

Abstract
N6-methyladenosine (m6A) methylation, which is modified by the METTL3/METTL14 complex, is a dominant internal modification in mammalian RNA and tightly linked to cancer progression. Here we reveal that METTL3-promoted cell migration, invasion, and epithelial-to-mesenchymal transition (EMT) are associated with expression and membrane localization of β-catenin (encoded by CTNNB1), as opposed to Wnt signaling activation in various types of cancer cells, including cervical, lung, and liver cancer. Specifically, METTL3 regulates the transcription, mRNA decay, translation, and subcellular localization of β-catenin. For CTNNB1 expression, METTL3 indirectly suppresses CTNNB1 transcription by stabilizing its transcription suppressor E2F1 mRNA; deposition of 5′UTR m6A in CTNNB1 promotes its decay in a content-dependent manner via YTHDF2 recognition; 5′UTR m6A in CTNNB1 suppresses its translation efficiency, whereas the global METTL3 level controls the canonical and non-canonical translation of CTNNB1, which is probably associated with the interaction between YTHDF1 and eIF4E1/eIF4E3. For β-catenin translocation, METTL3 represses membrane localization of β-catenin and its interaction with E-cadherin by downregulating c-Met kinase, leading to inhibition of cell motility. In vitro, in vivo, and clinical analyses confirm the essential role of β-catenin and its expression regulators in cancer cell dissemination. The findings not only expand our understanding of m6A modification and its roles in gene expression and subcellular localization of targets but also suggest that the METTL3/β-catenin axis might be a potential target to inhibit cancer metastasis.
Keywords: METTL3, β-catenin, E-cadherin, EMT, cancer cells
Graphical abstract
Introduction
N6-methyladenosine (m6A) is the most prevalent internal post-transcriptional modification in eukaryotic mRNAs and non-coding RNAs.1 This dynamic and reversible modification process is mediated by the “writer” proteins methyltransferase-like 3 and 14 (METTL3 and METTL14, respectively) and other cofactors, which transfer the methyl group from the donor S-adenosyl methionine (SAM) to target adenosine in the consensus m6A motif (GGAC), and the “eraser” proteins alkylation repair homolog protein 5 (ALKBH5) and fat mass and obesity-associated protein (FTO), which demethylate m6A.2,3 The subsequent outcome of m6A depends on the binding of different reader proteins to m6A. For example, the YT521-B homology (YTH) domain-containing readers YTHDF1/2/3 and YTHDC1/2 regulate mRNA splicing,4 translation,5 and decay.6 The heterogeneous nuclear ribonucleoproteins (HNRNPs) A2/B1 are associated with pri-mRNA processing,7 and insulin growth factor 2 (IGF2) mRNA-binding proteins (IGF2BPs, including IGF2BP1/2/3) promote mRNA stability and translation.8
Recent studies show that m6A methylation is critical for RNA metabolism and processing, such as transcription9, splicing,10,11 export,12,13 translation,5 and degradation.6 Therefore, perturbation of m6A modification in mRNA leads to dysregulation of many biological processes, including development of diseases such as cancer. For instance, METTL3 maintains pluripotency of mouse and human acute myeloid leukemia (AML) by regulating mRNA targets such as MYC.14 A recent study we conducted revealed that m6A is associated with biogenesis of microRNA (miRNA)15 and lncRNA,16 modulation of glycolysis,17 epithelial-to-mesenchymal transition (EMT),18 and chemoresistance19 of cancer cells. Gene-specific demethylation of the epidermal growth factor receptor (EGFR) and MYC by the dm6ACRISPR system can suppress proliferation of cancer cells.20 Further, the level of m6A in peripheral blood RNA has been reported to be a novel predictive biomarker for individuals with gastric cancer.21 These data indicate the critical role of m6A in cancer development.
Metastasis is the main cause of cancer lethality.22 EMT, which is due to loss of cell-cell adhesion, is frequently associated with E-cadherin (E-cad) downregulation, which is known to be the key regulator of cancer metastasis.23 β-Catenin (encoded by CTNNB1) is an intracellular signal transducer in the canonical WNT signaling pathway.24 It can also interact with the cytoplasmic domain of E-cad to suppress EMT of cancer cells.25 Tyrosine phosphorylation in the carboxyl terminus of β-catenin is critical for interaction between β-catenin and E-cad.25 Liu et al.26 have reported the relationship between β-catenin and m6A methylation, demonstrating that METTL3 regulates the Wnt/β-catenin pathway to promote proliferation of hepatoblastoma. Our recent study revealed that CTNNB1 mRNA can be modified by m6A methylation. Further, its methylation increases significantly during transforming growth factor β (TGF-β)-induced EMT of cancer cells,18 hinting at a potential role of CTNNB1 during EMT. Whether m6A methylation regulates the expression of β-catenin and whether β-catenin is involved in m6A-regulated EMT of cancer cells remains unknown.
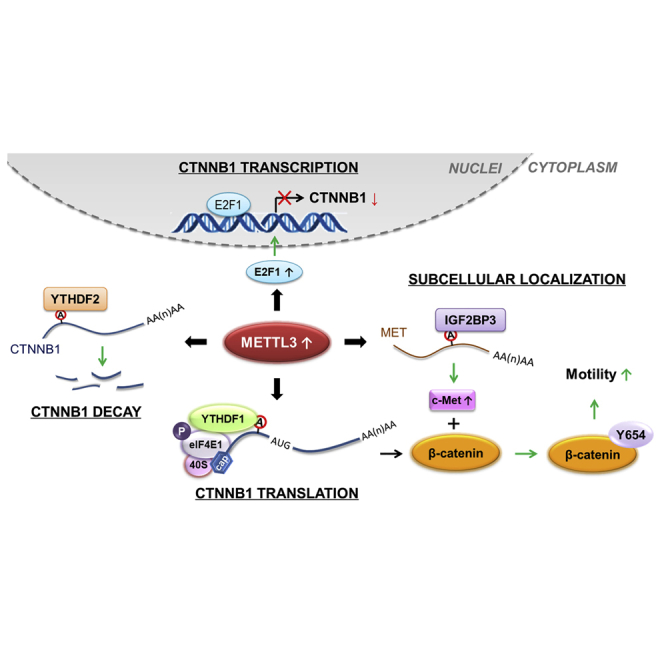
In this study, we demonstrate that β-catenin is critical for METTL3-regulated dissemination of cancer cells. Specifically, METTL3 can, on one hand, negatively modulate the transcription, mRNA decay, and translation efficiency of CTNNB1 mRNA. On the other hand, METTL3 can suppress the membrane localization of β-catenin by modulating expression of the c-Met kinase. Here we propose a comprehensive and novel pathway of METTL3-regulated EMT by modulating β-catenin expression and subcellular localization.
Results
Expression of β-catenin is associated with METTL3 in cancer cells
CTNNB1, which encodes β-catenin, was identified in our previous study as one of the m6A-modified targets involved in EMT (Figure 1A).18 To explore the potential role of CTNNB1 in cancer development, we first examined CTNNB1 expression in tumor and normal tissue. For cervical cancer, lung cancer, and liver cancer (Figures 1B–1D), the results showed lower CTNNB1 expression in tumor tissue than in normal tissue. In line with the oncogenic role of METTL3, higher METTL3 expression was found in tumor tissues (Figures 1B–1D). Kaplan-Meier analysis revealed that individuals with decreased CTNNB1 expression tended to have poorer survival (but there was no statical significance for cervical cancer and liver cancer) (Figures 1E–1G). These results suggest that CTNNB1 may play a role in cancer development.
Figure 1.

Expression of CTNNB1 is associated with METTL3 in cervical, lung, and liver cancers
(A) Overlap of 2-fold m6A expression changes in EMT cells and EMT-related functional genes (GEO: GSE112795). (B–D) Relative mRNA expression of CTNNB1 in Biewenga cervical cancer (B), Hou lung cancer (C), and Mas liver cancer (D) from Oncomine datasets. (E–G) Kaplan-Meier survival curves of overall survival (OS) based on CTNNB1 expression in individuals with cervical cancer with EMT (E), individuals with lung cancer (F), and individuals with liver cancer (G) from the TCGA DB. (H) Expression levels of CTNNB1 mRNA (left) and β-catenin protein (right) in HeLa cells silencing METTL3 were detected by quantitative real-time PCR and western blot, respectively. (I) Protein levels of β-catenin in HeLa and HeLa METTL3 knockdown cells (M3Mut/−) were detected by western blot (left) and analyzed quantitatively (right). Data in (H) and (I) are presented as mean ± SD from three independent experiments. Student’s t test; ∗p < 0.05; ∗∗p < 0.01; NS, not significant compared with control.
Expression of METTL3 positively correlating to β-catenin has been reported.26 However, we found that transiently silencing METTL3 upregulated the mRNA and protein levels of β-catenin in HeLa, H460, and Huh7 cells (Figures 1H, S1A, and S1B). Similar results were obtained in cells stably knocking down METTL3 (Figures 1I and S1C), suggesting that the expression of β-catenin was negatively associated with METTL3. Because knockdown of METTL3 resulted in an overall decrease of m6A methylation levels (Figure S1D), m6A methylation may be involved in METTL3-regulated expression of β-catenin.
Inhibition of β-catenin promotes EMT in METTL3 knockdown cells
Knockdown of METTL3 in HeLa, H460, and Huh7 cells showed decreased cell migration and invasion, as reported previously (Figures S2A and S2B),18 confirming the essential role of METTL3 in dissemination of cancer cells. To investigate whether β-catenin participates in METTL3-regulated EMT of cancer cells, METTL3 knockdown cells silencing β-catenin were used. Interestingly, knockdown of β-catenin (Figure S2C) significantly promoted cell migration (Figure 2A) and invasion (Figure 2B) of HeLa METTL3Mut/− and H460 sh-METTL3 cells. In addition, si-β-catenin attenuated upregulation of E-Cad and downregulation of FN in HeLa METTL3Mut/− and H460 sh-METTL3 cells (Figures 2C and 2D). Our data suggested that β-catenin, which was negatively regulated by METTL3, was involved in METTL3-regulated EMT of cancer cells.
Figure 2.

Inhibition of β-catenin promotes EMT in METTL3 knockdown cancer cells
(A) Wound healing of HeLa, HeLa METTL3Mut/−, H460 sh-NC, and H460 sh-METTL3 cells silencing β-catenin was recorded (left) and analyzed quantitatively (right). Scale bar, 100μm. (B) Cell invasion of HeLa, HeLa METTL3Mut/−, H460 sh-NC, and H460 sh-METTL3 cells silencing β-catenin was recorded (left) and analyzed quantitatively (right). Scale bar, 100μm. (C) Expression levels of CDH1 and FN1 mRNA in HeLa, HeLa METTL3Mut/−, H460 sh-NC, and H460 sh-METTL3 cells transfected with si-NC or si-β-catenin were detected by quantitative real-time PCR. (D)Protein levels of E-Cad and FN in HeLa and HeLa METTL3Mut/− cells were detected by western blot. Data are presented as mean ± SD from three independent experiments. Student’s t test; ∗p < 0.05, ∗∗p < 0.01.
