

Abstract
The evolution of antimicrobial resistance in Clostridioides difficile has markedly shaped its epidemiology and detrimentally impacted patient care. C. difficile exhibits resistance to multiple classes of antimicrobials, due to accumulation of horizontally acquired resistance genes and de novo mutations to drug targets. Particularly worrying is that declines in clinical success of firstline CDI antimicrobials coincide with the spread of strains that are more resistant to these drugs. Yet, there is still much to learn regarding the prevalence of genetic elements in clinical isolates, their molecular mechanisms, and the extent to which this information can be translated to develop molecular diagnostics that improve antimicrobial prescribing and antimicrobial stewardship approaches for CDI. Thus, this perspective discusses current understanding and knowledge gaps of antimicrobial resistance mechanisms in C. difficile, emphasizing on CDI therapies.
Keywords: Antibiotic treatment failure, susceptibility testing, molecular diagnostics
THE IMPACT OF ANTIMICROBIAL RESISTANCE IN CDI
Clostridioides difficile infection (CDI) is a leading cause of antimicrobial-associated diarrhea in hospitalized elderly patients. For over 40 years, metronidazole and vancomycin have been the firstline therapies, while fidaxomicin, approved in 2011, has been largely used to treat recurrent disease (rCDI). Today, the antimicrobial therapeutic model for CDI has changed, where the 2021 IDSA/SHEA and ESCMID guidelines recommend fidaxomicin as the drug of choice, vancomycin as an alternative, and metronidazole only if the other two options are unavailable [1,2]. Prior to the 2021 antimicrobial therapy guidelines, approximately 20% or more patients experience rCDI and 45–65% of these patients experience successive recurrent episodes [3]. Nonetheless, it is generally unclear how resistance to CDI antimicrobials influences treatment outcomes and the onset of rCDI (Figure 1). This is astonishing, since in other infections resistance is a common reason why antimicrobials fail. However, CDI treatment outcomes are not normally explained in the context of antimicrobial resistance (AMR), since anaerobic susceptibility testing of patient isolates is not routinely performed as part of the diagnostic work-up for CDI. Increasing reports of resistance to traditional and new CDI antibiotics warrant reevaluation of this view. C. difficile has also evolved resistance to fidaxomicin. In our view, the diagnostic work-up for CDI could be revolutionized by integrating rapid molecular diagnostics to identify AMR mechanisms in C. difficile. However, there is a fundamental need to delineate resistance mechanisms in C. difficile, in terms of their prevalence among clinical strains, impact on treatment responses, and effects on C. difficile pathophysiology. In Figure 2 and Table 1, we summarize the current knowledge of AMR mechanisms in C. difficile, with an emphasis on antimicrobial options for CDI. In this opinion article, we address the critical knowledge gaps in current understanding of genetically encoded resistance to antimicrobials in C. difficile, with the view of evaluating whether this information can be harnessed for molecular diagnostics and epidemiological surveillance, which could benefit prescribing practices and antimicrobial stewardship policies.
Figure 1.
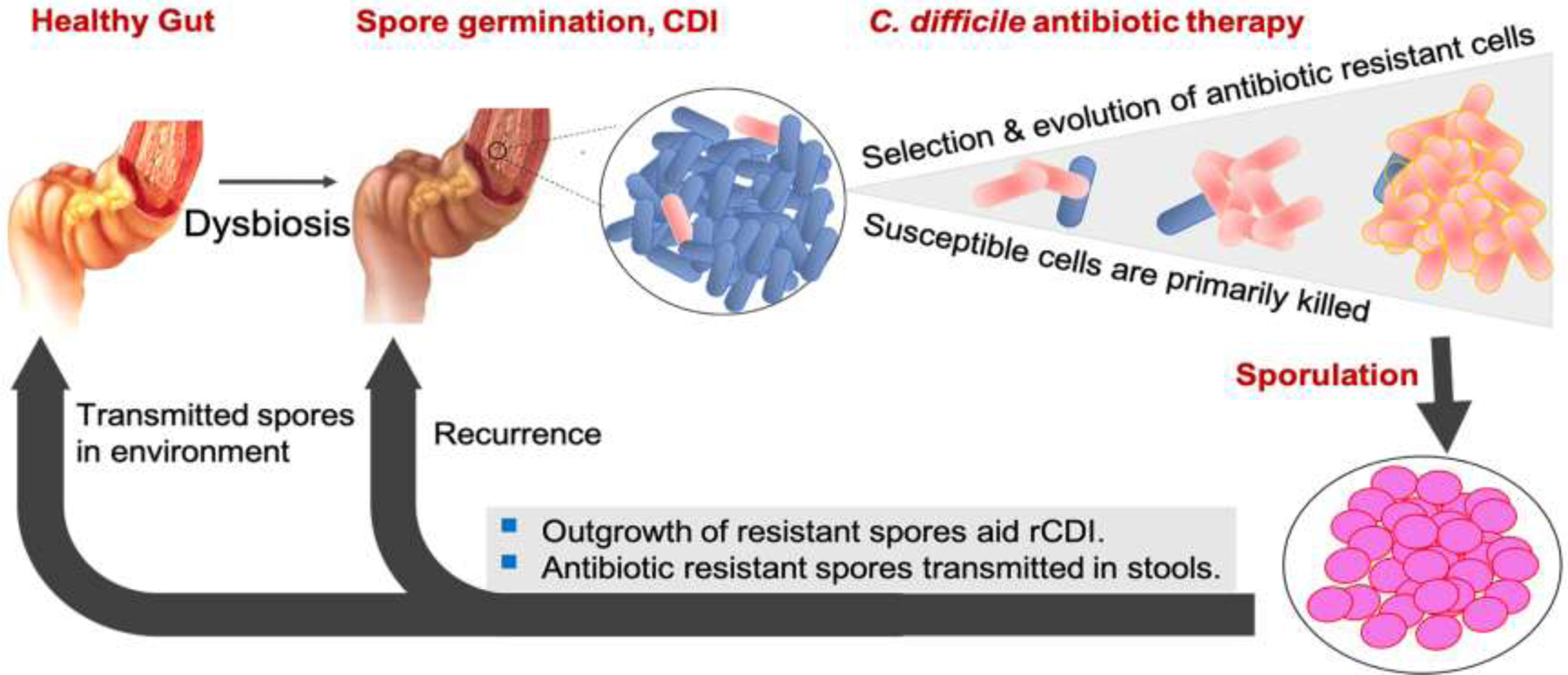
A Conceptual model demonstrating how genetic resistance to firstline antimicrobials could influence CDI recurrence. Resistant cells, including low-level resistant mutants, formed by spontaneous mutations could be selectively advantaged, even in drugs that achieve high luminal concentrations (e.g., fidaxomicin, vancomycin and rifaximin); low-level resistant mutants might be fitter than their wild type counterparts in niches with sub-physiological concentrations of drug. Sporulation by surviving low-level resistant cells increases the risk for recurrent disease. Drug-resistant spores transmitted in patient stools have an even higher likelihood of causing recurrence or acquiring further mutations that cause higher-level resistance and/or compensate for fitness costs. Strains with high-level resistance are better able to survive physiological concentrations of drug.
Figure 2. Laboratory and clinically associated mechanisms of resistance to firstline anti-C. difficile antimicrobials.
