


Abstract
Contact-dependent growth inhibition is a mechanism of interbacterial competition mediated by delivery of the C-terminal toxin domain of CdiA protein (CdiA–CT) into neighboring bacteria. The CdiA–CT of enterohemorrhagic Escherichia coli EC869 (CdiA–CTEC869) cleaves the 3′-acceptor regions of specific tRNAs in a reaction that requires the translation factors Tu/Ts and GTP. Here, we show that CdiA–CTEC869 has an intrinsic ability to recognize a specific sequence in substrate tRNAs, and Tu:Ts complex promotes tRNA cleavage by CdiA–CTEC869. Uncharged and aminoacylated tRNAs (aa-tRNAs) were cleaved by CdiA–CTEC869 to the same extent in the presence of Tu/Ts, and the CdiA–CTEC869:Tu:Ts:tRNA(aa-tRNA) complex formed in the presence of GTP. CdiA–CTEC869 interacts with domain II of Tu, thereby preventing the 3′-moiety of tRNA to bind to Tu as in canonical Tu:GTP:aa-tRNA complexes. Superimposition of the Tu:GTP:aa-tRNA structure onto the CdiA–CTEC869:Tu structure suggests that the 3′-portion of tRNA relocates into the CdiA–CTEC869 active site, located on the opposite side to the CdiA–CTEC869 :Tu interface, for tRNA cleavage. Thus, CdiA–CTEC869 is recruited to Tu:GTP:Ts, and CdiA–CT:Tu:GTP:Ts recognizes substrate tRNAs and cleaves them. Tu:GTP:Ts serves as a reaction scaffold that increases the affinity of CdiA–CTEC869 for substrate tRNAs and induces a structural change of tRNAs for efficient cleavage by CdiA–CTEC869.
Graphical Abstract
Graphical Abstract.
Model of tRNA cleavage by CdiA–CT in the presence of translation factors Tu/Ts and GTP. The Tu:GTP:Ts complex acts as a scaffold for tRNA cleavage by CdiA–CT.
INTRODUCTION
The contact-dependent growth inhibition (CDI) system is a mechanism of interbacterial competition that is widely observed in Gram-negative bacteria and common in pathogenetic proteobacteria (1–5). CDI requires direct physical contact between bacteria and is mediated by specific receptors and the delivery of toxin proteins. Toxin delivery into neighboring bacteria induces growth inhibition and is occasionally fatal. In Escherichia coli, the genes responsible for CDI are cdiA, cdiB and cdiI (1). CdiA, which adopts a large filamentous structure, is attached to the cell surface via its N-terminus and contains a receptor-binding domain (RBD) and a C-terminal toxin domain (CdiA–CT) (4,6,7). CdiB is an outer membrane protein through which CdiA is secreted. When the RBD of CdiA binds to the receptor of neighboring bacteria, CdiA autoproteolytically cleaves the CdiA–CT, which then translocates into the target cells. CdiI is an immunity protein that protects cells from self-intoxication by forming a tight CdiA–CT:CdiI complex (8,9). Accordingly, the growth of non-isogenic bacteria without the corresponding CdiI is inhibited by CdiA–CT. CdiA–CT toxins are highly polymorphic and include various types of toxin modules with pore-forming potential, RNase activity targeting the 16S rRNA or tRNA (tRNase), or DNase activity. Most E. coli CdiA–CT toxins have nuclease activity (2).
Cytosolic factors are sometimes required for the activity of CdiA–CT toxin (10–16). CdiA toxin from E. coli 563, CdiA–CTEC563, is a potent tRNA anticodon nuclease (1,10) that requires o-acetylserine sulfhydrylase A (CysK) for its RNase activity in vivo and in vitro (10,12,13). CysK interacts with the C-terminal Gly-Tyr-Gly-Ile motif of CdiA–CTEC563 and increases the tRNase activity of CdiA–CTEC563 by increasing its thermostability and promoting interactions with substrate tRNAs (10,11,15). Recently, it was shown that CdiAs from E. coli NC101 (CdiA–CTNC101), EC869 (CdiA–CTEC869), and Klebsiella pneumoniae 342 (CdiA–CTKp342) all cleave the 3′ regions of the acceptor stems of specific tRNAs (14–17). CdiA–CTEC869 cleaves the 3′-acceptor stems of tRNAGln and tRNAAsn between positions 71 and 72 (15); CdiA–CTNC101 cleaves tRNAGlu, tRNAAsp and others between positions 72 and 73 (14); and CdiA–CTKp342 cleaves tRNAIle (16). In vitro, EF-Tu and EF-Ts (hereafter simply Tu and Ts, respectively) together with GTP promote cleavage of tRNA by these CdiA–CT toxins. In vivo, tsf mutants in the coiled-coil domain of Ts are resistant to inhibition by these CdiA–CTs (14–16). Although CdiA–CTNC101 and CdiA–CTKp342 exhibit low similarity at the primary amino acid sequence level, their structures are homologous and adopt the Barnase/EndoU/Colicin/RelE (BECR) fold, suggesting that the similar structures of CdiA–CTs arose through convergent evolution (16). On the other hand, the structures of the corresponding immunity proteins CdiIs are diverse.
In protein translation, GTP-bound Tu (Tu:GTP) delivers aminoacyl-tRNAs (aa-tRNAs) to the ribosome A-site (18). When a codon–anticodon match occurs on the A-site, GTP is hydrolyzed to GDP by Tu, and GDP-bound Tu (Tu:GDP) is released from the ribosome (19,20). Ts binds Tu:GDP, displaces GDP, and recycles Tu, thus acting as a guanine nucleotide exchange factor (GEF) for Tu (21,22). Translation factors have functions beyond protein synthesis, e.g. in the replication of Qβ phage RNA by Qβ replicase, which consists of a phage-encoded RNA-dependent RNA polymerase (RdRp: β-subunit) along with host-derived Tu, Ts, and ribosomal protein S1 (23,24). In Qβ replicase, Tu and Ts function as chaperones (25) and RNA elongation cofactors that facilitate the separation of the template and growing RNAs during replication (26). The chaperone function of Tu was described (27–29), but its detailed mode of action has remained enigmatic.
When Tu, Ts and GTP were first described to be required for cleavage of the 3′-acceptor regions of specific tRNAs by CdiA–CTEC869, it was proposed that CdiA–CTEC869 recognizes an aa-tRNA complexed with Tu:GTP (15). The same scenario was proposed for CdiA–CTNC101, which also requires Tu, Ts, and GTP for specific tRNA cleavage (14). However, in these studies, the authors only tested cleavage of uncharged tRNAs, not aa-tRNAs, and neither the effect of the aminoacyl status of tRNA nor the kinetics of tRNA cleavage in the presence of translation factors has been well examined. A subsequent study showed that CdiA–CTKp342 cleaves uncharged tRNAIle, rather than Ile-tRNAIle, in the presence of translation factors in vitro, and that the closely related E. coli 3006 CdiA–CT, CdiA–CTEC3006, whose tRNase activity does not require Tu and Ts, cleaves uncharged tRNAIle between positions 70 and 71, but not Ile-tRNAIle (16). Therefore, unanswered questions remain about the mechanism underlying substrate tRNA recognition by these CdiA–CTs targeting the 3′-acceptor region of tRNAs, their specificity, and the functions of translation factors in the enhancement of tRNA cleavage by the above-mentioned CdiA–CTs (17).
In this study, we analyzed the CdiA toxin from enterohemorrhagic E. coli EC869, CdiA–CTEC869 (15). Our biochemical and structural analyses collectively suggest that tRNA cleavage by CdiA–CTEC869 in the presence of Tu, Ts, and GTP proceeds via the CdiA–CTEC869:Tu:GTP:Ts:tRNA complex. CdiA–CTEC869 is recruited to Tu:GTP:Ts and forms CdiA–CTEC869:Tu:GTP:Ts complex, which recognizes tRNA and aa-tRNA. Tu:GTP:Ts in the complex serves as a reaction scaffold that facilitates tRNA cleavage by CdiA–CTEC869 by increasing the affinity of CdiA–CTEC869 for the tRNA substrate and inducing a structural change of the 3′-portion of the tRNA with assistance from Ts in the complex.
MATERIALS AND METHODS
Plasmid construction
Synthetic DNA containing the cdiA-CT/cdiI module from enterohemorrhagic E. coli EC869 was purchased from Eurofins, Japan. The nucleotide sequence of the cdiA-CT/cdiIEC869 module is provided in Supplementary Table S1. For overexpression of the CdiA–CT/CdiIEC869 complex in E. coli, the cdiA-CT/cdiIEC869 module was PCR amplified and cloned between the NdeI and XhoI sites of pET15L, yielding pET15L-CdiA–CT/CdiIEC869. To generate plasmids for expression of the inactive CdiA–CT (H281A or ΔC5) mutant proteins, the mutations were introduced by PCR into the CdiA–CTEC869 coding region, and the resultant PCR fragments were cloned into the NdeI and XhoI sites of pET15L, yielding the plasmids pET15L-CdiA–CTEC869_H281A and pET15L-CdiA–CTEC869_ΔC5. For expression of CdiIEC869, the CdiI gene was also PCR amplified and cloned between the NdeI and XhoI sites of pET15L and pET15sumo, yielding plasmids pET15L-CdiIEC869 and pET15sumo-CdiIEC869, respectively. pET15sumo was designed to express proteins fused with an N-terminal Sumo-tag carrying the N-terminal hexahistidine (His6) sequence.
The DNA fragment containing the cdiA-CT/cdiIEC869 module was cloned between the NdeI and HindIII sites of pBAD33 (ATCC87402), yielding pBAD33_CdiAIEC869. pBAD33_CdiAIEC869 variants with a mutation in the CdiA–CT coding region were prepared by PCR. pBAD33_CdiAEC869_H281A and pBAD33_CdiAEC869_ΔC5 were constructed by PCR amplification of the CdiA coding regions of pET15L-CdiA–CTEC869_H281A and pET15L-CdiA–CTEC869_ΔC5, which were then cloned between the NdeI and HindIII sites of pBAD33. Expression plasmids for E. coli Tu and Ts were described previously (25). A gene for single chain Tu-Ts was designed as previously described (30). A DNA fragment encoding Tu, Ts or Tu-Ts was cloned into the NdeI and XhoI sites of pET15sumo, yielding plasmids pET15sumo-EF-Tu, pET15sumo-EF-Ts and pET15sumo-EF-Tu-Ts, respectively. The E. coli glutaminyl-tRNA synthase gene was PCR amplified from E. coli genomic DNA and cloned between the NdeI and XhoI sites of pET22b, yielding pET22EcGlnRS.
A synthetic DNA containing the E. coli tRNAGln gene harboring the U1G mutation, driven by the T7 RNA promoter, was purchased from Eurofins and cloned between the EcoRI and HindIII sites of pUC19, yielding pT7EctRNAGlnG1A72. Other plasmids for in vitro transcription of tRNAGln variants, precursor tRNAGln variants, and tRNATrp were purchased from Eurofins, and the DNA sequences used for in vitro transcription are listed in Supplementary Table S1. The oligonucleotide primers used for the PCR cloning and mutations are listed in Supplementary Table S2.
Protein purification
For expression of CdiA–CT/CdiIEC869, E. coli Rosetta1 cells (Novagen-Merck Millipore) were transformed with pET15L-CdiA–CT/CdiIEC869 and cultured until the OD600 reached ∼0.6. Protein expression was induced by addition of 1 mM of isopropyl-β-d-thiogalactopyranoside (IPTG), and culture was continued for 2 h at 37°C. For expression of CdiIEC869, CdiA–CTEC869_H281A, and CdiA–CTEC869_ΔC5, cells were transformed with pET15L-CdiIEC869, pET15L-CdiA–CTEC869_H281A and pET15L-CdiA–CTEC869_ΔC5. For expression of Tu, Ts and their variants, E. coli BL21(DE3) was transformed with pET22EF-Tu, pET22-Ts and their variants, respectively, and protein expression was induced by addition of 0.1 mM IPTG followed by culture for 20 h at 20°C. The harvested cells were sonicated in Ni-Buffer [50 mM Tris–Cl, pH 7.0, 500 mM NaCl, 10% (v/v) glycerol, 20 mM imidazole, 5 mM β-mercaptoethanol] supplemented with 0.1 mM PMSF (phenylmethylsulfonyl fluoride) and 50 μg/ml lysozyme. The lysates were centrifuged at 30 000 × g and 4°C for 1 h, and the supernatants were applied to a Ni-NTA column (QIAGEN, Japan). The column was washed with Ni-Buffer and the proteins were eluted with Ni-Buffer supplemented with 400 mM imidazole. The proteins were then applied to a Hi-Trap Heparin or Hi-Trap Q column (GE Healthcare, Japan). Finally, the proteins were purified on a Hi-Load 16/60 Superdex 200 column (GE Healthcare, Japan), equilibrated with buffer containing 25 mM Tris–Cl, pH 7.0, 150 mM NaCl, and 10 mM β-mercaptoethanol, and concentrated. For the purification of proteins without the His6-tag, SUMO (small ubiquitin-related modifier)-tagged proteins carrying His6 at their N-termini were expressed and purified by Ni-NTA chromatography as described above. The SUMO tags were removed from the proteins by digestion at 4°C overnight with Ulp1 (Ubl-specific protease 1). Subsequently, the SUMO-tag–free proteins were purified by applying to Ni-NTA column chromatography, and the flow-through fractions were collected. The proteins were further purified as described above.
