

Abstract
Hepatoblastoma is the most common liver cancer in children, and the aggressive subtype often has a poor prognosis and lacks effective targeted therapy. Although aggressive hepatoblastoma (HB) is often accompanied by abnormally high expression of the transcription factor c-Myc, the underlying mechanism remains unclear. In this study, we found that mitochondrial fragmentation was enhanced by c-Myc overexpression in human aggressive HB tissues and was associated with poor prognosis. Then, a mouse model resembling human HB was established via hydrodynamic injection of c-Myc plasmids. We observed that liver-specific knockout of the mitochondrial fusion molecule MFN1 or overexpression of mitochondrial fission molecule DRP1 promoted the occurrence of c-Myc-driven liver cancer. In contrast, when MFN1 was overexpressed in the liver, tumor formation was delayed. In vitro experiments showed that c-Myc transcriptionally upregulated the expression of DRP1 and decreased MFN1 expression through upregulation of miR-373-3p. Moreover, enhanced mitochondrial fragmentation significantly promoted aerobic glycolysis and the proliferation of HB cells by significantly increasing reactive oxygen species (ROS) production and activating the RAC-alpha serine/threonine-protein kinase (AKT)/mammalian target of rapamycin (mTOR) and nuclear factor κB (NF-κB) pathways. Taken together, our results indicate that c-Myc-mediated mitochondrial fragmentation promotes the malignant transformation and progression of HB by activating ROS-mediated multi-oncogenic signaling.
Keywords: hepatoblastoma, mitochondrial fragmentation, Drp1, Mfn1, c-Myc, ROS
Graphical abstract
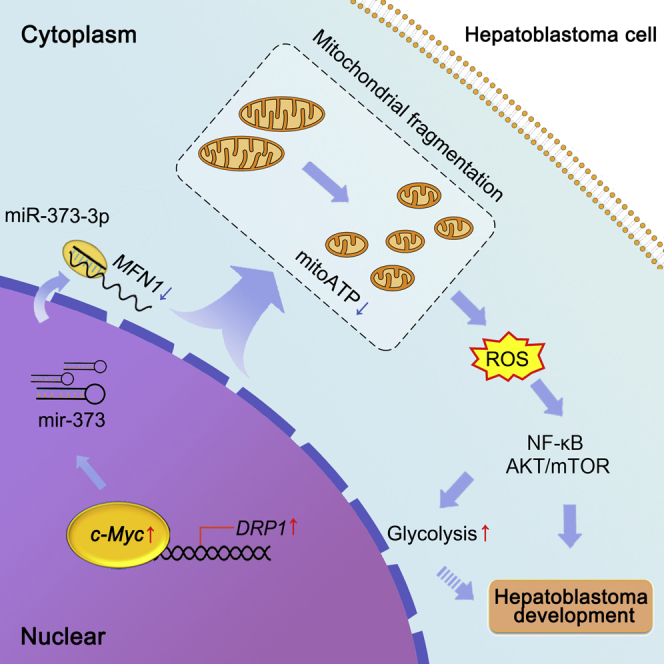
Mitochondrial fragmentation was enhanced by c-Myc in human HB and was associated with poor prognosis. Liver-specific MFN1 knockout or DRP1 overexpression promoted the occurrence of c-Myc-driven liver tumor. Mechanically, c-Myc decreased MFN1 and transcriptionally upregulated DRP1. Moreover, enhanced mitochondrial fragmentation promoted HB cell proliferation through ROS/AKT/mTOR and ROS/NF-κB pathways.
Introduction
Hepatoblastoma (HB) is the most common pediatric liver cancer diagnosed in infants and young children (it is extremely rare in adults), which presents unique parenting challenges.1 The incidence of HB has been increasing worldwide over the last several decades, partially due to the increased survival of infants with low birth weight and of premature infants.2,3 Although surgery combined with neoadjuvant chemotherapy significantly improves the overall survival rate of patients with HB, approximately 30% of patients still show relatively poor outcomes.4,5 Therefore, there is an urgent need to explore the underlying mechanism of HB with poor prognosis and pave the way for the development of novel therapeutic strategies.
c-Myc serves as a “master regulator” of multiple cellular processes, including proliferation, stemness, and metabolism.6,7 Previous studies have also shown that c-Myc plays a crucial role in HB tumorigenesis.8, 9, 10 Cairo et al. have indicated that the immature subtype HB (C2 subtype) is characterized by specific overexpression of the c-Myc gene.11 Similarly, transcriptomic analysis has further identified a persistently high expression level of c-Myc in HB with the poorest prognosis.12 However, the exact mechanism by which c-Myc exhibits oncogenic activity in HB remains poorly understood.
Mitochondria are highly dynamic organelles that constantly fuse and divide.13 Mitochondrial dynamics are mainly regulated by several molecules, such as dynamin-related protein 1 (DRP1), mitofusin 1 (MFN1), and MFN2, which are frequently deregulated in various human solid malignant neoplasms.14,15 Chen et al. identified that MFN1 is a leading downregulated candidate in triple-negative breast cancer and is closely associated with poor prognosis of patients.16 Lee et al. demonstrated that increased DRP1 expression indicates poor prognosis in patients with castration-resistant prostate cancer.17 Furthermore, a growing body of evidence suggests that inhibition of mitochondrial fission prevents the proliferation and metastasis of many types of human cancer cells.18,19 However, whether deregulation of mitochondrial dynamics is involved in the malignant transformation of normal cells, the most critical step in tumor occurrence, is far from clear.
It is now widely accepted that cancer cells exhibit aberrant redox homeostasis with increased reactive oxygen species (ROS) production.20,21 Numerous lines of evidence supported that ROS is an important signaling molecule and plays a critical role in both cell proliferation and apoptosis resistance. For example, high levels of ROS in cancer cells promote the activation of many oncogenic signaling pathways, such as phosphatidylinositol 3-kinase-protein kinase B/RAC-alpha serine/threonine-protein kinase (PI3K-PKB/AKT), nuclear factor κB (NF-κB), and mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK).21 Importantly, we and others have shown that mitochondrial fragmentation induced by DRP1 or deletion of MFN1/2 is associated with increased ROS levels in tumor cells.22, 23, 24 Nevertheless, the roles of mitochondrial fragmentation and ROS in HB tumorigenesis remain to be elucidated.
Here, transmission electron microscopy (TEM) was employed to identify changes in mitochondrial structure in aggressive HB. MFN1flox/flox mice and hydrodynamic injection technology were used to systematically investigate the role of mitochondrial fragmentation in c-Myc-driven HB-like liver tumors. We also explored the underlying mechanism in vitro using human HB cell lines.
Results
Mitochondrial morphology is deregulated in aggressive HB with c-Myc overexpression
To identify changes in the cellular or organelle structure in HB with high expression levels of c-Myc, human HB tissues were subjected to TEM. As shown in Figures 1A and 1B, mitochondria in HB tissues with high c-Myc expression were much smaller than those in HB tissues with low c-Myc expression. With respect to the length distribution of mitochondria, the short (<0.5 μm) mitochondria were abundant in the c-Myc high expression group (Figure 1C). Consistently, the mitochondrial population was increased in the c-Myc high expression group, while the total area occupied by mitochondria per unit area of 120 μm2 was decreased compared with the c-Myc low expression group (Figures S1A and S1B). These changes indicated the occurrence of mitochondrial fragmentation in HB with a high expression of c-Myc. To further confirm this phenomenon, the differentially expressed genes (DEGs) between the c-Myc high expression group and the c-Myc low expression group were annotated using two public datasets (GEO: GSE133039 and GSE75271). As expected, the DEGs were enriched in the mitochondrial-membrane-organization-related Gene Ontology (GO) terms (Figure 1D).
Figure 1.

Mitochondrial fragmentation is induced by c-Myc overexpression in hepatoblastoma and is associated with poor prognosis
(A) Representative TEM images of mitochondrial morphology in hepatoblastoma (HB) tissues with high (n = 5) or low (n = 5) expression levels of c-Myc. Scale bars: 1 μm. Mitochondria and nucleus were pseudo-colored green and blue. (B and C) The average area (B) and length (C) distributions of mitochondria were analyzed from five random fields (120 μm2) approximately equaling the low magnification images of TEM in each group samples. (D) Cellular component terms and biological process terms enriched by the differentially expressed genes between HB tissues with high and low c-Myc expression. (E) Representative confocal microscopy images of mitochondria in Huh6 cells with or without c-Myc knockdown. Scale bars: 10 μm. shc-Myc-1 and shc-Myc-2, shRNA against c-Myc; shctrl, control shRNA. (F) Morphology distribution of mitochondria was analyzed in Huh6 cells with different treatment as indicated. (G) Representative immunohistochemical (IHC) staining images of c-Myc, Mfn1, and Drp1 in HB tissues with high (n = 25) and low (n = 25) c-Myc expression. Scale bar: 50 μm. (H and I) Kaplan-Meier curve analysis of overall survival (OS) and disease-free survival (DFS) in HB patients by the expression of Drp1 and Mfn1. Data are expressed as mean ± SD. ∗p < 0.05, ∗∗p < 0.01.
