

Abstract
Acute pancreatitis (AP) is a common digestive disease without specific treatment, and its pathogenesis features multiple deleterious amplification loops dependent on translation, triggered by cytosolic Ca2+ ([Ca2+]i) overload; however, the underlying mechanisms in Ca2+ overload of AP remains incompletely understood. Here we show that microRNA-26a (miR-26a) inhibits pancreatic acinar cell (PAC) store-operated Ca2+ entry (SOCE) channel expression, Ca2+ overload, and AP. We find that major SOCE channels are post-transcriptionally induced in PACs during AP, whereas miR-26a expression is reduced in experimental and human AP and correlated with AP severity. Mechanistically, miR-26a simultaneously targets Trpc3 and Trpc6 SOCE channels and attenuates physiological oscillations and pathological elevations of [Ca2+]i in PACs. MiR-26a deficiency increases SOCE channel expression and [Ca2+]i overload, and significantly exacerbates AP. Conversely, global or PAC-specific overexpression of miR-26a in mice ameliorates pancreatic edema, neutrophil infiltration, acinar necrosis, and systemic inflammation, accompanied with remarkable improvements on pathological determinants related with [Ca2+]i overload. Moreover, pancreatic or systemic administration of an miR-26a mimic to mice significantly alleviates experimental AP. These findings reveal a previously unknown mechanism underlying AP pathogenesis, establish a critical role for miR-26a in Ca2+ signaling in the exocrine pancreas, and identify a potential target for the treatment of AP.
Keywords: noncoding RNA, store-operated calcium entry channels, transient receptor potential canonical channels, pancreatic acinar cell, inflammation, endoplasmic reticulum stress, autophagy, mouse models, targeted therapy
Graphical abstract
Du and colleagues identified microRNA-26a as a crucial checkpoint for calcium overload in pancreatic acinar cells, the central event that induces acute pancreatitis, a common digestive disease without specific treatment, providing a previously unknown mechanism underlying acute pancreatitis pathogenesis and a novel therapeutic option for this disease.
Introduction
Acute pancreatitis (AP) is an increasingly common digestive disease characterized by abnormal intracellular digestive enzyme activation that can lead to pancreatic necrosis, systemic inflammation, organ failure, and death,1, 2, 3 but remains without specific and targeted treatment.4 It is imperative to understand the underlying pathophysiological mechanisms and develop therapeutic strategies to halt disease progression.
Calcium (Ca2+) signaling is crucial to the function of pancreatic acinar cells (PACs) and regulates ductal epithelial cells, immune cells, and most other cells,5, 6, 7 but is sabotaged by toxins that induce AP through cytosolic Ca2+ ([Ca2+]i) overload.8,9 Physiological Ca2+ signaling in PACs is characterized by repetitive, oscillatory spikes of [Ca2+]i predominantly in the apical pole, to coordinate stimulus-metabolism and stimulus-secretion coupling.5 Toxins that induce AP cause sustained, global increases in [Ca2+]i, predominantly dependent on Ca2+ entry from the external milieu, the central upstream event that induces AP through mitochondrial injury, impaired autophagy, intracellular zymogen activation, endoplasmic reticulum (ER) stress, and inflammasome activation amplifying pathology.8, 9, 10 Experimental AP is mitigated by inhibition of store-operated Ca2+ entry (SOCE) through transient receptor potential cation channel canonical 3 (TRPC3),11,12 ORAI calcium release-activated calcium modulator 1 (ORAI1),13,14 TRP melastatin 2 channels,15 or increased activity of SOCE-associated regulatory factor (SARAF), which reduces TRPC and ORAI1 channel activation by stromal interaction molecule 1 (STIM1).16 SOCEs have been widely considered as attractive therapeutic targets for the treatment of AP.12, 13, 14 The functional importance and therapeutic implication of Ca2+ channels have been well documented. It has not been determined, however, how the dynamics of Ca2+ channel expression operates in AP, and whether this expression could serve as a pathophysiological mechanism for Ca2+ overload.
MicroRNAs (miRNAs) constitute a class of endogenous short noncoding RNAs that mediate post-transcriptional gene silencing.17 MiRNAs play critical roles in diverse biological and physiological processes, and thus have become promising therapeutic targets to treat numerous human diseases in both preclinical and early clinical trials.18,19 Specifically, miRNAs have been shown to regulate Ca2+ signaling in cardiomyocytes,20, 21, 22 neurons,23 and immune system,24 and have been implicated in heart diseases and neurodegenerative disorders. However, there has been no study reporting the involvement of miRNAs in AP-associated Ca2+ overload, albeit a few clues have suggested the potential of miRNAs in AP pathogenesis, diagnosis, and therapy.25,26 It is of interest to investigate the potential pathophysiological role of miRNAs in Ca2+ overload and explore new strategies for AP treatment.
MiR-26a is highly expressed in various human tissues27 and regulates numerous physiological and pathological processes.28 MiR-26a is often reduced in cancer,27,29,30 obesity,31 and metabolic disorders,32, 33, 34, 35 and has emerged as a promising therapeutic target for these diseases.36 However, the role of miR-26a in the exocrine pancreas, as well as in AP pathogenesis, is not known.
Here, we report that miR-26a functions as a crucial inhibitor of Ca2+ overload in PACs and prevents AP development. We find that the major SOCEs are post-transcriptionally induced in PACs during AP. Of these SOCEs, Trpc3 and Trpc6 are directly targeted by miR-26a, the levels of which are inversely proportional to severity of experimental and human AP. MiR-26a deficiency enhances while its overexpression or inhibition of its targeted SOCEs alleviates [Ca2+]i overload, ATP loss, and necrosis in PACs. MiR-26a deficiency exacerbates while its overexpression (generic or PAC-specific overexpression) or pancreatic or systemic administration of an miR-26a mimic markedly ameliorates severity of AP along with pancreatic autophagy impairment, ER stress, and inflammasome activation. Our results demonstrate that miR-26a regulates Ca2+ signaling and its sequelae cellular events in PACs with major effects on Ca2+ overload in AP, and identify the therapeutic potential of miR-26a for AP treatment.
Results
SOCE protein but not mRNA expression is induced in PACs and AP
To investigate the potential cause for pathogenic Ca2+ overload in AP, we measured changes in levels of key SOCE channels (Orai1, Trpc3, and Trpc6) and regulators (Stim1, Saraf) in experimental AP. Repetitive supramaximal stimulation with cerulein, a cholecystokinin (CCK) analogue, causes a sustained rise in intracellular Ca2+ levels and induces experimental AP (CER-AP) in rodents,37 which is the most widely used model resembling human pancreas histopathology.38 Therefore, in our study, CER-AP was induced in wild-type (WT) mice by repeated intraperitoneal injections of cerulein. Western blotting showed levels of SOCE proteins were significantly elevated in CER-AP pancreata (Figure 1A), while corresponding mRNA levels remained unchanged, except Orai1 (Figure 1B), in typical CER-AP (Figures S1A–S1C). Taurolithocholic acid 3-sulfate disodium (TLCS) also induces global transient oscillations and long-lasting Ca2+ elevations, and thus triggers cell injury/death and activation of inflammatory pathways.39 Elevation of SOCE proteins but not mRNAs was confirmed in another clinically relevant model (Figures 1C and 1D), in which TLCS was retrogradely infused into the pancreatic duct to induce TLCS-AP representing human biliary etiology (Figures S1D–S1F).38 These results suggest that a post-transcriptional increase in Ca2+ entry proteins may be a general feature of AP.
Figure 1.

SOCE proteins are increased in PACs and experimental AP
(A and B) Expression of proteins (A) and mRNAs (B) of SOCE channels and regulators in CER-AP pancreata (seven cerulein injections) 12 h after disease induction. (C and D) Expression of proteins (C) and mRNAs (D) of SOCE channels and regulators in TLCS-AP pancreata (5 mM TLCS) 6 h after disease induction. (E and F) Expression of proteins (E) and mRNAs (F) of SOCE channels and regulators in isolated mouse PACs stimulated with CCK (100 nM) for 1 h. (G and H) Expression of proteins (G) and mRNAs (H) of SOCE channels and regulators in isolated mouse PACs stimulated with TLCS (500 μM) for 30 min. Data are from 6–13 mice per group, shown as mean ± SEM.
To ascertain whether changes in SOCE proteins occur in PACs, we stimulated freshly isolated PACs from WT mice with CCK for 1 h. We found levels of Orai1, Trpc3, Trpc6, Stim1, and Saraf in PACs were markedly elevated by CCK (Figure 1E), while their mRNA levels remained unchanged (Figure 1F). These changes were confirmed in PACs exposed to TLCS for 30 min, further indicating a rapid post-transcriptional induction mechanism (Figures 1G and 1H). Taken together, these results suggest that SOCE channels can be post-transcriptionally regulated by distinct AP insults both in vivo and in vitro, which might contribute to Ca2+ overload and AP pathogenesis.
MiR-26a directly targets Trpc3 and Trpc6
MiRNAs mediate post-transcriptional gene silencing and function as prominent regulators of stress responses.17 Therefore, we explored the potential of miRNAs to regulate the post-transcriptional induction of the aforementioned SOCE channels. We used the TargetScan algorithm to search for evolutionarily conserved miRNAs that could target SOCE genes (Orai1, Stim1, Saraf, Trpc3, and Trpc6; Table S1). We identified that both human and mouse Trpc3 and Trpc6 harbor one predicted miR-26a binding site (Figure 2A), suggesting strong evolutionary conservation. Moreover, the predicted miR-26a target sites are located near the 5′ end of the 3′ UTR of these mRNAs, consistent with effective targeting.40 MiR-26a is highly expressed in human tissues27 and has been implicated in diabetes,31,33 liver diseases,32 and oncogenesis.27 In particular, miR-26a attenuates ER stress32 and inflammation,41,42 key features of AP;8, 9, 10,43 miR-26a also enhances autophagy,35 which is impaired in AP.10 Therefore, miR-26a was chosen for further analysis.
Figure 2.