For isolation of CdiA–CTEC869, the purified CdiA–CT/CdiIEC869 protein complex was denatured on a Ni-NTA column equilibrated with buffer containing 8 M urea, 500 mM NaCl, and 25 mM Tris–Cl, pH 8.0 (31–33). Denatured CdiIEC869 protein was removed from the column by washing the column with the denaturing buffer. The denatured CdiA–CTEC869 protein was refolded with a stepwise gradient of urea (6, 4, 2, 0.5 and 0 M) on the column, and the renatured CdiA–CTEC869 protein was eluted from the column with Ni-Buffer supplemented with 300 mM imidazole. The purified CdiA–CTEC869 protein was then applied to a Hi-Trap Heparin column (GE Healthcare, Japan) and further purified on a Hi-Load 16/60 Superdex 200 column (GE Healthcare, Japan), equilibrated with 25 mM Tris–Cl, pH 7.0, 150 mM NaCl, and 10 mM β-mercaptoethanol. To prepare nucleotide-free wild-type Tu, His84Ala mutant Tu and sg–Tu–Ts, the purified proteins were incubated for 20 min at 37°C in Mg2+-free buffer containing 25 mM Tris–Cl, pH 6.8, 150 mM NaCl, 10 mM β-mercaptoethanol, and 10 mM EDTA, as described (34). The samples were then purified by gel filtration on a Hi-Load 16/60 Superdex 200 column (GE Healthcare, Japan) in the same buffer without Mg2+.
tRNA preparation
For preparation of tRNAGln transcript variants starting with A1, C1 or U1, the corresponding precursor tRNAs (pre-tRNAs) with 5′-leader sequences were synthesized by T7 RNA polymerase using plasmid linearized by FokI digestion. The 5′-leader RNA was processed in vitro by RNase P, which consists of the M1 RNA and C5 protein (33). The pre-tRNA transcript was processed in a mixture containing 100 nM M1 RNA, 100 nM C5 protein, 50 mM Tris–Cl, pH 7.4, 10 mM MgCl2 and 100 mM NH4Cl at 37°C overnight, followed by phenol extraction and isopropanol precipitation. The processed mature tRNAs were dissolved in a buffer containing 20 mM Tris–Cl, pH 7.4 and 200 mM NaCl, applied to a Resource Q column (GE Healthcare, Japan), and separated by a linear NaCl gradient (0.2–1.0 M) in the buffer. tRNAs were ethanol-precipitated, rinsed, and dried. The tRNAfMet and tRNATrp transcripts were also prepared as described above (33). tRNAGln G1 variants were synthesized using T7 RNA polymerase and purified as described above.
In vitro tRNA cleavage assay
The standard CdiA–CTEC869 tRNase activity assay was conducted at 37°C in reaction buffer containing 20 mM Tris–Cl, pH 7.4, 150 mM NaCl, 5 mM MgCl2, 10 mM β-mercaptoethanol, 100 μg/ml bovine serum albumin (BSA), 1 μM tRNAGln transcript (or its variant), 0.1 μM CdiA–CTEC869, 0.1 μM Tu, 0.1 μM Ts and 1 mM GTP. Reactions were quenched by addition of an equal volume of formamide gel-loading buffer [95% (v/v) formamide, 0.02% (w/v) SDS, 0.02% (w/v) bromophenol blue, 0.01% (w/v) xylene cyanol] at the indicated time points, and the tRNAs were separated by 10% (w/v) polyacrylamide gel electrophoresis (PAGE) under denaturing conditions. The gels were stained with ethidium bromide, and tRNA band intensities were quantified by the Image Lab software (Bio-Rad, version 3.0). The specific conditions for each assay of tRNA cleavage by CdiA–CTEC869 are clearly described in either the text or figure legends.
Whole-mass analysis of tRNAs
tRNAGln, tRNATrp, and tRNAfMet transcripts were cleaved by CdiA–CT in the presence of translation factors and GTP, and the cleaved products were subjected to 10% (w/v) PAGE under denaturing conditions. A total of 10 pmol of each tRNA was subjected to reverse phase (ODS) capillary liquid chromatography (LC) coupled to nano electrospray (ESI)/mass spectrometry (MS) on a linear ion trap-Orbitrap hybrid mass spectrometer (LTQ Orbitrap XL; Thermo Fisher Scientific) as described previously (35,36). RNAs were scanned in a negative polarity mode and detected by a linear ion trap device. The scanning range was set to 600–2000. A multiply-charged ESI spectrum was deconvoluted by ProMass HR for Xcalibur (Novatia, LLC) to calculate the average molecular mass.
In vivo toxicity assay
E. coli strain MG1655 (NIG, Japan; ME7986) was transformed with pBAD33_CdiA–CTEC869 its variants, inoculated into LB containing 50 μg/ml chloramphenicol and 1% (w/v) glucose, and cultured at 37°C. The toxicities of CdiA–CTEC869 and its variants were evaluated by spot assay. Specifically, overnight LB cultures were serially diluted, 3-μl aliquots of the dilutions were spotted on LB agar plates containing 50 μg/ml chloramphenicol supplemented with 1% (w/v) arabinose or 1% (w/v) glucose, and the plates were incubated overnight at 37°C.
Preparation of aa-tRNAs and LC/MS spectrometry
E. coli strain MG1655 was transformed with pBAD33_CdiA–CTEC869 or control pBAD33, inoculated in LB containing 50 μg/ml chloramphenicol and 1% (w/v) glucose, and cultured overnight at 37°C. The overnight cultures were diluted to an OD660 of 0.02 in LB (4 ml) containing 50 μg/ml chloramphenicol and cultured at 37°C until the OD660 reached 0.3, and then 0.02% (w/v) arabinose was added. After a 10-min incubation, the cells were harvested and suspended in buffer containing 50 mM NaOAc, pH 5.0, 0.5 mM EDTA, and 0.2 M NaCl. RNA was extracted under acidic conditions using phenol saturated with 300 mM NaOAc, pH 5.2, followed by isopropyl alcohol precipitation. The precipitated RNA was dissolved in 100 μl of 300 mM NaOAc, pH 5.2. aa-tRNAs were chemically acetylated by addition of acetic anhydride (32,33,37) and then ethanol-precipitated and rinsed with 70% cold ethanol. The RNA was dissolved in cold buffer containing 50 mM NaOAc, pH 5.0, 0.5 mM EDTA, and 0.2 M NaCl, and loaded onto a 100-μl Q-Sepharose F.F. (GE Healthcare, Japan) column. The resin was washed with buffer containing 50 mM NaOAc, pH 5.0, 0.5 mM EDTA and 0.3 M NaCl. Finally, the tRNA fraction was eluted with buffer containing 50 mM NaOAc, pH 5.0, 0.5 mM EDTA and 0.6 M NaCl, ethanol-precipitated, and rinsed with 70% (v/v) ethanol. The acetylated aa-tRNAs were digested with RNase One Ribonuclease (Promega, Japan) at 37°C for 60 min in a 25-μl reaction mixture containing 25 mM NH4Oac and 2.5 units enzyme. The digests were subjected to an LC/MS analysis using a Q Exactive Hybrid Quadrupole–Orbitrap Mass Spectrometer (Thermo Fisher Scientific) equipped with a Dionex UltiMate 3000 LC System (Thermo Fisher Scientific) and an InertSustain C18 column (5 μm, 2.1 × 250 mm, GL Sciences), as described (32,33,38).
Gel-shift assay
tRNAGln was uniformly 32P-labeled with α-32P UTP (PerkinElmer, Japan, 3,000 Ci/mmol) using an in vitro transcription kit (Promega, Japan) and then gel-purified. The 32P-labeled tRNAGln (10 000 cpm) was incubated in 10 μl of a solution containing 50 mM Tris–Cl, pH 7.4, 10 mM MgCl2, 50 mM NaCl, 10 mM β-mercaptoethanol, 10% (v/v) glycerol and various amounts of CdiA–CTEC869_H281A, Tu, or single-chain Tu-Ts (sg–Tu–Ts), at 37°C for 10 min. The solutions were then cooled on ice. The solutions were separated by 6% (w/v) native acrylamide gel electrophoresis (1 × TBE) at room temperature (∼25°C), as described (39). 32P-labeled bands were quantified using a BAS-5000 imager (FujiFilm, Japan).
Northern blotting
E. coli tRNA mixtures used for the in vitro cleavage assay by CdiA–CTEC869 were prepared from JM109tr as described (31). The RNA fraction was deacylated by incubation in 1.8 M Tris–Cl, pH 8.0, for 2 h at 37°C and separated by Hi-Load 16/10 Q-Sepharose HP chromatography, and then the tRNA fractions were pooled and ethanol-precipitated. RNAs with or without CdiA–CTEC869 treatment were separated on a 10% (w/v) polyacrylamide gel containing 7 M urea and then transferred to a Hybond-N + membrane (GE Healthcare, Japan) by a Trans-Blot SD semi-dry cell (Bio-Rad, Japan). Hybridization was carried out overnight at 60°C in PerfectHyb Hybridization Solution (Toyobo, Japan). The membrane was washed three times with buffer containing 2 × standard saline citrate (SSC) and 0.1% SDS for 5 min and then washed twice with buffer containing 0.1 × SSC and 0.1% (w/v) SDS for 10 min at 60°C. The oligonucleotide sequences used as specific probes for tRNAs are listed in Supplementary Table S2.
Pull-down assay
Interactions between CdiA–CTEC869 and translation factors in the presence of tRNA/aa-tRNA were analyzed by a pull-down assay. Nucleotide-free Tu was preincubated with a 10-fold excess of GTP for 15 min at 37°C in buffer A (50 mM Tris–Cl, pH 7.0, 50 mM NH4Cl, 10 mM MgCl2 and 5 mM β-mercaptoethanol) as described previously (34). To prepare Gln-tRNAGln, tRNAGln was aminoacylated by GlnRS in buffer containing 30 mM Tris–HCl, pH 7.4, 15 mM MgCl2, 25 mM KCl, 5 mM DTT, 4 mM ATP and 2 mM glutamine as described previously (40). After incubation for 1 h at 37°C, RNA was extracted under cold acidic conditions using phenol saturated with 300 mM NaOAc, pH 5.2, followed by isopropyl alcohol precipitation. The precipitated RNA was dissolved in 300 mM NaOAc, pH 5.2, and purified using a NAP5 column (Amersham Biosciences, Japan), followed by isopropyl alcohol precipitation and a rinse with cold 70% (v/v) ethanol. The precipitated RNA was dissolved in 20 mM NaOAc, pH 5.2 and used in the pull-down assay.
Hexahistidine-tagged catalytic dead CdiA–CTEC869_H281A (5 μM) was mixed with an equimolar amount of translation factors in the presence of an equimolar amount of tRNA/aa-tRNA in buffer A containing 1 mM GTP. The solution (200 μl volume) was incubated for 5 min at 37°C and placed on ice. To this solution, 150 μl of Ni-NTA resin equilibrated with wash buffer (25 mM Tris–Cl, pH 7.0, 100 mM NaCl, 5 mM MgCl2, 5 mM NH4Cl, 5 mM β-mercaptoethanol, 10 mM imidazole, and 1 mM GTP) was added, and the mixture was incubated at 4°C for 60 min. The mixture was loaded onto a poly-prep chromatography column (Bio-Rad, Japan). The Ni-NTA resin was washed with 10 column volumes of wash buffer and eluted with the same buffer containing 300 mM imidazole. The fractions were separated by 12% (w/v) SDS PAGE to visualize proteins or 10% (w/v) polyacrylamide gel containing 7 M urea to visualize tRNA.