To explore the direct effects of c-Myc on mitochondrial morphology, c-Myc was knocked down in two HB cell lines, Huh6 and HepG2 (Figure S1C), which were reported to express high levels of c-Myc.11,12 Mitotracker Green staining analysis demonstrated that fragmented mitochondria were significantly decreased in HB cells with c-Myc knockdown (Figures 1E, 1F, S1D, and S1E). Immunohistochemical staining analysis in a relatively larger cohort of 50 patients with HB also revealed that the expression of c-Myc was negatively associated with the expression of mitochondrial fusion protein Mfn1 and was positively associated with the mitochondrial fission protein Drp1 in human HB tissues. However, the expression levels of other mitochondrial dynamic regulators (Mfn2, Opa1, and Fis1) were not significantly changed (Figures 1G and S1F). Consistently, c-Myc knockdown significantly decreased Drp1 expression, increased Mfn1 expression, and then inhibited mitochondrial fragmentation and the proliferation of HB cells. Restorative expression of c-Myc reversed this effect (Figures S1G–S1K). Furthermore, patients with HB with a low expression of Drp1 or a high expression of Mfn1 had a significantly better overall survival and disease-free survival than those with a high expression of Drp1 or a low expression of Mfn1 (Figures 1H and 1I).
Mitochondrial fragmentation promotes the formation of c-Myc-driven HB-like liver tumors in vivo
To evaluate whether mitochondrial fragmentation plays a key role in c-Myc-driven HB, MFN1 liver-specific knockout mice (Alb-Cre; MFN1flox/flox; Figure S2A) were generated. Two loxP recombination sites flank the exon encoding the canonical G-1 GTPase motif. When exposed to Cre recombinase, an excision of this critical exon and a frameshift occurs, thus preventing the formation of a functional protein.25 Then, the c-Myc plasmid along with the Sleeping Beauty (SB) transposon were delivered into mouse hepatocytes by hydrodynamic tail vein injection. Mouse livers were harvested at 4 weeks post-injection, and the deletion of MFN1 or the overexpression of DRP1 was confirmed in tumor tissues by immunohistochemistry (IHC) (Figures S2B and S2C). As shown in Figures 2A and S2D, Alb-Cre; MFN1flox/flox mice developed much larger tumors than MFN1flox/flox mice. Consistently, the deletion of MFN1 also resulted in a shorter survival time and a higher liver/body weight ratio (Figures 2B and 2C). Similar results were also observed in wild-type (WT) mice hydrodynamically injected with c-Myc, DRP1 plasmids, and SB transposons when compared with mice injected with c-Myc, SB transposons, and pT3 empty vectors as control (Figures 2A–2C and S2D). Histological analysis revealed that all tumors developed from c-Myc injection displayed HB-like characteristics (Figure 2D). Moreover, MFN1 knockout or DRP1 overexpression elevated the percentage of proliferating cell nuclear antigen (PCNA)(+) cells and cells with fragmented mitochondria but decreased the mtDNA content (Figures 2D–2G and S2E).
Figure 2.

Mitochondrial fragmentation caused by MFN1 knockout or DRP1 overexpression promotes tumorigenesis of c-Myc-driven HB in mice
(A) Representative gross images of livers from MFN1flox/flox (n = 12) or Alb-Cre; MFN1flox/flox (n = 12) mice injected with c-Myc/SB plasmids (left panel) or from WT mice injected with c-Myc/pT3 (n = 12) or c-Myc/DRP1 (n = 12) plasmids (right panel). MFN1flox/flox, MFN1flox/flox mice hydrodynamically injected with c-Myc plasmid and SB transposon; MFN1 LKO, MFN1 liver-specific knockout mice hydrodynamically injected with c-Myc plasmid and SB transposon; PT3, wild-type mice hydrodynamically injected with c-Myc plasmid and PT3-EF1α empty vector, as well as SB transposon; DRP1, wild-type mice hydrodynamically injected with c-Myc plasmid and PT3-EF1α-DRP1 construct, as well as SB transposon; w.p.i., weeks post-injection. (B and C) Survival curve (B) and liver/body weight ratio (C) of the mice with treatment as indicated. (D) Representative images of hematoxylin and eosin (H&E) and PCNA staining in mouse liver tumors. Scale bar: 50 μm. (E) Percentage of PCNA-positive cells in mouse liver tumors. (F and G) Representative TEM images of mitochondria in mouse liver tumors. Scale bars: 1 μm. Mitochondria and nucleus were pseudo-colored green and blue. The length distribution of mitochondria was analyzed. (H and I) Representative 18F-FDG micro-PET/CT images and analysis in mice. A higher intensity of red color indicates a higher 18F-FDG uptake. (J) Lactate production was measured in mouse tumor tissues. ∗p < 0.05, ∗∗p < 0.01.
Since previous studies have shown that both c-Myc and fragmented mitochondria induced a metabolic shift from oxidative phosphorylation to aerobic glycolysis, which thus facilitate tumor progression,26, 27, 28 we then first employed the Seahorse XF Real-Time ATP Rate Assay to evaluate the effect of c-Myc on aerobic glycolysis in HB cells (Figures S2F and S2G). Our data showed that c-Myc knockdown significantly decreased the total ATP and glycoATP production rates but promoted mitoATP production rates in HB cells, implying that the aerobic glycolysis was involved in c-Myc-driven HB-like liver tumors. Moreover, positron emission tomography (PET) scan analysis demonstrated that the uptake of 18F-FDG was significantly increased in MFN1 knockout and DRP1 overexpression groups when compared with that in the control groups (Figures 2H and 2I). Consistently, lactate levels were also elevated in the MFN1 knockout and DRP1 overexpression groups (Figure 2J).
Inhibition of mitochondrial fragmentation delays the formation of c-Myc-driven HB-like liver tumors in vivo
To further verify the hypothesis that mitochondrial fragmentation promotes c-Myc-driven HB, we hydrodynamically injected the MFN1 plasmids together with the c-Myc plasmids as well as the SB transposase into the mouse liver. Interestingly, overexpression of MFN1 significantly delayed tumorigenesis induced by c-Myc. Indeed, although all mice injected with pT3/c-Myc exhibited visible tumors, MFN1/c-Myc-injected mice displayed a nearly normal liver appearance as well as a nearly normal liver/body weight ratio at 4 weeks after injection (Figures 3A, 3C, and S3B). Over the long term (8 weeks after injection), pT3/c-Myc-injected mice developed a lethal burden of liver tumors. In contrast, MFN1/c-Myc-injected mice exhibited much less tumor burden, a decreased liver/body weight ratio, and prolonged survival time (Figures 3A–3C and S3B). Upon histological analysis, only small tumor nodules were occasionally visible on the liver of MFN1/c-Myc-injected mice at 4 weeks after injection (Figure 3D). IHC staining analysis demonstrated that the overexpression of MFN1 in the tumor lesions significantly inhibited the proliferation of tumor cells, as revealed by PCNA staining (Figures 3D, 3E, and S3A). Additionally, overexpression of MFN1 obviously inhibited mitochondrial fragmentation and decreased lactate production but increased mtDNA content in c-Myc-driven liver tumors (Figures 3F–3H and S3C).
Figure 3.

Inhibition of mitochondrial fragmentation by MFN1 overexpression delays tumorigenesis of c-Myc-driven HB in mice
(A) Representative gross images of livers from WT mice injected with c-Myc/pT3 (n = 12) or c-Myc/MFN1 (n = 12) plasmids. PT3, wild-type mice hydrodynamically injected with c-Myc plasmid and PT3-EF1α empty vector, as well as SB transposon; MFN1, wild-type mice hydrodynamically injected with c-Myc plasmid and PT3-EF1α-MFN1 construct, as well as SB transposon; w.p.i., weeks post-injection. (B and C) Survival curve (B) and liver/body weight ratio (C) of the mice with treatment as indicated. (D) Representative images of H&E and PCNA staining in mouse liver tumors. Scale bar: 50 μm. (E) Percentage of PCNA-positive cells in mouse liver tumors. (F) Representative TEM images of mitochondria in mouse liver tumors. Scale bars: 1 μm. Mitochondria and nucleus were pseudo-colored green and blue. (G) The length distribution of mitochondria was analyzed. (H) Lactate production was measured in mouse tumor tissues. Data are expressed as the mean ± SD. ns, no significant; ∗p < 0.05, ∗∗p < 0.01.
Mitochondrial fragmentation promotes cell proliferation in HB in vitro
To explore the effects of mitochondrial fragmentation on human HB cell survival, DRP1 was knocked down and Mfn1 was overexpressed in Huh6 and HepG2 cells (Figures S4A and S4B). MitoTracker Green staining analysis demonstrated that mitochondria became obviously elongated in both HB cells after DRP1 knockdown or MFN1 overexpression when compared with those in control cells (Figures 4A, 4B, S4C, and S4D). 5-ethynyl-2'-deoxyuridine (EdU) incorporation assay revealed that HB cells with DRP1 knockdown or MFN1 overexpression showed much less EdU incorporation than the control cells (Figures 4C, 4D, S4G, and S4H). Moreover, DRP1 knockdown or MFN1 overexpression in HB cells significantly reduced the size of tumor spheroids, which is in agreement with the inhibitory effects of mitochondrial fragmentation (Figures 4E, 4F, S4E, and S4F). Next, we evaluated the effects of mitochondrial fragmentation on tumor formation in vivo by constructing subcutaneous xenograft tumor models. Our results showed that subcutaneous tumors developed from HB cells with DRP1 knockdown or MFN1 overexpression exhibited a significant decrease in growth capacity when compared with control tumors (Figures 4G–4I).
