

Abstract
Biological aging is a proposed mechanism through which social determinants drive health disparities. We conducted proof-of-concept testing of 8 DNA-methylation (DNAm) and blood-chemistry quantifications of biological aging as mediators of disparities in healthspan between Black and White participants in the 2016 wave of the Health and Retirement Study (n = 9,005). We quantified biological aging from 4 DNAm “clocks” (Horvath, Hannum, PhenoAge, and GrimAge clock), a DNAm pace-of-aging measure (DunedinPoAm), and 3 blood-chemistry measures (PhenoAge, Klemera-Doubal method biological age, and homeostatic dysregulation). We quantified Black-White disparities in healthspan from cross-sectional and longitudinal data on physical performance tests, self-reported limitations in activities of daily living, and physician-diagnosed chronic diseases, self-rated health, and survival. DNAm and blood-chemistry quantifications of biological aging were moderately correlated (Pearson’s r = 0.1–0.4). The GrimAge clock, DunedinPoAm, and all 3 blood-chemistry measures were associated with healthspan characteristics (e.g., mortality effect-size hazard ratios were 1.71–2.32 per standard deviation of biological aging) and showed evidence of more advanced/faster biological aging in Black participants than in White participants (Cohen’s d = 0.4–0.5). These measures accounted for 13%–95% of Black-White differences in healthspan-related characteristics. Findings suggest that reducing disparities in biological aging can contribute to building health equity.
Keywords: aging, aging clocks, biological aging, healthy aging, pace of aging, racial disparities
Abbreviation
- ADLs
activities of daily living
- CI
confidence interval
- DNAm
DNA methylation
- HR
hazard ratio
- HRS
Health and Retirement Study
- IRR
incidence rate ratio
- KDM
Klemera-Doubal method
- SD
standard deviation
- VBS
Venous Blood Study
Black Americans experience excess morbidity and premature mortality relative to White Americans (1). This health disparity is mediated by multiple chronic diseases affecting different organ systems throughout the body and reflects an etiology extending from the earliest stages of life across adulthood, encompassing social, economic, and environmental factors (2–4). Differences in health between Black and White Americans vary between geographic locations and have changed over time, indicating that these disparities are socially determined and that they are modifiable (5–7). A range of policies and programs have been proposed to mitigate health disparities (8–11). However, rigorous evaluation of impact is challenging (12). Interventions to address health disparities delivered to older adults may come too late to prevent chronic disease (4, 13), while interventions delivered to younger people require long follow-up intervals to establish impact (14). Methods are needed to monitor the effectiveness of interventions over timescales of years rather than decades.
Measures that quantify processes of biological aging may provide near-term measurements of long-term impacts. Biological aging is the gradual and progressive decline in system integrity with advancing chronological age (15). This process is now being studied as a modifiable root cause of many different chronic diseases (16–18). One hypothesis advanced to explain Black-White health disparities across a range of diseases is that social and material stresses experienced by Black Americans act to accelerate biological aging, referred to as “weathering” (19, 20). In epidemiologic studies, Black Americans show more advanced biological aging than White Americans of the same chronological age (21–23). If advanced biological aging is a mediator of health disparities, then quantifications of biological aging could be used to monitor intervention impacts.
Many methods are proposed for quantifying biological aging from several biological levels of analysis (24, 25). Agreement between measures is often poor; there is no gold standard (26–28). Measures based on analysis of blood-chemistry and DNA-methylation (DNAm) data have received the most attention to date. We conducted proof-of-concept testing of 8 blood-chemistry and DNAm methods for quantifying biological aging as mediators of Black-White disparities in healthy aging. We analyzed deficits in physical functioning, limitations in activities of daily living (ADLs), chronic disease morbidity, and mortality in a national sample of US older adults in the Health and Retirement Study (HRS). Previous studies have documented Black-White differences in several of the measures of aging we analyzed (21, 22, 29–31). However, few studies have tested whether these differences could account for Black-White health disparities (21, 23), and none (to our knowledge) have specifically considered disparities in healthspan characteristics. Our analysis builds on an initial report of differences in DNAm measures of biological aging between Black and White adults in the HRS (30) in 3 ways. First, we analyze measures of biological aging derived from DNAm data together with measures derived from blood-chemistry data. Second, we compare the different measures of biological aging by testing associations with healthspan-related characteristics and mortality. Third, we quantify the fractions of Black-White differences in healthspan-related characteristics and mortality that are accounted for by biological-aging measures. Figure 1 provides a conceptual overview of our analysis.
Figure 1.
Conceptual overview of a study designed to test 8 measures of biological aging as mediators of Black-White healthspan disparities, Health and Retirement Study (HRS), 1992–2018. We analyzed data from 9,005 older adult participants in the HRS who provided blood-chemistry and/or DNA-methylation data during the 2016 measurement wave. We quantified biological aging from blood-chemistry and DNA-methylation data using published algorithms: the PhenoAge (55), Klemera-Doubal method biological age (56), and homeostatic dysregulation (57) methods for blood-chemistry data; the DNA-methylation “clocks” proposed by Horvath (36), Hannum et al. (58), Levine et al. (55), and Lu et al. (47); and the DunedinPoAm DNA methylation pace of aging (37). We analyzed Black-White disparities in healthspan from cross-sectional and longitudinal data on physical performance tests, self-reported activities of daily living limitations and physician-diagnosed chronic conditions, self-rated health, and survival. The analysis tested mediation of Black-White disparities in healthspan phenotypes by measures of biological aging.
METHODS
Sample
The HRS is a nationally representative longitudinal survey of US residents aged ≥50 years and their spouses that has been fielded every 2 years since 1992. A new cohort of 51- to 56-year-olds and their spouses is enrolled every 6 years to maintain representativeness of the US population over 50 years of age. Response rates over all waves of the HRS range from 81% to 91% (32). As of the most recent data release (February 2021), the HRS included data collected from 42,515 individuals in 26,600 households (33). We linked HRS data curated by the RAND Corporation (Santa Monica, California) with new data collected as part of the HRS’s 2016 Venous Blood Study (VBS) (33, 34). Analysis included participants aged 50–90 years at the time of the blood draw for whom data were also collected on prevalent chronic disease, ADLs, and/or physical functioning (n = 9,005). A comparison of our analysis sample with the full HRS sample is provided in Web Table 1 (available at https://doi.org/10.1093/aje/kwab281).
Measures
Biological aging.
There is no gold-standard measure of biological aging (24). Many methods based on different biological levels of analysis have been proposed. Current state-of-the-art methods use machine learning to sift large numbers of candidate markers and parameterize algorithms that predict aging-related parameters, including chronological age, mortality risk, and rate of decline in system integrity. Algorithms are developed in reference data sets and applied in new data sets to test hypotheses. We analyzed several different methods because each method uses different assumptions to develop a measure of the latent construct of biological aging. As we and others have shown, the different methods do not all measure the same aspects of the aging process (26, 28). Comparative analysis is therefore essential to interpretation. We focused on algorithms developed for blood-chemistry analytes routinely measured in clinical settings and DNAm marks included in commercial arrays, and algorithms which have received substantial attention in the research literature. Measures are described and their source publications are cited in Table 1. Detailed descriptions of the measures are provided in Web Appendix 1.
