

Abstract
UbiA prenyltransferase domain-containing protein-1 (UBIAD1) is responsible for the biosynthesis of menaquinone-4 (MK-4), a cofactor for extrahepatic carboxylation of vitamin K-dependent (VKD) proteins. Genetic variations of UBIAD1 are mainly associated with Schnyder corneal dystrophy (SCD), a disease characterized by abnormal accumulation of cholesterol in the cornea. Results from in vitro studies demonstrate that SCD-associated UBIAD1 mutations are defective in MK-4 biosynthesis. However, SCD patients do not exhibit typical phenotypes associated with defects of MK-4 or VKD carboxylation. Here, we coupled UBIAD1’s biosynthetic activity of MK-4 with VKD carboxylation in HEK293 cells that stably express a chimeric VKD reporter protein. The endogenous Ubiad1 gene in these cells were knocked out by CRISPR-Cas9-mediated genome editing. The effect of UBIAD1 mutations on MK-4 biosynthesis and VKD carboxylation were evaluated in Ubiad1-deficient reporter cells by determining the production of MK-4 or by measuring the efficiency of reporter-protein carboxylation. Our results show that the hot-spot mutation N102S has a moderate impact on MK-4 biosynthesis (retained ~82% activity) but does not affect VKD carboxylation. However, the G186R mutation significantly affected both MK-4 biosynthesis and VKD carboxylation. Other mutations exhibit varying degrees of effects on MK-4 biosynthesis and VKD carboxylation. These results are consistent with in vivo results obtained from gene knock-in mice and SCD patients. Our findings suggest that UBIAD1’s MK-4 biosynthetic activity does not directly correlate with the phenotypes of SCD patients. The established cell-based assays in this study provide a powerful tool for the functional studies of UBIAD1 in a cellular milieu.
Keywords: UBIAD1, Schnyder corneal dystrophy, Menaquinone, Vitamin K-dependent carboxylation, Cholesterol
Introduction
Schnyder corneal dystrophy (SCD) is a rare autosomal dominant inherited disorder characterized by an abnormal accumulation of cholesterol and phospholipids in the cornea leading to progressive vision loss [1]. However, the SCD phenotype does not directly correlate with the patient’s serum cholesterol or lipids levels [2–4]. Rather, it is a local metabolic defect of cholesterol and lipids in the cornea [4, 5]. Genetic studies have shown that SCD is associated with mutations in the gene encoding of UbiA prenyltransferase domain-containing protein-1 (UBIAD1) that is mapped to human chromosome 1p34.1-p36 [5–7]. The UbiA superfamily of prenyltransferase catalyzes the transfer of hydrophobic polyprenyl groups to a variety of aromatic acceptor molecules to generate ubiquinones [8], menaquinones [9], vitamin E [10], and structural lipids [11]. To date, near thirty UBIAD1 mutations have been identified in SCD patients [12–16].
UBIAD1–mediated cornea cholesterol accumulation has been proposed to occur via insufficient removal of cholesterol from the cornea [5]. Mutations of UBIAD1 altered its interaction with apolipoprotein E, a protein that is involved in cholesterol metabolism [17–19], resulting in decreased cholesterol removal from the cornea. An alternative explanation for UBIAD1-associated SCD is the overproduction of cholesterol. Recent studies suggest that SCD-associated UBIAD1 mutations bind to HMG-CoA reductase (HMGCR), the rate-limiting enzyme in cholesterol biosynthesis, which protects it from endoplasmic reticulum-associated degradation (ERAD) [20–24]. Thus, the stabilization of HMGCR in the ER by UBIAD1 mutations results in increased production of cholesterol in the cornea.
UBIAD1 is also the prenyltransferase responsible for the biosynthesis of menaquinones (vitamin K2) [25]. K vitamins are a group of 2-methyl-1,4-naphthoquinone derivatives, which include phylloquinone (vitamin K1), menaquinones (vitamin K2), and menadione (vitamin K3) [26, 27]. Menaquinones differ from phylloquinone in that the side chain at the 3-position comprises a number of repeating isoprenyl units (referred to as MK-n) rather than the semi-saturated phytyl chain. Vitamin K is a cofactor for the posttranslational modification of vitamin K-dependent (VKD) proteins involved in blood coagulation, vascular calcification, bone metabolism and other important physiological processes [28, 29]. It is generally accepted that vitamin K1 is responsible for the carboxylation of coagulation factors in the liver, while vitamin K2 is essential for the extrahepatic carboxylation of other VKD proteins, such as matrix Gla protein and osteocalcin. Defects of VKD carboxylation have been associated with both bleeding and non-bleeding disorders.
Genetic studies show that homozygous knockout of the Ubiad1 gene (Ubiad1−/−) in mice caused embryonic lethality, and the Ubiad1−/− embryonic stem cells failed to synthesize MK-4 [30]. Oral administration of MK-4 was unable to rescue the Ubiad1−/− mouse embryos; however, their embryonic lifespans were extended to term, suggesting that UBIAD1 plays a pivotal role in embryonic development by synthesizing vitamin K2. Using an Ubiad1 mutant zebrafish model, Hegarty et al. reported that UBIAD1 is required to generate MK-4 for the maintenance of vascular endothelial cell survival and development [31]. Cell-based studies show that UBIAD1 and MK-4 play an essential role in vascular cell differentiation and calcification [32]. Results from in vitro studies demonstrate that all naturally occurring SCD-associated UBIAD1 mutations are defective in MK-4 biosynthesis [24, 33, 34]. Nevertheless, gene knockin mice expressing SCD-associated UBIAD1 mutations were born at expected Mendelian ratios and appear normal [21, 35]. Additionally, results from the homozygous deletion of the Ubiad1 gene in knockin mice expressing ERAD-resistant HMGCR suggest that the embryonic lethality of Ubiad1 deficiency results from the interruption of the mevalonate pathway rather than from the reduced synthesis of MK-4 [20]. These studies raise questions of how SCD-associated UBIAD1 mutations affect MK-4 biosynthesis and VKD carboxylation, and whether they contribute to SCD pathogenesis.
In this study, we coupled the biosynthesis of MK-4 by UBIAD1 with VKD carboxylation in human embryonic kidney 293 (HEK293) cells to explore the effect of genetic variation of UBIAD1 on MK-4 production and VKD carboxylation. First, we compared the efficiency of K vitamins in supporting VKD carboxylation in HEK293 cells expressing a chimeric coagulation factor as a reporter protein. We then knocked out the endogenous UBIAD1 gene in the reporter cells by CRISPR-Cas9-mediated genome editing and explored how UBIAD1 mutations affect MK-4 supported VKD carboxylation. As an alternative approach, we determined the production of MK-4 directly from UBIAD1-deficient cells that express different UBIAD1 mutations using the conventional HPLC assay. Furthermore, by blocking the mevalonate pathway with statins, we studied how UBIAD1 mutations affect MK-4 biosynthesis using geranylgeranyl diphosphate (GGPP). Our results suggest that UBIAD1 mutations differentially affect MK-4 biosynthesis and VKD carboxylation.
Results
Vitamin K-dependent carboxylation supported by different forms of vitamin K
Vitamin K exists in three main forms (Figure 1A). Vitamin K1, presented in green leafy vegetables, is the main dietary source of vitamin K. Vitamin K2 is found in fermented food or produced by intestinal microbiota, while vitamin K3 is the synthetic form of the vitamin. To evaluate the efficiency of K vitamins as cofactors for VKD carboxylation in the native milieu, we determined the carboxylation efficiency of a chimeric reporter protein FIXgla-PC in HEK293 cells [36] using different forms of vitamin K as the substrate. Our results show that when menaquinone-4 (MK-4), the most common form of vitamin K2 in animal products, was used as vitamin K2, all three forms of vitamin K can efficiently support VKD carboxylation of the reporter protein (Figure 1B). Next, we compared the efficiency of VKD carboxylation supported by menaquinones with different isoprenyl side chains. Results in Figure 1C show that both MK-4 and MK-7 can efficiently support VKD carboxylation. However, menaquinone, which harbors less than 4 isoprenyl units, has a lower capability for reporter-protein carboxylation. In general, these results agreed with previous results obtained from an in vitro study, except that vitamin K3 was unable to support VKD carboxylation in the in vitro assay [37]. We reasoned that the carboxylation activity of vitamin K3 in our cell-based study results from the conversion of vitamin K3 to MK-4 by UBIAD1 [25].
Figure 1. Vitamin K-dependent carboxylation in HEK293 cells supported by different forms of vitamin K.
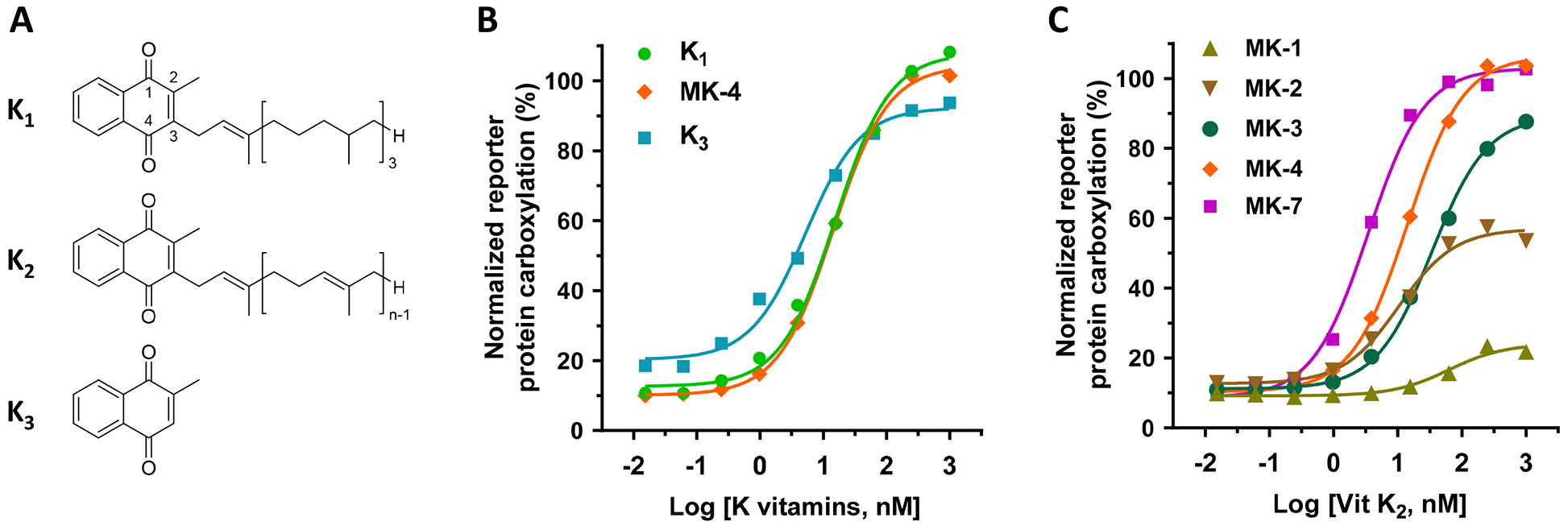
(A) Core structures of K vitamins. K1, vitamin K1 (phylloquinone); K2, vitamin K2 (menaquinones); K3, vitamin K3 (menadione). (B) VKD carboxylation in FIXgla-PC/HEK293 reporter cells supported by K1, menaquinone-4 (MK-4), and K3. FIXgla-PC/HEK293 reporter cells were incubated with a cell culture medium containing increasing concentrations (0.015 nM to 1000 nM) of K vitamins for 24 hours. Carboxylation activity was evaluated by determining the carboxylated reporter-protein FIXgla-PC using ELISA. (C) VKD carboxylation supported by menaquinones in FIXgla-PC/HEK293 reporter cells. FIXgla-PC/HEK293 reporter cells were cultured with increasing concentrations of menaquinones with 1 (MK-1), 2 (MK-2), 3 (MK-3), 4 (MK-4), or 7 (MK-7) isoprene units as the side chain at the 3-position. Reporter-protein FIXgla-PC carboxylation was determined as described in the legend of Figure 1B.