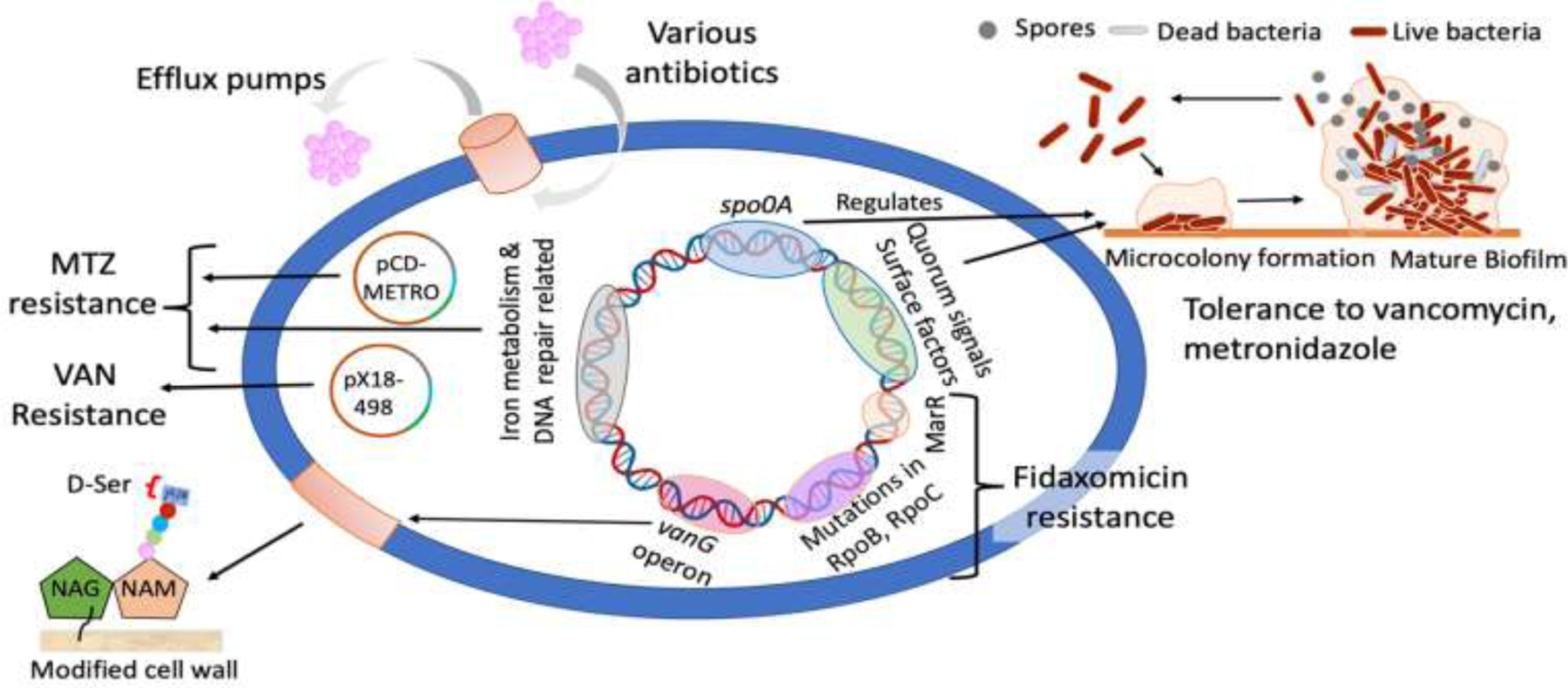
Clinical or laboratory studied mechanisms are indicated below. Metronidazole resistance mechanisms can involve the plasmid pCD-METRO (clinical) and endogenous genes (e.g., iron and redox metabolism [laboratory]). Vancomycin resistance is either encoded by the plasmid px18–498 (clinical) or the vanGCd operon (clinical and laboratory) that modifies the peptidoglycan by replacing the terminal D-Ala with D-Ser. Mutations in rpoB and rpoC are responsible for the reduced susceptibility to fidaxomicin (clinical and laboratory), but a mutation to MarR homolog CD2212 is another putative factor (laboratory). Biofilms (laboratory) that are metabolically inactive may also provide resistance to metronidazole and vancomycin. Biofilm formation can be promoted by selection pressure from antimicrobials, from quorum sensing signals or controlled by spo0A regulatory mechanisms [69]. Efflux pumps (laboratory) are also active in C. difficile e.g., deletion of the ATP-binding cassette transporter CD2068 in C. difficile 630Δerm causes a modest (1.4-fold) reduction in the activity of metronidazole [70].
Table 1.
Mechanisms of resistance to antimicrobials recommended to treat CDI.a
| Antimicrobial | Mechanism(s) of resistance in clinical or laboratory strains | Reported impact on therapy and phenotype |
|---|---|---|
| Vancomycin | Clinical isolates | |
| Inhibits cell wall synthesis by binding Lipid II motif of D-Ala-D-Ala | Putative N-acetylmuramoyl-L-alanine amidase encoded on broad host range plasmid pX18–498 [48]; predicted to preserve cell wall integrity | Associated with reduced therapeutic response in patients; strains with pX18–498 had MICs=2 μg/ml versus 0.5 μg/ml without the plasmid |
| Mutations to VanSR cause constitutive transcription of vanGCd and production of modified Lipid II; the D-Ala-D-Ser modified Lipid II has ~7-fold educed binding of vancomycin [38]. Mutations in clinical isolates are Ser313Phe and Thr349lle in VanS and Thr115Ala in VanR. | Unknown clinical impact; strains of MICs 4–8 μg/ml, MBCs ~64 μg/ml; strains were responsive to autolysis inducing conditions | |
| bClinical isolate with mutations in VanR (Thr115Ala), SdaB (His18Ala) and GdpP (Phe43frameshift) | MIC=8 μg/ml; response to autolysis inducing conditions is unknown | |
| Laboratory generated mutants | ||
| Mutations in VanS (Arg314Leu or Gly319Asp) and TrkA (Gln26STOP) [38] | MIC=8–16 μg/ml, MBCs ≥1024 μg/ml; strains were much less responsive to autolysis inducing conditions | |
| Mutations in MurG (Pro108Leu), GdpP (Glu327Stop) and SdaB (ΔAla295) [45] | MIC=16 μg/ml | |
| Mutation in RpoC (Asp244Tyr) [45] | MIC=8 μg/ml | |
|
| ||
| Metronidazole | Identified in clinical isolates | |
| Free radicals from nitro group reduction damage DNA and proteins and deplete cellular thiols | Transferable plasmid pCD-METRO (exact resistance gene on the plasmid is unknown) [28] | Clinical isolate from patient with recurrent CDI, treated with metronidazole; MICs ≥ 8 μg/ml |
| Ala1018Val mutation in PFOR, predicted to be adjacent to binding domain for 4Fe-4S and thiamine pyrophosphate [20] | MICs 8–16 μg/ml; complementation with wild type PFOR increased susceptibility (MIC=4 μg/ml), but full susceptibility was not restored with complementation suggesting other mechanisms of resistance occurred in the strain | |
| Laboratory generated mutants | ||
| Evolved mutants showing mutations to FeoB1 (Glu38frameshift or Lys40frameshift), PFOR (Pro32Leu or Gln803Arg), XDH (synonymous change GAG-2070-GAA) and IscR (Lys51frameshift or Val76Ala) [20] | Experimental validation showed ΔfeoB1 deletion caused 4-fold decrease in susceptibility (MIC=1 μg/ml in ΔfeoB1 mutant); higher-level resistance (MICs=8 μg/ml) occurred by silencing of genes for PFOR, XDH or IscR in the ΔfeoB1 deletion mutant | |
| An evolved mutant with mutations in HemN (Tyr214frameshift), ThiH (Ser328Phe), PFOR (Gly423Glu) and GlyC (Ala229Thr) [22] | MIC=8–16 μg/ml; involvement of PFOR in the strain was experimentally validated; complementation with wild type PFOR did not restore full susceptibility [20] | |
|
| ||
| Fidaxomicin | Identified in clinical isolates | |
| Inhibits transcription by binding to RNA polymerase clamp domain | Mutations to RNAP β subunit (Val1143Gly and Val1143Asp) [56,57] | Clinical impact unknown, but strain with Val1143Gly mutation was isolated from a patient with recurrent CDI after fidaxomicin therapy; strains with Val1143Gly and Val1143Asp had MICs of 16 μg/ml and >64 μg/ml, respectively |
| Laboratory generated mutants | ||
| Evolved and engineered mutations to RNAP β subunit (Val1143Gly, Gln1073Arg, Val1143Asp, and Val1143Phe) [54,55] | MICs=2 to >64 μg/ml | |
| An evolved mutant with Phe117frameshift in homolog of transcriptional regulator MarR [45] | MIC=1 μg/ml (4-fold increase over wild type progenitor) | |
|
| ||
| Rifaximin | Identified in clinical isolates | |
| Inhibits transcription by binding to RNA polymerase β subunit (RpoB) | Spectrum of mutations in the rifamycin resistance determining region of RNAP β subunit (e.g., Ser488Tyr, Asp492Tyr, His502Asn/Tyr, Arg505Lys, Ser550Phe/Tyr) [62] | Resistance arose within 32 hours of rifaximin therapy for recurrent CDI; the isolates had mutations of His502Tyr or His502Tyr/Pro496Ser and rifampin MICs ≥32 μg/ml [67] (rifaximin MICs are typically >1024 μg/ml against rifamycin-resistant mutants) |
|
| ||
| Tetracyclines (tigecycline) | Identified in clinical isolates | |
| Inhibits protein synthesis by binding to 30S ribosomal subunit | Ribosomal protection proteins e.g., Tet(M), Tet(W) found on various mobile genetic elements. TetM is the most common resistance mechanism; these mechanisms do not confer resistance to tigecycline | Clinical outbreaks and evolution of tetracycline resistance in agriculture-associated ribotype ribotype 078 occur in response to tetracycline use; MIC ≥16 μg/ml [68] |
Proteins abbreviated above
