

Abstract
Background:
Urine and plasma biomarker testing for lysosomal storage disorders by liquid chromatography mass spectrometry (LC-MS) currently requires multiple analytical methods to detect the abnormal accumulation of oligosaccharides, mucopolysaccharides, and glycolipids. To improve clinical testing efficiency, we developed a single LC-MS method to simultaneously identify disorders of oligosaccharide, mucopolysaccharide, and glycolipid metabolism with minimal sample preparation.
Methods:
We created a single chromatographic method for separating free glycans and glycolipids in their native form, using an amide column and high pH conditions. We used this glycomic profiling method both in untargeted analyses of patient and control urines using LC ion-mobility high-resolution MS (biomarker discovery), and targeted analyses of urine, serum, and dried blood spot samples by LC-MS/MS (clinical validation).
Results:
Untargeted glycomic profiling revealed twenty biomarkers that could identify and subtype mucopolysaccharidoses. We incorporated these with known oligosaccharide and glycolipid biomarkers into a rapid test that identifies at least 27 lysosomal storage disorders, including oligosaccharidoses, mucopolysaccharidoses, sphingolipidoses, glycogen storage disorders, and congenital disorders of glycosylation and de-glycosylation. In a validation set containing 115 urine samples from patients with lysosomal storage disorders, all were unambiguously distinguished from normal controls, with correct disease subtyping for 88% (101/115) of cases. Glucosylsphingosine was reliably elevated in dried blood spots from Gaucher disease patients with baseline resolution from galactosylsphingosine.
Conclusion:
Glycomic profiling by liquid chromatography mass spectrometry identifies a range of lysosomal storage disorders. This test can be used in clinical evaluations to rapidly focus a diagnosis, as well as to clarify or support additional gene sequencing and enzyme studies.
Keywords: Lysosomal storage disorders, Inborn errors of metabolism, Glycomics, Metabolomics, Biomarker discovery, Liquid chromatography mass spectrometry
1. Introduction
Lysosomal storage disorders (LSDs) encompass deficiencies of acid hydrolases arising from primary enzyme deficiencies or, more rarely, defects in activator proteins, lysosomal membrane transporters, or proteins involved in post-translational enzyme modification. Regardless of primary etiology, the resulting deficiency of one or more enzymes leads to intracellular accumulation of sugar-containing substrates in the broad categories of oligosaccharides, mucopolysaccharides, and glycolipids, which in turn damage tissues and organs and ultimately lead to clinical signs and symptoms. There is significant clinical overlap between the LSDs, with typical features including developmental and cognitive regression, organomegaly, and skeletal abnormalities. Early diagnosis and therapeutic intervention can significantly improve clinical outcome [1], with some disorders (e,g., Pompe disease and mucopolysaccharidosis type 1) detected by newborn screening [2–5] via multiplex enzyme assay and confirmed by a combination of gene sequencing, enzyme assay, and targeted biomarker measurements. However, most LSDs are not included in newborn screening panels, and many patient evaluations are still prompted by clinical or family history. For these patients, the workup may begin with biochemical tests for oligosaccharides and mucopolysaccharides in urine, and for certain glycolipids (e.g., galactosylsphingosine) in blood (plasma or dried blood spots [DBS]) [6]. Importantly, this approach requires a separate test for each of the three major compound classes. Positive results of metabolic testing must be confirmed by additional studies involving gene sequencing or enzyme testing, or both [7]. In many cases, the clinical evaluation instead begins with genetic testing through gene panels or exome or genome sequencing [8], with results often requiring additional biochemical correlation including testing for evidence of abnormal substrate accumulation [9,10].
A variety of simple tests can be used to detect abnormal substrate accumulations (e.g., urinary mucopolysaccharides) in patients undergoing clinical evaluation for a possible LSD, with mass spectrometry rapidly replacing traditional methods such as thin-layer chromatography, spectrophotometry, and electrophoresis. Matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) MS has been used, but does not separate isomeric species [11]. As applied to comprehensive metabolomic profiling, liquid chromatography high-resolution MS detects thousands of compounds and has successfully identified a number of inherited metabolic disorders [12–15], but with poor detection of LSDs [14,16]. More focused LC-MS/MS methods have successfully identified oligosaccharidoses by measuring branched-chain sugars [17–19], mucopolysaccharidoses (MPS) via large, linear, acidic sugars [20–23], and sphingolipidoses via glycolipids, or sugar-containing lipids, including the separation of the isomeric species glucosylsphingosine and galactosylsphingosine [24–26]. However, the structural diversity and resulting need for separate analytic conditions precludes the combination of these methods into a single, comprehensive analysis. Furthermore, testing for mucopolysaccharidoses in urine requires extra steps including derivatization [21,27], depolymerization [20], or digestion [22,23], resulting in conditions incompatible with other methods for detecting LSDs. While oligosaccharides [17] and glycolipids [24,25] can be analyzed without derivatization, their analysis still requires distinct methods that favor sugar- or lipid-based molecules, respectively. In an important advancement, naturally occurring, small glycosaminoglycan fragments have been detected in patients with mucopolysaccharidosis and successfully used for identifying and subtyping mucopolysaccharidoses without further digestion, although derivatization was still required [21,27].
Given that many LSDs have overlapping symptoms and require laboratory testing for diagnosis, the development of a LC-MS method to simultaneously detect a wide range of LSD biomarkers would improve testing efficiency and performance for patients undergoing clinical evaluation. Such a method requires that (1) the disease-associated biomarkers are detectable in their native form without further processing, and (2) the analytic conditions support the simultaneous measurement of glycans in patients with oligosaccharidosis or mucopolysaccharidosis, as well as glycolipids in patients with sphingolipidosis. Notably, despite the vast structural differences between biomarkers for the three disease classes, nearly all have sugar moieties as a common feature. This supports the creation of a chromatographic method for profiling metabolites with sugar moieties—i.e., a glycomic profiling method—that can detect the glycosaminoglycan fragments previously identified in derivatized form [21,27], reveal any additional glycans specific for identifying mucopolysaccharidoses, and separate both glycans and glycolipids while resolving clinically important isomers.
Here, we describe a LC-MS strategy that detects an extensive range of free glycan and glycolipid biomarkers for the identification of patients with one of a broad range of LSDs. We first used an untargeted glycomic profiling approach to discover biomarkers of mucopolysaccharidoses and identified twenty native biomarkers in urine that could identify and subtype mucopolysaccharidoses. We then combined these with known glycan and glycolipid biomarkers into a targeted LC-MS/MS method for clinical validation in urine, serum, and dried blood spots. Using Z-score analysis to reveal abnormal biomarker abundances, we show accurate detection in urine for a wide range of LSDs in the categories of oligosaccharidoses, mucopolysaccharidoses, glycogen storage disorders, and congenital disorders of glycosylation and de-glycosylation. We then show detection of glycolipid biomarkers that characterize sphingolipidoses in serum and dried blood spots, with baseline resolution of glucosylsphingosine from galactosylsphingosine. By extending disease coverage to LSDs, glycomic profiling represents a necessary analytical addition for clinical metabolomics platforms and paves the way for future investigations of glycan and glycolipid metabolism.
