

Abstract
Background
The onset and mechanisms of endothelial‐to‐mesenchymal transition (EndMT) in mitral valve (MV) leaflets following myocardial infarction (MI) are unknown, yet these events are closely linked to stiffening of leaflets and development of ischemic mitral regurgitation. We investigated whether circulating molecules present in plasma within days after MI incite EndMT in MV leaflets.
Methods and Results
We examined the onset of EndMT in MV leaflets from 9 sheep with inferior MI, 8 with sham surgery, and 6 naïve controls. Ovine MVs 8 to 10 days after inferior MI displayed EndMT, shown by increased vascular endothelial cadherin/α‐smooth muscle actin–positive cells. The effect of plasma on EndMT in MV endothelial cells (VECs) was assessed by quantitative polymerase chain reaction, migration assays, and immunofluorescence. In vitro, post‐MI plasma induced EndMT marker expression and enhanced migration of mitral VECs; sham plasma did not. Analysis of sham versus post‐MI plasma revealed a significant drop in the Wnt signaling antagonist sFRP3 (secreted frizzled‐related protein 3) in post‐MI plasma. Addition of recombinant sFRP3 to post‐MI plasma reversed its EndMT‐inducing effect on mitral VECs. RNA‐sequencing analysis of mitral VECs exposed to post‐MI plasma showed upregulated FOXM1 (forkhead box M1). Blocking FOXM1 reduced EndMT transcripts in mitral VECs treated with post‐MI plasma. Finally, FOXM1 induced by post‐MI plasma was downregulated by sFRP3.
Conclusions
Reduced sFRP3 in post‐MI plasma facilitates EndMT in mitral VECs by increasing the transcription factor FOXM1. Restoring sFRP3 levels or inhibiting FOXM1 soon after MI may provide a novel strategy to modulate EndMT in the MV to prevent ischemic mitral regurgitation and heart failure.
Keywords: endothelial‐to‐mesenchymal transition, ischemic mitral regurgitation, mitral valve, myocardial infarction, Wnt signaling
Subject Categories: Endothelium/Vascular Type/Nitric Oxide, Cell Signalling/Signal Transduction, Ischemia
Nonstandard Abbreviations and Acronyms
- EBM
endothelial basal media
- EndMT
endothelial‐to‐mesenchymal transition
- FOXM1
forkhead box M1
- IMR
ischemic mitral regurgitation
- MV
mitral valve
- qPCR
quantitative polymerase chain reaction
- sFRP3
secreted frizzled‐related protein 3
- SMAD
Sma genes and the Drosophila Mad, Mothers against decapentaplegic
- TGFβ
transforming growth factor beta
- VE
vascular endothelial
- VEC
valve endothelial cell
- α‐SMA
α‐smooth muscle actin
Clinical Perspective
What Is New?
Circulating factors in plasma prevent mitral valve (MV) endothelial cells from undergoing endothelial‐to‐mesenchymal transition (EndMT); within days after myocardial infarction (MI), plasma levels of sFRP3 (secreted frizzled‐related protein 3) fall, and EndMT in the MV begins.
Replenishing sFRP3 levels in post‐MI plasma prevents EndMT in MV endothelial cells by blocking Wnt/β‐catenin, blocking induction of FOXM1 (forkhead box M1), and blocking nuclear localization of Slug, a transcription factor required for EndMT.
This new pathway links sFRP3/β‐catenin signaling to FOXM1 and EndMT in the MV.
What Are the Clinical Implications?
EndMT in the MV portends an early onset of maladaptive changes in the MV in response to MI.
Reduced levels of plasma sFRP3 in post‐MI sheep were positively correlated with MI surface area and negatively correlated with left ventricular ejection fraction.
Replenishing plasma sFRP3 might be a novel therapeutic approach to prevent maladaptive EndMT to reduce ischemic mitral regurgitation and heart failure.
Ischemic mitral regurgitation (IMR) complicates recovery from myocardial infarction (MI), doubles mortality after MI, and is a major factor increasing heart failure. 1 , 2 , 3 Postinfarction left ventricular (LV) remodeling displaces the papillary muscles that tether the mitral valve (MV) leaflets; this restricts valve closure 4 , 5 and creates a postinfarct mismatch between leaflet surface area and the enlarged left ventricle. Increased MV leaflet thickness, stiffness, and fibrosis further contribute to progressive IMR. 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 Valve tethering and LV volume overload without MI induce adaptive MV growth with associated endothelial‐to‐mesenchymal transition (EndMT). 15 , 16 , 17 , 18 , 19 In contrast, MI induces excessive EndMT with overexpression of transforming growth factor beta (TGFβ) and collagen deposition over 2 to 6 months. 7 , 9 , 19 This excessive EndMT and collagen deposition is a substrate for fibrosis 20 that contributes to MV leaflet thickening and in turn reduces leaflet ability to form a tight seal 21 needed to prevent mitral regurgitation. These maladaptive pathways in the MV can be modified pharmacologically with losartan therapy over 2 months, which allows increased leaflet surface area while limiting fibrosis. 22
An understanding of the earliest inciting events is needed to determine how to prevent these maladaptive changes in the MV. To embark on this, we set out to determine if circulating molecules present in plasma within days after MI play a role in inciting EndMT.
We used an ovine inferior MI model to determine how soon EndMT begins in the MV after inferior MI and if the plasma would induce EndMT and fibrosis in mitral valve endothelial cells (VECs). We hypothesized that early after MI, circulating factors, released as a consequence of myocardial damage, would directly impact the MV endothelium and initiate responses that become maladaptive over time. Using a cytokine array and ELISA, we found sFRP3 (secreted frizzled‐related protein 3) reduced in post‐MI compared with sham‐operated and naïve plasma controls. sFRP3 is an inhibitor of Wnt signaling, which was previously shown to inhibit epithelial‐to‐mesenchymal transition. 23 Wnt signaling is known to promote EndMT in concert with TGFβ through coordinated action of nuclear localized Smad (Small Mothers Against Decapentaplegic) and Lef/β‐catenin to promote transcription of shared target genes. 24 These interactions, however, have not been studied in the context of the MV. We used bulk RNA sequencing to identify genes and pathways altered by 8‐ to 10‐day post‐MI plasma. This revealed significantly increased expression of the transcription factor FOXM1 (forkhead box M1) in mitral VECs treated with post‐MI plasma. FOXM1 is associated with TGFβ‐induced EndMT, 25 , 26 fibrosis, 27 and the Wnt pathway, 28 but its involvement in the development of IMR has not been described before. Immunostaining showed a stronger FOXM1 signal in MVs of animals with inferior MI compared with sham‐operated and naïve controls. In vitro, blocking FOXM1 pharmacologically inhibited expression of EndMT‐ and fibrosis‐related mRNA transcripts. Moreover, adding sFRP3 to post‐MI plasma blocked the increase in FOXM1 expression. Our findings suggest a potential therapeutic role for sFRP3 to limit EndMT and profibrotic processes in MV leaflets and thereby prevent the development of IMR. We also identified FOXM1 as a driver of MI‐associated EndMT, which points to a second therapeutic target to minimize IMR.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Experimental Animals
For animal studies, a minimum of n=6 for each group was selected for sample size based on previous studies. 7 , 9 , 17 , 22 For in vitro experiments using ovine plasma, a minimum of n=3 was used to provide biological replicates for statistical analysis. Twenty‐three adult Polypay sheep (weight >44 kg) were studied: 9 had inferior MI; 8 underwent sham surgery including thoracotomy without MI creation; 6 had immediate excision of hearts under general anesthesia to avoid any potential inflammation induced by thoracotomy (naïve controls). Under general anesthesia with isoflurane and oxygen, inferior MI was created by ligation of the second‐third obtuse marginal branches of the left circumflex coronary artery. 29 , 30 Animals were cared for, with pre‐MI and pre‐euthanasia blood sampling over 10±3 days and were euthanized following thoracotomy for final data collection and tissue harvest. This study conformed to National Institutes of Health animal care guidelines and received institutional Animal Care Committee approval. The approval reference number is 2003N000183.
Echocardiography
Comprehensive 2‐ and 3‐dimensional trans‐epicardial echocardiography was performed using a commercially available ultrasound system (iE33/X5‐1 transducer; Philips, Andover, MA) before and after MI at day 0 and before euthanasia. Three‐dimensional full‐volume images were collected with multibeat (4–6 beat) acquisition under electrocardiographic gating. Two‐dimensional data were analyzed with Syngo Dynamics (Siemens, Erlangen, Germany), and 3‐dimensional data were analyzed with 4‐dimensional LV analysis version 3.1.2 (TomTec, Unterschlessheim, Germany) for LV volume. 10 , 31 Left ventricular ejection fraction (LVEF) was calculated as ([end‐diastolic LV volume] – [end‐systolic LV volume])/(end‐diastolic LV volume). MI size was measured using ImageJ version 1.48 (National Institutes of Health, Bethesda, MD) by planimetry of the visually determined MI endocardial surface area of the open harvested heart and its ratio to total LV endocardial surface area.
Blood Sampling and Plasma Extraction
Equal volumes of ovine heparinized peripheral blood and sterile PBS were mixed and added to Ficoll‐Paque PLUS Media (GE Healthcare, No. GE17‐1440‐02) in 4:3 ratio. The mix was centrifuged for 30 minutes at 700g with no brake. The top layer, enriched for plasma, was collected, and stored at −80 °C.
Tissue Harvesting
The heart was harvested immediately after euthanasia, the left atrium opened, and the LV wall dissected starting from the anterior lateral commissure in a sterile manner, with irrigation of precooled PBS. Both MV leaflets were excised and immediately submerged in a solution of 5% heat‐inactivated fetal bovine serum, 4% penicillin/streptomycin/amphotericin B, 1% l‐glutamine, and 0.2% gentamycin sulfate in endothelial basal media (EBM)‐2 (Lonza Inc; No. CC‐3156) and kept on ice until processed, typically within 2 hours of harvest.
MV Cellular Analysis by Flow Cytometry
Ovine MV sections of the anterior leaflet (AL2L, AL1, AL2R) and posterior leaflet (P3a, P3b, P2b, P1a, and P1b) were minced and digested for 30 minutes at 37 °C with Liberase (Millipore Sigma; No. 5401020001) to prepare a single‐cell suspension of MV cells. 7 , 9 , 22 The cell suspension was fixed using Flow Cytometry Fixation Buffer (R&D Systems, Minneapolis, MN; No. FC004) and labeled in Flow Cytometry Permeabilization/Wash Buffer I (R&D Systems; No. FC005) for 45 minutes (100 000 cells/100 µL buffer I). Anti–VE‐cadherin antibody (1:50; Aviva Systems Biology, San Diego, CA; NO. ARP60108_P050) and anti–α‐smooth muscle actin (α‐SMA) antibody [E184] (1:5000; Abcam, Cambridge, England; No. ab209435).
Cell Culture
Ovine mitral VEC and ovine carotid artery endothelial cells, isolated previously, 32 were grown on 1% gelatin‐coated dishes in EBM‐2 medium (Lonza Inc, Morristown, NJ; No. CC‐3156) supplemented with 10% heat‐inactivated fetal bovine serum, and 2 ng/mL basic fibroblast growth factor (Millipore Sigma, St. Louis, MO; No. 11123149001).
EndMT Assay
Ovine mitral VECs and carotid artery endothelial cells were plated on 1% gelatin‐coated plates at 30 000 cells/cm2. After 24 hours, growth media was replaced with ovine post‐MI, sham, or naïve plasma, each at the same volume. Cells were analyzed 24 to 96 hours later as indicated.
Reagents and Inhibitors
See Table S1.
Cytokine Array
Eighteen circulating cytokines were quantified using Ovine Cytokine Array C1 (RayBiotech, Norcross, GA; No. AAO‐CYT‐1‐2). Positive control signals on each array were used for normalization following the manufacturer’s instructions.
ELISA Analysis
Concentrations of sFRP3 and TGFβ2 in ovine plasma samples were measured according to the manufacturer’s instructions using an ELISA kit available from RayBiotech (No. ELO‐sFRP3‐1, and No. ELH‐TGFb2‐1), respectively.
