

Abstract
Intratumoral heterogeneity is caused by genomic instability and phenotypic plasticity, but how these features co-evolve remains unclear. SOX10 is a neural crest stem cell (NCSC) specifier and candidate mediator of phenotypic plasticity in cancer. We investigated its relevance in breast cancer by immunophenotyping 21 normal breast and 1860 tumour samples. Nuclear SOX10 was detected in normal mammary luminal progenitor cells, the histogenic origin of most TNBCs. In tumours, nuclear SOX10 was almost exclusive to TNBC, and predicted poorer outcome amongst cross-sectional (p = 0.0015, hazard ratio 2.02, n = 224) and metaplastic (p = 0.04, n = 66) cases. To understand SOX10’s influence over the transcriptome during the transition from normal to malignant states, we performed a systems-level analysis of co-expression data, de-noising the networks with an eigen-decomposition method. This identified a core module in SOX10’s normal mammary epithelial network that becomes rewired to NCSC genes in TNBC. Crucially, this reprogramming was proportional to genome-wide promoter methylation loss, particularly at lineage-specifying CpG-island shores. We propose that the progressive, genome-wide methylation loss in TNBC simulates more primitive epigenome architecture, making cells vulnerable to SOX10-driven reprogramming. This study demonstrates potential utility for SOX10 as a prognostic biomarker in TNBC and provides new insights about developmental phenotypic mimicry—a major contributor to intratumoral heterogeneity.
Subject terms: Breast cancer, Tumour heterogeneity
Introduction
Effective management of triple-negative breast cancer (TNBC) remains a significant challenge worldwide. These tumours lack expression of oestrogen and progesterone receptors (ER/PR) and HER2, hence are not indicated for treatment with classical molecular-targeted agents. Chemotherapy remains the most reliable systemic treatment option, producing durable responses in ~60% of patients, while the other ~40% typically present with lung, liver and/or brain metastases within 5 years1–3. Second-line chemotherapy can temporarily stabilise metastatic disease but is rarely curative, so these patients endure a heavy treatment burden for no lasting benefit. Efforts to develop alternative treatments have been hampered by molecular and cellular variability between, and within, individual tumours. Intra-tumoural heterogeneity (ITH) directly increases the probability of relapse because it diversifies the substrate for clonal selection4–7. It has been proposed that to further improve the prognosis for TNBC patients, we need to develop agents that target the drivers of heterogeneity itself8.
TNBCs are characterised by defective DNA repair, mitotic spindle dysfunction, chromosomal aberrations, and a mutation rate around 13 times that of other breast tumours4,5. Genomic instability is a key driver of ITH, however only some cases can be explained by the selection of individual driver mutations9, and other sources of heterogeneity are coming to light10–12. For example, cellular heterogeneity is influenced by the differentiation state of the normal cellular precursor(s)13, which in TNBC is thought to be the luminal progenitor (LP) cell14–17.
ITH is also driven by phenotypic plasticity—the dynamic reprogramming of cell state in response to extrinsic stimuli10,11. Cancer cell state transitions can be de-differentiating (the loss of lineage commitment and acquisition of stem cell features) and/or trans-differentiating (assuming the state of another cell type)18. Compared to genomic and histogenic sources of ITH, how tumour cells invoke this capability is poorly understood, and yet potentially more ominous for the patient, as cell state transitions can be induced by treatment via heritable-epigenetic change. In controlled experimental conditions, drug-tolerant TNBC cell states can be averted by epigenome remodelling inhibitors19–23, suggesting these agents might reduce rates of relapse if used clinically8,11. However, epigenetic therapies have genome-wide effects, so our ability to use them rationally requires a deeper understanding of the epigenome-driven features of treatment-refractory human tumours8.
SOX10 is a transcription factor that was recently implicated in phenotypic plasticity in experimental models of TNBC24. It is first expressed in embryonic neural crest stem cells (NCSCs), where its self-reinforcing gene regulatory module facilitates multipotency and cell migration, orchestrating the embryo patterning process25–28. Once patterning is complete, SOX10 is silenced in all NCSC descendants except glial and melanocyte progenitors; and is nascently induced in ectoderm-derived epithelial progenitor cells of the salivary, lacrimal, and mammary glands29–33. In the mouse, Sox10 is an obligate requirement for mammary gland development. Its expression marks gland repopulating potential in the basal (myoepithelial) compartment, while Sox10+ luminal cells represent the committed progenitor fraction29. Functional studies have shown that Sox10 is one of several fate specifiers that regulates the equilibrium between mammary stem cell (MaSC) and LP states29,32.
In NCSCs where the genome is unmethylated and accessible, SOX10 facilitates a mesenchymal, migratory state, whereas its function in adult tissues is influenced by the tissue-specific growth factor milieu and lineage-specific DNA methylation. Remarkably, ectopic expression of SOX10 reprogrammed postnatal fibroblasts with multipotency and migration capabilities equivalent to NCSCs, providing they were also exposed to chromatin unpacking agents and early morphogens (DNA methylation and histone deacetylase inhibitors plus Wnt activation)34. This established that with the erasure of lineage-specific epigenetic marks and appropriate extrinsic cues, SOX10 can recreate its ‘default’ regulatory circuit and that this is sufficient to phenocopy NCSCs.
SOX10 expression in human breast cancer is associated with TN, basal-like, metaplastic and neural progenitor-like phenotypes4,35–39. In transgenic mouse mammary tumour cells, it promoted invasiveness, expression of mammary stem/progenitor, EMT and NCSC genes and the repression of epithelial differentiation genes24. These findings suggest that SOX10 could mediate de-differentiation in TNBC; but the relevance is unclear, particularly given there are no available inhibitors of SOX10 itself. We explored the significance of SOX10 in breast cancer development and progression by immunophenotyping histologically normal breast tissue, and large breast tumour sample cohorts. To understand its contribution to phenotypic plasticity and identify drivers of this capability, we performed systems-level analysis to map SOX10’s regulatory circuit in the broader TNBC transcriptional network.
Results
SOX10 is expressed in luminal progenitor cells of the human mammary gland
Functional studies have shown that SOX10 marks stem and luminal progenitor (LP) cells of the mouse mammary gland29,32, but its expression pattern in the human breast has not been established. Therefore, we performed immunohistochemical (IHC) analysis of 19 histologically normal reduction mammoplasty (RM) samples using a validated antibody (Supplementary Fig. 1a and Supplementary Table 1). SOX10 was detected in nuclei of ductal and lobular epithelia, with individual terminal ducto-lobular units (TDLUs) exhibiting either basal-restricted or combined baso-luminal expression (Fig. 1a). Compared to ducts, lobules were more likely to exhibit luminal compartment expression of SOX10 (Fig. 1b), consistent with a role in lobulogenesis. Indeed, TDLUs with basal-restricted SOX10 expressed high levels of luminal cytokeratins (CK)8/18, while TDLUs with dual-compartment SOX10 had low CK8/18. This was evident even in neighbouring structures of the same specimen (Fig. 1c and Supplementary Fig. 1b).
Fig. 1. SOX10 is expressed in basal and luminal progenitor cells of the human mammary gland.

a Representative SOX10 IHC analysis of reduction mammoplasty (RM) samples. Some terminal ducto-lobular units (TDLUs) had exclusive basal compartment expression (i) while others had expression in both basal and luminal compartments (ii). b (i) Analysis of SOX10 expression in ducts vs lobules of RM samples from 19 donors (whole sections). (ii) SOX10 expression in lobules was heterogeneous and more likely to occur in the luminal compartment (Mann–Whitney p = 0.011; n = 102 ducts and 102 lobules; median ± 95% confidence interval shown). c Representative immunofluorescent staining of SOX10 and CK8/18. Circled lobules and isolated cells (arrows) exhibited reciprocal expression of SOX10 (green) and CK8/18 (red) in structures with either (i) dual compartment (ii) or basal-restricted SOX10 expression. d IHC analysis of SOX10, c-kit, ER and Ki67 in serial RM sections. The three magnified regions represent major SOX10 staining patterns: (i) dual compartment, heterogeneous; (ii) dual compartment, homogeneous; and (iii) basal-restricted. Luminal SOX10 expression was directly associated with c-kit and inversely associated with ER, with no obvious relationship to Ki67 (e.g., cell cluster indicated with an arrow). e SOX10 mRNA levels in FACS-sorted human mammary epithelial cell (hMEC) subtypes15. Differentiation markers were analysed for comparison: basal markers CK14 and CK5; luminal progenitor (LP) markers KIT and ELF5; and markers enriched in mature luminal (ML) cells: CK18 and ESR1 (isolates with significantly different marker levels according to paired ANOVA tests are indicated and colour-coded: ****p < 0.00001; ***p < 0.0001; **p < 0.001). Data shown were means ± standard error of the mean from three donors. f Average methylation beta-values of SOX10 probes in FACS-sorted hMEC samples (DNAme), aligned with histone modification signals in a published ChIP-seq dataset42: H3K4me3, H3K27ac (activating) and H3K27me3 (repressive). Data were represented to scale on human chromosome 22. TSS transcription start site, UTR untranslated region. Indistinct = negative for CD45 (hematopoietic cells), CD31 (endothelia), CD140b (fibroblasts), EpCAM and CD49f (epithelia).
IHC analysis of serial sections showed SOX10+ luminal cells lacked ER and were positive for the LP marker c-Kit, with no obvious relationship to proliferation marker Ki67 (Fig. 1d). We also analysed SOX10 mRNA in a published dataset from FACS-sorted human mammary epithelial cells (hMECs)15. SOX10 levels were similar to established LP markers ELF5 and KIT: highest in EpCAM + /CD49f + LP cells, moderate in the EpCAM-/CD49f + basal compartment (myoepithelia and mammary stem cells (MaSCs)) and low in EpCAM + /CD49f- mature luminal (ML) cells (Fig. 1e).
SOX10 is epigenetically regulated in mouse mammary gland40,41, so we investigated this in human tissue. We isolated hMECs from two fresh RM samples using FACS with antibodies against CD49f and EpCAM, then performed high-density DNA methylation array profiling. SOX10 was hypomethylated in LP and basal samples (p < 1.0E−06; Fig. 1f). Consistently, analysis of hMEC chromatin immunoprecipitation sequencing (ChIP-seq) data from six independent RM samples42 showed the SOX10 locus is enriched with activating (H3K4me3, H3K27ac) and depleted of repressive H3K27me3 marks in LP and basal samples (Fig. 1f).
SOX10 is associated with poor clinical outcomes in TNBC
Analysis of TCGA, METABRIC and ICGC breast tumour datasets43–45 showed SOX10 mRNA is expressed almost exclusively in TNBC, with a bimodal distribution suggesting distinct SOX10 positive and negative (+/−) subgroups (Fig. 2a and Supplementary Fig. 2a). Consistent with other data39, SOX10 mRNA is highest amongst TNBCs classified as ‘basal-like, immune-suppressed’ (BLIS), though we noted that expression was heterogeneous amongst TNBC subtypes classified by gene expression profile (e.g. 23% of ‘basal-like, immune-activated’ (BLIA) TNBCs also had SOX10 levels in the top quartile; Supplementary Fig. 2b). In terms of genomic drivers of SOX10 expression in breast cancer, copy-number (CN) amplification or gain at the SOX10 locus was evident in ~20% of TNBCs (Fig. 2b) and was associated with higher mRNA levels in both METABRIC and TCGA datasets (Fisher’s Exact p ≤ 0.001). Analysis of TCGA HM450k methylation array data indicated that SOX10 is frequently hypomethylated in TNBC (Fig. 2b) and that this correlates strongly with expression (Fig. 2c and Figs. S2c, d), but does not extend to adjacent genes on chromosome 22 (Fig. 2d). Hence, like normal basal and luminal progenitor cells, gene-specific hypomethylation also underpins SOX10 expression in a subset of TNBCs, and in some cases, this appears to be reinforced by clonally selected CN gains.
