

Abstract
Cell signals for growth factors depend on the mechanical properties of the extracellular matrix (ECM) surrounding the cells. Microtubule acetylation is involved in the transforming growth factor (TGF)-β-induced myofibroblast differentiation in the soft ECM. However, the mechanism of activation of α-tubulin acetyltransferase 1 (α-TAT1), a major α-tubulin acetyltransferase, in the soft ECM is not well defined. Here, we found that casein kinase 2 (CK2) is required for the TGF-β-induced activation of α-TAT1 that promotes microtubule acetylation in the soft matrix. Genetic mutation and pharmacological inhibition of CK2 catalytic activity specifically reduced microtubule acetylation in the cells cultured on a soft matrix rather than those cultured on a stiff matrix. Immunoprecipitation analysis showed that CK2α, a catalytic subunit of CK2, directly bound to the C-terminal domain of α-TAT1, and this interaction was more prominent in the cells cultured on the soft matrix. Moreover, the substitution of alanine with serine, the 236th amino acid located at the C-terminus, which contains the CK2-binding site of α-TAT1, sig-nificantly abrogated the TGF-β-induced microtubule acetylation in the soft matrix, indicating that the successful binding of CK2 and the C-terminus of α-TAT1 led to the phosphorylation of serine at the 236th position of amino acids in α-TAT1 and regulation of its catalytic activity. Taken together, our findings provide novel insights into the molecular mechanisms underlying the TGF-β-induced activation of α-TAT1 in a soft matrix.
Keywords: α-tubulin acetyltransferase 1, Casein kinase 2, Extracellular matrix, Microtubule acetylation, TGF-β,
INTRODUCTION
Microtubules are composed of α/β-tubulin heterodimers that coordinate diverse cellular functions including mitosis, motility, polarization, and intracellular transport (1). Being highly dynamic structures, microtubules continuously undergo polymerization and depolymerization via interactions with various motor and microtubule-associated proteins. In addition, post-translational modifications including acetylation, polyglycylation, polyglutamylation, and tyrosination/detyrosination of microtubules are essential modulators of microtubule-related functions (2). Acetylation of microtubules occurs at the lysine 40 (K40) residue of α-tubulin, a marker for stable and long-lived microtubules, that regulates cellular cargo transport, gene expression, migration, and adhesion (2, 3). The level of tubulin acetylation is regulated by the balance among the activities of histone deacetylase 6 (HDAC6), NAD-dependent deacetylase sirtuin 2 (SIRT2), and α-tubulin acetyltransferase 1 (α-TAT1/MEC-17), a major acetyltransferase (4). Since the discovery of α-TAT1, several studies have focused on clarifying its structural characterization to understand its fundamental role (5). Moreover, lysine 56 and 146 (K56 and 146) are close to the acetyl-CoA binding site, suggesting that these residues might act as mediators for the transfer of acetyl groups to α-tubulin (6). Although the N-terminal domain of α-TAT1 as a catalytic subunit is comparatively well known, the role of its C-terminal domain remains elusive. Phosphorylation at serine 237 (S237) of α-TAT1 by TGF-β-activated kinase 1 (TAK1) increases the acetyltransferase activity upon TGF-β stimulation (7). Additionally, adenosine mo-nophosphate-activated protein kinase phosphorylates α-TAT1 under oxidative stress, resulting in high levels of microtubule acetylation (8). These results imply that α-TAT1 activation may be controlled via phosphorylation by an upstream kinase.
Microtubule acetylation is closely associated with various diseases, including cancer, fibrotic diseases, and neurodegenerative diseases, such as Parkinson’s and Alzheimer’s diseases (9-11). Proinflammatory cytokines are important in the pathophysiology of various diseases (12). Interestingly, TGF-β, reactive oxygen species, and lipopolysaccharides act as inducers of inflammation signaling and commonly increase the acetylation of microtubules (7, 8, 13). Incident increase in microtubule acetylation by inflammation signaling may offer a fertile environment to break out of pathogenesis, resulting in the loss of neuronal cell structure and function. Therefore, targeting microtubule acetylation induced by inflammatory signals in normal tissues may be a new strategy for overcoming neurodegenerative diseases.