2. Materials and methods
2.1. Samples
Urines were obtained as residual, de-identified specimens from the Stanford Clinical Biochemical Genetics Laboratory from patients with an LSD established through genetic, enzymatic, or biochemical testing. Two independent sample sets were used and independently analyzed. First, for the discovery and subsequent verification of mucopolysaccharidosis biomarkers, we analyzed urines from the Stanford Clinical Biochemical Genetics Laboratory from patients with mucopolysaccharidosis types 1 (n = 13), 2 (n = 35), 3a (n = 33), 3b (n = 2), 4a (n = 18), 4b (n = 2), 6 (n = 9), and 7 (n = 1). No samples from MPS 3c and MPS 3d were available. Negative controls (<6 months of age, n = 25; >2 years of age, n = 31) were residual urines originally submitted for amino acid or organic acid analysis with no evidence of a metabolic abnormality. Second, for clinical test validation, we analyzed residual urines from the Stanford Clinical Biochemical Genetics Laboratory, as well as urines provided by Dr. Tim Wood (Greenwood Genetic Center), and residual mucopolysaccharidosis proficiency testing samples from the College of American Pathologists (CAP), which are comprised of authentic patient samples. The samples from Dr. Tim Wood were de-identified, residual urines from oligosaccharidosis patients obtained with permission for use as positive controls in clinical test validation and were not used for the mucopolysaccharidosis biomarker discovery and verification study. The samples used in the clinical validation study encompassed mucopolysaccharidosis (types 1 [n = 5], 2 [n = 14], 3 [n = 20], 3a [n = 5], 3b [n = 3], 4 [n = 11], 4a [n = 4], 4b [n = 1], 6 [n = 6], and 7 [n = 3]), oligosaccharidosis (Aspartylglucosaminuria [n = 4], alpha-Fucosidosis [n = 5], GM1-gangliosidosis [n = 10], Sandhoff disease [n = 3], alpha-Mannosidosis [n = 2], beta-Mannosidosis [n = 1], Sialidosis [n = 4], ML2 [n = 1], ML3 [n = 1], Schindler disease [n = 2], Galactosialidosis [n = 1], and free sialic acid storage disease [n = 1]), N-Glycanase 1 deficiency (NGLY1; n = 5), Mannosyl-oligosaccharide glucosidase 1 deficiency-congenital disorder of glycosylation (MOGS-CDG; n = 2), Pompe disease (n = 1), and negative controls (≤2 years of age, n = 17; >2 years of age, n = 26). Because all urine samples were stored without patient identifiers, treatment status and the number of unique patients are unknown. For the glycolipid studies, residual proficiency testing samples were used from the European Research Network for evaluation and improvement of screening, Diagnosis, and treatment of Inherited disorders of Metabolism (ERNDIM), Special Assays in Serum (SAS) scheme 2020, which included glucosylsphingosine (Gaucher disease), lyso-Gb3 (Fabry disease), and lyso-SM (Niemann Pick disease) spiked into human serum. In addition, dried blood spot samples from five Gaucher disease patients and one Gaucher disease carrier were obtained as residual, de-identified specimens from the Stanford Clinical Biochemical Genetics Laboratory, but no ages were available. These samples were stored frozen for at least 5 years. Negative control dried blood spots (n = 20) were obtained as residual, de-identified samples originally submitted for amino acids analysis and stored for less than 2 months from the date of collection. For all samples collected for testing at Stanford Clinical Labs, the use of residual, de-identified specimens following clinical testing at our institution does not require additional institutional review board (IRB) approval and consent is waived. Once in our lab, all samples were stored below −30 °C.
2.2. Materials
Sialic acid, carbohydrates, and sugar alcohols were purchased from Sigma-Aldrich, USA. Hex3-hexNAc2 and N-acetylglucosamine-asparagine (glcNAc-asn) were purchased from Omicronbio, South Bend, IN. Maltotetraose and glucose tetrasaccharide were purchased from Carbosynth, San Diego, CA. All glycolipids were purchased from Cayman Chem, MI, except for galactosylsphingosine-d5, which was purchased from Avanti Polar Lipids, AL. Nanosep 3 kDa ultrafiltration devices were purchased from Pall, NY. All solvents were LC-MS grade (Honeywell Burdick & Jackson, VWR, USA).
2.3. Sample processing
For both untargeted and targeted analyses, urine samples (10–50 μL; limited by sample availability) were ultrafiltered using Nanosep 3 kDa devices at 17,000 ×g for 15 min, and the filtrates were diluted 1:6 with equal volumes of water and acetonitrile into a 96-well plate for analysis. Patient and control samples were always processed together to circumvent batch effects, but no internal standards were used. Separately, urine creatinine concentration was determined using an AU480 Chemistry Analyzer (Beckman Coulter, Brea, CA) following manufacturer’s instructions, and used for subsequent data normalization during clinical test validation. For glycolipid analyses, serum samples (50 μL) were deproteinized at a 1:5 ratio with methanol containing 125 nmol/L each of glucosylsphingosine-13c6 and galactosylsphingosine-d5, vortexed for 1 min, and centrifuged at 17,000 ×g for 15 min. Supernatants were transferred into a 96-well plate for analysis. Dried blood spots (one 3 mm punch) were extracted with 100 μL of methanol containing 125 nmol/L each of glucosylsphingosine-13c6 and galactosylsphingosine-d5, sonicated for 15 min, and centrifuged at 17,000 ×g for 5 min. Supernatants were transferred into a 96-well plate for analysis.
2.4. LC-MS analyses
The same LC conditions were used for both targeted and untargeted studies. A BEH Amide 2.1 × 50 mm 1.7 μm column (Waters, MA, USA) was used for separations. Mobile phase A was 0.2 mM ammonium formate in water adjusted to pH 10.5 and mobile phase B was acetonitrile. At a flow rate of 0.6 mL/min, the LC gradient started at 93%B, then ramped to 90%B at 1 min, 80%B at 2 min, 55%B at 4.6 min, 10%B at 5.2 min, 5%B at 5.9 min, and re-equilibrated at initial conditions from 6 to 9 min. The column temperature was 55 °C. Two microliters of sample was injected.
Untargeted analyses were performed with an Acquity H-Class UPLC system (Waters, MA, USA) coupled to a Vion ion-mobility quadrupole time of flight MS (IMS-qTOF MS) (Waters, MA, USA). Positive and negative electrospray ionization (ESI) were used in sensitivity mode. Profile-mode data was acquired from 60 to 1600 m/z over a scan time of 0.2 s with Data Independent Acquisition (DIA) and ion mobility (High Definition MSe). Targeted analyses were performed on an Acquity H-Class UPLC system coupled to a TQ-S Micro tandem quadrupole MS (Waters, MA, USA). Positive and negative ESI were used with rapid polarity switching. Multiple reaction monitoring (MRM) was used at unit resolution. Detailed methods are described in Table S1.
2.5. Bioinformatics
For data obtained by IMS-qTOF MS, run alignment and peak picking were performed using Progenesis (Waters, MA, USA). Discriminantion features were selected using the Orthogonal Projections to Latent Structures Discriminant Analysis (OPLS-DA) S-plot analysis in the EZ-Info module of Progenesis and the Top Importances output from random-Forest [28] implemented in R (version 3.5). Ion features were searched for possible identifications using Unifi software (Waters, MA, USA). Final ion feature annotations were made using accurate mass and insilico fragmentation analysis using Unifi, and biological relevance to each disorder. Glycans were annotated without specificity to branching, position of the sulfate (s), or location of the non-reducing end.
For targeted LC-MS/MS data analysis of urine samples in the validation set, peak areas were normalized to creatinine concentration. Analyte Z-scores were computed and aggregated into an overall Z-score profile for each sample, with biomarker abundances relative to a reference population of patients without an LSD (n = 20–30; see above). Gender was not used for stratification. Based on our experience with clinical oligosaccharidosis and mucopolysaccharidosis testing, we have observed that patients ≤2 years of age have overall higher levels of endogenous glycans. Accordingly, different reference populations were used for patients <2 and >2 years of age. For each compound, Z-scores were calculated using means and standard deviations of the appropriate reference population. Generally, Z-scores <2 were considered normal and >5 abnormal, and Z-scores between 2 and 5 were considered indeterminate. Results were interpreted by considering the overall profile of Z-scores in a patient run, rather than any single abnormality alone, in arriving at a diagnosis. Statistical analyses were performed using Excel (Microsoft 365) and R.
3. Results
3.1. Chromatographic strategy
To build a glycomic profiling method, we first incorporated the previously described oligosaccharide [17,29] and glycolipid [24,25] biomarkers into a targeted MS/MS analysis, and optimized LC conditions using a high-pH ammonium formate mobile phase with the Amide column to separate each compound based on its sugar group [30]. Selection of the final chromatographic method, a 9-min gradient program (detailed in Methods), was based on detection of the known biomarkers, with particular focus on resolving the isomers glucosylsphingosine (Gaucher disease) from galactosylsphingosine (Krabbe disease) and maltotetraose from glucose tetrasaccharide (glc4; Pompe disease). Fig. 1a shows a composite of representative extracted ion chromatograms for all detected glycan and glycolipid biomarkers and their associated disorders, with peaks normalized to maximal peak intensity for each sample. Resolution of isomers is demonstrated here and detailed in Figs. S1 and S2. Of note, the chromatography resolved many oligosaccharides with shared motifs, such as n-acetylhexosamine-asparagine (hexNAc-asn) and fucose-hexNAc-asn, minimizing interference from in-source fragmentation. Peak heights of disease specific biomarkers from abnormal urine samples were generally above one million counts.
Fig. 1.
