

Abstract
Breast cancer stem(-like) cells (BCSCs) have been found to be responsible for therapeutic resistance and disease relapse. BCSCs are difficult to eradicate due to their high resistance to conventional treatments and high plasticity. Functionalised nanoparticles have been investigated as smart vehicles to transport across various barriers and increase the interaction of therapeutic agents with cancer cells, as well as BCSCs. In this review, we discuss the different characteristics of BCSCs, and challenges to tackle BCSCs at cellular and molecular levels. The mechanisms of action and physicochemical properties of the current BCSC targeting agents are also covered. We will focus on the rational design and recent advances of “Nano + Nano” or single tumour targeting nanoparticle systems loaded with dual or multiple agents to kill all cancer cells including BCSCs. These cocktail therapies include the combination of a chemotherapy agent with a BCSC-specific inhibitor, a phytochemical agent or RNA based therapy. Given the heterogeneity of breast tumour tissue, targeting both BCSCs and bulk breast cancer cells simultaneously with multiple agents holds great promise in eliminating breast cancer. The future research needs to focus on overcoming various barriers in the ‘clinical translation’ of BCSC-targeting nanomedicines to cure breast cancer, which requires a significant multidisciplinary effort.
Breast cancer stem(-like) cells (BCSCs) have been found to be responsible for therapeutic resistance and disease relapse.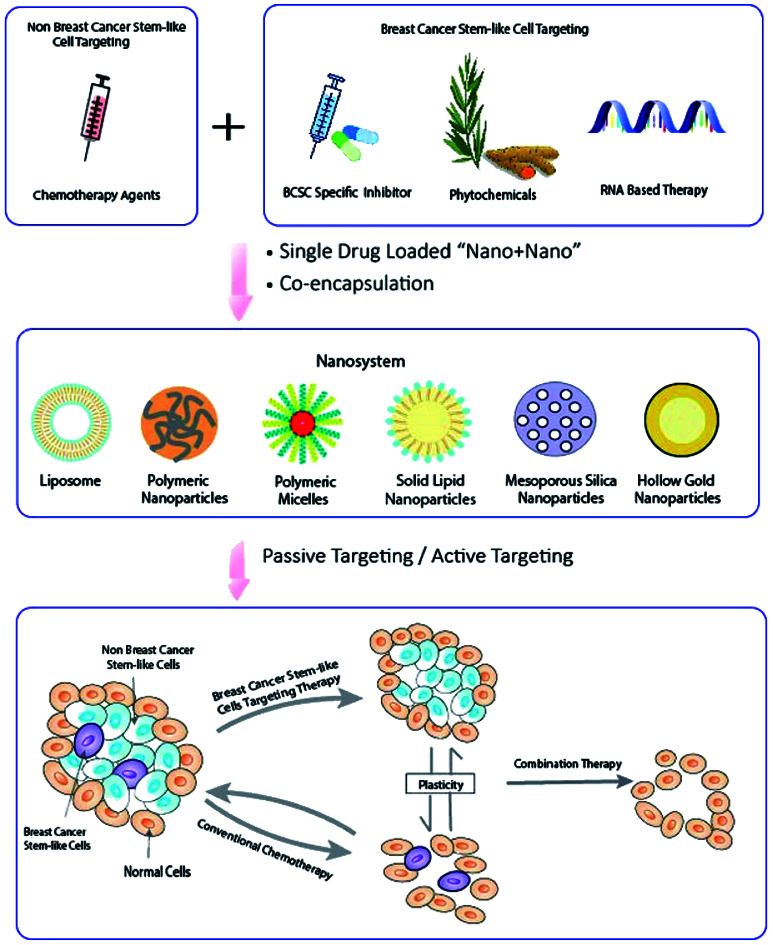
1. Introduction
Breast cancer is the most commonly diagnosed cancer and the leading cause of cancer death in women worldwide.1 In 2018, over 2 million new cases were diagnosed globally, with over 626 000 deaths. Breast cancer can be classified according to the histological expression of three important receptors, namely estrogen receptor (ER), progesterone receptor (PR) and human epidermal growth factor receptor 2 (HER2).2 However this simple classification is far from representing the vast heterogeneity of the disease.3 Molecular classification into five subtypes based on genetic profiling (Table 1) has arguably been the closest stratification that resembles the complexity of the disease. The development of hormone therapy and targeted therapy significantly improves the survival rate and forms integral parts of current breast cancer treatment in addition to the traditional surgery, radiation therapy and chemotherapy.4 However, many breast cancer patients still develop into metastases, therapeutic resistance or disease relapse. Emerging evidences have shown that a subgroup of breast cancer cells, known as breast cancer stem-like cells (BCSCs), play a crucial role in these processes.5,6 Similar to normal stem cells, BCSCs have the ability to self-renew, proliferate and generate multi-lineage differentiation7,8 (Fig. 1). Nevertheless, they exhibit high level of cell plasticity and are lack of a stem cell hierarchy.9 Eradication of BCSCs presents one of the most promising strategies, and yet one of the biggest hurdles in current anti-breast cancer therapy.
Molecular classification of breast cancer and examples of cell lines3,10.
| Subtypes | ER | PR | HER2 | Examples of cell lines |
|---|---|---|---|---|
| Luminal A | + | +/− | − | MCF-7, BT483, T47D, MDA-MB-134 |
| Luminal B | + | +/− | + | BT474, ZR-75, BSMZ |
| HER2 | − | − | + | SKBR3, MDA-MB-453 |
| Basal (triple-negative) | − | − | − | MDA-MB-468, SUM190 |
| Claudin-low | − | − | − | BT549, MDA-MB-231, Hs578T, SUM1315 |
Fig. 1. BCSCs involved in tumour self-renew, differentiation and metastasis.

The presence of BCSCs and their heterogeneity suggest that a single anti-cancer agent is insufficient to target the entire cancerous population. Combination therapy, in which multiple therapeutic agents with different mechanisms of action administered together, has emerged as a new approach to target breast cancer, and is expected to enhance the therapeutic effect without additional toxicity.11 However, in practice, the combined effect of free drugs has deviated from the expectations, and a number of issues has raised that could hamper the clinical efficacy, for example, solubility, stability and pharmacokinetic issues.12 A meticulous design of the nano-drug delivery systems is one of the solutions to address these challenges.
Since the approval of first nanodrug Doxil (liposomal doxorubicin) by Food and Drug Administration (FDA) in 1995, nanotechnology has been studied extensively in drug delivery for over two decades.13 Nanoparticles utilise the differences in the physiological features between tumour and normal cells, and achieve specific tumour targeting mainly through two strategies: (1) passive targeting, via the Enhanced Permeability and Retention (EPR) effect, by which nanoparticles accumulate in solid tumours; and (2) active targeting, which increases cancer cell uptake by covering a ligand on the surface of nanoparticles to bind to a specific receptor or protein overexpressed on the tumour cells.14 Further advances in research enable the development of nanoparticles that are activated by either particular internal stimuli in the tumour microenvironment or external stimuli applied.15 Nanotechnology offers a range of distinctive advantages in anti-cancer therapy, including enhanced therapeutic efficacy with reduced systemic side effects, targeted drug delivery from tissue to organelle level, improved pharmaceutical properties, co-delivery of multiple drugs and facilitated intracellular delivery of biomacromolecules.16 Among those advantages, co-deliver combinational drug therapy is believed to improve therapeutic efficacy and overcome drug resistance, therefore, research in this field has increased exponentially in the last decade.12
In this review, the nature of cancer stem-like cells and challenges of eliminating BCSCs including the treatment resistance and plasticity of BCSCs is addressed, followed by the discussion of the rationales and principles of designing a co-delivery nanosystem, in which the multiple anti-cancer agents are either loaded into separate nanoparticles individually “Nano + Nano” or contained in a single nanocarrier (co-encapsulation).12 A particular focus is given to the presentation and discussion of studies available to date that co-deliver dual or multiple agents to target BCSCs using “Nano + Nano” and co-encapsulation systems.
2. Characteristics of BCSCs
BCSCs were first proposed by Al-Hajj et al. in 2003, which marks the first cancer stem-like cells reported in solid tumours following their discovery in acute myeloid leukemia in 1997.17,18 BCSCs can be identified through a set of surface markers. The two most commonly used are the cluster of differentiation 44 (CD44) and aldehyde dehydrogenase (ALDH). Al-Hajj pointed out that a small number of CD44+/CD24− cells were able to generate tumours; whereas 100-fold greater of other phenotype cells did not show any tumorigenicity.17 Similarly, Ginestier et al.19 claimed that although only representing 3% to 10% of the total population, a low number of 500 ALDH1+ cells were able to form tumours in 40 days; whereas, ALDH1− cells was not able to generate tumour after up to 34 weeks. Interestingly, there is only 0.1–1.2% overlapping between ALDH1+ and CD44+/CD24− BCSCs, indicating the two surface markers identify different BCSCs populations.19 Other surface markers in breast cancer include CD133 (prominin-1) and epithelial cell adhesion molecule (EpCAM) or CD326.
Aberrant regulation in several key signaling pathways has been recognized as another characteristics of BCSCs.20 Over activation of Notch, Wnt/Frizzled/β-catenin, Hippo and Hedgehog pathways are the main ones believed to be responsible for the tumour initiation and maintenance of stemness, such as self-renewal, proliferation and differentiation.21
3. Challenges of eliminating BCSCs
The proven significance of BCSCs, and the potential of treating breast cancer by targeting the roots of tumour initiation, maintenance and metastasis have attracted immense interest. However, an effective strategy to eliminate BCSCs is still lacking. The biggest challenges presented include treatment resistance and high plasticity of BCSCs.
3.1. Treatment resistance
Similar to other cancer stem-like cells, BCSCs are highly chemotherapy and radiotherapy resistant, and a number of mechanisms have been proposed.22 Firstly, majority of BCSCs exist predominantly in quiescent or resting state of cell cycle (G0), therefore inherently immune to chemotherapy drugs which targeting rapidly proliferating cells.23 Secondly, there is an overexpression of adenosine triphosphate (ATP)-binding cassette transporters (ABC transporters) that utilise the energy generated in ATP hydrolysis to pump chemotherapeutic agents out of the cell cross the membrane.20 ABC subfamily B member 1 (ABCB1), also known as multi-drug resistance protein 1 (MDR1), ABC subfamily C member 1 (ABCC1) and ABC subfamily G isoform 2 (ABCG2), also known as breast cancer resistance protein (BCRP) are the main ABC transporters in breast cancer causing chemotherapeutic resistance.24 Thirdly, BCSCs have an enhanced DNA damage repair ability and decreased apoptosis.25 B-cell lymphoma 2 (BCL-2) is a family of regulator proteins that control apoptosis through pro-apoptotic (Bax, Bad and Bak) and anti-apoptotic (BCL-2 proper, BCL-XL, MCL) proteins; overexpression of anti-apoptotic proteins contributes to the treatment resistance.26 Fourthly, BCSCs predominately locate in hypoxia area. Conventional photon radiotherapy damages the tumour cells through the generation of free radicals, such as reactive oxygen species (ROS). The hypoxia environment and enhanced ROS scavenging system (for example, glutathione) results in a low ROS and leads to radiotherapy resistance.25 In addition, hypoxia-inducible factors 1α and 2α (HIF1α and HIF2α) are closely related to the stemness and drug resistance of BCSCs.27 HIF1α plays a role in BCSCs proliferation, and more importantly maintain the quiescence state of BCSCs; whereas, HIF2α helps with BCSCs survival. Other mechanisms in drug resistance of BCSCs include the overexpression of ALDH, a family of detoxifying enzymes, that prevents cells from oxidative insults and detoxifies toxic intermediates of chemotherapy drugs;28 and the deregulation of phosphatidylinositol 3-kinase (PI3K) pathway.29
3.2. Plasticity of BCSCs
While some believed BCSCs reside at the apex of hierarchy, and differentiate into bulk non-stem cells in a unidirectional manner, emerging evidence have supported that BCSCs are not a static state and they preserve a high plasticity in which BCSCs and bulk cancer cells are interconvertible.30,31 Transforming growth factor (TGF)-β signaling, overexpression of transcription factors Slug and Sox9, radiation therapy all showed the capability to induce the generation of BCSCs from non-stem cells.32,33
High level of plasticity also exists within BCSC population itself between the mesenchymal-like and epithelial-like states.34 Mesenchymal-like BCSCs are characterised as CD44+/CD24−, and are more quiescent, invasive and located at the tumour invasive front. Whereas, epithelial-like BCSCs, characterised as ALDH+, are more proliferative and located centrally in the more hypoxia area of the tumour.34 BCSCs are able to interchange between proliferative epithelial-like and quiescent mesenchymal-like states in the processes of epithelial–mesenchymal transition (EMT) and mesenchymal–epithelial transition (MET).33,35 The regulation of those processes is poorly understood, although tumour microenvironment (for example TGF-β signaling36 and interleukin-6 secretion37) is believed to be linked to the transition.
