anal. Chem. 2017, 89, 13326–13333. 10.1021/acs.analchem.7b03506. [DOI] [PubMed] [Google Scholar]
- Fassler R.; Edinger N.; Rimon O.; Reichmann D. Defining Hsp33’s Redox-Regulated Chaperone Activity and Mapping Conformational Changes on Hsp33 Using Hydrogen-Deuterium Exchange Mass Spectrometry. J. Visualized Exp. 2018, 136, 57806–57806. 10.3791/57806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clouser A. F.; Baughman H. E.; Basanta B.; Guttman M.; Nath A.; Klevit R. E. Interplay of Disordered and Ordered Regions of a Human Small Heat Shock Protein Yields an Ensemble of ‘Quasi-Ordered’ States. eLife 2019, 8, e50259 10.7554/eLife.50259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgescauld F.; Wales T. E.; Engen J. R. Hydrogen Deuterium Exchange Mass Spectrometry Applied to Chaperones and Chaperone-Assisted Protein Folding. Expert Rev. Proteomics 2019, 16, 613–625. 10.1080/14789450.2019.1633920. [DOI] [PubMed] [Google Scholar]
- Singh A. K.; Balchin D.; Imamoglu R.; Hayer-Hartl M.; Hartl F. U. Efficient Catalysis of Protein Folding by Groel/Es of the Obligate Chaperonin Substrate Metf. J. Mol. Biol. 2020, 432, 2304–2318. 10.1016/j.jmb.2020.02.031. [DOI] [PubMed] [Google Scholar]
- Imamoglu R.; Balchin D.; Hayer-Hartl M.; Hartl F. U. Bacterial Hsp70 Resolves Misfolded States and Accelerates Productive Folding of a Multi-Domain Protein. Nat. Commun. 2020, 11, 365. 10.1038/s41467-019-14245-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandova V.; Vankova P.; Durech M.; Houser J.; Kavan D.; Man P.; Muller P.; Trcka F. Hspa1a Conformational Mutants Reveal a Conserved Structural Unit in Hsp70 Proteins. Biochim. Biophys. Acta, Gen. Subj. 2020, 1864, 129458. 10.1016/j.bbagen.2019.129458. [DOI] [PubMed] [Google Scholar]
- Trcka F.; Durech M.; Vankova P.; Chmelik J.; Martinkova V.; Hausner J.; Kadek A.; Marcoux J.; Klumpler T.; Vojtesek B.; Muller P.; Man P. Human Stress-Inducible Hsp70 Has a High Propensity to Form Atp-Dependent Antiparallel Dimers That Are Differentially Regulated by Cochaperone Binding. Mol. Cell Proteomics 2019, 18, 320–337. 10.1074/mcp.RA118.001044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel A.; McAvoy C. Z.; Lam V.; Liang F. C.; Kroon G.; Miaou E.; Griffin P.; Wright P. E.; Shan S. O. A Disorder-to-Order Transition Activates an Atp-Independent Membrane Protein Chaperone. J. Mol. Biol. 2020, 432, 166708. 10.1016/j.jmb.2020.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese A. N.; Schiffrin B.; Watson M.; Karamanos T. K.; Walko M.; Humes J. R.; Horne J. E.; White P.; Wilson A. J.; Kalli A. C.; Tuma R.; Ashcroft A. E.; Brockwell D. J.; Radford S. E. Inter-Domain Dynamics in the Chaperone Sura and Multi-Site Binding to Its Outer Membrane Protein Clients. Nat. Commun. 2020, 11, 2155. 10.1038/s41467-020-15702-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coufalova D.; Remnant L.; Hernychova L.; Muller P.; Healy A.; Kannan S.; Westwood N.; Verma C. S.; Vojtesek B.; Hupp T. R.; Houston D. R. An Inter-Subunit Protein-Peptide Interface That Stabilizes the Specific Activity and Oligomerization of the Aaa+ Chaperone Reptin. J. Proteomics 2019, 199, 89–101. 10.1016/j.jprot.2019.02.012. [DOI] [PubMed] [Google Scholar]
- Ghode A.; Gross L. Z. F.; Tee W. V.; Guarnera E.; Berezovsky I. N.; Biondi R. M.; Anand G. S. Synergistic Allostery in Multiligand-Protein Interactions. Biophys. J. 2020, 119, 1833–1848. 10.1016/j.bpj.2020.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J. Y.; Qu C. X.; Li R. R.; Yang F.; Yu X.; Tian Z. M.; Shen Y. M.; Cai B. Y.; Yun Y.; Sun J. P.; Chung K. Y. Structural Mechanism of the Arrestin-3/Jnk3 Interaction. Structure 2019, 27, 1162–1170. e3 10.1016/j.str.2019.04.002. [DOI] [PubMed] [Google Scholar]
- McPhail J. A.; Lyoo H.; Pemberton J. G.; Hoffmann R. M.; van Elst W.; Strating J.; Jenkins M. L.; Stariha J. T. B.; Powell C. J.; Boulanger M. J.; Balla T.; van Kuppeveld F. J. M.; Burke J. E. Characterization of the C10orf76-Pi4kb Complex and Its Necessity for Golgi Pi4p Levels and Enterovirus Replication. EMBO Rep. 2020, 21, e48441 10.15252/embr.201948441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitz S. D.; Hamelin D. J.; Hoffmann R. M.; Greenberg N.; Salloum G.; Erami Z.; Khalil B. D.; Shymanets A.; Steidle E. A.; Gong G. Q.; Nürnberg B.; Burke J. E.; Flanagan J. U.; Bresnick A. R.; Backer J. M. A Single Discrete Rab5-Binding Site in Phosphoinositide 3-Kinase B Is Required for Tumor Cell Invasion. J. Biol. Chem. 2019, 294, 4621–4633. 10.1074/jbc.RA118.006032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loving H. S.; Underbakke E. S. Conformational Dynamics of Ferm-Mediated Autoinhibition in Pyk2 Tyrosine Kinase. Biochemistry 2019, 58, 3767–3776. 10.1021/acs.biochem.9b00541. [DOI] [PubMed] [Google Scholar]
- Chen M.; Pan H.; Sun L.; Shi P.; Zhang Y.; Li L.; Huang Y.; Chen J.; Jiang P.; Fang X.; Wu C.; Chen Z. Structure and Regulation of Human Epithelial Cell Transforming 2 Protein. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 1027–1035. 10.1073/pnas.1913054117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anandapadamanaban M.; Masson G. R.; Perisic O.; Berndt A.; Kaufman J.; Johnson C. M.; Santhanam B.; Rogala K. B.; Sabatini D. M.; Williams R. L. Architecture of Human Rag Gtpase Heterodimers and Their Complex with Mtorc1. Science 2019, 366, 203–210. 10.1126/science.aax3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPherson D. J.; Mills C. L.; Ondrechen M. J.; Hardy J. A. Tri-Arginine Exosite Patch of Caspase-6 Recruits Substrates for Hydrolysis. J. Biol. Chem. 2019, 294, 71–88. 10.1074/jbc.RA118.005914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumpkin R. J.; Ahmad A. S.; Blake R.; Condon C. J.; Komives E. A. The Mechanism of Nedd8 Activation of Cul5 Ubiquitin E3 Ligases. Mol. Cell Proteomics 2021, 20, 100019. 10.1074/mcp.RA120.002414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumpkin R. J.; Baker R. W.; Leschziner A. E.; Komives E. A. Structure and Dynamics of the Asb9 Cul-Ring E3 Ligase. Nat. Commun. 2020, 11, 2866. 10.1038/s41467-020-16499-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faull S. V.; Lau A. M. C.; Martens C.; Ahdash Z.; Hansen K.; Yebenes H.; Schmidt C.; Beuron F.; Cronin N. B.; Morris E. P.; Politis A. Structural Basis of Cullin 2 Ring E3 Ligase Regulation by the Cop9 Signalosome. Nat. Commun. 2019, 10, 3814. 10.1038/s41467-019-11772-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook M.; Delbecq S. P.; Schweppe T. P.; Guttman M.; Klevit R. E.; Brzovic P. S. The Ubiquitin Ligase Ssph1 from Salmonella Uses a Modular and Dynamic E3 Domain to Catalyze Substrate Ubiquitylation. J. Biol. Chem. 2019, 294, 783–793. 10.1074/jbc.RA118.004247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison R. A.; Engen J. R. Conformational Insight into Multi-Protein Signaling Assemblies by Hydrogen-Deuterium Exchange Mass Spectrometry. Curr. Opin. Struct. Biol. 2016, 41, 187–193. 10.1016/j.sbi.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad F.; Patterson A.; Deveryshetty J.; Mattice J. R.; Pokhrel N.; Bothner B.; Antony E. Hydrogen–Deuterium Exchange Reveals a Dynamic DNA-Binding Map of Replication Protein A. Nucleic Acids Res. 2021, 49, 1455–1469. 10.1093/nar/gkaa1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J.; Chang M. R.; Stites R. E.; Wang Y.; Bruning J. B.; Pascal B. D.; Novick S. J.; Garcia-Ordonez R. D.; Stayrook K. R.; Chalmers M. J.; Dodge J. A.; Griffin P. R. Hdx Reveals the Conformational Dynamics of DNA Sequence Specific Vdr Co-Activator Interactions. Nat. Commun. 2017, 8, 923. 10.1038/s41467-017-00978-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrales-Guerrero L.; He B.; Refes Y.; Panis G.; Bange G.; Viollier P. H.; Steinchen W.; Thanbichler M. Molecular Architecture of the DNA-Binding Sites of the P-Loop Atpases Mipz and Para from Caulobacter Crescentus. Nucleic Acids Res. 2020, 48, 4769–4779. 10.1093/nar/gkaa192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J.; Cui K.; Shen L.; Shi J.; Li L.; You L.; Fang C.; Zhao G.; Feng Y.; Yang B.; Zhang Y. Crl Activates Transcription by Stabilizing Active Conformation of the Master Stress Transcription Initiation Factor. eLife 2019, 8, e50928. 10.7554/eLife.50928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastie K. M.; Liu T.; Li S.; King L. B.; Ngo N.; Zandonatti M. A.; Woods V. L. Jr.; de la Torre J. C.; Saphire E. O. Crystal Structure of the Lassa Virus Nucleoprotein-Rna Complex Reveals a Gating Mechanism for Rna Binding. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 19365–19370. 10.1073/pnas.1108515108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina E.; Villalobos P.; Hamilton G. L.; Komives E. A.; Sanabria H.; Ramírez-Sarmiento C. A.; Babul J. Intrinsically Disordered Regions of the DNA-Binding Domain of Human Foxp1 Facilitate Domain Swapping. J. Mol. Biol. 2020, 432, 5411–5429. 10.1016/j.jmb.2020.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey K. M.; Narang D.; Komives E. A. Prediction of the Presence of a Seventh Ankyrin Repeat in Iκbε; from Homology Modeling Combined with Hydrogen-Deuterium Exchange Mass Spectrometry (Hdx-Ms). Protein Sci. 2018, 27, 1624–1635. 10.1002/pro.3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narang D.; Chen W.; Ricci C. G.; Komives E. A. Rela-Containing Nfκb Dimers Have Strikingly Different DNA-Binding Cavities in the Absence of DNA. J. Mol. Biol. 2018, 430, 1510–1520. 10.1016/j.jmb.2018.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trelle M. B.; Ramsey K. M.; Lee T. C.; Zheng W.; Lamboy J.; Wolynes P. G.; Deniz A.; Komives E. A. Binding of Nfκb Appears to Twist the Ankyrin Repeat Domain of Iκbα. Biophys. J. 2016, 110, 887–95. 10.1016/j.bpj.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H. T.; Bobst C. E.; Iwig J. S.; Chivers P. T.; Kaltashov I. A.; Maroney M. J. Co(Ii) and Ni(Ii) Binding of the Escherichia Coli Transcriptional Repressor Rcnr Orders Its N Terminus, Alters Helix Dynamics, and Reduces DNA Affinity. J. Biol. Chem. 2018, 293, 324–332. 10.1074/jbc.RA117.000398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke J. E.; Karbarz M. J.; Deems R. A.; Li S.; Woods V. L.; Dennis E. A. Interaction of Group Ia Phospholipase A2 with Metal Ions and Phospholipid Vesicles Probed with Deuterium Exchange Mass Spectrometry. Biochemistry 2008, 47, 6451–6459. 10.1021/bi8000962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudlur A.; Zeraik A. E.; Hirve N.; Rajanikanth V.; Bobkov A. A.; Ma G.; Zheng S.; Wang Y.; Zhou Y.; Komives E. A.; Hogan P. G. Calcium Sensing by the Stim1 Er-Luminal Domain. Nat. Commun. 2018, 9, 4536. 10.1038/s41467-018-06816-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazur S. J.; Gallagher E. S.; Debnath S.; Durell S. R.; Anderson K. W.; Miller Jenkins L. M.; Appella E.; Hudgens J. W. Conformational Changes in Active and Inactive States of Human Pp2cα Characterized by Hydrogen/Deuterium Exchange–Mass Spectrometry. Biochemistry 2017, 56, 2676–2689. 10.1021/acs.biochem.6b01220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houde D.; Berkowitz S. A. Conformational Comparability of Factor Ix–Fc Fusion Protein, Factor Ix, and Purified Fc Fragment in the Absence and Presence of Calcium. J. Pharm. Sci. 2012, 101, 1688–1700. 10.1002/jps.23064. [DOI] [PubMed] [Google Scholar]
- Skorupska A.; Bystranowska D.; Da̧browska K.; Ożyhar A. Calcium Ions Modulate the Structure of the Intrinsically Disordered Nucleobindin-2 Protein. Int. J. Biol. Macromol. 2020, 154, 1091–1104. 10.1016/j.ijbiomac.2020.03.110. [DOI] [PubMed] [Google Scholar]
- Seyfried N. T.; Atwood J. A. 3rd; Yongye A.; Almond A.; Day A. J.; Orlando R.; Woods R. J. Fourier Transform Mass Spectrometry to Monitor Hyaluronan-Protein Interactions: Use of Hydrogen/Deuterium Amide Exchange. Rapid Commun. Mass Spectrom. 2007, 21, 121–31. 10.1002/rcm.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperry J. B.; Huang R. Y. C.; Zhu M. M.; Rempel D. L.; Gross M. L. Hydrophobic Peptides Affect Binding of Calmodulin and Ca as Explored by H/D Amide Exchange and Mass Spectrometry. Int. J. Mass Spectrom. 2011, 302, 85–92. 10.1016/j.ijms.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranker A.; Robinson C. V.; Radford S. E.; Dobson C. M. Investigation of Protein Folding by Mass Spectrometry. FASEB J. 1996, 10, 93–101. 10.1096/fasebj.10.1.8566553. [DOI] [PubMed] [Google Scholar]
- Yang H.; Smith D. L. Kinetics of Cytochrome C Folding Examined by Hydrogen Exchange and Mass Spectrometry. Biochemistry 1997, 36, 14992–9. 10.1021/bi9717183. [DOI] [PubMed] [Google Scholar]
- Deng Y.; Zhang Z.; Smith D. L. Focus: H/D Exchange of Proteins in Solution Comparison of Continuous and Pulsed Labeling Amide Hydrogen Exchange/Mass Spectrometry for Studies of Protein Dynamics. J. Am. Soc. Mass Spectrom. 1999, 10, 675–84. 10.1016/S1044-0305(99)00038-0. [DOI] [PubMed] [Google Scholar]
- Deng Y.; Smith D. L. Rate and Equilibrium Constants for Protein Unfolding and Refolding Determined by Hydrogen Exchange-Mass Spectrometry. Anal. Biochem. 1999, 276, 150–60. 10.1006/abio.1999.4347. [DOI] [PubMed] [Google Scholar]
