

Abstract
Sparassis crispa polysaccharides have recently attracted considerable attention due to their excellent bioactivities. However, their extraction procedure is often tedious, time-consuming, and environmentally unfriendly; it even causes damage to their structures and reduces their bioactivities, all of which hinder their further development to some extent. Therefore, the ultrasonic-assisted enzymatic extraction (UAEE) technology was optimized to extract polysaccharides from Sparassis crispa (SCP) by the response surface methodology. The yields, physicochemical properties, and antioxidant activities of SCPs obtained from UAEE and conventional hot water extraction (HWE) were evaluated. According to the optimal parameters, the yield of SCPs extracted by UAEE reached up to 14.63%, which increased by 68.54% compared with that obtained from the conventional hot water extraction (HWE) method. Additionally, the UAEE methods affected the contents of the polysaccharides, molecular weights, and the molar percentage of the constituent monosaccharides of SCPs. SEM analysis indicated that the microstructures of the two SCPs were notably different. Antioxidant assays showed that both SCPs possessed good antioxidant activities against DPPH, ABTS˙+, and hydroxyl radicals in vitro. Additionally, the SCPs extracted by UAEE attenuated the HT22 cell neurotoxicity induced by H2O2 by the means of ameliorating cell viability, reducing extracellular LDH release, and decreasing the levels of intracellular ROS. These results provide scientific basis for the further investigation of SCPs as potential neuroprotective agents.
Extraction optimization, structural characterization, and neuroprotective effects of polysaccharides from Sparassis crispa.
1. Introduction
Sparassis crispa (S. crispa), belonging to the family of Sparassidaceae, is also known as cauliflower mushroom.1 It is widely distributed in the Pacific Northwest, eastern Asia, and Australia and has been very popular among consumers because of its unique taste and nutritional values.2,3 In recent years, S. crispa extracts have attracted more attention for their medicinal properties, such as antitumor,4 hematopoietic,5 anti-inflammatory,3 and hypocholesterolemic6 effects.
Modern research has demonstrated that the therapeutic effects of S. crispa can be attributed to its bioactive substances, including sesquiterpenoids, benzoate derivatives, phthalides, and polysaccharides.7–10 Among these compounds, polysaccharides have been found to be valuable functional components, which is mainly due to their bioactivities. For instance, the oral administration of polysaccharides obtained from S. crispa (SCP) can suppress angiogenic and metastatic neoplasm in vivo.11 Hida et al. found that treatment with SCPs may modulate cytokine production in the spleen of mice through its stimulation of the Peyer's patches.12 The oral administration of polysaccharides extracted from cultured S. crispa showed obvious antitumor activity against the solid form of Sarcoma 180 in ICR mice with strong vascular dilation and hemorrhage reactions.4 However, previous studies mainly focused on the bioactivities of polysaccharides obtained from S. crispa and paid little attention to their extraction method, which greatly influences the yield, chemical structure, and bioactivity of the polysaccharides.13 Therefore, an efficient extraction method for S. crispa polysaccharides is required.
Conventionally, the extraction methods of polysaccharides, such as immersion, heating or boiling, and refluxing, often require a long time, waste too much solvents, require a high temperature, decrease the yield of polysaccharides, and even lead to the loss of some pharmacological activities.14 To overcome these problems, various new technologies for polysaccharide extraction have been successfully developed, such as supercritical fluid extraction (SFE), infrared-assisted extraction (IAE), microwave-assisted extraction (MAE), ultrasonic-assisted extraction (UAE), and enzyme-assisted extraction (EAE).13,15 Among these technologies, UAE has received great attention because of its high efficiency. The acoustic cavitation in UAE can destroy cell walls, reduce particle sizes, and enhance the contact between solvents and active compounds.16 Moreover, compared with the traditional extraction methods, UAE has other advantages such as lower cost, lower temperature, and increase in the extraction yield with stable biological properties.17 Besides, the EAE method is considered as an efficient and effective approach for polysaccharide extraction as some specific enzymes (cellulase and proteases) can break the plant cell wall, hydrolyze the cytoderm, and then easily release the intracellular polysaccharides.18,19 Recently, many studies have reported that a combination of UAE and enzymatic treatment could improve the polysaccharide extraction from plants. To date, ultrasonic-assisted enzymatic extraction (UAEE) has been performed to efficiently extract polysaccharides from Atratylodes macrocephala,20Lycium barbarum,19 dragon fruit peel,18 and dandelion leaves.21 Liao et al. compared two polysaccharide extraction methods, namely, UAEE and EAE from Corbicula fluminea, and found that the yield obtained from the UAEE method applied for 32 min was higher than that of the EAE method applied for 4 h; besides, the polysaccharides extracted by UAEE had lower molecular weights and higher superoxide radical scavenging activity.22 Wang et al. obtained higher polysaccharides yield (14.05%) from dandelion leaves by using UAEE compared with EAE (9.84%) and UAE (8.84%) under the same enzyme concentration and extraction time.21 However, there are few reports on UAEE for polysaccharide extraction from S. crispa.
Several polysaccharides have been reported to possess neuroprotective effects in vitro, possibly due to their good antioxidant activities, such as polysaccharides obtained from Lonicera japonica Thunb.,23Lycium barbarum L.,24 and Amanita caesarea.25 In addition, a recent study reported that polysaccharides from S. crispa exhibited protective effects against glutamate-induced toxicity in differentiated PC12 cells,26 indicating that S. crispa polysaccharides are potential agents for intervention of neurodegenerative diseases. In this study, UAEE procedure was applied to efficiently obtain the SCPs and the extraction conditions were optimized by using response surface method (RSM). Subsequently, mouse hippocampal HT22 cell oxidative stress model was used to evaluate the potential neuroprotective benefits.
2. Materials and methods
2.1. Materials and reagents
Dried S. crispa powder was provided by Shanghai Hexian Mushroom Biotechnology Co., Ltd. The cellulase enzyme (15 000 U g−1) was purchased from Sinopharm Chemical Reagent Co., Ltd. 1,1-Diphenyl-2-picrylhydrazyl (DPPH), 2′-azinobis-(3-ethylbenzthiazoline-6-sulphonate) (ABTS), ascorbic acid (Vc), 2′,7′-dichlorofluorescein diacetate (DCFH-DA), and (±)-6-hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (Trolox) were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Dulbecco's modified essential medium (DEME), fetal bovine serum (FBS), Penicillin G, and streptomycin were purchased from Gibco (Grand Island, NY, USA). Cell Counting kit-8 (CCK8 kit) and LDH cytotoxicity assay kits were purchased from Beyotime Biotechnology (Shanghai, China). All other chemical reagents used in this study were of analytical grade.
