

Summary
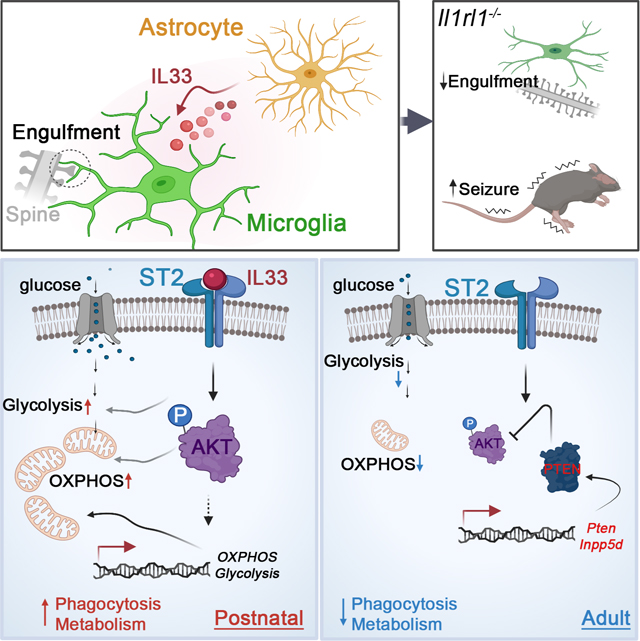
To accommodate the changing needs of the developing brain, microglia must undergo substantial morphological, phenotypic and functional reprogramming. Here we examined whether cellular metabolism regulates microglial function during neurodevelopment. Microglial mitochondria bioenergetics correlated with and was functionally coupled to phagocytic activity in the developing brain. Transcriptional profiling of microglia with diverse metabolic profiles revealed an activation signature wherein the interleukin (IL)-33 signaling axis associated with phagocytic activity. Genetic perturbation of IL-33 or its receptor ST2 led to microglial dystrophy, impaired synaptic function and behavioral abnormalities. Conditional deletion of Il33 from astrocytes or Il1rl1, encoding ST2, in microglia increased susceptibility to seizures. Mechanistically, IL-33 promoted mitochondrial activity and phagocytosis in an AKT dependent manner. Mitochondrial metabolism and AKT activity were temporally regulated in vivo. Thus, a microglia-astrocyte circuit mediated by the IL33-ST2-AKT signaling axis supports microglial metabolic adaptation and phagocytic function during early development, with implications for neurodevelopmental and neuropsychiatric disorders.
eTOC blurb
Microglia are capable of diverse functions to accommodate the changing needs of the central nervous system during development. He et al. define a microglia-astrocyte circuit mediated by the IL33-ST2-AKT signaling axis that supports microglial metabolic adaptation and phagocytic function during early development, with implications for neurodevelopmental and neuropsychiatric disorders.
Graphical Abstract
Introduction
Microglia, the predominant immune cells residing in brain parenchymal, have emerged as a critical player in regulating the patterning and wiring of the neural circuits in early development and modulating brain homeostasis throughout life (Colonna and Butovsky, 2017; Li and Barres, 2018; Thion et al., 2018). In the developing brain, microglia actively participate in complex neurodevelopmental programs via phagocytosing supernumerary synaptic materials and thereby fine-tuning brain connectivity (Hong et al., 2016). In the adult brain, microglia constantly survey synapses in an activity-dependent manner and play a functionally dynamic role in synaptic plasticity and cognitive function (Morris et al., 2013). Dysregulated phagocytic function of microglia could result in excessive or insufficient synaptic pruning, which may lead to neurodevelopmental and neuropsychiatric disorders, including autism and schizophrenia (Druart and Le Magueresse, 2019; Edmonson et al., 2016). To execute those diverse functions, microglia have to undergo substantial morphological, phenotypic and functional reprogramming, which is orchestrated coordinately to accommodate the changing needs of the brain. Despite recent progresses in mapping transcriptional heterogeneity of microglia (Hammond et al., 2019; Li et al., 2019; Masuda et al., 2019), how these transcriptional changes are induced and how they modify microglia function remains largely unknown. Furthermore, the cell-intrinsic signaling circuits and the cell-extrinsic stimuli that instruct developmental programs of microglia have not been well elucidated.
Cellular metabolism has been tightly linked to immune activation and effector function (Borst et al., 2019). However, it remains unknown whether microglia are metabolically programmed to fit them for the changing needs of the brain at different phases of neurodevelopment. Some emerging data suggest that metabolic changes may induce microglial dysfunction in neurodegenerative diseases; e.g., as a cell-extrinsic mechanism, chronic exposure to amyloid-β results in broad defects in energy metabolism in microglia in Alzheimer’s disease (AD) (Baik et al., 2019). Impaired anabolic and energetic metabolism and dysregulated lipid metabolism has been implicated in microglial dysfunction during neurodegeneration and chronic inflammation (Gao et al., 2017; Nugent et al., 2020; Ulland et al., 2017).
Despite these substantial advances, we still have an incomplete understanding of how cellular metabolism regulates microglial function during neurodevelopment. Here, combining transcriptomic analysis, metabolic profiling and perturbation studies, we found that microglial bioenergetics functionally coupled to their phagocytic capacity in the developing brain. Comprehensive gene expression analysis revealed a common gene program correlating with microglia metabolic and phagocytic activity. IL-33-ST2 signaling drove the pro-phagocytic state of microglia by regulating metabolic programs. Genetic perturbation of IL-33 and ST2 in vivo led to microglial dysfunction and dystrophy, accompanied with impaired synaptic function and behavioral abnormalities. Conditional deletion of IL-33 from astrocyte or ST2 on microglia resulted in increased susceptibility to seizures. Mechanistically, the IL-33-ST2 axis promoted microglia phagocytosis and energetic metabolism in an AKT-dependent manner. IL-33-ST2 signaling promoted, whereas PTEN or SHIP-1 restrained AKT activation, counter regulating microglia engulfment and energetic metabolism. Mitochondrial metabolism and AKT activity were temporally regulated in vivo, being central to coordinate microglial functional state during neurodevelopment. Our work thus identifies a microglia-astrocyte circuit mediated by IL33-ST2-AKT axis that supports microglial metabolic adaptation and phagocytic function during early brain development with implications for neurodevelopmental and neuropsychiatric disorders.
Results
Microglial mitochondria bioenergetics is tightly linked to their phagocytic activity in the developing brain
To explore whether the functional state of microglia is reflected in their cellular metabolism, we compared the phagocytic activity of microglia with varying metabolic profiles from the postnatal brain, when microglia exhibit the highest heterogeneity (Hammond et al., 2019; Li et al., 2019). We flow sorted cortical microglia into mitochondrial activity high (ΔΨm-high, top 10%) and low (ΔΨm-low, bottom 10%) fractions using the mitochondrial potential-sensitive dye TMRM (Dykens and Stout, 2001)(Figure 1A). When incubated with pH-sensitive fluorescent synaptosomes, ΔΨm-high microglia engulfed significantly more compared to ΔΨm-low subsets (Figure 1B). Consistently, ΔΨm-high subsets exhibited a higher level of surface CD63 (Figure S1A), a phagocytic marker. In addition, ΔΨm-high microglia were transcriptionally different from ΔΨm-low subsets by upregulating hallmark genes associated with microglial activation, including Apoe, Igf1, Tlr2 and Lilrb4a (Deczkowska et al., 2018)(Figure 1C). Importantly, the above observations were independent on the cell isolation methods (Figures S1A-S1B) or the brain regions analyzed (Figure S1C). Thus, microglia with high mitochondrial activity identifies microglia subsets with an enhanced phagocytic capacity and a transcriptional activation profile.
Figure 1. Mitochondrial bioenergetics and phagocytic activity are functionally coupled in cortical microglia during neurodevelopment.

(A) Schematic strategy to sort low-ΔΨm and high-ΔΨm microglia subsets for analyses.
(B) Representative FACS plot and quantification of cell fraction that has engulfed synaptosome in indicated ΔΨm microglia subsets (n = 8).
(C) qPCR analyses of selected genes in indicated ΔΨm microglia subsets isolated from P9 cortices (n = 9 mice).
(D) Schematic strategy to enrich Phagolo and Phagohi microglia subsets.
(E-F) Quantification TMRM (E) and MitoTracker Green (F) staining in indicated microglia subsets (n = 7).
(G) Oxygen consumption rate of indicated microglia subsets analyzed by Mito Stress test assay (n = 7, representative of 2 experiments).
(H) qPCR analyses of selected genes in indicated microglia subsets isolated from P10 cortices (n = 6–12).
(I) Schematic strategy to enrich microglia that are more potent to engulf neuronal debris in vivo. Bottom: representative images of sorted GFP+ and GFP− microglia isolated from P10 mice carrying Tau-GFP (Scale bar = 10μm). GFP signals were observed inside microglia.
(J) Heatmap depicting the relative expression of genes involved in bioenergetic metabolism from Tau-GFP+ and Tau-GFP− microglia. All genes listed are differentially expressed between two genotypes with FDR < 0.05.
(K) Relative engulfment capacity, basal and stressed OCR and ΔΨm of BV2 cells transduced with Cas9-GFP only or Cas9-GFP-sgRNAs targeting metabolic enzymes and transporters. All values are normalized to the mean value of Cas9-GFP-Empty group. Data is pooled from at least 2 experiments and all changes are significant with p < 0.05.
Statistics: mean ± SEM (B, E and F), mean ± SD (G); unpaired student’s t test (B, E, F and G), paired student’s t test (C and H), one-way ANOVA test (K); *p<0.05, **p<0.01, ***p<0.001.
To further establish a functional coupling between microglial effector states and their metabolic profiles, we compared the energetic status of microglia subsets differing in phagocytic capacity. We sorted phago-high and phago-low microglia by the relative expression of surface TLR2 and LILRB4 (Figure 1D, Figure S1D) and confirmed a higher phagocytic activity in freshly sorted Phagohi microglia (TLR2hiLILRB4hi) compared to Phagolo (TLR2loLILRB4lo) subset (Figure S1E) ex vivo. Surface staining revealed a higher level of CD63 in Phagohi microglia from the cortex (Figure S1F), which was similarly observed in other brain regions (Figure S1G). Measuring multiple parameters reflecting mitochondrial functions, we found that the Phagohi microglia showed remarkably higher mitochondrial mass and mitochondrial membrane potential (Figures 1E–1F), and elevated levels of mitochondrial proteins TOM20 and COXIV (Figures S1H-S1I). Consistently, Phagohi microglia exhibited greater basal oxygen consumption rate (OCR) and maximal respiratory capacity (MRC) compared to Phagolo subsets (Figure 1G), suggesting enhanced mitochondrial fitness in pro-phagocytic microglia in vivo. Phagohi microglia also showed higher extracellular acidification (ECAR) (Figure S1K) and glucose uptake rate (Figure S1L), indicating high energy demands and increased glucose influx in these cells. As ΔΨm-high microglia, Phagohi microglia also upregulated canonical microglia activation genes including Apoe, Igf1, Csf1 and Lyz2 (Figure 1H). Notably, similar observations were made in Phagohi vs Phagolo microglia isolated by Dounce homogenization (Figures S1F, S1H-S1J). To further confirm the heightened metabolic activity in Phagohi microglia in vivo, we purified the “more potent” phagocytic microglia directly by sorting GFP+ cortical microglia from tauGFP mice, in which neurons express a GFP-fusion protein throughout the cell (Tucker et al., 2001), and in which only microglia that have engulfed neuronal materials are labeled with GFP (Figure 1I, Figure S1M). GFP+ microglia exhibited higher expression of canonical activation markers (Figure 1J), akin to Phagohi and ΔΨm-high subsets. In support of their energetic status, GFP+ microglia upregulated genes involved in multiple energetic pathways, including genes essential for OXPHOS, glucose catabolism and fatty acid oxidation (Figure 1J). Collectively, these data pinpoint a correlation between microglial metabolic activity and phagocytic function.
Next, we sought to investigate how mitochondrial biogenetics impact phagocytic activity in microglia. We treated primary microglia with inhibitors of electron transport chain (ETC, Oligomycin, Antimycin and Rotenone), glucose transporter (STF31) and mitochondrial pyruvate transporter (UK5099) for a short period, without affecting cell viability, and analyzed the effects of these inhibitors on synaptosome engulfment by microglia (Figure S1N). We found that inhibiting mitochondria-dependent energetic metabolism diminished the capacity of microglia to engulf synaptosomes (Figure S1O). To further determine whether mitochondrial biogenetics promotes microglial phagocytosis, we studied the effects of genetic perturbation of metabolic genes enriched in phagocytic microglia that we identified (Figure 1J). BV2 cells transduced with Cas9 and sgRNAs against Itgam (encoding CD11b) exhibited a rapid loss of CD11b protein expression (Figure S1P-S1Q), validating our system. We found that sgRNA deletion of mitochondrial pyruvate transporters, ETC I, II, IV and V components all diminished synaptosome uptake (Figure 1K). Although these cells exhibited varying effects on mitochondrial membrane potential depending on which ETC components were targeted (Figure S1R), all displayed decreased basal and maximal OCR (Figure 1K), confirming diminished mitochondrial respiration. Collectively, we demonstrate that mitochondrial bioenergetics is functionally coupled to the phagocytic activity of microglia, possibly reflecting metabolic adaptations that correspond to microglial functional states in vivo.
Identification of an IL33-dependent gene program reflecting microglia metabolic and phagocytic activity in the developing brain
To characterize the molecular states associated with varying metabolic and functional spectrum of microglia in vivo, we transcriptionally profiled ΔΨm-high vs ΔΨm-low and Tau-GFP+ vs Tau-GFP− microglia subsets isolated from P9–10 cortices (Table S1). ΔΨm-high microglia enriched key enzymes involved in multiple energetic pathways and gene programs associated with pro-phagocytic microglia, whereas homoeostatic microglia genes including Slc2a5, Tmem119 and Sall1 were downregulated in ΔΨm-high cells (Figure 2A). Similar transcriptional features were observed in Tau-GFP+ microglia compared to Tau-GFP− subsets (Figures S2A-S2B, Figure 2B). Thus, our analysis identified a common gene signature, named as MGact (Table S2), which was shared by metabolically active and pro-phagocytic microglia in the developing cortex. Interestingly, the MGact signature showed significant overlap with the molecular signature from microglia under aging and neurodegeneration (Gandal et al., 2018; Krasemann et al., 2017)(Table S2, Figure 2C), highlighting the functional relevance of metabolic reprogramming in pro-phagocytic microglia in various settings.
Figure 2. Identification of an IL33-dependent gene program that is associated with microglial function and activity in the developing brain.

(A) Volcano plot depicting transcriptional profiles of ΔΨmhi vs ΔΨmlo microglia subsets. Grey: all differentially expressed genes (fold change ≥ 1, adjusted p < 0.05); red: activation markers; green: homeostatic markers.
(B) Distribution of the relative expression of ΔΨmhi gene signature in Tau-GFP+ and Tau-GFP- microglia. (****p <10−12).
(C) Graphical representation of similarities between the microglia activation signature (MGact) and transcriptomic signatures from mouse AD model, aging and human psychiatric disorder module. Each line represents an overlap, and the thickness of lines is coded by the number of genes overlapped. Genes/connectivity associated with MGact signature is depicted in orange.
(D) t-SNE plot showing 4 microglial clusters from P9 and P28 cortices identified by scRNA-seq.
(E) Distribution of MGact expression among the 4 single cell clusters.