Figure 4.

Increasing mitochondrial fragmentation promotes cell proliferation in HB
(A) Representative confocal microscopy images of mitochondria in Huh6 cells with treatment as indicated. Scale bars: 10 μm. shctrl, control shRNA; shDRP1-1 and shDRP1-2, shRNA against DRP1; EV, empty vector; MFN1, expression vector encoding MFN1. (B) Morphology distribution of mitochondria was analyzed. (C and D) EdU assay was performed to evaluate proliferation ability of Huh6 cells with treatment as indicated. Scale bar: 50 μm. (E and F) High-magnification images and size of spheres formed on day 7 by Huh6 cells with treatment as indicated. Scale bar: 50 μm. (G) Tumor volume curves of subcutaneous xenograft tumor models developed from Huh6 cells with treatment as indicated. Tumor volume was measured using vernier calipers every 5 days from day 10 after subcutaneous implantation. (H) Images of tumors from sacrificed mice were obtained on day 30 after implantation. Tumor weights of different groups were compared. (I) Representative images of PCNA staining and the percentage of PCNA-positive cells in subcutaneous xenograft tumors. Scale bar: 50 μm. Data are expressed as the mean ± SD. ∗p < 0.05, ∗∗p < 0.01.
Mitochondrial fragmentation activates AKT/mTOR and NF-κB pathways by promoting ROS production in HB cells
Previous studies have demonstrated that mitochondrial fragmentation induces the production of intracellular ROS in human cancer cells, leading to the activation of several key signaling pathways, such as AKT/mammalian target of rapamycin (mTOR) and NF-κB.29,30 Therefore, we hypothesized that the ROS-mediated AKT/mTOR and NF-κB pathways may be involved in aggressive HB. Flow cytometry analysis demonstrated that DRP1 knockdown or MFN1 overexpression significantly reduced ROS production in HB cells with high c-Myc expression (Figure 5A). We then investigated whether AKT/mTOR and NF-κB signaling pathways were also affected by inhibiting mitochondrial fragmentation in HB cells. As shown in Figure 5B, DRP1 knockdown or MFN1 overexpression significantly decreased the levels of phosphorylated-AKT (p-AKT; Ser473), p-p70S6K (Thr389, a downstream target of mTOR), p-IKK (Ser176/180), nuclear p65, and increased the protein level of IκB-α in Huh6 cells. To further illustrate whether mitochondrial-fragmentation-mediated ROS production is involved in this process, H2O2 was used to stimulate intracellular ROS levels (Figure 5C). Western blotting analysis demonstrated that H2O2 treatment significantly increased the levels of p-AKT, p-p70S6K, p-IKK, and nuclear p65 and decreased the protein level of IκB-α. More importantly, the effects of DRP1 knockdown or MFN1 overexpression on the AKT/mTOR and NF-κB pathways were significantly reversed by treatment with H2O2 (Figure 5D).
Figure 5.

Mitochondrial fragmentation activates oncogenic signaling in HB cells by production of ROS
(A) Intracellular ROS production was measured by flow cytometry analysis in Huh6 and HepG2 cells with treatment as indicated. shctrl, control shRNA; shDRP1-1 and shDRP1-2, shRNA against DRP1; EV, empty vector; MFN1, expression vector encoding MFN1. (B) Western blotting images of Drp1, Mfn1, Akt, phosphorylated Akt (p-Akt, Ser473), p70S6k, p-p70S6k (Thr389), p-IKK (Ser176/180), IκB-α, and nuclear and cytosolic p65 in Huh6 cells with treatment as indicated. (C) Intracellular ROS production in Huh6 cells stably transfected with shDRP1-1, shDRP1-2, or MFN1 expression vector and then treated with 100 μM H2O2 for 12 h before cell harvest as indicated. (D) Western blotting images of Drp1, Mfn1, Akt, p-Akt (Ser473), p70S6k, p-p70S6k (Thr389), p-IKK (Ser176/180), IκB-α, and nuclear and cytosolic p65 in Huh6 with treatment as indicated. (E) Cell proliferation was analyzed by CCK-8 in Huh6 cells with treatment as indicated. LY2584702, P70S6K (downstream molecule of mTOR) inhibitor; Torin1, mTOR inhibitor; MK2206, Akt inhibitor; JSH-23, NF-κB inhibitor; siIκBα-1 and siIκBα-2, siRNA against IκBα. Data are expressed as the mean ± SD. ∗p < 0.05, ∗∗p < 0.01.
To determine whether ROS-mediated AKT/mTOR and NF-κB pathways have functional consequences in HB, the Cell Counting Kit-8 (CCK-8) assay was performed (Figure 5E). The data demonstrated that the H2O2 treatment significantly promoted the proliferation of HB cells with or without mitochondrial fragmentation. Moreover, treatment with the inhibitor of the AKT/mTOR pathway (LY2584702, MK2206, or Torin1) or the inhibitor of the NF-κB pathway (JSH-23) significantly inhibited ROS-stimulated proliferation of HB cells, while knockdown of IκBα promoted the proliferation of HB cells with DRP1 knockdown or MFN1 overexpression (Figures 5E and S5C). Meanwhile, glycolysis was also impeded by AKT/mTOR or NF-κB inhibition in HB cells but was enhanced by H2O2 treatment (Figures S5A and S5B). Taken together, these data indicate that mitochondrial-fragmentation-induced ROS production promotes HB cell proliferation through the AKT/mTOR and NF-κB signaling pathways.
c-Myc upregulates Drp1 and downregulates Mfn1 through different underlying mechanisms
Recently, chromatin immunoprecipitation sequencing has identified that c-Myc occupies the DRP1 promoter in lymphoma cells.31 Therefore, we attempted to confirm this result in HB cells using a luciferase reporter assay. As shown in Figure 6A, c-Myc knockdown significantly reduced the transcriptional activity of the DRP1 promoter in both Huh6 and HepG2 cells. Moreover, the 233 bp region between −244 and −11 in the promoter region of DRP1 was identified to be critical for transcriptional regulation by a series of truncated promoter constructs in Huh6 (Figure 6B). Consistently, c-Myc was predicted to bind to this specific region using the Jaspar program, although the canonical E-box was lacking (Figure 6C). Furthermore, site-directed mutagenesis analysis showed that the binding site was crucial for DRP1 transcription in Huh6 cells (Figure 6D). Additionally, chromatin immunoprecipitation (ChIP) assay further verified that c-Myc specifically bound to the WT DRP1 promoter, but not the mutant promotor, in Huh6 cells (Figure 6E). These results demonstrate that c-Myc directly binds to the DRP1 promoter, thus inducing DRP1 transcription.
Figure 6.

c-Myc upregulates miR-373-3p to downregulate MFN1 and transcriptionally upregulates Drp1
(A) Transcriptional activity of the DRP1 promoter was assessed by luciferase reporter assay in Huh6 cells following c-Myc knockdown. shctrl, control shRNA; shc-Myc, shRNA against c-Myc. (B) Transcriptional activities of serially truncated DRP1 promoter constructs were assessed by luciferase reporter assay in Huh6 cells. (C) The putative binding site for c-Myc was predicted by in silico analysis. (D) The transcriptional activity of the mutant DRP1 promoter was assessed using a luciferase reporter assay in Huh6 cells. (E) Plasmid-based ChIP-PCR analyses of c-Myc binding to the DRP1 promoter in Huh6 cells. WT, wild type; MU, mutant. (F) Venn diagram of the predicted miRNAs. Two classic databases were used to screen for miRNAs that could regulate MFN1. The yellow region contains 56 miRNAs screened from the TargetScan database, and the blue region contains 92 miRNAs screened from the miRDB database. The green region represents the intersection of the two databases, including five miRNAs with identical predictions. (G) Quantitative real-time reverse-transcription PCR (quantitative real-time RT-PCR) analyses of expression levels of the five predicted miRNAs in Huh6 cells treated as indicated. (H–K) Representative confocal microscopy images and analyses of mitochondrial morphology in Huh6 cells treated as indicated. Scale bars: 10μm. TSB-NC, negative control of miRNA target site blocker; TSB-MFN1, a target site blocker that selectively prevents miR-373-3p binding to the 3′ UTR of MFN1. (L and M) Cell proliferation was analyzed by CCK-8 in Huh6 cells with treatment as indicated. (N) Tumor volume curves of subcutaneous xenograft tumor models developed from Huh6 cells with treatment as indicated. Tumor volume was measured using vernier calipers every 5 days from day 10 after subcutaneous implantation. (O and P) Images of tumors from sacrificed mice were obtained on day 30 after injection. Tumor weights of different groups were compared. (Q) The percentage of PCNA-positive cells in subcutaneous xenograft tumors. Data are expressed as the mean ± SD. ∗p < 0.05, ∗∗p < 0.01.
