

Keywords: acute kidney injury, damage-associated molecular patterns
Abstract
Acute kidney injury (AKI) is a systemic inflammatory disease that contributes to remote organ failures. Multiple organ failure is the leading cause of death due to AKI, and lack of understanding of the mechanisms involved has precluded the development of novel therapies. Mitochondrial injury in AKI leads to mitochondrial fragmentation and release of damage-associated molecular patterns, which are known to active innate immune pathways and systemic inflammation. This review presents current evidence suggesting that extracellular mitochondrial damage-associated molecular patterns are mediators of remote organ failures during AKI that have the potential to be modifiable.
INTRODUCTION
Acute kidney injury (AKI), as defined by the Kidney Disease Improving Global Outcome guidelines, occurs in one in five hospitalized patients worldwide (1). The 90-day mortality rate of AKI is 25–50% depending on AKI severity and recovery (2–5), and there are no proven treatments for AKI beyond renal replacement therapy. Despite improvements in renal replacement therapy technology and implementation, the mortality of AKI remains unacceptably high. Interestingly, multiple organ failure is a leading cause of death due to AKI (2), and recent data have demonstrated that AKI is a systemic disease that contributes directly to remote organ injuries (6–10). Investigations into the mechanisms of multiple organ failure during AKI have great potential to lead to novel life-saving therapies.
Several inflammatory mediators have been implicated in remote organ injuries due to AKI. Hoke et al. (9) used the ischemia-reperfusion (IR) and bilateral nephrectomy models of AKI to show that circulating levels of multiple cytokines, including IL-6 and IL-8, increase in the circulation just hours after AKI and are associated with neutrophil infiltration in the lung. Klein et al. (11) showed later that treatment with antibodies directed against IL-6 prevented lung injury in these same AKI models, and similar findings have been demonstrated for TNF-α (9, 12). Liu et al. (10) found increased levels of IL-1β, KC, granulocyte colony-stimulating factor, and other chemokines in the brain in mice with IR-AKI, which was associated with increased neurovascular permeability and reduced locomotor activity. This type of inflammatory cross talk has also been found to occur between the kidney and many other remote organs, including the gut (13) and liver (6). Release of cytokines from the injured kidney and reduced kidney function are implicated in the systemic accumulation of inflammatory mediators. However, markers of microvascular leak in the lung and liver have been found to be increased in models of IR-AKI compared with bilateral nephrectomy (6, 7). This suggests that direct release of inflammatory mediators from the injured kidney may be the more dominant mechanism. Unfortunately, treatments targeting specific mediators have not been proven to be beneficial in human studies, and research into novel mechanisms that may be modifiable is imperative.
Mitochondria have emerged as a promising target to prevent the direct complications of kidney injury, as mitochondrial injury is a key pathological feature of AKI (14–16). We have recently shown the temporal changes in mitochondrial structure and function in sepsis-associated AKI (16). Mitochondrial injury is also implicated in multiple organ failure syndromes (17–19), yet the role of mitochondria in remote organ failure due to AKI has not received much attention. Mitochondrial damage leads to the release of mitochondrial DNA (mtDNA) and mitochondrial proteins, such as transcription factor A, mitochondria (TFAM), that act as damage-associated molecular patterns (DAMPs) outside the cell (18, 20). In this review, we discuss how mitochondrial injury in AKI may contribute to the systemic release of mitochondrial DAMPs (mtDAMPs) and how therapies targeting mitochondrial health and mtDAMPs have the potential to be lifesaving in patients suffering from AKI.
ALTERED MITOCHONDRIAL DYNAMICS AND mtDAMPs IN AKI
The Kidney Is Highly Susceptible to Mitochondrial Injury
The kidney is one of the most metabolically active organs in the body due to the high energy required for fluid and electrolyte transport (21). Therefore, a large pool of healthy mitochondria is necessary to produce the ATP required to support these vital processes. For this reason, the kidney has a higher mitochondria content than every other organ in the body except the heart (22). Unlike the heart, the baseline partial pressure of oxygen in the kidney is relatively low despite high renal blood flow and oxygen delivery due to high rates of oxygen consumption (23). The majority of kidney perfusion is directed to the cortex, with decreasing tissue oxygenation from the outer to inner medulla (24). The outer medullary zone is particularly vulnerable to hypoxia/ischemia due to high oxygen demand to support tubular transport with relatively low perfusion and oxygen delivery, resulting in mitochondrial injury. Therefore, it is not surprising that mitochondrial dysfunction is one of the earliest manifestations of cellular injury in AKI (14).
Altered Mitochondrial Dynamics in AKI
Renal tubular cells have thousands of mitochondria that are continuously fusing, dividing, and regenerating to maintain optimal mitochondrial energetics (21). These processes are referred to as mitochondrial dynamics. Mitochondrial fission (division) results in short mitochondrial spheres as visualized by electron microscopy, and fusion (union) results in elongated, filamentous structures (25). The process of mitochondrial fission is largely controlled by the GTPase dynamin-related protein 1 (Drp1) (26). Drp1 resides in the cytoplasm but localizes to the mitochondria after injury (15), and Drp1-mediated fission is believed to be triggered by decreased ATP production and increased reactive oxygen species (ROS) (27). Mitochondrial fusion is also controlled by GTPases with mitofusin 1 (Mfn1), mitofusin 2 (Mfn2), and optic atrophy 1 (OPA1) being the major proteins involved (21). Mfn1 and Mfn2 are responsible for tethering of outer membranes during fusion, and Opa1 is responsible for inner membrane fusion and maintaining cristae structural integrity (28). In in vivo and in vitro models of ischemic and nephrotoxic AKI, Brooks et al. (14) demonstrated that Drp1 activation and fission precede kidney tubular injury. This group has also shown that apoptotic proteins during cellular injury interact with Mfn1 and Mfn2 to suppress fusion (29). Overall, AKI has been characterized as having an increase in fission and a decrease in fusion in tubular mitochondria (21). Interestingly, similar alterations in mitochondrial dynamics have been found to develop in remote organs during AKI. Mice subjected to renal ischemia perfusion demonstrated significant increase in Drp1 and mitochondrial fission in cardiac myocytes along with cardiac dysfunction, mitigated by pretreatment with Drp1 inhibitor (30).