(F) Heatmap depicting the relative average expression of selective microglial activation genes that were enriched in Cluster 4, in comparison to other clusters.
(G) Heatmap depicting relative expression of gene sets induced by indicated cytokines. IL33 gene signature was significantly enriched in Cluster 4 (p < 3.1 × 10−12).
(H) t SNE visualization of the single microglia, colored by IL33 induced gene signature derived from primary mouse microglia in vitro.
(I) Distribution of the relative expression of IL33 gene signature in the 4 single cell clusters. IL-33 gene signature was defined as genes significantly induced by IL-33 in primary microglia.
(J) Representative images and quantification of microglia engulfment capacity as measured by lysosomal content within each microglia (CD68 immunoreactivity per cell) in the somatosensory cortex of WT and Il1rl1−/− mice at P9 (Scale bar = 50μm). Data is normalized to WT (n = 49, 52; pooled from 3–5 mice/group).
(K) Representative images and quantification of lysosome content of microglia in the somatosensory cortex of WT mice injected with IL-33 vs PBS (Scale bar = 50μm). Data is normalized to PBS group (n = 50,48; pooled from 6 mice/group).
(L) Representative 3D reconstructions of Iba1-stained microglia residing in the somatosensory cortex of WT or Il1rl1−/− mice at P17 (Scale bar = 10μm). Cellular volume is quantified on the right side (n = 50, 41; pooled from 3–5 mice/group).
(M) Quantification of microglia engulfment capacity measured by engulfment index (the ratio of engulfed synaptic debris volume to the total volume of microglia) of microglia in the somatosensory cortex of WT or Il1rl1−/− mice at P17. Data is normalized to WT (n = 49, 52; pooled from 3–5 mice/group).
(N) The percentage of microglia engulfed synaptosome at indicated conditions (n = 8). Statistics: middle line: mean, box edges: 25th and 75th percentiles, whiskers: extend to 5th to 95th percentile (B, E and I); middle line: mean (J-N); student’s t test (B and J-N), one-way ANOVA test (E, G and I); ***p<0.001, ****p <10−12.
To test whether the MGact signature is expressed by a distinguishable population in vivo and whether it is developmentally regulated, we analyzed cortical microglia from P9 and P28 mice by sc-RNA Seq. Unsupervised clustering and signature scoring revealed that, as compared to P28 microglia (mainly composing cluster 1), P9 cells (predominantly composing cluster 2,3,4) exhibited much higher expression of the MGact signature (Figures 2D–2E), suggesting a decline in microglial phagocytic activity and metabolism in the adult brain. Notably, Cluster 4, a small group of cells primarily from the P9 cortex, displayed the strongest enrichment of MGact signature, expressing a unique set of hallmark genes featured in pro-phagocytic microglia (Figures 2E–2F). Taken together, our results reveal that the MGact signature, a gene program shared by metabolically active and pro-phagocytic microglia, is expressed by a distinguishable population in the postnatal cortex.
We hypothesized that the observed varying metabolic and functional spectrum of microglia may be established by different cytokine signal inputs, sensed through the wide range of cytokine receptors that play a pivotal role in modulating microglial development and function (ElAli and Rivest, 2016). We projected multiple cytokine-induced gene signatures (Methods, Table S2) onto our microglial single-cell profiles. Consistent with previous studies, Cluster 1 microglia were enriched for genes induced by TGFβ, a key milieu cytokine maintaining adult microglia homeostasis (Butovsky et al., 2014), validating the robustness of our method. The pro-phagocytic microglia cluster (cluster 4) scored the highest for the gene signature activated by the IL-1 family member IL-33 (p < 3.1 × 10−12, Figure 2G). Indeed, exposure to IL-33 in vivo strongly induced microglial activation and elevated the expression of MGact genes (Figure S2F). To further validate this finding, we profiled microglia stimulated in the presence or absence of rIL-33 in vitro, generated a microglial gene signature induced by IL-33, and confirmed its enrichment in Cluster 4 cells (p < 3.1 × 10−12, Figures 2H–2I, Table S3). Moreover, Cluster 4 also exhibited a significant enrichment of genes induced by IL-33 in hippocampal microglia in vivo (Nguyen et al., 2020) (Figures S2G-S2H).
smFISH, quantitative PCR and flow analysis revealed that Il1rl1, encoding the IL-33 receptor ST2, was exclusively expressed by microglia in the cerebral cortex (Figures S2I-S2K), as previously reported (Vainchtein et al., 2018). IL-33 co-localized with GFAP (Figure S2L), suggesting IL-33 was predominantly expressed by astrocytes, corroborating previously published RNA-seq dataset (Figure S2M) (Zeisel et al., 2018). Moreover, cortical IL-33 expression started from the 2nd week after birth, when microglia undergo morphological remodeling and phagocytose synaptic debris (Figures S2N-S2O).
Il1rl1−/− microglia had decreased lysosomal content, as measured by the lysosomal marker CD68 per Iba-1+ cell volume (Figure 2J), indicating diminished phagocytic ability. Conversely, cortical microglia from mice receiving rIL-33 exhibited increased lysosomal content (Figure 2K). IMARIS-based three dimensional (3D) morphometric measurement revealed that cortical microglia from Il1rl1−/− and Il33−/− mice exhibited dystrophic morphology with reduced cellular volumes and surface areas (Figure 2L, Figures S2P, S2R-S2T). Microglia dystrophy was also observed in the hippocampus of Il1rl1−/− mice (Figure S2Q). Super resolution imaging and 3D reconstruction revealed PSD-95 synaptic puncta localized to CD68+ lysosomes within microglia (Figures S2U-S2V), indicating synaptic engulfment by microglia in vivo. We observed a lower amount of PSD-95 synaptic puncta within cortical microglia processes and soma from Il1rl1−/− or Il33−/− mice at postnatal day 14–20, the time window of synaptic refinement (Filipello et al., 2018)(Figure 2M, Figures S2W-S2X). Conversely, rIL-33 administration promoted synaptic engulfment by cortical microglia ex vivo (Figure 2N, Figure S2Z) and in vivo (Figure S2Y). Taken together, our data demonstrate that IL-33-ST2 signaling drives a unique pro-phagocytic state of microglia, and its perturbation leads to microglial dysfunction and dystrophy in the developing brain.
Il1rl1 deficiency leads to impaired synaptic function and increased susceptibility to seizures
To gain mechanistic insights into how microglial function is regulated by IL-33-ST2, we analyzed WT and Il1rl1−/− microglia from P9 cortex by sc-RNA Seq. Unsupervised clustering partitioned the cells into 5 clusters, and the composition of each cluster was comparable between WT and Il1rl1−/− (Figure 3A, Figures S3A-S3B), suggesting that microglia heterogeneity remained unaltered in Il1rl1−/− mice. However, Il1rl1−/− microglia exhibited significantly lower expression of genes involved in lysosome organization and phagosome maturation (Figure S3C), consistent with the dysfunction of Il1rl1−/− microglia (Figure 2). We noted reduced expression of the IL-33 induced gene program in Il1rl1−/− microglia and the effect was more profound in Cluster 4, reminiscent of active microglia with an enrichment of IL-33 signature (Figures 3C–3D), suggesting that loss of Il1rl1 preferentially impacted microglia that would otherwise be “primed” by IL-33.
Figure 3. The IL-33-ST2 axis fine-tunes microglial function and regulates synaptic connectivity in vivo.

(A) tSNE visualization of microglia isolated from WT and Il1rl1−/− cortex at P9. Single cells are colored by the partitioning into 5 clusters or genotype.
(B) tSNE visualization of the single microglia, colored by IL-33 induced gene signature.
(C) Distribution of IL-33 induced gene signature expression in the 5 clusters from WT and Il1rl1−/− microglia.
(D) Heatmap depicting the relative average expression of selected IL-33 induced genes in the 5 single cell clusters identified in (A).
(E) Representative images showing staining for Synaptophysin (cyan) and PSD-95 (red) in the somatosensory cortex of WT and Il1rl1−/− mice at P17 (Scale bar =10μm). Quantification of colocalized pre-and postsynaptic puncta is shown on the right.
(F) Three-chamber test evaluating sociability of WT and Il1rl1−/− mice. Left: social preference index; right: the total object investigating time.
(G) Representative EEG recordings in Il1rl1−/− and WT mice.
(H) Seizure severity score following kainic acid injection to WT and Il1rl1−/− mice at P28-P35. Onset of seizure is quantified on the right (n = 7, 10).
(I) LTP induction in the CA1 area as measured using field recordings in the brain slices from WT and Il1rl1−/− mice. Summary of normalized LTP between 50 and 60 min of the recording is plotted on the right (pooled from 4–6 brain slices from 3 experiments).
Statistics: middle line: mean, box edges: 25th and 75th percentiles, whiskers: extend to 5th to 95th percentile (C); mean ± SEM (E, F and H); middle line: mean (H-I); student’s t test (E, F, H and I), one-way ANOVA test (C), two-way ANOVA test (F); ns: not significant, *p<0.05, ***p<0.001.
We found an increase in the number of synapses in the cortex of Il1rl1−/− animals than in littermate controls, as revealed by increased density of co-localized pre- and post-synaptic puncta (Synaptophysin and PSD-95), which was phenocopied in Il33−/− mice (Figure 3E). Furthermore, Il1rl1−/− mice exhibited substantial behavioral deficits, mirroring those found in neurodevelopmental disorders. In the three-chamber social interaction test, while WT mice showed a preference to interact with the social partner rather than the empty cage, Il1rl1−/− mice did not show such a preference (Figure 3F), indicating impaired sociability in Il1rl1−/− mice. In a standard tube test between Il1rl1−/− and WT animals, Il1rl1−/− mice showed a dramatic defect, losing ∼70% of the matches and eventually being pushed out of the tube by non-cage mate WT mice (Figure S3D). We noticed that Il1rl1−/− mice occasionally exhibited seizures during handling in routine husbandry procedures (Video S1). Electroencephographic (EEG) recordings revealed that some of the adult Il1rl1−/− mice (2 out of 8 tested) displayed electrographic seizures when moved into a new cage (Figure 3G). However, spontaneous seizures were never observed in Il1rl1−/− mice housed in their home cages (Figures S3E-S3F). To further confirm the hyperexcitatory activity of neural network in Il1rl1−/− mice, we measured their susceptibility to seizures after administration of kainic acid, a neuroexcitatory amino acid agonist. Il1rl1−/− mice were more susceptible to kainic acid, evident by reduced latency to seizure onset and increased seizure severity (Figure 3H). Conditional deletion of Il33 from astrocyte and deletion of Il1rl1 from microglia/monocyte phenocopied this finding (Figures S3G-S3H). Repeated electroconvulsive seizures “saturate” synapses and in turn decrease synaptic plasticity (Reid and Stewart, 1997). Indeed, Il1rl1−/− brains exhibited a dramatic defect in the induction of long-term-potentiation (LTP) (Figure 3I), a reflection of synaptic plasticity, suggesting that Il1rl1−/− mice were defective in synaptic transmission and plasticity. Collectively, our data demonstrate that lack of IL-33-ST2 leads to synaptic abnormalities and behavioral defects.
IL-33-ST2-AKT axis supports microglial mitochondrial metabolism and phagocytosis
Having established a functional link between cellular biogenetics and phagocytic activity, the latter supported by IL-33/ST2 in vitro and in vivo, we then asked which aspects of energetic metabolisms, if any, are regulated by IL-33-ST2 in microglia. IL-33 induced the upregulation of enzymatic genes involved in both glucose metabolism and mitochondrial OXPHOS in primary microglia in vitro, suggesting that carbon metabolism from glucose via glycolysis/TCA cycle and mitochondrial OXPHOS is enhanced by exogenous IL-33 (Figure 4A). In accordance with this, freshly isolated microglia from rIL-33 administered mice exhibited enhanced glucose uptake rate (Figure 4B). Similar observations were made in rIL-33 stimulated BV2 cells in vitro (Figures S4A-4B). IL-33 increased mitochondrial membrane potential (ΔΨm), as measured by TMRM fluorescence intensity in purified microglia in vitro in a dose dependent manner (Figure 4C). Moreover, microglia from mice that received rIL-33 exhibited higher ΔΨm, whereas Il1rl1−/− microglia exhibited diminished ΔΨm compared to WT cells (Figures 4D–4E). The total mitochondrial mass, however, remained unaffected in Il1rl1−/− microglia (Figure S4C), indicating that mitochondrial activity rather than mitochondrial biogenesis is regulated by IL-33-ST2. Extracellular flux analysis revealed a higher oxygen consumption rate in microglia isolated from mice injected with IL-33 compared to PBS (Figure 4F). Conversely, Il1rl1−/− microglia exhibited reduced basal and maximal OCR compared to WT cells (Figure 4G). Whereas exogenous IL-33 enhanced glycolytic activity in microglia (Figure S4D), EACR remained unaltered in Il1rl1−/− microglia (Figure S4E). This suggests that under the physiological condition when microglia are not overactivated by exogenous IL-33, it is the mitochondrial activity rather than glycolysis that relies on IL-33-ST2 signaling in vivo.
Figure 4. IL33-dependent mitochondrial bioenergetics promotes microglia engulfment.

(A) Heatmap depicting the relative expression of selective metabolic genes induced by IL-33 in primary microglia.
(B) 2-NBDG uptake by cortical microglia isolated from PBS and IL-33 administered mice at P9 (n = 6, 9).
(C) Quantification of TMRM MFI in primary rat microglia stimulated with 10ng/mL and 100ng/mL IL-33 for 12hrs (n = 7).
(D) TMRM staining on cortical microglia isolated from P9 mice injected with PBS and IL33 (n = 6).
(E) TMRM staining in cortical microglia isolated from WT and Il1rl1−/− mice at P9 (n = 7, 4).
(F) OCR of microglia under indicated conditions analyzed by Mito Stress test assay (n =12, representative of 2 experiments).
(G) Basal and stressed OCR of cortical microglia isolated from WT and Il1rl1−/− brains at P9 (n =12).
(H) Representative FACS plots and quantifications of engulfed synaptosome percentage in BV2 cells under indicated conditions (n = 11–30, pooled from 4 experiments).
(I) Engulfment capacity of WT and Il1rl1−/− BMDM as measured by engulfed synaptosome percentage at indicated conditions (n = 6, 6, 17, 16; pooled from 2 experiments).
Statistics: mean ± SEM (B, D, E, H and I); middle line: mean (C); mean ± SD (F and G); student’s t test (B, D, E, F and G), one-way ANOVA test (C, H and I); ***p<0.001.
To determine whether mitochondrial function, regulated by IL-33, is crucial to microglial phagocytosis, we treated BV2 cells with multiple metabolic inhibitors and analyzed the engulfment capacity (Figure S4F). Seahorse analysis confirmed that UK5099 and STF31 inhibited IL-33 induced-elevations in OXPHOS, and that ETC inhibitors decreased mitochondria-dependent oxygen consumption (Figure S4G). When fed with pHrodo-Red conjugated synaptosomes, IL-33 stimulated cells engulfed more synaptosomes, and inhibition of mitochondria-dependent energetic metabolism abrogated this effect (Figure 4H). Furthermore, genetic perturbation of Ndufb3, Ndufc1 and Mpc2 led to defective engulfment of synaptosomes of BV2 cells in response to IL-33 (Figure S4H).