MiR-26a targets TRPC3 and TRPC6 and is reduced in AP
(A) Schematic of human TRPC3 and TRPC6 and mouse Trpc3 and Trpc6 3′ UTRs. Locations of the predicted miR-26a binding sites are indicated. (B) Relative luciferase activity in HEK293T cells transfected with reporter constructs containing the WT or mutated (Mut) 3′ UTR of target genes and co-transfected with either miR-26a mimic (miR-26a) or negative control (NC). (C) Protein levels of Trpc3 and Trpc6 in PACs of 26a DKO, Hprt Tg, and control mice. (D and E) Levels of miR-26a in the pancreata with CER-AP (7 cerulein injections) at 12 h (D) and TLCS-AP (5 mM TLCS) at 24 h (E). (F and G) Levels of miR-26a in mouse PACs stimulated with CCK (100 nM) for 1 h (F) and TLCS (500 μM) for 30 min (G). (H and I) Expression of miR-26a (H) and representative immunohistochemical staining (scale bar, 50 μm; magnification ×200) of TRPC3 and TRPC6 (I) in AP and normal pancreata (Ctrl). Black outlines: higher magnification of the indicated pancreata area in the low magnification images by black squares (scale bars, 20 μm; magnification ×400). TRPC3 and TRPC6-positive area were quantified per five high-powered fields with ImageJ software. (J) Expression of miR-26a in AP pancreata of patients with sterile or infected necrosis. (K and L) Serum levels of miR-26a in CER-AP (K) and TLCS-AP (L) from (D and E), respectively. (M) Level of plasma miR-26a in healthy volunteers (left) and AP patients on admission (middle) and at 72 h of hospitalization (right). (N) Heapmap depicting Spearman correlation between admission miR-26a and routine clinical biomarkers for severity assessment. Data are from 6–13 mice per group in (D)–(G), n = 10 (AP) and n = 7 (Ctrl) in (H) and (I), n = 6 (sterile necrosis) and n = 4 (infected necrosis) in (J), n = 27 (healthy volunteers) and n = 85 (AP patients) in (M), and shown as mean ± SEM.
To directly test whether miR-26a targets Trpc3 and Trpc6, we generated luciferase constructs harboring WT or mutated miR-26a binding sites and found miR-26a significantly repressed luciferase activity of both WT constructs (Figure 2B). Mutation of the predicted miR-26a binding sites within Trpc3 or Trpc6 abolished inhibition by miR-26a (Figures 2B, S2A, and S2B), indicating direct interaction of miR-26a with these sites. Next, we determined the effect of miR-26a on Trpc3 and Trpc6 expression. Trpc3 and Trpc6 were markedly increased in PACs of miR-26a double-knockout (26a DKO) mice, but decreased in miR-26a overexpression (Hprt Tg) PACs (Figures 2C, S3A, and S3B),34 compared with their WT littermates. These data collectively demonstrate that miR-26a directly and simultaneously targets Trpc3 and Trpc6.
MiR-26a expression is reduced in murine and human AP
To determine if induction of SOCE proteins in AP is consequent on miR-26a reduction and confirm whether these changes occur in AP pathogenesis, we initially measured miR-26a expression in murine AP. MiR-26a was significantly decreased in murine CER-AP and TLCS-AP pancreata (Figures 2D and 2E). This decrease was further verified in murine PACs treated with CCK or TLCS (Figures 2F and 2G). The reciprocal expression of miR-26a and Trpc3/6 in mouse AP indicates a causal role of miR-26a in increased SOCE proteins and AP development.
We then extended the relevance of our experimental findings on pancreatic miR-26a/TRPCs axis to AP patients and disease severity. Samples of normal human pancreata were obtained from patients undergoing resectional surgery for left-sided or small unobstructing pancreatic tumors,44 while samples of AP pancreata were obtained from patients undergoing open necrosectomy or pseudocyst drainage procedures (Table S2). miR-26a levels were significantly reduced in AP compared with normal pancreata (Figure 2H), while TRPC3 and TRPC6 proteins were markedly elevated (Figure 2I). miR-26a levels were >50% lower in the pancreata of patients with infected than sterile necrosis (Figure 2J; Table S3), suggesting a negative correlation between pancreatic miR-26a expression and AP severity.
Next, we determined the circulating miR-26a levels in AP mice and patients. Circulating miR-26a levels were significantly decreased in CER-AP and TLCS-AP (Figures 2K and 2L). Similarly, plasma miR-26a levels were markedly reduced in human AP on the day of hospital admission compared with healthy controls and significantly restored circa 72 h thereafter (Figure 2M; Table S4). Plasma miR-26a levels were significantly and inversely correlated with serum urea, creatinine (CREA), lactate dehydrogenase (LDH), and hematocrit (HCT), routine biomarkers indicative of AP severity (Figure 2N). Taken together, these data reveal a consistent reduction of miR-26a expression in both murine and human AP that allows escape of SOCE protein translation from this shared checkpoint.
MiR-26a attenuates Ca2+ signaling and overload in PACs
Given the significance of TRPCs in the pancreas, we assessed the effect of miR-26a on Ca2+ responses in PACs. Acetylcholine (ACh) at quasi-physiological concentrations evokes Ca2+ oscillations in PACs, essential to enzyme secretion.5 Freshly isolated PACs from 26a DKO and Hprt Tg mice as well as their WT littermates were treated with 50 nM ACh and [Ca2+]i changes monitored in real time by confocal microscopy. The frequency and average peak amplitude of [Ca2+]i oscillations elicited by ACh were significantly increased in 26a DKO (Figure 3A) and markedly decreased in Hprt Tg PACs (Figure 3B), compared with WT. Supra-physiological concentrations of CCK or the pancreatitis toxin TLCS at increasing concentrations induce increasingly prolonged [Ca2+]i elevations in PACs, causing premature intracellular activation of zymogens.45 MiR-26a deficiency augmented the peak amplitude and prolonged [Ca2+]i elevations (Figures 3C and 3E), whereas miR-26a overexpression reduced both parameters induced by 10 nM CCK or 200 μM TLCS (Figures 3D and 3F), compared with WT. Similar changes were seen at higher toxin concentrations, which typically induce sustained [Ca2+]i elevations. MiR-26a deficiency substantially increased the peak amplitude, period, and area under the curve of the [Ca2+]i plateau (>2-fold) induced by 100 nM CCK or 500 μM TLCS compared with WT (Figures 3G and 3I), whereas miR-26a overexpression reduced the [Ca2+]i plateau by over 50% (Figures 3H and 3J). These results demonstrate that miR-26a is capable of modulating physiological and pathological Ca2+ responses in PACs.
Figure 3.

MiR-26a attenuates Ca2+ responses in PACs
(A–J) Representative traces and response areas (200–800 s) of Ca2+ oscillations or sustained Ca2+ plateaus induced by ACh (50 nM) (A and B), CCK (10 nM) (C and D), TLCS (200 μM) (E and F), CCK (100 nM) (G and H), or TLCS (500 μM) (I and J) in PACs isolated from 26a DKO (A, C, E, G, and I), Hprt Tg (B, D, F, H, and J), and control mice. (K and L) Representative traces and response areas (200–800 s) of sustained Ca2+ plateaus induced with 500 μM TLCS in isolated WT (K) and 26a DKO (L) PACs pre-treated with HEPES (Ctrl), Trpc3 inhibitor (Pyr3, 3 μM), Trpc6 inhibitor (SAR7334, 50 nM), or both for 30 min. Data are shown as mean ± SEM. All experiments were from three or more independent repeats which yielded a sum of 30–40 cells per group.
Trpc3 and Trpc6 inhibition parallels effects of miR-26a in PACs
Next, we tested whether the effect of miR-26a on Ca2+ responses is associated with Trpc3 and Trpc6. Ryr3 has been widely utilized to selectively and effectively inhibit TRPC3 at a concentration of 3 μM.12 However, the effect of TRPC6 inhibitor SAR7334 has not yet been explored in PACs. Therefore, we initially determined the potential toxicity of SAR7334 in PACs, with a working concentration from 5 nM to 100 nM.46 It showed that 100 nM SAR7334 induced PAC death (Figures S4A and S4B), and we thus chose 50 nM for further analysis. Sustained [Ca2+]i elevations induced by 500 μM TLCS were substantially diminished by the Trpc3 inhibitor Pyr3 and Trpc6 inhibitor SAR7334 in both WT (Figure 3K) and 26a DKO PACs (Figure 3L). Moreover, simultaneous blockade of Trpc3 and Trpc6 had a stronger inhibitory effect on TLCS-induced [Ca2+]i elevations than inhibition of either channel alone (Figures 3K and 3L).
Pathological Ca2+ elevations result in diminished ATP production and necrosis, both hallmarks of AP.47 A marked fall of ATP in WT and even greater decrease in 26a DKO PACs was induced by 500 μM TLCS, losses moderately reduced by Pyr3 treatment (Figure S4C); changes in necrotic cell death pathway activation were consistent with these findings (Figure S4D). In contrast, miR-26a overexpression prevented ATP loss (Figure S4E) and necrosis (Figure S4F) induced by TLCS. Intriguingly, administration of Pyr3 had no effect on TLCS-induced ATP loss and necrosis in Hprt Tg PACs (Figures S4E and S4F), suggesting functional redundancy between miR-26a overexpression and Trpc inhibition. Taken together, these results strengthen the link between miR-26a, Trpc3, Trpc6, and Ca2+ responses in AP pathogenesis.
MiR-26a deficiency exacerbates CER-AP and TLCS-AP
We next examined the in vivo role of miR-26a in experimental AP, which are dependent on dysregulated Ca2+ signaling.11, 12, 13, 14, 15, 16,48,49 In CER-AP, 26a DKO mice had greater pancreatic injury than WT (Figure 4A), although the pancreatic histopathology was similar in both genotypes without cerulein treatment. Histopathological analyses found 2-fold increases in edema, inflammation, necrosis, and total histopathology score (combined overall severity score of these parameters), in 26a DKO mice (Figure 4B). MiR-26a deficiency elevated serum amylase, lipase, and interleukin-6 (IL-6; Figure 4C), and augmented expression of pancreatic gene markers associated with inflammation (Asc, Casp1, Nlrp3, Il-1b, and Il-6) and oxidative (Hmox1) and ER (Bip, Chop, and Eif2s1) stress over those in WT (Figures 4D–4F and S5A). Of note, the levels of Trpc3 and Trpc6 proteins were markedly increased in 26a DKO pancreata (Figure 4G).
Figure 4.

MiR-26a deficiency exacerbates CER-AP and TLCS-AP
(A–G) Representative images (H&E; scale bar, 50 μm; magnification ×200) for pancreatic histopathology (A) and corresponding scores for edema, inflammatory cell infiltration, acinar cell necrosis, and their sum value (B), serum amylase, lipase, and IL-6 levels (C), pancreatic mRNA markers for inflammation (D), oxidation (E), and ER stress (F), and pancreatic protein levels of Trpc3 and Trpc6 (G) from 26a DKO and their control littermates in CER-AP (7 cerulein injections) 12 h after disease induction. (H–M) Pancreatic histopathology changes (H&E; scale bar, 50 μm; magnification ×200) (H) and corresponding scores for edema, inflammatory cell infiltration, acinar cell necrosis, and their sum value (I), serum amylase, lipase, and IL-6 levels (J), and pancreatic mRNA markers for inflammation (K), oxidation (L), and ER stress (M) from 26a DKO and their control littermates in TLCS-AP (3 mM) 12 h after disease induction. Data are from six to eight mice per group, shown as mean ± SEM.
In TLCS-AP, miR-26a deficiency significantly exacerbated pancreatic damage (Figures 4H and 4I), serum indices of AP (Figure 4J), and the pancreatic gene markers of inflammation, oxidation, and ER stress (Figures 4K–4M and S5B), recapitulating the full range of findings in CER-AP.
Taken together, these results demonstrate that miR-26a deficiency exacerbates the severity of AP through loss of miR-26a-mediated regulation of TRPCs, inflammation, and cell death.