- Englander S. W.; Mayne L.; Kan Z. Y.; Hu W. Protein Folding-How and Why: By Hydrogen Exchange, Fragment Separation, and Mass Spectrometry. Annu. Rev. Biophys. 2016, 45, 135–152. 10.1146/annurev-biophys-062215-011121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura C.; Beppu T. Review Folding of Apomyoglobin: Analysis of Transient Intermediate Structure During Refolding Using Quick Hydrogen Deuterium Exchange and Nmr. Proc. Jpn. Acad., Ser. B 2017, 93, 10–27. 10.2183/pjab.93.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W.; Kan Z. Y.; Mayne L.; Englander S. W. Cytochrome C Folds through Foldon-Dependent Native-Like Intermediates in an Ordered Pathway. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 3809–3814. 10.1073/pnas.1522674113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Liu Z.; Creagh J.; Zheng R.; McDonald T. V. Physical and Functional Interaction Sites in Cytoplasmic Domains of Kcnq1 and Kcne1 Channel Subunits. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H212–H222. 10.1152/ajpheart.00459.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysling S.; Kristensen K. K.; Larsson M.; Beigneux A. P.; GåRdsvoll H.; Fong L. G.; Bensadouen A.; Jørgensen T. J.; Young S. G.; Ploug M. The Acidic Domain of the Endothelial Membrane Protein Gpihbp1 Stabilizes Lipoprotein Lipase Activity by Preventing Unfolding of Its Catalytic Domain. eLife 2016, 5, e12095 10.7554/eLife.12095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysling S.; Kristensen K. K.; Larsson M.; Kovrov O.; Bensadouen A.; Jørgensen T. J.; Olivecrona G.; Young S. G.; Ploug M. The Angiopoietin-Like Protein Angptl4 Catalyzes Unfolding of the Hydrolase Domain in Lipoprotein Lipase and the Endothelial Membrane Protein Gpihbp1 Counteracts This Unfolding. eLife 2016, 5, e20958. 10.7554/eLife.20958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dornan G. L.; Dalwadi U.; Hamelin D. J.; Hoffmann R. M.; Yip C. K.; Burke J. E. Probing the Architecture, Dynamics, and Inhibition of the Pi4kiiiα/Ttc7/Fam126 Complex. J. Mol. Biol. 2018, 430, 3129–3142. 10.1016/j.jmb.2018.07.020. [DOI] [PubMed] [Google Scholar]
- Benhaim M. A.; Mangala Prasad V.; Garcia N. K.; Guttman M.; Lee K. K. Structural Monitoring of a Transient Intermediate in the Hemagglutinin Fusion Machinery on Influenza Virions. Sci. Adv. 2020, 6, eaaz8822 10.1126/sciadv.aaz8822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Shu Q.; Rempel D. L.; Frieden C.; Gross M. L. Continuous and Pulsed Hydrogen-Deuterium Exchange and Mass Spectrometry Characterize Csge Oligomerization. Biochemistry 2015, 54, 6475–81. 10.1021/acs.biochem.5b00871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Shu Q.; Frieden C.; Gross M. L. Deamidation Slows Curli Amyloid-Protein Aggregation. Biochemistry 2017, 56, 2865–2872. 10.1021/acs.biochem.7b00241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabareesan A. T.; Udgaonkar J. B. Pathogenic Mutations within the Disordered Palindromic Region of the Prion Protein Induce Structure Therein and Accelerate the Formation of Misfolded Oligomers. J. Mol. Biol. 2016, 428, 3935–3947. 10.1016/j.jmb.2016.08.015. [DOI] [PubMed] [Google Scholar]
- Renawala H. K.; Chandrababu K. B.; Topp E. M. Fibrillation of Human Calcitonin and Its Analogs: Effects of Phosphorylation and Disulfide Reduction. Biophys. J. 2021, 120, 86–100. 10.1016/j.bpj.2020.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M. M.; Rempel D. L.; Du Z.; Gross M. L. Quantification of Protein-Ligand Interactions by Mass Spectrometry, Titration, and H/D Exchange: Plimstex. J. Am. Chem. Soc. 2003, 125, 5252–3. 10.1021/ja029460d. [DOI] [PubMed] [Google Scholar]
- Tu T.; Drǎgußanu M.; Petre B. A.; Rempel D. L.; Przybylski M.; Gross M. L. Protein-Peptide Affinity Determination Using an H/D Exchange Dilution Strategy: Application to Antigen-Antibody Interactions. J. Am. Soc. Mass Spectrom. 2010, 21, 1660. 10.1016/j.jasms.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopper E. D.; Pittman A. M.; Tucker C. L.; Campa M. J.; Patz E. F. Jr.; Fitzgerald M. C. Hydrogen/Deuterium Exchange- and Protease Digestion-Based Screening Assay for Protein-Ligand Binding Detection. Anal. Chem. 2009, 81, 6860–6867. 10.1021/ac900854t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pancsa R.; Raimondi D.; Cilia E.; Vranken W. F. Article Early Folding Events, Local Interactions, and Conservation of Protein Backbone Rigidity. Biophys. J. 2016, 110, 572–583. 10.1016/j.bpj.2015.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimondi D.; Orlando G.; Pancsa R.; Khan T.; Vranken W. F. Exploring the Sequence-Based Prediction of Folding Initiation Sites in Proteins. Sci. Rep. 2017, 7, 8826. 10.1038/s41598-017-08366-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsirigotaki A.; Papanastasiou M.; Trelle M. B.; Jorgensen T. J.; Economou A. Analysis of Translocation-Competent Secretory Proteins by Hdx-Ms. Methods Enzymol. 2017, 586, 57–83. 10.1016/bs.mie.2016.09.028. [DOI] [PubMed] [Google Scholar]
- Uversky V. N.; Oldfield C. J.; Dunker A. K. Intrinsically Disordered Proteins in Human Diseases: Introducing the D2 Concept. Annu. Rev. Biophys. 2008, 37, 215–46. 10.1146/annurev.biophys.37.032807.125924. [DOI] [PubMed] [Google Scholar]
- Uversky V. N. A Decade and a Half of Protein Intrinsic Disorder: Biology Still Waits for Physics. Protein Sci. 2013, 22, 693–724. 10.1002/pro.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Lee R.; Buljan M.; Lang B.; Weatheritt R. J.; Daughdrill G. W.; Dunker A. K.; Fuxreiter M.; Gough J.; Gsponer J.; Jones D. T.; Kim P. M.; Kriwacki R. W.; Oldfield C. J.; Pappu R. V.; Tompa P.; Uversky V. N.; Wright P. E.; Babu M. M. Classification of Intrinsically Disordered Regions and Proteins. Chem. Rev. 2014, 114, 6589–631. 10.1021/cr400525m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babinchak W. M.; Surewicz W. K. Liquid-Liquid Phase Separation and Its Mechanistic Role in Pathological Protein Aggregation. J. Mol. Biol. 2020, 432, 1910–1925. 10.1016/j.jmb.2020.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nwanochie E.; Uversky V. N. Structure Determination by Single-Particle Cryo-Electron Microscopy: Only the Sky (and Intrinsic Disorder) Is the Limit. Int. J. Mol. Sci. 2019, 20, 4186. 10.3390/ijms20174186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu C. C.; Buehler M. J.; Tarakanova A. The Order-Disorder Continuum: Linking Predictions of Protein Structure and Disorder through Molecular Simulation. Sci. Rep. 2020, 10, 2068. 10.1038/s41598-020-58868-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra G. Emerging Role of Mass Spectrometry-Based Structural Proteomics in Elucidating Intrinsic Disorder in Proteins. Proteomics 2021, 21, 2000011. 10.1002/pmic.202000011. [DOI] [PubMed] [Google Scholar]
- Keppel T. R.; Weis D. D. Analysis of Disordered Proteins Using a Simple Apparatus for Millisecond Quench-Flow H/D Exchange. Anal. Chem. 2013, 85, 5161–5168. 10.1021/ac4004979. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Zhang A.; Xiao G. Improved Protein Hydrogen/Deuterium Exchange Mass Spectrometry Platform with Fully Automated Data Processing. Anal. Chem. 2012, 84, 4942–4949. 10.1021/ac300535r. [DOI] [PubMed] [Google Scholar]
- Mori S.; Van Zijl P. C. M.; Shortle D. Measurement of Water-Amide Proton Exchange Rates in the Denatured State of Staphylococcal Nuclease by a Magnetization Transfer Technique. Proteins: Struct., Funct., Genet. 1997, 28, 325–332. . [DOI] [PubMed] [Google Scholar]
- Del Mar C.; Greenbaum E. A.; Mayne L.; Englander S. W.; Woods V. L. Jr. Structure and Properties of Alpha-Synuclein and Other Amyloids Determined at the Amino Acid Level. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 15477–15482. 10.1073/pnas.0507405102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters B. T. Empirical Method to Accurately Determine Peptide-Averaged Protection Factors from Hydrogen Exchange Ms Data. Anal. Chem. 2017, 89, 1049–1053. 10.1021/acs.analchem.6b03908. [DOI] [PubMed] [Google Scholar]
- Keppel T. R.; Howard B. A.; Weis D. D. Mapping Unstructured Regions and Synergistic Folding in Intrinsically Disordered Proteins with Amide H/D Exchange Mass Spectrometry. Biochemistry 2011, 50, 8722–8732. 10.1021/bi200875p. [DOI] [PubMed] [Google Scholar]
- Rob T.; Liuni P.; Gill P. K.; Zhu S.; Balachandran N.; Berti P. J.; Wilson D. J. Measuring Dynamics in Weakly Structured Regions of Proteins Using Microfluidics-Enabled Subsecond H/D Exchange Mass Spectrometry. Anal. Chem. 2012, 84, 3771–9. 10.1021/ac300365u. [DOI] [PubMed] [Google Scholar]
- Rist W.; Rodriguez F.; Jørgensen T. J.; Mayer M. P. Analysis of Subsecond Protein Dynamics by Amide Hydrogen Exchange and Mass Spectrometry Using a Quenched-Flow Setup. Protein Sci. 2005, 14, 626–32. 10.1110/ps.041098305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keppel T. R.; Sarpong K.; Murray E. M.; Monsey J.; Zhu J.; Bose R. Biophysical Evidence for Intrinsic Disorder in the C-Terminal Tails of the Epidermal Growth Factor Receptor (Egfr) and Her3 Receptor Tyrosine Kinases. J. Biol. Chem. 2017, 292, 597–610. 10.1074/jbc.M116.747485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svejdal R. R.; Dickinson E. R.; Sticker D.; Kutter J. P.; Rand K. D. Thiol-Ene Microfluidic Chip for Performing Hydrogen/Deuterium Exchange of Proteins at Subsecond Time Scales. Anal. Chem. 2019, 91, 1309–1317. 10.1021/acs.analchem.8b03050. [DOI] [PubMed] [Google Scholar]
- Wilson D. J.; Konermann L. A Capillary Mixer with Adjustable Reaction Chamber Volume for Millisecond Time-Resolved Studies by Electrospray Mass Spectrometry. Anal. Chem. 2003, 75, 6408–6414. 10.1021/ac0346757. [DOI] [PubMed] [Google Scholar]
- Lento C.; Zhu S.; Brown K. A.; Knox R.; Liuni P.; Wilson D. J. Time-Resolved Electrospray Ionization Hydrogen-Deuterium Exchange Mass Spectrometry for Studying Protein Structure and Dynamics. J. Visualized Exp. 2017, 122, 55464. 10.3791/55464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S.; Khatun R.; Lento C.; Sheng Y.; Wilson D. J. Enhanced Binding Affinity Via Destabilization of the Unbound State: A Millisecond Hydrogen-Deuterium Exchange Study of the Interaction between P53 and a Pleckstrin Homology Domain. Biochemistry 2017, 56, 4127–4133. 10.1021/acs.biochem.7b00193. [DOI] [PubMed] [Google Scholar]
- Cieplak-Rotowska M. K.; Tarnowski K.; Rubin M.; Fabian M. R.; Sonenberg N.; Dadlez M.; Niedzwiecka A. Structural Dynamics of the Gw182 Silencing Domain Including Its Rna Recognition Motif (Rrm) Revealed by Hydrogen-Deuterium Exchange Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2018, 29, 158–173. 10.1007/s13361-017-1830-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beveridge R.; Phillips A. S.; Denbigh L.; Saleem H. M.; Macphee C. E.; Barran P. E. Relating Gas Phase to Solution Conformations: Lessons from Disordered Proteins. Proteomics 2015, 15, 2872–2883. 10.1002/pmic.201400605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarczewska A.; Kozłowska M.; Dobryszycki P.; Kaus-Drobek M.; Dadlez M.; Ożyhar A. Insight into the Unfolding Properties of Chd64, a Small, Single Domain Protein with a Globular Core and Disordered Tails. PLoS One 2015, 10, e0137074 10.1371/journal.pone.0137074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson G. R.; Perisic O.; Burke J. E.; Williams R. L. The Intrinsically Disordered Tails of Pten and Pten-L Have Distinct Roles in Regulating Substrate Specificity and Membrane Activity. Biochem. J. 2016, 473, 135–144. 10.1042/BJ20150931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell J. L.; Trible R. P.; Emert-Sedlak L. A.; Weis D. D.; Lerner E. C.; Applen J. J.; Sefton B. M.; Smithgall T. E.; Engen J. R. Functional Characterization and Conformational Analysis of the Herpesvirus Saimiri Tip-C484 Protein. J. Mol. Biol. 2007, 366, 1282–93. 10.1016/j.jmb.2006.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcsisin S. R.; Narute P. S.; Emert-Sedlak L. A.; Kloczewiak M.; Smithgall T. E.; Engen J. R. On the Solution Conformation and Dynamics of the Hiv-1 Viral Infectivity Factor. J. Mol. Biol. 2011, 410, 1008–22. 10.1016/j.jmb.2011.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatalska A.; Dzhindzhev N. S.; Dadlez M.; Glover D. M. Interaction Interface in the C-Terminal Parts of Centriole Proteins Sas6 and Ana2. Open Biol. 2020, 10, 200221. 10.1098/rsob.200221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts V. A.; Pique M. E.; Hsu S.; Li S. Combining H/D Exchange Mass Spectrometry and Computational Docking to Derive the Structure of Protein-Protein Complexes. Biochemistry 2017, 56, 6329–6342. 10.1021/acs.biochem.7b00643. [DOI] [PubMed] [Google Scholar]