RNA‐Sequencing Analysis: Library Preparation, Sequencing, and Quality Control of Fastq Files
Ovine mitral VECs were plated on 1% gelatin‐coated plates at 10 000 cells/cm2. After 24 hours, EBM‐B was replaced with ovine post‐MI or naïve plasma (n=3, plasma from 3 different sheep for each). After 24 hours, RNA was isolated using RNeasy Mini Kit (Qiagen, Valencia, CA; No. 74104). RNA integrity was assessed on an Agilent Bioanalyzer 2100 using a Nano 6000 assay kit (Agilent Technologies, Santa Clara, CA; No. 5067‐1511). All samples displayed an RNA integrity number of 10. Library preparation was performed using the TruSeq Stranded mRNA LT Sample Prep Kit (Illumina Inc., San Diego, CA; Part No. 15031047). Sequencing was performed on the Illumina HiSeq 2000 (paired‐end 150 bp) using the TruSeq SBS Kit v3‐HS (Illumina Inc.). A quality check of the raw sequencing data was performed using FastQC 33 (version 0.11.5) and MultiQC 34 (version 1.5) programs.
Mapping to the Sheep Genome
Aligning reads using STAR 35 (version 2.5.2b) was performed in 2 steps: (1) creating genome indices using Ovis_aries. Oar_v3.1 genome assembly (GCA_000298735.1) and Ovis_aries. Oar_v3.1.99.gtf annotation. We obtained the reference genome and the reference annotation for the sheep from the Ensembl database; and (2) read alignment. The alignments of fastq files produced random order with respect to their position in the reference genome. The Binary Alignment/Map files therefore were sorted using Samtools 36 (version 1.9).
Assessing Alignment Quality
Assessing alignment quality and mapping statistics were performed by ready Log.final.out files and uniquely mapped read. In addition to the STAR‐specific summary, we also obtained quality metrics using Qualimap 37 , 38 and RNA‐SeQC. 39 To perform subsetting and visualization of the alignment using a genome browser on the Binary Alignment/Map file, an index was generated using Samtools 36 (version 1.9) and Integrative Genomics Viewer was used to assess the quality of the alignment.
Counting Reads and Overall Count QC Metrics
The counts of reads were used as an input for featureCounts 40 (version 2.0.0) tool to get the gene counts. FeatureCounts output file (count matrix) was then used to extract the read count, genomic coordinates, and the length of the gene. Finally, MultiQC 34 (version 1.5) was run on the files from (1) FastQC, (2) Log.final.out files from STAR, and (3) summary file from featurecounts outputs from our workflow.
Differential Expression Analysis
DESeq2 41 (version 1.26.0) was used for estimation of variance‐mean dependence in count data and test for differential expression based on a model using the negative binomial distribution. The DESeq2 package was run using R program (Bioconductor version: Release [3.10]).
Hierarchical Clustering and Heat Maps
Function heatmap2 in the R package ggplot2 or GENE‐E R (Bioconductor version: Release [3.10]) package was used to compute the hierarchical clustering and the associated heat maps. The Pearson correlation method was used for a pairwise correlation matrix between items and then converted as a distance matrix. Clustering was calculated on the resulting distance matrix. The average linkage method implemented the average distance to compute the distance matrix. For the heatmap visualization, the log2 expression values were used.
Volcano Plots
The EnhancedVolcano package in R was used to generate the volcano plots. Adjusted P value (>0.05) and absolute log2‐fold change (<1) were used to illustrate the differentially regulated genes.
Principal Component Analysis
The Factoextra package (version v1.0.7) in R was used to extract and visualize the results of multivariate analyses. First, we generated the eigenvalues (scree plot) and after inspecting the components, the corresponding plot was generated.
Z Score Calculation for Activity of Canonical Pathways
Significantly regulated genes were evaluated to predict the activity of pathways (Z score) using Ingenuity Pathway Analysis (Qiagen). The significant values for the canonical pathways were computed by Fisher’s exact test right tailed.
Immunohistochemistry and Immunofluorescence Staining
Serial sections of 6 μm were cut from the frozen central portion of MV anterior leaflets, AL2. Cryosections were fixed in −20 °C acetone, blocked with 0.3% hydrogen peroxidase, and incubated with Protein Block Serum‐Free (Dako; #X0909). Sections were then incubated with rabbit antihuman FOXM1 polyclonal antibody (Novus Biologicals, Littleton, CO; No. NBP1‐30961) diluted 1:300 or anti‐Ki67 (Abcam; No. 15580) diluted 1:50 in 5% normal horse serum (Vector Laboratories, Burlingame, CA; No. S‐2000) for 90 minutes at room temperature, followed by 45 minutes incubation with antirabbit secondary antibody (LSAB Kit; Dako; No. K0675). The streptavidin peroxidase method was performed for each staining, and the reaction was visualized with a 3‐amino‐9‐ethylcarbazol substrate (AEC Substrate Chromogen, Dako; No. K3464). Sections were counterstained with Gill’s No. 3 Hematoxylin (Sigma‐Aldrich, St. Louis, MO; No. GHS316). For quantification, 3‐amino‐9‐ethylcarbazol substrate (AEC)‐positive cells (red reaction product) and total nuclei (blue) per high‐power field (400× magnification) were counted for 10 fields per section and expressed as AEC‐positive cells/nuclei.
For immunofluorescence staining of MV leaflets, sections were fixed in −20 °C acetone and blocked with 3% BSA (Sigma‐Aldrich; No. A9576) diluted in PBS containing 0.1% Tween. Double immunofluorescence staining was performed using anti‐FOXM1 (1:300, Novus Biologicals; #NBP1‐30961), anti‐α‐SMA (1:150, Dako; No. M0851), anti‐CD31 (1:50, Novus Biologicals; No. NB10001642), and anti‐β‐Catenin (1:40, Abcam; No. ab22656), overnight at 4 °C. Secondary conjugated antibodies were Alexa Fluor anti‐rabbit 647, Alexa Fluor anti‐rabbit 488, Alexa Fluor anti‐rabbit 594 (1:500, Fisher Scientific, MA, USA). Sections were counterstained with 4',6‐diamidino‐2‐phenylindole ‐ DAPI (Fisher Scientific, No. R37606). Images were analyzed using the imaging software NIS‐Elements AR (Advanced Research) 3.1 (Nikon Instruments).
For immunofluorescence staining of mitral VECs, cells were fixed with 4% PFA for 20 minutes and washed with ice‐cold PBS 3 times prior to staining. Cells were incubated overnight at 4 °C with 0.2 µg/mL of primary antibodies: anti–vascular endothelial (VE) cadherin (Invitrogen; No. 36‐1900), anti‐α‐SMA (Millipore Sigma; No. A5228), anti‐Slug (Santa Cruz, No. Sc‐166476), and anti‐FOXM1 (Abcam; No. ab207298). Dilutions were made in PBS containing 5% BSA and 0.1% Tween‐20. Cells were washed 3 times with ice‐cold PBS before incubation with secondary antibodies for 2 hours at room temperature. Secondary antibodies included Alexa Fluor 488 goat anti‐rabbit IgG (H+L) and Alexa Fluor 546 donkey anti‐rabbit IgG (H+L), which were diluted 1:1000 in PBS. Cells were washed 3 times with ice‐cold PBS and mounted using ProLong Gold Antifade with DAPI (Cell Signaling; No. 8961). Confocal imaging was conducted using ZEISS LSM 880 (ZEISS, Oberkochen, Germany).
Quantitative Polymerase Chain Reaction
Total cellular RNA was extracted using RNeasy Micro extraction kit (Qiagen;, No. 74004). Reverse transcriptase reactions were performed using an iScript Reverse Transcription Supermix for RT‐qPCR (Bio‐Rad Laboratories, Hercules, CA; No. 1708840). Quantitative polymerase chain reaction (qPCR) was performed using Kapa Sybr Fast ABI Prism 2x qPCR Master Mix (KAPA BioSystems, Wilmington, DE; No. KK4604). Amplification was carried out in a QuantStudio 6 Flex Real‐Time PCR System (ThermoFisher Scientific, Waltham, MA; No. 4485694). RPS9 was used as a housekeeping gene expression reference. Fold increases in gene expression were calculated according to 2 delta cycle time method, 42 with each amplification reaction performed in duplicate. At least 3 biological replicates were included in each analysis. Primer pairs used in this study are listed in Table S2.
Immunoblotting
Cells were lysed in 1× cell lysis buffer (Cell Signaling Technology, Danvers, MA; No. 9803) supplemented with 1× Protease/Phosphatase Inhibitor Cocktail (Cell Signaling Technology; No. 5872). Protein concentration was determined using a DC Protein Assay (Bio‐Rad Laboratories; No. 5000116); 15 µg of protein were loaded on 4% to 20% Criterion TGX Precast Midi Protein Gel (Bio‐Rad Laboratories; No. 5671094). Transfer was conducted using Trans‐Blot Turbo Transfer System (Bio‐Rad Laboratories; No. 1704150); 0.2 µm nitrocellulose membrane ready to assemble transfer packs were used (Bio‐Rad Laboratories; No. 1704271). Proteins were detected using antibodies from Cell Signaling Technology: anti–β‐catenin (No. 9562) and anti–phospho‐β‐catenin (No. 9566). All signals were detected by Clarity Western ECL Substrate (Bio‐Rad Laboratories; No. 1705061) using the ChemiDoc Imaging System (Bio‐Rad Laboratories; No. 7001401). The density of the bands was quantified by Fiji‐ImageJ.
Cellular Migration Assay
Mitral VECs were treated with sham and MI plasma for 24 hours to induce EndMT. The cells were trypsinized, and 20 000 cells in EBM‐2 supplemented with 2% FBS were placed in the upper chamber of 6.5 mm Transwell polycarbonate membranes with 8.0 µm pores (Corning; No. 3422). The lower chambers contained EBM‐2 media with 10% FBS to act as chemoattractant. Cells were allowed to migrate for 12 hours at 37 °C. Cells that migrated through the pores were fixed with methanol and stained with Eosin‐Y, Azure A, and Methylene Blue for visualization and quantification using Three Step Stain Set (Thermo Scientific; No. 22‐050‐272). Cells from 6 different fields in each well were counted in each assay.
Statistical Analysis
Because of the small number of observations, we used the nonparametric Mann‐Whitney U test for single comparisons. For multiple‐group comparisons, 1‐way ANOVA was conducted and followed by a Dunn’s, Sidak’s or Tukey’s post hoc analysis. All data were analyzed using Excel (Microsoft, Redmond, WA) and Prism 8.4.3 (GraphPad Software, La Jolla, CA).
Results
EndMT Observed in Mitral Valve Leaflets Within 8 to 10 Days Following MI
To determine if EndMT begins within days after inferior MI, we analyzed single‐cell suspensions from collagenase‐digested anterior and posterior MV leaflets from 23 animals (Figure 1) by flow cytometry. The percentage of endothelial cells (VE‐cadherin+) coexpressing α‐SMA, indicating EndMT, was significantly higher in post‐MI MVs compared with sham (mean±SD; 9.79%±6.01% versus 1.1%±2.5%) and naïve leaflets (0.1%±0.2%) (Figure 2A). The colored symbols represent results from individual sheep, with corresponding plasma from each sheep indicated by the same color in all subsequent experiments. Using immunofluorescence, we showed CD31, an endothelial marker, colocalized with α‐SMA, a mesenchymal marker, along the endothelium and extending into the interstitium of MV leaflets taken from animals 8 to 10 days after MI (Figure 2B). The flow cytometry and immunostaining results demonstrate onset of EndMT in MV leaflets as early as 8 to 10 days after inferior MI.
Figure 1. Experimental design for animals in this study.