MiR-26a overexpression prevents CER-AP and TLCS-AP
Next, we investigated the consequences of miR-26a overexpression in CER-AP. Compared with WT littermate controls, Hprt Tg mice had reduced pancreatic tissue damage (Figure 5A), histopathology scores (Figure 5B), and serum amylase, lipase, and IL-6 levels (Figure 5C) in CER-AP. Levels of mRNA marker for inflammation, oxidative and ER stress (Figures 5D–5F and S6A), and levels of Trpc3 and Trpc6 proteins (Figure 5G) were reduced in the pancreata of Hprt Tg mice. Notably, levels of serum amylase, IL-6, and pancreatic (mRNA) Asc, Nlrp3, Bip, and Chop were comparable in CER-AP and control Hprt Tg mice (Figures 5C, 5D, and 5F). Expression of PAC markers (mRNA) Amy2, Cpa1, and Rbpjl, which have been shown to be reduced by CER,50 was preserved in Hprt Tg mice (Figure S6B), further supporting a potent protection of miR-26a.
Figure 5.

MiR-26a overexpression ameliorates CER-AP and TLCS-AP
(A–G) Representative images (H&E; scale bar, 50 μm; magnification ×200) for pancreatic histopathology (A) and corresponding scores for edema, inflammatory cell infiltration, acinar cell necrosis, and their sum value (B), serum amylase, lipase, and IL-6 levels (C), pancreatic mRNA markers for inflammation (D), oxidation (E), and ER stress (F), and pancreatic protein levels of Trpc3 and Trpc6 (G) from Hprt Tg and their control littermates in CER-AP (seven cerulein injections) 12 h after disease induction. (H–M) Pancreatic histopathology changes (H&E; scale bar, 50 μm; magnification ×200) (H) and corresponding scores for edema, inflammatory cell infiltration, acinar cell necrosis, and their sum value (I), serum amylase, lipase, and IL-6 levels (J), and pancreatic mRNA markers for inflammation (K), oxidation (L), and ER stress (M) from Hprt Tg and their control littermates in TLCS-AP (5 mM) 24 h after disease induction. Data are from five to eight mice per group, shown as mean ± SEM.
In TLCS-AP, miR-26a overexpression decreased pancreatic injury (Figure 5H), histopathology scores (Figure 5I), elevations of serum amylase and lipase (Figure 5J), and inductions of pancreatic gene markers of inflammation (Figure 5K), and oxidative (Figure 5L) and ER stress (Figures 5M and S6C). Particularly, the histopathology scores (Figure 5I), serum lipase levels (Figure 5J), and mRNA levels of inflammatory (Figure 5K), oxidative (Figure 5L), and ER stress markers (Figure 5M) remained similar in TLCS-AP and control Hprt Tg mice. Together, these results strongly suggest that miR-26a overexpression has a marked protective effect on the likelihood and severity of AP, which is reproducible and model independent.
MiR-26a inhibits inflammation, ER stress, and impaired autophagy in AP
We determined the effect of miR-26a on early global gene expression in CER-AP by RNA sequencing (RNA-seq). Pancreata from Hprt Tg mice and their WT littermates were collected 5 h after the first of a total of four cerulein injections. We analyzed the transcriptome datasets using a self-organizing map (SOM) machine-learning technique,51 which enables a detailed evaluation of the transcriptomic data, taking into account the multidimensional nature of gene regulation. This yielded distinct SOM spots in Hprt Tg mice and their WT littermates (Figure 6A; Table S5). An analogous result was obtained for differences between Hprt Tg and WT mice, both with CER-AP (Figure 6B). The spot associated with immune responses in WT littermates (highlighted within rectangle in Figure 6B) disappeared in Hprt Tg mice, indicating an inflammatory signature modulated by miR-26a.
Figure 6.

MiR-26a overexpression ameliorates pancreatic inflammation, autophagy, and ER stress in experimental AP
(A) SOM patterns show different mRNA signature enrichments by RNA-seq analysis with spot areas representing clusters of upregulated (red) and downregulated (blue) expression of genes in the pancreata of WT and Hprt Tg mice with CER-AP (four cerulein injections) 4 h after disease induction compared with control. Each map depicts the average SOM profile across three mice per group. (B) SOM portrait of gene expression signature enrichment analysis depicting differences between WT and Hprt Tg in CER-AP derived from (A). (C and D) Chord diagrams displaying top 10 GO biological processes (C) and KEGG pathways (D) for WT versus Hprt Tg in CER-AP derived from (A). (E and F) Heatmaps demonstrating inflammation (E) and ER stress (F) gene expression in WT versus Hprt Tg with or without CER-AP derived from (A). (G) Heatmap of qRT-PCR analysis of six inflammation and three ER stress genes expressed in WT versus Hprt Tg pancreata with or without CER-AP (7 cerulein injections) 12 h after the first cerulein or saline injection. (H and I) Levels of protein markers for autophagy (P62 and LC3) and inflammasome activation (Pro Casp-1 and Casp-1 p10) in WT versus Hprt Tg pancreata (H) from same mice as in (G) and WT versus 26a DKO pancreata (I) from same mice as in Figures 4A–4G.
Comparison of the transcriptome data from WT with versus without CER-AP and Hprt Tg with versus without CER-AP identified 4,284 and 4,635 differentially expressed genes (DEGs; >2-fold, p < 0.05) respectively (Figures S7A–S7E; Tables S6 and S7). These two datasets of DEGs were analyzed using Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG). The analyses showed that Hprt Tg mice had significantly attenuated immune responses during CER-AP (Figures 6C and 6D), consistent with the SOM analysis (Figures 6A and 6B). Expression of 366 genes was downregulated and 269 upregulated in Hprt Tg mice compared with their WT littermates (Table S8), the largest proportion associated with chemotaxis and immune responses; 42 immune response genes exhibited significantly less expression (Figure 6E; Table S9), as did ER stress genes in Hprt Tg mice (Figure 6F; Table S10). The effects of miR-26a on inflammation and ER stress were verified by qRT-PCR of Hprt Tg (Figure 6G) and 26a DKO pancreata (Figure S7F). Moreover, western blotting revealed increases in markers for impaired autophagy (P62 and LC3) and NLRP3-inflammasome activation (Caspase-1), characteristic features of AP associated with Ca2+ overload,10 were consistently reduced in Hprt Tg mice (Figure 6H), but increased in 26a DKO mice (Figure 6I), compared with their respective WT littermates.
Altogether, the transcriptome dynamics suggest miR-26a mediates modular organization and substantial transcriptional reprogramming, further supporting the protective role of miR-26a in AP.
MiR-26a in PACs, but not myeloid cells, ameliorates pancreatic injury and systemic inflammation in CER-AP
Next, we further clarified which cellular source of miR-26a is responsible for the above protective effects observed in mouse AP models. Given the mighty suppression of miR-26a on AP-related inflammation, we initially determined the potential contribution of immune cells by utilizing our recently established myeloid cell-specific miR-26a overexpression (Lyz Tg) mice.52 Intriguingly, Lyz Tg mice and their WT littermate controls exhibited a similar phenotype in CER-AP, as shown by comparable localized pancreatic injury (Figure 7A), histopathology scores (Figure 7B), serum amylase and lipase levels (Figure 7C), and the expression of inflammatory genes (Il-6 and Il-1b, Figure 7D). Lung myeloperoxidase (Figure 7E) and histopathological scoring (Figure 7F) of the lung tissues were also nearly identical in both genotypes. These data argue against a protective role for miR-26a in myeloid cells, including macrophages, in AP pathogenesis.
Figure 7.

Overexpression of miR-26a in PACs, but not myeloid cells, mitigates pancreatic injury and systemic inflammation in CER-AP
(A–F) Effects of myeloid cell-specific miR-26a overexpression (Lyz Tg) on CER-AP. Representative images (H&E; scale bar, 50 μm; magnification ×200) for pancreatic histopathology (A) and corresponding scores for edema, inflammatory cell infiltration, PAC necrosis, and their sum value (B), serum amylase and lipase (C), pancreatic mRNA markers for inflammation (D), as well as lung myeloperoxidase (MPO) activity (E) and lung alveolar membrane thickening score (F) from Lyz Tg and their WT littermates in CER-AP (50 μg/kg/h × 7 cerulein) at 12 h after disease induction. (G–N) Effects of PAC-specific miR-26a overexpression on CER-AP. Schematic showing intraperitoneal administration of tamoxifen-induced miR-26a overexpression and cerulein administration regimens (G). Representative images (H&E; scale bar, 50 μm; magnification ×200) for pancreatic histopathology (H) and corresponding scores for edema, inflammatory cell infiltration, PAC necrosis, and their sum value (I), serum amylase, lipase, and IL-6 levels (J), pancreatic mRNA markers for oxidation (K), ER stress (L), and inflammasome activation (M), and Trpc3 and Trpc6 protein levels, impaired autophagy, and inflammasome activation (N) from Ela-Tg mice and their WT littermates in CER-AP. All experiments had six to nine mice in each group except for those in western blotting analysis (n = 3).
To directly demonstrate the significance of miR-26a in PACs, we then generated PAC-specific miR-26a overexpression (Ela-Tg) mice by utilizing a tamoxifen-inducible cela1-Cre (Figure S8A), which has been widely used in AP research by inducing specific transgene expression in PACs.53, 54, 55, 56 As expected, the expression of miR-26a in PACs was increased in Ela-Tg mice treated with tamoxifen (Figure S8B). We next examined the role of miR-26a in CER-AP (Figure 7G). Along with induced miR-26a expression (Figure S8C), Ela-Tg mice had lower pancreatic tissue damage (Figure 7H), histopathology scores (Figure 7I), serum amylase, lipase, and IL-6 levels (Figure 7J) compared with their WT littermate controls. Accordingly, levels of mRNA markers for oxidative stress, ER stress, and inflammation (Figures 7K–7M and S8D), and protein markers of inflammasome activation and autophagy impairment (Figure 7N), as well as Trpc3 and Trpc6 protein levels (Figure 7N), were reduced in the pancreata of Ela-Tg mice. Moreover, TdT-mediated dUTP nick-end labeling (TUNEL) staining revealed that CER-AP WT mice had a small proportion of apoptotic PACs, and it was decreased in CER-AP Ela-Tg mice (Figure S8E), in agreement with the notion that necrosis, but not apoptosis, is the major mode of PAC death in CER-AP.57 Similarly, a few infiltrations of CD3+ and CD4+ T cells was exhibited in CER-AP WT mice, but reduced in Ela-Tg mice, in line with their AP phenotypes (Figure S8F). These results strongly suggest that altered expression of miR-26a in PACs, but not myeloid cells, may be the primary contributor to AP development.