2.2. Ultrasonic-assisted enzymatic extraction (UAEE) of the polysaccharides
The powder of S. crispa (30 g) was twice extracted with 95% ethanol at 60 °C for 6 h each time to remove the alcohol-soluble substances, such as pigments, lipids, and oligosaccharides by using a Soxhlet set. After being air dried at 60 °C for 12 h, the defatted powder (1 g) was mixed with distilled water at a given solid–liquid ratio (range from 1 : 10 g mL−1 to 1 : 50 g mL−1) and fixed pH of 5 in a flask.19,21 Then, the mixtures were conducted at the designed time (range from 10 min to 50 min), ultrasonication power (range from 180 W to 420 W), and cellulase concentration (range from 0.4% to 1.2%) in an ultrasonic cleaner (KQ-600KDB, Kunshan Ultrasound Instruments Co., Ltd., Kunshan, China) at 55 °C.18 After extraction by the UAEE procedure, the mixtures were centrifuged at 5000 rpm for 10 min to obtain the supernatant. Afterwards, the residues were re-suspended with distilled water and centrifugation was repeated thrice, as mentioned above, to recover the residual polysaccharides. Then, the incorporated supernatant was concentrated by using a rotary evaporator and precipitated with four volumes of 80% (v/v) ethanol at 4 °C overnight. The precipitate was collected by centrifugation at 5000 rpm for 10 min, and washed by anhydrous ethanol and acetone. Then, the crude S. crispa polysaccharides (SCP) were obtained after the freeze-drying process. The extraction yield of SCPs (%) was calculated with the following equation:
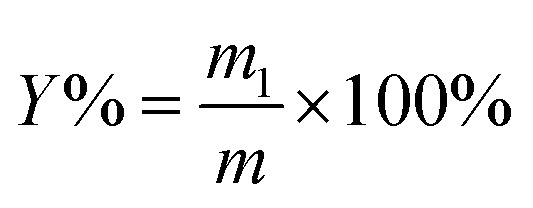 |
1 |
where m1 (g) represents the weight of dried SCPs and m (g) is the pretreated sample weight.
2.3. Experimental design and modeling
Based on the preliminary single-factor experiments, response surface methodology (RSM) was adopted to further evaluate the effects of independent variables (ultrasonication time X1, ultrasonication power X2, solid–liquid ratio X3, and cellulase concentration X4) on the yield of SCP. A four-factor, three-level Box–Behnken Design (BBD) was used to optimize the combined effects (shown in Table 1). The response value in each trial was an average of the triplicates.
Box-Behnken (BBD) design of four variables and response values.
| Factors | Units | Symbols | Level of factors | ||
|---|---|---|---|---|---|
| −1 | 0 | 1 | |||
| Ultrasonication time | min | X 1 | 30 | 40 | 50 |
| Ultrasonication power | W | X 2 | 240 | 300 | 360 |
| Solid–liquid ratio | g mL−1 | X 3 | 1 : 35 | 1 : 40 | 1 : 45 |
| Cellulase concentration | % | X 4 | 0.6 | 0.8 | 1.0 |
| Run | X 1 | X 2 | X 3 | X 4 | Yield of SCPs (%) | |
|---|---|---|---|---|---|---|
| Actual | Predicted | |||||
| 1 | −1 | −1 | 0 | 0 | 10.22 | 10.31 |
| 2 | 0 | 1 | 0 | −1 | 11.86 | 11.73 |
| 3 | 0 | 1 | 0 | 1 | 13.43 | 13.53 |
| 4 | −1 | 0 | 1 | 0 | 11.97 | 11.98 |
| 5 | 1 | 0 | 1 | 0 | 12.57 | 12.68 |
| 6 | 0 | 0 | −1 | 1 | 13.46 | 13.51 |
| 7 | 0 | −1 | 0 | −1 | 13.27 | 13.12 |
| 8 | 0 | 0 | 0 | 0 | 14.75 | 14.72 |
| 9 | 1 | 0 | −1 | 0 | 13.15 | 13.08 |
| 10 | −1 | 0 | 0 | −1 | 11.08 | 11.10 |
| 11 | −1 | 1 | 0 | 0 | 12.09 | 12.25 |
| 12 | 1 | 0 | 0 | −1 | 12.45 | 12.49 |
| 13 | −1 | 0 | 0 | 1 | 11.85 | 11.74 |
| 14 | 0 | −1 | 1 | 0 | 13.49 | 13.44 |
| 15 | 0 | 1 | −1 | 0 | 13.79 | 13.77 |
| 16 | 0 | 0 | 0 | 0 | 14.65 | 14.72 |
| 17 | 0 | −1 | −1 | 0 | 12.37 | 12.42 |
| 18 | 0 | 0 | −1 | −1 | 12.80 | 12.96 |
| 19 | 0 | 0 | 1 | 1 | 13.09 | 13.05 |
| 20 | 1 | −1 | 0 | 0 | 13.05 | 13.02 |
| 21 | 0 | −1 | 0 | 1 | 11.67 | 11.75 |
| 22 | 1 | 1 | 0 | 0 | 11.45 | 11.47 |
| 23 | −1 | 0 | −1 | 0 | 12.01 | 11.85 |
| 24 | 0 | 1 | 1 | 0 | 12.59 | 12.48 |
| 25 | 0 | 0 | 0 | 0 | 14.85 | 14.72 |
| 26 | 0 | 0 | 0 | 0 | 14.65 | 14.72 |
| 27 | 0 | 0 | 0 | 0 | 14.72 | 14.72 |
| 28 | 0 | 0 | 1 | −1 | 13.08 | 13.15 |
| 29 | 1 | 0 | 0 | 1 | 12.37 | 12.29 |
To predict the optimal parameters of UAEE of SCP, the data obtained from BBD were fitted to a second-order polynomial model for establishing the relationship between the independent variables and the responses. The equation was expressed as follows:
 |
2 |
Y represents the response variables, A0, Ai, Aii, and Aij are the regression coefficients for the intercept, linear, quadratic, and interactive coefficients, respectively. Xi and Xj are the independent variables.
2.4. Conventional hot water extraction (HWE) of the SCPs
The hot water extraction of SCPs was performed by the method described by Chen with a slight modification.27 In brief, the dried S. crispa powder was pretreated with 95% ethanol as mentioned above in the UAEE method. Then, the defatted powder was extracted with distilled water (1 : 40, w/v) at 90 °C for 6 h. After centrifuging at 5000 rpm for 10 min, the extract was concentrated, precipitated with ethanol, and lyophilized following the same procedures in section 2.2.
2.5. Characterization of the physicochemical properties of the SCPs
2.5.1. Determination of the chemical composition
The phenol–sulfuric acid method,28 Coomassie brilliant blue method,29 and m-hydroxydiphenyl method30 were applied to estimate the contents of sugar, protein, and uronic acid, respectively.
2.5.2. Monosaccharide composition analysis
The monosaccharide composition of SCPs was evaluated according to a previous method31 with slight modification. Briefly, SCPs (2 mg) were hydrolyzed with 2 M trifluoroacetic acid (2 mL) at 110 °C for 3 h in a sealed ampoule bottle. Subsequently, the hydrolysates were co-distilled with methanol under reduced pressure to fully remove TFA. Finally, the residues were re-dissolved in deionized water and then filtered by a 0.22 μm nylon membrane. The filtrates were detected using HPAEC-PAD (Thermo Scientific) and determined by comparing the retention time with those of the standards, including glucose, galactose, fucose, mannose, and fructose.