4. Principles of designing a co-delivery nanoparticle system
Conventional chemotherapy and radiotherapy may show initial response by killing bulk breast cancer cells, but the high treatment resistance and cell plasticity of BCSCs suggests that a single chemotherapy agent alone is not sufficient to eliminate the entire tumour tissue. Combination of two or more agents with different mechanism of actions presents the future research direction in breast cancer treatment. A number of factors need to be considered when designing an effective combined therapy.
4.1. Choice of medications
4.1.1. Criteria of drug combination
In clinic, co-delivery of two or more agents in the treatment is aimed to ultimately achieve superior therapeutic effect with minimal toxicity. Therefore, the drugs selected need to have:12,38 (1) different mechanism of actions; (2) no overlapping toxicity; (3) no cross-resistance; and (4) are able to target both BCSCs and non-BCSCs simultaneously.
4.1.2. The mechanisms of action of BCSC specific inhibitors in combination therapy
Simultaneous targeting of both BCSCs and non-BCSCs is the most essential factor in choosing a drug combination, since any BCSCs survived the treatments or new BCSCs interconverted from non-BCSCs are able to self-renew and differentiate, and finally lead to disease relapse, progress and distant metastases. The treatment resistance to conventional chemotherapeutic agents signifies that at least one of the compounds in the combination therapy has to show specific inhibition against BCSCs.
Currently, targeting of BCSCs is mainly achieved through four strategies.20,39,40 Firstly, inhibit the mechanisms that cause treatment resistance (Section 3.1).20 Secondly, target intrinsic self-renewal pathways of BCSCs. Thirdly, through the interference of the tumour microenvironment.20 Hypoxia, elevated level of cytokines (such as interleukin −8 and −6), tumour-associated macrophages, T cells and extracellular matrix have all been found to either promote or maintain the stemness. The fourth strategy is to target the reprogrammed metabolic pathway in BCSCs.40 Various metabolic pathways are altered in cancer cells in order to adapt to the abnormal growth needs. Aerobic glycolysis, mitochondrial metabolism, redox reaction and lipid metabolism are all potential BCSCs targets. BCSCs targeting strategies were well summarised in previous reviews, and interested readers can refer to original articles for details.20,39,40 A BCSCs specific inhibitor can act on one or more of those four aspects, Table 2 summaries a list of cancer stem cell specific inhibitors that have been used in breast cancer research, and their mechanism of actions have been proved in either breast cancer or other cancer stem cells. Table 3 shows their chemical structured properties.
Examples of cancer stem cell including BCSC specific inhibitors used in breast cancer research and their mechanism of actions.
| BCSCs specific inhibitor | Mechanism of actions | Reference |
|---|---|---|
| Targeting treatment resistance | ||
| Lapatinib | Inhibit MDR1 and BCRP | 41 |
| Salinomycin | Inhibit MDR1; induce apoptosis | 42 and 43 |
| Tamoxifen | Inhibit P-glycoprotein; inhibit BCRP | 44 and 45 |
| Thioridazine | Induce G1 arrest | 46 |
| All-trans retinoic acid | Arrest cell cycle through upregulation of cyclin-dependent kinase inhibitors and downregulation of cell-cycle progression activators; induce differentiation | 47 and 48 |
| Curcumin | Induce apoptosis by regulating Bcl-2 family proteins | 26 |
| Quinacrine | Activate pro-apoptotic Bax proteins | 49 |
| Parthenolide | Induce caspase-mediated apoptosis; activate pro-apoptotic P53; increase reactive oxygen species | 50 and 51 |
| Targeting intrinsic self-renew pathways | ||
| Salinomycin | Inhibit Wnt/β-catenin pathways | 42 and 43 |
| Verteporfin | Inhibit NF-κB, Wnt pathways | 52 |
| 8-Hydroxyquinoline | Inhibit NF-κB pathway | 53 |
| Curcumin | Inhibit Wnt/β-catenin, PI3k and sonic hedgehog pathways | 54 |
| Cyclopamine | Inhibit sonic hedgehog pathway | 55 |
| GANT 61 | Inhibit hedgehog pathway | 56 |
| Thioridazine | Inhibit STAT3 pathway | 46 |
| Parthenolide | Inhibit NF-κB signaling | 51 |
| Metformin | Inhibit NF-κB and STAT3 pathways | 57 |
| All-trans retinoic acid | Inhibit mitogen-activated protein kinase signaling pathways | 58 |
| Targeting tumour microenvironment | ||
| Camptothecin | Inhibit HIF-1α activity | 59 |
| Targeting metabolic pathways | ||
| Salinomycin | Inhibit mitochondrial oxidative phosphorylation | 42 |
| Metformin | Inhibit mitochondrial complex I | 60 |
Chemical structures, solubility and octanol/water partition coefficient (log P) or calculated log P (clog P) of the drugs used to target BCSCs, and the nanostructures as delivery systems.
| Name | Drug structure | Physiochemical propertiesa | Nanostructures |
|---|---|---|---|
| 8-Hydroxyquinoline |
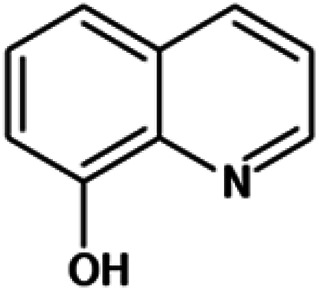
|
Insoluble in water, freely soluble in alcohol, acetone and chloroform | Mesoporous silica79 |
| log P 2.02 | |||
| All-trans retinoic acid (vitamin A acid) |
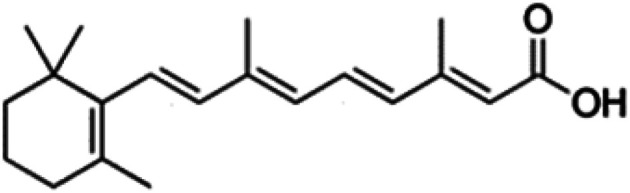
|
Insoluble in water, soluble in dimethyl sulfoxide (DMSO) | Liposome,74 polymeric nanoparticle38 |
| log P 6.3 | |||
| Camptothecin |
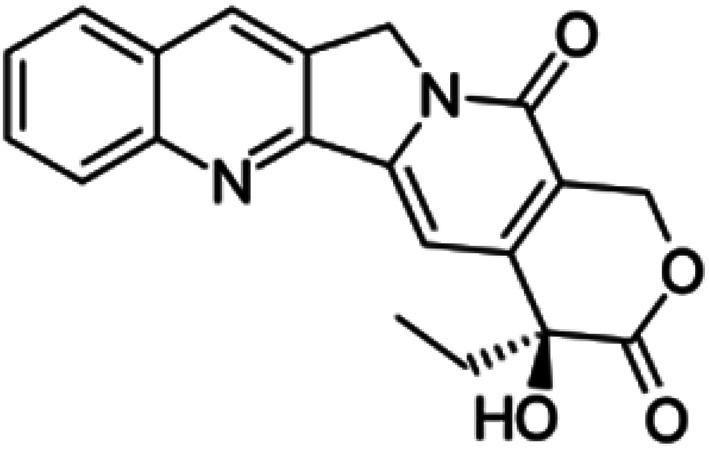
|
Insoluble in water, soluble in DMSO, methanol, and acetic acid | Drug–drug conjugate82 |
| log P 1.74 | |||
| Curcumin |
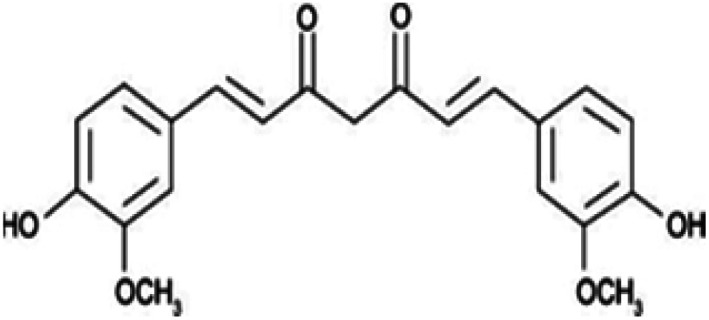
|
Insoluble in water, soluble in alcohol, acetic acid | Polymeric nanoparticle,66,83 polymeric micelle84,85 |