Casein kinase 2 (CK2) is ubiquitously expressed and highly conserved in eukaryotic cells. It is composed of two catalytic subunits (α and α’) and two regulatory β-subunits that form a hetero-tetrameric structure (14). Accumulating evidence suggests that CK2 is involved in a wide variety of cellular processes including cell cycle, apoptosis, and signal transduction (15). CK2 is also associated with the regulation of microtubule reorganization. CK2 catalytic subunits (α and α’) bind to tubulin and modulate microtubule dynamics and stabilization (16). Moreover, CK2 directly interacts with HDAC6 and phosphorylates S458, resulting in the activation of its deacetylase activity (17). Although the role of CK2 in microtubule function remains controversial, these findings suggest that CK2 is involved in the regulation of microtubule cytoskeletal dynamics.
We have previously reported that microtubule acetylation of fibroblasts cultured on a soft matrix that mimics normal tissue rigidity is specifically increased upon TGF-β stimulation. However, the molecular mechanism by which α-TAT1 is activated under soft matrix conditions remains unknown. In this study, we investigated the α-TAT1 activation mechanism by using a soft matrix that mimics the mechanical properties of normal tissues. We found that the C-terminus of α-TAT1 interacts with CK2α. Point mutation of putative phosphorylation sites by CK2 further revealed that the serine residue in the 236th amino acid likely regulates α-TAT1 activation via phosphorylation by CK2 in the soft matrix. Collectively, our results suggested that CK2 targeting can be considered a key strategy for microtubule acetylation-targeted therapies for diseases affecting soft tissues such as the brain.
RESULTS
Inhibition of CK2 impairs microtubule acetylation induced by TGF-β on the soft matrix
To investigate the molecular mechanisms by which TGF-β can induce microtubule acetylation of fibroblasts in a soft matrix, we first screened the kinases involved in the TGF-β-induced microtubule acetylation of fibroblasts cultured on a soft matrix using diverse pharmacological inhibitors and found that the TGF-β-induced microtubule acetylation was effectively inhibited by 4,5,6,7-tetrabromobenzotriazole (TBB), a CK2 inhibitor (Fig. 1A).
Fig. 1.

Casein kinase 2 (CK2) is required for the transforming growth factor (TGF)-β-induced microtubule acetylation on the soft matrix. (A, B) Mouse embryonic fibroblasts (MEFs) were seeded on fibronectin (10 μg/ml)-coated ∼0.5 kPa polyacrylamide gel (PAG) in a plastic culture dish with or without TGF-β (2 ng/ml), 4,5,6,7-tetrabromobenzotriazole (TBB) (10 μM), and blebbistatin (10 μM). After 8 h of incubation, protein lysates obtained from individual conditions were subjected to western blotting with the indicated antibodies. (C, D) Immunocytochemistry of MEFs seeded on fibronectin-coated ∼0.5 kPa PAG and coverslips with or without TGF-β (2 ng/ml) and TBB (10 μM) for 8 h. Graphs show the projected cell area of 30 cells randomly incubated on ∼0.5 kPa PAGs and coverslips. Statistical significance was determined via one-way analysis of variance (ANOVA) with Tukey’s post hoc test. Projected cell area; one-way ANOVA, F3,116 = 75.84. ***P < 0.005, n.s. not significant. Scale bar, 25 μm. For each value, measurements were made on 30 randomly photographed cells. (E) Wild-type (WT) and CK2α knockout (KO) MEFs were seeded on 0.5 kPa PAG in a plastic culture dish upon TGF-β stimulation for 8 h. (F) Measurement of projected cell area of WT and CK2α KO MEFs on ∼0.5 kPa PAGs (n = 30, each group). Statistical signifi-cance was determined via one-way ANOVA with Tukey’s post hoc test. Projected cell area; one-way ANOVA, F3,116 = 42.02. ***P < 0.005, n.s. not significant. For each value, measurements were made on 30 randomly photographed cells.
To test the inhibitory effect of TBB on microtubule acetylation based on the stiffness of the matrix, cells were treated with TBB and TGF-β under various matrix mechanical conditions. As shown in Fig. 1B, the inhibition of CK2 catalytic activity by TBB notably downregulated the level of microtubule acetylation in cells cultured on a 0.5 kPa matrix. Cells cultured with blebbistatin, a myosin inhibitor, showed significant inhibition of microtubule acetylation after TBB treatment. The phosphorylation status of the signal transducer and activator of tran-scription 3 (STAT3) was determined to confirm the CK2 catalytic activity induced by TGF-β (15). Moreover, the formation of acetylated-α-tubulin along with elongated cells induced by TGF-β was inhibited by TBB treatment on 0.5 kPa polyacrylamide hydrogel (PAG) (Fig. 1C). However, it only changed the cell morphology, but there was no change in the level of acetylated α-tubulin on the stiff matrix (Glass) (Fig. 1D). These results suggest that the requirement of CK2 for microtubule acetylation is dependent on the tensional status of cell-matrix interactions.