2.5.3. Molecular weight analysis
The molecular weight (Mw) of the SCPs was determined using high performance gel permeation chromatography (HPGPC) on a HPLC instrument (Waters 1525, USA) with a Ultrahydrogel™ Linear tandem column (300 mm × 7.8 mm, Waters, USA) and refractive index detector (Waters 2410, USA). The column was eluted with 0.1 M NaNO3 at a flow rate of 0.8 mL min−1 and maintained at a temperature of 35 °C. Dextran standards (2700, 9750, 135 030, 300 600, and 2 000 000 Da) were used for the calibration curve.
2.5.4. Scanning electron microscopy (SEM)
The lyophilized SCPs were mounted on a metal stub and coated with gold using an ion sputter coater (E-1010, Hitachi, Japan). Then, all the specimens were observed by a scanning electron microscope (NPE-218, Nova NanoSEM, USA) under high vacuum at an accelerating voltage of 10.0 kV and the images were photographed at 3000× magnification.
2.6. In vitro antioxidant activity
2.6.1. DPPH radical scavenging activity
The ability of SCPs to scavenge 1,1-diphenyl-2-picrylhydrazyl (DPPH) radicals was measured using a previous method13 with a slight modification. The SCP sample was dissolved in distilled water into a series of concentrations (0.0625–5 mg mL−1). 3 mL sample and 1 mL freshly prepared DPPH solution (0.2 mM in ethanol) were mixed and vortexed vigorously. The mixture was incubated at room temperature for 30 min in the dark. Then, the absorbance of sample was determined at 517 nm using an ultraviolet-visible spectrophotometer. Vitamin C was used as the positive control. The DPPH radical scavenging ability (%) was calculated using the following formula:
 |
3 |
where A0 is the absorbance of the blank (ethanol instead of the sample), Ai is the absorbance of the polysaccharide sample, and Aj is the absorbance of the sample control (ethanol instead of DPPH solution).
2.6.2. ABTS˙+ radical-scavenging activity
The ABTS˙+ radical-scavenging activity of the SCPs was determined as described by Hajji et al.32 The ABTS˙+ stock solution was prepared by mixing equal volumes of ABTS solution (7.00 mM) and potassium persulphate (2.45 mM). The mixture was incubated for 16 h at 25 °C in dark. To obtain the reaction solution, the ABTS˙+ stock solution was diluted with PBS (pH 7.4) to an absorbance of 0.700 (±0.02) at 734 nm. Then, the reaction solution was added to the SCP sample at given concentrations (0–5.0 mg mL−1) and the mixture was incubated at 25 °C for 6 min in dark. The absorbance at 734 nm was read by a micro-plate reader (ELx800, BioTek, USA). Vitamin C was used as the positive control. The ABTS˙+ radical scavenging ability was calculated according to the following equation:
 |
4 |
where A0 is the absorbance of the blank (distilled water instead of the sample), Ai is the absorbance of the polysaccharide sample, and Aj is the absorbance of the sample control (PBS instead of the reaction solution).
2.6.3. Hydroxyl radical scavenging activity
The hydroxyl radical-scavenging activity of the SCPs was detected by an improved Fenton-type reaction.33 Briefly, 1 mL sample of different concentrations (0–5.0 mg mL−1) was mixed with 1 mL FeSO4 (9 mM), 1 mL H2O2 (9 mM), and 1 mL salicylic acid (9 mM). Then, the mixtures were reacted at 37 °C for 30 min. Finally, the absorbance of the mixture was recorded at 562 nm. Vitamin C was used as the positive control. The radical scavenging ability of HO˙ was calculated as follows:
 |
5 |
where A0 is the absorbance of the blank (distilled water instead of the sample), Ai is the absorbance of the polysaccharide sample, and Aj is the absorbance of the sample control (distilled water instead of H2O2).
2.7. Neuroprotective assay
2.7.1. Cell culture
The immortalized mouse hippocampal cell line HT22 was obtained from Shanghai Zeye Company. The cells were cultured in Dulbecco's modified Eagle medium (DMEM) with 10% FBS, 100 U mL−1 penicillin, 100 g mL−1 streptomycin and incubated at 37 °C in a humidified incubator supplied with 5% CO2.
2.7.2. Assessment of the neuroprotective effects
The protective effect of the SCPs against H2O2-induced HT22 cell neurotoxicity was assessed by using the CCK8 kit according to the manufacturer's instructions. Briefly, the HT22 cells were seeded in a 96-well plate at a density of 5 × 103 cells per well. After 24 h incubation at 37 °C, different concentrations of the SCP solution (0–200 μg mL−1) or the positive control Trolox (50 μM) were added for 12 h. Then, 400 μM H2O2 was added and co-cultured for another 12 h. Subsequently, 10 μL CCK-8 solutions was added into each well and incubated for 2 h at 37 °C. The absorbance at 450 nm was measured with a micro-plate reader (BioTek instruments, USA). The HT22 cells incubated with DMEM were regarded as the negative control, while the cells treated with only H2O2 served as the model group.
2.7.3. Determination of lactate dehydrogenase (LDH)
Lactate dehydrogenase (LDH) was determined by a colorimetric assay following the manufacturer's protocol. Briefly, the HT22 cells were plated in 96-well plates for 24 h. After experimental treatment, the medium was removed and the cells were reacted with 2,4-dinitrophenylhydrazine for 1 h. For analysis, the supernatant was collected and centrifuged at 800 rpm. The absorbance was measured at the wavelength of 490 nm by using a micro-plate reader.
2.7.4. Intracellular ROS measurement
The HT22 cells were seeded into 24-well plates and pretreated with different concentrations of the SCPs for 12 h, then co-incubated with H2O2 (400 μM) for 12 h. The redox status of the HT22 cells was measured by using DCFH-DA for 20 min at 37 °C in dark. After treatment, all the cells were washed with PBS three times and the fluorescence intensity was determined using a microplate reader with excitation at 488 nm and emission at 525 nm.
2.8. Statistical analysis
All the experiments were carried out in triplicate. The data were analyzed by Duncan's multiple tests, and compared with analysis of variance (ANOVA). All the values were expressed as the mean ± standard deviation (SD) with P < 0.05 indicating the statistical significance. Statistical analyses were conducted with SPSS ver.17.0, professional edition.
3. Results and discussion
3.1. Single factor experimental analysis
3.1.1. Ultrasonication time
In the present experiment, the effect of ultrasonication time on the yield of the polysaccharides is shown in Fig. 1A. The ultrasonication time was set at various extraction times (10, 20, 30, 40, and 50 min), and the other extraction parameters were set as follow: ultrasonication power, 300 W; solid–liquid ratio, 1 : 40 g mL−1; cellulase concentration, 0.8%. As shown in Fig. 1A, the yield of the SCPs continued to increase in the range of 10 to 40 min, reached the critical point at 40 min, and then the curve began to decline. Generally speaking, there should be a positive correlation between the yield of the polysaccharides and extraction time to a certain extent.20 But longer irradiation time may cause degradation and consequently a decreased extraction yield.21 Therefore, 40 min was selected as the suitable extraction time for RSM design.
Fig. 1. Effects of ultrasonication time (A), ultrasonication power (B), solid–liquid ratio (C), and cellulase concentration (D) on the extraction yield of the SCPs (%).

