

Abstract
The ligand binding to protein and host–guest interactions are ubiquitous for molecular recognition. In drug design, the ligand binding to the active site of proteins is influenced by the charge density distribution and the electrostatic interactions of ligands and the nearby amino acids of the protein. The charge density analyses of ligand–protein complexes need accurate positions of hydrogen atoms and their valence electron distribution and the fine structure of proteins. Such information cannot be obtained from the conventional protein X-ray crystallography analysis in the resolution range of 1.5 to 3.5 Å. This can be realized from QM/MM based structure and charge density analysis of estrogens with the estrogen receptor. The charge density properties such as electron density, Laplacian of electron density and electrostatic properties of estrogens in the presence of active site amino acid residues have been determined and compared with the isolated estrogen molecules from theory and experimental. The present study reveals the chemical bonding nature of estrogen molecules and the strength of the intermolecular interactions in the active site of estrogen receptor, and also the importance of π⋯π interactions between the estrogens and Phe404 amino acid residue and protonation state of His524 amino acid residue have been identified using electrostatic potential maps. The difference in the electrostatic potential map of estrogens displays the hormone dependent actions of estrogen receptor. This method is very helpful to derive the charge density distribution of macromolecules to understand their biological recognition and interactions.
The ligand binding to protein and host–guest interactions are ubiquitous for molecular recognition.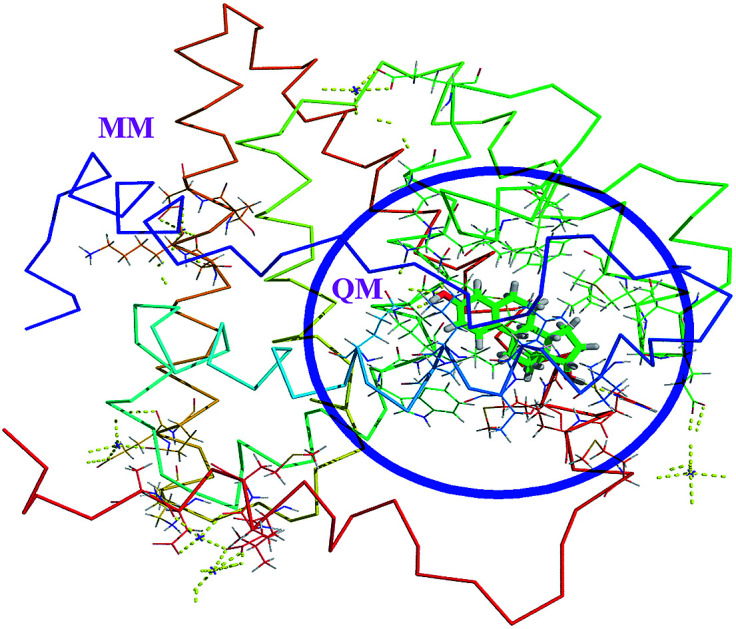
Introduction
In medicinal chemistry, the structure activity relationship and molecular recognition of drugs in the receptor at an electronic level is very much important for pharmacological drug design. The molecular recognition process is subject to intermolecular interactions such as steric and also the complementary electrostatic interactions.1,2 The intermolecular interactions are well characterized by the charge density analysis from X-ray diffraction techniques. The small molecule charge density analysis at high resolution is a plausible one. The charge density gives information about the bonding and the topology of electron density of intermolecular interactions and the strength of interactions which are requisite for molecular recognition process.3–5 In protein structures, the position of hydrogen atoms and their valence electron distributions are very important to understand the physical and chemical properties, because the hydrogen atoms are essential for the intermolecular interactions and the stability of the structure. To model the charge density distribution of molecules high resolution X-ray diffraction intensity data (1.0 Å) is required. As in the case of crystals of small molecules, the charge density analysis from the sub-atomic resolution X-ray diffraction data allows an understanding of the topological properties of electron densities of molecules. Such kinds of experimental charge density analysis have also been performed for the crystals of ligand–protein complexes, which gives the information about the topological and electrostatic properties of ligand–protein complexes.6–12 In recent years, the charge density analyses of ligand–protein complexes have been reported at sub-atomic resolution such as Crambin (0.54 Å),6 antifreeze protein (0.62 Å),7 human aldose reductase (0.66 Å),8 rubredoxin (0.69 Å),9 syntenin (0.73 Å)10 and lysozyme (0.65 Å).11 Interestingly, Hirano and co-workers reported the charge density analysis of metallo proteins at an ultra-high resolution (0.48 Å).12 This is possible because of the advent of advanced X-ray diffractometers and the availability of synchrotron radiation sources, which attains sub-atomic resolution.13,14 However, the protein structures at sub-atomic resolution are very much limited due to the low quality of the crystals, disorder, solvent effects and also the limitation of synchrotron radiation sources. To overcome these shortcomings, several methods are proposed to derive the electron density distribution at high resolution itself, namely invariom refinement, theoretical structure factor from the wave function and QM/MM methods.15–17 The present study explores the charge density distribution of ligand molecules in the binding pocket as a QM region and the remaining part of the amino acids as the MM region. The present charge density study of ligand–protein complexes is the model analysis of the binding pocket of estrogen receptors using QM/MM energy minimization coupled with the QTAIM study.
Estrogens are the hormones, which are essential for the development and maintenance of female reproductive system, responsible for the secondary female sex characteristics. And also it is present in the other tissues like bone, liver, uterus, skin and brain. The major estrogens are estrone (E1), estradiol (E2) and estriol (E3). These estrogens exert their function by binding to the estrogen receptors (ER). In the agonist position, the hormone molecule binds with the Helix3 (H3), H6 and H11 and closes the ligand binding domain (LBD); this could be the result of activation or repression of the genes.18–25 The activated complex then binds to the DNA, i.e., estrogen response elements and then cell dimerization occurs. In some cases, in the presence of DNA genetic errors leads to the rapid cell proliferation and also the genotoxic effect of estrogen metabolites damages the DNA causing cellular transformation and tumor cell growth.26–30 However, the exact mechanism of the estrogen in the initiation of breast cancer still to be elucidated. To elucidate the exact mechanism and the role of estrogen molecules in the breast cancer, the knowledge of the intermolecular interactions, electronic structure and the electrostatic potential need to be understood. The experimental electron density analysis of several hormone molecules such as estrone, 17β-estradiol, 17α-estradiol, estriol and synthetic steroid hormone molecules such as genistein and diethylstilbestrol are reported from crystallographic studies. The present study is mainly focused to understand the charge density distribution of estrone, 17β-estradiol and estriol molecules and their interactions with the active site amino acids of estrogen receptor. The charge density study on the above said estrogen–receptor complexes have been performed from the QM/MM calculation coupled31 with the Bader's quantum theory of atoms in molecules.32 The detailed topological properties of electron density and the electrostatic properties of these estrogen molecules have been discussed.
Materials and methods
The QM/MM method
The hybrid QM/MM methods were proposed and developed by Warshel and Levitt.31 In the QM/MM approach, the ligand and protein molecule are separated by two domains. The ligand molecule associated with the small region (active site) of protein in the ligand–protein complex is considered as QM region and the remaining part of the complex is considered as MM region (Fig. 1). The QM region was treated by quantum mechanical methods, whereas the MM region was treated by MM force fields. These two regions are connected through the electrostatic interactions. Then the Hamiltonian of the hybrid QM/MM is expressed as,33,34
| H = HQM + HMM + HQM/MM | 1 |
Fig. 1. The QM/MM scheme of estrogens. The ligand containing active site amino acids are considered as QM region. The remaining amino acids and solvent molecules are considered as MM region.

The effective Hamiltonian consists of three terms, first one is the QM region, the second the MM region and last one is the interaction region. The interaction term is the interaction between the MM point charges with electrons in the QM system and also interaction between MM charges with the QM nuclei. In which, the interactions are not covalent one, but in which the Hamiltonian is the sum of electrostatic and Lennard Jones potential.