A previous report has shown that m6A modification regulates β-catenin to promote proliferation of hepatoblastoma via activation of Wnt signaling.26 Here, we surprisingly found that knockdown of METTL3 did not affect the Wnt signaling pathway because neither nuclear localization of β-catenin (Figure S2D) nor expression of downstream genes of the Wnt pathway, such as MYC, CD44, AXIN2, and CCND1 (Figure S2E), was affected in HeLa cells. These results suggested that there was an unidentified regulatory mechanism in METTL3-regulated EMT mediated by β-catenin.
METTL3 negatively regulates transcription, mRNA stability, and translation of CTNNB1
CTNNB1 mRNA can be m6A methylated in its 5′UTR in HeLa cells, and its methylation level increased during EMT (Figure 3A; GEO: GSE112795). m6A-RNA immunoprecipitation (RIP)-qPCR results confirmed more than 3-fold m6A-enriched CTNNB1 mRNA in HeLa cells, and this enrichment decreased significantly in HeLa METTL3Mut/− cells (Figure 3B). m6A-enriched CTNNB1 mRNA was also observed in H460 and Huh7 cells, whereas the knockdown of METTL3 decreased enrichment (Figure S3A). Further, overexpression of the demethylase ALKBH5 can significantly decrease m6A methylation of CTNNB1 mRNA in HeLa cells (Figure 3C). Our data confirmed reversible m6A modification of CTNNB1 mRNA in cancer cells.
Figure 3.

METTL3 regulates the transcription, stability, and translation of CTNNB1 mRNA in HeLa cells
(A) m6A peaks were enriched in 5′UTR of CTNNB1 mRNA, as seen by m6A RIP-seq data (GEO: GSE112795). Squares mark the m6A peaks in HeLa cells (control [Con]) and cells undergoing EMT. (B) m6A RIP-qPCR analysis of CTNNB1 mRNA in Con and METTL3Mut/− HeLa cells. Enrichment of CTNNB1 mRNA in m6A RIP samples (IP) was normalized to IgG and sample input. (C) m6A RIP-qPCR analysis of CTNNB1 mRNA in HeLa cells transiently overexpressing empty vector pcDNA3 (Con) or pcDNA3-ALKBH5 (o/e A5). (D) Expression levels of CTNNB1 mRNA in HeLa and HeLa METTL3Mut/− cells were detected by quantitative real-time PCR. (E) Expression levels of CTNNB1 mRNA in HeLa cells overexpressing the empty vector PPB (mock), PPB-METTL3 (M3 wild type [WT]) and PPB-METTL3 mutant (M3 mutant) were detected by quantitative real-time PCR. (F) Expression levels of CTNNB1 pre-mRNA (pre-CTNNB1) in HeLa and HeLa METTL3Mut/− cells were detected by quantitative real-time PCR. (G) HeLa and HeLa METTL3Mut/− cells were co-transfected with the pGL3-Basic-CTNNB1-Fluc reporter and pRL-TK plasmids for 24 h. Results are presented as the ratios between the activity of the reporter plasmid and pRL-TK. (H) Half-life of CTNNB1 mRNA in HeLa and HeLa METTL3Mut/− cells were detected by quantitative real-time PCR. (I and J) Ribosomal profiling of HeLa and HeLa METTL3Mut/− cells (I) and the expression level of CTNNB1 mRNA in 40S, 60S, 80S, and polysome fractions (J). Data are presented as mean ± SD from three independent experiments. Student’s t test; ∗p < 0.05, ∗∗p < 0.01.
We next investigated the effects of m6A methylation of CTNNB1 on its expression. Quantitative real-time PCR results showed upregulation of CTNNB1 mRNA in HeLa METTL3Mut/−, H460 sh-METTL3, and Huh7 sh-METTL3 cells (Figures 3D and S3B). Overexpression of METTL3, but not the catalytically inactive METTL3 mutant DA (D395A),27 can significantly decrease CTNNB1 mRNA in HeLa cells (Figure 3E), indicating an m6A-dependent correlation between METTL3 and β-catenin expression.
We further evaluated the mechanisms of how METTL3 regulated expression of β-catenin. First, the precursor mRNA (pre-mRNA) level of CTNNB1 increased significantly in METTL3 knockdown HeLa, H460, and Huh7 cells (Figures 3F and S3C), and its splicing rate was not varied (Figure S3D), suggesting that the increased level of CTNNB1 pre-mRNA may be due to transcription activation. A dual-luciferase assay confirmed this suggestion and showed increased promoter activity of CTNNB1 in METTL3 knockdown cells (Figures 3G and S3E). Second, a fractionation assay suggested that knockdown of METTL3 did not affect the subcellular localization of CTNNB1 (Figure S2F). Third, the mRNA stability of CTNNB1 increased significantly in METTL3 knockdown HeLa, H460, and Huh7 cells (Figures 2H and S3G). Fourth, knockdown of METTL3 significantly increased the occurrence of CTNNB1 mRNA in translation initiation (40S) and active polysomes (>80S) of HeLa cells, as seen by comparing the ribosomal profiles of control and METTL3Mut/− HeLa cells (Figures 3I and 3J). Fifth, the protein stability of β-catenin was not affected by METTL3 knockdown (Figure S3H). These results suggested that METTL3 negatively regulated the transcription, mRNA stability, and translation of CTNNB1 in cancer cells.
E2F1 is involved in METTL3-suppressed transcription of CTNNB1
We evaluated the potential mechanisms responsible for METTL3-regulated transcription of CTNNB1 in cancer cells. We tested previously reported transcription factors (TFs) of CTNNB1, including the activators FOXP3,28 ZNF191,29 ZBP-89,30 and PWWP2A31 and the suppresser PEG3,32 but none of them matched our observations in control and METTL3 knockdown cells (Figure 4A). To explore potential TFs of CTNNB1, ENCODE chromatin immunoprecipitation sequencing (ChIP-seq) data from ChIPBase33 and CistromeMap34 were analyzed. Among 48 TFs identified by CistromeMap and 57 TFs identified by ChIPBase, 5 TFs (CTCF, E2F1, GABPA, MYC, and YY1) overlapped between two databases (Figure S4A). Among them, E2F1 was identified to be the TF involved in METTL3-regulated transcription of CTNNB1 because its expression varied according to METTL3 level (Figures 4B and S4B).
Figure 4.

E2F1 is involved in METTL3-suppressed transcription of CTNNB1
(A and B) Expression levels of reported TFs (A) and predicted TFs (B) of CTNNB1 in HeLa and HeLa METTL3Mut/− cells were detected by quantitative real-time PCR. (C) Binding between E2F1 and the promoter of CTNNB1 in HeLa, HeLa METTL3Mut/−, H460 sh-NC, and H460 sh-METTL3 cells was detected by ChIP-qPCR. (D and E) Expression levels of CTNNB1 mRNA (D) and β-catenin (E) in HeLa and H460 cells silencing E2F1 were detected by quantitative real-time PCR and western blot, respectively. (F) Schematic of the pGL3 reporter, which contains the CTNNB1 promoter before the F-LUC gene. Sequences of E2F1 binding motifs site 1 and site 2 in the CTNNB1 promoter are listed. The 100% conserved sequences of E2F1 binding motifs are highlighted, and their corresponding mutations pGL3-Mut1 and pGL3-Mut2 (CG to AA) are marked in red. (G) Binding between E2F1 and pGL3-WT, pGL3-Mut1, or pGL3-Mut2 in HeLa cells was analyzed by ChIP-qPCR. (H) Expression of FLUC mRNA in HeLa cells transfected with WT and mutated pGL3 reporters (Mut1 and Mut2) was detected by quantitative real-time PCR, which was normalized to the resistant gene AmpR of the pGL3 reporter. (I) Expression levels of CTNNB1 in HeLa (Con) and HeLa METTL3Mut/− (M3Mut/−) cells silencing E2F1 were detected by quantitative real-time PCR; (J) Protein expression levels of E2F1 in HeLa, HeLa M3Mut/−, H460 sh-NC, and H460 sh-METTL3 cells was detected by western blot. (K) m6A RIP-qPCR analysis of E2F1 mRNA in Con and M3Mut/− HeLa cells. Enrichment of E2F1 mRNA in m6A RIP samples (IP) was normalized to IgG and sample input. (L) Half-life of E2F1 mRNA in HeLa and HeLa M3Mut/− cells was detected by quantitative real-time PCR. (M and N) Correlation between METTL3 and E2F1 in individuals with cervical cancer (M) and individuals with lung cancer (N) from the TCGA DB. (O) Proposed mechanism of E2F1 regulating CTNNB1 transcription. Data in (A)–(L) are presented as mean ± SD from three independent experiments. Student’s t test; ∗∗p < 0.01, ∗∗∗p < 0.001.
ChIP-qPCR results confirmed that E2F1 can bind to the promoter region of CTNNB1 (Figure 4C). Downregulating E2F1 in HeLa and H460 cells increased CTNNB1 mRNA and protein levels (Figures 4D, 4E, and S4C), suggesting that E2F1 may be a transcription suppressor for CTNNB1. Assays on a dual-luciferase reporter derived from the CTNNB1 promoter confirmed this suggestion, showing increased FLUC mRNA in cells silencing E2F1 (Figure S4D). To further verify the interaction between E2F1 and the CTNNB1 promoter, two potential E2F1-binding motifs (Figure S4E) in the CTNNB1 promoter region were mutated (Figure 3F). After mutation, ChIP-qPCR showed weaker interactions between E2F1 and CTNNB1 promoters from pGL3-Mut1 and pGL3-Mut2 (Figure 3G). In addition, upregulation of FLUC mRNA expression of pGL3-Mut1 and pGL3-Mut2 was observed (Figure 3H), indicating that both mutations promoted FLUC transcription. Our results confirmed the critical role of E2F1 in transcription of CTNNB1.
We then questioned whether E2F1 was involved in METTL3-regulated transcription of CTNNB1. The results showed that silencing E2F1 could no longer increase CTNNB1 levels in HeLa METTL3Mut/− cells (Figure 4I), which could be due to downregulation of E2F1 in METTL3-knockdown cells (Figure 4J). Our previous m6A-seq data showed m6A modification peaks on E2F1 mRNA around the stop codon (Figure S4F), which was in line with recent reports.35,36 We further confirmed that there were reversible m6A modifications in E2F1 mRNA (Figure 4K). We determined that m6A methylation of E2F1 may regulate the mRNA stability of E2F1 because a shorter mRNA half-live was observed in HeLa METTL3Mut/− cells (Figure 4L). Accordingly, a positive correlation between E2F1 and METTL3 was observed in cervical cancer and lung cancer tissues (Figures 4M and 4N). Our results suggested that METTL3 regulated E2F1 expression by stabilizing E2F1 mRNA and, therefore, indirectly suppressed transcription of CTNNB1 (Figure 4O).