3.1.2. Ultrasonication power
The change in the yield of the SCPs with different ultrasonication power ranging from 180 to 420 W is shown in Fig. 1B, while the other parameters were set to the following conditions: ultrasonication time, 40 min; solid–liquid ratio, 1 : 40 g mL−1; cellulase concentration, 0.8%. The result revealed that the maximum yield of the SCPs was observed when the ultrasonication power was 300 W and then, a higher ultrasonication power decreased the yield. It has been demonstrated that cavitation intensity controlled by ultrasonication power assists in the release of the entrapped polysaccharides from the matrix.22 But a very high ultrasonication power generates abundant microscopic bubbles, which subsequently inhibit the cavitation.34 Thus, 300 W was selected as the optimal ultrasonication power.
3.1.3. Solid–liquid ratio
The extraction yield is influenced by the water to raw material ratio; different raw materials have different ratios. In this study, the ratios of water to material were set to 1 : 10, 1 : 20, 1 : 30, 1 : 40, and 1 : 50 g mL−1 to evaluate the effect of the ratio on the SCPs' extraction yield. Other parameters were set as follows: ultrasonication time, 40 min; ultrasonication power, 300 W; cellulase concentration, 0.8%. Fig. 1C reveals that the SCPs' yield increased continuously with increasing solid–liquid ratio until the peak value of 1 : 40 g mL−1. However, a further increase in the water to material ratio resulted in a decrease in the SCPs' yield, which might be due to the higher water to material ratio, possibly leading to a lower density and viscosity, thereby increasing the dilution of the extracts in the solvent.35 Therefore, 1 : 40 g mL−1 was selected as the optimal ratio for RSM experiments.
3.1.4. Cellulase concentration
It has been shown that the primary structure of a purified polysaccharide obtained from S. crispa is a 6-branched 1,3-beta-glucan.7,36 On the other hand, cellulase is enzyme that hydrolyzes the β-1,4 linkages in cellulose, which is the main component of plant cell walls. Previous studies have found that the use of cellulase can be an efficient means for polysaccharide extraction.18,21 As shown in Fig. 1D, different cellulase concentrations (0.4%, 0.6%, 0.8%, 1.0%, and 1.2%) were adopted in the extraction procedure. Other parameters were set as follows: ultrasonication time, 40 min; ultrasonication power, 300 W; solid–liquid ratio, 1 : 40 g mL−1. The results indicated that the yield increased significantly until the enzyme volume was 0.8%, after which the yield began to decrease slightly, which is in accordance with the previous study.20 Therefore, the cellulase concentration of 0.8% was selected as the optimal enzyme concentration for the following RSM experiment.
3.2. Optimization of the procedure by RSM
3.2.1. Statistical analysis and model fitting
Based on the single-factor tests, there were a total number of 29 runs for optimizing the four individual variables. The four autonomous variables including ultrasonication time (X1), ultrasonication power (X2), solid–liquid ratio (X3), and cellulase concentration (X4) are exhibited in Table 1. The yields of the SCPs range from 10.22% to 14.75% and the experimental results were analyzed by multiple regression analysis. The yield of SCPs was described by the following second-order polynomial eqn (6):
 |
6 |
where X1, X2, X3, and X4 are the ultrasonication time, ultrasonication power, solid–liquid ratio, and cellulase concentration, respectively. Y represents the predicted yield of SCP.
Analysis of variance (ANOVA) for the fitted quadratic polynomial model is summarized in Table 2. The P-value was regarded as an index for verifying the significance of each coefficient, wherein a smaller P-value indicates a more significant corresponding coefficient.37 As shown in Table 2, the higher F-value (160.69) and lower P-value (P < 0.0001) suggested that the regression model was of highly statistical significance. Moreover, the linear coefficients (X1, X3, and X4), the quadratic term coefficients (X12, X22, X32, and X42), as well as the cross-product coefficients (X1X2, X1X4, X2X3, X2X4, and X3X4) were significant terms with small P-values (P < 0.05). The other terms coefficients were not significant (P > 0.05). The lack of fit P-value was 0.1481, indicating that the lack of fit was not significant relative to the pure error. It means that the equation is adequate to predict the variations.38 The value of R2 reflects the proportion of variation in the response attributed to the model rather than to random error.20 The determination coefficient (R2 = 0.9938) and the adjusted determination coefficient (Radj2 = 0.9876) demonstrated that the actual values were found to fit the predicted values well. Moreover, the coefficient of variation (C.V.) was 1.01%, which clearly revealed that the experimental values were dependable and precise. Moreover, the diagnostics of model adequacy also implied that the response surface model could precisely evaluate the relevance between the predicted value and the actual data (Fig. S1†).
ANOVA for response surface quadratic model for the yield of SCPs.
| Source | Sum of squares | df | Mean square | F value | Probe > F | Significance |
|---|---|---|---|---|---|---|
| Model | 37.97 | 14 | 2.71 | 160.69 | <0.0001 | a |
| X 1 | 2.81 | 1 | 2.81 | 166.21 | <0.0001 | a |
| X 2 | 0.11 | 1 | 0.11 | 6.62 | 0.0221 | b |
| X 3 | 0.053 | 1 | 0.053 | 3.14 | 0.0983 | |
| X 4 | 0.15 | 1 | 0.15 | 8.63 | 0.0108 | b |
| X 1 X 2 | 3.02 | 1 | 3.02 | 178.97 | <0.0001 | a |
| X 1 X 3 | 0.073 | 1 | 0.073 | 4.30 | 0.0570 | |
| X 1 X 4 | 0.18 | 1 | 0.18 | 10.70 | 0.0056 | a |
| X 2 X 3 | 1.34 | 1 | 1.34 | 79.65 | <0.0001 | a |
| X 2 X 4 | 2.51 | 1 | 2.51 | 148.72 | <0.0001 | a |
| X 3 X 4 | 0.10 | 1 | 0.10 | 6.13 | 0.0267 | b |
| X 1 2 | 20.90 | 1 | 20.90 | 1238.14 | <0.0001 | a |
| X 2 2 | 8.83 | 1 | 8.83 | 522.94 | <0.0001 | a |
| X 3 2 | 1.83 | 1 | 1.83 | 108.18 | <0.0001 | a |
| X 4 2 | 6.82 | 1 | 6.82 | 404.32 | <0.0001 | a |
| Residual | 0.24 | 14 | 0.017 | |||
| Lack of fit | 0.21 | 10 | 0.021 | 3.03 | 0.1481 | |
| Pure error | 0.028 | 4 | 6.88 × 10−3 | |||
| Core total | 38.20 | 28 | ||||
| R 2 | 0.9938 | |||||
| Adj-R2 | 0.9876 | |||||
| Pred-R2 | 0.9674 | |||||
| Adeq precision | 47.20 | |||||
| C.V.% | 1.01 |
P < 0.01.
P < 0.05.
3.2.2. Response surface analysis
To describe the interaction between the dependent variables, three-dimensional (3D) response surfaces were constructed and the results are shown in Fig. 2 (contour plots shown in Fig. S2†). In this experiment, the two independent variables were exhibited by 3D surface plot whereas other variables were fixed at 0 level. As shown in Fig. 2A, the yield of the SCPs increased when the ultrasonication power increased from 240 W to 302.78 W and the extraction time increased from 30 min to 41.24 min, respectively. On extending these ranges, the SCPs' yield declined by several degrees. Likewise, the effect of interactions between the other two variables on the yield of the SCPs are also illustrated (Fig. 2B and F). By analyzing the result above by the Design-Expert software, the optimum values of the variables were obtained under the following optimal conditions: ultrasonication time of 41.24 min, ultrasonication power of 302.78 W, solid–liquid ratio of 1 : 38.86 g mL−1, and cellulase concentration of 0.81%.