VanSR (CD630_1625, CD630_1624), VanSR two component system upstream of vanGCd operon; TrkA (CD630_0697), Trk system potassium uptake protein; SdaB (CD630_3222), L-serine deaminase; GdpP (CD630_3659), cyclic-di-AMP phosphodiesterase; MurG (CD630_2725), peptidoglycan glycosyltransferase; PFOR (CD630_2682), pyruvate: ferredoxin oxidoreductase; FeoB1 (CD630_1479), ferrous iron transporter; XDH (CD630_3177), xanthine dehydrogenase; IscR (CD630_1278), iron sulfate cluster regulator; HemN (CD630_2464), oxygen independent coproporphyrinogen III oxidase; ThiH (CD630_1705), thiamine biosynthesis enzyme; GlyC (CD630_2630), glycerol-3-phosphate dehydrogenase; MarR (CD630_2212), multiple antibiotic resistance regulator homolog; RNAP (RNA Polymerase); RpoB (CD630_0066), RNA polymerase subunit beta; RpoC (CD630_0067), RNA polymerase subunit beta’; Tet(M), Tetracycline resistance protein TetM; Tet(W), Tetracycline resistance protein TetW.
Unpublished data by the authors.
ACQUIRED RESISTANCE TO NON-CDI ANTIMICROBIALS
Almost all major classes of antimicrobials can induce CDI, but ampicillin, amoxicillin, cephalosporins, clindamycin and fluoroquinolones pose higher risk [4]. Although spores are naturally refractory to most antimicrobials, C. difficile has acquired resistance determinants to several classes of CDI causing antimicrobials [5,6]. This makes C. difficile adept at taking advantage of selection pressures imposed by antimicrobials commonly used in hospital and community settings, including in agriculture. In hospitals, this is best exemplified by the antimicrobials lincomycin, clindamycin, cephalosporins and fluoroquinolones. In the late 1980s and early 1990s, clindamycin’s use in U.S. hospitals was associated with outbreaks of clindamycin-resistant strains of the REA (Restriction Endonuclease Analysis) group J that can include Ribotype 001 [7]. Clindamycin resistance in clinical strains of C. difficile is primarily due to the erythromycin resistance methylase (ErmB), which methylates the 23S rRNA and prevents binding of clindamycin and related members of the macrolide-lincosamide-streptogramin (MLS) class of antimicrobials. The ermB gene is encoded on transposons Tn5398, Tn6194, Tn6215 and Tn6218 [5]. Possession of these mobile elements acts as a reservoir for acquisition of other resistance genes. Indeed, Tn6218-like mobile elements were also shown to encode determinants that conferred resistance to multiple unrelated antimicrobials e.g., cfr-like 23S rRNA methyltransferases, matE (multidrug and toxic compound extrusion) and aacA-aphD aminoglycoside resistance determinants) [5]. These observations signify that horizontal gene transfer is integral to both the evolution of multidrug resistance in C. difficile, permitting it to respond to various antimicrobial selection pressures, and the maintenance of a reservoir for the intra and inter-species spread of AMR genes in the intestinal ecosystem.
C. difficile is also intrinsically resistant to some β-lactam antimicrobials, such as cephalosporins [8,9], which are commonly prescribed drugs. Recent studies implicate an endogenous class D β-lactamase, BlaCDD (encoded by CD630_04580), in intrinsic resistance to some β-lactam antimicrobials since the enzyme hydrolyzes the β-lactam ring of different sub-types of β-lactam antimicrobials (including penicillins, cephalosporins and monobactams) [8,9]. However, C. difficile shows varying levels of susceptibility to β-lactams, e.g., MICs for ampicillin, imipenem, ceftriaxone and aztreonam are 4, 4, 64, 2048 μg/mL, respectively) and deletion of CD630_04580 either had no effect on MICs or reduced MICs by 2–4-fold [8,9]. This suggests that a combination of BlaCDD and other β-lactamases, and/or differences in the affinities of β-lactams to C. difficile penicillin-binding proteins (PBPs), might account for variations in susceptibilities. Furthermore, it is debatable whether blaCDD is expressed at reduced levels [8] or highly expressed [9], and thus research is needed to define how C. difficile regulates its β-lactamase(s) and their involvement in β-lactam resistance and cell physiology. Since carbapenems effectively bind to multiple PBPs, particularly PBPs 1, 2 and 3, and are more resistant to β-lactamases, including BlaCDD, they are used to treat severe infections, including Gram-negative infections. C. difficile is still mostly susceptible to carbapenems, but resistance has emerged among various ribotypes, particularly ribotype 017 that is the main C. difficile lineage strain in Asia [10]. Imipenem resistance in isolates of RT017 (MIC >32 μg/mL) was associated with mutations near the transpeptidase domains of PBP1 (Ala555Thr) and PBP3 (Tyr721Ser), suggesting they decrease β-lactam affinity [10]. In other organisms, such as Streptococcus pneumoniae, mutations within PBPs are a leading cause of resistance to β-lactams [11]. RT017 strains also carry a fifth PBP (PBP5) on a mobile element, which is suggested to facilitate de novo mutations causing high-level imipenem resistance [10]. Further research is needed to understand the impact of PBP mutations on C. difficile pathophysiology, including the cellular function of PBP5 in facilitating resistance development. Interestingly, cephamycins (analogs of cephalosporins) inhibit sporulation in C. difficile by binding to the PBP SpoVD, a homolog of PBP4; cephamycins preferably inhibit PBP4 in bacteria [12]. However, the clinical utility of cephamycins for CDI is debatable [13]. Nonetheless, structural insights into C. difficile PBPs, such as SpoVD, could lead to novel therapeutics for CDI.
It has been established that widespread use of fluoroquinolones enabled the global spread fluoroquinolone-resistant epidemic ribotype 027 [14,15]. A Thr82Ile mutation in DNA Gyrase A, a Type II topoisomerase for DNA supercoiling, is the most common substitution in these fluoroquinolone-resistant strains [14,15]. This mutation does not appear to impose a fitness cost and paradoxically may enhance fitness [16,17], implying that fluoroquinolone resistance is unlikely to disadvantage C. difficile in the absence of selection pressure. Hence, observed reductions in CDI rates following fluoroquinolone restriction policies [18,19] may not be driven by fitness costs.