Crystallization and structural determination of Tu:CdiA–CT:CdiI complex
For crystallization of the Tu:CdiA–CT:CdiIEC869 ternary complex, 36 μM CdiA–CT:CdiI complex was mixed with an equimolar amount of Tu and incubated at 4°C for 30 min. A 1-μl aliquot of the protein mixture solution was mixed with 1.0 μl of reservoir solution containing 100 mM HEPES–KOH, pH 7.5, 2% (w/v) Tacsimate, pH 7.0, and 22% (w/v) PEG3350, and the crystals were generated by the hanging-drop vapor diffusion method at 20°C. The crystals were flash-cooled in 1.1 × concentrated reservoir solution, supplemented with 20% (v/v) ethylene glycol as a cryoprotectant. Data sets were collected on beamline 17A at the Photon Factory at KEK, Japan. The data were indexed, integrated, and scaled with XDS (41). The crystal belongs to space group C121 with one CdiA–CT:CdiIEC869 :Tu complex in the asymmetric unit cell. The initial phase was determined by the molecular replacement method, using the structure of E. coli Tu (PDB ID: 4PC3) as the search model. Although the electron densities corresponding to Tu were visible, the electron density corresponding to CdiA–CT:CdiI was not. We modeled CdiA–CT and CdiI structures using AlphaFold 2 (42). Molecular replacement using the modeled CdiA–CT and CdiI structures as search models improved the electron densities corresponding to CdiA–CT:CdiI. The structure was refined with phenix.refine (43), and manually modified with Coot (44). Finally, the structure was model-built and refined to an R factor of 23.7% (Rfree = 27.8%) at 3.41 Å resolution. The modeled CdiA–CT contains residues 175–285 (numbered from Val1 of the VENN peptide motif), and the modeled CdiIEC869 contains residues 3–179. The modeled Tu contains residues 10–393. The details of the crystallographic data collection and refinement statistics are provided in Table 1.
Table 1.
Data collection and refinement statistics
| Tu:CdiA–CT:CdiI | |
|---|---|
| Data collection | |
| Space group | C 1 2 1 |
| Cell dimensions | |
| a, b, c (Å) | 101.93, 102.76, 88.14 |
| α, β, γ (°) | 90, 111.863, 90 |
| Wavelength (Å) | 0.98000 |
| Resolution (Å)a | 49.76–3.40 (3.52–3.40) |
| R sym a | 0.308 (2.134) |
| I / σIa | 7.1 (1.0) |
| CC 1/2a | 0.994 (0.386) |
| Completeness (%)a | 98.4 (89.0) |
| Redundancya | 7.5 (6.7) |
| Refinement | |
| Resolution (Å) | 35.6–3.413 |
| No. reflections | 86153 |
| R work / Rfree (%) | 23.68 / 27.79 |
| No. atoms | |
| Protein | 5190 |
| Ligand | - |
| Water | - |
| B-factors (Å2) | |
| Protein | 111.13 |
| Ligand | - |
| Water | - |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.004 |
| Bond angles (°) | 1.04 |
aValues in parentheses are for the highest resolution shell.
RESULTS
Translation factors promote tRNA cleavage by CdiA–CTEC869
The specific tRNA cleavage reaction catalyzed by the C-terminal domain of CdiA from enterohemorrhagic E. coli EC869 (CdiA–CTEC869; hereafter simply CdiA–CT) requires translation elongation factors Tu and Ts as well as GTP, although GTP hydrolysis is not required (15).
To confirm the requirement of translation factors and GTP for tRNA cleavage by CdiA–CT, we prepared highly purified recombinant Tu, Ts and CdiA–CT by several chromatography steps (Supplementary Figure S1). Because Tu and Ts form Tu:Ts heterodimer (45), Tu (or Ts) was purified with great care to avoid the contamination of Ts (or Tu) in the preparations. We then tested the tRNA cleavage by CdiA–CT in the presence of translation factors using the tRNAGln transcript as the substrate in vitro. Under standard test conditions (pH 7.4), the tRNA cleavage level was increased in the presence of Tu, Ts, and GTP (Figure 1A). tRNA cleavage was suppressed by the prior incubation of CdiA–CT with its immunity protein, CdiIEC869, confirming that the observed tRNA cleavage was caused by CdiA–CT (Figure 1B).
Figure 1.

tRNA cleavage by CdiA–CTEC869 in the presence of translation factors and GTP. (A) tRNAGln cleavage by CdiA–CT is dependent on Tu, Ts and GTP under neutral pH conditions (pH = 7.4). tRNAGln (1 μM) was incubated at 37°C with 0.1 μM CdiA–CT in the presence of Tu (0.1 μM), Ts (0.1 μM), and GTP (1 mM), and the tRNA was separated by 10% (w/v) PAGE under denaturing conditions. The fraction of cleaved tRNA was quantified at the indicated time points. (B) tRNAGln cleavage is blocked by CdiIEC869, an immunity protein against CdiA–CTEC869. CdiA–CT was preincubated with an equimolar amount of CdiIEC869 before the reaction and then subjected to the cleavage reaction for 60 min at 37°C. (C) Steady-state kinetics of tRNAGln cleavage by CdiA–CTEC869 in the presence or absence of Tu, Ts, and GTP. The initial velocities of tRNAGln cleavage by CdiA–CTEC869 were measured at various concentrations of tRNAGln (2–16 μM). Tu (0.1 μM), Ts (0.1 μM) and GTP (1 mM) were added to CdiA–CTEC869 (0.1 μM) to calculate the initial velocities of tRNA cleavage by CdiA–CTEC869 in the presence of translation factors at pH 7.4. (D) CdiA–CT activity is pH-dependent and elevated under lower pH conditions. The cleavage of tRNAGln by CdiA–CT alone (open bars) and by CdiA–CTEC869 in the presence of Tu, Ts, and GTP (hatched bars) is shown under various pH conditions. The tRNAGln transcript (1.0 μM) was incubated for 10 min at 37°C with 0.2 μM of CdiA–CT in the presence or absence of Tu, Ts, and GTP. The Y-axis in the graph represents cleavage (%) at 10 min. For pH ranges from 5.3 to 6.9 and 7.3 to 8.5, 20 mM MES and 20 mM Tris–Cl buffers were used, respectively. The tRNA was separated by 10% (w/v) PAGE under denaturing conditions, and the fraction of the cleaved tRNA was quantified. (E) tRNAGln cleavage by CdiA–CT under acidic conditions (pH 5.3) is still, but to a lesser extent, dependent on the presence of Tu, Ts, and GTP. tRNAGln (1.0 μM) was incubated with 0.05 μM CdiA–CTEC869 in the presence of increasing amounts of Tu, Ts (0, 0.1 and 0.3 μM each), and GTP (1 mM) at 37°C. The bars in the graphs are SDs of more than three independent experiments, and the data are presented as mean values ± SD.
In these analyses, even in the absence of translation factors and GTP, CdiA–CT could cleave tRNAGln to some extent in vitro (Figure 1A, B). We analyzed the steady-state kinetics of tRNA cleavage by CdiA–CT with or without Tu, Ts, and GTP. In the absence of Tu, Ts, and GTP, the apparent Km value of tRNAGln was 24.5 ± 10.2 μM, and kcat was 0.0068 ± 0.002 s–1. On the other hand, in the presence of Tu, Ts and GTP, the apparent Km value of tRNAGln was 14.4 ± 4.1 μM, and kcat was 0.020 ± 0.003 s–1 (Figure 1C). Although it is unclear what the apparent Km value of tRNAGln in the presence of Tu:GTP:Ts represents in this analysis, these results imply that a putative Tu:GTP:Ts complex increases the overall catalytic efficiency of processing catalyzed by CdiA–CT.
As described above, CdiA–CT alone could cleave tRNA. Thus, the reaction conditions used for the measurement of tRNA cleavage by CdiA–CT were evaluated. Under lower pH conditions (i.e. below 6.9) in the presence of Tu, Ts and GTP, CdiA–CT cleaved tRNA more efficiently than under neutral or basic pH conditions (Figure 1D). Furthermore, even in the absence of Tu, Ts and GTP, under more acidic conditions (pH 5.7 and 5.3), CdiA–CT alone could cleave tRNA to almost the same extent as it could in the presence of Tu, Ts, and GTP (Figure 1D). To examine the promotion of tRNA cleavage by CdiA–CT in the presence of translation factors under acidic conditions, we evaluated the effects of Tu, Ts, and GTP on tRNA cleavage by CdiA–CT. The tRNA cleavage level by CdiA–CT at pH 5.3 was still elevated, but to a lesser extent, when Tu, Ts and GTP were present (Figure 1E). Together with the analysis of the substrate specificity of CdiA–CT using various tRNAs, described below (Figure 2), these observations suggest that the substrate specificity is governed by CdiA–CT itself and that the translation factors and GTP efficiently enhance tRNA cleavage by CdiA–CT at neutral pH. The mechanism underlying the increase in enzymatic activity of CdiA–CT under acidic conditions remains elusive. Under acidic conditions, the structure of the 3′-acceptor region of tRNA might be altered, allowing CdiA–CT to more efficiently cleave tRNAs as described below. Alternatively, the catalytic process might be accelerated by protonation of key catalytic residues, although the precise mechanism for such an effect remains unknown.
Figure 2.

Recognition elements in tRNA for cleavage by CdiA–CTEC869. (A) Nucleotide sequence and secondary cloverleaf structure of E. coli tRNAGln and tRNAAsn. The mutations introduced into the tRNAGln transcript are depicted on the left. (B) Time course of the cleavage of tRNAGln variants in (A) by CdiA–CTEC869 under neutral pH conditions (pH 7.4). tRNAGln transcript variants (1.0 μM) were incubated at 37°C with 0.2 μM CdiA–CTEC869 in the presence of Tu (0.2 μM), Ts (0.2 μM) and GTP (1 mM). The fraction of cleaved tRNA was quantified at the indicated time points as in (A). (C) Relative cleavage of tRNAGln variants by CdiA–CT in the presence of Tu, Ts, and GTP as in (B), at 37°C. Cleavage level of wild-type tRNAGln at 10 min was taken as 1.0. The error bars in the graphs represent SDs of at least three independent experiments, and the data are presented as mean values ± SD. (D) Comparison of nucleotide compositions at positions 1, 72, and 73 of 20 kinds of tRNAs in E. coli. (E) Cleavage of total tRNAs prepared from E. coli by CdiA–CT in vitro. Total tRNA mixtures (0.5 μg) were incubated at 37°C for 60 min with 0.5 μM CdiA–CTEC869 (20 μl reaction solution) in the presence of Tu (0.5 μM), Ts (0.5 μM) and GTP (1 mM). Cleavage of specific tRNAs was detected by northern blotting using DNA oligonucleotides specific for the corresponding tRNAs (tRNAAla, tRNAAsn, tRNAGln, tRNATrp, tRNAfMet and tRNAmMet).
Substrate tRNA recognition by CdiA–CTEC869in vitro
CdiA–CT reportedly cleaves tRNAGln and tRNAAsn between positions 71 and 72 (15). To clarify CdiA–CT recognition sites in the substrate tRNAs, we prepared tRNAGln transcript variants (Figure 2A) based on comparison with the tRNAAsn sequence, and then their cleavage by CdiA–CT was examined in vitro. Under standard conditions (pH 7.4), in the presence of Tu, Ts and GTP, mutant tRNAGln in which the discriminator G73 was replaced with A73 or U73 could not be efficiently cleaved by CdiA–CT (Figure 2B, C), suggesting that the discriminator at position 73 is a strong determinant of CdiA–CT recognition of substrate tRNAs. A tRNAGln variant with a G1–C72 base pair at the top of the acceptor stem was also inefficiently cleaved. On the other hand, tRNAGln variants with mis-pairings (A1–A72, G1–A72, U1–U72, U1–G72, U1–C72 or C1–A72) at the top of the acceptor stem were cleaved as efficiently as wild-type tRNAGln (Figure 2C). On the other hand, tRNAGln variants with the G1–U72 or A1–U72 mutation were less efficiently cleaved. Collectively, these observations suggest that the presence of the G73 discriminator, a mismatch, a weaker base pair at the top of the acceptor helix, and pyrimidine and purine bases at positions 1 and 72, respectively, are important for the cleavage of specific tRNAs by CdiA–CT. We also examined cleavage of tRNAGln variants by CdiA–CT alone under lower pH conditions (pH 5.7). The substrate specificity of CdiA–CT (Supplementary Figure S2B) was essentially identical to that observed under standard conditions (pH 7.4) in the presence of Tu, Ts, and GTP (Figure 2C). These results support the idea that CdiA–CT itself has an intrinsic ability to recognize and select specific tRNA substrates.