Computational details
The initial structure of hormone molecules estrone, 17β-estradiol and estriol are drawn using ChemDraw software. Then these structures were optimized using B3LYP method35 with 6-311G** basis set36 in Gaussian03 software.37 To perform docking study, the human estrogen receptor protein ligand binding domain has been downloaded from the RCSB PDB with the PDB id 3ERT with the resolution of 1.9 Å.38 During the protein preparation, all the crystallographic water molecules were retained; the hydrogen atom and kollman charges were added to the protein using AutoDockTools. The grid size of dimension 70 × 62 × 74 Å has been constructed with the grid spacing of 0.375 Å. Further, the Lamarckian algorithm has been used to perform the flexible molecular docking of estrogen molecules with the estrogen receptor-α (ERα) using AutoDock 4.2 package.39 Among the different conformers obtained, the lowest docked conformer has been chosen for further QM/MM study. The QM/MM study of ERα with the estrogen molecule complexes were performed using AmberTools14 (ref. 40–42) with the help of Amberff14SB force filed.43 The active site amino acid residues Glu353, Leu387, Leu391, Met388, Leu428, Ile424, Arg394, Met421, Gly521, Leu525, His524, Leu384, Leu346, Phe404 along with the estrogen molecule were considered as the QM region to achieve better approximation; whereas, the other amino acid residues and solvents were treated as MM region using MM force fields. The complex has been immersed in the solvent using TIP3P water box44,45 at 8 Å and 3 Na+ ions has been added to this mesh to neutralize the charges of the whole system. The QM cutoff was fixed as in the non-bonded cutoff region at 8 Å. The minimization of entire system has been performed for the 2000 steps, in which the first 500 steps the steepest descent minimization, after 500 steps the minimization was switched to steepest descent to conjugate gradient method. During the entire minimization, the SHAKE algorithm has not introduced and as for the interaction terms the Ewald and particle mesh Ewald methods46 were used for the long range electrostatics. The QM restraint has been applied for the Glu535, Arg394, His524 and HOH2 molecules with the restraint weight equal to 1 for the entire QM/MM minimization. The semi-empirical PM3 method was used for the QM calculations, whereas, for the MM region Amberff14SB force filed was used.
Results and discussion
Molecular docking studies
To explore the estrogenic activity of estrogen molecules such as estrone, 17β-estradiol and estriol (Fig. 2), the docking analysis has been carried out with the ERα using AutoDock software39 and the corresponding binding free energy of 10 different conformers are given in Table 1. Among the different conformers obtained, the lowest docked energy conformer of estradiol (conformer-2), estrone (conformer-2) and estriol (conformer-1) has been selected for the analysis of intermolecular interaction and QM/MM calculations. The active site amino acid residues interact with the three hormone molecules are found to be same and are: Glu353, Leu387, Leu391, Met388, Leu428, Ile424, Arg394, Met421, Gly521, Leu525, His524, Leu384, Leu346 and Phe404. All the three molecules bind in the cavity of Helix H3, H5 and H6 and forms hydrogen bonding interactions with the Glu353, Arg394, His524 and HOH2.38 The intermolecular interactions of estrogen molecules with the estrogen receptor are shown in Table 2.
Fig. 2. The chemical structure and energy minimized structure with atom numbering scheme of estrogens.

Binding free energy (kcal mol−1) of estrogens from molecular docking analysis.
| Conformers | 17β-Estradiol | Estriol | Estrone |
|---|---|---|---|
| 1 | −9.40 | −8.28 | −9.20 |
| 2 | −9.40 | −8.35 | −9.20 |
| 3 | −9.40 | −8.35 | −9.20 |
| 4 | −9.38 | −8.32 | −9.20 |
| 5 | −9.40 | −8.30 | −8.62 |
| 6 | −9.40 | −7.56 | −9.20 |
| 7 | −9.39 | −8.29 | −9.20 |
| 8 | −9.40 | −8.34 | −9.20 |
| 9 | −9.40 | −8.21 | −9.21 |
| 10 | −9.39 | −8.35 | −9.20 |
The intermolecular interactions of estrogen and ERα complex.
| Amino acid residue | 17β-Estradiol | Distance (Å) | Estrone | Distance (Å) | Estriol | Distance (Å) |
|---|---|---|---|---|---|---|
| Hydrogen bonding interactions | ||||||
| Glu353 | H(6)⋯Glu353(OE2) | 1.7 | H(6)⋯Glu353(OE2) | 1.7 | H(6)⋯Glu353(OE2) | 2.4 |
| H(6)⋯Glu353(OE1) | 2.5 | |||||
| Arg394 | O(1)⋯Arg394(2HH2) | 2.2 | O(1)⋯Arg394(2HH2) | 2.1 | O(1)⋯Arg394(2HH2) | 2.2 |
| His524 | H(3)⋯His524 (ND1) | 2.2 | O(2)⋯His524(HE2) | 2.5 | O(2)⋯His524 (HE2) | 1.7 |
| Gly521 | — | — | — | — | H(3)⋯Gly521(O) | 2.5 |
| HOH2 | O(1)⋯HOH(H1) | 2.6 | O(1)⋯HOH(H1) | 2.8 | O(1)⋯HOH(H1) | 2.0 |
| Water-bridge bond interactions | ||||||
| HOH2 | HOH2(O)⋯Arg394(2HH1) | 1.9 | HOH2(O)⋯Arg394(1HH1) | 2.6 | HOH2(H2)⋯Glu353(OE2) | 2.0 |
| HOH2(O)⋯Arg394(2HH2) | 2.9 | HOH2(O)⋯Arg394(2HH2) | HOH2(O)⋯Arg394(1HH1) | 2.6 | ||
| HOH2(H2)⋯Glu353(OE1) | 2.7 | HOH2(H2)⋯Glu353(OE2) | 2.0 | HOH2(H1)⋯Leu387(O) | 2.0 | |
| HOH2(H1)⋯Leu387(O) | 3.1 | HOH2(H1)⋯Leu387(O) | 2.0 | |||
| Hydrophobic interactions | ||||||
| Glu353 | H(5)⋯Glu353(OE2) | 2.6 | H(5)⋯Glu553(OE1) | 3.9 | H(5)⋯Glu553(OE1) | 1.6 |
| Leu391 | C(5)⋯Leu391(H07) | 3.4 | C(5)⋯Leu391(H07) | 3.1 | C(5)⋯Leu391(H07) | 3.3 |
| C(5)⋯Leu391(H06) | 2.7 | C(5)⋯Leu391(H06) | 2.9 | C(5)⋯Leu391(H06) | 3.7 | |
| C(5)⋯Leu391(H03) | 2.3 | |||||
| C(5)⋯Leu391(HN) | 3.1 | |||||
| Leu387 | O(1)⋯Leu387(H01) | 3.4 | O(1)⋯Leu387(H01) | 3.4 | O(1)⋯Leu387(H01) | 3.5 |
| O(1)⋯Leu387(H03) | 3.0 | H(5)⋯Leu387(O) | 3.7 | |||
| Met388 | C(9)⋯Met388(H01) | C(9)⋯Met388(H01) | 2.6 | C(9)⋯Met388(H05) | 2.4 | |
| H(10A)⋯Met388(SD) | C(9)⋯Met388(H04) | 2.9 | C(9)⋯Met388(H04) | 2.7 | ||
| H(10A)⋯Met388(CG) | C(10)⋯Met388(H06) | 2.8 | C(9)⋯Met388(H04) | 2.5 | ||
| C(10)⋯Met388(H07) | C(10)⋯Met388(H07) | 3.6 | C(10)⋯Met388(H02) | 2.8 | ||
| H(9A)⋯Met388(CE) | 2.7 | C(10)⋯Met388(H07) | 2.4 | |||
| H(9A)⋯Met388(CG) | 3.1 | C(10)⋯Met388(H04) | 3.1 | |||
| H(10A)⋯Met388(CA) | 3.0 | H(9A)⋯Met388(CG) | 2.5 | |||
| H(10A)⋯Met388(CG) | 2.8 | H(9A)⋯Met388(SD) | 3.5 | |||
| H(10A)⋯Met388(CB) | 3.0 | |||||
| Arg394 | O(1)⋯Arg394(NH2) | 3.1 | O(1)⋯Arg394(NH2) | 2.4 | O(1)⋯Arg394(NH2) | 2.6 |
| Met421 | H(16B)⋯Met421(CE) | 2.9 | H(16B)⋯Met421(CE) | 3.0 | H(16)⋯Met421(CE) | |
| H(16B)⋯Met421(SD) | 3.1 | H(16B)⋯Met421(SD) | 2.4 | H(16)⋯Met421(SD) | ||
| H(17A)⋯Met421(SD) | 2.6 | H(16A)⋯Met421(SD) | 3.1 | C(17)⋯Met421(H01) | ||
| H(17A)⋯Met421(CG) | 2.2 | O(2)⋯Met421(H07) | 2.7 | H(17A)⋯Met421(CE) | ||
| Ile424 | H(17B)⋯Ile424(CD1) | 2.3 | H(17B)⋯Ile424(CD1) | 2.9 | H(16B)⋯Ile424(CD1) | 2.2 |
| Gly521 | H(16A)⋯Gly521(O) | H(16A)⋯Gly521(O) | 3.1 | O(3)⋯Gly521(H01) | 2.7 | |
| C(16)⋯Gly521(H01) | C(16)⋯Gly521(H07) | 2.9 | O(3)⋯Gly521(H03) | 2.3 | ||