- Killoran R. C.; Sowole M. A.; Halim M. A.; Konermann L.; Choy W.-Y. Conformational Characterization of the Intrinsically Disordered Protein Chibby: Interplay between Structural Elements in Target Recognition. Protein Sci. 2016, 25, 1420–1429. 10.1002/pro.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorosz J.; Hyltoft Kristensen L.; Aduri N. G.; Mirza o.; Lousen R.; Bucciarelli s.; Mehta V.; Sellés-Baiget S.; Øie Solbak S. M.; Bach A.; Mesa p.; Alcon Hernandez p.; Montoya G.; Nguyen T. T. N.; Rand K. D.; Boesen t.; Gajhede M. Molecular Architecture of the Jumonji C Family Histone Demethylase Kdm5b. Sci. Rep. 2019, 9, 4019. 10.1038/s41598-019-40573-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X.; Yokom A. L.; Wang C.; Young L. N.; Youle R. J.; Hurley J. H. Ulk Complex Organization in Autophagy by a C-Shaped Fip200 N-Terminal Domain Dimer. J. Cell Biol. 2020, 219, e201911047. 10.1083/jcb.201911047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandacaru S. C.; do Vale L. H. F.; Vahidi S.; Xiao Y.; Skinner O. S.; Ricart C. A. O.; Kelleher N. L.; de Sousa M. V.; Konermann L. Characterizing the Structure and Oligomerization of Major Royal Jelly Protein 1 (Mrjp1) by Mass Spectrometry and Complementary Biophysical Tools. Biochemistry 2017, 56, 1645–1655. 10.1021/acs.biochem.7b00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su M. Y.; Morris K. L.; Kim D. J.; Fu Y.; Lawrence R.; Stjepanovic G.; Zoncu R.; Hurley J. H. Hybrid Structure of the Raga/C-Ragulator Mtorc1 Activation Complex. Mol. Cell 2017, 68, 835–846. e3 10.1016/j.molcel.2017.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodie N. I.; Popov K. I.; Petrotchenko E. V.; Dokholyan N. V.; Borchers C. H. Conformational Ensemble of Native A-Synuclein in Solution as Determined by Short-Distance Crosslinking Constraint-Guided Discrete Molecular Dynamics Simulations. PLoS Comput. Biol. 2019, 15, e1006859 10.1371/journal.pcbi.1006859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trelle M. B.; Pedersen S.; Østerlund E. C.; Madsen J. B.; Kristensen S. R.; Jørgensen T. J. D. An Asymmetric Runaway Domain Swap Antithrombin Dimer as a Key Intermediate for Polymerization Revealed by Hydrogen/Deuterium-Exchange Mass Spectrometry. Anal. Chem. 2017, 89, 616. 10.1021/acs.analchem.6b02518. [DOI] [PubMed] [Google Scholar]
- Zhu S.; Shala A.; Bezginov A.; Sljoka A.; Audette G.; Wilson D. J. Hyperphosphorylation of Intrinsically Disordered Tau Protein Induces an Amyloidogenic Shift in Its Conformational Ensemble. PLoS One 2015, 10, e0120416 10.1371/journal.pone.0120416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaldmäe M.; Leppert A.; Chen G.; Sarr M.; Sahin C.; Nordling K.; Kronqvist N.; Gonzalvo-Ulla M.; Fritz N.; Abelein A.; Laín S.; Biverstål H.; Jörnvall H.; Lane D. P.; Rising A.; Johansson J.; Landreh M. High Intracellular Stability of the Spidroin N-Terminal Domain in Spite of Abundant Amyloidogenic Segments Revealed by in-Cell Hydrogen/Deuterium Exchange Mass Spectrometry. FEBS J. 2020, 287, 2823–2833. 10.1111/febs.15169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantazatos D.; Kim J. S.; Klock H. E.; Stevens R. C.; Wilson I. A.; Lesley S. A.; Woods V. L. Jr. Rapid Refinement of Crystallographic Protein Construct Definition Employing Enhanced Hydrogen/Deuterium Exchange Ms. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 751–6. 10.1073/pnas.0307204101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler M. L.; McPhail J. A.; Jenkins M. L.; Masson G. R.; Rutaganira F. U.; Shokat K. M.; Williams R. L.; Burke J. E. Using Hydrogen Deuterium Exchange Mass Spectrometry to Engineer Optimized Constructs for Crystallization of Protein Complexes: Case Study of Pi4kiiib with Rab11. Protein Sci. 2016, 25, 826–839. 10.1002/pro.2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trabjerg E.; Kartberg F.; Christensen S.; Rand K. D. Conformational Characterization of Nerve Growth Factor-Beta Reveals That Its Regulatory Pro-Part Domain Stabilizes Three Loop Regions in Its Mature Part. J. Biol. Chem. 2017, 292, 16665–16676. 10.1074/jbc.M117.803320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saikusa K.; Nagadoi A.; Hara K.; Fuchigami S.; Kurumizaka H.; Nishimura Y.; Akashi S. Mass Spectrometric Approach for Characterizing the Disordered Tail Regions of the Histone H2a/H2b Dimer. Anal. Chem. 2015, 87, 2220. 10.1021/ac503689w. [DOI] [PubMed] [Google Scholar]
- Deredge D. J.; Huang W.; Hui C.; Matsumura H.; Yue Z.; Moënne-Loccoz P.; Shen J.; Wintrode P. L.; Wilks A. Ligand-Induced Allostery in the Interaction of the Pseudomonas Aeruginosa Heme Binding Protein with Heme Oxygenase. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 3421. 10.1073/pnas.1606931114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischer A.; Brehm M. A.; Machha V. R.; Moon-Tasson L.; Benson L. M.; Nelton K. J.; Leger R. R.; Obser T.; Martinez-Vargas M.; Whitten S. T.; Chen D.; Pruthi R. K.; Bergen H. R.; Cruz M. A.; Schneppenheim R.; Auton M. Evidence for the Misfolding of the A1 Domain within Multimeric Von Willebrand Factor in Type 2 Von Willebrand Disease. J. Mol. Biol. 2020, 432, 305–323. 10.1016/j.jmb.2019.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betts G. N.; van der Geer P.; Komives E. A. Structural and Functional Consequences of Tyrosine Phosphorylation in the Lrp1 Cytoplasmic Domain. J. Biol. Chem. 2008, 283, 15656–15664. 10.1074/jbc.M709514200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Eyles S. J.; Thompson L. K. Hydrogen Exchange of Chemoreceptors in Functional Complexes Suggests Protein Stabilization Mediates Long-Range Allosteric Coupling. J. Biol. Chem. 2019, 294, 16062–16079. 10.1074/jbc.RA119.009865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda M.; Nishimura Y. Extended String Binding Mode of the Phosphorylated Transactivation Domain of Tumor Suppressor P53. J. Am. Chem. Soc. 2014, 136, 14143–14152. 10.1021/ja506351f. [DOI] [PubMed] [Google Scholar]
- Kacirova M.; Kosek D.; Kadek A.; Man P.; Vecer J.; Herman P.; Obsilova V.; Obsil T. Structural Characterization of Phosducin and Its Complex with the 14–3-3 Protein. J. Biol. Chem. 2015, 290, 16246–60. 10.1074/jbc.M115.636563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papanastasiou M.; Koutsogiannaki S.; Sarigiannis Y.; Geisbrecht B. V.; Ricklin D.; Lambris J. D. Structural Implications for the Formation and Function of the Complement Effector Protein Ic3b 1 Hhs Public Access. J. Immunol. 2017, 198, 3326–3335. 10.4049/jimmunol.1601864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson E. R.; Jurneczko E.; Nicholson J.; Hupp T. R.; Zawacka-Pankau J.; Selivanova G.; Barran P. E. The Use of Ion Mobility Mass Spectrometry to Probe Modulation of the Structure of P53 and of Mdm2 by Small Molecule Inhibitors. Front. Mol. Biosci. 2015, 2, 39. 10.3389/fmolb.2015.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirrone G. F.; Emert-Sedlak L. A.; Wales T. E.; Smithgall T. E.; Kent M. S.; Engen J. R. The Membrane-Associated Conformation of Hiv-1 Nef Investigated with Hydrogen Exchange Mass Spectrometry at a Langmuir Monolayer Hhs Public Access. Anal. Chem. 2015, 87, 7030–7035. 10.1021/acs.analchem.5b01725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdi K.; Salladini E.; O’Brien D. P.; Brier S.; Chenal A.; Yacoubi I.; Longhi S. Structural Disorder and Induced Folding within Two Cereal, Aba Stress and Ripening (Asr) Proteins. Sci. Rep. 2017, 7, 15544. 10.1038/s41598-017-15299-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dembinski H. E.; Wismer K.; Vargas J. D.; Suryawanshi G. W.; Kern N.; Kroon G.; Dyson H. J.; Hoffmann A.; Komives E. A. Functional Importance of Stripping in Nfκb Signaling Revealed by a Stripping-Impaired Iκbα Mutant. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 1916–1921. 10.1073/pnas.1610192114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vera I. M. S.; Zheng J.; Novick S.; Shang J.; Hughes T.; Brust R.; Munoz-Tello P.; Gardner W. J.; Marciano D. P.; Kong X.; Griffin P. R.; Kojetin D. J. Synergistic Regulation of Coregulator/Nuclear Receptor Interaction by Ligand and DNA Hhs Public Access. Structure 2017, 25, 1506–1518. 10.1016/j.str.2017.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusinga F. I.; Weis D. D. Soft Interactions and Volume Exclusion by Polymeric Crowders Can Stabilize or Destabilize Transient Structure in Disordered Proteins Depending on Polymer Concentration. Proteins: Struct., Funct., Genet. 2017, 85, 1468–1479. 10.1002/prot.25307. [DOI] [PubMed] [Google Scholar]
- Giladi M.; Khananshvili D. Hydrogen-Deuterium Exchange Mass-Spectrometry of Secondary Active Transporters: From Structural Dynamics to Molecular Mechanisms. Front. Pharmacol. 2020, 11, 70. 10.3389/fphar.2020.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadas O.; Jenkins M. L.; Dornan G. L.; Burke J. E. Using Hydrogen-Deuterium Exchange Mass Spectrometry to Examine Protein-Membrane Interactions. Methods Enzymol. 2017, 583, 143–172. 10.1016/bs.mie.2016.09.008. [DOI] [PubMed] [Google Scholar]
- Calabrese A. N.; Radford S. E. Mass Spectrometry-Enabled Structural Biology of Membrane Proteins. Methods 2018, 147, 187–205. 10.1016/j.ymeth.2018.02.020. [DOI] [PubMed] [Google Scholar]
- Duc N. M.; Du Y.; Thorsen T. S.; Lee S. Y.; Zhang C.; Kato H.; Kobilka B. K.; Chung K. Y. Effective Application of Bicelles for Conformational Analysis of G Protein-Coupled Receptors by Hydrogen/Deuterium Exchange Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2015, 26, 808–817. 10.1007/s13361-015-1083-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M. J.; Guttman M.; Atkins W. M. Conformational Dynamics of P-Glycoprotein in Lipid Nanodiscs and Detergent Micelles Reveal Complex Motions on a Wide Time Scale. J. Biol. Chem. 2018, 293, 6297–6307. 10.1074/jbc.RA118.002190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkle P. S.; Gotfryd K.; Cuendet M. A.; Leth-Espensen K. Z.; Gether U.; Loland C. J.; Rand K. D. Substrate-Modulated Unwinding of Transmembrane Helices in the Nss Transporter Leut. Sci. Adv. 2018, 4, eaar6179 10.1126/sciadv.aar6179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikary S.; Deredge D. J.; Nagarajan A.; Forrest L. R.; Wintrode P. L.; Singh S. K. Conformational Dynamics of a Neurotransmitter:Sodium Symporter in a Lipid Bilayer. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E1786–e1795. 10.1073/pnas.1613293114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson G. R.; Burke J. E.; Williams R. L.. Methods in the Study of Pten Structure: X-Ray Crystallography and Hydrogen Deuterium Exchange Mass Spectrometry; Humana Press, 2016; Vol. 1388, pp 215–230. [DOI] [PubMed] [Google Scholar]
- Komolov K. E.; Du Y.; Duc N. M.; Betz R. M.; Rodrigues J. P. G. L. M.; Leib R. D.; Patra D.; Skiniotis G.; Adams C. M.; Dror R. O.; Chung K. Y.; Kobilka B. K.; Benovic J. L. Structural and Functional Analysis of a B2-Adrenergic Receptor Complex with Grk5. Cell 2017, 169, 407–421. e16 10.1016/j.cell.2017.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirrone G. F.; Vernon B. C.; Kent M. S.; Engen J. R. Hydrogen Exchange Mass Spectrometry of Proteins at Langmuir Monolayers. Anal. Chem. 2015, 87, 7022–9. 10.1021/acs.analchem.5b01724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebling C. M.; Morgan C. R.; Stafford D. W.; Jorgenson J. W.; Rand K. D.; Engen J. R. Conformational Analysis of Membrane Proteins in Phospholipid Bilayer Nanodiscs by Hydrogen Exchange Mass Spectrometry. Anal. Chem. 2010, 82, 5415–5419. 10.1021/ac100962c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker C. H.; Morgan C. R.; Rand K. D.; Engen J. R.; Jorgenson J. W.; Stafford D. W. A Conformational Investigation of Propeptide Binding to the Integral Membrane Protein Γ-Glutamyl Carboxylase Using Nanodisc Hydrogen Exchange Mass Spectrometry. Biochemistry 2014, 53, 1511–20. 10.1021/bi401536m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostelic M. M.; Ryan A. M.; Reid D. J.; Noun J. M.; Marty M. T. Expanding the Types of Lipids Amenable to Native Mass Spectrometry of Lipoprotein Complexes. J. Am. Soc. Mass Spectrom. 2019, 30, 1416–1425. 10.1007/s13361-019-02174-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens C.; Shekhar M.; Borysik A. J.; Lau A. M.; Reading E.; Tajkhorshid E.; Booth P. J.; Politis A. Direct Protein-Lipid Interactions Shape the Conformational Landscape of Secondary Transporters. Nat. Commun. 2018, 9, 4151. 10.1038/s41467-018-06704-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reading E.; Hall Z.; Martens C.; Haghighi T.; Findlay H.; Ahdash Z.; Politis A.; Booth P. J. Interrogating Membrane Protein Conformational Dynamics within Native Lipid Compositions. Angew. Chem., Int. Ed. 2017, 56, 15654–15657. 10.1002/anie.201709657. [DOI] [PubMed] [Google Scholar]
- Li S.; Lee S. Y.; Chung K. Y. Conformational Analysis of G Protein-Coupled Receptor Signaling by Hydrogen/Deuterium Exchange Mass Spectrometry. Methods Enzymol. 2015, 557, 261–78. 10.1016/bs.mie.2014.12.004. [DOI] [PubMed] [Google Scholar]