Contribution of UBIAD1 to vitamin K-dependent carboxylation
To confirm the above hypothesis, and to assess the contribution of UBIAD1 to VKD carboxylation, we coupled UBIAD1’s MK-4 biosynthetic activity with VKD carboxylation in HEK293 cells for reporter-protein carboxylation using vitamin K3 as the substrate (Figure 2A). We assumed that vitamin K3 would not be able to support VKD carboxylation when UBIAD1 is knocked out in HEK293 cells. To test this hypothesis, we deployed CRISPR-Cas9-mediated genome editing to knockout the Ubiad1 gene in FIXgla-PC/HEK293 reporter cells. We first tested the genome-editing efficacy of the 6 optimized gRNAs sequences from the GeCKO library [38] (Figure 2B). The plasmid carrying Cas9 and gRNA was transiently expressed in FIXgla-PC/HEK293 cells, and the UBIAD1 knockout was evaluated by measuring the carboxylation of FIXgla-PC using vitamin K3 as the substrate. Results in Figure 2C show that compared with the non-targeting gRNA (control), these UBIAD1 specific gRNAs knocked down 50% - 95% activity. The most efficient gRNA (gRNA-3) was selected for establishing the UBIAD1-knockout stable cell line. The selected positive UBIAD1-knockout cell colony expressed an undetectable UBIAD1 protein (Figure 2D) and was unable to carboxylate the reporter-protein when incubated with vitamin K3 (Figure 2E). Exogenous expression of UBIAD1 in UBIAD1-knockout cells restored carboxylation activity (Figure 2E), suggesting that UBIAD1 is required for vitamin K3-supported carboxylation. Compared with the endogenous UBIAD1, exogenously expressed UBIAD1 displayed a similar efficiency for supporting VKD carboxylation (Figure 1B and Figure 5B as shown below). Together, these results suggest that the coupled VKD carboxylation system in the UBIAD1-knockout cells was intact; only the function of UBIAD1 was specifically ablated.
Figure 2. Characterization of CRISPR-Cas9-mediated UBIAD1 knockout in HEK293 reporter cells.
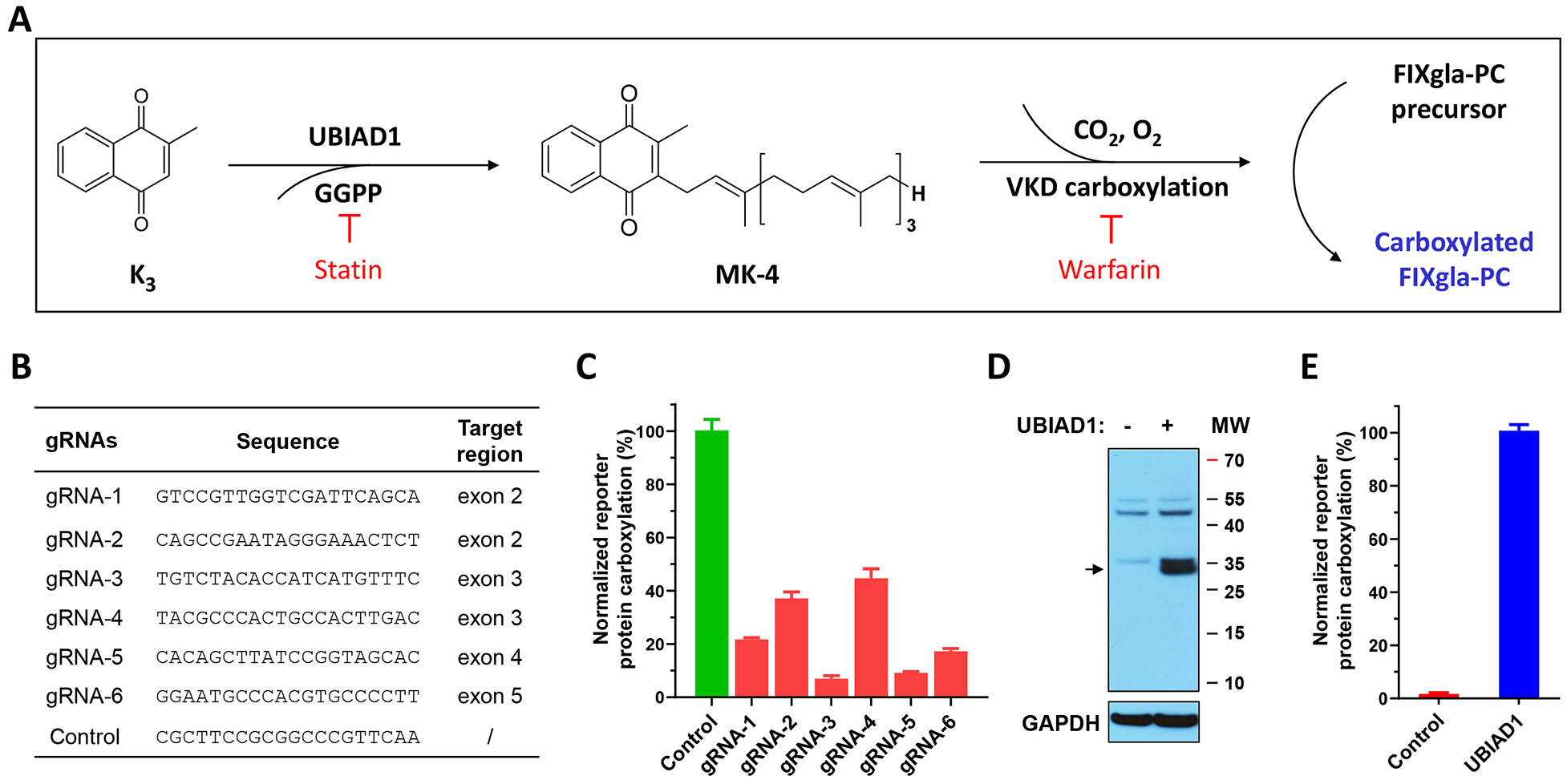
(A) Schematic diagram of coupling the conversion of vitamin K3 to MK-4 by UBIAD1 and VKD carboxylation of FIXgla-PC. Statin, the mevalonate pathway inhibitor, blocks the availability of GGPP and therefore inhibits MK-4 production. Warfarin, the most commonly prescribed oral anticoagulant, blocks vitamin K redox cycling and therefore inhibits VKD carboxylation. (B) The gRNA sequences targeting Ubiad1 and the corresponding targeting regions for CRISPR-Cas9-mediated UBIAD1 knockout. (C) The efficiency of UBIAD1 knockout by different gRNAs in HEK293 reporter cells. Cas9 and individual gRNA were transiently co-transfected into FIXgla-PC/HEK293 reporter cells. Transfected cells were incubated with a cell culture medium containing 1 μM vitamin K3 for 48 hours. Reporter protein carboxylation was determined as described in the legend of Figure 1B. The carboxylation activity of the control (non-targeting) gRNA was normalized to 100%. Data are presented as mean ± SD of three independent experiments (n=3). (D) Immunoblotting analysis of wild-type HEK293 cells and HEK293 cells with UBIAD1 knocked out. The whole-cell lysate was loaded to SDS-PAGE for immunoblotting assay. GAPDH was used as the loading control. The endogenous UBIAD1 band is indicated by an arrowhead. (E) VKD carboxylation activity of UBIAD1 knockout cells (control) and the cells transiently expressing exogenous UBIAD1. The carboxylation activity of cells transiently expressing exogenous UBIAD1 was normalized to 100%. Data are presented as mean ± SD of three independent experiments (n=3).
Functional study of UBIAD1 using VKD carboxylation reporter assay
It has been shown that both vitamin K1 and K2, but not K3, can directly function as cofactors for VKD carboxylation [37], and that vitamin K3 can be converted to MK-4 by UBIAD1 [25]. To explore the possibility of using vitamin K3-supported carboxylation in HEK293 cells as a readout for the functional study of UBIAD1, we incubated UBIAD1-deficient reporter cells with increasing concentrations of different forms of vitamin K, and determined the reporter-protein carboxylation. Consistent with our previous observation [39], results in Figure 3A show that both vitamin K1 and MK-4 can efficiently support VKD carboxylation in the absence of UBIAD1. However, vitamin K3 fails to carboxylate the reporter protein in UBIAD1-knockout cells. These results support the notion that vitamin K1 and MK-4 can directly function as cofactors for VKD carboxylation, while vitamin K3 needs to be converted to MK-4 by UBIAD1 in order to be active.
Figure 3. Vitamin K-dependent carboxylation in UBIAD1-deficient HEK293 reporter cells supported by different forms of vitamin K.