| log P 3.29 | |||
| Cyclopamine |
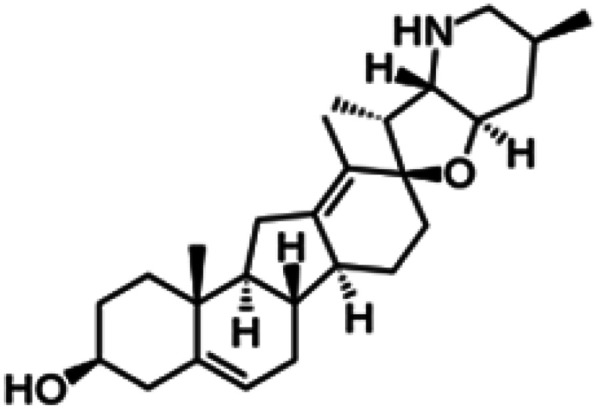
|
Insoluble in water, soluble in DMSO, ethanol, dimethyl formamide (DMF) and methanol | Polymeric nanoparticle55 |
| clog P 3.5 | |||
| GANT 61 |
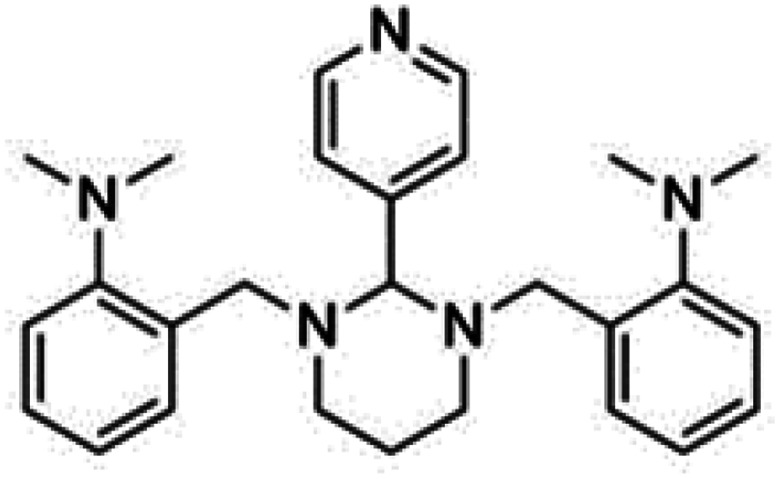
|
Insoluble in water, soluble in DMSO and ethanol | Polymeric nanoparticle86 |
| clog P 4.2 | |||
| Irinotecan |
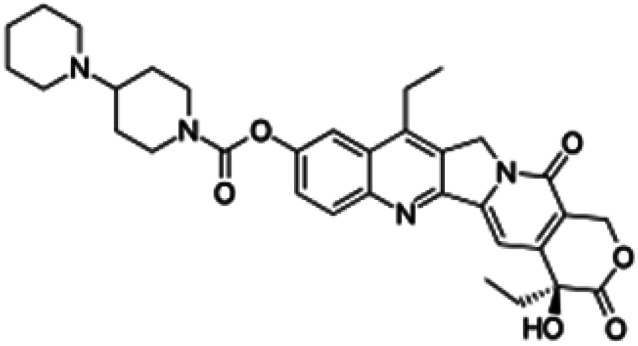
|
Soluble in water, soluble in DMSO | PLGA-based polymeric nanoparticles87 |
| clog P 3 | |||
| Parthenolide |
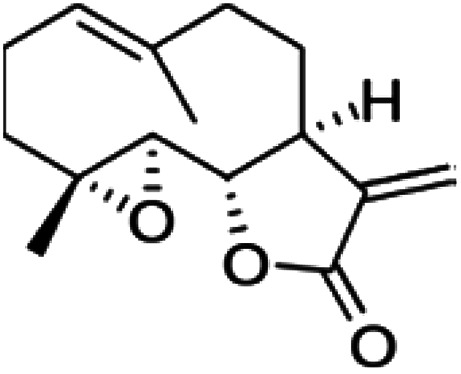
|
Insoluble in water, soluble in DMSO, ethanol | Liposomes88 |
| clog P 2.3 | |||
| Quinacrine |
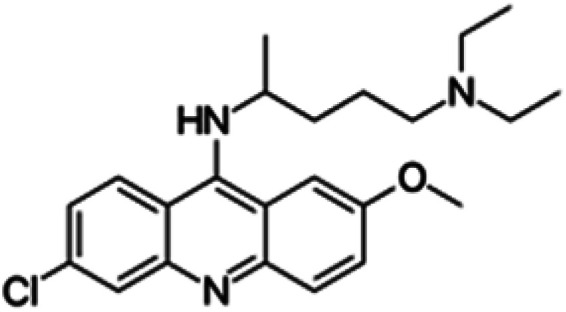
|
Sparingly soluble in water, slightly soluble in ethanol, insoluble in alcohol, benzene, chloroform and ether | Liposomes49 |
| log P 5.75 | |||
| Salinomycin |
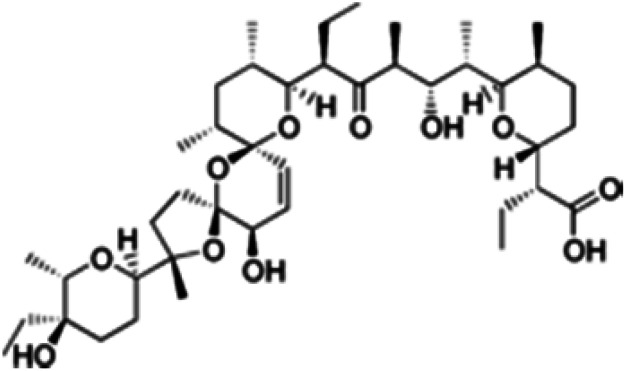
|
Insoluble in water, soluble in ethanol, DMSO and DMF | Polymeric micelles,89 PLGA-based polymeric nanoparticle,70,90 liposomes71 |
| log P 8.53 | |||
| Staurosporine |
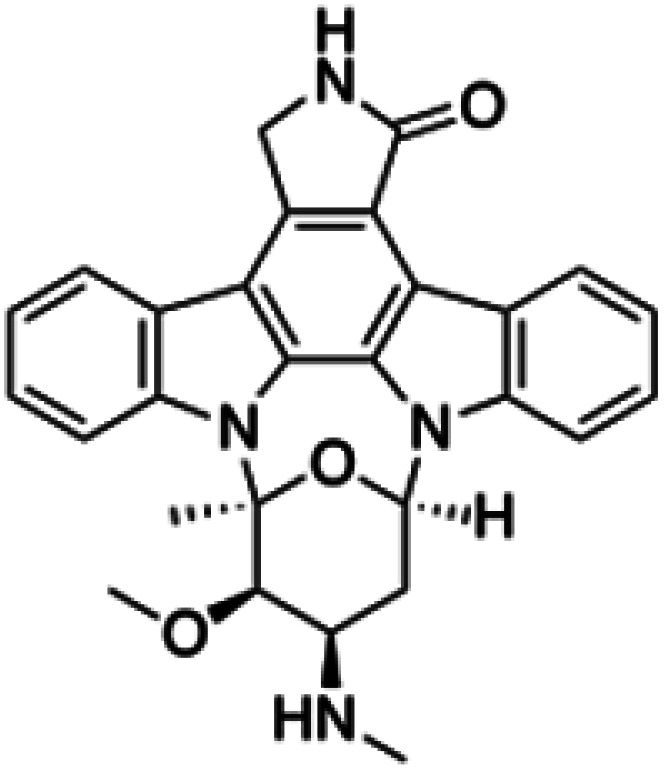
|
Insoluble in water, soluble in DMSO and DMF, slightly soluble in chloroform, methanol and acetone | Polymeric micelle91 |
| clog P 3.2 | |||
| Tamoxifen |
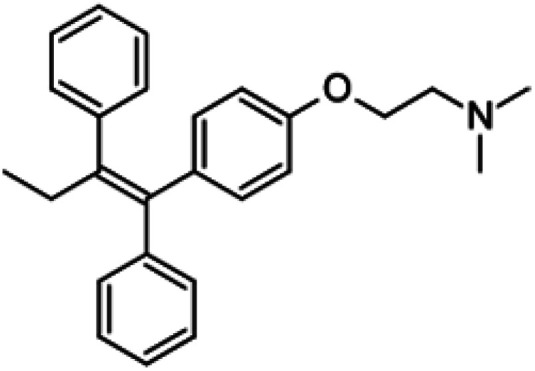
|
Insoluble in water, soluble in ethanol and DMSO | Liposomes92 |
| log P 6.30 | |||
| Thioridazine |
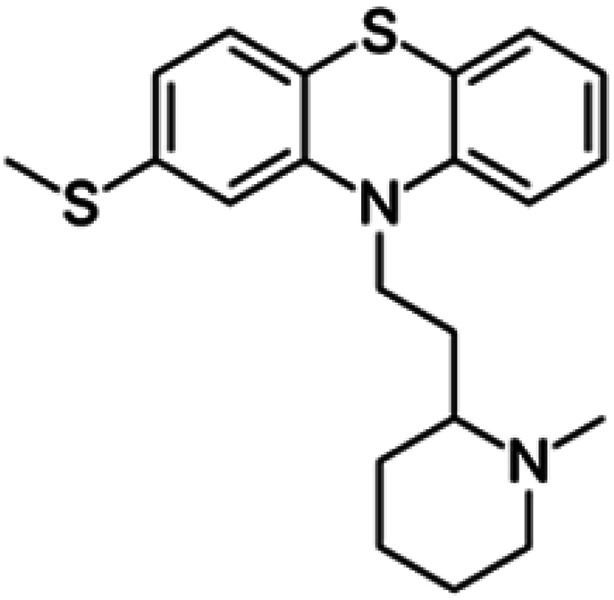
|
Insoluble in water, soluble in alcohol, chloroform, ether, and freely soluble in dehydrated alcohol | Polymeric micelles93,94 |
| log P 5.90 | |||
| Verteporfin |
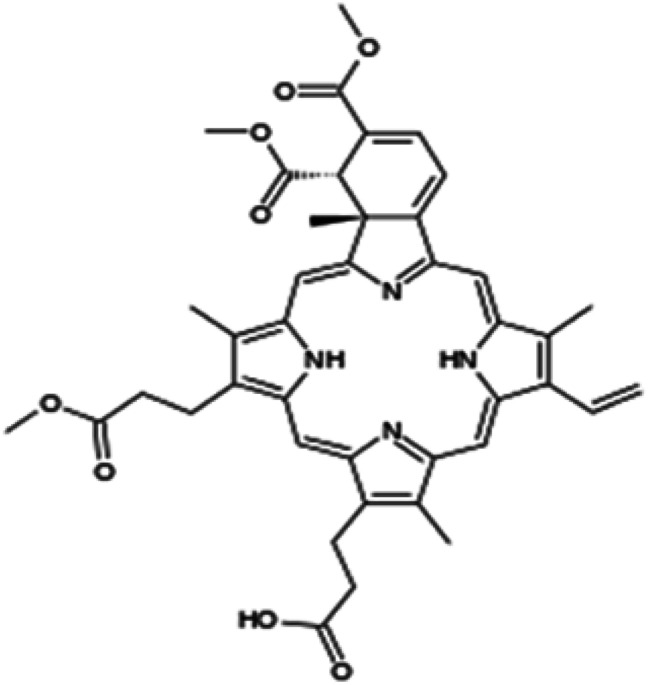
|
Insoluble in water, soluble in DMSO and DMF | PLGA-based nanoparticle52 |
| log P 2.1 |
Data are adopted from PubChem (https://www.pubchem.ncbi.nlm.nih.gov) or DrugBank (http://www.drugbank.ca).
4.2. Combined therapeutic effects
The combination of two or multiple agents do not always result in superior efficacy than the sum of agents administered individually. Combination index (CI), a quantitative measure of the drug interactions at a given effect level, is commonly used to determine the overall effect and is calculated as:61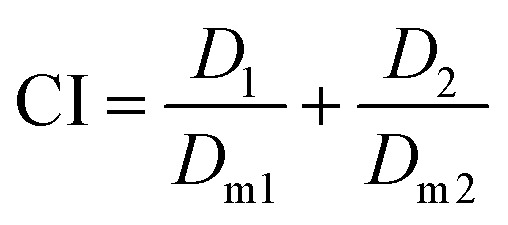 where; D1 and D2 are the dose of drug 1 and drug 2 in combination formulation to achieve a median effect, for example, the half maximal inhibitory concentration (IC50), medium effective dose (ED50) or medium lethal dose (LD50); Dm1 and Dm2 are the dose of drug 1 and drug 2 in single drug formulations, administered separately, to achieve the same effect.
where; D1 and D2 are the dose of drug 1 and drug 2 in combination formulation to achieve a median effect, for example, the half maximal inhibitory concentration (IC50), medium effective dose (ED50) or medium lethal dose (LD50); Dm1 and Dm2 are the dose of drug 1 and drug 2 in single drug formulations, administered separately, to achieve the same effect.
The therapeutic effect of the combination therapy could be equal to the sum of the monotherapy of each individual agent (additive effect, CI = 1), or it can be greater (synergistic effect, CI < 1) or smaller (antagonism, CI > 1) than the sum of monotherapy;62 an ideal design of a co-delivery system requires a synergistic or addictive effect (CI ≤ 1). For example, when doxorubicin and dihydroartemisinin combined together, they showed a synergistic effect (CI < 1) towards all tested breast cancer cell lines (MCF-7, MDA-MB-231 and T-47D), except antagonism (CI = 1.08) at high dose (20 μM of doxorubicin and 40 μM of dihydroartemisinin) in MDA-MB-231.63
Recently, Mitragotri's research team found that the currently used in vitro cytotoxicity parameters, such as CI and IC50 values, suffer from unpredictable translation to in vivo synergy quantification of the combination therapies.64 Instead, the drug combinations with high in vitro dose–response Hill coefficient showed the strongest correlation with a high in vivo anti-tumour response. Hill coefficient (m) is determined according to the Hill equation: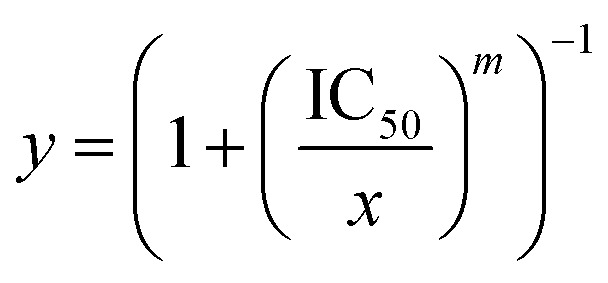 where; y is the fractional inhibition of the cancer cells, and x is the drug concentration.
where; y is the fractional inhibition of the cancer cells, and x is the drug concentration.
4.3. Dosing scheduling in combination therapy
The sequence of two drug administered is a crucial factor in combination therapy. Oliveras-Ferraros et al. demonstrated that the interaction between paclitaxel and gemcitabine in breast cancer cell lines is schedule-dependent.65 Synergism was only observed in most cell lines when paclitaxel was administered before gemcitabine, and addictive effect or even antagonism was observed when paclitaxel and gemcitabine were administered simultaneously or paclitaxel was administered after gemcitabine. The importance of dose sequencing can be explained by the modulation of drug resistance. For example, when curcumin is first released from the delivery system, it inhibits P-glycoprotein, which is responsible for the multi-drug resistance of doxorubicin; thus subsequently increase the intracellular level of doxorubicin and result in increased cytotoxicity.66 Same principle applies to RNA therapy. When combined with a chemotherapy agent, RNA usually binds to the surface of nanoparticles via electrostatic interaction, and gets released earlier than chemotherapy drugs.67,68 This sequential release ensures down-regulation of genes involved in drug resistance first, and improves the cell sensitivity to subsequent chemotherapeutic drugs.
