

Abstract
A method is presented for the direct transformation of a ketone to the corresponding reduced alkyl chloride or bromide. The process involves the reaction of a ketone trityl hydrazone with t-BuOCl to give a diazene that readily collapses to the α-chlorocarbinyl radical, reduction of which by a hydrogen atom source gives the alkyl chloride product. The use of N-bromosuccinimide provides the corresponding alkyl bromide. This unique transformation provides a reductive halogenation that complements Barton’s redox-neutral vinyl halide synthesis.
Keywords: Hydrazones, Radical Reactions, Chlorination, Bromination, Diastereoselectivity
Graphical Abstract
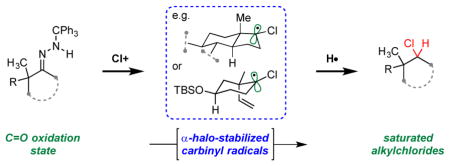
Halogen first, then hydrogen: Trityl hydrazones may be transformed by action of halonium ion sources to α-chloro and α-bromo carbinyl radicals, reduction of which affords the corresponding alkyl chlorides and alkyl bromides. Notably, this unique transformation is able to efficiently construct homoallylic and neopentyl chlorides and provides a reductive hydrazone halogenation complementary to Barton’s
The transformation of an alcohol to a chloride, one of the most basic reactions in organic chemistry, can present unanticipated difficulties when encountered in a complex molecule. In the course of studies toward the synthesis of N-methylwelwitindolinone B isothiocyanate (1, Figure 1), we attempted to convert the C-13 hydroxyl of 2 to the required, inverted chloride.[1] Unfortunately, the neopentyl and homoallylic nature of the alcohol conspired to trigger a skeletal rearrangement, thereby foiling the planned synthetic route.[2] Others have also observed difficulties, in completely different systems, in making alkyl chlorides by Walden inversion of suitably activated precursors.[3] Given the limitations of this transformation, combined with the prevalence of natural products[4] having a chloride attached to an sp3-hybridized carbon, we set forth to devise a fundamentally different solution for the synthesis of such chlorides.[5] We report here the realization of a method for the overall reductive transformation of ketones to alkyl halides.
Figure 1.

Welwitindolinone B and potential precursors.
The reductive chlorination method was conceived to provide a conceptually new way for the installation of a chlorine atom in high-value compounds. Rather than introducing the chloride through nucleophilic displacement of an activated alcohol, with the attendant difficulties and complications noted above, the idea was to introduce the chlorine first and then, through the generation of a reactive intermediate, add a hydrogen atom. This concept was expected to be realized through the use of hydrazone chemistry (Figure 2a).[6–8] The reaction of a trityl hydrazone with a chloronium source was expected to give a chlorodiazene (5),[9] which on thermolysis would extrude N2 to generate a trityl radical and the sought after reactive intermediate, α-chlorocarbinyl radical 6. Provided the thermolysis were carried out in the presence of a hydrogen atom donor, then diastereoselective hydrogen abstraction would give the desired chloroalkane product. Introduction of chlorine and generation and reduction of the reactive intermediate were envisioned though a single synthetic maneuver. Realized, this transformation provides a reductive chlorination from the C=O oxidation state, a method that complements Barton-type vinyl halide synthesis (Figure 2b).[10–11]
Figure 2.

Synthesis of organochlorides from hydrazone precursors.