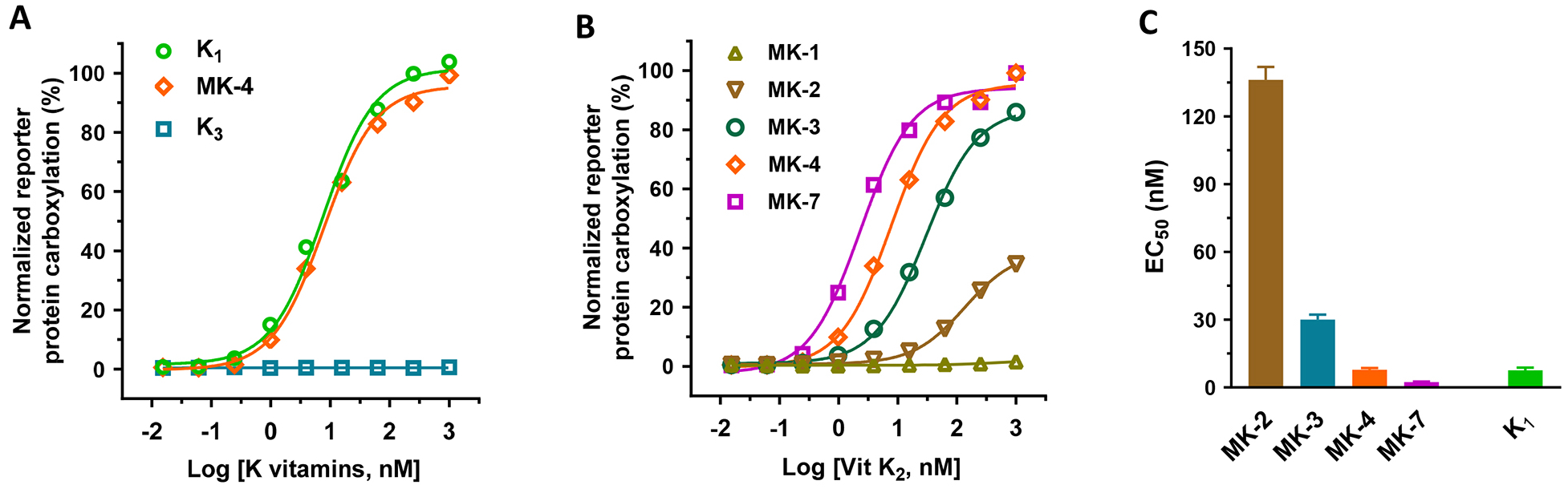
(A and B) VKD carboxylation in UBIAD1-deficient FIXgla-PC/HEK293 reporter cells using (A) vitamin K1, MK-4, and vitamin K3 or (B) menaquinones as the substrates. Reporter cells were incubated with a cell culture medium containing increasing concentrations of K vitamins. Reporter-protein carboxylation was determined as described in the legend of Figure 1B. (C) The half-maximal stimulation concentration (EC50) of K vitamins as determined from Figure 3A and 3B using GraphPad software. Data are presented as mean ± SD.
To further clarify the effect of the side chain of menaquinones on VKD carboxylation, we compared the carboxylation efficiency of different menaquinones in UBIAD1-deficient cells, as was done in the intact HEK293 cells (Figure 1C). Our result showed that the carboxylation efficiency of menaquinones increases with an increase of side chain isoprenyl units at the 3-position (Figure 3B). It is worth noting, that in contrast to the result shown in Figure 1C, MK-1 was unable to support VKD carboxylation in UBIAD1-deficient cells, even at higher concentrations, suggesting that the carboxylation activity of MK-1 in Figure 1C results from the conversion of MK-1 to MK-4 [40] in the intact HEK293 cells. Additionally, MK-7 appears to display improved efficiency for supporting VKD carboxylation than vitamin K1 (EC50 2.3 nM vs 7.6 nM) (Figure 3C).
When UBIAD1 synthesizes MK-4 from vitamin K3, it uses GGPP as the source for the geranylgeranyl side chain at the 3-position [25] (Figure 2A). Since GGPP is a product of the mevalonate pathway, inhibitors of the mevalonate pathway are supposed to affect the production of MK-4 [34, 41]. Results in Figure 4A show that simvastatin and atorvastatin (mevalonate pathway inhibitors) have a dose-response inhibition of VKD carboxylation when vitamin K3 is used as the substrate. This dose-response inhibition of the mevalonate pathway is similar to warfarin, an oral anticoagulant that inhibits the redox cycling of vitamin K. The inhibition appears to be specific to the conversion of vitamin K3 to MK-4, as these inhibitors do not affect vitamin K1-supported carboxylation (Figure 4B). Additionally, when the mevalonate pathway is blocked by simvastatin or atorvastatin, UBIAD1 can employ the exogenously added GGPP in the cell culture medium to synthesize MK-4 and thus support VKD carboxylation (Figure 4C). Furthermore, UBIAD1 can use GPP, FPP, or GGPP as the side chain source to synthesize MK-2, MK-3 or MK-4, respectively, and in turn support VKD carboxylation (Figure 4D), which agrees with results from previous in vitro studies [34, 37]. We assumed that the activity difference between GPP, FPP, and GGPP results from the capability of different menaquinone-supported VKD carboxylation (Figure 3) mechanisms rather than the capability of UBIAD1 using different isoprenyl side chains for synthesizing menaquinones. Together, these results suggested that our cell-based VKD carboxylation reporter system would be a useful tool for assessing the biological functions of UBIAD1.
Figure 4. Effect of mevalonate pathway inhibitors on VKD carboxylation.
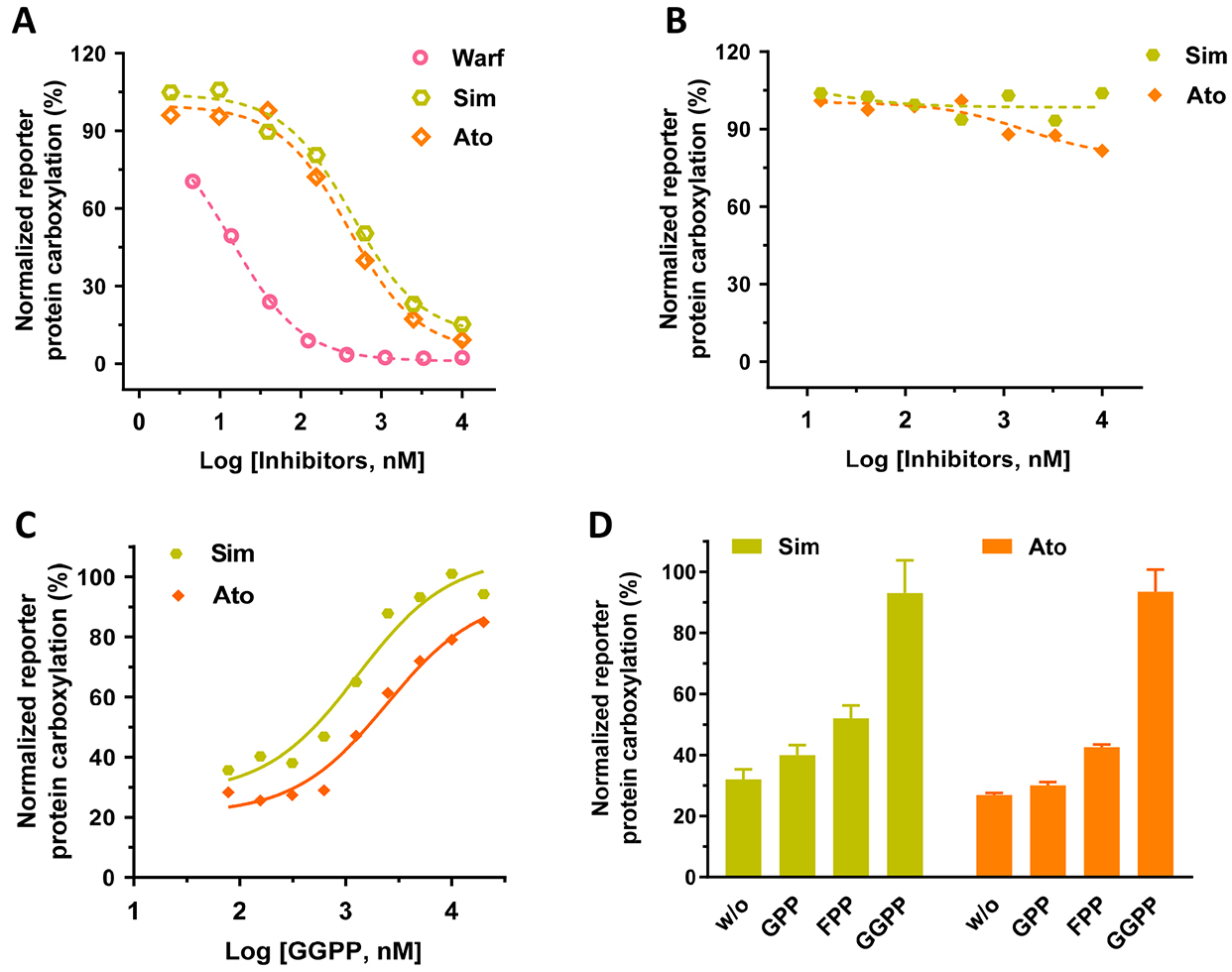
(A and B) Inhibition of VKD carboxylation by simvastatin (Sim), atorvastatin (Ato), or warfarin (Warf) in FIXgla-PC/HEK293 reporter cells when vitamin K3 (A) or vitamin K1 (B) were used as the substrate. Warfarin was used as a control for the inhibition of VKD carboxylation. Reporter cells were incubated with a cell culture medium containing 1 μM vitamin K3 (A) or 5 μM vitamin K1 (B) and increasing concentrations of the inhibitors for 24 hours. (C) The dose-dependent response of GGPP to VKD carboxylation when the endogenous GGPP production is blocked by simvastatin (Sim) or atorvastatin (Ato). FIXgla-PC/HEK293 reporter cells were incubated with cell culture medium containing increasing concentrations of GGPP and 10 μM atorvastatin (Ato) or simvastatin (Sim). (D) Conversion of vitamin K3 to MK-2, MK-3, and MK-4 by UBIAD1 using GPP, FPP, and GGPP as the side chain substrates, respectively. FIXgla-PC/HEK293 cells were incubated with cell culture medium containing 1 μM vitamin K3, 10 μM atorvastatin or simvastatin, and 20 μM GPP, FPP, or GGPP. Reporter-protein carboxylation was determined as described in the legend of Figure 1B. Carboxylation activity of GGPP was normalized to 100%. Data are presented as mean ± SD of three independent experiments (n=3).
Effect of UBIAD1 mutations on MK-4 biosynthesis and VKD carboxylation
In vitro studies show that SCD-associated UBIAD1 mutations dramatically affect MK-4 biosynthesis [24, 33, 34]. However, no obvious phenotypes of MK-4 deficiency have been associated with SCD patients. To explore the effect of UBIAD1 mutations on MK-4 biosynthesis and VKD carboxylation, we selected naturally-occurring mutations and alanine-scanning mutations in the three conserved domains in UBIAD1 as used in previous studies [33, 34, 42]. We transiently expressed the selected mutants in the UBIAD1-deficient reporter cells and determined their efficiency for supporting reporter-protein carboxylation using vitamin K3 as the substrate. Our results showed that, at a fixed vitamin K concentration, most UBIAD1 mutations have a comparable activity to the wild-type enzyme (Figure 5A). However, the R54A, L67Q, G186R, D236A, and D240A mutants decreased UBIAD1 activity to 30% - 85%. Interestingly, several UBIAD1 mutants even displayed a higher activity than the wild-type enzyme.
