

Abstract
The function of ATP binding cassette protein A1 (ABCA1) is central to cholesterol mobilization. Reduced ABCA1 expression or activity is implicated in Alzheimer's disease (AD) and other disorders. Therapeutic approaches to boost ABCA1 activity have yet to be translated successfully to the clinic. The risk factors for AD development and progression, including comorbid disorders such as type 2 diabetes and cardiovascular disease, highlight the intersection of cholesterol transport and inflammation. Upregulation of ABCA1 can positively impact APOE lipidation, insulin sensitivity, peripheral vascular and blood–brain barrier integrity, and anti-inflammatory signaling. Various strategies towards ABCA1-boosting compounds have been described, with a bias toward nuclear hormone receptor (NHR) agonists. These agonists display beneficial preclinical effects; however, important side effects have limited development. In particular, ligands that bind liver X receptor (LXR), the primary NHR that controls ABCA1 expression, have shown positive effects in AD mouse models; however, lipogenesis and unwanted increases in triglyceride production are often observed. The longstanding approach, focusing on LXRβ vs. LXRα selectivity, is over-simplistic and has failed. Novel approaches such as phenotypic screening may lead to small molecule NHR modulators that elevate ABCA1 function without inducing lipogenesis and are clinically translatable.
KEY WORDS: Alzheimer's disease, Cardiovascular disease, Cholesterol, Drug discovery, Liver X receptor, Nuclear hormone receptor, Type 2 diabetes
Graphical abstract
Boosting ABCA1 with small molecules that act at nuclear hormone receptors produces pleiotropic beneficial effects in the brain and periphery and, therefore, represents a promising therapeutic strategy for Alzheimer's disease.

1. Introduction
Alzheimer's disease (AD) is the most prevalent form of dementia in the US and worldwide, and it is currently the sixth leading cause of death in the US1,2. Age is the greatest risk factor for AD, which will translate to an increase in disease prevalence with increasing life expectancy globally: current estimates indicate the number of diagnosed cases in the US increasing from six million to fourteen million by 2050. The widespread prevalence of AD is contrasted by the dearth of available treatments. There are five FDA-approved small molecule drugs: donepezil, rivastigmine, and galantamine inhibit the acetylcholinesterase enzyme; memantine antagonizes the NMDA glutamate receptor; and a combination donepezil–memantine pill performs both functions. However, these treatments provide only temporary symptomatic improvement. They do not prevent, slow the progression, or alter the fatal prognosis of AD3,4. The AD clinical trial failure rate (99.6% in the decade from 2002 to 2012) is the highest of any disease state, and there is an urgent need to discover and develop effective new therapies5, 6, 7. The extremely high risk and cost of failure has led pharmaceutical companies to divest of AD drug development before Phase 2 proof-of-concept and pivotal Phase 3 clinical trials, which likely has led to premature abandonment of promising therapeutic strategies.
Diagnosis of AD rests on specialized tests for cognitive function combined with post-mortem histological analysis for hallmark AD pathology: a) extracellular amyloid plaques formed from amyloid-β (Aβ) peptide fragments and b) intracellular neurofibrillary tangles (NFT) composed of atypically phosphorylated tau protein. Inhibition of Aβ and tau peptide aggregation dominated early therapeutic approaches to AD, and the approval of aducanumab, in June 2021, is likely to revive these and other approaches targeting the removal of hallmark pathology. Although almost all disease-modifying AD therapeutics have failed in clinical trials, many targeted at Aβ have reported significant reductions in Aβ in these trials. This disconnect between attenuated Aβ without improved cognitive function has been explained by the need to treat patients in the prodromal stage of the disease. Longitudinal imaging studies have estimated that Aβ and NFT pathology develop up to three decades before symptoms, with brain atrophy (diminished hippocampal volume) and neuronal hypofunction (reduced glucose metabolism) preceding clinical onset by roughly ten years8. Unfortunately, the move “to treat before clinical symptoms” is stymied by the lack of predictive diagnostics. In 2020, the Lancet Commission on “Dementia prevention, intervention, and care” concluded that imaging techniques for hallmark Aβ and tau pathology (and blood tests for Aβ) have not reached clinical significance for predicting cognitive decline9. Indeed, it was concluded that most cognitively normal people with brain and/or plasma biomarkers for AD do not develop dementia within a clinically relevant timeframe.
Aducanumab, like other antibodies targeted at Aβ, dose-dependently reduced brain Aβ, as visualized by PET imaging. The FDA approval of aducanumab for prodromal AD, under the agency's accelerated approval pathway, requires substantial evidence of effect on an intermediate biomarker (in this case Aβ) and reasonable likelihood of a meaningful clinical benefit, which the FDA hopes will be achieved in post-market clinical trials. The approval of aducanumab is highly controversial because of unproven efficacy and direct contradiction of the Lancet Commission's findings on predictive biomarkers; however, it will likely maintain focus on clearance of Aβ as a clinical endpoint.
Many people develop a heavy Aβ burden but never suffer from cognitive decline10. In addition, the brains of patients who suffer cognitive decline and are diagnosed with AD show a wide range of pathologies post-mortem, including Lewy bodies, microinfarcts, and TDP-43 aggregates; some do not show a heavy Aβ or NFT burden11, 12, 13. The acronym ADRD (AD and related dementia) conveniently describes the disease from which the majority of those with dementia will die. Cognition declines with age, and ADRD may represent an accelerated version of normal aging phenomena driven by idiosyncratic loss of neural reserve14. Extensive ongoing research aims to define lifestyle, clinical, and physiological factors that underlie the susceptibility or, conversely, the resilience to dementia during aging15. Recent genome-wide association study (GWAS) reports highlighted pathways, such as lipid metabolism and immune signaling in the liver, distinct from traditional AD-associated genes and pathology that are associated with resilience to clinical dementia16,17.
In contrast to the difficulty of assessing pathological progression toward AD prior to diagnosis, there are documented risk factors for clinical AD, which are readily measurable. These risk factors include innate genetic traits, such as APOE4 (apolipoprotein E ε4 genotype), unmodifiable risk factors, such as aging, and modifiable environmental or behavioral factors that can be quantified in a clinical setting (e.g., blood pressure). As detailed above, targeting a drug to an amyloid plaque has yet to produce a robust clinical cognitive benefit, whereas neutralizing a risk factor could provide a profound therapeutic impact on ADRD development and progression. This paradigm has been used successfully in cardiovascular disease (CVD) with low-density lipoprotein (LDL) lowering statins and anti-hypertensive medications reducing risk and improving disease-free survival.
The Lancet Commission report on risk factors for ADRD stated that reducing the dementia risk created from diseases such as type 2 diabetes (T2D) and obesity would have a significant impact on healthy aging with cognition intact9. In recent years, appreciation has grown for the role of other pathophysiological factors, such as insulin resistance, neuroinflammation, and dyslipidemia, leading to ADRD progression; thus, new drug strategies should consider these facets of the disease18, 19, 20, 21. Chronic metabolic diseases, including T2D and CVD, represent a growing health burden driven by increasing obesity. T2D is a significant risk factor for comorbidity with ADRD22,23, with dementia risk paralleling the duration and severity of T2D24. Specifically, insulin resistance, impaired glucose metabolism, mitochondrial dysfunction, inflammation, dyslipidemia, and impaired cholesterol mobilization may be common underlying pathogenic promoters of dementia in T2D and ADRD. T2D is a major risk factor for CVD and recent findings demonstrate that risk factors contributing to CVD, including high LDL-cholesterol and blood pressure, also increase risk of developing AD25,26. Insulin resistance contributes to AD pathogenesis even in patients without overt diabetes27. In addition, the major genetic risk factor for ADRD, APOE4, shows significant association with T2D in several studies and contributes to increased LDL cholesterol levels that drive CVD progression28,29.
In this review article, we will demonstrate that boosting ATP binding cassette A1 (ABCA1) levels fulfills many of the objectives introduced above (Fig. 1). Evidence suggests that increased ABCA1 activity is beneficial for CVD, T2D, and ADRD. Moreover, several mechanisms link ABCA1 function to Aβ clearance and to mechanisms associated with heightened risk associated with APOE4. Therapeutic approaches to increasing levels of ABCA1 in the brain will be discussed along with results from studies in preclinical models of AD.
Figure 1.

Proposed beneficial roles of ABCA1 in AD, ADRD, and comorbidities. ABCA1 mediates cellular cholesterol efflux, such that increased ABCA1 expression would be expected to produce several direct and indirect beneficial effects via an enhancement of efflux activity. These effects would provide therapeutic efficacy both in the brain and in peripheral tissues.
2. ADRD and risk factors
2.1. Late onset AD versus rare familial AD (FAD)
ADRD is a disease of aging: incidence increases nearly twenty-fold from ages 65 to 90, and roughly one in every three over the age of 90 suffer from dementia30,31. Mild cognitive impairment (MCI) can be seen as a normal consequence of aging; however, not all MCI transitions to ADRD. Oxidative damage to DNA, lipids, and proteins accumulates with aging against a background of weakened repair and clearance mechanisms32,33, leading to attenuation of essential cellular functions, particularly in metabolism and mitochondrial function34. With the exception of FAD, caused by inherited mutations in amyloid precursor protein (APP) or presenilin (PSEN) 1 or 2 genes that account for ≤1% of all cases, clinical diagnosis comes during the seventh decade of life or later35.
Nevertheless, FAD transgenic (FAD-Tg) mice that overexpress these mutant forms of human APP, PSEN1, or PSEN2 alone or in combination, remain dominant in AD drug discovery, because FAD-Tg mice develop amyloid neuropathology similar to that seen in AD patients36, 37, 38, 39, 40, 41, 42, 43, 44. FAD-Tg mice are generally studied as young adults (at 3–12 months of age) and do not reproduce NFT pathology, nor do the FAD-Tg mice that also express mutations in the human gene responsible for tau protein expression (e.g., 3xTg mice) faithfully model human AD45, 46, 47, 48, 49, 50. This inability to reproduce the complexity of human AD in mouse models has been discussed extensively in other recent reviews51,52. The heavy focus on amyloid mutations in these models naturally biased AD drug development toward agents (e.g., aducanumab and several others) that could reverse the development of amyloid pathology in these mice, which, as discussed above, have not shown beneficial clinical effects.
FAD-Tg models still play a key role in AD drug discovery because some can model aspects of AD beyond amyloid pathology, such as the altered metabolism and mitochondrial function that are central to late-onset AD. They can also replicate some of the effects of AD risk factors, such as sex and other genetic variations52. Meanwhile, additional models are being generated and characterized using the vast amount of genomic and proteomic data that has rapidly become available over the past decade53. The recent overall trend in ADRD research has been to emphasize factors that drive the early stages of the disease, even before Aβ and NFT pathology appear. Better understanding of these factors, improved modeling of their pathophysiology in animal species, and development of therapeutic agents to target them may ultimately yield disease-modifying therapies for late-onset dementia, if appropriate clinical biomarkers can be found. In the following pages, we detail these key factors that promote ADRD onset and progression.
2.2. Genetic risk factors and APOE
Dozens of loci linked to AD development have been identified by GWAS16,54. These rare, risk-driving polymorphisms include variants in microglial genes TREM2 and ABCA7. TREM2 (trigger receptor expressed on myeloid cells 2) protein mediates the microglial stress response and the R47H missense mutation significantly increases AD risk55,56. ABCA7 plays a role in cholesterol transport and microglial phagocytosis, and loss-of-function mutations at this locus also enhance AD risk57,58. Both the understanding of the physiologic implications of these mutations and their occurrence are low44; for example, the frequency of TREM2∗R47H is <0.5% in Caucasians and even lower in Chinese and African-American populations59, 60, 61, 62, 63, 64. The interaction of these rare genetic variants with other AD risk factors is poorly understood.
Conversely, the APOE4 allele is much more common. The protein encoded by this gene, APOE (34 kDa, 299 amino acids), is required for the formation of lipoprotein particles in the brain65. The critical function of transporting cholesterol and phospholipids throughout the central nervous system (CNS) is performed by these particles, which share similarities to high-density lipoprotein particles (HDL) in the periphery. Across human APOE isoforms, the only amino acids that differ are residues 112 and 15865. The E2, E3 and E4 isoforms contain Cys/Cys, Cys/Arg, and Arg/Arg mutations of these residues, respectively. Despite these small differences, APOE genotype profoundly affects AD pathogenesis in a dose and allele-dependent manner66. Although the frequency of APOE4 allele is less than 14% in the global human population, the allele frequency in AD patients is 40%67. Carriers of one APOE4 allele have a ∼50% chance of developing AD, whereas in homozygotes the risk is >90%. Moreover, compared to non-carriers (mean age of onset = 84), APOE4 heterozygotes and homozygotes develop AD roughly 8 and 16 years earlier, respectively68.
APOE4 is a “loss-of-function” isoform: restoration of function to that of APOE2 or APOE3 would ameliorate AD pathology in APOE4 carriers68. This outcome holds true even if the alternative “gain of toxic function” hypothesis holds true. A proposed unique feature of the APOE4 isoform is increased intrinsic disorder, termed the “molten globule” state69,70. Most proteins contain intrinsically disordered regions, and these proteins are often stabilized by protein–protein interactions with scaffold proteins and chaperones. In the absence of stabilization, these proteins, including APOE4, are more susceptible to proteolytic degradation. Consequently, there is less APOE4 than APOE3 available for apolipoprotein formation; APOE4-containing lipoproteins are poorly lipidated and are less stable71, 72, 73, 74.
The structural and functional APOE isoform-specific differences contribute to profound pathologic effects of APOE4. Thus, therapeutic approaches that correct APOE4-mediated pathologic mechanisms or render neutral the risk relative to APOE3 should be prioritized. This conclusion is amplified by a recent observation on one rare APOE variant with a significant impact on AD risk. As part of a study on an FAD population in Colombia, a woman was identified who developed MCI in her seventies, compared to a mean of 45 years old for her relatives; protection in this patient was derived from being homozygous for an ultra-rare APOE-Christchurch variant characterized by a single, R136S amino acid substitution75. Reduced NFT pathology and neurodegeneration were observed, despite high Aβ levels characteristic of FAD being present76. This case emphasizes the profound role of APOE in modulating neurodegeneration and progression to dementia in AD. Several studies have demonstrated an interplay of female sex with APOE4 in accentuating AD risk. In one meta-analysis, female APOE3/E4 heterozygotes exhibited odds ratios ranging from 2 to 4, compared to the odds ratio for male APOE3/E4 heterozygotes of <1.567. Subsequent studies have demonstrated significant, but more modest, effects of sex on APOE4-mediated AD risk77,78.
2.3. Connectedness of APOE, T2D, CVD, and ADRD
Outside the CNS, two major chronic conditions—T2D and CVD—strongly drive risk of AD. T2D and CVD share close metabolic links, and their influence on AD is similarly driven by these metabolic factors. Epidemiological studies, systematic reviews, and meta-analyses have firmly established T2D as a risk factor for AD and related dementias, even after correction for underlying drivers such as obesity or physical inactivity24,79,80. Changes to glucose homeostasis and insulin signaling significantly impact the brain. Excess insulin may compete with Aβ peptides for degradation by insulin-degrading enzyme81. The connection between insulin resistance and AD pathogenesis is further bolstered by shared perturbations in cellular metabolism, inflammation secondary to deposition of advanced glycation end-products, and direct vascular damage82, 83, 84. The links between CVD pathology and AD risk are similarly multifaceted. Elevations in cholesterol and blood pressure promote atherosclerosis in cerebral vessels, resulting in stiff, narrow vasculature that reduces cerebral perfusion and precipitates Aβ and NFT pathology formation85,86. This hypoperfusion can be exacerbated, chronically, by heart arrhythmias, or, acutely and severely, by myocardial infarctions. Sudden brain ischemia causes immediate neuronal loss while also enhancing ongoing AD pathological processes87,88.
Of particular interest is that T2D29, CVD89, TBI90, and even behavioral risk factors, such as smoking status91 and exercise92, drive AD risk in a synergistic fashion when combined with the APOE4 allele. APOE4 correlates with increased plasma LDL levels, which may partially explain its connection with T2D/CVD in AD risk93, 94, 95. In some studies, relative risks increased from 1.5 to 2 for single risk factors up to 5–10 when APOE4 is present. The magnitude of this synergism highlights the importance of understanding the molecular basis of APOE4-mediated deficits and of the interactions among APOE4, other risk factors, and dementia.
There are two major pathophysiologic commonalities that tie together the biological traits of APOE4 and aging, comorbid conditions such as TBI, CVD, and T2D, and even lifestyle factors such as obesity and smoking: disrupted lipid homeostasis and chronically heightened inflammation. This conclusion is supported by GWAS evidence that has associated loci relating to cholesterol metabolism and immune function with AD and dementia17,96,97. Drug candidates that impact the intersection of these two pathological mediators would provide impactful, pleiotropic efficacy. This consideration has led us and others to suggest the ATP-binding cassette family member A1 (ABCA1) protein as a compelling therapeutic target for AD.
3. Roles of ABCA1 in health and disease
3.1. ABCA1 and its broad physiological roles
ABCA1 is expressed ubiquitously throughout the human body, with peripheral levels highest in liver hepatocytes98. In the CNS, ABCA1 is found in neurons, astrocytes, and microglia99. Human ABCA1, consisting of 2261 amino acids, is an integral membrane protein that utilizes ATP to transport cholesterol, phospholipids, and other lipid molecules to apolipoprotein carriers100, 101, 102. In the plasma membrane and in intracellular organelles, including endosomes and lysosomes, ABCA1 associates with cholesterol-rich membrane domains103, where it may serve to protect cells from excessive and potentially cytotoxic accumulation of free cholesterol. ABCA1 interacts with many of the protein components of HDL, namely APOA1, APOA2, APOA4, APOC1–3, and APOE104, particularly when these apolipoproteins are minimally lipidated105. ABCA1 is uniquely suited to add lipid to lipid-poor apolipoproteins, whereas other ABC family members (e.g., ABCG1/G4) and cholesterol transporters (e.g., SR-B1) interact with already-lipidated forms103,106. In addition to this putative direct transfer function, ABCA1 has many indirect functions resulting from its influence on cellular cholesterol homeostasis. These effects, which are depicted in Fig. 1 and elaborated in greater detail in the following sections, help explain why ABCA1 represents such a promising target for therapeutics that might broadly attenuate pathophysiologic processes shared between ADRD and its many underlying risk factors.
3.2. ABCA1 and CNS lipid transport
Cholesterol homeostasis is vital to CNS function. The brain contains 2% of the total mass of an adult human yet holds nearly 20% of the total cholesterol107,108. Most brain cholesterol resides stably in specialized myelin membranes, but roughly 30% is found in cellular membranes of neurons and glial cells, where it actively moves or is metabolized within and between cells108. Importantly, CNS and peripheral cholesterol pools are separated by the blood–brain barrier (BBB); thus, all CNS cholesterol is synthesized, transported, and recycled in situ. In adult brains, cholesterol synthesis and turnover occurs at a low, yet nonzero, rate109. Although neurons and astrocytes both express cholesterol synthetic enzymes110,111, neurons exhibit functional dependence on astrocytes for cholesterol delivery112.
CNS cholesterol transport occurs via formation of APOE-containing lipoproteins (Fig. 2). Although APOE can be, and often is, synthesized by other cell types113,114, astrocytes serve as the primary source of brain lipoproteins115. Newly-synthesized APOE receives cholesterol and phospholipids via ABCA1, then is secreted into brain parenchyma as discoidal lipoproteins. Cells with “excess” cholesterol complete additional lipid transfer to promote maturation of this lipoprotein particle, which can ultimately be internalized by cells “deficient” in cholesterol, a process mediated by proteins such as LDL receptor or LDL receptor-related protein 1 (LRP1)116.
Figure 2.

Brain cholesterol transport by ABCA1 and APOE. ABCA1 transports cholesterol out of cells to promote lipidation of secreted APOE to form lipoprotein particles (1), which are either internalized by cholesterol-deficient cells or transported across the BBB to maintain brain cholesterol homeostasis (2). Poorly lipidated APOE4 (compared to APOE3 or APOE2) is prone to degradation, disrupting this homeostasis and contributing to AD pathology (3). Internal CNS control of cholesterol homeostasis is regulated by neuronal expression of CYP46A1 enzyme, which converts excess cholesterol to 24-hydroxycholesterol (4). This form can cross the BBB (unlike cholesterol itself), and it acts as an endogenous agonist of LXR to promote expression of the cholesterol transport machinery. This endogenous control mechanism is disrupted in AD, predisposing brain cells to cholesterol overload. Small molecule ABCA1 inducers may elicit therapeutic effects against AD by boosting cholesterol transport, particularly via increased APOE lipidation in APOE4 carriers.
Neuronal cholesterol homeostasis is under tight control to allow adequate function and growth while preventing accumulation of cytotoxic free cholesterol. Internalization of lipoproteins via LRP1 is a critical source of cholesterol for neurite outgrowth, synaptogenesis, and remodeling117; but LRP1 also exerts negative feedback to limit intracellular cholesterol concentrations118. Lipoprotein receptors expressed at the BBB can promote cholesterol egress from the CNS119. A second efflux mechanism occurs via CYP46A1, an enzyme that converts cholesterol to 24-hydroxycholesterol (24HC), which, unlike cholesterol itself, is BBB-permeable120. 24HC is also an endogenous agonist of the liver X receptor (LXR) that drives transcription of key genes such as ABCA1, ABCG1, and APOE (Fig. 2).
3.3. Restoration of brain cholesterol homeostasis by boosting ABCA1
CNS cholesterol homeostasis is a critical and tightly regulated process dependent on APOE and strongly influenced by APOE isoform. As stated above, APOE4 represents a loss-of-function allele relative to the common, risk neutral APOE3 variant. Human plasma and cerebrospinal fluid121, along with brain parenchyma of APOE targeted-replacement mice122, exhibit isoform-dependent concentrations of APOE (APOE2 > E3 > E4). A similar isoform dependence occurs for APOE-promoted cholesterol efflux from CNS cells123. In human iPSC-derived CNS models, APOE4 broadly disrupts cholesterol metabolism124. Together, these deficits associated with APOE4 lead to smaller lipoproteins that contain less cholesterol125,126. In fact, APOE3-containing lipoproteins carry up to three times as much cholesterol per gram of protein as their APOE4-containing counterparts127.
