

Abstract
Mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1), which plays a crucial role in the nuclear factor-kappa B (NF-κB) activation signaling pathway as a paracaspase, is a new target for immunomodulatory and antitumor drugs. Here, novel inhibitors that target MALT1 allosteric sites were identified by virtual screening FDA-approved drug databases. Paliperidone, a compound that binds to the allosteric site of MALT1, is investigated. An in vitro study found that the proteolytic activity of MALT1 substrate cleavage was blocked by paliperidone. Meanwhile, the MALT1 proteolytic activity was reversible, as demonstrated by the partial recovery of the MALT1 substrate cleavage following compound wash out. The docking analysis of the interaction of MALT1 and paliperidone suggested that two hydrogen bonds formed in the allosteric pocket of MALT1. MALT1 and paliperidone achieved a good equilibrium, as demonstrated by 100 ns molecular dynamic (MD) simulations conducted with the program Gromacs. However, the catalytically active site of the MALT1 complex with paliperidone remained in an inactive conformation. Thus, paliperidone, a noncompetitive and allosteric inhibitor, was screened through in silico and in vitro methods. This study will be of significance for the development of effective and selective drugs that can treat MALT1-driven cancer or autoimmune diseases.
Paliperidone was screened as an effective and selective drug that can treat MALT1-driven cancer or autoimmune diseases.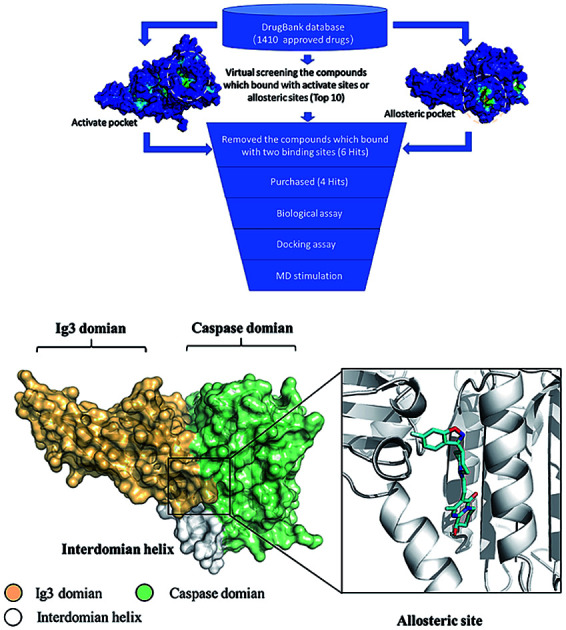
Introduction
Mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1), which is also called paracaspase 1, plays a crucial role in the activation of the NF-κB signaling pathway. The MALT1 comprises a putative caspase, death, and several immunoglobulin (Ig)-like domains. MALT1 is a cysteine-dependent, aspartate-specific protease that can cleave limited substrates, including CYLD,1 A20,2 and RelB,3 which are negative regulators of the NF-κB pathway. Therefore, MALT1 is an important part of the immune response in which the downstream of NF-κB is activated and the transcription of the growth-promoting cytokine interleukin-2 is induced in lymphocytes.4 Recent studies have demonstrated that the crucial role of MALT1 in the survival of the activated B-cell subtype of diffuse-large B-cell lymphoma (ABC-DLBCL);5 the constitutive MALT1 paracaspase activity is a common feature of ABC-DLBCL cells.6 Therefore, MALT1 is an important therapeutic target for the treatment of MALT lymphoma.
The inhibitors of MALT1 can effectively inhibit its intracellular activation, which is useful for immunomodulation and lymphoma treatment.7–9 The catalytically active sites of MALT1 have sequence conservation with a caspase family. Some effective MALT1 inhibitor drugs have been reported.10,11 MI-2, which targets the catalytically active sites of MALT1, significantly inhibits ABC-DLBCL tumors in vivo and in vitro.12 The inhibitor Z-VRPR-FMK, which covalently binds to Cys464 of the catalytically active sites of MALT1, may be helpful in the treatment of MALT1-related diseases.13
An allosteric site in the interface between the caspase and the Ig3 domains of MALT1 (this site is opposite the caspase active site) is another effective inhibitor combination mode. Given that this combination is non-covalent, allosteric inhibitors have the advantage of non-competition and reversibility. Phenothiazine derivatives, including mepazine, thioridazine, and promazine, target the allosteric site of MALT1 to prevent the rearrangement of MALT1 from the inactive state to the activated state and to inhibit the proteolytic cleavage of the substrate.14 MLT-747 and MLT-748 act as potent allosteric inhibitors by blocking the proteolytic activity of MALT1 and rescues the protein deficiency through increasing the stability of the MALT1-W580S mutant.15 The non-covalent binding of these allosteric inhibitors to MALT1 can be reversed through wash out, and such reversal provides an opportunity for regulatory treatment.