| C(16)⋯Gly521(H0\2) | C(17)⋯Gly521(H01) | 3.1 | H(3)⋯Gly521(O) | |||
| Phe404 | C(10)⋯Phe404(H06) | 2.5 | C(10)⋯Phe404(H06) | 2.8 | C(10)⋯Phe404(H06) | |
| H(10A)⋯Phe404(CE1) | 2.5 | H(10A)⋯Phe404(CE1) | 3.1 | H(10A)⋯Phe404(CE1) | ||
| Leu525 | C(12)⋯Leu525(H06) | 2.7 | C(18)⋯Leu525(H09) | 3.4 | O(2)⋯Leu525(H01) | 1.9 |
| H(12A)⋯Leu525(CD2) | 2.8 | O(2)⋯Leu525(H09) | O(2)⋯Leu525(H09) | 2.8 | ||
| O(2)⋯Leu525(H09) | 2.0 | O(2)⋯Leu525(H08) | H(4)⋯Leu525(CD2) | 2.8 | ||
| H(3)⋯Leu525(HN) | H(15)⋯Leu525(CD1) | 2.6 | ||||
| Leu384 | C(18)⋯Leu384(H06) | 3.1 | C(9)⋯Leu384(H05) | 2.2 | H(14)⋯Leu384(CD1) | 2.7 |
| H(18C)⋯Leu384(CD2) | 3.5 | H(9A)⋯Leu384(CD1) | 2.3 | C(14)⋯Leu384(H05) | 2.9 | |
| Leu346 | C(9)⋯Leu346(H08) | 2.7 | H(11B)⋯Leu346(CD2) | 2.7 | C(18)⋯Leu346(H03) | 3.1 |
| H(9B)⋯Leu346(CD1) | 2.7 | C(11)⋯Leu346(H03) | 2.6 | C(18)⋯Leu346(H02) | ||
| H(14)⋯Leu346(CD1) | 3.1 | H(11B)⋯Leu346(CG) | 3.1 | |||
| H(11B)⋯Leu346(CB) | 3.3 | |||||
| H(12A)⋯Leu346(CD1) | 2.8 | |||||
| C(12)⋯Leu346(H10) | 3.2 | |||||
Estrone–ERα interactions
In E1 molecule, the hydroxyl group (OH) of aromatic A-ring forms strong hydrogen bonding interactions with the Glu353 (OE1, OE2 atoms) residue at the distance of 1.7, 2.5 Å. Similarly, the O-atom of hydroxyl group of A-ring form hydrogen bonding interaction with the Arg394 residue at the distance of 2.1 Å. In the D-ring, the O(2) atom forms hydrogen bonding interaction with the NH group of His524 amino acid at the distance of 2.5 Å. The E1 molecule also forms several hydrophobic interactions with the other amino acid residues and create the hydrophobic groove. And, the O-atom in the D-ring also forms hydrogen bonding interaction with the active site water molecule HOH2 at the distance of 2.8 Å. This water molecule acts as a bridge and forming a link between the ligand and ERα as ligand–HOH2–Arg394, ligand–HOH2–Glu353 at the distances of 2.6 and 2.0 Å respectively (Table 2). Apart from these interactions, several hydrophobic interactions also stabilize the ligand binding cavity in the ERα.
17β-Estradiol–ERα interactions
In the A-ring of the E2 molecule, the hydroxyl group H(6) atom forms strong hydrogen bonding interaction with the Glu353 at the distance of 1.7 Å. The O-atom in the hydroxyl group forms strong hydrogen bonding interaction with the Arg394 residue at the distance of 2.2 Å. In addition to this, the O-atom forms strong hydrogen bonding interaction with HOH2 molecule at the distance of 2.6 Å, in which the water molecule acts as a bridge between ligand–HOH2–Arg394, Glu353 amino acid residues. The H(3) atom of D-ring of the molecule forms strong hydrogen bonding interaction with the His524 residue (ND1 atom) at the distance of 2.5 Å. In the active site of estrogen receptor, two protonation state of His524 residue is observed. The two isoforms of His524 residue rotated appropriately to form stable interaction with the estrogens. In the case of E2 molecule, the estradiol molecule act as a hydrogen bond donor and His524 is an acceptor. Whereas for the E1 and E3 molecules, the ligand molecule acts as hydrogen bond acceptor and His524 residue is a donor. In addition to this, the aromatic ring of the E2 molecule forms π⋯π aromatic T-shaped interactions with the Phe404 amino acid residue at the distance of 4.83 Å; this is a very important interaction for the estrogenic activity. Apart from this, the A-ring of the molecule forms π⋯alkyl interactions with the amino acid residues Ala350, Met387, Met388, Met391, and the B-ring of the molecule forms π⋯alkyl interaction with the Met388, Met391 and Phe404 residues. This interaction is not observed between E1, E3 molecules with ERα. The estrogen molecules are highly hydrophobic in nature, hence they form several hydrophobic interactions with the active site residues of ERα and forms strong hydrophobic groove in the active site.
Estriol–ERα interactions
In the estriol molecule, the hydroxyl group H(6) atom of A-ring forms hydrogen bonding interaction with the Glu353 residue at the distance of 2.4 Å, the distance between the donor and acceptor is very large compared with the E1 and E2 molecules. The ligand–water–amino acid interactions (water molecule acts as a bridge) also found for the estriol molecule. Hence, this water molecule interaction is very essential for the stabilizing the active site of the ERα. In addition to this, the D-ring hydroxyl group connected at the C(16) position forms hydrogen bonding interaction with the Gly521 residue at the distance of 2.5 Å. The D-ring hydroxyl group in the C(15) position forms hydrogen bonding interaction with the His524 residue at the distance of 1.7 Å. This interaction is considered to be very strong interaction on compared with all other interactions.
QM/MM interactions
After the QM/MM minimization, the important intermolecular interactions between the estrogen molecules with the ERα have been compared with the docking studies and are presented in Table 3. The QM/MM minimized estrogen receptor complex and the corresponding interactions are shown in Fig. 3. The hydrogen bonding interactions between the hydroxyl group of aromatic A-ring of E1 and E2 molecules with the Glu353 residue is not altered (1.7 Å) and are remains same before and after the QM/MM minimization, whereas in E3 molecule this distance reduces to 1.8 Å. The hydrogen bonding interaction between the Arg394 residue with the O-atom of the hydroxyl group of aromatic ring of E1 and E2 molecules are 2.2 and 2.1 Å, respectively; these values are found slightly varying, notably in E1 it decreases to 1.9 Å, whereas in E3 molecule, it increases to 2.6 Å. Similarly, the His524 hydrogen bonding interactions also shows some difference for all molecules. In E1, the H-bond distance decreases from 2.5 to 1.8 Å; in E2 molecule, the variation is found to be small (2.2 to 2.3 Å), whereas in E3 molecule, similar trend has been observed, the variation is 1.7 to 1.9 Å. This kind of small variation also found for water (HOH2) molecule, which involved estrogen–HOH2–ERα interactions. This is trend is same in all estrogen–ERα complexes. The T-shaped π⋯π aromatic interaction in the E2 molecule also slightly decreases from 4.83 to 4.79 Å. This indicates that, after the QM/MM minimization, the intermolecular interactions between the estrone and estradiol molecules with ERα not much altered only slight variation in the interaction distances is observed, whereas in the case of estriol molecule, large variation has been observed. This indicates that the E1 and E2 molecules are more stable in the active site of estrogen receptor. However, among all hormone molecules, the E2 molecule shows the highest binding affinity towards ERα.