Reduced lipidation of APOE4 produces broad functional consequences. Lipidation of APOE alters protein morphology to expose residues used for receptor binding and prevents toxic aggregation of unlipidated APOE128,129. Poorly lipidated APOE4 is unstable and, hence, susceptible to proteolytic degradation leading to generation of neurotoxic fragments73,130, and less efficient clearance of toxic Aβ species131. Overloading membrane cholesterol in neurons reproduces AD phenotypes, including Aβ overproduction and disrupted axonal transport132. In brain tissue from human AD patients, APOE4 is associated with heightened Aβ deposition, soluble oligomeric Aβ (oAβ) levels, and amyloid plaque pathology133, 134, 135, 136, in addition to elevated tau pathology137,138.
Based on the evidence elaborated above, a primary hypothesis for therapeutically boosting ABCA1 is relatively straightforward: increasing cholesterol efflux to APOE, via induction of ABCA1, will restore lipidation of APOE4 and correct other AD-related phenotypes. Genetic knockout of Abca1 in mice dramatically reduces brain APOE lipidation and secretion and disturbs cholesterol homeostasis139, 140, 141, 142. Murine ABCA1 overexpression enhances lipidation and reduces aggregation of APOE143,144. Treating mice with LXR agonist, elaborated in detail below, likewise raises APOE protein levels via ABCA1 action145. Increased lipidated APOE, particularly improvement in APOE4 lipidation, plays a significant role in clearance and detoxification of Aβ species146, 147, 148.
Moreover, ABCA1 induction may be beneficial for AD cholesterol homeostasis regardless of APOE4 status, as AD brain samples reveal plasma membrane cholesterol enrichment with disease progression149. Endogenous control mechanisms that respond to excess cholesterol to promote ABCA1 expression are dysfunctional in AD patients: both circulating levels of 24HC and neuronal CYP46A1 expression are reduced150,151. Consequently, cholesterol efflux capacity in AD patients is reduced by 30%, independent of APOE genotype152. Studies of genetic polymorphisms in ABCA1 have revealed increased or decreased risk of AD, possibly in a sex-dependent manner, associated with certain variants that alter cholesterol transport capacity153, 154, 155. One specific loss-of-function mutation, ABCA1∗N1800H, increased AD risk fourfold156. Together, these data strongly support the hypothesis that enhancement of ABCA1 expression and function could restore CNS cholesterol homeostasis and reverse AD phenotypes.
A new type of microglia, lipid droplet-accumulating microglia, resembling foamy macrophages in atherosclerotic lesions, has recently been shown to accumulate in the aging brain and to be associated with neurodegenerative disease157, 158, 159. Intracellular lipid droplets, containing glycerolipids and cholesterol, are markers of inflammation. The compromised cholesterol trafficking of astrocytes underlies the detrimental effect of APOE4 on lipid metabolism and lipid trafficking between astrocytes and neurons126. In astrocytes and neurons from APOE3 and APOE4 knock-in mice, APOE4-expressing astrocytes are also less able to metabolize fatty acids and both to transport and to internalize fatty acids from neurons, a process mediated by APOE-lipid particles (Fig. 3)160. The APOE4-induced defects in lipid transport and metabolism in neurons and glial cells leads to accumulation of lipid droplets, lipotoxicity, and decreased mitochondrial function; which can be reversed by ABCA1 activation. Similar observations on astrocyte-neuron dysfunction were made when neurons were subject to excitotoxicity induced by NMDA161.
Figure 3.

APOE, ABCA1, and inflammation. (A) Neuronal NMDA stimulation increased fatty acid and triglycerides (TGs) leading to decreased mitochondrial respiration, lipid peroxidation, and increased reactive oxygen species (ROS); in turn leading to lipotoxicity and neuronal death. Transport of fatty acids, TGs and lipid peroxidation products (LPPs) from neurons to astrocytes by lipidated APOE rescued neurons by lysosomal catabolism of fatty acids, storage in lipid droplets, and use for mitochondrial oxidative phosphorylation; which was accompanied by transcriptional upregulation of LXR and NRF2 target genes. (B) APOE4 neurons showed 36% lower APOE, reduced neurite branching, elevated fatty acids and TGs, and decreased mitochondrial function and glucose metabolism. APOE4 astrocytes were less efficient at transporting lipids and fatty acids from neurons and at fatty acid catabolism and energy conversion, containing fragmented mitochondria and elevated TGs.
3.4. ABCA1 and peripheral lipid transport
Although APOE and cholesterol cannot cross the blood–brain barrier, peripheral lipid transport plays an important role in AD pathogenesis. It has been proposed that the protective role of ABCA1 in AD may derive from enhancing plasma HDL, APOA1, and APOE, equally as from direct CNS actions162,163. This importance of peripheral actions is often neglected when evaluating AD drug development strategies. Plasma lipoprotein metabolism is more varied than in the CNS, in which APOE dominates. Several lipoprotein particles contribute to vascular cholesterol deposition, inflammation, and atherogenesis to drive atherosclerosis164, 165, 166. Conversely, HDL particles are anti-atherogenic. HDL facilitates removal of cholesterol from vascular walls and peripheral tissues via reverse cholesterol transport (RCT), thereby preventing formation of cytotoxic oxidized lipid species and reducing inflammation and atherogenic lesion formation167. ABCA1 contributes to formation of particles termed pre-βHDL101,106,168, and ABCA1/ABCG1 promote formation of mature HDL particles169. Reduced plasma cholesterol efflux capacity, which is directly tied to ABCA1 and ABCG1 expression, synergizes with other components of metabolic syndrome to promote atherogenesis170. Pathological changes that occur during atherogenesis, such as oxidation of LDL, downregulate ABCA1 expression171,172. Tangier disease, characterized by ABCA1 loss-of-function mutations, leads to premature atherosclerosis173, and ABCA1 variants that increase AD risk also have been associated with T2D and CVD risk174, 175, 176. Increased peripheral atheroprotection via ABCA1 induction would promote cerebral vascular health, preserving BBB integrity and function177,178, and, ultimately, protect against AD.
3.5. ABCA1 and insulin resistance
Insulin resistance plays a key role in the brain during AD pathogenesis. AD brains exhibit altered insulin-related gene expression and diminished phosphorylation of AKT and GSK3β179, even in patients not diagnosed with T2D180. Altered glucose homeostasis (i.e., cerebral hypometabolism) in neurons predates cognitive decline in AD patients8. Synaptic plasticity relies on insulin signaling181,182, and insulin protects neurons from oAβ-induced toxicity183. Several large, long-term cohort studies demonstrated an association of use of metformin, an insulin sensitizing drug, with a significantly reduced risk of developing MCI or dementia in patients with diabetes184, 185, 186. Two small, short-term pilot studies with metformin in non-diabetic patients with MCI or early AD showed improvements in cognitive performance187,188, but long-term data are lacking. A larger Phase 2/3 trial is currently enrolling patients with MCI to study the effect of metformin on progression to AD (NCT04098666). Similarly, intranasal insulin improved cognition in pilot studies in early-stage AD patients189. However, Phase 3 clinical trials with anti-diabetes agents rosiglitazone and pioglitazone demonstrated no efficacy in AD patients190, although this may be associated with insufficient bioavailability191. Thus, the description of AD as “type 3 diabetes” is an oversimplification192; however, therapeutic strategies that improve insulin sensitivity hold promise in AD. Boosting ABCA1 expression represents one such strategy. Tangier disease causes disrupted insulin homeostasis193; and ABCA1 deletion in pancreatic β-cells and insulin target tissues reduces insulin release and sensitivity, respectively194, 195, 196, 197. Patients with pre-diabetes or T2D exhibit reduced adipose and liver ABCA1 expression and diminished cholesterol efflux capacity198,199. These studies support pharmacological enhancement of ABCA1 as a therapeutic strategy to correct deficits in cholesterol transport and insulin resistance.
3.6. Inflammation: Role of ABCA1 and implications for ADRD
An increasing body of evidence suggests that a sustained, inappropriate inflammatory response is a driving mechanism behind AD pathology that underlies connections between peripheral risk factors and AD27,200, 201, 202, 203, 204. The neural reserve or cognitive resilience in individuals that develop Aβ pathology without cognitive decline may be associated with resilience to neuroinflammation205,206. Inflammation in the CNS and periphery is strongly associated with AD. A meta-analysis combining forty individual studies revealed higher plasma levels of cytokines (TNFα, IL1β, IL6, IL12, IL18, and TGFβ) in AD patients versus healthy controls207. These cytokines may cross the BBB, inducing neuroinflammatory responses208, or damage the BBB itself209, further enhancing immune cell infiltration and reducing glucose transport into the brain210,211. Microglia and astrocytes are the major mediators of immune response in the CNS. Microglial phagocytosis of toxic Aβ species is hypothesized to be beneficial during early AD pathogenesis212. However, as disease progresses, microglial phagocytosis may be overwhelmed, without diminishing cytokine production213,214, resulting in more immune cells being recruited to sites of inflammation, where they produce additional cytokines200. Proinflammatory cytokines increase brain insulin resistance215, damage synapses216, and induce neuronal death217. Neuroinflammation, particularly in response to Aβ pathology, is exacerbated in the presence of APOE4218, 219, 220, 221, 222.
Murine genetic deletion of brain Abca1 enhances neuroinflammation and astrogliosis223. In humans with ABCA1 mutations, plasma levels and immune cell expression of cytokines are elevated compared to healthy controls224. This pro-inflammatory profile associated with diminished ABCA1 expression stems from excess free membrane cholesterol, which promotes mobilization of inflammatory mediators, such as toll-like receptors, to lipid rafts225, 226, 227. Independent of cholesterol mobilization, interaction of ABCA1 with APOA1 activates JAK2/STAT3 to suppress proinflammatory cytokine production228. Thus, pharmacologic enhancement of ABCA1 expression may attenuate pro-inflammatory states associated with AD pathogenesis.
4. Pharmacological approaches to boasting ABCA1 for ADRD therapy
4.1. Overview
The broad therapeutic efficacy expected from pharmacological enhancement of ABCA1 expression and function has led to numerous therapeutic strategies; however, no candidate has been successfully translated to clinical use. A brief overview will be presented, with a more detailed analysis of LXR agonists.
4.2. Apolipoprotein peptide mimetics
HDL peptide mimetics were developed that enhanced HDL levels and prevented atherogenic lesion formation229,230. It was later determined that these peptides promoted ABCA1-mediated cholesterol efflux231. The many apolipoprotein mimetics232, can be grouped into two classes: APOA1 and APOE mimetics233. The initial model for APOA1 mimetics was a peptide named 18A (containing 18 amino-acids)234 that forms an amphipathic helix mimicking the structure of APOA1. Modifications of this peptide led to compounds with atheroprotective properties in mice235, 236, 237. In AD models, D-4F enhanced APOE lipidation by ABCA1 and was anti-inflammatory and pro-cognitive238, 239, 240. APOE-mimetic peptides include: COG1410, consisting of APOE residues 138–149241; COG112, consisting of APOE residues 133–149242; Ac-hE-18A-NH2, comprised of 18A linked to APOE residues 141–150243; and CS-2563, consisting of the modified C-terminal APOE domain (aa238–266)244,245. Only CS-2563 directly stimulates ABCA1-mediated cholesterol efflux. Despite anti-inflammatory and neuroprotective effects in models of neurodegeneration, these mimetics share poor CNS bioavailability. A smaller peptide of only five amino acids, termed CN-105, demonstrated improved brain bioavailability in humans and was reported to reduce Aβ pathology and cognitive deficits in APOE4-expressing FAD mice246,247. This peptide is in Phase 2A clinical trials for intracerebral hemorrhage (NCT03168581) and post-operative cognitive decline (NCT03802396).
4.3. Small molecule approaches to ABCA1 induction
Humans express 48 unique nuclear hormone receptors (NHRs) that share a common ancestral gene and, therefore, possess common structural motifs that include an N-terminal transcriptional transactivation domain, a conserved DNA-binding domain, and a C-terminal ligand-binding domain248,249. Some of these receptors are well-characterized and have been exploited clinically (e.g., estrogen receptor α; ERα), while a handful still possess the designation of “orphan receptor” because an endogenous ligand has yet to be identified250. ABCA1 expression is regulated primarily by LXR, retinoid X receptors (RXR), and peroxisome-proliferator activated receptors (PPAR). These represent the most well-studied therapeutic targets for ABCA1 induction by small molecules. Additional, non-NHR approaches are briefly summarized below:
-
(1)
cAMP analogs, PKA activators, phosphodiesterase inhibitors, and adenosine A2A receptor agonists are all designed to enhance the cAMP/PKA pathway, to increase ABCA1 expression and cholesterol efflux for anti-atherogenic and anti-inflammatory effects251,252.
-
(2)
Anti-hypertensive agents and targets have also been widely explored in AD; mechanisms include ABCA1-mediated cholesterol mobilization. Angiotensin II promotes intracellular cholesterol accumulation by reducing ABCA1 transcription via NHR downregulation, promoting foam cell formation and hampering glucose-stimulated insulin secretion from pancreatic β cells253,254, indicating beneficial roles for angiotensin-converting enzyme inhibitors and angiotensin-receptor blockers. Calcium channel blockers are also used clinically for hypertension and CVD: two have been shown to increase ABCA1 expression: nifedipine, which activated PPARγ via an ERK1/2-dependent mechanism at a clinically relevant concentration255, and verapamil, which stimulated ABCA1 promoter activation independent from LXR, albeit at supratherapeutic concentrations of 10–30 μmol/L256.
-
(3)
AMP-activated protein kinase (AMPK) is a major metabolic regulator257, which provides antioxidant functions, plays a role in CVD, and is an indirect target of T2D drug metformin258. Two studies showed that AMPK agonists increased Abca1 and/or Abcg1 mRNA and cholesterol efflux to APOA1, although they disagreed on whether this effect was LXR-dependent or -independent259,260.
-
(4)
Glucagon-like peptide-1 (GLP1) is a short peptide hormone classically associated with simulating insulin release and attenuating glucagon release to lower plasma glucose261. GLP1 receptor agonists and inhibitors of the GLP1-degrading enzyme are used clinically in T2D. GLP1 is neuroprotective and GLP1 therapeutics were shown to upregulate cholesterol transporters, enhance cholesterol efflux, and reduced proinflammatory cytokine release in cell models262, although the mechanism is not fully defined263, 264, 265. Liraglutide, a GLP1 analog, is currently in a Phase 2 trial of patients with mild AD266.
-
(5)
The clinical use of the vitamin niacin (nicotinic acid) in hyperlipidemia and hypertriglyceridemia has been decreasing over the years: niacin increases HDL and reduces levels of LDL and VLDL267. Niacin influences a vast number of biological mechanisms, one of which is activation of the G-protein coupled receptor GPR109A268. Niacin increases ABCA1 expression via multiple pathways269, 270, 271.
-
(6)
CYP46A1 catalyzes the conversion of cholesterol to 24HC, which permits BBB efflux and activates LXR endogenously272. Increasing CYP46A1 activity to promote ABCA1 activity and restore CNS cholesterol homeostasis has been proposed in AD273. The HIV drug efavirenz induces CYP46A1 at a dose below that used in HIV patients and is in clinical trial for AD (NCT03706885). CYP46A1 expression is brain-specific, thereby minimizing peripheral side effects; however, direct activators of CYP46A1 may interact with other CYPs because of structural similarity274,275. Efavirenz analogs and CYP46A1 activators are in early development276.
-
(7)
Histone deacetylase inhibitors (HDACi) have been studied extensively in preclinical and early clinical trials in AD. HDACs are epigenetic erasers modulating histone-mediated chromatin control and transcriptional activation. HDACs have many non-histone protein substrates, which have often been found to mediate observed activity. Pan-HDACi trichostatin A upregulates ABCA1 and ABCG1 expression and stimulates cellular cholesterol efflux via induction of PPARγ277, 278, 279. Similarly, Class I HDACi were shown to promote astrocytic APOE secretion and upregulate ABCA1 expression280.
4.4. Nuclear hormone receptor signaling: RXR
RXRs function as heterodimeric binding partners not only with LXRs, but also with all other class II nuclear hormone receptors (NR1 subfamily), which include PPAR, retinoic acid receptors (RAR), constitutive androstane receptors, pregnane X receptor, farnesoid X receptor (FXR), vitamin D receptor, and thyroid hormone receptors281. Most of these heterodimers function as permissive pairs, meaning that ligand activation of either RXR or its binding partner elicits similar effects282. Profiling of binding sites across the genome illustrated two key phenomena associated with this function: first, that most sites of LXR–DNA binding are shared with RXR but only a fraction of RXR binding sites are shared with LXR; and second, that these whole-genome binding profiles are quite different across cell types283. RXR agonists have been shown to increase ABCA1 expression284,285, but they also elicit lipogenic side effects. The prototypical RXR agonist bexarotene demonstrated this combination, with induction of ABCA1/ABCG1 in certain tissues, but not others, that was accompanied by increased hepatic lipogenesis286, 287, 288. Additionally, a landmark study demonstrated enhanced cholesterol transport, Aβ clearance, and cognition in FAD mice289, but follow-up studies were unable to fully replicate these results290, 291, 292, 293, 294. In humans, bexarotene had no efficacy in AD clinical trials and increased TGs295,296. Recent reports described novel RXR agonists that boost ABCA1 expression and improve pathology in FAD mice, without TG-related liver effects297,298. Based on the many heterodimer interactions of RXR, biased agonism to induce ABCA1 in certain cells and tissues while avoiding hepatic lipogenesis appears feasible, possibly by selectively activating RXR–FXR or RXR–RAR heterodimers in preference to RXR-LXRα299, 300, 301.
4.5. Nuclear hormone receptor signaling: PPAR
PPARs function via heterodimer formation with RXR and are activated endogenously by oxidized fatty acids. The three PPAR isoforms perform overlapping yet distinct functions compared with each other and with LXR isoforms. PPARα is most prominent in fatty acid metabolism, PPARγ in glucose metabolism and anti-inflammation, and the less well-studied PPARβ/δ in fatty acid metabolism and anti-inflammation302. All three isoforms have been shown to modulate ABCA1 expression levels303,304, although this effect may require LXRα. PPARγ/RXR dimers have been proposed to control LXRα transcription by direct promoter binding284. Various PPAR agonists have been used clinically in T2D and CVD. Gemfibrozil and other PPARα-selective agonists increase ABCA1 activity, improve HDL and TG levels in preclinical and clinical studies, and demonstrate efficacy in mouse AD models305, 306, 307. Rosglitazone and pioglitazone, used clinically in T2D, failed to show efficacy in AD clinical trials190. Recently-developed PPARγ agonists continue to show positive effects on ABCA1 expression308,309, and a PPARβ/δ agonist that increases ABCA1 has also been described310. Various PPAR ligands have been described that interact with other NHRs311,312. Furthermore, selective PPAR modulators (SPPARMs) have been explored313,314.
4.6. Nuclear hormone receptor signaling: LXR
LXR is the primary NHR target for pharmacologic induction of ABCA1 expression. Humans express two isoforms: LXRα that is highly expressed in liver, small intestine, and adipose, and LXRβ that demonstrates ubiquitous expression, including brain tissue106,315, 316, 317. Endogenous oxysterols, e.g., 24HC, activate LXR, which acts via LXR response elements consisting of two direct repeats of a consensus sequence (i.e., ATTGCA) occurring in the promoter regions of dozens of genes. LXR–DNA binding results in transactivation or transrepression of various target genes related to cholesterol transport (e.g., ABCA1) and synthesis, glucose metabolism, and inflammatory signaling318,319. A final set of LXR target genes consists of the machinery that controls TG synthesis, both via direct transcriptional activation and through upregulation of sterol-response element binding protein 1c (SREBP1c)320. The earliest synthetic LXR agonist, T0901317 (T0), elicited potent responses in all of these transcriptional functions, demonstrating phenotypic improvements in mouse models, while also provoking TG overproduction and steatohepatitis321, 322, 323, 324, 325, 326, 327.
Treatment of Lxra–/– and Lxrb–/– mice with T0 demonstrated that hypertriglyceridemia was dependent on LXRα328; thus, development of LXRβ-selective agonists was prioritized. The first such “selective” agonist was GW3965; however, this compound ultimately was not phenotypically selective as it increased TGs in mouse models329,330. Groups from Wyeth331,332, Merck333, Tokyo New Drug Research Laboratories334, 335, 336, Vitae337,338, and Bristol–Myers Squibb339, 340, 341 developed compounds highly selective for LXRβ in receptor binding assays. BMS-852927 was originally described as an LXRβ-selective partial agonist; and indeed, several LXR ligands show partial agonist activity at LXRα339, 340, 341, 342.
The results of development and characterization of LXR ligands in preclinical AD animal model studies are provided in Table 1. To summarize, early agonists T0 and GW3965 have been studied in multiple FAD models at doses ranging from 2.5 to 50 mg/kg/day and for treatment durations ranging from 6 days to 24 weeks, analyzed via a host of behavioral, immunohistochemical, and biochemical techniques. Despite markedly varied treatment paradigms, these two compounds consistently increased ABCA1 and APOE expression, reduced Aβ and inflammatory marker levels, and improved cognitive performance. Inclusion of ABCA1 KO mice in some studies revealed that many of the observed therapeutic effects were dependent on ABCA1 expression. Newer, LXRβ-selective ligands have primarily been tested in mouse models of atherosclerosis, but those evaluated for preclinical AD efficacy have demonstrated promising CNS pharmacokinetic–pharmacodynamic profiles and similar effects on AD-related pathology.
Table 1.
