

SUMMARY
Cataract, a clouding of the eye lens from protein precipitation, affects millions of people every year. The lens proteins, the crystallins, show extensive post-translational modifications (PTMs) in cataractous lenses. The most common PTMs, deamidation and oxidation, promote crystallin aggregation; however, it is not clear precisely how these PTMs contribute to crystallin insolubilization. Here, we report six crystal structures of the lens protein, γS-crystallin (γS): one of the wild-type and five of deamidated γS variants, from three to nine deamidation sites, after sample aging. The deamidation mutations do not change the overall fold of γS; however, increasing deamidation leads to accelerated disulfide bond formation. Addition of deamidated sites progressively destabilized protein structure and the deamidated variants display an increased propensity for aggregation. These results suggest the deamidated variants are useful as models for accelerated aging; the structural changes observed provide support for redox activity of γS-crystallin in the lens.
Graphical Abstract
eTOC Blurb
To mimic the accumulation of deleterious post-translational modifications in the eye lens, Norton-Baker et al. investigate a series of cataract-related variants of the lens protein γS-crystallin with progressively more deamidation sites. Increased disulfide bonding and aggregation suggest that both surface charge and increased dynamics impact cataract formation by deamidated crystallins.
INTRODUCTION
The eye lens is a unique environment in the human body, hosting some of the most long-lived proteins at extremely high concentrations (Andley, 2007). In order to establish and maintain transparency, the lens fiber cells lose much of their cellular machinery during early development via a combination of autophagy and lipase-mediated destruction of organelles (Morishita and Mizushima, 2016; Morishita et al., 2021). In adulthood, the lens undergoes very little protein turnover (Lynnerup et al., 2008). Crystallins are the predominant protein species in the lens, comprising over 90% of the dry mass and reaching concentrations above 400 mg/mL (Bloemendal et al., 2004; Vendra et al., 2016; Wistow, G. J.; Piatigorsky, 1988). Crystallins were originally characterized by their transparency and presence in the “crystalline lens,” and are divided into two superfamilies, α-crystallins and βγ-crystallins (Slingsby et al., 2013). The α-crystallins are related to small heat shock proteins and serve as molecular chaperones that offset aggregation (Horwitz, 1992). The βγ-crystallins are considered structural proteins; they form short-range ordered arrangements and increase the refractive index of the lens, focusing light onto the retina (Serebryany and King, 2014). In order to maintain lens transparency, crystallins need to remain stable over the entire human lifetime. During aging, however, modifications accumulate in the eye lens proteins from exposure to reactive oxygen species, ultraviolet light, and lens contamination with non-native molecules and metal ions (Rocha et al., 2021). Due to the presumed loss of structural stability concomitant with these modifications, eye lens proteins aggregate and form large, light-scattering masses in the lens, leading to cataract. Cataract is a leading cause of blindness worldwide, especially in middle- and low-income countries, where surgical intervention is often less accessible (Lee and Afshari, 2017; Liu et al., 2017).
Post-translational modifications (PTMs) of crystallins in the form of deamidation, oxidation, methylation, and truncation have all been observed in aged lenses, with deamidation reported to be one of the most prevalent (Dasari et al., 2009; Lampi et al., 1998; Srivastava et al., 2017; Wilmarth et al., 2006). Deamidation replaces an amide with a carboxylic acid, transforming glutamine or asparagine to glutamic acid/iso-glutamic acid or aspartic acid/iso-aspartic acid, respectively, with the possibility of racemization at each site (Geiger and Clarke, 1987; Lampi et al., 2014). To date, studies have focused on the Asp and Glu products, rather than any isomers, epimers, or crosslinked products. Although these additional products have been detected in long-lived proteins in the lens and brain (Friedrich et al., 2021a, 2021b; Fujii et al., 2016; Serra et al., 2016; Tao and Julian, 2014), detection techniques are still under development and studies on these variants are impeded due to the difficulty of producing proteins with these modifications. Even for surface-exposed residues, deamidation can lead to changes in conformational dynamics and an increase in aggregation propensity (Flaugh et al., 2006; Forsythe et al., 2019; Lampi et al., 2001; Pande et al., 2015; Ray et al., 2016; Takata et al., 2008; Vetter et al., 2020). Additionally, PTMs appear to be interdependent; Vetter et al. found that increased deamidation results in an increase in aggregation induced by oxidation. (Vetter et al., 2020). It is hypothesized that an increase in conformational flexibility in structurally destabilized proteins can lead to solvent exposure of key modifiable residues. Deamidation and disulfide crosslinking are both reported with higher prevalence in cataractous lenses than normal lenses (Hanson et al., 1998; Hooi et al., 2012; Lapko et al., 2002; Takemoto and Boyle, 2000; Wilmarth et al., 2006).
Deamidation has been studied in both β- and γ-crystallins. In β-crystallins, deamidation appeared to alter the dimer structure and led to a less compact fold in βB1 and βA3 (Lampi et al., 2001; Takata et al., 2007). Similar results were found with βB2, with the dimer destabilized by deamidation (Lampi et al., 2006). In the γ-crystallins, γD was destabilized by deamidations at the interface between the two domains of the monomer, although the prevalence of naturally-occurring deamidation at these buried sites in vivo is not clear. (Flaugh et al., 2006; Hanson et al., 1998; Searle et al., 2005). However, a number of single-site deamidation variants of surface asparagines on γD did not show any significant changes in structure or stability (Guseman et al., 2021). In contrast, deamidated variants of γS have been reported to have attractive protein-protein interactions, leading to increased propensity for dimerization and aggregation (Forsythe et al., 2019; Pande et al., 2015; Ray et al., 2016; Vetter et al., 2020). Although Vetter et al. and Pande et al. investigated 3- and 4-site deamidation variants (Pande et al., 2015; Vetter et al., 2020), respectively, most of the previous studies have focused on crystallin variants with only one or two deamidation sites. This study explores the effects of progressive accumulation of deamidations, evaluating both the moderate deamidation that likely occurs in highly aged lenses as well as more extreme deamidation as a means of assessing the tolerance of γS to major surface charge modification.
Due to the long-lived nature of the crystallins, the lens requires mechanisms of protection and repair to cope with decades of stress. Anti-oxidant compounds, notably glutathione, play vital roles in balancing the redox state of the lens (Michael and Bron, 2011). Crystallin proteins themselves also display adaptations that aid in longevity. Although they have historically been considered purely structural proteins, it has recently been suggested that members of the βγ-crystallin family have chemical functionality in metal binding and disulfide exchange in the lens (Roskamp et al., 2020; Serebryany et al., 2018). In particular, γS-crystallin (Figure 1A), with its high cysteine content compared to other γ-crystallins, has been proposed to fulfill an oxidoreductase-like role (Roskamp et al., 2020). γS-crystallin contains a cysteine tetrad (C23, C25, C27, C83) in its N-terminal domain (Figure 1B), with C23, C25, and C27 exhibiting unusually high solvent exposure. Solvent-exposed cysteines would appear to be disadvantageous for a long-lived protein. Indeed, the dimer of γS-crystallin, formed by the intermolecular disulfide linkage of C25 and C25 of two γS-crystallin monomers, is less stable and more aggregation-prone than the monomer (Thorn et al., 2019). The functional advantage of these solvent-accessible cysteines in γS-crystallin is still under investigation, although they have been demonstrated to play a critical role in metal interactions. A number of divalent cations, including Zn2+, Ni2+, Co2+ and Cu2+, cause aggregation of γS, and the mutation of C23/25/27 dramatically affects this metal-induced aggregation. For Zn2+, Ni2+, Co2+, the mutation of these cysteines to serines prevented aggregation (Roskamp et al., 2019). However, for Cu2+, the removal of C23/25/27 increased the susceptibility of γS to Cu2+-induced oxidation and aggregation, suggesting that these residues buffer against Cu2+-induced oxidative damage (Roskamp et al., 2019, 2020). Similarly, the cysteines of γD-crystallin have also been implicated in metal-induced aggregation (Fernández-Silva et al., 2020; Quintanar et al., 2016). Furthermore, similar solvent-exposed cysteines in γD-crystallin have been shown to undergo dynamic disulfide exchange, transferring disulfide bonds in an oxidoreductase-like role (Serebryany et al., 2018).