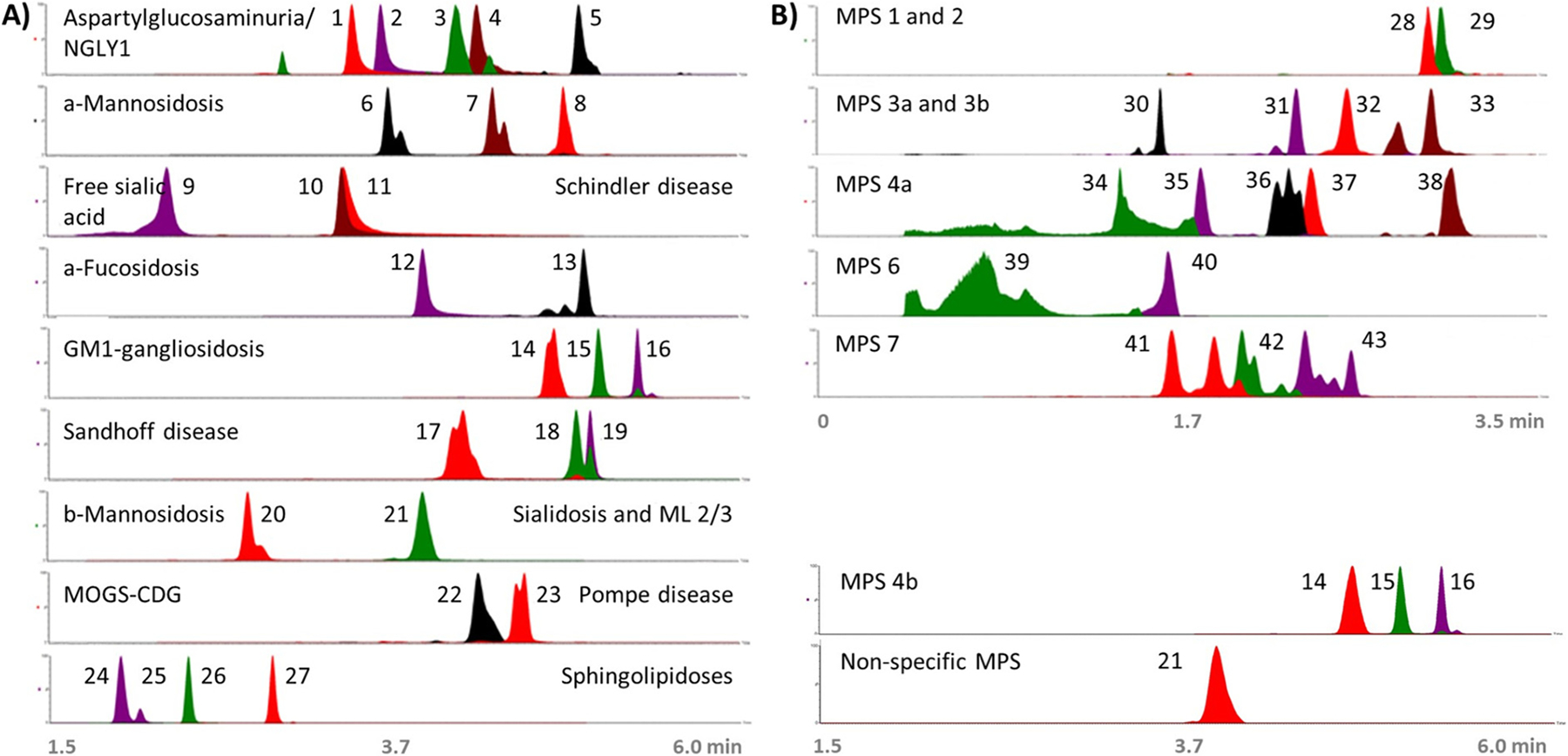
Chromatography of characteristic glycan and glycolipid biomarkers for each LSD. Merged chromatograms (retention time [x-axis] versus relative peak intensity [y-axis]) from targeted LC-MS/MS showing representative peaks of characteristic disease biomarkers. Oligosaccharidoses and mucopolysaccharidoses biomarkers were taken from positive urine samples and sphingolipidoses biomarkers were taken from ERNDIM serum samples. For galactosylsphingosine, an authentic standard was spiked into an ERNDIM sample. (A) Biomarkers of oligosaccharidoses and sphingolipidoses, and (B) biomarkers of mucopolysaccharidoses, as follows: 1) neuAC-hex-hexNAc, 2) hexNAc-asn, 3) neuAC-hex3-hexNAc2, 4) hex-hexNAc-asn, 5) hex2-hexNAc2-asn, 6) hex2-hexNAc, 7) hex3-hexNAc, 8) hex4-hexNAc, 9) sialic acid, 10) hexNAc-serine, 11) hexNAc-threonine, 12) fuc-hexNAc-asn, 13) fuc-hexNAc2-hex3, 14) hex3-hexNAc2, 15) hex4-hexNAc2, 16) hex5-hexNAc3, 17) hex2-hexNAc2, 18) hex3-hexNAc3, 19) hex3-hexNAc4, 20) man-hexNAc, 21) neuAC-hex3-hexNAc2, 22) glc3-man, 23) glc4, 24) glucosylsphingosine, 25) galactosylsphingosine standard, 26) lyso-sphingomyelin, 27) lyso-Gb3, 28) UA-hexNAc-s-UA-hexNAc-s, 29) UA-hexNAc-s-UA, 30) hexN-s, 31) hexN-s-UA, 32) hexNAc-s-UA-hexNAc-UA, 33) MPS 813, 34) hexNAc-s, 35) hex-s-hexNAc-s, 36) neuAC-hex-hexNAc-s, 37) hexNAc-s-UA-hexNAc-s, 38) hex3-hexNAc2-s, 39) hexNAc-s, 40) hexNAc-disulfate, 41) UA-hexNAc-s, 42) UA-hexNAc, 43) hexNAc-UA-hexNAc. Abbreviations: asparagine (asn); fucose (fuc); globotriaosylsphingosine (gb3); glucose (glc); hexose (hex); hexN (hexosamine); n-acetylhexosamine (hexNAc); n-acetylneuraminic acid (neuAC); mannose (man); sulfate (s); uronic acid (UA).
3.2. Discovery of native glycomic signatures for subtyping mucopolysaccharidoses
Next, we sought to identify biomarkers of mucopolysaccharidoses that could be detected in their native form and ultimately incorporated into a larger glycomic panel. Using the same chromatographic strategy described above, we performed untargeted glycomic profiling on 113 mucopolysaccharidosis urines, and analyzed the data for biomarkers that could (1) differentiate mucopolysaccharidosis samples from negative controls and (2) further distinguish the mucopolysaccharidosis subtype. From this, we selected twenty compounds for annotation, insilico fragmentation analysis, and incorporation into a targeted clinical test (Table 1). Ultimately, we annotated nineteen of these as glycans, with fifteen annotated as glycosaminoglycan fragments. No database matches were found for MPS813, but two of its fragments, 175.0245 m/z and 259.0418 m/z, were found in other glycosaminoglycan annotations, suggesting that this compound has glycan motifs. Fig. S3 contains chromatograms, fragmentation spectra, and isotopic patterns all collected in high-resolution accurate mass with ion-mobility separation to support the biomarker annotations. Of note, two baseline resolved isomers that helped distinguish MPS 4a from MPS 6 were annotated as hexNAc-s. The collision cross section (CCS) values and spectra were indistinguishable (Fig. S4). Consistent with previous reports, biomarkers for GM1-gangliosidosis were seen in MPS 4b samples [31], and neuAC-hex3-hexNAc2, a marker for Sialidosis, ML2/3, and Galactosialidosis, was seen in the majority of mucopolysaccharidosis samples [17].
Table 1.
Urinary biomarkers of mucopolysaccharidoses. Untargeted glycomic profiling of 113 mucopolysaccharidosis urines revealed 20 biomarkers for subtyping mucopolysaccharidoses. The table gives representative data for observed retention time, m/z, and CCS values. We compared annotations to biomarkers from previously published underivatized or derivatized methods: s - reported by Fuller et al. [27] or Saville et al. [21] for the same MPS; d - reported by Fuller et al. or Saville et al. for a different MPS; p - reported by Piraud et al. [17]; r – reported by Ramsay et al. [19] Biomarkers without a footnote indicate an unreported biomarker-disease association. Abbreviations: collision cross section in square Angstroms (CCS (Å2)); hexose (hex); uronic acid (UA); hexosamine (hexN); n-acetylhexosamine (hexNAc); n-acetylneuraminic acid (neuAC); dermatan sulfate (DS); heparan sulfate (HS); keratan sulfate (KS).