In vivo studies of LXR agonists in preclinical AD models.
| Compd. | Animal system | Key finding | Ref. |
|---|---|---|---|
Merck 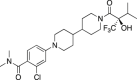
|
Wistar Han Rats s.c. 4 days. Pharmacokinetics (PK) and peripheral lipids. | Rats: Plasma and liver TG not significantly increased by compound or GW3965. CL = 87 mL/min/kg; t1/2 = 2.9 h; F (%) = 71%; PPB = 98.2% | Stachel et al., 2016333 |
| Tg2576 FAD-Tg mice s.c. 3 weeks. PK, open field test (OFT), brain ABCA1, apoE, and Aβ. | Mice: PK comparable to rat. Increased ABCA1 and apoE. Decreased soluble Aβ. Reduced locomotion back to WT control. | ||
| Rhesus monkey p.o. 2-weeks. PK, CSF apoE and Aβ, peripheral lipids. | Rhesus monkey: (CL = 18 mL/min/kg; t1/2 = 2.0 h; F (%) = 77%; PPB = 99.3%). 3-fold increased apoE. CSF Aβ levels increased. No change in liver fat content. | ||
Vitae 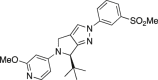
|
C57BL/6 mice 3 mg/kg p.o. 4 h. PK and brain ABCA1. | LXRβ EC50 = 38 nmol/L; 77% Emax; LXRα EC50 = 166 nmol/L; 83% Emax Mouse 4 h: Plasma 1075 nmol/L, brain = 1018 nmol/L; ABCA1 induction = 3.1× |
Tice et al., 2016338 |
| Sprague–Dawley rats p.o. 4 h (5 mg/kg) or 5 days (1, 3, or 10 mg/kg qd). Brain ABCA1, CSF and cerebral Aβ40/Aβ42. |
Rat 4 h: Plasma = 1215 nmol/L, brain = 2282 nmol/L, ABCA1 induction = 2.8× Rat 5 day: Plasma = 187–2800 nmol/L, Brain = 641–5360 nmol/L, ABCA1 induction = 5–9×; no significant changes in Aβ. |
||
T0901317 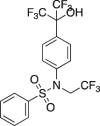
|
APP23 FAD-Tg mice p.o. 6 days. Measured Aβ. | Increased ABCA1; Aβ40 and Aβ42 significantly decreased. | Koldamova et al., 2005324 |
| APP23 mice p.o. 20–25 days. Cortical and hippocampal Aβ, ABCA1 and apoE, and inflammatory markers (mRNA). | Increased ABCA1 and apoE; significant reduction in insoluble Aβ with no effect on soluble Aβ. Inflammatory gene expression significantly decreased. | Lefterov et al., 2007346 | |
| Tg2576 mice p.o. 7 days. Contextual fear conditioning and contextual memory tasks (CFT). Hippocampal and cortical apoE and ABCA1 (mRNA), Aβ, and APP; plasma Aβ. | Complete reversal of cognitive deficits in CFT. Increased hippocampal ABCA1 and apoE. Significant Aβ42 reduction. No significant effects on full-length APP or processed APP. No significant effect on plasma Aβ. | Riddell et al., 2007347 | |
| APP23 mice p.o. 4 months with high-fat diet (HFD). Morris water maze (MWM) memory task. Cortical and hippocampal Aβ, apoE, ABCA1. | Reversal of MWM deficits caused by HFD. Reversal of increased insoluble Aβ due to HFD. Reduction of soluble Aβ beyond control diet levels. Increased ABCA1 and apoE. | Fitz et al., 2010348 | |
| APP23 mice p.o. 7 weeks. MWM. Drug levels, forebrain Aβ, ABCA1, and apoE. | No significant change in MWM performance. Brain drug level 5 nmol/g vs. blood 2 nmol/mL. Soluble and insoluble Aβ reduced; ABCA1 and apoE increased. | Terwel et al., 2011349 | |
| APPswe/PS1 e9 (APP/PS1) mice p.o. 2 months. NOR and object location task. Brain and serum cholesterol profiles, hippocampal and cortical Aβ, ABCA1, and apoE. | Improved cognitive performance. Increased cholesterol precursor levels. Increased ABCA1 and apoE. No change in Aβ plaque burden | Vanmierlo et al., 2011350 | |
| APP/PS1 mice p.o. 30 days. MWM. Hippocampal and cortical Aβ, GFAP, CD11b, ABCG1, apoJ, and inflammatory markers. Nucleus basalis ChAT. | Improved MWM performance. Increased ABCG1 and apoJ, increased cholinergic neurons. Reduced astrocytosis and microgliosis, reduced COX2 and iNOS, reduced total Aβ. | Cui et al., 2012351 | |
| APP23 mice p.o. 50 days. Radial arm water maze (RWM) and CFT. Hippocampal and cortical Aβ, ABCA1, apoE; interstitial fluid Aβ and apoE. | Cognitive performance restored. No change in soluble or insoluble Aβ, but reduced Aβ in ISF. Increased ABCA1 and apoE, increased lipidation of ISF apoE. | Fitz et al., 2014352 | |
| APP/PS1ΔE9/ABCA1+/−/APOE-TR mice p.o. 28 days. Novel object recognition (NOR) and CFT. Cortical Aβ, ABCA1 and apoE; apoE lipidation (native PAGE). | Increased ABCA1; increased apoE lipidation without change in total apoE. Significant reduction of soluble oAβ in APOE4 but not APOE3 mice, no change in Aβ plaques. CFT improvement in APOE4 but not APOE3 mice; no change in NOR. | Carter et al., 2017353 | |
GW3965 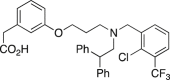
|
Tg2576 mice p.o. 4 months (Aβ) or 6 days (CFT). | Improved contextual memory, reduced Aβ plaque and peptide. | Jiang et al., 2008148 |
| APP/PS1 mice p.o. 8 or 24 weeks. NOR and Morris water maze (MWM). Cortical and hippocampal Aβ, ABCA1 and apoE. | Increased ABCA1 and apoE; increased CSF apoE lipidation. Reduced amyloid plaques and shifted Aβ from insoluble to soluble pool. Improved NOR and MWM performance. | Donkin et al., 2010145 | |
| Tg2576 mice p.o. 2 weeks. Odor habituation task, electrophysiology, whole-brain Aβ. | Enhanced odor habituation. Restored odor-evoked neural activity circuits. Reduced soluble and insoluble Aβ. | Wesson et al., 2011354 | |
| APP/PS1 (±ABCA1 KO) p.o. 8 weeks. Cortical, hippocampal, and CSF apoE and apoA1. | Cortical apoE and apoA1, hippocampal apoA1, and CSF apoA1 increased. Hippocampal and CSF apoE unchanged. | Stukas et al., 2012355 | |
| 3xTg FAD mice p.o. 12 weeks. MWM. Hippocampal and cortical Aβ, NFT, ABCA1, apoE; dentate gyrus nestin and pHH3. | Improved MWM performance. Soluble/insoluble Aβ and p-tau unchanged. Increased ABCA1 and apoE. Astro- and microgliosis reduced to WT baseline. Increased neurogenesis. | Sandoval-Hernandez et al., 2015356 | |
| APP/PS1 mice p.o. 9 days. CFT. Hippocampal and cortical ABCA1, ABCG1, apoE, inflammatory markers, and Aβ. | CFT performance restored to level of WT control. ABCA1/G1 and apoE increased. Non-significant reductions in Aβ. Iba1 significantly reduced; TNFα and others non-significant. | Skerrett et al., 2015357 | |
| 3xTg mice p.o. 6 days. MWM, hippocampal Aβ, gene methylation, and synaptic proteins (JMN). Hippocampal GFAP/Iba1, LRP1, and lectin staining (Neuro Lett). | MWM retention restored to WT baseline; learning unaffected. No change in Aβ; PSD95 and synapsin-1 increased; and DNA methylation of synaptic genes decreased. Reduced GFAP and altered morphology, no change in microglia; increased LRP1; decreased vascular tortuosity with reduced perivascular Aβ. | Sandoval-Hernandez et al., 2016 (split across two articles)358,359 |
Two LXRβ agonists, BMS-852927 and LXR-623, entered human clinical trials, with both compounds raising ABCA1 and ABCG1 expression levels342,343; however, CNS adverse events were observed with LXR-623; and peripheral side effects, including neutropenia and increased TG and LDL levels, were observed with BMS-852927. The findings with BMS-852927 are consistent with the differences between rodent and human lipid physiology that will need to be addressed in future LXR-based drug development efforts. Specifically, rodents lack the plasma cholesterol ester transfer protein (CETP) that humans express, and LXR produces a distinct response on inducible degrader of LDL (IDOL) protein in rodents versus humans342,344,345.
4.7. LXR-based drug discovery strategies for boosting ABCA1
Approaches to modulate LXR should be informed by the clinically successful modulation of other NHRs, notably ER. Selective estrogen receptor modulators (SERMs) are clinically important ERα ligands that deliver diverse pharmacology that is tissue-selective360, 361, 362. Selective androgen receptor modulators have not reached the clinic, but are drugs of abuse in sports because of muscle and bone building effects, purportedly without side effects associated with steroids363. SPPARMs have been introduced above. A similar concept has also been applied to label some LXR ligands as SLIMs (selective liver x receptor modulators)364,365. For example, SR9238, described as a liver-selective LXR agonist, could be classed as a SLIM366,367. The clinical relevance of NHR selective modulators rests on: a) enhancement of beneficial transcriptional events; without b) induction of transcriptional events associated with adverse effects. In this transcriptional context, the terms agonist and antagonist have little value. The transcriptional output of the NHR transcriptional complex depends on the cellular context (availability and binding of coregulators) and the ligand (differential stabilization of complexes)368. For example, IMB-808 binds both LXR isoforms, but, in contrast to T0, cholesterol efflux genes are selectively induced over lipogenic genes because IMB-808 and T0 recruit different coregulators369.
LXRs bind to LXR response elements that occur in the promoter regions of dozens of genes leading to transactivation or transrepression of genes related to (A) cholesterol transport (e.g., ABCA1, APOE) and glucose metabolism; and (B) the cellular lipogenic machinery that controls triglyceride synthesis, both via direct transcriptional activation and through upregulation of SREBP1c318,319. Biochemical measurement of ligand binding to truncated ligand-binding domains (LBD) of LXR does not reflect the influence of a ligand on transcriptional output, which includes expression of ABCA1 and hundreds of other genes327,370, 371, 372, 373. Specifically, ABCA1 and SREBP1c expression require distinct combinations of nuclear coregulator displacement/recruitment. LXR target genes are not controlled identically via the stabilization/derepression mechanism. ABCA1 is controlled via derepression, in which LXR–/– mice exhibit higher gene expression than LXR+/+ mice at baseline, whereas SREBF1 (gene encoding SREBP1) is controlled by the classical nuclear receptor model of receptor recruitment to the DNA promoter upon ligand binding, with basal expression in LXR+/+ ≥ LXR–/−372. Thus, unique ligands that are equipotent for LXRα and LXRβ LBD-binding in vitro can elicit unique transcriptional and phenotypic responses in complex biological systems.
A recent seminal paper describes the challenge for LXR ligand design: specifically, this work was focused on nonlipogenic ABCA1 inducers using in vivo phenotypic outputs of intestinal ABCA1 versus plasma triglycerides as binary ligand descriptors for multivariate statistical correlation with: i) H/D-exchange mass spectrometry; surface plasmon resonance binding to coregulators; LXR–LBD affinity; LXR transactivation; and ii) computational modeling374. One clear observation from hydrogen–deuterium exchange and surface plasmon resonance data is that the almost identical ligand binding pockets of LXRα and LXRβ are not an insurmountable barrier to design of LXR ligands with selective pharmacology: different chemical structures stabilized overlapping but nonidentical regions of LXR. More lipogenic ligands stabilized helix-12 (H12) and coactivator peptide binding (Fig. 4); whereas stabilization of the H3/H5 interface and corepressor peptide binding may bias towards ABCA1 inducers. As a corollary to this work, simply optimizing ligand affinity for LXRβ–LBD versus LXRα–LBD will result in LXRβ selectivity, but not necessarily nonlipogenic ABCA1 inducers. Structures for a variety of LXR complexes are shown (including one in a heterodimeric complex with RXR bound to DNA) and compared with ER complexes showing similarity between NHR architecture, ligand binding, and coregulator recruitment.
Figure 4.

NHR and ligand structures (H12 orange; H3 yellow; H11 blue; ligand red; coregulator silver). (A) ERα in “antagonist conformation” with SERM raloxifene displacing H12 (PDB 2JFA). (B) ERα in “agonist conformation” with TTC-352 inducing closure, stabilizing H12, and binding the coactivator NCOA2 (PDB 7JHD). (C) Merck Comp9 LXR agonist (LXRβ: PDB 5HJP). (D) LXR “inverse agonist” (LXRβ: PDB 6K9M)375. (E) GSK3986 (LXRα; PDB 2ACL with NCOA1 bound): showing similarity with ER agonist conformation forming AF-2; and similar binding poses with both LXR isoforms and with agonists and inverse agonists. (F) LXRβ:RXRα heterodimer bound to DR-4 DNA element including LBDs, coregulator, and DNA-binding domains (4NQA)376: although containing more components of the transcriptional complex, intrinsically disordered N-terminal domains (containing AF-1) are absent. Tissue selectivity of NR ligands is widely believed to result from cell-specific coregulator expression370; however, the ligand is dominant as the linchpin in allosteric communication between coregulators and DNA377.
A final complexity in designing LXR ligands is the extensive cross-talk that occurs among NHRs, such that activation of one may amplify or depress the expression and/or functional output of another283,378, 379, 380, 381. In addition to cross-talk, cross-reactivity between LXR and other NHRs is not uncommon. T0 is used as a benchmark LXR agonist; however, it has off-target activity at FXR382. The LXR agonist GSK2033 binds to PPARs366. Fibrates, used successfully to treat hypertriglyceridemia, bind both PPARα and LXRs, with inhibition of lipogenesis and SREBP1c expression attributed to “antagonist” action at LXR383. GSK2033, described as the first potent LXR antagonist (IC50 = 31.8 nmol/L for LXRβ)76, also engages glucocorticoid receptor, pregnane X receptor, and FXR109.
4.8. Phenotypic drug discovery strategies
Phenotypic drug discovery for nonlipogenic ABCA1 inducers is likely to identify some LXR ligands that act as SLIMs and others that regulate NHRs, either by direct binding or by feedback modulation. Given the poor correlation of phenotype with binding affinity (for LXRα versus LXRβ) and the complexity of NHR feedback, phenotypic screening is the logical approach to discover nonlipogenic ABCA1 inducers. A target-agnostic approach that prioritizes phenotype allows for identification of compounds that engage novel targets or, potentially, multiple targets to produce the desired effect. Notably, both PPAR agonist E3317 and the HDACi trichostatin A, described above, were identified as ABCA1 inducers via phenotypic approaches277,309.
A number of phenotypic screening efforts have been reported focusing on APOE transcription or secretion as the primary readout, some with ABCA1 upregulation as a secondary readout384, 385, 386. However, in one case, the counterscreen was designed to remove ABCA1 inducers387. The perceived lipogenic risk associated with LXR agonists led Pfizer, Astra Zeneca, and others to screen CCF-STTG1 astrocytoma cells to identify hits that increase APOE, counterscreening to triage LXR ligands, although subsequent validation of resulting compounds in animal models is lacking384, 385, 386,388,389. We recently described a luciferase-based phenotypic strategy employing an ABCA1 promoter to identify ABCA1 boosting compounds and a SREBF1 promoter to counterscreen against lipogenesis in HepG2 cells. This approach yielded multiple compounds with ABCA1-boosting, non-lipogenic profiles both in vitro and in vivo390. Chemical optimization resulted in a lead compound that had beneficial effects in an obesogenic mouse model of T2D. The broad metabolic effects observed prompted further investigation, revealing a binding profile of full agonist activity at LXRβ and partial agonist activity at LXRα, with weak antagonism of PPAR and RXR isoforms391. This strategy did not preclude LXR ligands from being identified; indeed, based on the promoter sequence, it was biased towards LXR ligands. Furthermore, this ABCA1-inducing LXR agonist was not only nonlipogenic, but also reduced triglycerides in the obesogenic mouse model.
5. Conclusions
AD and related metabolic conditions are among the top causes of morbidity and mortality in the United States and worldwide, with existing therapies demonstrating minimal efficacy at reversing the progression of or enhancing survival from AD. Cholesterol metabolism and transport in both the CNS and periphery are central to the pathophysiology of AD and related diseases such as T2D and CVD, impacting such processes as Aβ and NFT production and deposition, atherosclerosis, inflammatory signaling, and insulin resistance. Because of this critical importance of cholesterol homeostasis in these conditions, boosting expression or function of the primary cholesterol transport protein ABCA1 has been proposed as a therapeutic target. Drug development efforts to enhance ABCA1 have focused on nuclear hormone receptor—particularly, liver X receptor—agonists. These agonists have demonstrated promising results in preclinical AD models; however, their development and translation to the clinic has been hampered by an inability to avoid undesirable effects on triglyceride biogenesis. Recent research in LXR biology, as well as experience from drug discovery at other NHRs, suggests that the central paradigms that have driven LXR/ABCA1-based drug development are overly simplistic. In particular, the focus on developing agonists selective for LXRβ vs. LXRα isoforms will not yield non-lipogenic ABCA1 inducers as was previously thought. Rather, future medicinal chemistry efforts should strive to produce selective receptor modulators (e.g., SLIMs or SPPARMs described above) that elicit only a small fraction of biological effects regardless of activity in receptor binding assays. Phenotypic, instead of target-based, drug discovery approaches are well-suited to approach this challenge; indeed, several in vitro and early preclinical screens using various phenotypic assays have yielded numerous promising development candidates.
Acknowledgments
Cutler T. Lewandowski was supported by NIH T32AG57468 (USA) and American Heart Association 20PRE35150022 (USA) and is a trainee in the University of Illinois Medical Scientist Training Program (USA). Additional funding was provided through the UICentre for Drug Discovery as supported by the National Center for Advancing Translational Sciences, NIH UL1TR002003 (USA).
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Author contributions
Cutler T. Lewandowski wrote the original draft, produced figures and tables, and contributed to revisions. Megan S. Laham produced tables and figures and contributed to revisions. Gregory R.J. Thatcher provided supervision, acquired research funding, and contributed to revisions.
Conflicts of interest
Gregory R. J. Thatcher is an inventor on patents owned by the University of Illinois. The other authors have no competing interests or relationships to disclose.