Next, we compared the nucleotide compositions at positions 1, 72 and 73 of all tRNAs from E. coli (Figure 2D, Supplementary Figure S3). Among the 20 kinds of isoaccepting tRNAs, tRNAGln and tRNAAsn have G73 and the weak U1–A72 base pair. Thus, both tRNAGln and tRNAAsn can be efficiently cleaved by CdiA–CT, as described above. tRNATrp has a weak A1–U72 base pair and G73, and initiator tRNAfMet has a C1–A72 mismatch but A73. We investigated whether tRNATrp and tRNAfMet (Supplementary Figure S4A) could be cleaved by CdiA–CT. Total E. coli tRNA was treated with CdiA–CT under the standard conditions (pH 7.4) in the presence of Tu, Ts, and GTP, and tRNA cleavage was detected by northern hybridization. The results revealed that in addition to tRNAGln and tRNAAsn, tRNATrp and tRNAfMet were also cleaved in vitro (Figure 2E). Analysis of the cleavage sites in tRNAfMet and tRNATrp revealed that both tRNAfMet and tRNATrp were cleaved between positions 71 and 72 as tRNAGln yielding terminal 2′-3′-cyclic phosphate ends (Supplementary Figure S4B–D). While tRNATrp was cleaved almost as efficiently as tRNAGln, tRNAfMet was less efficiently cleaved than tRNAGln by CdiA–CT in the presence of translation factors in vitro (Figure 2E, Supplementary Figure S4E).
Specific aa-tRNAs are reduced by CdiA–CTEC869 expression in vivo
Our in vitro analyses suggested that CdiA–CT targets several tRNAs, including tRNAGln, tRNAAsn, tRNATrp and tRNAfMet (Figure 2E). The cleavage of tRNAs results in a shortage of aa-tRNAs and ultimately inhibits protein synthesis in the cell.
We analyzed the change in aa-tRNA levels upon expression of CdiA–CT in E. coli. CdiA–CT was expressed using the pBAD arabinose-inducible protein expression system. CdiA–CT expression in E. coli was toxic and suppressed cell growth on agar plates and in liquid media (Figure 3A, B). After induction of CdiA–CT in E. coli, we prepared aa-tRNAs under acidic conditions to prevent their deacylation. Immediately afterward, the α-NH2 groups of the aminoacyl bonds of aa-tRNAs were chemically acetylated by acetic anhydride, converting aa-tRNAs to stable acetyl aa-tRNAs (Ac-aa-tRNAs) (37). Finally, Ac-aa-tRNAs were hydrolyzed with RNase I, and the amounts of Ac-aa-A76 were quantified by LC/MS (Figure 3C) (32,33,38). Thus, the cellular levels of each aa-tRNA could be inferred from the amount of the corresponding Ac-aa-A76 fragment.
Figure 3.

Changes in aa-tRNA levels upon CdiA–CTEC869 induction in vivo. (A) Growth inhibition by CdiA–CTEC869 induction. Overnight cultures of E. coli MG1655 transformed with pBAD33 (control) and pBAD33_CdiA–CTEC869 were serially diluted, and the dilutions were spotted on LB agar plates containing 50 μg/ml chloramphenicol and supplemented with 1% (w/v) arabinose (lower panel) or 1% (w/v) glucose (upper panel). (B) Growth curves of E. coli MG1655 transformed with pBAD33 and pBAD33_CdiA–CTEC869. Induction of CdiA–CTEC869 suppresses the growth of E. coli harboring pBAD33_CdiA–CTEC869 in LB containing 50 μg/ml chloramphenicol. When the OD660 reached ∼0.3, arabinose was added (final concentration 0.02%) to the medium, and the culture was continued at 37°C. The arrow in the graph indicates the point of CdiA–CTEC869 induction by arabinose addition. (C) Schematic diagram of quantification and comparison of relative amounts of each aa-tRNA by LC/MS. (D) LC/MS analysis of RNase I–digested fragments of Ac-aa-tRNAs (or formyl-methionyl-tRNAfMet, fMet-A76) prepared from aa-tRNAs from E. coli with (pBAD33_CdiA–CT) or without (pBAD33, control) induction of CdiA–CTEC869. The amount of each Ac-aa-A76 derived from aa-tRNA prepared from cells with or without CdiA–CTEC869 induction is expressed as the amount relative to Ac-Ala-A76 in cells with or without induction of CdiA–CTEC869, respectively. (E) Change of relative amounts of each aa-tRNA (or fMet-tRNAfMet) in E. coli after CdiA–CTEC869 induction. The relative amounts of each aa-tRNA in E. coli with CdiA–CTEC869 induction were normalized against the amount of the corresponding aa-tRNA in E. coli without induction of CdiA–CTEC869. Error bars represent SDs of more than three independent experiments, and the data are presented as mean values ± SD. (F) Cleavage of each tRNA in E. coli was also evaluated by northern blotting of tRNAs (0.125 μg) prepared from E. coli with (+) or without (-) induction of CdiA–CTEC869.
Because tRNAAla is not cleaved by CdiA–CT in vitro (Figure 2E) or in vivo, as described below, the relative amounts of individual aa-tRNAs with or without induction of CdiA–CT expression were normalized against the relative amount of Ala-tRNAAla with or without induction of CdiA–CT expression, respectively (Supplementary Figure S5). LC/MS analyses of RNase I–digested RNAs prepared from E. coli in which CdiA–CT expression had been induced demonstrated that the amounts of Ac-Gln-A76 (m/z = 438.17), Ac-Asn-A76 (m/z = 424.16), and Ac-Trp-A76 (m/z = 496.19) were reduced to <1–5% of those detected when CdiA–CT was not induced (Figure 3D, E). Thus, the amounts of Gln-tRNAGln, Asn-tRNAAsn, and Trp-tRNATrp in cells were decreased by the action of CdiA–CT. Although the amount of Ac-Met-A76 (m/z = 441.15) was not altered by induction of CdiA–CT, the amount of formyl-Met-A76 (fMet-A76, m/z = 427.14) was reduced to ∼40% of that in the absence of CdiA–CT induction (Figure 3D, E). Thus, the amount of the initiator N-formyl-Met-tRNAfMet was decreased by the action of CdiA–CT. These results are consistent with the in vitro data showing that in addition to tRNAGln and tRNAAsn, tRNATrp and tRNAfMet were cleaved (Figure 2E). Furthermore, we prepared tRNAs from E. coli in which CdiA–CT expression had been induced and performed northern blots to monitor tRNA cleavage. Consistent with the in vitro analyses (Figure 2E) and in vivo aa-tRNA level analyses (Figure 3E), tRNAGln, tRNAAsn, tRNATrp and tRNAfMet were cleaved by the action of CdiA–CT in vivo (Figure 3F).
The reduction of the amounts of fMet-tRNAfMetin vivo was moderate (∼40%) relative to the reductions in the levels of Gln-tRNAGln, Asn-tRNAAsn and Trp-tRNATrp (Figure 3E). Consistent with this result, cleavage of intracellular tRNAfMet was not as efficient as that of tRNAGln, tRNAAsn, and tRNATrp (Figures 2E, 3F, Supplementary Figure S4E), because tRNAfMet carries a less favorable A residue at position 73 (Figure 2D). The unique C1–A72 mismatch at the top of the acceptor stem of tRNAfMet would be the main determinant of the cleavage by CdiA–CT. Taken together, these observations indicate that tRNAGln, tRNAAsn and tRNATrp are the primary targets of CdiA–CT. When these specific tRNAs are cleaved, protein synthesis should be blocked.
Both uncharged and aminoacylated tRNAs are cleaved by CdiA–CTEC869 in the presence of translation factors
In living cells, more than ∼80% of tRNAs are aminoacylated, and the small amount of uncharged tRNAs resides in the ribosome E-site (46). The affinity between aa-tRNA and Tu:GTP is stronger than that between uncharged tRNA and Tu:GTP (47). Hence, we investigated whether aa-tRNA is more efficiently cleaved than tRNA (i.e. uncharged tRNA) by CdiA–CT in the presence of Tu, Ts, and GTP in vitro.
To this end, before CdiA–CT–mediated cleavage in the presence of Tu, Ts and GTP, tRNAGln was aminoacylated by glutamine-tRNA synthetase. Unexpectedly, prior aminoacylation of tRNAs did not significantly increase the tRNA cleavage level by CdiA–CT, irrespective of whether Tu, Ts, and GTP were present or absent (Figure 4A, left and middle graphs, Supplementary Figure S6). Thus, the aminoacyl moieties of substrate tRNAs are not required for the enhancement of tRNA cleavage by CdiA–CT in the presence of Tu, Ts, and GTP in vitro. To further verify this observation, instead of using wild-type Tu, a Tu variant with Ala substitution of His66 in domain I (H66A) was used in the assays. His66 interacts with the side chain of the aminoacyl moiety of aa-tRNAs in the Tu:GTP:aa-tRNA complex (48). The His66Ala mutation of Tu neither affected in vitro cleavage of Gln-tRNAGln (Figure 4A, right) nor that of deacylated tRNAGln as described below. These results indicate that both, aa-tRNAs and uncharged tRNAs, are targets of CdiA–CT in vivo. These results also imply that the enhancement of tRNA and aa-tRNA cleavage by CdiA–CT in the presence of translation factors does not proceed via Tu:GTP:aa-tRNA complex as described below.
Figure 4.

Involvement of RNA-binding residues in Tu or the coiled-coil domain of Ts. (A) Time courses of tRNAGln and Gln-tRNAGln cleavage by CdiA–CTEC869 in the presence (left) or absence (middle) of Tu, Ts, and GTP at 37°C. For cleavage of Gln-tRNAGln, 1.0 μM of tRNAGln was aminoacylated by GlnRS (1.0 μM) for 60 min before cleavage by CdiA–CTEC869 (0.1 μM) in the absence or presence of translation factors (0.1 μM each) and GTP (1 mM) at pH 7.4. Under this condition, tRNAGln was aminoacylated to the level of 925 pmol/A260. Time courses of Gln-tRNAGln cleavage by CdiA–CTEC869 in the presence of wild-type or H66A mutant Tu (plus Ts, and GTP) (right). (B) Interaction of aa-tRNA with Tu (PDB ID: 1OB2). Domains I, II, and III of Tu are colored cyan, yellow, and green, respectively. Mutated residues are colored red. tRNA is shown as a gray stick model. The nucleotide numbers of tRNA are circled, and the site cleaved by CdiA–CTEC869 is depicted by an arrow. (C) Relative tRNAGln cleavage by CdiA–CTEC869 at 10 min in the presence of a Tu variant, Ts, and GTP. The tRNAGln transcript (1.0 μM) was incubated at 37°C for 10 min with 0.2 μM CdiA–CTEC869 in the presence of a Tu variant (0.2 μM), Ts (0.2 μM), and GTP (1 mM). (D) Crystal structure of the Tu:Ts complex (50). Tu and Ts are colored yellow and green, respectively. The coiled-coil domain in Ts is colored orange. (E) Relative tRNAGln cleavage by CdiA–CTEC869 at 10 min in the presence of Tu, a Ts variant, and GTP. The tRNAGln transcript (1.0 μM) was incubated at 37°C for 10 min with 0.2 μM CdiA–CTEC869 in the presence of Tu (0.2 μM), a Ts variant (0.2 μM), and GTP (1 mM). The tRNAGln cleavage level by CdiA–CTEC869 in the presence of Tu, Ts, and GTP was defined as 1.0 in (C) and (E). The bars in the graphs are SDs of more than three independent experiments, and the data are presented as mean values ± SD.