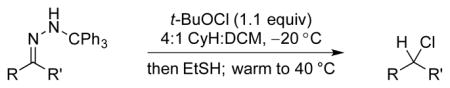
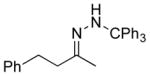
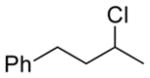
Feasibility of the above concept was evaluated using hydrazone 11a, available through condensation of trityl hydrazide and benzylacetone (74%).[12] Treatment of a solution of 11a in THF with t-BuOCl (1.1 equiv) at −20 °C followed by addition of an excess of EtSH and warming to room temperature afforded the desired product of reductive chlorination in 37% yield (NMR).[13] Modest yields in these early reactions were balanced significantly by the product of apparent hydrolysis of the starting hydrazone, benzylacetone. Mechanistic considerations suggested that the apparent “hydrolysis product” likely arises by way of peroxychloroalkane intermediate 14, the product of O2 capture by the α-chlorocarbinyl radical (Figure 3). Support for this hypothesis was obtained by carrying out the thermolysis in the absence of a reducing agent and placing it under an oxygen balloon prior to warming to room temperature. The major product of the reaction under these conditions was benzylacetone (57%, NMR), with no evidence of chloroalkane 13a. On the other hand, scrupulous exclusion of air through two freeze-pump-thaw cycles completely eliminated formation of ketone 10 in the reaction product. Variable temperature NMR experiments provided an understanding of the thermal requirements for the different steps of the reaction.[14–15] A −78 °C sample of hydrazone 11a and t-BuOCl was examined by NMR in a probe pre-cooled −30 °C. After 10 minutes had elapsed, a reaction was observed, and the starting hydrazone was found to be fully consumed. The resulting putative chlorodiazene 12 was found to persist as the temperature was increased from −30 °C to −10 °C. Upon further warming above −10 °C, diazene 12 decomposed to give a mixture of products. With the sequence of reagent addition and temperature control guided by the above study, as well as careful O2 exclusion, the reaction was optimized to furnish 13a in 82% isolated yield (Table 1, entry 1). A brief screen of chloronium ion sources and H-atom donors offered no improvement over t-BuOCl and EtSH, with N-chlorosuccinimide yielding none of the desired chloride. Less odorous, high molecular weight thiols were examined briefly as hydrogen atom donors, but found to give less satisfactory results.
Figure 3.

Optimization of reaction parameters. [a] Reactions performed in THF; NMR yield.
Table 1.
Reductive chlorination of trityl hydrazones.
| |||
|---|---|---|---|
| entry | hydrazone | chloride | yield[a] |
| 1 11a |
|
|
13a 82% |
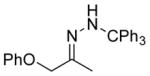
| 2 11b |
|
|
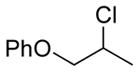
13b 85%[b] |
| 3 11c |
|
|
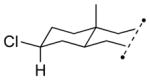
13c 57% (3.4:1) |
| 4 11d |
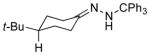
|
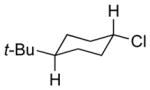
|
13d 69% (2.9:1) |
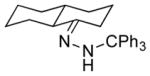
| 5 11e |
|
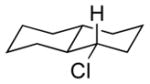
|
13e 71% (1.3:1) |
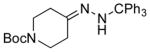
| 6 11f |
|
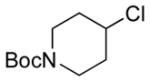
|
13f 56%[b] |
| 7 11g |
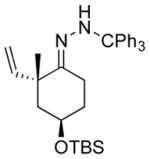
|
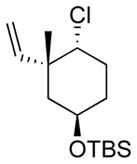
|
13g 50%[c] |
| 8 11h |
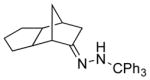
|
|
13h 83% (21:1) |
| 9 11i |
|
|
13i 70% (2.6:1) |
| 10 11j |
|
|
13j 67%[d] |
Isolated yields; diastereomeric ratio (dr) determined by 1H NMR of purified chlorides and indicated in parentheses.
NMR yield.
Isolated yield for diastereomer shown (major), 2.8:1 crude dr.
Lithiated hydrazone treated with dichloramine-T.