Table 1.
Measures of Biological Aging Included in an Analysis of Black-White Disparities in Biological Aging, Health and Retirement Study, 1992–2018
| Measure ofBiological Aging |
First Author, Year
(Reference No.) |
Criterion Used to
Develop Measure |
Interpretation ofMeasure’s Values |
|---|---|---|---|
| Blood-Chemistry Measures a | |||
| PhenoAge | Levine, 2018 (55) | Mortality | Age at which average mortality risk in NHANES III matches the mortality risk predicted by the blood-chemistry + chronological age algorithm |
| KDM biological age | Klemera, 2006 (56) | Chronological age | Age at which average physiology in NHANES III matches the physiology of the participant |
| Homeostatic dysregulation | Cohen, 2013 (57) | Deviation from healthy youth | Log biomarker Mahalanobis distance of participant from young, healthy NHANES III participants |
| DNA-Methylation Measures b | |||
| First-generation DNA-methylation clocks | |||
| Horvath clock | Horvath, 2013 (36) | Chronological age | Age predicted by DNA methylation |
| Hannum clock | Hannum, 2013 (58) | Chronological age | Age predicted by DNA methylation |
| Second-generation DNA-methylation clocks | |||
| PhenoAge clock | Levine, 2018 (55) | Blood-chemistry PhenoAge | Age at which average mortality risk in NHANES III matches the mortality risk predicted by the PhenoAge algorithm |
| GrimAge clock | Lu, 2019 (47) | Mortality | Age at which average mortality risk in the Framingham Heart Study offspring cohort matches predicted mortality risk |
| Pace of aging | |||
| DunedinPoAm pace of aging | Belsky, 2020 (37) | Change in 18 system-integrity biomarkers over 12 years of follow-up | Years of physiological decline experienced per 1 year of calendar time over the recent past. The expected value of DunedinPoAm in midlife adults is 1. Values greater than 1 indicate accelerated aging; values less than 1 indicate slowed aging. |
Abbreviations: KDM, Klemera-Doubal method; InCHIANTI, Invecchiare in Chianti; NHANES III, Third National Health and Nutrition Examination Survey.
a All algorithms were parameterized using data from NHANES III and included the following blood-chemistry measurements: albumin, alkaline phosphatase, creatinine, C-reactive protein (log), glucose, white blood cell count, lymphocyte percentage, mean corpuscular volume, and red cell distribution width. The PhenoAge and KDM biological-age algorithms additionally included chronological age. The NHANES III training sample is majority White but includes an oversample of Black Americans. Blood-chemistry measures were calculated using code available on GitHub (60) according to published methods. For analysis, PhenoAge and KDM biological age were differenced from chronological age to calculate biological-age advancement values.
b DNA-methylation measures were developed from analysis of genomewide DNA methylation measured on Illumina Infinium HumanMethylation 27K and 450K BeadChip arrays (Illumina, Inc., San Diego, California) in a range of different data sets. The Horvath clock was developed from analysis of 82 different data sets. The Hannum clock was developed from analysis of research volunteers at the University of California, San Diego (La Jolla, California), the University of Southern California (Los Angeles, California), and West China Hospital (Chengdu, China). The PhenoAge clock was developed from analysis of NHANES III data and the InCHIANTI Study (Tuscany region, Italy). The GrimAge clock was developed from analysis of the Framingham Heart Study (Framingham, Massachusetts) offspring cohort. The DunedinPoAm pace-of-aging measure was developed from analysis of the Dunedin Study (Dunedin, New Zealand). The InCHIANTI, Framingham, and Dunedin Study cohorts are mostly or entirely White European. DNA-methylation measures were calculated by the Health and Retirement Study investigators. For analysis, DNA-methylation clocks were residualized on chronological age to calculate biological-age advancement values.
We computed blood-chemistry measures using the R package (R Foundation for Statistical Computing, Vienna, Austria) BioAge (35). We obtained data on DNAm measures of biological aging from the HRS (30). Clock measures can also be computed with the software hosted by the Horvath Lab (Dr. Steve Horvath, University of California, Los Angeles) (36). Dunedin Pace of Aging methylation (DunedinPoAm) can be computed using the GitHub code published by Belsky et al. (37).
For analysis, we converted measures of biological age (blood-chemistry PhenoAge, Klemera-Doubal biological age, homeostatic dysregulation, PhenoAge clock, Horvath clock, Hannum clock, GrimAge clock, and DunedinPoAm pace of aging) to measures of biological-age advancement by fitting regressions of biological-age measures on chronological age and computing residual values. The literature commonly refers to these residuals as “age acceleration.” We instead use “age advancement” to distinguish measurements like the clocks—which compute a difference between biological age and chronological age at a single point in time—from pace-of-aging measurements that quantify how fast a person is aging. No residualization was applied to homeostatic dysregulation and DunedinPoAm, as these measures already quantify deviation from the expected sample norm.
Healthspan-related characteristics.
Healthspan is the portion of life lived free of disease and disability. We measured healthspan-related characteristics from performance-test measurements of functional impairment administered by trained interviewers, participant reports about ADL limitations, self-rated health, physician-diagnosed chronic conditions, and HRS follow-up for mortality status through 2019. Measures are described in detail in Table 2 and Web Table 2.
Table 2.