4.4. The role of dose ratio in combination therapy
Dose ratio plays an important role in maximizing the treatment efficacy in combination therapy. Various ratios of the same drug combination were found to lead to different or even opposite treatment outcomes. Tao et al.69 investigated the combination effect of disulfiram and doxorubicin at different weight ratio on breast cancer cell lines. In MDA-MB-231 cells, a synergistic effect was observed when the weight ratio of disulfiram/doxorubicin was at 1 : 2 and 1 : 1; whereas an addictive effect and antagonism were recorded when the ratio was at 1 : 0.2 and 1 : 0.5 respectively. In MCF-7 cells, all above mentioned weight ratios resulted in synergism, except an addictive effect at 1 : 0.5. The different effects resulted from varying dose ratio in a combined therapy were also demonstrated between salinomycin and docetaxel,70 and salinomycin and doxorubicin.71
4.5. Nanoparticle systems and delivery approaches to target BCSCs
4.5.1. Nano-structures
Nanosized drug delivery system as the novel approach in ‘cocktail’ anti-cancer therapy, has shown superiority over the traditional combination of free drug solutions. For instance, while no synergistic effect occurred when doxorubicin and paclitaxel were mixed in their free form, synergism was observed when the two drugs were combined into a single nanosystem at a weight ratio (doxorubicin/paclitaxel) of 5 : 1 and 3 : 3.72 Nanoparticles can be made of either natural or synthetic materials, and are classified into groups such as lipid-based nanoparticles, polymeric nanoparticles (nanospheres), polymeric micelles, dendrimers, and inorganic nanoparticles73 (Fig. 2A). Lipid-based nanoparticles, including liposomes and solid lipid nanoparticles, are biocompatible with enhanced cellular uptake and capable of delivering both hydrophobic and hydrophilic drugs, therefore, are commonly used in anti-BCSCs.49,67,71,74,75 Poly(lactic-co-glycolic acid) (PLGA) based drug delivery systems are the most attractive polymeric nanoparticles,47,66,76 because of their well-established biocompatibility, biodegradation and the ability to accommodate a large range of drug molecules by controlling the physiochemical parameters of the nanoparticle.77 Polymeric micelles, formed from self-assembling of amphiphilic block copolymers, are easy to prepare and highly stable in vitro and in vivo.78 Inorganic nanoparticles derived from their macromolecule counterparts, have various material properties and can exhibit fluorescence, and responsiveness to near-infrared radiation (NIR) and magnet.73 Hollow gold nanoparticles68 was used to deliver drugs to BCSCs triggered by NIR; and mesoporous silica nanoparticle was utilised to target breast cancer cells due to the favourable stability and biocompatibility.79
Fig. 2. Types of nanoparticles employed in anti-BCSCs combination therapies (A); and their delivery approaches (B): (a) “Nano + Nano” administered either sequentially or simultaneously; and (b) co-encapsulation for synchronised delivery.

Most of the nanoparticles have distinct advantages of improving solubility of poorly soluble drugs and prolonged circulation times.80 As shown in Table 3, drugs used to target BCSCs are poorly soluble with high lipophilicity therefore nanoparticles enable the formulation of these drugs as injections, optimisation of the pharmacokinetic profile on top of the passive tumour targeting ability. The choice of nano structures depends on the physiochemical properties of the two drugs loaded, stability, particle size and biocompatibility of the nanostructure, and the cost of manufacturing.80,81
4.5.2. Functionalisation of nanoparticles
Passive targeting, based on the abovementioned plain nanostructures, allows the accumulation of nanoparticles at the tumour site; functionalisation of nanoparticles enhance the cellular uptake via the interaction between the ligands on the surface and the receptors overexpressed on BCSCs.
A number of BCSCs surface markers have been explored, and CD44 is arguably the most commonly studied targets. Hyaluronic acid (HA), a natural component of extracellular matrix, is one of the main ligands bind to CD44, and thus often conjugated to the surface of nanoparticles for specific BCSCs targeting.95 HA functionalised lipoid has been synthesized to modify PLGA nanoparticles for co-delivery of paclitaxel and curcumin which synergistically decreased the mammosphere formation of BCSC and inhibited the growth of both non-BCSCs and BCSCs on MCF7 xenografted models.83 Alternatively, PLGA was coupled with HA via a bio-reducible disulfide linker to form HA-SS-PLGA, which was then formulated with Pluronic F127 and chitosan to co-deliver two topoisomerase inhibitors-doxorubicin and irinotecan.55 CD44 targeting had also been achieved through the use of chitosan,96 anti-CD44 antibody97 and anti-CD44 aptamers.98 Ligands have also been used to target other BCSC markers, for instance, anti-CD133 monoclonal antibody was conjugated to the nanoparticles via the maleimide group of polylactide–polyethylene glycol (PEG)–maleimide99 and EpCam aptamers were coated on PLGA-PEG-COOH nano-polymersomes via carbodiimide reaction.100
4.5.3. Delivery approach in combination therapy
For delivery, two drugs can be loaded into separate nanoparticles (“Nano + Nano” approach) to be administered either simultaneously or sequentially,12 or co-loaded into a single nanoparticle system (co-encapsulation approach) (Fig. 2B). Examples are listed in Tables 4 and 5.
Examples of combining a conventional chemotherapeutic agent and a BCSCs specific inhibitor.
| BCSCs inhibitor | Chemotherapy agent | Delivery system | Targeting strategy |
|---|---|---|---|
| “Nano + Nano” approach | |||
| Parthenolide | Vinorelbine | PEGylated liposome88 | Passive targeting |
| Thioridazine | Doxorubicin | Polymeric micelles with mixed copolymers93 | Passive targeting |
| Salinomycin | Paclitaxel | Polymeric micelles: poly(ethylene glycol)-block-poly(ε-caprolactone) diblock copolymer89 | Passive targeting to BCSCs and active targeting to non-BCSCs (somatostatin) |
| 8-Hydroxyquinoline | Docetaxel | Mesoporous silica core coated with lipid bilayer and HA79 | Active targeting to BCSCs (CD44) and passive targeting to non-BCSCs |
| Salinomycin | Paclitaxel | HA-PLGA nanospheres76 | Active targeting to BCSCs (CD44) and passive targeting to non-BCSCs |
| Co-encapsulation approach | |||
| Salinomycin | Docetaxel | Polymeric nanoparticles: PLGA/d-alpha-tocopherol polyethylene glycol 1000 succinate70 | Passive targeting |
| Salinomycin | Doxorubicin | Cross-linked multilamellar liposome vesicles71 | Passive targeting |
| Verteporfin | Paclitaxel | PLGA nanosphere52 | Passive targeting |
| Tamoxifen | Daunorubicin | PEGylated liposomes92 | Passive targeting |
| Camptothecin | Floxuridine | Drug–drug conjugate82 | Passive targeting |
| Quinacrine | Daunorubicin | Dequalinium-PEG-liposomes49 | Passive targeting and mitochondrial targeting |
| Irinotecan | Doxorubicin | PLGA nanosphere87 | Active targeting (CD44) |
| Staurosporine | Epirubicin | Polymeric micelle: PEG-b-poly(aspartate) copolymers91 | Passive targeting and pH sensitive |
| Salinomycin | Paclitaxel | Redox-sensitive drug conjugate103 | Active targeting (CD44) and redox-sensitive |
| Thioridazine | Paclitaxel and PD-1/PDL-1 inhibitor (HY19991) | Micelle in liposome nanodevice94 | Passive targeting and pH and matrix metalloproteinase (MMP) sensitive |
Examples of combining a chemotherapeutic agent with a phytochemical.
| Phytochemicals | Therapeutic agents | Delivery system | BCSCs targeting strategy | Combination strategy |
|---|---|---|---|---|
| Curcumin | Paclitaxel | PLGA based nanoparticle83 | Active targeting (CD44) | Co-encapsulation |
| Paclitaxel | Polymeric micelle; PEG-benzoic imine-poly(g-benzyl-laspartate)-b-poly(1-vinylimidazole) triblock copolymer84 | Passive targeting AND pH-sensitive | Co-encapsulation | |
| Paclitaxel | Nano-egg: oligosaccharides of hyaluronan (oHA)-histidine-menthone 1,2-glycerol ketal micelle with inorganic calcium and phosphate ions108 | Active targeting (CD44) AND pH-sensitive | Co-encapsulation | |
| Doxorubicin | Polymeric nanoparticle: monomethoxy-PEG-b-PLGA-b-P (l-glutamic acid) polymer66 | Passive targeting AND pH-sensitive | Co-encapsulation | |
| GANT 61 | Polymeric nanoparticles: poly lactic-co-glycolic acid based86 | Passive targeting | Co-encapsulation | |
| Icariin | Polymeric micelles: polymer oligomeric HA-hydrazone-folic acid-biotin85 | Active targeting (CD44) and pH-sensitive | Co-encapsulation | |
| All-trans retinoic acid | Vinorelbine | Liposomes74 | Passive targeting | Nano + Nano |
| Doxorubicin | PEG-block-polylactide based nanoparticle47 | Passive targeting | Co-encapsulation | |
| Cyclopamine | Doxorubicin | PLGA nanoparticle55 | Active targeting (CD44) and redox-sensitive | Co-encapsulation |
It is worth noting that despite the importance of dose scheduling as previously discussed (Section 4.3), most “Nano + Nano” administered two pharmaceutical agents simultaneously. Concurrent administration of two agents targeting BCSCs and non-BCSCs separately ensures both subgroups of cancer cells are killed at the same time, leaving no window for the differentiation and interconversion between the cells. In addition, since the two agents possess different mechanism of actions, the possibility of causing an overlapping toxicity is low. Sequential deliveries are also utilised in other studies. In this regard, “Nano + Nano” has the advantages of offering flexibility in dose scheduling.75 Li et al.74 reported that sequential administration of all-trans-retinoic-acid containing liposome and vinorelbine liposome with a 48 h interval induced the differentiation of BCSCs, and thus improved the sensitivity of tumour cells to vinorelbine. The sequential releasing of drugs in co-encapsulation is more challenging and can be achieved through judicious design of the delivery system.66,68,94 For example, by utilising different degree of electrostatic interactions, drug with weaker interaction force gets released faster;66 or through the response to the stimuli in the tumour microenvironment, such as pH94 and NIR.68
“Nano + Nano” provides an easy manipulation of the drug ratios administered, however, this approach is unable to manage the site specific delivery and maintain the pre-determined synergistic ratio of two physio-chemically and pharmacokinetically different drugs in vivo. When two free drugs are administered systematically, they undergo different mechanism of metabolism and elimination, resulting in diverse plasma half-lives, biodistributions and the final drug concentrations reaching the tumour sites.101 In this context, the co-encapsulation approach displays the advantage of synchronising the pharmacokinetics of different agents, co-localising the two drugs into the same tumour site, and maintaining a predetermined synergistic drug ratio, results in an enhanced efficacy. For example, co-delivery of salinomycin and doxorubicin by co-encapsulation with liposomes showed 2-fold more efficacy than the “Nano-salinomycin + Nano-doxorubicin” approach in elimination of BSCS and cancer cells.71 Nevertheless, physically loading multi-agents at a pre-determined dose into one nanosystem is often problematic due to different pharmacokinetics and carrier-drug affinity.102 In order to overcome this problem, the interaction between drugs and the nanosystem needs to be thoroughly investigated and designed. Covalent binding of drugs to nanocarrier or non-covalent loading using cyclodextrin derivatives are two of the examples to address this issue.102
5. Applications of dual- or multi-drug loaded nanoparticles to target BCSCs
The aforementioned principles of designing a co-delivery system are followed and provide a valuable foundation in current scientific research in targeting BCSCs. Because of the chemo-resistance, combination of two chemotherapeutic agents does not seem to be a promising strategy. In order to achieve synergistic killing, conventional chemotherapeutic agent is usually combined with a BCSCs specific inhibitor, a phytochemicals or a RNA based therapy (Tables 4–6).
Examples of combining one chemotherapeutic agent with a RNA based therapy.
| RNA duplexes | Gene targets | Chemotherapeutic agent | Delivery system | BCSCs targeting strategy |
|---|---|---|---|---|
| siRNA | ABCG2, BCL2 | Doxorubicin | Niosome116 | Passive targeting |
| Clusterin, glucose-regulated protein 78 (GRP78) | Camptothecin | Liposomes67 | Passive targeting | |
| miRNA | miRNA-200c | Paclitaxel | Solid lipid nanoparticles75 | Passive targeting |
| miRNA-21 | Doxorubicin | Hollow gold nanoparticle68 | NIR-responsive | |
| shRNA | NF-κB | Doxorubicin | Mixed micelle system: carbamate-mannose modified poly(ethylenimine)117 | Passive targeting |
5.1. Combination of conventional chemotherapeutic agent and a BCSCs specific inhibitor
BCSCs specific inhibitors target a specific characteristic that is unique to BCSCs as previously summarised (Table 2). Some BCSCs specific inhibitors, for example salinomycin and all-trans retinoic acid, show great toxicity towards BCSCs, but have little effect against non-BCSCs cells.47,89 Combination of a conventional chemotherapeutic agent and a BCSCs specific inhibitor is commonly investigated in BCSCs targeting (Table 4).
5.1.1. “Nano + Nano” approach
Passive targeting
The basic strategy in “Nano + Nano” is to mix two single-agent nanoparticles and target tumour tissue via passive targeting. Liu et al.88 investigated this approach over a decade ago using a combination of vinorelbine stealthy liposomes and parthenolide stealthy liposome. Free parthenolide (10 μM) or parthenolide stealthy liposome (10 μM) alone displayed 10% to 15% inhibition against BCSCs, and less inhibition against non-BCSCs; whereas the inhibitory effect of vinorelbine was stronger against non-BCSCs than BCSCs. Combination of vinorelbine and parthenolide liposomes showed a great sensitising effect, and an inhibition of approximately 90% was observed in BCSCs population. Co-delivery of thioridazine mixed micelle and doxorubicin mixed micelle was studied by Ke et al., in which thioridazine was used as a BCSCs specific inhibitor to target dopamine receptor overexpressed on BCSCs, and to augment the anti-proliferative effect of doxorubicin.93 Adding thioridazine containing micelle to doxorubicin micelle significantly reduced cell viability in both BT474 and MCF-7 cells in vitro, and showed highest tumour volume reduction in vivo. The mixed micelle system also remarkably reduced the side effects of both drugs. For example, free thioridazine (5 mg kg−1) caused uncontrollable movement or coma in mice, and such toxic effects were not observed when thioridazine was administered in micelles at the same dose. Similarly, doxorubicin (5 mg kg−1) containing mixed micelle system caused significantly lower loss in body weight comparing with formulations containing free doxorubicin.