Figure 1. Sites of deamidation of γS-crystallin.
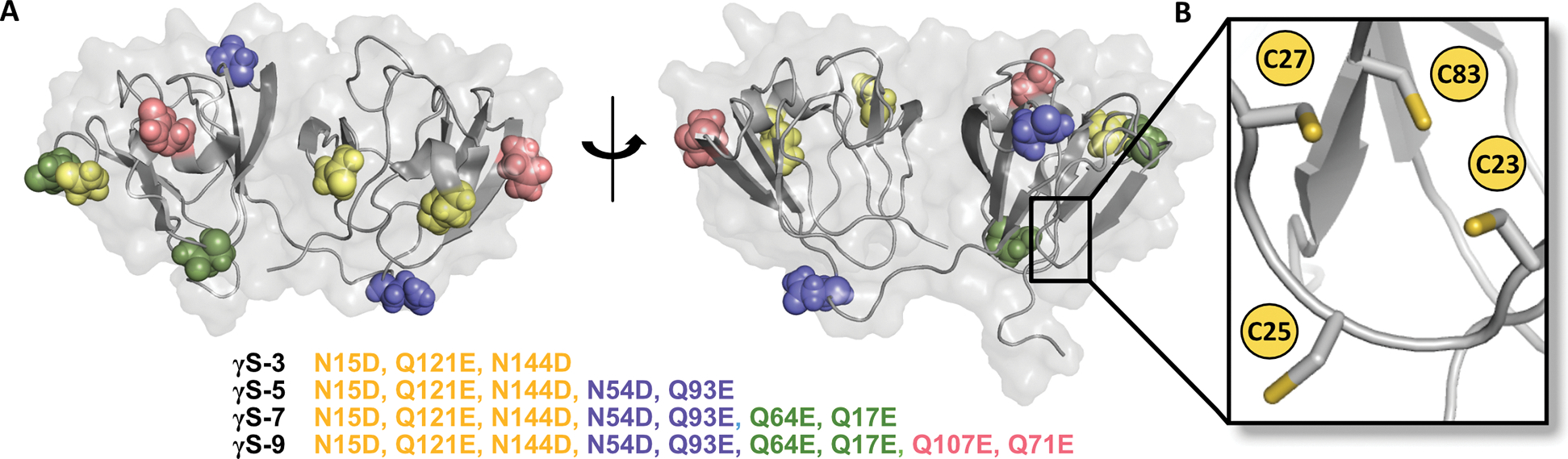
(A) Sites of mutagenesis in the 3-site (γS-3, yellow), 5-site (γS-5, blue), 7-site (γS-7, green), and 9-site deamidation variants (γS-9, pink). (Structure from PDB: 2M3T) (B) Magnified view of the cysteine tetrad located in the N-terminal domain.
In this study, we investigated the relationship between deamidation and disulfide bond formation in γS-crystallin. Crystallins have been shown to collect multiple PTMs; in order to mimic this natural aging process on a compressed timescale, we expressed four deamidated γS-crystallin variants that emulate moderate-to-extreme examples of deamidation, with 3, 5, 7, and 9 sites of deamidation at surface residues. (Figure 1A). With each addition of more deamidated sites, the variants demonstrated lower structural stability. Furthermore, the deamidated variants showed an increased aggregation propensity compared to wild-type γS-crystallin (γS-wt). Estimation of the second osmotic virial coefficient (A2) from small angle X-ray scattering data showed an increase in repulsive interaction forces with increasing deamidation, suggesting aggregation propensity is governed by competing effects from hydrophobic exposure during unfolding and electrostatic repulsion. Crystal structures were determined for the full-length, monomeric γS-wt as well as the deamidated variants at different time points in sample age. Although the deamidation mutations did not cause significant changes to the overall protein fold, these structures revealed that increased deamidation does lead to an increased propensity for disulfide bond formation, indicating accelerated oxidative aging compared to the wild type. An intramolecular disulfide bond between C23 and C27 formed first, with a second dimerizing bond at C25 occurring in increasingly aged samples. Additionally, the structural changes after sample aging give evidence of the role of γS-crystallin as a mediator of oxidation in the eye lens, exchanging deleterious cross-linking disulfide bonds for stable internal disulfide bonds.
RESULTS
γS deamidated variants are less stable and more aggregation prone than wild-type
We investigated the stability and aggregation propensity of the deamidated variants compared to γS wild-type (γS-wt). Thermal unfolding curves were determined by differential scanning fluorimetry (DSF), and the midpoint of the unfolding temperatures (Tm) were calculated for each variant (Figure 2A). The percent unfolded was calculated via fitting of the averaged fluorescence intensity data (Figure S1) to a Boltzmann function as described by Wright et al. (Wright et al., 2017). The thermal stability progressively decreased with cumulative deamidations from a Tm of 76.79 ± 0.06 °C for γS-wt to 73.91 ± 0.07 °C, 72.26 ± 0.01 °C, 71.62 ± 0.02 °C and 70.45 ± 0.04 °C for γS-3, γS-5, γS-7, and γS-9, respectively (Figure 2B). A linear relationship emerges from the comparison of the Tm and the calculated net charge at neutral pH for each variant, with increased charge leading to decreased stability in these variants. Calculations of net charge were performed with the Prot pi protein tool (https://www.protpi.ch/Calculator/ProteinTool).
Figure 2, related to Figure S1. Stability and thermal aggregation of γS wild-type and each variant.