To verify the functional role of CK2 in the TGF-β-induced microtubule acetylation in the soft matrix, we generated a CK2α (catalytic subunit of CK2) knockout cell line (CK2α KO) using the clustered regularly interspaced palindromic repeat-caspase 9 (CRISPR-Cas9) system (Supplementary Fig. 1A-C). In the wild-type (WT) cells, microtubule acetylation is markedly downregulated on the soft matrix upon TGF-β stimulation in CK2α KO cells compared with that on the stiff matrix (Fig. 1E). Furthermore, CK2α KO mouse embryonic fibroblasts (MEFs) showed a significant reduction in the projected cell area upon TGF-β stimulation of the soft matrix (Fig. 1F). Taken together, these results show that the catalytic activity of CK2 is required for the TGF-β-induced microtubule acetylation in cells cultured on a soft matrix.
CK2 specifically interacts with α-TAT1 under low tension status
To provide insight into the underlying mechanism by which CK2 regulates microtubule acetylation, we first performed immunoprecipitation (IP) experiments using the cells transfected with Flag-CK2α and green fluorescent protein (GFP)-α-TAT1 to demonstrate the association between CK2α and α-TAT1. Fig. 2A and B show that CK2α can interact with α-TAT1. As shown in Fig. 1B, the downregulation of microtubule acetylation by CK2α inhibitor was predominant in cells on the soft matrix (0.5 kPa) and under low tension status of the cell-matrix interaction (blebbistatin) (Fig. 1B). We examined whether the binding of CK2α and α-TAT1 is dependent on the tensional status of the cell. Compared with mock-treated cells, the binding of CK2α and α-TAT1 was predominant under blebbistatin treatment (Fig. 2C). Cellular localization of α-TAT1 and CK2α by immunostaining also showed that Flag-CK2α was dispersed in the cytosol, whereas α-TAT1 was localized in the tubules and its colocalization was rarely partial. Interestingly, both proteins in the cells cultured with blebbistatin exhibited colocalization with an elongated cell axis (Fig. 2D). Taken together, these results indicate that the interaction of CK2α and α-TAT1 occurs under the low tension status of cells, such as in the soft matrix.
Fig. 2.

CK2α strongly binds to α-tubulin acetyltransferase 1 (α-TAT1) on the soft matrix. (A, B) Co-immunoprecipitation (Co-IP) assay was performed to analyze the interaction between Flag-CK2α and green fluorescent protein (GFP)-α-TAT1 in HEK293T cells. (C) Co-IP assay was performed to analyze the interaction between Flag-CK2α and GFP-α-TAT1 with or without blebbistatin in WT MEFs. (D) Cellular localization of Flag-CK2α and GFP-α-TAT1 in MEFs with or without blebbistatin was observed via immunocytochemistry using anti-FLAG antibodies. White arrows indicate the colocalization of Flag-CK2α and GFP-α-TAT1. Scale bars: 25 μm and 5 μm (cropped image). Graphs show the fluorescence intensities of Flag-CK2α (red) and GFP-α-TAT1 (green).
CK2α binds to the C-terminal domain of α-TAT1 to regulate its acetyltransferase activity
Next, we generated truncated mutants of α-TAT1 to dissect the functional domain of the α-TAT1-CK2α interaction (Fig. 3A). For α-TAT1, we constructed two truncated mutants: one was the C-terminal-deleted mutant (GFP-α-TAT1DC) that retained its catalytic activity, and the other was a catalytic domain-deleted mutant that only had a C-terminal domain (GFP-α-TAT1DN). Co-immunoprecipitation (Co-IP) assay showed that CK2α specifically interacted with the C-terminal domain of α-TAT1 (Fig. 3B).
Fig. 3.