Figure 5. Effect of UBIAD1 mutations on VKD carboxylation.
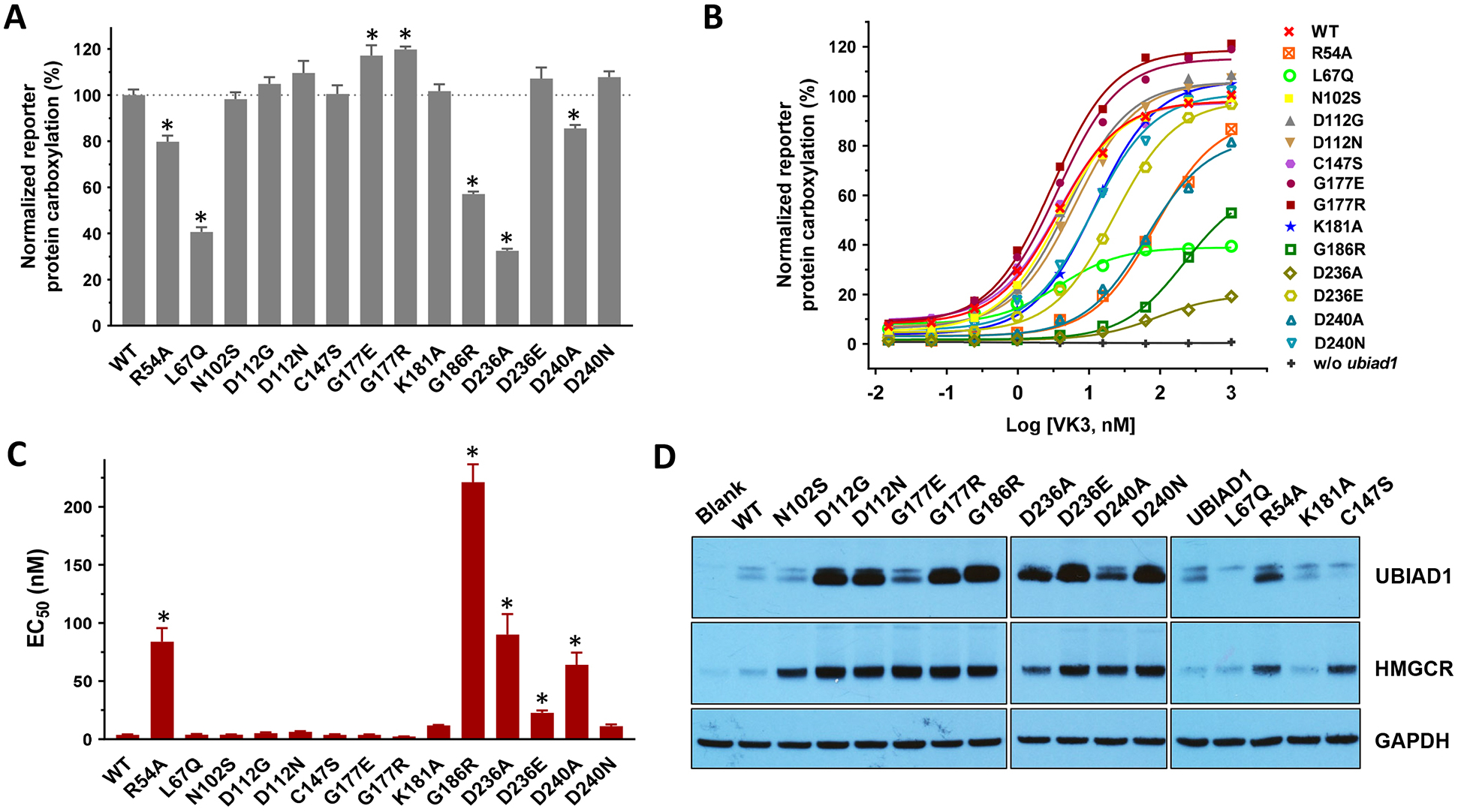
(A and B) UBIAD1 or its variants was transiently expressed in UBIAD1-deficient FIXgla-PC/HEK293 reporter cells. Transfected cells were incubated with a cell culture medium containing 1 μM vitamin K3 (A) or increasing concentrations of vitamin K3 (B) for 24 hours. Wild-type UBIAD1 activity was normalized to 100%. Data are presented as mean ± SD of three independent experiments (n=3). * T-test, P<0.001, compared with the wild-type UBIAD1 (WT). (C) The EC50 values of vitamin K3 for UBIAD1 variants were determined using GraphPad software. Data are presented as mean ± SD. * T-test, P<0.01, compared with the wild-type UBIAD1. (D) Protein expression levels of UBIAD1 variants and HMGCR were determined by western blot analysis. GAPDH was used as the loading control.
To better understand how UBIAD1 mutations might affect MK-4 biosynthesis and VKD carboxylation, we determined the half-maximal stimulation concentration (EC50 or apparent Km) of vitamin K3 for the selected UBIAD1 mutations. Results in Figure 5B show that except for R54A, L67Q, G186R, D236A, and D240A, most mutant proteins can reach equivalent wild-type UBIAD1 activity at higher vitamin K3 concentrations, which is consistent with the results seen in Figure 5A. However, the EC50 of vitamin K3 varies significantly among these mutations (Figure 5C and Table 1). For example, compared with the wild-type enzyme, the D236E mutant is fully active at a saturated vitamin K concentration. However, the EC50 value of vitamin K3 for D236E increased 6-fold (3.7 nM vs. 22.6 nM, Table 1). While the G186R mutant retained ~57% activity, the EC50 value of vitamin K3 increased 60-fold (3.7 nM vs. 221 nM), suggesting that this mutation significantly decreased the affinity of vitamin K3 with respect to the enzyme. Additionally, our results showed that mutating residue G177 to either a positively charged residue (G177R) or a negatively charged residue (G177E) does not affect the enzymatic activity or the EC50 value of vitamin K3, suggesting that this residue may not be involved in MK-4 biosynthesis.
Table 1.
Effect of UBIAD1 mutations on VKD carboxylation and MK-4 biosynthesis
| Vitamin K-dependent carboxylation | MK-4 synthesis, % | SCD-associate mutation [24] | ||
|---|---|---|---|---|
| Activity ± SD, % | EC50 ± SD, nM | |||
| WT | 100 ± 2.4 | 3.7 ± 0.4 | 100 | - |
| R54A | 79.8 ± 2.6 | 84 ± 11.6 | 47.3 ± 5 | - |
| L67Q | 40.7 ± 2 | 3.8 ± 0.8 | 27.9 ± 2.3 | - |
| N102S | 98.2 ± 3 | 3.9 ± 0.3 | 82.2 ± 17 | Yes |
| D112G | 104.8 ± 3 | 5.1 ± 0.8 | 92.5 ± 3.3 | Yes |
| D112N | 109.6 ± 5.2 | 6.4 ± 0.6 | 110.3 ± 10.8 | Yes |
| C147S | 100.5 ± 3.7 | 3.7 ± 0.6 | 106.1 ± 11.2 | - |
| G177E | 117.1 ± 4.5 | 3.7 ± 0.5 | 123.3 ± 10.9 | Yes |
| G177R | 119.8 ± 1.2 | 3.0 ± 0.3 | 125.9 ± 12.9 | Yes |
| K181A | 101.7 ± 3 | 11.9 ± 0.4 | 62.3 ± 4.8 | - |
| G186R | 57.1 ± 1.1 | 221.1 ± 15.4 | 8.2 ± 6.1 | Yes |
| D236A | 32.5 ± 0.9 | 90 ± 17.6 | 3.3 ± 1.9 | - |
| D236E | 107.1 ± 4.9 | 22.6 ± 2.2 | 26.6 ± 5.4 | Yes |
| D240A | 85.6 ± 1.4 | 64 ± 10.5 | 2.6 ± 2.3 | - |
| D240N | 107.8 ± 2.5 | 11.2 ± 1.5 | 26.9 ± 8.1 | Yes |
Note: The activity of vitamin K-dependent carboxylation was obtained at 1 μM vitamin K3. The EC50 value was obtained using vitamin K3 concentrations from 0.015nM to 1000 nM. MK-4 synthetic activity was determined using 1 μM COT-vitamin K.
It has been reported that UBIAD1 mutants are stabilized and intracellularly accumulated, which protects the ERAD of HMGCR [33]. To explore how this might affect MK-4 biosynthesis and VKD carboxylation, we transiently expressed the selected UBIAD1 mutations in our reporter cell line and examined the protein levels of UBIAD1 and HMGCR by western blot analysis. Results in Figure 5D show that, compared with the wild-type enzyme, most UBIAD1 mutations were dramatically stabilized and inhibited ERAD of HMGCR. However, this intracellular accumulation of UBIAD1 mutants appears not to be directly correlated with their VKD carboxylation activities or the EC50 of vitamin K3. For example, the G177R mutant has a significantly higher protein level than that of G177E. However, these two mutants exhibit a comparable VKD carboxylation activity and EC50 value to vitamin K3 (Figure 5 and Table 1). On the other hand, the D236A and D236E mutants have similar intracellular protein accumulations. However, the D236E mutant is fully active, while D236A only displays 32% activity. Additionally, the EC50 value of vitamin K3 for the D236E mutant is 4-fold lower than that of D236A (Table 1).
Next, we directly determined the production of MK-4 by the UBIAD1 mutants in UBIAD1-deficient cells using the conventional HPLC assay [39] (Figure 6A and 6B). Our results showed that, compared with their effect on VKD carboxylation (Figure 5A), several UBIAD1 mutations exhibited a dramatic effect on MK-4 biosynthesis (Figure 6C). For example, the K181A, D236E, and D240N mutants had no effect on VKD carboxylation activity at a saturated vitamin K concentration; however, they decreased MK-4 biosynthesis activity by 40–80%. This suggests that the residual MK-4 biosynthetic activities of these UBIAD1 mutants are sufficient to support VKD carboxylation. Nevertheless, the MK-4 biosynthesis activity of these mutants seem not to have a direct correlation to their intracellular protein accumulation (Figure 5D).
Figure 6. Effect of UBIAD1 mutations on MK-4 biosynthesis.