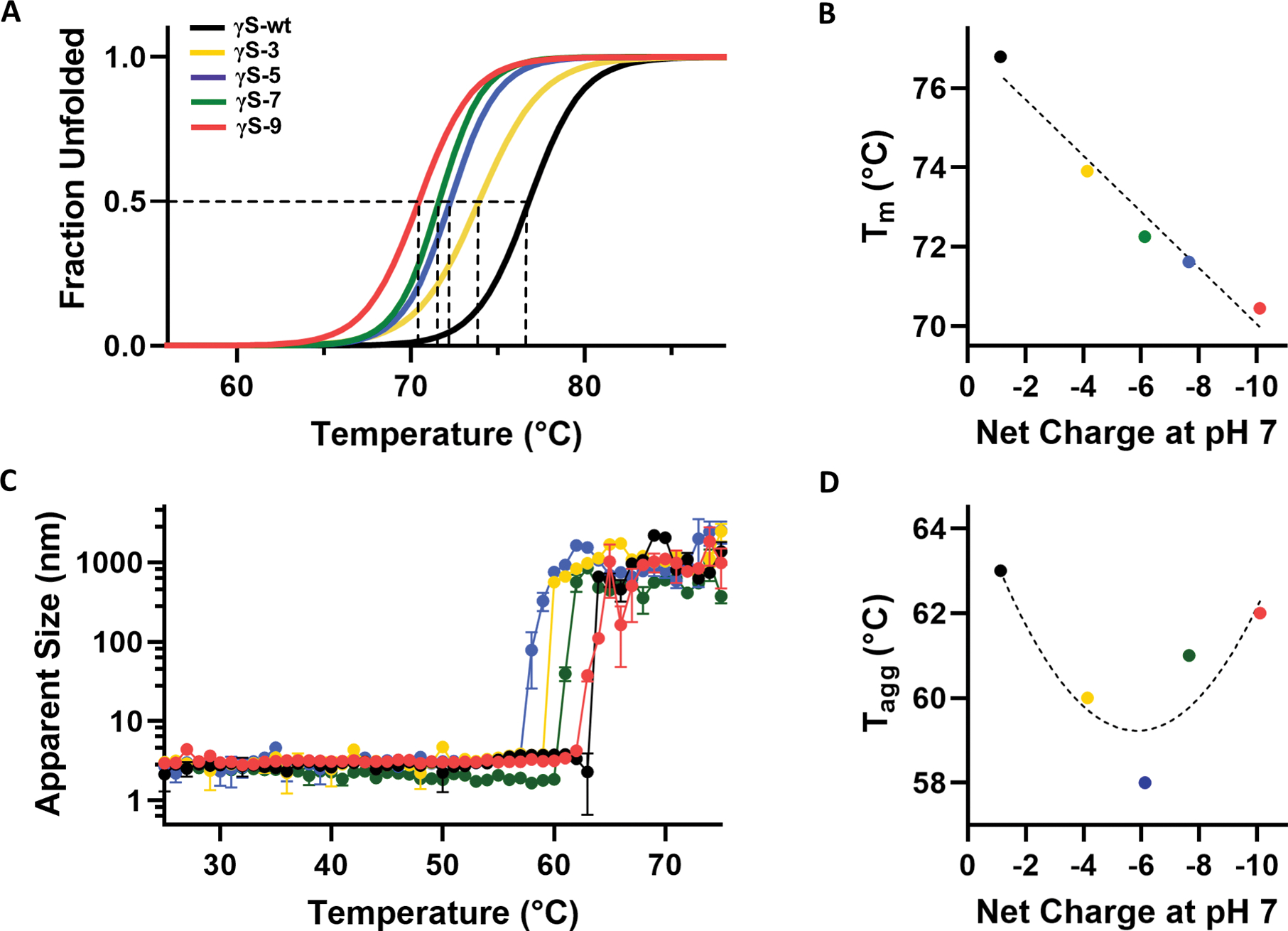
(A) Differential scanning fluorimetry (DSF) was used to determine the percent unfolded as a function of temperature for γS-wt (black), γS-3 (yellow), γS-5 (blue), γS-7 (green), and γS-9 (pink). (B) The midpoint temperature of the thermal unfolding (Tm) of γS-wt and each variant plotted against the net charge of the protein at neutral pH. (C) Dynamic light scattering (DLS) measurements of γS-wt and each variant to monitor thermally induced aggregation of γS wild-type and each variant. Measurements were performed in triplicate. Data are represented as mean ± SD. (D) The temperature of aggregation onset (Tagg) plotted against the net charge of the protein at neutral pH.
Thermally induced aggregation was monitored with dynamic light scattering (DLS). All proteins appeared monomeric at the starting temperature of 25 °C with diameters of 2–4 nm and remained monomeric until the rapid formation of large, insoluble aggregates at the aggregation onset temperature, Tagg (Figure 2C). The values of Tagg were 63 °C for γS-wt and 60 °C, 58 °C, 61 °C, and 62 °C for γS-3, γS-5, γS-7, and γS-9, respectively. All deamidated variants showed a lower Tagg compared to γS-wt; however, while Tagg for γS-3 and γS-5 decreases with successive deamidations, for γS-7 and γS-9, the trend reverses (Figure 2D). We hypothesized that the repulsive effects from the high net charge of γS-7 and γS-9 begin to offset the minor destabilization produced by deamidations, resisting aggregation.
Increasing repulsive interactions from deamidation
In order to assess interprotein interactions, we aimed to determine the osmotic second virial coefficient (A2). A2 characterizes the resultant force of individual interparticle interactions and has been used as a predictive measure for protein solubility, crystallization, aggregation, and liquid-liquid phase separation (Qin and Zhou, 2019). We used small angle X-ray scattering (SAXS), which yields information on protein size and shape (form factor) as well as interprotein interactions (structure factor) (Table 1). The SAXS profiles show that the overall fold of the protein is maintained among the variants (Figure 3A). The form factor stays consistent among variants as well as among concentrations, with the profile showing no shifts from concentration effects up to 40 mg/mL (Figure S2). However, at low scattering vectors, q, deviations from ideality are seen that reflect the interprotein interactions (Figure 3B). The Guinier plot for an ideal, non-interacting particle is linear at low q. Deviations from linearity are observed to be positive for the γS-wt and γS-3 and negative for γS-7 and γS-9. Solving for the structure factor at q=0 as a function of concentration allows for the calculation of A2 from the slope of the linear regression (Figure 3C). We found in the low ionic strength buffer used in these studies, the A2 values for γS-wt and γS-3 were in the attractive regime at −0.59 and −0.56 ×10−4 mol mL g−2, within the estimated error of each other. For γS-5, the value approaches ideality, with a value of +0.15 ×10−4 mol mL g−2. For γS-7 and γS-9, repulsive forces are observed, with positive A2 values of +1.45 and +2.40 ×10−4 mol mL g−2, respectively (Figure 3C). Despite the increased repulsive forces, thermally-induced aggregation (Figure 2D) was still enhanced for all deamidated variants, indicating aggregation propensity is influenced by both protein-protein interactions and structure destabilization (Figure 2B).
Table 1.
SAXS Data Collection and Structural Parameters
| γS-wt | γS-3 | γS-5 | γS-7 | γS-9 | |
|---|---|---|---|---|---|
|
|
|||||
| Energy (keV) | 10 | 10 | 10 | 10 | 10 |
| Detector | PILATUS 2M | PILATUS 2M | PILATUS 2M | PILATUS 2M | PILATUS 2M |
| Detector distance (m) | 2.0 | 2.0 | 2.0 | 2.0 | 2.0 |
| q range (Å−1) | 0.0094 – 0.4354 | 0.0094 – 0.4354 | 0.0094 – 0.4354 | 0.0094 – 0.4354 | 0.0094 – 0.4354 |
| Exposure time (s) | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 |
| Frames | 33 | 33 | 33 | 33 | 33 |
| Temperature (°C) | 20 | 20 | 20 | 20 | 20 |
| Concentration (mg/mL) | 8.7 – 40.4 | 8.6 – 40.8 | 8.6 – 40.3 | 15.8 – 37.8 | 17.1 – 41.0 |
| Rg (Å) from Guinier of lowest concentration | 18.5 ± 0.2 | 18.7 ± 0.6 | 18.1 ± 0.4 | 17.4 ± 0.3 | 15.0 ± 0.3 |
Figure 3, related to Figure S2. Small angle x-ray scattering (SAXS) analysis to determine A2.