C-terminal domain of α-TAT1 is essential for the regulation of its acetyltransferase activity. (A) Schematic representation of GFP-α-TAT1WT (1-421 aa), GFP-α-TAT1DC (1-211 aa) and GFP-α-TAT1DN (190-421 aa). (B) HEK293T cells were transfected with the indicated plasmids, and Co-IP was performed with anti-mouse IgG and anti-Flag antibodies. (C) Co-IP assay shows the binding affinities of Flag-CK2α and GFP-α-TAT1 by overexpression of GFP-α-TAT1DC and GFP-α-TAT1DN. (D) α-TAT1 KO MEFs were transfected with GFP-C1, GFP-α-TAT1WT, GFP-α-TAT1DC, and GFP-α-TAT1DN and treated with TGF-β and bleb-bistatin. Lysates from cells were subjected to western blotting with the indicated antibodies. (E) WT MEFs were overexpressed with Myc-empty vector or α-TAT1DN-Myc and treated with blebbistatin and/or TGF-β. Next, the cells were fixed and immunostained with anti-Myc and anti-acetylated α-tubulin anti-bodies. The number of cells having dendrites whose lengths were more or less than two-times the lengths of their cell bodies was counted. For each value, measurements were made on 50 randomly photographed cells. White arrows indicate the long tail-like dendrites. Scale bar, 50 μm.
Given these results, we expected that the C-terminal domain of α-TAT1 (GFP-α-TAT1DN) would act as a dominant-negative inhibitor by competing with WT α-TAT1 for binding to CK2α. Transient overexpression of the GFP-α-TAT1DN construct sig-nificantly inhibited the interaction between α-TAT1WT and CK2α by approximately 60%, but the N-terminal domain of α-TAT1 (GFP-α-TAT1DC) did not affect this interaction (Fig. 3C).
The C-terminal domain of α-TAT1 functions as a regulatory region via phosphorylation of TAK1 (7). Thus, we speculated that the interaction of CK2α with the C-terminus of α-TAT1 enables the control of its acetyltransferase activity. GFP-α-TAT1WT, GFP- α-TAT1DC, and GFP-α-TAT1DN constructs were overexpressed in the α-TAT1 KO MEFs, and the level of microtubule acetylation induced by TGF-β was analyzed. Western blotting results showed that GFP-α-TAT1DC expression significantly reduced microtubule acetylation even though CK2 activity was increased by TGF-β (based on the level of STAT3 phosphorylation) (Fig. 3D), suggesting that the C-terminal domain of α-TAT1 controls its acetyltransferase activity via interaction with CK2α.
Next, we examined whether the overexpression of the C-terminal domain of α-TAT1 causes a decrease in microtubule acetylation upon TGF-β stimulation, as the C-terminal domain of α-TAT1 acts as a dominant-negative inhibitor by competing with α-TAT1 for binding to CK2α. Cells overexpressing the Myc-tagged C-terminal of α-TAT1 (α-TAT1DN-Myc) in WT MEFs showed shortened dendritic extensions with a lack of microtubule acetylation upon TGF-β stimulation compared to that in the mock-transfected cells under blebbistatin treatment (Fig. 3E). Taken together, these results indicate that the interaction of CK2α with the C-terminal domain of α-TAT1 and is crucial for the regulation of α-TAT1 acetyltransferase activity by TGF-β in the soft matrix.
Phosphorylation of α-TAT1 at S236 by CK2 is essential for the acetyltransferase activity
We found that the C-terminus of α-TAT1 contains various putative phosphorylation sites (Supplementary Fig. 2). We attempted to determine the putative phosphorylation sites that are critical for the regulation of microtubule acetylation on the soft matrix. First, we divided the C-terminus of α-TAT1 into three clusters (Cluster 1: 315-421 aa, Cluster 2: 261-314 aa, and Cluster 3: 211-260 aa) based on the closely located putative phosphorylation sites and generated truncated mutants (Fig. 4A). Although the level of microtubule acetylation was decreased in α-TAT1 KO MEFs expressing all truncated mutants compared with that observed in WT α-TAT1-expressing cells, the expression levels of GFP-α-TAT1DC1, and GFP-α-TAT1DC1,2 likely increased microtubule acetylation by TGF-β stimulation. Notably, overexpression of GFP-α-TAT1DC(DC1-3) in α-TAT1 KO MEFs failed to increase microtubule acetylation (Fig. 4A). Based on this result, we expected that the putative phosphorylation sites in Cluster 3 might be responsible for the regulation of α-TAT1 acetyltransferase activity induced by TGF-β under soft matrix conditions.
Fig. 4.