Active targeting
The surface of each or both of the two single-agent nanoparticles can be modified with a specific ligand to allow more specific tumour cellular uptake via active targeting. Somatostatin receptors are overexpressed in bulk of tumour tissue compared with normal tissue, and octreotide, an analog of endogenous somatostatin, was consequently utilised as an active targeting ligand.89 Paclitaxel, a conventional chemotherapeutic agent, was loaded into octreotide-modified poly(ethylene glycol)-block-poly(ε-caprolactone) (PEG-b-PCL) micelles to actively target bulk breast cancer cells via somatostatin receptors; and salinomycin, with over 100 fold cytotoxicity against BCSCs than paclitaxel,104 was loaded into a non-modified PEG-b-PCL micelles to target BCSCs via passive targeting. In vitro, cytotoxicity of paclitaxel micelle was shown to be enhanced by octreotide conjugation through increased cellular uptake. The combination treatment of octreotide modified paclitaxel micelle and salinomycin-micelle showed better inhibition on tumour growth than single drug treatment, and the mixture of non-modified paclitaxel-micelle and salinomycin-micelle.
HA is one of the most commonly used ligand to target CD44 on BCSCs. Wang et al.79 fabricated a mesoporous silica nanoparticle coated with supported lipid bilayer (MSS), which combines the feature of mesoporous silica and liposomes. HA was conjugated to MSS and loaded with 8-hydroxyquinoline to actively targeting BCSCs, whereas docetaxel was loaded into MSS to passively targeting bulk breast cancer cells. The specific binding of HA to CD44 increased the internalisation of HA-MSS, enhanced 8-hydroxyquinoline release into cytoplasm and thus enhanced tumour growth inhibition. Combination of docetaxel-MSS plus 8-hydroxyquinoline-loaded HA-MSS produced superior anti-tumour effect than docetaxel plus 8-hydroxyquinoline, as a result of the active and passive targeting delivery. Comparing with fast disease relapse upon treatment discontinuation in docetaxel group, docetaxel-MSS plus 8-hydroxyquinoline loaded HA-MSS did not show any relapse, signifying the importance of combining docetaxel with a BCSCs specified inhibitor. In another study, HA conjugated PLGA nanoparticles was designed to deliver salinomycin to BCSCs via CD44 receptors, along with paclitaxel-loaded PLGA nanoparticles to target non-BCSCs.76 The combination with HA conjugation demonstrated a remarkable 88.9% in vitro cytotoxicity, compared with 47.5% without HA conjugation.
5.1.2. Co-encapsulation approach
Passive targeting
In a recent study, co-delivery of docetaxel and salinomycin at a pre-determined synergistic ratio (1 : 1) was achieved by loading both drugs in a nanoparticle consists of poly lactide-co-glycolide/d-alpha-tocopherol polyethylene glycol, and target breast cancer cells via passive targeting.70 The synergism between the two drugs was evident as the IC50 of nanoparticle co-delivering docetaxel and salinomycin was significantly lower than IC50 of two single-drug loaded nanoparticles administered together in non-BCSCs (3.1 nM and 5.2 nM, respectively) and BCSCs (1.4 nM and 4.2 nM, respectively). In vivo, the synergistic drug ratio was maintained for 24 h, and the number and size of tumour was significantly smaller with docetaxel and salinomycin co-loaded nanoparticles than that with single nanoparticle or mixture of free drugs (p < 0.05). Similarly, combining salinomycin and doxorubicin in a cross-linked multilamellar liposome vesicles (cMLVs) was able to coordinate the pharmacokinetics of the two drugs, and cMLVs(Doxorubicin+Salinomycin) demonstrated a 2-fold more toxicity than single-drug cMLVs or mixture of cMLV(Doxorubicin) and cMLV(Salinomycin), confirming the synergistic effect of co-localised delivery of doxorubicin and salinomycin.71
Verteporfin, a porphyrin-based photosensitiser, was found to be effective against both BCSCs and bulk tumour mass through the suppression of NF-κB and Wnt pathways, and was incorporated with paclitaxel into polymeric nanoparticles.52 Although the nanosystem has a lower dose of paclitaxel and verteporfin (0.5 mg kg−1 and 3.2 mg kg−1, respectively) than free-drug (1 mg kg−1 and 9 mg kg−1, respectively), it showed a significantly reduced tumour size and tumour weight in triple negative breast cancer patient-derived xenografts.
Camptothecin is a chemotherapeutic agent that inhibits DNA topoisomerase I; it has been studied as an anti-BCSCs agent due to its inhibition on HIF-1α.59 Zhang et al.82 developed an amphiphilic drug–drug conjugate, in which two molecules each of camptothecin and floxuridine was linked together via multivalent pentaerythritol ester linkage, and loaded with a third agent lovastatin (LCF-NC). Lovastatin is one of the statins showed synergistic anti-cancer effect with other chemotherapeutic agents. While camptothecin and floxuridine conjugate reduced BCSCs cell viability to 47.7%, loading of lovastatin reduced the BCSCs cell viability further to 22.7%, indicating a potential synergistic activity of the three drugs. In vivo, LCF NCs showed 85.2% reduction in lung metastases, and no relapse 30 days after treatment.
Mitochondria is an essential organelle in regulating the apoptosis of cells.49 Zhang et al.49 proposed a mitochondrial targeting liposome by decorating the liposome surface with dequalinium that accumulates in mitochondria. Daunorubicin and resistant modulator quinacrine were loaded into the liposome to target both BCSCs and non-BCSCs. The resulting mitochondrial targeting liposomal system induced apoptosis in MCF-7 cancer stem cells by almost 6-fold than PBS control. In vivo, the mitochondria targeting daunorubicin and quinacrine liposomes showed the greatest tumour inhibition ratio (79.3%) in relapsed tumour arising from cancer stem cells.
Active targeting
Irinotecan, an analogue of camptothecin, is one of the two camptothecin derivatives approved for clinical use with improved solubility. A nanoparticle consists of PLGA, Pluronic F127, chitosan and HA were developed to deliver doxorubicin and irinotecan together.87 Interestingly, mixing of two free drugs did not show significant improvement in the cytotoxicity, which was then enhanced up to 500 times by the delivery system. Furthermore, conjugation of HA to the nanoparticle showed an IC50 of 42.1 nM on MDA-MB-231, which is 419.1 times smaller than non-HA coated nanoparticle.
Stimuli-responsive targeting
Tumour microenvironment presents with unique features that allow the development of stimuli-responsive nanoparticles. Simultaneous delivery of two or more pharmaceutical agents in a stimuli-responsive nanosystem further enhanced the specificity of tumour targeting and anti-tumour cytotoxicity. pH-sensitive nanoparticles are designed to explore the pH gradient between tumour and normal tissue.105 Tumour tissue is more acidic (pH 6.5) than normal tissue (pH 7.4) due to accumulation of lactic acid from the increased aerobic glycolysis and reduced oxidative phosphorylation. A pH-sensitive micelle was constructed by connecting epirubicin to PEG-b-poly(aspartate) copolymers via hydrazine links with staurosporine loaded into the core;91 epirubicin was released by the cleavage of hydrazine bond at low endosomal pH. In epirubicin-sensitive breast cancer cells, staurosporine and epirubicin co-loaded micelles demonstrated the highest cytotoxicity, with IC50 5-fold lower than free epirubicin plus staurosporine or free staurosporine plus epirubicin micelle. In epirubicin-resistant breast tumours, co-delivery of both drugs in micelles demonstrated superior cytotoxicity both in vitro and in vivo, possibly due to the strong inhibition of staurosporine against ABC transporters, that prevents the efflux of epirubicin and circumvent the epirubicin resistance. Nevertheless, the exact effect of staurosporine on BCSCs is unclear. Some studies suggested that it may increase cancer stem-like cells (CD44+/CD24−) in breast cancer through the upregulation of Mucin 1 and EpCAM.106 Further study is required before staurosporine can be included in the anti-BCSCs treatments.
A redox-sensitive salinomycin prodrug nanosystem was fabricated by conjugating hydrophobic salinomycin to a Vit E derivative d-α-tocopheryl succinate via cystamine linkages.103 The amphiphilic conjugate was designed to serve as a drug carrier with or without HA modification, and loaded with chemotherapeutic agent paclitaxel. The tumour cells have elevated level of glutathione concentration (∼10 mM), and cystamine linkage in the system is cleaved to release salinomycin at tumour tissue. A redox-sensitive release of both drugs was observed, with 92.5% of paclitaxel and 93.3% of salinomycin released from the drug conjugate in the presence of glutathione at 48 h, compared with 60.3% and 13.2% respectively in the absence of glutathione. The paclitaxel loaded prodrug system improved the bioavailability of free salinomycin and paclitaxel, and showed synergised cytotoxicity on breast cancer cells. The addition of HA coating exhibited much more potent cytotoxicity on breast cancer cells and spheroids, due to improved cellular uptake and penetration via HA and CD44 interaction.
A recent development of a multi-stimuli triggered delivery system utilised the abundance of matrix metalloproteinase (MMPs) and low pH at the tumour tissue.94 A two steps synthesis was designed: chemotherapy agent paclitaxel was first loaded into a pH-responsive copolymer micelle, which was then loaded into a matrix metalloproteinase (MMPs) sensitive liposomes with programmed cell death protein (PD-1)/programmed death-ligand 1 (PD-L1) inhibitor HY19991 and an anti-BCSC agent thioridazine. At tumour tissue, the abundance of MMP triggers the dissociation of outer layer, and release HY19991 to interact with PD-L1 on the surface of tumour, and thioridazine to diffuse into tumour cells. pH-sensitive paclitaxel-loaded micelle enters into tumour cells via endocytosis, and release paclitaxel at low pH in endosome or lysosomes. The complex demonstrated a tumour-specific targeting, and increased intratumoral paclitaxel, thioridazine and HY19991 concentrations by 7.31, 7.23 and 3.65 times, respectively. The resulting multi-delivery system had a tumour inhibiting rate of 93.45%, and reduced the number of lung metastasis by 97.64%.
5.2. Combination of a therapeutic agent and a phytochemical
Comparing with conventional chemotherapeutic agents, naturally occurring compounds, such as phytochemicals, are widely available in nature, have big range of safety profiles, and often have the ability to target multiple pathways.107 Therefore, they have gained extensive interest in anti-cancer therapy, including targeting cancer stem cells (Table 5).
5.2.1. Curcumin
Curcumin is a phytochemical extracted from the dried rhizomes of Curcuma longa L. (turmeric), and is a well-known anti-oxidant and anti-inflammatory agent.109 Curcumin is a popular anti-BCSCs due to its action against multiple pathways and gene expressions involved in tumour survival and proliferation.107 However, the clinical application of curcumin as an anti-cancer drug is far from optimum, mainly due to its poor water solubility, fast metabolism and rapid clearance from the body.109 Combination of curcumin and paclitaxel in a HA-modified hybrid nanoparticles prolonged the circulation of both hydrophobic agents and enhanced tumour accumulation.83 While both drugs have low solubility in water and short blood circulation time, HA conjugated NPs exhibited a more controlled release up to 132 h. The tumour inhibition was highest with HA conjugated nanoparticles loaded with curcumin and paclitaxel (67.5%), even higher than the sum of the inhibition rates of paclitaxel-loaded (17.7%) and curcumin-loaded (23.8%) HA-conjugated nanoparticles. Combination of curcumin and paclitaxel was also investigated in other two studies, where a pH-sensitive system based on poly(ethylene glycol)-benzoic imine-poly(g-benzyl-laspartate)-b-poly(1-vinylimidazole) (mPEG-PBLA-PVIm, PPBV) triblock copolymer and a pH-sensitive mineralized micelle (“nano-egg”) were used as the delivery systems, respectively.84,108 Both nanosystems significantly improved the cellular uptake of paclitaxel and curcumin into the tumour, and significantly reduced ALDH+ cell population. Apart from paclitaxel, co-delivery of curcumin with other chemotherapeutic agents, for example, doxorubicin66 and other phytochemicals, for example, icariin,85 had shown improved cytotoxicity against BCSCs.