Drug repurposing or drug repositioning is performed to identify new uses for existing approved drugs.16 Drug repositioning also has important commercial value; it expands the market and discovers novel uses for compounds in a short time and with low financial risks. Successful examples include thalidomide,17 sildenafil,18 bupropion,19 and fluoxetine,20 which are currently used for purposes that are beyond their initially approved therapeutic indications. Approaches that combine traditional in vitro screening with computational screening (in silico) methods are being increasingly used to discover novel medicine.21
This study mainly aimed to develop selective and reversible novel MALT1 allosteric inhibitors that are listed in the FDA-approved drug database by using an in silico–in vitro approach. Paliperidone, a new and effective drug, was discovered through in vitro experiments and computer methods, such as virtual screening, molecular docking, and molecular dynamics. The method adopted in this work could be utilized as a fast and visible strategy for accelerating research on the treatment and prevention of MALT1-driven cancer or autoimmune diseases.
Methods
MALT1 protein structure preparation
The amino acid sequence of MALT1 was collected from UniProtKB/Swiss-Prot (Q9UDY8.1). A model of MALT1 was generated from its crystal structure (PDB ID 6F7I),15 which was used for the virtual screening of the inhibitor that targeted the allosteric site. This structure was then rebuilt by homology modeling through the SWISS-MODEL website (https://swissmodel.expasy.org/)22–24 because some residues in the crystal structure were not located (missing loops in amino acids 325–336, 466–481, 495–503, and 717–728). A composite scoring function was performed through the QMEAN server to estimate the global and local absolute qualities. The crystal structure of human MALT1 (caspase domain) in complex with an irreversible peptidic inhibitor (PDB ID 3V4O)13 was rebuilt and analyzed.
Virtual screening
The FDA-approved drug database (updated in February 2018) contains 1410 compounds and is freely available (http://zinc.docking.org/catalogs/dbfda/). Raccoon is a graphical interface that converts ligands to PDBQT format, which can be identified by virtual screening software.25 The program AutoDock 4.2,26 which uses a computational docking software that is based on an empirical free energy force field and the rapid Lamarckian genetic algorithm search method, was then applied for virtual screening. We screened the compounds that bind with the allosteric and catalytically active sites. Rigid receptor and flexible ligands were assumed in the docking simulations. A 50 Å × 50 Å × 50 Å grid was defined on the protein structure to generate a grid map. Hydrogen bond interactions were analyzed using the software LigPlot+.
Chemistry
Compounds, such as lurasidone, paliperidone, mitoxantrone, and risperidone, were purchased from Yuanye Biotech or Meilun. PMA/ionomycin was purchased from Beyotime Biotechnology. Anti-CYLD antibody was purchased from Cell Signaling Technology. Anti-β-actin antibody and HRP-labeled goat anti-rabbit or goat anti-mouse secondary antibody were purchased from TransGen Biotech.
Immunoblotting assay of CYLD cleavage by MALT1
Jurkat cells were purchased from ATCC and cultured in RPMI1640 supplemented with 10% FBS and 5% CO2 at 37 °C. Cells at 2 × 106 were treated with the indicated concentration of compounds for 12 h and stimulated with 50 ng mL−1 PMA and 1 μM ionomycin for 2 h. After treatment, cells were lysed by a RIPA lysis buffer (50 mM Tris–HCl, 0.1% Triton X-100, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, and protease inhibitors; pH 7.5), fractioned by SDS-PAGE, transferred to a PVDF membrane, and blocked. Subsequently, the PVDF membrane was incubated with the primary antibody (Anti-CYLD or β-actin antibody was diluted with 3% BSA) for 1 h. After being incubated, the PVDF membrane was fully washed and incubated with anti-HRP antibody for 1 h. Images were captured with chemiluminescence and X-ray photo film and then quantified with Image J.
Washout experiments
Jurkat cells at 2 × 106 were incubated with paliperidone for 12 h. After treatment, the cells were collected and washed with PBS. After being washed, the cells were cultured with a complete medium for 2 h and then stimulated with PMA/ionomycin for 2 h. After stimulation, the cells were collected and lysed with the RIPA lysis buffer. Lysates were collected for immunoblot assay with CYLD and β-actin as previously described.
Molecular dynamic simulation
MD simulations were performed with Gromacs version 2018 (ref. 27) with the CHARMM36 all-atom force field (March 28, 2019).28 The paliperidone initial conformation was screened from the result of molecular docking. The PRODRG Server was adopted for topology generation of compound.29 The protein was centered in separate cubic boxes and solvated by using the SPC216 water model.30 Fourteen additional NA ions were added to the system to automatically achieve elector-neutrality because the protein had a total charge of −14.000e. We relaxed the structure through a process called energy minimization to ensure a reasonable starting structure in terms of geometry and solvent orientation. Convergence was achieved at a maximum force of less than 1000 kJ mol−1 nm−1 in any atom. Equilibration was conducted in two phases, namely, NVT ensemble (constant number of particles, volume, and temperature) and NPT ensemble (constant number of particles, pressure, and temperature), for 100 ps until the system became well-equilibrated. Following equilibration, MD simulations were conducted for 100 ns under the same conditions as above. System stability and differences in the trajectories, root mean square deviation (RMSD), and root mean square fluctuations (RMSF) that were converted to B-factor and the minimum atom distances were calculated by the tools available in the Gromacs package. The trajectory of the protein was displayed, animated, and analyzed by the visualization program Pymol (The PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC).
Statistical analysis
Data are shown as mean SD. Statistical significance was evaluated through unpaired two-tailed Student's t-test. P value <0.05 was considered significant.