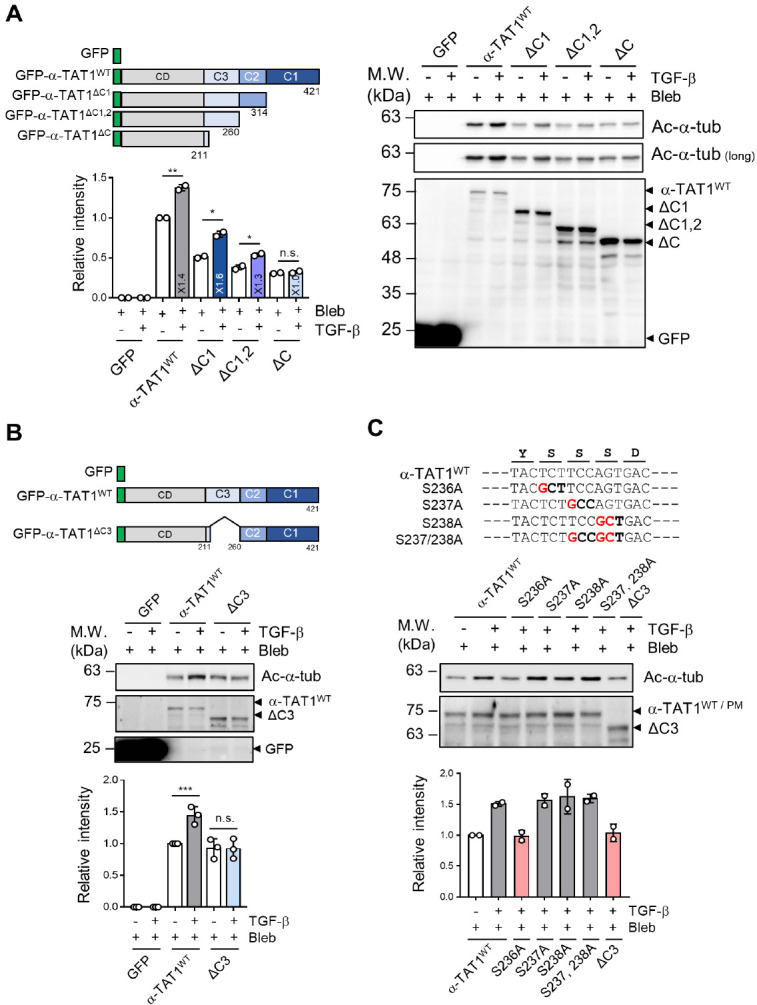
Phosphorylation of α-TAT1 at S236 regulates the TGF-β-induced microtubule acetylation. (A, B) The C-terminal domain (190-421 aa) is divided into three clusters, and each cluster and mutants with deleted Cluster 3 region are indicated. α-TAT1 KO MEFs were transfected with the indicated plasmids and treated with blebbistatin and/or TGF-β for 8 h. Graphs show the relative band intensity of acetylated-α-tubulin. Biological significance of the acetylated α-tubulin levels in α-TAT1 KO MEFs by TGF-β was assessed using Student’s t-test and compared with that of the control. *P < 0.05, **P < 0.01, ***P < 0.005, n.s. not significant. (C) α-TAT1 and its mutants, in which the conserved serine residues (S) (S236, S237, and S238) were mutated to alanine (A) (S236A, S237A, S238A and S237/238A). α-TAT1 KO MEFs were transfected with the indicated mutants, and the level of acetylated-α-tubulin upon TGF-β stimulation was determined.
After speculating that the overall reduction in microtubule acetylation in the samples that expressed every C-terminal deleted construct of α-TAT1 (Fig. 3D) was due to structural problems or low binding affinity with polymerized microtubules, we generated another α-TAT1 mutant by deleting the Cluster 3 region alone (GFP-α-TAT1DC3). Interestingly, α-TAT1 KO MEFs expressing GFP-α-TAT1DC3 did not induce microtubule acetylation upon TGF-β stimulation and did not affect the basal level of acetyltransferase activity (Fig. 4B). α-TAT1 with deleted Cluster 3 maintained its binding affinity to polymerized microtubules compared with the WT α-TAT1. However, GFP-α-TAT1DC1 dramatically reduced the microtubule binding affinity to ∼65%, which was consistent with a decrease in the basal level of microtubule acetylation when Cluster 1 was deleted (Supplementary Fig. 3). These results show that Cluster 3 (211-260 aa) of α-TAT1 may be required for the regulatory function of its acetyltransferase activity following TGF-β stimulation.