Fig. 2. Expression of SOX10 in human breast cancer.

a Bimodal expression of SOX10 in TNBC compared to other breast cancers (nonTNBC) in the METABRIC cohort. b Frequency of copy-number alterations (CNAs) and DNA hypomethylation affecting SOX10 in TNBC and nonTNBC compared to the archetypal SOX10 + malignancy, melanoma (SKCM; TCGA datasets). c Correlation between SOX10 methylation and expression (normalised RNAseq counts) in SKCM, TNBC and nonTNBC (Spearman correlation coefficients (r) and p values are shown; derived from TCGA data). d Proportions of TNBC and nonTNBC cases with hypomethylation at each probe across the SOX10 locus (as defined in (b)). e Representative IHC showing SOX10-neg, heterogeneous and nuclear-positive (+) TNBCs. Tumours with absent or very weak nuclear staining in ≥50% of tumour cells were classified as SOX10-negative, while those with any one of replicate TMA cores exhibiting moderate-strong nuclear staining in <50% OR weak-moderate nuclear staining in ≥50% of tumour cells were classified as heterogeneous (see also Supplementary Fig. 2h). Survival curves of heterogeneous and negative categories overlapped (Supplementary Fig. 2j) and hence are grouped together here. f Kaplan–Meier analysis of the relationship between SOX10 nuclear positivity and breast cancer-specific survival (BCSS) in cross-sectional TNBCs. Log-rank test p value and hazard ratio (HR) are shown (95% confidence interval). g Kaplan–Meier analysis of the relationship between SOX10 nuclear positivity and BCSS in TNBCs classified as metaplastic breast cancers. Gehan–Breslow–Wilcoxon test p value shown. h SOX10 expression in brain-metastatic TNBC and matching brain metastases (BrM), compared to the frequency in cross-sectional TNBCs (Chi-square p value shown).
Analysing published cell line gene expression and methylation array datasets46,47 and our cell line bank48,49, we found that in contrast to tumours, TNBC cell lines express very low to undetectable levels of SOX10, and the SOX10 gene is hypermethylated (Fig. S2e, f). shRNA-mediated depletion of SOX10 in one of the few positive lines (HCC1569) resulted in 100% cell death within a few passages (Supplementary Fig. 2g).
Next, we performed IHC studies to investigate the prognostic significance of SOX10 expression at the protein level. Surveying a large, cross-sectional cohort of invasive primary breast tumours from Australia and the UK (n = 1330), we detected SOX10 almost exclusively in tumour cell nuclei of TN cases (Fig. 2e; see Supplementary Table 2 for cohort characteristics). Approximately 38% of TNBCs were classified as SOX10+, and another 11.5% exhibited heterogeneous staining (see Fig. 2e and Supplementary Fig. 2h for scoring thresholds). SOX10 positivity was associated with histologic features typical of this group, such as high grade, metaplastic and medullary morphology, pushing margins and a larger size at diagnosis (Supplementary Table 2). Similar, though statistically weaker trends were found between these variables and heterogeneous SOX10 staining (Supplementary Fig. 2i).
Rather than a simple correlate of the TN phenotype, SOX10 positivity stratified TNBC-specific survival in both univariate (Fig. 2f and Supplementary Fig. 2j) and multivariate regression analyses, with a prognostic value greater than clinicopathologic indicators used in current clinical practice: tumour size, grade, and the density of tumour-infiltrating lymphocytes (TILs) (hazard ratio 1.8-2.5; p = 0.02–0.002; Supplementary Table 2). Increased propensity for brain metastasis is one of the factors underlying premature death in TNBC, so we also analysed patient-matched pairs of primary TNBCs and brain metastases (n = 19 pairs). Compared to cross-sectional TNBCs, SOX10 was over-represented in brain-metastatic cases, with SOX10 status concordant in ~90% of matching brain tumours (Fig. 2h). Consistent with previous reports37,50, we also detected nuclear SOX10 in an independent cohort of metaplastic breast cancers (MBC; Asia-Pacific Metaplastic Breast Cancer consortium51). Compared to cross-sectional cases, SOX10 staining was more heterogeneous in MBCs, and was not associated with TN status (Supplementary Fig. 2k); but was prognostic amongst MBCs with a TN phenotype (Fig. 2g).
Considering all our IHC study findings, we concluded that strong nuclear expression of SOX10 is associated with TNBC progression.
SOX10’s TNBC regulatory module confers transcriptomic similarity to NCSCs
To investigate the basis of SOX10’s association with poor patient outcomes, we compared the expression profiles of TNBCs expressing high versus low levels of SOX10 mRNA and found that SOX10high tumours were significantly enriched with the expression of mesenchymal, neural, and glial development genes (Supplementary Fig. 3 and Tables S3, S4).
We then mapped SOX10’s regulatory neighbourhood within the breast cancer transcriptome using weighted gene co-expression network analysis (WGCNA). This approach quantifies co-variation in gene expression across a biological sample set to identify genes with highly coordinated regulation, which is indicative of functional relatedness52,53. We built a network from TCGA breast cancer RNAseq data (n = 919 cases) and validated it with datasets from METABRIC (n = 1278, expression array) and ICGC (n = 342, RNAseq). In this model, all genes expressed above a background threshold are connected (12,588 genes, 12,5882 connections). The connection between each gene pair is based on a weighted correlation coefficient, and unsupervised clustering can reveal groups of genes with a high probability of co-functionality (modular transcription programmes). The module eigengene (ME) is a centroid calculated for each module in each sample that represents both module expression and net connection strength.
WGCNA partitioned ~20% of expressed genes into eight consensus modules that align with established hallmarks of breast cancer; for example, an ER/FOXA1-driven module expressed in luminal tumours, and a mitotic instability module in basal-like and luminal-B tumours (Table 1, Fig. 3a, Tables S5–S8 and Supp File 2). The remaining ~80% of genes were not linked to any one module. SOX10 was identified as one of the most interconnected genes in the ‘green’ module, which has a hierarchical structure (Fig. S4a, b) and is predominantly expressed in high-grade TNBCs (Supplementary Fig. 4c). In this module, SOX10’s co-expression profile was highly similar to genes implicated in Wnt signalling, neuroglial differentiation and embryo patterning (Fig. 3b). We named it the SOXE-module and ascribed ‘multipotency’ as its primary ontology, as the member gene list is enriched with developmental phenotypes, and includes all three SOXE family members (SOX8/9/10) and embryonic stem cell genes (LMO4, POU5F1) (Fig. 3c and Supplementary Table 9).
Table 1.
Key features of eight predominant gene co-expression modules extracted by WGCNA.
| Modules | Major functional ontologiesa | Signalling pathwaysa/intrinsic activatorsb | Size (no. genes) | Top ten hub genes (Highest kWithin; see Supplementary Table 5) | |
|---|---|---|---|---|---|
| Tumour-centric | Blue | Mitotic instability | FOXM1, MYBL2 | 1239 | TPX2, BUB1, CEP55, HJURP, NCAPH, KIF4A, KIF2C, CCNB2, NCAPG, FOXM1 |
| Green | Multipotency (SOXE) | Wnt signalling | 487 | ROPN1, SFRP1, FOXC1, RGMA, GABRP, CHST3, MAML2, APCN, ROPN1B, SOX10 | |
| Brown | Primary cilium | ER, FOXA1 | 1008 | FOXA1, MLPH, ESR1, AGR3, XBP1, THSD4, GATA3, CA12, PRR15, ZMYND10 | |
| Tumour-stromal | Magenta | ECM-1 (structural) | FBN1, RUNX2 | 186 | COL5A2, COL1A2, COL3A1, COL5A1, COL6A3, FAP, THBS2, COL1A1, LUM, VCAN |
| Black | ECM2 (regulatory) | – | 207 | OLFML1, RECK, FSTL1, DCN, MSRB3, ECM2, CCDC80, TCF4, ZEB1, GLT8D2 | |
| Red | Fatty acid metabolism | PPARγ | 274 | DIA1R, PDE2A, LHFP, LDB2, ARHGEF15, S1PR1, SDPR, EBF1, CD34, ERG | |
| Tan | Type-I IFN response | STAT1, IRF9 | 33 | IFIT3, OAS2, CMPK2, IFI44L, IFI44, IFIT1, MX1, OASL, IFIT2, RSAD2 | |
| Stromal | Yellow | Adaptive immunity (TILs) | CD40L, CD40, IFNγ, IRF1 | 712 | SASH3, IL2RG, CD53, PTPN7, CD48, CD2, CD3E, ARHGAP9, CD5, CD3D, SIT1, SH2D1A |
ECM extracellular matrix.
aGene set enrichment analysis (GSEA) of all BRCA genes ranked according to module eigengene correlation (Supplementary Table 9).
bIngenuity pathways analysis upstream regulator prediction (p ≤ 1.0E-07) based on kWithin values for module genes.
Fig. 3. SOX10’s regulatory network is associated with multipotency, cell migration and poor prognosis in TNBC.

a Relative expression of eight predominant transcription modules in human breast tumours, according to the PAM50 subtype (TCGA dataset). b SOXE-module co-expression profile similarity matrix, clustered to highlight genes with very highly coordinated expression. The similarity is based on cosine distance and has a maximum value of 1. SOX10 mapped to one of six module sub-clusters, the members of which are shown to the right of the matrix. See also Supplementary Fig. 4a, b. c Summary of results from unsupervised gene set enrichment analysis of the breast cancer transcriptome after ordering transcripts according to their correlations with SOXE-module expression (denoted by the ME value, TCGA dataset). d Tile plot showing overlapping expression of SOXE-module representatives. For each protein, significant co-expression with ≥2 other module members is indicated by a Fisher’s exact test result (*p < 0.05; ***p < 0.001; ****p < 0.0001). Refer to Supplementary Table 1 for scoring criteria. e IHC staining of representative SOXE-module nodes in serial sections from the same tumour. f Proportional expression of all eight modules (coloured as for (a)) in TNBCs annotated with PAM50 and TNBC subtypes (METABRIC dataset; LAR luminal androgen receptor-like, MES mesenchymal, BLIS basal-like immune-suppressed, BLIA basal-like immune-activated39). g Kaplan–Meier analysis of METABRIC TNBCs expressing different proportions of the three predominant TNBC modules. BCSS breast cancer-specific survival. ME fraction thresholds for classifying cases as high or low were 0.33 for SOXE/blue and 0.1 for yellow.
IHC analysis of six other module members confirmed that their co-expression in TNBC holds true at the protein level (Fig. 3d), with staining often observed in the same cells within individual tumour-rich tissue cores (Fig. 3e). Consistent with the defining features of TNBCs—de-differentiation, genomic instability, high mitotic index and the presence of TILs—TNBCs express variable proportions of primarily three modules: green (SOXE), blue (mitotic instability) and yellow (TILs) (Fig. 3f). Kaplan–Meier analysis showed that cases expressing high levels of both SOXE and mitotic instability modules had shorter survival compared to those with predominant expression of one or the other, while co-expression of the yellow module was associated with better prognosis, consistent with the protective effect of TILs in TNBC54 (Fig. 3g and Supplementary Fig. 4d).