Involvement of Tu amino acid residues in tRNA cleavage
To evaluate whether the amino acid residues in Tu that are involved in the interactions with aa-tRNA in Tu:GTP:aa-tRNA complexes (Figure 4B) also participate in the promotion of tRNA cleavage by CdiA–CT, we prepared variant Tu proteins (Supplementary Figure S7A) and monitored the increase/decrease in tRNA cleavage by CdiA–CT in the presence of these variants (plus Ts and GTP). Ala substitutions of Phe218, Glu259 or Arg262 in domain II which interacts with the 3′-acceptor region of aa-tRNAs in Tu:GTP:aa-tRNA decreased the level of tRNA cleavage by CdiA–CT to that observed in the absence of Tu (Figure 4C). The Ala mutation of Arg288, which interacts with the 5′-phosphate of the tRNA in Tu:GTP:aa-tRNA, also decreased tRNA cleavage to the level observed in the absence of Tu (Figure 4C). However, Ala substitution of His66 in domain I, which interacts with the side chain of the aminoacyl moiety of an aa-tRNA in Tu:GTP:aa-tRNA, did not affect tRNA cleavage by CdiA–CT (Figure 4C). Mutations in Arg318 and His319 in domain III, which interact with the TΨC loop of tRNA, decreased the level of tRNA cleavage by CdiA–CT to ∼60% to that observed in the presence of wild-type Tu. Ala mutation of His84 in domain I, a catalytic residue of the GTPase center, increased the level of tRNA cleavage by CdiA–CT (Figure 4C). Tu with the H84A mutation might easily adopt the proper GTP-bound conformation required for enhancement of tRNA cleavage by CdiA–CT. Although the H84A mutation increased the tRNA cleavage level by CdiA–CT, GTP and Ts were still required for efficient cleavage of tRNA by CdiA–CT (Supplementary Figure S8). Ala mutation of Arg58 in domain I, which interacts with the phosphate of the nucleotide at position 2 of tRNA, decreased the RNA cleavage level by CdiA–CT to ∼60% of that in the presence of wild-type Tu (Figure 4C).
It should be noted that Tu mutations at residues (Phe218, Glu259, or Arg262) in domain II reduced the tRNA cleavage level by CdiA–CT (Figure 4C) in the presence of translation factors, while mutation of His66 in domain I did not. As described below, in the determined structure of CdiA–CT:Tu, these residues (Phe218, Glu259, or Arg262) in domain II of Tu are involved in the interaction between CdiA–CT and Tu, while His66 is not. Thus, the domain II mutations would decrease the tRNA cleavage level by CdiA–CT via reducing the interaction between Tu and CdiA–CT, rather than by decreasing the binding affinity of aa-tRNA (tRNA) to Tu. This may explain why tRNA cleavage by CdiA–CT in the presence of translation factors is independent of the aminoacyl status of the substrate tRNA (Figure 4A) and is not affected by the H66A mutation in domain I of Tu (Figure 4A, C).
Involvement of the Ts coiled-coil domain in tRNA cleavage
Ts is a GEF for GDP-bound Tu that displaces the GDP and recycles Tu to the GTP form. Hence, we investigated whether the presence of excess Tu and GTP might promote tRNA cleavage by CdiA–CT, in which case Ts would no longer be required for promotion of cleavage. However, excess Tu (2.0 μM, 10-fold molar excess relative to 0.2 μM CdiA–CT) did not promote tRNA cleavage by CdiA–CT without Ts to the level observed in the presence of 0.2 μM Tu and 0.2 μM Ts (Supplementary Figure S9). Thus, Ts has a direct role in promoting tRNA cleavage by CdiA–CT together with Tu, rather than participating indirectly via its GEF function.
A recent study showed that the coiled-coil domain of Ts is required for the promotion of tRNA cleavage by CdiA–CT in vitro and that the Arg219Pro and Ala202Glu mutations in the coiled-coil domain inhibit the CdiA–CTEC869–mediated growth inhibition pathway in vivo (15). Notably, the coiled-coil domain contains several basic residues, Lys207, Lys209, Lys214, Arg219, Lys221 and Lys222 (Figure 4D, PDB ID: 4PC2). To evaluate the involvement of these basic residues in the promotion of tRNA cleavage by CdiA–CT, we prepared Ts variants (Supplementary Figure S7B). We examined the promotion of tRNA cleavage by CdiA–CT in the presence of the Ts variants (plus Tu and GTP). Triple Ala mutations at Arg219, Lys221 and Lys222 of Ts decreased the tRNA cleavage level by CdiA–CT to the same extent as Ala202Glu, Arg219Pro, or the coiled-coil deletion mutant (EF-Ts_ΔCC) (Figure 4E). The Lys207Ala–Lys209Ala and Lys214Glu Ts mutants also decreased tRNA cleavage level by CdiA–CT to ∼50% of the level in the presence of wild-type Ts. These results suggest that the basic residues in the coiled-coil domain of Ts are important for tRNA cleavage by CdiA–CT in the presence of translation factors and that the coiled-coil domain of Ts is involved in the interaction with tRNA during the reaction.
Tethering of Ts to Tu promotes tRNA cleavage by CdiA–CTEC869
The results described above suggest that both Tu and Ts are likely to interact with tRNA during the tRNA cleavage by CdiA–CT, and we assumed that the Tu:GTP:Ts complex and CdiA–CT would interact with substrate tRNA during cleavage of substrate tRNAs.
We examined whether artificial tethering of Ts to Tu promotes tRNA cleavage by CdiA–CT. To this end, we fused Ts with Tu and prepared single-chain Tu-Ts (sg–Tu–Ts) (Figure 5A). sg–Tu–Ts was previously demonstrated to be functional in the Qβ replicase system (49). We then tested the promotion of tRNA cleavage by CdiA–CT in the presence of sg–Tu–Ts and GTP. In the presence of GTP, the sg–Tu–Ts increased the tRNA cleavage level by CdiA–CT to a very similar extent as in the presence of separate Tu and Ts (Figure 5B, C). By contrast, mutant sg–Tu–Ts lacking the coiled-coiled domain of Ts (sg–Tu–Ts_ΔCC) did not promote tRNA cleavage by CdiA–CT (Figure 5B, C). To evaluate the tRNA-binding affinities of, CdiA–CT, Tu, and sg–Tu–Ts, we performed gel-shift assays at room temperature (∼25°C). We used catalytically inactive CdiA–CT with H281A mutation, CdiA–CT (H281A), instead of using wild-type CdiA–CT, because tRNA could be cleaved during the pre-incubation of tRNA with wild-type CdiA–CT. sg–Tu–Ts efficiently interacted with tRNA, whereas sg–Tu–Ts_ΔCC did not (Figure 5D, E). The estimated Kd value of tRNA for sg–Tu–Ts was 4.7 μM, and the Kd value of tRNA for sg–Tu–Ts_ΔCC was >>30 μM (Figure 5E). The Kd value of tRNA for Tu was >>30 μM under the same condition. The Kd value of tRNA for mutant CdiA–CT (H281A) was >30 μM. sg–Tu–Ts is functionally equivalent to the Tu:Ts complex in terms of enhancement of tRNA cleavage by CdiA–CT in vitro (Figure 5B, C). These results suggest that the Tu:Ts complex promotes the tRNA cleavage activity of CdiA–CT in the presence of GTP, and that Ts interacting with Tu has a direct role in enhancement of tRNA cleavage by CdiA–CT.
Figure 5.

Formation of CdiA–CT:Tu:Ts:aa-tRNA (tRNA) in the presence of GTP. (A) Schematic diagram of single-chain Tu-Ts (sg–Tu–Ts). The stop codon of Ts gene (tsf) was changed to a histidine codon (CAT), and the Ts gene is followed by Tu (tufB) gene in the same direction. (B) sg–Tu–Ts promotes tRNAGln cleavage by CdiA–CT. tRNAGln (1 μM) was incubated at 37°C with 0.1 μM CdiA–CT in the presence of Tu (0.1 μM), Ts (0.1 μM) or sg–Tu–Ts (0.1 μM) and GTP (1 mM). The fraction of cleaved tRNA was quantified at the indicated time points. (C) Relative tRNAGln cleavage by CdiA–CT at 5 min in the presence of Tu and Ts or sg–Tu–Ts (sg–Tu–Ts_ΔCC) and GTP at 37°C as in (B). The tRNAGln cleavage level by CdiA–CTEC869 in the presence of wild-type Tu, Ts and GTP was defined as 1.0. (D) Gel-shifts of tRNAGln by various amounts of CdiA–CT, Tu, sg–Tu–Ts, and sg–Tu–Ts_ΔCC (0–30 μM). The two complexes observed in the binding of sg–Tu–Ts to tRNA represent alternative binding modes. (E) tRNA fraction bound to each protein in (D). (F) A model of tRNA docking onto the Tu:GDPNP:Ts complex (50). Tu and Ts are colored yellow and green, respectively. The coiled-coil domain of Ts is colored orange and the tRNA is colored gray. The Tu:GTP:aa-tRNA structure (PDB ID: 1OB2) was superimposed onto the structure of Tu:GDPNP:Ts (PDB ID: 4PC7). For clarity, Tu:GTP in Tu:GTP:aa-tRNA is omitted. (G) His-tagged CdiA–CT was mixed with Tu or Tu and Ts in the presence of tRNA or aa-tRNA and GTP, and the mixture was loaded onto Ni-NTA column. The column was washed, and finally, CdiA–CT was eluted from the column. The quaternary complex of CdiA–CT:Tu:Ts:tRNA(aa-tRNA) was formed. Proteins were visualized by SDS PAGE stained with CBB, and tRNA was visualized by PAGE stained with ethidium bromide. (H) His-tagged Tu was mixed with aa-tRNA or tRNA in the absence (left) or presence (middle) of Ts and GTP, and the mixture was loaded onto a Ni-NTA column. The column was washed, and finally, Tu was eluted from the column. His-tagged sg–Tu–Ts was also mixed with aa-tRNA or tRNA in the same manner, and sg–Tu–Ts was eluted from the column (right). Proteins and RNA were visualized as in (G). The error in the graphs in (C) and (E) represent SDs of at least three independent experiments, and data are presented as average values ± SD.
In the Tu:GTP:Ts structure (50) (PDB ID: 4PC7), the tRNA-binding regions in Tu domains II and III are available for interactions with the acceptor stem of the tRNA, and the Tu binding sites for Ts and tRNA are not mutually exclusive. Previously, the structure of the Tu:GTP:aa-tRNA ternary complex (PDB ID: 1OB2) was superposed onto that of the Tu:GTP:Ts ternary complex, and the Tu:GTP:Ts:aa-tRNA complex was modeled (Figure 5F) (50). In the model, the coiled-coil domain of Ts is proximal to the elbow region of tRNA, explaining how Ts in Tu:GTP:Ts contributes to the interaction with aa-tRNA (tRNA).
Formation of the CdiA–CT:Tu:Ts:aa-tRNA(tRNA) complex
To obtain further mechanistic insight into the requirement of both Tu and Ts in efficient cleavage of tRNAs by CdiA–CT, we performed pull-down assays to investigate whether Tu, Ts, and CdiA–CT simultaneously interact with tRNAGln (or Gln-tRNAGln) (Figure 5G, Supplementary Figure S10).
Under the condition where aa-tRNA, but not uncharged tRNA, was pulled down with Tu in the presence of GTP (Figure 5H, left), CdiA–CT alone did not significantly pull-down tRNA or aa-tRNA (Figure 5G, left). Even in the presence of Tu and GTP, CdiA–CT did not pull-down tRNA or aa-tRNA, but did pull-down Tu (Figure 5G, middle), indicating that CdiA–CT:Tu:GTP:aa-tRNA (tRNA) is not formed. While only aa-tRNA, but not tRNA, was pulled down with Tu and Ts (or sg–Tu–Ts) (Figure 5H, middle and right), both tRNA and aa-tRNA were pulled down with CdiA–CT in the presence of Tu, Ts, and GTP with almost the same efficiency, together with Tu and Ts (Figure 5G, right). The interaction with tRNA was enhanced in the presence of GTP (Supplementary Figure S10B) and was dependent on the coiled-coil domain of Ts (Supplementary Figure S10C). Similar results were also obtained when sg–Tu–Ts and sg–Tu–Ts_ΔCC were used for pull-down assays. tRNA was pulled down with CdiA–CT and sg–Tu–Ts, but not with CdiA–CT and sg–Tu–Ts_ΔCC (Supplementary Figure S10D). Further, in the absence of tRNA, CdiA–CT pulled down Tu, but not Ts (15) (Supplementary Figure S10A), suggesting that CdiA–CT interacts with Tu even in the absence of tRNA.
Altogether, these observations suggest that binding of CdiA–CT to Tu:GTP:Ts cooperatively increases the aa-tRNA (tRNA) binding affinity and the quaternary complex of CdiA–CT:Tu:Ts:aa-tRNA(tRNA) can be efficiently formed in the presence of GTP. Notably, formation of the quaternary complex, CdiA–CT:Tu:Ts:aa-tRNA(tRNA), is independent of the aminoacyl status of tRNAs.
Structure of CdiA–CT:CdiIEC869 in complex with Tu
To determine the molecular basis of the interaction between CdiA–CT and Tu, we crystallized CdiA–CT:CdiIEC869 in complex with Tu and determined the structure (Table 1, Supplementary Figure S11), as the CdiA–CT:CdiIEC869 complex interacts more tightly with Tu than CdiA–CT in vitro (Supplementary Figure S10A).