Curcumin not only showed inhibition against BCSCs; due to its wide anti-tumour actions, it has also been used to target the non-BCSCs along with another BCSCs specific inhibitor. GANT61 is a Hedgehog pathway inhibitor, and was combined with curcumin in a PLGA based nanoparticle system.86 However, although the GANT61 and curcumin combined nanosystem showed significant inhibition on the self-renewal property of BCSCs, and enhanced cytotoxicity profile than the individual GANT61 PLGA and curcumin PLGA, it failed to show any superiority compare with the mixture of free GANT61 and curcumin.
5.2.2. All-trans retinoic acid
All-trans retinoic acid (ATRA) is an active metabolite of vitamin A that belongs to ‘retinoid family’.110 ATRA stealth liposomes modified with PEG was given as a co-therapy with a cytotoxic agent, vinorelbine stealth liposomes, to prevent the relapse from BCSCs.74 The co-delivery system showed a strong inhibition in tumour volume in vivo (91.6 ± 3.6%) at day 38, through the induction of BCSCs differentiation and cell arresting at G0/G1 phase in mitosis. Sun et al.47 co-loaded ATRA and doxorubicin into poly(ethylene glycol)-block-polylactide (PEG-b-PLA) by a single emulsion method. The resultant combination nanoparticle significantly reduced tumour initiating activity of BCSCs, and was most effective inhibiting BCSCs and BCCs in vitro and in vivo with a synergistic effect (c.i. < 1).
5.2.3. Cyclopamine
Cyclopamine (11-deoxojervine) is a naturally occurring steroidal alkaloid first isolated from Veratrum grandiflorum and Veratrum californicum.111 It exhibits anti-cancer activity through the inhibition of sonic Hedgehog signaling pathway. Cyclopamine is hydrophobic, and was combined with hydrophilic doxorubicin in a HA coated amphipathic and redox-responsive polymer nanoparticle based on HA-cystamine-PLGA.55 The release of cyclopamine and doxorubicin was significantly accelerated in the reducing environment, with 66.9% doxorubicin and 74.6% cyclopamine released in 24 h, compared with 33.7% doxorubicin and 40.3% cyclopamine in 24 h under physiological conditions. The redox-sensitive delivery system showed the most potent reduction in tumorsphere formation, and greatest inhibition of tumour growth in vivo.
5.3. Combination of chemotherapeutic agent and RNA interference-based therapy
Identified by Craig Mello and Andrew Fire in 1998, RNA-interference (RNAi) is a fundamental cellular process in many eukaryotes that inhibits gene expression or translation by silencing mRNA molecules.112,113 In anti-cancer therapy, RNAi is powerful in downregulating carcinogenic gene expression through the delivery of small RNA duplexes, including short interfering RNAs (siRNAs), microRNAs (miRNAs), short hairpin RNAs (shRNAs) and Dicer substrate RNAs (dsiRNAs).112 RNAi-based therapy holds great promise, however, delivery of small RNA duplexes to target cells in vivo faces a chain of hurdles, including degradation by serum nuclease, renal and reticuloendothelial system clearances, ‘off-target’ effects and poor cellular uptake.114 In order to improve the bioavailability of RNA duplexes, chemical modification of the genes, virus-based, nanoparticle-based systems, and conjugation of genes to a delivery system have all be explored.114 Co-delivery of RNA-related oncogene interference and a chemotherapeutic drug using nanocarriers could not only enhance the site specific delivery of genes, but also achieved a synergistic antitumour effect with reduced side-effects of chemotherapy agents.115 Because of a high diversity exists in the mechanism of RNA-based interferences, the selection of a suitable drug and RNA pair is vital in achieving a satisfactory clinical effect in the combination therapy (Table 6).
5.3.1. Combination of a chemotherapy agent with siRNA
siRNA in a combination therapy is often responsible for the downregulation of genes involved in drug resistance. Sun et al.116 developed a cationic niosomes to deliver siRNA to downregulate the expression of ABCG2 and BCL2 in tumour cells, and ultimately to resensitise breast cancer cells, including BCSCs, to doxorubicin. Niosomes incorporated with siABCG2 and siBCL2 at 50 nM siRNA significantly reduced corresponding mRNA expression by 60% and 65%, while free siRNA did not significantly change the mRNA expression. Comparing with free doxorubicin, co-delivery of doxorubicin and siABCG2 and siBCL2 in cationic noisome is 32 times and 236 times more toxic towards BCSCs and non-BCSCs respectively; the IC50 of doxorubicin containing nanoparticle with siABCG2 or siBCL2 was 90.9% and 60.5% less than free doxorubicin. The results confirmed that delivery of siABCG2 and siBCL2 indeed inhibited ABC efflux pump and caused suppression of anti-apoptosis.
Overexpression of clusterin (CLU) and glucose-regulated protein 78 (GRP78) in BCSCs is associated with cancer cell survival and chemo-resistance.67 1,2-Dioleoyloxy-3-trimethylammoniumpropane (DOTAP) containing cationic liposome co-loaded with camptothecin and siRNA targeting GLU or GRP78 released siGLU or siGRP78 first to improve the cell sensitivity to camptothecin. Compared to free camptothcin, combination with siGLU increased transfection efficiency by 4.1- and 5.9-fold in BCSCs and MCF-7 cells respectively; in addition, combination with siGRP78 increased transfection efficiency by 4.4- and 6.4-fold in BCSCs and MCF-7 respectively.
5.3.2. Combination of a chemotherapy agent with miRNA
The aberrant expression of miRNAs is widely recognised in cancers, and is involved in self-renew and proliferation of cancer stem cells.75 miRNA-200c had been proven to restore the chemo-sensitivity to microtubule-targeting agents, such as paclitaxel.75 miRNA-200c level is significantly downregulated in BCSCs. A DOTAP containing solid lipid nanoparticles (SLN) was formulated to deliver miRNA-200c, mixed together with nanostructured lipid carrier (NLC) loaded with paclitaxel. SLN protected miRNA-200c from degradation and showed effective release in 12 h after cellular uptake. IC50 values for NLC loaded paclitaxel after treated with SLN loaded miRNA-200c was significantly lower than control group (0.28 ± 0.06 μg mL−1 and 2.06 ± 0.13 μg mL−1, respectively), indicating miRNA-200c resensitised paclitaxel when delivered by SLN.
High expression of miRNA-21 was found to be associated with pluripotency of BCSCs and drug resistance.68 miRNA-21 inhibitor (miRNA-21i), a specific antisense oligonucleotide, and doxorubicin were formulated into NIR responsive hollow gold nanoparticle to achieve a sequential delivery. miRNA-21i was first released into cytoplasm to reduce the stemness of BCSCs. 4 h later, BCSCs became sensitive to doxorubicin, and release of doxorubicin was triggered by NIR. While simultaneous administration of doxorubicin and miRNA-21i resulted a 45% cell death, the time-staggered miRNA-21i and doxorubicin delivery led to a higher cellular killing rate of 69%.
5.3.3. Combination of a chemotherapy agent with shRNA
shRNA is located at the upstream of RNAi pathway and is activated to siRNA through nuclear processing.112 shRNA is more stable, more effective at gene silencing and more specific at targeting than siRNA.117 However, its delivery requires a plasmid vector.117 NF-κB targeting shRNA was delivered to BCSCs using carbamate-mannose modified poly(ethylenimine) (CMP) as a non-viral gene vector. The delivery system reduced the BCSCs population via apoptotic mechanism, and inhibited cell migration and invasion. Co-delivery of NF-κB targeting shRNA and doxorubicin mixed micelle showed a synergistic effect and enhanced inhibitory effect than doxorubicin mixed micelles at all concentrations.
6. Perspectives of anti-breast cancer nanomedicines: challenges in translation from benchtop to bedside
Despite the tremendous effort in the field of nanomedicine for tackling cancer, and more recently, in eradicating cancer stem cells, the success in clinical translation has been limited with only few products, mainly liposomes, being approved for clinical use, and furthermore, these nanomedicines have only reduced toxicity profile.118 One of the fundamental reasons is believed to be the low targeted delivery efficiency of the nanoparticles, with only 0.7% (median value) of the administered nanoparticles reaching tumours in patients.119 Principle for passive targeted delivery has been universally designed based on the EPR effect observed in solid tumours,118 and the EPR effect acts as the prerequisite for active targeting. The extent of EPR effect is largely influenced by the heterogeneities in tumours including type, size, perfusion and other factors, resulting in significant inter- and intra-patient variations.120 In addition, while almost all studies focus on intercellular extravasation via EPR effect, an alternative transcellular extravasation may play a more important role in delivering nanoparticles.119 Another main reason for the lack of clinical translation of anti-tumour nanoparticles is the over-manifestation of EPR effect in the commonly used preclinical murine models than in human, due to the different relative tumour size portion, developing rate and more importantly physiopathology,121 resulting in misleading preclinical data.
In order to increase the clinical translation, new strategies are proposed to enhance the patient response to nanomedicines, including selection of patient/cancer type; reduction of particle size of nanomedicines (to 60 nm); modulation of the stromal transport barriers from the extracellular matrix components.122 Patient-derived xenografts have be proposed to be a better preclinical model for cancer research, which more closely simulates the complexity and heterogeneity of human tumours.123 Notably, among various tumour types, breast cancer has been highly ranked to manifest significant EPR effect in patients due to the high blood perfusion in tumours.120
Compared with the single drug loaded nanoparticles, it is even more challenging in the rational design of nanoparticle-based combination therapy. Apart from the sequence of delivery and the drug ratio, there is a need of careful evaluation of formulation synesthetic performance before translation begins, including drug release, and pharmacokinetics of the agents, and correlation of in vitro cytotoxicity and in vivo efficacy.64 Future studies will focus on translation of nanoparticles in which the biology, pathophysiology and the genetics of the disease needs to be understood and used to guide the formulation design. For instance, the existence and plasticity of BCSCs, and circulating BCSCs (Fig. 1) could be important targeting areas.124 Therefore, a united effort from drug delivery scientists, clinical oncologists, biologists and wider disciplines is required to eradicate breast cancer.