The SOXE-module represents the shift from a luminal progenitor to an NCSC-like state
Ontology analysis showed that the SOXE-module includes genes typically expressed in differentiating glia, cardiomyocytes, and odontoblasts, which all descend from NCSCs. In fact, developmental genes comprised a large proportion of SOXE-module hubs (genes with the highest network connectivity and centrality values; Fig. 4a and Supplementary Table 10), hence representing points of maximal module vulnerability. These include cell-fate regulators ELF5, FOXC1 and SOX10; Wnt/β-catenin signalling genes SFRP1, MAML2 and TRIM29; and embryonic cell migration and neuronal development genes RGMA, ROPN1, ROPN1B, MID1 and APCN.
Fig. 4. The SOXE-module drives the transition from normal mammary epithelial stem/progenitor to NCSC-like phenotypic states.

a Influence of SOXE-module genes over network architecture and information flow. kWithin: intramodular ‘connectivity’ based on weighted correlations with all other module genes; Eigencentrality: considers the connectivity of each node’s nearest neighbours as an indicator of ‘local influence’; Betweenness centrality: ‘conductivity’ based on each node’s position along the shortest paths between other nodes (genes with high betweenness are information conduits). Key hub genes are indicated (see Supplementary Table 10 for the full dataset). b Chick (ch.)NCSC and neural crest (NC) terms genesets are largely independent of each other and from the SOXE-module. c Correlations between SOXE-ME values and NCSC genesets (singscore values) in TNBC (n = 106 TCGA cases with tumour cellularity ≥0.6). Correlation coefficients (r) and p values are shown. d GSEA using three TNBC gene expression datasets (ICGC, METABRIC, TCGA). Normalised enrichment scores (NES) and corrected p values (q) shown. e Overlap between members of the SOXE-module and SOX10’s normal breast module (from de novo module identification on n = 97 TCGA normal breast samples; Supplementary Table 12). Generic ontology enrichment results are summarised (full GO term lists in Supplementary Table 13). f Comparison of network structure and information flow metrics (as for (a)) between shared and SOXE-module-exclusive genes. Groups were compared using Mann–Whitney tests (**p = 2.4E-03; ***p = 5.6E-04). Boxes show the 10–90th percentiles and median, with whiskers extending to the minimum and maximum values. Mean is indicated with ‘+’. g Model depicting the mammary epithelial progenitor gene regulatory network core being sustained through transformation and rewired as the SOXE-module in TNBC. Shared hub genes are listed.
To directly investigate if the SOXE-module is associated with NCSC phenotypic mimicry, as has been reported for Sox10 in mouse mammary tumour cells24, we performed expression and enrichment analyses using two independent genesets: (1) 308 genes represented in at least two of the 78 terms matching ‘neural crest’ in the gene ontology database (‘NC terms’); and (2) transcripts specific to migratory, Sox10+ NCSCs in chick embryos (‘ch.NCSC’; n = 200 genes)55, representing Sox10’s most primitive transcription programme (Supplementary Table 11). Except for SOX10, SOX8 and LMO4, there is minimal overlap between the SOXE-module and these genesets (Fig. 4b), but their expression is strongly correlated (Fig. 4c). This was confirmed by geneset enrichment analysis (GSEA; Fig. 4d). Hence, the SOXE-module confers transcriptomic similarity to NCSCs.
Since several SOXE-module genes (e.g. SOX10, SOX9, LGR6 and ELF5) are key regulators of normal hMEC states56, we hypothesised that the SOXE-module might evolve from the deregulation of a lineage differentiation programme expressed in TNBC’s normal cellular precursors. Module preservation analysis using RNAseq data from TCGA normal breast samples indicated that the SOXE-module does not exist as an interconnected unit in the normal breast transcriptome (Supplementary Fig. 4e). But after performing de novo WGCNA module identification on this dataset (Supplementary Table 12), we found that SOX10’s normal breast module overlaps with the TNBC-specific SOXE-module significantly more than expected by chance (Fig. 4e; 109 shared genes, Chi-square p = 2.8E−26).
Both ‘normal-exclusive’ and ‘shared’ genes were enriched with epithelial differentiation ontologies, with cell adhesion distinctly over-represented in the shared set (Fig. 4e and Supplementary Table 13). According to network influence metrics, the shared genes were significantly more important to the SOXE-module than SOXE-exclusive genes (Fig. 4f and Supplementary Fig. 4f). This suggests that while SOXE-exclusive genes are primarily responsible for conferring NCSC-like attributes, genes ‘inherited’ from TNBC’s normal precursors are comparatively more important to the SOXE-module’s regulatory structure. Together, these data suggest that SOXE-module and its associated NCSC-like phenotype arise because a core set of epithelial differentiation and adhesion genes becomes rewired during TNBC development (Fig. 4g).
Genomic and epigenomic determinants of the NCSC-like transcriptional shift in TNBC
To address the central question of what drives this transcriptomic shift, we analysed case-matched gene copy-number (CN), RNAseq and WGCNA data (TCGA cases). Candidate module drivers were defined as those for which both CN and expression correlated significantly with SOXE-ME values. About 182 genes met these criteria (130 gains and 52 losses), of which 140 (77%) are part of large chromosomal alterations: 6p21-22 (gained/amplified in 56.7% of TNBC cases), 8q22-24 (gained/amplified in 78.7%), 9q34 (lost in 59.6%) (Supplementary Fig. 5a). SOXE-module genes were over-represented amongst the positively correlated genes (25/130 (19.2%) and had increased CN and expression in SOXEhigh TNBC; ChiSq p = 9.7E−31; Fig. 5a). However, network influence metrics for these 25 were no higher than other module genes (Fig. 5b). Hence, the SOXE-module may be augmented by increased CN of some of its component genes, but this seemed unlikely to be an early or dominant driver of module evolution.
Fig. 5. The SOXE-module is driven by the erosion of lineage-specific epigenetic marks.

a Decision tree for identifying candidate copy-number alteration (CNA) drivers of the SOXE-module. Of 17,694 genes with case-matched GISTIC, RNAseq and WGCNA data, CN, and expression of 130 correlated with the SOXE-module in TNBC, including 25 SOXE-module nodes. b Network influence metrics for SOXE-module nodes coloured according to candidate CN driver status (intramodular connectivity (kWithin), local influence (Eigencentrality) and conductivity (betweenness centrality) defined in Fig. 4a). Boxes show the 10–90th percentiles and median, with whiskers extending to the minimum and maximum values. Mean is indicated with ‘+’. No significant differences by ordinary ANOVA test. c Relationship between SOXE-module levels and mutation signatures in ICGC TNBCs (COSMIC v2 SigProfiler and HRDetect on n = 74 ICGC TNBCs)45. Associations are depicted according to the correlation between SOXE-ME values and signature event count (y-axis); and by the significance of average SOXE-ME differences between ICGC TNBCs with low (quartile-1) vs higher (quartile 2–4) signature burden. d t-Distributed stochastic neighbour embedding (t-SNE) visualisation of genome methylation profile similarities amongst cases in the BRCA-TCGA 450k methylation array dataset. Panels are coloured according to PAM50 intrinsic subtype, SOXE-ME values or global median methylation-b values. Circled cases are epigenetically divergent, basal-like TNBCs that express high levels of the SOXE-module and have eroded methylomes. e Correlation analysis summary showing relationships between SOXE-ME values and region-specific methylation (n = 75 TCGA TNBCs, tumour cellularity ≥0.6; n = 215,323 probes after quality filtering); ****p < 1.0E-07. CGI CpG island, IGR intergenic region, TSS transcription start site, UTR untranslated region. Solo-WCpGW: consensus sequence for late-replicating loci demethylated via replicative senescence. f Unsupervised clustering of the BRCA-TCGA 450k methylation dataset according to ME correlation. Data shown were minimum correlation coefficients of ME values versus gene-averaged methylation-b data from promoter region probes (TSS1500, TSS200 and 5′UTR). Of three clusters inversely correlated with SOXE-module expression, two (a, b) were enriched with developmental ontologies (Supplementary Table 14). g Network influence metrics for SOXE-module genes in the hypomethylated clusters versus other SOXE-module genes, as for (b). Ordinary ANOVA p values: *p < 0.05; **p < 0.01; ***p < 0.001; ns not significant.
Next, we investigated whether mutational processes that shape the breast cancer genome could be involved. To this end, we utilised case-matched mutational signature and WGCNA data for the ICGC cohort 45,57. There were direct relationships between the SOXE-module and overall mutation burden (substitutions and small insertion-deletion (indels)), as well as specific signatures of genome instability (rearrangement sigs (RS)3 and RS5), homologous recombination (HR)-directed repair of double-strand DNA breaks (DSBs) and genome editing (sig-3: HR deficiency; HRDetect; sig13: APOBEC; Fig. 5c).
APOBEC activity and DSB repair are both indirectly demethylating. For example, 5-methyl cytosine (5mC) loss occurs because of APOBEC-mediated genome editing and/or during the repair of edited bases, and DSB repair has been causally linked to the progressive loss of 5mC during cellular ageing58,59. Therefore, we hypothesised that the evolution of the SOXE-module in TNBC may be related to epigenetic dysregulation. Consistent with this idea, the 105 CN-driven SOXE-module correlates (i.e., those not part of the SOXE-module itself; Fig. 5a) were enriched with a transcription factor, chromatin remodelling and DNA repair genes (Fisher’s Exact p < 0.001). Furthermore, visualising SOXE-module strength relative to the overall methylome profile using t-SNE showed that SOXE-ME values were highest in the most epigenetically divergent tumours (Fig. 5d).
To investigate this further, we then correlated SOXE-ME values with probe-level methylation data directly, in the following regional categories: CpG islands (CGIs), CGI shores, shelves or open sea regions at transcription start site (TSS) regions, untranslated regions (UTRs), gene bodies or intergenic regions (IGRs). We also quantified methylation at ‘solo-WCpGW’ sites at late-replicating, heterochromatic loci, which act as a biomarker of replicative senescence60 and are hypomethylated in breast tumours compared to hMECs (Supplementary Fig. 5b). There was no relationship with solo-WCpGW sites (Supplementary Fig. 5c), but there was a striking inverse correlation between SOXE-ME values and genome-wide promoter methylation; particularly at CGI shores, the substrate for lineage-specific methylation in adult tissues (Fig. 5e and Supplementary Fig. 5c). These data indicate that SOXE-module expression and connectivity are directly proportional to promoter demethylation in TNBC (Fig. 5e). There was no such relationship with any other module in TNBC (Supplementary Fig. 5d).
Having established that SOXE-module levels correspond with loss of tissue-specific 5mC marks, we then built a correlation matrix from ME and genome-wide promoter methylation data (TCGA) and performed unsupervised clustering to look for evidence of epigenetic control. The SOXE-module had a distinct promoter methylation signature—three clusters of genes that are hypomethylated when SOXE-module strength is highest, of which two were enriched with developmental ontologies (Fig. 5f and Supplementary Table 14). Only 10% of these correspond to SOXE-module genes, but this 10% is enriched with hub genes (Fig. 5g), suggesting a higher level of epigenetic control over module structure and information flow. We then used GSEA to test the enrichment of the SOXE-associated promoter methylome with NCSC genesets. Like the transcriptome (Fig. 4d), the methylation landscape associated with the SOXE-module was also enriched with NCSC genes (NC terms: normalised enrichment score (NES) −1.5; q = 6.0E−03; Ch.NCSC: NES −1.3; q = 3.6E−02).