Considering that the expression of Mfn1 was decreased in c-Myc-overexpressed HB, we next determined whether microRNAs (miRNAs) were involved in this process. Among the five miRNAs that were predicted to regulate Mfn1 using TargetScan and miRDB databases (Figure 6F), only miR-373-3p was significantly decreased when c-Myc was knocked down (Figure 6G) and was able to repress the expression of Mfn1 in Huh6 cells (Figures S6A and S6D). Moreover, the miR-373-3p inhibitor significantly enhanced the expression of Mfn1 and mitochondrial fusion but decreased the proliferation of Huh6 cells (Figures 6H, 6J, 6L, S6B, and S6C). Similarly, treatment with a target site blocker (TSB), which selectively prevents miR-373-3p binding to the 3′ UTR of MFN1, blocked the inhibitory effect of the miR-373-3p mimic on Mfn1 expression and mitochondrial fusion and further impeded miR-373-3p-mimic-induced proliferation of HB cells (Figures 6I, 6K, 6M, and S6D). Importantly, our data also demonstrated that the TSB treatment significantly increased the expression of Mfn1 in tumors developed from Huh6 and suppressed tumor growth (Figures 6N–6Q, S6E, and S6F).
Discussion
In the present study, we aimed to reveal the mechanism underlying the poor prognosis of patients with HB with high levels of c-Myc. Several key findings have been obtained. First, c-Myc overexpression induced mitochondrial fragmentation by transcriptionally activating DRP1 and downregulating Mfn1 in an miR-373-3p-dependent manner. Second, mitochondrial fragmentation significantly promoted the tumorigenesis of c-Myc-driven HB-like liver tumors and was associated with the prognosis of patients with HB. Third, mitochondrial fragmentation induced ROS production to promote aerobic glycolysis and the proliferation of HB cells by activating the AKT/mTOR and NF-κB pathways. Our findings suggest that signaling pathways involving ROS may be potential therapeutic targets against aggressive HB.
Previous studies have reported that c-Myc is frequently overexpressed in HB.32,33 Moreover, it has been reported that c-Myc induced HB-like tumors in mice strikingly resembling the immature subtype of human HB.11,34 Additionally, c-Myc downregulation in HB cells impairs tumorigenesis in vivo.11 From this point of view, c-Myc appears to be an ideal therapeutic target for HB. However, there is no specific active site of c-Myc for small molecules to target, and it is barely possible to develop therapeutic drugs since c-Myc is mainly located in the nucleus.35 Therefore, the identification of targetable downstream molecules of c-Myc in HB is warranted.
It is widely accepted that abnormally aggravated mitochondrial fragmentation is a common characteristic of human tumors and is associated with a poor prognosis.36, 37, 38, 39 Similar to our results, previous studies have shown that silencing DRP1 or the overexpression of MFN1/2 resulted in a metabolic shift from aerobic glycolysis to oxidative phosphorylation and significantly suppressed the proliferation and metastasis of tumor cells.40,41 Since aerobic glycolysis (also known as the Warburg effect) is a key metabolic hallmark of tumor cells and facilitates tumor progression, it is reasonable to believe that mitochondrial-fragmentation-induced mitochondrial dysfunction in HB-like liver tumor cells may also enhance the glycolysis and thus promote the survival of tumor cells.
In the present study, we found that overexpression of c-Myc significantly induced mitochondrial fragmentation in HB. Several previous studies have partially supported this finding. For example, HoSeo et al. identified that the mitochondrial fission factor (MFF) is a novel transcriptional target of c-Myc in prostate cancer tissues.42 Moreover, it has been identified by ChIP sequencing that c-Myc occupies the DRP1 promoter in lymphoma cells, although the exact binding site has not been reported.31 Here, we have further shown that the region between −97 and −86 of the DRP1 promoter is critical for c-Myc binding. miR-373 has been identified as an oncogene in previous studies, as miR-373 promotes the proliferation and tumorigenesis of transformed cells.43 More importantly, the expression of miR-373-3p was reported to be directly controlled by c-Myc.44 In the present study, we uncovered a new function of miR-373 in mediating the expression of Mfn1 and mitochondrial fusion. This seems to explain why miR-373 was previously thought to be a hypoxia-related miRNA,45 since mitochondrial fusion is inhibited under hypoxia.46
A moderately elevated level of ROS has been observed in cancer cells compared with in normal cells.29 The increase in ROS production contributes to tumorigenesis by sustaining heightened mitogenic signaling, such as the PI3K/AKT and NF-κB signaling cascades.30 Moreover, approximately 90% of intracellular ROS are generated from the mitochondria. A series of studies have shown that deregulation of mitochondrial fission and fusion significantly influences mitochondrial functions, such as ROS production.23,47 On the other hand, ROS are also generated as a result of c-Myc activation.48 One proposed mechanism is the c-Myc-dependent induction of p53, some of which target gene-encoding proteins that regulate ROS.49 In addition, it has been estimated that approximately half of the ROS generated by c-Myc overexpression in human fibroblasts can be accounted for by the induction of CYP2C9.49 Our results indicate that mitochondrial fragmentation induced by c-Myc overexpression contributes to ROS production and subsequently promotes cell proliferation by activating the AKT/mTOR and NF-κB pathways. More importantly, these results also indicate that antioxidant reagents are promising choices for the treatment of aggressive HB subtypes.
It is noteworthy that only based on histopathologic assessment, it is not easy to distinguish human hepatocellular carcinoma (HCC) from epithelial HB if no information on the patient's age and gross histomorphology is available.50,51 Similarly, the pathological type of c-Myc-mediated mouse liver tumors has been controversial in previous studies, indicating that c-Myc-expressing mice developed both HB- and HCC-like tumors.10,52 In the present study, c-Myc-induced liver tumors were independently examined by two pathologists. Most of them were more like HB but less like HCC. Since c-Myc has been shown to induce the stem-cell-like state to favor the onset of tumorigenesis,53 we assumed that the different expression levels of c-Myc, different feeding environments, and other unidentified factors may affect the differentiation of tumor cells and thus pathological types (HB or HCC) in different laboratories. Therefore, several potential (but not perfect) biomarkers for the differential diagnosis between HB and HCC may help. Among them, β-catenin was reported to be mainly accumulated in the nucleus of poorly differentiated human HB cells but was localized in the membranous of fetal HB and HCC cells.54,55 Delta-like 1 homology (DLK1) and insulin-like growth factor 2 (IGF2) were demonstrated to be uniquely overexpressed in HB tissues.56,57
In conclusion, our findings demonstrate that mitochondrial fragmentation is frequently enhanced in c-Myc-overexpressed HB and plays a critical role in promoting the development of HB by increasing ROS production and subsequently activating the AKT/mTOR and NF-κB signaling pathways. Our findings suggest that targeting aberrant mitochondrial dynamics or particular pathways relevant to ROS may be a promising strategy for the treatment of aggressive HB.
Materials and methods
Cell culture and tissue collection
The HB cell lines HepG2 and Huh6 were purchased from the National Collection of Authenticated Cell Cultures (Shanghai, China). Cells were cultured in DMEM (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Sangon Biotech, Shanghai, China) and 1% penicillin-streptomycin (Solarbio, Beijing, China). All HB cell lines were authenticated using short tandem repeat DNA testing at the Fourth Military Medical University (FMMU) Center for DNA Typing. Formalin-fixed paraffin-embedded tissue samples from 50 patients with HB were collected from the Department of Pathology of Xijing Hospital affiliated with the Fourth Military Medical University (FMMU, Xi'an, Shaanxi, China) and Xi'an Children's Hospital (Xi'an, Shaanxi, China). This study was approved by the Ethics Committee of the FMMU. Written informed consent was obtained from all participants. The clinical characteristics of the patients with HB are listed in Table S1. All participants provided written informed consent. This study was approved by the Ethics Committee of the Fourth Military Medical University (permission number: KY20173189–1; date issued: 2017-03-06).
Experimental animals
MFN1flox/flox mice with loxP sites flanking exon 4 of MFN1 were generously gifted by Professor Yongzhan Nie (Department of Digestive Diseases, Xijing Hospital, FMMU). Alb-Cre mice were purchased from Shanghai Model Organisms Center (Shanghai, China). Mice with liver-specific knockout of MFN1 were generated by crossing MFN1flox/flox mice with Alb-Cre mice under specific pathogen-free (SPF) conditions at the laboratory animal center of FMMU. The animal study was conducted in compliance with the guidelines of the Animal Care Committee of the FMMU. The genotypes of mice were evaluated by multiplex PCR and agarose gel electrophoresis, as previously described.25 All mice used in this study were euthanized via an overdose inhalation of carbon dioxide in a CO2 chamber (CO2 displacement rate equivalent to 20% of the chamber volume/min). All animal experimental procedures were approved by the Animal Care Committee of FMMU (permission number: IACUC–20190902; date issued: 2019-09-01).