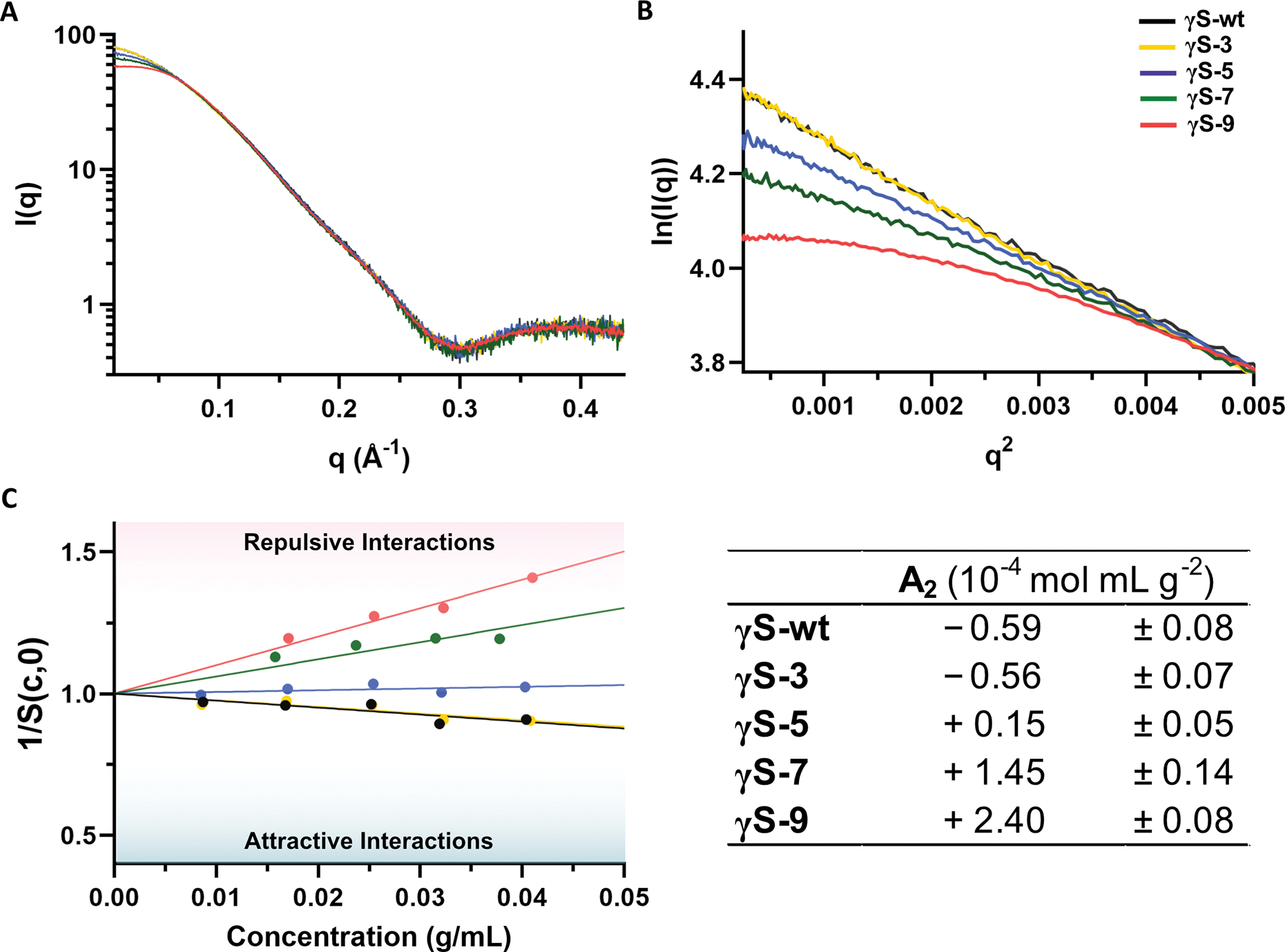
(A) SAXS profiles for γS-wt (black), γS-3 (yellow), γS-5 (blue), γS-7 (green), and γS-9 (pink) at approximately 40 mg/mL. (B) Guinier plot of the scattering profiles of each variant at approximately 40 mg/mL. For both (A) and (B), the profile for γS-7 was scaled to normalize for concentration. (C) Left: The reciprocal of the structure factor at q = 0 as a function of concentration. Right: Second virial coefficient (A2) values calculated from the slope of the linear regressions.
Crystal structure of monomeric wild-type γS-crystallin
We report the first crystal structure of full-length, monomeric wild-type human γS-crystallin. Descriptive and crystallographic parameters for the structures discussed in this paper are summarized in Table 2. The space group was found to be P21 with an asymmetric unit containing two γS-crystallin monomers. In contrast to the previously reported disulfide-bonded dimeric structure (Thorn et al., 2019), these monomers interact only through non-covalent contacts between the N-terminal domains. The structure was solved at a resolution of 2.0 Å and the coordinates have been deposited in the PDB (PDB ID: 7N36). As with other reported γS-crystallin structures, it shows two highly symmetric domains comprising the two double Greek key motifs characteristic of the βγ-crystallins (Figure 4A). Structural alignments of the Cα atoms show this novel structure has high similarity with both the γS-wt NMR structure (PDB: 2M3T) and the crystal structure of the C-terminal domain of γS-wt (PDB: 1HA4), with RMSDs of 1.78 and 1.03 Å, respectively. These RMSDs were derived by the alignment of chain A of 7N36 with the entire 2M3T structure using the lowest energy state in the ensemble and chain A of 1HA4 (Kingsley et al., 2013; Purkiss et al., 2002). The regions of structural difference between 2M3T and 7N36 are mostly localized to the C-terminal domain, with the loop containing K154 and K155 as well as the linker region between the domains showing the maximum difference (Figure S3). Notably, in all the crystal structures reported here, these are regions that show extensive dynamics, with high B-factors and/or multiple conformations. Likewise, in comparison to disulfide dimerized γS-crystallin structure (PDB: 6FD8), we observe very little structural difference in protein fold, consistent with the analysis by Thorn et al., with a Cα RMSD of 0.59 Å, derived from the alignment of chain A of 7N36 and chain A of 6FD8. In both the NMR structure (2M3T) and this reported structure, no disulfide bonds, either intra- or intermolecular, are observed (Figure 4B).
Table 2.