Finally, we investigated direct demethylation processes as potential enablers of SOXE-module formation by cross-referencing SOXE-ME values from our three WGCNA datasets (TCGA, ICGC, METABRIC) against the expression of demethylases in the EpiFactors database61. There were direct associations with APOBEC3A/3B cytosine deaminases and TET1 (Supplementary Fig. 5e). TET dioxygenase enzymes catalyse the first step of 5mC demethylation and are involved in processes requiring cell states to be reset or adjusted, such as methylome erasure in preimplantation embryos, and epigenetic plasticity in brain regions that facilitate learning and memory. TET1 is a maintenance demethylase that prevents methylation from spreading from silenced loci, particularly at CGI shores62,63. It has been causally implicated in TNBC metastasis64 and our findings suggest this may be at least partly due to reinforcement of the SOXE-module.
In summary, the SOXE-module’s dominance over the TNBC transcriptome is directly proportional to APOBEC activity, DSB repair and TET1 expression, which are all demethylating. Of all methylation domains across the genome, the module is most strongly correlated with hypomethylated promoter CGI shores—the substrate for lineage-specific methylation. Kim et al. showed that the minimal genetic requirements for reprogramming postnatal fibroblasts with an NCSC identity are SOX10 expression and the erasure of previous epigenetic memory34. We postulate that progressive erosion of the epigenome in SOX10+ tumour-initiating cells simulates these conditions, driving NCSC-like reprogramming and poor clinical outcomes in SOX10 + TNBCs (Fig. 6).
Fig. 6. Model summarising the study findings.
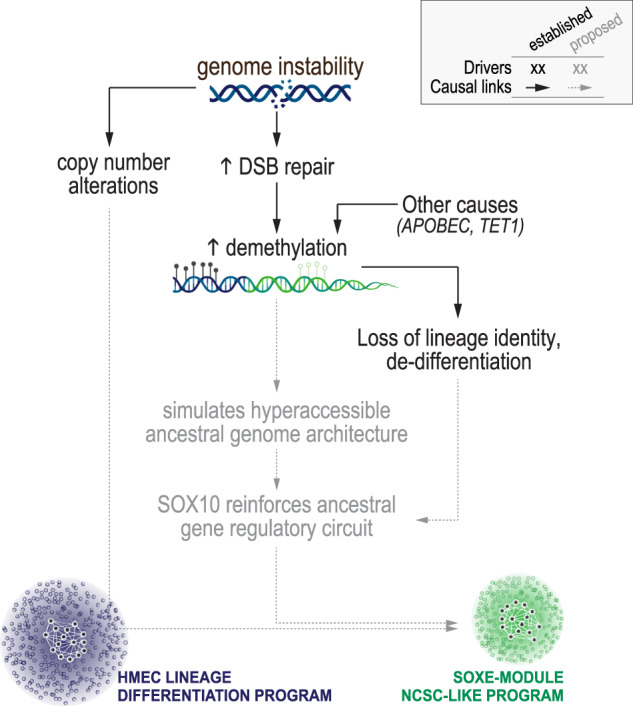
Proposed links between established drivers of TNBC progression, epigenome erosion and the emergence of a neural crest-like transcriptional programme in de-differentiated TNBCs.
Discussion
Heterogeneity has emerged as a major bottleneck to effective sub-classification and treatment of cancer, and TNBC is no exception. Post-treatment relapse occurs through clonal expansion of cells with pre-existing, advantageous mutations, but also cell state changes brought about by adaptive epigenetic remodelling—a phenomenon that unites the ‘cancer stem cell’ and ‘epigenetic progenitor’ models of cancer65. The intrinsic plasticity of TNBC is problematic because existing therapies cannot eradicate a shifting target. Early evidence implies that blocking this capability with epigenetic therapy may improve treatment efficacy, but this will require a deeper understanding of how phenotypic plasticity evolves66. TNBC exhibits genome-wide hypomethylation, which evidently drives de-differentiation by destroying the state-defining epigenetic barcode of its normal cellular precursor, the LP cell14–17,65,67. Differential methylation at certain genomic loci is prognostic in TNBC22, and myriad studies have helped to decipher the mechanistic contributions of individual writers, readers, and erasers of epigenetic marks, but the phenotypic manifestations of genome-wide 5mC loss have not been extensively studied.
Consistent with functional analysis of Sox10 in experimental mice29,32, our human tumour network studies show that SOX10’s TNBC-specific regulatory module confers similarity to highly plastic NCSCs. We traced a cluster of super-connected SOXE-module genes back to the tissue-resident mammary stem and progenitor cells and found that in contrast to the normal breast where it was associated with epithelial lineage differentiation, in TNBC this core was connected to Wnt signalling, neuroglial differentiation and embryo patterning genes. Critically, we found that expression of the SOXE-module amongst TNBCs was proportional to overall transcriptional similarity to Sox10+ migratory NCSCs from chick embryos55, despite there being minimal direct overlap in member genes. We also identified SOXE-module hub genes as points of maximum network vulnerability as candidate therapeutic targets. In support of this approach, two of these—BBOX1 and BCL11A—have already been validated as such in TNBC68–72.
To better understand the evolution of NCSC-like transcriptional reprogramming, we investigated potential links to the established drivers of TNBC development—genomic instability, large-scale CNAs, and defective DNA repair. We identified several processes that correlate significantly with the SOXE-module eigengene (DSB repair, APOBEC and TET1 activity, which are all demethylating); but most discernibly, the loss of lineage-specific methylation marks at CGI shores. Several mechanisms have been postulated to contribute to widespread methylome erosion in cancer, including DSB repair58,59 and reduced availability of 5mC substrates through metabolic reprogramming73. Accepting that there are probably multiple contributing factors in any individual tumour, our findings nevertheless suggest that NCSC-like reprogramming occurs concomitantly with epithelial de-programming in TNBC. The gene regulatory networks that operate in NCSCs are amongst the most evolutionarily conserved in vertebrates25,74. We postulate that when the broadly open chromatin landscape of the early embryo is simulated in epigenetically eroded tumours, dominant fate specifiers like SOX10 may recreate their ancestral regulatory circuits by default.
In summary, our data indicate that the extent of promoter methylation loss in SOX10+ breast tumours correlates with their transcriptomic similarity to NCSCs—the earliest developmental cell state programmed by SOX10 activity and one synonymous with migration, multipotency and phenotypic plasticity. We propose that during TNBC development, progressive erosion of the epigenome drives de-differentiation while simultaneously making cells vulnerable to NCSC-like reprogramming. Broadly, these findings support preclinical data19–23 on the potential for epigenetic modulators to combat phenotypic plasticity in TNBC.
Methods
Human tissue samples (also see Table 2)
Table 2.
Biological resources.
| Resource | Source, identifier and relevant citations | Related figure(s) |
|---|---|---|
| Tissue samples | ||
| Histologically normal breast FFPE whole sections | The Brisbane breast bank48 | 1a–e |
| Fresh RM surgical samples | The Brisbane breast bank48,76 | 1f, Supp-1b, Supp-6a |
| Australian BC series, FFPE TMA sections & clinical data | Pathology Qld & The Brisbane breast bank48,89 | 2e, f, 3d–e, Supp-2h-k |
| UK breast cancer series, FFPE TMA sections & clinical data | Nottingham Breast Cancer Research Centre90,91 | 2e, f, Supp-2h-k |
| Metaplastic tumour series, FFPE sections & clinical data | Asia-Pacific MBC consortium51,92 | 2g |
| Patient-matched primary TNBCs and brain metastases | Pathology Qld & The Brisbane breast bank48,89 | 2h |
| Cancer cell lines | ||
| 293 T | ATCC® CRL-3216™ | Supp-1a, Supp-2g |
| MDA-MB-435S | ATCC® HTB-129™ | Supp-1a, Supp-2e, Supp-2g |
| HCC38 | ATCC® CRL-2314™ | Supp-2e |
| HCC1569 | ATCC® CRL-2330™ | Supp-2e, Supp-2g |
| Primary melanoma cells (D41, D05) | Dr. Chris Schmidt, QIMR Berghofer77 | Supp-2e |
| TaqMan gene expression assays | ||
| SOX10 | ThermoFisher, Hs00366918_m1 | Supp-2e |
| RPL13A | ThermoFisher, Hs03043885_g1 | Supp-2e |
| shRNA sequences | ||
| SOX10_1 | Sigma-Aldrich TRCN0000018984 | Supp-1a, Supp-2g |
| SOX10_2 | Sigma-Aldrich TRCN0000018987 | Supp-1a, Supp-2g |
| SOX10_3 | Sigma-Aldrich TRCN0000018988 | Supp-1a, Supp-2g |
| Non-targeted negative control (NTNC) | Sigma-Aldrich SHC002 | Supp-1a, Supp-2g |
Supp supplementary.
This study involved immuno-detection of SOX10 and other biomarkers in the following human tissue cohorts:
Reduction mammoplasty (RM) samples: obtained in collaboration with Dr William Cockburn (Wesley Hospital, Brisbane) and the Royal Brisbane and Women’s Hospital (RBWH) Plastics Unit. Nineteen RM specimens were used for IHC and IF analysis, and two for methylation arrays. Age, parity and menopausal status of these patients were unknown. 30% of cases showed fibrocystic change and 10% presented with columnar cell lesions (histopathology review by SRL).
- Clinically annotated, primary breast tumour samples:
- A cross-sectional primary breast tumour cohort comprising samples from Australia (treated by the RBWH Breast Unit) and the UK (Nottingham University Hospital), from patients treated in the mid-1980s to mid-1990s. Tumour blocks were sampled as 0.6 mm cores in tissue microarrays (TMAs). For baseline characteristics see Supplementary Table 2.
- Metaplastic carcinomas (Asia-Pacific Metaplastic Breast Cancer Consortium (whole sections).
Patient-matched primary TNBC and brain metastases (n = 19 pairs). Tumour blocks were sampled as 1.0 mm cores in TMAs.
Ethics approval
Human research ethics approval was obtained from the Royal Brisbane and Women’s Hospital (2005000785), The University of Queensland (HREC/2005/022) and North West Greater Manchester Central Health (15/NW/0685). Written patient consent to use tissue for research purposes was obtained where required under the conditions of these approvals and all samples were de-identified in the analytical database. This study complies with the World Medical Association Declaration of Helsinki.
Immunohistochemistry (IHC)
Formalin-fixed, paraffin-embedded (FFPE) tissue samples or TMAs were sectioned, deparaffinised, subjected to antigen retrieval and chromogenically stained as described in ref. 75 and detailed in Supplementary Table 1. Slides were scanned using the Aperio ScanScope T2 digital scanning system at 40x magnification. TMA images were segmented using Spectrum software (Aperio), and high-resolution images of individual cores were extracted and scored by two experienced observers in a blinded fashion (hidden metadata tags corresponding to TMA position were used to link clinical and sample data). Digital image files were scored according to the criteria set out in the legends to Figs. 2e and S2h.
Immunofluorescence (IF)
FFPE RM tissue sections (Table 2) were sectioned, deparaffinised, subjected to antigen retrieval and stained as described in ref. 76 (Supplementary Table 1). Briefly, primary antibodies diluted in tris-buffered saline (TBS) were incubated on tissue sections for 1 h at room temperature, washed in TBS then incubated with secondary antibodies for 30 min in the dark. To minimise tissue autofluorescence, slides were stained with SUDAN Black for 20 min in the dark (Sigma #S-2380), then washed (0.1% TBS-Tween (30 min), TBS (10 min). Slides were mounted using Vectashield (Vecta Labs) with DAPI (Sigma-Aldrich), cover-slipped, sealed and imaged on a Carl Zeiss MicroImaging system using Axio Vision LE version 4.8.2 (PerkinElmer).