Results and discussion
Definition of the binding sites for the virtual screening and validation of the simulation method
We initiated a program to discover MALT1 inhibitors as a potential therapy for lymphoma and autoimmune diseases following the reported MALT1 paracaspase activity and its central role in immune response. Unlike catalytic site inhibitors, the allosteric inhibitors of MALT1 are mostly reversible, accompanied by stable proteins, and do not block catalytically active sites (Fig. 1A). Then, the allosteric site located between the catalytic and Ig3 domains was selected for virtual screening.
Fig. 1. The structures of MALT1 with inhibitors. (A) The surface structure of MALT1 was colored by different regions. Cartoon representations of MALT1 in complex with MLT-747 (PDB ID 6F7I) (down) and with Z-Val-Arg-Pro-Arg-CH2 (PBD ID:3V4O) (up). The inhibitor MLT-747 or Z-Val-Arg-Pro-Arg-CH2 was shown by stick. (B) Superposition of conformation of MLT-747 in crystal structure and in molecular docking results. The structure of MALT1 was shown by white cartoon, MLT-747 was shown in yellow (crystal) and orange (docking) stick.

Previously reported allosteric site binding inhibitor MLT-747 was re-docked into the corresponding region of MALT1 to verify the effectiveness of the docking method. The MALT1 model was a crystal structure with allosteric inhibitor MLT-747 (PDB ID:6F7I) and refined through homology modeling. Fig. 1B shows the alignment of the crystal structure and the docked structure of the MALT1–inhibitor complexes. The superposition of the compounds did not show any significant difference between the initial and docked structures; the RMSD value was 0.32 Å. The energy score of the inhibitor MLT-747 in the allosteric site was −14.13 kcal mol−1. Therefore, it demonstrated that this docking method was feasible for virtual screening in the development of novel inhibitors.
Identification of potential inhibitors by structure-based virtual screening
In accordance with the virtual screening simulation conducted with AutoDock 4.2, the 1410 compounds from the FDA database were scored and ranked on the basis of the estimated free energy of binding, which is an important indicator of compound binding efficiency. The predicted complexes were optimized and ranked according to the empirical scoring function, ScreenScore, which estimated the binding free energy of the ligand receptor complex. Each docking operation screened 10 runs for the protein–ligand complex that were advantageous for docking; each docking had 150 preferred conformations. Table S1† lists the docking poses and the relative docking energies of the top 10 compounds with energy scores ranging from −18.55 kcal mol−1 to −13.03 kcal mol−1. We analyzed the structure and properties of this top 10 compounds of the allosteric site binding compounds. We also screened the catalytically active site of MALT1 as a reference (Table S2†). Gentamicin, natamycin, tobramycin, and maraviroc had low binding energy scores with the catalytically active and allosteric sites. Therefore, they also interacted with the catalytically active site of MALT1 and were thus not utilized in subsequent study as the specific inhibitors of the allosteric site. Lapatinib was not studied because it is a known leukemia medicine. Ultimately, in consideration of commercial availability, four compounds that bind to allosteric sites, namely, lurasidone, paliperidone, risperidone, and mitoxantrone, were purchased for experimental testing (Table 1). Fig. 2 shows the workflow for developing the allosteric site inhibitor of MALT1.