Mitochondrial Fragmentation Into mtDAMPs
Altered mitochondrial dynamics in AKI, mediated by fission and fusion proteins, not only disrupt mitochondrial function and ATP production but also lead to mitochondrial fragmentation. Knockout of Mfn2 and increase in Drp1 expression are both associated with mitochondrial fragmentation, and pharmacological inhibition of Drp1 or siRNA targeting Drp1 has been shown to ameliorate it (14, 31). Electron microscopy of kidney cells has revealed that mitochondrial fragmentation is one of the earliest events postinjury, occurring in nearly 40% of proximal tubule cells immediately following an ischemic event (14). Many mitochondrial fragments function as DAMPs that activate innate immune pathways in the cytoplasm and outside the cell (19). The best-described extracellular mtDAMP is mtDNA, which has been implicated in inflammation in the heart (32, 33), lung (8, 19, 34), and liver (35, 36) via Toll-like receptor 9 (TLR9). For example, Oka et al. (32) showed that mtDNA accumulation in DNase II-deficient mice activated TLR9 and induced dilated cardiomyopathy, and injection of mtDNA into mice and rats leads to increases in serum cytokines, pulmonary edema, and neutrophil infiltration in the lung (19, 34). Similarly, mtDNA caused increases in pro-IL-1β transcript in the liver along with an increase in serum alanine aminotransferase in vivo (36). Interestingly, mtDNA is much more sensitive to damage compared with nuclear DNA. This suggests that there may be a biological role for mtDNA in alarming the cell of an injurious process before irreversible nuclear DNA damage (18, 37). Other extracellular mtDAMPs include TFAM (38, 39), ROS (40), cytochrome c (41), and N-formyl peptide (42), which have all been implicated in cell death or organ failure syndromes.
mtDAMPs Enter the Cytoplasm Through the Permeability Transition Pore
For mtDAMPs to engage innate immune receptors, they need to escape the mitochondrial membrane and enter the cytoplasm. This is believed to be accomplished through the permeability transition pore (PTP) of the inner and outer mitochondrial membrane. Opening of the PTP allows the passage of molecules up to 1,500 Da to escape the mitochondrial membrane (43), and mitochondrial proteins have been shown to accumulate outside the mitochondrial membrane during PTP opening (44). The PTP is activated by increases in ROS and calcium influx (45), which are known to occur in multiple forms of AKI.
Cell Death Leads to the Extracellular Release of mtDAMPs
mtDAMPs need to enter the extracellular space to potentiate systemic inflammation directly. Necrotic cell death, as seen in many forms of AKI, can release cytoplasmic mtDAMPs into the extracellular space. Recently, a type of programmed necrosis referred to as “necroptosis” has become an intense area of interest in AKI and has been implicated in extracellular mtDAMP accumulation (46, 47). Necroptosis is triggered by receptor-interacting protein kinase 1 and 3 upregulation, which activates the executor protein mixed lineage kinase 1, leading to a rapid increase in cellular permeability and release of cellular contents (48). Linkermann et al. (49, 50) demonstrated that these kinases are activated after IR-AKI, and similar findings were shown in a mouse model of contrast-induced injury. Especially relevant to this review are the findings by Zhao et al. (51), who discovered that necroptosis is activated in the kidney and remotely in the lung in a renal allograft model of AKI in rats. Importantly, in vitro and in vivo studies have shown that extracellular mitochondria and mtDAMPs are increased with necroptosis (46, 52).
Clearance of mtDAMPs
mtDAMPs activate multiple innate immune pathways within the cytoplasm, such as cGAS/STING (37) and inflammasomes (36, 53, 54). These are critical aspects of the innate immune response to pathogens, but a sophisticated mechanism to clear these components is necessary to prevent overwhelming inflammation and cell death. mtDAMP clearance is accomplished by a process referred to as mitophagy, a type of selective autophagy that sequesters whole mitochondria and mitochondrial components into autophagasomes (55). These autophagosomes eventually fuse with lysosomes to allow degradation, preventing the release of mtDAMPs into the extracellular space. During kidney injury, ROS, hypoxia, ATP depletion, and the energy-sensing kinases mammalian target of rapamycin (mTOR) and AMP-associated protein kinase are believed to promote mitophagy by promoting expression of the mitophagy proteins phosphatase and tensin homolog deleted on chromosome 10 (PTEN)-induced putative kinase protein 1 and Parkin (56–58). Mitophagy is considered an important component of mitochondrial quality control to remove damaged and dysfunctional mitochondria from the cell (21, 59, 60). There is well-established literature on the role of autophagy in in vitro and in vivo models of IR-AKI and cisplatin-induced AKI, and mice with impaired autophagy demonstrate more profound kidney injury (61–64). Evidence suggests that increased injury in the setting of impaired autophagy is due to the accumulation of mtDAMPs. Nakahira et al. (53) discovered that autophagy inhibition led to an accumulation of mtDNA in the cytoplasm with subsequent inflammasome activation, and Oka and colleagues (32) demonstrated that mtDNA fragments that escape autophagy increase inflammation via TLR9. In addition to these, studies specifically assessing mitophagy and demonstrating a protective role in AKI are growing (21, 59, 60, 65–67).
EXTRACELLULAR mtDAMPs CONTRIBUTING TO ORGAN FAILURES DURING AKI
The Kidney as a Source of Extracellular mtDAMPs
Strong evidence exists for mtDAMPs accumulating in the extracellular space during AKI. Whitaker et al. (68) showed that mtDNA accumulates in the urine just 10 min after kidney IR, and the amount of urinary mtDNA was predictive of the degree of kidney injury. This same group of investigators showed similar results for urinary levels of the mitochondrial protein ATP synthase-β (69). Tsuji and colleagues (17) discovered that mtDNA is increased in the circulation 4 h after septic AKI, and we (8) have recently shown that mtDNA and TFAM are increased in the plasma and lung following IR-AKI. However, there is not definite evidence yet to indicate that kidney cells are the source of these mtDAMPs. It is possible that they are released by leukocytes or other injured tissues.