In the structure of CdiA–CT:CdiIEC869 in complex with Tu, the CdiA–CT toxin interacts with domain II of Tu on one side and with CdiI on the other side (Figure 6A). The CdiA–CT toxin domain consists of an N-terminal three-stranded anti-parallel β-sheet (β1- β3) and a C-terminal three-stranded anti-parallel β-sheet (β4 - β6). These two β-sheets are flanked by two α-helices (α2 and α3). The N-terminal sheet interacts with the C-terminal sheet in a parallel orientation via β3 and β6, and the N-terminal and C-terminal sheets together wrap around α3 (Figure 6B). The CdiIEC869 protein consists of an N-terminal six-stranded mixed β-sheet (β2–β3−β6−β5−β4−β9; β4−β9 is parallel) and a C-terminal six-stranded mixed β-sheet (β7–β10–β11–β14–β13–β12; β7–β10 is parallel). These two are facing each other, thus adopting a β-sandwich structure (Figure 6C). The β-sandwich structure is supported by the surrounding anti-parallel β-sheet (β1–β8) and α-helix (α2).
Figure 6.

Structure of CdiA–CT:CdiIEC869 complexed with Tu. (A) Stereoview of the structure of the CdiA–CT:CdiI:Tu complex. CdiA–CTEC869, CdiIEC869 and Tu are colored cyan, magenta, and yellow, respectively. The modeled CdiA–CTEC869 contains residues 175–285 (numbered from Val1 of the VENN peptide motif, Supplementary Figure S13A), and the modeled CdiIEC869 contains residues 3–179. The modeled Tu contains residues 10–393. (B) Stereoview of CdiA–CTEC869. (C) Stereoview of CdiIEC869. (D) CdiA–CTEC869 adopts the BECR fold as observed in other CdiA–CTs targeting the 3′-acceptor region of tRNAs. CdiA–CTNC101 (blue) from E. coli NC101 (14), CdiA–CTKp342 (yellow) from Klebsiella pneumoniae, and CdiA–CTEC3006 (magenta) from E. coli 3006 (16). (E), (F) Interaction between CdiA–CT (cyan) and domain II of Tu (yellow). (G) Superimposition of the structure of CdiA–CTEC869:Tu onto that of CdiA–CTNC101:Tu (14). For clarity, the structures of domain I of Tu in both complex structures are omitted. CdiA–CTEC869 and domains II/III of Tu in the structure of CdiA–CTEC869:Tu are colored cyan and yellow, respectively. CdiA–CTNC101 and domains II/III of Tu in the structure of CdiA–CTNC101:Tu are colored blue and orange, respectively.
The overall structure of the CdiA–CT toxin domain is topologically homologous to those of other CdiA toxin domains from E. coli 3006 (CdiA–CTEC3006), E. coli NC101 (CdiA–CTNC101) (14) and Klebsiella pneumoniae 342 (CdiA–CTKp342) and adopts the Barnase/EndoU/Colicin/RelE (BECR) fold (16) (Figure 6D). All of these CdiA–CTs cleave the acceptor region of tRNAs (17). Although the structure of CdiA–CTEC869 is homologous to those of other CdiA–CTs with the BECR fold, the structures of the corresponding CdiIs are not homologous (Supplementary Figure S12A) (16). The interactions between CdiA–CTEC869 and CdiIEC869 are mainly mediated by hydrogen bonds (Supplementary Figure S12B, C). As described below, mutations His216 and His281 in CdiA–CT reduced the toxic activity of CdiA–CT in vivo, indicating that they are residues involved in catalysis and suggesting that CdiI inhibits the activity of CdiA–CT through masking the catalytic site of CdiA–CT, thereby preventing the 3′-portion of tRNA from binding to the catalytic pocket of CdiA–CT.
In this structure, Tu adopts the GDP-bound open form structure, although no GDP was bound to Tu. α3 of CdiA–CT is sandwiched between the β-barrel of the domain II of Tu and the β-sheet of CdiA–CT (Figure 6A). CdiA–CT extensively interacts with the domain II of Tu (Supplementary Figure S12D, E). In particular, Phe218 and Glu259 interact with CdiA–CT. Phe218 in Tu stacks with the side chain of Met237 and also interacts with Tyr220, Leu221 and Tyr284 in CdiA–CT through hydrophobic interactions, thereby stabilizing the CdiA–CT:Tu interaction (Figure 6E). Glu259 in Tu forms a hydrogen bond with the Nϵ atom of the side chain of Gln235 (Figure 6F). Arg262 in Tu might also interact with the N-terminal region of CdiA–CT, although the N-terminal region of CdiA–CT is unclear in the present structure. Reduced cleavage of tRNA by CdiA–CT in the presence of Tu mutants, such as Phe218Ala and Glu259Ala (Figure 4C), would be due to the reduced interaction between Tu and CdiA–CT, rather than reduced binding of aa-tRNA (tRNA) to Tu.
The structure of CdiA–CTEC869:Tu was superposed onto that of CdiA–CTNC101:Tu (14) (Figure 6G). Although CdiA–CTNC101 and CdiA–CTEC869 exhibit low similarity at the primary amino acid sequence level, they adopt the BECR fold (Figure 6D), and both interact with domain II of Tu (16). The shorter α helix (α2) in CdiA–CTNC101, which corresponds to α3 of CdiA–CTEC869, is more snugly accommodated in the space between the β-barrel of Tu domain II and the β-sheet of CdiA–CTNC101. Further, the N-terminal β-strands (β1a) in CdiA–CTNC101 additionally constitute a β-sheet with the β-strands of domain II of Tu, but the corresponding interactions are not observed between CdiA–CTEC869 and domain II of Tu. Thus, the two structures differ in terms of interaction modes of CdiA–CTs and Tu domain II, resulting in dissimilar position and orientation of the six-stranded ß-sheet surfaces containing the catalytic site of CdiA–CT, relative to the surface of Tu domain II (Figure 6G). These differences may be the cause for the differential effects the translation factors exert on tRNA cleavage by CdiA–CTEC869 versus CdiA–CTNC101.
tRNA docking onto the CdiA–CT:Tu:Ts complex
We superimposed domains II and III of Tu in CdiA–CT:Tu complex structure onto those in the Tu:GTP:Ts:aa-tRNA complex and aa-tRNA was modeled onto the CdiA–CT:Tu:GTP:Ts structure (Figure 7A). In the superimposition, the 3′-acceptor part of the aa-tRNA sterically clashes with CdiA–CT. Thus, it is likely that the 3′-end of the aa-tRNA would adopt an alternative conformation to allow cleavage of the aa-tRNA between positions 71 and 72 by CdiA–CT (Figure 7B, C).
Figure 7.

Recognition of the 3′-acceptor region of tRNA by CdiA–CTEC869. (A) A model of tRNA docking onto the CdiA–CT:Tu:GTP:Ts structure. CdiA–CT, Tu, and Ts are colored cyan, yellow, and green, respectively. The coiled-coil domain of Ts is colored orange. tRNA is shown in a stick model (gray). Domains II and III of Tu in the CdiA–CT:Tu complex structure were superimposed onto those in the Tu:GTP:Ts:aa-tRNA complex (Figure 5F), and the CdiA–CT:Tu:GTP:Ts:aa-tRNA model structure was constructed. (B) Close-up view of interactions of 3′-acceptor region of tRNA with CdiA–CT and Tu in (A). The 3′ portion of the tRNA sterically clashes with CdiA–CT. The arrowhead indicates the relocation of 3′-portion of tRNA into the catalytic site for tRNA cleavage. (C) The electrostatic surface potential of CdiA–CT. The positively charged area (blue) resides on the opposite side to the interface between Tu Domain II and CdiA–CT. (D) Comparison of structures of CdiA–CTEC869 and CdiA–CTKp342 from Klebsiella pneumoniae. CdiA–CTEC869 and CdiA–CTKp342 are colored cyan and magenta, respectively. The key amino acid residues required for toxicity and tRNase activity in CdiA–CTKp342 (K157, Y160, R252 and T255) are shown as stick models (16). (E) Effects of mutations on toxicity of CdiA–CTEC869 expressed in E. coli MG1665, as in Figure 3A. LB agar plates containing 50 μg/ml chloramphenicol and supplemented with 0.5% (w/v) arabinose (right panel: +Arabinose) or with 1% (w/v) glucose (left panel: + Glucose). (F) Growth curves of E. coli MG1655 transformed with pBAD33, pBAD33_CdiA–CTEC869 and its variants in LB containing 50 μg/ml chloramphenicol and supplemented with 1% (w/v) arabinose.
The electrostatic potential of the surface area of CdiA–CT in the CdiA–CT:Tu complex showed that the positively charged residues are clustered on the opposite side of the interface between CdiA–CT and Tu (Figure 7C). At the reaction stage of the CdiA–CT–mediated cleavage of tRNA between positions 71 and 72, the α2 helix in CdiA–CT may unwind the top of the acceptor stem of tRNA, and the loop between β1 and β2 would change its orientation, causing the 3′-end of tRNA substrate to relocate to the positively charged area of CdiA–CT (Figure 7C). The structure of CdiA–CTEC869 is homologous to that of CdiA–CT from K. pneumoniae 342 CdiA–CTKp342 (Figure 6D), with an RMSD of 4.3 Å for 68 structurally equivalent residues (calculated by the Dali server (51); RMSD calculated with Cα atoms). In CdiA–CTKp342, Lys157Ala, Tyr160Ala, Arg252Ala and Thr255Ala mutations decrease its toxicity and tRNase activity in vivo (16), and these residues are predicted to be involved in catalysis as well as substrate recognition. The residues His216 and Tyr220 in CdiA–CTEC869 would correspond to Lys157 and Tyr160 in CdiA–CTKp342, respectively (Figure 7D). These residues would be proximal to the 3′-acceptor region of tRNA at the reaction stage (Figure 7B, C).
We examined whether the residues that contribute to the interaction with the 3′ region of the tRNA or catalysis are involved in the toxicity of CdiA–CTEC869 in E. coli. Mutation of His281 to alanine (H281A) or deletion of the C-terminal five amino acids (ΔC5, including H281; Supplementary Figure S13) decreased CdiA–CTEC869 toxicity when expressed in E. coli (Figure 7E, F). The substitution of His216 to Ala (H216A) or Arg271 to Ala (R271A) also decreased CdiA–CTEC869 toxicity. Arg263Ala and Tyr220Ala mutations did not decrease the toxicity significantly in liquid culture medium, but slightly attenuated the toxicity on agar plates (Figure 7E, F). These results are in line with the model that the 3′-acceptor region of tRNA interacts with the positively charged surface of CdiA–CT in cleavage-competent CdiA–CT:Tu:GTP:Ts:tRNA complexes. Although the catalytic residues involved in cleavage of tRNA between positions 71 and 72 have not been precisely identified, His216 and His281 are located proximal to the cleavage site in tRNA (Figure 7B, C) and might be involved in the catalysis.
DISCUSSION
CdiA–CTEC869 has been reported to specifically cleave tRNAGln and tRNAAsn in a reaction promoted by Tu, Ts, and GTP (Figure 1A) (15). In a recently proposed model of the requirement of translation factors for tRNA cleavage by CdiA–CTEC869 (14,15), CdiA–CT recognizes specific tRNAs in the context of the Tu:GTP:aa-tRNA complex, and Ts stimulates an increase in the steady-state level of Tu:GTP:aa-tRNA via formation of Tu:GTP:Ts:aa-tRNA complex, thereby facilitating recognition of Tu:GTP:aa-tRNA by CdiA–CTEC869 (14,52,53).
In this study, we showed that CdiA–CTEC869 itself has an intrinsic ability to select substrate tRNAs and cleave tRNATrp and tRNAfMet, in addition to the previously identified substrates tRNAGln and tRNAAsn, in vitro and in vivo (Figures 2E, 3E, F, Supplementary Figure S5). At neutral pH, both Tu and Ts are required for efficient tRNA cleavage by CdiA–CTEC869 (Figure 1). We also showed that the enhancement of tRNA cleavage by CdiA–CTEC869 in the presence of translation factors is independent of the aminoacyl status of tRNA, and that CdiA–CTEC869 cleaves uncharged tRNA and aa-tRNA to the same extent in vitro (Figure 4A, Supplementary Figure S6). In living cells, more than ∼80% of tRNAs are aminoacylated, and a small amount of uncharged tRNAs resides in the ribosome E-site (46). It is likely that primarily cleavage of aa-tRNAs, and to a smaller extent also cleavage of uncharged tRNAs, contribute to the CDI by CdiA–CTEC869in vivo.