- Du Y.; Duc N. M.; Rasmussen S. G. F.; Hilger D.; Kubiak X.; Wang L.; Bohon J.; Kim H. R.; Wegrecki M.; Asuru A.; Jeong K. M.; Lee J.; Chance M. R.; Lodowski D. T.; Kobilka B. K.; Chung K. Y. Assembly of a Gpcr-G Protein Complex. Cell 2019, 177, 1232–1242. e11 10.1016/j.cell.2019.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien D. P.; Hourdel V.; Chenal A.; Brier S. Hydrogen/Deuterium Exchange Mass Spectrometry for the Structural Analysis of Detergent-Solubilized Membrane Proteins. Methods Mol. Biol. 2020, 2127, 339–358. 10.1007/978-1-0716-0373-4_22. [DOI] [PubMed] [Google Scholar]
- Rey M.; Mrázek H.; Pompach P.; Novák P.; Pelosi L.; Brandolin G.; Forest E.; Havlíček V.; Man P. Effective Removal of Nonionic Detergents in Protein Mass Spectrometry, Hydrogen/Deuterium Exchange, and Proteomics. Anal. Chem. 2010, 82, 5107–5116. 10.1021/ac100171m. [DOI] [PubMed] [Google Scholar]
- Morgan C. R.; Hebling C. M.; Rand K. D.; Stafford D. W.; Jorgenson J. W.; Engen J. R. Conformational Transitions in the Membrane Scaffold Protein of Phospholipid Bilayer Nanodiscs*. Mol. Cell. Proteomics 2011, 10, M111.010876. 10.1074/mcp.M111.010876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barclay L. A.; Wales T. E.; Garner T. P.; Wachter F.; Lee S.; Guerra R. M.; Stewart M. L.; Braun C. R.; Bird G. H.; Gavathiotis E.; Engen J. R.; Walensky L. D. Inhibition of Pro-Apoptotic Bax by a Noncanonical Interaction Mechanism. Mol. Cell 2015, 57, 873–886. 10.1016/j.molcel.2015.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson C. J.; Das M.; Jayaraman S.; Gursky O.; Engen J. R. Effects of Disease-Causing Mutations on the Conformation of Human Apolipoprotein a-I in Model Lipoproteins. Biochemistry 2018, 57, 4583–4596. 10.1021/acs.biochem.8b00538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redhair M.; Clouser A. F.; Atkins W. M. Hydrogen-Deuterium Exchange Mass Spectrometry of Membrane Proteins in Lipid Nanodiscs. Chem. Phys. Lipids 2019, 220, 14–22. 10.1016/j.chemphyslip.2019.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia R.; Martens C.; Shekhar M.; Pant S.; Pellowe G. A.; Lau A. M.; Findlay H. E.; Harris N. J.; Tajkhorshid E.; Booth P. J.; Politis A. Hydrogen-Deuterium Exchange Mass Spectrometry Captures Distinct Dynamics Upon Substrate and Inhibitor Binding to a Transporter. Nat. Commun. 2020, 11, 6162. 10.1038/s41467-020-20032-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reading E.; Ahdash Z.; Fais C.; Ricci V.; Wang-Kan X.; Grimsey E.; Stone J.; Malloci G.; Lau A. M.; Findlay H.; Konijnenberg A.; Booth P. J.; Ruggerone P.; Vargiu A. V.; Piddock L. J. V.; Politis A. Perturbed Structural Dynamics Underlie Inhibition and Altered Efflux of the Multidrug resistance Pump Acrb. Nat. Commun. 2020, 11, 5565. 10.1038/s41467-020-19397-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corey R. A.; Ahdash Z.; Shah A.; Pyle E.; Allen W. J.; Fessl T.; Lovett J. E.; Politis A.; Collinson I. Atp-Induced Asymmetric Pre-Protein Folding as a Driver of Protein Translocation through the Sec Machinery. eLife 2019, 8, e41803 10.7554/eLife.41803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehmood S.; Domene C.; Forest E.; Jault J.-M. Dynamics of a Bacterial Multidrug Abc Transporter in the Inward- and Outward-Facing Conformations. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 10832–10836. 10.1073/pnas.1204067109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopcho N.; Chang G.; Komives E. A. Dynamics of Abc Transporter P-Glycoprotein in Three Conformational States. Sci. Rep. 2019, 9, 15092. 10.1038/s41598-019-50578-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Liu Z.; Creagh J.; Zheng R.; McDonald T. V. Physical and Functional Interaction Sites in Cytoplasmic Domains of Kcnq1 and Kcne1 Channel Subunits. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H212–h222. 10.1152/ajpheart.00459.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauseman Z. J.; Harvey E. P.; Newman C. E.; Wales T. E.; Bucci J. C.; Mintseris J.; Schweppe D. K.; David L.; Fan L.; Cohen D. T.; Herce H. D.; Mourtada R.; Ben-Nun Y.; Bloch N. B.; Hansen S. B.; Wu H.; Gygi S. P.; Engen J. R.; Walensky L. D. Homogeneous Oligomers of Pro-Apoptotic Bax Reveal Structural Determinants of Mitochondrial Membrane Permeabilization. Mol. Cell 2020, 79, 68–83. e7 10.1016/j.molcel.2020.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim X. X.; Chandramohan A.; Lim X. Y.; Bag N.; Sharma K. K.; Wirawan M.; Wohland T.; Lok S. M.; Anand G. S. Conformational Changes in Intact Dengue Virus Reveal Serotype-Specific Expansion. Nat. Commun. 2017, 8, 14339. 10.1038/ncomms14339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K. K.; Lim X. X.; Tantirimudalige S. N.; Gupta A.; Marzinek J. K.; Holdbrook D.; Lim X. Y. E.; Bond P. J.; Anand G. S.; Wohland T. Infectivity of Dengue Virus Serotypes 1 and 2 Is Correlated with E-Protein Intrinsic Dynamics but Not to Envelope Conformations. Structure 2019, 27, 618–630. e4 10.1016/j.str.2018.12.006. [DOI] [PubMed] [Google Scholar]
- Wijesinghe K. J.; Urata S.; Bhattarai N.; Kooijman E. E.; Gerstman B. S.; Chapagain P. P.; Li S.; Stahelin R. V. Detection of Lipid-Induced Structural Changes of the Marburg Virus Matrix Protein Vp40 Using Hydrogen/Deuterium Exchange-Mass Spectrometry. J. Biol. Chem. 2017, 292, 6108–6122. 10.1074/jbc.M116.758300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy R. E.; Samal A. B.; Vlach J.; Mas V.; Prevelige P. E.; Saad J. S. Structural and Biophysical Characterizations of Hiv-1 Matrix Trimer Binding to Lipid Nanodiscs Shed Light on Virus Assembly. J. Biol. Chem. 2019, 294, 18600–18612. 10.1074/jbc.RA119.010997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulma M.; Dadlez M.; Kwiatkowska K. Insight into the Structural Dynamics of the Lysenin During Prepore-to-Pore Transition Using Hydrogen-Deuterium Exchange Mass Spectrometry. Toxins 2019, 11, 462. 10.3390/toxins11080462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidari Z.; Chrisman I. M.; Nemetchek M. D.; Novick S. J.; Blayo A.-L.; Patton T.; Mendes D. E.; Diaz P.; Kamenecka T. M.; Griffin P. R.; Hughes T. S. Definition of Functionally and Structurally Distinct Repressive States in the Nuclear Receptor PPARv. Nat. Commun. 2019, 10, 5825. 10.1038/s41467-019-13768-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trabjerg E.; Abu-Asad N.; Wan Z.; Kartberg F.; Christensen S.; Rand K. D. Investigating the Conformational Response of the Sortilin Receptor Upon Binding Endogenous Peptide- and Protein Ligands by Hdx-Ms. Structure 2019, 27, 1103–1113. e3 10.1016/j.str.2019.04.006. [DOI] [PubMed] [Google Scholar]
- Redhair M.; Hackett J. C.; Pelletier R. D.; Atkins W. M. Dynamics and Location of the Allosteric Midazolam Site in Cytochrome P4503a4 in Lipid Nanodiscs. Biochemistry 2020, 59, 766–779. 10.1021/acs.biochem.9b01001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treuheit N. A.; Redhair M.; Kwon H.; McClary W. D.; Guttman M.; Sumida J. P.; Atkins W. M. Membrane Interactions, Ligand-Dependent Dynamics, and Stability of Cytochrome P4503a4 in Lipid Nanodiscs. Biochemistry 2016, 55, 1058–69. 10.1021/acs.biochem.5b01313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson K. W.; Mast N.; Hudgens J. W.; Lin J. B.; Turko I. V.; Pikuleva I. A. Mapping of the Allosteric Site in Cholesterol Hydroxylase Cyp46a1 for Efavirenz, a Drug That Stimulates Enzyme Activity. J. Biol. Chem. 2016, 291, 11876–11886. 10.1074/jbc.M116.723577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabilane C. S.; Nguyen P. N.; Hernandez R. V.; Nirudodhi S.; Duong M.; Maier C. S.; Narayanaswami V. Mechanism of Lipid Binding of Human Apolipoprotein E3 by Hydrogen/Deuterium Exchange/Mass Spectrometry and Fluorescence Polarization. Protein Pept. Lett. 2016, 23, 404–413. 10.2174/0929866523666160223122257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson O.; Lindvall M.; Obici L.; Ekström S.; Lagerstedt J. O.; Del Giudice R. Structure Dynamics of Apoa-I Amyloidogenic Variants in Small Hdl Increase Their Ability to Mediate Cholesterol Efflux. J. Lipid Res. 2021, 62, 100004. 10.1194/jlr.RA120000920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L.; Hernandez R. V.; Tran T. N.; Nirudodhi S.; Beck W. H. J.; Maier C. S.; Narayanaswami V. Ordered Opening of Ldl Receptor Binding Domain of Human Apolipoprotein E3 Revealed by Hydrogen/Deuterium Exchange Mass Spectrometry and Fluorescence Spectroscopy. Biochim. Biophys. Acta, Proteins Proteomics 2018, 1866, 1165–1173. 10.1016/j.bbapap.2018.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chetty P. S.; Mayne L.; Lund-Katz S.; Englander S. W.; Phillips M. C. Helical Structure, Stability, and Dynamics in Human Apolipoprotein E3 and E4 by Hydrogen Exchange and Mass Spectrometry. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 968–973. 10.1073/pnas.1617523114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A. K.; Jagielnicki M.; McIntire W. E.; Purdy M. D.; Dharmarajan V.; Griffin P. R.; Yeager M. A Steric “Ball-and-Chain” Mechanism for Ph-Mediated Regulation of Gap Junction Channels. Cell Rep. 2020, 31, 107482. 10.1016/j.celrep.2020.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Günther S.; Deredge D.; Bowers A. L.; Luchini A.; Bonsor D. A.; Beadenkopf R.; Liotta L.; Wintrode P. L.; Sundberg E. J. Il-1 Family Cytokines Use Distinct Molecular Mechanisms to Signal through Their Shared Co-Receptor. Immunity 2017, 47, 510–523. e4 10.1016/j.immuni.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey M.; Forest E.; Pelosi L. Exploring the Conformational Dynamics of the Bovine Adp/Atp Carrier in Mitochondria. Biochemistry 2012, 51, 9727–35. 10.1021/bi300759x. [DOI] [PubMed] [Google Scholar]
- Varki A.Essentials of Glycobiology, 3rd ed.; The Consortium of Glycobiology, La Jolla, CA ; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, 2015. [Google Scholar]
- Mariño K.; Bones J.; Kattla J. J.; Rudd P. M. A Systematic Approach to Protein Glycosylation Analysis: A Path through the Maze. Nat. Chem. Biol. 2010, 6, 713–23. 10.1038/nchembio.437. [DOI] [PubMed] [Google Scholar]
- Leymarie N.; Zaia J. Effective Use of Mass Spectrometry for Glycan and Glycopeptide Structural Analysis. Anal. Chem. 2012, 84, 3040–3048. 10.1021/ac3000573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Flaherty R.; Trbojević-Akmačić I.; Greville G.; Rudd P. M.; Lauc G. The Sweet Spot for Biologics: Recent Advances in Characterization of Biotherapeutic Glycoproteins. Expert Rev. Proteomics 2018, 15, 13–29. 10.1080/14789450.2018.1404907. [DOI] [PubMed] [Google Scholar]
- Huang R. Y. C.; Chen G. Higher Order Structure Characterization of Protein Therapeutics by Hydrogen/Deuterium Exchange Mass Spectrometry. Anal. Bioanal. Chem. 2014, 406, 6541–6558. 10.1007/s00216-014-7924-3. [DOI] [PubMed] [Google Scholar]
- Lee J. Y.; Kim J. Y.; Park G. W.; Cheon M. H.; Kwon K. H.; Ahn Y. H.; Moon M. H.; Lee H. J.; Paik Y. K.; Yoo J. S. Targeted Mass Spectrometric Approach for Biomarker Discovery and Validation with Nonglycosylated Tryptic Peptides from N-Linked Glycoproteins in Human Plasma. Mol. Cell Proteomics 2011, 10, M111.009290 10.1074/mcp.M111.009290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesimer M.; Sheehan J. K. Mass Spectrometric Analysis of Mucin Core Proteins. Methods Mol. Biol. (N. Y., NY, U. S.) 2012, 842, 67–79. 10.1007/978-1-61779-513-8_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu R.; Zacharias L.; Wooding K. M.; Peng W.; Mechref Y. Glycoprotein Enrichment Analytical Techniques: Advantages and Disadvantages. Methods Enzymol. 2017, 585, 397–429. 10.1016/bs.mie.2016.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzihradszky K. F. Characterization of Protein N-Glycosylation. Methods Enzymol. 2005, 405, 116–38. 10.1016/S0076-6879(05)05006-8. [DOI] [PubMed] [Google Scholar]
- Riley N. M.; Malaker S. A.; Driessen M. D.; Bertozzi C. R. Optimal Dissociation Methods Differ for N- and O-Glycopeptides. J. Proteome Res. 2020, 19, 3286–3301. 10.1021/acs.jproteome.0c00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aboufazeli F.; Kolli V.; Dodds E. D. A Comparison of Energy-Resolved Vibrational Activation/Dissociation Characteristics of Protonated and Sodiated High Mannose N-Glycopeptides. J. Am. Soc. Mass Spectrom. 2015, 26, 587–595. 10.1007/s13361-014-1070-1. [DOI] [PubMed] [Google Scholar]
- Oberholtzer J. C.; Englander S. W.; Horwitz A. F. Hydrogen-Bonded Conformation of Hyaluronate Oligosaccharide Fragments in Aqueous Solution. FEBS Lett. 1983, 158, 305–309. 10.1016/0014-5793(83)80601-2. [DOI] [Google Scholar]
- Guttman M.; Scian M.; Lee K. K. Tracking Hydrogen/Deuterium Exchange at Glycan Sites in Glycoproteins by Mass Spectrometry. Anal. Chem. 2011, 83, 7492–7499. 10.1021/ac201729v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R. Y. C.; Hudgens J. W. Effects of Desialylation on Human A1-Acid Glycoprotein–Ligand Interactions. Biochemistry 2013, 52, 7127–7136. 10.1021/bi4011094. [DOI] [PubMed] [Google Scholar]