Cluster 3 had three putative serine phosphorylation sites (236, 237, and 238), which were highly conserved between the mouse and human species (Supplementary Fig. 4). To determine which serine residue is critical for the induction of microtubule acetylation by TGF-β, we constructed α-TAT1 mutants by substituting S236, S237, and S238 residues with alanine (S236A, S237A, and S238A, respectively) via site-directed muta-genesis. Western blotting results showed that TGF-β suppressed the acetylation of microtubules when they expressed the GFP-α-TAT1 S236A mutant instead of other alanine mutants (Fig. 4C), suggesting that phosphorylation of S236 of α-TAT1 promotes microtubule acetylation upon TGF-β stimulation.
DISCUSSION
α-TAT1 was first identified as a major acetyltransferase over a decade ago; however, the regulatory mechanism of its enzymatic activity is still not fully understood. In this study, we demonstrated that CK2α binds to the C-terminal domain of α-TAT1. Moreover, the substitution of serine at the 236th position, one of the serine residues that may be phosphorylated by CK2, at the C-terminus of α-TAT1 with alanine did not cause microtubule acetylation. Although we could not confirm whether CK2 directly phosphorylated S236 of α-TAT1, these results suggest the possibility of CK2 controlling the activity of α-TAT1.
CK2 is well known for its intracellular functions through the phosphorylation of proteins related to actin rearrangement (18, 19). For example, phosphorylation of cortactin by CK2 leads to the formation of an actin-related protein 2/3 complex, which regulates actin-based invadopodia formation for tumor progression (18). However, in the case of a soft matrix, actin-related signaling is not abundant due to the lack of integrin-mediated actomyosin activity; instead, microtubule lattices function as signaling mediators (20, 21). Meanwhile, the regulatory mechanism of microtubule dynamics in the soft matrix is well defined. In this study, we demonstrated that the requirement of CK2 for microtubule acetylation is limited to the soft matrix. Inhibition of CK2 activity with TBB caused changes in the cell morphology on stiff matrix but did not affect the level of microtubule acetylation, whereas treatment with TBB completely downregulated microtubule acetylation in the cells cultured on a soft matrix. Therefore, our results suggest that CK2 may be a critical regulator of microtubule dynamics by controlling α-TAT1 activity in a soft matrix.
α-TAT1 stochastically enters the microtubule ends to transfer the acetyl group to α-tubulin. Coombes et al. demonstrated that α-TAT1 enters the microtubule lumen through the microtubule ends and bends or breaks in the lattice by mechanical shearing (22). Additionally, the binding affinity between α-TAT1 and its binding sites is critical for determining its mobility within the microtubule lumen. Kalebic et al. suggested that the autoacetylation of α-TAT1 at K56, 146, 210, and 221 is essential for its catalytic activity (23). Although we propose that the acetyltransferase activity of α-TAT1 is also controlled by phosphorylation of S236 at the C-terminal domain, the mechanism by which the phosphorylation of α-TAT1 could increase microtubule acetylation remains elusive. We suppose that the phosphorylation of α-TAT1 by CK2 occurs in the cytosol or outside the microtubules considering the localization of CK2. Therefore, it could be possible that the phosphorylation of α-TAT1 allows it to increase the binding affinity with the microtubule lattice, which needs to be elucidated.
Although the role of microtubule acetylation in neurodegenerative diseases, such as Alzheimer’s and Parkinson’s diseases, remains controversial, the level of microtubule acetylation has been reported to increase in oxidative stress environments, such as neuroinflammation (11, 24, 25). In addition, this increase is required for breast cancer progression and involved in forming an active tumor microenvironment through the modulation of myofibroblast differentiation (26, 27). Microtubules function as a track where vesicle trafficking can be modulated. Acetylated microtubules preferentially recognize the kinesin-1 motor protein, thereby promoting the exocytosis of exosomes and various cytokines through polarized vesicle trafficking caused by the enhanced binding and motility of kinesin (28). These findings suggest the potential of modulating microtubule acetylation for breast cancer and neurodegenerative disease therapy. The pharmacological inhibitors of α-TAT1 have been identified, but they are yet to be fully explored (29). Therefore, α-TAT1 activity can be effectively controlled by using the CK2 inhibitor in a soft matrix mimicking the stiffening of normal tissues; this result is meaningful because it serves as a basis for developing a new bypass strategy to control microtubule acetylation.