| Biomarker number | Annotation (with probable origin) | Predominant MPS association | Formula | Retention Time (min) | Adduct | Expected m/z | Observed m/z | Mass error (ppm) | Observed CCS (Å2) |
|---|---|---|---|---|---|---|---|---|---|
| #1 d | hexNAc-s-UA-hexNAc-s-UA (DS, HS) | 1, 2, 3 | C28H44N2O29S2 | 3.0 | M−2H | 467.0662 | 467.0666 | 0.9 | 317 |
| #2 s | UA-s-hexNAc-UA (DS, HS) | 2, 1, 3 | C20H31NO21S | 3.2 | M−H | 652.1036 | 652.1004 | −4.9 | 224 |
| #3 s | hexN-s-UA (HS) | 3a, 1 or 2 | C12H21NO14S | 2.4 | M−H | 434.0610 | 434.0592 | − 4.1 | 226 |
| #4 | hexN-s (HS) | 3a, 1 or 2 | C6H13NO8S | 1.7 | M−H | 258.0289 | 258.0272 | −6.6 | 144 |
| #5 s | hexNAc-s-UA-hexNAc-UA (HS) | 3b | C28H44N2O26S | 2.6 | M−H | 855.1830 | 855.1802 | −3.3 | 266 |
| #6 | MPS813 | 3 | 3.0 | 813.1683 | 247 | ||||
| #7 | hex-s-hexNAc-s (KS) | 4a | C14H25NO17S2 | 1.9 | M−2H | 270.5209 | 270.5205 | −1.6 | 129 |
| #8 | neuAC-hex-hexNAc-s (KS) | 4a | C25H42N2O22S | 2.3 | M−H | 753.1877 | 753.1844 | − 4.4 | 245 |
| #9 s | hexNAc-s (KS) | 4a | C8H15NO9S | 1.5 | M−H | 300.0395 | 300.0387 | −2.6 | 154 |
| #10 | hex3-hexNAc2-s (KS) | 4b, 4a | C34H58N2O29S | 3.1 | M−H | 989.2773 | 989.2727 | − 4.7 | 285 |
| #11 | hexNAc-s-UA-hexNAc-s | 4a or 6 | C22H36N2O23S2 | 2.4 | M−2H | 379.0502 | 379.0500 | −0.7 | 288 |
| #12 pr | hex3-hexNAc2 (KS) | 4b | C34H58N2O26 | 4.8 | M+H | 911.3351 | 911.3335 | −1.7 | 273 |
| #13 r | hex4-hexNAc2 (KS) | 4b | C40H68N2O31 | 5.0 | M+H | 1073.3879 | 1073.3881 | 0.2 | 295 |
| #14 pr | hex5-hexNAc3 (KS) | 4b | C54H91N3O41 | 5.3 | M+H | 1438.5201 | 1438.5193 | −0.5 | 346 |
| #15 s | hexNAc-s (DS) | 6 | C8H15NO9S | 0.7 | M−H | 300.0395 | 300.0394 | −0.2 | 158 |
| #16 s | hexNAc-disulfate (DS) | 6 | C8H15NO12S2 | 1.6 | M−2H | 189.4945 | 189.4936 | −4.8 | 217 |
| #17 d | hexNAc-s-UA (DS) | 7 | C14H23NO15S | 1.8 | M−H | 476.0716 | 476.0715 | −0.1 | 198; 191 |
| #18 | hexNAc-UA (DS) | 7 | C14H23NO12 | 2.0 | M−H | 396.1148 | 396.1144 | −0.9 | 181 |
| #19 | hexNAc-UA-hexNAc (DS) | 7 | C22H36N2O17 | 2.4 | M−H | 599.1941 | 599.1924 | −2.9 | 235 |
| #20 p | neuAC-hex3-hexNAc2 | MPS | C45H75N3O34 | 3.9 | M+H | 1202.4305 | 1202.4310 | 0.4 | 312 |
To provide further support for these annotations, we next examined them for biological plausibility. Both dermatan sulfate (DS) and heparan sulfate (HS) contain uronic acids (UA), with hexNAc and hexosamine (hexN) specific to dermatan sulfate and heparan sulfate, respectively. Keratan sulfate (KS) contains hexose (hex), n-acetylhexosamine (hexNAc), and n-acetylneuraminic acid (neuAC, sialic acid). In agreement, uronic acid subunits were predominately found in MPS 1, 2, 3a, and 7; hexosamine was mainly found in MPS 3a; and hexose and n-acetylneuraminic acid were only present in MPS 4. However, one uronic acid-containing fragment, HexNAc-s-UA-hexNAc-s, was elevated in MPS 4a even though uronic acid is not a component of keratan sulfate. Uronic acid subunits were more frequently found in disorders of uronidase deficiency (MPS 1 and 7); n-acetylhexosamine was seen in n-acetylglucosaminidase deficiency (MPS 3b) but not sulfamidase deficiency (MPS 3a); the biomarkers unique for n-acetylgalactosamine-6-sulfatase deficiency (MPS 4a) were all sulfated and not elevated in beta-galactosidase deficiency (MPS 4b); and two distinct hexNAc-s isomers were found, each likely reflecting the defective, site-specific sulfatase that differentiates MPS 4a from MPS 6.
3.3. Verifying the untargeted findings for identifying mucopolysaccharidoses by targeted LC-MS/MS
To verify the biomarkers of mucopolysaccharidoses from our untargeted studies and evaluate their suitability for a targeted clinical assay, we incorporated the mass transitions of each biomarker (Table 1) into a targeted LC-MS/MS panel (Table S1) and re-analyzed the same samples used for biomarker discovery. Fig. 1b shows representative peaks for each biomarker. Fig. 2 shows the resulting peak area data, confirming all mucopolysaccharidosis biomarker associations established in the untargeted discovery study (Table 1). We saw unambiguous biomarker elevations for MPS 3b (Fig. 2, boxplot 5), MPS 4a (Fig. 2, boxplots 7 and 8), MPS 6 (Fig. 2, boxplot 15), and MPS 7 (Fig. 2, boxplots 17–19). However, we saw that some biomarkers were less specific or shared by more than one disease subtype: HexNAc-s-UA-hexNAc-s-UA (Fig. 2, boxplot 1) was predominately elevated in MPS 1 with slight elevations in MPS 2; UA-s-hexNAc-UA (Fig. 2, boxplot 2) was elevated in both MPS 1 and 2; and MPS 3 biomarkers were less specific (Fig. 2, boxplots 3, 4, and 6).
Fig. 2.
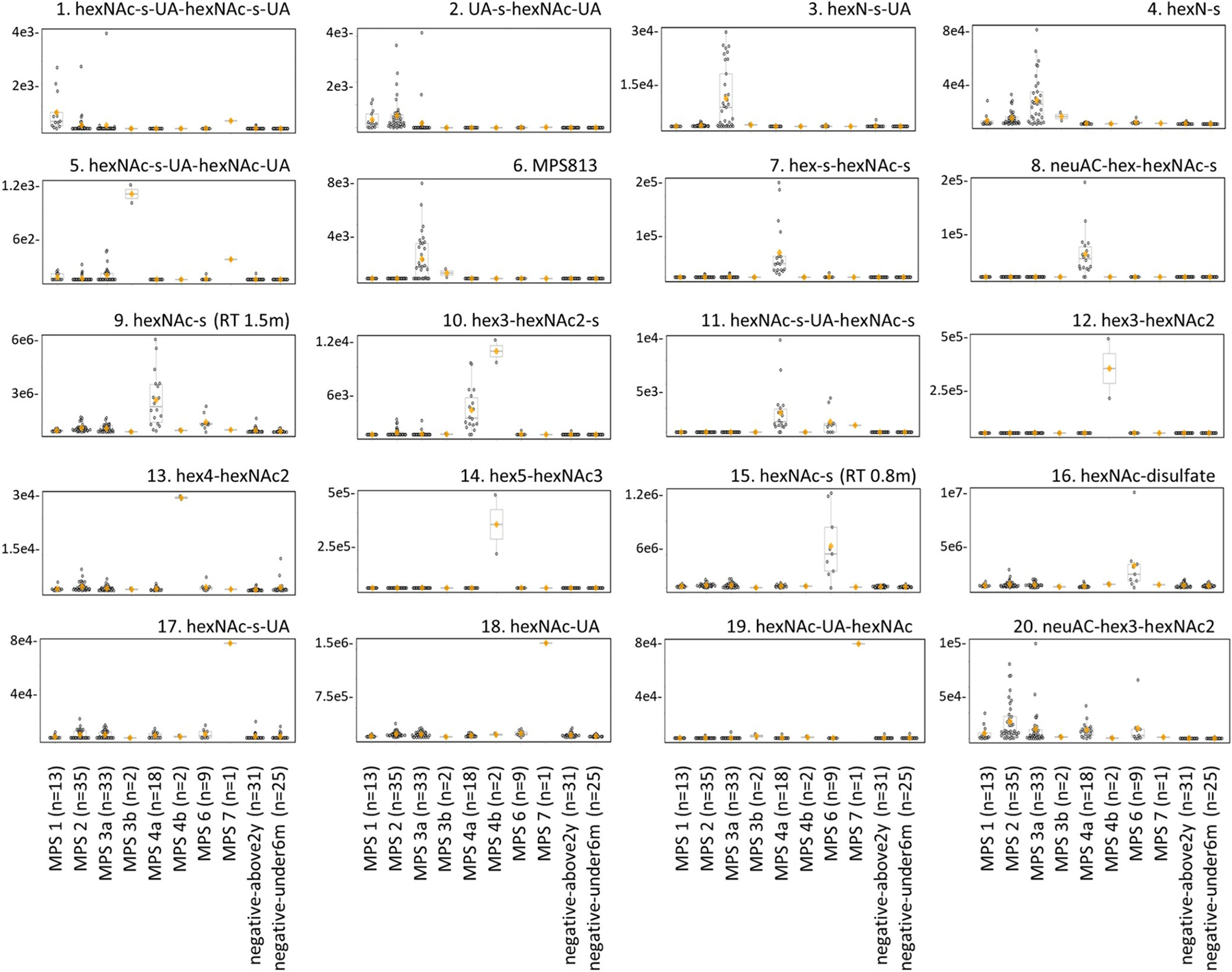
Verification of urinary biomarker findings for mucopolysaccharidoses by LC-MS/MS. Mucopolysaccharidosis urine samples (n = 113) used for biomarker discovery were re-analyzed using the targeted LC-MS/MS panel. The boxplots show biomarker peak areas (y-axis) with individual data points and are grouped by each condition (x-axis). Each dot represents one sample, the orange diamond indicates the group mean, and the horizontal divider inside the boxplot indicates the group median. Ages used for the negative population were under 6 months of age (negative-under6m) and above 2 years of age (negative-above2y). Retention time, RT.