The intermolecular interactions of estrogens-ERα after QM/MM minimization.
| Amino acid residue⋯17β-estradiol | Distance (Å) | Amino acid residue⋯estrone | Distance (Å) | Amino acid residue⋯estriol | Distance (Å) |
|---|---|---|---|---|---|
| H(6)⋯Glu353(OE2) | 1.7 | H(6)⋯Glu353(OE2) | 1.7 | H(6)⋯Glu353(OE2) | 2.4 |
| 1.7 | 1.7 | 1.8 | |||
| H(6)⋯Glu353(OE1) | 2.5 | ||||
| 3.1 | |||||
| O(1)⋯Arg394(2HH2) | 2.2 | O(1)⋯Arg394(2HH2) | 2.1 | O(1)⋯Arg394(2HH2) | 2.2 |
| 1.9 | 1.9 | 2.6 | |||
| H(3)⋯His524 (ND1) | 2.2 | O(2)⋯His524 (HE2) | 2.5 | O(2)⋯His524 (HE2) | 1.7 |
| 2.3 | 2.3 | 1.9 | |||
| H(3)⋯Gly521(O) | 2.5 | ||||
| O(1)⋯HOH2(H1) | 2.6 | O(1)⋯HOH2(H1) | 2.8 | O(1)⋯HOH2(H1) | 2.0 |
| 2.8 | 2.8 | 2.4 | |||
| HOH2(O)⋯Arg394(2HH1) | 1.9 | HOH2(O)⋯Arg394(1HH1) | 2.6 | HOH2(O)⋯Arg394(1HH1) | 2.6 |
| 1.8 | 1.8 | 2.6 | |||
| HOH2(H2)⋯Glu353(OE1) | 2.7 | HOH2(H2)⋯Glu353(OE2) | 2.0 | HOH2(H2)⋯Glu353(OE2) | 2.0 |
| 3.0 | 2.6 | 1.8 | |||
| HOH2(H1)⋯Leu387(O) | 3.1 | HOH2(H1)⋯Leu387(O) | 2.0 | HOH2(H1)⋯Leu387(O) | 2.0 |
| 3.1 | 3.1 | 2.7 |
Fig. 3. The intermolecular interactions of (a) 17β-estradiol (b) estrone (c) estriol molecules with estrogen receptor after QM/MM minimization.

Molecular conformation
The geometry of estrogen molecules in the gas phase, docking structure (lifted from the active site of docking), QM/MM minimization were compared. In all estrogen molecules, in bond lengths, bond angles and torsion angles, there is not much variation has been found between the gas phase and the active site structure of estrogens obtained from the molecular docking. Whereas, the conformation of the estrogen molecules after QM/MM minimization, it has been largely varied (Table S1†). In E1 molecule, the C(17)–C(16)–C(15)–O(2) torsion angle of D-ring in the gas phase is −175.4°, which decreases to 145.2° in both molecular docking and QM/MM minimized structure; this indicates that the bonds are exhibit trans conformation in the gas phase, whereas in the active site (when the molecules present) and the torsional angles of these bonds are altered to anticlinal conformation this facilitates to form intermolecular interactions with the nearby amino acids present in the active site of estrogen receptor. In the case of E2 and E3 molecule, the torsion angle of C(18)–C(13)–C(15)–O(2) bond of the five membered D-ring shows a considerable variation and exhibit anticlinal conformation. The above results reveal that the conformation of D-ring of the three estrogens has been largely altered in docking and QM/MM minimized structures compared to the gas phase structure, this confirm that the five membered D-ring of the estrogen molecules play an important role in the active site of ERα as it strongly interact with the active site amino acids.
Charge density analysis
The Bader's quantum theory of atoms in molecules is a powerful tool to describe the charge density distribution of chemical bonds (QTAIM).32 From the charge density analysis, we can understand the chemical bonding nature, atomic interactions, reactivity and stability of molecular systems. From the Bader's QTAIM theory, the point where the first derivative of electron density vanishes is called critical point (CP) in that bond. A bond critical point (bcp), r,c is classified based on its rank and signature. The local curvatures are expanded as nine partial second derivatives in three dimensional space, this forms the Hessian matrix of ρ(r). The diagonalization of Hessian matrix gives the second derivative of electron density ∇2ρ(r), which provides the measure of local charge concentration. If ∇2ρ(r) > 0, the charges are locally depleted; if ∇2ρ(r) < 0, the charges are locally concentrated. The Laplacian of electron density at the bond critical point gives the measure of charge concentration/depletion at the bcp. The single point energy calculation has been performed for all estrogen molecules along with the amino acid residues around 4 Å has been included in the calculation to provide the polarization effect. This is the exact situation as in the protein environment with the ligand molecule, i.e., biologically active environment. The wave function obtained from the above calculations, the topological properties of estrogen molecules in the active site environment has been determined. The topological parameters such as electron density, Laplacian of electron density of estrogen molecules have been determined from the QM/MM minimization, which present in the active site amino acids environment. These charge density parameters are compared with the corresponding reported charge density parameters of estrogen molecules.47–50
Electron density
The electron density of QM/MM minimized structure of estrogen molecules are compared with the reported experimental and gas phase studies. The calculated electron density ρbcp(r) values of all the bonds agree with the reported values (Table S2†). The electron densities of C–C bonds of A, B, C rings of estrogens are not much altered for the QM/MM minimized molecules on compare with the reported experimental47–50 and the gas phase structures. Whereas, for the D-ring of E1 and E2 molecules, the electron density of C–C bonds has been varied considerably, which are relatively high on compare with E3 molecule. In E1, the maximum deviation of electron density value of C–C bonds are ∼0.2 eÅ−3, whereas for E2 molecule the difference is ∼0.1 eÅ−3; this variation may be due to the intermolecular interactions with the His524 residue; and the charges of these bonds are drifted. In the case of QM/MM minimized E3 molecule, all the C–C bonds only exhibit considerable variation on compared with the reported experimental structure.50 The ρbcp(r) value of C–O bonds in the E1 and E2 molecules are showing some difference (∼0.1 eÅ−3) when compare with the experimental values. In E1 molecule, the C(15) O(2) bond has high electron density (2.771 eÅ−3), whereas for E3 molecule, the electron density is 1.764 eÅ−3 and the difference is very small (∼0.07 eÅ−3) when compare with the experimental values. The above results confirm that, the intermolecular interactions in the active site residues are not much altered the electron density values of estrogen molecules.
Laplacian of electron density
The Laplacian of electron density of all the bonds of estrogen molecules determined from the QM/MM minimized structure are showing large difference when compare with the corresponding the experimental values (Table S2†).47–50 From the Laplacian of electron density, the charges in the A-ring are more concentrated than all other rings of the estrogen molecules, which indicate the π-character of the A-ring. On comparing the QM/MM minimized structures, the Laplacian of electron density of C–C bonds are almost equal for all the molecules, whereas the Laplacian of C–C bonds of the D-ring decreases on compare with the experimental and gas phase DFT studies, the difference is 0.02 eÅ−5. This variation may be due to the intermolecular interactions of D-ring with the neighboring amino acids present in the active site. The Laplacian of electron density of C–O bond of all the molecules are almost equal and comparable with the respective experimental values. In E1 molecule, the Laplacian of C O bond shows some discrepancy with the reported experimental values, this is attributed to the large difference in eigen values of this bond.51–55 The λ3 value is lower for all the bonds in the molecule except the C O bonds. And the λ3 value in theory is two times higher than the experimental value and λ1, λ2 values are almost equal in both theory and experiment; the contribution of λ3 to the Laplacian value leads to have small Laplacian of electron density for this bond. Among all the bonds, the C O bond is highly polarized and its bcp position is 16.03% away from the midpoint. The bond ellipticity values of aromatic A-ring of estrogens exhibit high ellipticity than all other rings in the molecule and the value is 0.22, which indicates that the π-electrons are fully delocalized and also the A-ring of the molecules are showing high electron density and high negative Laplacian values at the bcp.