Pancreatic and systemic administration of miR-26a ameliorates AP
In view of the evident protective role of miR-26a, we sought to validate the therapeutic potential of miR-26a in murine AP. We first tested pancreas-specific miR-26a restoration by pancreatic ductal injection of an miR-26a mimic via mini-pump, shortly after infusion of 5 mM TLCS (Figure 8A). Pancreatic miR-26a expression was over 10-fold greater after the mimic than after vehicle (Figure 8B), but remained unchanged in other organs such as the liver and kidney (Figures S9A and S9B). The miR-26a mimic significantly reduced all sequelae of TLCS-AP, including pancreatic injury (Figure 8C) and histopathological scores (Figure 8D), preventing any detectable increase in serum indices (Figure 8E).
Figure 8.

Pancreatic and systemic administration of miR-26a mimic ameliorates AP
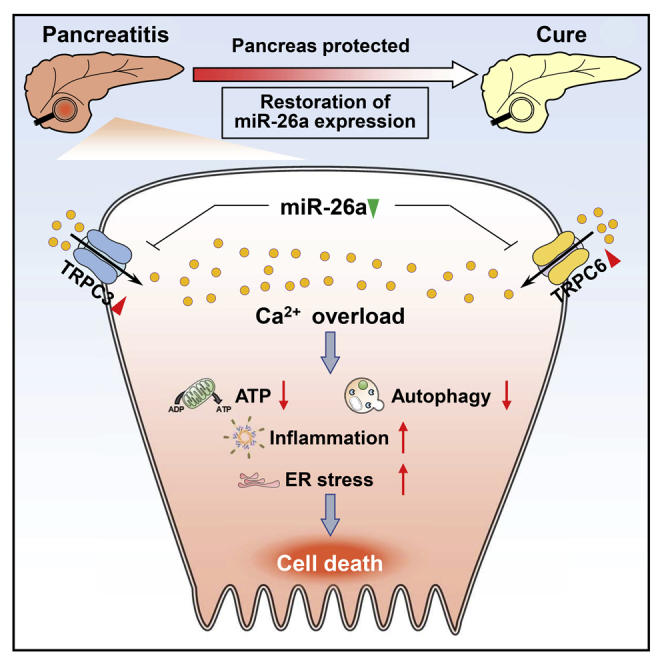
(A) Schematic showing pancreatic ductal TLCS and miR-26a mimic administration regimens. (B) Levels of miR-26a in the pancreata with or without TLCS-AP. (C–E) Representative images (H&E; scale bar, 50 μm; magnification ×200) of pancreatic histopathology (C) and corresponding scores for edema, inflammatory cell infiltration, cell necrosis, and their sum value (D), serum amylase, lipase, and IL-6 levels (E) in TLCS-AP with or without miR-26a mimic. (F) Schematic showing intraperitoneal cerulein and miR-26a mimic administration regimens. (G) Levels of miR-26a in the pancreata with or without CER-AP. (H–J) Representative images (H&E; scale bar, 50 μm; magnification ×200) of pancreatic histopathology (H) and corresponding scores for edema, inflammatory cell infiltration, cell necrosis, and their sum value (I) as well as serum amylase and lipase levels (J). (K) Levels of Trpc3, Trpc6, autophagy (P62 and LC3), and inflammasome activation (Pro Casp-1 and Casp-1 p10) proteins in CER-AP pancreata with or without miR-26a mimic. (L) Summary diagram of the critical role of miR-26a in PAC [Ca2+]i overload and its downstream pathological pathways, highlighting the therapeutic potential of miR-26a in AP. Data are from seven to nine mice per group, shown as mean ± SEM.
As an miR-26a mimic could become a common treatment for human AP of multiple etiologies, and to approximate more widely applicable routes of administration (e.g., intravenous), we tested the mimic in another model, CER-AP, by intraperitoneal administration. The mimic was injected 3.5 h after the first cerulein injection and CER-AP assessed at 24 h (Figure 8F), which resulted in >2-fold increase in pancreatic miR-26a expression (Figure 8G). This increase offset the comparable 50% suppression of miR-26a in CER-AP and TLCS-AP (Figures 2D and 2E), providing an excellent model to characterize the effects of miR-26a restoration in AP. Mice treated with the miR-26a mimic and its vehicle showed no discernible clinically relevant changes in histological or hematological parameters (Figures S10A and S10B). Strikingly, mice that received the mimic had significantly diminished parameters of AP (Figures 8H–8J), including decreased expression of pancreatic gene markers for inflammation, oxidative and ER stress (Figures S10C–S10E), reduced protein markers of inflammasome activation and autophagy impairment, and downregulated Trpc3 and Trpc6 levels (Figure 8K). Taken together, these results provide proof of principle for miR-26a as a potentially effective therapeutic for human AP.
Discussion
Here we report a previously unknown mechanism underlying AP pathogenesis and identify miR-26a as an intrinsic checkpoint for Ca2+ signaling and overload in PACs (Figure 8L). We found that SOCE proteins were increased in the pancreata of AP mice and humans caused by pathological Ca2+ signaling,9,38 as well as in isolated PACs treated with different AP inducers for a short period of time. These results define for the first time, to our knowledge, a dynamic profile of SOCE proteins during AP initiation and progression. Due to the lack of reliable experimental systems in long-term PAC culture in vitro, it is difficult to directly determine whether increased SOCE proteins are the cause of Ca2+ overload observed in AP. However, a strong correlation among levels of SOCE proteins, [Ca2+]i concentration, and disease progression has been indicated in other systems, such as proteinuric kidney disease.58 Importantly, we demonstrated that Trpc3/6-targeting miR-26a dramatically attenuates physiological and pathological Ca2+ responses, supporting a causal role of increased SOCE proteins in Ca2+ overload. These findings represent a general feature of AP and indicate a post-transcriptional regulation mechanism for Ca2+-handling protein underlying Ca2+ overload in AP pathogenesis.
In this study, we have demonstrated that miR-26a directly targets both Trpc3 and Trpc6 to reduce their expression, restricting Ca2+ influx and placing limits on SOCEs. This protective function is diminished rapidly in AP through suppression of miR-26a expression, resulting in escape and upregulation of SOCE protein expression, Ca2+ influx, and Ca2+ overload, inducing AP.8, 9, 10, 11, 12, 13, 14, 15, 16,43,48,49 These findings in two classical models of AP, with confirmation of miR-26a suppression and SOCE protein upregulation in human AP, underline the critical role of Ca2+ overload in the pathogenesis of AP. While overexpression of miR-26a did not wholly block but decreased neurohumoral Ca2+ signaling and toxic Ca2+ overload, it did significantly and consistently ameliorate the pathological sequelae of experimental AP, as did the miR-26a mimic. In line with the implications of miRNAs in either AP progression or Ca2+ signaling,59,60 our work provides the first evidence, to our knowledge, that a unique miRNA robustly modulates Ca2+ overload and AP pathogenesis on the basis of data obtained at the cellular, animal, and human levels. Interestingly, the regulation of Trpc3/6 by miR-26a has recently been reported in other systems,42,61 suggesting the significance of this prevalent regulatory mode in pathology. In addition, it is of importance to confirm the expression of pancreatic miR-26a in a larger cohort of patients with AP.
Each of the key SOCE proteins TRPC3, TRPC6, ORAI1, and STIM1 participate in Ca2+ entry,11, 12, 13, 14,62 while SARAF, through interaction with STIM1, mediates Ca2+-dependent channel inactivation.16 Our identification of TRPC6 as a channel regulated by miR-26a, integral to Ca2+ entry in PACs and upregulated in AP, expands the range of Ca2+ channels implicated in AP. Heteromultimerisation of TRPC3 with TRPC1 and separately TRPC6 with TRPC4 facilitates their cooperation with ORAI1 and STIM1 in an interdependent manner. The overall impact of upregulation of these channels is largely unrestrained by SARAF, however, as SARAF interaction with STIM1 is unstable in AP.14 Additionally, TRPC3 and TRPC6 operate in STIM1-independent modes, not curtailed by SARAF-STIM1 interaction.15 Moreover, the miR-26a suppression we found in AP leads to the loss of protective functional redundancy between miR-26a expression and Trpc3 inhibition. In the face of multiple mechanisms of injury, the attraction of an miR-26a mimic is simultaneous targeting of multiple Ca2+ channels, providing augmented inhibition of SOCE over that achieved by single-channel inhibition. In addition to SOCE-mediated calcium entry, release of Ca2+ from internal stores, mediated by inositol-1,4,5-trisphosphate (IP3), phospholipase C, ryanodine receptors, and IP3 receptors, etc.,63, 64, 65 also contribute to Ca2+ overload and AP development. It is of interest for future study to expand the potential role of miR-26a in intracellular Ca2+ channels and related Ca2+ influx.
MiR-26a is expressed comprehensively across vertebrates34 and human tissues,27 but, as shown here, is a target for decreased expression and/or degradation in AP, followed by loss of control of SOCE protein expression and a gamut of transcriptional responses that contribute to amplification loops in AP.9 MiR-26a could therefore have clinical utility as an early circulating prognostic biomarker in AP.25 We have shown the extent to which variation in the expression of this one miRNA can change the whole pancreas transcriptome. MiR-26a deficiency increased, whereas overexpression decreased, transcription of gene complexes integral to inflammation, oxidative stress, ER stress, and impaired autophagy, critical processes in AP.8, 9, 10,43 Inflammation, oxidative stress, ER stress, and autophagy are downstream pathological pathways of Ca2+ overload in the progression of AP,8,66 and it thus is reasonable that these effects of miR-26a may be associated with its suppression on TRPC3/6. Besides, certain Ca2+-independent mechanism(s) might contribute to these effects of miR-26a. Indeed, accumulating evidence suggests a direct regulatory role of miR-26a in inflammation, ER stress, and autophagy. For example, miR-26a can directly target the cytokine Il-6,41 as well as the ER stress mediator eukaryotic translation initiation factor 2, subunit 1 alpha (Eif2s1) and the negative regulators of autophagy dual specificity phosphatase 4 and 5 (Dusp4 and Dusp5), as in the liver.32,35 The molecular mechanisms underlying miR-26a′s function in these Ca2+ overload-related pathways await further investigation. Integrative omics approaches would be useful to explore possible crosstalk between these actions of miR-26a in disease. Under these circumstances, it is conceivable that a small change in miR-26a expression may result in a large physiological effect. Indeed, a minor decrease in miR-26a expression (about 2-fold) remarkably aggravated the development of AP, whereas a modest increase in miR-26a expression by genetic and pharmacological interventions was sufficient to prevent all sequelae of experimental AP. Furthermore, we generated PAC- and myeloid cell-specific miR-26a overexpression mice, respectively. Intriguingly, overexpression of miR-26a in PACs, but not myeloid cells, recapitulated the protective effects induced by global miR-26a overexpression in CER-AP. Of note, PAC-specific overexpression of miR-26a in our study was obtained by utilizing Ela-cre mice that express Cre under the clea1 promoter. Although Ela-cre mice have been broadly used to control PAC-specific transgene expression,53, 54, 55, 56,67 recent evidence suggests that clea1 is also expressed in other cell types, including T cells.68 In these circumstances, the possibility of synergistic effect of miR-26a overexpression in PACs and other cells, such as T cells, on AP pathogenesis would be critically evaluated in the future.