We then asked whether augmentation of mitochondrial metabolism could rescue the engulfment defect in Il1rl1−/− cells. We tested Oltipraz, known to promote mitochondrial reparation via activating PGC-1α/NRF2 (Chamoto et al., 2017). We confirmed that Oltipraz significantly increased mitochondrial membrane potential in primary microglia (Figures S4I). Oltipraz treatment enhanced synaptosome engulfment in Il1rl1−/− macrophage, although not to the same extend as WT cells, in response to IL-33 (Figure 4I), suggesting that increasing mitochondrial metabolism by Oltipraz is sufficient to partially restore the engulfment defect in Il1rl1−/− cells. Together, these data illustrate that mitochondrial activity sustained by IL-33-ST2 supports microglia phagocytosis in vitro and in vivo.
We next sought to dissect the underlying mechanisms by which IL-33 regulates the metabolic and phagocytic activity of microglia. IL-33 induced genes were enriched in gene ontology (GO) terms related to cellular metabolism and immune activation (Figure S5A). In addition, we found GO term of PI3K complex, was also overrepresented in IL-33 induced gene signature. Cellular metabolic programs, especially mitochondrial OXPHOS and glycolytic flux, are coordinated by energy-sensing mechanisms involving the activation of AKT, which acts as a coupler of extracellular stimuli and convergent effector of PI3K and mTOR (Linke et al., 2017). In line with this, we found IL-33 stimulation induced rapid phosphorylation of AKT, NDRG1, ribosomal-S6 and mTOR in BV2 cells (Figure 5A, Figure S5B). Induction of AKT and mTOR phosphorylation in microglia by IL-33 was further validated in vivo (Figures 5B–5C, Figure S5C). To corroborate the functional relevance of PI3K/mTOR/AKT pathway in microglia, we treated BV2 cells with multiple inhibitors for a short period without affecting cell viability to avoid any possibly deleterious effects. Inhibition of AKT (by Triciribine) and PI3K (by LY294002), or dual inhibition of mTORC1/mTORC2 (AZD8055, PP242), confirmed by reduced AKTS473 and S6 phosphorylation in response to IL-33, all robustly curtailed the uptake of pH-sensitive fluorescent synaptosomes at baseline and enhanced by IL-33 (Figures 5D–5E, Figure S5D). In contrast, rapamycin, at a wide-range of tested concentrations that selectively abrogated S6 phosphorylation, did not reduce AKTS473 phosphorylation, or synaptosome uptake (Figures 5D–5E), demonstrating that AKT activation, by PI3K and mTORC2 but not mTORC1, bridges IL33-ST2 signal to elevated microglia phagocytosis in response to IL-33. Furthermore, inhibiting AKT diminished baseline OCR and maximal respiratory capacity, which were otherwise enhanced by IL-33 (Figure 5F). Collectively, these results demonstrate that augmented energetic metabolism and enhanced phagocytosis sustained by IL33-ST2 signaling depends on AKT activation.
Figure 5. AKT mediates IL-33-enhanced bioenergetics and morphological remodeling to promote synaptosome engulfment activity.

(A) Immunoblots of p-AKT, pS6, pNDRG1 and p-mTOR in BV2 from indicated time points. (B-C) Representative FACS plots (B) and geometric mean fluorescence intensities (C) of p-AKT (T308) and p-AKT (S473) in cortical microglia isolated from P9 mice injected with PBS and IL-33 (n = 3–5).
(D) Relative phagocytic activity of BV2 cells under indicated conditions. Data is normalized to the vehicle group (n = 47, 61, 9, 10, 20, 45, 9; pooled from 4 experiments).
(E) Immunoblots of p-AKT, total AKT and p-S6 under indicated conditions.
(F) OCR of BV2 cells under indicated conditions analyzed by Mito Stress test assay (n = 10, representative of 2 experiments).
(G) MFI of TMRM staining in BV2 cells transduced with sgRNAs targeting Inpp5d and Pten (n = 8, 11, 11; representative of 3 experiments).
(H) OCR of BV2 cells transduced with sgRNAs targeting Inpp5d and Pten analyzed by Mito Stress test assay (n = 24, 15, 8; representative of 2 experiments).
(I) Relative fraction of cells engulfed synaptosomes in BV2 transduced with sgRNAs targeting Inpp5d and Pten. Data is normalized to the empty vector group (n = 22, 11, 20; representative of 5 experiments).
(J-K) Phalloidin (white) and DAPI (blue) staining on BV2 cells transduced with sgRNAs targeting Inpp5d and Pten (Scale bar = 20μm). Surface area of Phalloidin staining is enumerated in (K) (n = 105, 56, 127; representative of 2 experiments).
(L-M) Phalloidin (white) and DAPI (blue) on BV2 cells under indicated conditions (Scale bar= 20μm). Surface area of Phalloidin staining is quantified in (M) (n = 65, 46, 57, 58; representative of 2 experiments).
Statistics: mean ± SEM (C, D, G and I); middle line: mean (K and M); mean ± SD (F and H); student’s t test (C), one-way ANOVA test (D, F, G, H, I, K and M); * p<0.05, **p<0.01,
***p<0.001.
We asked if AKT functions as a signaling hub being central to regulate microglial metabolism and phagocytosis. We tested the effects of AKT activation on microglial phgocytosis by CRISPR/Cas9 mediated perturbation of Pten and Inpp5d, which encodes phosphoinositide phosphatases (PTEN and SHIP-1) that dephosphorylate PI(3,4,5)P3 and counterbalance PI3K activity (Figure S5E) (Lee et al., 1999; Lioubin et al., 1996). Intracellular staining of pAKTS473 and pAKTT308 confirmed markedly enhanced AKT activation in Pten and Inpp5d perturbed BV2 cells (Figures S5F-S5G). Similar to IL-33 stimulation, activation of AKT by perturbing Pten and Inpp5d resulted in increased mitochondrial membrane potential, baseline and maximal respiratory capacity and lactate production rate (Figures 5G–5H, Figure S5H), indicative of augmented mitochondrial activity and glycolysis. Similar effects were observed in AKT-overexpressing cells (Figures S5I-S5K). When fed with pHrodo Red-conjugated synaptosomes, Pten and Inpp5d deficient BV2 cells engulfed more synaptosomes (Figure 5I), phenocopying exogenous IL-33 stimulation and AKT overexpression (Figure S5L). This pro-phagocytic effect relied on mitochondrial function, as addition of ETC inhibitors reversed the phenotype of Pten deficient and AKT overexpressing BV2 cells (Figures S5M-S5N). In contrast to dystrophic morphology of Il1rl1−/− microglia, Pten and Inpp5d deficient cells exhibited enlarged cellular surface area with additional formation of filopodia-like structure (Figures 5J–5K), which might act as phagocytic tentacles and facilitate phagocytosis (Kress et al., 2007). We then asked whether IL-33 stimulation could directly induce microglial morphological changes in an AKT-dependent manner. Despite to a less extend compared to Pten deletion, IL-33 treatment induced a significant increase in the cellular surface area of BV2 cells, and the effect was completely abrogated by inhibiting AKT activation (Figures 5L–5M). Therefore, our data demonstrate that AKT functions as a pivotal effector downstream of IL33-ST2 in microglia, being central to impinge on microglial metabolism and phagocytosis.
Mitochondrial metabolism and AKT activity are regulated in a temporal manner in order to adapt microglia functional activity in the developing brain
Given that microglia undergo substantial morphological and functional changes over time during postnatal development (Colonna and Butovsky, 2017; Hammond et al., 2018), we reasoned that microglia are metabolically programmed to distinct states to accommodate their changing needs of functional machinery in the developing brain. To test this, we leveraged our sc-RNA seq profiles of cortical microglia from different ages and inferred a microglia developmental trajectory by ordering all microglia along a pseudo-temporal path based on their expression profiles and identified coordinated transcriptional waves that were sequentially activated or suppressed as microglia progress through the developmental path (Figure 6A, Figures S6A-S6B). The trajectory recapitulated temporal maturation of microglial phenotype from the embryonic to the adult brain (Matcovitch-Natan et al., 2016). Postnatal microglia acquired a transient phagocytic gene program with a transient upregulation of genes associated with lysosome organization and phagosome maturation, whereas genes associated with environment sensing were induced later along the pseudo temporal trajectory (Figure 6A, Figure S6C, Table S4). This transient phagocytic window was accompanied by a remarkable metabolically active state with an enrichment of genes involved in energetic metabolism, including oxidative phosphorylation and beta-oxidation (Figures 6A-6B), suggesting a temporal coupling between phagocytic activity and cell-intrinsic metabolic program during neurodevelopment. Measurement of multiple parameters associated with bioenergetic activity revealed significant reductions in mitochondrial membrane potential, mitochondrial mass and mitochondrial network in P28 microglia compared to P9 (Figures 6C-6D, Figure S6D), indicative of declined mitochondrial activity in the adult microglia. Furthermore, extracellular flux analysis confirmed both restrained mitochondrial respiration and reduced glycolysis in P28 microglia compared to P9 (Figure 6E, Figures S6E). Collectively, our data demonstrate metabolic reprogramming of microglia by restraining bioenergetics as microglia switch their functional states during neurodevelopment.
Figure 6. Mitochondrial bioenergetics and AKT activity of microglia is temporally regulated during neurodevelopment in vivo.

(A) Gene expression dynamics along the inferred pseudo-temporal trajectory defined by Monocle (Methods). Averaged gene expression profiles of gene groups along the trajectory (left). Heatmap of relative average expression of genes within each group along the trajectory (middle). Representative genes and top terms from gene-set enrichment analysis for each group (right). Bar graph at the bottom represents the age of single cells.
(B) Average expression of genes involved in mitochondria function related GO terms along the pseudo-temporal trajectory. Curve reflects a LOESS fit of single cell expression; color bar indicates relative expression along pseudo-time; β Oxidation: GO: GO:0006635; OXPHOS: GO:0006119.
(C) Quantification TMRM and MitoTracker Green in cortical microglia at indicated ages.
(D) MitoTracker Red CMXRos (red) and DAPI (blue) staining in cortical microglia at indicated ages. Quantification of mitochondria content (relative covering area normalized to nuclear area) is plotted on the right (n = 37, 50; representative of 2 experiments).
(E) OCR of P9 and P28 microglia analyzed by Mito Stress test assay (n = 10, 15).
(F) Inferring expression dynamics of Pten and Inpp5d along the pseudotime trajectory.
(G) Representative FACS profiles showing intracellular staining of p-AKT (T308) and p-AKT (S473) in P9 and P28 microglia.
(H) MFI of p-AKTT308 and p-AKTS473 staining in P9 and P28 microglia (n = 9, 10; representative of 2 experiments).
(I) TMRM staining in cortical microglia isolated from WT and Ptenf/f::CX3CR1-Cre+/− mice at P28 (n = 4, 3).
(J) Representative FACS profiles and quantification of relative engulfment capacity of cortical microglia isolated from WT and Ptenf/f::CX3CR1-Cre+/− mice at P28 (n = 4, 3).
(K) Representative 3D reconstructions of Iba1-stained microglia residing in somatosensory cortex of WT and Ptenf/f::CX3CR1-Cre+/− animals at P28 (Scale bar = 10μm).
(L) Quantification of cellular volume and surface area of cortical microglia from WT and Ptenf/f::CX3CR1-Cre+/− mice at P28 (n = 157, 80, pooled from 3 mice/group).
Statistics: mean ± SEM (C, H and J); middle line: mean (D and L); mean ± SD (E); student’s t test; * p<0.05, **p<0.01, ***p<0.001.
Although endogenous IL-33 started to express when microglia are highly metabolically active, we and others found that IL-33 expression still persists in the adult brain (Gadani et al., 2015), suggesting additional cell-intrinsic mechanisms that restrain cellular metabolism in adult microglia. Our trajectory analysis revealed an induction of genes that antagonize catabolic and biosynthetic processes (Figure 6A), among which were Pten and Inpp5d (Figure 6F), suggesting AKT activity was also temporally regulated during neurodevelopment. Indeed, we found P9 microglia exhibited higher levels of phosphorylation of AKT than that of P28 (Figure 6H). To directly test how AKT activation impacts microglia function in vivo, we conditionally depleted Pten from microglia/monocyte. In contrast to the microglial dysfunction observed in Il1rl1−/− mice, Pten deficient microglia exhibited a remarkable increase in mitochondrial membrane potential and enhanced capacity to engulf synaptosomes (Figures 6I-6J). Furthermore, cell morphology analysis revealed an activation phenotype of microglia in Ptenflox/flox::CX3CR1-Cre+/− mice, evident by an increase in cellular volume and surface, and CD68 levels (Figures 6K-6L, Figure S6F). Taken together, our data suggest that mitochondrial metabolism and AKT activity is temporally regulated to adapt the functional and phenotypic switch of microglia in the developing brain.
Discussion
Microglia are not only pivotal during CNS development, when they sculpt developing synaptic circuits and fine-tune synaptic connectivity, but also responsible for brain homeostasis throughout life, during which they survey surrounding axons and invading pathogens. Using comprehensive single-cell RNA sequencing analysis of microglia across different developmental stages, we and others found microglia could acquire distinct transcriptional programs to support parallel development of the brain. However, how these transcriptional programs are related to corresponding functional states and how they are regulated coordinately during development is not clear. Our study demonstrates how environmental cues and cell-intrinsic mechanisms are coordinated to meet the distinct metabolic demands and thereby execute varying tasks in the developing and adult brain. Metabolic reprogramming of microglia is achieved by the integration of a multitude of signals, both intrinsic and environmental: (1) glia-derived IL-33 promotes microglial metabolism and phagocytosis to facilitate synapse engulfment in the developing brain; (2) AKT activity is dampened by upregulating Pten and Inpp5d to transit into metabolically quiescent phase in adult microglia.
Changes in energy metabolism have been reported in many neurodevelopmental and neuropsychiatric disorders. For example, mitochondrial dysfunction, caused by mutations of mitochondrial DNAs, decreased activity of mitochondrial electron transport chain complexes or reduced expression of mitochondrial genes, has been described in autism spectrum (Rossignol and Frye, 2012) and schizophrenia (Rajasekaran et al., 2015). Our study suggests that perturbing bioenergetic metabolism in microglia could lead to impaired synaptic function and behavioral defects. Therefore, it stands to reason that dysfunctional neuronal network observed in neurodevelopmental disorders might originate, at least in part, from metabolic and mitochondrial dysregulation in microglia. Genomic alterations in IL33, IL1RL1 and IL1RAP (co-receptor that binds to ST2 to mediate IL33 signaling) have been described in a wide range of neurodevelopmental and neurodegenerative disorders (Iossifov et al., 2012; Pinto et al., 2014), and our study may provide underlying cellular and molecular mechanisms. Interestingly, variants in the PTEN and INPP5D loci are associated with autism (Buxbaum et al., 2007) and Alzheimer’s disease (Lambert et al., 2013). Considering AKT activity being central to regulate metabolic adaptation and functional switch in microglia, it will be interesting to investigate the specific role of PTEN and INPP5D in microglia in the pathogenesis of neurodevelopmental diseases.
In summary, we demonstrate that microglia phagocytic activity was functionally coupled to its bioenergetic metabolisms. We show that mitochondrial metabolism and AKT activity were temporally regulated by integrating both cell-intrinsic and cell-extrinsic environment and developmental cues to adapt the functional and phenotypic switch of microglia in the developing brain. Our work, therefore, illustrates that metabolic programming acts in parallel to transcriptional remodeling to enforce microglia functional state and demonstrate a key role of IL-33-ST2-AKT dependent metabolic adaptations to promote microglial phagocytosis during early brain development with implications for neurodevelopmental and neuropsychiatric disorders.