Measurement of Healthspan-Related Characteristics and Mortality, Health and Retirement Study, 1992–2018
| Outcome Measure | No. of Participants | Description | |
|---|---|---|---|
|
VBS
Sample |
VBS DNAm
Sample |
||
| Functional impairment | 3,621 | 1,596 | Functional impairments were measured from tests of lung function (peak expiratory flow), grip strength, gait speed, and balance. The tests are described in detail in Web Table 1. We classified test scores at the 30th percentile of the HRS distribution or lower as indicating functional impairment. Percentiles for grip strength were calculated separately for men and women. We calculated numbers of functional impairments for participants providing data on at least 3 of the 4 tests. Counts were winsorized at a value of 3 (HRS VBS sample: mean = 1.13 (SD, 1.08); VBS DNAm sample: mean = 1.17 (SD, 1.09)). |
| No. of ADL limitations | 8,198 | 3,598 | Participants were asked whether they had difficulty performing 6 ADLs: dressing, eating (such as cutting up one’s food), bathing and showering, getting into and out of bed, using the toilet, and walking across a room. We summed the number of activities for which participants reported having difficulty and coded the resulting ADL score as 0, 1, 2, or ≥3 ADL limitations (VBS sample: mean = 0.30 (SD, 0.75); VBS DNAm sample: mean = 0.31 (SD, 0.78)). For longitudinal analysis, we counted any new ADL difficulties reported at the HRS 2018 assessment (VBS sample: mean = 0.19 (SD, 0.65); VBS DNAm sample: mean = 0.20 (SD, 0.67)). |
| Chronic conditions | 8,196 | 3,597 | Chronic disease diagnoses were ascertained from participants’ reports about whether a physician had ever diagnosed them with hypertension, type 2 diabetes, cancer (excluding minor skin cancer), chronic lung disease, heart problems (heart attack, coronary heart disease, angina, congestive heart failure), and/or stroke. Counts of chronic disease diagnoses were winsorized at a value of 3 (VBS sample: mean = 1.45 (SD, 1.04); VBS DNAm sample: mean = 1.48 (SD, 1.04)). For longitudinal analysis, we counted any new diagnoses reported at the 2018 HRS assessment (VBS sample: mean = 0.14 (SD, 0.39); VBS DNAm sample: mean = 0.15 (SD, 0.39)). |
| Self-rated health | 8,191 | 3,595 | Participants were asked to rate their own health on a scale of 1 to 5 (1 = excellent, 5 = poor; VBS sample: mean = 2.88 (SD, 1.03); DNAm sample: mean = 2.89 (SD, 1.03)). For longitudinal analysis, we calculated the difference in self-rated health between waves by subtracting the HRS 2016-wave rating from the 2018-wave rating (possible range, −4 to 4; VBS sample: mean = 0.04 (SD, 0.82); VBS DNAm sample: mean = 0.04 (SD, 0.81)). |
| Mortality | 8,198 | 3,598 | Mortality follow-up was conducted through 2019. During follow-up, 237 deaths were recorded in the full VBS sample, of which 123 were recorded for participants in the VBS DNAm sample. |
Abbreviations: ADLs, activities of daily living; DNAm, DNA methylation; HRS, Health and Retirement Study; SD, standard deviation; VBS, Venous Blood Study.
Analysis
Our primary analysis tested associations of biological-aging measures with healthspan-related characteristics. We conducted analysis of prevalent functional impairments, ADL limitations, chronic conditions, and current self-rated health based on data collected in the 2016 wave of the HRS. We conducted longitudinal analysis of incident ADL limitations, incident chronic conditions, and changes in self-rated health using data from the 2016 and 2018 waves. (We did not conduct longitudinal analysis of performance-test measures because these data are collected from participants at every other wave, so no follow-up was available.) Analysis of mortality was based on the most recent ascertainment of mortality status by the HRS.
We used Poisson regression to estimate incidence rate ratios (IRRs) for associations of biological aging with counts of functional impairments, ADL limitations, and chronic conditions. We used linear regression to estimate standardized effect sizes (Pearson’s r) for continuous measures of self-rated health. We used Cox proportional hazards regression to estimate mortality hazard ratios (HRs). Effect sizes for biological-aging measures were denominated in standard deviation (SD) units.
For analysis of the full HRS VBS sample and the VBS DNAm subsample (VBS DNAm), which were designed to represent the US population aged 50 years or more, we applied probability sampling weights to generate estimates for this population. For analyses of subsamples of Black and White older adults, we included covariate adjustment for chronological age, sex, and region of residence.
We tested mediation of Black-White disparities in biological aging using a regression-based approach as described by Valeri and VanderWeele (38). We used the R package CMAverse (39) to estimate direct and indirect effects and proportions mediated. We calculated 95% confidence intervals (CIs) using bootstrapping to obtain standard error estimates. We tested the robustness of mediation results to potential exposure-mediator interactions following the approaches outlined by Valeri and VanderWeele (38, 40). Details of this analysis are provided in Web Appendices 2 and 3 and Web Figure 1.
RESULTS
We conducted analysis using 2 sets of biological-aging measures. We analyzed blood-chemistry measures of biological aging in 9,005 HRS participants included in the VBS (41% male; 74% White and 18% Black; age range, 50–90 years; mean age = 69 (SD, 9) years). We analyzed DNAm measures of aging in 3,928 VBS participants who were included in the VBS DNAm subsample (42% male; 75% White and 17% Black; age range, 50–90 years; mean age = 70 (SD, 9) years). We first tested associations of biological-aging measures with healthspan-related characteristics. Next, we tested Black-White disparities in biological aging. Finally, we conducted mediation analysis for measures demonstrating 1) associations of more advanced/faster biological aging with healthspan-related characteristics and 2) more advanced/faster biological aging in Black participants as compared with White participants (41).
Comparison of blood-chemistry and DNAm measures of aging
Participants’ biological-age values were correlated with their chronological ages (blood-chemistry PhenoAge: r =0.76; Klemera-Doubal method (KDM) biological age: r =0.30; DNAm clocks: r = 0.72–0.83). Participants’ homeostatic dysregulation values indicated greater dysregulation relative to young, healthy adults (log Mahalanobis distance: mean =3.94 SD units (SD, 0.92)). Participants’ DunedinPoAm values indicated that they were aging 7% faster than the rate expected for adults in early midlife (i.e., 1 year of biological change per chronological year; mean =1.07 (SD, 0.09)). For further analysis, PhenoAge, KDM biological age, and the DNAm clocks were converted to age residuals.
Biological-aging measures were varied in their correlations with one another. Correlations among different blood-chemistry measures of aging were strong (r > 0.7). Correlations among different DNAm measures of aging were weak to moderate (r < 0.7). Correlations between DNAm measures and blood-chemistry measures were varied. Horvath clock age residuals were not correlated with blood-chemistry measures of aging (r < 0.1); Hannum clock age residuals were weakly correlated with blood-chemistry measures (r = 0.1–0.2); and correlations were somewhat stronger for PhenoAge clock residuals (r = 0.2–0.3), GrimAge clock residuals (r = 0.3–0.4), and DunedinPoAm (r = 0.2–0.3). Correlations and scatterplots are shown in Web Figures 2 and 3.
Associations of biological-aging measures with healthspan-related characteristics and mortality
Participants with more advanced/faster biological aging had poorer outcomes for all healthspan-related characteristics and increased risk of mortality. In blood-chemistry analysis, participants with more advanced PhenoAge and KDM biological age and greater homeostatic dysregulation showed increased risk of prevalent functional impairments, prevalent and incident ADL limitations, and prevalent and incident chronic conditions; poorer self-rated health and more negative change in self-rated health; and increased hazard of mortality. PhenoAge advancement effect sizes per SD were as follows: for functional impairments and prevalent and incident ADL limitations and chronic conditions, IRR = 1.19–1.54; for self-rated health, r = 0.34 for level and r = 0.09 for change; and for mortality, HR = 1.85 (95% CI: 1.66, 2.06). Effect sizes were similar for KDM biological age and homeostatic dysregulation measures.