7. Conclusion
BCSCs play a major role in tumour growth, progression and metastasis. Due to their resistance to conventional chemotherapeutic agents and radiotherapy, and their high plasticity, the eradication of BCSCs is extremely difficult and presents one of the greatest challenges in anti-cancer therapy. In this review, the studies of co-delivery nanosystems to target BCSCs available to date were summarised. The combined drug delivery has the potential to target both BCSCs and non-BCSCs simultaneously and exhibit a synergistic effect. However, the selection of drug combination, drug ratio and the sequence of drug administration and design of the nano drug delivery system are all crucial factors to consider. The co-encapsulation system is generally more superior than the “Nano + Nano” approach, since it can synchronise the pharmacokinetics of two different agents and achieve synergistic effect. Although it is difficult to load drugs with pre-determined ratio and difficult to achieve sequential release in the co-encapsulation system, these problems can be overcome by judicious design of the nanosystem. Given breast cancer may manifest significant EPR effect in patients, the combination of one chemotherapeutic agent with a BCSCs specific inhibitor, phytochemical agent or RNA based therapy that demonstrated positive preclinical results may hold promise to cure breast cancer. In the future interdisciplinary studies of anti-BCSCs therapy need to focus on the clinical translation.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
References
- Bray F. Ferlay J. Soerjomataram I. Siegel R. L. Torre L. A. Jemal A. Ca-Cancer J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- Anampa J. Makower D. Sparano J. A. BMC Med. 2015;13:195. doi: 10.1186/s12916-015-0439-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X. Cheng H. Bai Z. Li J. J. Cancer. 2017;8:3131–3141. doi: 10.7150/jca.18457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majeed W. Aslam B. Javed I. Khaliq T. Muhammad F. Ali A. et al. . Asian Pac. J. Cancer Prev. 2014;15:3353–3358. doi: 10.7314/APJCP.2014.15.8.3353. [DOI] [PubMed] [Google Scholar]
- Rodriguez C. E. Berardi D. E. Abrigo M. Todaro L. B. Bal de Kier Joffe E. D. Fiszman G. L. et al. . J. Cell. Biochem. 2018;119:1381–1391. doi: 10.1002/jcb.26298. [DOI] [PubMed] [Google Scholar]
- Rodriguez D. Ramkairsingh M. Lin X. Kapoor A. Major P. Tang D. Rodriguez D. Ramkairsingh M. Lin X. Kapoor A. Major P. Tang D. Cancer. 2019;11:1028. doi: 10.3390/cancers11071028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawood S. Austin L. Cristofanilli M. Oncology. 2014;28:1101–1107. [PubMed] [Google Scholar]
- Geng S. Q. Alexandrou A. T. Li J. J. Cancer Lett. 2014;349:1–7. doi: 10.1016/j.canlet.2014.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampayo R. G. and Bissell M. J., in Advances in Cancer Research, Academic Press Inc., 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holliday D. L. Speirs V. Breast Cancer Res. 2011;13:215. doi: 10.1186/bcr2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C. Tang Z. Tian H. Chen X. Adv. Drug Delivery Rev. 2016;98:64–76. doi: 10.1016/j.addr.2015.10.021. [DOI] [PubMed] [Google Scholar]
- Zhang R. X. Wong H. L. Xue H. Y. Eoh J. Y. Wu X. Y. J. Controlled Release. 2016;240:489–503. doi: 10.1016/j.jconrel.2016.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventola C. L. Pharm. Ther. 2017;42:742–755. [PMC free article] [PubMed] [Google Scholar]
- Bertrand N. Wu J. Xu X. Y. Kamaly N. Farokhzad O. C. Adv. Drug Delivery Rev. 2014;66:2–25. doi: 10.1016/j.addr.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi M. Ghasemi A. Zangabad P. S. Rahighi R. Basri S. Mirshekari H. et al. . Chem. Soc. Rev. 2016;45:1457–1501. doi: 10.1039/C5CS00798D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J. Kantoff P. W. Wooster R. Farokhzad O. C. Nat. Rev. Cancer. 2017;17:20–37. doi: 10.1038/nrc.2016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hajj M. Wicha M. S. Benito-Hernandez A. Morrison S. J. Clarke M. F. Proc. Natl. Acad. Sci. U. S. A. 2003;100:6890. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet D. Dick J. E. Nat. Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- Ginestier C. Hur M. H. Charafe-Jauffret E. Monville F. Dutcher J. Brown M. et al. . Cell Stem Cell. 2007;1:555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomeras S. Ruiz-Martínez S. Puig T. Molecules. 2018;23(9):2193. doi: 10.3390/molecules23092193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dandawate P. R. Subramaniam D. Jensen R. A. Anant S. Semin. Cancer Biol. 2016;40–41:192–208. doi: 10.1016/j.semcancer.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto-Vila M. Takahashi R. U. Usuba W. Kohama I. Ochiya T. Int. J. Mol. Sci. 2017;18(12):2574. doi: 10.3390/ijms18122574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. Liu X. Jiang Z. Li L. Cui Z. Gao Y. Kong D. Liu X. Oncol. Lett. 2016;12:1355–1360. doi: 10.3892/ol.2016.4757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shima H. Yamada A. Ishikawa T. Endo I. Gland Surg. 2017;6:82–88. doi: 10.21037/gs.2016.08.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause M. Dubrovska A. Linge A. Baumann M. Adv. Drug Delivery Rev. 2017;109:63–73. doi: 10.1016/j.addr.2016.02.002. [DOI] [PubMed] [Google Scholar]
- Zhou Q.-M. Sun Y. Lu Y.-Y. Zhang H. Chen Q.-L. Su S.-B. Cancer Cell Int. 2017;17:84. doi: 10.1186/s12935-017-0453-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schöning J. P. Monteiro M. Gu W. Clin. Exp. Pharmacol. Physiol. 2017;44:153–161. doi: 10.1111/1440-1681.12693. [DOI] [PubMed] [Google Scholar]
- Pan Q. Li Q. Liu S. Ning N. Zhang X. Xu Y. Stem Cells. 2015;33:2085–2092. doi: 10.1002/stem.2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F. Zhao J. Hu Y. Zhou Y. Guo R. Bai J. Zhang S. Zhang H. Zhang J. Oncol. Rep. 2016;36:356–364. doi: 10.3892/or.2016.4799. [DOI] [PubMed] [Google Scholar]
- Chaffer C. Brueckmann I. Scheel C. Kaestli A. Wiggins P. Rodrigues L. Brooks M. Proc. Natl. Acad. Sci. U. S. A. 2011;108:7950–7955. doi: 10.1073/pnas.1102454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zomer A. Ellenbroek S. I. J. Ritsma L. Beerling E. Vrisekoop N. Van Rheenen J. Stem Cells. 2013;31:602–606. doi: 10.1002/stem.1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F. Zhou K. Gao L. Zhang B. Li W. Yan W. Song X. Yu H. Wang S. Yu N. Jiang Q. Oncol. Lett. 2016;12:3059–3065. doi: 10.3892/ol.2016.5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa B. Ribeiro A. Paredes J. Adv. Exp. Med. Biol. 2019;1139:83–103. doi: 10.1007/978-3-030-14366-4_5. [DOI] [PubMed] [Google Scholar]
- Liu S. Cong Y. Wang D. Sun Y. Deng L. Liu Y. Martin-Trevino R. Shang L. McDermott S. P. Landis M. D. Hong S. Adams A. D'Angelo R. Ginestier C. Charafe-Jauffret E. Clouthier S. G. Birnbaum D. Wong S. T. Zhan M. Chang J. C. Wicha M. S. Stem Cell Rep. 2014;2:78–91. doi: 10.1016/j.stemcr.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo M. Brooks M. Wicha M. Curr. Pharm. Des. 2015;21:1301–1310. doi: 10.2174/1381612821666141211120604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G. Quan Y. Wang W. Fu Q. Wu J. Mei T. Br. J. Cancer. 2012;106:1512–1519. doi: 10.1038/bjc.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lliopoulos D. Hirsch H. Wang G. Struhl K. Proc. Natl. Acad. Sci. U. S. A. 2011;108:1397–1402. doi: 10.1073/pnas.1018898108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun R. Liu Y. Li S. Y. Shen S. Du X. J. Xu C. F. Cao Z. T. Bao Y. Zhu Y. H. Li Y. P. Yang X. Z. Wang J. Biomaterials. 2015;37:405–414. doi: 10.1016/j.biomaterials.2014.10.018. [DOI] [PubMed] [Google Scholar]
- Yang F. Xu J. Tang L. Guan X. Cell. Mol. Life Sci. 2017;74:951–966. doi: 10.1007/s00018-016-2334-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagust P. De Luxán-Delgado B. Parejo-Alonso B. Sancho P. Front. Pharmacol. 2019;10:203. doi: 10.3389/fphar.2019.00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun S. Y. Kwon Y. S. Nam K. S. Kim S. Y. Biomed. Pharmacother. 2015;72:37–43. doi: 10.1016/j.biopha.2015.03.009. [DOI] [PubMed] [Google Scholar]
- Dewangan J. Srivastava S. Rath S. K. Tumor Biol. 2017;39 doi: 10.1177/1010428317695035. [DOI] [PubMed] [Google Scholar]
- Naujokat C. Steinhart R. J. Biomed. Biotechnol. 2012;2012:950658. doi: 10.1155/2012/950658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto Y. Tsukahara S. Imai Y. Sugimoto Y. Ueda K. Tsuruo T. Mol. Cancer Ther. 2003;2:105–112. [PubMed] [Google Scholar]
- Bekaii-Saab T. S. Perloff M. D. Weemhoff J. L. Greenblatt D. J. von Moltke L. L. Biopharm. Drug Dispos. 2004;25:283–289. doi: 10.1002/bdd.411. [DOI] [PubMed] [Google Scholar]
- Tegowski M. Fan C. Baldwin A. S. J. Biol. Chem. 2018;293:15977–15990. doi: 10.1074/jbc.RA118.003719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun R. Liu Y. Li S. Y. Shen S. Du X. J. Xu C. F. Cao Z. T. Bao Y. Zhu Y. H. Li Y. P. Yang X. Z. Wang J. Biomaterials. 2015;37:405–414. doi: 10.1016/j.biomaterials.2014.10.018. [DOI] [PubMed] [Google Scholar]
- Nguyen P. H. Giraud J. Staedel C. Chambonnier L. Dubus P. Chevret E. Bœuf H. Gauthereau X. Rousseau B. Fevre M. Soubeyran I. Belleannée G. Evrard S. Collet D. Mégraud F. Varon C. Oncogene. 2016;35:5619–5628. doi: 10.1038/onc.2016.87. [DOI] [PubMed] [Google Scholar]
- Zhang L. Yao H. Yu Y. Zhang Y. Li R. Ju R. Biomaterials. 2012;33:565–582. doi: 10.1016/j.biomaterials.2011.09.055. [DOI] [PubMed] [Google Scholar]
- Gunn E. J. Williams J. T. Huynh D. T. Iannotti M. J. Han C. Barrios F. J. Kendall S. Glackin C. A. Colby D. A. Kirshner J. Leuk. Lymphoma. 2011;52:1085–1097. doi: 10.3109/10428194.2011.555891. [DOI] [PubMed] [Google Scholar]
- Guzman M. L. Rossi R. M. Karnischky L. Li X. Peterson D. R. Howard D. S. Jordan C. T. Blood. 2005;105:4163–4169. doi: 10.1182/blood-2004-10-4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulaiman A. McGarry S. El-Sahli S. Li L. Chambers J. Phan A. Côte M. Cron G. O. Alain T. Le Y. Lee S. H. Liu S. Figeys D. Gadde S. Wang L. Mol. Cancer Ther. 2019;18:1755–1764. doi: 10.1158/1535-7163.MCT-18-0873. [DOI] [PubMed] [Google Scholar]
- Zhou J. Zhang H. Gu P. Margolick J. B. Yin D. Zhang Y. Breast Cancer Res. Treat. 2009;115:269–277. doi: 10.1007/s10549-008-0072-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. Wang X. Xie C. Zhu J. Meng Y. Chen Y. Li Y. Jiang Y. Yang X. Wang S. Chen J. Zhang Q. Geng S. Wu J. Zhong C. Zhao Y. Anticancer. Drugs. 2018;29:1. doi: 10.1097/CAD.0000000000000572. [DOI] [PubMed] [Google Scholar]
- Hu K. Zhou H. Liu Y. Liu Z. Liu J. Tang J. Li J. Zhang J. Sheng W. Zhao Y. Wu Y. Chen C. Nanoscale. 2015;7:8607–8618. doi: 10.1039/C5NR01084E. [DOI] [PubMed] [Google Scholar]
- Kurebayashi J. Koike Y. Ohta Y. Saitoh W. Yamashita T. Kanomata N. Moriya T. Cancer Sci. 2017;108:918–930. doi: 10.1111/cas.13205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch H. A. Iliopoulos D. Struhl K. Proc. Natl. Acad. Sci. U. S. A. 2013;110:972–977. doi: 10.1073/pnas.1221055110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsy M. Albert L. Tobias M. E. Murali R. Jhanwar-Uniyal M. Anticancer Res. 2010;30:4915–4920. [PubMed] [Google Scholar]