Limitations of study
We show conditional deletion of Il1rl1 from microglia and Il33 from astrocytes leads to increased susceptibility to kainic acid induced seizures. However, CX3CR1-Cre also targets monocytes (Yona et al., 2013)and GFAP-Cre affects radial glia, which will result in genomic recombination in neurons (Garcia et al., 2004). Additional Cre lines that can specifically delete Il33 from astrocytes and Il1rl1 from microglial cells are needed to fully address cell type specific role of IL-33/ST2 signaling pathway in neurodevelopment and seizure susceptibility in vivo.
START★METHODS
RESOURCE AVAILABILITY
Lead contact
All information and requests for further resources and reagents should be directed to and will be fulfilled by the lead contact: Vijay Kuchroo (vkuchroo@rics.bwh.harvard.edu).
Materials availability
All requests for resources and reagents should be directed to and will be fulfilled by the lead contact. This study did not generate new unique reagents.
Data and code availability
The bulk RNA-Seq reported in Figure 1 and 2, as weel as the single-cell RNA-Seq data reporped in Figure 2 and Figure 3 have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PE anti-mouse/human CD11b Antibody (clone M1/70) | Biolegend | Cat# 101208, RRID: AB_312791 |
| FITC anti-mouse/human CD11b Antibody (clone M1/70) | Biolegend | Cat# 101206, RRID: AB_312789 |
| Brilliant Violet 421™ anti-mouse/human CD11b (clone M1/70) | Biolegend | Cat# 101236, RRID: AB_11203704 |
| APC anti-mouse CD45.2 Antibody (clone 104) | Biolegend | Cat# 109814, RRID:AB_389211 |
| PE anti-mouse CD45.2 (clone 104) | Biolegend | Cat# 109808, RRID:AB_313445 |
| FITC anti-mouse CD45.2 (clone 104) | Biolegend | Cat# 109806, RRID:AB_313443 |
| PE/Cyanine7 anti-mouse CD45.2 (clone 104) | Biolegend | Cat# 109830, RRID:AB_1186098 |
| APC/Cyanine7 anti-mouse CD45 Antibody (Clone 30-F11) | Biolegend | Cat# 103116, RRID:AB_312981 |
| PE/Cyanine7 anti-mouse CD45 Antibody (clone 30-F11) | Biolegend | Cat# 103114, RRID:AB_312979 |
| CD282 (TLR2) Monoclonal Antibody (6C2), PE | eBioscience | Cat# 12-9021-82, RRID:AB_466230 |
| CD282 (TLR2) Monoclonal Antibody (6C2) | eBioscience | Cat# 11-9021-82, RRID:AB_465440 |
| Alexa Fluor® 647 anti-mouse CD85k (gp49 Receptor) Antibody | Biolegend | Cat# 144906, RRID:AB_2562044 |
| Alexa Fluor® 647 Mouse Anti-Akt (pS473) Clone M89-61 | BD Biosciences | Cat# 561670, RRID:AB_10896328 |
| Phospho-mTOR (Ser2448) Monoclonal Antibody (MRRBY), PE | eBioscience | Cat# 12-9718-41, RRID:AB_2572723 |
| PE Mouse Anti-Akt (pT308) Clone J1-223.371 | BD Biosciences | Cat# 558275, RRID:AB_2225329 |
| Phospho-Akt (Thr308) (244F9) Rabbit mAb | Cell Signaling Technology | Cat# 4056, RRID:AB_331163 |
| Phospho-Akt (Ser473) (D9E) | Cell Signaling | Cat# 4060, RRID:AB_2315049 |
| XP® Rabbit mAb | Technology | |
| Pho spho -NDRG1 (Thr346) (D98G11) XP® Rabbit mAb | Cell Signaling Technology | Cat# 5482, RRID:AB_10693451 |
| Histone H3 (D2B12) XP® Rabbit mAb (ChIP Formulated) | Cell Signaling Technology | Cat# 4620, RRID:AB_1904005 |
| Phospho-mTOR (Ser2448) Antibody | Cell Signaling Technology | Cat# 2971, RRID:AB_330970 |
| mTOR (7C10) Rabbit mAb antibody | Cell Signaling Technology | Cat# 2983, RRID:AB_2105622 |
| Akt (pan) (C67E7) Rabbit mAb | Cell Signaling Technology | Cat# 4691, RRID:AB_915783 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7074, RRID:AB_2099233 |
| Rat anti Mouse CD68 antibody, (clone FA-11) | Serotec | Cat# MCA1957, RRID:AB_322219) |
| Rabbit anti-Synaptophysin (SP11) | Abcam | Cat# ab16659, RRID:AB_443419 |
| Rabbit anti-Iba1 | Wako | Cat# 019-19741, RRID:AB_839504 |
| Goat anti-Iba1 | Abcam | Cat# ab5076, RRID:AB_2224402 |
| Mouse anti-PSD95 | Chemicon | Cat# MAB1596, RRID:AB_2092365 |
| Mouse IL-33 Antibody | R&D Systems | Cat# AF3626, RRID:AB_884269 |
| Purified anti-P2RY12 Antibody | Biolegend | Cat# 848001, RRID:AB_2650633 |
| Cy™2 AffiniPure Donkey Anti-Goat IgG (H+L) | Jackson Immunoresearch | Cat# 705-225-147, RRID:AB_2307341 |
| Cy™3 AffiniPure Donkey Anti-Mouse IgG (H+L) | Jackson Immunoresearch | Cat# 715-165-150, RRID:AB_2340813 |
| Cy™3 AffiniPure Donkey Anti-Goat IgG (H+L) | Jackson Immunoresearch | Cat# 705-165-147, RRID:AB_2307351 |
| Cy™5 AffiniPure Donkey Anti-Mouse IgG (H+L) | Jackson Immunoresearch | Cat# 715-175-151, RRID:AB_2340820 |
| Cy™2 AffiniPure Donkey Anti-Rabbit IgG (H+L) | Jackson Immunoresearch | Cat# 711-225-152, RRID:AB_2340612 |
| Cy™3 AffiniPure Donkey Anti-Rabbit IgG (H+L) | Jackson Immunoresearch | at# 711-165-152, RRID:AB_2307443 |
| Proteins, Chemicals and Kits | ||
| MitoTracker™ Deep Red FM - Special Packaging | ThermoFisher Scientific | Cat# M22426 |
| Image-iT™ TMRM | ThermoFisher Scientific | Cat# I34361 |
| 2-NBDG (2-(N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)Amino)-2-Deoxyglucose) | ThermoFisher Scientific | Cat# N13195 |
| Alexa Fluor™ 488 Phalloidin | ThermoFisher Scientific | Cat# A12379 |
| Alexa Fluor™ 594 Phalloidin | ThermoFisher Scientific | Cat# A12381 |
| MitoTracker™ Green FM - Special Packaging | ThermoFisher Scientific | Cat# M7514 |
| MitoTracker™ Red CMXRos - Special Packaging | ThermoFisher Scientific | Cat# M7512 |
| DAPI | Sigma-Aldrich | Cat# D9542 |
| Kainic acid monohydrate | Sigma-Aldrich | Cat# K0250-10MG |
| pHrodo™ iFL Green Microscale Protein Labeling Kit | ThermoFisher Scientific | Cat# P36015 |
| pHrodo™ iFL Red Microscale Protein Labeling Kit | ThermoFisher Scientific | Cat# P36014 |
| Seahorse XF Cell Mito Stress Test Kit | Agilent | Cat# 103015-100 |
| HEPES (1 M) | ThermoFisher Scientific | Cat# 15630080 |
| D-(+)-Glucose solution | Sigma-Aldrich | Cat# G8769-100ML |
| Percoll Plus | Sigma-Aldrich | Cat# GE17-5445-01 |
| PicoPure RNA Isolation Kit | Thermo Fisher Scientific | Cat# KIT0204 |
| iScript cDNA Synthesis Kit | Bio-Rad | Cat# 1708891 |
| iTaq Universal SYBR Green Supermix | Bio-Rad | Cat# 1725121 |
| TCL buffer (QIAGEN) | Qiagen | Cat# 1031576 |
| 2-Mercaptoethanol | Sigma-Aldrich | Cat# M6250 |
| Nextera XT Sample Preparation Kit | Illumina | Cat# FC-131-1096 |
| DMEM/F-12, GlutaMAX™ supplement | ThermoFisher Scientific | Cat# 10565042 |
| DMEM, high glucose | ThermoFisher Scientific | Cat# 11965126 |
| GlutaMAX™ Supplement | ThermoFisher Scientific | Cat# 35050061 |
| Trypsin-EDTA (0.05%), phenol red | ThermoFisher Scientific | Cat# 25300120 |
| Penicillin-Streptomycin Mixture | Lonza | Cat# 17-602E |
| Sodium Pyruvate Solution (100 mM) | Lonza | Cat# 13-115E |
| N-acetyl cysteine | Sigma | Cat# A9165 |
| N-2 Supplement (100X) | Gibco | Cat# 17502048 |
| Recombinant Mouse M-CSF Protein | R&D Systems | Cat# 416-ML-050 |
| Recombinant Mouse IL-33 | Biolegend | Cat# 580506 |
| UK-5099 | Sigma-Aldrich | Cat# PZ0160-5MG |
| STF-31 | Selleckchem | Cat# S7931 |
| Oligomycin | Agilent | Cat# 103015-100 |
| Antimycin | Agilent | Cat# 103015-100 |
| Rotenone | Agilent | Cat# 103015-100 |
| AZD8055 | Selleckchem | Cat# S1555 |
| Ly294002 | Selleckchem | Cat# S1105 |
| Triciribine | Selleckchem | Cat# S1117 |
| Torkinib (PP242) | Selleckchem | Cat# S2218 |
| Rapamycin (Sirolimus) | Selleckchem | Cat# S1039 |
| Oltipraz | Sigma-Aldrich | Cat# O9389-5MG |
| ProlongGold Antifade reagent | ThermoFisher Scientific |
Cat# P36930 |
| O.C.T. Compound | Electron Microscopy Sciences | Cat# 62550-01 |
| Triton X-100 | Sigma-Aldrich | Cat# T8787 |
| Normal Donkey Serum | Jackson ImmunoResearch |
Cat# 017-000-001 |
| Papain Dissociation System | Worthington | Cat# LK003150 |
| PBS, pH 7.4 | ThermoFisher Scientific |
Cat# 10010023 |
| 20X Bolt™ MOPS SDS Running Buffer | ThermoFisher Scientific |
Cat# B0001 |
| PageRuler™ Prestained Protein Ladder | ThermoFisher Scientific |
Cat# 26617 |
| Bolt™ 4 to 12%, Bis-Tris | ThermoFisher Scientific |
Cat# NW04120BOX |
| Bolt™ Transfer Buffer (20X) | ThermoFisher Scientific |
Cat# BT00061 |
| Power Blotter Pre-cut Membranes and Filters, PVDF, regular size | ThermoFisher Scientific | Cat# PB9320 |
| 4X Bolt™ LDS Sample Buffer | ThermoFisher Scientific |
Cat#B0008 |
| Western Blotting Filter Paper, 0.83 mm thick, 7 × 8.4 cm | ThermoFisher Scientific | Cat# 84783 |
| Pierce™ ECL Western Blotting Substrate | ThermoFisher Scientific | Cat# 32209 |
| RIPA Lysis and Extraction Buffer | ThermoFisher Scientific |
Cat# 89900 |
| Paraformaldehyde solution 4% in PBS | Santa Cruz | Cat# sc-281692 |
| Sucrose | Sigma-Aldrich | Cat# S0389 |
| cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail | Roche | Cat# 4693159001 |
| Buffer TCL (125 mL) | QIAGEN | Cat# 1031576 |
| B-Mercaptoethanol | Sigma-Aldrich | Cat# M3148-100ML |
| Recombinant RNase Inhibitor | Takara | Cat# 2313B |
| Deoxynucleotide (dNTP) Solution Mix | New England Biolabs | Cat# N0447L |
| Maxima H Minus Reverse Transcriptase (200 U/uL) | Thermo Fisher Scientific | Cat# EP0753 |
| KAPA HiFi HotStart ReadyMix PCR Kit | Roche | Cat# KK2602 |
| Agencourt RNAClean (RNA SPRI beads) | Beckman Coulter | Cat# A63987 |
| Nuclei EZ Prep | Sigma-Aldrich | Cat# NUC101-1KT |
| AMPureXP SPRI (DNA SPRI beads) | Beckman Coulter | Cat# A63881 |
| Nextera XT DNA Library Preparation Kit | Illumina | Cat# FC-131-1096 |
| NextSeq 500 high output kit V2, 75 cycles | Illumina | Cat# FC-404-2005 |
| 10X Chromium Single Cell 30 Kit | 10X Genomics | Cat# 120237 |
| Oligonucleotides | ||
| sgRNA sequences | See Table S5 | NA |
| qPCR primer sequences | See Table S5 | NA |
| Experimental Model: Strains | ||
| Mouse: C57BL/6J | Jackson Laboratory | JAX:000664, RRID:IMSR_JAX:000664 |
| Mouse: B6J.B6N(Cg)-Cx3cr1tm1.1(cre)Jung/J | Jackson Laboratory | JAX:025524, RRID:IMSR_JAX:025524 |
| Mouse: B6;FVB-Tg(Aldh1l1-EGFP/Rpl10a)JD133Htz/J | Jackson Laboratory | JAX:030248, RRID:IMSR_JAX:030248 |
| Mouse: B6.129S4(Cg)-Mapttm1(EGFP)Klt/J | Jackson Laboratory | JAX:029219, RRID:IMSR_JAX:029219 |
| Mouse: B6.Cg-Tg(Gfap-cre)73.12Mvs/J | Jackson Laboratory | JAX:012886, RRID:IMSR_JAX:012886 |
| Mouse: ilirl1tmIAnnm | (Townsend et al., 2000) | N/A |
| Mouse: Il33−/− | (Oboki et al., 2010) | N/A |
| Mouse: Il1rl1 flox | (Chen et al., 2015) | N/A |
| Mouse: Il33 flox | (Chen et al., 2015) | N/A |
| Mouse: C;129S4-Ptentm1Hwu/J | (Groszer et al., 2001) | JAX:004597, RRID:IMSR_JAX:004597 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GSE127449 |
| Software and Algorithms | ||
| ImageJ | (Schneider et al., 2012) | https://imagej.nih.gov/ij/ |
| Imaris | Bitplane | https://imaris.oxinst.com/ |
| GraphPad Prism 8 | GraphPad Software | https://www.graphpad.com/scientificsoftware/prism/ |
| FlowJo | TreeStar | https://www.flowjo.com/solutions/flowjo |
| RSEM version 1.2.8 | (Li and Dewey, 2011) | http ://deweylab. github. io/RSEM/ |
| edgeR version 3.5.2 | (Robinson et al., 2010) | https://gist.github.com/jdblischak/11384914 |
| Seurat version 2.3.3 | (Satija et al., 2015) | https://github.com/satijalab/seurat |
| 10x Cell Ranger version 1.2 | 10X Genomics |
https://support.10xgenomics.com/singlecell/software/overview/welcome
|
| Monocle version 2.8.0 | (Qiu et al., 2017) | http://cole-trapnell-lab.github.io/monocle-release/ |
METHOD DETAILS
Animals
ST2-deficient mice (Il1rl1tm1Anjm)(Townsend et al., 2000), ST2 flox mice (Il1rl1f/f)(Chen et al., 2015) and Il33 flox mice (Il33f/f)(Chen et al., 2015) on a mixed genetic background were generously provided by Dr. R. Lee, and were further backcrossed for at least 10 generations onto the B6 genetic background. Il33−/− mice (Oboki et al., 2010) were kindly provided by Dr. Susumu Nakae. Ptenf/f mice (Groszer et al., 2001) were generously provided by Dr. Hong Wu. GFAP-Cre (Garcia et al., 2004)mice were a generous gift from Dr. Quintana Francisco. Mice with a specific deletion of IL33 in astrocytes were generated by crossing GFAP-Cre with Il33f/f mice. Mice with a specific deletion of Il1rl1 and Pten on microglia/monocyte were generated by crossing CX3CR1-Cre with Il1rl1f/f mice or Ptenf/f mice, respectively. CX3CR1-Cre (B6J.B6N(Cg)-Cx3cr1tm1.1(cre)Jung/J, JAX 025524), Aldh1l1-GFP (B6;FVB-Tg(Aldh1l1-EGFP/Rpl10a)JD133Htz/J, JAX 030248) and Tau-GFP (B6.129S4(Cg)-Mapttm1(EGFP)Klt/J, JAX 029219) mice were purchased from Jackson Laboratories. All mice were housed in our specific-pathogen-free facilities at Brigham and Women’s Hospital, Harvard Medical School or Westlake University. For all the experiments, littermates that were either homozygous or heterozygous for the wild-type allele were used as controls. For in vivo IL-33 treatment, postnatal day 5 mice were intraperitoneally injected with IL-33 (Biolegend, 100ng) every other day and microglial phenotype was analyzed on postnatal day 9. The dose of IL-33 used in the current study was comparable to that reported previously (Nguyen et al., 2020; Vainchtein et al., 2018). All experiments were conducted in accordance with animal protocols approved by the Harvard Medical Area Standing Committee on Animals, BWH IACUC or Westlake University IACUC.