Fresh reduction mammoplasty (RM) tissue processing and fluorescence-activated cell sorting (FACS)
RM samples were processed, and single-cell suspensions were prepared as previously described (Table 2 and refs. 48,76). Briefly, tissue was cut into small pieces (~5 mm3) and digested overnight with agitation at 37 °C in DMEM-F12 (Gibco), foetal bovine serum ((FBS), 5%, Gibco), antibiotic/antimycotic (Gibco), Amphotericin B (2.5 μg/mL, Gibco), collagenase type I-A (200 U/mL, Sigma-Aldrich) and Hyaluronidase I-S (100 U/mL, Sigma-Aldrich). Epithelial organoids were obtained by centrifugation (80 × g, 1 min), then dissociated to single-cell suspensions for 5–10 min in TrypLE (Gibco), followed by Dispase (5 mg/mL, Gibco) and DNAse-I (100 ug/mL, Invitrogen). Enzymatic activity was quenched in ice-cold Hank’s Balanced Salt Solution ((HBSS), Gibco) with 2% FBS and cells were filtered through a 40-μm cell strainer (BD Falcon).
Cell concentration and viability were determined using a Countess® automated counter (Invitrogen) with trypan blue and adjusted to 2.0E6/mL. Single-cell suspensions (typically 30–60 mL) were labelled for 10 min on ice with SytoxTM green (Invitrogen) plus a cocktail of fluorescent antibody conjugates to discriminate hMEC subsets (negatively gated, non-epithelial ‘lineage’ markers: CD31, CD45, CD140b; positively gated hMEC markers: CD49f, EpCAM—see Supplementary Table 1 and Supplementary Fig 6a). Samples were washed (80×g, 2 min) and then resuspended in cold HBSS + 2% FBS. For robust fluorescence compensation and gating of specific hMEC populations, we also tested in parallel small samples stained with isotype control antibodies, and ‘fluorescence minus one’ negative controls (samples from which one of the main conjugates was omitted). Fluorescence data acquisition, gate placement and sorting were performed on a BD FACS Aria II instrument with FACSDiva software (v6.1.3; QIMR Berghofer). Sorted cells were collected on ice before being pelleted (80×g, 2 min) and snap-frozen at −70 °C.
Methylation array profiling and ChIP-seq meta-analysis
DNA was extracted from FACS-sorted hMEC samples using the QIAGEN AllPrep DNA/RNA mini kit, with bisulphite conversion using the EZ DNA methylation Kit (Zymo Research) following the manufacturer’s protocol with modification for Illumina methylation arrays. Bisulphite-converted DNA was amplified and hybridised to Infinium methylationEPIC 850k beadchips (Illumina) according to the manufacturer’s protocol. Arrays were scanned on an iScan, and data were processed using GenomeStudio (Illumina) with BMIQ array normalisation to derive average methylation beta-values.
Histone modification ChIP-seq data were obtained from Pellacani et al.42. Bigwig format files were retrieved from www.epigenomes.ca, and the mean signal/bin was plotted across the region chr22:38365030-38396083 for each histone mark in each cell type.
Analysis of SOX10 expression in cell lines
MDA-MB-435, HCC1569 and HCC38 cells were from the American Type Cell Culture Collection (ATCC; (Table 2); authenticated in our laboratory and cultured according to ATCC recommendations49. D41 and D05 melanoma cells were selected from the primary melanoma cell line bank of Dr Chris Schmidt and Prof Nick Hayward (QIMR Berghofer) based on having high and low baseline SOX10 expression, respectively77. Cells were routinely cultured at 37 °C in a humidified atmosphere with 5% CO2 and routinely screened for mycoplasma. RNA and protein were extracted from cells in the exponential phase of growth using standard Trizol and RIPA buffer methods78. SOX10 mRNA was quantified relative to RPL13A as previously described (ref. 79 and Table 2). For Western analysis (MDA-MB-435, HCC1569, HCC38 cells), protein lysates (30 μg) were resolved by SDS-PAGE then SOX10 and β-actin were detected using standard chemiluminescence (Supplementary Table 1).
Stable-shRNA knockdown of SOX10 in breast cancer cell lines
Three pre-validated SOX10-targeted shRNA constructs, and a non-targeting negative control (NTNC) construct (pLKO.1), were purchased from Sigma-Aldrich (Table 2). Plasmid DNA was isolated from overnight bacterial cultures, then lentiviral particles were produced by triple transient transfection of HEK-293T (human embryonic kidney) packaging cells with one of the four transfer plasmids (pLKO.1-puro; 2 μg), together with companion plasmids encoding lentiviral packaging and replication elements (2 μg pHR’8.2ΔR + 0.25 μg pCMV-VSV-G; donated by Dr Wei Shi, QIMR Berghofer). Virus-containing supernatants (in target cell media) were then collected over the following two days and filtered (0.45 μm). MDA-MB-435 target cells were seeded at 3.1 × 104/cm2 in six-well plates, then after 24–48 h (at ~50% confluence), cells were infected with filtered viral supernatants, supplemented with 1 mg/mL polybrene (Sigma-Aldrich) for 24 h. Stably transduced cells were then selected with 1 μg/mL puromycin (Sigma-Aldrich) for 2 weeks to eliminate uninfected cells.
Datasets and processing
TCGA level-3 normalised RNAseq data ('rnaseqv2 illuminahiseq rnaseqv2 unc edu Level 3 RSEM genes normalised data.data.txt') from the Data Analysis Center Firehose (http://firebrowse.org/) were used for all single-gene analyses (Supplementary Figs. 2a, 5e; test group stratification for Supplementary Fig. 3; SOX10 heatstrips in Fig. 3a and Supplementary Fig. 6a, c). Scaled estimate columns of the 'rnaseqv2 illuminahiseq rnaseqv2 unc edu Level 3 RSEM genes data.data.txt' were used for all other algorithmic analyses.
For methylation datasets, TCGA level-3 Illumina HM450k data were downloaded from the National Cancer Institute Genomics Data Commons (GDC) data portal (https://portal.gdc.cancer.gov/) and processed using the ChAMP package80. We applied the champ.filter function to remove problematic probes (those mapping to X/Y chromosomes, mapping to multiple locations, located near an SNP and non-CG probes). Filtered data were normalised using the champ.norm function, according to the Beta-Mixture Quantile (BMIQ) algorithm; is an intra-sample normalisation procedure that corrects the bias of type-2 probe values.
Level-4 GISTIC-2 copy-number data for TCGA cases were downloaded from the Data Analysis Center Firehose (http://firebrowse.org/) and used for correlative analyses with no further processing. To apply tumour purity cutoffs (TCGA cases), we used a consensus measurement of four different purity estimation methods81.
With permission from the METABRIC data access committee, normalised Illumina HT 12 expression array data were downloaded from the European Genome-phenome Archive (EGAD00010000210-211). For the ICGC RNAseq dataset, normalised data were downloaded as supplementary data45 and used with no further processing. Mutational signature data (COSMIC, v2 SigProfiler) were downloaded as raw event counts from ref. 45 and HRDetect probability scores for these cases from ref. 57.
Differential expression analysis of SOX10-high and -low TNBCs (Supplementary Fig. 3)
To characterise the transcriptomic phenotype associated with SOX10 expression in TNBC, we performed differential expression analysis of SOX10-high versus SOX10-low (median split) TCGA and METABRIC datasets using limma82 (differential expression was defined by a corrected p value cutoff of 0.01).
Ontology enrichment analyses
GO term enrichment analysis was performed using the Generic GO term finder hosted by Princeton University (Lewis-Sigler Institute for Integrative Genomics; https://go.princeton.edu). Gene set enrichment analysis (GSEA) was performed using the Prerank function of GenePattern83 using 1000 permutations. For Supplementary Fig 3, GSEA inputs comprised differentially expressed genes (q ≤ 0.01) ranked by fold-change in each dataset. The input for all other GSEA experiments was whole transcriptome gene lists ranked by a Spearman correlation coefficient. Biological process genesets (Gene Ontology v7.2; gene set size 15-500) were mined for unsupervised analyses and neural crest genesets for supervised analyses (Supplementary Table 11). Datasets and ranking metrics are indicated in the respective Figure legends. Normalised enrichment scores (NES) and corrected p values are reported. GeneGo (Metacore® Clarivate Analytics) and Ingenuity® Pathway Analysis (Ingenuity) were also used to analyse pre-ranked gene lists. REVIGO84 was used to resolve semantic redundancy and identify major themes amongst the enriched terms.
Weighted gene co-expression network analysis (WGCNA)—module identification and validation
WGCNA is a powerful network analysis tool that identifies groups of transcripts (modules) that fluctuate in a highly coordinated fashion, implying co-functionality52,53. First, it iteratively correlates the expression of every pair of transcripts in a test dataset, producing an adjacency matrix. It then converts this to a topological overlap matrix that reflects net connection weight, accounting for both direct connections and the impacts of shared neighbours. In this study, we created ‘signed’ networks, which reflect the overall topological overlap considering both positive and negative correlations. Dynamic module identification and characterisation (derivation of network metrics, sample eigengene values and module preservation in orthogonal datasets, see below) were performed in the R coding environment, and publication-quality figures were prepared from raw datasets using GraphPad Prism or Clustergrammer (Table 2).
Modules were identified using the TCGA RNAseq (n = 919 samples after quality filtering) and validated using METABRIC (n = 1278; expression array; Supplementary Fig 6b–d). A consensus set of eight modules was determined according to satisfactory concordance between these two orthogonal networks and a third was generated from the ICGC dataset (n = 342; RNAseq). We further validated the eight consensus modules using preservation analysis on a third breast cancer expression dataset. For normal breast samples, WGCNA was performed independently on TCGA normal breast samples (n = 97 after quality filtering).
Standard WGCNA outputs include the following (raw data in Supplementary Tables 5–11):
Module eigengene (ME): a theoretical gene that is the most strongly connected to all other genes in the module and hence represents net module expression and connectivity. Mathematically, the first principal component of each module’s adjacency matrix.
-
Module membership and connectivity: Each gene is ascribed k values describing modular and network connectivity (kTotal, kWithin and kOut). These continuous variables are amenable to integrated analysis of overlapping transcriptional programmes, utilising the granularity in expression datasets rather than levelling it as is done when assigning fixed phenotypes or categories. kME correlation and kME p values describe how tightly individual genes are linked to all other genes within each module.
To identify hub genes (Supplementary Fig. 6e), additional network connectivity and influence measures were calculated for each node in the SOXE-module topological overlap matrix using igraph toolkit functions in R:
betweenness centrality: betweenness(graph, v = V(graph), directed = FALSE, weights = NULL, nobigint = TRUE, normalised = FALSE).
eigencentrality: eigencentrality(graph, directed = FALSE, scale = TRUE, weights = NULL, options = arpack defaults).
Finally, we used community detection algorithms85,86 to examine the substructure of the SOXE-module (MATLAB 2020a), using the adjacency matrix as input. This revealed a hierarchical, sub-modular organisation, and consistently discriminated two partitions (59 and 41% of nodes each). To identify the module ‘control centre’ and hub genes as points of structural vulnerability, submodule assignment was cross-referenced against clustered Cosine similarity data (Fig. 3b, Clustergrammer87) with the same input (Supplementary Fig. 4).