Extracellular mtDAMPs Contribute to Systemic Inflammation and Remote Organ Failures
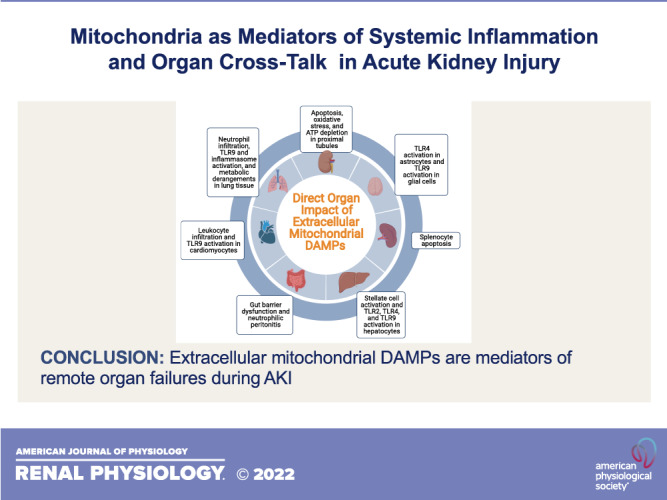
Increases in extracellular mtDAMPs are associated with detrimental changes in almost all organ systems, as shown in Fig. 1. Direct endothelial damage, activation of inflammatory pathways, and noninflammatory mechanisms have all been implicated as potential mechanisms of these changes. In a series of elegant experiments, Sun et al. (70) showed that mtDAMPs isolated from human tissues directly increase human endothelial cell permeability in vitro, which is an important pathological feature of multiple organ failure syndromes (71). In this study, mtDAMPs isolated from the liver were cocultured with various types of endothelial cells, and transendothelial electrical resistance was measured as a marker of permeability. mtDAMPs increased permeability in both EA.hy926 cells and human pulmonary artery endothelial cells. However, there was less of an impact on human pulmonary microvascular endothelial cells.
Figure 1.

Mechanisms of direct organ injury by extracellular mitochondrial damage-associated molecular patterns (DAMPs). TLR, Toll-like receptor. [Image created with Biorender.com.]
Regarding inflammatory pathways, extracellular mtDAMPs are well-described activators of inflammasomes and innate immune receptors, including NF-κB (72); NOD-, LRR- and pyrin domain-containing protein 3 (53, 72); and TLRs (17, 73) that lead to leukocyte activation and cytokine production. In a seminal study, Zhang et al. (19) injected healthy rats with mtDAMPs, which led to neutrophil activation and lung injury as demonstrated by histology and an increase in neutrophils in bronchoalveolar lavage fluid (BALF). Cytokine levels were also increased in BALF, along with an increase in BALF albumin and the lung wet-to-dry ratio, which is consistent with capillary leak. Finally, they showed that mtDAMPs cause neutrophils to release IL-8 in vitro. Intraperitoneal injection of mtDNA in mice has also been shown to cause lung injury by histology and increased BALF protein levels, which were attenuated in TLR9 knockout mice (34). Extracellular TFAM and N-formyl peptide have been implicated as inducers of inflammatory lung injury via dendritic cell activation and MAPK pathways (20, 39, 74). TLR9-mediated inflammation via mtDNA has also been found to contribute to the development of heart failure (32) and liver failure (35), and extracellular mtDAMPs have been shown to contribute to cytokine production in the gut (75) and in neuronal cells (76).
Interestingly, there is also evidence implicating noninflammatory mechanisms in remote organ dysfunction due to mtDAMPs in AKI. We have recently shown that intraperitoneal injection of extracellular mtDAMPs isolated from kidney tissue altered lung metabolism in a pattern consistent with mitochondrial dysfunction (8). Similarly, Tsuji et al. (17) showed that mtDAMP administration altered ATP production in the kidney, and Sumida et al. (30) showed altered mitochondrial dynamics develop in the heart after IR-AKI along with an upregulation of TNF-α. Interestingly, therapies aimed at improving mitochondrial dynamics in the latter study improved cardiac function but had no impact on TNF-α expression, indicating that these mitochondrial changes function independently of inflammatory pathways. Finally, human studies have shown that an increase in circulating mtDAMPs is associated with several multiple organ failure syndromes (77–80). Circulating mtDAMPs have also been found in patients with AKI (46).
THERAPEUTIC OPPORTUNITIES
With the increasing evidence for the role of mitochondrial injury and mtDAMPs in the pathophysiology of AKI and remote organ injury, therapeutic approaches targeting them are an area of active investigation. These are shown in Fig. 2 and further discussed below.
Figure 2.

Therapeutic strategies to target mitochondrial injury and mitochondrial damage-associated molecular patterns (mtDAMPS) in remote organ injury in acute kidney injury (AKI). AICAR, 5-Aminoimidazole-4-carboxamide ribonucleotide; Mdivi-1, mitochondrial division inhibitor; mTOR, mammalian target of rapamycin; NFP, N-formyl peptide; TFAM, transcription factor A, mitochondria. [Image created with Biorender.com.]
Preventing Mitochondrial Fragmentation Through Fission Inhibition
Targeted interventions to prevent fission and fusion imbalance during kidney injury would be predicted to prevent the mitochondrial fragmentation that leads to the release of mtDAMPs. This strategy has been successful in multiple preclinical models using a pharmacological inhibitor of Drp1 called mitochondrial division inhibitor (Mdivi-1). Brooks et al. (14) showed that Mdivi-1, given before injury, prevented mitochondrial fragmentation after cisplatin and IR renal injury with attenuation of cytochrome c release and kidney tubular apoptosis. In a rat model of rhabdomyolysis, Tang et al. (81) showed that Mdivi-1 after the induction of injury did not reduce Drp-1 expression; however, it did ameliorate mitochondrial fragmentation and AKI by preventing Drp-1 translocation to the mitochondria. Finally, Sumida and colleagues (30) showed that Mdivi-1 prevented cardiac injury after kidney IR when given as a pretreatment before IR as well as a delayed treatment after IR, which suggests that Mdivi-1 could be used to prevent other remote organ failures.