Flow cytometry and microglia isolation
Mice were sacrificed by CO2 asphyxiation, followed by transcardial perfusion with cold PBS. The brain regions of interest were dissected and dissociated using the Worthington Papain kit (for mitochondrial staining, Seahorse experiments and primary microglia culture), with 20 min enzymatic digestion, followed by manual trituration and filtering through a 70μm cell strainer to remove clumps. Importantly, cells were kept on ice or at 4°C at all times except for enzymatic digestion. For experiments with epitope sensitive to enzymatic digestion (for isolation of Phagohi and Phagolo microglia), brains were dissociated with collagenase D (2.5 mg/ml; Roche Diagnostics) and DNaseI (1 mg/ml; Sigma). For gene expression analysis, brains were homogenized in ice-cold HBSS supplemented with 15mM HEPES and 0.5% glucose by 5 gentle strokes in a 7 mL glass Dounce homogenizer. Myelin debris were removed by density gradient separation; pellet was resuspended in 40% Percoll and centrifuged in 800G for 20 min at 4°C. Isolated microglia were labeled with a combination of conjugated antibodies, and FACS analysis of the microglial profile was performed by gating CD11bintCD45int cells. Appropriate IgG isotype controls were used for all staining. For intracellular staining of phospho-AKT, cells were fixed and permeabilized using the BD™ Phosflow Fix Buffer and Perm/Wash Buffer Set, followed by staining with anti-pAKTT308 and anti-pAKTS473 antibodies. Dead cells were excluded with 7-AAD or Fixable Viability Dye eFluor 506. To isolate neurons and astrocytes from postnatal mice, brains were dissociated using the Worthington Papain kit with 20 min enzymatic digestion, followed by manual trituration and filtering through a 100μm cell strainer to remove clumps. The resulting single cell suspension was stained with a combination of conjugated antibodies. Neurons and astrocytes were sorted from P9 brains of Aldh1l1-GFP mice and gated as Thy1+GFP−CD45− and GFP+CD45−, respectively. Flow cytometry was performed on LSR II or Fortessa (BD Biosciences)), and data were analyzed with FlowJo Software (TreeStar). Sorting was performed with the Aria A (BD Biosciences).
Assessment of mitochondrial membrane potential and mitochondrial mass
To assess microglial mitochondrial membrane potential, 50,000 freshly sorted microglia or BV2 cells were incubated at 37 °C with Tetramethylrhodamine, methyl ester (TMRM, Thermo Fisher Scientific) at a final concentration of 100 nM for 30 min. At this concentration, mitochondrial dye accumulation is largely dependent on the mitochondrial membrane potential, without forming aggregates in cell membranes and minimal interaction with membrane proteins. After incubation, the cell suspension was diluted with ice-cold PBS and TMRM fluorescence was immediately acquired with a BD LSRII instrument. Similarly, to assess microglia mitochondrial mass, 50,000 freshly sorted microglia were incubated at 37 °C with MitoTracker Green FM at a final concentration of 20nM for 30min, and then washed once with ice-cold PBS. MitoTracker Green fluorescence was immediately acquired with a BD LSRII instrument.
Synaptosome purification and labeling
Synaptosomes were purified by sucrose gradient from the adult mouse brains as previously described (Dunkley et al., 2008). To obtain pHrodo-conjugated synaptosomes, synaptosomes purified from the wild-type adult brains were incubated with pHrodo red or pHrodo green, succinimidyl ester (Invitrogen) in 0.1 M sodium carbonate (pH 9.0) for 2h at room temperature with gentle agitation. Unbound pHrodo dye was washed out with multiple rounds of centrifugation and pHrodo-conjugated synaptosomes were re-suspended with PBS containing 5% DMSO for subsequent freezing.
Ex vivo microglial phagocytosis assay
Fresh sorted microglia were recovered in DMEM/F-12 (Thermo Fisher Scientific, 11320033) supplemented with 2% FBS, 100U/ml penicillin, 100ug/ml streptomycin, 5 ug/ml N-acetyl cysteine and 1X N-2 Supplement (Gibco). 50,000 microglia were plated onto U-bottom 96 well plate, followed by incubation with 5μl of pHrodo-dye conjugated synaptosomes at 37°C for 3hrs. Phagocytosis was stopped by transferring the suspensions onto ice. Microglial phagocytosis was analyzed by flow cytometry by assessing pH-dependent particle fluorescence within cells. The eating gate was determined using an unfed control, and confirmed by sorting an aliquot of the sample into non-eating and eating populations by FACS followed by microscopic assessment.
Lentivirus Production and Transduction
sgRNAs were inserted into Lenti-Cas9-gRNA-GFP vector (Addgene, #124770) and co-transferred into 293T cells with packaging plasmids psPAX2 and pMD2.G using PolyJet (Signagen Laboratories). Supernatant was harvested at 48h and 72h and concentrated by ultracentrifuge. BV2 cells were spin infected with lentiviruses with Polybrene (4μg/ml, Sigma). 7–10 days after infection, GFP+ BV2 cells were flow sorted for further expansion and subsequent analysis. All cell lines were assayed as a polyclonal population.
Measurement of Real-Time ECAR and OCR
Real-time extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) were measured using a Seahorse XF96 Extracellular Flux Analyzer (Seahorse Bioscience) according to the manufacturer’s instructions, with minor modifications. Briefly, 2 × 105 freshly sorted microglia or 2 × 104 BV2 cells were plated on the PDL-coated XF96 cell culture microplate and preincubated at 37°C for minimum of 30min in the absence of CO2 in assay medium (XF base medium containing 1 mM pyruvate, 2 mM L-glutamine, and 25 mM glucose). OCR and ECAR were measured under basal conditions, and after the addition of the following drugs: 0.75 μM oligomycin, 1μM flurorcarbonyl cyanide phenylhydrazon (FCCP) and 0.5μM rotenone + 0.5μM antimycin A (Seahorse Bioscience) as indicated.
Primary Microglia Culture
Primary mouse microglia were enriched and cultured from brains of postnatal day 2–7 mice, as previously described (Bohlen et al., 2017). Briefly, brains were digested enzymatically with papain then mechanically dissociated to generate a single cell suspension, followed by density gradient separation with 40% Percoll. Cell suspensions were then positively selected for CD45 expression by CD45 magnetic beads (Miltenyi) using the MACS system. CD45 purified mouse microglia were cultured in PDL-coated plates in a defined culture media containing DMEM/F-12 (Thermo Fisher Scientific, 11320033), 100U/ml penicillin, 100ug/ml streptomycin, 5 ug/ml N-acetyl cysteine, 1X N-2 Supplement (Gibco), 10ng/ml M-CSF (R&D) and 2% heat inactivated FCS (Sigma). Primary rat microglia were enriched and cultured from P7-P12 brains. Brains were dissected and transferred to an ice-cold Dounce homogenizer (Wheaton) with ice-cold PBS supplemented with 0.5g/L glucose and 10mM HEPES. Tissues were subjected to three successive rounds of 3 −5 gentle strokes of the homogenizer piston and centrifuged, followed by density gradient separation with 40% Percoll. Cell suspensions were then positively selected for CD11b expression by incubating with mouse anti-Rat CD11b (Biorad) antibody, followed by anti-mouse IgG magnetic beads (Miltenyi). CD11b purified rat microglia were cultured in PDL-coated plates in a defined culture media containing DMEM/F-12 (Thermo Fisher Scientific, 11320033), 100U/ml penicillin, 100ug/ml streptomycin, 5 ug/ml N-acetyl cysteine, 5 μg/ml N-acetyl cysteine, 5 μg/ml insulin, 100 μg/mL apo-transferrin, and 100 ng/mL sodium selenite, and 20ng/ml M-CSF (R&D).
For IL-33 stimulation and compound treatment assays, microglia at DIV 5–9 (days in vitro) were stimulated with 200ng/ml IL-33 (Biolegend) for 3hrs. Microglia were then enzymatically detached and analyzed by FACS.
For Oltipraz treatment, primary rat microglia at DIV 9–11 were stimulated with 0.5μM Oltipraz (#O9389, Sigma) for 12hrs. Microglia were then stained with MitoTracker GreenFM or TMRM for FACS analysis.
For ex vivo synaptosome phagocytosis assay, microglia from various stimulation conditions were incubated with 5μl of pHrodo-dye -conjugated synaptosomes for 1–3hrs.
RNA FISH with Hybridization Chain Reaction (HCR) Amplification
FISH probes were designed and ordered from Molecular Instruments. Snap-frozen brain sections were incubated with wash buffer (20% formamide, 2× SSC) for 30mins at room temperature and hybridized with HCR initiator tagged FISH probes in hybridization buffer (20% formamide, 10% dextran sulfate, 2× SSC) overnight at 37 °C. Following hybridization, samples were washed twice with wash buffer, 30mins per wash, and incubated with 1× PBS for 2hrs at 37°C. Subsequently, samples were incubated with 1× PBS for at least 6hrs at room temperature. Before HCR amplification, hybridized samples were pre-incubated with amplification buffer (10% dextran sulfate, 5× SSC, 0.1% Tween 20) for 30 mins. To initiate amplification, HCR hairpin stocks (Alexa 456 and Alexa 647 fluorophores) at 3 μM were snap-cooled by heating to 95°C for 90 seconds, and leaving to cool at room temperature for 30 mins. Gelled samples were then incubated with HCR hairpins diluted to 60 nM in amplification buffer for 3hrs at room temperature. After amplification, gels were washed with 5× SSCT (5× SSC, 0.1% Tween 20) twice with one hour per wash.
Immunohistochemistry
Animals were perfused with PBS followed by 4% paraformaldehyde. Brains were dissected out, post-fixed for 6h in 4% paraformaldehyde at 4 °C and transferred to 30% sucrose for 24 h. 30–40 μm tissue sections were cut by Leica cryostats and collected into PBS. Free floating brain sections containing desired brain regions were permeabilized at RT in 5% Normal Donkey Serum and 0.3% Triton X-100 for 1 h, followed by primary antibody overnight at 4°C. Sections were stained with the appropriate Cyanine dyes conjugated secondary antibodies and DAPI, and mounted with Fluorsave Mounting Medium (Millipore). For confocal, all images were acquired using Zeiss LSM710 or Olympus FV3000 confocal microscope, and Imaris was used to perform background subtraction and thresholding.
In vivo Synapse Engulfment and Lysosome Content Quantification
Microglial engulfment analysis was performed similarly as previously described (Schafer et al., 2014). Brain sections were stained with Iba1 and PSD-95. Z-stacks encompassing entire microglia were collected on a Zeiss laser scanning confocal microscope with a 60x objective using 0.2 μm z steps; laser power and gain were consistent across all experiments. For each hemisphere, at least 10 fields were imaged. Subsequent images were processed and quantified using Imaris software (Bitplane) to create 3D volume surface renderings. Surface rendered images were used to determine the volume of the microglia and of all synaptic materials (PSD95 signal). To measure the volume of engulfed synaptic materials, any PSD95 signals that were not within the microglia volume were subtracted from the image using the mask function. The remaining engulfed/internal fluorescence was surface rendered using parameters previously determined for all PSD95 and total volume of engulfed/internal PSD95 was calculated. Engulfment index determined by volume of internalized PSD95 (μm3) / volume of microglial cell (μm3) was used as a measure of engulfment ability. For quantification of microglial lysosome content, the volume of CD68 signals that were within the microglia volume was calculated and the volume of rendering CD68 (μm3) / volume of microglia cell (μm3) was calculated.
Synapse quantifications
3–5 animals/genotype/age were stained with pre- (Synaptophysin) and post-synaptic (PSD95) marker pairs. Three independent coronal sections per each mouse, which contain the somatosensory cortex were used for analyses. Confocal images were acquired at 60× magnification on LSM710 microscope, and synapse puncta (determined by pre- and post-synaptic protein colocalization) was analyzed using an ImageJ script developed by the Eroglu lab (PunctaAnalyzer plugin (Risher et al., 2014)). Image quality was checked by repeating analyses after 90° rotation of one channel to verify that co-localization was not due to random chance. To calculate % of WT co-localization, co-localized puncta values for WT were averaged, then all image values were converted to % of the calculated WT average.
Cell culture
BV2 cells were originally obtained from Oleg Butovsky (Brigham and Women’s Hospital) and expanded in DMEM supplemented with 10% FBS, sodium pyruvate, penicillin/streptomycin, and GlutaMAX (Thermo Fisher Scientific). HEK293T cells (ATCC) were cultured in DMEM supplemented with 10% FBS, sodium pyruvate, penicillin/streptomycin, and GlutaMAX. All cells were maintained in a humidified incubator containing 10% CO2 at 37 °C. To prepare bone marrow-derived macrophages (BMDMs), femurs were removed and flushed with PBS. Cells were counted and plated at 3×105 cells/24 well plate in RPMI supplemented with GlutaMAX, penicillin/streptomycin, non-essential amino acids, pyruvate, 10% heat inactivated fetal bovine serum and 20ng/ml MCSF. Cells were cultured for 7 days before use.
To examine the effect of IL33, metabolic inhibitor and PI3K/AKT inhibitors on mitochondria function and synaptosome engulfment, BV2 cells were pre-treated with STF-31 (5μM, Selleckchem, #S7931), UK5099 (10μM, Sigma, #PZ0160-5MG), Oligomycin (0.2μM, Seahorse Bioscience, #103015-100), Antimycin (0.1μM, Seahorse Bioscience, #103015-100), Rotenone (0.2μM, Seahorse Bioscience, #103015-100), Triciribine (2μM, Selleckchem, #S1117), Ly2940024 (4μM, Selleckchem, # S1105), AZD8055 (2μM, Selleckchem, # S1555) or PP242 (2.5μM, Selleckchem, # S2218) for 1h, followed by stimulation with 100ng/mL IL33 for additional 5hrs. IL33 and the drugs were present in culture medium for subsequent metabolic analysis and synaptosome engulfment assay.