Taken together, we have demonstrated through this research that the acetylation of microtubules increased in soft matrix can be effectively blocked through downregulation of CK2 activity. Thus, it is expected to present a new treatment method for malignant tumors or brain diseases such as Alzheimer’s disease occurring in a mechanical and soft matrix environment.
MATERIALS AND METHODS
Materials and methods are available in the supplemental material.
SUPPLEMENTARY MATERIALS
ACKNOWLEDGEMENTS
This research was supported by grants from the National Research Foundation of Korea (NRF) funded by the Korean government (MSIT) (NRF-2020R1A2C2007389) and the Chung-Ang University Research Grant (2020).
Footnotes
CONFLICTS OF INTEREST
The authors have no conflicting interests.
REFERENCES
- 1.Garcin C, Straube A. Microtubules in cell migration. Essays Biochem. 2019;63:509–520. doi: 10.1042/EBC20190016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Westermann S, Weber K. Post-translational modifications regulate microtubule function. Nat Rev Mol Cell Biol. 2003;4:938–947. doi: 10.1038/nrm1260. [DOI] [PubMed] [Google Scholar]
- 3.Balabanian L, Berger CL, Hendricks AG. Acetylated microtubules are preferentially bundled leading to enhanced kinesin-1 motility. Biophys J. 2017;113:1551–1560. doi: 10.1016/j.bpj.2017.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Janke C, Montagnac G. Causes and consequences of microtubule acetylation. Curr Biol. 2017;27:R1287–R1292. doi: 10.1016/j.cub.2017.10.044. [DOI] [PubMed] [Google Scholar]
- 5.Al-Bassam J, Corbett KD. Alpha-tubulin acetylation from the inside out. Proc Natl Acad Sci U S A. 2012;109:19515–19516. doi: 10.1073/pnas.1217594109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taschner M, Vetter M, Lorentzen E. Atomic resolution structure of human alpha-tubulin acetyltransferase bound to acetyl-CoA. Proc Natl Acad Sci U S A. 2012;109:19649–19654. doi: 10.1073/pnas.1209343109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shah N, Kumar S, Zaman N, et al. TAK1 activation of alpha-TAT1 and microtubule hyperacetylation control AKT signaling and cell growth. Nat Commun. 2018;9:1696. doi: 10.1038/s41467-018-04121-y.a82d270420fc4d0b91e4bda9d9eca3d2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mackeh R, Lorin S, Ratier A, et al. Reactive oxygen species, AMP-activated protein kinase, and the transcription cofactor p300 regulate alpha-tubulin acetyltransferase-1 (alphaTAT-1/MEC-17)-dependent microtubule hyperacetylation during cell stress. J Biol Chem. 2014;289:11816–11828. doi: 10.1074/jbc.M113.507400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ko P, Choi JH, Song S, et al. Microtubule acetylation controls MDA-MB-231 breast cancer cell invasion through the modulation of endoplasmic reticulum stress. Int J Mol Sci. 2021;22:6018. doi: 10.3390/ijms22116018.ffbed288f7f946da92cafb0e9b8fa9a5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.You E, Huh YH, Kwon A, et al. SPIN90 depletion and microtubule acetylation mediate stromal fibroblast activation in breast cancer progression. Cancer Res. 2017;77:4710–4722. doi: 10.1158/0008-5472.CAN-17-0657. [DOI] [PubMed] [Google Scholar]
- 11.Cappelletti G, Calogero AM, Rolando C. Microtubule acetylation: a reading key to neural physiology and degeneration. Neurosci Lett. 2021;755:135900. doi: 10.1016/j.neulet.2021.135900. [DOI] [PubMed] [Google Scholar]
- 12.Friedrich M, Pohin M, Powrie F. Cytokine networks in the pathophysiology of inflammatory bowel disease. Immunity. 2019;50:992–1006. doi: 10.1016/j.immuni.2019.03.017. [DOI] [PubMed] [Google Scholar]
- 13.Wang B, Rao YH, Inoue M, et al. Microtubule acetylation amplifies p38 kinase signalling and anti-inflammatory IL-10 production. Nat Commun. 2014;5:3479. doi: 10.1038/ncomms4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Graham KC, Litchfield DW. The regulatory beta subunit of protein kinase CK2 mediates formation of tetrameric CK2 complexes. J Biol Chem. 2000;275:5003–5010. doi: 10.1074/jbc.275.7.5003. [DOI] [PubMed] [Google Scholar]