Atomic charges and volume
Atomic charges were derived from the integration of atomic basin by the zero flux surfaces based on the Bader's quantum theory of atoms in molecules.32,56 From the atomic charges, the electrophilic and nucleophilic attack and possible hydrogen bonds i.e., donor/acceptor nature of the atoms can be well understood. The AIM derived charges56 and Mulliken Population Analysis57 (MPA) charges of estrogens isolated molecule in gas phase and QM/MM minimized structure are shown in Table S3.† Here, in the QM/MM minimized structure, the atomic charges (MPA and AIM) of almost all the atoms are slightly increased when compared to the isolated gas phase molecule. The difference is more pronounced in the case of polar groups and the corresponding linked atoms. The calculated MPA charges are similar to the monopole charges derived from the multipolar refinement. The aromatic hydroxyl group connected to the carbon atom (C(6)) of all the molecules exhibit high positive charge 0.5e and the corresponding bonded hydrogen atom H(6) also exhibit high positive charge, this due to the polarization of carbon and hydrogen atoms towards high electronegative oxygen atom O(1) (MPA: −0.37e, AIM: −1.07e). This trend also found in the QM/MM minimized structure, in which the charges are found to be increased and the difference is 0.1e, when compared to isolated molecule in the gas phase. This confirms that the polarization effect of amino acid residues which are well included in the calculation. The high negative charge of the O(1) atom act as a acceptor to form hydrogen bonding interaction with the Arg394 H atom (0.48e) and H atom of the water molecule (0.59e), whereas the positive charge of the H(6) atom act as a donor and attracts towards the Glu353 residue O-atoms (−1.18e). Similarly for E2 and E3 molecules, the high positive hydrogen atom H(3) (0.54e) of D-ring attracts towards the electronegative N-atom (−1.04e) of His524 residue. In E1 molecule, the C O group charges are very highly negative compared to all atoms in the molecule, the corresponding AIM charge of C(15) atom is 1.0e and the AIM charge of O(2) atom is −1.11e, this charges are slightly decreases for the QM/MM minimized structure. In E2 and E3 molecules, the charges of –OH group connected to the D-ring exhibits high polarization towards the carbon and hydrogen atoms which is high on compared to the A-ring –OH group charges. On comparing the gas phase and the QM/MM minimized structure of E2 molecule, in the latter case the charges of atoms have been increased largely; whereas the charges of E1 and E3 molecules are decreases, this may be due to involvement of their intermolecular interaction. The methyl group C(18) atom exhibits high negative charge and the corresponding hydrogen atoms typically carry neutral or positive charge.
The atomic volume is defined as the volume bounded by an electron density distribution and the interatomic surfaces of the atom.32 The atomic volume of estrogens for isolated molecule and for the QM/MM minimized structures are calculated (Table S3†) based on the density value of 0.01 au using AIM theory, these are good agreement with the experimentally reported structures. The carbon atom has volume range from 6 to 13 Å3 and hydrogen atoms have volume range from 2 to 8 Å3. The atomic volumes of the polar oxygen atoms has high volume compared to all other atoms in the molecule and the corresponding bonded hydrogen atom has less volume when compared to other hydrogen atoms in the molecule, because these hydrogen atoms are involving in intermolecular interactions. This effect reduces the volume approximately half of its original volume. Similar trend also found for H(3), H(4), H(6) atoms and this effect is more pronounced in the corresponding donor/acceptor atoms. The decreased volume and increased charge of hydrogen atoms is the criteria for hydrogen bonding expressed by the Koch and Popelier.58
Topological analysis of electron density of estrogens–ERα interactions
The intermolecular interactions between the estrogens with the estrogen receptor are analyzed using the AIM theory.32 The AIM theory is a powerful tool to analyze the strength of intermolecular interactions and the type of interactions to be classified which are important for the molecular recognition.59,60 This is a new method to identify and calculate the strength of the intermolecular interactions between ligand and protein complex computationally. In the active site of estrogen receptor, the estrogens form important hydrogen bonding interactions with Arg394, Glu353, His524 residues and the water molecule HOH2. A cp search has been performed for the estrogens and the interacting active site residues which are forming hydrogen bonding interactions, found bond path and viral path32,61 (Table 4). The deformation of electron density and the Laplacian of electron density maps of estrogens are generated for all the important interactions and are shown in Fig. 4(a) and (b). In estrogens, the O-atom of hydroxyl group of A-ring has two lone pairs act as an acceptor forming hydrogen bonding interaction with the H-atom of Arg394 residue and the H-atoms of water molecule (HOH2). Here, the lone pair positions of O-atom are aligned in the direction of hydrogen bonding interactions with the residues. The lone pair positions of O-atoms are clearly visible and are shown in Fig. 4b. From the CP analysis, the electron density, Laplacian of electron density of these interactions have been determined. Among all the molecules, E2 molecule shows high electron density 0.203 eÅ−3 and positive Laplacian 2.734 eÅ−5 for O(1)⋯Arg394(2HH2) interactions. Similarly, the H-atom of hydroxyl group has positive charge (0.56 e) act as a donor forming hydrogen bonding interaction with the Glu353 residue; the lone pair position of O-atoms of Glu353 is visible (Fig. 4). In the Glu353 residue, the one lone pair of O-atom aligned in the direction of H-bond interactions with the estrogens and the other lone pair aligned in the direction of H atom of water molecule HOH2. The E3 molecule possess high electron density (0.289 eÅ−3) and positive Laplacian value (3.068 eÅ−5) for H(6)⋯Glu353(OE2) interaction. Similarly, the D-ring of the estrogens form interaction with the His524 residue and their corresponding CP has been found for all molecules, which displays the high electron density and positive Laplacian value (Table 4).
Topological properties of electron density of hydrogen bonding interactions for estrogens and ERα complex.