RESISTANCE TO CDI ANTIMICROBIALS
Metronidazole
Metronidazole is a nitro-group-containing drug that is bioreductively activated within cells, producing free radicals that damage cellular components, including DNA and metalloclusters of proteins, and deplete cellular low-molecular weight thiols [20,21]. Metronidazole resistance was first reported in the early 2000s, but only recently have resistance mechanisms been elucidated. This is partly due to the inaccurate depiction of metronidazole resistance as an unstable or heterogenous phenotype [22–24]. Rather, recent work has reported that the biological cofactor heme was required to reproducibly detect metronidazole-resistant C. difficile [25,26]. Indeed, when susceptibility testing agars lacked heme, or when it was photo-decomposed in agars, then metronidazole-resistant strains appeared to be susceptible [25,26]. After this discovery it was shown that C. difficile strains with metronidazole MICs of ≥1 μg/mL were more prone to cause the failure of metronidazole therapy in adult patients diagnosed with CDI [27]. Resistance breakpoints for metronidazole are: >2 μg/mL per the European Committee on Antimicrobial Susceptibility Testing (EUCAST) and ≥32 μg/mL per the Clinical and Laboratory Standards Institute (CLSI). In our opinion since the physiological concentration of metronidazole is on average 9.3 ± 7.5 μg/g wet weight of watery stools, then the CLSI breakpoint may not properly reflect the concentrations of metronidazole in the colon of CDI patients.
Over the last decade genetic mechanisms of metronidazole resistance have been described. Recently, plasmid mediated resistance was reported, involving a high copy number plasmid (pCD-METRO) isolated in a ribotype 020 strain from a patient who failed metronidazole therapy. pCD-Metro was found in ~3.8% of ~585 strains studied and occurred in ribotypes 027, 010, and 020 from different European countries [28]. So far, it is unknown which gene on pCD-METRO confers resistance. Also, resistance mediated by pCD-METRO is not dependent on heme [26], indicating that other mechanisms are likely to be responsible for heme-dependent resistance in most clinical strains. To understand chromosomally mediated resistance, a mutator non-toxigenic strain was constructed [20]. Evolutionary experiments with this mutator identified that mutations to pyruvate:ferredoxin oxidoreductase (PFOR), iron sulfur cluster regulator (IscR) and xanthine dehydrogenase (XDH) conferred resistance in cells deficient in cellular iron, following inactivation of the ferrous iron transporter (FeoB1) [20]. These genetic changes were predicted to impair the reduction of metronidazole to reactive species within cells [20]. Metronidazole-resistant clinical strains (ribotype 027) have also been reported with mutations to catalytic domains of PFOR [20], but complementation of these strains with the wild type gene did not completely restore susceptibility to metronidazole in the presence of heme (Table 1). This implied that other mechanisms contribute to metronidazole resistance in C. difficile. A genome-wide association study revealed that non-synonymous mutations (Tyr130Ser, Tyr130Cys) in C. difficile NimB (CD1459) and a SNP within the gene’s promoter region were associated with strains exhibiting reduced susceptibility to metronidazole; 1501 isolates from the MODIFY I and II clinical trials of bezlotoxumab were examined in this study [29]. Nim proteins are thought to inactivate metronidazole to an amino derivative, bypassing the formation of the antimicrobial’s reactive species [30]. Although NimB is common among C. difficile genomes, experimental evidence is needed to establish if it can confer resistance to metronidazole in C. difficile.
Vancomycin
The glycopeptide vancomycin binds with high affinity to D-Ala-D-Ala of lipid-II at the C-terminal pentapeptide, thus inhibiting peptidoglycan synthesis and assembly, which weakens the cell wall and causes cells to eventually undergo autolysis. Like metronidazole, the clinical success of vancomycin also declined [31–34]. Interestingly, C. difficile isolates from 1984 to 2003 were associated with a vancomycin MIC90 of 1 μg/ml, compared to an MIC90 of 4 μg/ml for isolates from 2011 to 2012 [35]. This suggests that there has been an increase in strains that are less susceptible to vancomycin. Yet there is no direct correlation between poorer therapeutic outcomes and vancomycin-resistant strains, with MICs of 4–16 μg/mL (i.e., EUCAST breakpoint of >2 μg/mL). It is widely thought that low-level vancomycin-resistant strains (MICs=4–16 μg/mL), are less likely to cause treatment failure since the fecal concentrations of vancomycin (i.e., ~100–1000-fold MIC90 [1 μg/ml]), should inhibit the growth of resistant strains (MIC=4–16 μg/ml) [27,36]. However, three aspects complicate the interpretation of MIC data of individual strains in relation to vancomycin treatment failure: a) drug concentration at site of infection; b) antimicrobial tolerance mechanisms (Eagle effect and resistance to autolysis); and c) the microbiome. The actual concentrations of vancomycin along the length of the colon are unknown, as well as whether drug deposition is affected by fulminant disease [37]. In our view, MICs alone do not always predict survivability, especially in a complex dynamic environment such as the gastrointestinal tract. Additionally, less studied phenotypes such as reduced autolytic responses [38] and ‘Eagle effect resistance’ [39] (a paradoxical phenomenon where bacteria grow above bactericidal concentrations), might enable C. difficile to survive in physiological concentrations of vancomycin (Table 1 and Figure 3). It has been suggested that higher therapeutic doses of vancomycin should be used (i.e., 500 mg instead of 125 mg, four times/day) [37]. However, it is possible that this could enhance collateral damage to the gut microbiota and promote overgrowth of vancomycin-resistant Enterococci (VRE) that is associated with more severe CDI [40]. In our view, these factors suggest that evolution of low-level resistance in C. difficile should not be negated, as either a potential driver for increasing recurrence or as a step toward higher level resistance.
Figure 3. Conceptual models of strains that survive physiological vancomycin by adopting the Eagle resistance or reduced response to autolysis.

Their survival in physiological concentrations of drug could be related to: (A) a paradoxical phenomenon known as the Eagle effect, whereby strains that are susceptible, as based on MICs, grow above the MBC; or (B) diminished cell lysis under autolysis inducing conditions (autolysis is measured as a loss of optical density over time in Triton-X 100 buffer). Such approaches can be used to characterize strains that survive physiological concentrations of vancomycin.
Vancomycin resistance mechanisms in enterococci are well-documented, involving modification of the terminal D-Ala with either D-Lac or D-Ser [41]. High-level resistance conferred by D-Ala-D-Lac is encoded by vanA and vanB gene clusters, whereas low-level resistance is caused by D-Ala-D-Ser encoded by vanC, vanE and vanG gene clusters. C. difficile isolates encode a vanG-type gene cluster (vanGCd) that is cryptic in vancomycin-sensitive strains [38,42]. vanGCd does not cause resistance in susceptible strains, possibly due to cells favoring use of peptidoglycan precursors ending in the dipeptide D-Ala-D-Ala [43]. Recently, it was described that constitutive expression of vanGCd occurred in vancomycin-resistant clinical strains (MICs=4–8 μg/mL) and laboratory generated mutants (MICs=8–16 μg/mL), which carried mutations in the VanSR two-component system that regulates vanGCd [38]. A Thr115Ala mutation in VanR was common in clinical strains from different geographic regions and molecular modelling suggested it locked phosphorylated VanR into its DNA binding conformation, making it more prone to induce vanGCd transcription. Mutations in the VanS sensor occurred in a conserved region that affected the phosphatase function of VanS and presumably increased cellular levels of phosphorylated of VanR. Interestingly, lab mutants containing a stop codon in trkA, a potassium transporter (i.e., Gln26STOP in TrkA; Table 1), were less responsive to autolysis and survived physiological concentrations of vancomycin (i.e., MBCs of ≥1024 μg/mL). It is likely that the loss of TrkA enabled the accumulation of organic osmoprotectants that protect against cell lysis. Although TrkA mutations have not been reported in clinical strains to date, the idea that autolysis resistant strains exhibit enhanced survival in physiological concentrations of vancomycin points to a mechanism for drug tolerance and recurrence by strains with vancomycin susceptible MICs or low-level resistance to vancomycin. Indeed, vancomycin-intermediate Staphylococcus aureus showing reduced autolysis can survive in vancomycin concentrations of ≥32-fold higher their MICs [44]. Based on these findings, Figure 3B describes a model whereby low-level vancomycin-resistant C. difficile survive physiological concentrations of the drug, by being less responsive to autolysis conditions. Other in vitro vancomycin-resistant mutants (MIC=16 μg/mL) were reported [45]. One such lab-generated mutant had a mutation of Asp244Tyr in RNA polymerase β’ subunit, while the other mutant carried mutations of ΔAla295 in L-serine deaminase (SdaB), Glu327Stop in cyclic-di-AMP phosphodiesterase (CD630_36590; predicted to be GdpP with 59% homology to S. aureus GdpP) and Pro108Leu in peptidoglycan glycosyltransferase (MurG). In unpublished observations, the authors identified a clinical isolate with mutations of ΔAla296 in SdaB, ΔLeu443 in GdpP, and Thr115Ala in VanR (Table 1). This might suggest that mutations to SdaB and GdpP are clinically relevant resistance mechanisms, but this will require experimental validation. We speculate that inactivation of SdaB in C. difficile increases the cellular pool of L-serine, enabling the VanT serine racemase of vanGCd to make D-Serine for Lipid-II-D-Ala-D-Ser biosynthesis. In bacteria, GdpP affects several processes including osmotic regulation, cell wall homeostasis, and biofilm development [46]. Since its deletion confers tolerance to cell wall acting antimicrobials [47], it is possible that loss of GdpP in vancomycin-resistant C. difficile also promotes tolerance to vancomycin, but this will need to be confirmed experimentally.