References
- 1.Alzheimer’s A. 2020 Alzheimer's disease facts and figures. Alzheimers Dement. 2020;16:391–460. [Google Scholar]
- 2.Alzheimer's A. 2016 Alzheimer's disease facts and figures. Alzheimers Dement. 2016;12:459–509. doi: 10.1016/j.jalz.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 3.Briggs R., Kennelly S.P., O'Neill D. Drug treatments in Alzheimer's disease. Clin Med (Lond) 2016;16:247–253. doi: 10.7861/clinmedicine.16-3-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yiannopoulou K.G., Papageorgiou S.G. Current and future treatments for Alzheimer's disease. Ther Adv Neurol Disord. 2013;6:19–33. doi: 10.1177/1756285612461679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cummings J.L., Morstorf T., Zhong K. Alzheimer's disease drug-development pipeline: few candidates, frequent failures. Alzheimer's Res Ther. 2014;6:37. doi: 10.1186/alzrt269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berk C., Sabbagh M.N. Successes and failures for drugs in late-stage development for Alzheimer's disease. Drugs Aging. 2013;30:783–792. doi: 10.1007/s40266-013-0108-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cummings J., Lee G., Ritter A., Sabbagh M., Zhong K. Alzheimer's disease drug development pipeline: 2020. Alzheimers Dement (N Y) 2020;6:e12050. doi: 10.1002/trc2.12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang H.F., Shen X.N., Li J.Q., Suckling J., Tan C.C., Wang Y.J., et al. Clinical and biomarker trajectories in sporadic Alzheimer's disease: a longitudinal study. Alzheimers Dement (Amst) 2020;12:e12095. doi: 10.1002/dad2.12095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Livingston G., Huntley J., Sommerlad A., Ames D., Ballard C., Banerjee S., et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet. 2020;396:413–446. doi: 10.1016/S0140-6736(20)30367-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bennett D.A., Schneider J.A., Arvanitakis Z., Kelly J.F., Aggarwal N.T., Shah R.C., et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology. 2006;66:1837–1844. doi: 10.1212/01.wnl.0000219668.47116.e6. [DOI] [PubMed] [Google Scholar]
- 11.Negash S., Wilson R.S., Leurgans S.E., Wolk D.A., Schneider J.A., Buchman A.S., et al. Resilient brain aging: characterization of discordance between Alzheimer's disease pathology and cognition. Curr Alzheimer Res. 2013;10:844–851. doi: 10.2174/15672050113109990157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu L., Tasaki S., Schneider J.A., Arfanakis K., Duong D.M., Wingo A.P., et al. Cortical proteins associated with cognitive resilience in community-dwelling older persons. JAMA Psychiatry. 2020;77:1172–1180. doi: 10.1001/jamapsychiatry.2020.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu L., Petyuk V.A., Gaiteri C., Mostafavi S., Young-Pearse T., Shah R.C., et al. Targeted brain proteomics uncover multiple pathways to Alzheimer's dementia. Ann Neurol. 2018;84:78–88. doi: 10.1002/ana.25266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Graham E.K., James B.D., Jackson K.L., Willroth E.C., Boyle P., Wilson R., et al. Associations between personality traits and cognitive resilience in older adults. J Gerontol B Psychol Sci Soc Sci. 2021;76:6–19. doi: 10.1093/geronb/gbaa135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Legdeur N., Badissi M., Carter S.F., de Crom S., van de Kreeke A., Vreeswijk R., et al. Resilience to cognitive impairment in the oldest-old: design of the EMIF-AD 90+ study. BMC Geriatr. 2018;18:289. doi: 10.1186/s12877-018-0984-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jansen I.E., Savage J.E., Watanabe K., Bryois J., Williams D.M., Steinberg S., et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat Genet. 2019;51:404–413. doi: 10.1038/s41588-018-0311-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dumitrescu L., Mahoney E.R., Mukherjee S., Lee M.L., Bush W.S., Engelman C.D., et al. Genetic variants and functional pathways associated with resilience to Alzheimer's disease. Brain. 2020;143:2561–2575. doi: 10.1093/brain/awaa209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsuzaki T., Sasaki K., Tanizaki Y., Hata J., Fujimi K., Matsui Y., et al. Insulin resistance is associated with the pathology of Alzheimer disease: the Hisayama study. Neurology. 2010;75:764–770. doi: 10.1212/WNL.0b013e3181eee25f. [DOI] [PubMed] [Google Scholar]
- 19.Bolos M., Perea J.R., Avila J. Alzheimer's disease as an inflammatory disease. Biomol Concepts. 2017;8:37–43. doi: 10.1515/bmc-2016-0029. [DOI] [PubMed] [Google Scholar]
- 20.Heneka M.T., Carson M.J., El Khoury J., Landreth G.E., Brosseron F., Feinstein D.L., et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol. 2015;14:388–405. doi: 10.1016/S1474-4422(15)70016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vieira M.N.N., Lima-Filho R.A.S., De Felice F.G. Connecting Alzheimer's disease to diabetes: underlying mechanisms and potential therapeutic targets. Neuropharmacology. 2018;136:160–171. doi: 10.1016/j.neuropharm.2017.11.014. [DOI] [PubMed] [Google Scholar]
- 22.Vagelatos N.T., Eslick G.D. Type 2 diabetes as a risk factor for Alzheimer's disease: the confounders, interactions, and neuropathology associated with this relationship. Epidemiol Rev. 2013;35:152–160. doi: 10.1093/epirev/mxs012. [DOI] [PubMed] [Google Scholar]
- 23.Li J., Cesari M., Liu F., Dong B.R., Vellas B. Effects of diabetes mellitus on cognitive decline in patients with Alzheimer disease: a systematic review. Can J Diabetes. 2017;41:114–119. doi: 10.1016/j.jcjd.2016.07.003. [DOI] [PubMed] [Google Scholar]
- 24.Chatterjee S., Peters S.A., Woodward M., Mejia Arango S., Batty G.D., Beckett N., et al. Type 2 diabetes as a risk factor for dementia in women compared with men: a pooled analysis of 2.3 million people comprising more than 100,000 cases of dementia. Diabetes Care. 2016;39:300–307. doi: 10.2337/dc15-1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Santos C.Y., Snyder P.J., Wu W.C., Zhang M., Echeverria A., Alber J. Pathophysiologic relationship between Alzheimer's disease, cerebrovascular disease, and cardiovascular risk: a review and synthesis. Alzheimers Dement (Amst) 2017;7:69–87. doi: 10.1016/j.dadm.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sabia S., Fayosse A., Dumurgier J., Schnitzler A., Empana J.P., Ebmeier K.P., et al. Association of ideal cardiovascular health at age 50 with incidence of dementia: 25 year follow-up of Whitehall II cohort study. BMJ. 2019;366:l4414. doi: 10.1136/bmj.l4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jayaraman A., Pike C.J. Alzheimer's disease and type 2 diabetes: multiple mechanisms contribute to interactions. Curr Diab Rep. 2014;14:476. doi: 10.1007/s11892-014-0476-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Irie F., Fitzpatrick A.L., Lopez O.L., Kuller L.H., Peila R., Newman A.B., et al. Enhanced risk for Alzheimer disease in persons with type 2 diabetes and APOE epsilon4: the cardiovascular health study cognition study. Arch Neurol. 2008;65:89–93. doi: 10.1001/archneurol.2007.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peila R., Rodriguez B.L., Launer L.J., Honolulu-Asia Aging S. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: the Honolulu-Asia aging study. Diabetes. 2002;51:1256–1262. doi: 10.2337/diabetes.51.4.1256. [DOI] [PubMed] [Google Scholar]
- 30.Kukull W.A., Higdon R., Bowen J.D., McCormick W.C., Teri L., Schellenberg G.D., et al. Dementia and Alzheimer disease incidence: a prospective cohort study. Arch Neurol. 2002;59:1737–1746. doi: 10.1001/archneur.59.11.1737. [DOI] [PubMed] [Google Scholar]
- 31.Gardner R.C., Valcour V., Yaffe K. Dementia in the oldest old: a multi-factorial and growing public health issue. Alzheimer's Res Ther. 2013;5:27. doi: 10.1186/alzrt181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kritsilis M., Rizou S.V., Koutsoudaki P.N., Evangelou K., Gorgoulis V.G., Papadopoulos D. Ageing, cellular senescence and neurodegenerative disease. Int J Mol Sci. 2018;19:2937. doi: 10.3390/ijms19102937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liguori I., Russo G., Curcio F., Bulli G., Aran L., Della-Morte D., et al. Oxidative stress, aging, and diseases. Clin Interv Aging. 2018;13:757–772. doi: 10.2147/CIA.S158513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bishop N.A., Lu T., Yankner B.A. Neural mechanisms of ageing and cognitive decline. Nature. 2010;464:529–535. doi: 10.1038/nature08983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bekris L.M., Yu C.E., Bird T.D., Tsuang D.W. Genetics of Alzheimer disease. J Geriatr Psychiatry Neurol. 2010;23:213–227. doi: 10.1177/0891988710383571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oakley H., Cole S.L., Logan S., Maus E., Shao P., Craft J., et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Games D., Adams D., Alessandrini R., Barbour R., Berthelette P., Blackwell C., et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 38.Duff K., Eckman C., Zehr C., Yu X., Prada C.M., Perez-tur J., et al. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383:710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- 39.Holcomb L., Gordon M.N., McGowan E., Yu X., Benkovic S., Jantzen P., et al. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- 40.Hsiao K., Chapman P., Nilsen S., Eckman C., Harigaya Y., Younkin S., et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 41.Radde R., Bolmont T., Kaeser S.A., Coomaraswamy J., Lindau D., Stoltze L., et al. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006;7:940–946. doi: 10.1038/sj.embor.7400784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mucke L., Masliah E., Yu G.Q., Mallory M., Rockenstein E.M., Tatsuno G., et al. High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jankowsky J.L., Slunt H.H., Ratovitski T., Jenkins N.A., Copeland N.G., Borchelt D.R. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng. 2001;17:157–165. doi: 10.1016/s1389-0344(01)00067-3. [DOI] [PubMed] [Google Scholar]
- 44.Lewandowski C.T., Maldonado Weng J., LaDu M.J. Alzheimer's disease pathology in APOE transgenic mouse models: the who, what, when, where, why, and how. Neurobiol Dis. 2020;139:104811. doi: 10.1016/j.nbd.2020.104811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oddo S., Caccamo A., Shepherd J.D., Murphy M.P., Golde T.E., Kayed R., et al. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 46.Oddo S., Caccamo A., Kitazawa M., Tseng B.P., LaFerla F.M. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer's disease. Neurobiol Aging. 2003;24:1063–1070. doi: 10.1016/j.neurobiolaging.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 47.Jackson R.J., Rudinskiy N., Herrmann A.G., Croft S., Kim J.M., Petrova V., et al. Human tau increases amyloid beta plaque size but not amyloid beta-mediated synapse loss in a novel mouse model of Alzheimer's disease. Eur J Neurosci. 2016;44:3056–3066. doi: 10.1111/ejn.13442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jankowsky J.L., Zheng H. Practical considerations for choosing a mouse model of Alzheimer's disease. Mol Neurodegener. 2017;12:89. doi: 10.1186/s13024-017-0231-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lippi S.L.P., Smith M.L., Flinn J.M. A novel hAPP/htau mouse model of Alzheimer's disease: inclusion of APP with tau exacerbates behavioral deficits and zinc administration heightens tangle pathology. Front Aging Neurosci. 2018;10:382. doi: 10.3389/fnagi.2018.00382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saito T., Mihira N., Matsuba Y., Sasaguri H., Hashimoto S., Narasimhan S., et al. Humanization of the entire murine Mapt gene provides a murine model of pathological human tau propagation. J Biol Chem. 2019;294:12754–12765. doi: 10.1074/jbc.RA119.009487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mckean N.E., Handley R.R., Snell R.G. A review of the current mammalian models of Alzheimer's disease and challenges that need to be overcome. Int J Mol Sci. 2021;22:13168. doi: 10.3390/ijms222313168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tai L.M., Maldonado Weng J., LaDu M.J., Brady S.T. Relevance of transgenic mouse models for Alzheimer's disease. Prog Mol Biol Transl Sci. 2021;177:1–48. doi: 10.1016/bs.pmbts.2020.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vitek M.P., Araujo J.A., Fossel M., Greenberg B.D., Howell G.R., Rizzo S.J.S., et al. Translational animal models for Alzheimer's disease: an Alzheimer's Association Business Consortium Think Tank. Alzheimers Dement (N Y) 2020;6:e12114. doi: 10.1002/trc2.12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Van Cauwenberghe C., Van Broeckhoven C., Sleegers K. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet Med. 2016;18:421–430. doi: 10.1038/gim.2015.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ulland T.K., Colonna M. TREM2—a key player in microglial biology and Alzheimer disease. Nat Rev Neurol. 2018;14:667–675. doi: 10.1038/s41582-018-0072-1. [DOI] [PubMed] [Google Scholar]
- 56.Jonsson T., Stefansson H., Steinberg S., Jonsdottir I., Jonsson P.V., Snaedal J., et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med. 2013;368:107–116. doi: 10.1056/NEJMoa1211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aikawa T., Holm M.L., Kanekiyo T. ABCA7 and pathogenic pathways of Alzheimer's disease. Brain Sci. 2018;8 doi: 10.3390/brainsci8020027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Steinberg S., Stefansson H., Jonsson T., Johannsdottir H., Ingason A., Helgason H., et al. Loss-of-function variants in ABCA7 confer risk of Alzheimer's disease. Nat Genet. 2015;47:445–447. doi: 10.1038/ng.3246. [DOI] [PubMed] [Google Scholar]
- 59.Guerreiro R., Wojtas A., Bras J., Carrasquillo M., Rogaeva E., Majounie E., et al. TREM2 variants in Alzheimer's disease. N Engl J Med. 2013;368:117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ma J., Zhou Y., Xu J., Liu X., Wang Y., Deng Y., et al. Association study of TREM2 polymorphism rs75932628 with late-onset Alzheimer's disease in Chinese Han population. Neurol Res. 2014;36:894–896. doi: 10.1179/1743132814Y.0000000376. [DOI] [PubMed] [Google Scholar]
- 61.Miyashita A., Wen Y., Kitamura N., Matsubara E., Kawarabayashi T., Shoji M., et al. Lack of genetic association between TREM2 and late-onset Alzheimer's disease in a Japanese population. J Alzheimers Dis. 2014;41:1031–1038. doi: 10.3233/JAD-140225. [DOI] [PubMed] [Google Scholar]
- 62.Jin S.C., Carrasquillo M.M., Benitez B.A., Skorupa T., Carrell D., Patel D., et al. TREM2 is associated with increased risk for Alzheimer's disease in African Americans. Mol Neurodegener. 2015;10:19. doi: 10.1186/s13024-015-0016-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mehrjoo Z., Najmabadi A., Abedini S.S., Mohseni M., Kamali K., Najmabadi H., et al. Association study of the TREM2 gene and identification of a novel variant in exon 2 in Iranian patients with late-onset Alzheimer's disease. Med Princ Pract. 2015;24:351–354. doi: 10.1159/000430842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang P., Guo Q., Zhou Y., Chen K., Xu Y., Ding D., et al. Lack of association between triggering receptor expressed on myeloid cells 2 polymorphism rs75932628 and late-onset Alzheimer's disease in a Chinese Han population. Psychiatr Genet. 2018;28:16–18. doi: 10.1097/YPG.0000000000000188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mahley R.W., Rall S.C., Jr. Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet. 2000;1:507–537. doi: 10.1146/annurev.genom.1.1.507. [DOI] [PubMed] [Google Scholar]
- 66.Corder E.H., Saunders A.M., Strittmatter W.J., Schmechel D.E., Gaskell P.C., Small G.W., et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 67.Farrer L.A., Cupples L.A., Haines J.L., Hyman B., Kukull W.A., Mayeux R., et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA. 1997;278:1349–1356. [PubMed] [Google Scholar]
- 68.Liu C.C., Liu C.C., Kanekiyo T., Xu H., Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9:106–118. doi: 10.1038/nrneurol.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Morrow J.A., Hatters D.M., Lu B., Hochtl P., Oberg K.A., Rupp B., et al. Apolipoprotein E4 forms a molten globule. A potential basis for its association with disease. J Biol Chem. 2002;277:50380–50385. doi: 10.1074/jbc.M204898200. [DOI] [PubMed] [Google Scholar]
- 70.Dong L.M., Weisgraber K.H. Human apolipoprotein E4 domain interaction. Arginine 61 and glutamic acid 255 interact to direct the preference for very low density lipoproteins. J Biol Chem. 1996;271:19053–19057. doi: 10.1074/jbc.271.32.19053. [DOI] [PubMed] [Google Scholar]
- 71.Morrow J.A., Segall M.L., Lund-Katz S., Phillips M.C., Knapp M., Rupp B., et al. Differences in stability among the human apolipoprotein E isoforms determined by the amino-terminal domain. Biochemistry. 2000;39:11657–11666. doi: 10.1021/bi000099m. [DOI] [PubMed] [Google Scholar]
- 72.de Chaves E.P., Narayanaswami V. Apolipoprotein E and cholesterol in aging and disease in the brain. Future Lipidol. 2008;3:505–530. doi: 10.2217/17460875.3.5.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tamboli I.Y., Heo D., Rebeck G.W. Extracellular proteolysis of apolipoprotein E (apoE) by secreted serine neuronal protease. PLoS One. 2014;9:e93120. doi: 10.1371/journal.pone.0093120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rohn T.T. Proteolytic cleavage of apolipoprotein E4 as the keystone for the heightened risk associated with Alzheimer's disease. Int J Mol Sci. 2013;14:14908–14922. doi: 10.3390/ijms140714908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Arboleda-Velasquez J.F., Lopera F., O'Hare M., Delgado-Tirado S., Marino C., Chmielewska N., et al. Resistance to autosomal dominant Alzheimer's disease in an APOE3 Christchurch homozygote: a case report. Nat Med. 2019;25:1680–1683. doi: 10.1038/s41591-019-0611-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wardell M.R., Brennan S.O., Janus E.D., Fraser R., Carrell R.W. Apolipoprotein E2-Christchurch (136 Arg→Ser). New variant of human apolipoprotein E in a patient with type III hyperlipoproteinemia. J Clin Invest. 1987;80:483–490. doi: 10.1172/JCI113096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Altmann A., Tian L., Henderson V.W., Greicius M.D., Alzheimer’s Disease Neuroimaging Initiative I Sex modifies the APOE-related risk of developing Alzheimer disease. Ann Neurol. 2014;75:563–573. doi: 10.1002/ana.24135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Neu S.C., Pa J., Kukull W., Beekly D., Kuzma A., Gangadharan P., et al. Apolipoprotein E genotype and sex risk factors for Alzheimer disease: a meta-analysis. JAMA Neurol. 2017;74:1178–1189. doi: 10.1001/jamaneurol.2017.2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ott A., Stolk R.P., van Harskamp F., Pols H.A., Hofman A., Breteler M.M. Diabetes mellitus and the risk of dementia: the Rotterdam study. Neurology. 1999;53:1937–1942. doi: 10.1212/wnl.53.9.1937. [DOI] [PubMed] [Google Scholar]
- 80.Bellou V., Belbasis L., Tzoulaki I., Middleton L.T., Ioannidis J.P.A., Evangelou E. Systematic evaluation of the associations between environmental risk factors and dementia: an umbrella review of systematic reviews and meta-analyses. Alzheimers Dement. 2017;13:406–418. doi: 10.1016/j.jalz.2016.07.152. [DOI] [PubMed] [Google Scholar]
- 81.Farris W., Mansourian S., Chang Y., Lindsley L., Eckman E.A., Frosch M.P., et al. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A. 2003;100:4162–4167. doi: 10.1073/pnas.0230450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kellar D., Craft S. Brain insulin resistance in Alzheimer's disease and related disorders: mechanisms and therapeutic approaches. Lancet Neurol. 2020;19:758–766. doi: 10.1016/S1474-4422(20)30231-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cai Z., Liu N., Wang C., Qin B., Zhou Y., Xiao M., et al. Role of RAGE in Alzheimer's disease. Cell Mol Neurobiol. 2016;36:483–495. doi: 10.1007/s10571-015-0233-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hughes T.M., Craft S. The role of insulin in the vascular contributions to age-related dementia. Biochim Biophys Acta. 2016;1862:983–991. doi: 10.1016/j.bbadis.2015.11.013. [DOI] [PubMed] [Google Scholar]
- 85.Roher A.E., Tyas S.L., Maarouf C.L., Daugs I.D., Kokjohn T.A., Emmerling M.R., et al. Intracranial atherosclerosis as a contributing factor to Alzheimer's disease dementia. Alzheimers Dement. 2011;7:436–444. doi: 10.1016/j.jalz.2010.08.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wingo A.P., Fan W., Duong D.M., Gerasimov E.S., Dammer E.B., Liu Y., et al. Shared proteomic effects of cerebral atherosclerosis and Alzheimer's disease on the human brain. Nat Neurosci. 2020;23:696–700. doi: 10.1038/s41593-020-0635-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zetterberg H., Mortberg E., Song L., Chang L., Provuncher G.K., Patel P.P., et al. Hypoxia due to cardiac arrest induces a time-dependent increase in serum amyloid beta levels in humans. PLoS One. 2011;6:e28263. doi: 10.1371/journal.pone.0028263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Beach T.G., Wilson J.R., Sue L.I., Newell A., Poston M., Cisneros R., et al. Circle of Willis atherosclerosis: association with Alzheimer's disease, neuritic plaques and neurofibrillary tangles. Acta Neuropathol. 2007;113:13–21. doi: 10.1007/s00401-006-0136-y. [DOI] [PubMed] [Google Scholar]