X-Ray Data Collection and Refinement Statistics
| γS-wt | γS-3 | γS-5 | γS-7 | γS-9A | γS-9B | |
|---|---|---|---|---|---|---|
|
| ||||||
| PDB | 7N36 | 7N37 | 7N38 | 7N39 | 7N3A | 7N3B |
| Wavelength (Å) | 0.9762 | 0.9763 | 0.9763 | 0.9763 | 0.9762 | 0.9763 |
| Resolution range | 46.89 – 2.00 | 27.62 – 1.30 | 39.90 – 1.22 | 40.27 – 1.56 | 43.74 – 1.50 | 58.87 – 2.09 |
| (2.07 – 2.00) | (1.35 – 1.30) | (1.27 – 1.22) | (1.62 – 1.56) | (1.55 – 1.50) | (2.17 – 2.09) | |
| Space group | P21 | P212121 | P212121 | P22121 | P21 | P22121 |
| Unit cell | ||||||
| a, b, c (Å) | 52.8, 60.6, 56.0 | 29.5, 62.6, 79.6 | 29.3, 62.7, 79.8 | 49.2, 76.6, 94.7 | 29.8, 57.7, 44.9 | 48.7, 75.8, 93.4 |
| α, β, γ (°) | 90.0, 117.5, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 103.3, 90.0 | 90.0, 90.0, 90.0 |
| Total reflections | 146421 (14703) | 473244 (46020) | 528084 (20734) | 682592 (67123) | 160193 (16077) | 270089 (26342) |
| Unique reflections | 20807 (2056) | 36161 (3477) | 41328 (2350) | 51461 (5051) | 23517 (2292) | 20969 (2008) |
| Multiplicity | 7.0 (7.2) | 13.1 (13.2) | 12.8 (8.8) | 13.3 (13.3) | 6.8 (6.9) | 12.9 (13.1) |
| Completeness (%) | 97.42 (97.07) | 97.57 (95.18) | 93.52 (54.14) | 99.61 (99.00) | 98.40 (97.03) | 99.73 (98.14) |
| Mean I/sigma(I) | 16.29 (2.37) | 27.64 (2.00) | 30.24 (2.32) | 27.17 (1.76) | 13.85 (1.24) | 13.98 (1.43) |
| Wilson B-factor | 39.6 | 17.0 | 15.6 | 26.2 | 25.4 | 39.0 |
| Rmerge | 0.06225 (0.7879) | 0.04061 (1.275) | 0.03872 (0.8968) | 0.04674 (1.609) | 0.05397 (1.855) | 0.1236 (1.642) |
| Rmeas | 0.06731 (0.8493) | 0.04234 (1.325) | 0.04035 (0.9517) | 0.04869 (1.674) | 0.05855 (2.004) | 0.1288 (1.708) |
| Rpim | 0.02531 (0.3145) | 0.01175 (0.3582) | 0.01117 (0.3052) | 0.01345 (0.4546) | 0.02239 (0.752) | 0.03573 (0.4662) |
| CC½ | 0.999 (0.782) | 1.000 (0.795) | 1.000 (0.715) | 0.999 (0.753) | 0.999 (0.734) | 0.998 (0.674) |
| CC* | 1.000 (0.937) | 1.000 (0.941) | 1.000 (0.913) | 1.000 (0.927) | 1.000 (0.920) | 1.000 (0.898) |
|
| ||||||
| Reflections used in refinement | 20797 (2056) | 36160 (3478) | 41328 (2348) | 51460 (5048) | 23388 (2286) | 20969 (2008) |
| Reflections used for R-free | 962 (101) | 1811 (172) | 2077 (108) | 2650 (256) | 1147 (108) | 1083 (99) |
| Rwork | 0.1891 (0.2478) | 0.1451 (0.2398) | 0.1464 (0.2219) | 0.1934 (0.3036) | 0.1953 (0.4619) | 0.1884 (0.2879) |
| Rfree | 0.2199 (0.3105) | 0.1793 (0.3169) | 0.1792 (0.2817) | 0.2127 (0.3241) | 0.2237 (0.5012) | 0.2396 (0.3641) |
| Number of nonhydrogen atoms | 3030 | 1731 | 1830 | 3442 | 1578 | 3058 |
| macromolecules | 2912 | 1521 | 1588 | 3130 | 1468 | 2935 |
| ligands | - | 29 | 15 | 5 | - | - |
| solvent | 118 | 181 | 227 | 307 | 110 | 123 |
| Protein residues | 348 | 174 | 174 | 348 | 173 | 348 |
| RMS bonds (Å) | 0.006 | 0.014 | 0.010 | 0.006 | 0.008 | 0.007 |
| RMS angles (°) | 0.88 | 1.30 | 1.07 | 0.81 | 0.99 | 0.94 |
| Ramachandran | ||||||
| favored (%) | 96.8 | 98.84 | 98.26 | 96.51 | 95.91 | 95.06 |
| allowed (%) | 3.20 | 1.16 | 1.74 | 3.49 | 3.51 | 3.49 |
| outliers (%) | 0 | 0 | 0 | 0 | 0.58 | 1.45 |
| Rotamer outliers (%) | 0.98 | 1.83 | 1.76 | 3.02 | 3.21 | 3.25 |
| Clashscore | 5.67 | 3.62 | 5.43 | 4.26 | 6.00 | 6.90 |
| Average B-factor | 43.59 | 24.73 | 20.92 | 35.75 | 36.02 | 45.70 |
| macromolecules | 43.52 | 23.09 | 19.06 | 34.81 | 35.41 | 45.69 |
| ligands | - | 54.04 | 29.52 | 72.73 | - | - |
| solvent | 45.35 | 33.90 | 33.35 | 44.68 | 44.25 | 46.06 |
Figure 4, related to Figure S3 and S7. Crystal structures and electron density maps of the cysteine loop for aged γS wild-type and each variant.
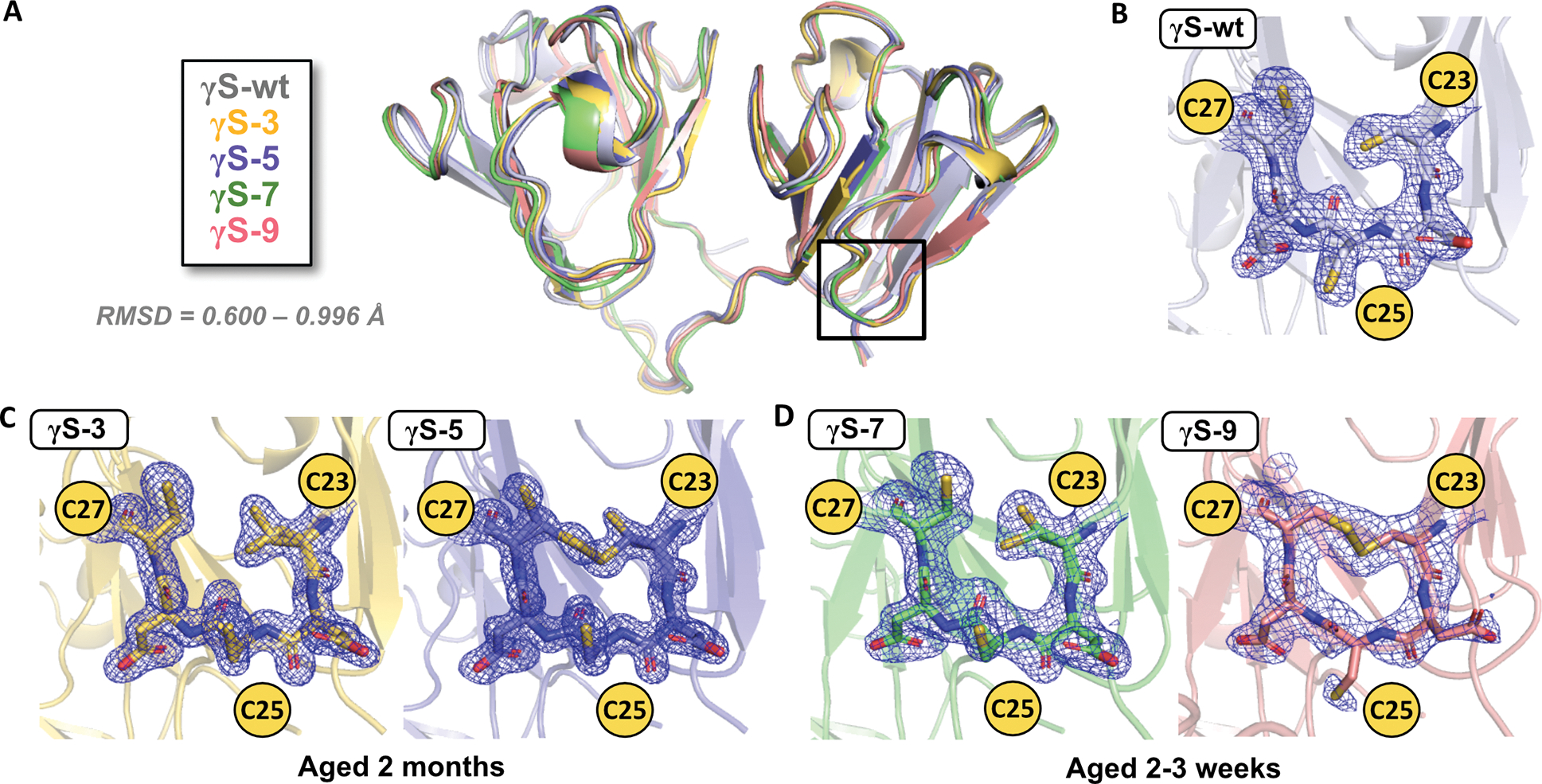
(A) Alignment of the structures of γS-wt (PDB: 7N36, gray), γS-3 (PDB: 7N37, yellow), γS-5 (PDB: 7N38, blue), γS-7 (PDB: 7N39, green), and γS-9B (PDB: 7N3B, pink). All structures have Cα RMSDs less than 1 Å compared to γS-wt, PDB: 7N36. Magnified views of the cysteine loop region with 2FO-FC (contoured at 1σ) electron density maps for (B) γS-wt aged 1.5 months, (C) γS-3 and γS-5 aged 2 months and (D) γS-7 and γS-9B aged 2–3 weeks.