Plasmid construction and cell transfection
Plasmids pT3-EF1α and pT3-EF1α-c-Myc and the pCMV/SB transposon were kindly provided by Dr. Xin Chen (University of California, San Francisco, CA, USA). The full-length mouse DRP1 or MFN1 fragment was amplified from the cDNA of mouse liver and then cloned into the pT3-EF1α plasmid using the Gateway system (ThermoFisher Scientific, Waltham, MA, USA). For overexpression of Mfn1 in HB cells, the coding sequence of MFN1 was amplified from cDNA derived from HepG2 cells using primers containing restriction sites of BamHI and EcoRV and then cloned into the pcDNA 3.1 expression vector. The primers used are shown in Table S2. All plasmids were amplified and purified using the EndoFree Plasmid Maxi kit (TIANGEN, Beijing, China). To generate small hairpin RNA (shRNA) expression vectors, the shRNA containing specific sequences targeting the c-Myc or Drp1 mRNA sequence was cloned into the pSilencer 3.1-H1 neo vector (Ambion, Austin, TX, USA). The specific mRNA sequences used are shown in Table S2. The miRNA/miR mimic and inhibitor were synthesized by Shanghai GenePharma (Shanghai, China), and the sequences are listed in Table S2. The TSB was designed, synthesized, and verified by QIAGEN (Hilden, Germany). All transfections were performed using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. Cell lines stably expressing shDRP1, MFN1, or shc-Myc in HepG2 and Huh6 cells were established by G418 (Sigma, St. Louis, MO, USA) treatment after transfection. For the in vivo TSB treatment, subcutaneous xenograft tumor models were established using Huh6 cells. Once tumors reached approximately 200 mm3, 25 mg/kg−1 of TSB-MFN1 or control TSB-NC was subcutaneously injected in the vicinity of the tumors every day.
Hydrodynamic transfection
To establish a murine HB tumor model, hydrodynamic injection of plasmid DNA was performed on 6-week-old male C57BL/6J mice as previously described.58 In brief, 20 μg plasmids encoding the gene(s) of interest along with the SB transposase in a ratio of 25:1 were diluted in 2 mL saline for each mouse. The above-mentioned solution was then filtered and injected into the lateral tail vein of mice for 5–7 s.
Real-time ATP rate assay
The XF Real-Time ATP Rate Assay kit (Agilent Seahorse Bioscience, North Billerica, MA, USA) was used to measure cell function by kinetic quantification of ATP production, and the data were acquired and analyzed using the Agilent XFe96 Extracellular Flux Analyzer according to the manufacturer's instructions. Briefly, Huh6 cells were seeded at a density of 1.0 × 104 cells/well, while HepG2 cells were seeded at a density of 2.0 × 104 cells/well. Cells were incubated with DMEM medium overnight. The XFe96 sensor cartridge was hydrated, and the Seahorse XF Calibrant was equilibrated in a non-CO2 incubator overnight. Prior to the assay, cells were washed twice with pre-warmed Seahorse XF DMEM medium supplemented with 10 mM glucose, 2 mM L-glutamine, and 1 mM pyruvate and then incubated in 180 μL medium at 37°C without CO2 for 60 min. The real-time ATP production rate was measured at 37°C in an XFe96 extracellular flux analyzer, which was previously calibrated using the Seahorse XF Calibrant. During the run, cells received 1.5 μM oligomycin (ATP synthase inhibitor) from port A and a mixture of antimycin A and rotenone (0.5 μM) from port B to inhibit the activity of complexes I and III, respectively. Data were collected using Agilent Seahorse Wave 2.6.1 desktop software and exported to GraphPad Prism v.8.3 for analysis.
Lactic acid content detection
Lactic acid content was detected using a lactic acid assay kit (Solarbio, Beijing, China) according to the manufacturer's instructions. In brief, 0.1 g tissue or 5 × 104 cells in 1 mL extraction reagent I was sonicated on ice. The homogenate was centrifuged at 12,000 × g for 10 min at 4°C. Supernatant of 0.8 mL was mixed with 0.15 mL extraction reagent Ⅱ and centrifuged at 12,000 × g for 10 min at 4°C. The supernatant was collected for detection. The working solution was diluted to standard solutions of different concentrations (2.5, 1.25, 0.625, 0.3125, 0.15625, and 0.078 μmol/mL). The absorbance at 570 nm was then measured using a microplate reader (BioTek, New York, NY, USA). The lactic acid content was calculated according to a standard curve, which was depicted based on standard substances. All results were normalized based on the tissue mass or total protein amounts of cells.
Glucose uptake detection
Glucose uptake was detected using a glucose assay kit (Solarbio, Beijing, China) according to the manufacturer's instructions. In brief, 5 × 104 cells in 1 mL sterile water were sonicated on ice and then heated for 10 min in boiling water. The homogenate was then centrifuged, and the supernatant was collected for detection. The absorbance at 505 nm was measured using a microplate reader (BioTek, New York, NY, USA). Glucose uptake was calculated based on the absorbance of the standard solution. All results were normalized based on the total protein content of cells.
Detection of ROS
Cellular ROS was detected using the fluorescent probe DCFH-DA (Beyotime Biotechnology, Shanghai, China) as previously described.23 Briefly, DCFH-DA was diluted with DMEM to a final concentration of 10 μM. HB cells were then resuspended in DCFH-DA solution at 1 × 105 cells/mL and incubated for 20 min at 37°C in the dark. Finally, the samples were analyzed using a flow cytometer (Beckman, Fullerton, CA, USA).
Mitochondrial imaging by electron microscopy and confocal microscopy
Conventional TEM analysis was performed as previously described.59 In brief, tumor tissues from HB patients and mouse models were fixed with 4% glutaraldehyde. The samples were post-fixed in OsO4, dehydrated in alcohol, and embedded in araldite. Ultrathin sections were stained with uranyl acetate and lead citrate and examined using a Tecnai G2 electron microscope (FEI, Hillsboro, OR, USA) at 8,200 and 16,500 magnifications. The fluorescent dye MitoTracker Green FM (Invitrogen, Carlsbad, CA, USA) was used to monitor mitochondrial morphology with excitation/emission set at 490 and 516 nm in living cells rinsed with serum-free media in a 5% CO2 and 37°C conditions, as described previously.59 Then z stack images (with 0.2 μm increments) of mitochondria from the cells were taken using a Nikon A1 plus laser scanning confocal microscope (Nikon, Melville, NY, USA) with a 60 × oil-immersion objective (numerical aperture 1.4) for Huh6 or a 100 × oil-immersion objective (numerical aperture 1.4) for HepG2. For morphometric analysis, the number, length, and area of mitochondria were measured using ImageJ software (NIH, Bethesda, MD, USA) together with a plugin named Mitochondria Analyzer.60 The mean form factor indicates the mean mitochondrial morphology per cell. The mitochondria are round when the index is 1, and the morphology becomes longer when the index increases. According to the index of the mean form factor, the fragmented form of mitochondria per cell is defined between 1 and 1.5, the intermediate form is defined between 1.5 and 2.5, and the elongated form is defined as greater than 2.5. The number of cells with different forms of mitochondria was counted, and the proportions were calculated.
Quantitative RT-PCR, western blotting, IHC, and hematoxylin and eosin
RNA extraction, cDNA synthesis, quantitative RT-PCR (qRT-PCR), and western blotting were performed as previously described.61 The primer sequences used are shown in Table S2. mtDNA content was evaluated as previously described.61 Primer sequences are listed in Table S2. The relative mtDNA content was calculated according to the ratio of mtDNA copy number to nuclear DNA copy number. HB tissues were processed for IHC as previously described.62 The intensity (0, none; 1, faint yellow; 2, yellow; 3, brown) and the proportion of positive cells (0, 0%–9%; 1, 10%–25%; 2, 26%–50%; 3, 51%–75%; 4, 76%–100%) were determined within five random microscopic visual fields per slide by two independent pathologists blinded to the clinical data. IHC staining was scored (0–12) by multiplying the percentage of positive cells by the intensity. The primary antibodies used in this study are listed in Table S3.
Collection of public datasets and bioinformatic analysis
Two public datasets of mRNA expression data derived from HB tissues (GEO: GSE13303963 and GSE7527164) were downloaded from the Gene Expression Omnibus database (Table S4) and analyzed. HB patients were ranked according to the expression level of c-Myc in each cohort. Patients in the top 25% and bottom 25% were defined as c-Myc-high and c-Myc-low, respectively. DEGs between the two groups were calculated using an independent shrinkage t test. An unweighted gene set enrichment analysis was performed using the GeneTrail 3.0 web service.65
Cell proliferation assay
Cell proliferation was quantified using the CCK-8 (Beyotime, Shanghai, China) and EdU incorporation assays. For the CCK-8 assay, HB cells were seeded in a 96-well plate and incubated at 37 °C. CCK-8 solution (10 μL) was added to each well, followed by incubation at 37°C for 2 h. Absorbance at 450 nm was measured using a microplate reader (BioTek, New York, NY, USA). The EdU incorporation assay (Ribobio, Guangzhou, China) was performed as previously described.66 The results were evaluated using a fluorescent microscope (Olympus, Tokyo, Japan).