Neural crest genesets
Geneset-1 (NC terms) comprises 308 genes represented in at least two of the 78 terms matching ‘neural crest’ and ‘human’ in the gene ontology database (http://geneontology.org). Geneset-2 (ch.NCSC) comprises the top 200 transcripts statistically over-represented in Sox10+ chick neural crest cells compared to all other embryo cells (fold-change 3.9–23.3; false discovery rate 9.3E−03–1.0E−15)55 (Supplementary Table 11). The ch.NCSC gene set represents genes coordinately expressed with Sox10 in a stem cell state hence was also suiTable-for network analyses (see below). We used the singscore algorithm88 to score RNAseq datasets against the neural crest genesets at the individual sample level.
Breast cancer methylation data analyses
Methylation beta-values were derived from TCGA level-3 Illumina HM450k data as outlined above. Beta-values for all probes corresponding to TSS1500, TSS200 and 5′UTR regions in each sample were first normalised to correct for their bimodal distribution (median absolute deviation (MAD): Pβ – median(Pβ – median(Rβ)); where P = probe in the promoter region and R = all probes in promoter region). After filtering out genes with >2 missing probes and those for which >2% of samples were missing data, the final dataset included average MAD-normalised promoter methylation beta-values for 4482 genes (determined from a total of 518 samples with complete clinical annotation). Pairwise Spearman correlations were then calculated between each promoter region and each module eigengene across the sample cohort. Unsupervised hierarchical clustering of correlation values was performed in R using the Flashclust package based on the Euclidean distance method. Clusters were visualised and validated with the cluster package, using the Silhouette coefficient to confirm distinct clusters. To generate t-distributed stochastic neighbour embedding (t-SNE) plots, we used the Rtsne package (https://cran.r-project.org/web/packages/Rtsne/) on normalised beta methylation values, with 5000 iterations and a perplexity parameter of 40.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Acknowledgements
We thank the many thousands of patients who have donated tissue for cancer research, and clinical staff who facilitate biobanking, particularly the Brisbane Breast Bank and Pathology Queensland. We acknowledge the support of Metro North Hospital and Health Services for the collection of the clinical subject data and clinical subject materials. We are grateful to Dr Lynne Reid and Clay Winterford for valuable contributions; Dr Katia Nones (QIMR Berghofer) who supervised XMDL; Dr Chris Schmidt (QIMR Berghofer) and Prof. Alex Swarbrick (Garvan Institute) for donating cell lines; Dr William Cockburn and clinical staff (Wesley Hospital) for normal breast tissue collections; Drs. Nic Waddell and Olga Kondrashova (QIMR Berghofer) for supportive data analyses; and Drs. Juliet French (QIMR Berghofer) and Delphine Merino (Olivia Newton-John Cancer Research Institute) for critical feedback. This study makes use of data generated by the Molecular Taxonomy of Breast Cancer International Consortium, funded by Cancer Research UK, and the British Columbia Cancer Agency Branch. It was funded by NHRMC programme awards to S.R.L., G.C.-T. and K.K.K. (APP1017028 and APP1113867), NHRMC project grants to PTS (APP1080985 and APP1164770) and an Australian Leadership Award to A.R.
Author contributions
Conception and design: J.M.S., X.M.D.L., K.N., A.R., D.V.N., P.T.S. and S.R.L. Data collection/contribution: J.M.S., X.M.D.L., K.N., A.R., A.H., A.E.M.R., M.L., A.C.V., J.R.K., A.J.D., M.M., E.K., P.K.-d.C., I.G., F.A.-E., J.M.W.G., C.O., K.K.K., J.B., G.C.-T., A.R.G., E.A.R., I.O.E., D.V.N. and P.T.S. Data analysis: J.M.S., X.M.D.L., K.N., A.R., A.H., S.L., D.V.N. Manuscript drafting: J.M.S., A.E.M.R., D.V.N., P.T.S. and S.R.L. All authors read and approved the final manuscript.
Data availability
Published datasets used in this paper are outlined in Table 3. Network data generated by the study are also outlined in Table 3, and available as supplementary data. Raw DNA methylation array data for FACS-sorted normal breast epithelial cell subsets are available from the Gene Expression Omnibus (GSE199579; Table 3).
Table 3.
software, code, and published datasets.
| ResRource | Source, identifier and relevant citations | Related figure(s) | Related table(s) |
|---|---|---|---|
| Software packages and code | |||
| ChAMP | https://bioconductor.org/packages/release/bioc/html/ChAMP.html80 | 5d–f | – |
| Clustergrammer | https://maayanlab.cloud/clustergrammer/87 | 3b | Supp-10 |
| Community detection algorithms | Refs. 85,86 | Supp-4a | – |
| Epifactors database | https://epifactors.autosome.ru61 | Supp-5e | – |
| FACSDiva™ | BD Biosciences, licensed | 1f, Supp-6a | – |
| FCS Express (v7) | De Novo Software, licensed | 1f, Supp-6a | – |
| GSEAPreranked | https://genepattern.org83 | 3c, 4d, 5f, Supp-3 | 1, Supp-4, Supp-9 |
| Ingenuity Pathways Analysis (IPA) | Ingenuity, licensed | – | 1 |
| MATLAB | Mathworks, licensed | Supp-4a | Supp-10 |
| Princeton Generic GO term finder | https://go.princeton.edu93 | 5a | Supp-13, 14 |
| Prism (v8.4.3) | GraphPad, licensed | Multiple | S2 |
| R package, Cluster | https://cran.r-project.org/web/packages/cluster/index.html | 5f | – |
| R package, FlashClust | https://cran.r-project.org/web/packages/flashClust/index.html | 5f, g | Supp-14 |
| R package, Limma | https://www.bioconductor.org/packages/release/bioc/html/limma.html | Supp-3 | Supp-3 |
| R package, t-SNE | https://CRAN.R-project.org/package=Rtsne | 5d | – |
| R package, WGNCA | https://cran.r-project.org/web/packages/WGCNA/index.html52,53 | Multiple | Multiple |
| REVIGO | http://revigo.irb.hr | Supp-3 | Supp-4 |
| Singscore | https://www.bioconductor.org/packages/release/bioc/html/singscore.html88 | 4c | – |
| SPSS | IBM, licensed | – | Supp-2 |
| Tableau desktop (2020.4) | Tableau, licensed | 4a | – |
| Published datasets | |||
| Cell line expression data | https://www.ebi.ac.uk/arrayexpress47 (E-TABM-157) | Supp-2e, f | – |
| Cell line expression, CNA and methylation datasets | https://www.ncbi.nlm.nih.gov/gds46 (GSE42944; GSE48216) | Supp-2e, f | – |
| Chicken embryo neural crest gene set | Ref. 55, Supplementary Table 1 | 4b–d | Supp-11 |
| Gene ontology resource | http://geneontology.org | – | Supp-11 |
| Genomic locations of solo-WCpGW sites | Ref. 60 | Supp-5c | – |
| hMEC ChIP-seq data | www.epigenomes.ca; ref. 42 | 1f | – |
| hMEC gene expression array data | Gene expression omnibus, https://www.ncbi.nlm.nih.gov/geo/ (GSE16997); and ref. 15 (Tables S5–8) | 1e | – |
| Human reference genome NCBI build 37 (GRCh37/hg19) | UCSC Genome Browser https://genome.ucsc.edu | 2d, Supp-5a | – |
| ICGC gene expression data | Ref. 45, Supplementary Table 7 | – | Supp-8 |
| ICGC HRDetect scores | Ref. 57, Supplementary Table 3b | 5c | – |
| ICGC mutational signatures (COSMIC, v2 SigProfiler) | Ref. 45, Supplementary Table 21B, S21E | 5c | – |
| Illumina Infinium Omni2.5 array data | https://www.ncbi.nlm.nih.gov/geo/ (GSE199579) | 1f, Supp-5b | – |
| METABRIC gene expression & clinical data | EGAD00010000210, EGAD00010000211, EGAS00000000083; EGA portal, via data access committee43 | 2a, 3f, g, Supp-3, Supp-4c, d | Supp-4, Supp-7 |
| MetaCore | https://portal.genego.com | Supp-3 | Supp-4 |
| SOXE-module network metrics | This paper | 4a, f, 5b, g | Supp-10 |
| TCGA clinicopathologic annotation | Ref. 94 | 2a–d, 3a | – |
| TCGA gene copy-number data | Gistic2.Level_4; TCGA Data Analysis Center Firehose44 https://gdac.broadinstitute.org | 2b, 5a, b, Supp-5a | – |
| TCGA gene-level methylation data | Preprocess/meth.by_min_expr_corr; TCGA Data Analysis Center Firehose44 https://gdac.broadinstitute.org | 2b, c | – |
| TCGA Illumina HiSeq RNASeq-v2 RSEM level-3 normalised datasets | illuminahiseq_rnaseqv2-RSEM_genes_normalized (MD5); TCGA Data Analysis Center Firehose44 https://gdac.broadinstitute.org | 2a, c | Supp-4 |
| TCGA Illumina HiSeq RNASeq-v2 RSEM level-3 raw counts | TCGA Data Analysis Center Firehose44 https://gdac.broadinstitute.org | 3a, S3 | Supp-3, 5, 6, 9, 10, 12, 13 |
| TCGA probe-level methylation data | Humanmethylation_450; TCGA Data Analysis Center Firehose44 https://gdac.broadinstitute.org | 5d–f, Supp-5b–d | – |
| Triple-negative breast cancer subtypes (Burstein et al) | Ref. 39, Supplementary Table 19 | 3f, Supp-2b | – |
| Tumour purity for TCGA cases | Supp data-1 (CPE metric) & infinium metric, refs. 81,95 | Multiple | – |
| WGCNA ME dataset, ICGC cases | This paper | Multiple | Supp-8 |
| WGCNA ME dataset, METABRIC cases | This paper | Multiple | Supp-7 |
| WGCNA ME dataset, TCGA normal cases | This paper | Multiple | Supp-12 |
| WGCNA ME dataset, TCGA tumour cases | This paper | Multiple | Supp-6 |
| WGCNA mod membership dataset (TCGA cohort) | This paper | Multiple | Supp-5 |
Supp supplementary.
Code availability
This study used published code and/or publicly available tools (see Table 3).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Jodi M. Saunus, Email: j.saunus@uq.edu.au
Sunil R. Lakhani, Email: s.lakhani@uq.edu.au
Supplementary information
The online version contains supplementary material available at 10.1038/s41523-022-00425-x.