Promoting Mitophagy and Inhibiting Necroptosis to Prevent mtDAMP Release
Promoting mitophagy via mTOR inhibitors such as rapamycin, given as a pretreatment, has been shown to mitigate IR and nephrotoxic kidney injury with cisplatin (82, 83). However, mTOR inhibition may prevent cell growth during the recovery phases of AKI, limiting its clinical use (57). Mitophagy induction may also be limited by the recent finding that the mitophagy protein PTEN-induced putative kinase protein 1 promotes necroptosis (84); thus therapeutics promoting mitophagy may need to be combined with inhibitors of necroptosis. Recently, a pharmacological inhibitor of necroptosis, necrostatin-1, has shown promise in preventing kidney injury in IR, nephrotoxic, and septic models when given at the time of or before injury (50, 85–87). Necroptosis has also been observed in remote organs in animal models of AKI. Interestingly, necrostatin-1 prevented lung injury in a kidney allograft model when given at the time of grafting (51). While the exact mechanism of this protective effect is not known, prevention of necroptosis in kidney cells with a reduction in the release of extracellular mtDAMPs is highly plausible.
Therapies Directly Targeting mtDAMPs
Finally, interventions targeting mtDAMPs directly may mitigate downstream inflammatory effects. Pretreatment with systemic DNase 1 targeting extracellulIar DNA, including mtDNA, was shown to mitigate endothelial inflammation in a coronary artery bypass model (88). Systemic DNase also decreased liver injury due to acetaminophen in mice when given after the induction of liver injury (89). In a recent study of ventilator-associated pneumonia in rats, Simmons et al. (90) used inhaled DNase, given before and after injury, to target alveolar mtDNA and prevent lung injury. They also showed that patients with ventilator-associated pneumonia had significantly higher levels of mtDNA DAMPs in serum and BAL. Similar results were demonstrated in a lung allograft model with bronchial DNase instillation at the time of grafting (73). Importantly, inhaled DNase has an excellent safety profile and is already Federal Drug Administration approved in the treatment of cystic fibrosis and, hence, an attractive therapeutic option.
CONCLUSIONS
In conclusion, the kidney has a higher mitochondrial content than all other organs except the heart. The kidney is also uniquely prone to hypoxic injury, which makes it a likely site of mitochondrial damage and mtDAMP formation and release. PTP opening, as seen in AKI, allows mtDAMPs to escape the mitochondrial membrane and enter the cytoplasm. Impaired mitophagy to clear these mtDAMPS and subsequent cell death allow these DAMPs to accumulate in the extracellular space and in the circulation. Circulating mtDAMPs are well-described mediators of systemic inflammation resulting in multiple organ failures, which is the leading cause of death due to AKI. Investigations into novel pharmacological therapies targeting mitochondrial health and mtDAMPs have great potential to improve AKI as well as remote organ injury.
GRANTS
This work was supported by Veterans Affairs Merit Award BX002175 (to P.S.), National Institutes of Health (NIH) Grant R01DK107852 (to P.S.), Veterans Affairs Award CDA-2 IK2BX004338-01 (to M.H.), and the University of Alabama at Birmingham-University of California-San Diego O’Brien Center (NIH Grant P30DK079337).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
P.S. conceived and designed research; M.H. prepared figures; M.H. and P.S. drafted manuscript; M.H. and P.S. edited and revised manuscript; M.H. and P.S. approved final version of manuscript.
REFERENCES
- 1.Wang HE, Muntner P, Chertow GM, Warnock DG. Acute kidney injury and mortality in hospitalized patients. Am J Nephrol 35: 349–355, 2012. doi: 10.1159/000337487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chertow GM, Christiansen CL, Cleary PD, Munro C, Lazarus JM. Prognostic stratification in critically ill patients with acute renal failure requiring dialysis. Arch Intern Med 155: 1505–1511, 1995. [PubMed] [Google Scholar]
- 3.Kellum JA, Sileanu FE, Bihorac A, Hoste EA, Chawla LS. Recovery after acute kidney injury. Am J Respir Crit Care Med 195: 784–791, 2017. doi: 10.1164/rccm.201604-0799OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mehta RL, Pascual MT, Gruta CG, Zhuang S, Chertow GM. Refining predictive models in critically ill patients with acute renal failure. J Am Soc Nephrol 13: 1350–1357, 2002. doi: 10.1097/01.ASN.0000014692.19351.52. [DOI] [PubMed] [Google Scholar]
- 5.Uchino S, Bellomo R, Morimatsu H, Morgera S, Schetz M, Tan I, Bouman C, Macedo E, Gibney N, Tolwani A, Doig GS, Oudemans van Straaten H, Ronco C, Kellum JA; Beginning and Ending Supportive Therapy for the Kidney (B.E.S.T. Kidney) Investigators. External validation of severity scoring systems for acute renal failure using a multinational database. Crit Care Med 33: 1961–1967, 2005. doi: 10.1097/01.CCM.0000172279.66229.07. [DOI] [PubMed] [Google Scholar]
- 6.Golab F, Kadkhodaee M, Zahmatkesh M, Hedayati M, Arab H, Schuster R, Zahedi K, Lentsch AB, Soleimani M. Ischemic and non-ischemic acute kidney injury cause hepatic damage. Kidney Int 75: 783–792, 2009. doi: 10.1038/ki.2008.683. [DOI] [PubMed] [Google Scholar]
- 7.Hassoun HT, Grigoryev DN, Lie ML, Liu M, Cheadle C, Tuder RM, Rabb H. Ischemic acute kidney injury induces a distant organ functional and genomic response distinguishable from bilateral nephrectomy. Am J Physiol Renal Physiol 293: F30–F40, 2007. doi: 10.1152/ajprenal.00023.2007. [DOI] [PubMed] [Google Scholar]