Finally, we explored the therapeutic potential of miR-26a in AP. Entry of miR-26a into the pancreas after intraperitoneal administration in severe CER-AP demonstrated effective target engagement, essential to drug development. This was further validated in a more severe TLCS-AP in which miR-26a was infused via pancreatic duct shortly after TLCS. In view of the rapid loss of miR-26a in PACs and AP, an miR-26a mimic is likely to require early administration in human AP for maximum effect. Notably, both Trpc3 and Trpc6 are broadly expressed in many tissues,69 and the administration of exogenous miR-26a mimic may lead to its biodistribution into other organs beyond the pancreas. Intriguingly, the expression of miR-26a in the organs that we examined appeared to be unchanged post administration of the miR-26a mimic. Therefore, it is likely that the miR-26a/TRPC regulatory axis in the pancreas, rather than other tissues, predominantly accounts for the beneficial role of miR-26a in AP pathogenesis, although the possibility of contributions from other organs warrants future study. Additionally, it would be interesting to investigate the potential protective role of miR-26a in AP under comorbid obesity or metabolic disorder conditions as well as in chronic pancreatitis.
In summary, this work demonstrates that miR-26a can robustly modulate PAC Ca2+ overload and AP pathogenesis based on data obtained at the cellular, animal, and human levels. These findings reveal a previously unknown pathophysiological mechanism of surveilling Ca2+ overload and highlights the potential for clinical translation of miR-26a for AP management.
Materials and methods
Human studies
All human studies were conducted according to the principles of the Declaration of Helsinki, and approved by the Institutional Review Board and Biomedical Ethics Committee of West China Hospital, Sichuan University (2017, no. 456; 2020, no. 196). The biobanking procedures were certified by the China Human Genetic Resources Management Office as a part of West China Biobanks (2016, no. 406). Fully informed consent was obtained from all patients prior to sampling of blood and/or pancreatic tissues. The procedures for clinical data collection were described in our previous studies70 and data were stored in an electronical database (Microsoft Access).
Human blood sample collection
Samples of peripheral venous blood were taken from patients diagnosed with AP according to the revised Atlanta classification,71 within 72 h after the onset of abdominal pain. Patients were excluded from blood sampling if they had chronic pancreatitis, pancreatic tumor, trauma, or pregnancy as etiologies or any advanced pre-existing comorbidities.70 Patients who died were also excluded. Plasma samples of fasted healthy volunteers were retrieved from the West China Biobanks.
Blood samples collected in ethylenediaminetetraacetic acid (EDTA) tubes were processed following a protocol we previously described.72 For AP patients, routine blood (Sysmex XN-9000) and biochemical (Cobas8000) indices were determined using automatic analyzers.
Human pancreata samples collection
Small (<1 cm2 × 1 mm) samples of viable pancreas were taken from patients undergoing surgery for symptomatic pseudocysts complicating AP or for infected pancreatic necrosis. Normal pancreatic samples (<1 cm2 × 1 mm) were taken from patients undergoing surgery for left-sided or small unobstructing pancreatic tumors following Liverpool's protocols.44
Animals
C57BL/6 background, 10- to 12-week-old, male WT or genetically modified mice were used throughout the study. Mice were fed a standard laboratory chow, water ad libitum, under a 12-h light-dark cycle (lights on from 8 a.m. to 8 p.m.) at constant temperature (22°C). All animal experiments were conducted according to the protocols approved by the Institutional Animal Care and Use Committee of West China Hospital, Sichuan University.
Generation of generic 26a DKO or overexpression mice
Generation of 26a double knockout (DKO)31 and transgenic miR-26a (Hprt Tg)34 mice was previously described.
Generation of PAC-specific miR-26a overexpression mice
Generation of Tg (CAG-Neo-STOPfl-Mir26a1) mice has been described by us previously.34 We generated a cela1-cre driver line consisting of a tamoxifen-inducible CreERT2 recombinase to construct Ela-CRE mice. PAC-specific miR-26a overexpression (Ela-Tg) mice were generated by crossing Tg (CAG-Neo-STOPfl-Mir26a1) mice with Ela-CRE mice. Ela-Tg and littermate Tg (CAG-Neo-STOPfl-Mir26a1) mice were used for experiments. To induce ectopic miR-26a expression, Ela-Tg mice, as well as their WT littermates, were treated with tamoxifen (T5648, Sigma-Aldrich, St. Louis, MO) before experiments. Tamoxifen was dissolved at a final concentration of 20 mg/mL in corn oil. Two- to 3-month-old mice received intraperitoneal injection of tamoxifen (100 mg/kg) once per day for five consecutive days. Induction of experimental AP in these mice began 1 day after the last injection tamoxifen.
Generation of myeloid cell-specific miR-26a overexpression mice
Myeloid cell-specific miR-26a overexpression (Lyz Tg) mice were generated by crossing Tg (CAG-Neo-STOPfl-Mir26a1) mice with Lyz-CRE mice as described previously.52 Lyz Tg and littermate Tg (CAG-Neo-STOPfl-Mir26a1) mice were used for experiments.
Induction of experimental AP models
These protocols were performed by following our previously publications.13,49,73,74 Mice received four, seven, or 12 intraperitoneal injections of cerulein (50 μg/kg; catalog number 6264, Tocris Bioscience; Ellisville, MO) at hourly intervals to induce hyperstimulation AP (CER-AP), while control mice received saline injections of the same volume at the same intervals.13,49,73,74 Taurolithocholic acid 3-sulfate disodium (TLCS; 3 or 5 mM; T0512, Sigma-Aldrich, St. Louis, MO) was retrogradely infused (5 μL/min for 10 min) into the pancreatic duct via a mini-pump (Pump33DDS, Harvard Apparatus; Boston, MA) to induce AP (TLCS-AP).13,49,73 Mice were anesthetized with isoflurane immediately prior and during the infusion TLCS or saline.
Administration of miR-26a mimic
The miR-26a mimic and its negative control without chemical modification were synthesized (Xi'an XiRQCON Biotechnology, Xi’an, China), and then prepared freshly with its transfection reagent Entranster-in vivo (18668, Engreen, Beijing, China) before systemic or pancreatic administration to WT mice. Briefly, miR-26a mimic and its negative control were diluted with endotoxin-free water to 1 μg/μL, and then an equal volume of transfection reagent was added and mixed thoroughly. After incubation at room temperature for 15 min, the transfection reagent-miR-26a/negative control mixture was injected intraperitoneally or injected into the pancreatic duct of mice. For CER-AP, the miR-26a mimic (2 mg/kg) was intraperitoneally administered at 3.5 h after the first cerulein injection. For TLCS-AP, miR-26a mimic (0.8 mg/kg) was retrogradely infused via the pancreatic duct 3 min after TLCS (5 mM). Control mice received systemic or pancreatic application of negative control (vehicle) at the same volume and time regimen.
Isolation of primary PACs
Mouse PACs were freshly isolated by collagenase digestion as described previously.49 After culling the mice, the pancreas was immediately removed, washed twice in HEPES solution (pH 7.3; 140 mM NaCl, 4.7 mM KCl, 10 mM HEPES, 10 mM glucose, 1 mM CaCl2, and 1.13 mM MgCl2), followed by injections of 250 U collagenase (C9697, Sigma-Aldrich, St. Louis, MO) into the main pancreatic duct and surrounding tissue to maximize homogeneous digestion. After incubation at 37°C for up to 17 min, the pancreas was placed into a 50-mL centrifuge tube with 3–5 mL of HEPES and quickly pipetted, a process repeated several times until the solution became clear. The solution of digested tissue was passed through a 70-μm cell filter to obtain dispersed PACs, followed by centrifugation at 260 g × 2 min twice. The final pellet was resuspended in HEPES and PACs were used within 4 h of isolation.
Ca2+ measurement in PACs
Ca2+ measurement in PACs was performed as described previously with minor modifications.49 PACs were loaded with Fluo-4 AM (5 μM; F14201, Thermo Fisher Scientific, Waltham, MA), followed by incubation with ACh (50 μM; A7000, Sigma-Aldrich), CCK (10 and 100 nM without O-sulfation, which would otherwise increase activity for approximately × 100; C2175, Sigma-Aldrich),75 or TLCS (200 and 500 μM) with or without Trpc3 inhibitor Pyr3 (3 μM; P0032, Sigma-Aldrich), Trpc6 inhibitor SAR7334 (50 nM; HY-15699, MedChem Express, Shanghai, China), or both during imaging. After incubation, the cells were centrifuged (260 × g × 1 min) and resuspended in fresh HEPES. The [Ca2+]i concentration was determined by confocal microscopy (Nikon, N-STORM & A1; excitation 488 nm, emission 500–550 nm) in a flow chamber perfused with HEPES-based extracellular solution. The series of images were recorded at 256 × 256 pixel resolution with two consecutive frames (time interval 2 s) averaged. Fluorescence was plotted as F/F0, where F0 was an averaged fluorescence signal collected from the first 10 baseline images, and samples or average traces were presented on graphs. For quantitative analysis of Ca2+ responses, areas under individual traces (the increase over baseline) recorded between 200 and 800 s were calculated and averaged to be presented with mean ± SEM.
Cell culture
HEK293T cells (CRL3216, ATCC, Rockville, MD) were cultured in Dulbecco's modified Eagle’s medium (Gibco, Grand Island, NY) supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin-streptomycin (Gibco), grown in a 37°C incubator with 5% CO2.
In vitro necrosis assays
Necrotic cell death activation was determined as a proportion of cells stained with propidium iodide (PI; 1 μM; P1304MP, Invitrogen, Carlsbad, CA) contrasted with total nuclei (Hoechst 33342; 10 μg/mL; H1399, Invitrogen) representing the total number of cells. Fluorescence of PI (excitation 488 nm, emission 630–693 nm) and Hoechst 33342 (excitation 364 nm, emission 405–450 nm) was detected by an epi-fluorescent confocal microscope (ZEISS AX10 imager A2/AX10 cam HRC, Heidelberg, Germany). Images were made of 10 separate fields from each experiment, repeated in triplicate or more.
Western blotting
Total proteins of pancreatic tissue and PACs were extracted using RIPA lysis buffer (89901, Thermo Fisher Scientific) with protease inhibitors (11873580001, Roche, Basel, Switzerland) and phosphatase inhibitors (B15001, Bimake, Houston, TX) and determined by Bradford assay (500-0205, Bio-Rad, Hercules, CA). The following antibodies were used: anti-Caspase-1 (sc-56036, Santa Cruz Biotechnology, San Lucas, CA), anti-ORAI1 (ab59330, Abcam, Cambridge, England), anti-SARAF (PA5-77334, Thermo Fisher Scientific), anti-TRPC3 (A7742, ABclonal, Wuhan, China), anti-LC3A/B (12741, Cell Signaling Technology, Danvers, MA), anti-STIM1 (5668, Cell Signaling Technology, Danvers, MA), anti-P62 (18420-1-AP, ProteintechGroup, Wuhan, China), anti-TRPC6 (18236-1-AP, ProteintechGroup), and anti-tubulin (10094-1-AP, ProteintechGroup).