In DNAm analysis, effect sizes were largest for the GrimAge clock (for functional impairments and prevalent and incident ADL limitations and chronic conditions, IRR = 1.20–1.39 per SD; for self-rated health, r = 0.32 for 2016 level and r = 0.08 for change; for mortality, HR = 2.32 per SD (95% CI: 1.82, 2.96)). Effect sizes were somewhat smaller for the PhenoAge and Hannum clocks. For the Horvath clock, effect sizes were smaller and often not statistically different from 0 at the α = 0.05 level. For DunedinPoAm pace of aging, which measures how rapidly a person is aging at the time of measurement, effect sizes were smaller than those for the GrimAge clock and somewhat larger than those for the other clocks (for functional impairments and prevalent and incident ADL limitations and chronic conditions, IRR = 1.14–1.27 per SD; for self-rated health, r = 0.22 for level and r = 0.06 for change; for mortality, HR = 1.71 per SD (95% CI: 1.36, 2.14)). Effect sizes are shown in Figure 2 and Web Tables 3 and 4.
Figure 2.

Effect sizes for associations of 8 biological-aging measures with healthspan-related characteristics, Health and Retirement Study, 1992–2018. The graph shows effect sizes for associations of biological-aging measures with healthspan characteristics measured cross-sectionally at the 2016 wave of the Health and Retirement Study (left column: functional impairments (A), limitations in activities of daily living (ADLs) (C), chronic disease diagnoses (E), and self-rated health (G)) and with healthspan-related characteristics and mortality measured longitudinally through the end of the 2018 wave (right column: mortality (B), incident ADL limitations (D), incident chronic disease diagnoses (F), and change in self-rated health (H)). For functional impairment, prevalent and incident chronic conditions, and prevalent and incident ADL limitations, effect sizes are incidence rate ratios (IRRs) for 1–standard-deviation (SD) increases in biological-aging measures estimated from Poisson regression. For cross-sectional self-rated health and change in self-rated health, effect sizes are Pearson r’s for 1-SD increases in biological-aging measures estimated from linear regression. For mortality, effect sizes are hazard ratios (HRs) for 1-SD increases in biological-aging measures estimated from Poisson regression. Cross-sectional measures are shown in the left-hand column; longitudinal measures are shown in the right-hand column. Error bars show 95% confidence intervals. For cross-sectional measures, sample sizes (n’s) for the Venous Blood Study (VBS) and VBS DNA methylation (DNAm) samples were 3,721 and 1,676, respectively, for functional impairment; 8,484 and 3,785, respectively, for ADL limitations; 8,482 and 3,784, respectively, for chronic disease diagnoses; and 8,476 and 3,782, respectively, for self-rated health. For longitudinal measures, sample sizes for the VBS and VBS DNAm samples were 8,484 and 3,785, respectively, for mortality; 7,491 and 3,327, respectively, for ADL limitations; 7,497 and 3,329, respectively, for chronic conditions; and 7,488 and 3,326, respectively, for self-rated health.
Racial disparities in healthspan-related characteristics and mortality
Black older adults showed deficits in healthspan-related characteristics and increased risk of mortality as compared with White older adults.
In the HRS VBS sample, Black participants more often demonstrated functional impairments than White participants (IRR = 1.25, 95% CI: 1.15, 1.35) and reported more ADL limitations (IRR = 1.91, 95% CI: 1.75, 2.10) and chronic conditions (IRR = 1.26, 95% CI: 1.20, 1.32) and poorer self-rated health (Cohen’s d = 0.33, 95% CI: 0.28, 0.39). Over follow-up, Black participants were more likely to report incident ADL limitations (IRR = 1.49, 95% CI: 1.31, 1.69) and declines in self-rated health (d = 0.11, 95% CI: 0.06, 0.15). Black-White differences in mortality risk were in the expected direction but not statistically different from 0 at the α = 0.05 threshold (HR = 1.36, 95% CI: 0.97, 1.91). Black-White differences were in the opposite direction for incident chronic conditions (IRR = 0.93, 95% CI: 0.79, 1.11). Effect sizes were similar in the VBS DNAm subsample. Results of both comparisons are reported in Web Table 5.
Black-White disparities in biological aging
Black older adults showed more advanced biological aging than White older adults.
In blood-chemistry analysis, Black participants showed more advanced biological aging than White participants across all 3 measures (Cohen’s d range = 0.35–0.50). In DNAm analysis, the GrimAge clock and the DunedinPoAm pace-of-aging measure indicated more advanced/faster biological aging in Black participants than in White participants (GrimAge clock: d = 0.36 (95% CI: 0.25, 0.48); DunedinPoAm: d = 0.38 (95% CI: 0.24, 0.51)). In contrast, the first-generation Horvath and Hannum clocks and the PhenoAge clock did not (Horvath clock: d = −0.02 (95% CI: −0.10, 0.07); Hannum clock: d = −0.37 (95% CI: −0.49, –0.26); PhenoAge clock: d = 0.08 (95% CI: −0.04, 0.20)). Distributions of biological-aging measures for Black and White participants are shown in Figure 3.
Figure 3.
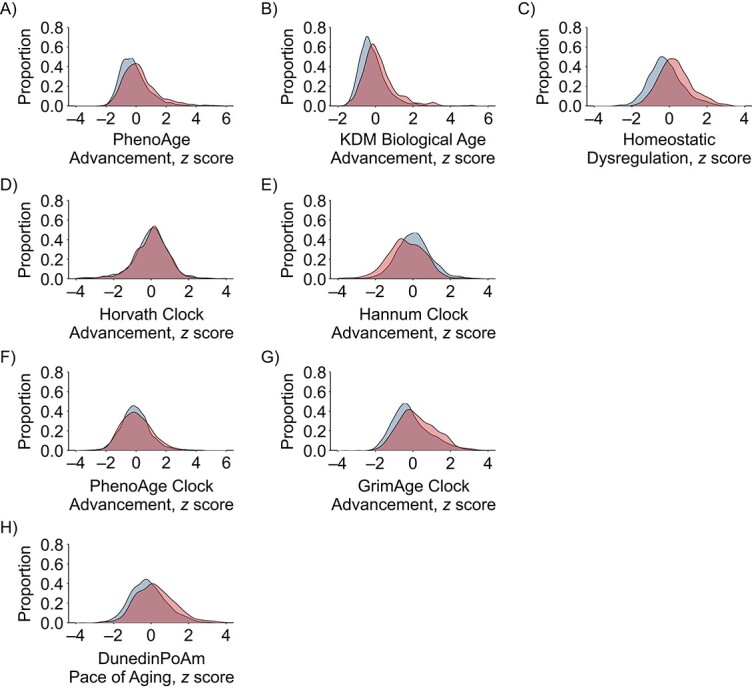
Distributions of 8 biological-aging measures in Black and White participants, Health and Retirement Study, 1992–2018. The graph shows the probability density functions of biological-age advancement for PhenoAge (A), Klemera-Doubal method biological age (B), homeostatic dysregulation (C), Horvath clock advancement (D), Hannum clock advancement (E), PhenoAge clock advancement (F), GrimAge clock advancement (G), and DunedinPoAm pace of aging (H). The distribution of biological-age advancement measures is shown in blue for White participants and in red for Black participants. For blood-chemistry PhenoAge, Klemera-Doubal method (KDM) biological age, and the DNA-methylation clocks, values are biological-age advancements (i.e., the difference between measured biological age and chronological age). For homeostatic dysregulation, values capture blood-chemistry deviation from the norm in a healthy sample. For DunedinPoAm, values are pace of aging (i.e., years of physiological decline experienced per 1 year of calendar time over the recent past). To allow comparison across measures, biological-aging values were standardized to mean = 0 and standard deviation = 1 in the full Venous Blood Study sample. Densities were adjusted for sampling weights. Sampling-weight–adjusted Cohen’s d values are reported in Web Table 9.