- 89.Bown C.W., Liu D., Osborn K.E., Gupta D.K., Mendes L.A., Pechman K.R., et al. Apolipoprotein E genotype modifies the association between cardiac output and cognition in older adults. J Am Heart Assoc. 2019;8:e011146. doi: 10.1161/JAHA.118.011146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mayeux R., Ottman R., Maestre G., Ngai C., Tang M.X., Ginsberg H., et al. Synergistic effects of traumatic head injury and apolipoprotein-epsilon 4 in patients with Alzheimer's disease. Neurology. 1995;45:555–557. doi: 10.1212/wnl.45.3.555. [DOI] [PubMed] [Google Scholar]
- 91.Durazzo T.C., Mattsson N., Weiner M.W., Alzheimer’s Disease Neuroimaging I. Interaction of cigarette smoking history with APOE genotype and age on amyloid level, glucose metabolism, and neurocognition in cognitively normal elders. Nicotine Tob Res. 2016;18:204–211. doi: 10.1093/ntr/ntv075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jensen C.S., Simonsen A.H., Siersma V., Beyer N., Frederiksen K.S., Gottrup H., et al. Patients with Alzheimer's disease who carry the APOE epsilon4 allele benefit more from physical exercise. Alzheimers Dement (N Y) 2019;5:99–106. doi: 10.1016/j.trci.2019.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bennet A.M., Di Angelantonio E., Ye Z., Wensley F., Dahlin A., Ahlbom A., et al. Association of apolipoprotein E genotypes with lipid levels and coronary risk. JAMA. 2007;298:1300–1311. doi: 10.1001/jama.298.11.1300. [DOI] [PubMed] [Google Scholar]
- 94.Xu M., Zhao J., Zhang Y., Ma X., Dai Q., Zhi H., et al. Apolipoprotein E gene variants and risk of coronary heart disease: a meta-analysis. BioMed Res Int. 2016;2016:3912175. doi: 10.1155/2016/3912175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.El-Lebedy D., Raslan H.M., Mohammed A.M. Apolipoprotein E gene polymorphism and risk of type 2 diabetes and cardiovascular disease. Cardiovasc Diabetol. 2016;15:12. doi: 10.1186/s12933-016-0329-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kunkle B.W., Grenier-Boley B., Sims R., Bis J.C., Damotte V., Naj A.C., et al. Genetic meta-analysis of diagnosed Alzheimer's disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet. 2019;51:414–430. doi: 10.1038/s41588-019-0358-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Misra A., Chakrabarti S.S., Gambhir I.S. New genetic players in late-onset Alzheimer's disease: findings of genome-wide association studies. Indian J Med Res. 2018;148:135–144. doi: 10.4103/ijmr.IJMR_473_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wellington C.L., Walker E.K., Suarez A., Kwok A., Bissada N., Singaraja R., et al. ABCA1 mRNA and protein distribution patterns predict multiple different roles and levels of regulation. Lab Invest. 2002;82:273–283. doi: 10.1038/labinvest.3780421. [DOI] [PubMed] [Google Scholar]
- 99.Kim W.S., Guillemin G.J., Glaros E.N., Lim C.K., Garner B. Quantitation of ATP-binding cassette subfamily—a transporter gene expression in primary human brain cells. Neuroreport. 2006;17:891–896. doi: 10.1097/01.wnr.0000221833.41340.cd. [DOI] [PubMed] [Google Scholar]
- 100.Fitzgerald M.L., Mendez A.J., Moore K.J., Andersson L.P., Panjeton H.A., Freeman M.W. ATP-binding cassette transporter A1 contains an NH2-terminal signal anchor sequence that translocates the protein's first hydrophilic domain to the exoplasmic space. J Biol Chem. 2001;276:15137–15145. doi: 10.1074/jbc.m100474200. [DOI] [PubMed] [Google Scholar]
- 101.Oram J.F. HDL apolipoproteins and ABCA1: partners in the removal of excess cellular cholesterol. Arterioscler Thromb Vasc Biol. 2003;23:720–727. doi: 10.1161/01.ATV.0000054662.44688.9A. [DOI] [PubMed] [Google Scholar]
- 102.Dean M., Hamon Y., Chimini G. The human ATP-binding cassette (ABC) transporter superfamily. J Lipid Res. 2001;42:1007–1017. [PubMed] [Google Scholar]
- 103.Oram J.F., Heinecke J.W. ATP-binding cassette transporter A1: a cell cholesterol exporter that protects against cardiovascular disease. Physiol Rev. 2005;85:1343–1372. doi: 10.1152/physrev.00005.2005. [DOI] [PubMed] [Google Scholar]
- 104.Remaley A.T., Stonik J.A., Demosky S.J., Neufeld E.B., Bocharov A.V., Vishnyakova T.G., et al. Apolipoprotein specificity for lipid efflux by the human ABCAI transporter. Biochem Biophys Res Commun. 2001;280:818–823. doi: 10.1006/bbrc.2000.4219. [DOI] [PubMed] [Google Scholar]
- 105.Francis G.A., Knopp R.H., Oram J.F. Defective removal of cellular cholesterol and phospholipids by apolipoprotein A-I in Tangier disease. J Clin Invest. 1995;96:78–87. doi: 10.1172/JCI118082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Frambach S., de Haas R., Smeitink J.A.M., Rongen G.A., Russel F.G.M., Schirris T.J.J. Brothers in arms: ABCA1- and ABCG1-mediated cholesterol efflux as promising targets in cardiovascular disease treatment. Pharmacol Rev. 2020;72:152–190. doi: 10.1124/pr.119.017897. [DOI] [PubMed] [Google Scholar]
- 107.Dietschy J.M., Turley S.D. Cholesterol metabolism in the brain. Curr Opin Lipidol. 2001;12:105–112. doi: 10.1097/00041433-200104000-00003. [DOI] [PubMed] [Google Scholar]
- 108.Mahley R.W. Central nervous system lipoproteins: ApoE and regulation of cholesterol metabolism. Arterioscler Thromb Vasc Biol. 2016;36:1305–1315. doi: 10.1161/ATVBAHA.116.307023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Andersson M., Elmberger P.G., Edlund C., Kristensson K., Dallner G. Rates of cholesterol, ubiquinone, dolichol and dolichyl-P biosynthesis in rat brain slices. FEBS Lett. 1990;269:15–18. doi: 10.1016/0014-5793(90)81107-y. [DOI] [PubMed] [Google Scholar]
- 110.Nieweg K., Schaller H., Pfrieger F.W. Marked differences in cholesterol synthesis between neurons and glial cells from postnatal rats. J Neurochem. 2009;109:125–134. doi: 10.1111/j.1471-4159.2009.05917.x. [DOI] [PubMed] [Google Scholar]
- 111.Valdez C.M., Smith M.A., Perry G., Phelix C.F., Santamaria F. Cholesterol homeostasis markers are localized to mouse hippocampal pyramidal and granule layers. Hippocampus. 2010;20:902–905. doi: 10.1002/hipo.20743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhang J., Liu Q. Cholesterol metabolism and homeostasis in the brain. Protein Cell. 2015;6:254–264. doi: 10.1007/s13238-014-0131-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Uchihara T., Duyckaerts C., He Y., Kobayashi K., Seilhean D., Amouyel P., et al. ApoE immunoreactivity and microglial cells in Alzheimer's disease brain. Neurosci Lett. 1995;195:5–8. doi: 10.1016/0304-3940(95)11763-m. [DOI] [PubMed] [Google Scholar]
- 114.Xu Q., Bernardo A., Walker D., Kanegawa T., Mahley R.W., Huang Y. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J Neurosci. 2006;26:4985–4994. doi: 10.1523/JNEUROSCI.5476-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pitas R.E., Boyles J.K., Lee S.H., Foss D., Mahley R.W. Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochim Biophys Acta. 1987;917:148–161. doi: 10.1016/0005-2760(87)90295-5. [DOI] [PubMed] [Google Scholar]
- 116.Rebeck G.W., Reiter J.S., Strickland D.K., Hyman B.T. Apolipoprotein E in sporadic Alzheimer's disease: allelic variation and receptor interactions. Neuron. 1993;11:575–580. doi: 10.1016/0896-6273(93)90070-8. [DOI] [PubMed] [Google Scholar]
- 117.Orth M., Bellosta S. Cholesterol: its regulation and role in central nervous system disorders. Cholesterol. 2012;2012:292598. doi: 10.1155/2012/292598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.El Asmar Z., Terrand J., Jenty M., Host L., Mlih M., Zerr A., et al. Convergent signaling pathways controlled by LRP1 (receptor-related protein 1) cytoplasmic and extracellular domains limit cellular cholesterol accumulation. J Biol Chem. 2016;291:5116–5127. doi: 10.1074/jbc.M116.714485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zlokovic B.V., Deane R., Sagare A.P., Bell R.D., Winkler E.A. Low-density lipoprotein receptor-related protein-1: a serial clearance homeostatic mechanism controlling Alzheimer's amyloid beta-peptide elimination from the brain. J Neurochem. 2010;115:1077–1089. doi: 10.1111/j.1471-4159.2010.07002.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Moutinho M., Nunes M.J., Rodrigues E. Cholesterol 24-hydroxylase: brain cholesterol metabolism and beyond. Biochim Biophys Acta. 2016;1861:1911–1920. doi: 10.1016/j.bbalip.2016.09.011. [DOI] [PubMed] [Google Scholar]
- 121.Cruchaga C., Kauwe J.S., Nowotny P., Bales K., Pickering E.H., Mayo K., et al. Cerebrospinal fluid APOE levels: an endophenotype for genetic studies for Alzheimer's disease. Hum Mol Genet. 2012;21:4558–4571. doi: 10.1093/hmg/dds296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Riddell D.R., Zhou H., Atchison K., Warwick H.K., Atkinson P.J., Jefferson J., et al. Impact of apolipoprotein E (ApoE) polymorphism on brain ApoE levels. J Neurosci. 2008;28:11445–11453. doi: 10.1523/JNEUROSCI.1972-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Minagawa H., Gong J.S., Jung C.G., Watanabe A., Lund-Katz S., Phillips M.C., et al. Mechanism underlying apolipoprotein E (ApoE) isoform-dependent lipid efflux from neural cells in culture. J Neurosci Res. 2009;87:2498–2508. doi: 10.1002/jnr.22073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lin Y.T., Seo J., Gao F., Feldman H.M., Wen H.L., Penney J., et al. APOE4 causes widespread molecular and cellular alterations associated with Alzheimer's disease phenotypes in human iPSC-derived brain cell types. Neuron. 2018;98:1141–1154. doi: 10.1016/j.neuron.2018.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Boehm-Cagan A., Bar R., Harats D., Shaish A., Levkovitz H., Bielicki J.K., et al. Differential effects of apoE4 and activation of ABCA1 on brain and plasma lipoproteins. PLoS One. 2016;11:e0166195. doi: 10.1371/journal.pone.0166195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhao J., Davis M.D., Martens Y.A., Shinohara M., Graff-Radford N.R., Younkin S.G., et al. APOE epsilon4/epsilon4 diminishes neurotrophic function of human iPSC-derived astrocytes. Hum Mol Genet. 2017;26:2690–2700. doi: 10.1093/hmg/ddx155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Fu Y., Zhao J., Atagi Y., Nielsen H.M., Liu C.C., Zheng H., et al. Apolipoprotein E lipoprotein particles inhibit amyloid-beta uptake through cell surface heparan sulphate proteoglycan. Mol Neurodegener. 2016;11:37. doi: 10.1186/s13024-016-0099-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chen J., Li Q., Wang J. Topology of human apolipoprotein E3 uniquely regulates its diverse biological functions. Proc Natl Acad Sci U S A. 2011;108:14813–14818. doi: 10.1073/pnas.1106420108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hubin E., Verghese P.B., van Nuland N., Broersen K. Apolipoprotein E associated with reconstituted high-density lipoprotein-like particles is protected from aggregation. FEBS Lett. 2019;593:1144–1153. doi: 10.1002/1873-3468.13428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Munoz S.S., Garner B., Ooi L. Understanding the role of ApoE fragments in Alzheimer's disease. Neurochem Res. 2019;44:1297–1305. doi: 10.1007/s11064-018-2629-1. [DOI] [PubMed] [Google Scholar]
- 131.Tai L.M., Mehra S., Shete V., Estus S., Rebeck G.W., Bu G., et al. Soluble apoE/Abeta complex: mechanism and therapeutic target for APOE4-induced AD risk. Mol Neurodegener. 2014;9:2. doi: 10.1186/1750-1326-9-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Marquer C., Laine J., Dauphinot L., Hanbouch L., Lemercier-Neuillet C., Pierrot N., et al. Increasing membrane cholesterol of neurons in culture recapitulates Alzheimer's disease early phenotypes. Mol Neurodegener. 2014;9:60. doi: 10.1186/1750-1326-9-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tai L.M., Bilousova T., Jungbauer L., Roeske S.K., Youmans K.L., Yu C., et al. Levels of soluble apolipoprotein E/amyloid-beta (Abeta) complex are reduced and oligomeric Abeta increased with APOE4 and Alzheimer disease in a transgenic mouse model and human samples. J Biol Chem. 2013;288:5914–5926. doi: 10.1074/jbc.M112.442103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hashimoto T., Serrano-Pozo A., Hori Y., Adams K.W., Takeda S., Banerji A.O., et al. Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid beta peptide. J Neurosci. 2012;32:15181–15192. doi: 10.1523/JNEUROSCI.1542-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Koffie R.M., Hashimoto T., Tai H.C., Kay K.R., Serrano-Pozo A., Joyner D., et al. Apolipoprotein E4 effects in Alzheimer's disease are mediated by synaptotoxic oligomeric amyloid-beta. Brain. 2012;135:2155–2168. doi: 10.1093/brain/aws127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hoglund K., Kern S., Zettergren A., Borjesson-Hansson A., Zetterberg H., Skoog I., et al. Preclinical amyloid pathology biomarker positivity: effects on tau pathology and neurodegeneration. Transl Psychiatry. 2017;7:e995. doi: 10.1038/tp.2016.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Koriath C., Lashley T., Taylor W., Druyeh R., Dimitriadis A., Denning N., et al. ApoE4 lowers age at onset in patients with frontotemporal dementia and tauopathy independent of amyloid-beta copathology. Alzheimers Dement (Amst) 2019;11:277–280. doi: 10.1016/j.dadm.2019.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Farfel J.M., Yu L., De Jager P.L., Schneider J.A., Bennett D.A. Association of APOE with tau-tangle pathology with and without beta-amyloid. Neurobiol Aging. 2016;37:19–25. doi: 10.1016/j.neurobiolaging.2015.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wahrle S.E., Jiang H., Parsadanian M., Legleiter J., Han X., Fryer J.D., et al. ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. J Biol Chem. 2004;279:40987–40993. doi: 10.1074/jbc.M407963200. [DOI] [PubMed] [Google Scholar]
- 140.Karasinska J.M., Rinninger F., Lutjohann D., Ruddle P., Franciosi S., Kruit J.K., et al. Specific loss of brain ABCA1 increases brain cholesterol uptake and influences neuronal structure and function. J Neurosci. 2009;29:3579–3589. doi: 10.1523/JNEUROSCI.4741-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hirsch-Reinshagen V., Zhou S., Burgess B.L., Bernier L., McIsaac S.A., Chan J.Y., et al. Deficiency of ABCA impairs apolipoprotein E metabolism in brain. J Biol Chem. 2004;279:41197–41207. doi: 10.1074/jbc.M407962200. [DOI] [PubMed] [Google Scholar]
- 142.Hirsch-Reinshagen V., Maia L.F., Burgess B.L., Blain J.F., Naus K.E., McIsaac S.A., et al. The absence of ABCA1 decreases soluble ApoE levels but does not diminish amyloid deposition in two murine models of Alzheimer disease. J Biol Chem. 2005;280:43243–43256. doi: 10.1074/jbc.M508781200. [DOI] [PubMed] [Google Scholar]
- 143.Rawat V., Wang S., Sima J., Bar R., Liraz O., Gundimeda U., et al. ApoE4 alters ABCA1 membrane trafficking in astrocytes. J Neurosci. 2019;39:9611–9622. doi: 10.1523/JNEUROSCI.1400-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wahrle S.E., Jiang H., Parsadanian M., Kim J., Li A., Knoten A., et al. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J Clin Invest. 2008;118:671–682. doi: 10.1172/JCI33622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Donkin J.J., Stukas S., Hirsch-Reinshagen V., Namjoshi D., Wilkinson A., May S., et al. ATP-binding cassette transporter A1 mediates the beneficial effects of the liver X receptor agonist GW3965 on object recognition memory and amyloid burden in amyloid precursor protein/presenilin 1 mice. J Biol Chem. 2010;285:34144–34154. doi: 10.1074/jbc.M110.108100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Fitz N.F., Cronican A.A., Saleem M., Fauq A.H., Chapman R., Lefterov I., et al. Abca1 deficiency affects Alzheimer's disease-like phenotype in human ApoE4 but not in ApoE3-targeted replacement mice. J Neurosci. 2012;32:13125–13136. doi: 10.1523/JNEUROSCI.1937-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lefterov I., Fitz N.F., Cronican A., Lefterov P., Staufenbiel M., Koldamova R. Memory deficits in APP23/Abca1+/– mice correlate with the level of Aβ oligomers. ASN Neuro. 2009;1:e00006. doi: 10.1042/AN20090015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Jiang Q., Lee C.Y., Mandrekar S., Wilkinson B., Cramer P., Zelcer N., et al. ApoE promotes the proteolytic degradation of Aβ. Neuron. 2008;58:681–693. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Cutler R.G., Kelly J., Storie K., Pedersen W.A., Tammara A., Hatanpaa K., et al. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer's disease. Proc Natl Acad Sci U S A. 2004;101:2070–2075. doi: 10.1073/pnas.0305799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bogdanovic N., Bretillon L., Lund E.G., Diczfalusy U., Lannfelt L., Winblad B., et al. On the turnover of brain cholesterol in patients with Alzheimer's disease. Abnormal induction of the cholesterol-catabolic enzyme CYP46 in glial cells. Neurosci Lett. 2001;314:45–48. doi: 10.1016/s0304-3940(01)02277-7. [DOI] [PubMed] [Google Scholar]
- 151.Bretillon L., Siden A., Wahlund L.O., Lutjohann D., Minthon L., Crisby M., et al. Plasma levels of 24S-hydroxycholesterol in patients with neurological diseases. Neurosci Lett. 2000;293:87–90. doi: 10.1016/s0304-3940(00)01466-x. [DOI] [PubMed] [Google Scholar]
- 152.Yassine H.N., Feng Q., Chiang J., Petrosspour L.M., Fonteh A.N., Chui H.C., et al. ABCA1-mediated cholesterol efflux capacity to cerebrospinal fluid is reduced in patients with mild cognitive impairment and Alzheimer's disease. J Am Heart Assoc. 2016;5:e002886. doi: 10.1161/JAHA.115.002886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Teresa J.C., Fernado C., Nancy M.R., Gilberto V.A., Alberto C.R., Roberto R.R. Association of genetic variants of ABCA1 with susceptibility to dementia: (SADEM study) Metab Brain Dis. 2020;35:915–922. doi: 10.1007/s11011-020-00577-4. [DOI] [PubMed] [Google Scholar]
- 154.Wollmer M.A., Streffer J.R., Lutjohann D., Tsolaki M., Iakovidou V., Hegi T., et al. ABCA1 modulates CSF cholesterol levels and influences the age at onset of Alzheimer's disease. Neurobiol Aging. 2003;24:421–426. doi: 10.1016/s0197-4580(02)00094-5. [DOI] [PubMed] [Google Scholar]
- 155.Sundar P.D., Feingold E., Minster R.L., DeKosky S.T., Kamboh M.I. Gender-specific association of ATP-binding cassette transporter 1 (ABCA1) polymorphisms with the risk of late-onset Alzheimer's disease. Neurobiol Aging. 2007;28:856–862. doi: 10.1016/j.neurobiolaging.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 156.Nordestgaard L.T., Tybjaerg-Hansen A., Nordestgaard B.G., Frikke-Schmidt R. Loss-of-function mutation in ABCA1 and risk of Alzheimer's disease and cerebrovascular disease. Alzheimers Dement. 2015;11:1430–1438. doi: 10.1016/j.jalz.2015.04.006. [DOI] [PubMed] [Google Scholar]
- 157.Jaitin D.A., Adlung L., Thaiss C.A., Weiner A., Li B., Descamps H., et al. Lipid-associated macrophages control metabolic homeostasis in a Trem2-dependent manner. Cell. 2019;178:686–698.e14. doi: 10.1016/j.cell.2019.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Marschallinger J., Iram T., Zardeneta M., Lee S.E., Lehallier B., Haney M.S., et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat Neurosci. 2020;23:194–208. doi: 10.1038/s41593-019-0566-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Jung E.S., Mook-Jung I. New microglia on the block. Cell Metab. 2020;31:664–666. doi: 10.1016/j.cmet.2020.03.015. [DOI] [PubMed] [Google Scholar]
- 160.Qi G., Mi Y., Shi X., Gu H., Brinton R.D., Yin F. ApoE4 impairs neuron-astrocyte coupling of fatty acid metabolism. Cell Rep. 2021;34:108572. doi: 10.1016/j.celrep.2020.108572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ioannou M.S., Jackson J., Sheu S.H., Chang C.L., Weigel A.V., Liu H., et al. Neuron–astrocyte metabolic coupling protects against activity-induced fatty acid toxicity. Cell. 2019;177:1522–1535.e14. doi: 10.1016/j.cell.2019.04.001. [DOI] [PubMed] [Google Scholar]
- 162.Koldamova R., Fitz N.F., Lefterov I. ATP-binding cassette transporter A1: from metabolism to neurodegeneration. Neurobiol Dis. 2014;72 Pt A:13–21. doi: 10.1016/j.nbd.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Tang Q., Wang F., Yang J., Peng H., Li Y., Li B., et al. Revealing a novel landscape of the association between blood lipid levels and Alzheimer's disease: a meta-analysis of a case-control study. Front Aging Neurosci. 2019;11:370. doi: 10.3389/fnagi.2019.00370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Feingold K.R. In: Endotext. Feingold K.R., Anawalt B., Boyce A., Chrousos G., de Herder W.W., Dungan K., et al., editors. MDText.com, Inc.; South Dartmouth: 2000. Introduction to lipids and lipoproteins. [Google Scholar]