- Kong L.; Huang C. C.; Coales S. J.; Molnar K. S.; Skinner J.; Hamuro Y.; Kwong P. D. Local Conformational Stability of Hiv-1 Gp120 in Unliganded and Cd4-Bound States as Defined by Amide Hydrogen/Deuterium Exchange. J. Virol. 2010, 84, 10311–21. 10.1128/JVI.00688-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houde D.; Demarest S. J. Fine Details of Igf-1r Activation, Inhibition, and Asymmetry Determined by Associated Hydrogen /Deuterium-Exchange and Peptide Mass Mapping. Structure 2011, 19, 890–900. 10.1016/j.str.2011.03.014. [DOI] [PubMed] [Google Scholar]
- Plummer T. H. Jr.; Elder J. H.; Alexander S.; Phelan A. W.; Tarentino A. L. Demonstration of Peptide:N-Glycosidase F Activity in Endo-Beta-N-Acetylglucosaminidase F Preparations. J. Biol. Chem. 1984, 259, 10700–4. 10.1016/S0021-9258(18)90568-5. [DOI] [PubMed] [Google Scholar]
- Wagner N. D.; Huang Y.; Liu T.; Gross M. L. Post-HDX Deglycosylation of Fc Gamma Receptor IIIa Glycoprotein Enables HDX Characterization of Its Binding Interface with IgF. J. Am. Soc. Mass Spectrom. 2021, 32, 1638–1643. 10.1021/jasms.1c00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comamala G.; Madsen J. B.; Voglmeir J.; Du Y.-M.; Jensen P. F.; Østerlund E. C.; Trelle M. B.; Jørgensen T. J. D.; Rand K. D. Deglycosylation by the Acidic Glycosidase Pngase H+ Enables Analysis of N-Linked Glycoproteins by Hydrogen/Deuterium Exchange Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2020, 31, 2305–2312. 10.1021/jasms.0c00258. [DOI] [PubMed] [Google Scholar]
- Wang T.; Cai Z. P.; Gu X. Q.; Ma H. Y.; Du Y. M.; Huang K.; Voglmeir J.; Liu L. Discovery and Characterization of a Novel Extremely Acidic Bacterial N-Glycanase with Combined Advantages of Pngase F and A. Biosci. Rep. 2014, 34, e00149 10.1042/BSR20140148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo R. R.; Comamala G.; Yang H. H.; Gramlich M.; Du Y. M.; Wang T.; Zeck A.; Rand K. D.; Liu L.; Voglmeir J. Discovery of Highly Active Recombinant Pngase H(+) Variants through the Rational Exploration of Unstudied Acidobacterial Genomes. Front. Bioeng. Biotechnol. 2020, 8, 741. 10.3389/fbioe.2020.00741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reusch D.; Tejada M. L. Fc Glycans of Therapeutic Antibodies as Critical Quality Attributes. Glycobiology 2015, 25, 1325–1334. 10.1093/glycob/cwv065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houde D.; Arndt J.; Domeier W.; Berkowitz S.; Engen J. R. Characterization of Igg1 Conformation and Conformational Dynamics by Hydrogen/Deuterium Exchange Mass Spectrometry. Anal. Chem. 2009, 81, 2644–2651. 10.1021/ac802575y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houde D.; Peng Y.; Berkowitz S. A.; Engen J. R. Post-Translational Modifications Differentially Affect Igg1 Conformation and Receptor Binding*. Mol. Cell. Proteomics 2010, 9, 1716–1728. 10.1074/mcp.M900540-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar R.; Esfandiary R.; Bishop S. M.; Samra H. S.; Middaugh C. R.; Volkin D. B.; Weis D. D. Correlations between Changes in Conformational Dynamics and Physical Stability in a Mutant Igg1Mab Engineered for Extended Serum Half-Life. MAbs 2015, 7, 84–95. 10.4161/19420862.2014.985494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T. F.; Sazinsky S. L.; Houde D.; DiLillo D. J.; Bird J.; Li K. K.; Cheng G. T.; Qiu H.; Engen J. R.; Ravetch J. V.; Wittrup K. D. Engineering Aglycosylated Igg Variants with Wild-Type or Improved Binding Affinity to Human Fc Gamma Riia and Fc Gamma Riiias. J. Mol. Biol. 2017, 429, 2528–2541. 10.1016/j.jmb.2017.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang A.; Hu P.; MacGregor P.; Xue Y.; Fan H.; Suchecki P.; Olszewski L.; Liu A. Understanding the Conformational Impact of Chemical Modifications on Monoclonal Antibodies with Diverse Sequence Variation Using Hydrogen/Deuterium Exchange Mass Spectrometry and Structural Modeling. Anal. Chem. 2014, 86, 3468–75. 10.1021/ac404130a. [DOI] [PubMed] [Google Scholar]
- Fang J.; Richardson J.; Du Z.; Zhang Z. Effect of Fc-Glycan Structure on the Conformational Stability of Igg Revealed by Hydrogen/Deuterium Exchange and Limited Proteolysis. Biochemistry 2016, 55, 860–868. 10.1021/acs.biochem.5b01323. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Shah B.; Richardson J. Impact of Fc N-Glycan Sialylation on Igg Structure. mAbs 2019, 11, 1381–1390. 10.1080/19420862.2019.1655377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves K.; Cryar A.; Cowen S.; Ashcroft A. E.; Quaglia M. Mass Spectrometry Characterization of Higher Order Structural Changes Associated with the Fc-Glycan Structure of the Nistmab Reference Material, Rm 8761. J. Am. Soc. Mass Spectrom. 2020, 31, 553–564. 10.1021/jasms.9b00022. [DOI] [PubMed] [Google Scholar]
- Kuhne F.; Bonnington L.; Malik S.; Thomann M.; Avenal C.; Cymer F.; Wegele H.; Reusch D.; Mormann M.; Bulau P. The Impact of Immunoglobulin G1 Fc Sialylation on Backbone Amide H/D Exchange. Antibodies 2019, 8, 49. 10.3390/antib8040049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose R. J.; van Berkel P. H.; van den Bremer E. T.; Labrijn A. F.; Vink T.; Schuurman J.; Heck A. J.; Parren P. W. Mutation of Y407 in the Ch3 Domain Dramatically Alters Glycosylation and Structure of Human Igg. MAbs 2013, 5, 219–28. 10.4161/mabs.23532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyama M.; Hashii N.; Tsukimura W.; Osumi K.; Harazono A.; Tada M.; Kiyoshi M.; Matsuda A.; Ishii-Watabe A. Effects of Terminal Galactose Residues in Mannose A1–6 Arm of Fc-Glycan on the Effector Functions of Therapeutic Monoclonal Antibodies. MAbs 2019, 11, 826–836. 10.1080/19420862.2019.1608143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klontz E. H.; Trastoy B.; Deredge D.; Fields J. K.; Li C.; Orwenyo J.; Marina A.; Beadenkopf R.; Günther S.; Flores J.; Wintrode P. L.; Wang L. X.; Guerin M. E.; Sundberg E. J. Molecular Basis of Broad Spectrum N-Glycan Specificity and Processing of Therapeutic Igg Monoclonal Antibodies by Endoglycosidase S2. ACS Cent. Sci. 2019, 5, 524–538. 10.1021/acscentsci.8b00917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen P. F.; Larraillet V.; Schlothauer T.; Kettenberger H.; Hilger M.; Rand K. D. Investigating the Interaction between the Neonatal Fc Receptor and Monoclonal Antibody Variants by Hydrogen/Deuterium Exchange Mass Spectrometry*. Mol. Cell. Proteomics 2015, 14, 148–161. 10.1074/mcp.M114.042044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters B. T.; Jensen P. F.; Larraillet V.; Lin K.; Patapoff T.; Schlothauer T.; Rand K. D.; Zhang J. Conformational Destabilization of Immunoglobulin G Increases the Low Ph Binding Affinity with the Neonatal Fc Receptor. J. Biol. Chem. 2016, 291, 1817–1825. 10.1074/jbc.M115.691568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L.; Liu T.; Gross M. L.; Huang Y. Recognition of Human Igg1 by Fcγ Receptors: Structural Insights from Hydrogen–Deuterium Exchange and Fast Photochemical Oxidation of Proteins Coupled with Mass Spectrometry. Biochemistry 2019, 58, 1074–1080. 10.1021/acs.biochem.8b01048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlandi C.; Deredge D.; Ray K.; Gohain N.; Tolbert W.; DeVico A. L.; Wintrode P.; Pazgier M.; Lewis G. K. Antigen-Induced Allosteric Changes in a Human Igg1 Fc Increase Low-Affinity Fcγ Receptor Binding. Structure 2020, 28, 516–527. e5 10.1016/j.str.2020.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rincon Pabon J. P.; Kochert B. A.; Liu Y.-H.; Richardson D.; Weis D. D. Protein A Does Not Induce Allosteric Structural Changes in an Igg1 Antibody During Binding. J. Pharm. Sci. 2021, 110, 2355. 10.1016/j.xphs.2021.02.027. [DOI] [PubMed] [Google Scholar]
- Song H.; Olsen O. H.; Persson E.; Rand K. D. Sites Involved in Intra- and Interdomain Allostery Associated with the Activation of Factor Viia Pinpointed by Hydrogen-Deuterium Exchange and Electron Transfer Dissociation Mass Spectrometry. J. Biol. Chem. 2014, 289, 35388–96. 10.1074/jbc.M114.614297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloem E.; van den Biggelaar M.; Wroblewska A.; Voorberg J.; Faber J. H.; Kjalke M.; Stennicke H. R.; Mertens K.; Meijer A. B. Factor Viii C1 Domain Spikes 2092–2093 and 2158–2159 Comprise Regions That Modulate Cofactor Function and Cellular Uptake. J. Biol. Chem. 2013, 288, 29670–29679. 10.1074/jbc.M113.473116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda S.; Zhang J.; Yang L.; Anand G. S.; Ding J. L. Molecular Interaction between Natural Igg and Ficolin--Mechanistic Insights on Adaptive-Innate Immune Crosstalk. Sci. Rep. 2015, 4, 3675. 10.1038/srep03675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calmettes C.; Yu R. H.; Silva L. P.; Curran D.; Schriemer D. C.; Schryvers A. B.; Moraes T. F. Structural Variations within the Transferrin Binding Site on Transferrin-Binding Protein B, Tbpb. J. Biol. Chem. 2011, 286, 12683–12692. 10.1074/jbc.M110.206102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielkopf C. S.; Ghosh M.; Anand G. S.; Brown S. H. J. Hdx-Ms Reveals Orthosteric and Allosteric Changes in Apolipoprotein-D Structural Dynamics Upon Binding of Progesterone. Protein Sci. 2019, 28, 365–374. 10.1002/pro.3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D.; Zhang P.; Li F.; Chi L.; Zhu D.; Zhang Q.; Chi L. Recognition of N-Glycoforms in Human Chorionic Gonadotropin by Monoclonal Antibodies and Their Interaction Motifs. J. Biol. Chem. 2015, 290, 22715–22723. 10.1074/jbc.M115.657072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Shannessy D. J.; Somers E. B.; Albone E.; Cheng X.; Park Y. C.; Tomkowicz B. E.; Hamuro Y.; Kohl T. O.; Forsyth T. M.; Smale R.; Fu Y. S.; Nicolaides N. C. Characterization of the Human Folate Receptor Alpha Via Novel Antibody-Based Probes. Oncotarget 2011, 2, 1227–43. 10.18632/oncotarget.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji C.; Wei G. Deglycosylation Induces Extensive Dynamics Changes in A-Amylase Revealed by Hydrogen/Deuterium Exchange Mass Spectrometry. Rapid Commun. Mass Spectrom. 2013, 27, 2625–30. 10.1002/rcm.6732. [DOI] [PubMed] [Google Scholar]
- Lee S. M.; Jeong Y.; Simms J.; Warner M. L.; Poyner D. R.; Chung K. Y.; Pioszak A. A. Calcitonin Receptor N-Glycosylation Enhances Peptide Hormone Affinity by Controlling Receptor Dynamics. J. Mol. Biol. 2020, 432, 1996–2014. 10.1016/j.jmb.2020.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar A.; Wintrode P. L. Effects of Glycosylation on the Stability and Flexibility of a Metastable Protein: The Human Serpin A(1)-Antitrypsin. Int. J. Mass Spectrom. 2011, 302, 69–75. 10.1016/j.ijms.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton C.; Li X. S.; Li S. P.; Flaherty B.; Sison L.; Lu Q.; Yeung B.; Wu S. L. Impact of Glycosylation on the Comparability of the Higher-Order Structures in Idursulfase by Hydrogen-Deuterium Exchange Mass Spectrometry. Anal. Chem. 2020, 92, 8306–8314. 10.1021/acs.analchem.0c00661. [DOI] [PubMed] [Google Scholar]
- Wang A. L.; Zhou Y.; Palmieri M. J.; Hao G. G. Hydrogen Deuterium Exchange Reveals Changes to Protein Dynamics of Recombinant Human Erythropoietin Upon N- and O- Desialylation. J. Pharm. Biomed. Anal. 2018, 154, 454–459. 10.1016/j.jpba.2018.02.060. [DOI] [PubMed] [Google Scholar]
- Danwen Q.; Code C.; Quan C.; Gong B.-J.; Arndt J.; Pepinsky B.; Rand K. D.; Houde D. Investigating the Role of Artemin Glycosylation. Pharm. Res. 2016, 33, 1383–1398. 10.1007/s11095-016-1880-x. [DOI] [PubMed] [Google Scholar]