References
- 1.Fulford LG, et al. Basal-like grade III invasive ductal carcinoma of the breast: patterns of metastasis and long-term survival. Breast Cancer Res. 2007;9:R4. doi: 10.1186/bcr1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prat A, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast cancer Res. 2010;12:R68. doi: 10.1186/bcr2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Symmans WF, et al. Long-term prognostic risk after neoadjuvant chemotherapy associated with residual cancer burden and breast cancer subtype. J. Clin. Oncol. 2017;35:1049–1060. doi: 10.1200/JCO.2015.63.1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gao R, et al. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nat. Genet. 2016;48:1119–1130. doi: 10.1038/ng.3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Y, et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014;512:155–160. doi: 10.1038/nature13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yates LR, et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat. Med. 2015;21:751–759. doi: 10.1038/nm.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang F, et al. Intratumor heterogeneity predicts metastasis of triple-negative breast cancer. Carcinogenesis. 2017;38:900–909. doi: 10.1093/carcin/bgx071. [DOI] [PubMed] [Google Scholar]
- 8.Lin B, et al. Modulating cell fate as a therapeutic strategy. Cell Stem Cell. 2018;23:329–341. doi: 10.1016/j.stem.2018.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nguyen DX, Bos PD, Massagué J. Metastasis: from dissemination to organ-specific colonization. Nat. Rev. Cancer. 2009;9:274–284. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- 10.Gupta PB, Pastushenko I, Skibinski A, Blanpain C, Kuperwasser C. Phenotypic plasticity: driver of cancer initiation, progression, and therapy resistance. Cell Stem Cell. 2019;24:65–78. doi: 10.1016/j.stem.2018.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hinohara K, Polyak K. Intratumoral heterogeneity: more than just mutations. Trends Cell Biol. 2019;29:569–579. doi: 10.1016/j.tcb.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bell CC, Gilan O. Principles and mechanisms of non-genetic resistance in cancer. Br. J. Cancer. 2020;122:465–472. doi: 10.1038/s41416-019-0648-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Granit RZ, et al. Regulation of cellular heterogeneity and rates of symmetric and asymmetric divisions in triple-negative breast cancer. Cell Rep. 2018;24:3237–3250. doi: 10.1016/j.celrep.2018.08.053. [DOI] [PubMed] [Google Scholar]
- 14.Keller PJ, et al. Defining the cellular precursors to human breast cancer. Proc. Natl Acad. Sci. USA. 2012;109:2772–2777. doi: 10.1073/pnas.1017626108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lim E, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat. Med. 2009;15:907–913. doi: 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- 16.Molyneux G, et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell. 2010;7:403–417. doi: 10.1016/j.stem.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 17.Proia TA, et al. Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate. Cell Stem Cell. 2011;8:149–163. doi: 10.1016/j.stem.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chaffer CL, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl Acad. Sci. USA. 2011;108:7950–7955. doi: 10.1073/pnas.1102454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hinohara K, et al. KDM5 histone demethylase activity links cellular transcriptomic heterogeneity to therapeutic resistance. Cancer Cell. 2018;34:939–953 e9. doi: 10.1016/j.ccell.2018.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Risom T, et al. Differentiation-state plasticity is a targetable resistance mechanism in basal-like breast cancer. Nat. Commun. 2018;9:3815. doi: 10.1038/s41467-018-05729-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flavahan, W. A., Gaskell, E. & Bernstein, B. E. Epigenetic plasticity and the hallmarks of cancer. Science. 357, eaal2380 (2017). [DOI] [PMC free article] [PubMed]
- 22.Stirzaker C, et al. Methylome sequencing in triple-negative breast cancer reveals distinct methylation clusters with prognostic value. Nat. Commun. 2015;6:5899. doi: 10.1038/ncomms6899. [DOI] [PubMed] [Google Scholar]
- 23.Deblois, G. et al. Epigenetic switch-induced viral mimicry evasion in chemotherapy resistant breast cancer. Cancer Discov. 10, 1312–1329 (2020). [DOI] [PubMed]
- 24.Dravis C, et al. Epigenetic and transcriptomic profiling of mammary gland development and tumor models disclose regulators of cell state plasticity. Cancer Cell. 2018;34:466–482 e6. doi: 10.1016/j.ccell.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu N, Strobl-Mazzulla PH, Bronner ME. Epigenetic regulation in neural crest development. Dev. Biol. 2014;396:159–168. doi: 10.1016/j.ydbio.2014.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Southard-Smith EM, Kos L, Pavan WJ. Sox10 mutation disrupts neural crest development in Dom Hirschsprung mouse model. Nat. Genet. 1998;18:60–64. doi: 10.1038/ng0198-60. [DOI] [PubMed] [Google Scholar]
- 27.Kim J, Lo L, Dormand E, Anderson DJ. SOX10 maintains multipotency and inhibits neuronal differentiation of neural crest stem cells. Neuron. 2003;38:17–31. doi: 10.1016/S0896-6273(03)00163-6. [DOI] [PubMed] [Google Scholar]
- 28.McKeown SJ, Lee VM, Bronner-Fraser M, Newgreen DF, Farlie PG. Sox10 overexpression induces neural crest-like cells from all dorsoventral levels of the neural tube but inhibits differentiation. Dev. Dyn. 2005;233:430–444. doi: 10.1002/dvdy.20341. [DOI] [PubMed] [Google Scholar]
- 29.Dravis C, et al. Sox10 regulates stem/progenitor and mesenchymal cell states in mammary epithelial cells. Cell Rep. 2015;12:2035–2048. doi: 10.1016/j.celrep.2015.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Z, et al. FGF signaling activates a Sox9-Sox10 pathway for the formation and branching morphogenesis of mouse ocular glands. Development. 2014;141:2691–2701. doi: 10.1242/dev.108944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Athwal HK, et al. Sox10 regulates plasticity of epithelial progenitors toward secretory units of exocrine glands. Stem Cell Rep. 2019;12:366–380. doi: 10.1016/j.stemcr.2019.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo W, et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell. 2012;148:1015–1028. doi: 10.1016/j.cell.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mertelmeyer S, et al. The transcription factor Sox10 is an essential determinant of branching morphogenesis and involution in the mouse mammary gland. Sci. Rep. 2020;10:17807. doi: 10.1038/s41598-020-74664-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim YJ, et al. Generation of multipotent induced neural crest by direct reprogramming of human postnatal fibroblasts with a single transcription factor. Cell Stem Cell. 2014;15:497–506. doi: 10.1016/j.stem.2014.07.013. [DOI] [PubMed] [Google Scholar]
- 35.Ivanov SV, et al. Diagnostic SOX10 gene signatures in salivary adenoid cystic and breast basal-like carcinomas. Br. J. Cancer. 2013;109:444–451. doi: 10.1038/bjc.2013.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Panaccione A, Guo Y, Yarbrough WG, Ivanov SV. Expression profiling of clinical specimens supports the existence of neural progenitor-like stem cells in basal breast cancers. Clin. Breast Cancer. 2017;17:298–306 e7. doi: 10.1016/j.clbc.2017.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cimino-Mathews A, et al. Neural crest transcription factor Sox10 is preferentially expressed in triple-negative and metaplastic breast carcinomas. Hum. Pathol. 2013;44:959–965. doi: 10.1016/j.humpath.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jamidi, S. K. et al. SOX10 as a sensitive marker for triple negative breast cancer. Histopathology77, 936–948 (2020). [DOI] [PubMed]
- 39.Burstein MD, et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. 2015;21:1688–1698. doi: 10.1158/1078-0432.CCR-14-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu N, Strobl-Mazzulla PH, Simoes-Costa M, Sanchez-Vasquez E, Bronner ME. DNA methyltransferase 3B regulates duration of neural crest production via repression of Sox10. Proc. Natl Acad. Sci. USA. 2014;111:17911–17916. doi: 10.1073/pnas.1318408111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Strobl-Mazzulla PH, Bronner ME. A PHD12-Snail2 repressive complex epigenetically mediates neural crest epithelial-to-mesenchymal transition. J. Cell Biol. 2012;198:999–1010. doi: 10.1083/jcb.201203098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pellacani D, et al. Analysis of normal human mammary epigenomes reveals cell-specific active enhancer states and associated transcription factor networks. Cell Rep. 2016;17:2060–2074. doi: 10.1016/j.celrep.2016.10.058. [DOI] [PubMed] [Google Scholar]
- 43.Curtis C, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.TCGA. Cancer Genome Atlas Network: Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nik-Zainal S, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016;534:47–54. doi: 10.1038/nature17676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Daemen A, et al. Modeling precision treatment of breast cancer. Genome Biol. 2013;14:R110. doi: 10.1186/gb-2013-14-10-r110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Neve RM, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McCart Reed AE, et al. The Brisbane breast bank. Open J. Bioresour. 2018;5:5. doi: 10.5334/ojb.33. [DOI] [Google Scholar]
- 49.Saunus, J. M. et al. Multidimensional phenotyping of breast cancer cell lines to guide preclinical research. Breast Cancer Res. Treat. 167, 289–301 (2018). [DOI] [PubMed]
- 50.Qi J, et al. SOX10 - A novel marker for the differential diagnosis of breast metaplastic squamous cell carcinoma. Cancer Manag. Res. 2020;12:4039–4044. doi: 10.2147/CMAR.S250867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McCart Reed AE, et al. Phenotypic and molecular dissection of metaplastic breast cancer and the prognostic implications. J. Pathol. 2019;247:214–227. doi: 10.1002/path.5184. [DOI] [PubMed] [Google Scholar]
- 52.Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005;4:Article17. doi: 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- 53.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinforma. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Denkert C, et al. Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: a pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol. 2018;19:40–50. doi: 10.1016/S1470-2045(17)30904-X. [DOI] [PubMed] [Google Scholar]
- 55.Simoes-Costa M, Tan-Cabugao J, Antoshechkin I, Sauka-Spengler T, Bronner ME. Transcriptome analysis reveals novel players in the cranial neural crest gene regulatory network. Genome Res. 2014;24:281–290. doi: 10.1101/gr.161182.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pellacani D, Tan S, Lefort S, Eaves CJ. Transcriptional regulation of normal human mammary cell heterogeneity and its perturbation in breast cancer. EMBO J. 2019;38:e100330. doi: 10.15252/embj.2018100330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Davies H, et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 2017;23:517–525. doi: 10.1038/nm.4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hayano, M. et al. DNA break-induced epigenetic drift as a cause of mammalian aging. Preprint at bioRxiv10.1101/808659 (2019).
- 59.Yang, J.-H. et al. Erosion of the Epigenetic Landscape and Loss of Cellular Identity as a Cause of Aging in Mammals. BioRxiv preprint: 10.1101/808642. (2019).