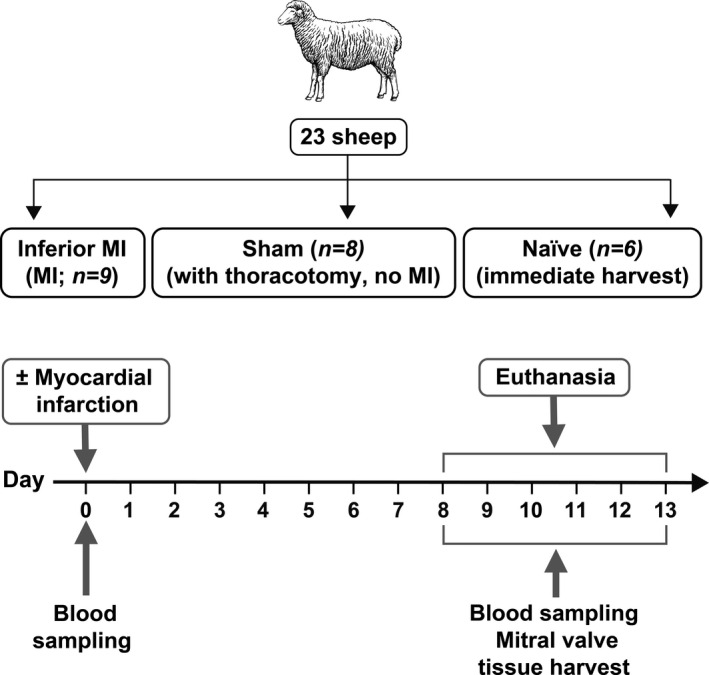
The inferior MI group consisted of 5 female and 4 male sheep; the sham group consisted of 3 female and 5 male sheep, and the naïve group consisted of 4 female and 2 male sheep. MI indicates myocardial infarction.
Figure 2. MV EndMT detected at 8 to 10 days following MI.
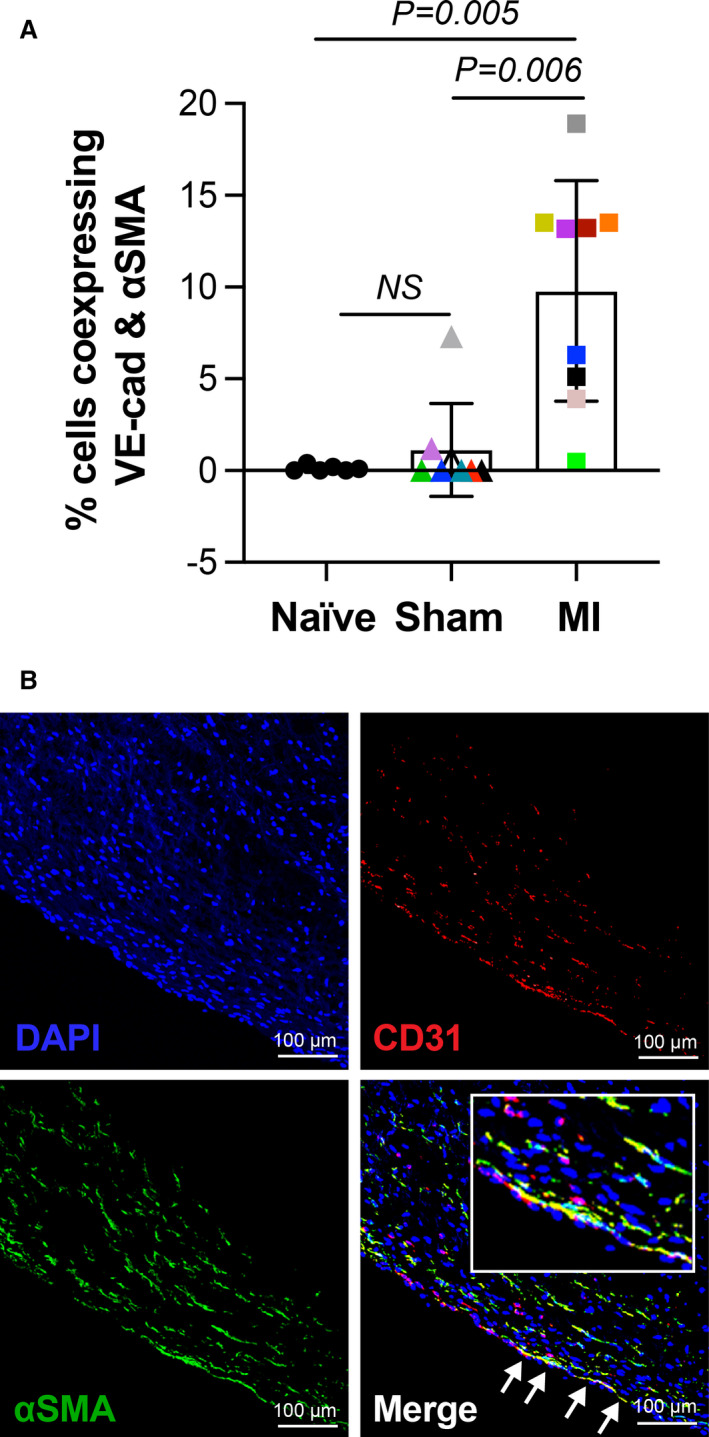
A, Single‐cell suspensions of MV leaflets from animals with naïve, sham, and inferior MI were analyzed for coexpression of VE‐cadherin and α‐SMA by flow cytometry. Values represent mean±SD, subjected to Kruskal‐Wallis (P=0.001). Post hoc Dunn’s test was used for multiple comparisons test indicating the presented adjusted P values. MV leaflets were obtained from 11 male and 12 female sheep. We found no significant difference in EndMT in male (3.24±4.46) vs female (6±7.11; P=0.4327). B, Immunofluorescent staining of MV leaflet from animal with MI shows coexpression of CD31 and α‐SMA along the endothelium, indicated by white arrows (scale bar: 100 µm). α‐SMA indicates α‐smooth muscle actin; EndMT, endothelial‐to‐mesenchymal transition; MI, myocardial infarction; MV, mitral valve; and VE, vascular endothelial.
Post‐MI Plasma Can Induce EndMT in Primary Mitral Valve ECs
We hypothesized that circulating molecules present in post‐MI plasma are responsible for initiating EndMT and may stimulate further profibrotic cellular transitions. To test this, mitral VECs were exposed to ovine whole blood plasma drawn from sheep 8 to 10 days after inferior MI (n=6), sham (n=3), or naïve (n=2) for 24 hours, lysed for RNA extraction and analyzed by qPCR. Figure 3A summarizes the experimental design. Mitral VECs treated with 8‐ to 10‐day post‐MI plasma showed significantly reduced expression of the endothelial marker VE‐cadherin, increased expression of α‐SMA, and increased expression of the EndMT‐driving transcription factor Slug (also known as Snail2), consistent with EndMT (Figure 3B). mRNA transcripts for all 3 TGFβ isoforms were also increased in mitral VECs treated with MI plasma (Figure 3C), supporting our hypothesis that MI plasma contains factors that can induce fibrosis. Ovine carotid artery endothelial cells, which do not undergo TGFβ‐mediated EndMT in vitro, 32 were tested in the same manner. There were no notable changes in the tested markers (Figure S1A, and S1B), indicating that post‐MI plasma has a specific effect on mitral VECs.
Figure 3. MI plasma‐induced EndMT in mitral VECs.
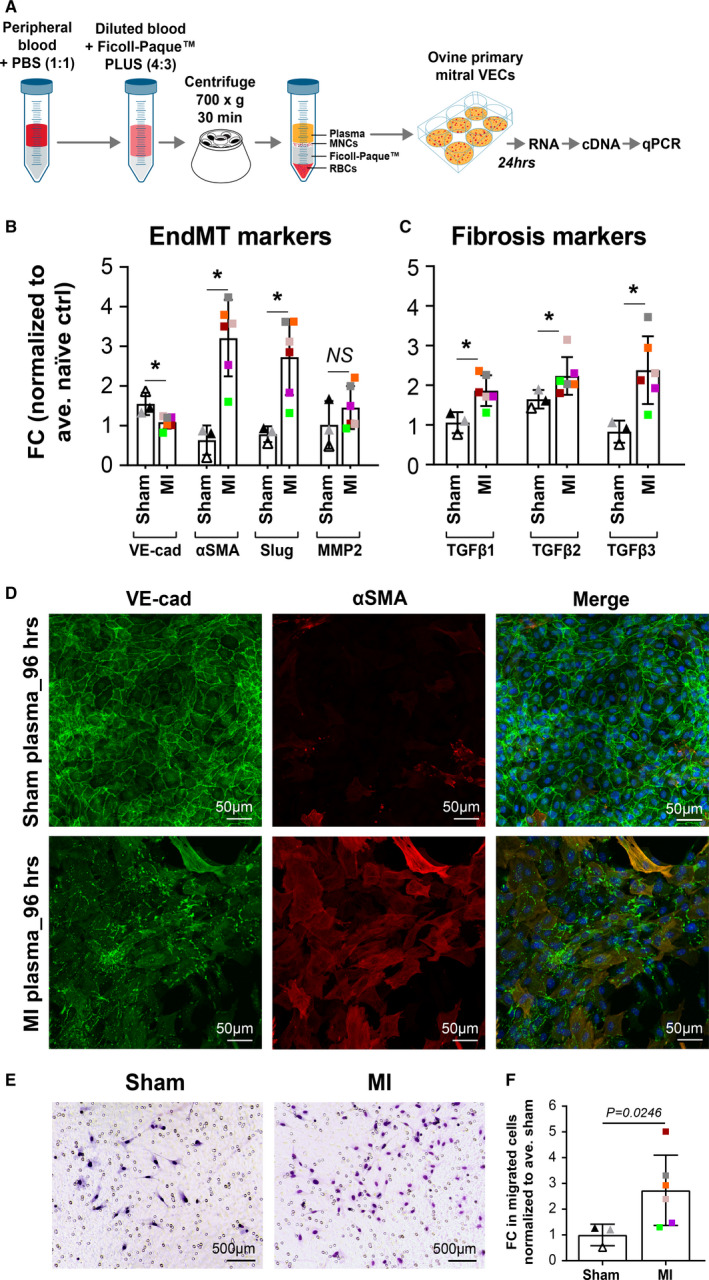
A, Experimental steps from plasma isolation to qPCR analysis. B and C, Ovine mitral VECs were exposed to naïve (n=2), sham (n=3), and MI (n=6) plasma for 24 hours before qPCR. All values were normalized to average naïve value. Data represent mean±SD of 3 independent assays. P values were calculated using nonparametric Mann‐Whitney test (*P<0.05). D, Expression of VE‐cadherin (green) and α‐SMA (red) visualized in mitral VECs treated with sham or post‐MI plasma for 96 hours (scale bar: 50 µm). E, Ovine mitral VECs treated with sham (n=3) or MI plasma (n=6) for 24 hours to induce EndMT were allowed to migrate across Transwell membranes toward media containing 10% FBS for 12 hours. Images represent each treatment group (scale bar: 500 µm). F, Migrated cells in 6 different fields were counted; graph shows mean±SD values of 3 independent assays. P values were calculated using nonparametric Mann‐Whitney test. α‐SMA indicates α‐smooth muscle actin; EndMT, endothelial‐to‐mesenchymal transition; MI, myocardial infarction; MV, mitral valve; and VEC, valve endothelial cell.
To visualize morphologic changes in mitral VECs exposed to 8‐ to 10‐day post‐MI plasma, cells were treated with sham or MI plasma for 48 and 96 hours, then costained with anti–VE‐cadherin and anti–α‐SMA antibodies. Cells treated with sham plasma for 96 hours showed expected VE‐cadherin localization at cell‐cell borders and no α‐SMA (Figure 3D, top row). Cells treated with post‐MI plasma for 96 hours showed coexpression of VE‐cadherin and α‐SMA in a subset of cells that displayed a mesenchymal (spindle‐shaped) rather than endothelial (cobblestone) morphology (Figure 3D, bottom row). Similar staining patterns were seen emerging in cells exposed to sham or post‐MI plasma for 48 hours (Figure S1C). In summary, after 96 hours in post‐MI plasma, mesenchymal morphology in a subset of cells was apparent, VE‐cadherin was dispersed from cell‐cell junctions and the number of α‐SMA+ cells was increased compared with sham‐treated mitral VECs.
Increased migration is a hallmark of endothelial cells undergoing EndMT. Therefore, we examined migration of mitral VECs treated with sham plasma or 8‐ to 10‐day post‐MI plasma for 24 hours. Cells were removed from culture dishes and allowed to migrate for 12 hours across a Transwell membrane toward EBM supplemented with 10% FBS. 43 Mitral VECs treated with 8‐ to 10‐day post‐MI plasma migrated significantly more than those treated with sham plasma (Figure 3E). Cells in 6 different fields for each condition were counted and normalized to sham plasma (Figure 3F). These results further demonstrate that 8‐ to 10‐day post‐MI plasma induces mitral VECs to undergo EndMT.