5′UTR m6A methylation suppressed the mRNA stability of CTNNB1
m6A-RIP-seq showed m6A peaks in 5′UTR CTNNB1 mRNA ranging from chr20:48,600,490–48,600,630 (Figure 3A). m6A-RIP-qPCR using fragmented RNA confirmed the suggestion that only the 5′UTR (not CDS or the 3′UTR) of CTNNB1 mRNA was significantly enriched in the m6A-IP sample (Figure 5A). We hypothesized that 5′UTR m6A of CTNNB1 might be associated with its mRNA stability. To confirm this, we constructed two reporters containing the CTNNB1 5′UTR before the CTNNB1-CDS and F-Luc genes, called the CDS reporter and F-Luc reporter, respectively (Figure 5B). In HeLa METTL3Mut/− cells, the mRNA (Figure 5C) and protein (Figures 5D and 5E) levels of CDS and F-Luc reporters were upregulated. We determined that knockdown of METTL3 could also increase the half-life of CTNNB1-CDS (Figure 5F) or F-Luc (Figure 5G), which was similar to the endogenous CTNNB1 mRNA, suggesting the importance of the CTNNB1 5′UTR for its stability.
Figure 5.

5′UTR m6A methylation destabilizes CTNNB1 mRNA
(A) m6A RIP-qPCR analysis of CTNNB1 mRNA in Con or M3Mut/− HeLa cells using fragmented RNA. (B) Schematic of the CDS reporter and F-Luc reporter, which contains the CTNNB1 5′UTR before CTNNB1 CDS or reporter genes. (C) The CDS reporter and F-Luc reporter were transfected into Con and M3Mut/− HeLa cells, respectively. Expression levels of reporter mRNA were detected by quantitative real-time PCR: CTNNB1 mRNA for the CDS reporter, normalized to endogenous CTNNB1 and GAPDH mRNA levels, and F-LUC mRNA for the F-Luc reporter, normalized to R-LUC mRNA. (D) The CDS reporter was transfected into Con or M3Mut/− HeLa cells for 48 h. Expression levels of exogenous β-catenin (FLAG) were detected by western blot. Band intensities of β-catenin were analyzed using ImageJ and are listed at the bottom of the target bands. (E) The F-Luc reporter was transfected into Con or M3Mut/− HeLa cells for 48 h. A dual-luciferase assay was performed to measure F-Luc production, which was normalized to R-Luc levels. (F) The CDS reporter was transfected into Con or M3Mut/− HeLa cells for 48 h and then treated with Act-D for the indicated times. The expression levels of CTNNB1 mRNA were detected by quantitative real-time PCR. (G) The F-Luc reporter was transfected into Con or M3Mut/− HeLa cells for 48 h and then treated with Act-D for the indicated times. Expression levels of F-LUC mRNA were detected by quantitative real-time PCR. (H) Schematic of m6A sites within CTNNB1 mRNA. Mutations of m6A sites in the CTNNB1 5′UTR are marked in red. (I) WT and mutated CDS reporters were transfected into HeLa cells. Expression levels of exogenous β-catenin (FLAG) were detected by western blot. Band intensities of FLAG-β-catenin were analyzed using ImageJ and are listed at the bottom of the target bands. (J) WT and mutated F-Luc reporters were transfected into HeLa cells. A dual-luciferase assay was performed to measure F-Luc production, which was normalized to R-Luc levels. (K) Schematic of m6A insertions within CTNNB1 mRNA. Insertions of continued m6A motifs in the CTNNB1 5′UTR are marked in orange. (L) WT and CDS reporters with inserted m6A motifs were transfected into HeLa cells. Expression levels of exogenous β-catenin (FLAG) were detected by western blot. Band intensities of FLAG-β-catenin were analyzed using ImageJ and are listed at the bottom of the target bands. (M) WT and F-Luc reporters with inserted GGAC motifs were transfected into HeLa cells. A dual-luciferase assay was performed to measure F-Luc production, which was normalized to R-Luc levels. (N) YTHDF2-RIP analysis of CTNNB1 mRNA in Con and M3Mut/− HeLa cells. (O) CDS reporters were transfected into Con and M3Mut/− HeLa cells. YTHDF2 RIP analysis of reporter mRNA was performed. (P) F-Luc reporters were transfected into Con and M3Mut/− HeLa cells. YTHDF2-RIP analysis of reporter mRNA was performed. (Q) Expression levels of β-catenin in HeLa cells silencing YTHDF2 were detected by western blot. (R) Expression levels of CTNNB1 mRNA in HeLa cells silencing YTHDF2 were detected by quantitative real-time PCR, (S) Con or M3Mut/−- HeLa cells were transfected with si-NC or si-YTHDF2 and then treated with CHX for the indicated time. Expression of CTNNB1 mRNA was detected by quantitative real-time PCR. (T) Mechanism of m6A regulating the stability of CTNNB1 mRNA. Data are presented as mean ± SD from three independent experiments. Student’s t test; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To evaluate the potential roles of m6A in CTNNB1 stability, three m6A motifs in the CTNNB1 5′UTR, identified by m6A-RIP-seq, were mutated (GGAC to GGCC), called Mut-1/2/3, corresponding to the S1/S2/S3 sites in the CDS and F-Luc reporters (Figure 5H). m6A enrichment of each mutant was detected, showing significant decreases in m6A levels after mutation (Figure S5A). Quantitative real-time PCR (Figure S5B) and western blot (Figures 5I and 5J) results showed enhanced expression of the double mutation Mut-1/2 in the CDS and F-Luc reporters. It is important to note that single mutation of CTNNB1-CDS or the F-Luc reporter showed limited effects on expression. This indicated that sites 1 and 2 of the m6A motif in the 5′UTR can synergistically regulate the stability of CTNNB1 mRNA.
To further verify the suppressed effect of the m6A methylated 5′UTR on CTNNB1 expression, three or five continued m6A methylation motifs (GGAC) were inserted into the CTNNB1 5′UTR named Insert-3 and Insert-5, respectively (Figure 5K). Increased m6A methylation of Insert-3 was confirmed (Figure S5A). Western blot and dual-luciferase assays showed that the increased GGAC motifs could decrease the protein expression of the CTNNB1-CDS (Figure 5L) and F-Luc (Figure 5M) reporters. The observed reductions were independent of the protein stability of mutants because the control, Mut-1/2, and Insert-3 had comparable half-lives in a protein stability analysis (Figure S5C). Accordingly, mRNA expression of Insert-3 and Insert-5 decreased significantly compared with the control (Figure S5D). These results confirmed that the m6A deposition in the CTNNB1 5′UTR was responsible for METTL3-regulated expression and mRNA stability of CTNNB1.
The m6A reader YTHDF2, associated with mRNA degradation, has been reported to interact with CTNNB1 in HEK293T cells.20 RIP-qPCR confirmed binding of YTHDF2 to endogenous CTNNB1 mRNA (Figure 5N), CTNNB1-CDS (Figure 5O), and F-Luc (Figure 5P) reporters in HeLa cells, and their interactions significantly decreased in HeLa METTL3Mut/− cells. Silencing YTHDF2 (Figure S5E) significantly increased protein (Figure 5Q) and mRNA (Figure 5R) expression of CTNNB1 because of inhibition of CTNNB1 mRNA degradation (Figure 5S). In addition, mutation of sites 1 and 2 in the 5′UTR stabilized its mRNA secondary structure according to structural predictions (Figure S5F). Our data indicated that methylation of sites 1 and 2 in the 5′UTR m6A motif destabilizes CTNNB1 mRNA via recruitment of YTHDF2 (Figure 5T).
YTHDF1 affects eIF4E1-mediated translation of CTNNB1
We further investigated the mechanisms of METTL3-regulated translation of CTNNB1. The results showed that the translation efficiency of CTNNB1 was significantly greater in METTL3-knockdown HeLa, H460, and Huh7 cells (Figure 6A). Similar observations were obtained using the CTNNB1-CDS and F-Luc reporters (Figure S6A). Further, mutation of the m6A motif enhanced, and insertion of m6A motifs annotated, its translation efficiency in the CDS and F-Luc reporters (Figure 5B), indicating inhibition of the m6A methylated 5′UTR in translation efficiency of CTNNB1 in cancer cells. The m6A reader YTHDF1 is commonly associated with mRNA translation.5 RIP-qPCR showed interaction between YTHDF1 and CTNNB1 mRNA in a METTL3-dependent manner (Figure 6C). Further, transient silencing of YTHDF1 upregulated β-catenin in control and METTL3Mut/− HeLa cells (Figure 6D) without affecting CTNNB1 mRNA levels (Figure S6B). These results suggested that 5′UTR m6A of CTNNB1 inhibited its translation efficiency and that YTHDF1 was involved in METTL3-regulated translation of CTNNB1.
Figure 6.

YTHDF1 affects eIF4E1-mediated translation of CTNNB1
(A) Translation efficiency of CTNNB1 in HeLa, HeLa M3Mut/−, H460 sh-NC, H460 sh-METTL3, Huh7 sh-NC, and Huh7 sh-METTL3 cells was calculated as the quotient of β-catenin protein production divided by mRNA abundance.5 (B) Translation efficiency of the CDS and F-Luc reporters with mutations in Con and M3Mut/− cells was analyzed. (C) Binding between YTHDF1 and CTNNB1 mRNA in Con and M3Mut/− HeLa cells was detected by RIP-qPCR. (D) Expression levels of β-catenin in Con and M3Mut/− HeLa cells silencing YTHDF1 were detected by western blot (left) and analyzed quantitatively by ImageJ (right). (E) RIP analysis of CTNNB1 mRNA in Con and M3Mut/− HeLa cells was performed with antibodies against eIF4E1 (4 E1) and eIF4E3 (4 E3), respectively. (F) Interactions between CTNNB1 mRNA (β-MS2bs) and eIF4E1 S209 or eIF4E3 in Con and M3Mut/− HeLa cells were detected by IP (left) and analyzed quantitatively (right). (G) RIP analysis of CTNNB1 mRNA in Con and M3Mut/− HeLa cells silencing YTHDF1 was performed with antibodies against eIF4E1 (4E1) and eIF4E3 (4E3), respectively. (H) Interactions between YTHDF1 and eIF4E1 or eIF4E3 in Con and M3Mut/− HeLa cells treated with RNase inhibitor (RRI) or RNase were detected by IP. (I) Interactions between 40S and eIF4E1 S209 or eIF4E3 in Con and M3Mut/− HeLa cells were detected by IP (left) and analyzed quantitatively (right). (J) Proposed mechanism of how YTHDF1 controls canonical and non-canonical translation of CTNNB1. Data are presented as mean ± SD from three independent experiments. Student’s t test; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
We hypothesized that 5′UTR m6A of CTNNB1 may participate in its translation initiation (Figure 2J). The eukaryotic translation initiation factors eIF4E1 and eIF4E3, competitively initiating canonical and non-canonical translation,37 respectively, were investigated. RIP-qPCR showed that eIF4E1 and eIF4E3 can interact with CTNNB1 mRNA (Figure 6E). Interestingly, binding of eIF4E1-CTNNB1 decreased but that of eIF4E3-CTNNB1 increased in METTL3Mut/− cells (Figure 6E). This suggested that eIF4E3-mediated non-canonical translation of CTNNB1 may be enhanced in METTL3Mut/− cells. To confirm this, we constructed a CTNNB1 overexpression plasmid with 2 × MS2bs (two MS2 binding loops in the CTNNB1 3′UTR that can interact with MS2 proteins) and performed co-immunoprecipitation (coIP) analysis by hemagglutinin (HA) antibody (Figure 6F). The results suggested that eIF4E3 was more abundant whereas p-S209-eIF4E1 was less abundant on CTNNB1 mRNA in METTL3Mut/− HeLa cells (Figures 6F and S6C). This indicated that knockdown of METTL3 promoted canonical translation but suppressed non-canonical translation of CTNNB1. This suggestion was supported by treating cells with the eIF4E1-specific cap-dependent inhibitor rapamycin,38 which showed a decrease in β-catenin in HeLa cells while being maintained in METTL3Mut/− HeLa cells (Figure S6D). This maintenance cannot be attributed to an increase in β-catenin protein stability because co-treating cells with the protein synthesis inhibitor cycloheximide (CHX) resulted in rapid disappearance of β-catenin (Figure S6D).