- 60.Zhou W, et al. DNA methylation loss in late-replicating domains is linked to mitotic cell division. Nat. Genet. 2018;50:591–602. doi: 10.1038/s41588-018-0073-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Medvedeva YA, et al. EpiFactors: a comprehensive database of human epigenetic factors and complexes. Database. 2015;2015:bav067. doi: 10.1093/database/bav067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jin C, et al. TET1 is a maintenance DNA demethylase that prevents methylation spreading in differentiated cells. Nucleic Acids Res. 2014;42:6956–6971. doi: 10.1093/nar/gku372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Putiri EL, et al. Distinct and overlapping control of 5-methylcytosine and 5-hydroxymethylcytosine by the TET proteins in human cancer cells. Genome Biol. 2014;15:R81. doi: 10.1186/gb-2014-15-6-r81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Good CR, et al. TET1-mediated hypomethylation activates oncogenic signaling in triple-negative breast cancer. Cancer Res. 2018;78:4126–4137. doi: 10.1158/0008-5472.CAN-17-2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Feinberg AP, Ohlsson R, Henikoff S. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006;7:21–33. doi: 10.1038/nrg1748. [DOI] [PubMed] [Google Scholar]
- 66.Wahl GM, Spike BT. Cell state plasticity, stem cells, EMT, and the generation of intra-tumoral heterogeneity. NPJ Breast Cancer. 2017;3:14. doi: 10.1038/s41523-017-0012-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Visvader JE, Stingl J. Mammary stem cells and the differentiation hierarchy: current status and perspectives. Genes Dev. 2014;28:1143–1158. doi: 10.1101/gad.242511.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liao C, Zhang Q. BBOX1 promotes triple-negative breast cancer progression by controlling IP3R3 stability. Mol. Cell Oncol. 2020;7:1813526. doi: 10.1080/23723556.2020.1813526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liao C, et al. Identification of BBOX1 as a therapeutic target in triple-negative breast cancer. Cancer Discov. 2020;10:1706–1721. doi: 10.1158/2159-8290.CD-20-0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhu L, Pan R, Zhou D, Ye G, Tan W. BCL11A enhances stemness and promotes progression by activating Wnt/beta-catenin signaling in breast cancer. Cancer Manag. Res. 2019;11:2997–3007. doi: 10.2147/CMAR.S199368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Errico A. Genetics: BCL11A-targeting triple-negative breast cancer? Nat. Rev. Clin. Oncol. 2015;12:127. doi: 10.1038/nrclinonc.2015.10. [DOI] [PubMed] [Google Scholar]
- 72.Khaled WT, et al. BCL11A is a triple-negative breast cancer gene with critical functions in stem and progenitor cells. Nat. Commun. 2015;6:5987. doi: 10.1038/ncomms6987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Saggese, P. et al. Metabolic regulation of epigenetic modifications and cell differentiation in cancer. Cancers 12, 3788 (2020). [DOI] [PMC free article] [PubMed]
- 74.Simoes-Costa M, Bronner ME. Establishing neural crest identity: a gene regulatory recipe. Development. 2015;142:242–257. doi: 10.1242/dev.105445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Saunus JM, et al. Integrated genomic and transcriptomic analysis of human brain metastases identifies alterations of potential clinical significance. J. Pathol. 2015;237:363–378. doi: 10.1002/path.4583. [DOI] [PubMed] [Google Scholar]
- 76.Johnston RL, et al. High content screening application for cell-type specific behaviour in heterogeneous primary breast epithelial subpopulations. Breast Cancer Res. 2016;18:18. doi: 10.1186/s13058-016-0681-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pavey S, et al. Microarray expression profiling in melanoma reveals a BRAF mutation signature. Oncogene. 2004;23:4060–4067. doi: 10.1038/sj.onc.1207563. [DOI] [PubMed] [Google Scholar]
- 78.Momeny M, et al. Heregulin-HER3-HER2 signaling promotes matrix metalloproteinase-dependent blood-brain-barrier transendothelial migration of human breast cancer cell lines. Oncotarget. 2015;6:3932–3946. doi: 10.18632/oncotarget.2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vargas AC, et al. Gene expression profiling of tumour epithelial and stromal compartments during breast cancer progression. Breast Cancer Res. Treat. 2012;135:153–165. doi: 10.1007/s10549-012-2123-4. [DOI] [PubMed] [Google Scholar]
- 80.Tian Y, et al. ChAMP: updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics. 2017;33:3982–3984. doi: 10.1093/bioinformatics/btx513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Aran D, Sirota M, Butte AJ. Systematic pan-cancer analysis of tumour purity. Nat. Commun. 2015;6:8971. doi: 10.1038/ncomms9971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ritchie ME, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. Usa. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Supek F, Bosnjak M, Skunca N, Smuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE. 2011;6:e21800. doi: 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Blondel VD, Guillaume J-L, Lambiotte R, Lefebvre E. Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp. 2008;2008:P10008. doi: 10.1088/1742-5468/2008/10/P10008. [DOI] [Google Scholar]
- 86.Lambiotte, R., Delvenne, J. C. & Barahona, M. IEEE Trans. Netw. Sci. Eng.1, 76-90 10.1109/TNSE.2015.2391998 (2014).
- 87.Fernandez NF, et al. Clustergrammer, a web-based heatmap visualization and analysis tool for high-dimensional biological data. Sci. Data. 2017;4:170151. doi: 10.1038/sdata.2017.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Foroutan M, et al. Single sample scoring of molecular phenotypes. BMC Bioinforma. 2018;19:404. doi: 10.1186/s12859-018-2435-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kalita-de Croft P, et al. Clinicopathologic significance of nuclear HER4 and phospho-YAP(S(127)) in human breast cancers and matching brain metastases. Ther. Adv. Med. Oncol. 2020;12:1758835920946259. doi: 10.1177/1758835920946259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tarek MA, et al. SPAG5 as a prognostic biomarker and chemotherapy sensitivity predictor in breast cancer: a retrospective integrated genomic transcriptomic and protein analysis. Lancet Oncol. 2016;17:1004–1018. doi: 10.1016/S1470-2045(16)00174-1. [DOI] [PubMed] [Google Scholar]
- 91.Tarek MA, et al. Association of Sperm-Associated Antigen 5 and Treatment Response in Patients With Estrogen Receptor–Positive Breast Cancer. JAMA Network Open. 2020;3:e209486. doi: 10.1001/jamanetworkopen.2020.9486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kalaw E, et al. Metaplastic breast cancers frequently express immune checkpoint markers FOXP3 and PD-L1. Br J Cancer. 2020;123:1665–1672. doi: 10.1038/s41416-020-01065-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Boyle EI, et al. GO::TermFinder–open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics. 2004;20:3710–3715. doi: 10.1093/bioinformatics/bth456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liu J, et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell. 2018;173:400–416.e11. doi: 10.1016/j.cell.2018.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zheng, X., Zhang, N., Wu, H. J. & Wu, H. Estimating and accounting for tumor purity in the analysis of DNA methylation data from cancer studies. Genom Biol 18, 10.1186/s13059-016-1143-5 (2017). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Published datasets used in this paper are outlined in Table 3. Network data generated by the study are also outlined in Table 3, and available as supplementary data. Raw DNA methylation array data for FACS-sorted normal breast epithelial cell subsets are available from the Gene Expression Omnibus (GSE199579; Table 3).
Table 3.
software, code, and published datasets.
| ResRource | Source, identifier and relevant citations | Related figure(s) | Related table(s) |
|---|---|---|---|
| Software packages and code | |||
| ChAMP | https://bioconductor.org/packages/release/bioc/html/ChAMP.html80 | 5d–f | – |
| Clustergrammer | https://maayanlab.cloud/clustergrammer/87 | 3b | Supp-10 |
| Community detection algorithms | Refs. 85,86 | Supp-4a | – |
| Epifactors database | https://epifactors.autosome.ru61 | Supp-5e | – |
| FACSDiva™ | BD Biosciences, licensed | 1f, Supp-6a | – |
| FCS Express (v7) | De Novo Software, licensed | 1f, Supp-6a | – |
| GSEAPreranked | https://genepattern.org83 | 3c, 4d, 5f, Supp-3 | 1, Supp-4, Supp-9 |
| Ingenuity Pathways Analysis (IPA) | Ingenuity, licensed | – | 1 |
| MATLAB | Mathworks, licensed | Supp-4a | Supp-10 |
| Princeton Generic GO term finder | https://go.princeton.edu93 | 5a | Supp-13, 14 |
| Prism (v8.4.3) | GraphPad, licensed | Multiple | S2 |
| R package, Cluster | https://cran.r-project.org/web/packages/cluster/index.html | 5f | – |
| R package, FlashClust | https://cran.r-project.org/web/packages/flashClust/index.html | 5f, g | Supp-14 |
| R package, Limma | https://www.bioconductor.org/packages/release/bioc/html/limma.html | Supp-3 | Supp-3 |
| R package, t-SNE | https://CRAN.R-project.org/package=Rtsne | 5d | – |
| R package, WGNCA | https://cran.r-project.org/web/packages/WGCNA/index.html52,53 | Multiple | Multiple |
| REVIGO | http://revigo.irb.hr | Supp-3 | Supp-4 |
| Singscore | https://www.bioconductor.org/packages/release/bioc/html/singscore.html88 | 4c | – |
| SPSS | IBM, licensed | – | Supp-2 |
| Tableau desktop (2020.4) | Tableau, licensed | 4a | – |
| Published datasets | |||
| Cell line expression data | https://www.ebi.ac.uk/arrayexpress47 (E-TABM-157) | Supp-2e, f | – |
| Cell line expression, CNA and methylation datasets | https://www.ncbi.nlm.nih.gov/gds46 (GSE42944; GSE48216) | Supp-2e, f | – |
| Chicken embryo neural crest gene set | Ref. 55, Supplementary Table 1 | 4b–d | Supp-11 |
| Gene ontology resource | http://geneontology.org | – | Supp-11 |
| Genomic locations of solo-WCpGW sites | Ref. 60 | Supp-5c | – |
| hMEC ChIP-seq data | www.epigenomes.ca; ref. 42 | 1f | – |
| hMEC gene expression array data | Gene expression omnibus, https://www.ncbi.nlm.nih.gov/geo/ (GSE16997); and ref. 15 (Tables S5–8) | 1e | – |
| Human reference genome NCBI build 37 (GRCh37/hg19) | UCSC Genome Browser https://genome.ucsc.edu | 2d, Supp-5a | – |
| ICGC gene expression data | Ref. 45, Supplementary Table 7 | – | Supp-8 |
| ICGC HRDetect scores | Ref. 57, Supplementary Table 3b | 5c | – |
| ICGC mutational signatures (COSMIC, v2 SigProfiler) | Ref. 45, Supplementary Table 21B, S21E | 5c | – |
| Illumina Infinium Omni2.5 array data | https://www.ncbi.nlm.nih.gov/geo/ (GSE199579) | 1f, Supp-5b | – |
| METABRIC gene expression & clinical data | EGAD00010000210, EGAD00010000211, EGAS00000000083; EGA portal, via data access committee43 | 2a, 3f, g, Supp-3, Supp-4c, d | Supp-4, Supp-7 |
| MetaCore | https://portal.genego.com | Supp-3 | Supp-4 |
| SOXE-module network metrics | This paper | 4a, f, 5b, g | Supp-10 |
| TCGA clinicopathologic annotation | Ref. 94 | 2a–d, 3a | – |
| TCGA gene copy-number data | Gistic2.Level_4; TCGA Data Analysis Center Firehose44 https://gdac.broadinstitute.org | 2b, 5a, b, Supp-5a | – |
| TCGA gene-level methylation data | Preprocess/meth.by_min_expr_corr; TCGA Data Analysis Center Firehose44 https://gdac.broadinstitute.org | 2b, c | – |
| TCGA Illumina HiSeq RNASeq-v2 RSEM level-3 normalised datasets | illuminahiseq_rnaseqv2-RSEM_genes_normalized (MD5); TCGA Data Analysis Center Firehose44 https://gdac.broadinstitute.org | 2a, c | Supp-4 |
| TCGA Illumina HiSeq RNASeq-v2 RSEM level-3 raw counts | TCGA Data Analysis Center Firehose44 https://gdac.broadinstitute.org | 3a, S3 | Supp-3, 5, 6, 9, 10, 12, 13 |
| TCGA probe-level methylation data | Humanmethylation_450; TCGA Data Analysis Center Firehose44 https://gdac.broadinstitute.org | 5d–f, Supp-5b–d | – |
| Triple-negative breast cancer subtypes (Burstein et al) | Ref. 39, Supplementary Table 19 | 3f, Supp-2b | – |
| Tumour purity for TCGA cases | Supp data-1 (CPE metric) & infinium metric, refs. 81,95 | Multiple | – |
| WGCNA ME dataset, ICGC cases | This paper | Multiple | Supp-8 |
| WGCNA ME dataset, METABRIC cases | This paper | Multiple | Supp-7 |
| WGCNA ME dataset, TCGA normal cases | This paper | Multiple | Supp-12 |
| WGCNA ME dataset, TCGA tumour cases | This paper | Multiple | Supp-6 |
| WGCNA mod membership dataset (TCGA cohort) | This paper | Multiple | Supp-5 |
Supp supplementary.
This study used published code and/or publicly available tools (see Table 3).