Fig. 2. Response surface plots showing the effects of ultrasonication time (X1), ultrasonication power (X2), solid–liquid ratio (X3), and cellulase concentration (X4) on the extraction yield of the SCPs (%).

To further examine the suitability of the model, practical extraction parameters were set as follows: ultrasonication time was 41 min, ultrasonication power was 300 W, solid–liquid ratio was 1 : 39 g mL−1, and cellulase concentration was 0.81%. Under these conditions, the actual yield of the SCPs was obtained to be 14.63 ± 0.57% (n = 3), which was close to the predicted value (14.76%). These results demonstrated that the response model was suitable and valid for the UAEE procedure of SCP.
3.3. Comparison of the SCPs extracted by UAEE and HWE
The comparison of UAEE and HWE was mainly conducted based on the yield of SCPs, chemical and sugar composition, molecular weight (Mw), and effects of the two extraction procedures on the microstructures with SEM.
3.3.1. Extraction yield and chemical composition
According to the extraction conditions of UAEE and HWE, the yield of the SCPs was 14.63 ± 0.57% and 8.68 ± 0.32% (shown in Table 3), respectively. Compared with the HWE procedure, the yield of UAEE increased by about 68.54%, while the extraction time and extraction temperature greatly reduced from 360 min to 41 min, and from 90 °C to 55 °C, respectively. As listed in Table 3, UAEE was better than HWE, which was characterized by the higher polysaccharide content (83.25 ± 2.85% vs. 79.98 ± 2.34%) and lower protein content (3.13 ± 0.09% vs. 4.77 ± 0.12%). These results might be attributed to the acoustic cavitation of ultrasound and specific hydrolysis of cellulose, both of which improve the extraction efficiency of biological polysaccharide.
Extraction yield and chemical composition of the SCPs extracted by UAEE and HWEa.
| Parameters | SCP-HWE | SCP-UAEE |
|---|---|---|
| Extraction yields (%) | 8.68 ± 0.32% | 14.63 ± 0.57% |
| Total polysaccharides (%) | 79.98 ± 2.34 | 83.25 ± 2.85 |
| Total uronic acids (%) | N.D. | N.D. |
| Proteins (%) | 4.77 ± 0.12 | 3.13 ± 0.09 |
N.D.: not detectable or lower than the limit of quantification.
3.3.2. Molecular weight and monosaccharide analysis of the SCPs
Generally, the bioactivities of polysaccharides are related to their molecular weight (Mw) and monosaccharide composition.39 Therefore, the effects of the two extraction methods on the Mw and constituent monosaccharides of SCPs are compared in Table 4 and Fig. S3 (see in the ESI†). As shown in Table 4, the three polysaccharide fractions (fraction 1, fraction 2, and fraction 3) were detected in SCP-HWE with Mw of 1.945 × 106 Da, 2.111 × 104 Da, and 1.753 × 103 Da, respectively. However, only two fractions (fraction1 and fraction 3) in SCP-UAEE were determined to be 1.567 × 106 Da and 7.662 × 103 Da, respectively. Fraction 1 (high Mw) and fraction 3 (lower Mw) were the dominant peaks, which were much different from that of the polysaccharides (Mw 7.5 × 104 Da) isolated from S. crispa.26 In addition, an obvious degradation of fraction 1 was detected in the HPGPC chromatograms of SCP-UAEE. Indeed, the Mw of fraction 2 in SCP-UAEE could not be precisely measured due to the relatively poor column efficiency and the co-elution of numerous molecules with different Mw from 15 min to 20 min. The results indicated that the UAEE method could degrade and convert higher Mw fraction into lower Mw fractions.
Molecular weights and monosaccharide constitutions of SCPs.
| SCP-HWE | SCP-UAEE | |
|---|---|---|
| Mw × 10 4 (Da) | ||
| Fraction 1 | 194.5 | 156.7 |
| Fraction 2 | 2.111 | — |
| Fraction 3 | 0.1753 | 0.7662 |
| Monosaccharide (molar percentages, %) | ||
| Glucose (Glc) | 75.18 | 66.96 |
| Galactose (Gal) | 19.29 | 23.57 |
| Fucose (Fuc) | 4.32 | 6.92 |
| Mannose (Manmbn) | 1.21 | 2.55 |
Furthermore, the monosaccharide constituents of SCPs are depicted in Table 4 and Fig. S3 (in the ESI†). The results showed that the constituent monosaccharides of SCP-HWE and SCP-UAEE were similar, which were determined as Glc, Gal, Fuc, and Man with molar percentages of 75.18 : 19.29 : 4.32 : 1.21 and 66.96 : 23.57 : 6.92 : 2.55, respectively. The major compositional monosaccharide (Glc) in SCPs was similar as that in some previous studies.36,40 Interestingly, Gal, Fuc, and Man were first reported in the present study, which might be attributed to the environment, varieties, climate, growing methods, etc. These results suggest that the extraction methods had little effect on the types of monosaccharide composition, while they can significantly affect the molar percentage. Similar results were also reported before in the other plant polysaccharide with different extraction methods.35,39
3.3.3. Microstructural analysis of the SCPs
The microstructures of the SCPs powder were investigated by using SEM analysis and the images are exhibited in Fig. 3. The results showed that the SCPs extracted by HWE possessed a larger lamelliform morphology with some small holes and exhibited a smooth and dense appearance. However, the polysaccharides extracted by UAEE contained plenty of powders and loose surfaces with some bubble-like holes and interspaces. Compared to conventional extraction by hot water, ultrasound-assisted or enzymatic-assisted extraction had a drastic effect on the cell walls of the tissues in breaking down the cell structure,41 which may explain the high extraction efficiency by UAEE. Also, similar results were reported by Wu42 and Yin.38 Besides, the SCPs obtained from the two different extraction methods possessed similar typical absorption peaks, which indicated that the UAEE procedure had no effect on the polysaccharide functional groups (shown in Fig. S3†).
Fig. 3. The scanning electron microstructures of SCPs extracted by HWE (A) and UAEE (B).

3.4. Antioxidant activity assays
In this study, the antioxidant activity of the SCPs was determined by using three complementary tests, namely DPPH radical scavenging activity, ABTS˙+ radical-scavenging activity, and hydroxyl radical scavenging activity.
As shown in Fig. 4A, as the concentration of SCPs increased from 0.0625 to 5.0 mg mL−1, the DPPH radical scavenging activity of SCP-UAEE and SCP-HWE gradually increased in a concentration-dependent manner and reached 88.99% and 80.62% at 5.0 mg mL−1, respectively. In addition, the DPPH radical scavenging activities of UAEE at different concentrations were always higher than those of HWE, even if the activities of both were lower than those of Vc. Also, the antioxidant activity of the SCPs was higher than that of the polysaccharides extracted from different natural sources, such as bamboo shoots,14 cassia seed,35 and Inonotus hispidus,43 which suggested that the SCPs possessed remarkable antioxidant activity.