Mediation analysis
Black-White differences in healthspan-related characteristics and mortality risk were partly mediated by biological-aging measures.
In blood-chemistry analysis, more advanced PhenoAge and KDM biological age and elevated homeostatic dysregulation in Black as compared with White participants mediated 18%–45% of Black-White differences in healthspan-related characteristics and 60%–95% of the Black-White difference in mortality risk. In DNAm analysis, more advanced GrimAge clock and faster DunedinPoAm pace of aging in Black as compared with White participants mediated 9%–92% of Black-White differences in healthspan-related characteristics and 52%–80% of the Black-White difference in mortality risk. The Horvath, Hannum, and PhenoAge clocks did not indicate more advanced aging in Black as compared with White participants and were not included in mediation analysis. Incident chronic conditions were more common in White as compared with Black older adults and were therefore not included in mediation analysis. For all other healthspan-related characteristics, mediation was stronger in analysis of longitudinal change as compared with cross-sectional differences. Mediation model results for the blood-chemistry PhenoAge and DNAm GrimAge clocks and DunedinPoAm pace of aging are reported in Table 3. Effect-size estimates for Black-White differences in healthspan-related characteristics before and after adjustment for biological-aging measures are reported in Web Table 5. Complete mediation analysis results are reported in Web Tables 6 and 7.
Table 3.
Results of a Mediation Analysis of Black-White Disparities in Healthspan-Related Characteristics, Health and Retirement Study, 1992–2018a
| Measure of Biological Aging | ||||||
|---|---|---|---|---|---|---|
| Outcome | PhenoAge | GrimAge Clock | DunedinPoAm | |||
| Estimate | 95% CI | Estimate | 95% CI | Estimate | 95% CI | |
| Cross-Sectional Measures b | ||||||
| Functional impairments | ||||||
| Indirect effect | 0.04 | 0.03, 0.05 | 0.08 | 0.06, 0.10 | 0.06 | 0.04, 0.08 |
| Total effect | 0.22 | 0.15, 0.29 | 0.18 | 0.09, 0.31 | 0.17 | 0.06, 0.29 |
| Proportion mediated | 0.19 | 0.43 | 0.34 | |||
| No. of ADL limitations | ||||||
| Indirect effect | 0.12 | 0.09, 0.14 | 0.10 | 0.07, 0.14 | 0.08 | 0.05, 0.11 |
| Total effect | 0.63 | 0.52, 0.74 | 0.59 | 0.37, 0.80 | 0.59 | 0.41, 0.75 |
| Proportion mediated | 0.18 | 0.16 | 0.13 | |||
| No. of chronic conditions | ||||||
| Indirect effect | 0.07 | 0.05, 0.08 | 0.06 | 0.04, 0.07 | 0.04 | 0.03, 0.06 |
| Total effect | 0.23 | 0.19, 0.25 | 0.22 | 0.16, 0.27 | 0.22 | 0.16, 0.28 |
| Proportion mediated | 0.29 | 0.26 | 0.19 | |||
| Self-rated health | ||||||
| Indirect effect | 0.10 | 0.08, 0.12 | 0.10 | 0.08, 0.13 | 0.07 | 0.05, 0.09 |
| Total effect | 0.33 | 0.27, 0.40 | 0.28 | 0.20, 0.37 | 0.28 | 0.20, 0.37 |
| Proportion mediated | 0.29 | 0.37 | 0.24 | |||
| Longitudinal Measures c | ||||||
| Mortality | ||||||
| Indirect effect | 0.16 | 0.12, 0.21 | 0.26 | 0.18, 0.35 | 0.16 | 0.09, 0.25 |
| Total effect | 0.27 | −0.22, 0.57 | 0.32 | −0.25, 0.81 | 0.31 | −0.27, 0.85 |
| Proportion mediated | 0.60 | 0.80 | 0.52 | |||
| Change in no. of ADL limitations | ||||||
| Indirect effect | 0.07 | 0.04, 0.09 | 0.10 | 0.05, 0.15 | 0.08 | 0.04, 0.14 |
| Total effect | 0.39 | 0.18, 0.56 | 0.10 | −0.23, 0.45 | 0.11 | −0.25, 0.35 |
| Proportion mediated | 0.17 | 0.92 | 0.75 | |||
| Change in no. of chronic conditions | ||||||
| Total effect | −0.07 | −0.27, 0.07 | −0.19 | −0.44, 0.12 | −0.18 | −0.46, 0.11 |
| Change in self-rated health | ||||||
| Indirect effect | 0.02 | 0.01, 0.03 | 0.02 | 0.01, 0.03 | 0.02 | 0.01, 0.03 |
| Total effect | 0.10 | 0.06, 0.15 | 0.10 | 0.03, 0.16 | 0.10 | 0.03, 0.17 |
| Proportion mediated | 0.18 | 0.22 | 0.22 | |||
Abbreviations: ADLs activities of daily living; CI, confidence interval; DNAm, DNA methylation; HRS, Health and Retirement Study; VBS, Venous Blood Study.
a The table shows parameter estimates and proportion-mediation calculations from mediation analysis. Results are presented for analysis of different healthspan-related characteristics and mortality in the table rows. For each outcome, there are 3 sections of results corresponding to the PhenoAge blood-chemistry measure (left), the GrimAge DNA methylation clock (center), and the DunedinPoAm pace of aging (right). For each outcome-aging measure pair, the analysis estimated results from the mediation model under the assumption that associations between biological-aging measures and healthspan-related characteristics/mortality were the same in the Black and White subsamples (i.e., no exposure-mediator interaction). Indirect effect estimates represent the portion of the Black-White disparity mediated through the biological-aging measure. Total effect estimates represent the Black-White healthspan disparity. The proportion mediated was computed as a ratio of the indirect effect to the total effect. Total effect estimates varied between the first section of results (for blood-chemistry PhenoAge) and the second 2 sections (GrimAge DNA methylation clock and DunedinPoAm) because of the difference in sample size. Differences in total effect estimates between the final 2 sections are due to rounding within the CMAverse R package (39).
b Mediation analysis of cross-sectional data collected in the 2016 wave of the HRS. Sample sizes (n) for the HRS VBS and VBS DNAm samples were 3,621 and 1,596, respectively, for functional impairment; 8,197 and 3,598, respectively, for ADL limitations; 8,195 and 3,597, respectively, for chronic conditions; and 8,190 and 3,595, respectively, for self-rated health.
c Mediation analysis of longitudinal data in which measures of biological aging were collected in the 2016 wave of the HRS and mortality, incident ADL limitations and chronic conditions, and changes in self-rated health were measured from baseline (2016) through follow-up in the 2018 data collection wave. Sample sizes (n) for the HRS VBS and VBS DNAm samples were 8,197 and 3,598, respectively, for mortality; 8197 and 3598, respectively, for ADL limitations; 7,229 and 3,164, respectively, for chronic conditions; and 7,228 and 3,163, respectively, for self-rated health.