RNA extraction and qRT-PCR
The total RNA of pancreatic tissues and PACs was extracted using TRIzol reagent (TR118, MRC, Cincinnati, OH) as described previously.31 The total RNA from mouse serum and human AP plasma of AP was extracted using TRIzol LS (TS120, MRC) according to the manufacturer's protocol. The reverse transcription of RNA (1–2 μg) to cDNA was conducted using M-MLV Reverse Transcriptase (28025021, Thermo Fisher Scientific). qRT-PCR was done using SYBR Green PCR Master Mix (208052, Qiagen, Nasdaq, NY) with specific primers. Primers used are provided in Table S11.
ATP measurements
Cellular ATP was assayed using the ATP Assay Kit (S0026, Beyotime, Shanghai, China) according to the manufacturer's instructions.
Luciferase reporter assay
3′ UTR sequences of TRPC3 and TRPC6 were generated by PCR with specific primers and cloned into psiCHECK-2 vector, which contains a Renilla luciferase open reading frame (ORF) and a constitutively expressed firefly luciferase gene. The predicted miR-26a binding site was mutated using Blunting Kination Ligation Kit (6127A, TaKaRa, Kyoto, Japan). HEK293T cells were cultured in 96-well plates and co-transfected with 200 ng of the indicated 3′ UTR luciferase reporter vectors with miR-26a mimic (40 nM) or its negative control using Attractene reagent (301005, Qiagen). Activities of firefly and Renilla luciferase were analyzed using the Dual-Luciferase Reporter Assay System (E1910, Promega, Madison, WI) at 24 h after transfection. Primers used are provided in Table S12.
RNA-seq analysis
RNA-seq analysis was performed by the Bejing Novogene Company. Sequencing libraries were generated using NEBNext Ultra RNA Library Prep Kit (E7530L, Ipswich, MA) for Illumina (NEB) following manufacturer's recommendations, and index codes were added to attribute sequences to each sample. Downstream analysis was performed using a combination of programs including Bowtie2, Tophat2, HTseq, and Cufflink. RNA-seq data were deposited into NCBI Sequence Read Archive (SRA) database and can be accessed via the BioProject accession number: PRJNA782201.
Bioinformatic analysis environment
All bioinformatic analyses were performed within the R environment (version 3.6.2, https://www.r-project.org/).
SOMs
Genes with zero or constant expression were removed from the gene-centric expression data, which was log2-transformed and quantile normalized. The resulting matrix of DEGs and their origins were used as input for the unsupervised, SOM neural network building using the oposSOM Bioconductor package (https://bioconductor.org/packages/release/bioc/html/oposSOM.html, version 2.4.0) with a map space of 40 × 40.51 To investigate associations between the sample phenotype and the SOM metagenes, sample groups were provided as group labels.
Functional and pathway enrichment analysis
Functional and pathway enrichment analysis of the DEGs was conducted using clusterProfiler Bioconductor package (https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html, version 3.14.3). GO and KEGG analyses for the DEGs were conducted. The Benjamini and Hochberg method was used to adjust the p values and <0.05 was significant.
H&E staining and histopathology
Fresh pancreas was collected and immersed in 10% neutral-buffered formalin for fixation, followed by paraffin embedment. Then they were cut into 4-μm-thick sections and stained with hematoxylin-eosin (H&E) for morphological analyses. The pancreatic (edema, inflammatory cell infiltration, and PAC necrosis) histopathology scores were evaluated as previously reported.49,76
For all experimental groups, 10 random fields of each pancreatic slide were graded by two independent blinded observers at magnification of 200×. Severity of pancreatic injury was defined by the extent of edema (scored as 0–4 where 0 = absent, 1 = focally increased between lobules, 2 = diffusely increased between lobules, 3 = acini disrupted, 4 = acini separated), inflammatory cell infiltration (scored as 0–4 where 0 = absent, 1 = around ductal margins in ducts, 2 = in <50% of the lobules of the parenchyma, 3 = in 50%–75% of the lobules of the parenchyma, 4 = in > 75% of the lobules of the parenchyma), and PAC necrosis (scored as 0–4 where 0 = absent, 1 = < 5% periductal necrosis, 2 = 5%–20% focal parenchymal necrosis, 3 = 20%–50% diffuse parenchymal necrosis, 4 = > 50% diffuse parenchymal necrosis), and overall histopathology score calculated as the sum of individual scores.76
Immunohistochemistry staining
Pancreatic sections were incubated with anti-TRPC3 and TRPC6 antibodies, followed by binding with horseradish peroxidase-conjugated secondary antibodies, and detected with a DAB Kit (ZLI-9017, Zsbio, Beijing, China) and counterstained with hematoxylin.
TUNEL staining
TUNEL assays were performed on paraffin sections according to the manufacturer's instructions (A113, Vazyme; Nanjing, China). Briefly, sections were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; 62248, Thermo Fisher Scientific), and then digital images were obtained using an epi-fluorescent confocal microscope (ZEISS AX10 imager A2/AX10 cam, HRC, Heidelberg, Germany).
Determination of serum amylase, lipase, and IL-6
Serum amylase (C016) and lipase (A054) were measured by respective specific kits from Nanjing Jiancheng Bioengineering Institute. Serum IL-6 was measured by Quantikine ELISA from R&D Systems (M600B, Minneapolis, MN).
Measurement of myeloperoxidase
Lung myeloperoxidase was tested as previously reported.49
Statistical analysis
Independent data are presented as mean ± SEM in box or violin plots with dot plots representing individual values, while continuous variables are depicted as line graphs. Statistical analysis was performed with GraphPad Prism 8 software (version 8.0.1) using two-tailed unpaired Student's t test with a p value of < 0.05 considered significant.
Acknowledgments
We thank the staff from the Experimental Animal Center (Xiaoting Chen and Xijing Yang), Laboratory of Clinical Pathology (Li, Fei Chen, and Chunjuan Bao), and Core Research Facilities (Jie Zhang, Lin Bai, Zhen Yang, Jinkui Pi, Yu Ding, and Sisi Wu) of West China Hospital for their continuous support. This work was supported by the National Natural Science Foundation of China (81970561, 92157205, and 91540113 to X.F.; 82172986 to Y.T.; 81973632 to Wei Huang; 81774120 to Q.X.; 81800575 to T.L.; 82100682 to N.S.); the Ministry of Science and Technology (2018ZX09201018-005 to X.F.); the 1.3.5 Project for Disciplines of Excellence, West China Hospital, Sichuan University (ZYJC18049 to X.F.; ZYJC18005 to Q.X.); the National Clinical Research Center for Geriatrics, West China Hospital, Sichuan University (Z20191005 to X.F.); the China Postdoctoral Science Foundation (2019M660239 to N.S.); the National Cancer Institute (5R01CA139158 to Wendong Huang); the National Science Center of Poland (Narodowe Centrum Nauki [NCN], 2019/33/B/NZ3/02578 to P.E.F.); HOMING Program of the Foundation for Polish Science (Fundacja na rzecz Nauki Polskiej [FNP], HOMING/2017-4/31 to P.E.F.; HOMING/2017-3/23 to M.A.J.) co-financed by the European Union under the European Regional Development Fund; the UK National Institute for Health Research (NIHR) ICAT Award (R.M.); and the NIHR Senior Investigator Award (R.S.).
Author contributions
X.F., Wei Huang, Y.T., R.S., and W.D. conceptualized and designed the experiments. X.F., Wei Huang, Y.T., and R.S. supervised the study. X.F., Wei Huang, T.Y., and Q.X. obtained funding. W.D. with the help of G.L., N.S., D.T., and S.L. carried out animal models and molecular biology experiments. P.E.F., M.A.J., and W.D. with help of L.Y. performed calcium measurements. D.T., S.L., Xinyue Zhu, X.S., S.Z., T.L., and Xiaofeng Zheng. were involved in certain parts of the experimental work. J.Z. provided SOM and heatmaps. N.S. and Q.X. provided human samples and clinical data. X.F., Wei Huang, Y.T., and R.S. wrote the manuscript. Wendong Huang provided critical mouse materials. R.M., D.N.C., and P.-O.B. critically revised the manuscript. All authors read and approved the final version of the manuscript.
Declaration of interest
Wei Huang has received research support and/or funding from Cypralis, Farsight, and Corvidia (all funds to the West China Hospital of Sichuan University). R.S. has received research support and/or funding from Calcimedica, Cypralis, GlaxoSmithKline, MSD/Merck, and Novartis; has been a consultant for AbbVie, Calcimedica, Cypralis, Eagle Pharmaceuticals, Novartis, and Takeda (all funds to the University of Liverpool); and is collaborating in the IMI2 TransBioLine project with multiple public and private institutions, including Janssen, Lilly, MSD/Merck, Novartis, Pfizer, Roche, and Sanofi-Aventis.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.01.033.
Contributor Information
Robert Sutton, Email: r.sutton@liverpool.ac.uk.
Yan Tian, Email: tyfxh@163.com.
Wei Huang, Email: dr_wei_huang@scu.edu.cn.
Xianghui Fu, Email: xfu@scu.edu.cn.