Plasmid-mediated reduction of vancomycin susceptibility has recently been reported in isolates (MIC=2 μg/mL) from patients failing to respond to vancomycin therapy [48]. The plasmid, pX18–498, is a large broad host range plasmid with 51 ORFs, including a gene encoding a putative N-acetylmuramoyl-L-alanine-amidase, a peptidoglycan remodeling enzyme. Conjugation of pX18–498 into a vancomycin-susceptible strain conferred decreased susceptibility to vancomycin. Possible clinical relevance of pX18–498 was also demonstrated, as mice infected with C. difficile-pX18–498 and treated with vancomycin had >1 log greater bioburdens than counterpart mice infected with an isogenic strain lacking the plasmid. This study [48] certainly raises the question of whether there are niche-specific concentrations of vancomycin that favor colonization and survival with low-level resistant mutants and whether there are interactions between determinants on pX18–498 and the core genome.
Biofilm mediated resistance to vancomycin and metronidazole.
Laboratory studies suggest that biofilms might play a role in vancomycin and metronidazole resistance in C. difficile. Subinhibitory concentrations of vancomycin and metronidazole were shown to enhance biofilm formation [49,50]. In general, within biofilms, cells have reduced metabolism and are more tolerant to antimicrobials; C. difficile biofilms tolerate high concentrations of metronidazole (10–100 μg/mL) [51] and vancomycin (20 μg/mL) [50] that are bactericidal to planktonic cells.
Fidaxomicin
The narrow-spectrum antimicrobial fidaxomicin binds to the RNA polymerase (RNAP) clamp, inhibiting the initiation step of transcription of DNA into RNA [52]. Since its approval for treatment of CDI by the U.S. Food and Drug Administration in 2011, fidaxomicin has been mainly used for rCDI. Fidaxomicin-resistant C. difficile (MIC=16 μg/mL) was isolated from a patient with rCDI, following therapy with fidaxomicin [53]. Fidaxomicin resistance arises from mutations in RNAP, in RpoB (Gln1074Lys, Val1143Asp, Gly, Phe) and RpoC (Gln781Arg and Asp1127Glu, Asp237Tyr) [54,55]. Val1143Asp (MIC>64 μg/mL) and Val1143Gly (MIC=16 μg/mL) have also been reported in fidaxomicin-resistant clinical isolates [56,57] (Table 1). Mutations at position 1143 in RpoB affect the fitness and virulence of C. difficile, as was shown using recombinant isogenic strains [56]. The clinically occurring mutations of Val1143Asp and Val1143Gly showed reductions in overall growth, competitive fitness, and production of toxins A/B, when compared to their parental strain R20291. Virulence was also reduced in the hamster model of CDI. However, it is unknown whether fidaxomicin-resistant strains, with mutations in RNAP, will survive in physiological fidaxomicin (the fecal concentration of fidaxomicin is reported to be 1396 ± 1019 μg/g) [58]. Because fidaxomicin shows a narrow-spectrum of activity [59], it is possible that the rebounding microbiota could help mitigate the effect of mutants if they emerge during therapy, particularly those with fitness costs. However, if lessons can be learnt from other bacterial pathogens, it is that antimicrobials inhibiting a single drug target are prone to more rapid evolution of resistance. Furthermore, second site compensatory mutations could evolve that reinstate fitness and allow resistance alleles to become fixed. It is interesting that mutations to RNAP are linked to development of vancomycin resistance in S. aureus [60,61], by enhancing transcription of cell wall synthesis genes. This certainly raises the question of whether increased use of vancomycin and fidaxomicin might drive co-resistance to these drugs. Therefore, proactive surveillance of fidaxomicin resistance is warranted. Lab-evolved fidaxomicin-resistant mutants (MIC=16 μg/ml) were also found to carry a frameshift mutation in CD2212, a homolog of MarR (multiple antibiotic resistance regulator), but confirmation of the mutation’s role in fidaxomicin resistance requires molecular genetic validation [45].
Rifaximin and Tetracyclines
Alternate therapies for CDI include the rifamycin rifaximin and the tetracycline tigecycline [1,2]. Rifaximin is recommended as a follow-up therapy after initial treatment with vancomycin for rCDI. However, C. difficile has a mutation frequency of ~108 to rifaximin, in which high-level resistant mutants (e.g., MICs >1024 μg/mL) can arise without significant effects on in vitro or in vivo fitness [62]. Thus, it has been reported that rifaximin resistance can arise during CDI therapy, resulting in clinical failure of rifaximin (Table 1). Furthermore, rifaximin-resistant C. difficile is common in hospitals (i.e., rates of 29.1– 48.9%), which increases the risk for therapeutic failure [63]. In contrast to rifaximin, tigecycline has a lower rate of de novo resistance. Nonetheless, a recent meta-analysis showed that 20% of C. difficile human isolates are tetracycline-resistant [64]. Tetracycline-resistant C. difficile strains mainly encode the ribosomal protection protein tet(M) on conjugative Tn916-like elements. Because tigecycline has a higher affinity for the ribosome than older tetracyclines, it is active against strains bearing Tet(M). Experiments in E. coli show that mutations to Tet(M) can engender low-level tigecycline resistance [65]. High-level resistance to tigecycline is encoded by tetracycline destructases, i.e., Tet(X), that enzymatically inactivate tetracyclines. Recent discoveries of tet(X) orthologs on mobile elements in commensal, livestock and human isolates also affords a path for tigecycline resistance in C. difficile [66]. Because rifaximin and tigecycline are not firstline drugs, it is conceivable that susceptibility testing, and genome sequencing could be applied to improve selection of these therapies for rCDI.
CONCLUDING REMARKS
Over the last decade, knowledge of various mechanisms of resistance to firstline drugs by C. difficile now make it possible to explore the effect of resistance on treatment outcomes and epidemiology. For example, because fidaxomicin resistance typically maps to RNAP, mutations can readily be identified by PCR methods or genome sequencing. Similarly, PCR methods can be developed to identify vancomycin resistance mechanisms involving pX18–498 and VanSR mutations. However, further experimental validation and/or determining correlations with clinical failure is required for vancomycin and fidaxomicin resistance mechanisms. Regarding vancomycin, whether the ‘Eagle effect’ or reduced autolysis also contributes to C. difficile survival in high doses of the drug will also require further experimentation. It will also be crucial to learn lessons from declining efficacies of metronidazole and vancomycin to better employ these and other anti-C. difficile therapeutics. In this regard, whole-genome sequencing-based typing and comparative genomics on patient isolates alongside susceptibility testing results could be essential. There is also an important role to be played by evolutionary genomics (e.g., genome wide-association studies) on global strains to understand the epidemiology of resistance mechanisms and to discover evolving mechanisms that are associated with AMR phenotypes. This information can be harnessed to develop molecular diagnostics that improve therapeutic selection for CDI, including the use of future anti-C. difficile therapeutics, which are at various stages of pre-clinical and clinical development.