The capability of the reductive chlorination procedure was examined in a range of substrates, as shown in Table 1. The hydrazone of phenoxyacetone was converted to the corresponding chloride in 85% yield (entry 2). Diastereoselectivity in the reduction event displayed high substrate dependence. Substrates in which the hydrazone was part of a conformationally-locked six-membered ring favored axial hydrogen abstraction to give the equatorial chloride. Reductive chlorination of 11c and 11d gave a mixture of chlorides, favoring the equatorial chlorides by ca. 3:1 (entries 3 and 4). Diminished selectivity was observed for the reaction of the hydrazone of trans-1-decalone, wherein the three 1,3-diaxial interactions may disfavor axial hydrogen abstraction (entry 5). The neopentyl, homoallylic hydrazone 11g, comprising the cyclohexane core of welwitindolinone B, gave a 2.8:1 mixture of chloride diastereomers, from which the major component (13g) was isolated in 50% yield. Notably, this reductive chlorination occurs without 1,2-migration of the vinyl group, possibly reflecting the radical stabilizing effect of chlorine.[16]
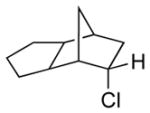
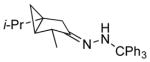
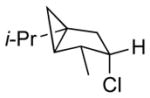
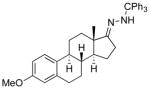
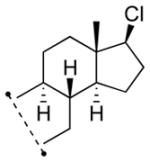
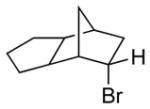
Among cyclopentanone-derived trityl hydrazones, the facial bias of the [2.2.1]-bridged system in 11h engendered high selectivity for the endo chloride 13h, isolated in 83% yield, with greater than 20:1 diastereoselectivity. The hydrazone of (−)-α-thujone (11i) gave a mixture of chlorides 13i in 70% yield, wherein hydrogen abstraction had taken place predominantly from the face opposite the α-methyl substituent.[17] The utility of this method was further demonstrated by the reductive chlorination of sterically encumbered hydrazone 11j, derived from O-Me estrone. The lower reactivity of hydrazone 11j to chlorination necessitated deprotonation followed by chlorination, achieved efficiently with dichloramine-T. The protocol afforded chloride 13j in good yield and high selectivity for the β-chloride shown, with hydrogen abstraction taking place anti to the adjacent C-13 methyl group.
The underlying concept of the reductive chlorination appeared transferrable to bromination. Thus, upon treatment of hydrazone 11a with N-bromosuccinimide (NBS) in place of t-BuOCl, followed by addition of EtSH and warming, it was converted to the expected reductive bromination product 16a (Table 2). Three other hydrazones were similarly subjected to the bromination conditions and gave the anticipated alkyl bromides in good yields. Of note, hydrogen abstraction by the α-bromocarbinyl radicals gave consistently lower diastereoselectivities than that observed for their α-chloro congeners, with apparent selectivity reversal in the bromination of trans-1-decalone (16e). The effect of the different halides on selectivity appears to parallel that observed for other free radical processes, including halogenations of alkanes. The slightly higher selectivity seen for reductive chlorination vs bromination comports with greater stabilization of the radical accorded by chlorine over bromine, as reflected by C-H bond dissociation energies of simple haloalkanes.[18]
Table 2.
Reductive bromination of trityl hydrazones.

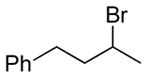
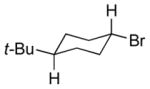
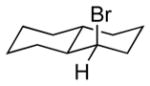
NBS solubilized in THF.
Isolated yields.
Diastereomeric ratio (dr) determined by 1H NMR of purified bromides.
In summary, we have disclosed a novel method for the conversion of ketones to the respective saturated alkyl chlorides. The key step involves chlorination of a ketone trityl hydrazone, which upon warming fragments to give an α-halo-stabilized carbinyl radical that is then reduced by EtSH to furnish the alkyl chloride product. The method is effective with a range of trityl hydrazones, and affords chloride products with stereoselectivities that may complement those available through ionic processes. The basic transformation was also successfully demonstrated for the synthesis of alkyl bromides.[19] The ability of this method to efficiently construct neopentyl alkyl chlorides from the carbonyl functional group provides a distinct tactic for the preparation of such halide-containing natural products, syntheses of which are of ongoing interest in our laboratories.