The properties and energy score of the potential MALT1 allosteric inhibitors.
| ZNIC ID | Chemical name | Structure | Molecular formula | log P | M wt | Energya (kcal mol−1) |
|---|---|---|---|---|---|---|
| ZINC000003794794 | Mitoxantrone |
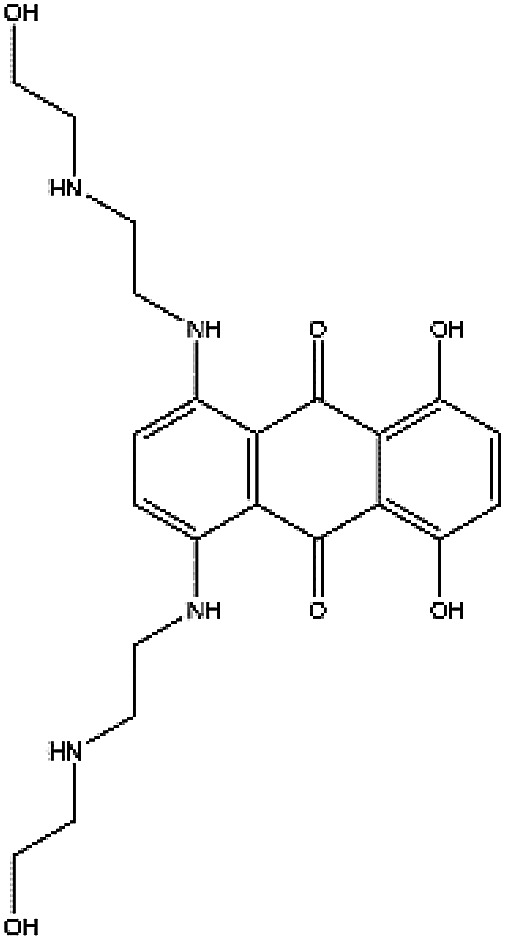
|
C22H28N4O6 | −0.139 | 444.488 | −14.35 |
| ZINC000003927822 | Lurasidone |
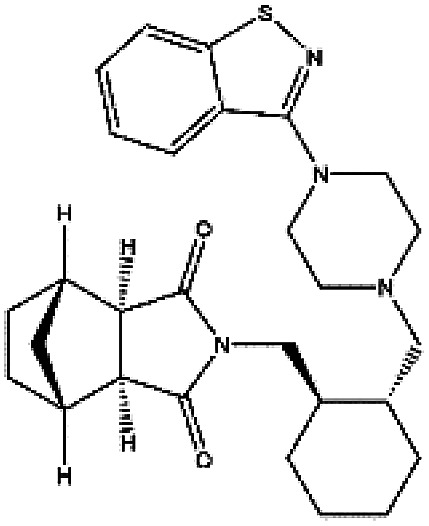
|
C28H36N4O2S | 4.256 | 492.682 | −13.56 |
| ZINC000001481956 | Paliperidone |
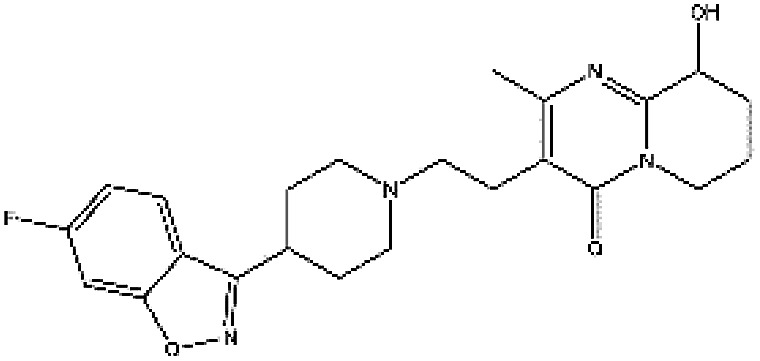
|
C23H27FN4O3 | 3.081 | 426.492 | −13.16 |
| ZINC000000538312 | Risperdal |
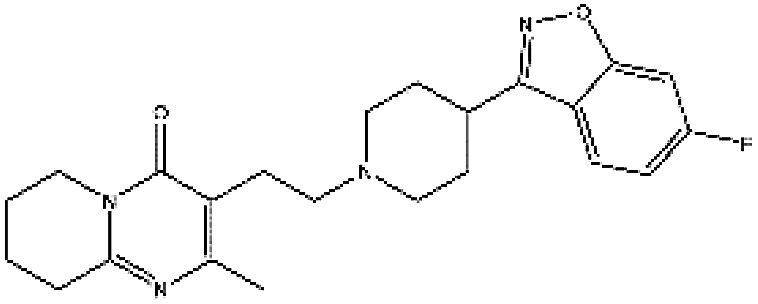
|
C23H27FN4O2 | 3.59 | 410.493 | −13.07 |
Estimated free energy of binding of virtual screening results by AutoDock program.
Fig. 2. Workflow for developing allosteric site inhibitors of MALT1.

Blockage of MALT1 substrate CYLD cleavage by paliperidone
MALT1 is an arginine-specific protease and can cleavage A20, Bcl10, RelB, and CYLD. The cleavage of CYLD was detected as previously reported to identify the anti-proteolysis activity of the inhibitor.31,32 After PMA/ion treatment for 2 h, MALT1 was activated followed by cleavage of substrate CYLD (Fig. 3A). The protein level of the full length of CYLD at 100 kD was decreased, and the cleaved CYLD at 40 kD was increased. The protein levels of the cleaved CYLD decreased with compound paliperidone treatment (Fig. 3B). Therefore, the compound paliperidone can decrease the proteolysis activation of MALT1.
Fig. 3. Compounds paliperidone blocked the cleavage of MALT1 substrate CYLD. (A) Immunoblots showed the full length and cleaved of CYLD in Jurkat cells under PMA/ionomycin treatment for 120 min. (B) Compounds paliperidone blocked the cleavage of CYLD. Jurkat cells were treated with paliperidone for 12 h and then stimulated with PMA/ionomycin for 120 min. Histogram showed the quantification of immunoblot that normalized with β-actin. Three independent experiments were adopted in per test group. Data was shown as mean ± SD, *P < 0.05.

Binding mode analysis of novel MALT1 inhibitor paliperidone
The binding pose was predicted through molecular docking and revealed the potential interaction between compound paliperidone and MALT1. Fig. 4A and B shows the chemical structures of MLT-747 and paliperidone. The docking results showed that paliperidone was bound to the same allosteric pocket as the MALT1 allosteric inhibitor MLT-747 (Fig. 4C and D) and formed hydrogen bond interactions with MALT1. Table 2 details the hydrogen-bond parameters of paliperidone and MLT-747 with MALT1.
Fig. 4. Comparison of the interaction of paliperidone–MALT1 complex and MLT-747–MALT1 complex. Chemical structures of MLT-747 (A) and paliperidone (B). The structure superposition between MLT-747 and paliperidone docked into the allosteric site of MALT1, the protein was shown by vacuum electrostatics potential (C), and cartoon (D). The inhibitors were shown in stick model representation with different colors of carbon skeleton: MLT-747 was shown in magenta and paliperidone was shown in yellow. Compared 2D interaction of MALT1 with MLT-747 (E) or paliperidone (F). Green dotted lines represented the hydrogen bonding.