Next, we wanted to determine whether YTHDF1 was associated with canonical and non-canonical translation of CTNNB1. After silencing YTHDF1, there was no significant difference in eIF4E1-CTNNB1 interaction between HeLa and METTL3Mut/− HeLa cells (compare Figure 6G with Figure 6E), indicating that YTHDF1 was associated with eIF4E1-mediated canonical translation of CTNNB1. Remarkably, the eIF4E3-CTNNB1 interaction remained significant after si-YTHDF1. This hints at a complicated and unknown mechanism of non-canonical translation of CTNNB1 (Figure 6G).
To explore how YTHDF1 regulated the translation, we performed coIP between YTHDF1 and eIF4E1/eIF4E3. The results revealed that YTHDF1 can interact with eIF4E1 and eIF4E3 in an RNA-independent manner (Figure 6H). Notably, YTHDF1 prefers binding to eIF4E1 rather than eIF4E3 in METTL3 knockdown cells, hinting that the global METTL3 level may affect canonical and non-canonical translation in cells, probably in a YTHDF1-regulated manner. To confirm this, we analyzed the interactions of eIF4E1-RPS6 (40S ribosome) and eIF4E3-RPS6. Interestingly, there was significant decrease in the p-S209-eIF4E1 and 40S ribosome but not the eIF4E3 and 40S ribosome interaction in METTL3Mut/− HeLa cells (Figure 6I), indicating that canonical translation mediated by eIF4E1 was suppressed in METTL3 knockdown cells. These results indicate that cellular METTL3 levels regulate canonical and non-canonical translation in cells, which is most likely associated with the interaction between YTHDF1 and free eIF4E1 or eIF4E3 (Figure 6J). However, the detailed mechanism requires further exploration, especially for METTL3-related non-canonical translation.
METTL3 regulates membrane localization of β-catenin via the c-Met kinase
Subcellular localization of β-catenin is critical for its functions: β-catenin interacts with cadherins at the cell membrane to regulate cell-cell adhesion, whereas cytoplasmic β-catenin transports to the nucleus to activate Wnt signaling.39 To investigate the potential roles of METTL3 in subcellular localization of β-catenin, a fractionation assay was performed. The results suggested that increasing amounts of β-catenin were located in the cell membrane fraction but not in the cytoplasmic or nuclear fractions of METTL3Mut/− HeLa cells (Figure 7A). E-Cad is the main interacting protein of β-catenin in the cell membrane. CoIP analysis showed increased interaction between β-catenin and E-Cad in METTL3Mut/− HeLa cells (Figure 7B). It indicates that knockdown of METTL3 can increase the membrane localization of β-catenin and its interaction with E-Cad.
Figure 7.

METTL3 regulates membrane localization of β-catenin via Met kinase
(A) Expression levels of β-catenin and E-Cad in membrane (Mem), cytoplasmic (Cyto), and nuclear (Nu) fractions from Con and M3Mut/− HeLa cells were detected by western blot (left) and analyzed quantitatively (right). (B) Interactions between β-catenin and E-Cad in Con and M3Mut/− HeLa cells were detected by IP (left) and analyzed quantitatively (right). (C) Expression levels of β-catenin and phosphorylated β-catenin in Con and M3Mut/− HeLa cells were detected by western blot (left). Percentages of phosphorylated β-catenin were analyzed quantitatively (right). (D) Expression levels of MET mRNA in HeLa, HeLa M3Mut/−, H460 sh-NC, H460 sh-METTL3, Huh7 sh-NC, and Huh7 sh-METTL3 cells were detected by quantitative real-time PCR. (E) Expression levels of c-Met and p-AKT S473 in HeLa, HeLa M3Mut/−, H460 sh-NC, H460 sh-METTL3, Huh7 sh-NC, and Huh7 sh-METTL3 cells were detected by western blot (left) and analyzed quantitatively (right). (F and G) Expression levels of β-catenin in Mem and Cyto fractions from HeLa cells silencing c-Met (F) and HeLa M3Mut/− cells overexpressing c-Met (G) were detected by western blot (left) and analyzed quantitatively (right). (H) m6A RIP-qPCR analysis of MET mRNA in Con or M3Mut/− HeLa cells. (I) Expression levels of MET mRNA in Con and HeLa M3Mut/− HeLa cells treated with 5μg/mL Act-D for the indicated time were detected by quantitative real-time PCR. (J) Expression levels of MET mRNA in HeLa cells silencing IGF2BP3 were detected by quantitative real-time PCR. (K) Con or M3Mut/− HeLa cells silencing IGF2BP3 were treated with 5μg/mL Act-D for the indicated time. Expression levels of MET mRNA were detected by quantitative real-time PCR. (L) Mechanism of m6A regulating cellular localization of β-catenin. Data are presented as mean ± SD from three independent experiments. Student’s t test; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Phosphorylation of β-catenin determines its subcellular localization: phosphorylation at Y654 by c-Met inhibits membrane-binding of β-catenin, whereas phosphorylation at S552 by p-S473-AKT promotes its nuclear transport and enhances its transcriptional activity.40, 41, 42 Our results showed that the relative level of p-Y654-β-catenin, but not p-S552-β-catenin, statistically decreased in METTL3Mut/− HeLa (Figure 7C), H460 sh-METTL3 (Figure S7A), and Huh7 sh-METTL3 (Figure S7B) cells. Accordingly, expression of c-Met, which phosphates β-catenin at Y654, was downregulated in METTL3 knockdown cells (Figures 7D and 7E). p-S473-AKT, which phosphates β-catenin at S552, showed no statistical difference (Figure 7E). These results suggested that METTL3-regulated subcellular localization of β-catenin was associated with its phosphorylation level, mediated by c-Met kinase. This suggestion was supported by silencing and recruitment assays, showing that β-catenin increased dramatically in the membrane fraction from HeLa cells silencing c-Met (Figure 7F), whereas HeLa METTL3Mut/− cells overexpressing c-Met can decrease membrane localization of β-catenin (Figures 7G and S7C). These results suggested that c-Met was associated with METTL3-suppressed membrane localization of β-catenin.
We further investigated the mechanism of METTL3-regulated c-Met expression. m6A-seq revealed significant enrichment of m6A modifications in MET mRNA in HeLa cells (Figure S7D), and m6A-qPCR confirmed these variable m6A modifications (Figure 7H). A mRNA stability assay revealed a shorter half-life of MET in METTL3 knockdown HeLa (Figure 7I), H460, and Huh7 cells (Figure S7E). IGF2BP3, which promotes stability of targets,8 has been reported to recognize MET. Silencing IGF2BP3 suppressed mRNA levels of MET in HeLa cells, whereas knockdown of METTL3 attenuated this affect (Figures 7J and S7F). si-IGF2BP3 in HeLa cells can promote MET mRNA decay, but it has no significant effect on MET mRNA stability in METTL3Mut/− HeLa cells (Figure 7K). Our results suggested that METTL3 indirectly suppressed the membrane localization of β-catenin by modulating c-Met levels (Figure 7L).
β-catenin regulated by METTL3 is associated with in vivo dissemination of cancer cells
We further investigated the potential roles of β-catenin in METTL3-regulated cancer progression in three animal cohorts. First, subcutaneous xenograft tumors of HeLa and METTL3Mut/− HeLa cells were analyzed. Immunohistochemistry (IHC) results showed that knockdown of METTL3 increased the expression of β-catenin and E-Cad but suppressed the expression of E2F1 and Met in HeLa cell xenografts (Figure 8A). Second, metastasized lung tumors and primary breast tumors were isolated from MMTV-PyMT mice. Quantitative real-time PCR (Figure 8B), western blot analysis (Figure 8C), and IHC (Figure 8D) showed enhanced expression of METTL3 and decreased expression of β-catenin in metastasized lung tumor tissues. Third, the breast cancer cell model BT549LMF3, which has high potential for lung metastasis, was used.18,43 BT549LMF3 cells exhibited decreased expression of β-catenin and E-Cad and increased expression of METTL3 and c-Met compared with the control group (Figures 8E and 8F). In addition, clinical data from an online database showed that mRNA expression of CTNNB1 was lower in metastasized lung cancer tissues than in non-metastasized lung cancer tissues (Figure 8G). Also, the expression of CTNNB1 in stage IV lung cancer tissue was significantly (p < 0.05) lower than in stage I–III lung cancer (Figure 8H). These results hint at potential roles of the METTL3/β-catenin axis in cancer metastasis.
Figure 8.

β-catenin and METTL3 are involved in in vivo dissemination of cancer cells
(A) IHC-stained (β-catenin, E-Cad, E2F1, and c-Met), paraffin-embedded sections obtained from HeLa and HeLa M3Mut/− cells. The scale bar represents 100 μm. (B) Expression levels of CTNNB1, CDH1, MET, and METTL3 mRNA in primary tumors and metastasized lung tumors isolated from MMTV-PyMT mice were detected by quantitative real-time PCR. (C) Protein levels of β-catenin, E-Cad, c-Met, and METTL3 in primary tumors and metastasized lung tumors isolated from MMTV-PyMT mice were detected by western blot analysis. (D) IHC-stained (METTL3 and β-catenin), paraffin-embedded sections obtained from primary tumors and metastasized lung tumors of MMTV-PyMT mice. The scale bar represents 50 μm. (E) Expression levels of CDH1, CTNNB1, METTL3, and MET in BT549F0 and BT549LMF3 cells were detected by quantitative real-time PCR. (F) Expression levels of E-Cad, β-catenin, METTL3, and c-Met in BT549F0 and BT549LMF3 cells were detected by western blot. (G) Relative mRNA expression of CTNNB1 in metastasized or non-metastasized lung cancers from Oncomine datasets. (H) Relative mRNA expression of CTNNB1 in stage I, II, III, and IV of lung cancer from Oncomine datasets. (I) Kaplan-Meier survival curves of OS based on E2F1 expression in individuals with lung cancer from the TCGA DB. (J) Kaplan-Meier survival curves of OS based on MET expression in individuals with lung cancer from the TCGA DB. Data in (B) and (E) are presented as mean ± SD from three independent experiments. Student’s t test; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 compared with Con. A representative from a total of three independent experiments is shown for (A) and (D).