HIGHLIGHTS.
Plasmid mediated resistances to metronidazole (pCD-METRO) and vancomycin (pX18–498) may be associated with reduced therapeutic efficacies.
Heme-dependent resistance to metronidazole found in most metronidazole-resistant C. difficile may be associated with clinical failure.
Resistance to autolysis and the ‘Eagle effect’ could mediate survival in physiological vancomycin.
Resistance testing should be a part of the diagnostic work-up for CDI.
Genomic surveillances could track C. difficile evolution to improve antimicrobial stewardship policies.
Footnotes
DECLARATIONS OF INTEREST
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1. Johnson S, Lavergne V, Skinner AM, Gonzales-Luna AJ, Garey KW, Kelly CP, Wilcox MH: Clinical Practice Guideline by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA): 2021 Focused Update Guidelines on Management of Clostridioides difficile Infection in Adults. Clin Infect Dis 2021, 73:755–757. Most recent IDSA/SHEA guidelines for management of CDI, in which metronidazole is not endorsed as a first-line drug, but as an alternate if vancomycin or fidaxomicin is not available. Fidaxomicin has been recommended as a first choice option ahead of vancomycin.
- 2. van Prehn J, Reigadas E, Vogelzang EH, Bouza E, Hristea A, Guery B, Krutova M, Noren T, Allerberger F, Coia J, et al.: European Society of Clinical Microbiology and Infectious Diseases: 2021 update on the treatment guidance document for Clostridioides difficile infection in adults. Clin Microbiol Infect 2021. The most recent ESCMID guidelines for management of CDI, similarly does not recommend metronidazole for the treatment of CDI when fidaxomicin or vancomycin is available.
- 3.Kelly CP: Can we identify patients at high risk of recurrent Clostridium difficile infection? Clin Microbiol Infect 2012, 18 Suppl 6:21–27. [DOI] [PubMed] [Google Scholar]
- 4.Leffler DA, Lamont JT: Clostridium difficile infection. N Engl J Med 2015, 372:1539–1548. [DOI] [PubMed] [Google Scholar]
- 5.O’Grady K, Knight DR, Riley TV: Antimicrobial resistance in Clostridioides difficile. Eur J Clin Microbiol Infect Dis 2021, 40:2459–2478. [DOI] [PubMed] [Google Scholar]
- 6.Wickramage I, Spigaglia P, Sun X: Mechanisms of antibiotic resistance of Clostridioides difficile. J Antimicrob Chemother 2021, 76:3077–3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson S, Samore MH, Farrow KA, Killgore GE, Tenover FC, Lyras D, Rood JI, DeGirolami P, Baltch AL, Rafferty ME, et al. : Epidemics of diarrhea caused by a clindamycin-resistant strain of Clostridium difficile in four hospitals. N Engl J Med 1999, 341:1645–1651. [DOI] [PubMed] [Google Scholar]
- 8.Toth M, Stewart NK, Smith C, Vakulenko SB: Intrinsic Class D beta-Lactamases of Clostridium difficile. mBio 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sandhu BK, Edwards AN, Anderson SE, Woods EC, McBride SM: Regulation and Anaerobic Function of the Clostridioides difficile beta-Lactamase. Antimicrob Agents Chemother 2019, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Isidro J, Santos A, Nunes A, Borges V, Silva C, Vieira L, Mendes AL, Serrano M, Henriques AO, Gomes JP, et al. : Imipenem Resistance in Clostridium difficile Ribotype 017, Portugal. Emerg Infect Dis 2018, 24:741–745. This paper describes mutations in penicillin binding protein (PBP) 1 and 3 associated with resistance to imipenem.
- 11.Dessen A, Mouz N, Gordon E, Hopkins J, Dideberg O: Crystal structure of PBP2x from a highly penicillin-resistant Streptococcus pneumoniae clinical isolate: a mosaic framework containing 83 mutations. J Biol Chem 2001, 276:45106–45112. [DOI] [PubMed] [Google Scholar]
- 12. Srikhanta YN, Hutton ML, Awad MM, Drinkwater N, Singleton J, Day SL, Cunningham BA, McGowan S, Lyras D: Cephamycins inhibit pathogen sporulation and effectively treat recurrent Clostridioides difficile infection. Nat Microbiol 2019, 4:2237–2245. This paper describes that cephamycins, which inhibit SpoVD a homolog of PBP4, could be potential treatments for recurrent CDI.
- 13.Wilcox MH: Caution is warranted in using cephamycin antibiotics against recurrent Clostridioides difficile infection. Nat Microbiol 2020, 5:236. [DOI] [PubMed] [Google Scholar]
- 14.Endres BT, Begum K, Sun H, Walk ST, Memariani A, Lancaster C, Gonzales-Luna AJ, Dotson KM, Basseres E, Offiong C, et al. : Epidemic Clostridioides difficile Ribotype 027 Lineages: Comparisons of Texas Versus Worldwide Strains. Open Forum Infect Dis 2019, 6:ofz013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He M, Miyajima F, Roberts P, Ellison L, Pickard DJ, Martin MJ, Connor TR, Harris SR, Fairley D, Bamford KB, et al. : Emergence and global spread of epidemic healthcare-associated Clostridium difficile. Nat Genet 2013, 45:109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wasels F, Kuehne SA, Cartman ST, Spigaglia P, Barbanti F, Minton NP, Mastrantonio P: Fluoroquinolone resistance does not impose a cost on the fitness of Clostridium difficile in vitro. Antimicrob Agents Chemother 2015, 59:1794–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vernon JJ, Wilcox MH, Freeman J: Effect of fluoroquinolone resistance mutation Thr-82-->Ile on Clostridioides difficile fitness. J Antimicrob Chemother 2019, 74:877–884. [DOI] [PubMed] [Google Scholar]
- 18.Lawes T, Lopez-Lozano JM, Nebot CA, Macartney G, Subbarao-Sharma R, Wares KD, Sinclair C, Gould IM: Effect of a national 4C antibiotic stewardship intervention on the clinical and molecular epidemiology of Clostridium difficile infections in a region of Scotland: a non-linear time-series analysis. Lancet Infect Dis 2017, 17:194–206. [DOI] [PubMed] [Google Scholar]
- 19.Tischendorf J, Brunner M, Knobloch MJ, Schulz L, Barker A, Wright MO, Lepak A, Safdar N: Evaluation of a successful fluoroquinolone restriction intervention among high-risk patients: A mixed-methods study. PLoS One 2020, 15:e0237987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Deshpande A, Wu X, Huo W, Palmer KL, Hurdle JG: Chromosomal Resistance to Metronidazole in Clostridioides difficile Can Be Mediated by Epistasis between Iron Homeostasis and Oxidoreductases. Antimicrob Agents Chemother 2020, 64. This paper describes an unusual mechanism of epistatic interaction between ferrous iron transporter FeoB1 and proteins involved in redox metabolism (PFOR, XDH and IscR). It describes that mutations to catalytic domains of PFOR can be found in metronidazole-resistant C. difficile, but this mechanism does not account for resistance that requires heme.
- 21.Kumar M, Adhikari S, Hurdle JG: Action of nitroheterocyclic drugs against Clostridium difficile. Int J Antimicrob Agents 2014, 44:314–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lynch T, Chong P, Zhang J, Hizon R, Du T, Graham MR, Beniac DR, Booth TF, Kibsey P, Miller M, et al. : Characterization of a stable, metronidazole-resistant Clostridium difficile clinical isolate. PLoS One 2013, 8:e53757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moura I, Monot M, Tani C, Spigaglia P, Barbanti F, Norais N, Dupuy B, Bouza E, Mastrantonio P: Multidisciplinary analysis of a nontoxigenic Clostridium difficile strain with stable resistance to metronidazole. Antimicrob Agents Chemother 2014, 58:4957–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pelaez T, Cercenado E, Alcala L, Marin M, Martin-Lopez A, Martinez-Alarcon J, Catalan P, Sanchez-Somolinos M, Bouza E: Metronidazole resistance in Clostridium difficile is heterogeneous. J Clin Microbiol 2008, 46:3028–3032. This paper describes that metronidazole resistance in C. difficile is unstable and such has presented issues to understand the genetic mechanism of resistance.