- 15.Borgo C, D'Amore C, Sarno S, Salvi M, Ruzzene M. Protein kinase CK2: a potential therapeutic target for diverse human diseases. Signal Transduct Target Ther. 2021;6:183. doi: 10.1038/s41392-021-00567-7.4e2233794ece4f208ef8e20378e774a9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lim AC, Tiu SY, Li Q, Qi RZ. Direct regulation of microtubule dynamics by protein kinase CK2. J Biol Chem. 2004;279:4433–4439. doi: 10.1074/jbc.M310563200. [DOI] [PubMed] [Google Scholar]
- 17.Watabe M, Nakaki T. Protein kinase CK2 regulates the formation and clearance of aggresomes in response to stress. J Cell Sci. 2011;124:1519–1532. doi: 10.1242/jcs.081778. [DOI] [PubMed] [Google Scholar]
- 18.Markwell SM, Ammer AG, Interval ET, et al. Cortactin phosphorylation by casein kinase 2 regulates actin-related protein 2/3 complex activity, invadopodia function, and tumor cell invasion. Mol Cancer Res. 2019;17:987–1001. doi: 10.1158/1541-7786.MCR-18-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xavier CP, Rastetter RH, Blomacher M, et al. Phosphorylation of CRN2 by CK2 regulates F-actin and Arp2/3 interaction and inhibits cell migration. Sci Rep. 2012;2:241. doi: 10.1038/srep00241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rhee S, Jiang H, Ho CH, Grinnell F. Microtubule function in fibroblast spreading is modulated according to the tension state of cell-matrix interactions. Proc Natl Acad Sci U S A. 2007;104:5425–5430. doi: 10.1073/pnas.0608030104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bouchet BP, Akhmanova A. Microtubules in 3D cell motility. J Cell Sci. 2017;130:39–50. doi: 10.1242/jcs.189431. [DOI] [PubMed] [Google Scholar]
- 22.Coombes C, Yamamoto A, McClellan M, et al. Mechanism of microtubule lumen entry for the alpha-tubulin acetyltransferase enzyme alphaTAT1. Proc Natl Acad Sci U S A. 2016;113:7176–7184. doi: 10.1073/pnas.1605397113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kalebic N, Martinez C, Perlas E, et al. Tubulin acetyltransferase alphaTAT1 destabilizes microtubules independently of its acetylation activity. Mol Cell Biol. 2013;33:1114–1123. doi: 10.1128/MCB.01044-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cartelli D, Ronchi C, Maggioni MG, Rodighiero S, Giavini E, Cappelletti G. Microtubule dysfunction precedes transport impairment and mitochondria damage in MPP+ -induced neurodegeneration. J Neurochem. 2010;115:247–258. doi: 10.1111/j.1471-4159.2010.06924.x. [DOI] [PubMed] [Google Scholar]
- 25.Mao CX, Wen X, Jin S, Zhang YQ. Increased acetylation of microtubules rescues human tau-induced microtubule defects and neuromuscular junction abnormalities in Drosophila. Dis Model Mech. 2017;10:1245–1252. doi: 10.1242/dmm.028316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.You E, Ko P, Jeong J, et al. Dynein-mediated nuclear translocation of yes-associated protein through microtubule acetylation controls fibroblast activation. Cell Mol Life Sci. 2020;77:4143–4161. doi: 10.1007/s00018-019-03412-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boggs AE, Vitolo MI, Whipple RA, et al. Alpha-tubulin acetylation elevated in metastatic and basal-like breast cancer cells promotes microtentacle formation, adhesion, and invasive migration. Cancer Res. 2015;75:203–215. doi: 10.1158/0008-5472.CAN-13-3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reed NA, Cai D, Blasius TL, et al. Microtubule acetylation promotes kinesin-1 binding and transport. Curr Biol. 2006;16:2166–2172. doi: 10.1016/j.cub.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 29.Kwon A, Lee GB, Park T, et al. Potent small-molecule inhibitors targeting acetylated microtubules as anticancer agents against triple-negative breast cancer. Biomedicines. 2020;8:338. doi: 10.3390/biomedicines8090338.01ce05e80d3a4662bc42777ba35107d4 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.