Sensitivity analysis
Standard mediation analysis assumes that the association between the mediator and the outcome is consistent across levels of exposure. We conducted sensitivity analysis to evaluate this assumption and to test the robustness of mediation results when this assumption was relaxed (Web Appendix 3). Overall, association magnitudes tended to be smaller for analysis of Black participants than for White participants, although tests of exposure-mediator interactions were mostly not statistically significant at the α = 0.05 level (Web Tables 3, 4, and 8). When we relaxed the mediation-analysis assumption that association magnitudes were the same for Black and White subsamples, mediation proportions were reduced. The magnitude of this reduction varied from near-0 to as much as 50% (see “exposure-mediator interaction” columns of Web Tables 6 and 7).
DISCUSSION
We investigated biological aging as a mediator of Black-White disparities in healthspan-related characteristics in the HRS. In the HRS VBS and VBS DNAm samples, Black participants experienced more functional impairments, more difficulties with ADLs, and higher incidence of ADL limitations over the course of follow-up; more chronic disease diagnoses, poorer self-rated health, and greater declines in self-rated health over follow-up; and increased risk of mortality as compared with White participants. Black participants also showed more advanced/faster biological aging based on the 3 blood-chemistry measures we analyzed, the DNAm GrimAge clock, and the DunedinPoAm pace-of-aging measure. In mediation analysis, these measures of more advanced/faster biological aging accounted for up to 95% of Black-White differences in healthspan-related characteristics and mortality. These findings are consistent with the weathering hypothesis that racially patterned determinants of health accelerate biological aging, contributing to Black-White disparities in healthspan (19, 20), and provide limited proof of concept for use of quantifications of biological aging in health disparities research.
Measures of biological aging have been suggested as surrogate endpoints for trials testing therapies to prolong healthy life span based on evidence that they predict aging-related changes in health, functioning, and mortality risk (42–44). No measures of biological aging have yet been tested in randomized clinical trials that include measurements of primary endpoints related to healthy life span. As a result, all fall short of the Food and Drug Administration’s criterion that validated surrogate endpoints be reliable predictors of clinical benefit (45).
Our findings offer mixed support for the measures we studied as candidate surrogate endpoints (i.e., proposed surrogates for which prediction of clinical benefit is not yet established) (45). Specifically, a criterion for a candidate surrogate endpoint is the robustness of associations between the candidate surrogate and primary outcomes across population subgroups. This robustness is not yet established for measures of biological aging, especially in the case of DNAm-based measures, for which algorithms were developed using data from mostly White European samples (e.g., see Levine (46)). Our analysis found mixed support for a hypothesis of consistent associations across Black and White older adults. Effect sizes tended to be smaller for analyses of Black as compared with White participants. However, the same Black-White differences in effect sizes were also observed for analysis of chronological age, indicating that differential precision of aging measures between Black and White participants was likely not the cause. Instead, our data may reflect that Black Americans are disproportionately subject to non–aging-related causes of disease and disability, such as injury or accidents, which would not be captured in measures of biological aging. Therefore, while our findings do not yet establish validity of biological-aging measures as candidate surrogate endpoints, they do support cautious interpretation of group differences in these measures of biological aging as evidence of disparities in healthy aging.
Our findings suggest guidance for future studies. The conceptual model guiding our study proposes that Black-White disparities in healthspan arise from faster/more advanced biological aging among Black Americans than among White Americans. In our analysis, biological-aging measurements from the Horvath, Hannum, and PhenoAge clocks did not fit this model; they showed no Black-White differences or showed differences in the opposite direction. These clocks may not be well-suited to characterizing Black-White disparities in healthy aging. The HRS result that PhenoAge clock values did not differ between Black and White participants contrasts with a report from the Women’s Health Initiative showing more advanced biological aging in Black women than in White women using this measure (23). Follow-up in additional cohorts is needed. Results also contribute evidence that first-generation DNAm clocks—developed to predict chronological age—are both less predictive of healthspan-related characteristics and less sensitive to exposures that shorten healthy life span, as compared with blood-chemistry–derived measures, newer DNAm clocks developed to predict mortality, and DunedinPoAm DNAm pace of aging (26, 28, 37, 47, 48). Future studies using DNAm to investigate biological aging as a mediator between risk exposures and healthy-aging phenotypes, especially in the context of health disparities, may be best served by a focus on second-generation DNAm clocks, especially the GrimAge clock, and DunedinPoAm pace-of-aging measures.
We acknowledge that this study had limitations. There is no gold standard measure of the construct of biological aging (49). Our conclusions regarding biological aging as a candidate mediator of health disparities could be specific to the measures we analyzed. However, consistent evidence across different biological substrates and measurement methods builds confidence that results do reflect aging processes. DNAm measures of aging may reflect variation in the white blood cell composition of samples from which DNA is extracted (50). The sensitivity of our results to this variation could not be tested because the HRS has not yet released whole-genome DNAm data or estimates of white blood cell proportions. Mortality selection may bias results toward the null. Many individuals born in the same years as the HRS participants whose data we analyzed will not have survived to the time of HRS data collection, especially Black Americans, who face shorter life expectancies than White Americans (51). If survivors aged more slowly than those who died at younger ages, our analysis could have underestimated Black-White differences in biological aging and healthspan. Therefore, our estimates of disparities are likely to have been conservative. Indirect-effect estimates in our mediation models are conditional on the assumption that there are no common causes of biological aging and healthspan-related characteristics omitted from the model. To the extent that these causes exist, our estimates of the proportion mediated may have been biased upwards. Finally, there is the possibility of detection and reporting bias. For example, racial disparities in chronic disease might be underestimated if White participants were more likely to be diagnosed due to greater access to health care (52, 53).
Within the bounds of these limitations, our findings have implications for future research and public health surveillance. More advanced/faster biological aging in Black HRS participants than in White participants and the potential role of these differences in mediating Black-White health disparities highlight the need for studies of when and how Black-White differences in biological aging arise. Life-course longitudinal studies are needed to establish when aging trajectories begin to diverge for Black and White Americans. Studies are also needed to identify life-course phenomena through which racism and socioeconomic resource differentials drive faster aging in Black Americans compared with White Americans. Deficits in maternal and perinatal health, social exclusion and victimization in young adulthood, occupational exposures during young adulthood and midlife, and lack of access to health care later in life all represent potential drivers of Black-White disparities in aging.