- 25. Wu X, Shen WJ, Deshpande A, Olaitan AO, Palmer KL, Garey KW, Hurdle JG: The Integrity of Heme Is Essential for Reproducible Detection of Metronidazole-Resistant Clostridioides difficile by Agar Dilution Susceptibility Tests. J Clin Microbiol 2021, 59:e0058521. This work describes that metronidazole resistance is not unstable, but rather requires the biological cofactor heme to be present in testing agars and protected from light that causes photo-decomposition of heme.
- 26. Boekhoud IM, Sidorov I, Nooij S, Harmanus C, Bos-Sanders I, Viprey V, Spittal W, Clark E, Davies K, Freeman J, et al. : Haem is crucial for medium-dependent metronidazole resistance in clinical isolates of Clostridioides difficile. J Antimicrob Chemother 2021, 76:1731–1740. This work also describes that heme is needed for reproducible detection of metronidazole resistant C. difficile that lacks pCD-METRO.
- 27. Gonzales-Luna AJ, Olaitan AO, Shen WJ, Deshpande A, Carlson TJ, Dotson KM, Lancaster C, Begum K, Alam MJ, Hurdle JG, et al. : Reduced Susceptibility to Metronidazole Is Associated With Initial Clinical Failure in Clostridioides difficile Infection. Open Forum Infect Dis 2021, 8:ofab365. This paper discribes a retrospective clinical study showing that an MIC breakpoint of ≥1 μg/ml was associated with higher risk for treatment failure for metronidazole. Strains were identified as resistant using light protected heme in biological agars.
- 28. Boekhoud IM, Hornung BVH, Sevilla E, Harmanus C, Bos-Sanders I, Terveer EM, Bolea R, Corver J, Kuijper EJ, Smits WK: Plasmid-mediated metronidazole resistance in Clostridioides difficile. Nat Commun 2020, 11:598. This paper describes the discovery of pCD-METRO and its association with metronidazole resistance and poor treatment response in a patient.
- 29.Zhao H, Nickle DC, Zeng Z, Law PYT, Wilcox MH, Chen L, Peng Y, Meng J, Deng Z, Albright A, et al.: Global Landscape of Clostridioides Difficile Phylogeography, Antibiotic Susceptibility, and Toxin Polymorphisms by Post-Hoc Whole-Genome Sequencing from the MODIFY I/II Studies. Infect Dis Ther 2021, 10:853–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carlier JP, Sellier N, Rager MN, Reysset G: Metabolism of a 5-nitroimidazole in susceptible and resistant isogenic strains of Bacteroides fragilis. Antimicrob Agents Chemother 1997, 41:1495–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wenisch C, Parschalk B, Hasenhundl M, Hirschl AM, Graninger W: Comparison of vancomycin, teicoplanin, metronidazole, and fusidic acid for the treatment of Clostridium difficile-associated diarrhea. Clin Infect Dis 1996, 22:813–818. [DOI] [PubMed] [Google Scholar]
- 32.Zar FA, Bakkanagari SR, Moorthi KMLST, Davis MB: A Comparison of Vancomycin and Metronidazole for the Treatment of Clostridium difficile-Associated Diarrhea, Stratified by Disease Severity. Clinical Infectious Diseases 2007, 45:302–307. [DOI] [PubMed] [Google Scholar]
- 33.Louie TJ, Peppe J, Watt CK, Johnson D, Mohammed R, Dow G, Weiss K, Simon S, John JF Jr., Garber G, et al. : Tolevamer, a novel nonantibiotic polymer, compared with vancomycin in the treatment of mild to moderately severe Clostridium difficile-associated diarrhea. Clin Infect Dis 2006, 43:411–420. [DOI] [PubMed] [Google Scholar]
- 34.Dudley MN, McLaughlin JC, Carrington G, Frick J, Nightingale CH, Quintiliani R: Oral bacitracin vs vancomycin therapy for Clostridium difficile-induced diarrhea. A randomized double-blind trial. Arch Intern Med 1986, 146:1101–1104. [PubMed] [Google Scholar]
- 35.Snydman DR, McDermott LA, Jacobus NV, Thorpe C, Stone S, Jenkins SG, Goldstein EJ, Patel R, Forbes BA, Mirrett S, et al. : U.S.-Based National Sentinel Surveillance Study for the Epidemiology of Clostridium difficile-Associated Diarrheal Isolates and Their Susceptibility to Fidaxomicin. Antimicrob Agents Chemother 2015, 59:6437–6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baines SD, O’Connor R, Freeman J, Fawley WN, Harmanus C, Mastrantonio P, Kuijper EJ, Wilcox MH: Emergence of reduced susceptibility to metronidazole in Clostridium difficile. J Antimicrob Chemother 2008, 62:1046–1052. [DOI] [PubMed] [Google Scholar]
- 37.Cunha BA, Sessa J, Blum S: Enhanced Efficacy of High Dose Oral Vancomycin Therapy in Clostridium difficile Diarrhea for Hospitalized Adults Not Responsive to Conventional Oral Vancomycin Therapy: Antibiotic Stewardship Implications. J Clin Med 2018, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shen WJ, Deshpande A, Hevener KE, Endres BT, Garey KW, Palmer KL, Hurdle JG: Constitutive expression of the cryptic vanGCd operon promotes vancomycin resistance in Clostridioides difficile clinical isolates. J Antimicrob Chemother 2020, 75:859–867. This paper describes vancomycin resistance mechanisms in C. difficile clinical isolates, showing involvement of mutations that upregulate the vanG cluster; it also reports lab mutants that were tolerant to autolysis and survived in phsyiological concentrations of vancomycin.
- 39. Jarrad AM, Blaskovich MAT, Prasetyoputri A, Karoli T, Hansford KA, Cooper MA: Detection and Investigation of Eagle Effect Resistance to Vancomycin in Clostridium difficile With an ATP-Bioluminescence Assay. Front Microbiol 2018, 9:1420. This paper describes the discovery that a paradoxical phenomenon known as the Eagle effect allowed C. difficile to grow at concentrations above the MIC and MBC of the drug.
- 40.Stevens VW, Khader K, Echevarria K, Nelson RE, Zhang Y, Jones M, Timbrook TT, Samore MH, Rubin MA: Use of Oral Vancomycin for Clostridioides difficile Infection and the Risk of Vancomycin-Resistant Enterococci. Clin Infect Dis 2020, 71:645–651. [DOI] [PubMed] [Google Scholar]
- 41.Ahmed MO, Baptiste KE: Vancomycin-Resistant Enterococci: A Review of Antimicrobial Resistance Mechanisms and Perspectives of Human and Animal Health. Microb Drug Resist 2018, 24:590–606. [DOI] [PubMed] [Google Scholar]
- 42.Sebaihia M, Wren BW, Mullany P, Fairweather NF, Minton N, Stabler R, Thomson NR, Roberts AP, Cerdeno-Tarraga AM, Wang H, et al. : The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat Genet 2006, 38:779–786. [DOI] [PubMed] [Google Scholar]
- 43.Ammam F, Meziane-Cherif D, Mengin-Lecreulx D, Blanot D, Patin D, Boneca IG, Courvalin P, Lambert T, Candela T: The functional vanGCd cluster of Clostridium difficile does not confer vancomycin resistance. Mol Microbiol 2013, 89:612–625. [DOI] [PubMed] [Google Scholar]
- 44.Howden BP, Davies JK, Johnson PD, Stinear TP, Grayson ML: Reduced vancomycin susceptibility in Staphylococcus aureus, including vancomycin-intermediate and heterogeneous vancomycin-intermediate strains: resistance mechanisms, laboratory detection, and clinical implications. Clin Microbiol Rev 2010, 23:99–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leeds JA, Sachdeva M, Mullin S, Barnes SW, Ruzin A: In vitro selection, via serial passage, of Clostridium difficile mutants with reduced susceptibility to fidaxomicin or vancomycin. J Antimicrob Chemother 2014, 69:41–44. [DOI] [PubMed] [Google Scholar]
- 46.Corrigan RM, Abbott JC, Burhenne H, Kaever V, Grundling A: c-di-AMP is a new second messenger in Staphylococcus aureus with a role in controlling cell size and envelope stress. PLoS Pathog 2011, 7:e1002217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Griffiths JM, O’Neill AJ: Loss of function of the gdpP protein leads to joint beta-lactam/glycopeptide tolerance in Staphylococcus aureus. Antimicrob Agents Chemother 2012, 56:579–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pu M, Cho JM, Cunningham SA, Behera GK, Becker S, Amjad T, Greenwood-Quaintance KE, Mendes-Soares H, Jones-Hall Y, Jeraldo PR, et al. : Plasmid Acquisition Alters Vancomycin Susceptibility in Clostridioides difficile. Gastroenterology 2021, 160:941–945 e948. This paper describes the first plasmid-mediated resistance to vancomycin in C. difficile. The plasmid was associated with isolates from patients that did not respond to vancomycin therapy. Using isogenic strains, with and without the plasmid, possession of the plasmid was reported to enhance survival during vancomycin therapy.