- Ranaweera A.; Ratnayake P. U.; Ekanayaka E. A. P.; Declercq R.; Weliky D. P. Hydrogen–Deuterium Exchange Supports Independent Membrane-Interfacial Fusion Peptide and Transmembrane Domains in Subunit 2 of Influenza Virus Hemagglutinin Protein, a Structured and Aqueous-Protected Connection between the Fusion Peptide and Soluble Ectodomain, and the Importance of Membrane Apposition by the Trimer-of-Hairpins Structure. Biochemistry 2019, 58, 2432–2446. 10.1021/acs.biochem.8b01272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia N. K.; Guttman M.; Ebner J. L.; Lee K. K. Dynamic Changes During Acid-Induced Activation of Influenza Hemagglutinin. Structure 2015, 23, 665–676. 10.1016/j.str.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M.; Garcia N. K.; Cupo A.; Matsui T.; Julien J. P.; Sanders R. W.; Wilson I. A.; Moore J. P.; Lee K. K. Cd4-Induced Activation in a Soluble Hiv-1 Env Trimer. Structure 2014, 22, 974–984. 10.1016/j.str.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale S.; Liu T.; Li S.; Wang Y.; Abelson D.; Fusco M.; Woods V. L. Jr.; Saphire E. O. Ebola Virus Glycoprotein Needs an Additional Trigger, Beyond Proteolytic Priming for Membrane Fusion. PLoS Neglected Trop. Dis. 2011, 5, e1395 10.1371/journal.pntd.0001395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghuvamsi P. V.; Tulsian N. K.; Samsudin F.; Qian X.; Purushotorman K.; Yue G.; Kozma M. M.; Hwa W. Y.; Lescar J.; Bond P. J.; MacAry P. A.; Anand G. S. Sars-Cov-2 S Protein:Ace2 Interaction Reveals Novel Allosteric Targets. eLife 2021, 10, e63646. 10.7554/eLife.63646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basore K.; Kim A. S.; Nelson C. A.; Zhang R.; Smith B. K.; Uranga C.; Vang L.; Cheng M.; Gross M. L.; Smith J.; Diamond M. S.; Fremont D. H. Cryo-Em Structure of Chikungunya Virus in Complex with the Mxra8 Receptor. Cell 2019, 177, 1725–1737. e16 10.1016/j.cell.2019.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcandalli J.; Fiala B.; Ols S.; Perotti M.; de van der Schueren W.; Snijder J.; Hodge E.; Benhaim M.; Ravichandran R.; Carter L.; Sheffler W.; Brunner L.; Lawrenz M.; Dubois P.; Lanzavecchia A.; Sallusto F.; Lee K. K.; Veesler D.; Correnti C. E.; Stewart L. J.; Baker D.; Loré K.; Perez L.; King N. P. Induction of Potent Neutralizing Antibody Responses by a Designed Protein Nanoparticle Vaccine for Respiratory Syncytial Virus. Cell 2019, 176, 1420–1431. e17 10.1016/j.cell.2019.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blais N.; Gagné M.; Hamuro Y.; Rheault P.; Boyer M.; Steff A.-M.; Baudoux G.; Dewar V.; Demers J.; Ruelle J.-L.; Martin D. Characterization of Pre-F-Gcn4t, a Modified Human Respiratory Syncytial Virus Fusion Protein Stabilized in a Noncleaved Prefusion Conformation. J. Virol. 2017, 91, e02437-16 10.1128/JVI.02437-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce M. G.; Zhang B.; Ou L.; Chen M.; Chuang G. Y.; Druz A.; Kong W. P.; Lai Y. T.; Rundlet E. J.; Tsybovsky Y.; Yang Y.; Georgiev I. S.; Guttman M.; Lees C. R.; Pancera M.; Sastry M.; Soto C.; Stewart-Jones G. B. E.; Thomas P. V.; Van Galen J. G.; Baxa U.; Lee K. K.; Mascola J. R.; Graham B. S.; Kwong P. D. Iterative Structure-Based Improvement of a Fusion-Glycoprotein Vaccine against Rsv. Nat. Struct. Mol. Biol. 2016, 23, 811–820. 10.1038/nsmb.3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impagliazzo A.; Milder F.; Kuipers H.; Wagner M. V.; Zhu X.; Hoffman R. M.; van Meersbergen R.; Huizingh J.; Wanningen P.; Verspuij J.; de Man M.; Ding Z.; Apetri A.; Kükrer B.; Sneekes-Vriese E.; Tomkiewicz D.; Laursen N. S.; Lee P. S.; Zakrzewska A.; Dekking L.; Tolboom J.; Tettero L.; van Meerten S.; Yu W.; Koudstaal W.; Goudsmit J.; Ward A. B.; Meijberg W.; Wilson I. A.; Radošević K. A Stable Trimeric Influenza Hemagglutinin Stem as a Broadly Protective Immunogen. Science 2015, 349, 1301. 10.1126/science.aac7263. [DOI] [PubMed] [Google Scholar]
- Torrents de la Peña A.; Julien J. P.; de Taeye S. W.; Garces F.; Guttman M.; Ozorowski G.; Pritchard L. K.; Behrens A. J.; Go E. P.; Burger J. A.; Schermer E. E.; Sliepen K.; Ketas T. J.; Pugach P.; Yasmeen A.; Cottrell C. A.; Torres J. L.; Vavourakis C. D.; van Gils M. J.; LaBranche C.; Montefiori D. C.; Desaire H.; Crispin M.; Klasse P. J.; Lee K. K.; Moore J. P.; Ward A. B.; Wilson I. A.; Sanders R. W. Improving the Immunogenicity of Native-Like Hiv-1 Envelope Trimers by Hyperstabilization. Cell Rep. 2017, 20, 1805–1817. 10.1016/j.celrep.2017.07.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Taeye S. W.; Ozorowski G.; Torrents de la Peña A.; Guttman M.; Julien J. P.; van den Kerkhof T. L.; Burger J. A.; Pritchard L. K.; Pugach P.; Yasmeen A.; Crampton J.; Hu J.; Bontjer I.; Torres J. L.; Arendt H.; DeStefano J.; Koff W. C.; Schuitemaker H.; Eggink D.; Berkhout B.; Dean H.; LaBranche C.; Crotty S.; Crispin M.; Montefiori D. C.; Klasse P. J.; Lee K. K.; Moore J. P.; Wilson I. A.; Ward A. B.; Sanders R. W. Immunogenicity of Stabilized Hiv-1 Envelope Trimers with Reduced Exposure of Non-Neutralizing Epitopes. Cell 2015, 163, 1702–1715. 10.1016/j.cell.2015.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders R. W.; van Gils M. J.; Derking R.; Sok D.; Ketas T. J.; Burger J. A.; Ozorowski G.; Cupo A.; Simonich C.; Goo L.; Arendt H.; Kim H. J.; Lee J. H.; Pugach P.; Williams M.; Debnath G.; Moldt B.; van Breemen M. J.; Isik G.; Medina-Ramírez M.; Back J. W.; Koff W. C.; Julien J. P.; Rakasz E. G.; Seaman M. S.; Guttman M.; Lee K. K.; Klasse P. J.; LaBranche C.; Schief W. R.; Wilson I. A.; Overbaugh J.; Burton D. R.; Ward A. B.; Montefiori D. C.; Dean H.; Moore J. P. Hiv-1 Vaccines. Hiv-1 Neutralizing Antibodies Induced by Native-Like Envelope Trimers. Science 2015, 349, aac4223 10.1126/science.aac4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M.; Lee K. K. A Functional Interaction between Gp41 and Gp120 Is Observed for Monomeric but Not Oligomeric, Uncleaved Hiv-1 Env Gp140. J. Virol. 2013, 87, 11462–11475. 10.1128/JVI.01681-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkerke H. P.; Williams J. A.; Guttman M.; Simonich C. A.; Liang Y.; Filipavicius M.; Hu S.-L.; Overbaugh J.; Lee K. K. Epitope-Independent Purification of Native-Like Envelope Trimers from Diverse Hiv-1 Isolates. J. Virol. 2016, 90, 9471. 10.1128/JVI.01351-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y.; Guttman M.; Davenport T. M.; Hu S. L.; Lee K. K. Probing the Impact of Local Structural Dynamics of Conformational Epitopes on Antibody Recognition. Biochemistry 2016, 55, 2197–213. 10.1021/acs.biochem.5b01354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon Y. D.; Pancera M.; Acharya P.; Georgiev I. S.; Crooks E. T.; Gorman J.; Joyce M. G.; Guttman M.; Ma X.; Narpala S.; Soto C.; Terry D. S.; Yang Y.; Zhou T.; Ahlsen G.; Bailer R. T.; Chambers M.; Chuang G. Y.; Doria-Rose N. A.; Druz A.; Hallen M. A.; Harned A.; Kirys T.; Louder M. K.; O’Dell S.; Ofek G.; Osawa K.; Prabhakaran M.; Sastry M.; Stewart-Jones G. B.; Stuckey J.; Thomas P. V.; Tittley T.; Williams C.; Zhang B.; Zhao H.; Zhou Z.; Donald B. R.; Lee L. K.; Zolla-Pazner S.; Baxa U.; Schön A.; Freire E.; Shapiro L.; Lee K. K.; Arthos J.; Munro J. B.; Blanchard S. C.; Mothes W.; Binley J. M.; McDermott A. B.; Mascola J. R.; Kwong P. D. Crystal Structure, Conformational Fixation and Entry-Related Interactions of Mature Ligand-Free Hiv-1 Env. Nat. Struct. Mol. Biol. 2015, 22, 522. 10.1038/nsmb.3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringe R. P.; Yasmeen A.; Ozorowski G.; Go E. P.; Pritchard L. K.; Guttman M.; Ketas T. A.; Cottrell C. A.; Wilson I. A.; Sanders R. W.; Cupo A.; Crispin M.; Lee K. K.; Desaire H.; Ward A. B.; Klasse P. J.; Moore J. P. Influences on the Design and Purification of Soluble, Recombinant Native-Like Hiv-1 Envelope Glycoprotein Trimers. J. Virol. 2015, 89, 12189–210. 10.1128/JVI.01768-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y.; Guttman M.; Williams J. A.; Verkerke H.; Alvarado D.; Hu S. L.; Lee K. K. Changes in Structure and Antigenicity of Hiv-1 Env Trimers Resulting from Removal of a Conserved Cd4 Binding Site-Proximal Glycan. J. Virol. 2016, 90, 9224–36. 10.1128/JVI.01116-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport T. M.; Guttman M.; Guo W.; Cleveland B.; Kahn M.; Hu S. L.; Lee K. K. Isolate-Specific Differences in the Conformational Dynamics and Antigenicity of Hiv-1 Gp120. J. Virol. 2013, 87, 10855–10873. 10.1128/JVI.01535-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner H. L.; Pallesen J.; Lang S.; Bangaru S.; Urata S.; Li S.; Cottrell C. A.; Bowman C. A.; Crowe J. E. Jr.; Wilson I. A.; Ward A. B. Potent Anti-Influenza H7 Human Monoclonal Antibody Induces Separation of Hemagglutinin Receptor-Binding Head Domains. PLoS Biol. 2019, 17, e3000139 10.1371/journal.pbio.3000139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauch E. M.; Bernard S. M.; La D.; Bohn A. J.; Lee P. S.; Anderson C. E.; Nieusma T.; Holstein C. A.; Garcia N. K.; Hooper K. A.; Ravichandran R.; Nelson J. W.; Sheffler W.; Bloom J. D.; Lee K. K.; Ward A. B.; Yager P.; Fuller D. H.; Wilson I. A.; Baker D. Computational Design of Trimeric Influenza-Neutralizing Proteins Targeting the Hemagglutinin Receptor Binding Site. Nat. Biotechnol. 2017, 35, 667–671. 10.1038/nbt.3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh R.; Lim X. X.; Raghuvamsi P. V.; Wu C.; Wong S. M.; Anand G. S. Uncovering Metastability and Disassembly Hotspots in Whole Viral Particles. Prog. Biophys. Mol. Biol. 2019, 143, 5–12. 10.1016/j.pbiomolbio.2018.12.006. [DOI] [PubMed] [Google Scholar]
- Houde D.; Berkowitz S. A.; Engen J. R. The Utility of Hydrogen/Deuterium Exchange Mass Spectrometry in Biopharmaceutical Comparability Studies. J. Pharm. Sci. 2011, 100, 2071–2086. 10.1002/jps.22432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkowitz S. A.; Engen J. R.; Mazzeo J. R.; Jones G. B. Analytical Tools for Characterizing Biopharmaceuticals and the Implications for Biosimilars. Nat. Rev. Drug Discovery 2012, 11, 527–540. 10.1038/nrd3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houde D.; Engen J. R. Conformational Analysis of Recombinant Monoclonal Antibodies with Hydrogen/Deuterium Exchange Mass Spectrometry. Methods Mol. Biol. 2013, 988, 269–289. 10.1007/978-1-62703-327-5_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar R.; Middaugh C. R.; Weis D. D.; Volkin D. B. Hydrogen-Deuterium Exchange Mass Spectrometry as an Emerging Analytical Tool for Stabilization and Formulation Development of Therapeutic Monoclonal Antibodies. J. Pharm. Sci. 2015, 104, 327–45. 10.1002/jps.24224. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Cui W.; Gross M. L. Mass Spectrometry for the Biophysical Characterization of Therapeutic Monoclonal Antibodies. FEBS Lett. 2014, 588, 308–17. 10.1016/j.febslet.2013.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houde D.; Berkowitz S. A.. Biophysical Characterization of Proteins in Developing Biopharmaceuticals; Elsevier, 2019. [Google Scholar]
- Gallagher D. T.; McCullough C.; Brinson R. G.; Ahn J.; Marino J. P.; Dimasi N. Structure and Dynamics of a Site-Specific Labeled Fc Fragment with Altered Effector Functions. Pharmaceutics 2019, 11, 546. 10.3390/pharmaceutics11100546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkitt W.; Domann P.; O’Connor G. Conformational Changes in Oxidatively Stressed Monoclonal Antibodies Studied by Hydrogen Exchange Mass Spectrometry. Protein Sci. 2010, 19, 826–835. 10.1002/pro.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo J.; Yan Q.; So C. K.; Soden T.; Lewis M. J.; Hu P. Understanding the Impact of Methionine Oxidation on the Biological Functions of Igg1 Antibodies Using Hydrogen/Deuterium Exchange Mass Spectrometry. Anal. Chem. 2016, 88, 9495–9502. 10.1021/acs.analchem.6b01958. [DOI] [PubMed] [Google Scholar]
- Hageman T.; Wei H.; Kuehne P.; Fu J.; Ludwig R.; Tao L.; Leone A.; Zocher M.; Das T. K. Impact of Tryptophan Oxidation in Complementarity-Determining Regions of Two Monoclonal Antibodies on Structure-Function Characterized by Hydrogen-Deuterium Exchange Mass Spectrometry and Surface Plasmon Resonance. Pharm. Res. 2019, 36, 24. 10.1007/s11095-018-2545-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo J.; Jin R.; Yan Q.; Sokolowska I.; Lewis M. J.; Hu P. Quantitative Analysis of Glycation and Its Impact on Antigen Binding. MAbs 2018, 10, 406–415. 10.1080/19420862.2018.1438796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei M.; Carcelen T.; Walters B. T.; Zamiri C.; Quan C.; Hu Y.; Nishihara J.; Yip H.; Woon N.; Zhang T.; Kao Y. H.; Schöneich C. Structure-Based Correlation of Light-Induced Histidine Reactivity in a Model Protein. Anal. Chem. 2017, 89, 7225–7231. 10.1021/acs.analchem.7b01457. [DOI] [PubMed] [Google Scholar]
- Gamage C. L. D.; Hageman T. S.; Weis D. D. Rapid Prediction of Deamidation Rates of Proteins to Assess Their Long-Term Stability Using Hydrogen Exchange-Mass Spectrometry. J. Pharm. Sci. 2019, 108, 1964–1972. 10.1016/j.xphs.2019.01.019. [DOI] [PubMed] [Google Scholar]
- Goswami D.; Zhang J.; Bondarenko P. V.; Zhang Z. Ms-Based Conformation Analysis of Recombinant Proteins in Design, Optimization and Development of Biopharmaceuticals. Methods 2018, 144, 134–151. 10.1016/j.ymeth.2018.04.011. [DOI] [PubMed] [Google Scholar]
- Pan L. Y.; Salas-Solano O.; Valliere-Douglass J. F. Antibody Structural Integrity of Site-Specific Antibody-Drug Conjugates Investigated by Hydrogen/Deuterium Exchange Mass Spectrometry. Anal. Chem. 2015, 87, 5669–76. 10.1021/acs.analchem.5b00764. [DOI] [PubMed] [Google Scholar]
- Shi X. E.; Wales T. E.; Elkin C.; Kawahata N.; Engen J. R.; Annis D. A. Hydrogen Exchange-Mass Spectrometry Measures Stapled Peptide Conformational Dynamics and Predicts Pharmacokinetic Properties. Anal. Chem. 2013, 85, 11185–8. 10.1021/ac403173p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocks B. B.; Bird G. H.; Walensky L. D.; Melanson J. E. Characterizing Native and Hydrocarbon-Stapled Enfuvirtide Conformations with Ion Mobility Mass Spectrometry and Hydrogen-Deuterium Exchange. J. Am. Soc. Mass Spectrom. 2021, 32, 753–761. 10.1021/jasms.0c00453. [DOI] [PubMed] [Google Scholar]
- Houde D.; Nazari Z. E.; Bou-Assaf G. M.; Weiskopf A. S.; Rand K. D. Conformational Analysis of Proteins in Highly Concentrated Solutions by Dialysis-Coupled Hydrogen/Deuterium Exchange Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2016, 27, 669–676. 10.1007/s13361-015-1331-7. [DOI] [PubMed] [Google Scholar]