Fig. 4. Antioxidant activity of SCPs extracted by UAEE and HWE. DPPH radical scavenging activity (A), ABTS radical scavenging activity (B), and hydroxyl radical scavenging activity (C).

Similar to the DPPH results, the SCPs extracted by two different methods exhibited remarkable radical scavenging effect against ABTS˙+ and the ability was positively correlated with the increasing concentration of the SCPs (0.0625–5.0 mg mL−1). At the concentration of 5.0 mg mL−1, the ABTS˙+ radical scavenging effect of SCP-UAEE, SCP-HWE, and Vc reached 84.02%, 80.70%, and 97.48%, respectively (Fig. 4B). Furthermore, SCP-UAEE and SCP-HWE also showed significant scavenging effects against hydroxyl radicals in a dose-dependent manner (Fig. 4C). Similarly, the scavenging activities towards the hydroxyl radicals of SCP-UAEE and SCP-HWE reached 75.48% and 63.55% at the concentration of 5.0 mg mL−1, respectively. Compared to the capacity of Vc (95.02%), the SCPs showed a relatively weak but significant potential of scavenging ability of hydroxyl radicals.
Many factors affect the antioxidant capacity of polysaccharides, including their monosaccharide compositions, structural configurations, molecular weights, and chain conformation.14,44 Generally, polysaccharides with lower molecular weights and higher contents of uronic acid have been found to possess stronger antioxidant capacity. In the present study, SCP-UAEE exhibited a better antioxidant activity than SCP-HWE in the three test systems, which was consistent with the degradation of molecular weight of the polysaccharide extracted by UAEE. Similar results were also confirmed in the previous study.45
3.5. Neuroprotective activity towards HT22 cells
Hydrogen peroxide (H2O2) is an endogenous source of hydroxyl free radicals commonly produced in cells. It can easily cross the cytomembrane and generate powerful radicals including singlet oxygen and hydroxyl radical. Thus, H2O2 is normally regarded as an inducer of oxidatively-damaged cells.46 A large number of studies have demonstrated that H2O2 can cause toxicity in various cells,47–49 including neurons. Emerging evidences have demonstrated that oxidative stress is one of critical causes for the progression of neurodegenerative diseases. Some plant polysaccharides, as excellent natural antioxidants, have exhibited potential protective effects on ROS-induced apoptosis in various neurodegenerative diseases.50 Herein, the HT22 cell model was employed to explore the protective effects of the SCPs on H2O2-induced oxidative impairment.
To determine the cytotoxic and neuroprotective effects of the SCPs on the HT22 cells, cell viability and release of lactate dehydrogenase were validated by the CCK8 assay and LDH assay, respectively. Water-soluble tetrazolium salt-8 (WST-8) is a compound similar to MTT and can be reduced by dehydrogenases in the mitochondria to produce orange-yellow formazan.51 In addition, WST-8 does not require further treatment as it produces a highly water-soluble formazan dye during the assay, making the CCK8 method more sensitive than the MTT assay.52 As shown in Fig. 5A, the results indicated that SCPs (10–200 μg mL−1, for 24 h) had no significant cytotoxicity. After 12 h of incubation with 400 μM H2O2, the cell viability decreased to 49.97% when compared with that of the untreated cells (P < 0.05, Fig. 5B). However, pretreatment with different concentrations of the SCPs for 12 h prior to H2O2 obviously reversed the death of HT22s cell in a dose-dependent manner (P < 0.05). On the other hand, the positive control (50 μM Trolox) herein also obviously protected the HT22 cells against H2O2-induced oxidative stress. These results indicated that the SCPs significantly increased the viability of HT22 cells exposed to H2O2.
Fig. 5. Neuroprotective effects of the SCPs on the cytotoxicity (A), viability in H2O2-induced HT22 cells (B), LDH release levels (C), and intracellular ROS accumulation (D). Different letters in each testing parameter represent the statistical significance among the groups (p < 0.05), n = 5.

LDH is a cytoplasmic enzyme that quickly releases into the cell culture media if the cell integrity is injured by oxidative stress.53 To some extent, LDH is another key indicator of cytotoxicity and the increased LDH activity in the surrounding environment is associated with cell necrosis.54 In this study, after the cells were exposed to H2O2 damage, LDH release in H2O2-treated cells was markedly higher than that in the control cells (P < 0.05). The pretreatment of the SCPs or Trolox significantly decreased the LDH levels when compared with the negative control group (P < 0.05) (Fig. 5C). The present studies correspond with the results reported by Hu et al., which revealed that the protein-rich extract from Bombyx batryticatus exhibited protective effects against H2O2-induced oxidative damage in PC12 cells.55 Similar results were also observed in the in vitro model in the case of different natural polysaccharides, such as Cantharellus cibarius,56Cantharellus cibarius,54 and Hemp seed.57
As a byproduct of oxidative phosphorylation, the overproduction of ROS may lead to breakage of the cell structure and damage the mitochondria.25 Oxidative stress is one of causative factors for neuronal cell death in neurodegenerative disease.58,59 Some natural polysaccharides, namely, Apios americana Medik flowers,48Sphallerocarpus gracilis50 as well as Morchella importuna,60 were verified as potent ROS scavengers in H2O2-induced neuroprotective cell models. In consideration of the superior antioxidant activities of the SCPs in vitro, we further evaluated whether the antioxidant of the SCPs was due to the modulation of intracellular ROS levels. As illustrated in Fig. 5D, the intracellular ROS levels stimulated by H2O2 were significantly inhibited by the SCPs in the concentration range of 50–200 μg mL−1 and Trolox in the concentration range of 50 μM in the HT22 cells, compared with that of the negative control group (P < 0.05).
These results indicated a significant neuroprotective effect of the SCPs on the HT22 cells against H2O2-induced cytotoxicity, which might be associated with its strong antioxidant activities.
4. Conclusion
Ultrasonic-assisted enzymatic extraction was optimized to extract the SCPs by RSM. The results showed that the optimal conditions were: ultrasonication time of 41 min, ultrasonication power of 300 W, with solid–liquid ratio of 1 : 39 g mL−1, and cellulase concentration of 0.81%. The yield of the SCPs with UAEE extraction yield was up to 14.63%, which has increased by about 68.54% compared with that of the conventional HWE extraction method. The results also showed that the extraction methods significantly affected the contents of polysaccharides, molecular weights and molar percentage of the constituent monosaccharides of SCPs. The SEM analysis illustrated that the microstructures of the two SCPs were notably different, which was likely due to a drastic effect on the cell walls, causing them to break down the cell structure by UAEE extraction. In addition, the two SCPs exhibited strong antioxidant activity against DPPH, ABTS, and hydroxyl radicals in vitro. It was also noted that the SCPs extracted by UAEE possessed the potential to protect the HT22 cells against H2O2-induced oxidative damage, and effectively ameliorate the cell viability, reduce extracellular LDH release, as well as decrease the levels of intracellular ROS in the injured cells. To conclude, these results proved that SCPs could be considered as a useful source of natural antioxidants and neuroprotective agents in functional foods. Therefore, further research on purification, structural characterization, and biological properties could be conducted.