Hydrogen bond parameters of MALT1 and MTL-747/paliperidone.
| Compounds | Donors atom | Receptor atom | Distances (Å)a |
|---|---|---|---|
| MTL-747 | Asn393:ND2 | MTL-747:O9 | 2.94 |
| MTL-747:N11 | Glu397:OE1 | 2.77 | |
| MTL-747:N25 | Glu397:OE1 | 3.17 | |
| Paliperidone | Asp716:N | Paliperidone:F16 | 2.91 |
| Paliperidone:O30 | Arg576:O | 3.03 |
The length of the hydrogen bonds.
Fig. 4C shows that MLT-747 was bound to the hydrophobic region between the caspase and Ig3 domains of MALT1. The methoxy-containing substituent formed a hydrogen bond interaction with residue Asn393, and the nitrogen of the central urea moiety formed a hydrogen bond interaction with residue Glu397 (Fig. 4E). Meanwhile, the hydroxyl group of paliperidone formed a hydrogen bond interaction with the carbonyl oxygen of residue Arg576, which was in the interhelix domain of MALT1. However, paliperidone was also bound to the lg3 domain of MALT1. That is, the residue Asp716 in the lg3 domain of MALT1 formed a hydrogen bond interaction with the fluorine atom in the fluoro-benzisoxazole moiety of paliperidone (Fig. 4F). These structural analyses revealed that paliperidone efficiently occupied the allosteric pocket as MLT-747 and thereby equally stabilized the inactive conformation.
Compound reversible combination with MALT1
The covalent binding of an active inhibitor, such as MI-2 and Z-VRPR-FMK, to Cys464 of MALT1 inhibits proteolytic activation. The reported allosteric inhibitor initiated a non-covalent interaction with the allosteric pocket and inhibited the proteolytic activation of MALT1. We performed a washout experiment in Jurkat cells to validate the reversible inhibition of MALT1 proteolytic activation by the proposed paliperidone. The Jurkat cells were incubated with paliperidone for 12 h. Afterward, the compounds were removed, and the cells were fully washed with PBS. Finally, the cells were incubated with a complete medium and then stimulated with PMA/ionomycin. The protein levels of the MALT1 substrate CYLD in the cells were detected, and the cleavage of CYLD was restored after the compound paliperidone was removed (Fig. 5). These results verified that the blockage of MALT1 proteolysis activation can be reversible after the inhibitors were removed. The inhibition of MALT1 proteolytic activation by paliperidone is therefore regulatable.
Fig. 5. The inhibition of MALT1 substrate CYLD Cleavage is reversible. Paliperidone was washed out with PBS and incubated with complete medium for 2 h before PMA/ionomycin treatment for 2 h. Immunoblots with anti-CYLD antibody showing the full length and cleavage of CYLD. Histogram showed quantification of immunoblot that normalized with β-actin. Data was shown as mean ± SD, *P < 0.05.

Retained stability of conformations of MALT1 and paliperidone during MD simulations
We performed 100 ns MD simulations by using Gromacs with an explicit solvent to gain insights into the energy change of the system after reaching equilibrium. The starting structure of protein was rebuilt by the aforementioned homology model. The initial conformation of paliperidone was obtained from the optimal pose of the molecular docking simulation. RMSD values are an important parameter in assessing the stability of a protein–ligand complex. Low RMSD values indicate stability of an equilibrated system. As shown in Fig. 6A, the MALT1 and paliperidone structures acquired good equilibrium in 100.0 ns, and their RMSD values were below 0.25 nm. Furthermore, the mean RMSF values (Fig. 6B) and B factors were calculated and added to the reference structure (B-factor structure) to estimate the structural flexibility. Then, we colored the B-factor structure file in accordance with the B factors and checked the flexible area (Fig. 6C). Fig. 6B shows that the overall stability can be maintained in the protein, especially the activity-related residues His415 and Cys464, except for the residue Glu500 site. Fig. 6C also supports this finding.
Fig. 6. Schematic drawing of the RMSD, RMSF, B-factor structure during the 100 ns MD simulations. (A) RMSD plots of MALT1 and paliperidone in 100 ns simulation. (B) RMSF plot of the residues of MALT1 in 100 ns simulation. The residues C464 and Q494 were shown as green triangle. The residues 491–498 in elbow loop were colored by pink. (C) Secondary structure of MALT1 was colored by B factors. Red represents a stable conformation, and blue represents a fluctuating conformation. Glu500 was shown by the stick model with cyan carbon skeleton. (D) Superposition of MALT1 in inactive state (cyan) and in active state (grey). The elbow loop of MALT1 was colored pink in inactive state and orange in active state.

A short elbow loop (residues 491–498) on loop L3 (residues 490–509) was formed in active state of the reported MALT1 but was not present in the inactive state of MALT1. This elbow loop contributes substantially to the stabilization of the active conformation, as it is engaged in a network of hydrogen bonds and charge–charge interactions with loop L2 (residues 463–485). Although the highly flexible residue Glu500 was also on loop L3, the elbow loop structure in an inactive state was stable (Fig. 6D). Perhaps the flexible swing of one residue is not sufficient to change the state of the catalytically active site.