Because we identified several factors that were associated with expression of β-catenin, we questioned the links between these factors and clinical development of cancer. For E2F1 and YTHDF2, which negatively regulated expression of CTNNB1, increased expression of E2F1 (Figure S8A) and YTHDF2 (Figure S8B) was observed in lung cancer tissue. For c-Met, which promotes cell motility by decreasing membrane localization of β-catenin, MET expression also increased in lung cancer tissue (Figure S8C). A Kaplan-Meier plot showed that increased E2F1 (Figure 8I) or increased MET (Figure 8J) expression in individuals with lung cancer is associated with poorer overall survival (OS). However, there was no significant difference in OS for individuals with lung cancer with decreased or increased METTL3 (Figure S8D). This might be due to the fact that METTL3 influenced the expression of many other downstream signal genes rather than CTNNB1. All of these data confirmed the positive roles of β-catenin expression regulators in in vivo progression in individuals with lung cancer. The animal experiments and the clinical dataset suggest critical functions of β-catenin in cancer development, especially in cancer metastasis.
Discussion
Increasing evidences shows that m6A methylation is associated with various biological functions and development of tumorigenesis.44,45 Here we report that METTL3, acting as an m6A methylase, negatively modulates expression and membrane localization of β-catenin, which forms a complex with E-Cad to act as an epithelial barrier.46 On one hand, METTL3 negatively modulated the transcription, mRNA decay, and translation of CTNNB1 mRNA via different regulators and mechanisms. In particular, METTL3 indirectly repressed CTNNB1 transcription by increasing the expression of its suppresser E2F1, 5′UTR m6A of CTNNB1 facilitated its decay via recruitment of YTHDF2, and METTL3 controlled canonical and non-canonical translation of CTNNB1 via YTHDF1. On the other hand, knockdown of METTL3 indirectly increased membrane localization of β-catenin by decreasing the c-Met-catalyzed p-Y654-β-catenin, leading to downregulation of cell motility (Figure 9). This negative regulation of β-catenin was essential for METTL3-regulated dissemination of cancer cells.
Figure 9.

Proposed mechanism of METTL3-regulated expression and localization of β-catenin
We report that METTL3 regulated transcription of CTNNB1. Various factors have been reported to regulate transcription of CTNNB1.47 Here we reveal that E2F1 is a novel TF of CTNNB1 and that METTL3 can negatively regulate its mRNA decay, leading to further suppression of CTNNB1 transcription. E2F1 acts as an oncogenic factor that positively associates with tumorigenesis, including tumor formation and EMT of cancer cells.48 However, the mechanisms involved in METTL3-stabilized E2F1 mRNA needed further investigation.
Regulation of 5′UTR m6A on mRNA stability has rarely been reported. We demonstrated that the m6A-methylated 5′UTR negatively regulated the mRNA stability of CTNNB1. Deposition of m6A on the 5′UTR promoted CTNNB1 mRNA decay in a content-dependent manner via the m6A reader YTHDF2. YTHDF2 can directly recognize over 3,000 cellular m6A-methylated RNA targets and promote mRNA decay through P body and other forms.6 Specifically, YTHDF2 can bind with RNase P/MRP (endoribonucleases) to elicit rapid degradation of YTHDF2-bound RNA.49 The essential role of YTHDF2 in m6A-regulated mRNA stability of CTNNB1 was shown, illustrating that YTHDF2 can directly bind to CTNNB1 mRNA and that si-YTHDF2 can increase the half-life of CTNNB1 mRNA. Further, we identified that GGAC motifs 1 and 2 are essential for m6A-regulated mRNA stability of CTNNB1 and that double mutation of them significantly stabilized endogenous CTNNB1 mRNA and reporter genes.
The m6A-regulated translation process has been studied for years, and the locations and deposition of m6A methylation on transcripts seem to be crucial for distinct translation regulations.50 The 5′UTR of mRNA can adopt elaborate RNA secondary and tertiary structures to regulate translation initiation in a cap-dependent or cap-independent manner.51 Meyer et al.52 report that 5′UTR m6A promotes cap-independent mRNA translation via recruitment of eIF3. Here we revealed two aspects of functions in m6A-regulated CTNNB1 translation. Firstly, 5′UTR m6A in CTNNB1 inhibited its translation, and increased 5′UTR GGAC content further suppressed the translation efficiency. Second, the global METTL3 level affected the binding affinity of YTHDF1 to eIF4E1 or eIF4E3 via an unknown mechanism. YTHDF1 prefers binding to eIF4E1 in METTL3 knockdown cells, which increases the occupancy of free eIF4E3 to targets to initiate non-canonical translation.53 This observation is in line with the tumor suppressor role of eIF4E3, which can also inhibit the oncogenic activity of eIF4E1.54 A similar regulation of the eIF4E1/eIF4E3 switch was observed in diffuse large B cell lymphoma (DLBCL) cells with knockdown of MNK.37 Our results suggest that YTHDF1 may act as an eIF4E1/eIF4E3 switch regulator in cells, according to cellular METTL3 levels. However, the link between YTHDF1, eIF4E1/eIF4E3 switch, and tumorigenesis needs further investigation.
How METTL3 regulated cancer cell metastasis via β-catenin was explored. In conflict with a previous report,26 we showed that Wnt signaling was not activated in METTL3 knockdown cells, according to nuclear β-catenin levels, downstream gene expression, p-S552-β-catenin, and p-S473-Akt levels.24 We propose that the dual biological functions of β-catenin, activation of Wnt signaling and regulation of cell adhesion, might be regulated by global METTL3/m6A levels. In cells with high m6A levels, such as cancer cells (commonly with high METTL3 expression), β-catenin on cancer cell dissemination mainly depends on activation of Wnt/β-catenin signaling.55,56 This is evidenced by the fact that the β-catenin inhibitor57 or si-β-catenin58,59 promoted expression of E-Cad, which is in line with our results (Figure 2D); however, this effect was attenuated in METTL3Mut/− cells. In cells with low m6A levels, such as METTL3 knockdown cells, β-catenin might exhibit a dominant effect on cell adhesion regulation rather than Wnt activation because the cell adhesion function of β-catenin seemed to be enhanced in METTL3Mut/− cells (Figure 7). Previous reports support the suggestion that METTL3Mut/− cells fail to perform EMT because of suppression of Snail18 and TGF-β,60 both of which can efficiently activate canonical Wnt signaling.61,62 Thus, it is reasonable to believe that Wnt/β-catenin signaling is suppressed or cannot be activated properly in METTL3Mut/− cells.
Here we revealed a novel mechanism of METTL3-regulated EMT, via modulating β-catenin expression and subcellular localization (Figure 8). In vitro and in vivo data confirmed the essential roles of METTL3/β-catenin in dissemination of cancer cells. Because a great number of genes are involved in EMT, we cannot exclude the possibility that METTL3 regulates cancer cell dissemination by indirectly targeting other genes. Our study not only expands our understanding of METTL3 regulating the expression and subcellular localization of targets via multiple mechanisms but also suggests that the METTL3/β-catenin axis might be a potential target for inhibition of cancer metastasis.
Materials and methods
Cell culture and transfection
Because complete knockout of METTL3 has been shown to cause cell lethality, with heterozygous knockout of METTL3Mut/−, a HeLa cell line was generated by CRISPR-Cas9 editing according to a published protocol.63 HeLa cells and METTL3Mut/− cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS) at 37°C in a 5% CO2 atmosphere. The stable cell lines Huh7 sh-NC, Huh7 sh-METTL3, H460 sh-NC, and H460 sh-METTL3 were generated by virus packaging. Huh7 sh-NC and Huh7 sh-METTL3 cells were maintained in DMEM, and H460 sh-NC and H460 sh-METTL3 cells were maintained in RPMI 1640 culture medium (Gibco, Carlsbad, CA, USA) supplemented with 10% FBS and 1μg/mL puromycin. For transfection, plasmids or small interfering RNA (siRNA) were transfected into cells using Lipofectamine 3000 reagent (Invitrogen Life Technology, USA), following the manufacturer’s instructions. The sequences of siRNA are listed in Table S1.
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) assay for m6A quantification
Detection of the global m6A level by LC-MS/MS assay was performed as in a previous study.17 Briefly, mRNA was extracted by oligo dT magnetic beads and digested by nuclease PI (0.5 U, Sigma). Digested mRNA was mixed with NH4HCO3 and alkaline phosphatase at 37°C for 2 h. After filtering, samples were separated by a C18 column (Agilent) and analyzed using the Agilent 6410 QQQ triple-quadrupole LC mass spectrometer. The m6A level was calculated by the ratio of m6A to A, based on calibration curves.
Wound healing assay
Cells were seeded and cultured until a 90% confluent monolayer was formed. Cells were then scratched with a sterile pipette tip and subjected treatments in FBS-free medium as indicated in the text. The migration distances of cells into the scratched area were measured in 10 randomly chosen fields under a microscope.
In vitro invasion assay
The Transwell assay was conducted using CytoSelect 24-well cell invasion assay kits. Briefly, polycarbonate filters (8-μm pore size, Corning) coated with 50% Matrigel (BD Biosciences, Bedford, MA, USA) were used to separate the upper and lower chambers. 5×104 cells in 200 μL culture medium (supplemented with 0.1% FBS) were added to the upper chamber, and 600 μL medium supplemented with 10% FBS was added to the lower chamber and served as a chemotactic agent. After 48-h incubation, the cells invading the lower chamber were fixed, stained, and counted under an upright microscope (5 fields per chamber).
RNA extraction and quantitative real-time PCR
RNA extraction with Trizol (Invitrogen) and quantitative real-time PCR were performed as described in our previous study.18 GAPDH served as a control for normalization. HPRT1 and 18S served as controls for normalization in the nuclear fraction and actinomycin D (Act-D)-treated samples, respectively. Primers for quantitative real-time PCR are listed in Table S2.