Mitochondrial staining and imaging
Freshly sorted microglia were cultured in μ-Slide 8 well glass bottom slides (ibidi GmbH) and incubated for 30 min at 37°C with MitoTracker™ Red CMXRos (100 nM, ThermoFisher). After two rinses with PBS, cells were fixed with 3.7% paraformaldehyde for 15 min. DAPI (1 μg/mL, Sigma) and Alexa Fluor™ 488 Phalloidin (150 nM, ThermoFisher) were used for nuclear and actin staining. Z-stack images were acquired and mitochondrial area was quantified in Fiji software.
qRT-PCR
Total RNA was isolated with RNeasy Plus Mini Kit (Qiagen) or PicoPure RNA Isolation Kit (Thermo fisher Scientific), and single-strand cDNA was synthesized with iScript™ cDNA Synthesis Kit (BioRad). Real-time PCR was performed using either SYBR Green real-time PCR master mix (BioRad) or TaqMan Fast Universal PCR Master Mix (Thermo fisher Scientific).
Hippocampal slice preparation
WT and Il1rl1−/− mice were euthanized with Isoflurane at 2 to 3 months of age. Brains were quickly removed and submerged in ice-cold oxygenated sucrose-replaced artificial cerebrospinal fluid (aCSF) cutting solution (206mM sucrose, 2mM KCl, 2 mM MgSO4, 1.25mM NaH2PO4, 1mM CaCl2, 1mM MgCl2, 26mM NaHCO3, 10mM D-glucose, pH 7.4, 315 mOsm). Transverse slices (350 μm thickness) from the middle portion of each hippocampus were cut with a vibroslicer. After sectioning, slices were incubated in aCSF (124mM NaCl, 2mM KCl, 2mM MgSO4, 1.25mM NaH2PO4, 2.5mM CaCl2, 26mM NaHCO3, 10mM D-glucose, pH 7.4, 310 mOsm), in which they were allowed to recover for at least 90 min before recording. A single slice was then transferred to the recording chamber and submerged beneath continuously perfusing aCSF that had been saturated with 95% O2 and 5% CO2. Slices were incubated in the recording chamber for 20 min before stimulation under room temperature (~ 26 °C).
Electrophysiology
Standard field excitatory postsynaptic potentials (fEPSP) and population spikes (PS) were recorded in the CA1 region of hippocampus. A bipolar stimulating electrode (FHC Inc., Bowdoin, ME) was placed in the Schaffer collaterals to deliver test and conditioning stimuli. Two borosilicate glass recording electrodes filled with aCSF were positioned in stratum radiatum and stratum pyramidale of CA1, 200–300 μm from the stimulating electrode. fEPSP (from stratum radiatum) and population spikes (from stratum pyramidale) in CA1 were induced by test stimuli at 0.05 Hz with an intensity that elicited a fEPSP amplitude of 40~50% of maximum. Test responses were recorded for 30–60 min prior to beginning the experiment, to ensure stability of the response. To induce LTP, two consecutive trains (1s) of stimuli at 100 Hz separated by 20 s, a protocol that induces LTP lasting ~1.5 hr in wild-type mice of this genetic background were applied to the slices. The field potentials were amplified 100× using Axon Instruments 200B amplifier and digitized with Digidata 1322A. The data were sampled at 10 kHz and filtered at 2 kHz. Traces were obtained by pClamp 9.2 and analyzed using the Clampfit 9.2.
Behavioral test
All behavioral analysis was performed in the Neurobehavioral Laboratory Core facility at Harvard NeuroDiscovery Center. All of the mice included for behavioral studies were still generally active, still gaining weight, had no signs of seizures (before, during, or after behavioral testing): they did not display any staring-freezing episodes, did not show labored breathing, and had no signs of delay in performing the behavioral tasks. One Il1rl1−/− mouse was eliminated from social interaction analysis due to staring-freezing behavior.
Three-chamber social interaction.
The three-chamber sociability test was performed in a rectangular arena of clear Plexiglas with dimensions 63 cm length × 40 cm width × 22 cm height. The test arena contained three equally sized chambers (40 cm length × 20 cm width × 22 cm height), and the outer left and right chambers each contained an inverted wire cup. During the habituation session, the test mouse was first placed in the center chamber and allowed to freely explore for 10 minutes, with the doorways into the two side chambers open. For the social interaction session, test mice were returned to the center chamber, an unfamiliar target mouse was placed under one of the two wire cups, an object under the second cup and test mice were again allowed to move freely among the three chambers. The position of the first target mouse was alternated and equally distributed between the left and right cups for both mutant and control groups. The position was not changed between the social interaction and social recognition sessions. The test operator was blinded to test mouse genotype and remained outside of the test room during all sessions. Test subject performance was recorded automatically using TopScan software (CleverSys, Inc.), and the following behaviors were scored: amount of time spent in close proximity (3 cm radius) of the wire cups, amount of time spent in each chamber of the three-chamber arena and latency to approach each wire cup.
Social dominance tube test.
Mice from different litters that are sex-matched and weight-matched are placed inside opposite ends of a white PVC pipe that is 30.5 cm in length, 2.5 cm internal diameter. Mice are released simultaneously at both ends of tube. Start sides are alternated between trials. Five consecutive trials are performed for each pair with a maximum time of 2 min per trial. The trial ends when one mouse backed out of the tube so that all four limbs were outside of tube. Matches lasting more than two minutes were excluded from analysis and scored as a draw. The latency of each trial is recorded as the percentage of trials won by each mouse. Mice are considered socially dominant if they win more than 50% of the trials. The pipe is cleaned with 70% ethanol between each trial.
EEG Implantation and Recording
5–6month old WT and ST2 KO mice were surgically implanted under isoflurane anesthesia prefabricated EEG/EMG headmounts (8201, Pinnacle Technology, Lawrence, KS). Briefly, mice were anesthetized with isoflurane (5% induction, 2% maintenance) and eye ointment applied to each eye. The headmount was first fixed to the skull and four stainless steel EEG screws (8209&8212, Pinnacle) were inserted through openings in the headmount. Silver epoxy was applied to ensure electrical connectivity between the electrodes and the headmount. After one or more days of post-surgical recovery, mice were monitored using a video/EEG system 24 hr per day for up to 2 weeks. To visualize stress/challenge induced seizures, mice were habituated in home cage for one day, then tethered to the recording system and placed into EEG monitoring chamber. Observers who were blinded to genotype visually analyzed the patterns.
KA Induced Seizure Assay
Kainate (KA) in saline was administered subcutaneously (18 mg/kg) into 5–6-week old mice. Seizure activity was scored every 10 min for 2h using a modified Racine scale (0, normal; 1, hypoactivity; 2, rigidity (extensive limbs with raised tail); 3, rearing with repetitive head/forepaw movements; 4, rearing and falling; 5, continuous rearing/falling; 6, generalized convulsions) (Racine, 1972).
Single-cell RNA-Seq
For droplet-based 3′ end massively parallel single-cell RNA sequencing (scRNA-seq), freshly sorted microglia were encapsulated into droplets, and libraries were prepared using Chromium Single Cell 3′ Reagent Kits v2 according to manufacturer’s protocol (10× Genomics). The generated scRNA-seq libraries were sequenced using a 75 cycle Nextseq 500 high output V2 kit. All RNA-seq data represent pooled data from at least two replicates.
Quantification and Statistical Analyses
Bulk RNA-Seq Processing and Analysis
1000 microglia from each condition were lysed in 10μl TCL buffer plus 1% Mercaptoethanol. Samples were processed with SMART-Seq2 (Picelli et al., 2013), reads were aligned to the mouse mm10 transcriptome using Tophat (Trapnell et al., 2009), and expression abundance TPM estimates were obtained using RSEM parameters (Li and Dewey, 2011). Differential gene expression analysis was performed using EdgeR package (Robinson et al., 2010)with default parameters.
Single-cell RNA-seq data analysis
Preprocessing, mapping and initial QC.
De-multiplexing, alignment to the mm10 mouse transcriptome and UMI-collapsing were performed using the Cellranger toolkit (version 1.2, 10X Genomics). We selected cells for downstream processing and filtered out low quality cells and cell doublets that had (1) a low number (<1000) of unique detected genes, and (2) a high mitochondrial content (6%) determined by the ratio of reads mapping to the mitochondria. Expression values were then internally normalized to 10,000 transcripts to create TPM-like values, and finally calculating log2(TPM+1) values.
Signature scoring and cell type assignment.
For a given set of genes, their average expression was defined as signature score. Z-scored relative expression values rather than actual expression values were used. Brain myeloid cell type-specific signatures (Friedman et al., 2018) were used to assign each cell to one of the following categories: microglia, macrophage and monocyte. Only microglia were kept for further analysis.
PCA, clustering, TSNE and differential gene analysis.
Data normalization, clustering and differential expression were performed using the Seurat R package (Satija et al., 2015) (version 2.3.4). Highly variable genes were computed using the ‘mean.var.plot’ function with setting parameters x.low.cutoff = 0.0125, x.high.cutoff = 5, y.cutoff = 0.18, fxn,x = expMean, fxn.y = logVarDivMean, and these highly variable genes were used to perform PCA. Principal components that can significantly distinguish cells were first selected by jackstraw analysis, and these principal components were used to perform t-SNE analysis through the run_tsne function of the Seurat package. The 7 PCs were included for subsequent analysis. We confirmed that the resulting analyses were not particularly sensitive to this exact choice.
The cells were clustered via Seurat’s FindClusters function, which optimizes a modularity function on a k-nearest-neighbour graph computed from the top eigenvectors.
Cluster-specific marker genes were identified by running the ‘find_all_markers’ function. To identify differentially expressed genes between two clusters or two genotypes, we used the ‘find.markers’ function with thresh.test = 0.25 and test.use = ’ poisson’.
Generation of gene signatures from the literature.
Cytokine signature were defined as genes that were significantly induced by cytokine treatment.
TGFβ activation signature was defined previously (Butovsky et al., 2014).
For the IL33 activation signature, we identified induced genes between IL33 treated and control eosinophils (Bouffi et al., 2013)(GSE43660), as genes significantly different under an FDR-corrected t-test (P<0.05) and that had a log2(fold-change in expression) >1.5 (IL33 over control).
For IFN-β activation signature, RNA Sequencing of microglia isolated from mice overexpressing IFN-β and controls (Deczkowska et al., 2017) (GSE98401) were downloaded. Significantly induced genes were determined as genes with (1) a mean fold-change ≥2 (AAV-IFN-β over control) and (2) P < 0.05.
For the IL33 activation signature in hippocampal microglia, we identified induced genes in hippocampal microglia after IL-33 versus PBS intracerebroventricular injection (Nguyen et al., 2020), as genes with (1) fold-change in expression >1.5 (IL-33 over control) and (2) adjusted P value < 0.01.
For IFNγ activation signature, transcriptional profiles of human fetal microglia treated with IFNγ for 1 hour (Rock et al., 2005)(GSE1432) were downloaded and analyzed. Significantly induced genes were determined as genes with (1) a mean fold-change ≥2 (IFNγ over control) and (2) P < 0.05.
For IL1 and IL6 activation signature, microarray profiles of human monocyte derived macrophages treated with IL1 and IL6 were analyzed (GSE8515). Significantly induced genes were defined as genes with (1) a mean fold-change ≥1.5 (cytokine over control) and (2) P < 0.05.
For IL4 and IL10 and IL15 activation signature, microarray profiles of human monocyte treated with IL4, IL10, or IL15 for 6 hours (Montoya et al., 2014)(GSE59184) were analyzed. Significantly induced genes were defined as genes with (1)a mean fold change ≥3 (cytokine over control) and (2) P < 0.01.
For IL17A activation signature, microarray profiles of human monocyte derived dendritic cells treated with IL17A for 12 days (Salvatore et al., 2015)(GSE53163) were analyzed. Significantly induced genes were defined as genes with (1) a mean fold change ≥16 (IL17A over control) and (2) P < 0.05.
For neurodegeneration associated microglia signature (GSE65067), genes significantly enriched in 5XFAD microglia (Keren-Shaul et al., 2017)were defined as (1) a mean fold change ≥2, (2) P < 0.05; ribosomal RNAs were removed from the resulting DEG list.
Psychiatric disorder immune signature was defined previously (Gandal et al., 2018).
Trajectory analysis.
To analyze the trajectory of microglia based on single cell RNA-seq expression data, we used Monocle v. 2.8.0 (Qiu et al., 2017), which provides a toolkit to order cells according to progressing along a learned trajectory. As input to Monocle’s Reversed Graph Embedding algorithm, we used all variable genes to order the cells along pseudo-time. To improve the quality of the trajectory, we called the reduceDimension function with max_components = 2 in Monocle2 to apply a dimensionality reduction to the input data set. We then ordered the cells using orderCells function. To test the robustness of the inferred trajectory, we evaluated the distribution of developmental ages, and expression of known differentiation markers and observed that the pseudotime matched well with the physiological development time and prior knowledge. To uncover transcriptional dynamics along the inferred development trajectory, we computed genes that significantly change as the cells make progress and cluster these genes by their pseudotemporal expression pattern.
Statistical analysis of qPCR, imaging and flow cytometry data flow cytometry data
Flow cytometric data were analyzed with FlowJo software (BD). Imaging data were anayzed with Fiji or IMARIS as indicated. Statistical analyses were performed with GraphPad Prism 8.0 software. When comparing two groups, an Student’s t test was performed. n case of multiple comparisons, we used a parametric one-way analysis of variance analysis (ANOVA) followed by Tukey’s pairwise comparisons and two-tailed Student’s t test. Where appropriate, a Wilcoxon matched-pairs signed rank test was used. All n and P values and statistical tests are indicated in figure legends. One Il1rl1−/− mouse was eliminated from social interaction analysis due to staring-freezing behavior.
Supplementary Material
Table S1. Transcriptional analysis of TMRM high vs TMRM low microglia subset and Tau-GFP positive vs Tau-GFP negative microglia, related to Figure 2.
Table S2. A compiled list of MGact gene signature, cell cytokine signatures and microglia associated signatures under neurological disease settings, related to Figure 2.
Table S3. Transcriptional analysis of IL33 treated microglia, related to Figure 2.
Table S4. A summary of gene modules defined by monocle 2, related to Figure 6.
Table S5. sgRNA and qPCR primer sequences, related to STAR Methods
Movie S1. Seizure event in Il1rl1−/− mice when placed into a new cage, related to Figure 3.
Highlights.
Phagocytic activity and bioenergetics is coupled in microglia during development
Astrocyte derived IL33 promotes microglial engulfment and bioenergetics via AKT
Loss of ST2 and IL33 results in microglial dysfunction and susceptibility to seizures
Microglial mitochondrial metabolism and AKT activity is temporally regulated in vivo
ACKNOWLEDGEMENTS
We thank I. Chiu, T. Hammond, A. Walker for discussions, D. Kozoriz, Y, J. Shi, A.R. Bialas and J Presumey for technical advice, W. Zhu and J. Xia for mouse genotyping, O. Butovsky and M. Collins for careful reading of the manuscript, and S. Riesenfeld for computational advice. This work was supported by NIH grants (to V.K.K), the Klarman Cell Observatory (to A.R.), National Natural Science Foundation of China (grant 32070953 to D.H.) and National Key R&D program of China (grant 2020YFA0804200 to D.H.) and the Education Foundation of Westlake University (to D.H.). A.R. is an Investigator of the Howard Hughes Medical Institute. D.H. was supported by the Crohn’s & Colitis Foundation post-doctoral research fellowship.