- 8.Hepokoski M, Wang J, Li K, Li Y, Gupta P, Mai T, Moshensky A, Alotaibi MC, Alexander LE, Malhotra A, Singh P. Altered lung metabolism and mitochondrial DAMPs in lung injury due to acute kidney injury. Am J Physiol Lung Cell Mol Physiol 320: L821–L831, 2021. doi: 10.1152/ajplung.00578.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoke TS, Douglas IS, Klein CL, He Z, Fang W, Thurman JM, Tao Y, Dursun B, Voelkel NF, Edelstein CL, Faubel S. Acute renal failure after bilateral nephrectomy is associated with cytokine-mediated pulmonary injury. J Am Soc Nephrol 18: 155–164, 2007. doi: 10.1681/ASN.2006050494. [DOI] [PubMed] [Google Scholar]
- 10.Liu M, Liang Y, Chigurupati S, Lathia JD, Pletnikov M, Sun Z, Crow M, Ross CA, Mattson MP, Rabb H. Acute kidney injury leads to inflammation and functional changes in the brain. J Am Soc Nephrol 19: 1360–1370, 2008. doi: 10.1681/ASN.2007080901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klein CL, Hoke TS, Fang WF, Altmann CJ, Douglas IS, Faubel S. Interleukin-6 mediates lung injury following ischemic acute kidney injury or bilateral nephrectomy. Kidney Int 74: 901–909, 2008. doi: 10.1038/ki.2008.314. [DOI] [PubMed] [Google Scholar]
- 12.White LE, Santora RJ, Cui Y, Moore FA, Hassoun HT. TNFR1-dependent pulmonary apoptosis during ischemic acute kidney injury. Am J Physiol Lung Cell Mol Physiol 303: L449–L459, 2012. doi: 10.1152/ajplung.00301.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park SW, Kim M, Kim JY, Ham A, Brown KM, Mori-Akiyama Y, Ouellette AJ, D'Agati VD, Lee HT. Paneth cell-mediated multiorgan dysfunction after acute kidney injury. J Immunol 189: 5421–5433, 2012. doi: 10.4049/jimmunol.1200581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brooks C, Wei Q, Cho SG, Dong Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest 119: 1275–1285, 2009. doi: 10.1172/JCI37829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Funk JA, Schnellmann RG. Accelerated recovery of renal mitochondrial and tubule homeostasis with SIRT1/PGC-1alpha activation following ischemia-reperfusion injury. Toxicol Appl Pharmacol 273: 345–354, 2013. doi: 10.1016/j.taap.2013.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y, Nourbakhsh N, Pham H, Tham R, Zuckerman JE, Singh P. Evolution of altered tubular metabolism and mitochondrial function in sepsis-associated acute kidney injury. Am J Physiol Renal Physiol 319: F229–F244, 2020. [Erratum in Am J Physiol Renal Physiol 320: F1019, 2021]. doi: 10.1152/ajprenal.00390.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsuji N, Tsuji T, Ohashi N, Kato A, Fujigaki Y, Yasuda H. Role of mitochondrial DNA in septic AKI via Toll-like receptor 9. J Am Soc Nephrol 27: 2009–2020, 2016. doi: 10.1681/ASN.2015040376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Q, Itagaki K, Hauser CJ. Mitochondrial DNA is released by shock and activates neutrophils via p38 map kinase. Shock 34: 55–59, 2010. doi: 10.1097/SHK.0b013e3181cd8c08. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464: 104–107, 2010. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Julian MW, Shao G, Vangundy ZC, Papenfuss TL, Crouser ED. Mitochondrial transcription factor A, an endogenous danger signal, promotes TNFalpha release via RAGE- and TLR9-responsive plasmacytoid dendritic cells. PLoS One 8: e72354, 2013. doi: 10.1371/journal.pone.0072354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bhargava P, Schnellmann RG. Mitochondrial energetics in the kidney. Nat Rev Nephrol 13: 629–646, 2017. doi: 10.1038/nrneph.2017.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Z, Ying Z, Bosy-Westphal A, Zhang J, Schautz B, Later W, Heymsfield SB, Muller MJ. Specific metabolic rates of major organs and tissues across adulthood: evaluation by mechanistic model of resting energy expenditure. Am J Clin Nutr 92: 1369–1377, 2010. doi: 10.3945/ajcn.2010.29885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nourbakhsh N, Singh P. Role of renal oxygenation and mitochondrial function in the pathophysiology of acute kidney injury. Nephron Clin Pract 127: 149–152, 2014. doi: 10.1159/000363545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haase VH. Mechanisms of hypoxia responses in renal tissue. J Am Soc Nephrol 24: 537–541, 2013. doi: 10.1681/ASN.2012080855. [DOI] [PubMed] [Google Scholar]
- 25.Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol 11: 872–884, 2010. doi: 10.1038/nrm3013. [DOI] [PubMed] [Google Scholar]
- 26.Pitts KR, McNiven MA, Yoon Y. Mitochondria-specific function of the dynamin family protein DLP1 is mediated by its C-terminal domains. J Biol Chem 279: 50286–50294, 2004. doi: 10.1074/jbc.M405531200. [DOI] [PubMed] [Google Scholar]
- 27.Ishimoto Y, Inagi R. Mitochondria: a therapeutic target in acute kidney injury. Nephrol Dial Transplant 31: 1062–1069, 2016. doi: 10.1093/ndt/gfv317. [DOI] [PubMed] [Google Scholar]
- 28.Santel A, Fuller MT. Control of mitochondrial morphology by a human mitofusin. J Cell Sci 114: 867–874, 2001. doi: 10.1242/jcs.114.5.867. [DOI] [PubMed] [Google Scholar]