- 49.Vuotto C, Moura I, Barbanti F, Donelli G, Spigaglia P: Subinhibitory concentrations of metronidazole increase biofilm formation in Clostridium difficile strains. Pathog Dis 2016, 74. [DOI] [PubMed] [Google Scholar]
- 50.Dapa T, Unnikrishnan M: Biofilm formation by Clostridium difficile. Gut Microbes 2013, 4:397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Semenyuk EG, Laning ML, Foley J, Johnston PF, Knight KL, Gerding DN, Driks A: Spore formation and toxin production in Clostridium difficile biofilms. PLoS One 2014, 9:e87757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin W, Das K, Degen D, Mazumder A, Duchi D, Wang D, Ebright YW, Ebright RY, Sineva E, Gigliotti M, et al. : Structural Basis of Transcription Inhibition by Fidaxomicin (Lipiarmycin A3). Mol Cell 2018, 70:60–71 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goldstein EJ, Citron DM, Sears P, Babakhani F, Sambol SP, Gerding DN: Comparative susceptibilities to fidaxomicin (OPT-80) of isolates collected at baseline, recurrence, and failure from patients in two phase III trials of fidaxomicin against Clostridium difficile infection. Antimicrob Agents Chemother 2011, 55:5194–5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Babakhani F, Gomez A, Robert N, Sears P: Killing kinetics of fidaxomicin and its major metabolite, OP-1118, against Clostridium difficile. J Med Microbiol 2011, 60:1213–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Babakhani F, Seddon J, Sears P: Comparative microbiological studies of transcription inhibitors fidaxomicin and the rifamycins in Clostridium difficile. Antimicrob Agents Chemother 2014, 58:2934–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kuehne SA, Dempster AW, Collery MM, Joshi N, Jowett J, Kelly ML, Cave R, Longshaw CM, Minton NP: Characterization of the impact of rpoB mutations on the in vitro and in vivo competitive fitness of Clostridium difficile and susceptibility to fidaxomicin. J Antimicrob Chemother 2018, 73:973–980. This paper describes mutations causing fidaxomicin resistance and their effect on fitness and virulence of C. difficile.
- 57.Schwanbeck J, Riedel T, Laukien F, Schober I, Oehmig I, Zimmermann O, Overmann J, Gross U, Zautner AE, Bohne W: Characterization of a clinical Clostridioides difficile isolate with markedly reduced fidaxomicin susceptibility and a V1143D mutation in rpoB. J Antimicrob Chemother 2019, 74:6–10. [DOI] [PubMed] [Google Scholar]
- 58.Louie TJ, Miller MA, Mullane KM, Weiss K, Lentnek A, Golan Y, Gorbach S, Sears P, Shue YK, Group OPTCS: Fidaxomicin versus vancomycin for Clostridium difficile infection. N Engl J Med 2011, 364:422–431. [DOI] [PubMed] [Google Scholar]
- 59.Finegold SM, Molitoris D, Vaisanen ML, Song Y, Liu C, Bolanos M: In vitro activities of OPT-80 and comparator drugs against intestinal bacteria. Antimicrob Agents Chemother 2004, 48:4898–4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Matsuo M, Hishinuma T, Katayama Y, Cui L, Kapi M, Hiramatsu K: Mutation of RNA polymerase beta subunit (rpoB) promotes hVISA-to-VISA phenotypic conversion of strain Mu3. Antimicrob Agents Chemother 2011, 55:4188–4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guerillot R, Goncalves da Silva A, Monk I, Giulieri S, Tomita T, Alison E, Porter J, Pidot S, Gao W, Peleg AY, et al. : Convergent Evolution Driven by Rifampin Exacerbates the Global Burden of Drug-Resistant Staphylococcus aureus. mSphere 2018, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Dang UT, Zamora I, Hevener KE, Adhikari S, Wu X, Hurdle JG: Rifamycin Resistance in Clostridium difficile Is Generally Associated with a Low Fitness Burden. Antimicrob Agents Chemother 2016, 60:5604–5607. This paper describes that mutations causing rifaximin resistance are not associated with fitness costs in vitro and in vivo, and this may explain why these mutants have become prevalent in the clinic.
- 63.Ng QX, Loke W, Foo NX, Mo Y, Yeo WS, Soh AYS: A systematic review of the use of rifaximin for Clostridium difficile infections. Anaerobe 2019, 55:35–39. [DOI] [PubMed] [Google Scholar]
- 64.Sholeh M, Krutova M, Forouzesh M, Mironov S, Sadeghifard N, Molaeipour L, Maleki A, Kouhsari E: Antimicrobial resistance in Clostridioides (Clostridium) difficile derived from humans: a systematic review and meta-analysis. Antimicrob Resist Infect Control 2020, 9:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Linkevicius M, Sandegren L, Andersson DI: Potential of Tetracycline Resistance Proteins To Evolve Tigecycline Resistance. Antimicrob Agents Chemother 2016, 60:789–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gasparrini AJ, Markley JL, Kumar H, Wang B, Fang L, Irum S, Symister CT, Wallace M, Burnham CD, Andleeb S, et al.: Tetracycline-inactivating enzymes from environmental, human commensal, and pathogenic bacteria cause broad-spectrum tetracycline resistance. Commun Biol 2020, 3:241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Carman RJ, Boone JH, Grover H, Wickham KN, Chen L: In vivo selection of rifamycin-resistant Clostridium difficile during rifaximin therapy. Antimicrob Agents Chemother 2012, 56:6019–6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dingle KE, Didelot X, Quan TP, Eyre DW, Stoesser N, Marwick CA, Coia J, Brown D, Buchanan S, Ijaz UZ, et al. : A Role for Tetracycline Selection in Recent Evolution of Agriculture-Associated Clostridium difficile PCR Ribotype 078. mBio 2019, 10. This paper describes different tetracycline resistance determinants found on mobile genetic elements, as well as genes encoding resistance determinants to several other classes of antimicrobials.
- 69.Ethapa T, Leuzzi R, Ng YK, Baban ST, Adamo R, Kuehne SA, Scarselli M, Minton NP, Serruto D, Unnikrishnan M: Multiple factors modulate biofilm formation by the anaerobic pathogen Clostridium difficile. J Bacteriol 2013, 195:545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ngernsombat C, Sreesai S, Harnvoravongchai P, Chankhamhaengdecha S, Janvilisri T: CD2068 potentially mediates multidrug efflux in Clostridium difficile. Sci Rep 2017, 7:9982. [DOI] [PMC free article] [PubMed] [Google Scholar]