Tumor spheroid cell culture
One hundred microliters of 10% Matrigel Matrix (BD Biosciences, San Jose, CA, USA) was added to a well of a 96-well chamber slide and allowed to solidify in a cell incubator for about 8 h. Then, 2,000 HB cells suspended in 0.1 mL 2% matrix diluted complete DMEM containing epidermal growth factor (EGF; 20 ng/mL), basic fibroblast growth factor (bFGF; 10 ng/mL), and B27 (1:50) were seeded and cultured at 37°C for about 1 week. Images of the cell spheroids per group were captured using a fluorescent microscope.
Subcutaneous tumor models
For the xenograft models, male BALB/c nude mice (4–6 weeks old) were randomly divided into two groups. For the development of tumor xenografts in nude mice, 1 × 107 Huh6 cells treated as indicated were injected into the flanks of nude mice (n = 6 in each group). Tumor length (L) and width (W) were measured using a Vernier caliper every 5 days, and tumor volume was calculated according to the formula (L × W2)/2. The transplanted mice were sacrificed 30 days later. Tumor nodules were photographed and weighed. It should be noted that the maximum tumor diameter was allowed to be within 1.5 cm, and the maximum tumor volume was allowed to be within 2,000 mm3.
18F-FDG micro-PET/CT imaging
Since tumors generated by hydrodynamic transfection often showed multiple nodules of varying sizes in vivo, xenograft models were also generated to perform a PET scan with 18F-FDG. Moreover, to avoid the influence of different tumor volumes on the uptake of 18F-FDG, varying amounts of tumor tissue mass (20–50 mg) induced by c-Myc hydrodynamic transfection were used to establish the xenograft models by subcutaneous implantation. Then, mice with comparable tumor volumes (approximately 1,500 mm3) were chosen to fast overnight and were then injected with approximately 3.7 MBq (100 μCi) of 18F-FDG through the tail vein. The scanning was started 60 min after tracer injection before the mice were anesthetized with 1.5%–2.5% isoflurane using a micro-PET/CT scanner (Mediso Medical Imaging Systems, Budapest, Hungary). All PET/CT images were processed and analyzed using Interview Fusion 1.0 (Mediso Medical Imaging Systems, Budapest, Hungary) software. For semiquantitative analysis, 3-dimensional regions of interest were carefully depicted and manually adjusted over the borders of the tumor. Mean 18F-FDG uptake was presented as % ID/g (percentage of injected dose per gram of tissue), which was calculated by dividing the tumor radioactivity by the injected dose. and compared between the different interventions.
miRNA target prediction
The miRNAs targeting MFN1 were predicted using the TargetScan67 and miRDB68 databases. Venn diagrams were constructed to illustrate overlapping candidate miRNAs from the two databases.
Luciferase reporter assay
Luciferase reporter constructs were prepared by cloning the DRP1 promoter region from the genomic DNA of HB cells into the pGL3-basic luciferase reporter vector (Promega, Madison, WI, USA). The constructs were transfected into Huh6 cells along with a plasmid containing the Renilla luciferase reporter gene, as previously described.66 Then, HB cells were lysed and analyzed using the Dual Luciferase Reporter Assay kit (Promega, Madison, WI, USA) according to the manufacturer's instructions. The firefly luciferase activity for a specific promoter construct was normalized to the Renilla luciferase activity. The relative light units were measured using a microplate reader (BioTek, New York, NY, USA).
Serially truncation and site-directed mutagenesis
Promoter sequences of DRP1 were obtained from the UCSC Genome Browser. Truncated regions of the DRP1 promoter were amplified and inserted into the pGL3-basic vector. Site-directed mutagenesis was performed using the QuikChange II Site-Directed Mutagenesis Kit (Agilent, Santa Clara, CA, USA) according to the manufacturer's instructions. The E-box was mutated (underlined) to CAAAAG (WT, CATGCG). The primers used for site-directed mutagenesis of the DRP1 promoter are listed in Table S2.
ChIP-PCR analysis
The ChIP assay was performed using an EZ-ChIP kit (Millipore Corporation, Billerica, MA, USA) according to the manufacturer's instructions. Briefly, anti-c-Myc antibody or isotype immunoglobulin G (IgG) was immunoprecipitated with chromatin of Huh6 cells transfected with the WT DRP1 reporter or the site-mutated (MU) DRP1 reporter. A product of 297 bp from the regulatory region of DRP1 was generated by PCR. Primer sequences are provided in Table S2.
Statistical analyses
The experiments were repeated three times, where appropriate. GraphPad Prism 8.3.0 software (GraphPad Software, La Jolla, CA, USA) was used for all statistical analyses, and p < 0.05 was considered significant. Unpaired t tests were used for comparisons between the two groups, where appropriate. Correlations between measured variables were tested using Spearman's rank correlation analyses.
Acknowledgments
The authors thank Prof. Xin Chen (University of California, San Francisco) for the generous gift of plasmids PT3-EF1α and pT3-EF1α-c-Myc and the pCMV/SB transposen. The authors thank Prof. Yongzhan Nie (Xijing Hospital, Fourth Military Medical University, Xi'an, China) for the generous gift of MFN1flox/flox. The authors are grateful to Prof. Zengshan Li (Xijing Hospital, Fourth Military Medical University, Xi'an, China) and Prof. Guangsheng Chen (Xian Children's Hospital, Xi'an, China) for histological analyses. We also thank the participants who generously participated in this study. This work was supported by grants from the National Natural Science Foundation of China (nos. 82172919, 81772935, 81972590, 81773177, and 82020108023) and the State Key Laboratory of Cancer Biology Project (CBSKL2019ZZ26 and CBSKL2019ZZ06).
Author contributions
All authors of this paper have directly participated in the planning, execution, or analyses of the study. D.W. and J.T. were involved in the acquisition, analyses, and interpretation of the data and the drafting of the manuscript. Q.Y. and S.G. were involved in bioinformatics analyses. Z.Y., D.W., and X.L. were involved in the analyses and interpretation of data. S.Y. was involved in tissue sample collection. J.W. was involved in 18F-FDG micro-PET/CT imaging. Y.Y. and J.X. were involved in securing research funding and acquiring, analyzing, and interpreting the data. J.A. and Q.H. were involved in the conception and design of the study, critically revised the manuscript, and supervised the overall study. All authors read and approved the final manuscript.
Declaration of interest
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.01.032.
Contributor Information
Jiaze An, Email: anchen@fmmu.edu.cn.
Qichao Huang, Email: huangqichao@fmmu.edu.cn.
Supplemental information
References
- 1.Rougemont A.-L., McLin V.A., Toso C., Wildhaber B.E. Adult hepatoblastoma: learning from children. J. Hepatol. 2012;56:1392–1403. doi: 10.1016/j.jhep.2011.10.028. [DOI] [PubMed] [Google Scholar]
- 2.Hubbard A.K., Spector L.G., Fortuna G., Marcotte E.L., Poynter J.N. Trends in international incidence of pediatric cancers in children under 5 Years of age: 1988-2012. JNCI Cancer Spectr. 2019;3:pkz007. doi: 10.1093/jncics/pkz007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heck J.E., Lee P.-C., Wu C.-K., Tsai H.-Y., Ritz B., Arah O.A., Li C.-Y. Gestational risk factors and childhood cancers: a cohort study in Taiwan. Int. J. Cancer. 2020;147:1343–1353. doi: 10.1002/ijc.32905. [DOI] [PubMed] [Google Scholar]
- 4.Aronson D.C., Weeda V.B., Maibach R., Czauderna P., Dall'Igna P., de Ville de Goyet J., Branchereau S., Perilongo G., Brock P., Zsiros J., et al. Microscopically positive resection margin after hepatoblastoma resection: what is the impact on prognosis? A Childhood Liver Tumours Strategy Group (SIOPEL) report. Eur. J. Cancer. 2019;106:126–132. doi: 10.1016/j.ejca.2018.10.013. [DOI] [PubMed] [Google Scholar]
- 5.Meyers R.L., Maibach R., Hiyama E., Häberle B., Krailo M., Rangaswami A., Aronson D.C., Malogolowkin M.H., Perilongo G., von Schweinitz D., et al. Risk-stratified staging in paediatric hepatoblastoma: a unified analysis from the Children's Hepatic tumors International Collaboration. Lancet Oncol. 2017;18:122–131. doi: 10.1016/S1470-2045(16)30598-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baluapuri A., Wolf E., Eilers M. Target gene-independent functions of MYC oncoproteins. Nat. Rev. Mol. Cel. Biol. 2020;21:255–267. doi: 10.1038/s41580-020-0215-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gabay M., Li Y., Felsher D.W. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med. 2014;4:a014241. doi: 10.1101/cshperspect.a014241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shachaf C.M., Kopelman A.M., Arvanitis C., Karlsson A., Beer S., Mandl S., Bachmann M.H., Borowsky A.D., Ruebner B., Cardiff R.D., et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature. 2004;431:1112–1117. doi: 10.1038/nature03043. [DOI] [PubMed] [Google Scholar]