Cytokine Array to Screen Post‐MI Plasma for Inciting Molecules Reveals sFRP3
To identify molecules within post‐MI plasma responsible for the induction of EndMT/fibrosis, we tested MI (n=6), sham (n=3), and naïve (n=4) plasma for 18 proinflammatory cytokines using an ovine‐specific cytokine array. Representative arrays incubated with MI and naïve plasma are shown in Figure 4A. The Wnt inhibitor sFRP3 was reduced in MI plasma compared with sham plasma; duplicates are outlined by the red boxes. Signal densities were quantified and normalized to naïve samples, which showed significant reduction of sFRP3 in MI plasma (Figure 4B). To verify our results, plasma samples were assayed for sFRP3 by ELISA. The measured concentration of sFRP3 was significantly higher in sham plasma compared with 8‐ to 10‐day post‐MI plasma (mean±SD; 187.44±57.31 ng/mL versus 46.72±23.2 ng/mL, respectively: Figure 4C). To determine if the drop in sFRP3 is associated with inferior MI and its detrimental impacts, sFRP3 levels were analyzed relative to infarct surface area and LVEF in animals with MI. sFRP3 in plasma of 8‐ to 10‐day inferior MI animals (colored boxes) showed a negative correlation with infarct surface area relative to the total LV endocardial surface area (R2=0.8183; P=0.0132; Figure 4D). LVEF, which indicates how well the heart is pumping, was measured in sheep with MI (n=6) by echocardiography. LVEF showed a strong positive correlation with plasma sFRP3 levels (R2=0.7864; P=0.0185; Figure 4E), consistent with a link between decreased circulating sFRP3 and MI‐induced heart dysfunction at this early time point.
Figure 4. sFRP3 is reduced in post‐MI plasma.
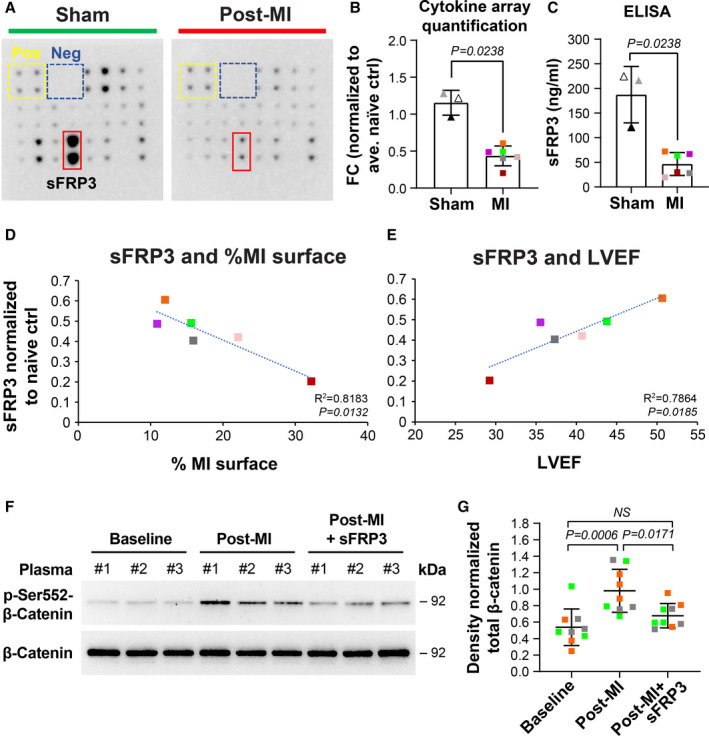
A, Representative images of ovine cytokine arrays exposed to ovine plasma. sFRP3 in sham (n=3) and MI (n=6) plasma were measured. Values from signal intensity in arrays using ImageJ software, normalized to the positive controls of each individual array (B) and ELISA (C) were quantified. Fold changes (FC) were calculated by normalizing values against naïve plasma. Mean±SD were graphed. P value was calculated using Mann‐Whitney test and revealed a significant difference in the mean of the 2 groups. Two‐sample Wilcoxon test revealed that there was no significant difference in the mean quantified sFRP3 when the observations were categorized based on the sex of the animals (P=0.4762). D, sFRP3 values MI (n=6) plasma were normalized to naïve (n=4), measured by cytokine array, and plotted against infarct size (normalized for heart size as infarct relative to total LV endocardial surface area) and (E) LVEF. The fitted regression lines in D and E showed a significant slope parameter with P=0.0132 and P=0.0185, respectively. F, Mitral VECs were treated with 3 individual ovine baseline (before MI), after MI, and sFRP3 supplemented post‐MI plasma for 10 minutes and lysates were subjected to western blot analysis. G, Band densities from 3 independent assays were quantified and subjected to 1‐way ANOVA with Tukey’s multiple comparisons test. α‐SMA indicates α‐smooth muscle actin; EndMT, endothelial‐to‐mesenchymal transition; FC, fold changes; LV, left ventricular; LVEF, left ventricular ejection fraction; MI, myocardial infarction; MMP, matrix metalloproteinase; MNCs, mononuclear cells; qPCR, quantitative polymerase chain reaction; RBCs, red blood cells; sFRP3, secreted frizzled‐related protein 3; TGFβ, transforming growth factor beta; VE, vascular endothelial; and VEC, valve endothelial cell.
We next asked if the reduced level of the Wnt antagonist sFRP3 in post‐MI plasma allows activation of β‐catenin in mitral VECs. To test this, we analyzed phosphorylation of β‐catenin at Ser 552 (p‐Ser‐552‐β‐catenin, the active form of β‐catenin) 44 in lysates from mitral VECs treated with 3 samples of baseline plasma, 3 post‐MI plasma samples, and the same post‐MI plasma samples supplemented with sFRP3 (25 ng/mL). All 3 post‐MI plasma samples increased phosphorylation of β‐catenin at Ser 552, indicating canonical Wnt activation. Addition of sFRP3 to post‐MI plasma reduced p‐Ser552‐β‐catenin to near baseline levels (Figure 4F). Quantification of western blot results from 3 independent assays, each with plasma from 3 different sheep, showed a significant increase in p‐Ser552‐β‐catenin in post‐MI plasma–treated mitral VECs that was reduced to baseline levels when recombinant sFRP3 was added to the post‐MI plasma (Figure 4G). In addition, coexpression of β‐catenin and α‐SMA was observed, suggesting activation of Wnt canonical pathway in MV leaflets at 8 to 10 days following MI (Figure S2). To investigate whether post‐MI plasma affects TGFβ signaling, we examined the lysates for effect of baseline and 8‐ to 10‐day post‐MI plasma±sFRP3 on activation of canonical (Smad‐related) and noncanonical (extracellular signal‐related kinase–related) TGFβ pathways. Post‐MI plasma samples had no significant effect on phosphorylation of Smad3 but significantly increased phosphorylation of extracellular signal‐related kinase compared with respective baseline controls. Addition of sFRP3 did not reduce extracellular signal‐related protein kinase or pSmad3 levels (Figure S3A through S3C). Collectively, these results indicate that the level of sFRP3 in plasma drops after MI, which allows activation of the canonical Wnt pathway and noncanonical TGFβ pathway in primary mitral VECs.
sFRP3 Inhibits EndMT Induced by Post‐MI Plasma
The decreased sFRP3 in plasma from MI animals prompted us to investigate if restoring the sFRP3 level in post‐MI plasma would block its ability to induce EndMT/fibrosis. First, we tested increasing doses of sFRP3 on mitral VEC proliferation and viability and found no significant changes (Figure S4A through S4C). Therefore, we supplemented post‐MI plasma (n=6) with 0, 250, or 1250 ng/mL recombinant sFRP3 10 minutes before adding the plasma to mitral VECs for 24 hours. ED50 for the recombinant sFRP3 was estimated to be 250 ng/mL according to the manufacturer. Cell lysates were analyzed by qPCR to assess expression of EndMT and profibrotic markers. Adding recombinant sFRP3 to post‐MI plasma increased VE‐cadherin and reduced α‐SMA (Figure 5A). Furthermore, TGFβ1‐3 mRNA transcripts were reduced at both sFRP3 concentrations (Figure 5B). Slug is a transcription factor that drives EndMT in response to TGFβ1 and TGFβ2. 45 , 46 Therefore, we examined the sub‐cellular localization of Slug in mitral VECs after treatment with naïve, sham, or post‐MI plasma, and with addition of recombinant sFRP3 (250 ng/mL) to post‐MI plasma. Slug was detected primarily in the cytoplasm of naïve and sham plasma‐treated cells but shifted to the nucleus in the majority of mitral VECs exposed to post‐MI plasma (Figure 5C). Addition of sFRP3 (250 ng/mL) to post‐MI plasma reduced nuclear and restored cytoplasmic localization of Slug. Cells with Slug+ nuclei in each treatment were quantified and normalized to the corresponding total number of nuclei (Figure 5D). These results confirm that adding sFRP3 to post‐MI plasma can block EndMT processes. Collectively, our results suggest that the decrease in sFRP3 in post‐MI plasma incites EndMT and fibrosis in mitral VECs, both of which are associated in vivo with impaired MV function and, in turn, heart function.
Figure 5. sFRP3 inhibits EndMT.
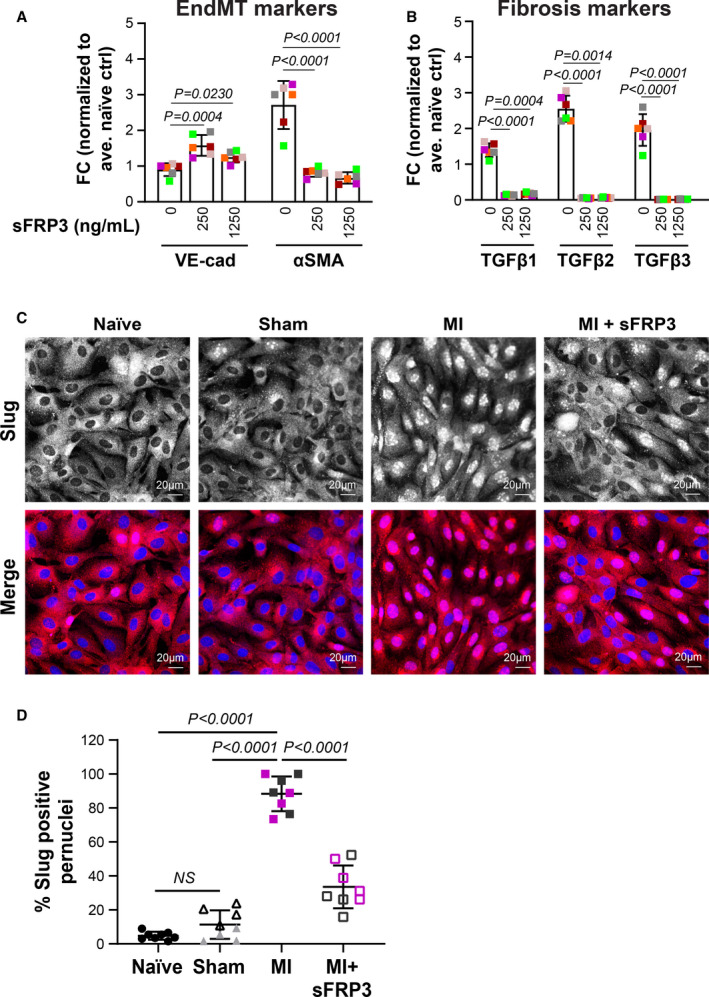
A and B, Mitral VECs were treated with MI plasma (n=6) supplemented with 0, 250, or 1250 ng/mL sFRP3 for 24 hours before qPCR. Mean±SD from three independent assays were graphed. P values were calculated using nonparametric Mann‐Whitney test. C, Mitral VECs were treated with post‐MI plasma±sFRP3 (250 ng/mL) for 24 hours before immunofluorescent staining using anti‐Slug (red). Naïve and sham plasma served as control. DAPI was used to stain nuclei (blue). Both black‐and‐white and colored merged images are shown to ease visualization (scale bar: 20 µm). D, Number of nuclei positive for Slug divided by total nuclei from 4 wells—duplicates incubated with 2 individual plasmas for each group of animals—were graphed. P values were calculated using 1‐way ANOVA with Sidak’s multiple comparisons test. EndMT indicates endothelial‐to‐mesenchymal transition; FC, fold changes; LVEF, left ventricular ejection fraction; MI, myocardial infarction; qPCR, quantitative polymerase chain reaction; sFRP3, secreted frizzled‐related protein 3; and VEC, valve endothelial cell.