| Bonds | ρ bcp(r) | ∇ 2 ρ(r) | λ 1 | λ 2 | λ 3 | V(r) | G(r) | H(r) | d 1 | d 2 | D |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Estrone | |||||||||||
| H(6)⋯Glu353(OE2) | 0.279 | 3.082 | −1.61 | −1.564 | 6.256 | −0.251 | 0.233 | −0.018 | 1.159 | 0.599 | 146.94 |
| O(1)⋯Arg394(2HH2) | 0.198 | 2.665 | −1.035 | −0.987 | 4.686 | −0.16 | 0.173 | 0.013 | 0.68 | 1.211 | 87.15 |
| O(1)⋯HOH2(H2) | 0.021 | 0.307 | −0.059 | −0.043 | 0.409 | −0.013 | 0.017 | 0.004 | 1.202 | 1.72 | 3.17 |
| HOH2(O1)⋯Arg394(2HH1) | 0.251 | 3.13 | −1.464 | −1.361 | 5.955 | −0.221 | 0.22 | −0.001 | 1.17 | 0.628 | 0.0477 |
| O(2)⋯His524(HE2) | 0.266 | 2.537 | −1.463 | −1.414 | 5.414 | −0.218 | 0.198 | −0.02 | 0.623 | 1.227 | 134.28 |
| 17β-Estradiol | |||||||||||
| H(6)⋯Glu353(OE2) | 0.269 | 3.026 | −1.527 | −1.483 | 6.036 | −0.239 | 0.225 | −0.014 | 0.605 | 1.164 | 138.75 |
| O(1)⋯Arg394(2HH2) | 0.203 | 2.734 | −1.071 | −1.028 | 4.832 | −0.166 | 0.179 | 0.013 | 1.206 | 0.676 | 90.64 |
| O(1)⋯HOH2(H2) | 0.027 | 0.382 | −0.081 | −0.069 | 0.532 | −0.017 | 0.022 | 0.005 | 1.67 | 1.162 | 4.4 |
| HOH2(O1)⋯Arg394(2HH1) | 0.258 | 3.22 | −1.536 | −1.4 | 6.156 | −0.231 | 0.228 | −0.003 | 0.622 | 1.165 | 131.02 |
| H(3)⋯His524(ND1) | 0.102 | 1.061 | −0.397 | −0.386 | 1.844 | −0.052 | 0.063 | 0.011 | 1.483 | 0.815 | 29.4 |
| Estriol | |||||||||||
| H(6)⋯Glu353(OE2) | 0.286 | 3.068 | −1.612 | −1.557 | 6.238 | −0.259 | 0.237 | −0.022 | 1.163 | 0.612 | 152.49 |
| O(1)⋯Arg394(2HH2) | 0.056 | 0.715 | −0.172 | −0.107 | 0.994 | −0.039 | 0.045 | 0.006 | 1.651 | 1.52 | 12.38 |
| O(1)⋯HOH2(H2) | 0.027 | 0.382 | −0.081 | −0.069 | 0.532 | −0.017 | 0.022 | 0.005 | 1.67 | 1.162 | 4.44 |
| HOH2(O1)⋯Arg394(2HH1) | 0.054 | 0.634 | −0.187 | −0.151 | 0.972 | −0.03 | 0.037 | 0.007 | 0.983 | 1.557 | 11.48 |
| H(4)⋯His524(ND1) | 0.195 | 2.798 | −0.954 | −0.889 | 4.641 | −0.162 | 0.179 | 0.017 | 0.725 | 1.214 | 85.87 |
| Glu353(OE1)⋯HOH2(H2) | 0.237 | 2.976 | −1.278 | −1.211 | 5.464 | −0.206 | 0.207 | 0.001 | 1.182 | 0.64 | 114.8 |
| O(3)⋯Gly521(O) | 0.042 | 0.538 | −0.138 | −0.124 | 0.8 | −0.027 | 0.033 | 0.006 | 1.101 | 1.549 | 8.1 |
Fig. 4. (a) The deformation density map of hydrogen bonding interactions of estrogens with estrogen receptor. (b) The Laplacian of electron density map of hydrogen bonding interactions between estrogens and estrogen receptor. Contours are drawn in a logarithmic scale, 3 × 2N eű5, where N = 2, 4 and 8 × 10n, n = −2, −1, 0, 1, 2. Solid blue lines and dotted red lines represent positive and negative contours, respectively.

The dissociation energy has been estimated for all interactions. Among all interactions, the E3 molecule has high dissociation energy of 152.5 kJ mol−1 for H(6)⋯Glu353(OE2) interaction. However, in E2 molecule all the important hydrogen bonding interactions exhibit high electron density, positive Laplacian value and |V|/G > 1 and H(r) < 0 indicates that all the important hydrogen bonding interactions are partial covalent bonding interactions. The less electron density, positive Laplacian value and |V|/G < 1 and H(r) > 0 indicates the closed shell type of interaction.62 The estimated dissociation energy is high for all the interactions for E2 molecule when compared to the respective interactions of E1 and E3 molecules indicate that the E2 molecule is more stable in the active site of the estrogen receptor; hence the hormone dependent actions are takes place. The relief map and gradient plot of the intermolecular hydrogen bonding interactions of E2 molecule with the amino acid residues are shown in Fig. 5.
Fig. 5. The relief map and gradient vector plot of hydrogen bonding interactions of 17β-estradiol and estrogen receptor-α.

Electrostatic potential and binding affinity
The electrostatic potential map of estrogen molecules for QM/MM minimized structure and the gas phase structure have been shown in Fig. 6. The ESP map is a potent tool to evaluate the reactive locations and chemical reactivity of the molecules.63 The positive regions in the ESP map is the electrophilic regions and negative regions are expected to where the nucleophilic attack takes place. The difference in the ESP regulates the biological activity of the estrogen receptor i.e., hormone dependent genes. The gas phase ESP map of estrogens, the electronegative regions of hydroxyl group of aromatic ring is found high when compare with the D-ring of all estrogen molecules. In E2 molecule, the large negative π-electron cloud is found above and below the aromatic ring, whereas in E1 and E3 molecule this π-electron cloud is found to be less. Notably, the estrogens in the active site of estrogen receptor, the hydroxyl group of aromatic ring acts as a donor for Glu353 residue, whereas it act as a acceptor for Arg394 residue. The π-electron in the aromatic ring form T-shaped π⋯π interaction with the Phe404 residue in the active site and it is shown in Fig. 7. The D-ring of the molecule forms intermolecular interaction with the His524 residue. In the His524 residue there are two protonation states are existing, either hydroxyl group of estrogen act as a donor or acceptor or His524 residue ND1 atom act as a hydrogen bond acceptor or donor, in the case of E1 molecule, the His524 ND1 atom only act as a acceptor. After the QM/MM minimization of estrogens along with the active site amino acid residues, the ESP map has been altered considerably. The electronegative regions of hydroxyl group of aromatic ring are reduced and extended in Arg394 residue facilitates to form hydrogen bonding interaction with the Arg394 residue and also the high electronegative region in the Glu353 residue extended to form hydrogen bonding interaction with the H atom of the hydroxyl group, this scenario is common for all the estrogens. For π⋯π interaction, the negative ESP observed around aromatic ring and the Phe404 residue. The π⋯π interaction between the aromatic ring and the Phe404 residue is only observed in E2 molecule after the QM/MM minimization, which indicates that relatively the E2 molecule is more stable in the active site of the estrogen receptor. In E3 molecule, the D-ring electronegative region is largely reduced and forms interactions with the His524 and Gly521 residue, which indicates that the hormone dependent action of E3 molecule is different from the E1 and E2 molecule. Based on the above discussion, it may be confirmed that for the hormone dependent actions, both A and D-rings of estrogens are contributes equally. However, the D-ring intermolecular interaction is not stable, if the hormone molecule approaches the estrogen receptor, because of the uncertainty of His524 residue orientation.
Fig. 6. Showing the electrostatic potential map of estrogens in gas phase and QM/MM minimized structure. The positive surface value is 0.7 eÅ−1 and the negative surface value is −0.05 eÅ−1.

Fig. 7. The ESP map showing the π⋯π interaction between 17β-estradiol molecule A ring and Phe404 amino acid residue of estrogen receptor-α.

Conclusion
This QM/MM calculation is the new method to understand the charge density distribution and electrostatic properties of ligand molecule in the active site of proteins. In this present study, the intermolecular interactions and its strength of three estrogen molecules with ERα have been well characterized by QM/MM based charge density analysis. From the electrostatic potential maps, the regions of donor and acceptor atoms, protonation state of His524 residue and π⋯π interactions are identified. The above study reveals the hormone dependent actions of estrogens are depends on the D-ring and the protonation state of His524 amino acid residue. In E2 molecule the His524 residue is more stable than all other molecules indicates that more binding affinity of E2 towards estrogen receptor-α. This theoretical study is a very useful method to understand the charge density distribution and electrostatic properties of ligands in the binding pocket of proteins and it is a complementary to the experimental charge density distribution from the high resolution diffraction data.
Conflicts of interest
The authors declare that there is no conflict of interest.
Supplementary Material
Acknowledgments
CK is grateful to UGC-RGNF for providing the Senior Research Fellowship (SRF) to carry out this Research work. The authors thank C-DAC, Bangalore for providing the GARUDA supercomputing facility.
Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra08607b
References
- Náray-Szabó G. Ferenczy G. G. Molecular Electrostatics. Chem. Rev. 1995;95:829–847. doi: 10.1021/cr00036a002. [DOI] [Google Scholar]
- Mladenovic M. Arnone M. Fink R. F. Engels B. Environmental effects on charge densities of biologically active molecules: do molecule crystal environments indeed approximate protein surroundings? J. Phys. Chem. B. 2009;113:5072–5082. doi: 10.1021/jp809537v. [DOI] [PubMed] [Google Scholar]
- Schmidt A. Lamzin V. S. Cell. Mol. Life Sci. 2007;64:1959–1969. doi: 10.1007/s00018-007-7195-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cachau R. E. Podjarny A. D. J. Mol. Recognit. 2005;18:193–202. doi: 10.1002/jmr.738. [DOI] [PubMed] [Google Scholar]
- Luger P. Fast electron density methods in the life sciences – A routine application in the future? Org. Biomol. Chem. 2007;5:2529–2540. doi: 10.1039/B706235D. [DOI] [PubMed] [Google Scholar]
- Jelsch C. Teeter M. M. Lamzin V. Pichon-Pesme V. Blessing R. H. Lecomte C. Accurate protein crystallography at ultra-high resolution: valence electron distribution in crambin. Proc. Natl. Acad. Sci. U. S. A. 2000;97:3171–3176. doi: 10.1073/pnas.97.7.3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko T.-P. Robinson H. Gao Y.-G. Cheng C.-H. C. DeVries A. L. Wang A. H.-J. The refined crystal structure of an eel pout type III antifreeze protein RD1 at 0.62-A resolution reveals structural microheterogeneity of protein and solvation. Biophys. J. 2003;84:1228–1237. doi: 10.1016/S0006-3495(03)74938-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podjarny A. Howard E. Mitschler A. Chevrier B. Lecomte C. Guillot B. Pichon-Pesme V. Jelsch C. X-ray crystallography at subatomic resolution. Europhys. News. 2002;33:113–117. doi: 10.1051/epn:2002401. [DOI] [Google Scholar]
- Bönisch H. Schmidt C. L. Bianco P. Ladenstein R. Ultrahigh-resolution study on Pyrococcus abyssi rubredoxin. I. 0.69 Å X-ray structure of mutant W4L/R5S. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2005;61:990–1004. doi: 10.1107/S090744490501293X. [DOI] [PubMed] [Google Scholar]
- Kang B. S. Devedjiev Y. Derewenda U. Derewenda Z. S. The PDZ2 domain of syntenin at ultra-high resolution: bridging the gap between macromolecular and small molecule crystallography. J. Mol. Biol. 2004;338:483–493. doi: 10.1016/j.jmb.2004.02.057. [DOI] [PubMed] [Google Scholar]
- Wang J. Dauter M. Alkire R. Joachimiak A. Dauter Z. Triclinic lysozyme at 0.65 Å resolution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2007;63:1254–1268. doi: 10.1107/S0907444907054224. [DOI] [PubMed] [Google Scholar]
- Hirano Y. Takeda K. Miki K. Charge-density analysis of an iron-sulfur protein at an ultra-high resolution of 0.48 Å. Nature. 2016;534:281–284. doi: 10.1038/nature18001. [DOI] [PubMed] [Google Scholar]
- Petrova T. Podjarny A. Protein crystallography at subatomic resolution. Rep. Prog. Phys. 2004;67:1565–1605. doi: 10.1088/0034-4885/67/9/R01. [DOI] [Google Scholar]
- Schmidt A. Lamzin V. S. Curr. Opin. Struct. Biol. 2002;12:698–703. doi: 10.1016/S0959-440X(02)00394-9. [DOI] [PubMed] [Google Scholar]
- Dittrich B. Hübschle C. B. Messerschmidt M. Kalinowski R. Girnt D. Luger P. The invariom model and its application: refinement of d,l-serine at different temperatures and resolution. Acta Crystallogr., Sect. A: Found. Crystallogr. 2005;61:314–320. doi: 10.1107/S0108767305005039. [DOI] [PubMed] [Google Scholar]
- Mebs S. Lüth A. Luger P. A simple procedure for the derivation of electron density based surfaces of drug–receptor complexes from a combination of X-ray data and theoretical calculations. Bioorg. Med. Chem. 2010;18:5965–5974. doi: 10.1016/j.bmc.2010.06.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel K. Magistrato A. Modeling anticancer drug-DNA interactions via mixed QM/MM molecular dynamics simulations. Org. Biomol. Chem. 2006;4:2507–2517. doi: 10.1039/B604263P. [DOI] [PubMed] [Google Scholar]
- Thomas C. Gustafsson J. Å. Nat. Rev. Cancer. 2011;11:597–608. doi: 10.1038/nrc3093. [DOI] [PubMed] [Google Scholar]
- Evans R. M. The steroid and thyroid hormone receptor superfamily. Science. 1988;240:889–895. doi: 10.1126/science.3283939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deroo B. J. Estrogen receptors and human disease. J. Clin. Invest. 2006;116:561–570. doi: 10.1172/JCI27987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourguet W. Germain P. Gronemeyer H. Trends Pharmacol. Sci. 2000;21:381–388. doi: 10.1016/S0165-6147(00)01548-0. [DOI] [PubMed] [Google Scholar]
- Berg J. M., Tymoczko J. L. and Stryer L., Transcriptional Activation and Repression Are Mediated by Protein–Protein Interactions, Biochemistry, W. H. Freeman, 2001 [Google Scholar]
- Brzozowski A. M. Pike A. C. Dauter Z. Hubbard R. E. Bonn T. Engström O. Ohman L. Greene G. L. Gustafsson J. A. Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- Yaşar P. Ayaz G. User S. D. Güpür G. Muyan M. Molecular mechanism of estrogen-estrogen receptor signaling. Reprod. Med. Biol. 2017;16:4–20. doi: 10.1002/rmb2.12006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooq A. Structural and Functional Diversity of Estrogen Receptor Ligands. Curr. Top. Med. Chem. 2015;15:1372–1384. doi: 10.2174/1568026615666150413154841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatson G. T. On the treatment of inoperable cases of carcinoma of the mamma: suggestions for a new method of treatment, with illustrative cases.1. Lancet. 1896;148:104–107. doi: 10.1016/S0140-6736(01)72307-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson B. E. Feigelson H. S. Carcinogenesis. 2000;21:427–433. doi: 10.1093/carcin/21.3.427. [DOI] [PubMed] [Google Scholar]
- Kitagishi Y. Kobayashi M. Matsuda S. Defective DNA repair systems and the development of breast and prostate cancer. Int. J. Oncol. 2013;42:29–34. doi: 10.3892/ijo.2012.1696. [DOI] [PubMed] [Google Scholar]
- Matta J. Morales L. Ortiz C. Adams D. Vargas W. Casbas P. Dutil J. Echenique M. Suárez E. Estrogen Receptor Expression Is Associated with DNA Repair Capacity in Breast Cancer. PLoS One. 2016;11:e0152422. doi: 10.1371/journal.pone.0152422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J. Wei X. Tufan T. Kuscu C. Unlu H. Farooq S. Demirtas E. Paschal B. M. Adli M. Recurrent mutations at estrogen receptor binding sites alter chromatin topology and distal gene expression in breast cancer. Genome Biol. 2018;19:190. doi: 10.1186/s13059-018-1572-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshel A. Levitt M. Theoretical studies of enzymic reactions: dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J. Mol. Biol. 1976;103:227–249. doi: 10.1016/0022-2836(76)90311-9. [DOI] [PubMed] [Google Scholar]
- Bader R. F. W., Atoms in Molecules; a quantum theory, Clarendon Press, 1994 [Google Scholar]
- Sherwood P., Hybrid Quantum Mechanics/Molecular Mechanics Approaches, in Modern Methods and Algorithms of Quantum Chemistry, ed. J. Grotend, Forschungszentrum Juelich, 2000 [Google Scholar]
- Gao J. Truhlar D. G. Quantum mechanical methods for enzyme kinetics. Annu. Rev. Phys. Chem. 2002;53:467–505. doi: 10.1146/annurev.physchem.53.091301.150114. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993;98:5648–5652. doi: 10.1063/1.464913. [DOI] [Google Scholar]