Supplementary Material
Acknowledgments
Financial support from the National Cancer Institute of the NIH (R01 CA101438) is gratefully acknowledged. Additionally, we generously thank Dr. Antoni Jurkiewicz for his NMR expertise and assistance.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.a) Bhat V, Rawal VH. Chem Commun. 2011;47:9705–9707. doi: 10.1039/c1cc13498a. [DOI] [PubMed] [Google Scholar]; b) Bhat V, MacKay JA, Rawal VH. Tetrahedron. 2011;67:10097–10104. doi: 10.1016/j.tet.2011.09.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Both Shea and Garg have noted similar challenges in converting an alcohol to the chloride. See: Cleary L, Pitzen J, Brailsford JA, Shea KJ. Org Lett. 2014;16:4460–4463. doi: 10.1021/ol5020043.Weires NA, Styduhar ED, Baker EL, Garg NK. J Am Chem Soc. 2014;136:14710–14713. doi: 10.1021/ja5087672.
- 3.a) Gorthey LA, Vairamani M, Djerassi C. J Org Chem. 1985;50:4173–4182. [Google Scholar]; b) Albone KS, Macmillan J, Pitt AR, Willis CL. Tetrahedron. 1986;42:3203–3214. [Google Scholar]; c) Snyder DC. J Org Chem. 1995;60:2638–2639. [Google Scholar]; d) Korakas P, Chaffee S, Shotwell JB, Duque P, Wood JL. Proc Natl Acad Sci USA. 2004;101:12054–12057. doi: 10.1073/pnas.0402274101. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Pluempanupat W, Chavasiri W. Tetrahedron Lett. 2006;47:6821–6823. [Google Scholar]
- 4.Gribble GW. Acc Chem Res. 1998;31:141–152.Gribble GW. Heterocycles. 2012;84:157–207.Acutumine S, King M, Herzon SB. Alkaloids Chem Biol. 2014;73:161–222. doi: 10.1016/B978-0-12-411565-1.00003-2.Chlorosulpholipids: Bedke DK, Vanderwal CD. Nat Prod Rep. 2011;28:15–25. doi: 10.1039/c0np00044b.Rhodomelaceae: Wang B-G, Gloer JB, Ji N-Y, Zhao J-C. Chem Rev. 2013;113:3632–3685. doi: 10.1021/cr9002215.
- 5.Synthesis of alkyl chlorides from vinyl halides: Iwasaki K, Wan KK, Oppedisano A, Crossley SWM, Shenvi RA. J Am Chem Soc. 2014;136:1300–1303. doi: 10.1021/ja412342g.King SM, Ma X, Herzon SB. J Am Chem Soc. 2014;136:6884–6887. doi: 10.1021/ja502885c.Lo JC, Gui J, Yabe Y, Pan CM, Baran PS. Nature. 2014;516:343–348. doi: 10.1038/nature14006.
- 6.For general reviews of hydrazone chemistry: Rogen BA, El-Awa A. “Hydrazine,” e-EROS Encyclopedia of Reagents for Organic Synthesis. 2014:1–7.Chamberlin AR, Sheppeck JE, Goess B, Lee C. “p-Toluenesulfonylhydrazide,” e-EROS Encyclopedia of Reagents for Organic Synthesis. 2007:1–11.Robinson B. Chem Rev. 1963;63:373–401.Robinson B. Chem Rev. 1969;69:227–250. doi: 10.1021/cr60262a003.
- 7.Baldwin has employed lithiated trityl hydrazones as umpoled synthons: Baldwin JE, Bottaro JC, Kolhe JN, Adlington RM. J Chem Soc, Chem Commun. 1984:22–23.Baldwin JE, Adlington RM, Bottaro JC, Jain AU, Kolhe JN, Perry MWD, Newington IM. J Chem Soc, Chem Commun. 1984:1095–1096.Baldwin JE, Adlington RM, Newington IM. J Chem Soc, Chem Commun. 1986:176–178.Baldwin JE, Adlington RM, Bottaro JC, Kolhe JN, Newington IM, Perry MWD. Tetrahedron. 1986;42:4235–4246.