RNA‐Sequencing of Mitral VECs Exposed to Post‐MI Plasma Reveals FOXM1
To broaden our understating of how post‐MI plasma affects mitral VECs, we analyzed the transcriptome by bulk RNA‐Seq. Mitral VECs were treated with naïve (n=3) or MI (n=3) plasma, each plasma from a different sheep, for 24 hours before RNA extraction and sequencing. Principal component analysis was used to visualize and investigate data clustering (Figure S5A). The hierarchical clustering and heat map of normalized read counts of 206 significantly regulated mRNAs shows the major differences in gene expression between naïve and post‐MI plasma‐treated mitral VECs (Figure 6A). Statistically significant differentially expressed genes are shown in red in the volcano plot (Figure S5B). The top 50 regulated genes are shown in Figure S5C. Ingenuity Pathway Analysis showed that many upregulated genes were related to the master transcription factor, FOXM1, which has been implicated as a driver of TGFβ‐induced EndMT 25 and induction of fibrosis. 27 , 47 Hierarchical clustering and heat map of log2‐fold change in 24 FOXM1‐related genes confirmed upregulation of those genes in mitral VECs treated with post‐MI plasma (Figure 6B). For further confirmation, mRNA levels of FOXM1 and Cyclin B1, a known direct target of FOXM1, were examined by qPCR; both were significantly increased in mitral VECs exposed to MI plasma for 24 hours (n=6; Figure 6C). Since FOXMI is known to play a role in cell‐cycle progression, cellular proliferation was assessed in vitro by cell counts after 24‐hour treatment with either sham (n=4) or post‐MI (n=5) plasma. Post‐MI plasma significantly increased proliferation of mitral VECs (P=0.0002; Figure 6D). In vivo, staining MV leaflets with anti–Ki‐67, a proliferating cell marker, revealed an increase in the number of Ki‐67+ cells in MVs from animals with MI when compared with sham and naïve controls (Figure S6A through S6D). Using fluorescence staining, we further confirmed the expression of Ki‐67 along the endothelium in MV from MI animals (Figure S6E). Next, we assessed the expression of FOXM1 on the disease‐prone atrial side of the MV 9 by immunohistochemical staining. FOXM1 was not evident in MVs of naïve and sham animals (Figure 6E and 6F). In contrast, MVs from animals with MI showed many FOXM1‐positive cells along the atrial side of the leaflet (Figure 6G). FOXM1‐positive cells were counted in MVs from each animal and normalized to the number of nuclei, which showed significantly higher FOXM1‐positive cells in animals with MI compared with sham (P=0.0349) and naïve (P=0.003; Figure 6H). Furthermore, double‐labeled immunofluorescence staining of MVs from inferior MI animals revealed FOXM1 co‐localized with CD31, and in some cells localized in the DAPI‐stained nuclei (Figure 6I). These results suggest that FOXM1 is increased in the MV endothelium within 10 days after inferior MI.
Figure 6. FOXM1 is increased in mitral VECs after MI.
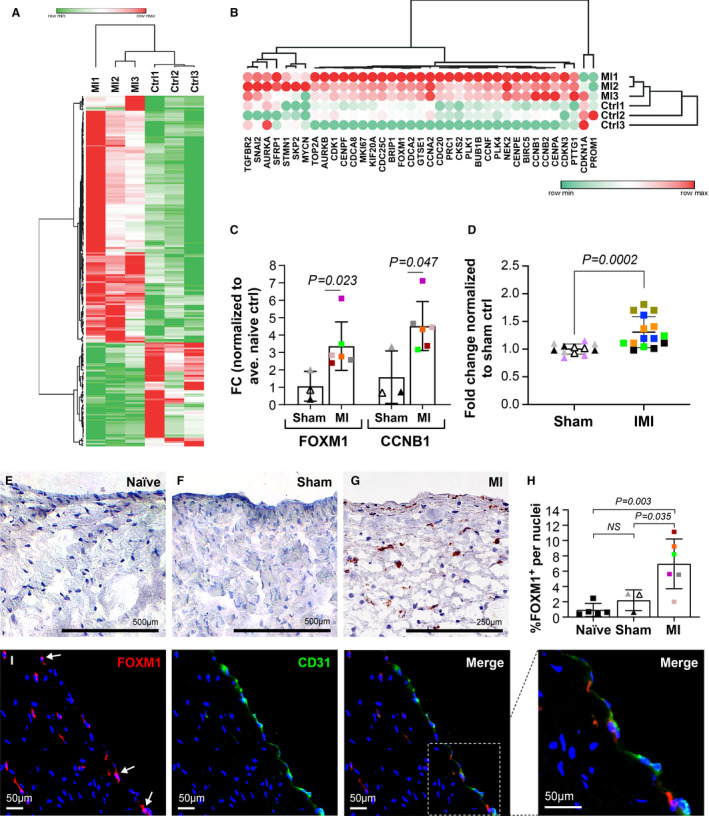
A, Hierarchical clustering and heat map of normalized read counts of 206 significantly regulated mRNA transcripts in mitral VECs treated with naïve (n=3) and post‐MI plasma (n=3). Red color represents higher expression and green color indicates the lower expression. One minus Pearson correlation metric was used for hierarchical clustering. B, Hierarchical clustering and heat map of log2 fold change of FOXM1‐related genes. Red color represents the higher expression and green color indicates the lower expression for each mRNA. One minus Pearson correlation metric was used for hierarchical clustering. C, Ovine mitral VECs were exposed to naïve (n=3), sham (n=3), and MI (n=6) plasma for 24 hours before qPCR analysis. All values were normalized to average naïve and graphed. Data represent mean±SD of 3 independent assays. P values were calculated using nonparametric Mann‐Whitney test. D, Equal numbers of ovine mitral VECs were seeded and treated with sham (n=4) or MI (n=5) plasma for 24 hours before cell count. Fold change was calculated using average number of cells in triplicates for all sham controls. Mean±SD from 2 independent assays were graphed. P values were calculated using nonparametric Mann‐Whitney test. E through G, Mitral valve tissue from animals with naïve (E), sham (F), and inferior MI (G) were stained for FOXM1 expression using immunohistochemistry (scale bar: 500 µm). H, Number of cells expressing FOXM1 divided by total nuclei from 10 fields per each individual section was graphed. P values were calculated using 1‐way ANOVA with Tukey’s multiple comparisons test. I, Post‐MI MV was costained with anti‐FOXM1 (red) and anti‐CD31 (green) using immunofluorescence staining and DAPI staining to visualize nuclei. Arrows indicate FOXM1+ nuclei. scale bar: 50 µm. α‐SMA indicates α‐smooth muscle actin; CCNB1, Cyclin B1; FC, fold changes; FOXM1, forkhead box M1; MI, myocardial infarction; MV, mitral valve; qPCR, quantitative polymerase chain reaction; sFRP3, secreted frizzled‐related protein 3; TGFβ, transforming growth factor beta; VE, vascular endothelial; and VEC, valve endothelial cell.
FOXM1 Increased by Post‐MI Plasma Contributes to EndMT and Can Be Modulated by sFRP3
With the observed increase in FOXM1 in MV leaflets 8 to 10 days following MI and in post‐MI plasma‐treated mitral VECs, and its known role in TGFβ‐mediated EndMT in other settings, 25 , 48 we speculated that FOXM1 might contribute to the onset of EndMT in MVs. Immunofluorescence staining revealed a shift of FOXM1 from cytoplasm to the nuclei of mitral VECs treated with post‐MI plasma compared with sham. Addition of the FOXM1 inhibitor Siomycin A—which has been shown to downregulate the transcriptional activity as well as the protein and mRNA abundance of FOXM1 49 , 50 —reduced FOXM1 nuclear localization (Figure 7A). Quantification showed a significant increase in FOXM1+ nuclei in post‐MI plasma‐treated mitral VECs, which was significantly reduced when Siomycin A was added (Figure 7B). To determine if the EndMT‐related genes induced by post‐MI plasma were affected by FOXM1 inhibition, mitral VECs were treated with post‐MI plasma±Siomycin A for 24 hours before qPCR analysis. Addition of Siomycin A to post‐MI plasma samples significantly reduced FOXM1 as well as α‐SMA, Slug, and TGFβ1‐3 in mitral VECs compared with those treated with post‐MI plasma without Siomycin A (Figure 7C). Given the inhibitory role of sFRP3 on EndMT induced by post‐MI plasma, we next asked if sFRP3 affects FOXM1. To test this, we measured FOXM1 mRNA levels in mitral VECs treated with post‐MI plasma (n=6) ±250 ng/mL sFRP3 for 24 hours. sFRP3 significantly reduced FOXM1 mRNA to below naïve plasma‐treated levels (dashed line, Figure 7D). We further examined the expression of FOXM1 in post‐MI plasma treated mitral VECs, in the absence and presence of sFRP3, by immunofluorescence. The increased number of cells with FOXM1‐positive nuclei in post‐MI plasma treated VECs was reduced by adding sFRP3 (Figure 7E and 7F). Taken together, our findings suggest that FOXM1 is required for induction of EndMT by post‐MI plasma and that sFRP3, a Wnt inhibitor, blocks induction of EndMT at least in part by inhibiting FOXM1 expression and reducing FOXM1 transcriptional activity because of the decrease in its nuclear localization.
Figure 7. FOXM1 contributes to EndMT and can be suppressed by sFRP3.
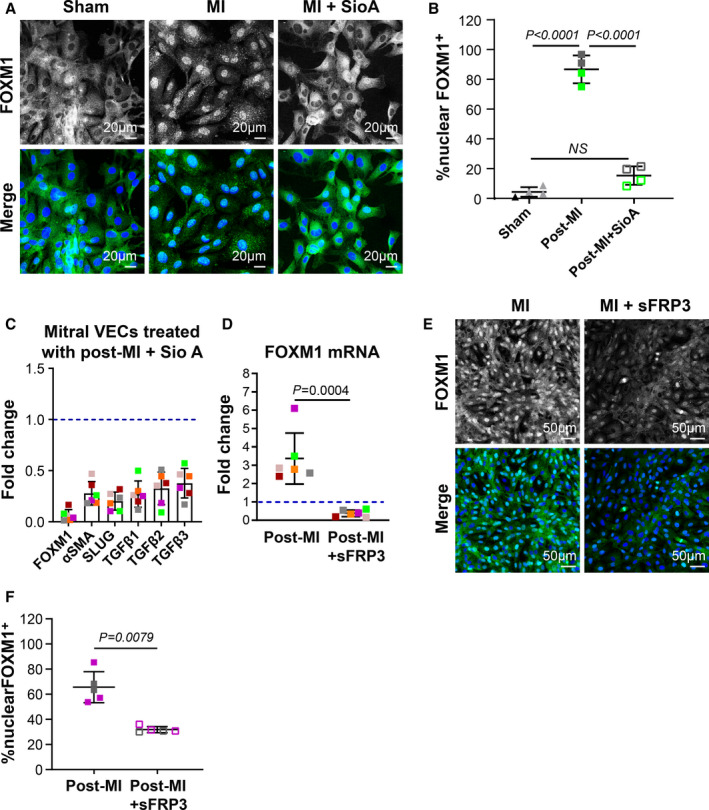
A, Mitral VECs were treated with sham, post‐MI plasma, and post‐MI plasma with Siomycin A (1 µmol/L) for 24 hours before immunofluorescence staining using FOXM1 antibody (green; scale bar: 20 µm). B, Percentage of the cells positive for nuclear FOXM1 per total nuclei from 4 independent immunofluorescence assays were graphed. Statistical analysis was conducted using 1‐way ANOVA with Tukey’s multiple comparisons test. C, Mitral VECs were treated with MI plasma (n=6) with and without Siomycin A (1 µmol/L) for 24 hours before qPCR analysis. Mean±SD from 3 independent assays were graphed. P values were calculated using the Wilcoxon signed‐rank test and were significant (P<0.05) for all tested genes. D, Mitral VECs were treated with MI plasma (n=6) supplemented with sFRP3 (250 ng/mL) for 24 hours before qPCR analysis. Mean±SD from 3 independent assays were graphed. P value was calculated using nonparametric Mann‐Whitney test. E, FOXM1 staining in mitral VECs following exposure to post‐MI plasma and post‐MI plasma supplemented with 250 ng/mL of sFRP3 for 24 hours. DAPI was used to stain nuclei (scale bar: 50 µm). F, Mean±SD of % cells positive for nuclear FOXM1 per total nuclei from 5 independent immunofluorescence assays with 2 individual post‐MI plasma were graphed. P values were calculated using Mann‐Whitney test. EndMT indicates endothelial‐to‐mesenchymal transition; FC, fold changes; FOXM1, forkhead box M1; MI, myocardial infarction; NS, not significant; qPCR, quantitative polymerase chain reaction; sFRP3, secreted frizzled‐related protein 3; SioA, Siomycin A; and VEC, valve endothelial cell.