- 29.Brooks C, Cho SG, Wang CY, Yang T, Dong Z. Fragmented mitochondria are sensitized to Bax insertion and activation during apoptosis. Am J Physiol Cell Physiol 300: C447–C455, 2011. doi: 10.1152/ajpcell.00402.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sumida M, Doi K, Ogasawara E, Yamashita T, Hamasaki Y, Kariya T, Takimoto E, Yahagi N, Nangaku M, Noiri E. Regulation of mitochondrial dynamics by dynamin-related protein-1 in acute cardiorenal syndrome. J Am Soc Nephrol 26: 2378–2387, 2015. doi: 10.1681/ASN.2014080750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gall JM, Wang Z, Liesa M, Molina A, Havasi A, Schwartz JH, Shirihai O, Borkan SC, Bonegio RG. Role of mitofusin 2 in the renal stress response. PLoS One 7: e31074, 2012. doi: 10.1371/journal.pone.0031074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, Akira S, Yamamoto A, Komuro I, Otsu K. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 485: 251–255, 2012. [Erratum in Nature 490: 292, 2012]. doi: 10.1038/nature10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sudakov NP, Apartsin KA, Lepekhova SA, Nikiforov SB, Katyshev AI, Lifshits GI, Vybivantseva AV, Konstantinov YM. The level of free circulating mitochondrial DNA in blood as predictor of death in case of acute coronary syndrome. Eur J Med Res 22: 1, 2017. doi: 10.1186/s40001-016-0241-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang L, Deng S, Zhao S, Ai Y, Zhang L, Pan P, Su X, Tan H, Wu D. Intra-peritoneal administration of mitochondrial DNA provokes acute lung injury and systemic inflammation via Toll-like receptor 9. Int J Mol Sci 17: 1425, 2016. doi: 10.3390/ijms17091425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He Y, Feng D, Li M, Gao Y, Ramirez T, Cao H, Kim SJ, Yang Y, Cai Y, Ju C, Wang H, Li J, Gao B. Hepatic mitochondrial DNA/Toll-like receptor 9/microRNA-223 forms a negative feedback loop to limit neutrophil overactivation and acetaminophen hepatotoxicity in mice. Hepatology 66: 220–234, 2017. doi: 10.1002/hep.29153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Imaeda AB, Watanabe A, Sohail MA, Mahmood S, Mohamadnejad M, Sutterwala FS, Flavell RA, Mehal WZ. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J Clin Invest 119: 305–314, 2009. doi: 10.1172/jci35958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA, Kaech SM, Smiley JR, Means RE, Iwasaki A, Shadel GS. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520: 553–557, 2015. doi: 10.1038/nature14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chang AL, Ulrich A, Suliman HB, Piantadosi CA. Redox regulation of mitophagy in the lung during murine Staphylococcus aureus sepsis. Free Radic Biol Med 78: 179–189, 2015. doi: 10.1016/j.freeradbiomed.2014.10.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chaung WW, Wu R, Ji Y, Dong W, Wang P. Mitochondrial transcription factor A is a proinflammatory mediator in hemorrhagic shock. Int J Mol Med 30: 199–203, 2012. doi: 10.3892/ijmm.2012.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Al-Mehdi AB, Pastukh VM, Swiger BM, Reed DJ, Patel MR, Bardwell GC, Pastukh VV, Alexeyev MF, Gillespie MN. Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci Signal 5: ra47, 2012. doi: 10.1126/scisignal.2002712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zager RA, Johnson AC, Hanson SY. Proximal tubular cytochrome c efflux: determinant, and potential marker, of mitochondrial injury. Kidney Int 65: 2123–2134, 2004. doi: 10.1111/j.1523-1755.2004.00638.x. [DOI] [PubMed] [Google Scholar]
- 42.Wenceslau CF, McCarthy CG, Szasz T, Goulopoulou S, Webb RC. Mitochondrial N-formyl peptides induce cardiovascular collapse and sepsis-like syndrome. Am J Physiol Heart Circ Physiol 308: H768–H777, 2015. doi: 10.1152/ajpheart.00779.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garcia N, Garcia JJ, Correa F, Chavez E. The permeability transition pore as a pathway for the release of mitochondrial DNA. Life Sci 76: 2873–2880, 2005. doi: 10.1016/j.lfs.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 44.Garcia N, Chavez E. Mitochondrial DNA fragments released through the permeability transition pore correspond to specific gene size. Life Sci 81: 1160–1166, 2007. doi: 10.1016/j.lfs.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 45.Briston T, Roberts M, Lewis S, Powney B, M Staddon J, Szabadkai G, Duchen MR. Mitochondrial permeability transition pore: sensitivity to opening and mechanistic dependence on substrate availability. Sci Rep 7: 10492, 2017. doi: 10.1038/s41598-017-10673-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sureshbabu A, Patino E, Ma KC, Laursen K, Finkelsztein EJ, Akchurin O, Muthukumar T, Ryter SW, Gudas L, Choi AM, Choi ME. RIPK3 promotes sepsis-induced acute kidney injury via mitochondrial dysfunction. JCI Insight 3: e2125, 2018. doi: 10.1172/jci.insight.98411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang S, Zhang C, Hu L, Yang C. Necroptosis in acute kidney injury: a shedding light. Cell Death Dis 7: e2125, 2016. doi: 10.1038/cddis.2016.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Linkermann A, Green DR. Necroptosis. N Engl J Med 370: 455–465, 2014. doi: 10.1056/NEJMra1310050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Linkermann A, Brasen JH, Darding M, Jin MK, Sanz AB, Heller JO, De Zen F, Weinlich R, Ortiz A, Walczak H, Weinberg JM, Green DR, Kunzendorf U, Krautwald S. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc Natl Acad Sci USA 110: 12024–12029, 2013. doi: 10.1073/pnas.1305538110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Linkermann A, Heller JO, Prokai A, Weinberg JM, De Zen F, Himmerkus N, Szabo AJ, Brasen JH, Kunzendorf U, Krautwald S. The RIP1-kinase inhibitor necrostatin-1 prevents osmotic nephrosis and contrast-induced AKI in mice. J Am Soc Nephrol 24: 1545–1557, 2013. [Erratum in J Am Soc Nephrol 25: 2942, 2014]. doi: 10.1681/ASN.2012121169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao H, Ning J, Lemaire A, Koumpa FS, Sun JJ, Fung A, Gu J, Yi B, Lu K, Ma D. Necroptosis and parthanatos are involved in remote lung injury after receiving ischemic renal allografts in rats. Kidney Int 87: 738–748, 2015. doi: 10.1038/ki.2014.388. [DOI] [PubMed] [Google Scholar]