3.4. Validation of glycan biomarkers on the targeted LC-MS/MS panel
To validate these biomarkers for incorporation into clinical testing, we analyzed previously untested, blinded LSD and negative control samples on the targeted LC-MS/MS panel, and generated Z-scores for all biomarkers in each sample. The resulting profiles are visually displayed as a heatmap in Fig. 3, with individual Z-scores detailed in Fig. S5. Features key to each of the disorders are summarized in Table 2, which can serve as an aide in interpretation. It is important to note that individual biomarkers may appear in more than one disorder (e.g., UA-s-hexNAc-UA in MPS types 1, 2, and 3), such that a diagnosis is suggested by overall patterns rather than any single analyte. In scoring our interpretations for purposes of test validation, we broadly interpreted results to account for all possible conditions.
Fig. 3.

Heatmap of characteristic urinary biomarker profiles. For clinical test validation, previously untested urine samples were analyzed on the targeted LC-MS/MS panel. For each individual sample (horizontal), Z-scores for each biomarker (vertical) were generated using peak areas normalized by creatinine concentration. Relative biomarker abundances are defined according to the color-scale, with white squares representing normal biomarker levels, red squares representing abnormal biomarker levels, and black squares representing the highest degree of abnormality. For samples without age information, Z-scores were generated using the reference statistics from the older-age reference population. Specific subtypes of MPS 3 and 4 were confirmed by genetic or enzymatic testing. For each age range, three representative negative samples are shown. A total of 43 negative samples were evaluated.
Table 2.
Characteristic patterns of biomarkers and associated diseases detected in urine (oligosaccharidoses, mucopolysaccharidoses, glycogen storage disorders, and congenital disorders of glycosylation and de-glycosylation) and serum (sphingolipidoses). Diseases are characterized by overall biomarker pattern rather than the presence of any single biomarker since individual biomarkers may be present in more than one disease.
| Disorder | Defective protein | Biomarker(s) |
|---|---|---|
| α-Mannosidosis | α-D-mannosidase | hex2-hexNAc, hex3-hexNAc, hex4-hexNAc |
| β-Mannosidosis | β-mannosidase | man-glcNAc |
| Aspartylglucosaminuria | Aspartylglucosaminidase | glcNAc-asn, neuAC-hex-hexNAc-asn, neuAC-hex2-hexNAc2-asn, hex2-hexNAc2-asn, hex-glcNAc-asn |
| NGLY1 deficiency | N-glycanase 1 | glcNAc-asn, neuAC-hex2-hexNAc2-asn, hex-glcNAc-asn |
| Schindler disease | α-N-acetylgalactosaminidase | galNAc-O-ser, galNAc-O-thr |
| Sialuria | Sialin | sialic acid |
| Fucosidosis | α-fucosidase | fuc-hexNAc-asn, fuc-hexNAc2-hex3 |
| GM1-gangliosidosis | β-galactosidase | hex3-hexNAc2, hex4-hexNAc2, hex5-hexNAc3 |
| Sandhoff disease | Hexosaminidase A and B | hex2-hexNAc2, hex3-hexNAc3, hex3-hexNAc4 |
| Galactosialidosis | Cathepsin A | hex3-hexNAc2, hex4-hexNAc2, hex5-hexNAc3, neuAC-hex3-hexNAc2 |
| ML 1 (Sialidosis) | Neuraminidase | neuAC-hex3-hexNAc2 |
| ML 2 (I-cell disease) | N-acetylglucosamine-1-phosphotransferase | neuAC-hex3-hexNAc2, hex3-hexNAc2, and general elevation of oligosaccharides and glycosaminoglycan fragments |
| ML 3 (Pseudo-Hurler polydystrophy) | N-acetylglucosamine-1-phosphotransferase | neuAC-hex3-hexNAc2 |
| Pompe disease | α-glucosidase | glc4 |
| MOGS-CDG | glucosidase 1 | glc3-man |
| MPS 1 (Hurler) | α-L-iduronidase | UA-hexNAc-S-UA-hexNAc-S, UA-hexNAc-S-UA, neuAC-hex3-hexNAc2 |
| MPS 2 (Hunter) | Iduronate 2-sulphatase | UA-hexNAc-S-UA, UA-hexNAc-S-UA-hexNAc-S, neuAC-hex3-hexNAc2 |
| MPS 3a (Sanfilippo A) | N-sulfoglucosamine sulfohydrolase | hexN-S-UA, hexN-S, MPS 3813, neuAC-hex3-hexNAc2 |
| MPS 3b (Sanfilippo B) | N-acetyl-α-D-glucosaminidase | hexNAc-S-UA-hexNAc-UA, neuAC-hex3-hexNAc2 |
| MPS 4a (Morquio A) | Galactosamine-6-sulfatase | hex-S-hexNAc-S, neuAC-hex-hexNAc-S, hexNAc-S (RT 1.5 min), hex3-hexNAc2-S, hexNAc-S-UA-hexNAc-S, neuAC-hex3-hexNAc2 |
| MPS 4b (Morquio B) | β-galactosidase | hex3-hexNAc2, hex4-hexNAc2, hex5-hexNAc3 |
| MPS 6 (Maroteaux-Lamy) | n-acetylgalactosamine-4-sulfatase | hexNAc-S (RT 0.7 min), hexNAc-disulfate, hexNAc-S-UA-hexNAc-S, hexNAc-S-UA-hexNAc-S, neuAC-hex3-hexNAc2 |
| MPS 7 (Sly) | β-glucuronidase | UA-hexNAc-S, UA-hexNAc, hexNAc-UA-hexNAc, neuAC-hex3-hexNAc2 |
| Gaucher disease | β-glucocerebrosidase | glucosylsphingosine |
| Krabbe disease | Galactosylceramidase | galactosylsphingosine |
| Fabry disease | α-galactosidase A | lyso-Gb3 |
| Niemann Pick disease types A and B | Sphingomyelinase | lyso-SM |
3.4.1. Mucopolysaccharidoses
Based on the disease profiles represented in Fig. 3 and Table 2, we unambiguously identified all mucopolysaccharidosis samples, regardless of subtype, as abnormal, largely based on the presence of neuAC-hex3-hexNAc2. Furthermore, we correctly subtyped all cases of MPS 3b, 4a, 4b, 6, and 7 based on characteristic, easily recognizable profiles based on biomarker abundances shown in Fig. 2. Subtyping for MPS 1, 2, and 3 was more difficult for some samples. With elevated hexNAc-s-UA-hexNAc-s-UA, we easily distinguished MPS 1 from MPS 2. However, minor to moderate elevations of heparan sulfate biomarkers in MPS 1 and 2 samples led to incorrect subtyping in 1/5 MPS 1 and 5/14 MPS 2 cases, which we mistook for MPS 3. Similarly, elevated hexNAc-s-UA-hexNAc-s-UA and UA-s-hexNAc-UA, likely from heparan sulfate, led to incorrect subtyping in 8/20 MPS 3 cases, which we mistook as MPS 1 or 2. Of note, we correctly subtyped all genetically or enzymatically confirmed MPS 3a samples.