Crystal structures of γS-crystallin deamidated variants
Five crystal structures have been determined for deamidated variants of γS-crystallin at various stages of sample aging. Sample age is the combined time of storage at 4 °C after resolubilization and time spent in the crystallization plate until the crystals were frozen for diffraction data collection. All samples were reduced with dithiothreitol (DTT) to establish a fully reduced starting point prior to aging and crystallization. We have confirmed with Ellman’s assay, mass spectrometry, and size-exclusion chromatography that this treatment reduces the disulfide bonds in all variants (data not shown). Given the short half-life of DTT (Stevens et al., 1959), we suspect very little reducing agent remains active during crystallization after several weeks, allowing the progression of cysteine oxidation. The γS deamidation variants crystallized in a variety of space groups (Table 2). The 3-site deamidation variant (γS-3) and the 5-site deamidation variant (γS-5), both aged for 2 months and grown in the same crystallization solution, were found in space group P212121 with a single monomer in the asymmetric unit. The structures for γS-3 and γS-5 were solved at resolution of 1.30 and 1.22 Å, respectively. The refined structures have been deposited in the PDB (PDB ID: 7N37 and 7N38). In both structures, a Mg2+ ion was identified, although in different locations. There were no direct interactions between the Mg2+ and any residues; interactions occurred through hydrogen bonding with Mg2+-coordinated waters. γS-crystallin has shown binding and aggregation in the presence of other divalent cations, including Cu2+, Zn2+, Ni2+, and Co2+, but shows no such interactions with Mg2+ (Roskamp et al., 2019, 2020).
The structures for γS-7 and γS-9B, both aged for 2 – 3 weeks and grown in similar crystallization conditions to each other, were solved at a resolution of 1.56 and 2.09 Å, respectively. They were both found to be in space group P22121 with two monomers in the asymmetric unit. The refined structures have been deposited in the PDB (PDB ID: 7N39 and 7N3B). An additional structure was solved of γS-9 from protein aged for 4 – 5 days (γS-9A). γS-9A crystallized in space group P21 with one monomer in the asymmetric unit and was solved at a resolution of 1.50 Å (PDB ID: 7N3A). In general, the deamidated variants grew crystals more readily, in multiple different conditions in the sparse screens, and more reproducibly than γS-wt. Other mutations in crystallins have been shown to enhance crystallization, notably R58H and R36S in γD-crystallin, which lead to the formation of crystal cataract (Pande et al., 2001).
γS-crystallin deamidation is not significantly detrimental to protein fold
Despite the enhanced crystallization propensity of the variants, the fold of γS-crystallin is remarkably resistant to change after extensive deamidation. Relative to the new γS-wt crystal structure (PDB: 7N36), the backbone fold is maintained in all deamidation variants, even for the highly deamidated γS-9. The Cα RMSD relative to the wild-type structure does not exceed 1 Å for any deamidated variant (Figure 4A). Additionally, the intramolecular disulfide bond observed in the γS-9B crystal structure does not seem to significantly affect protein fold, with a Cα RMSD of 1.19 Å in comparison to chain A of γS-9B to the γS-9A structure.
Deamidated γS-crystallin variants have a higher propensity for disulfide formation
Comparisons of different crystal structures can be challenging, as many factors related to crystallization or diffraction data collection, rather than the protein structure, can have substantial effects. Additionally, the rate of oxidation may be affected by the crystallization conditions. To this end, we limit our direct comparisons of crystal structures to γS-3 vs. γS-5 and γS-7 vs. γS-9B. These structures were determined from protein samples that were grown in identical or highly similar crystallization conditions and formed crystals in the same space group, with similar unit cells, and of comparable sizes. The data collection strategies were kept uniform so that radiation dose would also be consistent between samples.
Comparing crystal structures from samples aged for similar periods, we observe an increase in disulfide bond formation in variants with more deamidation. After aging for 2 months, the γS-3 structure has fully reduced cysteines (Figure 4C). In contrast, the γS-5 structure shows a mixed state for C23 and C27, with both reduced and disulfide-bonded species present (Figure 4C). Occupancy refinement in Phenix suggests that the disulfide-bonded configuration accounts for ~50–70%, with a range estimated due to the different occupancy calculation for the two cysteines, C23 and C27. The same trend is observed in samples of γS-7 and γS-9B aged 2–3 weeks. The γS-7 structure shows the cysteines to be fully reduced, while the γS-9B structure shows that C23 and C27 are fully oxidized, forming an intramolecular disulfide bond (Figure 4D). The fourth cysteine in the tetrad, C83, is not shown in these maps, but in all cases, it appears to be fully reduced. This may indicate that C83 is not amenable to disulfide bonding or that the C83 disulfide-bonded structure is unfavorable for crystallization, possibly due to large structural disruption from this bond.