Inactive conformation state of the catalytically active site of MALT1 in complex with allosteric inhibitor
In this study, we compared the conformation difference of MALT1 in active and inactive states. As Fig. 7A shows, catalytic dyad C464–H415 is exposed to the catalytically active site of MALT1 when the conformation is in the active state. This open state of the active pocket facilitates the entry of the substrates. However, in the inactive state, the catalytically active site of MALT1 is covered by the loop L3 (Fig. 7B). We found that Q494 plays a crucial role in protease active and inactive state transitions. In the active conformation of MALT1, the amide group of Q494 moves toward the dimer interface, thereby causing residues 491–498 to form a short elbow loop. On the contrary, the residues 491–498 lose the short elbow loop, and the S1 pocket is occupied by the side chain of Q494, which blocks access of potential substrates when MALT1 is in the inactive form. The distance between the oxygen atom of the carboxycarbonyl group on Q494 and the sulfur atom of the sulfhydryl group on C464 was changed from 4.13 Å in the active state (PDB ID 3V4O) to 10.54 Å in the inactive state (PDB ID 6F7I) (Fig. 7C). This movement causes changes between the nitrogen atom of the amide group on R465 and the oxygen atom of the amide group on Q494 (from 14.69 Å in the active state [PDB ID 3V4O] to 5.40 Å in the inactive state [PDB ID 6F7I]) (Fig. 7D). In addition, residue L480 on loop L2 exhibits a significant change in distance from L446 (from 14.24 Å in the active state [PDB ID 3V4O] to 3.38 Å in the inactive state [PDB ID 6F7I]) (Fig. 7E). Table 3 provides the detailed data. The abovementioned minimum distances between the atoms were calculated during the 100 ns MD simulation (Fig. 7F). We found that these distances in MALT1–paliperidone complex are consistent with the inactive conformation. Therefore, the catalytically active site of MALT1 in complex with an allosteric inhibitor remains in inactive conformation.
Fig. 7. Paliperidone maintained the inactive conformation of the catalytically active region of MALT1. Black circle represents the catalytically active region. MALT1 was shown by vacuum electrostatics potential. (A) The catalytically active site region is open in active state, (B) and is closed in inactive state. Superimposition of MALT1 in active state (green) with in inactive state (cyan). The distances of C464–Q494 (C), R465–Q494 (D) and L446–L480 (E) were displayed in yellow dotted lines. (F) The distance of C464–Q494, R465–Q494 and L446–L480 in MALT1–paliperidone complex during the 100 ns MD simulations.

Comparison of the selected distances in MALT1 catalytically active sites at the active/inactive conformational state.
| Protein state | PDB ID | Inhibitor properties | Inhibitor | C464:SG–Q494:Oa | R465:NH2–Q494:OE1a | L446:CD2–L480:CD1a |
|---|---|---|---|---|---|---|
| Active | 3V4O | Active inhibitor | Oligopeptide | 10.54 | 5.40 | 3.38 |
| Active | 4I1P | Active inhibitor | Oligopeptide | 10.77 | 5.91 | 3.47 |
| Inactive | 6F7I | Allosteric inhibitor | MLT-747 | 4.13 | 14.69 | 14.24 |
| Inactive | 6H4A | Allosteric inhibitor | MLT-748 | 3.51 | 15.19 | 15.73 |
| Inactive | 3V55 | — | — | 5.01 | 13.41 | 15.75 |
The length between two atoms (Å).
Conclusion
MALT1 inhibitors are potential immunomodulatory and antitumor drugs. The reported inhibitors bind with different regions in MALT1. Inhibitors that bind to the catalytically active site of MALT1 prevent cleavage of the substrate, whereas compounds that enter the allosteric site prevent the transition of MALT1 from inactive to active conformation. The catalytically active sites of MALT1 perform sequence conservation with the caspase family. The allosteric site binding inhibitor acts as a unique tool that affects the proteolytic activity of MALT1 without blocking the catalytically active site.
In this work, we developed selective and reversible MALT1 allosteric inhibitors from the FDA Approved Drugs Database using an in silico–in vitro approach. Paliperidone, a novel and effective compound, was screened by computer methods, such as virtual screening, molecular docking, and molecular dynamics. In vitro experiments confirmed that paliperidone blocks the cleavage of the MALT1 substrate CYLD. A washout experiment was performed in Jurkat cells to demonstrate that paliperidone reversibly inhibits MALT1 proteolytic activity. Therefore, paliperidone's inhibition of the proteolytic activity of MALT1 is regulatable. This finding is significant for the development of effective and selective drugs for the treatment of MALT1-driven cancer or autoimmune diseases.
Funding
The authors gratefully acknowledge financial support from the National Natural Science Foundation of China (No. 81603459). The 13th Five-Year Scientific Research Project of the Education Department of Jilin Province (No. 3D5196742412).
Conflicts of interest
The authors have no conflicts of interest with the content of this article.