3.4.2. Oligosaccharidoses
We next validated the detection of oligosaccharidoses. The biomarker profiles of the oligosaccharidosis samples were all consistent with previously described patterns [17,18,29] and nearly all abnormal peak areas were at least two logs higher than those in negative samples (Fig. 3). Thus, we unambiguously interpreted all samples correctly.
Closer examination of oligosaccharidosis patterns in Fig. 3 revealed several new insights. First, for Schindler disease (alpha-N-acetylgalactosaminidase deficiency), galNAc-threonine (Z ≈ 80) predominated over galNAc-serine (Z ≈ 20), and hex3-hexNAc3, a marker for Sandhoff disease, was also elevated. Second, in addition to the typical pattern for GM1-gangliosidosis, 3/10 GM1 samples had substantially elevated neuAC-hex3-hexNAc2, likely reflecting cases of actual Galactosialidosis. Finally, the inclusion of mucopolysaccharidosis biomarkers in oligosaccharidosis analysis revealed new glycomic differences between Sialidosis and ML2/3, which previously were identified together using neuAC-hex3-hexNAc2 [17]. ML2 and its milder allelic form, ML3, are characterized by deficient activities of multiple lysosomal enzymes and generalized glycan accumulation. In agreement, we saw substantial elevation of multiple oligosaccharidosis and mucopolysaccharidosis biomarkers in ML2, but not in Sialidosis and ML3.
3.4.3. NGLY1 deficiency
Along with oligosaccharidoses, we validated the detection of NGLY1 deficiency, a congenital disorder of de-glycosylation that shares biomarkers with Aspartylglucosaminuria [32]. Z-scores of NGLY1 biomarkers were generally below 30, while the corresponding Z-scores for Aspartylglucosaminuria generally exceeded 50 (Fig. 3). Using these significant differences in elevation, we unambiguously identified all NGLY1 deficiency samples.
3.4.4. Mannosyl-oligosaccharide glucosidase (MOGS) 1 deficiency and Pompe disease
We next evaluated the detection of two additional disorders with elevated glycans, MOGS-CDG, a congenital disorder of glycosylation characterized by increased urinary glucose3-mannose (glc3-man) [33], and Pompe disease, a glycogen storage disorder characterized by increased urinary glucose tetrasaccharide [34]. The two MOGS-CDG samples had significantly elevated glc3-man (Z > 50), which was not present in any other sample in this validation (Fig. 3), while the single Pompe disease sample had elevated glucose tetrasaccharide (Z = 11). Based on the baseline separation of the isomers, glc3-man from glucose tetrasaccharide, we clearly identified all samples. Glucose tetrasaccharide has also been shown to be elevated in glycogen storage disorders types 3, 4, and 9 [34]. Samples from these or other glycogen storage disorders or congenital disorders of glycosylation were not available for study, such that the overall specificity of biomarkers was not evaluated. Nevertheless, our analysis would be able to distinguish MOGS-CDG from other glycogen storage disorders based on the unambiguous detection of glc3-man.
3.5. Assessment of glycolipid detection on the targeted LC-MS/MS panel
In contrast to urine testing for mucopolysaccharidoses and oligosaccharidoses, sphingolipidoses are more readily detected in serum, plasma, or dried blood spots. To evaluate the detection of glycolipids in serum, we applied our same targeted glycomic profiling method to the analysis of residual proficiency testing survey serum samples, which contained glucosylsphingosine (Gaucher disease), lyso-Gb3 (Fabry disease), and lyso-SM (Niemann Pick disease) at target concentrations determined through proficiency testing survey results. Glycolipids at the following concentrations were easily detected: glucosylsphingosine at 0.6 nmol/L (normal: < 10.61 nmol/L [35]), lyso-SM at 3.75 nmol/L (normal: < 10.3 nmol/L [36]), and lyso-Gb3 at 12.5 nmol/L (normal: < 0.7 nmol/L [25]) (Fig. S6). Although lyso-Gb3 at the next lowest concentration in the survey (0.4 nmol/L) was not detected, typical lyso-Gb3 concentrations in Fabry disease patients range from 5 to 80 nmol/L [25]. While no Krabbe disease samples or proficiency testing materials were available for study, we were able to demonstrate baseline separation of galactosylsphingosine from glucosylsphingosine using a mixture of standards spiked into serum (Fig. S1). Finally, as an initial investigation of qualitative glycolipid detection in dried blood spots, we evaluated test performance for glucosylsphingosine in residual dried blood spots samples from five patients with Gaucher disease and one Gaucher disease carrier. Levels of glucosylsphingosine, expressed as peak area divided by internal standard peak area, were clearly elevated in Gaucher disease patients and easily distinguishable from carriers and negative controls (Fig. 4). In assessing matrix effects for both serum and dried blood spot samples, peak areas of the internal standards, glucosylsphingosine-13c6 and galactosylsphingosine-d5, remained steady. Taken together, the results indicate that the method is capable of qualitatively detecting glycolipids at pathologic concentrations.
Fig. 4.
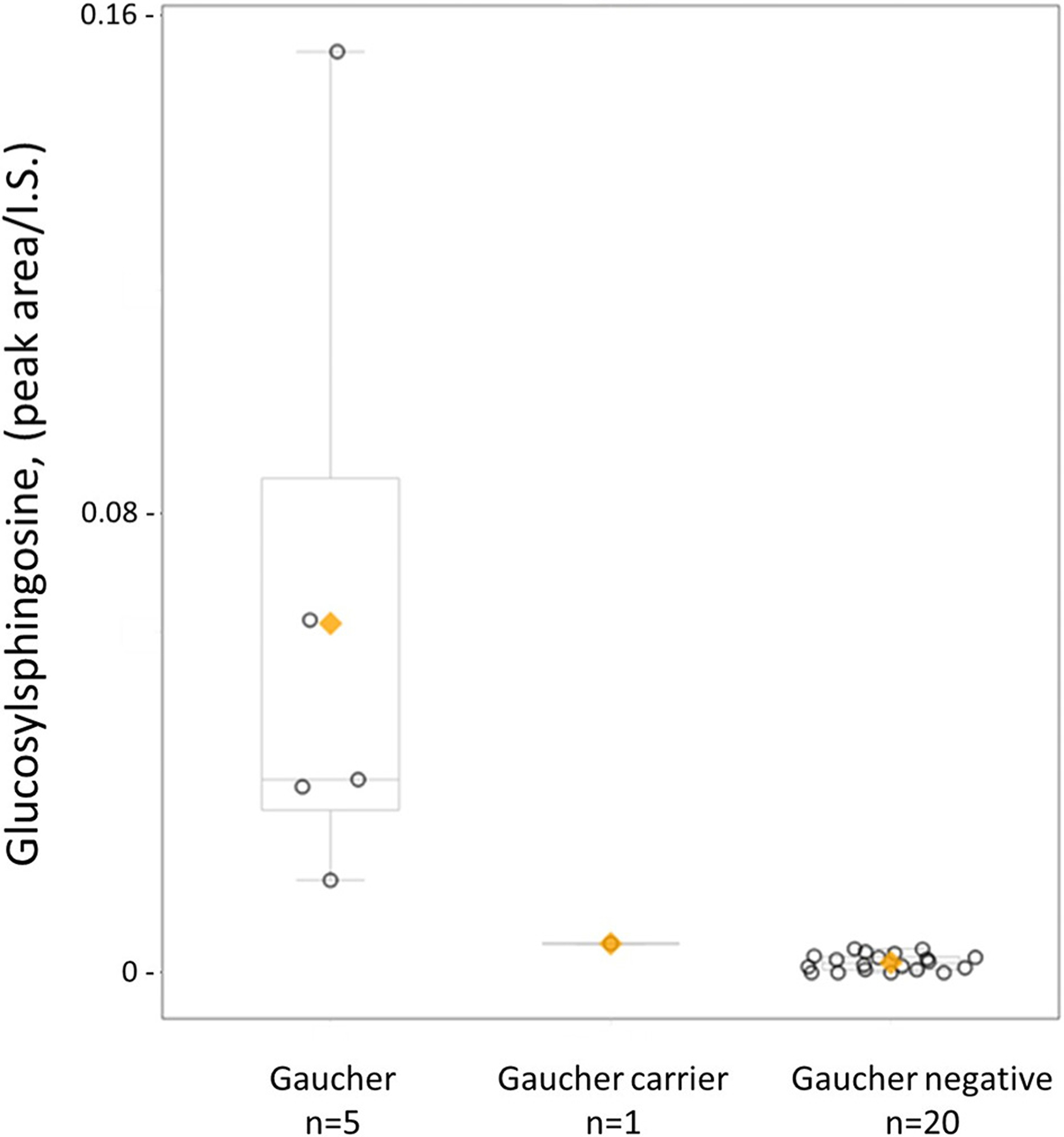
Detection of glucosylsphingosine extracted from dried blood spots. Samples from five patients with Gaucher disease, one Gaucher disease carrier, and twenty patients without Gaucher disease were analyzed using the targeted LC-MS/MS panel. Boxplots with individual datapoints show each sample’s peak area of glucosylsphingosine divided by the peak area of glucosylsphingosine-13c6, the internal standard (I.S.) (y-axis), and are grouped by each condition (x-axis). Each dot represents one sample, the orange diamond indicates the group mean, and the horizontal line inside the boxplot indicates the group median.