- Tian Y.; Huang L.; Ruotolo B. T.; Wang N. Hydrogen/Deuterium Exchange-Mass Spectrometry Analysis of High Concentration Biotherapeutics: Application to Phase-Separated Antibody Formulations. MAbs 2019, 11, 779–788. 10.1080/19420862.2019.1589850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora J.; Hickey J. M.; Majumdar R.; Esfandiary R.; Bishop S. M.; Samra H. S.; Middaugh C. R.; Weis D. D.; Volkin D. B. Hydrogen Exchange Mass Spectrometry Reveals Protein Interfaces and Distant Dynamic Coupling Effects During the Reversible Self-Association of an Igg1Monoclonal Antibody. MAbs 2015, 7, 525–539. 10.1080/19420862.2015.1029217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacob R. E.; Bou-Assaf G. M.; Makowski L.; Engen J. R.; Berkowitz S. A.; Houde D. Investigating Monoclonal Antibody Aggregation Using a Combination of H/Dx-Ms and Other Biophysical Measurements. J. Pharm. Sci. 2013, 102, 4315–29. 10.1002/jps.23754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R. Y.; Iacob R. E.; Krystek S. R.; Jin M.; Wei H.; Tao L.; Das T. K.; Tymiak A. A.; Engen J. R.; Chen G. Characterization of Aggregation Propensity of a Human Fc-Fusion Protein Therapeutic by Hydrogen/Deuterium Exchange Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2017, 28, 795–802. 10.1007/s13361-016-1452-7. [DOI] [PubMed] [Google Scholar]
- Lindner R.; Heintz U.; Winkler A. Applications of Hydrogen Deuterium Exchange (Hdx) for the Characterization of Conformational Dynamics in Light-Activated Photoreceptors. Front. Mol. Biosci. 2015, 2, 33. 10.3389/fmolb.2015.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintz U.; Schlichting I. Blue Light-Induced Lov Domain Dimerization Enhances the Affinity of Aureochrome 1a for Its Target DNA Sequence. eLife 2016, 5, e11860 10.7554/eLife.11860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhrke D.; Gourinchas G.; Müller M.; Michael N.; Hildebrandt P.; Winkler A. Distinct Chromophore-Protein Environments Enable Asymmetric Activation of a Bacteriophytochrome-Activated Diguanylate Cyclase. J. Biol. Chem. 2020, 295, 539–551. 10.1074/jbc.RA119.011915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz-Badur S.; Penner A.; Straß S.; von Horsten S.; Linne U.; Essen L. O. Structural Changes within the Bifunctional Cryptochrome/Photolyase Cracry Upon Blue Light Excitation. Sci. Rep. 2019, 9, 9896. 10.1038/s41598-019-45885-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assafa T. E.; Anders K.; Linne U.; Essen L. O.; Bordignon E. Light-Driven Domain Mechanics of a Minimal Phytochrome Photosensory Module Studied by Epr. Structure 2018, 26, 1534–1545. 10.1016/j.str.2018.08.003. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay A.; Van Eps N.; Eger B. T.; Rauscher S.; Yedidi R. S.; Moroni T.; West G. M.; Robinson K. A.; Griffin P. R.; Mitchell J.; Ernst O. P. A Novel Polar Core and Weakly Fixed C-Tail in Squid Arrestin Provide New Insight into Interaction with Rhodopsin. J. Mol. Biol. 2018, 430, 4102–4118. 10.1016/j.jmb.2018.08.009. [DOI] [PubMed] [Google Scholar]
- Etzl S.; Lindner R.; Nelson M. D.; Winkler A. Structure-Guided Design and Functional Characterization of an Artificial Red Light-Regulated Guanylate/Adenylate Cyclase for Optogenetic Applications. J. Biol. Chem. 2018, 293, 9078–9089. 10.1074/jbc.RA118.003069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Horsten S.; Straß S.; Hellwig N.; Gruth V.; Klasen R.; Mielcarek A.; Linne U.; Morgner N.; Essen L. O. Mapping Light-Driven Conformational Changes within the Photosensory Module of Plant Phytochrome B. Sci. Rep. 2016, 6, 34366. 10.1038/srep34366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann L.; Alexander N. S.; Sun W.; Zhang J.; Orban T.; Palczewski K. Hydrogen/Deuterium Exchange Mass Spectrometry of Human Green Opsin Reveals a Conserved Pro-Pro Motif in Extracellular Loop 2 of Monostable Visual G Protein-Coupled Receptors. Biochemistry 2017, 56, 2338–2348. 10.1021/acs.biochem.7b00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S.; Guttman M.; Leverenz R. L.; Zhumadilova K.; Pawlowski E. G.; Petzold C. J.; Lee K. K.; Ralston C. Y.; Kerfeld C. A. Local and Global Structural Drivers for the Photoactivation of the Orange Carotenoid Protein. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, E5567–74. 10.1073/pnas.1512240112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Williams T. D.; Schowen R. L.; Topp E. M. Characterizing Protein Structure in Amorphous Solids Using Hydrogen/Deuterium Exchange with Mass Spectrometry. Anal. Biochem. 2007, 366, 18–28. 10.1016/j.ab.2007.03.041. [DOI] [PubMed] [Google Scholar]
- Moorthy B. S.; Zarraga I. E.; Kumar L.; Walters B. T.; Goldbach P.; Topp E. M.; Allmendinger A. Solid-State Hydrogen-Deuterium Exchange Mass Spectrometry: Correlation of Deuterium Uptake and Long-Term Stability of Lyophilized Monoclonal Antibody Formulations. Mol. Pharmaceutics 2018, 15, 1–11. 10.1021/acs.molpharmaceut.7b00504. [DOI] [PubMed] [Google Scholar]
- Li Y.; Williams T. D.; Topp E. M. Effects of Excipients on Protein Conformation in Lyophilized Solids by Hydrogen/Deuterium Exchange Mass Spectrometry. Pharm. Res. 2008, 25, 259–67. 10.1007/s11095-007-9365-6. [DOI] [PubMed] [Google Scholar]
- Sophocleous A. M.; Topp E. M. Localized Hydration in Lyophilized Myoglobin by Hydrogen-Deuterium Exchange Mass Spectrometry. 2. Exchange Kinetics. Mol. Pharmaceutics 2012, 9, 727–33. 10.1021/mp2004093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AbouGhaly M. H. H.; Du J.; Patel S. M.; Topp E. M. Effects of Ionic Interactions on Protein Stability Prediction Using Solid-State Hydrogen Deuterium Exchange with Mass Spectrometry (Sshdx-Ms). Int. J. Pharm. 2019, 568, 118512. 10.1016/j.ijpharm.2019.118512. [DOI] [PubMed] [Google Scholar]
- Tukra R.; Gardner S.; Topp E. M. Effects of Temperature and Relative Humidity in D(2)O on Solid-State Hydrogen Deuterium Exchange Mass Spectrometry (Sshdx-Ms). Int. J. Pharm. 2021, 596, 120263. 10.1016/j.ijpharm.2021.120263. [DOI] [PubMed] [Google Scholar]
- Wilson N. E.; Topp E. M.; Zhou Q. T. Effects of Drying Method and Excipient on Structure and Stability of Protein Solids Using Solid-State Hydrogen/Deuterium Exchange Mass Spectrometry (Sshdx-Ms). Int. J. Pharm. 2019, 567, 118470. 10.1016/j.ijpharm.2019.118470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorthy B. S.; Schultz S. G.; Kim S. G.; Topp E. M. Predicting Protein Aggregation During Storage in Lyophilized Solids Using Solid State Amide Hydrogen/Deuterium Exchange with Mass Spectrometric Analysis (Sshdx-Ms). Mol. Pharmaceutics 2014, 11, 1869–79. 10.1021/mp500005v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar L.; Chandrababu K. B.; Balakrishnan S. M.; Allmendinger A.; Walters B.; Zarraga I. E.; Chang D. P.; Nayak P.; Topp E. M. Optimizing the Formulation and Lyophilization Process for a Fragment Antigen Binding (Fab) Protein Using Solid-State Hydrogen-Deuterium Exchange Mass Spectrometry (Sshdx-Ms). Mol. Pharmaceutics 2019, 16, 4485–4495. 10.1021/acs.molpharmaceut.9b00614. [DOI] [PubMed] [Google Scholar]
- Kabaria S. R.; Mangion I.; Makarov A. A.; Pirrone G. F. Use of Maldi-Ms with Solid-State Hydrogen Deuterium Exchange for Semi-Automated Assessment of Peptide and Protein Physical Stability in Lyophilized Solids. Anal. Chim. Acta 2019, 1054, 114–121. 10.1016/j.aca.2018.12.034. [DOI] [PubMed] [Google Scholar]
- Moussa E. M.; Singh S. K.; Kimmel M.; Nema S.; Topp E. M. Probing the Conformation of an Igg1Monoclonal Antibody in Lyophilized Solids Using Solid-State Hydrogen-Deuterium Exchange with Mass Spectrometric Analysis (Sshdx-Ms). Mol. Pharmaceutics 2018, 15, 356–368. 10.1021/acs.molpharmaceut.7b00696. [DOI] [PubMed] [Google Scholar]
- Moussa E. M.; Wilson N. E.; Zhou Q. T.; Singh S. K.; Nema S.; Topp E. M. Effects of Drying Process on an Igg1Monoclonal Antibody Using Solid-State Hydrogen Deuterium Exchange with Mass Spectrometric Analysis (Sshdx-Ms). Pharm. Res. 2018, 35, 12. 10.1007/s11095-017-2318-9. [DOI] [PubMed] [Google Scholar]
- Kammari R.; Topp E. M. Solid-State Hydrogen-Deuterium Exchange Mass Spectrometry (Sshdx-Ms) of Lyophilized Poly-D,L-Alanine. Mol. Pharmaceutics 2019, 16, 2935–2946. 10.1021/acs.molpharmaceut.9b00162. [DOI] [PubMed] [Google Scholar]
- Kammari R.; Topp E. M. Prehydration and the Reversibility of Solid-State Hydrogen-Deuterium Exchange. Mol. Pharmaceutics 2020, 17, 3541–3552. 10.1021/acs.molpharmaceut.0c00571. [DOI] [PubMed] [Google Scholar]
- Kammari R.; Topp E. M. Effects of Secondary Structure on Solid-State Hydrogen-Deuterium Exchange in Model A-Helix and B-Sheet Peptides. Mol. Pharmaceutics 2020, 17, 3501–3512. 10.1021/acs.molpharmaceut.0c00521. [DOI] [PubMed] [Google Scholar]
- Miyagi M.; Wan Q.; Ahmad M. F.; Gokulrangan G.; Tomechko S. E.; Bennett B.; Dealwis C. Histidine Hydrogen-Deuterium Exchange Mass Spectrometry for Probing the Microenvironment of Histidine Residues in Dihydrofolate Reductase. PLoS One 2011, 6, e17055 10.1371/journal.pone.0017055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cebo M.; Kielmas M.; Adamczyk J.; Cebrat M.; Szewczuk Z.; Stefanowicz P. Hydrogen-Deuterium Exchange in Imidazole as a Tool for Studying Histidine Phosphorylation. Anal. Bioanal. Chem. 2014, 406, 8013–8020. 10.1007/s00216-014-8218-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J.; Callahan K. L.; Borotto N. B.; Vachet R. W. Identifying Zn-Bound Histidine Residues in Metalloproteins Using Hydrogen-Deuterium Exchange Mass Spectrometry. Anal. Chem. 2014, 86, 766–773. 10.1021/ac4032719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran D. T.; Banerjee S.; Alayash A. I.; Crumbliss A. L.; Fitzgerald M. C. Slow Histidine H/D Exchange Protocol for Thermodynamic Analysis of Protein Folding and Stability Using Mass Spectrometry. Anal. Chem. 2012, 84, 1653–1660. 10.1021/ac202927p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Largy E.; Gabelica V. Native Hydrogen/Deuterium Exchange Mass Spectrometry of Structured DNA Oligonucleotides. Anal. Chem. 2020, 92, 4402–4410. 10.1021/acs.analchem.9b05298. [DOI] [PubMed] [Google Scholar]
- Taraban M. B.; Deredge D. J.; Smith M. E.; Briggs K. T.; Li Y.; Jiang Z. X.; Wintrode P. L.; Yu Y. B. Monitoring Dendrimer Conformational Transition Using (19) F and (1) H(2) O Nmr. Magn. Reson. Chem. 2019, 57, 861–872. 10.1002/mrc.4849. [DOI] [PubMed] [Google Scholar]
- Burkitt W.; O’Connor G. Assessment of the Repeatability and Reproducibility of Hydrogen/Deuterium Exchange Mass Spectrometry Measurements. Rapid Commun. Mass Spectrom. 2008, 22, 3893–3901. 10.1002/rcm.3794. [DOI] [PubMed] [Google Scholar]
- Hudgens J. W.; Gallagher E. S.; Karageorgos I.; Anderson K. W.; Filliben J. J.; Huang R. Y.; Chen G.; Bou-Assaf G. M.; Espada A.; Chalmers M. J.; Harguindey E.; Zhang H. M.; Walters B. T.; Zhang J.; Venable J.; Steckler C.; Park I.; Brock A.; Lu X.; Pandey R.; Chandramohan A.; Anand G. S.; Nirudodhi S. N.; Sperry J. B.; Rouse J. C.; Carroll J. A.; Rand K. D.; Leurs U.; Weis D. D.; Al-Naqshabandi M. A.; Hageman T. S.; Deredge D.; Wintrode P. L.; Papanastasiou M.; Lambris J. D.; Li S.; Urata S. Interlaboratory Comparison of Hydrogen-Deuterium Exchange Mass Spectrometry Measurements of the Fab Fragment of Nistmab. Anal. Chem. 2019, 91, 7336–7345. 10.1021/acs.analchem.9b01100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheff J. G.; Schriemer D. C. Toward Standardizing Deuterium Content Reporting in Hydrogen Exchange-Ms. Anal. Chem. 2014, 86, 11962–5. 10.1021/ac5034424. [DOI] [PubMed] [Google Scholar]
- Moroco J. A.; Engen J. R. Replication in Bioanalytical Studies with Hdx Ms: Aim as High as Possible. Bioanalysis 2015, 7, 1065–7. 10.4155/bio.15.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J.; Cao M. J.; Yu Y. Q.; Engen J. R. Accessing the Reproducibility and Specificity of Pepsin and Other Aspartic Proteases. Biochim. Biophys. Acta, Proteins Proteomics 2013, 1834, 1222–1229. 10.1016/j.bbapap.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorauer C.; Wrigley M. S.; Rincon Pabon J. P.; Watson M. J.; Mundorff C. C.; Weis D. D.; Guttman M., Rapid Assessment of Pepsin Column Activity for Reliable Hdx-Ms Studies. J. Am. Soc. Mass Spectrom. 2021. 10.1021/jasms.1c00080 [DOI] [PubMed] [Google Scholar]
- Wei H.; Ahn J.; Yu Y. Q.; Tymiak A.; Engen J. R.; Chen G. Using Hydrogen/Deuterium Exchange Mass Spectrometry to Study Conformational Changes in Granulocyte Colony Stimulating Factor Upon Pegylation. J. Am. Soc. Mass Spectrom. 2012, 23, 498–504. 10.1007/s13361-011-0310-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheff J. G.; Rey M.; Schriemer D. C. Peptide-Column Interactions and Their Influence on Back Exchange Rates in Hydrogen/Deuterium Exchange-Ms. J. Am. Soc. Mass Spectrom. 2013, 24, 1006–15. 10.1007/s13361-013-0639-4. [DOI] [PubMed] [Google Scholar]
- West G. M.; Pascal B. D.; Ng L. M.; Soon F. F.; Melcher K.; Xu H. E.; Chalmers M. J.; Griffin P. R. Protein Conformation Ensembles Monitored by Hdx Reveal a Structural Rationale for Abscisic Acid Signaling Protein Affinities and Activities. Structure 2013, 21, 229–35. 10.1016/j.str.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Goswami D.; Zhang Z.. In New Insight into Differences in Intrinsic HDX Rates at Different pH and Temperature. In 67th ASMS Conference on Mass Spectrometry and Allied Topics, 2019.