- Hariharan P. C. Pople J. A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta. 1973;28:213–222. doi: 10.1007/BF00533485. [DOI] [Google Scholar]
- Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Montgomery Jr J. A., Vreven T., Kudin K. N., Burant J. C., Millam J. M., Iyengar S. S., Tomasi J., Barone V., Mennucci B., Cossi M., Scalmani G., Rega N., Petersson G. A., Nakatsuji H., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Klene M., Li X., Knox J. E., Hratchian H. P., Cross J. B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Ayala P. Y., Morokuma K., Voth G. A., Salvador P., Dannenberg J. J., Zakrzewski V. G., Dapprich S., Daniels A. D., Strain M. C., Farkas O., Malick D. K., Rabuck A. D., Raghavachari K., Foresman J. B., Ortiz J. V., Cui Q., Baboul A. G., Clifford S., Cioslowski J., Stefanov B. B., Liu G., Liashenko A., Piskorz P., Komaromi I., Martin R. L., Fox D. J., Keith T., Al-Laham M. A., Peng C. Y., Nanayakkara A., Challacombe M., Gill P. M. W., Johnson B., Chen W., Wong M. W., Gonzalez C., and Pople J. A., Gaussian, Inc., Wallingford CT, 2004 [Google Scholar]
- Shiau A. K. Barstad D. Loria P. M. Cheng L. Kushner P. J. Agard D. A. Greene G. L. The Structural Basis of Estrogen Receptor/Coactivator Recognition and the Antagonism of This Interaction by Tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/S0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- Morris G. M. Huey R. Lindstrom W. Sanner M. F. Belew R. K. Goodsell D. S. Olson A. J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D. A., Ben-Shalom I. Y., Brozell S. R., Cerutti D. S., Cheatham III T. E., Cruzeiro V. W. D., Darden T. A., Duke R. E., Ghoreishi D., Gilson M. K., Gohlke H., Goetz A. W., Greene D., Harris R., Homeyer N., Izadi S., Kovalenko A., Kurtzman T., Lee T. S., LeGrand S., Li P., Lin C., Liu J., Luchko T., Luo R., Mermelstein D. J., Merz K. M., Miao Y., Monard G., Nguyen C., Nguyen H., Omelyan I., Onufriev A., Pan F., Qi R., Roe D. R., Roitberg A., Sagui C., Schott-Verdugo S., Shen J., Simmerling C. L., Smith J., Salomon-Ferrer R., Swails J., Walker R. C., Wang J., Wei H., Wolf R. M., Wu X., Xiao L., York D. M. and Kollman P. A., AMBER 2014, University of California, San Francisco, 2014 [Google Scholar]
- Wang J. Wang W. Kollman P. A. Case D. A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graphics Modell. 2006;25:247–260. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Wang J. Wolf R. M. Caldwell J. W. Kollman P. A. Case D. A. Development and testing of a general amber force field. J. Comput. Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Maier J. A. Martinez C. Kasavajhala K. Wickstrom L. Hauser K. E. Simmerling C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015;11:3696–3713. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrach M. F. Drossel B. Structure and dynamics of TIP3P, TIP4P, and TIP5P water near smooth and atomistic walls of different hydroaffinity. J. Chem. Phys. 2014;140:174501. doi: 10.1063/1.4872239. [DOI] [PubMed] [Google Scholar]
- Mark P. Nilsson L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A. 2001;105:9954–9960. doi: 10.1021/jp003020w. [DOI] [Google Scholar]
- Essmann U. Perera L. Berkowitz M. L. Darden T. Lee H. Pedersen L. G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. doi: 10.1063/1.470117. [DOI] [Google Scholar]
- Zhurova E. A. Matta C. F. Wu N. Zhurov V. V. Pinkerton A. A. Experimental and theoretical electron density study of estrone. J. Am. Chem. Soc. 2006;128:8849–8861. doi: 10.1021/ja061080v. [DOI] [PubMed] [Google Scholar]
- Parrish D. a Zhurova E. Kirschbaum K. Pinkerton a A. Experimental charge density study of estrogens: 17beta-estradiol urea. J. Phys. Chem. B. 2006;110:26442–26447. doi: 10.1021/jp065638x. [DOI] [PubMed] [Google Scholar]
- Zhurova E. A. Zhurov V. V. Chopra D. Stash A. I. Pinkerton A. A. 17α-estradiol·1/2 H2O: Super-structural ordering, electronic properties, chemical bonding, and biological activity in comparison with other estrogens. J. Am. Chem. Soc. 2009;131:17260–17269. doi: 10.1021/ja906057z. [DOI] [PubMed] [Google Scholar]
- Zhurova E. A. Zhurov V. V. Kumaradhas P. Cenedese S. Pinkerton A. A. Charge Density and Electrostatic Potential Study of 16α,17β-Estriol and the Binding of Estrogen Molecules to the Estrogen Receptors ER α and ER β. J. Phys. Chem. B. 2016;120:8882–8891. doi: 10.1021/acs.jpcb.6b05961. [DOI] [PubMed] [Google Scholar]
- Birkedal H. Madsen D. Mathiesen R. H. Knudsen K. Weber H. P. Pattison P. Schwarzenbach D. The charge density of urea from synchrotron diffraction data. Acta Crystallogr., Sect. A: Found. Crystallogr. 2004;60:371–381. doi: 10.1107/S0108767304015120. [DOI] [PubMed] [Google Scholar]
- Volkov A. Abramov Y. Coppens P. Gatti C. On the origin of topological differences between experimental and theoretical crystal charge densities. Acta Crystallogr., Sect. A: Found. Crystallogr. 2000;56(4):332–339. doi: 10.1107/S0108767300003202. [DOI] [PubMed] [Google Scholar]
- Volkov A. Li X. Koritsanszky T. Coppens P. Ab initio quality electrostatic atomic and molecular properties including intermolecular energies from a transferable theoretical pseudoatom databank. J. Phys. Chem. A. 2004;108:4283–4300. doi: 10.1021/jp0379796. [DOI] [Google Scholar]
- Volkov A. Abramov Y. A. Coppens P. Density-optimized radial exponents for X-ray charge-density refinement from ab initio crystal calculations. Acta Crystallogr., Sect. A: Found. Crystallogr. 2001;57:272–282. doi: 10.1107/S0108767300018547. [DOI] [PubMed] [Google Scholar]
- Rajalakshmi G. Hathwar V. R. Kumaradhas P. Topological analysis of electron density and the electrostatic properties of isoniazid: an experimental and theoretical study. Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater. 2014;70:331–341. doi: 10.1107/S2052520613033209. [DOI] [PubMed] [Google Scholar]
- Bader R. F. W. A Quantum Theory of Molecular Structure and Its Applications. Chem. Rev. 1991;91:893–928. doi: 10.1021/cr00005a013. [DOI] [Google Scholar]
- Mulliken R. S. Electronic population analysis on LCAO-MO molecular wave functions. I. J. Chem. Phys. 1955;23:1833–1840. doi: 10.1063/1.1740588. [DOI] [Google Scholar]
- Koch U. Popelier P. L. A. Characterization of C–H–O hydrogen bonds on the basis of the charge density. J. Phys. Chem. 1995;99:9747–9754. doi: 10.1021/j100024a016. [DOI] [Google Scholar]
- Politzer P. Murray J. S. The fundamental nature and role of the electrostatic potential in atoms and molecules. Theor. Chem. Acc. Theory Comput. Model. 2002;108:134–142. doi: 10.1007/s00214-002-0363-9. [DOI] [Google Scholar]
- Stalke D. Meaningful structural descriptors from charge density. Chem.–Eur. J. 2011;17:9264–9278. doi: 10.1002/chem.201100615. [DOI] [PubMed] [Google Scholar]
- Bader R. F. W. A bond path: a universal indicator of bonded interactions. J. Phys. Chem. A. 1998;102:7314–7323. doi: 10.1021/jp981794v. [DOI] [Google Scholar]
- Gatti C. Chemical bonding in crystals: new directions. Z. für Kristallogr. – Cryst. Mater. 2005;220:399–457. [Google Scholar]
- Desiraju G. R. Angew. Chem., Int. Ed. Engl. 1995;34:2311–2327. doi: 10.1002/anie.199523111. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.