To support these observations of the crystal structures, we also conducted a biochemical assay to evaluate free thiol content over the course of sample aging at room temperature. Ellman’s assay is a colorimetric method used to estimate sulfhydryl content in a sample using 5,5-dithio-bis-(2-nitrobenzoic acid) (DTNB). DTNB reacts with free thiols and releases TNB, which absorbs at 412 nm. In order to get a full understanding of the thiol content, including buried cysteines, the samples were denatured with 2% SDS prior to reaction with DTNB. The extinction coefficient of TNB in the buffer used was calibrated using DTT and was calculated to be 12968 M−1cm−1 (Figure S4). The rate of the reaction differed for each deamidation variant, with more deamidated sites leading to longer reaction times (Figure S5). All samples showed initial thiol content after purification was between 6.2 – 6.6 free thiols per protein molecule, consistent with the known 7 cysteines in γS (Figure S6). Measurements of thiol content were then collected over 18 days (Figure 5A) of aging at room temperature. In the first two days of aging, the free thiol content of the samples decreased more rapidly with more deamidated sites, with the thiol content decreasing to 98%, 96%, 93%, 92% and 84%, for γS-wt, γS-3, γS-5, γS-7, and γS-9, respectively. After the first 6 days of aging at room temperature, the rate of thiol loss slowed. After 18 days, the oxidation of the samples reached 60–70% of their starting thiol content. It should be noted that the rate of oxidation was observed to depend significantly on buffer aeration. Samples stored in degassed buffer after size-exclusion chromatography showed very little oxidation over several months, while samples stored in dialysis in stirring buffer oxidized extremely rapidly. In order to age samples in an equivalent manner, during purification the samples for these experiments were reduced with DTT, the DTT was removed via buffer exchange, then the samples were lyophilized. The lyophilized powders were resuspended in buffer from the same stock, achieving the same aeration for all samples.
Figure 5, related to Figure S5 and S6. Cysteine oxidation and dimerization/aggregation of aged samples.
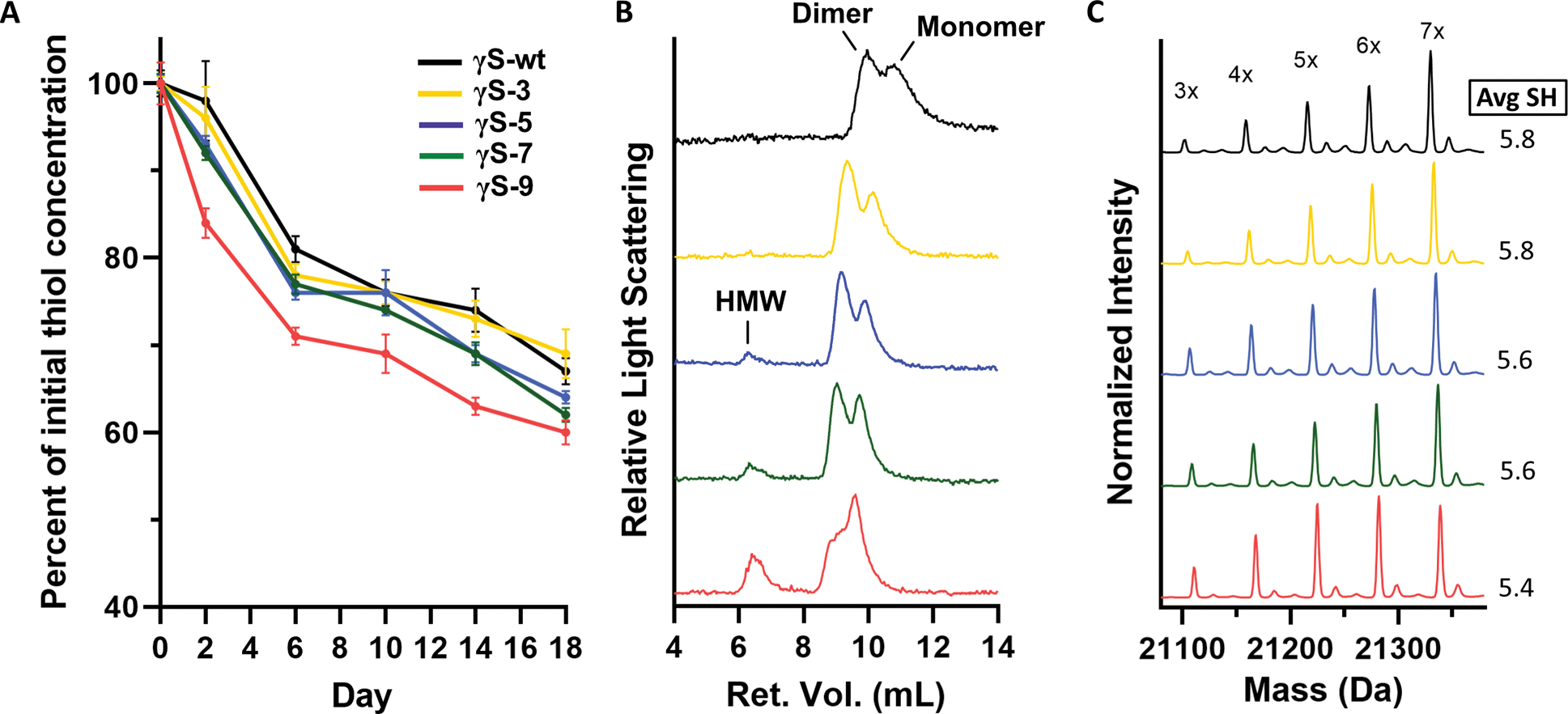
(A) Assessment of the decrease in free thiol content over time using Ellman’s assay for γS-wt (black), γS-3 (yellow), γS-5 (blue), γS-7 (green), and γS-9 (pink). Measurements were performed in triplicate. Data are represented as mean ± SD. (B) Size-exclusion chromatography in line with multiangle light scattering (SEC-MALS) conducted on each variant after aging for 1 month. High molecular weight (HMW) peaks are seen in γS-5, γS-7, γS-9. (C) Intact mass spectra of each aged variant after reaction with iodoacetamide (IAM) and subsequent reduction with dithiothreitol (DTT). The average free thiol content per protein molecule weighted by the peak intensity decreases with increasing deamidation.
The decrease in free thiol content as evaluated by Ellman’s assay may arise from the oxidation of cysteines, but also from the protein aggregation reducing the accessibility of thiol groups. We therefore conducted size-exclusion chromatography multi-angle light scattering (SEC-MALS) experiments to evaluate the formation of disulfide-bonded dimers as well as the formation of high molecular weight (HMW) aggregates. Figure 5B shows the results of SEC-MALS studies on γS-wt and the deamidation variants after aging for 1 month at room temperature. All samples contained both monomer and dimer, with the γS-3 and γS-5 appearing to form more dimer than γS-wt. These data suggest that deamidation promotes the formation of dimers. These dimers were confirmed to be disulfide bonded, as they were reduced by treatment with DTT (data not shown). However, for the γS-7 and γS-9, less dimer is present than in the γS-3 and γS-5 samples. Dimerization may be inhibited by their higher surface charge and the repulsive forces between molecules. Additionally, the dimer form of γS-wt is known to be less stable (Thorn et al., 2019), therefore the already deamidation-destabilized γS-7 and γS-9 may form HMW oligomers/aggregates upon dimerization. The increased propensity of deamidated variants to aggregation induced by oxidation has previously been shown (Vetter et al., 2020). We also detected HMW peaks in the γS-5, γS-7, γS-9 (Figure 5B). For γS-9, the SEC-MALS experiment also shows a shoulder of the monomer peak blending into the dimer peak, unlike the other traces. We hypothesize that this peak shape arises from a partially unfolded state.