Western blot analysis
Total cell lysates were collected as described previously.64 Primary antibodies used for immunoblotting included anti-β-catenin (8480S, Cell Signaling Technology), anti-E-Cad (ab1416, Abcam), anti-FN (sc-52331, Santa Cruz Biotechnology), anti-METTL3 (ab195352, Abcam), anti-E2F1 (ab179445, Abcam), anti-FLAG (MAB3118, Sigma-Aldrich), anti-ZEB1 (ABD53, Millipore), anti-YTHDF2 (17479-1-AP, Proteintech), anti-YTHDF1 (17479-1-AP, Proteintech), anti-eIF4E1 (11149-1-AP, Proteintech), anti-eIF4E1 S209 (9741T, Cell Signaling Technology), anti-eIF4E3 (17282-1-AP, Proteintech), anti-RPS6 (14823-1-AP, Proteintech), anti-HA (ab9110, Abcam), anti-β-catenin Y654 (ab59430, Abcam), anti-β-catenin S552 (5651T, Cell Signaling Technology), anti-Na+/K+-ATPase (BS1436, Bioworld), anti-H2A (ab11175, Abcam), anti-Met (sc-514149, Santa Cruz), and anti-phosphorylated AKT (p-AKT) (pSer473; SAB4504331, Sigma-Aldrich). The immunoblot results presented are representative of at least three independent experiments.
m6A RIP (MeRIP)-qPCR
MeRIP-qPCR was conducted according to a previous study with slight modifications.65 Briefly, total RNAs (200 μg) extracted by Trizol were used for IP by m6A antibody (Synaptic Systems) in IP buffer (150 mM NaCl, 0.1% NP-40, 10 mM Tris [pH 7.4], and 100 U RNase inhibitor) to obtain m6A-conjugated mRNAs. Control experiments were set up using immunoglobulin G (IgG). The m6A RNAs were immunoprecipitated by Dynabead Protein G (Thermo Fisher Scientific) and eluted twice by elution buffer (5 mM Tris-HCl [pH 7.5], 1 mM EDTA [pH 8.0], 0.05% SDS, and 20 mg/mL Proteinase K). m6A-IP RNAs were precipitated by ethanol, and the RNA concentration was measured with a Qubit RNA HS Assay Kit (Thermo Fisher Scientific). 2 ng of total RNA and m6A IP RNA was used as the template for quantitative real-time PCR. HPRT1 acted as an internal control of input because it did not contain m6A peaks from the m6A profiling data.6 For MeRIP using fragmented RNA, total RNA was fragmentated by fragmentation reagents (Thermo Fisher Scientific), following the manufacturer’s instructions.
Promoter activity assay/dual-luciferase reporter assay
The promoter activity/dual luciferase reporter assay was performed with the pGL3-Basic-CTNNB1-Fluc reporter, which contained the CTNNB1 promoter region before the F-Luc gene. For F-Luc reporter investigating effects of CTNNB1 5′UTR on mRNA stability, the CTNNB1 5′UTR was inserted between the promoter and F-Luc reporter gene. Cells were co-transfected with reporter and pRL-TK (as a control) plasmids for 48 h. The promoter activity/dual luciferase reporter assay was performed using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA) according to the manufacturer’s instructions. Results were calculated as the luciferase values of the reporter plasmid divided to pRL-TK.
Subcellular fractionation
Subcellular fractionation was performed according to a previous report.66 Briefly, 2 × 107 cells were pelleted and lysed by sonication on ice. The supernatant was centrifuged to obtain cytoplasmic proteins, and the pellet was saved to obtain nuclear proteins. After isolating cytoplasmic proteins, the supernatant was subjected to 1M–2M sucrose gradient centrifugation to separate membrane proteins. Concentrations of all protein fractions were measured by Bradford assay, and 60 μg total protein was loaded for western blot detection. To obtain cytoplasmic and nuclear RNA, cytoplasmic and nuclear fractions were mixed with Trizol (Invitrogen), followed by the RNA extraction process according to the protocol.18 CTNNB1 mRNA in cytoplasmic and nuclear RNA was detected by real-time PCR, where GAPDH and HPRT1 served as controls for normalization in the cytoplasmic and nuclear fraction, respectively.
RNA stability and protein stability assays
HeLa cells and METTL3Mut/− cells were seeded 1 day before treatment. 5 μg/mL Act-D or 100 μg/mL CHX was added to the cell culture medium for the indicated times for RNA stability and protein stability assays, respectively. For the RNA stability assay, cells were used to extract total RNA and detected by quantitative real-time PCR. For the protein stability assay, cells were lysed in 1× SDS loading dye and detected by western blot.
Polysome profiling
Polysome profiling was conducted according to a previous study.5 Briefly, 5 × 107 cells were incubated with 100 μg/mL CHX for 2 min at 37°C before harvest. Cells were washed with PBS-CHX and lysed in 300 μL lysis buffer (140 mM NaCl, 5 mM MgCl2, 10 mM Tris HCl [pH 8.0], 1% Triton X-100, 0.5% sodium deoxycholate, 0.4 U/μL RNase inhibitor, 20 mM DTT, 0.1 mg/mL CHX, 10 mM RVC, and 0.1% cocktail). Clear cell lysate was obtained by 16,000 × g centrifugation for 10 min and then digested by DNase for 10 min at room temperature. DNase-treated cell lysate was loaded into 10 mL 5%–50% sucrose solution and centrifuged at 170,000 × g for 2 h at 4°C. Ribosomal fractions were separated by gradient profiling Triax (Biocomp Instruments). RNAs in each polysome fraction were extracted by Trizol, and the RNA concentrations were determined using the Qubit RNA HS Assay Kit (Thermo Fisher Scientific).
Site-directed mutagenesis
Site-directed mutagenesis was performed using PrimerSTAR Max DNA polymerase (R045A, Takara). Primers used for mutagenesis are listed in Table S3. The 50-μL PCR reaction was set up as follows: 1× PrimerSTAR, 5 ng DNA template, 125 ng forward primer, and 125 ng reverse primer. The cycling steps were set up as follows: 30 s at 95°C, 27 cycles (15 s at 98°C, 30 s at 55°C, 1 min/kb of plasmid length at 72°C), and 20 min at 72°C. PCR products were then collected using a PCR purification kit (QIAGEN). 10 U/reaction of DpnI restriction enzyme (New England Biolabs) was added to purified PCR products and incubated at 37°C for 1 h. Half of the DpnI-treated DNA was transformed to DH5α cells. Positive clones were sent to process DNA sequencing to verify successful mutagenesis of target genes.
RIP-qPCR
Cells were UV cross-linked for 30 min before harvest. Clear lysate with 200 μg total protein (supplemented with protease inhibitors and RNase inhibitor) was incubated with antibody- or IgG-conjugated Dynabead Protein G (Thermo Fisher Scientific) for 3 h. RNAs pulled down by the antibody were eluted and dissolved in Trizol (Invitrogen). RNAs were precipitated by ethanol, and the RNA concentration was measured with a Qubit RNA HS Assay Kit (Thermo Fisher Scientific). 2 ng of total RNA and IP RNA was used as the template for quantitative real-time PCR.
CoIP assay
2 × 107 cells were pelleted and lysed in 400 μL RIPA buffer supplemented with protease inhibitors. Clear lysate was pre-cleaned using 20 μL Dynabead Protein G (Thermo Fisher Scientific) for 2 h at 4°C. Pre-cleaned cell lysate was further incubated with antibody- or IgG-conjugated Dynabead Protein G at 4°C. After overnight incubation, beads were rinsed three times withy RIPA buffer and boiled in 30 μL 1× SDS loading dye. The eluted samples were analyzed by western blot.
Establishment of lung metastasis (LM) of breast cancer (BC) cell models
Establishment of LM of BC cell models has been described previously18,43,67. In brief, first-generation LM cells were obtained from the lungs of immunodeficient mice (4 weeks old) after injection of 2×105 viable BT-549 cells into the lateral tail vein in a volume of 0.1 mL. Eight weeks later, the metastasized cells were isolated, primarily cultured with medium supplemented with 10% FBS, and called BT-549 LMF1. LMF2 and LMF3 BC cells were generated using the same method. BT-549 LMF3 cells were used for further investigation.
IHC
The xenograft slides were deparaffinized and rehydrated through an alcohol gradient, followed by antigen retrieval with sodium citrate buffer. Tumor sections were blocked with 5% normal goat serum (vector) with 0.1% Triton X-100 and 3% H2O2 in PBS for 60 min at room temperature and then incubated with primary antibodies at 4°C overnight. IHC staining was performed with horseradish peroxidase (HRP) conjugates using DAB detection. Images were taken with a Nikon microscope.
RNA sequencing (RNA-seq) and m6A sequencing (m6A-seq)
Total RNA was isolated from BT-549 and BT-549LMF3 cells and purified using the RNeasy Mini Kit (QIAGEN, Germany). Construction of the cDNA library with the Tru-seq mRNA sample preparation kit (Illumina, USA) was sequenced in Illumina platform. For m6A-seq, the RNA was treated with a chemical reagent for fragmentation. The fragmented RNA was incubated with an m6A antibody for IP according to the manufacturer’s instructions (MeRIP m6A Kit, Merck Millipore, USA). m6A-enriched RNA was used to generate the cDNA library in parallel with the input RNA and then sequenced using the HiSeq 2000 system (Illumina, USA). All sequencing reads were mapped to a reference human genome sequence (NCBI 36.1 [hg19] assembly) by TopHat (version 2.0.6). Sequence motifs enriched in m6A peaks were identified by HOMER, with m6A peaks as the target sequences and control peaks as the background.
Plasmids, mutagenesis, siRNA, and transfection
Plasmids for transient overexpression were as follows: pcDNA3-ALKBH5, PPB-METTL3, pcDNA3-β-catenin, pcDNA3-β-catenin 5′UTR, pcDNA3-β-catenin-MS2bs, and pHM6-MS2. siRNAs used for this study and primers for mutagenesis are listed in Tables S1 and S3, respectively. Transfection of plasmids and siRNAs was performed using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions.
Experimental animals and xenograft models
All animal experiments complied with the Zhongshan School of Medicine Policy on the Care and Use of Laboratory Animals. Four cohorts of animals were used in the present study.
First, a subcutaneous transplantation model was used to evaluate the growth of BT-549 LMF3 and BT-549 cells. Cells (5 × 106 per mouse, n = 5 for each group) were diluted in 200μL PBS +200 μL Matrigel (BD Biosciences) and injected subcutaneously into immunodeficient female mice. When tumor volumes in the BT-549 LMF3 group reached approximately 1,000 mm3, mice were sacrificed, and tumors were removed and weighed. Tumor volume was quantified using manual calipers every 3 days and calculated using the formula V = 1/2× larger diameter × (smaller diameter)2.
Second, a subcutaneous transplantation model was used to evaluate the metastasis potential of BT-549 LMF3 and BT-549 cells. Cells (5 × 106 per mouse, n = 5 for each group) were diluted in 200μL PBS +200 μL Matrigel (BD Biosciences) and injected subcutaneously into immunodeficient female mice. When the tumors of all mice reached about 100 mm3, mice were sacrificed, and tumors were isolated to measure the expression of targets.
Third, the in vivo LM model was established by injection with BT-549 and BT-549 LMF3 cells (1 × 106 per mouse, n = 5 for each group). Eight weeks after injection, the experiment was terminated, mice were sacrificed, and metastatic lung tumors were analyzed for the presence of metastatic tumors.