We also showed sg–Tu–Ts, which tethers Ts to Tu and mimics the Tu:Ts complex, promotes tRNA cleavage by CdiA–CTEC869 to the same extent as in the presence of separate Tu and Ts proteins (Figure 5B). sg–Tu–Ts interacts with tRNA with a higher affinity than Tu alone, and the coiled-coil domain of Ts contributes to the higher affinity for tRNA (Figure 5E). Furthermore, CdiA–CTEC869 interacts with the domain II of Tu (Figure 6A) where the 3′-portion of aa-tRNA binds in Tu:GTP:aa-tRNA ternary complexes. CdiA–CTEC869, Tu, and Ts form a complex with tRNA and aa-tRNA in the presence of GTP in vitro (Figure 5G), and CdiA–CT:Tu:GTP:Ts:aa-tRNA (tRNA) complex formation is dependent on the coiled-coil domain of Ts (Supplementary Figure S10C, D).
Considering that cleavage of uncharged tRNA and aa-tRNA by CdiA–CTEC869 is enhanced by translation factors to the same extent, but aa-tRNA interacts with Tu:GTP and Tu:GTP:Ts with a much higher affinity than uncharged tRNA (Figure 5H), it is unlikely that tRNA cleavage by CdiA–CT in the presence of translation factors proceeds via formation of the Tu:GTP:aa-tRNA(tRNA) or Tu:GTP:Ts:aa-tRNA(tRNA) complex. Indeed, CdiA–CTEC869 interacts with the domain II of Tu where 3′-portion of aa-tRNA binds in Tu:GTP:aa-tRNA complex (Figure 6A).
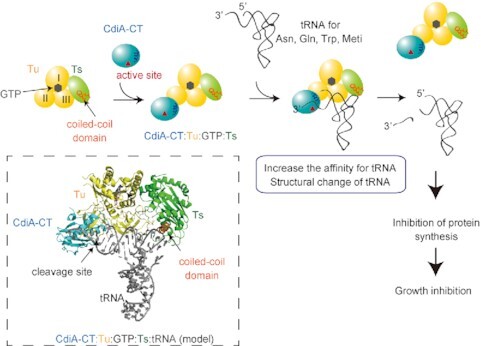
Thus, tRNA cleavage proceeds via the CdiA–CT:Tu:GTP:Ts:tRNA(aa-tRNA) complex (Figure 8). CdiA–CTEC869 delivered into cells is recruited to domain II of Tu in the Tu:GTP:Ts complex and forms CdiA–CT:Tu:GTP:Ts. Then, tRNA and aa-tRNA could be recognized by the CdiA–CT:Tu:GTP:Ts complex. This model explains why the enhancement of tRNA cleavage by CdiA–CT is unaffected by mutation of His66, but is reduced by other Tu mutations in domain II (Phe218Ala, Glu259Ala, and Arg262Ala). Mutations of these Tu residues in domain II would reduce the interaction between Tu and CdiA–CT and thus reduce the enhancement of tRNA cleavage by CdiA–CT in the presence of translation factors. The interaction between CdiA–CTEC869 and the Tu:GTP:Ts complex would increase the affinity of the CdiA–CT:Tu:GTP:Ts complex for aa-tRNA (and tRNA). The coiled-coil domain of Ts in CdiA–CT:Tu:GTP:Ts enhances the affinity of aa-tRNA (tRNA) toward the complex and anchors the aa-tRNA (tRNA) substrate onto the complex. tRNA anchoring would prevent ejection of the tRNA from the complex, as the tail domains of CCA-adding enzymes do (54,55), during the structural shift of the 3′-region to the productive form that enables catalysis by CdiA–CT. According to the mechanistic model, α2 in CdiA–CTEC869 unwinds the top of the acceptor stem of tRNAs and, together with a conformational change of the loop between β1 and β2, induce the relocation of the 3′-part of tRNA into the active site of CdiA–CTEC869 for tRNA cleavage (Figure 7B, C).
Figure 8.

Model of tRNA cleavage by CdiA–CTEC869 in the presence of translation factors. The Tu:GTP:Ts complex acts as a scaffold for tRNA cleavage by CdiA–CTEC869. First, CdiA–CTEC869 delivered into the cell is recruited to the Tu:GTP:Ts complex to form the CdiA–CT:Tu:GTP:Ts complex. Substrate aa-tRNA (or tRNA) is recognized by CdiA–CT:Tu:GTP:Ts and forms CdiA–CT:Tu:GTP:Ts:aa-tRNA(tRNA). Ts in the CdiA–CT:Tu:GTP:Ts complex increases the affinity of tRNA for the complex and induces a structural change in tRNA and/or CdiA–CT to promote productive catalysis by CdiA–CT. CdiA–CT might interact with Tu:GTP and form CdiA–CT:Tu:GTP, and aa-tRNA (tRNA) could be recognized by the complex. However, aa-tRNA (tRNA) cannot be cleaved by CdiA–CT due to the low affinity of aa-tRNA (tRNA) to the CdiA–CT:Tu:GTP complex. Association of Ts to CdiA–CT:Tu:GTP or association of CdiA–CT to Tu:GTP:Ts is required for aa-tRNA (tRNA) binding and cleavage by CdiA–CT. It is not clear whether CdiA–CT first associates with Tu:GTP and then Ts is recruited to form CdiA–CT:Tu:GTP:Ts or CdiA–CT associates with Tu:GTP:Ts to form CdiA–CT:Tu:GTP:Ts.
The model proposed here also explains why tRNAs with a weak base pair or mismatch at the top of the acceptor stem are favorable substrates (Figure 2), whereas the mechanism underlying the requirement for G73 and pyrimidine and purine bases at positions 1 and 72 remains elusive. Under acidic conditions, CdiA–CTEC869 cleaves its specific substrate tRNAs without assistance of translation factors (Figure 1D, Supplementary Figure S2). Although the precise underlying mechanism remains unknown, under acidic conditions, the structure of the 3′-acceptor stem of tRNA or CdiA–CT might be altered, allowing CdiA–CT to cleave tRNAs more efficiently. The precise dynamical mechanisms underlying the promotion of tRNA cleavage by CdiA–CT in the presence of translation factors and the mechanisms of tRNA cleavage under acidic conditions by CdiA–CT alone await further structural determination of CdiA–CT in complex with translation factors and tRNAs.
CdiA–CTEC869, CdiA–CTNC101, CdiA–CTEC3006 and CdiA–CTKp342 exhibit low similarity at the primary amino acid sequence level. However, the structures of CdiA–CTNC101, CdiA–CTKp342, CdiA–CTEC869 and CdiA–CTEC3006 are homologous and adopt the BECR fold (Figure 6D), which includes the structure of the RNase domain of colicin D (16,56,57). CdiA–CTEC869, CdiA–CTNC101, CdiA–CTEC3006 and CdiA–CTKp342 cleave specific tRNAs in their acceptor stems (14–17). CdiA–CTNC101 mainly cleaves tRNAAsp and tRNAGlu between nucleotide positions 72 and 73, CdiA–CTKp342 cleaves tRNAIle between nucleotide positions 71 and 72, and CdiA–CTEC3006 cleaves tRNAIle between positions 70 and 71. For both CdiA–CTNC101 and CdiA–CTKp342, as well as CdiA–CTEC869, specific tRNA cleavage is promoted in the presence of Tu, Ts, and GTP in vitro (14–16), and tsf mutants are resistant to growth inhibition by these CdiA–CTs. On the other hand, CdiA–CTEC3006 does not require Tu, Ts, or GTP for the cleavage of specific tRNAs, in vitro and possibly also in vivo (16). Notably, CdiA–CTEC3006 has an additional α-helical domain (α2, α3 and α 4) (Figure 6D). Perhaps the α-helical domain in CdiA–CTEC3006 interacts with the tRNA substrate together with the catalytic domain, allowing CdiA–CTEC3006 to efficiently cleave substrate tRNAs without the aid of translation factors (17).
As shown in this study, the cleavage of tRNAs by CdiA–CTEC869 is not affected by the aminoacyl status of substrate tRNA in vitro (Figure 4A); however, CdiA–CTKp342 and CdiA–CTEC3006 cleave the 3′-acceptor region of uncharged tRNAs rather than aa-tRNAs in vitro (16). CdiA–CTKp342 and CdiA–CTEC3006 target the small amount of uncharged tRNAs in the ribosome E-site (46) in vivo. The basic mechanism underlying tRNA cleavage by CdiA–CTEC869, CdiA–CTNC101 and CdiA–CTKp342 in the presence of translation factors seems to be conserved. However, the specificity for certain tRNA substrates, the individual cleavage sites in the target tRNAs, and the recognition of the aminoacyl status of tRNA 3′-end are determined by the properties of the respective CdiA–CT variants. While the binding site for the 3′-moiety of tRNA on CdiA–CTEC869 is tolerant to the aminoacylation status, the corresponding binding sites for the tRNA 3′ -moiety on CdiA–CTKp342 and CdiA–CTEC3006 are not. Thus, CdiA–CTKp342 and CdiA–CTEC3006 cleave uncharged tRNAs, but not aa-tRNAs (16). The detailed molecular basis of substrate recognition by these CdiA–CTs awaits further structural determination of CdiA–CTs in complex with translation factors and tRNAs. Comparative analyses of CdiA–CTs that cleave 3′-acceptor regions of specific tRNAs will provide detailed views of the mechanism and evolution of these CdiA toxin families.
DATA AVAILABILITY
Coordinates and structure factors for the crystal structure of CdiA–CT:CdiIEC869:Tu have been deposited to PDB under the accession number, 7VMC.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the beamline staff of BL-17A (KEK, Tsukuba) for technical assistance during data collection. We thank Yuka Fujimoto for assistance with plasmid construction and protein purifications.
Contributor Information
Jing Wang, Department of Computational Biology and Medical Sciences, Graduate School of Frontier Sciences, The University of Tokyo, Kashiwa, Chiba, 277-8562, Japan.
Yuka Yashiro, Department of Computational Biology and Medical Sciences, Graduate School of Frontier Sciences, The University of Tokyo, Kashiwa, Chiba, 277-8562, Japan.
Yuriko Sakaguchi, Department of Chemistry and Biotechnology, Graduate School of Engineering, The University of Tokyo, Bunkyo-ku, Tokyo 113-8656, Japan.
Tsutomu Suzuki, Department of Chemistry and Biotechnology, Graduate School of Engineering, The University of Tokyo, Bunkyo-ku, Tokyo 113-8656, Japan.
Kozo Tomita, Department of Computational Biology and Medical Sciences, Graduate School of Frontier Sciences, The University of Tokyo, Kashiwa, Chiba, 277-8562, Japan.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Grants-in-Aid for Scientific Research [18H03980 to K.T., 18H05272 to T.S.]; Grant-in-Aid for Scientific Research on Innovative Areas [26113002 to K.T.] from JSPS; Exploratory Research for Advanced Technology (ERATO) [JPMJER2002 to T.S.] grant from JST; Japan Foundation for Applied Enzymology (to K.T.); Toyo Suisan Foundation; Tojuro Iijima Foundation for Food Science and Technology; Naito Foundation; Kobayashi Foundation; Takahashi Industrial and Economic Research Foundation. Funding for open access charge: Grant-in-Aid for Scientific Research (A) from JSPS (to K.T.).
Conflict of interest statement. None declared.