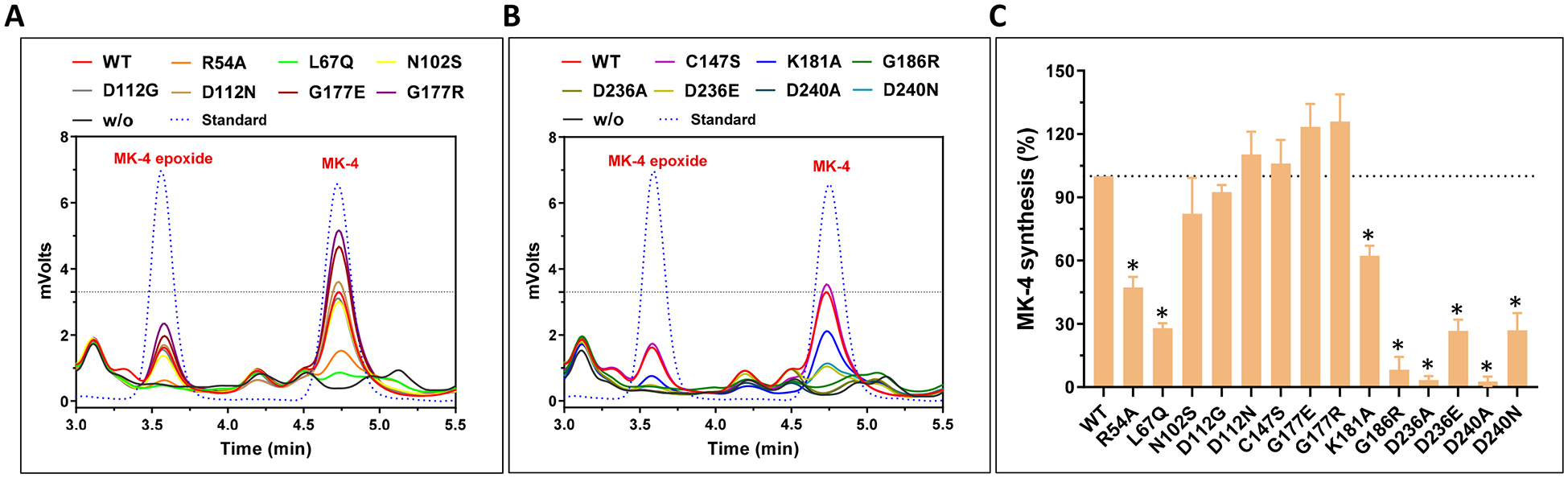
(A and B) UBIAD1 or its variants was transiently expressed in UBIAD1-deficient FIXgla-PC/HEK293 reporter cells. Transfected cells were incubated with a cell culture medium containing 1 μM COT-vitamin K for 24 hours. K vitamins were extracted from the treated cells for the conventional HPLC assay. Pure MK-4 and MK-4 epoxide were used as standards to define the retention time of these compounds in the HPLC chromatogram. (C) MK-4 biosynthetic activity of UBIAD1 and its variants were determined from peak areas of MK-4 and MK-4 epoxide in Figure 6A and 6B. Wild-type UBIAD1 activity was normalized to 100%. Data are presented as mean ± SD of three independent experiments (n=3). * T-test, P<0.01, compared with the wild-type UBIAD1 (WT).
Effect of UBIAD1 mutations on GGPP binding
It has been proposed that residues N102, D112, K181, D236, and D240 mediate the binding of Mg2+/isoprenyl phosphate groups to UBIAD1 for MK-4 synthesis, and that residue G186 is located in close proximity to the proposed active site [11, 33, 34, 43]. To explore whether our cell-based assays could be used for quantitative evaluation of the binding of GGPP to UBIAD1, we determined the EC50 value of GGPP for the selected UBIAD1 mutations. We transiently expressed these UBIAD1 mutations in UBIAD1-deficient reporter cells and inactivated the cell’s mevalonate pathway by simvastatin. Results in Figure 7 show that compared with the wild-type enzyme, most UBIAD1 mutations have a minor effect on the EC50 values of GGPP. However, the EC50 values of GGPP for K181A and G186R mutants increased 5-fold. Taken together with results seen in Figure 5, this demonstrated that the K181A mutation significantly affects GGPP binding, but has a minor effect on vitamin K3 binding. Whereas, the G186R mutant dramatically decreased the binding of both GGPP and vitamin K3 to UBIAD1. These results are consistent with the previous observations that K181 is involved in the isoprenyl phosphate group binding, and that G186 is located near the proposed active site.
Figure 7. Effect of UBIAD1 mutations on GGPP recognition.
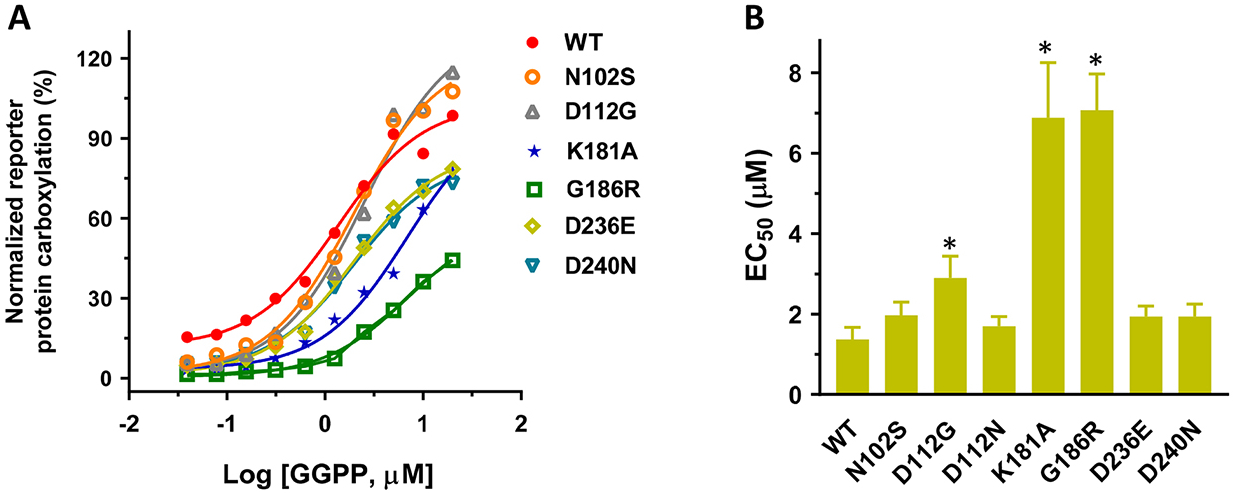
(A) UBIAD1 or the selected UBIAD1 mutants was transiently expressed in UBIAD1-deficient FIXgla-PC/HEK293 reporter cells. Transfected cells were incubated with a cell culture medium containing 10 μM atorvastatin, 1 μM vitamin K3, and increasing concentrations of GGPP for 24 hours. Reporter protein carboxylation was determined as described in the legend of Figure 1B. The EC50 values of GGPP for UBIAD1 variants were determined from Figure 7A using GraphPad software (B). Data are presented as mean ± SD. * T-test, P<0.01, compared with the wild-type UBIAD1 (WT).
Discussion
UBIAD1 is a prenyltransferase that is responsible for the biosynthesis of vitamin K2 [25], the cofactor for extrahepatic carboxylation of VKD proteins. Defects of vitamin K2 have been linked to osteoporosis and cardiovascular diseases [44–46]. However, genetic variations of UBIAD1 are mainly associated with SCD, a disease characterized by the abnormal accumulation of cholesterol in the cornea. Additionally, SCD patients do not exhibit typical phenotypes of vitamin K deficiency [14, 24]. Therefore, the connection between the biological function of UBIAD1 and the clinical consequences of UBIAD1 mutations remains elusive. The purpose of this study was to clarify the effect of UBIAD1 mutations on MK-4 biosynthesis and VKD carboxylation in a cellular milieu.
We utilized HEK293 cells that not only expressed a chimeric VKD reporter-protein, but also had the endogenous Ubiad1 gene knockout, to study the function of UBIAD1. As the main function of UBIAD1 is to synthesize vitamin K2, first, we compared the efficiency of different forms of vitamin K for supporting VKD carboxylation. Our results showed that vitamin K2, especially MK-4 and MK-7, has a comparable capability as vitamin K1 in supporting VKD carboxylation (Figure 3). Therefore, we surmised that the preference and efficacy of vitamin K1 and K2 for hepatic and extrahepatic carboxylation was mainly due to their bioavailability in different tissues [47]. It is also possible that vitamin K1 and K2 have different efficiencies in the carboxylation of structurally distinct VKD proteins [48] in the liver and extrahepatic tissues. Nevertheless, our results showed that the carboxylation efficiency of MK-7 is about 3-fold higher than that of MK-4. Despite the longer half-life of MK-7 in serum [49], results from our cell-based study are consistent with previous observations from healthy adults showing that MK-7 has a better efficacy than MK-4 for extrahepatic carboxylation of osteocalcin [50–52].
Next, we sought to evaluate how UBIAD1 mutations might affect MK-4 biosynthesis and VKD carboxylation. Results of this study indicate that most SCD-associated UBIAD1 mutations have a negligible effect on both MK-4 biosynthesis and VKD carboxylation (Figure 6 and Figure 5). For the hot-spot mutations at positions N102 and G177, only N102S had a moderate effect on MK-4 biosynthesis (retained 82% activity). These results are consistent with clinical observations that typical signs of vitamin K deficiency in SCD patients is not observed. It is also in line with results obtained from the homozygous Ubiad1 N100S (corresponding to N102S in human) knock-in mice showing that the production of MK-4 reduced by 50% compared with the wild-type animal [21]. As vitamin K can be efficiently recycled via the vitamin K cycle enzymes for VKD carboxylation [53], the moderate effect of the N102S mutation on MK-4 production appeared not to affect the final production of functional VKD proteins (Figure 5A). This is true for other UBIAD1 mutations, such as K181A, D236E, and D240N. This observation is in line with a recent clinical study highlighting that the VKD carboxylation of matrix Gla protein (extrahepatic carboxylation supported by MK-4) is normal in the cornea of SCD patients bearing the N102S mutation [54].
It is worth noting that our results reveal that mutations L67Q and G186R dramatically decreased VKD carboxylation (Figure 5) and MK-4 production (Figure 6), suggesting possible consequences for individuals bearing these two mutations. The L65Q mutation (equivalent to L67Q in humans) was identified in zebrafish that displayed cranial vascular hemorrhages [31]. Treatment of the mutant zebrafish with MK-4 rescued 58% of the vascular phenotype, whereas vitamin K1 treatment rescued only 6%, suggesting a pivotal role of MK-4 produced by UBIAD1 in zebrafish. The G186R mutation is a missense mutation found in an early-onset SCD family [55]. It has been reported, that in contrast to the homozygous Ubiad1 N100S knock-in mice, the homozygous Ubiad1 G184R (equivalent to G186R in humans) knock-in mice are embryonic lethal [22]. The authors concluded that the G184R mutation affects MK-4 synthesis more profoundly than that of N100S. This observation agrees with our results that G186R mutant dramatically affects both MK-4 production and VKD carboxylation compared with the N102S mutation (Table 1). Overall, results from our cell-based study agree with results from in vivo studies, and it is reasonable to assume that patients bearing the G186R mutation could have clinical phenotypes associated with the defects of MK-4.