Last, primary tumors and metastasized tumors in lungs isolated from MMTV-PyMT mice (n = 3) were isolated to analyze the expression of targets.
Database (DB) analysis
We used the Kaplan-Meier DB (http://kmplot.com/analysis/) to test OS of targets in individuals with lung cancer. The difference between survival curves was determined by log rank test; p < 0.05 was considered statistically significant. Correlations between CTNNB1 expression in cancer tissue and clinicopathological features were extracted from the TCGA DB and Oncomine DB (http://www.oncomine.org). The data were analyzed with a Pearson chi-square test.
Statistical analyses
Data are reported as mean ± SD from at least three independent experiments unless otherwise specified. For capillary tube formation and luciferase activity assays, data were derived from three independent experiments that were performed in duplicate. Data were analyzed by two-tailed unpaired Student’s t test between two groups and by one-way ANOVA followed by Bonferroni test for multiple comparisons. Statistical analysis was carried out using SPSS 16.0 for Windows. All statistical tests were two sided. A p value of less than 0.05 was considered statistically significant. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; NS, not significant.
Acknowledgments
This research was supported by the National Natural Science Foundation of China (Nos. 32161143017, 82173833, 82173126, 81973343, 81871994, and 81871943), the Fundamental Research Funds for the Central Universities (Sun Yat-sen University) (19ykzd24, 19ykpy130), the International Cooperation Project of the Science and Technology Planning Project of Guangdong Province, China (2021A0505030029), the Open Program of Shenzhen Bay Laboratory (No. SZBL202009051006), the Guangdong Provincial Key Laboratory of Chiral Molecule and Drug Discovery (2019B030301005), the Guangdong Basic and Applied Basic Research Foundation (No. 2020A1515010290), the China Postdoctoral Science Foundation (2018M643354, 2020T130751), the National Science Fund for Excellent Young Scholars (82022037), the Guangzhou Science and Technology Planning Program (201902020018), and the Guangdong Provincial Natural Science Foundation for Distinguished Young Scholars (2019B151502063).
Author contributions
Conception and design, J.L., H.W., J.C., and W.H.; acquisition of data, J.L., G.X., Y.T., Y.W., F.C., Y.L., and X.L.; analysis and interpretation of data, J.L., G.X., and H.W.; writing, review, and/or revision of the manuscript, J.L., H.W., W.L., J.C., W.H., and S.W.-N.A.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.01.019.
Contributor Information
Jie Cao, Email: czhongt@126.com.
Weiling He, Email: hewling@mail.sysu.edu.cn.
Hongsheng Wang, Email: whongsh@mail.sysu.edu.cn.
Supplemental information
References
- 1.Dubin D.T., Taylor R.H. The methylation state of poly A-containing messenger RNA from cultured hamster cells. Nucleic Acids Res. 1975;2:1653–1668. doi: 10.1093/nar/2.10.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jia G., Fu Y., Zhao X., Dai Q., Zheng G., Yang Y., Yi C., Lindahl T., Pan T., Yang Y.G., He C. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011;7:885–887. doi: 10.1038/nchembio.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zheng G., Zheng G., Dahl J.A., Niu Y., Fedorcsak P., Huang C.M., Li C.J., Vågbø C.B., Shi Y., Wang W.L., et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell. 2013;49:18–29. doi: 10.1016/j.molcel.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xiao W., Adhikari S., Dahal U., Chen Y.-S., Hao Y.-J., Sun B.-F., Sun H.-Y., Li A., Ping X.-L., Lai W.-Y., et al. Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol. Cell. 2016;61:507–519. doi: 10.1016/j.molcel.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 5.Wang X., Zhao B.S., Roundtree I.A., Lu Z., Han D., Ma H., Weng X., Chen K., Shi H., He C. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161:1388–1399. doi: 10.1016/j.cell.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang X., Lu Z., Gomez A., Hon G.C., Yue Y., Han D., Fu Y., Parisien M., Dai Q., Jia G., et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–120. doi: 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alarcon C.R., Goodarzi H., Lee H., Liu X., Tavazoie S., Tavazoie S.F. HNRNPA2B1 is a mediator of m(6)A-dependent nuclear RNA processing events. Cell. 2015;162:1299–1308. doi: 10.1016/j.cell.2015.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang H., Weng H., Sun W., Qin X., Shi H., Wu H., Zhao B.S., Mesquita A., Liu C., Yuan C.L., et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 2018;20:285–295. doi: 10.1038/s41556-018-0045-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patil D.P., Chen C.K., Pickering B.F., Chow A., Jackson C., Guttman M., Jaffrey S.R. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature. 2016;537:369–373. doi: 10.1038/nature19342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bartosovic M., Molares H.C., Gregorova P., Hrossova D., Kudla G., Vanacova S. N6-methyladenosine demethylase FTO targets pre-mRNAs and regulates alternative splicing and 3'-end processing. Nucleic Acids Res. 2017;45:11356–11370. doi: 10.1093/nar/gkx778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haussmann I.U., Bodi Z., Sanchez-Moran E., Mongan N.P., Archer N., Fray R.G., Soller M. m(6)A potentiates Sxl alternative pre-mRNA splicing for robust Drosophila sex determination. Nature. 2016;540:301–304. doi: 10.1038/nature20577. [DOI] [PubMed] [Google Scholar]
- 12.Fustin J.M., Doi M., Yamaguchi Y., Hida H., Nishimura S., Yoshida M., Isagawa T., Morioka M.S., Kakeya H., Manabe I., et al. RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell. 2013;155:793–806. doi: 10.1016/j.cell.2013.10.026. [DOI] [PubMed] [Google Scholar]
- 13.Roundtree I.A., Luo G.Z., Zhang Z., Wang X., Zhou T., Cui Y., Sha J., Huang X., Guerrero L., Xie P., et al. YTHDC1 mediates nuclear export of N(6)-methyladenosine methylated mRNAs. eLife. 2017;6 doi: 10.7554/eLife.31311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barbieri I., Tzelepis K., Pandolfini L., Shi J., Millán-Zambrano G., Robson S.C., Aspris D., Migliori V., Bannister A.J., Han N., et al. Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature. 2017;552:126–131. doi: 10.1038/nature24678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang H., Deng Q., Lv Z., Ling Y., Hou X., Chen Z., Dinglin X., Ma S., Li D., Wu Y., et al. N6-methyladenosine induced miR-143-3p promotes the brain metastasis of lung cancer via regulation of VASH1. Mol. Cancer. 2019;18:181. doi: 10.1186/s12943-019-1108-x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16.Wu Y., Yang X., Chen Z., Tian L., Jiang G., Chen F., Li J., An P., Lu L., Luo N., et al. m(6)A-induced lncRNA RP11 triggers the dissemination of colorectal cancer cells via upregulation of Zeb1. Mol. Cancer. 2019;18:87. doi: 10.1186/s12943-019-1014-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Z.H., Peng Y., Li J., Chen Z., Chen F., Tu J., Lin S., Wang H. N-6-methyladenosine regulates glycolysis of cancer cells through PDK4. Nat. Commun. 2020;11:2578. doi: 10.1038/s41467-020-16306-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin X., Chai G., Wu Y., Li J., Chen F., Liu J., Luo G., Tauler J., Du J., Lin S., et al. RNA m(6)A methylation regulates the epithelial mesenchymal transition of cancer cells and translation of Snail. Nat. Commun. 2019;10:2065. doi: 10.1038/s41467-019-09865-9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19.Chen Z.J., Wu L., Zhou J., Lin X., Peng Y., Ge L., Chiang C.M., Huang H., Wang H., He W. N6-methyladenosine-induced ERRγ triggers chemoresistance of cancer cells through upregulation of ABCB1 and metabolic reprogramming. Theranostics. 2020;10:3382–3396. doi: 10.7150/thno.40144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li J., Chen Z., Chen F., Xie G., Ling Y., Peng Y., Lin Y., Luo N., Chiang C.M., Wang H., et al. Targeted mRNA demethylation using an engineered dCas13b-ALKBH5 fusion protein. Nucleic Acids Res. 2020;48:5684–5694. doi: 10.1093/nar/gkaa269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ge L., Zhang N., Chen Z., Song J., Wu Y., Li Z., Chen F., Wu J., Li D., Li J., et al. Level of N6-methyladenosine in peripheral blood RNA: a novel predictive biomarker for gastric cancer. Clin. Chem. 2020;66:342–351. doi: 10.1093/clinchem/hvz004. [DOI] [PubMed] [Google Scholar]
- 22.Stuelten C.H., Parent C.A., Montell D.J. Cell motility in cancer invasion and metastasis: insights from simple model organisms. Nat. Rev. Cancer. 2018;18:296–312. doi: 10.1038/nrc.2018.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang J., Antin P., Berx G., Blanpain C., Brabletz T., Bronner M., Campbell K., Cano A., Casanova J., Christofori G., et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020;21:341–352. doi: 10.1038/s41580-020-0237-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang P., Yan R., Zhang X., Wang L., Ke X., Qu Y. Activating Wnt/beta-catenin signaling pathway for disease therapy: challenges and opportunities. Pharmacol. Ther. 2019;196:79–90. doi: 10.1016/j.pharmthera.2018.11.008. [DOI] [PubMed] [Google Scholar]
- 25.Nusse R., Clevers H. Wnt/beta-Catenin signaling, disease, and emerging Therapeutic modalities. Cell. 2017;169:985–999. doi: 10.1016/j.cell.2017.05.016. [DOI] [PubMed] [Google Scholar]
- 26.Liu L., Wang J., Sun G., Wu Q., Ma J., Zhang X., Huang N., Bian Z., Gu S., Xu M., et al. m(6)A mRNA methylation regulates CTNNB1 to promote the proliferation of hepatoblastoma. Mol. Cancer. 2019;18:188. doi: 10.1186/s12943-019-1119-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang P., Doxtader K.A., Nam Y. Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol. Cell. 2016;63:306–317. doi: 10.1016/j.molcel.2016.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pan D.Y., Zeng X.Q., Ma G.F., Gao J., Li N., Miao Q., Lian J.J., Zhou H., Xu L.L., Chen S.Y. Label-free quantitative proteomic analysis identifies CTNNB1 as a direct target of FOXP3 in gastric cancer cells. Oncol. Lett. 2018;15:7655–7660. doi: 10.3892/ol.2018.8277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu G., Jiang S., Wang C., Jiang W., Liu Z., Liu C., Saiyin H., Yang X., Shen S., Jiang D. Zinc finger transcription factor 191, directly binding to beta-catenin promoter, promotes cell proliferation of hepatocellular carcinoma. Hepatology. 2012;55:1830–1839. doi: 10.1002/hep.25564. [DOI] [PubMed] [Google Scholar]