- 9.Chow E.K.-H., Fan L.-l., Chen X., Bishop J.M. Oncogene-specific formation of chemoresistant murine hepatic cancer stem cells. Hepatology. 2012;56:1331–1341. doi: 10.1002/hep.25776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kress T.R., Pellanda P., Pellegrinet L., Bianchi V., Nicoli P., Doni M., Recordati C., Bianchi S., Rotta L., Capra T., et al. Identification of MYC-dependent transcriptional programs in oncogene-addicted liver tumors. Cancer Res. 2016;76:3463–3472. doi: 10.1158/0008-5472.CAN-16-0316. [DOI] [PubMed] [Google Scholar]
- 11.Cairo S., Armengol C., De Reyniès A., Wei Y., Thomas E., Renard C.-A., Goga A., Balakrishnan A., Semeraro M., Gresh L., et al. Hepatic stem-like phenotype and interplay of Wnt/beta-catenin and Myc signaling in aggressive childhood liver cancer. Cancer Cell. 2008;14:471–484. doi: 10.1016/j.ccr.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 12.Hooks K.B., Audoux J., Fazli H., Lesjean S., Ernault T., Dugot-Senant N., Leste-Lasserre T., Hagedorn M., Rousseau B., Danet C., et al. New insights into diagnosis and therapeutic options for proliferative hepatoblastoma. Hepatology. 2018;68:89–102. doi: 10.1002/hep.29672. [DOI] [PubMed] [Google Scholar]
- 13.Archer S.L. Mitochondrial dynamics--mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 2013;369:2236–2251. doi: 10.1056/NEJMra1215233. [DOI] [PubMed] [Google Scholar]
- 14.Tilokani L., Nagashima S., Paupe V., Prudent J. Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem. 2018;62:341–360. doi: 10.1042/EBC20170104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maycotte P., Marín-Hernández A., Goyri-Aguirre M., Anaya-Ruiz M., Reyes-Leyva J., Cortés-Hernández P. Mitochondrial dynamics and cancer. Tumour Biol. 2017;39 doi: 10.1177/1010428317698391. 1010428317698391. [DOI] [PubMed] [Google Scholar]
- 16.Chen L., Zhang J., Lyu Z., Chen Y., Ji X., Cao H., Jin M., Zhu J., Yang J., Ling R., et al. Positive feedback loop between mitochondrial fission and Notch signaling promotes survivin-mediated survival of TNBC cells. Cell Death Dis. 2018;9:1050. doi: 10.1038/s41419-018-1083-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee Y.G., Nam Y., Shin K.J., Yoon S., Park W.S., Joung J.Y., Seo J.K., Jang J., Lee S., Nam D., et al. Androgen-induced expression of DRP1 regulates mitochondrial metabolic reprogramming in prostate cancer. Cancer Lett. 2020;471:72–87. doi: 10.1016/j.canlet.2019.12.017. [DOI] [PubMed] [Google Scholar]
- 18.Ma J.T., Zhang X.Y., Cao R., Sun L., Jing W., Zhao J.Z., Zhang S.L., Huang L.T., Han C.B. Effects of dynamin-related protein 1 regulated mitochondrial dynamic changes on invasion and metastasis of lung cancer cells. J. Cancer. 2019;10:4045–4053. doi: 10.7150/jca.29756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yi T., Luo H., Qin F., Jiang Q., He S., Wang T., Su J., Song S., Qin X., Qin Y., et al. LncRNA LL22NC03-N14H11.1 promoted hepatocellular carcinoma progression through activating MAPK pathway to induce mitochondrial fission. Cell Death Dis. 2020;11:832. doi: 10.1038/s41419-020-2584-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hayes J.D., Dinkova-Kostova A.T., Tew K.D. Oxidative stress in cancer. Cancer Cell. 2020;38:167–197. doi: 10.1016/j.ccell.2020.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sabharwal S.S., Schumacker P.T. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles' heel? Nat. Rev. Cancer. 2014;14:709–721. doi: 10.1038/nrc3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prieto J., León M., Ponsoda X., Sendra R., Bort R., Ferrer-Lorente R., Raya A., López-García C., Torres J. Early ERK1/2 activation promotes DRP1-dependent mitochondrial fission necessary for cell reprogramming. Nat. Commun. 2016;7:11124. doi: 10.1038/ncomms11124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang Q., Zhan L., Cao H., Li J., Lyu Y., Guo X., Zhang J., Ji L., Ren T., An J., et al. Increased mitochondrial fission promotes autophagy and hepatocellular carcinoma cell survival through the ROS-modulated coordinated regulation of the NFKB and TP53 pathways. Autophagy. 2016;12:999–1014. doi: 10.1080/15548627.2016.1166318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang Q., Cao H., Zhan L., Sun X., Wang G., Li J., Guo X., Ren T., Wang Z., Lyu Y., et al. Mitochondrial fission forms a positive feedback loop with cytosolic calcium signaling pathway to promote autophagy in hepatocellular carcinoma cells. Cancer Lett. 2017;403:108–118. doi: 10.1016/j.canlet.2017.05.034. [DOI] [PubMed] [Google Scholar]
- 25.Chen H., McCaffery J.M., Chan D.C. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130:548–562. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 26.Ganapathy-Kanniappan S. Molecular intricacies of aerobic glycolysis in cancer: current insights into the classic metabolic phenotype. Crit. Rev. Biochem. Mol. Biol. 2018;53:667–682. doi: 10.1080/10409238.2018.1556578. [DOI] [PubMed] [Google Scholar]
- 27.Feng J., Li J., Wu L., Yu Q., Ji J., Wu J., Dai W., Guo C. Emerging roles and the regulation of aerobic glycolysis in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2020;39:126. doi: 10.1186/s13046-020-01629-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen H., Chan D.C. Mitochondrial dynamics in regulating the unique phenotypes of cancer and stem cells. Cell Metab. 2017;26:39–48. doi: 10.1016/j.cmet.2017.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gogvadze V., Orrenius S., Zhivotovsky B. Mitochondria in cancer cells: what is so special about them? Trends Cell Biol. 2008;18:165–173. doi: 10.1016/j.tcb.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 30.Moloney J.N., Cotter T.G. ROS signalling in the biology of cancer. Semin. Cel. Dev. Biol. 2018;80:50–64. doi: 10.1016/j.semcdb.2017.05.023. [DOI] [PubMed] [Google Scholar]
- 31.Agarwal E., Altman B.J., Ho Seo J., Bertolini I., Ghosh J.C., Kaur A., Kossenkov A.V., Languino L.R., Gabrilovich D.I., Speicher D.W., et al. Myc regulation of a mitochondrial trafficking network mediates tumor cell invasion and metastasis. Mol. Cell Biol. 2019;39 doi: 10.1128/MCB.00109-19. e00109–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Comerford S.A., Hinnant E.A., Chen Y., Bansal H., Klapproth S., Rakheja D., Finegold M.J., Lopez-Terrada D., O'Donnell K.A., Tomlinson G.E., Hammer R.E. Hepatoblastoma modeling in mice places Nrf2 within a cancer field established by mutant -catenin. JCI Insight. 2016;1:e88549. doi: 10.1172/jci.insight.88549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fujita N., Hayashi-Nishino M., Fukumoto H., Omori H., Yamamoto A., Noda T., Yoshimori T. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol. Biol. Cell. 2008;19:4651–4659. doi: 10.1091/mbc.E08-03-0312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goga A., Yang D., Tward A.D., Morgan D.O., Bishop J.M. Inhibition of CDK1 as a potential therapy for tumors over-expressing MYC. Nat. Med. 2007;13:820–827. doi: 10.1038/nm1606. [DOI] [PubMed] [Google Scholar]
- 35.Chen H., Liu H., Qing G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Target. Ther. 2018;3:5. doi: 10.1038/s41392-018-0008-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shi L., Liu J., Peng Y., Zhang J., Dai X., Zhang S., Wang Y., Liu J., Long J. Deubiquitinase OTUD6A promotes proliferation of cancer cells via regulating Drp1 stability and mitochondrial fission. Mol. Oncol. 2020;14:3169–3183. doi: 10.1002/1878-0261.12825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liang J., Yang Y., Bai L., Li F., Li E. DRP1 upregulation promotes pancreatic cancer growth and metastasis through increased aerobic glycolysis. J. Gastroenterol. Hepatol. 2020;35:885–895. doi: 10.1111/jgh.14912. [DOI] [PubMed] [Google Scholar]
- 38.Gao T., Zhang X., Zhao J., Zhou F., Wang Y., Zhao Z., Xing J., Chen B., Li J., Liu S. SIK2 promotes reprogramming of glucose metabolism through PI3K/AKT/HIF-1α pathway and Drp1-mediated mitochondrial fission in ovarian cancer. Cancer Lett. 2020;469:89–101. doi: 10.1016/j.canlet.2019.10.029. [DOI] [PubMed] [Google Scholar]