- Claesen J.; Burzykowski T. Computational Methods and Challenges in Hydrogen/Deuterium Exchange Mass Spectrometry. Mass Spectrom. Rev. 2017, 36, 649–667. 10.1002/mas.21519. [DOI] [PubMed] [Google Scholar]
- Iacob R. E.; Murphy J. P. 3rd; Engen J. R. Ion Mobility Adds an Additional Dimension to Mass Spectrometric Analysis of Solution-Phase Hydrogen/Deuterium Exchange. Rapid Commun. Mass Spectrom. 2008, 22, 2898–904. 10.1002/rcm.3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggertson M. J.; Fadgen K.; Engen J. R.; Wales T. E. Considerations in the Analysis of Hydrogen Exchange Mass Spectrometry Data. Methods Mol. Biol. 2020, 2051, 407–435. 10.1007/978-1-4939-9744-2_18. [DOI] [PubMed] [Google Scholar]
- Pascal B. D.; Willis S.; Lauer J. L.; Landgraf R. R.; West G. M.; Marciano D.; Novick S.; Goswami D.; Chalmers M. J.; Griffin P. R. Hdx Workbench: Software for the Analysis of H/D Exchange Ms Data. J. Am. Soc. Mass Spectrom. 2012, 23, 1512–21. 10.1007/s13361-012-0419-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan Z.-Y.; Ye X.; Skinner J. J.; Mayne L.; Englander S. W. Exms2: An Integrated Solution for Hydrogen-Deuterium Exchange Mass Spectrometry Data Analysis. Anal. Chem. 2019, 91, 7474. 10.1021/acs.analchem.9b01682. [DOI] [PubMed] [Google Scholar]
- Venable J. D.; Scuba W.; Brock A. Feature Based Retention Time Alignment for Improved Hdx Ms Analysis. J. Am. Soc. Mass Spectrom. 2013, 24, 642–645. 10.1007/s13361-012-0566-9. [DOI] [PubMed] [Google Scholar]
- Abzalimov R. R.; Kaltashov I. A. Extraction of Local Hydrogen Exchange Data from Hdx Cad Ms Measurements by Deconvolution of Isotopic Distributions of Fragment Ions. J. Am. Soc. Mass Spectrom. 2006, 17, 1543–1551. 10.1016/j.jasms.2006.07.017. [DOI] [PubMed] [Google Scholar]
- Palmblad M.; Buijs J.; Håkansson P. Automatic Analysis of Hydrogen/Deuterium Exchange Mass Spectra of Peptides and Proteins Using Calculations of Isotopic Distributions. J. Am. Soc. Mass Spectrom. 2001, 12, 1153–62. 10.1016/S1044-0305(01)00301-4. [DOI] [PubMed] [Google Scholar]
- Guttman M.; Weis D. D.; Engen J. R.; Lee K. K. Analysis of Overlapped and Noisy Hydrogen/Deuterium Exchange Mass Spectra. J. Am. Soc. Mass Spectrom. 2013, 24, 1906–12. 10.1007/s13361-013-0727-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q.; Easterling M. L.; Agar J. N. Resolving Isotopic Fine Structure to Detect and Quantify Natural Abundance- and Hydrogen/Deuterium Exchange-Derived Isotopomers. Anal. Chem. 2014, 86, 820–5. 10.1021/ac403365g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner R.; Lou X.; Reinstein J.; Shoeman R. L.; Hamprecht F. A.; Winkler A. Hexicon 2: Automated Processing of Hydrogen-Deuterium Exchange Mass Spectrometry Data with Improved Deuteration Distribution Estimation. J. Am. Soc. Mass Spectrom. 2014, 25, 1018–28. 10.1007/s13361-014-0850-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raval S.; Sarpe V.; Hepburn M.; Crowder D. A.; Zhang T.; Viner R.; Schriemer D. C. Improving Spectral Validation Rates in Hydrogen-Deuterium Exchange Data Analysis. Anal. Chem. 2021, 93, 4246. 10.1021/acs.analchem.0c05045. [DOI] [PubMed] [Google Scholar]
- Slysz G. W.; Baker C. A.; Bozsa B. M.; Dang A.; Percy A. J.; Bennett M.; Schriemer D. C. Hydra: Software for Tailored Processing of H/D Exchange Data from Ms or Tandem Ms Analyses. BMC Bioinf. 2009, 10, 162. 10.1186/1471-2105-10-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renard B. Y.; Kirchner M.; Steen H.; Steen J. A.; Hamprecht F. A. Nitpick: Peak Identification for Mass Spectrometry Data. BMC Bioinf. 2008, 9, 355. 10.1186/1471-2105-9-355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller D. E.; Prasannan C. B.; Villar M. T.; Fenton A. W.; Artigues A. Hdxfinder: Automated Analysis and Data Reporting of Deuterium/Hydrogen Exchange Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2012, 23, 425–9. 10.1007/s13361-011-0234-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salisbury J. P.; Liu Q.; Agar J. N. Qudex-Ms: Hydrogen/Deuterium Exchange Calculation for Mass Spectra with Resolved Isotopic Fine Structure. BMC Bioinf. 2014, 15, 403. 10.1186/s12859-014-0403-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltzberg D. J.; Broughton H. B.; Pellarin R.; Chalmers M. J.; Espada A.; Dodge J. A.; Pascal B. D.; Griffin P. R.; Humblet C.; Sali A. A Residue-Resolved Bayesian Approach to Quantitative Interpretation of Hydrogen-Deuterium Exchange from Mass Spectrometry: Application to Characterizing Protein-Ligand Interactions. J. Phys. Chem. B 2017, 121, 3493–3501. 10.1021/acs.jpcb.6b09358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou X.; Kirchner M.; Renard B. Y.; Kothe U.; Boppel S.; Graf C.; Lee C. T.; Steen J. A.; Steen H.; Mayer M. P.; Hamprecht F. A. Deuteration Distribution Estimation with Improved Sequence Coverage for Hx/Ms Experiments. Bioinformatics 2010, 26, 1535–41. 10.1093/bioinformatics/btq165. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Ramachandran P.; Kumar R.; Gross M. L. H/D Exchange Centroid Monitoring Is Insufficient to Show Differences in the Behavior of Protein States. J. Am. Soc. Mass Spectrom. 2013, 24, 450–453. 10.1007/s13361-012-0555-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan Z. Y.; Mayne L.; Sevugan Chetty P.; Englander S. W.. ExMS: Data Analysis for Hx-Ms Experiments. J. Am. Soc. Mass Spectrom. 2011, 22, DOI: 10.1007/s13361-011-0236-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrotchenko E. V.; Borchers C. H. Hdx Match Software for the Data Analysis of Top-Down Ecd-Ftms Hydrogen/Deuterium Exchange Experiments. J. Am. Soc. Mass Spectrom. 2015, 26, 1895–1898. 10.1007/s13361-015-1213-z. [DOI] [PubMed] [Google Scholar]
- Zhang Z. Complete Extraction of Protein Dynamics Information in Hydrogen/Deuterium Exchange Mass Spectrometry Data. Anal. Chem. 2020, 92, 6486–6494. 10.1021/acs.analchem.9b05724. [DOI] [PubMed] [Google Scholar]
- Engen J. R.; Smithgall T. E.; Gmeiner W. H.; Smith D. L. Identification and Localization of Slow, Natural, Cooperative Unfolding in the Hematopoietic Cell Kinase Sh3 Domain by Amide Hydrogen Exchange and Mass Spectrometry. Biochemistry 1997, 36, 14384–14391. 10.1021/bi971635m. [DOI] [PubMed] [Google Scholar]
- Deng Y.; Smith D. L. Identification of Unfolding Domains in Large Proteins by Their Unfolding Rates. Biochemistry 1998, 37, 6256–6262. 10.1021/bi972711o. [DOI] [PubMed] [Google Scholar]
- Deng Y.; Smith D. L. Hydrogen Exchange Demonstrates Three Domains in Aldolase Unfold Sequentially. J. Mol. Biol. 1999, 294, 247–58. 10.1006/jmbi.1999.3251. [DOI] [PubMed] [Google Scholar]
- Guttman M.; Lee K. K. Isotope Labeling of Biomolecules: Structural Analysis of Viruses by Hdx-Ms. Methods Enzymol. 2016, 566, 405–426. 10.1016/bs.mie.2015.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascal B. D.; Chalmers M. J.; Busby S. A.; Griffin P. R. Hd Desktop: An Integrated Platform for the Analysis and Visualization of H/D Exchange Data. J. Am. Soc. Mass Spectrom. 2009, 20, 601–10. 10.1016/j.jasms.2008.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajer P. G.; Bou-Assaf G. M.; Marshall A. G. Improved Sequence Resolution by Global Analysis of Overlapped Peptides in Hydrogen/Deuterium Exchange Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2012, 23, 1202–1208. 10.1007/s13361-012-0373-3. [DOI] [PubMed] [Google Scholar]
- Gessner C.; Steinchen W.; Bedard S.; Skinner J. J.; Woods V. L.; Walsh T. J.; Bange G.; Pantazatos D. P. Computational Method Allowing Hydrogen-Deuterium Exchange Mass Spectrometry at Single Amide Resolution. Sci. Rep. 2017, 7, 3789. 10.1038/s41598-017-03922-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner S. P.; Radou G.; Tuma R.; Houwing-Duistermaat J. J.; Paci E. Estimating Constraints for Protection Factors from Hdx-Ms Data. Biophys. J. 2019, 116, 1194–1203. 10.1016/j.bpj.2019.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmas R. E.; Borysik A. J. Hdxmodeller: An Online Webserver for High-Resolution Hdx-Ms with Auto-Validation. Commun. Biol. 2021, 4, 199. 10.1038/s42003-021-01709-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns-Hamuro L. L.; Hamuro Y.; Kim J. S.; Sigala P.; Fayos R.; Stranz D. D.; Jennings P. A.; Taylor S. S.; Woods V. L. Jr. Distinct Interaction Modes of an Akap Bound to Two Regulatory Subunit Isoforms of Protein Kinase a Revealed by Amide Hydrogen/Deuterium Exchange. Protein Sci. 2005, 14, 2982–92. 10.1110/ps.051687305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavan D.; Man P. Mstools—Web Based Application for Visualization and Presentation of Hxms Data. Int. J. Mass Spectrom. 2011, 302, 53–58. 10.1016/j.ijms.2010.07.030. [DOI] [Google Scholar]
- Nevin P.; Engen J. R.; Beuning P. J. Steric Gate Residues of Y-Family DNA Polymerases Dinb and Pol Kappa Are Crucial for Dntp-Induced Conformational Change. DNA Repair 2015, 29, 65–73. 10.1016/j.dnarep.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevin P.; Lu X.; Zhang K.; Engen J. R.; Beuning P. J. Noncognate DNA Damage Prevents the Formation of the Active Conformation of the Y-Family DNA Polymerases Dinb and DNA Polymerase K. FEBS J. 2015, 282, 2646–60. 10.1111/febs.13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerres N.; Steurer S.; Schlager S.; Bader G.; Berger H.; Caligiuri M.; Dank C.; Engen J. R.; Ettmayer P.; Fischerauer B.; Flotzinger G.; Gerlach D.; Gerstberger T.; Gmaschitz T.; Greb P.; Han B.; Heyes E.; Iacob R. E.; Kessler D.; Kölle H.; Lamarre L.; Lancia D. R.; Lucas S.; Mayer M.; Mayr K.; Mischerikow N.; Mück K.; Peinsipp C.; Petermann O.; Reiser U.; Rudolph D.; Rumpel K.; Salomon C.; Scharn D.; Schnitzer R.; Schrenk A.; Schweifer N.; Thompson D.; Traxler E.; Varecka R.; Voss T.; Weiss-Puxbaum A.; Winkler S.; Zheng X.; Zoephel A.; Kraut N.; McConnell D.; Pearson M.; Koegl M. Chemically Induced Degradation of the Oncogenic Transcription Factor Bcl6. Cell Rep. 2017, 20, 2860–2875. 10.1016/j.celrep.2017.08.081. [DOI] [PubMed] [Google Scholar]
- Das M.; Wilson C. J.; Mei X.; Wales T. E.; Engen J. R.; Gursky O. Structural Stability and Local Dynamics in Disease-Causing Mutants of Human Apolipoprotein a-I: What Makes the Protein Amyloidogenic?. J. Mol. Biol. 2016, 428, 449–462. 10.1016/j.jmb.2015.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison R. A.; Lu J.; Carrasco M.; Hunter J.; Manandhar A.; Gondi S.; Westover K. D.; Engen J. R. Structural Dynamics in Ras and Related Proteins Upon Nucleotide Switching. J. Mol. Biol. 2016, 428, 4723–4735. 10.1016/j.jmb.2016.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hourdel V.; Volant S.; O’Brien D. P.; Chenal A.; Chamot-Rooke J.; Dillies M. A.; Brier S. MEMHDX: An Interactive Tool to Expedite the Statistical Validation and Visualization of Large Hdx-Ms Datasets. Bioinformatics 2016, 32, btw420. 10.1093/bioinformatics/btw420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouyssie D.; Lesne J.; Locard-Paulet M.; Albigot R.; Burlet-Schiltz O.; Marcoux J. Hdx-Viewer: Interactive 3d Visualization of Hydrogen-Deuterium Exchange Data. Bioinformatics 2019, 35, 5331–5333. 10.1093/bioinformatics/btz550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau A. M.; Claesen J.; Hansen K.; Politis A. Deuteros 2.0: Peptide-Level Significance Testing of Data from Hydrogen Deuterium Exchange Mass Spectrometry. Bioinformatics 2021, 37, 270. 10.1093/bioinformatics/btaa677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumpkin R. J.; Komives E. A. Deca, a Comprehensive, Automatic Post-Processing Program for Hdx-Ms Data. Mol. Cell Proteomics 2019, 18, 2516–2523. 10.1074/mcp.TIR119.001731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puchała W.; Burdukiewicz M.; Kistowski M.; Dabrowska K. A.; Badaczewska-Dawid A. E.; Cysewski D.; Dadlez M. Hadex: An R Package and Web-Server for Analysis of Data from Hydrogen-Deuterium Exchange Mass Spectrometry Experiments. Bioinformatics 2020, 36, 4516. 10.1093/bioinformatics/btaa587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N.; Yu X.; Zhang X.; D’Arcy S. Hd-Explosion: Visualization of Hydrogen-Deuterium Exchange Data as Chiclet and Volcano Plots with Statistical Filtering. Bioinformatics 2021, 37, 1926. 10.1093/bioinformatics/btaa892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar R.; Manikwar P.; Hickey J. M.; Samra H. S.; Sathish H. A.; Bishop S. M.; Middaugh C. R.; Volkin D. B.; Weis D. D. Effects of Salts from the Hofmeister Series on the Conformational Stability, Aggregation Propensity, and Local Flexibility of an Igg1Monoclonal Antibody. Biochemistry 2013, 52, 3376–89. 10.1021/bi400232p. [DOI] [PubMed] [Google Scholar]
- Hageman T. S.; Weis D. D. Reliable Identification of Significant Differences in Differential Hydrogen Exchange-Mass Spectrometry Measurements Using a Hybrid Significance Testing Approach. Anal. Chem. 2019, 91, 8008–8016. 10.1021/acs.analchem.9b01325. [DOI] [PubMed] [Google Scholar]
- Weis D. D. Recommendations for the Propagation of Uncertainty in Hydrogen Exchange-Mass Spectrometric Measurements. J. Am. Soc. Mass Spectrom. 2021, 32, 1610. 10.1021/jasms.0c00475. [DOI] [PubMed] [Google Scholar]
- Craig P. O.; Lätzer J.; Weinkam P.; Hoffman R. M. B.; Ferreiro D. U.; Komives E. A.; Wolynes P. G. Prediction of Native-State Hydrogen Exchange from Perfectly Funneled Energy Landscapes. J. Am. Chem. Soc. 2011, 133, 17463–17472. 10.1021/ja207506z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L.; So P. K.; Yao Z. P. Protein Dynamics Revealed by Hydrogen/Deuterium Exchange Mass Spectrometry: Correlation between Experiments and Simulation. Rapid Commun. Mass Spectrom. 2019, 33, 83–89. 10.1002/rcm.8307. [DOI] [PubMed] [Google Scholar]
- Liu T.; Pantazatos D.; Li S.; Hamuro Y.; Hilser V. J.; Woods V. L. Jr. Quantitative Assessment of Protein Structural Models by Comparison of H/D Exchange Ms Data with Exchange Behavior Accurately Predicted by Dxcorex. J. Am. Soc. Mass Spectrom. 2012, 23, 43–56. 10.1007/s13361-011-0267-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris M. J.; Raghavan D.; Borysik A. J. Quantitative Evaluation of Native Protein Folds and Assemblies by Hydrogen Deuterium Exchange Mass Spectrometry (Hdx-Ms). J. Am. Soc. Mass Spectrom. 2019, 30, 58–66. 10.1007/s13361-018-2070-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaurs D.; Antunes D. A.; Papanastasiou M.; Moll M.; Ricklin D.; Lambris J. D.; Kavraki L. E. Coarse-Grained Conformational Sampling of Protein Structure Improves the Fit to Experimental Hydrogen-Exchange Data. Front. Mol. Biosci. 2017, 4, 13. 10.3389/fmolb.2017.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw R. T.; Marinelli F.; Faraldo-Gomez J. D.; Forrest L. R. Interpretation of Hdx Data by Maximum-Entropy Reweighting of Simulated Structural Ensembles. Biophys. J. 2020, 118, 1649–1664. 10.1016/j.bpj.2020.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borysik A. J. Simulated Isotope Exchange Patterns Enable Protein Structure Determination. Angew. Chem., Int. Ed. 2017, 56, 9396–9399. 10.1002/anie.201704604. [DOI] [PubMed] [Google Scholar]
- Markwick P. R. L.; Peacock R. B.; Komives E. A. Accurate Prediction of Amide Exchange in the Fast Limit Reveals Thrombin Allostery. Biophys. J. 2019, 116, 49–56. 10.1016/j.bpj.2018.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau A. M.; Jia R.; Bradshaw R. T.; Politis A. Structural Predictions of the Functions of Membrane Proteins from Hdx-Ms. Biochem. Soc. Trans. 2020, 48, 971. 10.1042/BST20190880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens C.; Shekhar M.; Lau A. M.; Tajkhorshid E.; Politis A. Integrating Hydrogen-Deuterium Exchange Mass Spectrometry with Molecular Dynamics Simulations to Probe Lipid-Modulated Conformational Changes in Membrane Proteins. Nat. Protoc. 2019, 14, 3183–3204. 10.1038/s41596-019-0219-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorthy B. S.; Iyer L. K.; Topp E. M. Mass Spectrometric Approaches to Study Protein Structure and Interactions in Lyophilized Powders. J. Visualized Exp. 2015, 2015, 52503. 10.3791/52503. [DOI] [PMC free article] [PubMed] [Google Scholar]