- 52.Maeda A, Fadeel B. Mitochondria released by cells undergoing TNF-alpha-induced necroptosis act as danger signals. Cell Death Dis 5: e1312, 2014. doi: 10.1038/cddis.2014.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AM. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12: 222–230, 2011. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, Wong J, Ding S, Seki E, Schnabl B, Hevener AL, Greenberg HB, Kisseleva T, Karin M. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560: 198–203, 2018. doi: 10.1038/s41586-018-0372-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yoshii SR, Mizushima N. Autophagy machinery in the context of mammalian mitophagy. Biochim Biophys Acta 1853: 2797–2801, 2015. doi: 10.1016/j.bbamcr.2015.01.013. [DOI] [PubMed] [Google Scholar]
- 56.Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 333: 1109–1112, 2011. doi: 10.1126/science.1201940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kaushal GP, Shah SV. Autophagy in acute kidney injury. Kidney Int 89: 779–791, 2016. doi: 10.1016/j.kint.2015.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell 40: 280–293, 2010. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Parikh SM, Yang Y, He L, Tang C, Zhan M, Dong Z. Mitochondrial function and disturbances in the septic kidney. Semin Nephrol 35: 108–119, 2015. doi: 10.1016/j.semnephrol.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tang C, Cai J, Yin XM, Weinberg JM, Venkatachalam MA, Dong Z. Mitochondrial quality control in kidney injury and repair. Nat Rev Nephrol 17: 299–318, 2021. doi: 10.1038/s41581-020-00369-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Declèves AE, Sharma K, Satriano J. Beneficial effects of AMP-activated protein kinase agonists in kidney ischemia-reperfusion: autophagy and cellular stress markers. Nephron Exp Nephrol 128: 98–110, 2014. doi: 10.1159/000368932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kimura T, Takabatake Y, Takahashi A, Kaimori JY, Matsui I, Namba T, Kitamura H, Niimura F, Matsusaka T, Soga T, Rakugi H, Isaka Y. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol 22: 902–913, 2011. doi: 10.1681/ASN.2010070705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Periyasamy-Thandavan S, Jiang M, Wei Q, Smith R, Yin XM, Dong Z. Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells. Kidney Int 74: 631–640, 2008. doi: 10.1038/ki.2008.214. [DOI] [PubMed] [Google Scholar]
- 64.Yang C, Kaushal V, Shah SV, Kaushal GP. Autophagy is associated with apoptosis in cisplatin injury to renal tubular epithelial cells. Am J Physiol Renal Physiol 294: F777–F787, 2008. doi: 10.1152/ajprenal.00590.2007. [DOI] [PubMed] [Google Scholar]
- 65.Ishihara M, Urushido M, Hamada K, Matsumoto T, Shimamura Y, Ogata K, Inoue K, Taniguchi Y, Horino T, Fujieda M, Fujimoto S, Terada Y. Sestrin-2 and BNIP3 regulate autophagy and mitophagy in renal tubular cells in acute kidney injury. Am J Physiol Renal Physiol 305: F495–F509, 2013. doi: 10.1152/ajprenal.00642.2012. [DOI] [PubMed] [Google Scholar]
- 66.Tang C, Han H, Liu Z, Liu Y, Yin L, Cai J, He L, Liu Y, Chen G, Zhang Z, Yin XM, Dong Z. Activation of BNIP3-mediated mitophagy protects against renal ischemia-reperfusion injury. Cell Death Dis 10: 677, 2019. doi: 10.1038/s41419-019-1899-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tang C, Han H, Yan M, Zhu S, Liu J, Liu Z, He L, Tan J, Liu Y, Liu H, Sun L, Duan S, Peng Y, Liu F, Yin XM, Zhang Z, Dong Z. PINK1-PRKN/PARK2 pathway of mitophagy is activated to protect against renal ischemia-reperfusion injury. Autophagy 14: 880–897, 2018. doi: 10.1080/15548627.2017.1405880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Whitaker RM, Stallons LJ, Kneff JE, Alge JL, Harmon JL, Rahn JJ, Arthur JM, Beeson CC, Chan SL, Schnellmann RG. Urinary mitochondrial DNA is a biomarker of mitochondrial disruption and renal dysfunction in acute kidney injury. Kidney Int 88: 1336–1344, 2015. doi: 10.1038/ki.2015.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Whitaker RM, Korrapati MC, Stallons LJ, Jesinkey SR, Arthur JM, Beeson CC, Zhong Z, Schnellmann RG. Urinary ATP synthase subunit beta is a novel biomarker of renal mitochondrial dysfunction in acute kidney injury. Toxicol Sci 145: 108–117, 2015. doi: 10.1093/toxsci/kfv038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sun S, Sursal T, Adibnia Y, Zhao C, Zheng Y, Li H, Otterbein LE, Hauser CJ, Itagaki K. Mitochondrial DAMPs increase endothelial permeability through neutrophil dependent and independent pathways. PLoS One 8: e59989, 2013. doi: 10.1371/journal.pone.0059989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pierrakos C, Karanikolas M, Scolletta S, Karamouzos V, Velissaris D. Acute respiratory distress syndrome: pathophysiology and therapeutic options. J Clin Med Res 4: 7–16, 2012. doi: 10.4021/jocmr761w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu G, Zhu Q, Zeng J, Gu X, Miao Y, Xu W, Lv T, Song Y. Extracellular mitochondrial DNA promote NLRP3 inflammasome activation and induce acute lung injury through TLR9 and NF-kappaB. J Thorac Dis 11: 4816–4828, 2019. doi: 10.21037/jtd.2019.10.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mallavia B, Liu F, Lefrançais E, Cleary SJ, Kwaan N, Tian JJ, Magnen M, Sayah DM, Soong A, Chen J, Saggar R, Shino MY, Ross DJ, Derhovanessian A, Lynch JP, Ardehali A, Weigt SS, Belperio JA, Hays SR, Golden JA, Leard LE, Shah RJ, Kleinhenz ME, Venado A, Kukreja J, Singer JP, Looney MR. Mitochondrial DNA stimulates TLR9-dependent neutrophil extracellular trap formation in primary graft dysfunction. Am J Respir Cell Mol Biol 62: 364–372, 2020. doi: 10.1165/rcmb.2019-0140OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang X, Wang T, Yuan ZC, Dai LQ, Zeng N, Wang H, Liu L, Wen FQ. Mitochondrial peptides cause proinflammatory responses in the alveolar epithelium via FPR-1, MAPKs, and AKT: a potential mechanism involved in acute lung injury. Am J Physiol Lung Cell Mol Physiol 315: L775–L786, 2018. doi: 10.1152/ajplung.00466.2017. [DOI] [PubMed] [Google Scholar]