Statins are the most common cholesterol-lowering medications to reduce the risk of cardiovascular diseases [56, 57]. They function by blocking the production of mevalonate, an essential intermediate in the synthesis of GGPP utilized in MK-4 production. Consistent with the previous study [34], our results showed that statins have a dose-response inhibition of VKD carboxylation (Figure 4A). Nevertheless, the inhibition potency of statins is about 30-fold lower than that of warfarin. As statins mainly target the biosynthesis of MK-4, it is reasonable that no bleeding disorders (defects of vitamin K1 associated with carboxylation of coagulation factors) have been reported in statin therapy [56, 58]. However, this result raises a concern of whether statin therapy could have side effects associated with MK-4 deficiency. It has been reported that MK-4 production was reduced by ~45% in the kidneys of atorvastatin-treated mice [59]. A clinical study showed that frequent statin users exhibit accelerated vascular calcification compared with less frequent users [60]. This statin-associated vascular calcification has been confirmed and associated with MK-4 deficiency only very recently [41]. The authors demonstrated that uncarboxylated osteocalcin (extrahepatic carboxylation supported by MK-4) was significantly elevated in statin users and positively correlated with coronary artery calcification in the group of statin users, but not in statin non-users. These results taken together suggest that statin therapy could result in MK-4 deficiency, and it could be beneficial to supplement treatment with MK-4 during the therapy.
The effect of SCD-associated UBIAD1 mutations on MK-4 biosynthesis has been extensively investigated [24, 33, 34]. Results from these studies suggest that all SCD-associated UBIAD1 mutations dramatically reduced MK-4 synthetic activity. These results differ from results obtained in our current study. Our explanation for this discrepancy is that previous studies normalized UBIAD1’s MK-4 biosynthetic activity by protein levels. As UBIAD1 is a multi-organelle localized protein and most UBIAD1 mutant proteins accumulated inside the cells [24, 33, 34], without knowing the linear response range of UBIAD1’s MK-4 biosynthetic activity to the protein levels, normalization of UBIAD1 activity by protein levels could artificially affect MK-4 biosynthetic activity. In this study, we determined the overall impact of UBIAD1 mutations on MK-4 production and VKD carboxylation without normalization by UBIAD1 protein levels. Our results are consistent with results obtained from in vivo studies, as discussed above. It also agrees with the in vitro data from previous studies prior to the data being normalized. Normalization of VKD carboxylation activity (Figure 5A) and MK-synthetic activity (Figure 6C) with protein levels (Figure 5D) significantly changed the overall impact patterns of these mutations (Supplementary Figure 1), as previously reported. Nevertheless, these results suggest that the effect of SCD-associated UBIAD1 mutations on MK-4 biosynthesis and VKD carboxylation does not directly correlate with the clinical phenotype of the visual loss in SCD patients.
A potential limitation of this study is that MK-4 biosynthesis and VKD carboxylation was measured using a coagulation factor as the reporter protein in HEK293 cells. Studies have shown that MK-4 is synthesized locally in different tissues by UBIAD1 using the precursor menadione, which is converted from dietary K vitamins during intestinal absorption and circulated to targeted tissues [25–27, 61, 62]. Additionally, extrahepatic VKD proteins are carboxylated locally in different tissues [63], which differ in vitamin K uptake and utilization [64, 65]. These tissues also have different abundances of vitamin K recycling enzymes [66]. Therefore, further studies are needed on the effect of UBIAD1 mutations on MK-4 biosynthesis and the VKD carboxylation of structure distinct extrahepatic VKD proteins in different cell types. The impact of UBIAD1 mutations on MK-4 biosynthesis in different cell types could have clinical consequences related to the extrahepatic carboxylation of VKD proteins, such as abnormalities in skeletal muscle and bone as observed in UBIAD1 knockout mice [20], which could be due to the uncarboxylation of osteocalcin or matrix Gla protein. The availability of clinical phenotypes associated with defects of MK-4-dependent extrahepatic carboxylation in SCD patients would be beneficial for clarifying the molecular mechanism of UBIAD1 mutations and their clinical consequences.
Materials and Methods
Reagents and cell lines –
Warfarin, vitamin K1, menadione, menaquinone-4 (MK-4), MK-7, MK-4 epoxide, geranyl pyrophosphate (GPP), farnesyl pyrophosphate (FPP), geranylgeranyl pyrophosphate (GGPP), polyethoxylated castor oil (Cremophor EL) and simvastatin were obtained from Sigma-Aldrich (St. Louis, MO). MK-1, MK-2, MK-3, and cyclooctatetraene (COT) derived vitamin K were synthesized based on previously described methods [39, 67, 68]. Stock solutions of K vitamins were prepared by using polyethoxylated castor oil. Atorvastatin was obtained from MedChemExpress LLC (Monmouth Junction, NJ). Xfect transfection reagent was obtained from Clontech Laboratories, Inc. (Mountain View, CA). Mouse anti-carboxylated factor IX gla domain (FIXgla) monoclonal antibody was acquired from Green Mountain Antibodies (Burlington, VT). The anti-HMGCR monoclonal antibody and anti-UBIAD1 polyclonal antibody were sourced from ThermoFisher Scientific (Waltham, MA), and the anti-GAPDH mouse monoclonal antibody from Proteintech Group, Inc. (Rosemont, IL). Horseradish peroxidase-conjugated sheep anti-human protein C (PC) was obtained from Affinity Biologicals, Inc. (Ancaster, ON, Canada). The ABTS (2,2’-Azino-bis (3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt) peroxidase substrate kit for the enzyme-linked immunosorbent assay (ELISA) was purchased from KPL Inc. (Gaithersburg, MD).
Human embryonic kidney 293 (HEK293) cells were acquired from ATCC (Manassas, VA). HEK293 cells stably expressing the reporter-protein FIXgla-PC (protein C with its Gla domain exchanged with that of factor IX) (FIXgla-PC/HEK293) were obtained, as previously described [36]. FIXgla-PC/HEK293 cells with their endogenous Ubiad1 gene knocked out were obtained by CRISPR-Cas9-mediated genome editing [38, 69]. HEK293 cells were cultured in DMEM/F12 medium (Gibco, Grand Island, NY) supplemented with 10% fetal bovine serum (Avantor, Radnor, PA).
DNA Manipulations and plasmids construction –
The cDNA encoding Ubiad1 was cloned into the mammalian expression vector pCI-neo (Promega, Madison, WI). The resulting vector pCI-neo-UBIAD1 was used as a template for the site-directed mutagenesis of UBIAD1 mutations. Naturally occurring and alanine scanning mutations of UBIAD1 were created by QuickChange site-directed mutagenesis. The nucleotide sequences of all the constructs were verified by DNA sequencing at Eton Bioscience Inc. (Research Triangle Park, NC).
VKD carboxylation as supported by different forms of vitamin K –
To determine the efficiency of VKD carboxylation supported by different forms of vitamin K, we cultured FIXgla-PC/HEK293 cells with a fixed concentration or with increasing concentrations of K vitamins for 24 hours. To explore the contribution of UBIAD1 to VKD carboxylation, we used FIXgla-PC/HEK293 cells with their Ubiad1 gene knocked out. The efficiency of FIXgla-PC carboxylation was determined by ELISA, as previously described [36]. When the serial dilution of K vitamins was used in the assay, we plotted the data using GraphPad software and determined the half-maximal stimulation concentration (EC50 or apparent Km).
Cell-based functional study of UBIAD1 and its mutants –
Functional analysis of UBIAD1 and its mutants were performed in FIXgla-PC/HEK293 cells with their endogenous Ubiad1 gene knocked out. This assay is based on the ability of the exogenously expressed UBIAD1 or its mutants to convert vitamin K3 to MK-4 in HEK293 cells to support the carboxylation of FIXgla-PC. Briefly, plasmid DNA of pCI-neo containing the cDNA of wild-type or mutant UBIAD1 was transiently expressed in the Ubiad1-deficient FIXgla-PC/HEK293 cells using the Xfect transfection reagent. At five hours post-transfection, the transfection medium was replaced by a complete culture medium containing 1 μM vitamin K3 or increasing concentrations of K vitamins as described in Figure legends. After incubation for 24 hours, the cell culture medium was collected and directly used for ELISA to determine the level of carboxylated FIXgla-PC, as previously described [36]. UBIAD1 activity was expressed as normalized carboxylated FIXgla-PC. Wild-type UBIAD1 activity was normalized to 100%. The EC50 values of vitamin K3 were determined when the serial dilution of vitamin K3 was used in the assay.
UBIAD1 activity assay using geranyl pyrophosphate as the substrate –
To determine the activity of UBIAD1 using different geranyl pyrophosphates as the substrate, we first incubated FIXgla-PC/HEK293 reporter cells with 10 μM atorvastatin or simvastatin to inhibit the production of the endogenous geranyl pyrophosphate. After incubation for 12 hours, we added a fixed concentration or increasing concentrations of isoprenyl side chain substrates to the cell culture medium. To determine the effect of UBIAD1 mutations on GGPP binding, we transiently expressed wild-type UBIAD1 or its mutants in UBIAD1-deficient FIXgla-PC/HEK293 cells. After 24 hours, we incubated the transfected cells with a medium containing 10 μM atorvastatin for 3 hours. We then incubated the cells with a medium containing 1 μM vitamin K3, 10 μM atorvastatin, and increasing concentrations of GGPP. The carboxylation efficiency of FIXgla-PC was determined by ELISA after 24 hours incubation.
MK-4 production determined directly from HEK293 cells using the conventional HPLC assay –
To determine the effect of UBIAD1 mutations on MK-4 production, we transiently expressed wild-type UBIAD1 or its mutants in UBIAD1-deficient FIXgla-PC/HEK293 cells. Transfected cells were incubated with 1 μM COT-vitamin K for 24 hours. Cells were washed by PBS before harvest, and K vitamins were extracted from the cell pellet. The production of MK-4 and MK-4 epoxide were determined using the conventional HPLC assay, as previously described [39]. In this assay, we used COT-vitamin K instead of vitamin K3 as the substrate, because COT-vitamin K is more efficient and has less cytotoxicity than vitamin K3, as observed previously [39].
Detection of UBIAD1 and its mutant proteins by immunoblotting –
Wild-type UBIAD1 or its mutant proteins were transiently expressed in UBIAD1-deficient HEK293 cells. Forty-eight hours post-transfection, cells were harvested and lysed in RIPA lysis buffer with 1X Roche cOmplete™ protease inhibitor cocktail. Proteins in the cell lysate were separated by SDS-PAGE and transferred onto the PVDF membrane. After blocking the membrane with 5% non-fat milk for 1 hour, protein bands were detected by the primary antibodies (anti-UBIAD1, anti-HMGCR, or anti-GAPDH) and the corresponding secondary antibodies conjugated with HRP. The protein bands were then visualized using Amersham ECL western blotting detection kit.