Our results suggest promise for the application of biological-aging measures for evaluating and monitoring Black-White disparities in healthy aging, particularly the GrimAge clock and DunedinPoAm pace of aging, with the caveat that these measures do not provide a complete summary of processes driving Black-White health disparities. These same measures can, in parallel, provide new outcome measures for evaluations of social policy experiments. A primary application of biological-aging measures within the emerging field of geroscience is to provide surrogate endpoints for extension of healthy life span (43, 54). Results from this study suggest they may also have utility in trials of interventions that aim to eliminate health disparities by repairing inequalities in social determinants of health.
Supplementary Material
ACKNOWLEDGMENTS
Author affiliations: Department of Epidemiology, Mailman School of Public Health, Columbia University, New York, New York, United States (Gloria H. Graf, Christopher L. Crowe, Daniel W. Belsky); Robert N. Butler Columbia Aging Center, Mailman School of Public Health, Columbia University, New York, New York, United States (Gloria H. Graf, Christopher L. Crowe, Meeraj Kothari, Dayoon Kwon, Daniel W. Belsky); Psychiatric Epidemiology Training Program, Mailman School of Public Health, Columbia University, New York, New York, United States (Christopher L. Crowe); Taub Institute for Research on Alzheimer’s Disease and the Aging Brain, College of Physicians and Surgeons, Columbia University, New York, New York, United States (Jennifer J. Manly, Indira C. Turney); Gertrude H. Sergievsky Center, College of Physicians and Surgeons, Columbia University, New York, New York, United States (Jennifer J. Manly, Indira C. Turney); Department of Neurology, College of Physicians and Surgeons, Columbia University, New York, New York, United States (Jennifer J. Manly, Indira C. Turney); and Department of Biostatistics, Mailman School of Public Health, Columbia University, New York, New York, United States (Linda Valeri).
This research was supported by the National Institute on Aging (grants R01AG061378 and R01AG066887), the Russell Sage Foundation (grant 1810-08987), and the Jacobs Foundation. C.L.C. was supported by a predoctoral fellowship from the National Institute of Mental Health (grant 5T32MH013043). D.W.B. is a fellow of the Canadian Institute for Advanced Research Child Brain Development Network.
Data are available from the US Health and Retirement Study (https://hrs.isr.umich.edu/data-products). Analysis code is available on GitHub (59).
D.W.B. is an inventor of a Duke University (Durham, North Carolina) and University of Otago (Dunedin, New Zealand) epigenetic aging algorithm, DunedinPACE, which has been licensed to TruDiagnostic (Lexington, Kentucky).
REFERENCES
- 1. Chowkwanyun M, Reed AL Jr. Racial health disparities and Covid-19—caution and context. N Engl J Med. 2020;383(3):201–203. [DOI] [PubMed] [Google Scholar]
- 2. Bagby SP, Martin D, Chung ST, et al. From the outside in: biological mechanisms linking social and environmental exposures to chronic disease and to health disparities. Am J Public Health. 2019;109(suppl 1):S56–S63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mehra R, Keene DE, Kershaw TS, et al. Racial and ethnic disparities in adverse birth outcomes: differences by racial residential segregation. SSM Popul Health. 2019;8:100417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jones NL, Gilman SE, Cheng TL, et al. Life course approaches to the causes of health disparities. Am J Public Health. 2019;109(suppl 1):S48–S55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brown AF, Ma GX, Miranda J, et al. Structural interventions to reduce and eliminate health disparities. Am J Public Health. 2019;109(suppl 1):S72–S78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Adler NE, Rehkopf DH. US disparities in health: descriptions, causes, and mechanisms. Annu Rev Public Health. 2008;29(1):235–252. [DOI] [PubMed] [Google Scholar]
- 7. Phelan JC, Link BG, Tehranifar P. Social conditions as fundamental causes of health inequalities: theory, evidence, and policy implications. J Health Soc Behav. 2010;51(1 suppl):S28–S40. [DOI] [PubMed] [Google Scholar]
- 8. Anderson AC, O’Rourke E, Chin MH, et al. Promoting health equity and eliminating disparities through performance measurement and payment. Health Aff (Millwood). 2018;37(3):371–377. [DOI] [PubMed] [Google Scholar]
- 9. Paskett E, Thompson B, Ammerman AS, et al. Multilevel interventions to address health disparities show promise in improving population health. Health Aff (Millwood). 2016;35(8):1429–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Butler M, McCreedy E, Schwer N, et al. Improving Cultural Competence to Reduce Health Disparities. (AHRQ Comparative Effectiveness Reviews, no. 170). Rockville, MD: Agency for Healthcare Research and Quality; 2016. (Report no. 16-EHC006-EF). [PubMed] [Google Scholar]
- 11. Agurs-Collins T, Persky S, Paskett ED, et al. Designing and assessing multilevel interventions to improve minority health and reduce health disparities. Am J Public Health. 2019;109(suppl 1):S86–S93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thornton RL, Glover CM, Cené CW, et al. Evaluating strategies for reducing health disparities by addressing the social determinants of health. Health Aff (Millwood). 2016;35(8):1416–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Braveman P, Barclay C. Health disparities beginning in childhood: a life-course perspective. Pediatrics. 2009;124(suppl 3):S163–S175. [DOI] [PubMed] [Google Scholar]
- 14. Moffitt TE, Belsky DW, Danese A, et al. The longitudinal study of aging in human young adults: knowledge gaps and research agenda. J Gerontol A Biol Sci Med Sci. 2017;72(2):210–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kirkwood TB. Understanding the odd science of aging. Cell. 2005;120(4):437–447. [DOI] [PubMed] [Google Scholar]
- 16. Kennedy BK, Berger SL, Brunet A, et al. Geroscience: linking aging to chronic disease. Cell. 2014;159(4):709–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barzilai N, Cuervo AM, Austad S. Aging as a biological target for prevention and therapy. JAMA. 2018;320(13):1321–1322. [DOI] [PubMed] [Google Scholar]
- 18. Bellantuono I. Find drugs that delay many diseases of old age. Nature. 2018;554(7692):293–295. [DOI] [PubMed] [Google Scholar]
- 19. Geronimus AT, Hicken M, Keene D, et al. “Weathering” and age patterns of allostatic load scores among blacks and whites in the United States. Am J Public Health. 2006;96(5):826–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Geronimus AT. The weathering hypothesis and the health of African-American women and infants: evidence and speculations. Ethn Dis. 1992;2(3):207–221. [PubMed] [Google Scholar]