4. Discussion
This study expands the clinical biochemical genetics toolkit by streamlining the detection of biomarkers for at least 27 different LSDs into a single analysis. It is based on two key, interlinked advancements, (1) an optimized high-pH method that can simultaneously separate underivatized glycans and glycolipids, including the resolution of clinically significant isomers, and (2) the discovery and detection of small, endogenous glycans from mucopolysaccharidosis patients without derivatization, digestion, or depolymerization. Based on these findings, we created and validated a targeted LC-MS/MS method to simultaneously test for LSDs in the categories of oligosaccharidoses, mucopolysaccharidoses, glycogen storage disorders, congenital disorders of glycosylation and de-glycosylation, and sphingolipidoses. In a typical clinical workflow, this test may be ordered because of clinical signs and symptoms suggestive of an LSD, with abnormal results followed by confirmatory enzyme or DNA testing, or both. Alternatively, in a workup beginning with DNA sequencing, this test can be used to assess the functional significance of variants identified. In either case, it provides a single, streamlined approach for simultaneously evaluating many different LSDs. Based on the chromatographic conditions, the method is likely to detect additional LSDs but the lack of available samples prevented further evaluation. It is important to distinguish this workflow from newborn screening, where disorders are identified by low enzyme activity and followed by genetic or targeted biomarker testing, or both. We did not evaluate the suitability of our method in the newborn screening setting.
By untargeted glycomic profiling of mucopolysaccharidosis samples, we detected 20 biomarkers of mucopolysaccharidoses in native form, including four previously reported oligosaccharides [17,19,29], eight glycosaminoglycan fragments previously reported in derivatized form [21], and eight that were previously unreported (Table 1 and Fig. S3). Of the eight newly discovered biomarkers, four significantly contributed to test specificity for MPS 4a (hex-s-hexNAc-s and neuAC-hex-hexNAc-s) and MPS 7 (hexNAc-s-UA and hexNAc-UA-hexNAc). In addition, six of the eight glycosaminoglycan fragments here had disease associations consistent with those previously seen using derivatization [21]: UA-s-hexNAc-UA (MPS 1 and 7), hexN-s-UA (MPS 3a), hexNAc-s-UA-hexNAc-UA (MPS 3b), hexNAc-disulfate (MPS 6), hexNAc-s (MPS 4a), and hexNAc-s (MPS 6), with the latter two now separated as distinct isomers in our method to help distinguish MPS 4a from MPS 6 (Fig. S4). However, two markers had different disease associations from those previously reported: hexNAc-s-UA, seen here in MPS 7 and previously reported for MPS 1, 2, and 4a, and hexNAc-s-UA-hexNAc-s-UA, seen in MPS 1 and previously reported for MPS 3d. Additionally, one marker for MPS 4b, hex2-hexNAc2-disulfate, was not found in our study, perhaps related to differences in experimental conditions including derivatization.
The ability to reliably measure glycolipids in the same run together with glycans facilitates the combined identification of mucopolysaccharidoses, oligosaccharidoses, sphingolipidoses, and likely additional disorders with biomarkers containing sugar moieties. In this setting, the separation of isomers is also critical and contributes to improved detection performance. For example, the separation of galactosylsphingosine from glucosylsphingosine is necessary to detect patients with Krabbe disease, since asymptomatic patients with early infantile Krabbe disease have galactosylsphingosine levels three times less than the glucosylsphingosine levels in unaffected individuals [26, 37]. While these isomers currently can be separated using dedicated methods [25,26], our approach’s ability to separate many glycolipids without needing a secondary reflex method or multiple individual methods will be valuable, particularly as glycolipid testing is increasingly integrated into the diagnostic process [9,24–26,38]. Although our work documents analytical separation and limits of detection, validation using additional patient samples will be necessary. In another example, the resolution of hexose tetrasaccharide (hex4) isomers is necessary to distinguish Pompe disease (glc4) from MOGS-CDG (glc3-man). This is exemplified by a recent case in our laboratory in which urine was submitted for testing without accompanying clinical history. Testing revealed a significant elevation of hex4 (ultimately annotated as glc3-man) at a retention time distinct from glc4 and maltotetraose (Fig. S2). Since no enzyme test for MOGS was available, our data provided the necessary supporting evidence for interpreting the sequencing results, which revealed a variant of unknown significance in the MOGS gene.
Glycomic profiling expands the reach of clinical metabolomics testing, which currently consists of two main arms, lipidomics and metabolomics. The principal detection of lipids, short peptides, and central carbon metabolic intermediates (e.g., amino acids, organic acids, acylcarnitines, monosaccharides, and their derivatives) enables the identification of a wide range of inborn errors of metabolism [12–14,39–42]. However, using lipidomics or metabolomics platforms, or both, recent biomarker discovery studies aiming to detect mucopolysaccharidoses did not report glycans outlined in this report [43–46] and recent evaluations of clinical metabolomic profiling platforms showed limited identification of LSDs [14,16]. Here, our strategy extends analyte coverage to more complex free glycans, showing that glycomics is the third, critical arm for clinical metabolomics testing.
Like other qualitative biomarker panels such as urine organic acids analysis, interpreting the results of a glycomic profile requires consideration of the overall pattern, rather than on any single biomarker abnormality. Interpretation requires familiarity with the diseases and their associated biomarker patterns, ideally considered along with the patient’s clinical history. The heatmap in Fig. 3 shows representative profiles, including normal variation, and, together with the summary patterns in Table 2, can serve as an aid in interpretation. In this study, the detection of highly specific biomarkers led to straightforward interpretations for all cases of oligosaccharidoses as well as MPS 3a, 3b, 4a, 4b, 6, and 7. Subtyping MPS 1, 2, and 3 cases was relatively more challenging because dermatan sulfate and heparan sulfate share individual sugar subunits. Mucopolysaccharidosis methods incorporating a derivatization step may yield higher specificity for distinguishing MPS 1, 2, and 3 [21].
Taken together, this report details a unified free glycan and glycolipid testing strategy for LSDs that rapidly provides supportive biomarker data for genetic and enzymatic testing results. As the field of clinical metabolomics expands, glycomic profiling also represents an important analytical addition to clinical metabolomics platforms and paves the way for future investigations of glycan and glycolipid metabolism.
Supplementary Material
Acknowledgements
The authors gratefully acknowledge Dr. Tim Wood (Greenwood Genetic Center) for providing positive samples.
Funding statement
No external funding sources.
Footnotes
Informed consent statement
The use of specimens for clinical test development does not require further institutional review board (IRB) approval at our institution. Informed consent was obtained or waived for samples submitted for clinical testing. All data was de-identified.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ymgme.2021.08.006.
Competing interests
The authors declare no competing interests.
Data availability statement
All biomarker data is provided in the manuscript and Supplementary Information.