V.K.K. has an ownership interest and is a member of the SAB of Celsius Therapeutics and Tizona Therapeutics. V.K.K.’s interests were reviewed and managed by the Brigham and Women’s Hospital and Partners Healthcare in accordance with their conflict-of-interest policies. A.R. is a co-founder and equity holder of Celsius Therapeutics, equity holder of Immunitas, and, until August, 2020, a SAB member of ThermoFisher Scientific, Syros Pharmaceuticals, Neogene Therapeutics, and Asimov. A.R. is an employee of Genentech Inc. ORR is an employee of Genentech. ORR is a co-inventor on patent applications filed by the Broad related to single cell genomics.
Footnotes
DECLARATION OF INTERESTS
All other authors declare that they have no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- Baik SH, Kang S, Lee W, Choi H, Chung S, Kim JI, and Mook-Jung I. (2019). A Breakdown in Metabolic Reprogramming Causes Microglia Dysfunction in Alzheimer’s Disease. Cell Metab 30, 493–507 e496. 10.1016/j.cmet.2019.06.005. [DOI] [PubMed] [Google Scholar]
- Bohlen CJ, Bennett FC, Tucker AF, Collins HY, Mulinyawe SB, and Barres BA (2017). Diverse Requirements for Microglial Survival, Specification, and Function Revealed by Defined-Medium Cultures. Neuron 94, 759–773 e758. 10.1016/j.neuron.2017.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst K, Schwabenland M, and Prinz M. (2019). Microglia metabolism in health and disease. Neurochem Int 130, 104331. 10.1016/j.neuint.2018.11.006. [DOI] [PubMed] [Google Scholar]
- Bouffi C, Rochman M, Zust CB, Stucke EM, Kartashov A, Fulkerson PC, Barski A, and Rothenberg ME (2013). IL-33 markedly activates murine eosinophils by an NF-kappaB-dependent mechanism differentially dependent upon an IL-4-driven autoinflammatory loop. J Immunol 191, 4317–4325. 10.4049/jimmunol.1301465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, et al. (2014). Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci 17, 131–143. 10.1038/nn.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum JD, Cai G, Chaste P, Nygren G, Goldsmith J, Reichert J, Anckarsater H, Rastam M, Smith CJ, Silverman JM, et al. (2007). Mutation screening of the PTEN gene in patients with autism spectrum disorders and macrocephaly. Am J Med Genet B Neuropsychiatr Genet 144B, 484–491. 10.1002/ajmg.b.30493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamoto K, Chowdhury PS, Kumar A, Sonomura K, Matsuda F, Fagarasan S, and Honjo T. (2017). Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent antitumor activity. Proc Natl Acad Sci U S A 114, E761–E770. 10.1073/pnas.1620433114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WY, Hong J, Gannon J, Kakkar R, and Lee RT (2015). Myocardial pressure overload induces systemic inflammation through endothelial cell IL-33. Proc Natl Acad Sci U S A 112, 7249–7254. 10.1073/pnas.1424236112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna M, and Butovsky O. (2017). Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu Rev Immunol 35, 441–468. 10.1146/annurev-immunol-051116-052358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deczkowska A, Keren-Shaul H, Weiner A, Colonna M, Schwartz M, and Amit I. (2018). Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 173, 1073–1081. 10.1016/j.cell.2018.05.003. [DOI] [PubMed] [Google Scholar]
- Deczkowska A, Matcovitch-Natan O, Tsitsou-Kampeli A, Ben-Hamo S, Dvir-Szternfeld R, Spinrad A, Singer O, David E, Winter DR, Smith LK, et al. (2017). Mef2C restrains microglial inflammatory response and is lost in brain ageing in an IFN-I-dependent manner. Nat Commun 8, 717. 10.1038/s41467-017-00769-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druart M, and Le Magueresse C. (2019). Emerging Roles of Complement in Psychiatric Disorders. Front Psychiatry 10, 573. 10.3389/fpsyt.2019.00573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunkley PR, Jarvie PE, and Robinson PJ (2008). A rapid Percoll gradient procedure for preparation of synaptosomes. Nat Protoc 3, 1718–1728. 10.1038/nprot.2008.171. [DOI] [PubMed] [Google Scholar]
- Dykens JA, and Stout AK (2001). Assessment of mitochondrial membrane potential in situ using single potentiometric dyes and a novel fluorescence resonance energy transfer technique. Methods Cell Biol 65, 285–309. 10.1016/s0091-679x(01)65018-0. [DOI] [PubMed] [Google Scholar]
- Edmonson CA, Ziats MN, and Rennert OM (2016). A Non-inflammatory Role for Microglia in Autism Spectrum Disorders. Front Neurol 7, 9. 10.3389/fneur.2016.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ElAli A, and Rivest S. (2016). Microglia Ontology and Signaling. Front Cell Dev Biol 4, 72. 10.3389/fcell.2016.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipello F, Morini R, Corradini I, Zerbi V, Canzi A, Michalski B, Erreni M, Markicevic M, Starvaggi-Cucuzza C, Otero K, et al. (2018). The Microglial Innate Immune Receptor TREM2 Is Required for Synapse Elimination and Normal Brain Connectivity. Immunity 48, 979–991 e978. 10.1016/j.immuni.2018.04.016. [DOI] [PubMed] [Google Scholar]
- Friedman BA, Srinivasan K, Ayalon G, Meilandt WJ, Lin H, Huntley MA, Cao Y, Lee SH, Haddick PCG, Ngu H, et al. (2018). Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep 22, 832–847. 10.1016/j.celrep.2017.12.066. [DOI] [PubMed] [Google Scholar]
- Gadani SP, Walsh JT, Smirnov I, Zheng J, and Kipnis J. (2015). The glia-derived alarmin IL-33 orchestrates the immune response and promotes recovery following CNS injury. Neuron 85, 703–709. 10.1016/j.neuron.2015.01.013. [DOI] [PubMed] [Google Scholar]
- Gandal MJ, Zhang P, Hadjimichael E, Walker RL, Chen C, Liu S, Won H, van Bakel H, Varghese M, Wang Y, et al. (2018). Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 362. 10.1126/science.aat8127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Vidal-Itriago A, Kalsbeek MJ, Layritz C, Garcia-Caceres C, Tom RZ, Eichmann TO, Vaz FM, Houtkooper RH, van der Wel N, et al. (2017). Lipoprotein Lipase Maintains Microglial Innate Immunity in Obesity. Cell Rep 20, 3034–3042. 10.1016/j.celrep.2017.09.008. [DOI] [PubMed] [Google Scholar]
- Garcia AD, Doan NB, Imura T, Bush TG, and Sofroniew MV (2004). GFAP-expressing progenitors are the principal source of constitutive neurogenesis in adult mouse forebrain. Nat Neurosci 7, 1233–1241. 10.1038/nn1340. [DOI] [PubMed] [Google Scholar]
- Groszer M, Erickson R, Scripture-Adams DD, Lesche R, Trumpp A, Zack JA, Kornblum HI, Liu X, and Wu H. (2001). Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science 294, 2186–2189. 10.1126/science.1065518. [DOI] [PubMed] [Google Scholar]
- Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, Walker AJ, Gergits F, Segel M, Nemesh J, et al. (2019). Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 50, 253–271 e256. 10.1016/j.immuni.2018.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond TR, Robinton D, and Stevens B. (2018). Microglia and the Brain: Complementary Partners in Development and Disease. Annu Rev Cell Dev Biol 34, 523–544. 10.1146/annurev-cellbio-100616-060509. [DOI] [PubMed] [Google Scholar]
- Hong S, Dissing-Olesen L, and Stevens B. (2016). New insights on the role of microglia in synaptic pruning in health and disease. Curr Opin Neurobiol 36, 128–134. 10.1016/j.conb.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee YH, Narzisi G, Leotta A, et al. (2012). De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299. 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, et al. (2017). A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290 e1217. 10.1016/j.cell.2017.05.018. [DOI] [PubMed] [Google Scholar]
- Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O’Loughlin E, Xu Y, Fanek Z, et al. (2017). The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 47, 566–581 e569. 10.1016/j.immuni.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress H, Stelzer EH, Holzer D, Buss F, Griffiths G, and Rohrbach A. (2007). Filopodia act as phagocytic tentacles and pull with discrete steps and a load-dependent velocity. Proc Natl Acad Sci U S A 104, 11633–11638. 10.1073/pnas.0702449104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC, Beecham GW, Grenier-Boley B, et al. (2013). Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 45, 1452–1458. 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JO, Yang H, Georgescu MM, Di Cristofano A, Maehama T, Shi Y, Dixon JE, Pandolfi P, and Pavletich NP (1999). Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell 99, 323–334. 10.1016/s0092-8674(00)81663-3. [DOI] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, and Barres BA (2018). Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol 18, 225–242. 10.1038/nri.2017.125. [DOI] [PubMed] [Google Scholar]
- Li Q, Cheng Z, Zhou L, Darmanis S, Neff NF, Okamoto J, Gulati G, Bennett ML, Sun LO, Clarke LE, et al. (2019). Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron 101, 207–223 e210. 10.1016/j.neuron.2018.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linke M, Fritsch SD, Sukhbaatar N, Hengstschlager M, and Weichhart T. (2017). mTORC1 and mTORC2 as regulators of cell metabolism in immunity. FEBS Lett 591, 3089–3103. 10.1002/1873-3468.12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lioubin MN, Algate PA, Tsai S, Carlberg K, Aebersold A, and Rohrschneider LR (1996). p150Ship, a signal transduction molecule with inositol polyphosphate-5-phosphatase activity. Genes Dev 10, 1084–1095. 10.1101/gad.10.9.1084. [DOI] [PubMed] [Google Scholar]
- Masuda T, Sankowski R, Staszewski O, Bottcher C, Amann L, Sagar, Scheiwe C, Nessler S, Kunz P, van Loo G, et al. (2019). Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 566, 388–392. 10.1038/s41586-019-0924-x. [DOI] [PubMed] [Google Scholar]
- Matcovitch-Natan O, Winter DR, Giladi A, Vargas Aguilar S, Spinrad A, Sarrazin S, Ben-Yehuda H, David E, Zelada Gonzalez F, Perrin P, et al. (2016). Microglia development follows a stepwise program to regulate brain homeostasis. Science 353, aad8670. 10.1126/science.aad8670. [DOI] [PubMed] [Google Scholar]
- Montoya D, Inkeles MS, Liu PT, Realegeno S, Teles RM, Vaidya P, Munoz MA, Schenk M, Swindell WR, Chun R, et al. (2014). IL-32 is a molecular marker of a host defense network in human tuberculosis. Sci Transl Med 6, 250ra114. 10.1126/scitranslmed.3009546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris GP, Clark IA, Zinn R, and Vissel B. (2013). Microglia: a new frontier for synaptic plasticity, learning and memory, and neurodegenerative disease research. Neurobiol Learn Mem 105, 40–53. 10.1016/j.nlm.2013.07.002. [DOI] [PubMed] [Google Scholar]
- Nguyen PT, Dorman LC, Pan S, Vainchtein ID, Han RT, Nakao-Inoue H, Taloma SE, Barron JJ, Molofsky AB, Kheirbek MA, and Molofsky AV (2020). Microglial Remodeling of the Extracellular Matrix Promotes Synapse Plasticity. Cell 182, 388–403 e315. 10.1016/j.cell.2020.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent AA, Lin K, van Lengerich B, Lianoglou S, Przybyla L, Davis SS, Llapashtica C, Wang J, Kim DJ, Xia D, et al. (2020). TREM2 Regulates Microglial Cholesterol Metabolism upon Chronic Phagocytic Challenge. Neuron 105, 837–854 e839. 10.1016/j.neuron.2019.12.007. [DOI] [PubMed] [Google Scholar]
- Oboki K, Ohno T, Kajiwara N, Arae K, Morita H, Ishii A, Nambu A, Abe T, Kiyonari H, Matsumoto K, et al. (2010). IL-33 is a crucial amplifier of innate rather than acquired immunity. Proc Natl Acad Sci U S A 107, 18581–18586. 10.1073/pnas.1003059107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S, Bjorklund AK, Faridani OR, Sagasser S, Winberg G, and Sandberg R. (2013). Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods 10, 1096–1098. 10.1038/nmeth.2639. [DOI] [PubMed] [Google Scholar]
- Pinto D, Delaby E, Merico D, Barbosa M, Merikangas A, Klei L, Thiruvahindrapuram B, Xu X, Ziman R, Wang Z, et al. (2014). Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet 94, 677–694. 10.1016/j.ajhg.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, and Trapnell C. (2017). Reversed graph embedding resolves complex single-cell trajectories. Nat Methods 14, 979–982. 10.1038/nmeth.4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine RJ (1972). Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol 32, 281–294. 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Rajasekaran A, Venkatasubramanian G, Berk M, and Debnath M. (2015). Mitochondrial dysfunction in schizophrenia: pathways, mechanisms and implications. Neurosci Biobehav Rev 48, 10–21. 10.1016/j.neubiorev.2014.11.005. [DOI] [PubMed] [Google Scholar]
- Reid IC, and Stewart CA (1997). Seizures, memory and synaptic plasticity. Seizure 6, 351–359. 10.1016/s1059-1311(97)80034-9. [DOI] [PubMed] [Google Scholar]
- Risher WC, Ustunkaya T, Singh Alvarado J, and Eroglu C. (2014). Rapid Golgi analysis method for efficient and unbiased classification of dendritic spines. PLoS One 9, e107591. 10.1371/journal.pone.0107591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock RB, Hu S, Deshpande A, Munir S, May BJ, Baker CA, Peterson PK, and Kapur V. (2005). Transcriptional response of human microglial cells to interferon-gamma. Genes Immun 6, 712–719. 10.1038/sj.gene.6364246. [DOI] [PubMed] [Google Scholar]
- Rossignol DA, and Frye RE (2012). Mitochondrial dysfunction in autism spectrum disorders: a systematic review and meta-analysis. Mol Psychiatry 17, 290–314. 10.1038/mp.2010.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvatore G, Bernoud-Hubac N, Bissay N, Debard C, Daira P, Meugnier E, Proamer F, Hanau D, Vidal H, Arico M, et al. (2015). Human monocyte-derived dendritic cells turn into foamy dendritic cells with IL-17A. J Lipid Res 56, 1110–1122. 10.1194/jlr.M054874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, and Regev A. (2015). Spatial reconstruction of single-cell gene expression data. Nat Biotechnol 33, 495–502. 10.1038/nbt.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Heller CT, and Stevens B. (2014). An engulfment assay: a protocol to assess interactions between CNS phagocytes and neurons. J Vis Exp. 10.3791/51482. [DOI] [PMC free article] [PubMed]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thion MS, Ginhoux F, and Garel S. (2018). Microglia and early brain development: An intimate journey. Science 362, 185–189. 10.1126/science.aat0474. [DOI] [PubMed] [Google Scholar]
- Townsend MJ, Fallon PG, Matthews DJ, Jolin HE, and McKenzie AN (2000). T1/ST2-deficient mice demonstrate the importance of T1/ST2 in developing primary T helper cell type 2 responses. J Exp Med 191, 1069–1076. 10.1084/jem.191.6.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, and Salzberg SL (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker KL, Meyer M, and Barde YA (2001). Neurotrophins are required for nerve growth during development. Nat Neurosci 4, 29–37. 10.1038/82868. [DOI] [PubMed] [Google Scholar]
- Ulland TK, Song WM, Huang SC, Ulrich JD, Sergushichev A, Beatty WL, Loboda AA, Zhou Y, Cairns NJ, Kambal A, et al. (2017). TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 170, 649–663 e613. 10.1016/j.cell.2017.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainchtein ID, Chin G, Cho FS, Kelley KW, Miller JG, Chien EC, Liddelow SA, Nguyen PT, Nakao-Inoue H, Dorman LC, et al. (2018). Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science 359, 1269–1273. 10.1126/science.aal3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, et al. (2013). Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38, 79–91. 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Transcriptional analysis of TMRM high vs TMRM low microglia subset and Tau-GFP positive vs Tau-GFP negative microglia, related to Figure 2.