Conflicts of interest
The authors declare no identifiable conflict of interest.
Supplementary Material
Acknowledgments
The work described in this article is supported by the National Key Research and Development Program of China (Project No. 2017YFC1601806) and Science and Technology Support Plan of Jiangsu Province (BE2018316).
Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01581d
References
- Bang S. Lee C. Ryu J. Li W. Koh Y.-S. Jeon J.-H. Lee J. Shim S. H. Arch. Pharmacal Res. 2018;41:823–829. doi: 10.1007/s12272-018-1054-y. [DOI] [PubMed] [Google Scholar]
- Bang S. Chae H.-S. Lee C. Choi H. G. Ryu J. Li W. Lee H. Jeong G.-S. Chin Y.-W. Shim S. H. J. Agric. Food Chem. 2017;65:6152–6157. doi: 10.1021/acs.jafc.7b02657. [DOI] [PubMed] [Google Scholar]
- Han J. M. Lee E. K. Gong S. Y. Sohng J. K. Kang Y. J. Jung H. J. J. Ethnopharmacol. 2019;231:10–18. doi: 10.1016/j.jep.2018.11.003. [DOI] [PubMed] [Google Scholar]
- Ohno N. Miura N. N. Nakajima M. Yadomae T. Biol. Pharm. Bull. 2000;23:866–872. doi: 10.1248/bpb.23.866. [DOI] [PubMed] [Google Scholar]
- Harada T. Miura N. Adachi Y. Nakajima M. Yadomae T. Ohno N. Biol. Pharm. Bull. 2002;25:931–939. doi: 10.1248/bpb.25.931. [DOI] [PubMed] [Google Scholar]
- Hong K. B. Hong S.-Y. Joung E. Y. Kim B. H. Bae S.-H. Park Y. Suh H. J. Int. J. Med. Mushrooms. 2015;17:965–975. doi: 10.1615/IntJMedMushrooms.v17.i10.60. [DOI] [PubMed] [Google Scholar]
- Tada R. Harada T. Nagi-Miura N. Adachi Y. Nakajima M. Yadomae T. Ohno N. Carbohydr. Res. 2007;342:2611–2618. doi: 10.1016/j.carres.2007.08.016. [DOI] [PubMed] [Google Scholar]
- Kodani S. Hayashi K. Hashimoto M. Kimura T. Dombo M. Kawagishi H. Biosc. Biotech. Biochem. 2009;73:228–229. doi: 10.1271/bbb.80595. [DOI] [PubMed] [Google Scholar]
- Woodward S. Sultan H. Y. Barrett D. K. Pearce R. B. J. Gen. Microbiol. 1993;139:153–159. doi: 10.1099/00221287-139-1-153. [DOI] [Google Scholar]
- Yoshikawa K. Kokudo N. Hashimoto T. Yamamoto K. Inose T. Kimura T. Biol. Pharm. Bull. 2010;33:1355–1359. doi: 10.1248/bpb.33.1355. [DOI] [PubMed] [Google Scholar]
- Yamamoto K. Kimura T. Sugitachi A. Matsuura N. Biol. Pharm. Bull. 2009;32:259–263. doi: 10.1248/bpb.32.259. [DOI] [PubMed] [Google Scholar]
- Hida T. H. Kawaminami H. Ishibashi K.-i. Miura N. N. Adachi Y. Ohno N. Int. J. Med. Mushrooms. 2013;15:525–538. doi: 10.1615/IntJMedMushr.v15.i6.20. [DOI] [PubMed] [Google Scholar]
- Wang K. Li M. Wen X. Chen X. He Z. Ni Y. Int. J. Biol. Macromol. 2018;114:1056–1063. doi: 10.1016/j.ijbiomac.2018.03.145. [DOI] [PubMed] [Google Scholar]
- Chen G. Bu F. Chen X. Li C. Wang S. Kan J. Int. J. Biol. Macromol. 2018;112:656–666. doi: 10.1016/j.ijbiomac.2018.02.013. [DOI] [PubMed] [Google Scholar]
- Chen Y. Yao F. Ming K. Wang D. Hu Y. Liu J. Molecules. 2016;21:1705. doi: 10.3390/molecules21121705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D. Y. Wan Y. Xu J. Y. Wu G. H. Li L. Yao X. H. Carbohydr. Polym. 2016;137:473–479. doi: 10.1016/j.carbpol.2015.11.016. [DOI] [PubMed] [Google Scholar]
- Raza A. Li F. Xu X. Tang J. Int. J. Biol. Macromol. 2017;94:335–344. doi: 10.1016/j.ijbiomac.2016.10.033. [DOI] [PubMed] [Google Scholar]
- Qian S. Fang X. Dan D. Diao E. Lu Z. RSC Adv. 2018;8:42145–42152. doi: 10.1039/C8RA06449K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. Gong G. Zhang J. Jia S. Li F. Wang Y. Wu S. Carbohydr. Polym. 2014;110:278–284. doi: 10.1016/j.carbpol.2014.03.040. [DOI] [PubMed] [Google Scholar]
- Pu J.-B. Xia B.-H. Hu Y.-J. Zhang H.-J. Chen J. Zhou J. Liang W.-Q. Xu P. Molecules. 2015;20:22220–22235. doi: 10.3390/molecules201219837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L. B. Li T. F. Liu F. C. Liu D. W. Xu Y. Q. Yang Y. Zhao Y. B. Wei H. Int. J. Biol. Macromol. 2019;126:846–856. doi: 10.1016/j.ijbiomac.2018.12.232. [DOI] [PubMed] [Google Scholar]
- Liao N. Zhong J. Ye X. Lu S. Wang W. Zhang R. Xu J. Chen S. Liu D. LWT–Food Sci. Technol. 2015;60:1113–1121. doi: 10.1016/j.lwt.2014.10.009. [DOI] [Google Scholar]
- Wang P. Liao W. Fang J. Liu Q. Yao J. Hu M. Ding K. Carbohydr. Polym. 2014;110:142–147. doi: 10.1016/j.carbpol.2014.03.060. [DOI] [PubMed] [Google Scholar]
- Zhou L. Liao W. Chen X. Yue H. Li S. Ding K. Carbohydr. Polym. 2018;195:643–651. doi: 10.1016/j.carbpol.2018.05.022. [DOI] [PubMed] [Google Scholar]
- Li Z. P. Chen X. Zhang Y. F. Liu X. Wang C. Y. Teng L. S. Wang D. Int. J. Biol. Macromol. 2019;121:29–37. doi: 10.1016/j.ijbiomac.2018.09.216. [DOI] [PubMed] [Google Scholar]
- Hu S. Wang D. Zhang J. Du M. Cheng Y. Liu Y. Zhang N. Wang D. Wu Y. Int. J. Mol. Sci. 2016;17:133. doi: 10.3390/ijms17020133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. Y. Xue Y. T. Carbohydr. Polym. 2019;206:179–186. doi: 10.1016/j.carbpol.2018.10.093. [DOI] [PubMed] [Google Scholar]
- Michel D. Gilles K. A. Hamilton J. K. Rebers P. A. Smith F. Anal. Chem. 1956;28:350–356. doi: 10.1021/ac60111a017. [DOI] [Google Scholar]
- Bradford M. M. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Blumenkrantz N. Asboe-Hansen G. Anal. Biochem. 1973;54:484–489. doi: 10.1016/0003-2697(73)90377-1. [DOI] [PubMed] [Google Scholar]