References
- [1].Schielen PCJI, Kemper EA, Gelb MH, Newborn screening for lysosomal storage diseases: a concise review of the literature on screening methods, therapeutic possibilities and regional programs, Int. J. Neonatal Screen 3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Matern D, et al. , Newborn screening for lysosomal storage disorders, Semin. Perinatol 39 (2015) 206–216. [DOI] [PubMed] [Google Scholar]
- [3].Wasserstein MP, et al. , The New York pilot newborn screening program for lysosomal storage diseases: report of the first 65,000 infants, Genet. Med 21 (2019) 631–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Burton BK, et al. , Newborn screening for lysosomal storage disorders in Illinois: the initial 15-month experience, J. Pediatr 190 (2017) 130–135. [DOI] [PubMed] [Google Scholar]
- [5].Elliott S, et al. , Pilot study of newborn screening for six lysosomal storage diseases using Tandem mass spectrometry, Mol. Genet. Metab 118 (2016) 304–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Piraud M, et al. , Contribution of tandem mass spectrometry to the diagnosis of lysosomal storage disorders, J. Inherit. Metab. Dis 41 (2018) 457–477. [DOI] [PubMed] [Google Scholar]
- [7].Filocamo M, Morrone A, Lysosomal storage disorders: molecular basis and laboratory testing, Hum. Genomics 5 (2011) 156–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zanetti A, et al. , Setup and validation of a targeted next-generation sequencing approach for the diagnosis of lysosomal storage disorders, J. Mol. Diagnostics 22 (2020) 488–502. [DOI] [PubMed] [Google Scholar]
- [9].Stiles AR, et al. , A comprehensive testing algorithm for the diagnosis of Fabry disease in males and females, Mol. Genet. Metab 130 (2020) 209–214. [DOI] [PubMed] [Google Scholar]
- [10].Fernández-Marmiesse A, et al. , Assessment of a targeted resequencing assay as a support tool in the diagnosis of lysosomal storage disorders, Orphanet J. Rare Dis 9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Fuller M, Laboratory diagnosis of lysosomal diseases: newborn screening to treatment, Clin. Biochem. Rev 41 (2020) 53–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Miller MJ, et al. , Untargeted metabolomic analysis for the clinical screening of inborn errors of metabolism, J. Inherit. Metab. Dis 38 (2015) 1029–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gertsman I, Gangoiti JA, Barshop BA, Validation of a dual LC–HRMS platform for clinical metabolic diagnosis in serum, bridging quantitative analysis and untargeted metabolomics, Metabolomics 10 (2014) 312–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Coene KLMM, et al. , Next-generation metabolic screening: targeted and untargeted metabolomics for the diagnosis of inborn errors of metabolism in individual patients, J. Inherit. Metab. Dis 41 (2018) 337–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Körver-Keularts IMLW, et al. , Fast and accurate quantitative organic acid analysis with LC-QTOF/MS facilitates screening of patients for inborn errors of metabolism, J. Inherit. Metab. Dis 41 (2018) 415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Alaimo JT, et al. , Integrated analysis of metabolomic profiling and exome data supplements sequence variant interpretation, classification, and diagnosis, Genet. Med (2020) 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Piraud M, et al. , Development of a new tandem mass spectrometry method for urine and amniotic fluid screening of oligosaccharidoses, Rapid Commun. Mass Spectrom 31 (2017) 951–963. [DOI] [PubMed] [Google Scholar]
- [18].Huang R, Cathey S, Pollard L, Wood T, UPLC-MS/MS analysis of urinary free oligosaccharides for lysosomal storage diseases: diagnosis and potential treatment monitoring, Clin. Chem 64 (2018) 1772–1779. [DOI] [PubMed] [Google Scholar]
- [19].Ramsay SL, Meikle PJ, Hopwood JJ, Clements PR, Profiling oligosaccharidurias by electrospray tandem mass spectrometry: quantifying reducing oligosaccharides, Anal. Biochem 345 (2005) 30–46. [DOI] [PubMed] [Google Scholar]
- [20].Auray-Blais C, et al. , UPLC-MS/MS detection of disaccharides derived from glycosaminoglycans as biomarkers of mucopolysaccharidoses, Anal. Chim. Acta 936 (2016) 139–148. [DOI] [PubMed] [Google Scholar]
- [21].Saville JT, McDermott BK, Fletcher JM, Fuller M, Disease and subtype specific signatures enable precise diagnosis of the mucopolysaccharidoses, Genet. Med 21 (2019) 753–757. [DOI] [PubMed] [Google Scholar]
- [22].Lawrence R, et al. , Disease-specific non–reducing end carbohydrate biomarkers for mucopolysaccharidoses, Nat. Chem. Biol 8 (2012) 197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tomatsu S, et al. , Newborn screening and diagnosis of mucopolysaccharidoses, Mol. Genet. Metab 110 (2013) 42–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Pettazzoni M, et al. , LC-MS/MS multiplex analysis of lysosphingolipids in plasma and amniotic fluid: a novel tool for the screening of sphingolipidoses and Niemann-Pick type C disease, PLoS One 12 (2017), e0181700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Polo G, et al. , Diagnosis of sphingolipidoses: a new simultaneous measurement of lysosphingolipids by LC-MS/MS, Clin. Chem. Lab. Med 55 (2017) 403–414. [DOI] [PubMed] [Google Scholar]
- [26].Turgeon CT, et al. , Measurement of psychosine in dried blood spots — a possible improvement to newborn screening programs for Krabbe disease, J. Inherit. Metab. Dis 38 (2015) 923–929. [DOI] [PubMed] [Google Scholar]
- [27].Fuller M, Rozaklis T, Ramsay SL, Hopwood JJ, Meikle PJ, Disease-specific markers for the mucopolysaccharidoses, Pediatr. Res 56 (2004) 733–738. [DOI] [PubMed] [Google Scholar]
- [28].Liaw A, Wiener M, Classification and regression by random forest, R News 2 (2002) 18–22. [Google Scholar]
- [29].Xia B, et al. , Oligosaccharide analysis in urine by MALDI-TOF mass spectrometry for the diagnosis of lysosomal storage diseases, Clin. Chem 59 (2013) 1357–1368. [DOI] [PubMed] [Google Scholar]
- [30].Fountain KJ, Hudalla CJ, Mccabe DR, Morrison D, Analysis of carbohydrates by ultra performance liquid chromatography and mass spectrometry, Waters Application Note, 2009https://www.waters.com/nextgen/xg/en/library/application-notes/2009/analysis-of-carbohydrates-by-uplc-and-mass-spectrometry.html.
- [31].Nickander KK, et al. , Oligosacchariduria profiles by MALDI-TOF mass spectrometry and post-analytical interpretation using multivariate pattern recognition software, Mol. Genet. Metab 120 (2017) S102–S103. [Google Scholar]
- [32].Hall PL, et al. , Urine oligosaccharide screening by MALDI-TOF for the identification of NGLY1 deficiency, Mol. Genet. Metab 124 (2018) 82–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].De Praeter CM, et al. , A novel disorder caused by defective biosynthesis of N-linked oligosaccharides due to glucosidase I deficiency, Am. J. Hum. Genet 66 (2000) 1744–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sluiter W, et al. , Rapid ultraperformance liquid chromatography-tandem mass spectrometry assay for a characteristic glycogen-derived tetrasaccharide in pompe disease and other glycogen storage diseases, Clin. Chem 58 (2012) 1139–1147. [DOI] [PubMed] [Google Scholar]
- [35].Revel-Vilk S, Fuller M, Zimran A, Value of glucosylsphingosine (Lyso-Gb1) as a biomarker in gaucher disease: a systematic literature review, Int. J. Mol. Sci 21 (2020) 1–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Welford RWD, et al. , Plasma lysosphingomyelin demonstrates great potential as a diagnostic biomarker for niemann-pick disease type C in a retrospective study, PLoS One 9 (2014) 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Polo G, et al. , Plasma and dried blood spot lysosphingolipids for the diagnosis of different sphingolipidoses: a comparative study, Clin. Chem. Lab. Med 57 (2019) 1863–1874. [DOI] [PubMed] [Google Scholar]
- [38].Maruyama H, et al. , Effectiveness of plasma lyso-Gb3 as a biomarker for selecting high-risk patients with Fabry disease from multispecialty clinics for genetic analysis, Genet. Med 21 (2019) 44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Almontashiri NAM, et al. , Clinical validation of targeted and untargeted metabolomics testing for genetic disorders: a 3 year comparative study, Sci. Rep 10 (2020) 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kennedy AD, et al. , Metabolomic profiling of human urine as a screen for multiple inborn errors of metabolism, Genet. Test. Mol. Biomark 20 (2016) 485–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Rhee EP, et al. , Variability of two metabolomic platforms in CKD, Clin. J. Am. Soc. Nephrol 14 (2019) 40–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Bonte R, et al. , Untargeted metabolomics-based screening method for inborn errors of metabolism using semi-automatic sample preparation with an UHPLC-orbitrap-MS platform, Metabolites 9 (2019) 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zerimech F, et al. , Urinary metabolic phenotyping of mucopolysaccharidosis type I combining untargeted and targeted strategies with data modeling, Clin. Chim. Acta 475 (2017) 7–14. [DOI] [PubMed] [Google Scholar]
- [44].Tebani A, et al. , Analysis of mucopolysaccharidosis type VI through integrative functional metabolomics, Int. J. Mol. Sci 20 (2019) 446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Tebani A, et al. , Unveiling metabolic remodeling in mucopolysaccharidosis type III through integrative metabolomics and pathway analysis, J. Transl. Med 16 (2018) 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Fu H, et al. , Serum global metabolomics profiling reveals profound metabolic impairments in patients with MPS IIIA and MPS IIIB, Metab. Brain Dis 32 (2017) 1403–1415. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All biomarker data is provided in the manuscript and Supplementary Information.