- 75.Hu Q, Ren H, Ren J, Liu Q, Wu J, Wu X, Li G, Wang G, Gu G, Guo K, Hong Z, Liu S, Li J. Released mitochondrial DNA following intestinal ischemia reperfusion induces the inflammatory response and gut barrier dysfunction. Sci Rep 8: 7350, 2018. [Erratum in Sci Rep 12: 2524, 2022]. doi: 10.1038/s41598-018-25387-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lin MM, Liu N, Qin ZH, Wang Y. Mitochondrial-derived damage-associated molecular patterns amplify neuroinflammation in neurodegenerative diseases. Acta Pharmacol Sin. In press. doi: 10.1038/s41401-022-00879-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Faust HE, Reilly JP, Anderson BJ, Ittner CA, Forker CM, Zhang P, Weaver BA, Holena DN, Lanken PN, Christie JD, Meyer NJ, Mangalmurti NS, Shashaty MG. Plasma mitochondrial DNA levels are associated with ARDS in trauma and sepsis patients. Chest 157: 67–76, 2020. doi: 10.1016/j.chest.2019.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Krychtiuk KA, Ruhittel S, Hohensinner PJ, Koller L, Kaun C, Lenz M, Bauer B, Wutzlhofer L, Draxler DF, Maurer G, Huber K, Wojta J, Heinz G, Niessner A, Speidl WS. Mitochondrial DNA and toll-like receptor-9 are associated with mortality in critically ill patients. Crit Care Med 43: 2633–2641, 2015. doi: 10.1097/CCM.0000000000001311. [DOI] [PubMed] [Google Scholar]
- 79.Nakahira K, Kyung SY, Rogers AJ, Gazourian L, Youn S, Massaro AF, Quintana C, Osorio JC, Wang Z, Zhao Y, Lawler LA, Christie JD, Meyer NJ, Mc Causland FR, Waikar SS, Waxman AB, Chung RT, Bueno R, Rosas IO, Fredenburgh LE, Baron RM, Christiani DC, Hunninghake GM, Choi AM. Circulating mitochondrial DNA in patients in the ICU as a marker of mortality: derivation and validation. PLoS Med 10: e1001577, 2013. doi: 10.1371/journal.pmed.1001577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Scozzi D, Cano M, Ma L, Zhou D, Zhu JH, O'Halloran JA, Goss C, Rauseo AM, Liu Z, Peritore V, Rocco M, Ricci A, Amodeo R, Aimati L, Ibrahim M, Hachem R, Kreisel D, Mudd PA, Kulkarni HS, Gelman AE. Circulating mitochondrial DNA is an early indicator of severe illness and mortality from COVID-19. JCI Insight 6: e143299, 2021. doi: 10.1172/jci.insight.143299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tang WX, Wu WH, Qiu HY, Bo H, Huang SM. Amelioration of rhabdomyolysis-induced renal mitochondrial injury and apoptosis through suppression of Drp-1 translocation. J Nephrol 26: 1073–1082, 2013. doi: 10.5301/jn.5000268. [DOI] [PubMed] [Google Scholar]
- 82.Alshaman R, Truong L, Oyekan A. Role of mechanistic target of rapamycin (mTOR) in renal function and ischaemia-reperfusion induced kidney injury. Clin Exp Pharmacol Physiol 43: 1087–1096, 2016. doi: 10.1111/1440-1681.12648. [DOI] [PubMed] [Google Scholar]
- 83.Wang Y, Tang C, Cai J, Chen G, Zhang D, Zhang Z, Dong Z. PINK1/Parkin-mediated mitophagy is activated in cisplatin nephrotoxicity to protect against kidney injury. Cell Death Dis 9: 1113, 2018. doi: 10.1038/s41419-018-1152-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T, Glass K, Owen CA, Mahmood A, Washko GR, Hashimoto S, Ryter SW, Choi AM. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest 124: 3987–4003, 2014. doi: 10.1172/JCI74985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dong W, Li Z, Chen Y, Zhang L, Ye Z, Liang H, Li R, Xu L, Zhang B, Liu S, Wang W, Li C, Shi W, Liang X. Necrostatin-1 attenuates sepsis-associated acute kidney injury by promoting autophagosome elimination in renal tubular epithelial cells. Mol Med Rep 17: 3194–3199, 2018. [DOI] [PubMed] [Google Scholar]
- 86.Linkermann A, Brasen JH, Himmerkus N, Liu S, Huber TB, Kunzendorf U, Krautwald S. Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int 81: 751–761, 2012. doi: 10.1038/ki.2011.450. [DOI] [PubMed] [Google Scholar]
- 87.Martin-Sanchez D, Fontecha-Barriuso M, Carrasco S, Sanchez-Niño MD, Mässenhausen AV, Linkermann A, Cannata-Ortiz P, Ruiz-Ortega M, Egido J, Ortiz A, Sanz AB. TWEAK and RIPK1 mediate a second wave of cell death during AKI. Proc Natl Acad Sci USA 115: 4182–4187, 2018. [Erratum in Proc Natl Acad Sci USA 115: E4731, 2018]. doi: 10.1073/pnas.1716578115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Weber C, Jenke A, Chobanova V, Yazdanyar M, Chekhoeva A, Eghbalzadeh K, Lichtenberg A, Wahlers T, Akhyari P, Paunel GA. Targeting of cell-free DNA by DNase I diminishes endothelial dysfunction and inflammation in a rat model of cardiopulmonary bypass. Sci Rep 9: 19249, 2019. doi: 10.1038/s41598-019-55863-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Araujo AM, Antunes MM, Mattos MS, Diniz AB, Alvarenga DM, Nakagaki BN, Carvalho E, Lacerda VA, Carvalho-Gontijo R, Goulart J, Mafra K, Freitas-Lopes MA, Oliveira HM, Dutra CM, David BA, Mendes Silva A, Quesniaux V, Ryffel B, Oliveira SC, Barber GN, Mansur DS, Cunha TM, Rezende RM, Oliveira AG, Menezes GB. Liver immune cells release type 1 interferon due to DNA sensing and amplify liver injury from acetaminophen overdose. Cells 7: 88, 2018. doi: 10.3390/cells7080088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Simmons JD, Freno DR, Muscat CA, Obiako B, Lee YL, Pastukh VM, Brevard SB, Gillespie MN. Mitochondrial DNA damage associated molecular patterns in ventilator-associated pneumonia: Prevention and reversal by intratracheal DNase I. J Trauma Acute Care Surg 82: 120–125, 2017. doi: 10.1097/ta.0000000000001269. [DOI] [PMC free article] [PubMed] [Google Scholar]