Supplementary Material
Acknowledgments:
This work was supported by the National Institutes of Health HL131690 to JKT and DWS. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations:
- COT
cyclooctatetraene
- ERAD
endoplasmic reticulum-associated degradation
- FIXgla-PC
protein C with its Gla domain exchanged with that of factor IX
- FPP
farnesyl pyrophosphate
- GGPP
geranylgeranyl diphosphate
- GPP
geranyl pyrophosphate
- HEK293
human embryonic kidney 293
- SCD
Schnyder corneal dystrophy
- UBIAD1
UbiA prenyltransferase domain-containing protein-1
- VKD
vitamin K-dependent
Footnotes
Conflict-of-Interest Disclosure: All authors declare no competing financial interests.
References
- 1.Weiss JS (2009) Schnyder corneal dystrophy, Curr Opin Ophthalmol. 20, 292–8. [DOI] [PubMed] [Google Scholar]
- 2.Bron AJ, Williams HP & Carruthers ME (1972) Hereditary crystalline stromal dystrophy of Schnyder. I. Clinical features of a family with hyperlipoproteinaemia, Br J Ophthalmol. 56, 383–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rodrigues MM, Kruth HS, Krachmer JH & Willis R (1987) Unesterified cholesterol in Schnyder’s corneal crystalline dystrophy, Am J Ophthalmol. 104, 157–63. [DOI] [PubMed] [Google Scholar]
- 4.Lisch W, Weidle EG, Lisch C, Rice T, Beck E & Utermann G (1986) Schnyder’s dystrophy. Progression and metabolism, Ophthalmic Paediatr Genet. 7, 45–56. [DOI] [PubMed] [Google Scholar]
- 5.Weiss JS, Kruth HS, Kuivaniemi H, Tromp G, White PS, Winters RS, Lisch W, Henn W, Denninger E, Krause M, Wasson P, Ebenezer N, Mahurkar S & Nickerson ML (2007) Mutations in the UBIAD1 gene on chromosome short arm 1, region 36, cause Schnyder crystalline corneal dystrophy, Invest Ophthalmol Vis Sci. 48, 5007–12. [DOI] [PubMed] [Google Scholar]
- 6.Shearman AM, Hudson TJ, Andresen JM, Wu X, Sohn RL, Haluska F, Housman DE & Weiss JS (1996) The gene for schnyder’s crystalline corneal dystrophy maps to human chromosome 1p34.1-p36, Hum Mol Genet. 5, 1667–72. [DOI] [PubMed] [Google Scholar]
- 7.Orr A, Dube MP, Marcadier J, Jiang H, Federico A, George S, Seamone C, Andrews D, Dubord P, Holland S, Provost S, Mongrain V, Evans S, Higgins B, Bowman S, Guernsey D & Samuels M (2007) Mutations in the UBIAD1 gene, encoding a potential prenyltransferase, are causal for Schnyder crystalline corneal dystrophy, PLoS One. 2, e685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ashby MN, Kutsunai SY, Ackerman S, Tzagoloff A & Edwards PA (1992) Coq2 Is a Candidate for the Structural Gene Encoding Para-Hydroxybenzoate - Polyprenyltransferase, Journal of Biological Chemistry. 267, 4128–4136. [PubMed] [Google Scholar]
- 9.Suvarna K, Stevenson D, Meganathan R & Hudspeth ME (1998) Menaquinone (vitamin K2) biosynthesis: localization and characterization of the menA gene from Escherichia coli, J Bacteriol. 180, 2782–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schledz M, Seidler A, Beyer P & Neuhaus G (2001) A novel phytyltransferase from Synechocystis sp PCC 6803 involved in tocopherol biosynthesis, Febs Lett. 499, 15–20. [DOI] [PubMed] [Google Scholar]
- 11.Li W (2016) Bringing Bioactive Compounds into Membranes: The UbiA Superfamily of Intramembrane Aromatic Prenyltransferases, Trends Biochem Sci. 41, 356–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nickerson ML, Kostiha BN, Brandt W, Fredericks W, Xu KP, Yu FS, Gold B, Chodosh J, Goldberg M, Lu DW, Yamada M, Tervo TM, Grutzmacher R, Croasdale C, Hoeltzenbein M, Sutphin J, Malkowicz SB, Wessjohann L, Kruth HS, Dean M & Weiss JS (2010) UBIAD1 mutation alters a mitochondrial prenyltransferase to cause Schnyder corneal dystrophy, PLoS One. 5, e10760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin BR, Frausto RF, Vo RC, Chiu SY, Chen JL & Aldave AJ (2016) Identification of the First De Novo UBIAD1 Gene Mutation Associated with Schnyder Corneal Dystrophy, J Ophthalmol. 2016, 1968493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sarosiak A, Udziela M, Sciezynska A, Ozieblo D, Wawrzynowska A, Szaflik JP & Oldak M (2018) Clinical diversity in patients with Schnyder corneal dystrophy-a novel and known UBIAD1 pathogenic variants, Graefes Arch Clin Exp Ophthalmol. 256, 2127–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gonzalvez M, Ho Wang Yin G, Gascon P, Denis D & Hoffart L (2018) Clinical and para-clinical description of a novel mutation for Schnyder dystrophy in a French family, J Fr Ophtalmol. 41, 920–925. [DOI] [PubMed] [Google Scholar]
- 16.Evans CJ, Dudakova L, Skalicka P, Mahelkova G, Horinek A, Hardcastle AJ, Tuft SJ & Liskova P (2018) Schnyder corneal dystrophy and associated phenotypes caused by novel and recurrent mutations in the UBIAD1 gene, BMC Ophthalmol. 18, 250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McGarvey TW, Nguyen TB & Malkowicz SB (2005) An interaction between apolipoprotein E and TERE1 with a possible association with bladder tumor formation, J Cell Biochem. 95, 419–28. [DOI] [PubMed] [Google Scholar]
- 18.Zhang WY, Gaynor PM & Kruth HS (1996) Apolipoprotein E produced by human monocyte-derived macrophages mediates cholesterol efflux that occurs in the absence of added cholesterol acceptors, J Biol Chem. 271, 28641–6. [DOI] [PubMed] [Google Scholar]
- 19.Kruth HS, Skarlatos SI, Gaynor PM & Gamble W (1994) Production of cholesterol-enriched nascent high density lipoproteins by human monocyte-derived macrophages is a mechanism that contributes to macrophage cholesterol efflux, J Biol Chem. 269, 24511–8. [PubMed] [Google Scholar]
- 20.Jo Y, Kim SS, Garland K, Fuentes I, DiCarlo LM, Ellis JL, Fu X, Booth SL, Evers BM & DeBose-Boyd RA (2020) Enhanced ER-associated degradation of HMG CoA reductase causes embryonic lethality associated with Ubiad1 deficiency, Elife. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jo Y, Hamilton JS, Hwang S, Garland K, Smith GA, Su S, Fuentes I, Neelam S, Thompson BM, McDonald JG & DeBose-Boyd RA (2019) Schnyder corneal dystrophy-associated UBIAD1 inhibits ER-associated degradation of HMG CoA reductase in mice, Elife. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang SY, Tang JJ, Xiao X, Qi W, Wu S, Jiang C, Hong J, Xu J, Song BL & Luo J (2019) Schnyder corneal dystrophy-associated UBIAD1 mutations cause corneal cholesterol accumulation by stabilizing HMG-CoA reductase, PLoS Genet. 15, e1008289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schumacher MM, Elsabrouty R, Seemann J, Jo Y & DeBose-Boyd RA (2015) The prenyltransferase UBIAD1 is the target of geranylgeraniol in degradation of HMG CoA reductase, Elife. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nickerson ML, Bosley AD, Weiss JS, Kostiha BN, Hirota Y, Brandt W, Esposito D, Kinoshita S, Wessjohann L, Morham SG, Andresson T, Kruth HS, Okano T & Dean M (2013) The UBIAD1 prenyltransferase links menaquinone-4 [corrected] synthesis to cholesterol metabolic enzymes, Hum Mutat. 34, 317–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakagawa K, Hirota Y, Sawada N, Yuge N, Watanabe M, Uchino Y, Okuda N, Shimomura Y, Suhara Y & Okano T (2010) Identification of UBIAD1 as a novel human menaquinone-4 biosynthetic enzyme, Nature. 468, 117–21. [DOI] [PubMed] [Google Scholar]
- 26.Shearer MJ & Newman P (2014) Recent trends in the metabolism and cell biology of vitamin K with special reference to vitamin K cycling and MK-4 biosynthesis, J Lipid Res. 55, 345–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shearer MJ & Okano T (2018) Key Pathways and Regulators of Vitamin K Function and Intermediary Metabolism, Annu Rev Nutr. 38, 127–151. [DOI] [PubMed] [Google Scholar]
- 28.Halder M, Petsophonsakul P, Akbulut AC, Pavlic A, Bohan F, Anderson E, Maresz K, Kramann R & Schurgers L (2019) Vitamin K: Double Bonds beyond Coagulation Insights into Differences between Vitamin K1 and K2 in Health and Disease, Int J Mol Sci. 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Azuma K & Inoue S (2019) Multiple Modes of Vitamin K Actions in Aging-Related Musculoskeletal Disorders, Int J Mol Sci. 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakagawa K, Sawada N, Hirota Y, Uchino Y, Suhara Y, Hasegawa T, Amizuka N, Okamoto T, Tsugawa N, Kamao M, Funahashi N & Okano T (2014) Vitamin K2 biosynthetic enzyme, UBIAD1 is essential for embryonic development of mice, PLoS One. 9, e104078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hegarty JM, Yang H & Chi NC (2013) UBIAD1-mediated vitamin K2 synthesis is required for vascular endothelial cell survival and development, Development. 140, 1713–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu S, Guo W, Han X, Dai W, Diao Z & Liu W (2016) Role of UBIAD1 in Intracellular Cholesterol Metabolism and Vascular Cell Calcification, PLoS One. 11, e0149639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jun DJ, Schumacher MM, Hwang S, Kinch LN, Grishin NV & DeBose-Boyd RA (2020) Schnyder corneal dystrophy-associated UBIAD1 is defective in MK-4 synthesis and resists autophagy-mediated degradation([S]), J Lipid Res. 61, 746–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hirota Y, Nakagawa K, Sawada N, Okuda N, Suhara Y, Uchino Y, Kimoto T, Funahashi N, Kamao M, Tsugawa N & Okano T (2015) Functional characterization of the vitamin K2 biosynthetic enzyme UBIAD1, PLoS One. 10, e0125737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dong F, Jin X, Boettler MA, Sciulli H, Abu-Asab M, Del Greco C, Wang S, Hu YC, Campos MM, Jackson SN, Muller L, Woods AS, Combs CA, Zhang J, Nickerson ML, Kruth HS, Weiss JS & Kao WW (2018) A Mouse Model of Schnyder Corneal Dystrophy with the N100S Point Mutation, Sci Rep. 8, 10219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tie JK, Jin DY, Straight DL & Stafford DW (2011) Functional study of the vitamin K cycle in mammalian cells, Blood. 117, 2967–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buitenhuis HC, Soute BA & Vermeer C (1990) Comparison of the vitamins K1, K2 and K3 as cofactors for the hepatic vitamin K-dependent carboxylase, Biochim Biophys Acta. 1034, 170–5. [DOI] [PubMed] [Google Scholar]