Supplementary Material
Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07036b
References
- Staal J. Driege Y. Bekaert T. Demeyer A. Muyllaert D. Van Damme P. Gevaert K. Beyaert R. T-cell receptor-induced JNK activation requires proteolytic inactivation of CYLD by MALT1. EMBO J. 2011;30:1742–1752. doi: 10.1038/emboj.2011.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coornaert B. Baens M. Heyninck K. Bekaert T. Haegman M. Staal J. Sun L. Chen Z. J. Marynen P. Beyaert R. T cell antigen receptor stimulation induces MALT1 paracaspase-mediated cleavage of the NF-kappaB inhibitor A20. Nat. Immunol. 2008;9:263–271. doi: 10.1038/ni1561. [DOI] [PubMed] [Google Scholar]
- Hailfinger S. Nogai H. Pelzer C. Jaworski M. Cabalzar K. Charton J.-E. Guzzardi M. Décaillet C. Grau M. Dörken B. Lenz P. Lenz G. Thome M. Malt1-dependent RelB cleavage promotes canonical NF-κB activation in lymphocytes and lymphoma cell lines. Proc. Natl. Acad. Sci. U. S. A. 2011;108:14596–14601. doi: 10.1073/pnas.1105020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAuley J. R. Bailey K. M. Ekambaram P. Klei L. R. Kang H. Hu D. Freeman T. J. Concel V. J. Hubel N. E. Lee J. L. Klei H. B. Cheng J. Sekar P. Bridwell R. E. Covic L. Lucas P. C. McAllister-Lucas L. M. MALT1 is a critical mediator of PAR1-driven NF-kappaB activation and metastasis in multiple tumor types. Oncogene. 2019 doi: 10.1038/s41388-019-0958-4. [DOI] [PubMed] [Google Scholar]
- Gehring T. Erdmann T. Rahm M. Grass C. Flatley A. O'Neill T. J. Woods S. Meininger I. Karayel O. Kutzner K. Grau M. Shinohara H. Lammens K. Feederle R. Hauck S. M. Lenz G. Krappmann D. MALT1 Phosphorylation Controls Activation of T Lymphocytes and Survival of ABC-DLBCL Tumor Cells. Cell Rep. 2019;29:873–888. doi: 10.1016/j.celrep.2019.09.040. [DOI] [PubMed] [Google Scholar]
- Nagel D. Spranger S. Vincendeau M. Grau M. Raffegerst S. Kloo B. Hlahla D. Neuenschwander M. Peter von Kries J. Hadian K. Dorken B. Lenz P. Lenz G. Schendel D. J. Krappmann D. Pharmacologic inhibition of MALT1 protease by phenothiazines as a therapeutic approach for the treatment of aggressive ABC-DLBCL. Cancer Cell. 2012;22:825–837. doi: 10.1016/j.ccr.2012.11.002. [DOI] [PubMed] [Google Scholar]
- Jaworski M. Thome M. The paracaspase MALT1: biological function and potential for therapeutic inhibition. Cell. Mol. Life Sci. 2016;73:459–473. doi: 10.1007/s00018-015-2059-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontan L. Qiao Q. Hatcher J. M. Casalena G. Us I. Teater M. Durant M. Du G. Xia M. Bilchuk N. Chennamadhavuni S. Palladino G. Inghirami G. Philippar U. Wu H. Scott D. A. Gray N. S. Melnick A. Specific covalent inhibition of MALT1 paracaspase suppresses B cell lymphoma growth. J. Clin. Invest. 2018;128:4397–4412. doi: 10.1172/JCI99436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T. Connolly P. J. Philippar U. Sun W. Cummings M. D. Barbay K. Gys L. Van Nuffel L. Austin N. Bekkers M. Shen F. Cai A. Attar R. Meerpoel L. Edwards J. Discovery and optimization of a series of small-molecule allosteric inhibitors of MALT1 protease. Bioorg. Med. Chem. Lett. 2019;29:126743. doi: 10.1016/j.bmcl.2019.126743. [DOI] [PubMed] [Google Scholar]
- Bardet M. Unterreiner A. Malinverni C. Lafossas F. Vedrine C. Boesch D. Kolb Y. Kaiser D. Gluck A. Schneider M. A. Katopodis A. Renatus M. Simic O. Schlapbach A. Quancard J. Regnier C. H. Bold G. Pissot-Soldermann C. Carballido J. M. Kovarik J. Calzascia T. Bornancin F. The T-cell fingerprint of MALT1 paracaspase revealed by selective inhibition. Immunol. Cell Biol. 2018;96:81–99. doi: 10.1111/imcb.1018. [DOI] [PubMed] [Google Scholar]
- Scott D. A. Hatcher J. M. Liu H. Fu M. Du G. Fontan L. Us I. Casalena G. Qiao Q. Wu H. Melnick A. Gray N. S. Quinoline and thiazolopyridine allosteric inhibitors of MALT1. Bioorg. Med. Chem. Lett. 2019;29:1694–1698. doi: 10.1016/j.bmcl.2019.05.040. [DOI] [PubMed] [Google Scholar]
- Fontan L. Yang C. Kabaleeswaran V. Volpon L. Osborne M. J. Beltran E. Garcia M. Cerchietti L. Shaknovich R. Yang S. N. Fang F. Gascoyne R. D. Martinez-Climent J. A. Glickman J. F. Borden K. Wu H. Melnick A. MALT1 small molecule inhibitors specifically suppress ABC-DLBCL in vitro and in vivo. Cancer Cell. 2012;22:812–824. doi: 10.1016/j.ccr.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesmann C. Leder L. Blank J. Bernardi A. Melkko S. Decock A. D'Arcy A. Villard F. Erbel P. Hughes N. Freuler F. Nikolay R. Alves J. Bornancin F. Renatus M. Structural determinants of MALT1 protease activity. J. Mol. Biol. 2012;419:4–21. doi: 10.1016/j.jmb.2012.02.018. [DOI] [PubMed] [Google Scholar]