- 21. Levine ME, Crimmins EM. Evidence of accelerated aging among African Americans and its implications for mortality. Soc Sci Med. 2014;118:27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Parker DC, Bartlett BN, Cohen HJ, et al. Association of blood chemistry quantifications of biological aging with disability and mortality in older adults. J Gerontol A Biol Sci Med Sci. 2020;75(9):1671–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu Z, Chen BH, Assimes TL, et al. The role of epigenetic aging in education and racial/ethnic mortality disparities among older US women. Psychoneuroendocrinology. 2019;104:18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ferrucci L, Gonzalez-Freire M, Fabbri E, et al. Measuring biological aging in humans: a quest. Aging Cell. 2020;19(2):e13080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jylhävä J, Pedersen NL, Hägg S. Biological age predictors. EBioMedicine. 2017;21:29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Belsky DW, Moffitt TE, Cohen AA, et al. Eleven telomere, epigenetic clock, and biomarker-composite quantifications of biological aging: do they measure the same thing? Am J Epidemiol. 2018;187(6):1220–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jansen R, LKM H, Verhoeven JE, et al. An integrative study of five biological clocks in somatic and mental health. Elife. 2021;10:e59479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li X, Ploner A, Wang Y, et al. Longitudinal trajectories, correlations and mortality associations of nine biological ages across 20-years follow-up. Elife. 2020;9:e51507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu Z, Chen X, Gill TM, et al. Associations of genetics, behaviors, and life course circumstances with a novel aging and healthspan measure: evidence from the Health and Retirement Study. PLoS Med. 2019;16(6):e1002827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Crimmins EM, Thyagarajan B, Levine ME, et al. Associations of age, sex, race/ethnicity and education with 13 epigenetic clocks in a nationally representative us sample: the Health and Retirement Study. J Gerontol A Biol Sci Med Sci. 2021;76(6):1117–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Horvath S, Gurven M, Levine ME, et al. An epigenetic clock analysis of race/ethnicity, sex, and coronary heart disease. Genome Biol. 2016;17(1):171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. HRS Staff . Sample Sizes and Response Rates. Ann Arbor, MI: Survey Research Center, Institute for Social Research, University of Michigan; 2017. [Google Scholar]
- 33. Health and Retirement Study . RAND HRS Longitudinal File 2018 (V1). (Public use data set). Ann Arbor, MI: Survey Research Center, Institute for Social Research, University of Michigan; 2021. [Google Scholar]
- 34. Crimmins EM, Faul JD, Thyagarajan B, et al. Venous Blood Collection and Assay Protocol in the 2016 Health and Retirement Study. Ann Arbor, MI: Survey Research Center, Institute for Social Research, University of Michigan; 2017. [Google Scholar]
- 35. Kwon D, Belsky DW. A toolkit for quantification of biological age from blood-chemistry and organ-function-test data: BioAge. Geroscience. 2021;43(6):2795–2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14(10):R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Belsky DW, Caspi A, Arseneault L, et al. Quantification of the pace of biological aging in humans through a blood test, the DunedinPoAm DNA methylation algorithm. Elife. 2020;9:e54870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Valeri L, Vanderweele TJ. Mediation analysis allowing for exposure–mediator interactions and causal interpretation: theoretical assumptions and implementation with SAS and SPSS macros. Psychol Methods. 2013;18(2):137–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shi B, Choirat C, Coull BA, et al. CMAverse: a suite of functions for reproducible causal mediation analyses. Epidemiology. 2021;32(5):e20–e22. [DOI] [PubMed] [Google Scholar]
- 40. Valeri L, Lin X, Vanderweele TJ. Mediation analysis when a continuous mediator is measured with error and the outcome follows a generalized linear model. Stat Med. 2014;33(28):4875–4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ward JB, Gartner DR, Keyes KM, et al. How do we assess a racial disparity in health? Distribution, interaction, and interpretation in epidemiological studies. Ann Epidemiol. 2019;29:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Justice JN, Ferrucci L, Newman AB, et al. A framework for selection of blood-based biomarkers for geroscience-guided clinical trials: report from the TAME Biomarkers Workgroup. Geroscience. 2018;40(5-6):419–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Justice JN, Kritchevsky SB. Aging and geroscience: putting epigenetic biomarkers to the test for clinical trials. Elife. 2020;9:e58592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kritchevsky SB, Justice JN. Testing the geroscience hypothesis: early days. J Gerontol A Biol Sci Med Sci. 2020;75(1):99–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. US Food and Drug Administration . Surrogate endpoint resources for drug and biologic development. https://www.fda.gov/drugs/development-resources/surrogate-endpoint-resources-drug-and-biologic-development. Published July 24, 2018. Accessed October 19, 2021.
- 46. Levine ME. Assessment of epigenetic clocks as biomarkers of aging in basic and population research. J Gerontol A Biol Sci Med Sci. 2020;75(3):463–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lu AT, Quach A, Wilson JG, et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging (Albany NY). 2019;11(2):303–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fiorito G, McCrory C, Robinson O, et al. Socioeconomic position, lifestyle habits and biomarkers of epigenetic aging: a multi-cohort analysis. Aging (Albany NY). 2019;11(7):2045–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cohen AA, Kennedy BK, Anglas U, et al. Lack of consensus on an aging biology paradigm? A global survey reveals an agreement to disagree, and the need for an interdisciplinary framework. Mech Ageing Dev. 2020;191:111316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mill J, Heijmans BT. From promises to practical strategies in epigenetic epidemiology. Nat Rev Genet. 2013;14(8):585–594. [DOI] [PubMed] [Google Scholar]
- 51. E A, J X, Kochanek KD. United States life tables, 2016. Natl Vital Stat Rep. 2019;68(4):1–66. [PubMed] [Google Scholar]
- 52. Virnig BA, Baxter NN, Habermann EB, et al. A matter of race: early-versus late-stage cancer diagnosis. Health Aff (Millwood). 2009;28(1):160–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Geiger HJ. Racial and ethnic disparities in diagnosis and treatment: a review of the evidence and a consideration of causes. In: Smedley BD, Stith AY, Nelson AR, eds. Unequal Treatment: Confronting Racial and Ethnic Disparities in Health Care. Washington, DC: National Academies Press; 2003:417–454. [PubMed] [Google Scholar]
- 54. Belsky DW, Huffman KM, Pieper CF, et al. Change in the rate of biological aging in response to caloric restriction: CALERIE biobank analysis. J Gerontol A Biol Sci Med Sci. 2017;73(1):4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Levine ME, Lu AT, Quach A, et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging (Albany NY). 2018;10(4):573–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Klemera P, Doubal S. A new approach to the concept and computation of biological age. Mech Ageing Dev. 2006;127(3):240–248. [DOI] [PubMed] [Google Scholar]
- 57. Cohen AA, Milot E, Yong J, et al. A novel statistical approach shows evidence for multi-system physiological dysregulation during aging. Mech Ageing Dev. 2013;134(3-4):110–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hannum G, Guinney J, Zhao L, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49(2):359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Graf GH, Crowe CL, Kothari M, et al. gogogoglo/BioAge_SDH. https://github.com/gogogoglo/BioAge_SDH. Accessed January 13, 2021.
- 60. Kwon D. dayoonkwon/BioAge. https://github.com/dayoonkwon/BioAge/. Accessed January 24, 2021.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.