- 30.Essien B.E., Sundaresan S., Ocadiz-Ruiz R., Chavis A., Tsao A.C., Tessier A.J., Hayes M.M., Photenhauer A., Saqui-Salces M., Kang A.J., et al. Transcription factor ZBP-89 drives a feedforward loop of beta-catenin expression in colorectal cancer. Cancer Res. 2016;76:6877–6887. doi: 10.1158/0008-5472.CAN-15-3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Link S., Spitzer R.M.M., Sana M., Torrado M., Völker-Albert M.C., Keilhauer E.C., Burgold T., Pünzeler S., Low J.K.K., Lindström I., et al. PWWP2A binds distinct chromatin moieties and interacts with an MTA1-specific core NuRD complex. Nat. Commun. 2018;9:4300. doi: 10.1038/s41467-018-06665-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang X., Yu Y., Yang H.W., Agar N.Y., Frado L., Johnson M.D. The imprinted gene PEG3 inhibits Wnt signaling and regulates glioma growth. J. Biol. Chem. 2010;285:8472–8480. doi: 10.1074/jbc.M109.069450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou K.R., Liu S., Sun W.J., Zheng L.L., Zhou H., Yang J.H., Qu L.H. ChIPBase v2.0: decoding transcriptional regulatory networks of non-coding RNAs and protein-coding genes from ChIP-seq data. Nucleic Acids Res. 2017;45:D43–D50. doi: 10.1093/nar/gkw965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qin B., Zhou M., Ge Y., Taing L., Liu T., Wang Q., Wang S., Chen J., Shen L., Duan X., et al. CistromeMap: a knowledgebase and web server for ChIP-Seq and DNase-Seq studies in mouse and human. Bioinformatics. 2012;28:1411–1412. doi: 10.1093/bioinformatics/bts157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zou D., Dong L., Li C., Yin Z., Rao S., Zhou Q. The m(6)A eraser FTO facilitates proliferation and migration of human cervical cancer cells. Cancer Cell Int. 2019;19:321. doi: 10.1186/s12935-019-1045-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Muller S., Bley N., Busch B., Glaß M., Lederer M., Misiak C., Fuchs T., Wedler A., Haase J., Bertoldo J.B., et al. The oncofetal RNA-binding protein IGF2BP1 is a druggable, post-transcriptional super-enhancer of E2F-driven gene expression in cancer. Nucleic Acids Res. 2020;48:8576–8590. doi: 10.1093/nar/gkaa653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Landon A.L., Muniandy P.A., Shetty A.C., Lehrmann E., Volpon L., Houng S., Zhang Y., Dai B., Peroutka R., Mazan-Mamczarz K., et al. MNKs act as a regulatory switch for eIF4E1 and eIF4E3 driven mRNA translation in DLBCL. Nat. Commun. 2014;5:5413. doi: 10.1038/ncomms6413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beretta L., Gingras A.C., Svitkin Y.V., Hall M.N., Sonenberg N. Rapamycin blocks the phosphorylation of 4E-BP1 and inhibits cap-dependent initiation of translation. EMBO J. 1996;15:658–664. [PMC free article] [PubMed] [Google Scholar]
- 39.Cui C., Zhou X., Zhang W., Qu Y., Ke X. Is beta-catenin a druggable target for cancer therapy? Trends Biochem. Sci. 2018;43:623–634. doi: 10.1016/j.tibs.2018.06.003. [DOI] [PubMed] [Google Scholar]
- 40.Schaefer K.N., Peifer M. Wnt/Beta-catenin signaling regulation and a role for biomolecular condensates. Dev. Cell. 2019;48:429–444. doi: 10.1016/j.devcel.2019.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Piedra J., Martínez D., Castaño J., Miravet S., Duñach M., García de Herreros A. Regulation of beta-catenin structure and activity by tyrosine phosphorylation. J. Biol. Chem. 2001;276:20436–20443. doi: 10.1074/jbc.M100194200. [DOI] [PubMed] [Google Scholar]
- 42.Stamos J.L., Weis W.I. The beta-catenin destruction complex. Cold Spring Harb Perspect. Biol. 2013;5:a007898. doi: 10.1101/cshperspect.a007898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu L., Chen Z., Lin X., Tian L., Su Q., An P., Li W., Wu Y., Du J., Shan H., et al. Inhibition of BRD4 suppresses the malignancy of breast cancer cells via regulation of Snail. Cell Death Differ. 2020;27:255–268. doi: 10.1038/s41418-019-0353-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang H., Weng H., Chen J. m(6. A modification in coding and non-coding RNAs: roles and Therapeutic implications in cancer. Cancer Cell. 2020;37:270–288. doi: 10.1016/j.ccell.2020.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.He L.E., Li H., Wu A., Peng Y., Shu G., Yin G. Functions of N6-methyladenosine and its role in cancer. Mol. Cancer. 2019;18:176. doi: 10.1186/s12943-019-1109-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harris T.J.C., Tepass U. Adherens junctions: from molecules to morphogenesis. Nat. Rev. Mol. Cell Biol. 2010;11:502–514. doi: 10.1038/nrm2927. [DOI] [PubMed] [Google Scholar]
- 47.Harris T.J.C., Peifer M. Decisions, decisions: beta-catenin chooses between adhesion and transcription. Trends Cell Biol. 2005;15:234–237. doi: 10.1016/j.tcb.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 48.Emanuele M.J., Enrico T.P., Mouery R.D., Wasserman D., Nachum S., Tzur A. Complex cartography: regulation of E2F transcription factors by cyclin F and ubiquitin. Trends Cell Biol. 2020:640–652. doi: 10.1016/j.tcb.2020.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Park O.H., Ha H., Lee Y., Boo S.H., Kwon D.H., Song H.K., Kim Y.K. Endoribonucleolytic cleavage of m(6)A-containing RNAs by RNase P/MRP complex. Mol. Cell. 2019;74:494–507 e8. doi: 10.1016/j.molcel.2019.02.034. [DOI] [PubMed] [Google Scholar]
- 50.Roundtree I.A., Evans M.E., Pan T., He C. Dynamic RNA modifications in gene expression regulation. Cell. 2017;169:1187–1200. doi: 10.1016/j.cell.2017.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leppek K., Das R., Barna M. Functional 5 ' UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat. Rev. Mol. Cell Biol. 2018;19:158–174. doi: 10.1038/nrm.2017.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Meyer K.D., Patil D.P., Zhou J., Zinoviev A., Skabkin M.A., Elemento O., Pestova T.V., Qian S.B., Jaffrey S.R. 5' UTR m(6)A promotes cap-independent translation. Cell. 2015;163:999–1010. doi: 10.1016/j.cell.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Volpon L., Osborne M.J., Culjkovic-Kraljacic B., Borden K.L.B. eIF4E3, a new actor in mRNA metabolism and tumor suppression. Cell Cycle. 2013;12:1159–1160. doi: 10.4161/cc.24566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Osborne M.J., Volpon L., Kornblatt J.A., Culjkovic-Kraljacic B., Baguet A., Borden K.L. eIF4E3 acts as a tumor suppressor by utilizing an atypical mode of methyl-7-guanosine cap recognition. P Natl. Acad. Sci. U S A. 2013;110:3877–3882. doi: 10.1073/pnas.1216862110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Corada M., Corada M., Nyqvist D., Orsenigo F., Caprini A., Giampietro C., Taketo M.M., Iruela-Arispe M.L., Adams R.H., Dejana E. The Wnt/beta-catenin pathway modulates vascular remodeling and specification by upregulating Dll4/Notch signaling. Dev. Cell. 2010;18:938–949. doi: 10.1016/j.devcel.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang S., Liu Y., Li M.Y., Ng C.S.H., Yang S.L., Wang S., Zou C., Dong Y., Du J., Long X., et al. FOXP3 promotes tumor growth and metastasis by activating Wnt/beta-catenin signaling pathway and EMT in non-small cell lung cancer. Mol. Cancer. 2017;16:124. doi: 10.1186/s12943-017-0700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liao S., Chen H., Liu M., Gan L., Li C., Zhang W., Lv L., Mei Z. Aquaporin 9 inhibits growth and metastasis of hepatocellular carcinoma cells via Wnt/beta-catenin pathway. Aging (Albany NY) 2020;12:1527–1544. doi: 10.18632/aging.102698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang B., Tang Z., Gong H., Zhu L., Liu X. Wnt5a promotes epithelial-to-mesenchymal transition and metastasis in non-small-cell lung cancer. Biosci. Rep. 2017;37 doi: 10.1042/BSR20171092. BSR20171092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen L.L., Gao G.X., Shen F.X., Chen X., Gong X.H., Wu W.J. SDC4 gene silencing favors human papillary Thyroid carcinoma cell apoptosis and inhibits epithelial mesenchymal transition via Wnt/beta-catenin pathway. Mol. Cells. 2018;41:853–867. doi: 10.14348/molcells.2018.0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li J., Chen F., Peng Y., Lv Z., Lin X., Chen Z., Wang H. N6-Methyladenosine regulates the expression and secretion of TGFbeta1 to affect the epithelial-mesenchymal transition of cancer cells. Cells. 2020;9:296. doi: 10.3390/cells9020296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang K., Wang X., Zhang H., Wang Z., Nan G., Li Y., Zhang F., Mohammed M.K., Haydon R.C., Luu H.H., et al. The evolving roles of canonical WNT signaling in stem cells and tumorigenesis: implications in targeted cancer therapies. Lab Invest. 2016;96:116–136. doi: 10.1038/labinvest.2015.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu X., Zhang M., Xu F., Jiang S. Wnt signaling in breast cancer: biological mechanisms, challenges and opportunities. Mol. Cancer. 2020;19:165. doi: 10.1186/s12943-020-01276-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shen B., Zhang J., Wu H., Wang J., Ma K., Li Z., Zhang X., Zhang P., Huang X. Generation of gene-modified mice via Cas9/RNA-mediated gene targeting. Cell Res. 2013;23:720–723. doi: 10.1038/cr.2013.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li J., Lam W.W., Lai T.W., Au S.W. Degradation of nuclear Ubc9 induced by listeriolysin O is dependent on K(+) efflux. Biochem. Biophys. Res. Commun. 2017;493:1115–1121. doi: 10.1016/j.bbrc.2017.09.051. [DOI] [PubMed] [Google Scholar]
- 65.Li Z., Weng H., Su R., Weng X., Zuo Z., Li C., Huang H., Nachtergaele S., Dong L., Hu C., et al. FTO plays an oncogenic role in acute myeloid leukemia as a N(6)-methyladenosine RNA demethylase. Cancer Cell. 2017;31:127–141. doi: 10.1016/j.ccell.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Debelec-Butuner B., Alapinar C., Ertunc N., Gonen-Korkmaz C., Yörükoğlu K., Korkmaz K.S. TNFalpha-mediated loss of beta-catenin/E-cadherin association and subsequent increase in cell migration is partially restored by NKX3.1 expression in prostate cells. PloS one. 2014;9 doi: 10.1371/journal.pone.0109868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Minn A.J., Gupta G.P., Siegel P.M., Bos P.D., Shu W., Giri D.D., Viale A., Olshen A.B., Gerald W.L., Massagué J. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.