- Schlauderer F. Lammens K. Nagel D. Vincendeau M. Eitelhuber A. C. Verhelst S. H. Kling D. Chrusciel A. Ruland J. Krappmann D. Hopfner K. P. Structural analysis of phenothiazine derivatives as allosteric inhibitors of the MALT1 paracaspase. Angew. Chem., Int. Ed. 2013;52:10384–10387. doi: 10.1002/anie.201304290. [DOI] [PubMed] [Google Scholar]
- Quancard J. Klein T. Fung S. Y. Renatus M. Hughes N. Israel L. Priatel J. J. Kang S. Blank M. A. Viner R. I. Blank J. Schlapbach A. Erbel P. Kizhakkedathu J. Villard F. Hersperger R. Turvey S. E. Eder J. Bornancin F. Overall C. M. An allosteric MALT1 inhibitor is a molecular corrector rescuing function in an immunodeficient patient. Nat. Chem. Biol. 2019;15:304–313. doi: 10.1038/s41589-018-0222-1. [DOI] [PubMed] [Google Scholar]
- Ashburn T. T. Thor K. B. Drug repositioning: identifying and developing new uses for existing drugs. Nat. Rev. Drug Discovery. 2004;3:673–683. doi: 10.1038/nrd1468. [DOI] [PubMed] [Google Scholar]
- Franks M. E. Macpherson G. R. Figg W. D. Thalidomide. Lancet. 2004;363:1802–1811. doi: 10.1016/S0140-6736(04)16308-3. [DOI] [PubMed] [Google Scholar]
- Goldstein I. Lue T. F. Padma-Nathan H. Rosen R. C. Steers W. D. Wicker P. A. Oral sildenafil in the treatment of erectile dysfunction. N. Engl. J. Med. 1998;338:1397–1404. doi: 10.1056/NEJM199805143382001. [DOI] [PubMed] [Google Scholar]
- Jorenby D. E. Leischow S. J. Nides M. A. Rennard S. I. Johnston J. A. Hughes A. R. Smith S. S. Muramoto M. L. Daughton D. M. Doan K. A controlled trial of sustained-release bupropion, a nicotine patch, or both for smoking cessation. N. Engl. J. Med. 1999;340:685–691. doi: 10.1056/NEJM199903043400903. [DOI] [PubMed] [Google Scholar]
- Steiner M. Steinberg S. Stewart D. Carter D. Berger C. Reid R. Grover D. Streiner D. Fluoxetine in the treatment of premenstrual dysphoria. N. Engl. J. Med. 1995;332:1529–1534. doi: 10.1056/NEJM199506083322301. [DOI] [PubMed] [Google Scholar]
- Ekins S. Williams A. J. Krasowski M. D. Freundlich J. S. In silico repositioning of approved drugs for rare and neglected diseases. Drug Discovery Today. 2011;16:298–310. doi: 10.1016/j.drudis.2011.02.016. [DOI] [PubMed] [Google Scholar]
- Biasini M. Bienert S. Waterhouse A. Arnold K. Studer G. Schmidt T. Kiefer F. Gallo Cassarino T. Bertoni M. Bordoli L. Schwede T. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:W252–W258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienert S. Waterhouse A. de Beer T. A. Tauriello G. Studer G. Bordoli L. Schwede T. The SWISS-MODEL Repository-new features and functionality. Nucleic Acids Res. 2017;45:D313–D319. doi: 10.1093/nar/gkw1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkert P. Biasini M. Schwede T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics. 2011;27:343–350. doi: 10.1093/bioinformatics/btq662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forli S. Huey R. Pique M. E. Sanner M. F. Goodsell D. S. Olson A. J. Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016;11:905–919. doi: 10.1038/nprot.2016.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M. Huey R. Lindstrom W. Sanner M. F. Belew R. K. Goodsell D. S. Olson A. J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantharam A. Kreutzberger A. J. B. Unraveling the mechanisms of calcium-dependent secretion. J. Gen. Physiol. 2019;151:417–434. doi: 10.1085/jgp.201812298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best R. B. Zhu X. Shim J. Lopes P. E. Mittal J. Feig M. Mackerell Jr A. D. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J. Chem. Theory Comput. 2012;8:3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuttelkopf A. W. van Aalten D. M. PRODRG: a tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- Berendsen H. Grigera J. Straatsma T. The missing term in effective pair potentials. J. Phys. Chem. 1987;91:6269–6271. doi: 10.1021/j100308a038. [DOI] [Google Scholar]
- Lim S. M. Jeong Y. Lee S. Im H. Tae H. S. Kim B. G. Park H. D. Park J. Hong S. Identification of beta-Lapachone Analogs as Novel MALT1 Inhibitors To Treat an Aggressive Subtype of Diffuse Large B-Cell Lymphoma. J. Med. Chem. 2015;58:8491–8502. doi: 10.1021/acs.jmedchem.5b01415. [DOI] [PubMed] [Google Scholar]
- Hachmann J. Snipas S. J. van Raam B. J. Cancino E. M. Houlihan E. J. Poreba M. Kasperkiewicz P. Drag M. Salvesen G. S. Mechanism and specificity of the human paracaspase MALT1. Biochem. J. 2012;443:287–295. doi: 10.1042/BJ20120035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.