Discussion
Our results reveal that EndMT begins in MV leaflets within 8 to 10 days after inferior MI, coincident with reduced sFRP3, a Wnt signaling inhibitor, in the peripheral blood plasma. This provides, for the first time, a link between the infarcted myocardium and the MV. sFRP3‐deficient post‐MI plasma‐increased phosphorylation of β‐catenin, nuclear‐localization of Slug, and EndMT markers in mitral VEC, all blocked by addition of recombinant sFRP3. Post‐MI plasma upregulated the transcription factor FOXM1, and this was also blocked by recombinant sFRP3. Endothelial FOXM1 was detected in MV leaflets 8 to 10 days following MI and in vitro, and inhibition of FOXM1 blocked EndMT. There is sufficient TGFβ2 (>2 ng/mL) present in post‐MI plasma (Figure S7A) to induce EndMT when sFRP3 levels fall and Wnt signaling is enabled. In support of this, TGFβ was able to induce FOXM1 expression and nuclear localization in mitral VECs cultured in vitro in standard growth media (Figure S7B and S7C). Based on this, we propose that when circulating sFRP3 levels fall within days after MI, the brake on Wnt signaling is released, allowing plasma TGFβ to initiate EndMT through upregulation of FOXM1 (Figure 8).
Figure 8. Proposed model of EndMT induction in MV by post‐MI plasma.
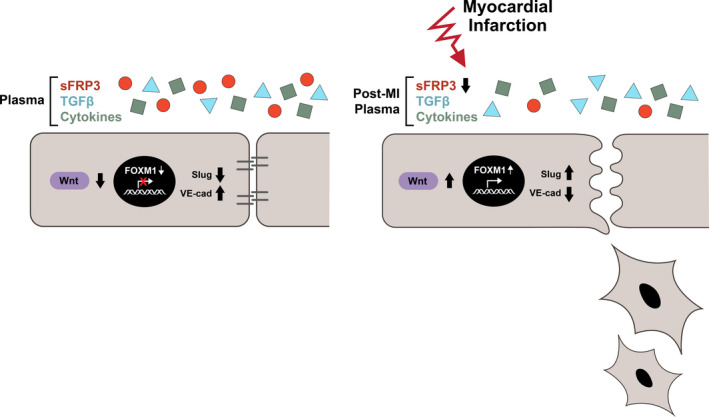
sFRP3‐deficient post‐MI plasma releases the brake on Wnt signaling, increases FOXM1 transcriptional activity, nuclear localization of Slug, which initiates EndMT within days after MI. EndMT indicates endothelial‐to‐mesenchymal transition; FOXM1, forkhead box M1; MI, myocardial infarction; MV, mitral valve; NS, not significant; sFRP3, secreted frizzled‐related protein 3; TGFβ, transforming growth factor beta; VE, vascular endothelial; and Wnt, wingless‐related integration site.
Our previous work uncovered increased EndMT and fibrosis associated with IMR at 2 months and 6 months following MI, but little was known about how soon these processes begin or the molecules that incite them. Here, we show onset of EndMT in MV leaflets as early as 8–10 days following MI, indicating that early intervention may be needed to prevent IMR from developing.
In vitro, post‐MI plasma‐treated mitral VECs transitioned to a mesenchymal morphology with a gradual breakdown of VE‐cadherin at endothelial cell‐cell borders, and showed increased phosphorylation of β‐catenin, nuclear localization of Slug, and EndMT markers, which could be blocked by addition of sFRP3. We conclude that high levels of sFRP3 prevent EndMT in MV endothelium by inhibiting canonical Wnt signaling. Wnt signaling in endocardial cushions is required for valvulogenesis 51 , 52 ; however, its function in post‐MI MV has not been previously investigated. Wnt‐mediated signaling is well established as a participant in maladaptive fibrotic responses in many tissues, including the heart. 53 Modulation of Wnt ligands and upregulation of sFRP family members sFRP1 and sFRP2 post‐MI have been implicated in LV remodeling, myocardial healing, and heart function, but less is known about sFRP3. 54 , 55 , 56 , 57 Upregulating sFRP1 in transgenic mice reduced infarct size and improved cardiac function, 58 suggesting Wnt inhibition may be helpful. Direct injection of recombinant sFRP2 in the infarcted myocardium of rats was reported to have antifibrotic effects and improve cardiac function. 59 In another study, sFRP5 was protective against the inflammatory response to acute ischemic cardiac injury. 60
We show here that levels of sFRP3 in plasma are negatively correlated with the size of MI and positively correlated with ejection fraction in sheep with inferior MI. Despite some caveats, reduction in LVEF is an indication for mitral regurgitation–associated MV repair surgery in clinical practice. 61 In a recent study analyzing 578 patients, risk associated with IMR was described as an interaction between IMR severity and myocardial infarct size. 62 This may suggest an underlying correlation between infarct size and the subsequent molecular changes that contribute to the development of IMR.
To explore potential mechanisms operating downstream of sFRP3, we show that sFRP3 blocks phosphorylation of β‐catenin at Ser552 in post‐MI plasma‐treated mitral VECs. The Ser552 phosphorylation site is associated with increased transcriptional activity of β‐catenin and activation of the canonical Wnt pathway. 44 Activation of Wnt/β‐catenin signaling was previously shown to be required during EndMT in adult aortic VECs, 63 and its inhibition has been suggested as an approach to target TGFβ signaling in pathogenesis of fibrotic diseases. 64 Next, we showed that sFRP3 decreased nuclear localization of Slug, a transcription factor required for EndMT initiation, 65 and increased VE‐cadherin expression in mitral VECs treated with post‐MI plasma. By suppressing transcriptional activation at the VE‐cadherin promoter, 66 which leads to disassembly of endothelial cell‐cell junctions, Slug regulates essential steps in EndMT.
RNA‐sequencing analysis of mitral VECs treated with post‐MI plasma revealed an upregulation of FOXM1 and FOXM1‐regulated genes. FOXM1 was detected along the MV endothelium in sheep that had undergone inferior MI 8 to 10 days before, consistent with the in vitro study. We also found increased expression and nuclear localization of FOXM1 in mitral VECs treated with 8‐ to 10‐day post‐MI plasma, both of which were decreased by sFRP3. Consistent with our findings in the post‐MI MV, a growing body of evidence has identified potential roles for FOXM1 in driving fibrosis 27 and induction and promotion of epithelial‐to‐mesenchymal transition. 47 , 67 A previous study implicated FOXM1 in TGFβ‐mediated epithelial‐to‐mesenchymal transition in breast cancer cells by showing FOXM1 increased the retention of SMAD (Sma genes and the Drosophila Mad, Mothers against decapentaplegic) 3 in the nuclei of TGFβ‐treated cancer cells. Conversely, loss of FOXM1 abolished TGFβ‐induced SMAD3/SMAD4. The authors also showed increased FOXM1/SMAD3 interaction upregulated Slug and promoted invasiveness of the breast cancer cells. 48 Other studies have shown that Wnt signaling increases β‐catenin stability and transcriptional activity via FOXM1. 28 , 68
Our results connect Wnt signaling to FOXMI in EndMT‐driven MV dysfunction for the first time by showing sFRP3 blocks the post‐MI plasma induced increase in FOXM1. We therefore suggest a new molecular pathway in MV endothelium wherein Wnt signaling, activated by the drop in plasma sFRP3 levels soon after MI, coordinates with TGFβ signaling to incite EndMT via upregulation of FOXM1. Additional studies are needed to gain a deeper understanding of the molecular interactions that regulate Wnt/TGFβ crosstalk in MV endothelium in the post‐MI setting, and to identify the cellular source of plasma sFRP3 and the mechanism by which sFRP3 levels drop after MI. These questions will require extensive investigation beyond the scope of this study.
Our study has some limitations. First, we used sheep as our animal model because the papillary muscle anatomy and well‐defined extracellular matrix layers of the MV leaflets are similar to human MV leaflets; however, this species is not amenable to genetic manipulations such as lineage tracing, which have been elegantly applied in mouse models of cardiovascular disease. While early studies suggested that EndMT contributed to increased cardiac myofibroblasts and onset of fibrosis, application of more specified lineage tracing modalities and markers find no evidence of EndMT. Instead, resident cardiac fibroblasts were shown to proliferate and become activated to a myofibroblastic state in models of cardiac injury such as pressure overload 69 and MI. 70 , 71 , 72 In contrast, lineage tracing has shown evidence for EndMT in atherosclerotic lesions and specifically in the fibrous caps, 73 and cells transitioning from endothelial to mesenchymal have been detected in human atherosclerotic plaques. 74 Single cell RNA sequencing of human aortic valves from patients with calcific aortic valve disease also showed evidence of EndMT in the thickened aortic valve leaflets. 75 These studies indicate that EndMT may contribute to specific types of cardiovascular disease but most likely not to myocardial fibrosis. Second, we studied a single time point—8 to 10‐days after MI—chosen as the starting point to identify early inciting events. EndMT may begin sooner, or conversely, 8 to 10 days may be too early to observe maladaptive EndMT. For example, CD45+/VE‐cadherin+/α‐SMA+ VECs were not detected in the 8‐ to 10‐day post–inferior MI MV‐derived cells, whereas such cells were found in MV endothelium at 6 months after inferior MI. 9
In summary, we provide in vivo evidence of EndMT in MV leaflets as early as 8 to 10 days after inferior MI, which in 26% to 38% 76 , 77 of humans predisposes to the development of IMR and heart failure. This early onset of EndMT is coincident with the fall in plasma sFRP3 levels following MI, which provides for the first time a link between infarcted myocardium and the MV. Mechanistically, we link reduced sFRP3 to upregulation of the transcription factor FOXM1, which drives EndMT. The early incitement of this molecular pathway may underlie the mechanisms by which the MV adapts after MI by increasing leaflet size to prevent IMR.
Conclusions
We propose that the drop in plasma sFRP3 within days after inferior MI could be used as a biomarker to predict development of mitral regurgitation and that restoration of sFRP3 to normal levels might provide a therapeutic strategy to prevent MR and the eventual heart failure that ensues.
Sources of Funding
The work was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under award number R01HL141917 to Drs Aikawa, Bischoff, and Levine; T32 HL007917; and R01HL136431 and R01HL147095 to Dr Aikawa.
Disclosures
None.
Supporting information
Tables S1–S2
Figures S1–S7
Acknowledgments
We thank Eric Engelbrecht in Timothy Hla’s lab for help with initial steps in the RNA‐sequencing data analysis.
Supplemental Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.121.023695
For Sources of Funding and Disclosures, see page 17.