Table S2. A compiled list of MGact gene signature, cell cytokine signatures and microglia associated signatures under neurological disease settings, related to Figure 2.
Table S3. Transcriptional analysis of IL33 treated microglia, related to Figure 2.
Table S4. A summary of gene modules defined by monocle 2, related to Figure 6.
Table S5. sgRNA and qPCR primer sequences, related to STAR Methods
Movie S1. Seizure event in Il1rl1−/− mice when placed into a new cage, related to Figure 3.
Data Availability Statement
The bulk RNA-Seq reported in Figure 1 and 2, as weel as the single-cell RNA-Seq data reporped in Figure 2 and Figure 3 have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PE anti-mouse/human CD11b Antibody (clone M1/70) | Biolegend | Cat# 101208, RRID: AB_312791 |
| FITC anti-mouse/human CD11b Antibody (clone M1/70) | Biolegend | Cat# 101206, RRID: AB_312789 |
| Brilliant Violet 421™ anti-mouse/human CD11b (clone M1/70) | Biolegend | Cat# 101236, RRID: AB_11203704 |
| APC anti-mouse CD45.2 Antibody (clone 104) | Biolegend | Cat# 109814, RRID:AB_389211 |
| PE anti-mouse CD45.2 (clone 104) | Biolegend | Cat# 109808, RRID:AB_313445 |
| FITC anti-mouse CD45.2 (clone 104) | Biolegend | Cat# 109806, RRID:AB_313443 |
| PE/Cyanine7 anti-mouse CD45.2 (clone 104) | Biolegend | Cat# 109830, RRID:AB_1186098 |
| APC/Cyanine7 anti-mouse CD45 Antibody (Clone 30-F11) | Biolegend | Cat# 103116, RRID:AB_312981 |
| PE/Cyanine7 anti-mouse CD45 Antibody (clone 30-F11) | Biolegend | Cat# 103114, RRID:AB_312979 |
| CD282 (TLR2) Monoclonal Antibody (6C2), PE | eBioscience | Cat# 12-9021-82, RRID:AB_466230 |
| CD282 (TLR2) Monoclonal Antibody (6C2) | eBioscience | Cat# 11-9021-82, RRID:AB_465440 |
| Alexa Fluor® 647 anti-mouse CD85k (gp49 Receptor) Antibody | Biolegend | Cat# 144906, RRID:AB_2562044 |
| Alexa Fluor® 647 Mouse Anti-Akt (pS473) Clone M89-61 | BD Biosciences | Cat# 561670, RRID:AB_10896328 |
| Phospho-mTOR (Ser2448) Monoclonal Antibody (MRRBY), PE | eBioscience | Cat# 12-9718-41, RRID:AB_2572723 |
| PE Mouse Anti-Akt (pT308) Clone J1-223.371 | BD Biosciences | Cat# 558275, RRID:AB_2225329 |
| Phospho-Akt (Thr308) (244F9) Rabbit mAb | Cell Signaling Technology | Cat# 4056, RRID:AB_331163 |
| Phospho-Akt (Ser473) (D9E) | Cell Signaling | Cat# 4060, RRID:AB_2315049 |
| XP® Rabbit mAb | Technology | |
| Pho spho -NDRG1 (Thr346) (D98G11) XP® Rabbit mAb | Cell Signaling Technology | Cat# 5482, RRID:AB_10693451 |
| Histone H3 (D2B12) XP® Rabbit mAb (ChIP Formulated) | Cell Signaling Technology | Cat# 4620, RRID:AB_1904005 |
| Phospho-mTOR (Ser2448) Antibody | Cell Signaling Technology | Cat# 2971, RRID:AB_330970 |
| mTOR (7C10) Rabbit mAb antibody | Cell Signaling Technology | Cat# 2983, RRID:AB_2105622 |
| Akt (pan) (C67E7) Rabbit mAb | Cell Signaling Technology | Cat# 4691, RRID:AB_915783 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7074, RRID:AB_2099233 |
| Rat anti Mouse CD68 antibody, (clone FA-11) | Serotec | Cat# MCA1957, RRID:AB_322219) |
| Rabbit anti-Synaptophysin (SP11) | Abcam | Cat# ab16659, RRID:AB_443419 |
| Rabbit anti-Iba1 | Wako | Cat# 019-19741, RRID:AB_839504 |
| Goat anti-Iba1 | Abcam | Cat# ab5076, RRID:AB_2224402 |
| Mouse anti-PSD95 | Chemicon | Cat# MAB1596, RRID:AB_2092365 |
| Mouse IL-33 Antibody | R&D Systems | Cat# AF3626, RRID:AB_884269 |
| Purified anti-P2RY12 Antibody | Biolegend | Cat# 848001, RRID:AB_2650633 |
| Cy™2 AffiniPure Donkey Anti-Goat IgG (H+L) | Jackson Immunoresearch | Cat# 705-225-147, RRID:AB_2307341 |
| Cy™3 AffiniPure Donkey Anti-Mouse IgG (H+L) | Jackson Immunoresearch | Cat# 715-165-150, RRID:AB_2340813 |
| Cy™3 AffiniPure Donkey Anti-Goat IgG (H+L) | Jackson Immunoresearch | Cat# 705-165-147, RRID:AB_2307351 |
| Cy™5 AffiniPure Donkey Anti-Mouse IgG (H+L) | Jackson Immunoresearch | Cat# 715-175-151, RRID:AB_2340820 |
| Cy™2 AffiniPure Donkey Anti-Rabbit IgG (H+L) | Jackson Immunoresearch | Cat# 711-225-152, RRID:AB_2340612 |
| Cy™3 AffiniPure Donkey Anti-Rabbit IgG (H+L) | Jackson Immunoresearch | at# 711-165-152, RRID:AB_2307443 |
| Proteins, Chemicals and Kits | ||
| MitoTracker™ Deep Red FM - Special Packaging | ThermoFisher Scientific | Cat# M22426 |
| Image-iT™ TMRM | ThermoFisher Scientific | Cat# I34361 |
| 2-NBDG (2-(N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)Amino)-2-Deoxyglucose) | ThermoFisher Scientific | Cat# N13195 |
| Alexa Fluor™ 488 Phalloidin | ThermoFisher Scientific | Cat# A12379 |
| Alexa Fluor™ 594 Phalloidin | ThermoFisher Scientific | Cat# A12381 |
| MitoTracker™ Green FM - Special Packaging | ThermoFisher Scientific | Cat# M7514 |
| MitoTracker™ Red CMXRos - Special Packaging | ThermoFisher Scientific | Cat# M7512 |
| DAPI | Sigma-Aldrich | Cat# D9542 |
| Kainic acid monohydrate | Sigma-Aldrich | Cat# K0250-10MG |
| pHrodo™ iFL Green Microscale Protein Labeling Kit | ThermoFisher Scientific | Cat# P36015 |
| pHrodo™ iFL Red Microscale Protein Labeling Kit | ThermoFisher Scientific | Cat# P36014 |
| Seahorse XF Cell Mito Stress Test Kit | Agilent | Cat# 103015-100 |
| HEPES (1 M) | ThermoFisher Scientific | Cat# 15630080 |
| D-(+)-Glucose solution | Sigma-Aldrich | Cat# G8769-100ML |
| Percoll Plus | Sigma-Aldrich | Cat# GE17-5445-01 |
| PicoPure RNA Isolation Kit | Thermo Fisher Scientific | Cat# KIT0204 |
| iScript cDNA Synthesis Kit | Bio-Rad | Cat# 1708891 |
| iTaq Universal SYBR Green Supermix | Bio-Rad | Cat# 1725121 |
| TCL buffer (QIAGEN) | Qiagen | Cat# 1031576 |
| 2-Mercaptoethanol | Sigma-Aldrich | Cat# M6250 |
| Nextera XT Sample Preparation Kit | Illumina | Cat# FC-131-1096 |
| DMEM/F-12, GlutaMAX™ supplement | ThermoFisher Scientific | Cat# 10565042 |
| DMEM, high glucose | ThermoFisher Scientific | Cat# 11965126 |
| GlutaMAX™ Supplement | ThermoFisher Scientific | Cat# 35050061 |
| Trypsin-EDTA (0.05%), phenol red | ThermoFisher Scientific | Cat# 25300120 |
| Penicillin-Streptomycin Mixture | Lonza | Cat# 17-602E |
| Sodium Pyruvate Solution (100 mM) | Lonza | Cat# 13-115E |
| N-acetyl cysteine | Sigma | Cat# A9165 |
| N-2 Supplement (100X) | Gibco | Cat# 17502048 |
| Recombinant Mouse M-CSF Protein | R&D Systems | Cat# 416-ML-050 |
| Recombinant Mouse IL-33 | Biolegend | Cat# 580506 |
| UK-5099 | Sigma-Aldrich | Cat# PZ0160-5MG |
| STF-31 | Selleckchem | Cat# S7931 |
| Oligomycin | Agilent | Cat# 103015-100 |
| Antimycin | Agilent | Cat# 103015-100 |
| Rotenone | Agilent | Cat# 103015-100 |
| AZD8055 | Selleckchem | Cat# S1555 |
| Ly294002 | Selleckchem | Cat# S1105 |
| Triciribine | Selleckchem | Cat# S1117 |
| Torkinib (PP242) | Selleckchem | Cat# S2218 |
| Rapamycin (Sirolimus) | Selleckchem | Cat# S1039 |
| Oltipraz | Sigma-Aldrich | Cat# O9389-5MG |
| ProlongGold Antifade reagent | ThermoFisher Scientific |
Cat# P36930 |
| O.C.T. Compound | Electron Microscopy Sciences | Cat# 62550-01 |
| Triton X-100 | Sigma-Aldrich | Cat# T8787 |
| Normal Donkey Serum | Jackson ImmunoResearch |
Cat# 017-000-001 |
| Papain Dissociation System | Worthington | Cat# LK003150 |
| PBS, pH 7.4 | ThermoFisher Scientific |
Cat# 10010023 |
| 20X Bolt™ MOPS SDS Running Buffer | ThermoFisher Scientific |
Cat# B0001 |
| PageRuler™ Prestained Protein Ladder | ThermoFisher Scientific |
Cat# 26617 |
| Bolt™ 4 to 12%, Bis-Tris | ThermoFisher Scientific |
Cat# NW04120BOX |
| Bolt™ Transfer Buffer (20X) | ThermoFisher Scientific |
Cat# BT00061 |
| Power Blotter Pre-cut Membranes and Filters, PVDF, regular size | ThermoFisher Scientific | Cat# PB9320 |
| 4X Bolt™ LDS Sample Buffer | ThermoFisher Scientific |
Cat#B0008 |
| Western Blotting Filter Paper, 0.83 mm thick, 7 × 8.4 cm | ThermoFisher Scientific | Cat# 84783 |
| Pierce™ ECL Western Blotting Substrate | ThermoFisher Scientific | Cat# 32209 |
| RIPA Lysis and Extraction Buffer | ThermoFisher Scientific |
Cat# 89900 |
| Paraformaldehyde solution 4% in PBS | Santa Cruz | Cat# sc-281692 |
| Sucrose | Sigma-Aldrich | Cat# S0389 |
| cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail | Roche | Cat# 4693159001 |
| Buffer TCL (125 mL) | QIAGEN | Cat# 1031576 |
| B-Mercaptoethanol | Sigma-Aldrich | Cat# M3148-100ML |
| Recombinant RNase Inhibitor | Takara | Cat# 2313B |
| Deoxynucleotide (dNTP) Solution Mix | New England Biolabs | Cat# N0447L |
| Maxima H Minus Reverse Transcriptase (200 U/uL) | Thermo Fisher Scientific | Cat# EP0753 |
| KAPA HiFi HotStart ReadyMix PCR Kit | Roche | Cat# KK2602 |
| Agencourt RNAClean (RNA SPRI beads) | Beckman Coulter | Cat# A63987 |
| Nuclei EZ Prep | Sigma-Aldrich | Cat# NUC101-1KT |
| AMPureXP SPRI (DNA SPRI beads) | Beckman Coulter | Cat# A63881 |
| Nextera XT DNA Library Preparation Kit | Illumina | Cat# FC-131-1096 |
| NextSeq 500 high output kit V2, 75 cycles | Illumina | Cat# FC-404-2005 |
| 10X Chromium Single Cell 30 Kit | 10X Genomics | Cat# 120237 |
| Oligonucleotides | ||
| sgRNA sequences | See Table S5 | NA |
| qPCR primer sequences | See Table S5 | NA |
| Experimental Model: Strains | ||
| Mouse: C57BL/6J | Jackson Laboratory | JAX:000664, RRID:IMSR_JAX:000664 |
| Mouse: B6J.B6N(Cg)-Cx3cr1tm1.1(cre)Jung/J | Jackson Laboratory | JAX:025524, RRID:IMSR_JAX:025524 |
| Mouse: B6;FVB-Tg(Aldh1l1-EGFP/Rpl10a)JD133Htz/J | Jackson Laboratory | JAX:030248, RRID:IMSR_JAX:030248 |
| Mouse: B6.129S4(Cg)-Mapttm1(EGFP)Klt/J | Jackson Laboratory | JAX:029219, RRID:IMSR_JAX:029219 |
| Mouse: B6.Cg-Tg(Gfap-cre)73.12Mvs/J | Jackson Laboratory | JAX:012886, RRID:IMSR_JAX:012886 |
| Mouse: ilirl1tmIAnnm | (Townsend et al., 2000) | N/A |
| Mouse: Il33−/− | (Oboki et al., 2010) | N/A |
| Mouse: Il1rl1 flox | (Chen et al., 2015) | N/A |
| Mouse: Il33 flox | (Chen et al., 2015) | N/A |
| Mouse: C;129S4-Ptentm1Hwu/J | (Groszer et al., 2001) | JAX:004597, RRID:IMSR_JAX:004597 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GSE127449 |
| Software and Algorithms | ||
| ImageJ | (Schneider et al., 2012) | https://imagej.nih.gov/ij/ |
| Imaris | Bitplane | https://imaris.oxinst.com/ |
| GraphPad Prism 8 | GraphPad Software | https://www.graphpad.com/scientificsoftware/prism/ |
| FlowJo | TreeStar | https://www.flowjo.com/solutions/flowjo |
| RSEM version 1.2.8 | (Li and Dewey, 2011) | http ://deweylab. github. io/RSEM/ |
| edgeR version 3.5.2 | (Robinson et al., 2010) | https://gist.github.com/jdblischak/11384914 |
| Seurat version 2.3.3 | (Satija et al., 2015) | https://github.com/satijalab/seurat |
| 10x Cell Ranger version 1.2 | 10X Genomics |
https://support.10xgenomics.com/singlecell/software/overview/welcome
|
| Monocle version 2.8.0 | (Qiu et al., 2017) | http://cole-trapnell-lab.github.io/monocle-release/ |