- 165.Goldstein J.L., Brown M.S. The LDL receptor. Arterioscler Thromb Vasc Biol. 2009;29:431–438. doi: 10.1161/ATVBAHA.108.179564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Schnitzler J.G., Hoogeveen R.M., Ali L., Prange K.H.M., Waissi F., van Weeghel M., et al. Atherogenic lipoprotein(a) increases vascular glycolysis, thereby facilitating inflammation and leukocyte extravasation. Circ Res. 2020;126:1346–1359. doi: 10.1161/CIRCRESAHA.119.316206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Rye K.A., Bursill C.A., Lambert G., Tabet F., Barter P.J. The metabolism and anti-atherogenic properties of HDL. J Lipid Res. 2009;50 Suppl:S195–S200. doi: 10.1194/jlr.R800034-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Segrest J.P., Jones M.K., De Loof H., Brouillette C.G., Venkatachalapathi Y.V., Anantharamaiah G.M. The amphipathic helix in the exchangeable apolipoproteins: a review of secondary structure and function. J Lipid Res. 1992;33:141–166. [PubMed] [Google Scholar]
- 169.Tall A.R., Yvan-Charvet L. Cholesterol, inflammation and innate immunity. Nat Rev Immunol. 2015;15:104–116. doi: 10.1038/nri3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Gall J., Frisdal E., Bittar R., Le Goff W., Bruckert E., Lesnik P., et al. Association of cholesterol efflux capacity with clinical features of metabolic syndrome: relevance to atherosclerosis. J Am Heart Assoc. 2016;5:e004808. doi: 10.1161/JAHA.116.004808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Zhu Y., Liao H., Xie X., Yuan Y., Lee T.S., Wang N., et al. Oxidized LDL downregulates ATP-binding cassette transporter-1 in human vascular endothelial cells via inhibiting liver X receptor (LXR) Cardiovasc Res. 2005;68:425–432. doi: 10.1016/j.cardiores.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 172.Shao B., Tang C., Sinha A., Mayer P.S., Davenport G.D., Brot N., et al. Humans with atherosclerosis have impaired ABCA1 cholesterol efflux and enhanced high-density lipoprotein oxidation by myeloperoxidase. Circ Res. 2014;114:1733–1742. doi: 10.1161/CIRCRESAHA.114.303454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Schaefer E.J., Santos R.D., Asztalos B.F. Marked HDL deficiency and premature coronary heart disease. Curr Opin Lipidol. 2010;21:289–297. doi: 10.1097/MOL.0b013e32833c1ef6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Villarreal-Molina M.T., Flores-Dorantes M.T., Arellano-Campos O., Villalobos-Comparan M., Rodriguez-Cruz M., Miliar-Garcia A., et al. Association of the ATP-binding cassette transporter A1 R230C variant with early-onset type 2 diabetes in a Mexican population. Diabetes. 2008;57:509–513. doi: 10.2337/db07-0484. [DOI] [PubMed] [Google Scholar]
- 175.Doosti M., Najafi M., Reza J.Z., Nikzamir A. The role of ATP-binding-cassette-transporter-A1 (ABCA1) gene polymorphism on coronary artery disease risk. Transl Res. 2010;155:185–190. doi: 10.1016/j.trsl.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 176.Jung D., Cao S., Liu M., Park S. A meta-analysis of the associations between the ATP-binding cassette transporter ABCA1 R219K (rs2230806) polymorphism and the risk of type 2 diabetes in Asians. Horm Metab Res. 2018;50:308–316. doi: 10.1055/a-0583-0201. [DOI] [PubMed] [Google Scholar]
- 177.Tai L.M., Thomas R., Marottoli F.M., Koster K.P., Kanekiyo T., Morris A.W., et al. The role of APOE in cerebrovascular dysfunction. Acta Neuropathol. 2016;131:709–723. doi: 10.1007/s00401-016-1547-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Bowman G.L., Kaye J.A., Quinn J.F. Dyslipidemia and blood–brain barrier integrity in Alzheimer's disease. Curr Gerontol Geriatr Res. 2012;2012:184042. doi: 10.1155/2012/184042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Steen E., Terry B.M., Rivera E.J., Cannon J.L., Neely T.R., Tavares R., et al. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—is this type 3 diabetes?. J Alzheimers Dis. 2005;7:63–80. doi: 10.3233/jad-2005-7107. [DOI] [PubMed] [Google Scholar]
- 180.Talbot K., Wang H.Y., Kazi H., Han L.Y., Bakshi K.P., Stucky A., et al. Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest. 2012;122:1316–1338. doi: 10.1172/JCI59903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.van der Heide L.P., Kamal A., Artola A., Gispen W.H., Ramakers G.M. Insulin modulates hippocampal activity-dependent synaptic plasticity in a N-methyl-d-aspartate receptor and phosphatidyl-inositol-3-kinase-dependent manner. J Neurochem. 2005;94:1158–1166. doi: 10.1111/j.1471-4159.2005.03269.x. [DOI] [PubMed] [Google Scholar]
- 182.Mielke J.G., Wang Y.T. Insulin, synaptic function, and opportunities for neuroprotection. Prog Mol Biol Transl Sci. 2011;98:133–186. doi: 10.1016/B978-0-12-385506-0.00004-1. [DOI] [PubMed] [Google Scholar]
- 183.De Felice F.G., Vieira M.N., Bomfim T.R., Decker H., Velasco P.T., Lambert M.P., et al. Protection of synapses against Alzheimer’s-linked toxins: insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc Natl Acad Sci U S A. 2009;106:1971–1976. doi: 10.1073/pnas.0809158106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Campbell J.M., Stephenson M.D., de Courten B., Chapman I., Bellman S.M., Aromataris E. Metformin use associated with reduced risk of dementia in patients with diabetes: a systematic review and meta-analysis. J Alzheimers Dis. 2018;65:1225–1236. doi: 10.3233/JAD-180263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Chin-Hsiao T. Metformin and the risk of dementia in type 2 diabetes patients. Aging Dis. 2019;10:37–48. doi: 10.14336/AD.2017.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Sluggett J.K., Koponen M., Bell J.S., Taipale H., Tanskanen A., Tiihonen J., et al. Metformin and risk of Alzheimer's disease among community-dwelling people with diabetes: a national case-control study. J Clin Endocrinol Metab. 2020;105:e963–e972. doi: 10.1210/clinem/dgz234. [DOI] [PubMed] [Google Scholar]
- 187.Luchsinger J.A., Perez T., Chang H., Mehta P., Steffener J., Pradabhan G., et al. Metformin in amnestic mild cognitive impairment: results of a pilot randomized placebo controlled clinical trial. J Alzheimers Dis. 2016;51:501–514. doi: 10.3233/JAD-150493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Koenig A.M., Mechanic-Hamilton D., Xie S.X., Combs M.F., Cappola A.R., Xie L., et al. Effects of the insulin sensitizer metformin in Alzheimer disease: pilot data from a randomized placebo-controlled crossover study. Alzheimer Dis Assoc Disord. 2017;31:107–113. doi: 10.1097/WAD.0000000000000202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Claxton A., Baker L.D., Hanson A., Trittschuh E.H., Cholerton B., Morgan A., et al. Long-acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer's disease dementia. J Alzheimers Dis. 2015;44:897–906. doi: 10.3233/JAD-141791. [DOI] [PubMed] [Google Scholar]
- 190.Gold M., Alderton C., Zvartau-Hind M., Egginton S., Saunders A.M., Irizarry M., et al. Rosiglitazone monotherapy in mild-to-moderate Alzheimer's disease: results from a randomized, double-blind, placebo-controlled phase III study. Dement Geriatr Cognit Disord. 2010;30:131–146. doi: 10.1159/000318845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Maeshiba Y., Kiyota Y., Yamashita K., Yoshimura Y., Motohashi M., Tanayama S. Disposition of the new antidiabetic agent pioglitazone in rats, dogs, and monkeys. Arzneimittelforschung. 1997;47:29–35. [PubMed] [Google Scholar]
- 192.de la Monte S.M. Type 3 diabetes is sporadic Alzheimers disease: mini-review. Eur Neuropsychopharmacol. 2014;24:1954–1960. doi: 10.1016/j.euroneuro.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Koseki M., Matsuyama A., Nakatani K., Inagaki M., Nakaoka H., Kawase R., et al. Impaired insulin secretion in four Tangier disease patients with ABCA1 mutations. J Atheroscler Thromb. 2009;16:292–296. doi: 10.5551/jat.e599. [DOI] [PubMed] [Google Scholar]
- 194.Key C.C., Liu M., Kurtz C.L., Chung S., Boudyguina E., Dinh T.A., et al. Hepatocyte ABCA1 deletion impairs liver insulin signaling and lipogenesis. Cell Rep. 2017;19:2116–2129. doi: 10.1016/j.celrep.2017.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Kruit J.K., Wijesekara N., Fox J.E., Dai X.Q., Brunham L.R., Searle G.J., et al. Islet cholesterol accumulation due to loss of ABCA1 leads to impaired exocytosis of insulin granules. Diabetes. 2011;60:3186–3196. doi: 10.2337/db11-0081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.de Haan W., Bhattacharjee A., Ruddle P., Kang M.H., Hayden M.R. ABCA1 in adipocytes regulates adipose tissue lipid content, glucose tolerance, and insulin sensitivity. J Lipid Res. 2014;55:516–523. doi: 10.1194/jlr.M045294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Tang C., Liu Y., Yang W., Storey C., McMillen T.S., Houston B.A., et al. Hematopoietic ABCA1 deletion promotes monocytosis and worsens diet-induced insulin resistance in mice. J Lipid Res. 2016;57:100–108. doi: 10.1194/jlr.M064303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Vincent V., Thakkar H., Aggarwal S., Mridha A.R., Ramakrishnan L., Singh A. ATP-binding cassette transporter A1 (ABCA1) expression in adipose tissue and its modulation with insulin resistance in obesity. Diabetes Metab Syndr Obes. 2019;12:275–284. doi: 10.2147/DMSO.S186565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Patel D.C., Albrecht C., Pavitt D., Paul V., Pourreyron C., Newman S.P., et al. Type 2 diabetes is associated with reduced ATP-binding cassette transporter A1 gene expression, protein and function. PLoS One. 2011;6:e22142. doi: 10.1371/journal.pone.0022142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Kinney J.W., Bemiller S.M., Murtishaw A.S., Leisgang A.M., Salazar A.M., Lamb B.T. Inflammation as a central mechanism in Alzheimer's disease. Alzheimers Dement (N Y) 2018;4:575–590. doi: 10.1016/j.trci.2018.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Yang Y., Song W. Molecular links between Alzheimer's disease and diabetes mellitus. Neuroscience. 2013;250:140–150. doi: 10.1016/j.neuroscience.2013.07.009. [DOI] [PubMed] [Google Scholar]
- 202.Huang N.Q., Jin H., Zhou S.Y., Shi J.S., Jin F. TLR4 is a link between diabetes and Alzheimer's disease. Behav Brain Res. 2017;316:234–244. doi: 10.1016/j.bbr.2016.08.047. [DOI] [PubMed] [Google Scholar]
- 203.Marottoli F.M., Katsumata Y., Koster K.P., Thomas R., Fardo D.W., Tai L.M. Peripheral inflammation, apolipoprotein E4, and amyloid-beta interact to induce cognitive and cerebrovascular dysfunction. ASN Neuro. 2017;9 doi: 10.1177/1759091417719201. 1759091417719201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Guillemot-Legris O., Masquelier J., Everard A., Cani P.D., Alhouayek M., Muccioli G.G. High-fat diet feeding differentially affects the development of inflammation in the central nervous system. J Neuroinflammation. 2016;13:206. doi: 10.1186/s12974-016-0666-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Heuer S.E., Neuner S.M., Hadad N., O'Connell K.M.S., Williams R.W., Philip V.M., et al. Identifying the molecular systems that influence cognitive resilience to Alzheimer's disease in genetically diverse mice. Learn Mem. 2020;27:355–371. doi: 10.1101/lm.051839.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Barroeta-Espar I., Weinstock L.D., Perez-Nievas B.G., Meltzer A.C., Siao Tick Chong M., Amaral A.C., et al. Distinct cytokine profiles in human brains resilient to Alzheimer's pathology. Neurobiol Dis. 2019;121:327–337. doi: 10.1016/j.nbd.2018.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Swardfager W., Lanctot K., Rothenburg L., Wong A., Cappell J., Herrmann N. A meta-analysis of cytokines in Alzheimer's disease. Biol Psychiatry. 2010;68:930–941. doi: 10.1016/j.biopsych.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 208.Banks W.A., Kastin A.J., Broadwell R.D. Passage of cytokines across the blood–brain barrier. Neuroimmunomodulation. 1995;2:241–248. doi: 10.1159/000097202. [DOI] [PubMed] [Google Scholar]
- 209.Huang X., Hussain B., Chang J. Peripheral inflammation and blood–brain barrier disruption: effects and mechanisms. CNS Neurosci Ther. 2021;27:36–47. doi: 10.1111/cns.13569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Marchetti L., Engelhardt B. Immune cell trafficking across the blood–brain barrier in the absence and presence of neuroinflammation. Vasc Biol. 2020;2:H1–H18. doi: 10.1530/VB-19-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Kalaria R.N., Harik S.I. Reduced glucose transporter at the blood–brain barrier and in cerebral cortex in Alzheimer disease. J Neurochem. 1989;53:1083–1088. doi: 10.1111/j.1471-4159.1989.tb07399.x. [DOI] [PubMed] [Google Scholar]
- 212.Wyss-Coray T., Yan F., Lin A.H., Lambris J.D., Alexander J.J., Quigg R.J., et al. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer's mice. Proc Natl Acad Sci U S A. 2002;99:10837–10842. doi: 10.1073/pnas.162350199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Hickman S.E., Allison E.K., El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci. 2008;28:8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Michelucci A., Heurtaux T., Grandbarbe L., Morga E., Heuschling P. Characterization of the microglial phenotype under specific pro-inflammatory and anti-inflammatory conditions: effects of oligomeric and fibrillar amyloid-beta. J Neuroimmunol. 2009;210:3–12. doi: 10.1016/j.jneuroim.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 215.Bomfim T.R., Forny-Germano L., Sathler L.B., Brito-Moreira J., Houzel J.C., Decker H., et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer's disease-associated Abeta oligomers. J Clin Invest. 2012;122:1339–1353. doi: 10.1172/JCI57256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Wang W.Y., Tan M.S., Yu J.T., Tan L. Role of pro-inflammatory cytokines released from microglia in Alzheimer's disease. Ann Transl Med. 2015;3:136. doi: 10.3978/j.issn.2305-5839.2015.03.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Combs C.K., Karlo J.C., Kao S.C., Landreth G.E. beta-Amyloid stimulation of microglia and monocytes results in TNFalpha-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J Neurosci. 2001;21:1179–1188. doi: 10.1523/JNEUROSCI.21-04-01179.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Maezawa I., Zaja-Milatovic S., Milatovic D., Stephen C., Sokal I., Maeda N., et al. Apolipoprotein E isoform-dependent dendritic recovery of hippocampal neurons following activation of innate immunity. J Neuroinflammation. 2006;3:21. doi: 10.1186/1742-2094-3-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Zhu Y., Nwabuisi-Heath E., Dumanis S.B., Tai L.M., Yu C., Rebeck G.W., et al. APOE genotype alters glial activation and loss of synaptic markers in mice. Glia. 2012;60:559–569. doi: 10.1002/glia.22289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Rodriguez G.A., Tai L.M., LaDu M.J., Rebeck G.W. Human APOE4 increases microglia reactivity at Abeta plaques in a mouse model of Abeta deposition. J Neuroinflammation. 2014;11:111. doi: 10.1186/1742-2094-11-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Tai L.M., Ghura S., Koster K.P., Liakaite V., Maienschein-Cline M., Kanabar P., et al. APOE-modulated Abeta-induced neuroinflammation in Alzheimer's disease: current landscape, novel data, and future perspective. J Neurochem. 2015;133:465–488. doi: 10.1111/jnc.13072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Tao Q., Ang T.F.A., DeCarli C., Auerbach S.H., Devine S., Stein T.D., et al. Association of chronic low-grade inflammation with risk of Alzheimer disease in ApoE4 carriers. JAMA Netw Open. 2018;1:e183597. doi: 10.1001/jamanetworkopen.2018.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Karasinska J.M., de Haan W., Franciosi S., Ruddle P., Fan J., Kruit J.K., et al. ABCA1 influences neuroinflammation and neuronal death. Neurobiol Dis. 2013;54:445–455. doi: 10.1016/j.nbd.2013.01.018. [DOI] [PubMed] [Google Scholar]
- 224.Bochem A.E., van der Valk F.M., Tolani S., Stroes E.S., Westerterp M., Tall A.R. Increased systemic and plaque inflammation in ABCA1 mutation carriers with attenuation by statins. Arterioscler Thromb Vasc Biol. 2015;35:1663–1669. doi: 10.1161/ATVBAHA.114.304959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Yvan-Charvet L., Welch C., Pagler T.A., Ranalletta M., Lamkanfi M., Han S., et al. Increased inflammatory gene expression in ABC transporter-deficient macrophages: free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation. 2008;118:1837–1847. doi: 10.1161/CIRCULATIONAHA.108.793869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Zhu X., Owen J.S., Wilson M.D., Li H., Griffiths G.L., Thomas M.J., et al. Macrophage ABCA1 reduces MyD88-dependent Toll-like receptor trafficking to lipid rafts by reduction of lipid raft cholesterol. J Lipid Res. 2010;51:3196–3206. doi: 10.1194/jlr.M006486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Ito A., Hong C., Rong X., Zhu X., Tarling E.J., Hedde P.N., et al. LXRs link metabolism to inflammation through Abca1-dependent regulation of membrane composition and TLR signaling. Elife. 2015;4:e08009. doi: 10.7554/eLife.08009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Tang C., Liu Y., Kessler P.S., Vaughan A.M., Oram J.F. The macrophage cholesterol exporter ABCA1 functions as an anti-inflammatory receptor. J Biol Chem. 2009;284:32336–32343. doi: 10.1074/jbc.M109.047472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Anantharamaiah G.M. Synthetic peptide analogs of apolipoproteins. Methods Enzymol. 1986;128:627–647. doi: 10.1016/0076-6879(86)28096-9. [DOI] [PubMed] [Google Scholar]
- 230.Luciani M.F., Denizot F., Savary S., Mattei M.G., Chimini G. Cloning of two novel ABC transporters mapping on human chromosome 9. Genomics. 1994;21:150–159. doi: 10.1006/geno.1994.1237. [DOI] [PubMed] [Google Scholar]
- 231.Xie Q., Zhao S.P., Li F. D-4F, an apolipoprotein A-I mimetic peptide, promotes cholesterol efflux from macrophages via ATP-binding cassette transporter A1. Tohoku J Exp Med. 2010;220:223–228. doi: 10.1620/tjem.220.223. [DOI] [PubMed] [Google Scholar]
- 232.White C.R., Garber D.W., Anantharamaiah G.M. Anti-inflammatory and cholesterol-reducing properties of apolipoprotein mimetics: a review. J Lipid Res. 2014;55:2007–2021. doi: 10.1194/jlr.R051367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Chernick D., Zhong R., Li L. The role of HDL and HDL mimetic peptides as potential therapeutics for Alzheimer's disease. Biomolecules. 2020;10:1276. doi: 10.3390/biom10091276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Anantharamaiah G.M., Jones J.L., Brouillette C.G., Schmidt C.F., Chung B.H., Hughes T.A., et al. Studies of synthetic peptide analogs of the amphipathic helix. Structure of complexes with dimyristoyl phosphatidylcholine. J Biol Chem. 1985;260:10248–10255. [PubMed] [Google Scholar]
- 235.Datta G., Chaddha M., Hama S., Navab M., Fogelman A.M., Garber D.W., et al. Effects of increasing hydrophobicity on the physical-chemical and biological properties of a class A amphipathic helical peptide. J Lipid Res. 2001;42:1096–1104. [PubMed] [Google Scholar]
- 236.Navab M., Anantharamaiah G.M., Reddy S.T., Hama S., Hough G., Grijalva V.R., et al. Oral D-4F causes formation of pre-beta high-density lipoprotein and improves high-density lipoprotein-mediated cholesterol efflux and reverse cholesterol transport from macrophages in apolipoprotein E-null mice. Circulation. 2004;109:3215–3220. doi: 10.1161/01.CIR.0000134275.90823.87. [DOI] [PubMed] [Google Scholar]
- 237.Navab M., Anantharamaiah G.M., Hama S., Garber D.W., Chaddha M., Hough G., et al. Oral administration of an Apo A-I mimetic peptide synthesized from d-amino acids dramatically reduces atherosclerosis in mice independent of plasma cholesterol. Circulation. 2002;105:290–292. doi: 10.1161/hc0302.103711. [DOI] [PubMed] [Google Scholar]
- 238.Chernick D., Ortiz-Valle S., Jeong A., Swaminathan S.K., Kandimalla K.K., Rebeck G.W., et al. High-density lipoprotein mimetic peptide 4F mitigates amyloid-beta-induced inhibition of apolipoprotein E secretion and lipidation in primary astrocytes and microglia. J Neurochem. 2018;147:647–662. doi: 10.1111/jnc.14554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Handattu S.P., Garber D.W., Monroe C.E., van Groen T., Kadish I., Nayyar G., et al. Oral apolipoprotein A-I mimetic peptide improves cognitive function and reduces amyloid burden in a mouse model of Alzheimer's disease. Neurobiol Dis. 2009;34:525–534. doi: 10.1016/j.nbd.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Buga G.M., Frank J.S., Mottino G.A., Hendizadeh M., Hakhamian A., Tillisch J.H., et al. D-4F decreases brain arteriole inflammation and improves cognitive performance in LDL receptor-null mice on a Western diet. J Lipid Res. 2006;47:2148–2160. doi: 10.1194/jlr.M600214-JLR200. [DOI] [PubMed] [Google Scholar]
- 241.Laskowitz D.T., McKenna S.E., Song P., Wang H., Durham L., Yeung N., et al. COG1410, a novel apolipoprotein E-based peptide, improves functional recovery in a murine model of traumatic brain injury. J Neurotrauma. 2007;24:1093–1107. doi: 10.1089/neu.2006.0192. [DOI] [PubMed] [Google Scholar]
- 242.Laskowitz D.T., Thekdi A.D., Thekdi S.D., Han S.K., Myers J.K., Pizzo S.V., et al. Downregulation of microglial activation by apolipoprotein E and apoE-mimetic peptides. Exp Neurol. 2001;167:74–85. doi: 10.1006/exnr.2001.7541. [DOI] [PubMed] [Google Scholar]
- 243.Datta G., Chaddha M., Garber D.W., Chung B.H., Tytler E.M., Dashti N., et al. The receptor binding domain of apolipoprotein E, linked to a model class A amphipathic helix, enhances internalization and degradation of LDL by fibroblasts. Biochemistry. 2000;39:213–220. doi: 10.1021/bi991209w. [DOI] [PubMed] [Google Scholar]