References
- 1. Grigioni F, Detaint D, Avierinos JF, Scott C, Tajik J, Enriquez‐Sarano M. Contribution of ischemic mitral regurgitation to congestive heart failure after myocardial infarction. J Am Coll Cardiol. 2005;45:260–267. doi: 10.1016/j.jacc.2004.10.030 [DOI] [PubMed] [Google Scholar]
- 2. Grigioni F, Enriquez‐Sarano M, Zehr KJ, Bailey KR, Tajik AJ. Ischemic mitral regurgitation: long‐term outcome and prognostic implications with quantitative Doppler assessment. Circulation. 2001;103:1759–1764. doi: 10.1161/01.CIR.103.13.1759 [DOI] [PubMed] [Google Scholar]
- 3. Bartko PE, Arfsten H, Heitzinger G, Pavo N, Spinka G, Kastl S, Prausmüller S, Strunk G, Mascherbauer J, Hengstenberg C, et al., Global regurgitant volume: approaching the critical mass in valvular‐driven heart failure. Eur Heart J Cardiovasc Imaging. 2019. doi: 10.1093/ehjci/jez170 [DOI] [PubMed] [Google Scholar]
- 4. Levine RA, Schwammenthal E. Ischemic mitral regurgitation on the threshold of a solution: from paradoxes to unifying concepts. Circulation. 2005;112:745–758. doi: 10.1161/CIRCULATIONAHA.104.486720 [DOI] [PubMed] [Google Scholar]
- 5. Otsuji Y, Handschumacher MD, Schwammenthal E, Jiang L, Song JK, Guerrero JL, Vlahakes GJ, Levine RA. Insights from three‐dimensional echocardiography into the mechanism of functional mitral regurgitation: direct in vivo demonstration of altered leaflet tethering geometry. Circulation. 1997;96:1999–2008. doi: 10.1161/01.CIR.96.6.1999 [DOI] [PubMed] [Google Scholar]
- 6. Nishino S, Watanabe N, Gi T, Kuriyama N, Shibata Y, Asada Y. Longitudinal evaluation of mitral valve leaflet remodeling after acute myocardial infarction: serial quantitation of valve geometry using real‐time 3‐dimensional echocardiography. Circ Cardiovasc Imaging. 2020;13:e011396. doi: 10.1161/CIRCIMAGING.120.011396 [DOI] [PubMed] [Google Scholar]
- 7. Dal‐Bianco JP, Aikawa E, Bischoff J, Guerrero JL, Hjortnaes J, Beaudoin J, Szymanski C, Bartko PE, Seybolt MM, Handschumacher MD, et al. Myocardial infarction alters adaptation of the tethered mitral valve. J Am Coll Cardiol. 2016;67:275–287. doi: 10.1016/j.jacc.2015.10.092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Debonnaire P, Al Amri I, Leong DP, Joyce E, Katsanos S, Kamperidis V, Schalij MJ, Bax JJ, Marsan NA, Delgado V. Leaflet remodelling in functional mitral valve regurgitation: characteristics, determinants, and relation to regurgitation severity. Eur Heart J Cardiovasc Imaging. 2015;16:290–299. doi: 10.1093/ehjci/jeu216 [DOI] [PubMed] [Google Scholar]
- 9. Bischoff J, Casanovas G, Wylie‐Sears J, Kim D‐H, Bartko PE, Guerrero JL, Dal‐Bianco JP, Beaudoin J, Garcia ML, Sullivan SM, et al. CD45 expression in mitral valve endothelial cells after myocardial infarction. Circ Res. 2016;119:1215–1225. doi: 10.1161/CIRCRESAHA.116.309598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chaput M, Handschumacher MD, Tournoux F, Hua L, Guerrero JL, Vlahakes GJ, Levine RA. Mitral leaflet adaptation to ventricular remodeling: occurrence and adequacy in patients with functional mitral regurgitation. Circulation. 2008;118:845–852. doi: 10.1161/CIRCULATIONAHA.107.749440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grande‐Allen KJ, Barber JE, Klatka KM, Houghtaling PL, Vesely I, Moravec CS, McCarthy PM. Mitral valve stiffening in end‐stage heart failure: evidence of an organic contribution to functional mitral regurgitation. J Thorac Cardiovasc Surg. 2005;130:783–790. doi: 10.1016/j.jtcvs.2005.04.019 [DOI] [PubMed] [Google Scholar]
- 12. Grande‐Allen KJ, Borowski AG, Troughton RW, Houghtaling PL, Dipaola NR, Moravec CS, Vesely I, Griffin BP. Apparently normal mitral valves in patients with heart failure demonstrate biochemical and structural derangements: an extracellular matrix and echocardiographic study. J Am Coll Cardiol. 2005;45:54–61. doi: 10.1016/j.jacc.2004.06.079 [DOI] [PubMed] [Google Scholar]
- 13. Saito K, Okura H, Watanabe N, Obase K, Tamada T, Koyama T, Hayashida A, Neishi Y, Kawamoto T, Yoshida K. Influence of chronic tethering of the mitral valve on mitral leaflet size and coaptation in functional mitral regurgitation. JACC Cardiovasc Imaging. 2012;5:337–345. doi: 10.1016/j.jcmg.2011.10.004 [DOI] [PubMed] [Google Scholar]
- 14. Beaudoin J, Dal‐Bianco JP, Aikawa E, Bischoff J, Guerrero JL, Sullivan S, Bartko PE, Handschumacher MD, Kim D‐H, Wylie‐Sears J, et al. Mitral leaflet changes following myocardial infarction: clinical evidence for maladaptive valvular remodeling. Circ Cardiovasc Imaging. 2017;10. doi: 10.1161/CIRCIMAGING.117.006512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bischoff J. Endothelial‐to‐mesenchymal transition. Circ Res. 2019;124:1163–1165. doi: 10.1161/CIRCRESAHA.119.314813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kovacic JC, Dimmeler S, Harvey RP, Finkel T, Aikawa E, Krenning G, Baker AH. Endothelial to mesenchymal transition in cardiovascular disease: JACC state‐of‐the‐art review. J Am Coll Cardiol. 2019;73:190–209. doi: 10.1016/j.jacc.2018.09.089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dal‐Bianco JP, Aikawa E, Bischoff J, Guerrero JL, Handschumacher MD, Sullivan S, Johnson B, Titus JS, Iwamoto Y, Wylie‐Sears J, et al. Active adaptation of the tethered mitral valve: insights into a compensatory mechanism for functional mitral regurgitation. Circulation. 2009;120:334–342. doi: 10.1161/CIRCULATIONAHA.108.846782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Balachandran K, Alford PW, Wylie‐Sears J, Goss JA, Grosberg A, Bischoff J, Aikawa E, Levine RA, Parker KK. Cyclic strain induces dual‐mode endothelial‐mesenchymal transformation of the cardiac valve. Proc Natl Acad Sci USA. 2011;108:19943–19948. doi: 10.1073/pnas.1106954108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Marsit O, Clavel MA, Côté‐Laroche C, Hadjadj S, Bouchard MA, Handschumacher MD, Clisson M, Drolet MC, Boulanger M‐C, Kim DH, et al. Attenuated mitral leaflet enlargement contributes to functional mitral regurgitation after myocardial infarction. J Am Coll Cardiol. 2020;75:395–405. doi: 10.1016/j.jacc.2019.11.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, et al. Endothelial‐to‐mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. doi: 10.1038/nm1613 [DOI] [PubMed] [Google Scholar]
- 21. Kunzelman KS, Quick DW, Cochran RP. Altered collagen concentration in mitral valve leaflets: biochemical and finite element analysis. Ann Thorac Surg. 1998;66:S198–S205. doi: 10.1016/S0003-4975(98)01106-0 [DOI] [PubMed] [Google Scholar]
- 22. Bartko PE, Dal‐Bianco JP, Guerrero JL, Beaudoin J, Szymanski C, Kim D‐H, Seybolt MM, Handschumacher MD, Sullivan S, Garcia ML, et al. Effect of losartan on mitral valve changes after myocardial infarction. J Am Coll Cardiol. 2017;70:1232–1244. doi: 10.1016/j.jacc.2017.07.734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zi X, Guo Y, Simoneau AR, Hope C, Xie J, Holcombe RF, Hoang BH. Expression of Frzb/secreted frizzled‐related protein 3, a secreted Wnt antagonist, in human androgen‐independent prostate cancer PC‐3 cells suppresses tumor growth and cellular invasiveness. Can Res. 2005;65:9762–9770. doi: 10.1158/0008-5472.CAN-05-0103 [DOI] [PubMed] [Google Scholar]
- 24. Guo X, Wang XF. Signaling cross‐talk between TGF‐beta/BMP and other pathways. Cell Res. 2009;19:71–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Song S, Zhang R, Cao W, Fang G, Yu Y, Wan Y, Wang C, Li Y, Wang Q. Foxm1 is a critical driver of TGF‐beta‐induced EndMT in endothelial cells through Smad2/3 and binds to the snail promoter. J Cell Physiol. 2019;234:9052–9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang X, Zhang L, Du Y, Zheng H, Zhang P, Sun Y, Wang Y, Chen J, Ding P, Wang N, et al. A novel FOXM1 isoform, FOXM1D, promotes epithelial‐mesenchymal transition and metastasis through ROCKs activation in colorectal cancer. Oncogene. 2017;36:807–819. doi: 10.1038/onc.2016.249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Penke LR, Speth JM, Dommeti VL, White ES, Bergin IL, Peters‐Golden M. FOXM1 is a critical driver of lung fibroblast activation and fibrogenesis. J Clin Investig. 2018;128:2389–2405. doi: 10.1172/JCI87631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang NU, Wei P, Gong A, Chiu W‐T, Lee H‐T, Colman H, Huang HE, Xue J, Liu M, Wang Y, et al. FOXM1 promotes β‐catenin nuclear localization and controls Wnt target‐gene expression and glioma tumorigenesis. Cancer Cell. 2011;20:427–442. doi: 10.1016/j.ccr.2011.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Llaneras MR, Nance ML, Streicher JT, Lima JA, Savino JS, Bogen DK, Deac RF, Ratcliffe MB, Edmunds LH Jr. Large animal model of ischemic mitral regurgitation. Ann Thorac Surg. 1994;57:432–439. doi: 10.1016/0003-4975(94)91012-X [DOI] [PubMed] [Google Scholar]
- 30. Kim DH, Morris B, Guerrero JL, Sullivan SM, Hung J, Levine RA. Ovine model of ischemic mitral regurgitation. Methods Mol Biol. 2018;1816:295–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Beaudoin J, Thai WE, Wai B, Handschumacher MD, Levine RA, Truong QA. Assessment of mitral valve adaptation with gated cardiac computed tomography: validation with three‐dimensional echocardiography and mechanistic insight to functional mitral regurgitation. Circ Cardiovasc Imaging. 2013;6:784–789. doi: 10.1161/CIRCIMAGING.113.000561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wylie‐Sears J, Levine RA, Bischoff J. Losartan inhibits endothelial‐to‐mesenchymal transformation in mitral valve endothelial cells by blocking transforming growth factor‐beta–induced phosphorylation of erk. Biochem Biophys Res Commun. 2014;446:870–875. doi: 10.1016/j.bbrc.2014.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fastqc AS. A quality control tool for high throughput sequence data. 2010.