Supplemental information
References
- 1.Petrov M.S., Yadav D. Global epidemiology and holistic prevention of pancreatitis. Nat. Rev. Gastroenterol. Hepatol. 2019;16:175–184. doi: 10.1038/s41575-018-0087-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garg P.K., Singh V.P. Organ failure due to systemic injury in acute pancreatitis. Gastroenterology. 2019;156:2008–2023. doi: 10.1053/j.gastro.2018.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hernandez G., Luo T., Javed T.A., Wen L., Kalwat M.A., Vale K., Ammouri F., Husain S.Z., Kliewer S.A., Mangelsdorf D.J. Pancreatitis is an FGF21-deficient state that is corrected by replacement therapy. Sci. Transl. Med. 2020;12:eaay5186. doi: 10.1126/scitranslmed.aay5186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mederos M.A., Reber H.A., Girgis M.D. Acute pancreatitis: a review. JAMA. 2021;325:382–390. doi: 10.1001/jama.2020.20317. [DOI] [PubMed] [Google Scholar]
- 5.Petersen O.H., Tepikin A.V. Polarized calcium signaling in exocrine gland cells. Annu. Rev. Physiol. 2008;70:273–299. doi: 10.1146/annurev.physiol.70.113006.100618. [DOI] [PubMed] [Google Scholar]
- 6.Maleth J., Hegyi P. Calcium signaling in pancreatic ductal epithelial cells: an old friend and a nasty enemy. Cell Calcium. 2014;55:337–345. doi: 10.1016/j.ceca.2014.02.004. [DOI] [PubMed] [Google Scholar]
- 7.Feske S., Wulff H., Skolnik E.Y. Ion channels in innate and adaptive immunity. Annu. Rev. Immunol. 2015;33:291–353. doi: 10.1146/annurev-immunol-032414-112212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saluja A., Dudeja V., Dawra R., Sah R.P. Early intra-acinar events in pathogenesis of pancreatitis. Gastroenterology. 2019;156:1979–1993. doi: 10.1053/j.gastro.2019.01.268. [DOI] [PubMed] [Google Scholar]
- 9.Barreto S.G., Habtezion A., Gukovskaya A., Lugea A., Jeon C., Yadav D., Hegyi P., Venglovecz V., Sutton R., Pandol S.J. Critical thresholds: key to unlocking the door to the prevention and specific treatments for acute pancreatitis. Gut. 2021;70:194–203. doi: 10.1136/gutjnl-2020-322163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Habtezion A., Gukovskaya A.S., Pandol S.J. Acute pancreatitis: a multifaceted set of organelle and cellular interactions. Gastroenterology. 2019;156:1941–1950. doi: 10.1053/j.gastro.2018.11.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim M.S., Hong J.H., Li Q., Shin D.M., Abramowitz J., Birnbaumer L., Muallem S. Deletion of TRPC3 in mice reduces store-operated Ca2+ influx and the severity of acute pancreatitis. Gastroenterology. 2009;137:1509–1517. doi: 10.1053/j.gastro.2009.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim M.S., Lee K.P., Yang D., Shin D.M., Abramowitz J., Kiyonaka S., Birnbaumer L., Mori Y., Muallem S. Genetic and pharmacologic inhibition of the Ca2+ influx channel TRPC3 protects secretory epithelia from Ca2+-dependent toxicity. Gastroenterology. 2011;140:2107–2115. doi: 10.1053/j.gastro.2011.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wen L., Voronina S., Javed M.A., Awais M., Szatmary P., Latawiec D., Chvanov M., Collier D., Huang W., Barrett J., et al. Inhibitors of ORAI1 prevent cytosolic calcium-associated injury of human pancreatic acinar cells and acute pancreatitis in 3 mouse models. Gastroenterology. 2015;149:481–492.e7. doi: 10.1053/j.gastro.2015.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Waldron R.T., Chen Y., Pham H., Go A., Su H.Y., Hu C., Wen L., Husain S.Z., Sugar C.A., Roos J., et al. The Orai Ca(2+) channel inhibitor CM4620 targets both parenchymal and immune cells to reduce inflammation in experimental acute pancreatitis. J. Physiol. 2019;597:3085–3105. doi: 10.1113/JP277856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fanczal J., Pallagi P., Gorog M., Diszhazi G., Almassy J., Madacsy T., Varga Á., Csernay-Biró P., Katona X., Tóth E., et al. TRPM2-mediated extracellular Ca(2+) entry promotes acinar cell necrosis in biliary acute pancreatitis. J. Physiol. 2020;598:1253–1270. doi: 10.1113/JP279047. [DOI] [PubMed] [Google Scholar]
- 16.Son A., Ahuja M., Schwartz D.M., Varga A., Swaim W., Kang N., Maleth J., Shin D.M., Muallem S. Ca(2+) influx channel inhibitor SARAF protects mice from acute pancreatitis. Gastroenterology. 2019;157:1660–1672. doi: 10.1053/j.gastro.2019.08.042. [DOI] [PubMed] [Google Scholar]
- 17.Leung A.K., Sharp P.A. MicroRNA functions in stress responses. Mol. Cell. 2010;40:205–215. doi: 10.1016/j.molcel.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rupaimoole R., Slack F.J. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017;16:203–222. doi: 10.1038/nrd.2016.246. [DOI] [PubMed] [Google Scholar]
- 19.Tian Y., Xu J., Du X., Fu X. The interplay between noncoding RNAs and insulin in diabetes. Cancer Lett. 2018;419:53–63. doi: 10.1016/j.canlet.2018.01.038. [DOI] [PubMed] [Google Scholar]
- 20.Aurora A.B., Mahmoud A.I., Luo X., Johnson B.A., van Rooij E., Matsuzaki S., Humphries K.M., Hill J.A., Bassel-Duby R., Sadek H.A., et al. MicroRNA-214 protects the mouse heart from ischemic injury by controlling Ca2⁺ overload and cell death. J. Clin. Invest. 2012;122:1222–1232. doi: 10.1172/jci59327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Finger F., Hoppe T. MicroRNAs meet calcium: joint venture in ER proteostasis. Sci. Signal. 2014;7:re11. doi: 10.1126/scisignal.2005671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Drawnel F.M., Wachten D., Molkentin J.D., Maillet M., Aronsen J.M., Swift F., Sjaastad I., Liu N., Catalucci D., Mikoshiba K., et al. Mutual antagonism between IP(3)RII and miRNA-133a regulates calcium signals and cardiac hypertrophy. J. Cell Biol. 2012;199:783–798. doi: 10.1083/jcb.201111095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harraz M.M., Eacker S.M., Wang X., Dawson T.M., Dawson V.L. MicroRNA-223 is neuroprotective by targeting glutamate receptors. Proc. Natl. Acad. Sci. U S A. 2012;109:18962–18967. doi: 10.1073/pnas.1121288109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malhotra N., Qi Y., Spidale N.A., Frascoli M., Miu B., Cho O., Sylvia K., Kang J. SOX4 controls invariant NKT cell differentiation by tuning TCR signaling. J. Exp. Med. 2018;215:2887–2900. doi: 10.1084/jem.20172021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang Y., Huang Q., Luo C., Wen Y., Liu R., Sun H., Tang L. MicroRNAs in acute pancreatitis: from pathogenesis to novel diagnosis and therapy. J. Cell Physiol. 2020;235:1948–1961. doi: 10.1002/jcp.29212. [DOI] [PubMed] [Google Scholar]
- 26.Dixit A.K., Sarver A.E., Yuan Z., George J., Barlass U., Cheema H., Sareen A., Banerjee S., Dudeja V., Dawra R., et al. Comprehensive analysis of microRNA signature of mouse pancreatic acini: overexpression of miR-21-3p in acute pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2016;311:G974–G980. doi: 10.1152/ajpgi.00191.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fu X., Meng Z., Liang W., Tian Y., Wang X., Han W., Lou G., Wang X., Lou F., Yen Y., et al. miR-26a enhances miRNA biogenesis by targeting Lin28B and Zcchc11 to suppress tumor growth and metastasis. Oncogene. 2014;33:4296–4306. doi: 10.1038/onc.2013.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li X., Pan X., Fu X., Yang Y., Chen J., Lin W. MicroRNA-26a: an emerging regulator of renal biology and disease. Kidney Blood Press Res. 2019;44:287–297. doi: 10.1159/000499646. [DOI] [PubMed] [Google Scholar]
- 29.Jiang Q., Isquith J., Zipeto M.A., Diep R.H., Pham J., Delos Santos N., Reynoso E., Chau J., Leu H., Lazzari E., et al. Hyper-editing of cell-cycle regulatory and tumor suppressor RNA promotes malignant progenitor propagation. Cancer Cell. 2019;35:81–94.e7. doi: 10.1016/j.ccell.2018.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Urabe F., Kosaka N., Sawa Y., Yamamoto Y., Ito K., Yamamoto T., Kimura T., Egawa S., Ochiya T. miR-26a regulates extracellular vesicle secretion from prostate cancer cells via targeting SHC4, PFDN4, and CHORDC1. Sci. Adv. 2020;6:eaay3051. doi: 10.1126/sciadv.aay3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu H., Du X., Xu J., Zhang Y., Tian Y., Liu G., Wang X., Ma M., Du W., Liu Y., et al. Pancreatic beta cell microRNA-26a alleviates type 2 diabetes by improving peripheral insulin sensitivity and preserving beta cell function. PLoS Biol. 2020;18:e3000603. doi: 10.1371/journal.pbio.3000603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu H., Tian Y., Tang D., Zou S., Liu G., Song J., Zhang G., Du X., Huang W., He B., et al. An endoplasmic reticulum stress-microRNA-26a feedback circuit in NAFLD. Hepatology. 2021;73:1327–1345. doi: 10.1002/hep.31428. [DOI] [PubMed] [Google Scholar]
- 33.Liu G., Du W., Xu H., Sun Q., Tang D., Zou S., Zhang Y., Ma M., Zhang G., Du X., et al. RNA G-quadruplex regulates microRNA-26a biogenesis and function. J. Hepatol. 2020;73:371–382. doi: 10.1016/j.jhep.2020.02.032. [DOI] [PubMed] [Google Scholar]
- 34.Fu X., Jin L., Wang X., Luo A., Hu J., Zheng X., Tsark W.M., Riggs A.D., Ku H.T., Huang W. MicroRNA-26a targets ten eleven translocation enzymes and is regulated during pancreatic cell differentiation. Proc. Natl. Acad. Sci. U S A. 2013;110:17892–17897. doi: 10.1073/pnas.1317397110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Han W., Fu X., Xie J., Meng Z., Gu Y., Wang X., Li L., Pan H., Huang W. MiR-26a enhances autophagy to protect against ethanol-induced acute liver injury. J. Mol. Med. (Berl) 2015;93:1045–1055. doi: 10.1007/s00109-015-1282-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lujambio A., Lowe S.W. The microcosmos of cancer. Nature. 2012;482:347–355. doi: 10.1038/nature10888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ward J.B., Sutton R., Jenkins S.A., Petersen O.H. Progressive disruption of acinar cell calcium signaling is an early feature of cerulein-induced pancreatitis in mice. Gastroenterology. 1996;111:481–491. doi: 10.1053/gast.1996.v111.pm8690215. [DOI] [PubMed] [Google Scholar]