- 8.Trityl hydrazides have been shown to provide entry to acyl radicals: Bath S, Laso NM, Lopez-Ruiz H, Quiclet-Sire B, Zard SZ. Chem Commun. 2003:204–205. doi: 10.1039/b210764c.
- 9.a) Moon MW. J Org Chem. 1972;37:383–385. [Google Scholar]; b) Moon MW. J Org Chem. 1972;37:386–390. [Google Scholar]; c) Wyman JM, Jochum S, Brewer M. Synth Commun. 2008;38:3623–3630. [Google Scholar]
- 10.Synthesis of vinyl halides from hydrazone precursors: Barton DHR, O’Brien RE, Sternhell S. J Chem Soc. 1962:470–476.Mori H, Wada S, Tsuneda K. Chem Pharm Bull. 1963;11:684–685. doi: 10.1248/cpb.11.1413.Mori H, Tsuneda K. Chem Pharm Bull. 1963;11:1413–1417. doi: 10.1248/cpb.11.1413.From N-silylhydrazones: Furrow ME, Myers AG. J Am Chem Soc. 2004;126:5436–5445. doi: 10.1021/ja049694s.
- 11.Reductive chlorinations: Brewer M. Tetrahedron Lett. 2006;47:7731–7733.Mundal DA, Lee JJ, Thomson RJ. J Am Chem Soc. 2008;130:1148–1149. doi: 10.1021/ja7101967.
- 12.Please see Supporting Information.
- 13.(a) Among the by-products were triphenylmethane and ethyl trityl sulphide. For a recent review regarding thiyl radicals, including the use of thiols as H-atom donors, see: Dénès F, Pichowicz M, Povie G, Renaud P. Chem Rev. 2014;114:2587–2693. doi: 10.1021/cr400441m.
- 14.Please see Supporting Information for selected 1H NMR spectra (Figure S1).
- 15.Studies by Malament and co-workers regarding stereospecific 1,4-additions of Cl2 gas to ketazines have shown that thermolyses (25 °C, 30 days) or photolyses (10 hours) of the resulting α, α′-dichloroazoalkanes in the presence of thiophenol produces significant quantities of radical coupling product relative to alkyl chloride: Malament DS, McBride JM. J Am Chem Soc. 1970;92:4586–4593.Nissim L, Malament DS. Israel J Chem. 1974;12:925–936.Levi N, Malament DS. J Chem Soc, Perkin Trans. 1976;2:1249–1256.
- 16.Selected references regarding homoallyl-homoallyl rearrangements: Castaing M, Pereyre M, Ratier M, Blum PM, Davies AG. J Chem Soc Perkin Trans. 1979;2:287–292.Beckwith ALJ, O’Shea DM. Tetrahedron Lett. 1986;27:4525–4528.Stork G, Mook R. Tetrahedron Lett. 1986:4529–4532.Toyota M, Wada T, Fukumoto K, Ihara M. J Am Chem Soc. 1998;120:4916–4925.Toyota M, Yokota M, Ihara M. Tetrahedron Lett. 1999;40:1551–1554.As an undesired side reaction: Chu-Moyer MY, Danishefsky SJ, Schulte GK. J Am Chem Soc. 1994;116:11213–11228.
- 17.The NOESY spectrum of 13i suggests that the stereochemistry of these chlorides has been misassigned in the following reference: Rees JC, Whittaker D. Org Magn Reson. 1981;15:363–369.
- 18.a) Furuyama S, Golden DM, Benson SW. J Am Chem Soc. 1969;91:7564–7569. [Google Scholar]; b) Tschuikow-Roux E, Paddison S. Int J Chem Kinet. 1987;19:15–24. [Google Scholar]; c) Tschuikow-Roux E, Salomon DR. J Phys Chem. 1987;91:699–702. [Google Scholar]; d) Miyokawa K, Tschuikow-Roux E. J Phys Chem. 1990;94:715–717. [Google Scholar]
- 19.In preliminary studies, we have found that lithiation of benzylacetone trityl hydrazone 11a followed by treatment with NFSI generated the corresponding fluoride in 33% yield (NMR, unoptimized).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.