- 34. Ewels P, Magnusson M, Lundin S, Kaller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32:3047–3048. doi: 10.1093/bioinformatics/btw354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. Star: ultrafast universal RNA‐seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 1000 Genome Project Data Processing Subgroup . The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Okonechnikov K, Conesa A, Garcia‐Alcalde F. Qualimap 2: advanced multi‐sample quality control for high‐throughput sequencing data. Bioinformatics. 2016;32:292–294. doi: 10.1093/bioinformatics/btv566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Garcia‐Alcalde F, Okonechnikov K, Carbonell J, Cruz LM, Gotz S, Tarazona S, Dopazo J, Meyer TF, Conesa A. Qualimap: evaluating next‐generation sequencing alignment data. Bioinformatics. 2012;28:2678–2679. doi: 10.1093/bioinformatics/bts503 [DOI] [PubMed] [Google Scholar]
- 39. DeLuca DS, Levin JZ, Sivachenko A, Fennell T, Nazaire MD, Williams C, Reich M, Winckler W, Getz G. RNA‐SeQC: RNA‐seq metrics for quality control and process optimization. Bioinformatics. 2012;28:1530–1532. doi: 10.1093/bioinformatics/bts196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liao Y, Smyth GK, Shi W. Featurecounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656 [DOI] [PubMed] [Google Scholar]
- 41. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schmittgen TD, Livak KJ. Analyzing real‐time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73 [DOI] [PubMed] [Google Scholar]
- 43. Paranya G, Vineberg S, Dvorin E, Kaushal S, Roth SJ, Rabkin E, Schoen FJ, Bischoff J. Aortic valve endothelial cells undergo transforming growth factor‐beta‐mediated and non‐transforming growth factor‐beta–mediated transdifferentiation in vitro. Am J Pathol. 2001;159:1335–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fang D, Hawke D, Zheng Y, Xia Y, Meisenhelder J, Nika H, Mills GB, Kobayashi R, Hunter T, Lu Z. Phosphorylation of beta‐catenin by AKT promotes beta‐catenin transcriptional activity. J Biol Chem. 2007;282:11221–11229. doi: 10.1074/jbc.M611871200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Medici D, Hay ED, Olsen BR. Snail and slug promote epithelial‐mesenchymal transition through beta‐catenin‐T‐cell factor‐4‐dependent expression of transforming growth factor‐beta3. Mol Biol Cell. 2008;19:4875–4887. doi: 10.1091/mbc.e08‐05‐0506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bolós V, Peinado H, Pérez‐Moreno MA, Fraga MF, Esteller M, Cano A. The transcription factor slug represses E‐cadherin expression and induces epithelial to mesenchymal transitions: a comparison with snail and E47 repressors. J Cell Sci. 2003;116:499–511. doi: 10.1242/jcs.00224 [DOI] [PubMed] [Google Scholar]
- 47. Balli D, Ustiyan V, Zhang Y, Wang IC, Masino AJ, Ren X, Whitsett JA, Kalinichenko VV, Kalin TV. Foxm1 transcription factor is required for lung fibrosis and epithelial‐to‐mesenchymal transition. EMBO J. 2013;32:231–244. doi: 10.1038/emboj.2012.336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xue J, Lin X, Chiu WT, Chen YH, Yu G, Liu M, Feng XH, Sawaya R, Medema RH, Hung MC, et al. Sustained activation of SMAD3/SMAD4 by FOXM1 promotes TGF‐β–dependent cancer metastasis. J Clin Investig. 2014;124:564–579. doi: 10.1172/JCI71104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Priller M, Pöschl J, Abrão L, von Bueren AO, Cho YJ, Rutkowski S, Kretzschmar HA, Schüller U. Expression of FoxM1 is required for the proliferation of medulloblastoma cells and indicates worse survival of patients. Clin Cancer Res. 2011;17:6791–6801. doi: 10.1158/1078-0432.CCR-11-1214 [DOI] [PubMed] [Google Scholar]
- 50. Radhakrishnan SK, Bhat UG, Hughes DE, Wang IC, Costa RH, Gartel AL. Identification of a chemical inhibitor of the oncogenic transcription factor Forkhead Box M1. Can Res. 2006;66:9731–9735. doi: 10.1158/0008-5472.CAN-06-1576 [DOI] [PubMed] [Google Scholar]
- 51. Bosada FM, Devasthali V, Jones KA, Stankunas K. Wnt/β‐catenin signaling enables developmental transitions during valvulogenesis. Development. 2016;143:1041–1054. doi: 10.1242/dev.130575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Alfieri CM, Cheek J, Chakraborty S, Yutzey KE. Wnt signaling in heart valve development and osteogenic gene induction. Dev Biol. 2010;338:127–135. doi: 10.1016/j.ydbio.2009.11.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mizutani M, Wu JC, Nusse R. Fibrosis of the neonatal mouse heart after cryoinjury is accompanied by Wnt signaling activation and epicardial‐to‐mesenchymal transition. J Am Heart Assoc. 2016;5:e002457. doi: 10.1161/JAHA.115.002457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Foulquier S, Daskalopoulos EP, Lluri G, Hermans KCM, Deb A, Blankesteijn WM. Wnt signaling in cardiac and vascular disease. Pharmacol Rev. 2018;70:68–141. doi: 10.1124/pr.117.013896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Moon J, Zhou H, Zhang LS, Tan W, Liu Y, Zhang S, Morlock LK, Bao X, Palecek SP, Feng JQ, et al. Blockade to pathological remodeling of infarcted heart tissue using a porcupine antagonist. Proc Natl Acad Sci USA. 2017;114:1649–1654. doi: 10.1073/pnas.1621346114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yang D, Fu W, Li L, Xia X, Liao Q, Yue R, Chen H, Chen X, An S, Zeng C, et al. Therapeutic effect of a novel Wnt pathway inhibitor on cardiac regeneration after myocardial infarction. Clin Sci. 2017;131:2919–2932. doi: 10.1042/CS20171256 [DOI] [PubMed] [Google Scholar]
- 57. Bastakoty D, Saraswati S, Joshi P, Atkinson J, Feoktistov I, Liu J, Harris JL, Young PP. Temporary, systemic inhibition of the WNT/β‐catenin pathway promotes regenerative cardiac repair following myocardial infarct. Cell Stem Cells Regen Med. 2016;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Barandon L, Couffinhal T, Ezan J, Dufourcq P, Costet P, Alzieu P, Leroux L, Moreau C, Dare D, Duplàa C. Reduction of infarct size and prevention of cardiac rupture in transgenic mice overexpressing FrzA. Circulation. 2003;108:2282–2289. doi: 10.1161/01.CIR.0000093186.22847.4C [DOI] [PubMed] [Google Scholar]
- 59. He W, Zhang L, Ni A, Zhang Z, Mirotsou M, Mao L, Pratt RE, Dzau VJ. Exogenously administered secreted frizzled related protein 2 (Sfrp2) reduces fibrosis and improves cardiac function in a rat model of myocardial infarction. Proc Natl Acad Sci USA. 2010;107:21110–21115. doi: 10.1073/pnas.1004708107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nakamura K, Sano S, Fuster JJ, Kikuchi R, Shimizu I, Ohshima K, Katanasaka Y, Ouchi N, Walsh K. Secreted frizzled‐related protein 5 diminishes cardiac inflammation and protects the heart from ischemia/reperfusion injury. J Biol Chem. 2016;291:2566–2575. doi: 10.1074/jbc.M115.693937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Grayburn PA, Smith RL II. Left ventricular ejection fraction in mitral regurgitation because of flail leaflet. Circ Cardiovasc Imaging. 2014;7:220–221. doi: 10.1161/CIRCIMAGING.114.001675 [DOI] [PubMed] [Google Scholar]
- 62. Cavalcante JL, Kusunose K, Obuchowski NA, Jellis C, Griffin BP, Flamm SD, Kwon DH. Prognostic impact of ischemic mitral regurgitation severity and myocardial infarct quantification by cardiovascular magnetic resonance. JACC Cardiovasc Imaging. 2020;13:1489–1501. doi: 10.1016/j.jcmg.2019.11.008 [DOI] [PubMed] [Google Scholar]
- 63. Zhong A, Mirzaei Z, Simmons CA. The roles of matrix stiffness and ß‐catenin signaling in endothelial‐to‐mesenchymal transition of aortic valve endothelial cells. Cardiovasc Eng Technol. 2018;9:158–167. doi: 10.1007/s13239-018-0363-0 [DOI] [PubMed] [Google Scholar]
- 64. Akhmetshina A, Palumbo K, Dees C, Bergmann C, Venalis P, Zerr P, Horn A, Kireva T, Beyer C, Zwerina J, et al. Activation of canonical Wnt signalling is required for TGF‐β–mediated fibrosis. Nat Commun. 2012;3:735. doi: 10.1038/ncomms1734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Niessen K, Fu Y, Chang L, Hoodless PA, McFadden D, Karsan A. Slug is a direct notch target required for initiation of cardiac cushion cellularization. J Cell Biol. 2008;182:315–325. doi: 10.1083/jcb.200710067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lopez D, Niu G, Huber P, Carter WB. Tumor‐induced upregulation of twist, snail, and slug represses the activity of the human VE‐cadherin promoter. Arch Biochem Biophys. 2009;482:77–82. doi: 10.1016/j.abb.2008.11.016 [DOI] [PubMed] [Google Scholar]
- 67. Yu CP, Yu S, Shi L, Wang S, Li ZX, Wang YH, Sun CJ, Liang J. FoxM1 promotes epithelial‐mesenchymal transition of hepatocellular carcinoma by targeting Snail. Mol Med Rep. 2017;16:5181–5188. doi: 10.3892/mmr.2017.7223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chen Y, Li YU, Xue J, Gong A, Yu G, Zhou A, Lin K, Zhang S, Zhang NU, Gottardi CJ, et al. Wnt‐induced deubiquitination FoxM1 ensures nucleus β‐catenin transactivation. EMBO J. 2016;35:668–684. doi: 10.15252/embj.201592810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Moore‐Morris T, Guimarães‐Camboa N, Banerjee I, Zambon AC, Kisseleva T, Velayoudon A, Stallcup WB, Gu Y, Dalton ND, Cedenilla M, et al. Resident fibroblast lineages mediate pressure overload‐induced cardiac fibrosis. J Clin Investig. 2014;124:2921–2934. doi: 10.1172/JCI74783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, Brody MJ, J. Lin S‐C, Aronow BJ, Tallquist MD, et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun. 2016;7:12260. doi: 10.1038/ncomms12260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Fu X, Khalil H, Kanisicak O, Boyer JG, Vagnozzi RJ, Maliken BD, Sargent MA, Prasad V, Valiente‐Alandi I, Blaxall BC, et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J Clin Investig. 2018;128:2127–2143. doi: 10.1172/JCI98215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zhang S, Li Y, Huang X, Liu K, Wang QD, Chen AF, Sun K, Lui KO, Zhou B. Seamless genetic recording of transiently activated mesenchymal gene expression in endothelial cells during cardiac fibrosis. Circulation. 2021;144:2004–2020. doi: 10.1161/CIRCULATIONAHA.121.055417 [DOI] [PubMed] [Google Scholar]
- 73. Newman AAC, Serbulea V, Baylis RA, Shankman LS, Bradley X, Alencar GF, Owsiany K, Deaton RA, Karnewar S, Shamsuzzaman S, et al. Multiple cell types contribute to the atherosclerotic lesion fibrous cap by PDGFRβ and bioenergetic mechanisms. Nat Metab. 2021;3:166–181. doi: 10.1038/s42255‐020‐00338‐8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Evrard SM, Lecce L, Michelis KC, Nomura‐Kitabayashi A, Pandey G, Purushothaman K‐R, d’Escamard V, Li JR, Hadri L, Fujitani K, et al. Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat Commun. 2016;7:11853. doi: 10.1038/ncomms11853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Xu K, Xie S, Huang Y, Zhou T, Liu M, Zhu P, Wang C, Shi J, Li F, Sellke FW, et al. Cell‐type transcriptome atlas of human aortic valves reveal cell heterogeneity and endothelial to mesenchymal transition involved in calcific aortic valve disease. Arterioscler Thromb Vasc Biol. 2020;40:2910–2921. doi: 10.1161/ATVBAHA.120.314789 [DOI] [PubMed] [Google Scholar]
- 76. Nishino S, Watanabe N, Kimura T, Enriquez‐Sarano M, Nakama T, Furugen M, Koiwaya H, Ashikaga K, Kuriyama N, Shibata Y. The course of ischemic mitral regurgitation in acute myocardial infarction after primary percutaneous coronary intervention: from emergency room to long‐term follow‐up. Circ Cardiovasc Imaging. 2016;9:e004841. doi: 10.1161/CIRCIMAGING.116.004841 [DOI] [PubMed] [Google Scholar]
- 77. Kumanohoso T, Otsuji Y, Yoshifuku S, Matsukida K, Koriyama C, Kisanuki A, Minagoe S, Levine RA, Tei C. Mechanism of higher incidence of ischemic mitral regurgitation in patients with inferior myocardial infarction: quantitative analysis of left ventricular and mitral valve geometry in 103 patients with prior myocardial infarction. J Thorac Cardiovasc Surg. 2003;125:135–143. doi: 10.1067/mtc.2003.78 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S2
Figures S1–S7