- 38.Cong L, Ran FA, Cox D, Lin SL, Barretto R, Habib N, Hsu PD, Wu XB, Jiang WY, Marraffini LA & Zhang F (2013) Multiplex Genome Engineering Using CRISPR/Cas Systems, Science. 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen X, Liu Y, Furukawa N, Jin DY, Paul Savage G, Stafford DW, Suhara Y, Williams CM & Tie JK (2021) A novel vitamin K derived anticoagulant tolerant to genetic variations of vitamin K epoxide reductase, J Thromb Haemost. 19, 689–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Okano T, Shimomura Y, Yamane M, Suhara Y, Kamao M, Sugiura M & Nakagawa K (2008) Conversion of phylloquinone (Vitamin K1) into menaquinone-4 (Vitamin K2) in mice: two possible routes for menaquinone-4 accumulation in cerebra of mice, J Biol Chem. 283, 11270–9. [DOI] [PubMed] [Google Scholar]
- 41.Zhelyazkova-Savova MD, Yotov YT, Nikolova MN, Nazifova-Tasinova NF, Vankova DG, Atanasov AA & Galunska BT (2021) Statins, vascular calcification, and vitamin K-dependent proteins: Is there a relation?, Kaohsiung J Med Sci. [DOI] [PubMed] [Google Scholar]
- 42.Wang X, Wang D, Jing P, Wu Y, Xia Y, Chen M & Hong L (2013) A novel Golgi retention signal RPWS for tumor suppressor UBIAD1, PLoS One. 8, e72015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang H, Levin EJ, Liu S, Bai Y, Lockless SW & Zhou M (2014) Structure of a membrane-embedded prenyltransferase homologous to UBIAD1, PLoS Biol. 12, e1001911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mandatori D, Pelusi L, Schiavone V, Pipino C, Di Pietro N & Pandolfi A (2021) The Dual Role of Vitamin K2 in “Bone-Vascular Crosstalk”: Opposite Effects on Bone Loss and Vascular Calcification, Nutrients. 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spronk HM, Soute BA, Schurgers LJ, Thijssen HH, De Mey JG & Vermeer C (2003) Tissue-specific utilization of menaquinone-4 results in the prevention of arterial calcification in warfarin-treated rats, J Vasc Res. 40, 531–7. [DOI] [PubMed] [Google Scholar]
- 46.Mansour AG, Hariri E, Daaboul Y, Korjian S, El Alam A, Protogerou AD, Kilany H, Karam A, Stephan A & Bahous SA (2017) Vitamin K2 supplementation and arterial stiffness among renal transplant recipients-a single-arm, single-center clinical trial, J Am Soc Hypertens. 11, 589–597. [DOI] [PubMed] [Google Scholar]
- 47.Shearer MJ & Newman P (2008) Metabolism and cell biology of vitamin K, Thromb Haemost. 100, 530–47. [PubMed] [Google Scholar]
- 48.Hao Z, Jin DY, Chen X, Schurgers LJ, Stafford DW & Tie JK (2021) gamma-Glutamyl carboxylase mutations differentially affect the biological function of vitamin K-dependent proteins, Blood. 137, 533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schurgers LJ, Teunissen KJ, Hamulyak K, Knapen MH, Vik H & Vermeer C (2007) Vitamin K-containing dietary supplements: comparison of synthetic vitamin K1 and natto-derived menaquinone-7, Blood. 109, 3279–83. [DOI] [PubMed] [Google Scholar]
- 50.Inaba N, Sato T & Yamashita T (2015) Low-Dose Daily Intake of Vitamin K(2) (Menaquinone-7) Improves Osteocalcin gamma-Carboxylation: A Double-Blind, Randomized Controlled Trials, J Nutr Sci Vitaminol (Tokyo). 61, 471–80. [DOI] [PubMed] [Google Scholar]
- 51.Theuwissen E, Cranenburg EC, Knapen MH, Magdeleyns EJ, Teunissen KJ, Schurgers LJ, Smit E & Vermeer C (2012) Low-dose menaquinone-7 supplementation improved extra-hepatic vitamin K status, but had no effect on thrombin generation in healthy subjects, Br J Nutr. 108, 1652–7. [DOI] [PubMed] [Google Scholar]
- 52.Sato T, Schurgers LJ & Uenishi K (2012) Comparison of menaquinone-4 and menaquinone-7 bioavailability in healthy women, Nutr J. 11, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tie JK & Stafford DW (2016) Structural and functional insights into enzymes of the vitamin K cycle, J Thromb Haemost. 14, 236–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sarosiak A, Ozieblo D, Udziela M, Vermeer C, Malejczyk J, Szaflik JP & Oldak M (2020) High expression of Matrix Gla Protein in Schnyder corneal dystrophy patients points to an active role of vitamin K in corneal health, Acta Ophthalmol. [DOI] [PubMed] [Google Scholar]
- 55.Weiss JS, Kruth HS, Kuivaniemi H, Tromp G, Karkera J, Mahurkar S, Lisch W, Dupps WJ Jr., White PS, Winters RS, Kim C, Rapuano CJ, Sutphin J, Reidy J, Hu FR, Lu DW, Ebenezer N & Nickerson ML (2008) Genetic analysis of 14 families with Schnyder crystalline corneal dystrophy reveals clues to UBIAD1 protein function, Am J Med Genet A. 146A, 271–83. [DOI] [PubMed] [Google Scholar]
- 56.Adhyaru BB & Jacobson TA (2018) Safety and efficacy of statin therapy, Nat Rev Cardiol. 15, 757–769. [DOI] [PubMed] [Google Scholar]
- 57.Cholesterol Treatment Trialists C, Mihaylova B, Emberson J, Blackwell L, Keech A, Simes J, Barnes EH, Voysey M, Gray A, Collins R & Baigent C (2012) The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials, Lancet. 380, 581–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Newman CB, Preiss D, Tobert JA, Jacobson TA, Page RL 2nd, Goldstein LB, Chin C, Tannock LR, Miller M, Raghuveer G, Duell PB, Brinton EA, Pollak A, Braun LT, Welty FK, American Heart Association Clinical Lipidology, L. M., Thrombosis Committee, a. J. C. o. t. C. o. A. T., Vascular, B., Council on, L., Cardiometabolic, H., Council on Cardiovascular Disease in the, Y., Council on Clinical, C. & Stroke, C. (2019) Statin Safety and Associated Adverse Events: A Scientific Statement From the American Heart Association, Arterioscler Thromb Vasc Biol. 39, e38–e81. [DOI] [PubMed] [Google Scholar]
- 59.Harshman SG, Shea MK, Fu X, Grusak MA, Smith D, Lamon-Fava S, Kuliopulos A, Greenberg A & Booth SL (2019) Atorvastatin Decreases Renal Menaquinone-4 Formation in C57BL/6 Male Mice, J Nutr. 149, 416–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Saremi A, Bahn G, Reaven PD & Investigators V (2012) Progression of vascular calcification is increased with statin use in the Veterans Affairs Diabetes Trial (VADT), Diabetes Care. 35, 2390–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hirota Y, Tsugawa N, Nakagawa K, Suhara Y, Tanaka K, Uchino Y, Takeuchi A, Sawada N, Kamao M, Wada A, Okitsu T & Okano T (2013) Menadione (vitamin K3) is a catabolic product of oral phylloquinone (vitamin K1) in the intestine and a circulating precursor of tissue menaquinone-4 (vitamin K2) in rats, J Biol Chem. 288, 33071–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thijssen HHW, DrittijReijnders MJ & Fischer MAJG (1996) Phylloquinone and menaquinone-4 distribution in rats: Synthesis rather than uptake determines menaquinone-4 organ concentrations, Journal of Nutrition. 126, 537–543. [DOI] [PubMed] [Google Scholar]
- 63.Danziger J (2008) Vitamin K-dependent proteins, warfarin, and vascular calcification, Clin J Am Soc Nephrol. 3, 1504–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ellis JL, Fu X, Karl JP, Hernandez CJ, Mason JB, DeBose-Boyd RA & Booth SL (2021) Multiple Dietary Vitamin K Forms Are Converted to Tissue Menaquinone-4 in Mice, J Nutr. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Suhara Y, Murakami A, Nakagawa K, Mizuguchi Y & Okano T (2006) Comparative uptake, metabolism, and utilization of menaquinone-4 and phylloquinone in human cultured cell lines, Bioorg Med Chem. 14, 6601–7. [DOI] [PubMed] [Google Scholar]
- 66.Wallin R, Cain D & Sane DC (1999) Matrix Gla protein synthesis and gamma-carboxylation in the aortic vessel wall and proliferating vascular smooth muscle cells--a cell system which resembles the system in bone cells, Thromb Haemost. 82, 1764–7. [PubMed] [Google Scholar]
- 67.Yoshimura H, Hirota Y, Soda S, Okazeri M, Takagi Y, Takeuchi A, Tode C, Kamao M, Osakabe N & Suhara Y (2020) Study on structure-activity relationship of vitamin K derivatives: Conversion of the naphthoquinone part into another aromatic ring and evaluation of their neuronal differentiation-inducing activity, Bioorg Med Chem Lett. 30, 127059. [DOI] [PubMed] [Google Scholar]
- 68.Xing H, Houston SD, Chen XJ, Ghassabian S, Fahrenhorst-Jones T, Kuo A, Murray CEP, Conn KA, Jaeschke KN, Jin DY, Pasay C, Bernhardt PV, Burns JM, Tsanaktsidis J, Savage GP, Boyle GM, De Voss JJ, McCarthy J, Walter GH, Burne THJ, Smith MT, Tie JK & Williams CM (2019) Cyclooctatetraene: A Bioactive Cubane Paradigm Complement, Chem-Eur J. 25, 2729–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tie JK, Carneiro JD, Jin DY, Martinhago CD, Vermeer C & Stafford DW (2016) Characterization of vitamin K-dependent carboxylase mutations that cause bleeding and nonbleeding disorders, Blood. 127, 1847–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.