- 244.Hafiane A., Bielicki J.K., Johansson J.O., Genest J. Novel apoE-derived ABCA1 agonist peptide (CS-6253) promotes reverse cholesterol transport and induces formation of prebeta-1 HDL in vitro. PLoS One. 2015;10:e0131997. doi: 10.1371/journal.pone.0131997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Bielicki J.K., Zhang H., Cortez Y., Zheng Y., Narayanaswami V., Patel A., et al. A new HDL mimetic peptide that stimulates cellular cholesterol efflux with high efficiency greatly reduces atherosclerosis in mice. J Lipid Res. 2010;51:1496–1503. doi: 10.1194/jlr.M003665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Guptill J.T., Raja S.M., Boakye-Agyeman F., Noveck R., Ramey S., Tu T.M., et al. Phase 1 randomized, double-blind, placebo-controlled study to determine the safety, tolerability, and pharmacokinetics of a single escalating dose and repeated doses of CN-105 in healthy adult subjects. J Clin Pharmacol. 2017;57:770–776. doi: 10.1002/jcph.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Krishnamurthy K., Cantillana V., Wang H., Sullivan P.M., Kolls B.J., Ge X., et al. ApoE mimetic improves pathology and memory in a model of Alzheimer's disease. Brain Res. 2020;1733:146685. doi: 10.1016/j.brainres.2020.146685. [DOI] [PubMed] [Google Scholar]
- 248.Zhang Z., Burch P.E., Cooney A.J., Lanz R.B., Pereira F.A., Wu J., et al. Genomic analysis of the nuclear receptor family: new insights into structure, regulation, and evolution from the rat genome. Genome Res. 2004;14:580–590. doi: 10.1101/gr.2160004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Kumar R., Thompson E.B. The structure of the nuclear hormone receptors. Steroids. 1999;64:310–319. doi: 10.1016/s0039-128x(99)00014-8. [DOI] [PubMed] [Google Scholar]
- 250.de Vera I.M.S. Advances in orphan nuclear receptor pharmacology: a new era in drug discovery. ACS Pharmacol Transl Sci. 2018;1:134–137. doi: 10.1021/acsptsci.8b00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Haidar B., Denis M., Krimbou L., Marcil M., Genest J., Jr. cAMP induces ABCA1 phosphorylation activity and promotes cholesterol efflux from fibroblasts. J Lipid Res. 2002;43:2087–2094. doi: 10.1194/jlr.m200235-jlr200. [DOI] [PubMed] [Google Scholar]
- 252.Bingham T.C., Fisher E.A., Parathath S., Reiss A.B., Chan E.S., Cronstein B.N. A2A adenosine receptor stimulation decreases foam cell formation by enhancing ABCA1-dependent cholesterol efflux. J Leukoc Biol. 2010;87:683–690. doi: 10.1189/jlb.0709513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Lyu J., Imachi H., Fukunaga K., Sato S., Ibata T., Kobayashi T., et al. Angiotensin II induces cholesterol accumulation and impairs insulin secretion by regulating ABCA1 in beta cells. J Lipid Res. 2018;59:1906–1915. doi: 10.1194/jlr.M085886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Sun L., Bian K. The nuclear export and ubiquitin–proteasome-dependent degradation of PPARgamma induced by angiotensin II. Int J Biol Sci. 2019;15:1215–1224. doi: 10.7150/ijbs.29741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Ishii N., Matsumura T., Kinoshita H., Fukuda K., Motoshima H., Senokuchi T., et al. Nifedipine induces peroxisome proliferator-activated receptor-gamma activation in macrophages and suppresses the progression of atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2010;30:1598–1605. doi: 10.1161/ATVBAHA.109.202309. [DOI] [PubMed] [Google Scholar]
- 256.Suzuki S., Nishimaki-Mogami T., Tamehiro N., Inoue K., Arakawa R., Abe-Dohmae S., et al. Verapamil increases the apolipoprotein-mediated release of cellular cholesterol by induction of ABCA1 expression via Liver X receptor-independent mechanism. Arterioscler Thromb Vasc Biol. 2004;24:519–525. doi: 10.1161/01.ATV.0000117178.94087.ba. [DOI] [PubMed] [Google Scholar]
- 257.Shirwany N.A., Zou M.H. AMPK in cardiovascular health and disease. Acta Pharmacol Sin. 2010;31:1075–1084. doi: 10.1038/aps.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Hawley S.A., Gadalla A.E., Olsen G.S., Hardie D.G. The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes. 2002;51:2420–2425. doi: 10.2337/diabetes.51.8.2420. [DOI] [PubMed] [Google Scholar]
- 259.Li D., Wang D., Wang Y., Ling W., Feng X., Xia M. Adenosine monophosphate-activated protein kinase induces cholesterol efflux from macrophage-derived foam cells and alleviates atherosclerosis in apolipoprotein E-deficient mice. J Biol Chem. 2010;285:33499–33509. doi: 10.1074/jbc.M110.159772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Kemmerer M., Wittig I., Richter F., Brune B., Namgaladze D. AMPK activates LXRalpha and ABCA1 expression in human macrophages. Int J Biochem Cell Biol. 2016;78:1–9. doi: 10.1016/j.biocel.2016.06.014. [DOI] [PubMed] [Google Scholar]
- 261.Graaf C., Donnelly D., Wootten D., Lau J., Sexton P.M., Miller L.J., et al. Glucagon-like peptide-1 and its class B G protein-coupled receptors: a long march to therapeutic successes. Pharmacol Rev. 2016;68:954–1013. doi: 10.1124/pr.115.011395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Mostafa A.M., Hamdy N.M., El-Mesallamy H.O., Abdel-Rahman S.Z. Glucagon-like peptide 1 (GLP-1)-based therapy upregulates LXR–ABCA1/ABCG1 cascade in adipocytes. Biochem Biophys Res Commun. 2015;468:900–905. doi: 10.1016/j.bbrc.2015.11.054. [DOI] [PubMed] [Google Scholar]
- 263.Wu Y.R., Shi X.Y., Ma C.Y., Zhang Y., Xu R.X., Li J.J. Liraglutide improves lipid metabolism by enhancing cholesterol efflux associated with ABCA1 and ERK1/2 pathway. Cardiovasc Diabetol. 2019;18:146. doi: 10.1186/s12933-019-0954-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Yao Y., Li Q., Wang W., Zhang J., Gao P., Xu Y. Glucagon-like peptide-1 modulates cholesterol homeostasis by suppressing the miR-19b-induced downregulation of ABCA1. Cell Physiol Biochem. 2018;50:679–693. doi: 10.1159/000494235. [DOI] [PubMed] [Google Scholar]
- 265.Lyu J., Imachi H., Fukunaga K., Sato S., Kobayashi T., Dong T., et al. Role of ATP-binding cassette transporter A1 in suppressing lipid accumulation by glucagon-like peptide-1 agonist in hepatocytes. Mol Metab. 2020;34:16–26. doi: 10.1016/j.molmet.2019.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Femminella G.D., Frangou E., Love S.B., Busza G., Holmes C., Ritchie C., et al. Evaluating the effects of the novel GLP-1 analogue liraglutide in Alzheimer's disease: study protocol for a randomised controlled trial (ELAD study) Trials. 2019;20:191. doi: 10.1186/s13063-019-3259-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Chai J.T., Digby J.E., Choudhury R.P. GPR109A and vascular inflammation. Curr Atheroscler Rep. 2013;15:325. doi: 10.1007/s11883-013-0325-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Offermanns S., Colletti S.L., Lovenberg T.W., Semple G., Wise A., AP I.J. International Union of Basic and Clinical Pharmacology. LXXXII: nomenclature and classification of hydroxy-carboxylic acid receptors (GPR81, GPR109A, and GPR109B) Pharmacol Rev. 2011;63:269–290. doi: 10.1124/pr.110.003301. [DOI] [PubMed] [Google Scholar]
- 269.Gaidarov I., Chen X., Anthony T., Maciejewski-Lenoir D., Liaw C., Unett D.J. Differential tissue and ligand-dependent signaling of GPR109A receptor: implications for anti-atherosclerotic therapeutic potential. Cell Signal. 2013;25:2003–2016. doi: 10.1016/j.cellsig.2013.06.008. [DOI] [PubMed] [Google Scholar]
- 270.Zhang L.H., Kamanna V.S., Ganji S.H., Xiong X.M., Kashyap M.L. Niacin increases HDL biogenesis by enhancing DR4-dependent transcription of ABCA1 and lipidation of apolipoprotein A-I in HepG2 cells. J Lipid Res. 2012;53:941–950. doi: 10.1194/jlr.M020917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Xu G.B., Yang L.Q., Guan P.P., Wang Z.Y., Wang P. Prostaglandin A1 inhibits the cognitive decline of APP/PS1 transgenic mice via PPARgamma/ABCA1-dependent cholesterol efflux mechanisms. Neurotherapeutics. 2019;16:505–522. doi: 10.1007/s13311-018-00704-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Petrov A.M., Pikuleva I.A. Cholesterol 24-hydroxylation by CYP46A1: benefits of modulation for brain diseases. Neurotherapeutics. 2019;16:635–648. doi: 10.1007/s13311-019-00731-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.van der Kant R., Langness V.F., Herrera C.M., Williams D.A., Fong L.K., Leestemaker Y., et al. Cholesterol metabolism Is a druggable axis that independently regulates tau and amyloid-beta in iPSC-derived Alzheimer's disease neurons. Cell Stem Cell. 2019;24:363–375.e9. doi: 10.1016/j.stem.2018.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Mast N., Norcross R., Andersson U., Shou M., Nakayama K., Bjorkhem I., et al. Broad substrate specificity of human cytochrome P450 46A1 which initiates cholesterol degradation in the brain. Biochemistry. 2003;42:14284–14292. doi: 10.1021/bi035512f. [DOI] [PubMed] [Google Scholar]
- 275.Mast N., Charvet C., Pikuleva I.A., Stout C.D. Structural basis of drug binding to CYP46A1, an enzyme that controls cholesterol turnover in the brain. J Biol Chem. 2010;285:31783–31795. doi: 10.1074/jbc.M110.143313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Mast N., Verwilst P., Wilkey C.J., Guengerich F.P., Pikuleva I.A. In vitro activation of cytochrome P450 46A1 (CYP46A1) by efavirenz-related compounds. J Med Chem. 2020;63:6477–6488. doi: 10.1021/acs.jmedchem.9b01383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Xu Y., Xu Y., Bao Y., Hong B., Si S. Identification of dehydroxytrichostatin A as a novel up-regulator of the ATP-binding cassette transporter A1 (ABCA1) Molecules. 2011;16:7183–7198. doi: 10.3390/molecules16097183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Gao Q., Wei A., Chen F., Chen X., Ding W., Ding Z., et al. Enhancing PPARgamma by HDAC inhibition reduces foam cell formation and atherosclerosis in ApoE deficient mice. Pharmacol Res. 2020;160:105059. doi: 10.1016/j.phrs.2020.105059. [DOI] [PubMed] [Google Scholar]
- 279.Van den Bossche J., Neele A.E., Hoeksema M.A., de Heij F., Boshuizen M.C., van der Velden S., et al. Inhibiting epigenetic enzymes to improve atherogenic macrophage functions. Biochem Biophys Res Commun. 2014;455:396–402. doi: 10.1016/j.bbrc.2014.11.029. [DOI] [PubMed] [Google Scholar]
- 280.Dresselhaus E., Duerr J.M., Vincent F., Sylvain E.K., Beyna M., Lanyon L.F., et al. Class I HDAC inhibition is a novel pathway for regulating astrocytic apoE secretion. PLoS One. 2018;13:e0194661. doi: 10.1371/journal.pone.0194661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Weikum E.R., Liu X., Ortlund E.A. The nuclear receptor superfamily: a structural perspective. Protein Sci. 2018;27:1876–1892. doi: 10.1002/pro.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Li D., Li T., Wang F., Tian H., Samuels H.H. Functional evidence for retinoid X receptor (RXR) as a nonsilent partner in the thyroid hormone receptor/RXR heterodimer. Mol Cell Biol. 2002;22:5782–5792. doi: 10.1128/MCB.22.16.5782-5792.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Boergesen M., Pedersen T.A., Gross B., van Heeringen S.J., Hagenbeek D., Bindesboll C., et al. Genome-wide profiling of liver X receptor, retinoid X receptor, and peroxisome proliferator-activated receptor alpha in mouse liver reveals extensive sharing of binding sites. Mol Cell Biol. 2012;32:852–867. doi: 10.1128/MCB.06175-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Chawla A., Boisvert W.A., Lee C.H., Laffitte B.A., Barak Y., Joseph S.B., et al. A PPAR gamma–LXR–ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol Cell. 2001;7:161–171. doi: 10.1016/s1097-2765(01)00164-2. [DOI] [PubMed] [Google Scholar]
- 285.Nishimaki-Mogami T., Tamehiro N., Sato Y., Okuhira K., Sai K., Kagechika H., et al. The RXR agonists PA024 and HX630 have different abilities to activate LXR/RXR and to induce ABCA1 expression in macrophage cell lines. Biochem Pharmacol. 2008;76:1006–1013. doi: 10.1016/j.bcp.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 286.Mukherjee R., Davies P.J., Crombie D.L., Bischoff E.D., Cesario R.M., Jow L., et al. Sensitization of diabetic and obese mice to insulin by retinoid X receptor agonists. Nature. 1997;386:407–410. doi: 10.1038/386407a0. [DOI] [PubMed] [Google Scholar]
- 287.Lalloyer F., Fievet C., Lestavel S., Torpier G., van der Veen J., Touche V., et al. The RXR agonist bexarotene improves cholesterol homeostasis and inhibits atherosclerosis progression in a mouse model of mixed dyslipidemia. Arterioscler Thromb Vasc Biol. 2006;26:2731–2737. doi: 10.1161/01.ATV.0000248101.93488.84. [DOI] [PubMed] [Google Scholar]
- 288.Lalloyer F., Pedersen T.A., Gross B., Lestavel S., Yous S., Vallez E., et al. Rexinoid bexarotene modulates triglyceride but not cholesterol metabolism via gene-specific permissivity of the RXR/LXR heterodimer in the liver. Arterioscler Thromb Vasc Biol. 2009;29:1488–1495. doi: 10.1161/ATVBAHA.109.189506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Cramer P.E., Cirrito J.R., Wesson D.W., Lee C.Y., Karlo J.C., Zinn A.E., et al. ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science. 2012;335:1503–1506. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Fitz N.F., Cronican A.A., Lefterov I., Koldamova R. Comment on “ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models”. Science. 2013;340 doi: 10.1126/science.1235809. 924-c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Price A.R., Xu G., Siemienski Z.B., Smithson L.A., Borchelt D.R., Golde T.E., et al. Comment on “ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models”. Science. 2013;340 doi: 10.1126/science.1234089. 924-d. [DOI] [PubMed] [Google Scholar]
- 292.Tesseur I., Lo A.C., Roberfroid A., Dietvorst S., Van Broeck B., Borgers M., et al. Comment on “ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models”. Science. 2013;340 doi: 10.1126/science.1233937. 924-e. [DOI] [PubMed] [Google Scholar]
- 293.Veeraraghavalu K., Zhang C., Miller S., Hefendehl J.K., Rajapaksha T.W., Ulrich J., et al. Comment on “ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models”. Science. 2013;340 doi: 10.1126/science.1235505. 924-f. [DOI] [PubMed] [Google Scholar]
- 294.Tai L.M., Koster K.P., Luo J., Lee S.H., Wang Y.T., Collins N.C., et al. Amyloid-beta pathology and APOE genotype modulate retinoid X receptor agonist activity in vivo. J Biol Chem. 2014;289:30538–30555. doi: 10.1074/jbc.M114.600833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Duvic M., Martin A.G., Kim Y., Olsen E., Wood G.S., Crowley C.A., et al. Phase 2 and 3 clinical trial of oral bexarotene (Targretin capsules) for the treatment of refractory or persistent early-stage cutaneous T-cell lymphoma. Arch Dermatol. 2001;137:581–593. [PubMed] [Google Scholar]
- 296.Cummings J.L., Zhong K., Kinney J.W., Heaney C., Moll-Tudla J., Joshi A., et al. Double-blind, placebo-controlled, proof-of-concept trial of bexarotene in moderate Alzheimer's disease. Alzheimer's Res Ther. 2016;8:4. doi: 10.1186/s13195-016-0173-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Ren G., Bao W., Zeng Z., Zhang W., Shang C., Wang M., et al. Retinoid X receptor alpha nitro-ligand Z-10 and its optimized derivative Z-36 reduce beta-amyloid plaques in Alzheimer's disease mouse model. Mol Pharm. 2019;16:480–488. doi: 10.1021/acs.molpharmaceut.8b00096. [DOI] [PubMed] [Google Scholar]
- 298.Yuan C., Guo X., Zhou Q., Du F., Jiang W., Zhou X., et al. OAB-14, a bexarotene derivative, improves Alzheimer's disease-related pathologies and cognitive impairments by increasing beta-amyloid clearance in APP/PS1 mice. Biochim Biophys Acta Mol Basis Dis. 2019;1865:161–180. doi: 10.1016/j.bbadis.2018.10.028. [DOI] [PubMed] [Google Scholar]
- 299.Singh A.B., Dong B., Kraemer F.B., Liu J. FXR activation promotes intestinal cholesterol excretion and attenuates hyperlipidemia in SR-B1-deficient mice fed a high-fat and high-cholesterol diet. Physiol Rep. 2020;8:e14387. doi: 10.14814/phy2.14387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Jiao Y., Lu Y., Li X.Y. Farnesoid X receptor: a master regulator of hepatic triglyceride and glucose homeostasis. Acta Pharmacol Sin. 2015;36:44–50. doi: 10.1038/aps.2014.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Ayaori M., Yakushiji E., Ogura M., Nakaya K., Hisada T., Uto-Kondo H., et al. Retinoic acid receptor agonists regulate expression of ATP-binding cassette transporter G1 in macrophages. Biochim Biophys Acta. 2012;1821:561–572. doi: 10.1016/j.bbalip.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 302.Tyagi S., Gupta P., Saini A.S., Kaushal C., Sharma S. The peroxisome proliferator-activated receptor: a family of nuclear receptors role in various diseases. J Adv Pharm Technol Res. 2011;2:236–240. doi: 10.4103/2231-4040.90879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Chinetti G., Lestavel S., Bocher V., Remaley A.T., Neve B., Torra I.P., et al. PPAR-alpha and PPAR-gamma activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat Med. 2001;7:53–58. doi: 10.1038/83348. [DOI] [PubMed] [Google Scholar]
- 304.Ogata M., Tsujita M., Hossain M.A., Akita N., Gonzalez F.J., Staels B., et al. On the mechanism for PPAR agonists to enhance ABCA1 gene expression. Atherosclerosis. 2009;205:413–419. doi: 10.1016/j.atherosclerosis.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Rubins H.B., Robins S.J., Collins D., Fye C.L., Anderson J.W., Elam M.B., et al. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. N Engl J Med. 1999;341:410–418. doi: 10.1056/NEJM199908053410604. [DOI] [PubMed] [Google Scholar]
- 306.Ferri N., Corsini A., Sirtori C., Ruscica M. PPAR-alpha agonists are still on the rise: an update on clinical and experimental findings. Expert Opin Investig Drugs. 2017;26:593–602. doi: 10.1080/13543784.2017.1312339. [DOI] [PubMed] [Google Scholar]
- 307.Chandra S., Pahan K. Gemfibrozil, a lipid-lowering drug, lowers amyloid plaque pathology and enhances memory in a mouse model of Alzheimer's disease via peroxisome proliferator-activated receptor alpha. J Alzheimers Dis Rep. 2019;3:149–168. doi: 10.3233/ADR-190104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Silva J.C., de Oliveira E.M., Turato W.M., Trossini G.H.G., Maltarollo V.G., Pitta M.G.R., et al. GQ-11: a new PPAR agonist improves obesity-induced metabolic alterations in LDLr–/– mice. Int J Obes (Lond) 2018;42:1062–1072. doi: 10.1038/s41366-018-0011-7. [DOI] [PubMed] [Google Scholar]
- 309.Wang X., Luo J., Li N., Liu L., Han X., Liu C., et al. E3317 promotes cholesterol efflux in macrophage cells via enhancing ABCA1 expression. Biochem Biophys Res Commun. 2018;504:68–74. doi: 10.1016/j.bbrc.2018.08.125. [DOI] [PubMed] [Google Scholar]
- 310.Oliver W.R., Jr., Shenk J.L., Snaith M.R., Russell C.S., Plunket K.D., Bodkin N.L., et al. A selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transport. Proc Natl Acad Sci U S A. 2001;98:5306–5311. doi: 10.1073/pnas.091021198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Chamberlain S., Gabriel H., Strittmatter W., Didsbury J. An exploratory Phase IIa study of the PPAR delta/gamma agonist T3D-959 assessing metabolic and cognitive function in subjects with mild to moderate Alzheimer's disease. J Alzheimers Dis. 2020;73:1085–1103. doi: 10.3233/JAD-190864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Beyer T.P., Schmidt R.J., Foxworthy P., Zhang Y., Dai J., Bensch W.R., et al. Coadministration of a liver X receptor agonist and a peroxisome proliferator activator receptor-alpha agonist in mice: effects of nuclear receptor interplay on high-density lipoprotein and triglyceride metabolism in vivo. J Pharmacol Exp Ther. 2004;309:861–868. doi: 10.1124/jpet.103.064535. [DOI] [PubMed] [Google Scholar]
- 313.Govindarajulu M., Pinky P.D., Bloemer J., Ghanei N., Suppiramaniam V., Amin R. Signaling mechanisms of selective PPARgamma modulators in Alzheimer's disease. PPAR Res. 2018;2018:2010675. doi: 10.1155/2018/2010675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Godoy J.A., Zolezzi J.M., Inestrosa N.C. INT131 increases dendritic arborization and protects against Abeta toxicity by inducing mitochondrial changes in hippocampal neurons. Biochem Biophys Res Commun. 2017;490:955–962. doi: 10.1016/j.bbrc.2017.06.146. [DOI] [PubMed] [Google Scholar]
- 315.Zhao C., Dahlman-Wright K. Liver X receptor in cholesterol metabolism. J Endocrinol. 2010;204:233–240. doi: 10.1677/JOE-09-0271. [DOI] [PubMed] [Google Scholar]
- 316.Hong C., Tontonoz P. Liver X receptors in lipid metabolism: opportunities for drug discovery. Nat Rev Drug Discov. 2014;13:433–444. doi: 10.1038/nrd4280. [DOI] [PubMed] [Google Scholar]
- 317.Jakobsson T., Treuter E., Gustafsson J.A., Steffensen K.R. Liver X receptor biology and pharmacology: new pathways, challenges and opportunities. Trends Pharmacol Sci. 2012;33:394–404. doi: 10.1016/j.tips.2012.03.013. [DOI] [PubMed] [Google Scholar]