- Cai G. Liu Y. Li X. Lu J. J. Agric. Food Chem. 2019;67:8029–8034. doi: 10.1021/acs.jafc.9b03234. [DOI] [PubMed] [Google Scholar]
- Hajji M. Hamdi M. Sellimi S. Ksouda G. Laouer H. Li S. M. Nasri M. Carbohydr. Polym. 2019;206:380–388. doi: 10.1016/j.carbpol.2018.11.020. [DOI] [PubMed] [Google Scholar]
- Zhang T. Xiang J. Zheng G. Yan R. Min X. J. Funct. Foods. 2018;41:19–24. doi: 10.1016/j.jff.2017.12.028. [DOI] [Google Scholar]
- Cheng Z. Zhang Y. Song H. Zhou H. Zhong F. Hu H. Feng Y. Int. J. Biol. Macromol. 2016;93:369–380. doi: 10.1016/j.ijbiomac.2016.08.059. [DOI] [PubMed] [Google Scholar]
- Wu D.-T. Liu W. Han Q.-H. Wang P. Xiang X.-R. Ding Y. Zhao L. Zhang Q. Li S.-Q. Qin W. Molecules. 2019;24:2817. doi: 10.3390/molecules24152817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T. BioMed Res. Int. 2013;2013:982317. doi: 10.1155/2013/982317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyen Uddin Pk M. Islam M. S. Pervin R. Dutta S. Talukder R. I. Rahman M. PloS One. 2019;14:e0209371. doi: 10.1371/journal.pone.0209371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin C. Fan X. Fan Z. Shi D. Gao H. Int. J. Biol. Macromol. 2018;111:446–454. doi: 10.1016/j.ijbiomac.2018.01.007. [DOI] [PubMed] [Google Scholar]
- Guo H. Yuan Q. Fu Y. Liu W. Su Y.-H. Liu H. Wu C.-Y. Zhao L. Zhang Q. Lin D.-R. Chen H. Qin W. Wu D.-T. Polymers. 2019;11:215. doi: 10.3390/polym11020215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Thi Nhu N. Oh Y.-K. Lee Y.-J. Lee Y.-C. Int. J. Mol. Sci. 2018;19 doi: 10.3390/ijms19051487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Z. Han X. Li J. Food Chem. 2011;127:1273–1279. doi: 10.1016/j.foodchem.2011.01.083. [DOI] [PubMed] [Google Scholar]
- Wu Z. Li H. Wang Y. Yang D. Tan H. Zhan Y. Yang Y. Luo Y. Chen G. Int. J. Biol. Macromol. 2019;135:1151–1161. doi: 10.1016/j.ijbiomac.2019.06.020. [DOI] [PubMed] [Google Scholar]
- Liu X. Hou R. L. Xu K. Q. Chen L. Wu X. P. Lin W. X. Zheng M. F. Fu J. S. Int. J. Biol. Macromol. 2019;123:468–476. doi: 10.1016/j.ijbiomac.2018.11.069. [DOI] [PubMed] [Google Scholar]
- Yang B. Wu Q. J. Luo Y. X. Yang Q. Chen G. J. Wei X. Y. Kan J. Q. Int. J. Biol. Macromol. 2019;134:631–644. doi: 10.1016/j.ijbiomac.2019.05.079. [DOI] [PubMed] [Google Scholar]
- Zhao X. Li B. Xue C. Sun L. J. Appl. Phycol. 2011;24:295–300. doi: 10.1007/s10811-011-9679-y. [DOI] [Google Scholar]
- Zhao Z. Y. Luan P. Huang S. X. Xiao S. H. Zhao J. Zhang B. Gu B. B. Pi R. B. Liu J. CNS Neurosci. Ther. 2013;19:163–169. doi: 10.1111/cns.12044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai B. N. Wan P. Chen H. Chen D. K. Chen X. Sun H. L. Pan J. Y. Int. J. Biol. Macromol. 2019;124:246–254. doi: 10.1016/j.ijbiomac.2018.11.154. [DOI] [PubMed] [Google Scholar]
- Chu Q. Chen M. Song D. X. Li X. X. Yang Y. Y. Zheng Z. H. Li Y. L. Liu Y. Y. Yu L. S. Hua Z. Zheng X. D. Int. J. Biol. Macromol. 2019;123:1115–1124. doi: 10.1016/j.ijbiomac.2018.11.078. [DOI] [PubMed] [Google Scholar]
- Guo Q. W. Xu L. L. Chen Y. Ma Q. Q. Santhanam R. K. Xue Z. H. Gao X. D. Chen H. X. Carbohydr. Polym. 2019;208:161–167. doi: 10.1016/j.carbpol.2018.12.049. [DOI] [PubMed] [Google Scholar]
- Guo J. Liu Q. Wang C. B. Shi J. P. Zhang J. Int. J. Biol. Macromol. 2019;129:1133–1139. doi: 10.1016/j.ijbiomac.2018.11.208. [DOI] [PubMed] [Google Scholar]
- Shu Q. Wei T. Lu H. Niu Y. Chen Q. Appl. Microbiol. Biotechnol. 2020;104:5053–5064. doi: 10.1007/s00253-020-10561-8. [DOI] [PubMed] [Google Scholar]
- Yang H. Lopina S. T. DiPersio L. P. Schmidt S. P. J. Mater. Sci. Mater. Med. 2008;19:1991–1997. doi: 10.1007/s10856-007-3278-0. [DOI] [PubMed] [Google Scholar]
- Akanda M. R. Kim M. J. Kim I. S. Ahn D. Tae H. J. Rahman M. M. Park Y. G. Seol J. W. Nam H. H. Choo B. K. Park B. Y. Cell. Mol. Neurobiol. 2018;38:497–505. doi: 10.1007/s10571-017-0496-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye P. Li P. Yang W. Zhao Y. Zhao Y. Sun K. Wang B. Chen Y. Mar. Drugs. 2019;17:188. doi: 10.3390/md17030188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M. Liu Y. He L. Yuan X. Peng W. Wu C. Oxid. Med. Cell. Longevity. 2019;2019:7897584. doi: 10.1155/2019/7897584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemieszek M. K. Nunes F. M. Cardoso C. Marques G. Rzeski W. Carbohydr. Polym. 2018;197:598–607. doi: 10.1016/j.carbpol.2018.06.038. [DOI] [PubMed] [Google Scholar]
- Wen Z. S. Xue R. Du M. Tang Z. Xiang X. W. Zheng B. Qu Y. L. Int. J. Biol. Macromol. 2019;135:203–211. doi: 10.1016/j.ijbiomac.2019.05.082. [DOI] [PubMed] [Google Scholar]
- Querfurth H. W. LaFerla F. M. N. Engl. J. Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- Grimm A. Eckert A. J. Neurochem. 2017;143:418–431. doi: 10.1111/jnc.14037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong C. Li Q. Chen C. Chen Z. Huang W. Biomed. Pharmacother. 2016;83:569–576. doi: 10.1016/j.biopha.2016.07.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.