- 39.Rodrigues T., Ferraz L.S. Therapeutic potential of targeting mitochondrial dynamics in cancer. Biochem. Pharmacol. 2020;182:114282. doi: 10.1016/j.bcp.2020.114282. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Z., Li T.E., Chen M., Xu D., Zhu Y., Hu B.Y., Lin Z.F., Pan J.J., Wang X., Wu C., et al. MFN1-dependent alteration of mitochondrial dynamics drives hepatocellular carcinoma metastasis by glucose metabolic reprogramming. Br. J. Cancer. 2020;122:209–220. doi: 10.1038/s41416-019-0658-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hagenbuchner J., Kuznetsov A.V., Obexer P., Ausserlechner M.J. BIRC5/Survivin enhances aerobic glycolysis and drug resistance by altered regulation of the mitochondrial fusion/fission machinery. Oncogene. 2013;32:4748–4757. doi: 10.1038/onc.2012.500. [DOI] [PubMed] [Google Scholar]
- 42.Seo J.H., Agarwal E., Chae Y.C., Lee Y.G., Garlick D.S., Storaci A.M., Ferrero S., Gaudioso G., Gianelli U., Vaira V., Altieri D.C. Mitochondrial fission factor is a novel Myc-dependent regulator of mitochondrial permeability in cancer. EBioMedicine. 2019;48:353–363. doi: 10.1016/j.ebiom.2019.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Voorhoeve P.M., le Sage C., Schrier M., Gillis A.J., Stoop H., Nagel R., Liu Y.P., van Duijse J., Drost J., Griekspoor A., et al. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell. 2006;124:1169–1181. doi: 10.1016/j.cell.2006.02.037. [DOI] [PubMed] [Google Scholar]
- 44.Cairo S., Wang Y., de Reyniès A., Duroure K., Dahan J., Redon M.J., Fabre M., McClelland M., Wang X.W., Croce C.M., Buendia M.A. Stem cell-like micro-RNA signature driven by Myc in aggressive liver cancer. Proc. Natl. Acad. Sci. U S A. 2010;107:20471–20476. doi: 10.1073/pnas.1009009107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crosby M.E., Kulshreshtha R., Ivan M., Glazer P.M. MicroRNA regulation of DNA repair gene expression in hypoxic stress. Cancer Res. 2009;69:1221–1229. doi: 10.1158/0008-5472.CAN-08-2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu X., Hajnóczky G. Altered fusion dynamics underlie unique morphological changes in mitochondria during hypoxia-reoxygenation stress. Cell Death Differ. 2011;18:1561–1572. doi: 10.1038/cdd.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sabouny R., Shutt T.E. Reciprocal regulation of mitochondrial fission and fusion. Trends Biochem. Sci. 2020;45:564–577. doi: 10.1016/j.tibs.2020.03.009. [DOI] [PubMed] [Google Scholar]
- 48.Vafa O., Wade M., Kern S., Beeche M., Pandita T.K., Hampton G.M., Wahl G.M. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol. Cell. 2002;9:1031–1044. doi: 10.1016/s1097-2765(02)00520-8. [DOI] [PubMed] [Google Scholar]
- 49.Ray S., Atkuri K.R., Deb-Basu D., Adler A.S., Chang H.Y., Herzenberg L.A., Felsher D.W. MYC can induce DNA breaks in vivo and in vitro independent of reactive oxygen species. Cancer Res. 2006;66:6598–6605. doi: 10.1158/0008-5472.CAN-05-3115. [DOI] [PubMed] [Google Scholar]
- 50.López-Terrada D., Alaggio R., de Dávila M.T., Czauderna P., Hiyama E., Katzenstein H., Leuschner I., Malogolowkin M., Meyers R., Ranganathan S., et al. Towards an international pediatric liver tumor consensus classification: proceedings of the Los Angeles COG liver tumors symposium. Mod. Pathol. 2014;27:472–491. doi: 10.1038/modpathol.2013.80. [DOI] [PubMed] [Google Scholar]
- 51.von Schweinitz D. Management of liver tumors in childhood. Semin. Pediatr. Surg. 2006;15:17–24. doi: 10.1053/j.sempedsurg.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 52.Xu P., Widmer G., Wang Y., Ozaki L.S., Alves J.M., Serrano M.G., Puiu D., Manque P., Akiyoshi D., Mackey A.J., et al. The genome of Cryptosporidium hominis. Nature. 2004;431:1107–1112. doi: 10.1038/nature02977. [DOI] [PubMed] [Google Scholar]
- 53.Poli V., Fagnocchi L., Fasciani A., Cherubini A., Mazzoleni S., Ferrillo S., Miluzio A., Gaudioso G., Vaira V., Turdo A., et al. MYC-driven epigenetic reprogramming favors the onset of tumorigenesis by inducing a stem cell-like state. Nat. Commun. 2018;9:1024. doi: 10.1038/s41467-018-03264-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Armengol C., Cairo S., Fabre M., Buendia M.A. Wnt signaling and hepatocarcinogenesis: the hepatoblastoma model. Int. J. Biochem. Cell Biol. 2011;43:265–270. doi: 10.1016/j.biocel.2009.07.012. [DOI] [PubMed] [Google Scholar]
- 55.Zhou S., Parham D.M., Yung E., Pattengale P., Wang L. Quantification of glypican 3, β-catenin and claudin-1 protein expression in hepatoblastoma and paediatric hepatocellular carcinoma by colour deconvolution. Histopathology. 2015;67:905–913. doi: 10.1111/his.12730. [DOI] [PubMed] [Google Scholar]
- 56.Luo J.H., Ren B., Keryanov S., Tseng G.C., Rao U.N., Monga S.P., Strom S., Demetris A.J., Nalesnik M., Yu Y.P., et al. Transcriptomic and genomic analysis of human hepatocellular carcinomas and hepatoblastomas. Hepatology. 2006;44:1012–1024. doi: 10.1002/hep.21328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dezso K., Halász J., Bisgaard H.C., Paku S., Turányi E., Schaff Z., Nagy P. Delta-like protein (DLK) is a novel immunohistochemical marker for human hepatoblastomas. Virchows Arch. 2008;452:443–448. doi: 10.1007/s00428-007-0571-8. [DOI] [PubMed] [Google Scholar]
- 58.Li L., Pilo G.M., Li X., Cigliano A., Latte G., Che L., Joseph C., Mela M., Wang C., Jiang L., et al. Inactivation of fatty acid synthase impairs hepatocarcinogenesis driven by AKT in mice and humans. J. Hepatol. 2016;64:333–341. doi: 10.1016/j.jhep.2015.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang Q., Li J., Xing J., Li W., Li H., Ke X., Zhang J., Ren T., Shang Y., Yang H., et al. CD147 promotes reprogramming of glucose metabolism and cell proliferation in HCC cells by inhibiting the p53-dependent signaling pathway. J. Hepatol. 2014;61:859–866. doi: 10.1016/j.jhep.2014.04.035. [DOI] [PubMed] [Google Scholar]
- 60.Chaudhry A., Shi R., Luciani D.S. A pipeline for multidimensional confocal analysis of mitochondrial morphology, function, and dynamics in pancreatic β-cells. Am. J. Physiol. Endocrinol. Metab. 2020;318 doi: 10.1152/ajpendo.00457.2019. E87–e101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sun X., Zhan L., Chen Y., Wang G., He L., Wang Q., Zhou F., Yang F., Wu J., Wu Y., et al. Increased mtDNA copy number promotes cancer progression by enhancing mitochondrial oxidative phosphorylation in microsatellite-stable colorectal cancer. Signal Transduct. Target. Ther. 2018;3:8. doi: 10.1038/s41392-018-0011-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ji L., Zhao Y., He L., Zhao J., Gao T., Liu F., Qi B., Kang F., Wang G., Zhao Y., et al. Deficiency attenuates diet-induced obesity and insulin resistance by promoting fatty acid oxidation and thermogenesis in Brown adipocytes. Adv. Sci. (Weinh) 2021;8:2002794. doi: 10.1002/advs.202002794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Carrillo-Reixach J., Torrens L., Simon-Coma M., Royo L., Domingo-Sàbat M., Abril-Fornaguera J., Akers N., Sala M., Ragull S., Arnal M., et al. Epigenetic footprint enables molecular risk stratification of hepatoblastoma with clinical implications. J. Hepatol. 2020;73:328–341. doi: 10.1016/j.jhep.2020.03.025. [DOI] [PubMed] [Google Scholar]
- 64.Sumazin P., Chen Y., Treviño L.R., Sarabia S.F., Hampton O.A., Patel K., Mistretta T.-A., Zorman B., Thompson P., Heczey A., et al. Genomic analysis of hepatoblastoma identifies distinct molecular and prognostic subgroups. Hepatology. 2017;65:104–121. doi: 10.1002/hep.28888. [DOI] [PubMed] [Google Scholar]
- 65.Gerstner N., Kehl T., Lenhof K., Müller A., Mayer C., Eckhart L., Grammes N.L., Diener C., Hart M., Hahn O., et al. GeneTrail 3: advanced high-throughput enrichment analysis. Nucleic Acids Res. 2020;48:W515–W520. doi: 10.1093/nar/gkaa306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang G., Wang Q., Huang Q., Chen Y., Sun X., He L., Zhan L., Guo X., Yin C., Fang Y., et al. Upregulation of mtSSB by interleukin-6 promotes cell growth through mitochondrial biogenesis-mediated telomerase activation in colorectal cancer. Int. J. Cancer. 2019;144:2516–2528. doi: 10.1002/ijc.31978. [DOI] [PubMed] [Google Scholar]
- 67.Agarwal V., Bell G.W., Nam J.-W., Bartel D.P. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4:e05005. doi: 10.7554/eLife.05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen Y., Wang X. miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020;48:D127–D131. doi: 10.1093/nar/gkz757. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.