REFERENCES
- 1. Aoki S.K., Diner E.J., de Roodenbeke C.T., Burgess B.R., Poole S.J., Braaten B.A., Jones A.M., Webb J.S., Hayes C.S., Cotter P.A. et al. A widespread family of polymorphic contact-dependent toxin delivery systems in bacteria. Nature. 2010; 468:439–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ruhe Z.C., Low D.A., Hayes C.S. Polymorphic toxins and their immunity proteins: diversity, evolution, and mechanisms of delivery. Annu. Rev. Microbiol. 2020; 74:497–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hayes C.S., Koskiniemi S., Ruhe Z.C., Poole S.J., Low D.A. Mechanisms and biological roles of contact-dependent growth inhibition systems. Cold Spring Harbor Perspect. Med. 2014; 4:a010025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ruhe Z.C., Low D.A., Hayes C.S. Bacterial contact-dependent growth inhibition. Trends Microbiol. 2013; 21:230–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Willett J.L., Ruhe Z.C., Goulding C.W., Low D.A., Hayes C.S. Contact-Dependent growth inhibition (CDI) and CdiB/CdiA two-partner secretion proteins. J. Mol. Biol. 2015; 427:3754–3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ruhe Z.C., Subramanian P., Song K., Nguyen J.Y., Stevens T.A., Low D.A., Jensen G.J., Hayes C.S. Programmed secretion arrest and receptor-triggered toxin export during antibacterial contact-dependent growth inhibition. Cell. 2018; 175:921–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ruhe Z.C., Wallace A.B., Low D.A., Hayes C.S. Receptor polymorphism restricts contact-dependent growth inhibition to members of the same species. Mbio. 2013; 4:e00480-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Aoki S.K., Pamma R., Hernday A.D., Bickham J.E., Braaten B.A., Low D.A. Contact-dependent inhibition of growth in escherichia coli. Science (New York, N.Y.). 2005; 309:1245–1248. [DOI] [PubMed] [Google Scholar]
- 9. Morse R.P., Nikolakakis K.C., Willett J.L.E., Gerrick E., Low D.A., Hayes C.S., Goulding C.W. Structural basis of toxicity and immunity in contact-dependent growth inhibition (CDI) systems. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:21480–21485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Diner E.J., Beck C.M., Webb J.S., Low D.A., Hayes C.S. Identification of a target cell permissive factor required for contact-dependent growth inhibition (CDI). Genes Dev. 2012; 26:515–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Johnson P.M., Beck C.M., Morse R.P., Garza-Sánchez F., Low D.A., Hayes C.S., Goulding C.W. Unraveling the essential role of CysK in CDI toxin activation. Proc. Nat. Acad. Sci. U.S.A. 2016; 113:9792–9797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kaundal S., Uttam M., Thakur K.G. Dual role of a biosynthetic enzyme, CysK, in contact dependent growth inhibition in bacteria. PLoS One. 2016; 11:e0159844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Benoni R., Beck C.M., Garza-Sánchez F., Bettati S., Mozzarelli A., Hayes C.S., Campanini B. Activation of an anti-bacterial toxin by the biosynthetic enzyme cysk: mechanism of binding, interaction specificity and competition with cysteine synthase. Sci. Rep. 2017; 7:8817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Michalska K., Gucinski G.C., Garza-Sanchez F., Johnson P.M., Stols L.M., Eschenfeldt W.H., Babnigg G., Low D.A., Goulding C.W., Joachimiak A. et al. Structure of a novel antibacterial toxin that exploits elongation factor tu to cleave specific transfer RNAs. Nucleic Acids Res. 2017; 45:10306–10320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jones A.M., Garza-Sánchez F., So J., Hayes C.S., Low D.A. Activation of contact-dependent antibacterial tRNase toxins by translation elongation factors. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E1951–E1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gucinski G.C., Michalska K., Garza-Sánchez F., Eschenfeldt W.H., Stols L., Nguyen J.Y., Goulding C.W., Joachimiak A., Hayes C.S. Convergent evolution of the Barnase/EndoU/Colicin/RelE (BECR) fold in antibacterial tRNase toxins. Structure. 2019; 27:1660–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang R.H. Ribotoxins kill cells by chopping off the head of tRNAs. Structure. 2019; 27:1613–1614. [DOI] [PubMed] [Google Scholar]
- 18. Agirrezabala X., Frank J Elongation in translation as a dynamic interaction among the ribosome, tRNA, and elongation factors EF-G and EF-Tu. Q. Rev. Biophys. 2009; 42:159–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schuette J.C., Murphy F.V., Kelley A.C., Weir J.R., Giesebrecht J., Connell S.R., Loerke J., Mielke T., Zhang W., Penczek P.A. et al. GTPase activation of elongation factor EF-Tu by the ribosome during decoding. EMBO J. 2009; 28:755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schmeing T.M., Voorhees R.M., Kelley A.C., Gao Y.G., Murphy F.V.t., Weir J.R., Ramakrishnan V. The crystal structure of the ribosome bound to EF-Tu and aminoacyl-tRNA. Science (New York, N.Y.). 2009; 326:688–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Miller D.L., Weissbach H. Interactions between the elongation factors: the displacement of GPD from the TU-GDP complex by factor ts. Biochem. Biophys. Res. Commun. 1970; 38:1016–1022. [DOI] [PubMed] [Google Scholar]
- 22. Gromadski K.B., Wieden H.J., Rodnina M.V. Kinetic mechanism of elongation factor Ts-catalyzed nucleotide exchange in elongation factor tu. Biochemistry. 2002; 41:162–169. [DOI] [PubMed] [Google Scholar]
- 23. Tomita K. Structures and functions of qβ replicase: translation factors beyond protein synthesis. Int. J. Mol. Sci. 2014; 15:15552–15570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Blumenthal T., Carmichael G.G. RNA replication: function and structure of Qbeta-replicase. Annu. Rev. Biochem. 1979; 48:525–548. [DOI] [PubMed] [Google Scholar]
- 25. Takeshita D., Tomita K. Assembly of Q{beta} viral RNA polymerase with host translational elongation factors EF-Tu and -Ts. Proc. Nat. Acad. Sci. U.S.A. 2010; 107:15733–15738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Takeshita D., Tomita K. Molecular basis for RNA polymerization by qβ replicase. Nat. Struct. Mol. Biol. 2012; 19:229–237. [DOI] [PubMed] [Google Scholar]
- 27. Kudlicki W., Coffman A., Kramer G., Hardesty B. Renaturation of rhodanese by translational elongation factor (EF) tu. Protein refolding by EF-Tu flexing. J. Biol. Chem. 1997; 272:32206–32210. [DOI] [PubMed] [Google Scholar]
- 28. Caldas T.D., El Yaagoubi A., Richarme G. Chaperone properties of bacterial elongation factor EF-Tu. J. Biol. Chem. 1998; 273:11478–11482. [DOI] [PubMed] [Google Scholar]
- 29. Suzuki H., Ueda T., Taguchi H., Takeuchi N. Chaperone properties of mammalian mitochondrial translation elongation factor tu. J. Biol. Chem. 2007; 282:4076–4084. [DOI] [PubMed] [Google Scholar]
- 30. Kita H., Cho J., Matsuura T., Nakaishi T., Taniguchi I., Ichikawa T., Shima Y., Urabe I., Yomo T. Functional qβ replicase genetically fusing essential subunits EF-Ts and EF-Tu with β-subunit. J. Biosci. Bioeng. 2006; 101:421–426. [DOI] [PubMed] [Google Scholar]
- 31. Yashiro Y., Yamashita S., Tomita K. Crystal structure of the enterohemorrhagic escherichia coli AtaT-AtaR toxin-antitoxin complex. Structure. 2019; 27:476–484. [DOI] [PubMed] [Google Scholar]
- 32. Zhang C., Yashiro Y., Sakaguchi Y., Suzuki T., Tomita K. Substrate specificities of escherichia coli ItaT that acetylates aminoacyl-tRNAs. Nucleic Acids Res. 2020; 48:7532–7544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yashiro Y., Sakaguchi Y., Suzuki T., Tomita K. Mechanism of aminoacyl-tRNA acetylation by an aminoacyl-tRNA acetyltransferase AtaT from enterohemorrhagic e. coli. Nat. Commun. 2020; 11:5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wieden H.J., Gromadski K., Rodnin D., Rodnina M.V Mechanism of elongation factor (EF)-Ts-catalyzed nucleotide exchange in EF-Tu - contribution of contacts at the guanine base. J. Biol. Chem. 2002; 277:6032–6036. [DOI] [PubMed] [Google Scholar]
- 35. Suzuki T., Ikeuchi Y., Noma A., Suzuki T., Sakaguchi Y. Mass spectrometric identification and characterization of RNA-modifying enzymes. Methods Enzymol. 2007; 425:211–229. [DOI] [PubMed] [Google Scholar]
- 36. Suzuki T., Yashiro Y., Kikuchi I., Ishigami Y., Saito H., Matsuzawa I., Okada S., Mito M., Iwasaki S., Ma D. et al. Complete chemical structures of human mitochondrial tRNAs. Nat. Commun. 2020; 11:4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Walker S.E., Fredrick K. Preparation and evaluation of acylated tRNAs. Methods. 2008; 44:81–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yashiro Y., Zhang C., Sakaguchi Y., Suzuki T., Tomita K. Molecular basis of glycyl-tRNA(Gly) acetylation by TacT from salmonella typhimurium. Cell Rep. 2021; 37:110130. [DOI] [PubMed] [Google Scholar]
- 39. Takeshita D., Yamashita S., Tomita K. Molecular insights into replication initiation by qβ replicase using ribosomal protein s1. Nucleic Acids Res. 2014; 42:10809–10822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jahn M., Rogers M.J., Söll D Anticodon and acceptor stem nucleotides in tRNAGln are major recognition elements for e. coli glutaminyl-tRNA synthetase. Nature. 1991; 352:258–260. [DOI] [PubMed] [Google Scholar]
- 41. Kabsch W xds. Acta Crystallogr. Sect. D-Biol. Crystallogr. 2010; 66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A. et al. Highly accurate protein structure prediction with alphafold. Nature. 2021; 596:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Afonine P.V., Grosse-Kunstleve R.W., Echols N., Headd J.J., Moriarty N.W., Mustyakimov M., Terwilliger T.C., Urzhumtsev A., Zwart P.H., Adams P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. Sect. D-Biol. Crystallogr. 2012; 68:352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of coot. Acta Crystallogr. Sect. D-Biol. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Furano A.V. Content of elongation factor tu in escherichia coli. Proc. Nat. Acad. Sci. U.S.A. 1975; 72:4780–4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Evans M.E., Clark W.C., Zheng G., Pan T. Determination of tRNA aminoacylation levels by high-throughput sequencing. Nucleic Acids Res. 2017; 45:e133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Janiak F., Dell V.A., Abrahamson J.K., Watson B.S., Miller D.L., Johnson A.E. Fluorescence characterization of the interaction of various transfer RNA species with elongation factor tu.gtp: evidence for a new functional role for elongation factor tu in protein biosynthesis. Biochemistry. 1990; 29:4268–4277. [DOI] [PubMed] [Google Scholar]
- 48. Chapman S.J., Schrader J.M., Uhlenbeck O.C. Histidine 66 in escherichia coli elongation factor tu selectively stabilizes aminoacyl-tRNAs. J. Biol. Chem. 2012; 287:1229–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fukano H., Tamotsu Z., Eiji S., Kimitsuna W., Teruyuki N. Genetically engineered active qbeta replicase in rabbit reticulocyte cell-free system: a fusion protein of EF-Tu and EF-Ts is functional as the subunit of qbeta replicase. J. Biosci. Bioeng. 2002; 93:20–24. [DOI] [PubMed] [Google Scholar]
- 50. Thirup S.S., Van L.B., Nielsen T.K., Knudsen C.R. Structural outline of the detailed mechanism for elongation factor Ts-mediated guanine nucleotide exchange on elongation factor tu. J. Struct. Biol. 2015; 191:10–21. [DOI] [PubMed] [Google Scholar]
- 51. Holm L., Rosenström P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010; 38:W545–W549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Burnett B.J., Altman R.B., Ferrao R., Alejo J.L., Kaur N., Kanji J., Blanchard S.C. Elongation factor ts directly facilitates the formation and disassembly of the Escherichia coli elongation factor Tu·GTP·aminoacyl-tRNA ternary complex. J. Biol. Chem. 2013; 288:13917–13928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Burnett B.J., Altman R.B., Ferguson A., Wasserman M.R., Zhou Z., Blanchard S.C. Direct evidence of an elongation factor-Tu/Ts·GTP·aminoacyl-tRNA quaternary complex. J. Biol. Chem. 2014; 289:23917–23927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tomita K., Yamashita S. Molecular mechanisms of template-independent RNA polymerization by tRNA nucleotidyltransferases. Front. Genet. 2014; 5:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yamashita S., Martinez A., Tomita K. Measurement of acceptor-TΨC helix length of tRNA for terminal A76-addition by A-adding enzyme. Structure. 2015; 23:830–842. [DOI] [PubMed] [Google Scholar]
- 56. Graille M., Mora L., Buckingham R.H., van Tilbeurgh H., de Zamaroczy M. Structural inhibition of the colicin d tRNase by the tRNA-mimicking immunity protein. EMBO J. 2004; 23:1474–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tomita K., Ogawa T., Uozumi T., Watanabe K., Masaki H. A cytotoxic ribonuclease which specifically cleaves four isoaccepting arginine tRNAs at their anticodon loops. Proc. Nat. Acad. Sci. U.S.A. 2000; 97:8278–8283. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coordinates and structure factors for the crystal structure of CdiA–CT:CdiIEC869:Tu have been deposited to PDB under the accession number, 7VMC.