- Bertozzi D. Marinello J. Manzo S. G. Fornari F. Gramantieri L. Capranico G. Mol. Cancer Ther. 2014;13:239–248. doi: 10.1158/1535-7163.MCT-13-0729. [DOI] [PubMed] [Google Scholar]
- Shi P. Liu W. Tala T. Wang H. Li F. Zhang H. Wu Y. Kong Y. Zhou Z. Wang C. Chen W. Liu R. Chen C. Cell Discov. 2017;3:17010. doi: 10.1038/celldisc.2017.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou T.-C. Talalay P. Trends Pharmacol. Sci. 1983;4:450–454. doi: 10.1016/0165-6147(83)90490-X. [DOI] [Google Scholar]
- Chou T.-C. Cancer Res. 2010;70:440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- Lu J.-J. and Wang Y.-T., Synergistic anti-cancer activity of the combination of dihydroartemisinin and doxorubicin in breast cancer cells, 2013 [DOI] [PubMed] [Google Scholar]
- Wu D. Pusuluri A. Vogus D. Krishnan V. Shields C. W. Kim J. Razmi A. Mitragotri S. J. Control. Release. 2020;323:36–46. doi: 10.1016/j.jconrel.2020.04.018. [DOI] [PubMed] [Google Scholar]
- Oliveras-Ferraros C. Vazquez-Martin A. Colomer R. De Llorens R. Brunet J. Menendez J. A. Int. J. Oncol. 2008;32:113–120. [PubMed] [Google Scholar]
- Yuan J. D. ZhuGe D. L. Tong M. Q. Lin M. T. Xu X. F. Tang X. Zhao Y. Z. Xu H. L. Artif. Cells, Nanomed., Biotechnol. 2018;46:302–313. doi: 10.1080/21691401.2017.1423495. [DOI] [PubMed] [Google Scholar]
- Samson A. A. S. Park S. Kim S. Y. Min D. H. Jeon N. L. Song J. M. J. Liposome Res. 2019;29:44–52. doi: 10.1080/08982104.2017.1420081. [DOI] [PubMed] [Google Scholar]
- Ren Y. Wang R. Gao L. Li K. Zhou X. Guo H. Liu C. Han D. Tian J. Ye Q. Hu Y. T. Sun D. Yuan X. Zhang N. J. Controlled Release. 2016;228:74–86. doi: 10.1016/j.jconrel.2016.03.008. [DOI] [PubMed] [Google Scholar]
- Tao X. Gou J. Zhang Q. Tan X. Ren T. Yao Q. Tian B. Kou L. Zhang L. Tang X. Biomater. Sci. 2018;6:1869–1881. doi: 10.1039/C8BM00271A. [DOI] [PubMed] [Google Scholar]
- Gao J. Liu J. Xie F. Lu Y. Yin C. Shen X. Int. J. Nanomedicine. 2019;14:9199–9216. doi: 10.2147/IJN.S230376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. J. Liu Y. Li S. Rohrs J. Zhang R. Zhang X. Wang P. Mol. Pharmacol. 2015;12:2811–2822. doi: 10.1021/mp500754r. [DOI] [PubMed] [Google Scholar]
- Liu Y. Fang J. Kim Y.-J. Wong M. K. Wang P. Mol. Pharm. 2014;11:1651–1661. doi: 10.1021/mp5000373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenthamara D. Subramaniam S. Ramakrishnan S. G. Krishnaswamy S. Essa M. M. Lin F. H. Qoronfleh M. W. Biomater. Res. 2019;23:1–29. doi: 10.1186/s40824-018-0153-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R.-J. Ying X. Zhang Y. Ju R.-J. Wang X.-X. Yao H.-J. Men Y. Tian W. Yu Y. Zhang L. Huang R.-J. Lu W.-L. J. Controlled Release. 2011;149:281–291. doi: 10.1016/j.jconrel.2010.10.019. [DOI] [PubMed] [Google Scholar]
- Liu J. Meng T. Yuan M. Wen L. Cheng B. Liu N. Huang X. Hong Y. Yuan H. Hu F. Int. J. Nanomedicine. 2016;11:6713–6725. doi: 10.2147/IJN.S111647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muntimadugu E. Kumar R. Saladi S. Rafeeqi T. Khan W. Colloids Surf., B. 2016;143:532–546. doi: 10.1016/j.colsurfb.2016.03.075. [DOI] [PubMed] [Google Scholar]
- Makadia H. K. Siegel S. J. Polymers. 2011;3:1377–1397. doi: 10.3390/polym3031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhaveri A. M. Torchilin V. P. Front. Pharmacol. 2014;5:77. doi: 10.3389/fphar.2014.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D. Huang J. Wang X. Yu Y. Zhang H. Chen Y. Liu J. Sun Z. Zou H. Sun D. Zhou G. Zhang G. Lu Y. Zhong Y. Biomaterials. 2013;34:7662–7673. doi: 10.1016/j.biomaterials.2013.06.042. [DOI] [PubMed] [Google Scholar]
- Eftekhari R. B. Maghsoudnia N. Samimi S. Zamzami A. Dorkoosh F. A. Pharm. Nanotechnol. 2019;7:90–112. doi: 10.2174/2211738507666190321112237. [DOI] [PubMed] [Google Scholar]
- Li B. B. Li Q. H. Mo J. X. Dai H. L. Front. Pharmacol. 2017;8:51. doi: 10.3389/fphar.2017.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N. Liang X. Gao C. Chen M. Zhou Y. Krueger C. J. Bao G. Gong Z. Dai Z. ACS Appl. Mater. Interfaces. 2018;10:29385–29397. doi: 10.1021/acsami.8b11723. [DOI] [PubMed] [Google Scholar]
- Yang Z. Sun N. Cheng R. Zhao C. Liu J. Tian Z. J. Mater. Chem. B. 2017;5:6762–6775. doi: 10.1039/C7TB01510K. [DOI] [PubMed] [Google Scholar]
- Yang Z. Sun N. Cheng R. Zhao C. Liu Z. Li X. Liu J. Tian Z. Biomaterials. 2017;147:53–67. doi: 10.1016/j.biomaterials.2017.09.013. [DOI] [PubMed] [Google Scholar]
- Liu M. Wang B. Guo C. Hou X. Cheng Z. Chen D. Drug Deliv. 2019;26:1002–1016. doi: 10.1080/10717544.2019.1669734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borah A. Pillai S. C. Rochani A. Palaninathan V. Nakajima Y. Maekawa T. Kumar S. Nanotechnology. 2020;31(18):185102. doi: 10.1088/1361-6528/ab6d20. [DOI] [PubMed] [Google Scholar]
- Wang H. Agarwal P. Zhao S. Xu R. Yu J. Lu X. He X. Biomaterials. 2015:6072–6078. doi: 10.1016/j.biomaterials.2015.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. Lu W.-L. Guo J. Du J. Li T. Wu J.-W. Wang G.-L. Wang J.-C. Zhang X. Zhang Q. J. Controlled Release. 2008;129:18–25. doi: 10.1016/j.jconrel.2008.03.022. [DOI] [PubMed] [Google Scholar]
- Zhang Y. Zhang H. Wang X. Wang J. Zhang X. Zhang Q. Biomaterials. 2012;33:679–691. doi: 10.1016/j.biomaterials.2011.09.072. [DOI] [PubMed] [Google Scholar]
- Muntimadugu E. Kumar R. Saladi S. Rafeeqi T. A. Khan W. Colloids Surf., B. 2016;143:532–546. doi: 10.1016/j.colsurfb.2016.03.075. [DOI] [PubMed] [Google Scholar]
- Zhang J. Kinoh H. Hespel L. Liu X. Quader S. Martin J. Chida T. Cabral H. Kataoka K. J. Controlled Release. 2017;264:127–135. doi: 10.1016/j.jconrel.2017.08.025. [DOI] [PubMed] [Google Scholar]
- Guo J. Zhou J. Ying X. Men Y. Li R. J. Zhang Y. Du J. Tian W. Yao H. J. Wang X. X. Ju R. J. Lu W. L. J. Pharm. Pharm. Sci. 2010;13:136–151. doi: 10.18433/j3p88z. [DOI] [PubMed] [Google Scholar]
- Ke X. Y. Lin Ng V. W. Gao S. J. Tong Y. W. Hedrick J. L. Yang Y. Y. Biomaterials. 2014;35:1096–1108. doi: 10.1016/j.biomaterials.2013.10.049. [DOI] [PubMed] [Google Scholar]
- Lang T. Liu Y. Zheng Z. Ran W. Zhai Y. Yin Q. Zhang P. Li Y. Adv. Mater. 2019;31:1–8. [Google Scholar]
- Mattheolabakis G. Milane L. Singh A. Amiji M. M. J. Drug Target. 2015;23:605–618. doi: 10.3109/1061186X.2015.1052072. [DOI] [PubMed] [Google Scholar]
- Rao W. Wang H. Han J. Zhao S. Dumbleton J. Agarwal P. ACS Nano. 2015;9:5725–5740. doi: 10.1021/nn506928p. [DOI] [PubMed] [Google Scholar]
- Al Faraj A. Shaik A. S. Al Sayed B. Halwani R. Al Jammaz I. Nanomedicine. 2016;11:31–46. doi: 10.2217/nnm.15.182. [DOI] [PubMed] [Google Scholar]
- Alshaer W. Hillaireau H. Vergnaud J. Mura S. Delomenie C. Sauvage F. Ismail S. Fattal E. J. Controlled Release. 2018;271:98–106. doi: 10.1016/j.jconrel.2017.12.022. [DOI] [PubMed] [Google Scholar]
- Swaminathan S. K. Roger E. Toti U. Niu L. Ohlfest J. R. Panyam J. J. Controlled Release. 2013;171(3):280–287. doi: 10.1016/j.jconrel.2013.07.014. [DOI] [PubMed] [Google Scholar]
- Alibolandi M. Ramezani M. Sadeghi F. Abnous K. Hadizadeh F. Int. J. Pharm. 2015;479:241–251. doi: 10.1016/j.ijpharm.2014.12.035. [DOI] [PubMed] [Google Scholar]
- Baabur-Cohen H. Vossen L. I. Krüger H. R. Eldar-boock A. Yeini E. Landa-Rouben N. Tiram G. Wedepohl S. Markovsky E. Leor J. Calderón M. Satchi-Fainaro R. J. Controlled Release. 2017;257:118–131. doi: 10.1016/j.jconrel.2016.06.037. [DOI] [PubMed] [Google Scholar]
- Chen C. Tao R. Ding D. Kong D. Fan A. Wang Z. Zhao Y. Eur. J. Pharm. Sci. 2017;107:16–23. doi: 10.1016/j.ejps.2017.06.030. [DOI] [PubMed] [Google Scholar]
- Liang D. S. Liu J. Peng T. X. Peng H. Guo F. Zhong H. J. Colloids Surf., B. 2018;172:506–516. doi: 10.1016/j.colsurfb.2018.08.063. [DOI] [PubMed] [Google Scholar]
- Gupta P. B. Onder T. T. Jiang G. Tao K. Kuperwasser C. Weinberg R. A. Lander E. S. Cell. 2009;138:645–659. doi: 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao W. W. Chan J. Farokhzad O. C. Mol. Pharmacol. 2010;7:1913–1920. doi: 10.1021/mp100253e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou N. Wang R. Zhang Y. Lei Z. Zhang X. Hu R. Li H. Mao Y. Wang X. Irwin D. M. Niu G. Tan H. J. Cancer. 2015;6:1049–1057. doi: 10.7150/jca.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dandawate P. R. Subramaniam D. Jensen R. A. Anant S. Semin. Cancer Biol. 2016:192–208. doi: 10.1016/j.semcancer.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D. Wang G. Song W. Zhang Q. J. Nanoparticle Res. 2015;17:1–10. doi: 10.1007/s11051-014-2856-6. [DOI] [Google Scholar]
- Mirzaei H. Shakeri A. Rashidi B. Jalili A. Banikazemi Z. Sahebkar A. Biomed. Pharmacother. 2017;85:102–112. doi: 10.1016/j.biopha.2016.11.098. [DOI] [PubMed] [Google Scholar]
- Siddikuzzaman C. G. Berlin Grace V. M. Immunopharmacol. Immunotoxicol. 2011;33:241–249. doi: 10.3109/08923973.2010.521507. [DOI] [PubMed] [Google Scholar]
- Iovine V. Mori M. Calcaterra A. Berardozzi S. Botta B. Curr. Pharm. Des. 2016;22:1658–1681. doi: 10.2174/1381612822666160112130157. [DOI] [PubMed] [Google Scholar]
- Bobbin M. L. Rossi J. J. Annu. Rev. Pharmacol. Toxicol. 2016;56:103–122. doi: 10.1146/annurev-pharmtox-010715-103633. [DOI] [PubMed] [Google Scholar]
- Fire A. Xu S. Montgomery M. K. Kostas S. A. Driver S. E. Mello C. C. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- Singh A. Trivedi P. Jain N. K. Artif. Cells, Nanomedicine, Biotechnol. 2018;46:274–283. doi: 10.1080/21691401.2017.1307210. [DOI] [PubMed] [Google Scholar]
- Wang M. Wang J. Li B. Meng L. Tian Z. Colloids Surf., B. 2017;157:297–308. doi: 10.1016/j.colsurfb.2017.06.002. [DOI] [PubMed] [Google Scholar]
- Sun M. Yang C. Zheng J. Wang M. Chen M. Svend Le D. Acta Biomater. 2015;28:171–182. doi: 10.1016/j.actbio.2015.09.029. [DOI] [PubMed] [Google Scholar]
- Ke X. Yang C. Cheng W. Yang Y. Y. Nanomedicine. 2018;14:405–414. doi: 10.1016/j.nano.2017.11.015. [DOI] [PubMed] [Google Scholar]
- Hua S. de Matos M. B. C. Metselaar J. M. Storm G. Front. Pharmacol. 2018;9:790. doi: 10.3389/fphar.2018.00790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm S. Tavares A. J. Dai Q. Ohta S. Audet J. Dvorak H. F. Chan W. C. W. Nat. Rev. Mater. 2016;1:1–12. [Google Scholar]
- Natfji A. A. Ravishankar D. Osborn H. Greco F. J. Pharm. Sci. 2017;106:3179–3187. doi: 10.1016/j.xphs.2017.06.019. [DOI] [PubMed] [Google Scholar]
- Danhier F. J. Controlled Release. 2016;244:108–121. doi: 10.1016/j.jconrel.2016.11.015. [DOI] [PubMed] [Google Scholar]
- Golombek S. K. May J. N. Theek B. Appold L. Drude N. Kiessling F. Lammers T. Adv. Drug Delivery Rev. 2018;130:17–38. doi: 10.1016/j.addr.2018.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murayama T. Gotoh N. Cell. 2019;8:621. doi: 10.3390/cells8060621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M. H. Imrali A. Heeschen C. Chinese J. Cancer Res. 2015;27:437–449. doi: 10.3978/j.issn.1000-9604.2015.04.08. [DOI] [PMC free article] [PubMed] [Google Scholar]