- 318.Laffitte B.A., Chao L.C., Li J., Walczak R., Hummasti S., Joseph S.B., et al. Activation of liver X receptor improves glucose tolerance through coordinate regulation of glucose metabolism in liver and adipose tissue. Proc Natl Acad Sci U S A. 2003;100:5419–5424. doi: 10.1073/pnas.0830671100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Zhu R., Ou Z., Ruan X., Gong J. Role of liver X receptors in cholesterol efflux and inflammatory signaling (review) Mol Med Rep. 2012;5:895–900. doi: 10.3892/mmr.2012.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Schultz J.R., Tu H., Luk A., Repa J.J., Medina J.C., Li L., et al. Role of LXRs in control of lipogenesis. Genes Dev. 2000;14:2831–2838. doi: 10.1101/gad.850400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Chisholm J.W., Hong J., Mills S.A., Lawn R.M. The LXR ligand T0901317 induces severe lipogenesis in the db/db diabetic mouse. J Lipid Res. 2003;44:2039–2048. doi: 10.1194/jlr.M300135-JLR200. [DOI] [PubMed] [Google Scholar]
- 322.Efanov A.M., Sewing S., Bokvist K., Gromada J. Liver X receptor activation stimulates insulin secretion via modulation of glucose and lipid metabolism in pancreatic beta-cells. Diabetes. 2004;53 Suppl 3:S75–S78. doi: 10.2337/diabetes.53.suppl_3.s75. [DOI] [PubMed] [Google Scholar]
- 323.Miao B., Zondlo S., Gibbs S., Cromley D., Hosagrahara V.P., Kirchgessner T.G., et al. Raising HDL cholesterol without inducing hepatic steatosis and hypertriglyceridemia by a selective LXR modulator. J Lipid Res. 2004;45:1410–1417. doi: 10.1194/jlr.M300450-JLR200. [DOI] [PubMed] [Google Scholar]
- 324.Koldamova R.P., Lefterov I.M., Staufenbiel M., Wolfe D., Huang S., Glorioso J.C., et al. The liver X receptor ligand T0901317 decreases amyloid beta production in vitro and in a mouse model of Alzheimer's disease. J Biol Chem. 2005;280:4079–4088. doi: 10.1074/jbc.M411420200. [DOI] [PubMed] [Google Scholar]
- 325.Baranowski M., Zabielski P., Blachnio-Zabielska A.U., Harasim E., Chabowski A., Gorski J. Insulin-sensitizing effect of LXR agonist T0901317 in high-fat fed rats is associated with restored muscle GLUT4 expression and insulin-stimulated AS160 phosphorylation. Cell Physiol Biochem. 2014;33:1047–1057. doi: 10.1159/000358675. [DOI] [PubMed] [Google Scholar]
- 326.Cui W., Sun Y., Wang Z., Xu C., Xu L., Wang F., et al. Activation of liver X receptor decreases BACE1 expression and activity by reducing membrane cholesterol levels. Neurochem Res. 2011;36:1910–1921. doi: 10.1007/s11064-011-0513-3. [DOI] [PubMed] [Google Scholar]
- 327.Pehkonen P., Welter-Stahl L., Diwo J., Ryynanen J., Wienecke-Baldacchino A., Heikkinen S., et al. Genome-wide landscape of liver X receptor chromatin binding and gene regulation in human macrophages. BMC Genom. 2012;13:50. doi: 10.1186/1471-2164-13-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Quinet E.M., Savio D.A., Halpern A.R., Chen L., Schuster G.U., Gustafsson J.A., et al. Liver X receptor (LXR)-beta regulation in LXRalpha-deficient mice: implications for therapeutic targeting. Mol Pharmacol. 2006;70:1340–1349. doi: 10.1124/mol.106.022608. [DOI] [PubMed] [Google Scholar]
- 329.Sparrow C.P., Baffic J., Lam M.H., Lund E.G., Adams A.D., Fu X., et al. A potent synthetic LXR agonist is more effective than cholesterol loading at inducing ABCA1 mRNA and stimulating cholesterol efflux. J Biol Chem. 2002;277:10021–10027. doi: 10.1074/jbc.M108225200. [DOI] [PubMed] [Google Scholar]
- 330.Quinet E.M., Savio D.A., Halpern A.R., Chen L., Miller C.P., Nambi P. Gene-selective modulation by a synthetic oxysterol ligand of the liver X receptor. J Lipid Res. 2004;45:1929–1942. doi: 10.1194/jlr.M400257-JLR200. [DOI] [PubMed] [Google Scholar]
- 331.Wrobel J., Steffan R., Bowen S.M., Magolda R., Matelan E., Unwalla R., et al. Indazole-based liver X receptor (LXR) modulators with maintained atherosclerotic lesion reduction activity but diminished stimulation of hepatic triglyceride synthesis. J Med Chem. 2008;51:7161–7168. doi: 10.1021/jm800799q. [DOI] [PubMed] [Google Scholar]
- 332.Quinet E.M., Basso M.D., Halpern A.R., Yates D.W., Steffan R.J., Clerin V., et al. LXR ligand lowers LDL cholesterol in primates, is lipid neutral in hamster, and reduces atherosclerosis in mouse. J Lipid Res. 2009;50:2358–2370. doi: 10.1194/jlr.M900037-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Stachel S.J., Zerbinatti C., Rudd M.T., Cosden M., Suon S., Nanda K.K., et al. Identification and in vivo evaluation of liver X receptor beta-selective agonists for the potential treatment of Alzheimer's disease. J Med Chem. 2016;59:3489–3498. doi: 10.1021/acs.jmedchem.6b00176. [DOI] [PubMed] [Google Scholar]
- 334.Koura M., Matsuda T., Okuda A., Watanabe Y., Yamaguchi Y., Kurobuchi S., et al. Design, synthesis and pharmacology of 1,1-bistrifluoromethylcarbinol derivatives as liver X receptor beta-selective agonists. Bioorg Med Chem Lett. 2015;25:2668–2674. doi: 10.1016/j.bmcl.2015.04.080. [DOI] [PubMed] [Google Scholar]
- 335.Matsuda T., Okuda A., Watanabe Y., Miura T., Ozawa H., Tosaka A., et al. Design and discovery of 2-oxochromene derivatives as liver X receptor beta-selective agonists. Bioorg Med Chem Lett. 2015;25:1274–1278. doi: 10.1016/j.bmcl.2015.01.047. [DOI] [PubMed] [Google Scholar]
- 336.Koura M., Yamaguchi Y., Kurobuchi S., Sumida H., Watanabe Y., Enomoto T., et al. Discovery of a 2-hydroxyacetophenone derivative as an outstanding linker to enhance potency and beta-selectivity of liver X receptor agonist. Bioorg Med Chem. 2016;24:3436–3446. doi: 10.1016/j.bmc.2016.05.048. [DOI] [PubMed] [Google Scholar]
- 337.Zheng Y., Zhuang L., Fan K.Y., Tice C.M., Zhao W., Dong C., et al. Discovery of a novel, orally efficacious liver X receptor (LXR) beta agonist. J Med Chem. 2016;59:3264–3271. doi: 10.1021/acs.jmedchem.5b02029. [DOI] [PubMed] [Google Scholar]
- 338.Tice C.M., Noto P.B., Fan K.Y., Zhao W., Lotesta S.D., Dong C., et al. Brain penetrant liver X receptor (LXR) modulators based on a 2,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole core. Bioorg Med Chem Lett. 2016;26:5044–5050. doi: 10.1016/j.bmcl.2016.08.089. [DOI] [PubMed] [Google Scholar]
- 339.Kirchgessner T.G., Martin R., Sleph P., Grimm D., Liu X., Lupisella J., et al. Pharmacological characterization of a novel liver X receptor agonist with partial LXRalpha activity and a favorable window in nonhuman primates. J Pharmacol Exp Ther. 2015;352:305–314. doi: 10.1124/jpet.114.219923. [DOI] [PubMed] [Google Scholar]
- 340.Kick E., Martin R., Xie Y., Flatt B., Schweiger E., Wang T.L., et al. Liver X receptor (LXR) partial agonists: biaryl pyrazoles and imidazoles displaying a preference for LXRbeta. Bioorg Med Chem Lett. 2015;25:372–377. doi: 10.1016/j.bmcl.2014.11.029. [DOI] [PubMed] [Google Scholar]
- 341.Kick E.K., Busch B.B., Martin R., Stevens W.C., Bollu V., Xie Y., et al. Discovery of highly potent liver X receptor beta agonists. ACS Med Chem Lett. 2016;7:1207–1212. doi: 10.1021/acsmedchemlett.6b00234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Kirchgessner T.G., Sleph P., Ostrowski J., Lupisella J., Ryan C.S., Liu X., et al. Beneficial and adverse effects of an LXR agonist on uman lipid and lipoprotein metabolism and circulating neutrophils. Cell Metab. 2016;24:223–233. doi: 10.1016/j.cmet.2016.07.016. [DOI] [PubMed] [Google Scholar]
- 343.Katz A., Udata C., Ott E., Hickey L., Burczynski M.E., Burghart P., et al. Safety, pharmacokinetics, and pharmacodynamics of single doses of LXR-623, a novel liver X-receptor agonist, in healthy participants. J Clin Pharmacol. 2009;49:643–649. doi: 10.1177/0091270009335768. [DOI] [PubMed] [Google Scholar]
- 344.Honzumi S., Shima A., Hiroshima A., Koieyama T., Ubukata N., Terasaka N. LXRalpha regulates human CETP expression in vitro and in transgenic mice. Atherosclerosis. 2010;212:139–145. doi: 10.1016/j.atherosclerosis.2010.04.025. [DOI] [PubMed] [Google Scholar]
- 345.Hong C., Marshall S.M., McDaniel A.L., Graham M., Layne J.D., Cai L., et al. The LXR–Idol axis differentially regulates plasma LDL levels in primates and mice. Cell Metab. 2014;20:910–918. doi: 10.1016/j.cmet.2014.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Lefterov I., Bookout A., Wang Z., Staufenbiel M., Mangelsdorf D., Koldamova R. Expression profiling in APP23 mouse brain: inhibition of Abeta amyloidosis and inflammation in response to LXR agonist treatment. Mol Neurodegener. 2007;2:20. doi: 10.1186/1750-1326-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Riddell D.R., Zhou H., Comery T.A., Kouranova E., Lo C.F., Warwick H.K., et al. The LXR agonist TO901317 selectively lowers hippocampal Abeta42 and improves memory in the Tg2576 mouse model of Alzheimer's disease. Mol Cell Neurosci. 2007;34:621–628. doi: 10.1016/j.mcn.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 348.Fitz N.F., Cronican A., Pham T., Fogg A., Fauq A.H., Chapman R., et al. Liver X receptor agonist treatment ameliorates amyloid pathology and memory deficits caused by high-fat diet in APP23 mice. J Neurosci. 2010;30:6862–6872. doi: 10.1523/JNEUROSCI.1051-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Terwel D., Steffensen K.R., Verghese P.B., Kummer M.P., Gustafsson J.A., Holtzman D.M., et al. Critical role of astroglial apolipoprotein E and liver X receptor-alpha expression for microglial Abeta phagocytosis. J Neurosci. 2011;31:7049–7059. doi: 10.1523/JNEUROSCI.6546-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Vanmierlo T., Rutten K., Dederen J., Bloks V.W., van Vark-van der Zee L.C., Kuipers F., et al. Liver X receptor activation restores memory in aged AD mice without reducing amyloid. Neurobiol Aging. 2011;32:1262–1272. doi: 10.1016/j.neurobiolaging.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 351.Cui W., Sun Y., Wang Z., Xu C., Peng Y., Li R. Liver X receptor activation attenuates inflammatory response and protects cholinergic neurons in APP/PS1 transgenic mice. Neuroscience. 2012;210:200–210. doi: 10.1016/j.neuroscience.2012.02.047. [DOI] [PubMed] [Google Scholar]
- 352.Fitz N.F., Castranio E.L., Carter A.Y., Kodali R., Lefterov I., Koldamova R. Improvement of memory deficits and amyloid-beta clearance in aged APP23 mice treated with a combination of anti-amyloid-beta antibody and LXR agonist. J Alzheimers Dis. 2014;41:535–549. doi: 10.3233/JAD-132789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Carter A.Y., Letronne F., Fitz N.F., Mounier A., Wolfe C.M., Nam K.N., et al. Liver X receptor agonist treatment significantly affects phenotype and transcriptome of APOE3 and APOE4 Abca1 haplo-deficient mice. PLoS One. 2017;12:e0172161. doi: 10.1371/journal.pone.0172161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Wesson D.W., Borkowski A.H., Landreth G.E., Nixon R.A., Levy E., Wilson D.A. Sensory network dysfunction, behavioral impairments, and their reversibility in an Alzheimer's beta-amyloidosis mouse model. J Neurosci. 2011;31:15962–15971. doi: 10.1523/JNEUROSCI.2085-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Stukas S., May S., Wilkinson A., Chan J., Donkin J., Wellington C.L. The LXR agonist GW3965 increases apoA-I protein levels in the central nervous system independent of ABCA1. Biochim Biophys Acta. 2012;1821:536–546. doi: 10.1016/j.bbalip.2011.08.014. [DOI] [PubMed] [Google Scholar]
- 356.Sandoval-Hernandez A.G., Buitrago L., Moreno H., Cardona-Gomez G.P., Arboleda G. Role of liver X receptor in AD pathophysiology. PLoS One. 2015;10:e0145467. doi: 10.1371/journal.pone.0145467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Skerrett R., Pellegrino M.P., Casali B.T., Taraboanta L., Landreth G.E. Combined liver X receptor/peroxisome proliferator-activated receptor gamma agonist treatment reduces amyloid beta levels and improves behavior in amyloid precursor protein/presenilin 1 mice. J Biol Chem. 2015;290:21591–21602. doi: 10.1074/jbc.M115.652008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Sandoval-Hernandez A.G., Hernandez H.G., Restrepo A., Munoz J.I., Bayon G.F., Fernandez A.F., et al. Liver X receptor agonist modifies the DNA ethylation profile of synapse and neurogenesis-related genes in the triple transgenic mouse model of Alzheimer's disease. J Mol Neurosci. 2016;58:243–253. doi: 10.1007/s12031-015-0665-8. [DOI] [PubMed] [Google Scholar]
- 359.Sandoval-Hernandez A.G., Restrepo A., Cardona-Gomez G.P., Arboleda G. LXR activation protects hippocampal microvasculature in very old triple transgenic mouse model of Alzheimer's disease. Neurosci Lett. 2016;621:15–21. doi: 10.1016/j.neulet.2016.04.007. [DOI] [PubMed] [Google Scholar]
- 360.Smith C.L., O'Malley B.W. Coregulator function: a key to understanding tissue specificity of selective receptor modulators. Endocr Rev. 2004;25:45–71. doi: 10.1210/er.2003-0023. [DOI] [PubMed] [Google Scholar]
- 361.Katzenellenbogen B.S., Choi I., Delage-Mourroux R., Ediger T.R., Martini P.G., Montano M., et al. Molecular mechanisms of estrogen action: selective ligands and receptor pharmacology. J Steroid Biochem Mol Biol. 2000;74:279–285. doi: 10.1016/s0960-0760(00)00104-7. [DOI] [PubMed] [Google Scholar]
- 362.Jordan V.C. Tamoxifen: catalyst for the change to targeted therapy. Eur J Cancer. 2008;44:30–38. doi: 10.1016/j.ejca.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Leaney A.E., Beck P., Biddle S., Brown P., Grace P.B., Hudson S.C., et al. Analysis of supplements available to UK consumers purporting to contain selective androgen receptor modulators. Drug Test Anal. 2021;13:122–127. doi: 10.1002/dta.2908. [DOI] [PubMed] [Google Scholar]
- 364.Bunay J., Fouache A., Trousson A., de Joussineau C., Bouchareb E., Zhu Z., et al. Screening for liver X receptor modulators: where are we and for what use?. Br J Pharmacol. 2021;178:3277–3293. doi: 10.1111/bph.15286. [DOI] [PubMed] [Google Scholar]
- 365.Viennois E., Mouzat K., Dufour J., Morel L., Lobaccaro J.M., Baron S. Selective liver X receptor modulators (SLiMs): what use in human health?. Mol Cell Endocrinol. 2012;351:129–141. doi: 10.1016/j.mce.2011.08.036. [DOI] [PubMed] [Google Scholar]
- 366.Griffett K., Burris T.P. Promiscuous activity of the LXR antagonist GSK2033 in a mouse model of fatty liver disease. Biochem Biophys Res Commun. 2016;479:424–428. doi: 10.1016/j.bbrc.2016.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Griffett K., Solt L.A., El-Gendy Bel D., Kamenecka T.M., Burris T.P. A liver-selective LXR inverse agonist that suppresses hepatic steatosis. ACS Chem Biol. 2013;8:559–567. doi: 10.1021/cb300541g. [DOI] [PubMed] [Google Scholar]
- 368.Gabbi C., Warner M., Gustafsson J.A. Action mechanisms of liver X receptors. Biochem Biophys Res Commun. 2014;446:647–650. doi: 10.1016/j.bbrc.2013.11.077. [DOI] [PubMed] [Google Scholar]
- 369.Li N., Wang X., Xu Y., Lin Y., Zhu N., Liu P., et al. Identification of a novel liver X receptor agonist that regulates the expression of key cholesterol homeostasis genes with distinct pharmacological characteristics. Mol Pharmacol. 2017;91:264–276. doi: 10.1124/mol.116.105213. [DOI] [PubMed] [Google Scholar]
- 370.Phelan C.A., Weaver J.M., Steger D.J., Joshi S., Maslany J.T., Collins J.L., et al. Selective partial agonism of liver X receptor alpha is related to differential corepressor recruitment. Mol Endocrinol. 2008;22:2241–2249. doi: 10.1210/me.2008-0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Wagner B.L., Valledor A.F., Shao G., Daige C.L., Bischoff E.D., Petrowski M., et al. Promoter-specific roles for liver X receptor/corepressor complexes in the regulation of ABCA1 and SREBP1 gene expression. Mol Cell Biol. 2003;23:5780–5789. doi: 10.1128/MCB.23.16.5780-5789.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Ramon-Vazquez A., de la Rosa J.V., Tabraue C., Lopez F., Diaz-Chico B.N., Bosca L., et al. Common and differential transcriptional actions of nuclear receptors liver X receptors alpha and beta in macrophages. Mol Cell Biol. 2019;39 doi: 10.1128/MCB.00376-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Torocsik D., Szanto A., Nagy L. Oxysterol signaling links cholesterol metabolism and inflammation via the liver X receptor in macrophages. Mol Aspects Med. 2009;30:134–152. doi: 10.1016/j.mam.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 374.Belorusova A.Y., Evertsson E., Hovdal D., Sandmark J., Bratt E., Maxvall I., et al. Structural analysis identifies an escape route from the adverse lipogenic effects of liver X receptor ligands. Commun Biol. 2019;2:431. doi: 10.1038/s42003-019-0675-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Chen Z., Chen H., Zhang Z., Ding P., Yan X., Li Y., et al. Discovery of novel liver X receptor inverse agonists as lipogenesis inhibitors. Eur J Med Chem. 2020;206:112793. doi: 10.1016/j.ejmech.2020.112793. [DOI] [PubMed] [Google Scholar]
- 376.Lou X., Toresson G., Benod C., Suh J.H., Philips K.J., Webb P., et al. Structure of the retinoid X receptor alpha–liver X receptor beta (RXRalpha–LXRbeta) heterodimer on DNA. Nat Struct Mol Biol. 2014;21:277–281. doi: 10.1038/nsmb.2778. [DOI] [PubMed] [Google Scholar]
- 377.de Vera I.M.S., Zheng J., Novick S., Shang J., Hughes T.S., Brust R., et al. Synergistic regulation of coregulator/nuclear receptor interaction by ligand and DNA. Structure. 2017;25:1506–1518. doi: 10.1016/j.str.2017.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Xu P., Zhai Y., Wang J. The role of PPAR and its cross-talk with CAR and LXR in obesity and atherosclerosis. Int J Mol Sci. 2018;19:1260. doi: 10.3390/ijms19041260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Ide T., Shimano H., Yoshikawa T., Yahagi N., Amemiya-Kudo M., Matsuzaka T., et al. Cross-talk between peroxisome proliferator-activated receptor (PPAR) alpha and liver X receptor (LXR) in nutritional regulation of fatty acid metabolism. II. LXRs suppress lipid degradation gene promoters through inhibition of PPAR signaling. Mol Endocrinol. 2003;17:1255–1267. doi: 10.1210/me.2002-0191. [DOI] [PubMed] [Google Scholar]
- 380.Yoshikawa T., Ide T., Shimano H., Yahagi N., Amemiya-Kudo M., Matsuzaka T., et al. Cross-talk between peroxisome proliferator-activated receptor (PPAR) alpha and liver X receptor (LXR) in nutritional regulation of fatty acid metabolism. I. PPARs suppress sterol regulatory element binding protein-1c promoter through inhibition of LXR signaling. Mol Endocrinol. 2003;17:1240–1254. doi: 10.1210/me.2002-0190. [DOI] [PubMed] [Google Scholar]
- 381.Xiao L., Xie X., Zhai Y. Functional crosstalk of CAR–LXR and ROR–LXR in drug metabolism and lipid metabolism. Adv Drug Deliv Rev. 2010;62:1316–1321. doi: 10.1016/j.addr.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 382.Houck K.A., Borchert K.M., Hepler C.D., Thomas J.S., Bramlett K.S., Michael L.F., et al. T0901317 is a dual LXR/FXR agonist. Mol Genet Metab. 2004;83:184–187. doi: 10.1016/j.ymgme.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 383.Thomas J., Bramlett K.S., Montrose C., Foxworthy P., Eacho P.I., McCann D., et al. A chemical switch regulates fibrate specificity for peroxisome proliferator-activated receptor alpha (PPARalpha) versus liver X receptor. J Biol Chem. 2003;278:2403–2410. doi: 10.1074/jbc.M209629200. [DOI] [PubMed] [Google Scholar]
- 384.Fan J., Zareyan S., Zhao W., Shimizu Y., Pfeifer T.A., Tak J.H., et al. Identification of a chrysanthemic ester as an apolipoprotein E inducer in astrocytes. PLoS One. 2016;11:e0162384. doi: 10.1371/journal.pone.0162384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Fan J., Zhao R.Q., Parro C., Zhao W., Chou H.Y., Robert J., et al. Small molecule inducers of ABCA1 and apoE that act through indirect activation of the LXR pathway. J Lipid Res. 2018;59:830–842. doi: 10.1194/jlr.M081851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Zhao W., Fan J., Kulic I., Koh C., Clark A., Meuller J., et al. Axl receptor tyrosine kinase is a regulator of apolipoprotein E. Mol Brain. 2020;13:66. doi: 10.1186/s13041-020-00609-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Finan G.M., Realubit R., Chung S., Lutjohann D., Wang N., Cirrito J.R., et al. Bioactive compound screen for pharmacological enhancers of apolipoprotein E in primary human astrocytes. Cell Chem Biol. 2016;23:1526–1538. doi: 10.1016/j.chembiol.2016.10.015. [DOI] [PubMed] [Google Scholar]
- 388.Seneviratne U., Huang Z., Am Ende C.W., Butler T.W., Cleary L., Dresselhaus E., et al. Photoaffinity labeling and quantitative chemical proteomics identify LXRbeta as the functional target of enhancers of astrocytic apoE. Cell Chem Biol. 2021;28:148–157.e7. doi: 10.1016/j.chembiol.2020.09.002. [DOI] [PubMed] [Google Scholar]
- 389.Tian L.W., Feng Y., Shimizu Y., Pfeifer T.A., Wellington C., Hooper J.N., et al. ApoE secretion modulating bromotyrosine derivative from the Australian marine sponge Callyspongia sp. Bioorg Med Chem Lett. 2014;24:3537–3540. doi: 10.1016/j.bmcl.2014.05.054. [DOI] [PubMed] [Google Scholar]
- 390.Ben Aissa M., Lewandowski C.T., Ratia K.M., Lee S.H., Layden B.T., LaDu M.J., et al. Discovery of nonlipogenic ABCA1 inducing compounds with potential in Alzheimer's disease and type 2 diabetes. ACS Pharmacol Transl Sci. 2021;4:143–154. doi: 10.1021/acsptsci.0c00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Lewandowski C.T., Khan M.W., BenAissa M., Dubrovskyi O., Ackerman-Berrier M., LaDu M.J., et al. Metabolomic analysis of a selective ABCA1 inducer in obesogenic challenge provides a rationale for therapeutic development. EBioMedicine. 2021;66:103287. doi: 10.1016/j.ebiom.2021.103287. [DOI] [PMC free article] [PubMed] [Google Scholar]