- 38.Yang X., Yao L., Fu X., Mukherjee R., Xia Q., Jakubowska M.A., Ferdek P.E., Huang W. Experimental acute pancreatitis models: history, current status, and role in translational research. Front. Physiol. 2020;11:614591. doi: 10.3389/fphys.2020.614591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Voronina S., Longbottom R., Sutton R., Petersen O.H., Tepikin A. Bile acids induce calcium signals in mouse pancreatic acinar cells: implications for bile-induced pancreatic pathology. J. Physiol. 2002;540:49–55. doi: 10.1113/jphysiol.2002.017525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grimson A., Farh K.K., Johnston W.K., Garrett-Engele P., Lim L.P., Bartel D.P. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol. Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jones M.R., Quinton L.J., Blahna M.T., Neilson J.R., Fu S., Ivanov A.R., Wolf D.A., Mizgerd J.P. Zcchc11-dependent uridylation of microRNA directs cytokine expression. Nat. Cell Biol. 2009;11:1157–1163. doi: 10.1038/ncb1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feng M., Xu D., Wang L. miR-26a inhibits atherosclerosis progression by targeting TRPC3. Cell Biosci. 2018;8:4. doi: 10.1186/s13578-018-0203-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee P.J., Papachristou G.I. New insights into acute pancreatitis. Nat. Rev. Gastroenterol. Hepatol. 2019;16:479–496. doi: 10.1038/s41575-019-0158-2. [DOI] [PubMed] [Google Scholar]
- 44.Murphy J.A., Criddle D.N., Sherwood M., Chvanov M., Mukherjee R., McLaughlin E., Booth D., Gerasimenko J.V., Raraty M.G., Ghaneh P., et al. Direct activation of cytosolic Ca2+ signaling and enzyme secretion by cholecystokinin in human pancreatic acinar cells. Gastroenterology. 2008;135:632–641. doi: 10.1053/j.gastro.2008.05.026. [DOI] [PubMed] [Google Scholar]
- 45.Raraty M., Ward J., Erdemli G., Vaillant C., Neoptolemos J.P., Sutton R., Petersen O.H. Calcium-dependent enzyme activation and vacuole formation in the apical granular region of pancreatic acinar cells. Proc. Natl. Acad. Sci. U S A. 2000;97:13126–13131. doi: 10.1073/pnas.97.24.13126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maier T., Follmann M., Hessler G., Kleemann H.W., Hachtel S., Fuchs B., Weissmann N., Linz W., Schmidt T., Löhn M., et al. Discovery and pharmacological characterization of a novel potent inhibitor of diacylglycerol-sensitive TRPC cation channels. Br. J. Pharmacol. 2015;172:3650–3660. doi: 10.1111/bph.13151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peng S., Gerasimenko J.V., Tsugorka T.M., Gryshchenko O., Samarasinghe S., Petersen O.H., Gerasimenko O.V. Galactose protects against cell damage in mouse models of acute pancreatitis. J. Clin. Invest. 2018;128:3769–3778. doi: 10.1172/JCI94714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Swain S.M., Romac J.M., Shahid R.A., Pandol S.J., Liedtke W., Vigna S.R., Liddle R.A. TRPV4 channel opening mediates pressure-induced pancreatitis initiated by Piezo1 activation. J. Clin. Invest. 2020;130:2527–2541. doi: 10.1172/JCI134111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang W., Cane M.C., Mukherjee R., Szatmary P., Zhang X., Elliott V., Ouyang Y., Chvanov M., Latawiec D., Wen L., et al. Caffeine protects against experimental acute pancreatitis by inhibition of inositol 1,4,5-trisphosphate receptor-mediated Ca2+ release. Gut. 2017;66:301–313. doi: 10.1136/gutjnl-2015-309363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prevot P.P., Augereau C., Simion A., Van den Steen G., Dauguet N., Lemaigre F.P., Jacquemin P. Let-7b and miR-495 stimulate differentiation and prevent metaplasia of pancreatic acinar cells by repressing HNF6. Gastroenterology. 2013;145:668–678. doi: 10.1053/j.gastro.2013.05.016. [DOI] [PubMed] [Google Scholar]
- 51.Loffler-Wirth H., Kalcher M., Binder H. oposSOM: R-package for high-dimensional portraying of genome-wide expression landscapes on bioconductor. Bioinformatics. 2015;31:3225–3227. doi: 10.1093/bioinformatics/btv342. [DOI] [PubMed] [Google Scholar]
- 52.Zhang W., Fu X., Xie J., Pan H., Han W., Huang W. miR-26a attenuates colitis and colitis-associated cancer by targeting the multiple intestinal inflammatory pathways. Mol. Ther. Nucleic Acids. 2021;24:264–273. doi: 10.1016/j.omtn.2021.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Coate K.C., Hernandez G., Thorne C.A., Sun S., Le T.D.V., Vale K., Kliewer S.A., Mangelsdorf D.J., et al. FGF21 is an exocrine pancreas secretagogue. Cell Metab. 2017;25:472–480. doi: 10.1016/j.cmet.2016.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ahuja M., Schwartz D.M., Tandon M., Son A., Zeng M., Swaim W., Eckhaus M., Hoffman V., Cui Y., Xiao B., et al. Orai1-mediated antimicrobial secretion from pancreatic acini shapes the gut microbiome and regulates gut innate immunity. Cell Metab. 2017;25:635–646. doi: 10.1016/j.cmet.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Luo Y., Yang Y., Liu M., Wang D., Wang F., Bi Y., Ji J., Li S., Liu Y., Chen R., et al. Oncogenic KRAS reduces expression of FGF21 in acinar cells to promote pancreatic tumorigenesis in mice on a high-fat diet. Gastroenterology. 2019;157:1413–1428.e1. doi: 10.1053/j.gastro.2019.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang H., Swidnicka-Siergiejko A.K., Daniluk J., Gaiser S., Yao Y., Peng L., Zhang Y., Liu Y., Dong M., Zhan X., et al. Transgenic expression of PRSS1(R122H) sensitizes mice to pancreatitis. Gastroenterology. 2020;158:1072–1082.e7. doi: 10.1053/j.gastro.2019.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mareninova O.A., Sung K.F., Hong P., Lugea A., Pandol S.J., Gukovsky I., Gukovskaya A.S. Cell death in pancreatitis: caspases protect from necrotizing pancreatitis. J. Biol. Chem. 2006;281:3370–3381. doi: 10.1074/jbc.M511276200. [DOI] [PubMed] [Google Scholar]
- 58.Moller C.C., Wei C., Altintas M.M., Li J., Greka A., Ohse T., Pippin J.W., Rastaldi M.P., Wawersik S., Schiavi S., et al. Induction of TRPC6 channel in acquired forms of proteinuric kidney disease. J. Am. Soc. Nephrol. 2007;18:29–36. doi: 10.1681/ASN.2006091010. [DOI] [PubMed] [Google Scholar]
- 59.Ma X., Conklin D.J., Li F., Dai Z., Hua X., Li Y., Xu-Monette Z.Y., Young K.H., Xiong W., Wysoczynski M., et al. The oncogenic microRNA miR-21 promotes regulated necrosis in mice. Nat. Commun. 2015;6:7151. doi: 10.1038/ncomms8151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang F., Ma D., Zhao W., Wang D., Liu T., Liu Y., Yang Y., Liu Y., Mu J., Li B., et al. Obesity-induced overexpression of miR-802 impairs insulin transcription and secretion. Nat. Commun. 2020;11:1822. doi: 10.1038/s41467-020-15529-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shen B., Mei M., Pu Y., Zhang H., Liu H., Tang M., Pan Q., He Y., Wu X., Zhao H. Necrostatin-1 attenuates renal ischemia and reperfusion injury via meditation of HIF-1alpha/mir-26a/TRPC6/PARP1 signaling. Mol. Ther. Nucleic Acids. 2019;17:701–713. doi: 10.1016/j.omtn.2019.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zheng H., Drumm B.T., Earley S., Sung T.S., Koh S.D., Sanders K.M. SOCE mediated by STIM and Orai is essential for pacemaker activity in the interstitial cells of Cajal in the gastrointestinal tract. Sci. Signal. 2018;11 doi: 10.1126/scisignal.aaq0918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bootman M.D., Berridge M.J., Roderick H.L. Calcium signalling: more messengers, more channels, more complexity. Curr. Biol. 2002;12:R563–R565. doi: 10.1016/s0960-9822(02)01055-2. [DOI] [PubMed] [Google Scholar]
- 64.Kelley G.G., Reks S.E., Ondrako J.M., Smrcka A.V. Phospholipase C(epsilon): a novel Ras effector. EMBO J. 2001;20:743–754. doi: 10.1093/emboj/20.4.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee H.C. Mechanisms of calcium signaling by cyclic ADP-ribose and NAADP. Physiol. Rev. 1997;77:1133–1164. doi: 10.1152/physrev.1997.77.4.1133. [DOI] [PubMed] [Google Scholar]
- 66.Gukovsky I., Li N., Todoric J., Gukovskaya A., Karin M. Inflammation, autophagy, and obesity: common features in the pathogenesis of pancreatitis and pancreatic cancer. Gastroenterology. 2013;144:1199–1209.e4. doi: 10.1053/j.gastro.2013.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ji B., Song J., Tsou L., Bi Y., Gaiser S., Mortensen R., Logsdon C. Robust acinar cell transgene expression of CreErT via BAC recombineering. Genesis. 2008;46:390–395. doi: 10.1002/dvg.20411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Karlsson M., Zhang C., Mear L., Zhong W., Digre A., Katona B., Sjöstedt E., Butler L., Odeberg J., Dusart P., et al. A single-cell type transcriptomics map of human tissues. Sci. Adv. 2021;7:eabh2169. doi: 10.1126/sciadv.abh2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kunert-Keil C., Bisping F., Kruger J., Brinkmeier H. Tissue-specific expression of TRP channel genes in the mouse and its variation in three different mouse strains. BMC Genom. 2006;7:159. doi: 10.1186/1471-2164-7-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shi N., Liu T., de la Iglesia-Garcia D., Deng L., Jin T., Lan L., Zhu P., Hu W., Zhou Z., Singh V., et al. Duration of organ failure impacts mortality in acute pancreatitis. Gut. 2020;69:604–605. doi: 10.1136/gutjnl-2019-318241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Banks P.A., Bollen T.L., Dervenis C., Gooszen H.G., Johnson C.D., Sarr M.G., Tsiotos G.G., Vege S.S. Classification of acute pancreatitis—2012: revision of the Atlanta classification and definitions by international consensus. Gut. 2013;62:102–111. doi: 10.1136/gutjnl-2012-302779. [DOI] [PubMed] [Google Scholar]
- 72.Liu T., Huang W., Szatmary P., Abrams S.T., Alhamdi Y., Lin Z., Greenhalf W., Wang G., Sutton R., Toh C.H. Accuracy of circulating histones in predicting persistent organ failure and mortality in patients with acute pancreatitis. Br. J. Surg. 2017;104:1215–1225. doi: 10.1002/bjs.10538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mukherjee R., Mareninova O.A., Odinokova I.V., Huang W., Murphy J., Chvanov M., Javed M.A., Wen L., Booth D.M., Cane M.C., et al. Mechanism of mitochondrial permeability transition pore induction and damage in the pancreas: inhibition prevents acute pancreatitis by protecting production of ATP. Gut. 2016;65:1333–1346. doi: 10.1136/gutjnl-2014-308553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ou X., Cheng Z., Liu T., Tang Z., Huang W., Szatmary P., Zheng S., Sutton R., Toh C.H., Zhang N., et al. Circulating histone levels reflect disease severity in animal models of acute pancreatitis. Pancreas. 2015;44:1089–1095. doi: 10.1097/MPA.0000000000000416. [DOI] [PubMed] [Google Scholar]
- 75.Chandra R., Liddle R.A. Cholecystokinin. Curr. Opin. Endocrinol. Diabetes Obes. 2007;14:63–67. doi: 10.1097/MED.0b013e3280122850. [DOI] [PubMed] [Google Scholar]
- 76.Zhou X., Xue C. Ghrelin inhibits the development of acute pancreatitis and nuclear factor kappaB activation in pancreas and liver. Pancreas. 2009;38:752–757. doi: 10.1097/MPA.0b013e3181a86b74. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.