We next aimed for a more detailed understanding of the disulfide state of aged samples. Samples aged for 1.5 months were denatured in 8 M urea to solubilize HMW aggregates. The denatured samples were then reacted with iodoacetamide (IAM), which modifies free thiol groups and imparts a mass shift of +57 for each addition. The samples were subsequently treated with DTT to reduce the dimer and any internal disulfide bonds. Figure 5C shows the deconvoluted intact mass spectra for aged γS-wt and the deamidated variants. All samples show a distribution of IAM mass additions, from 3 up to 7 additions, with 7 being the total number of cysteines present in γS. As IAM adds to free thiols and not to disulfide-bonded sulfurs, the number of IAM additions indicates the number of thiols per protein molecule. A calculation of the weighted average for each variant reveals a decrease in the number of thiols with increasing deamidation, with 5.8, 5.8, 5.6, 5.6, and 5.4 for γS-wt, γS-3, γS-5, γS-7, and γS-9, respectively.
Disulfide bonding in the cysteine loop during sample aging
Because no significant structural disruption to the protein fold is observed in the deamidated variants, we propose that they may serve as more crystallizable models for the wild-type protein to gain atomic and near-atomic resolution structures at varying time points. Structural changes over time in the deamidated variants may give insight into the aging behavior of the wild-type protein, but in an accelerated timeframe. In this study, γS-9 was crystallized at two time points of sample age: several days and several weeks. Recently, we reported another crystal structure of γS-9 that was aged for several months prior to crystallization in a silicon chip. That structure was solved via a fixed-target serial crystallographic approach using microcrystals to a resolution of 3.0 Å (referred to here as γS-9C, PDB: 7NJE) (Norton-Baker et al., 2021). The comparison of these three structures allows us to observe the change in cysteine oxidation over time (Figure 6). In the earliest time point structure, γS-9A, the protein is monomeric and all three cysteines appear to be mostly reduced, with minor partial occupancy of a C23-C27 disulfide bond (Figure 6A). After several weeks, as discussed above, γS-9B has a full intramolecular disulfide between C23 and C27 and the protein remains monomeric (Figure 6B). After several months, γS-9C contains a second disulfide bond between C25 and C25 of another γS-9, forming a dimer (Figure 6C). Interestingly, the orientation of C25 shifts after the formation of the C23-C27 disulfide bond. In γS-9A, the side chain of C25 projects out from the ring and has a high calculated solvent exposed surface area (SASA) of 85 Å2. Similarly, γS-wt shows fully reduced cysteines with a SASA of 82 Å2 calculated for C25. In γS-9B, after the formation of the C23-C27 disulfide, C25 flips and the SASA is lowered to 30 Å2. SASA values were calculated by AREAIMOL in the CCP4 package (Lee and Richards, 1971; Winn et al., 2011).
Figure 6. Disulfide bond formation in the cysteine loop over the course of aging.
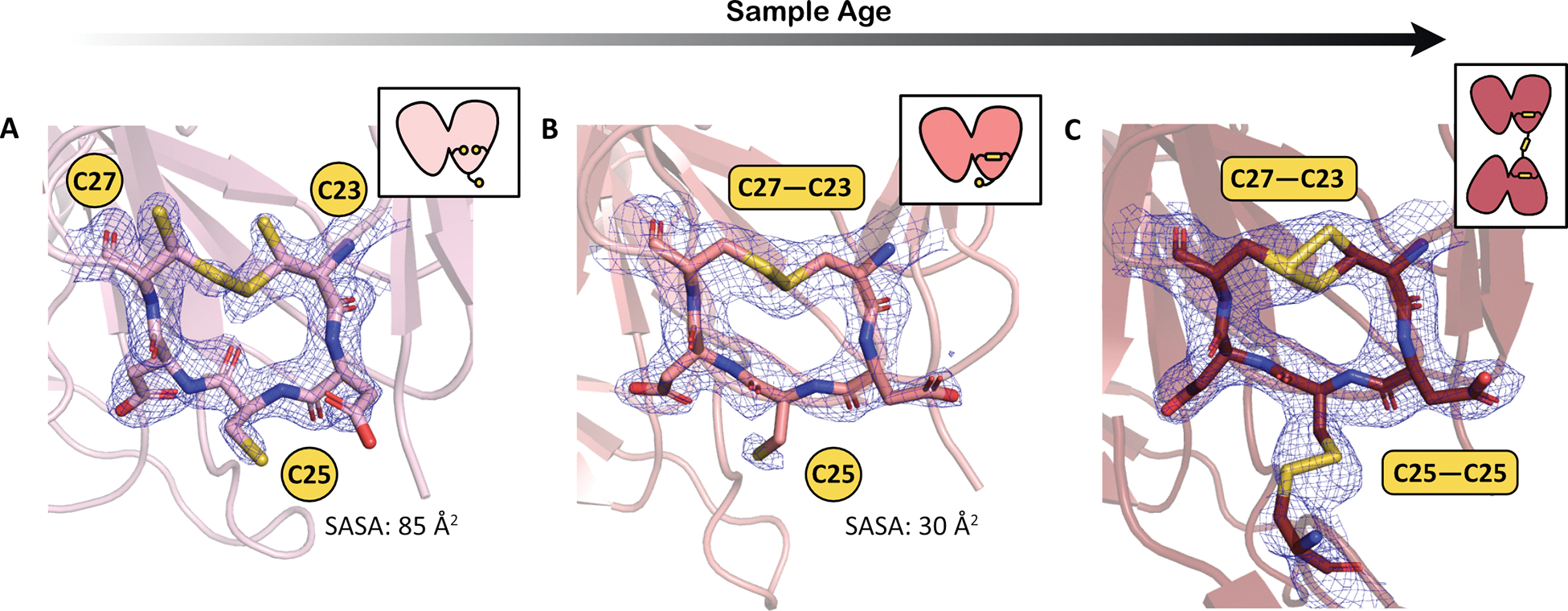
Magnified views of the cysteine loop region with 2FO-FC (contoured at 1σ) electron density maps for (A) γS-9 aged for several days (γS-9A, PDB: 7N3A), (B) γS-9 aged for several weeks (γS-9B, PDB: 7N3B), and (C) γS-9 aged for several months (γS-9C, PDB: 7NJE). Insets show a depiction of the γS-9 structures indicating their oligomeric states (either monomeric or dimeric) with intramolecular and intermolecular disulfide bonds in the cysteine loop shown. The solvent accessible surface area (SASA) for C25 is lower after disulfide bond formation between C23 and C27.
DISCUSSION
Deamidation accelerates oxidative aging
In comparing structures of samples of γS-wt and deamidation variants aged for time periods ranging from several days to several months, we observe an increased propensity for disulfide bonding with increased deamidation. The duration of sample aging of γS-wt, γS-3 and γS-5 was similar, between 1.5 – 2 months; however, only γS-5 shows partial occupancy of disulfide bonded C23-C27. At an earlier aging timepoint, γS-7 and γS-9 can be compared after several weeks aging; γS-7 remains fully reduced, while γS-9 shows a full disulfide bond between C23-C27. Based on this trend, we propose that deamidation accelerates the process of oxidative aging, possibly by increasing conformational flexibility, which then increases solvent exposure of the cysteine residues. Additional experiments using Ellman’s assay and mass spectrometry after thiol blocking with iodoacetamide (IAM) on aged samples also supported the trend of increased oxidation with increased deamidation (Figure 5). Other studies have linked deamidation with an increase in conformational dynamics and oxidation, even in regions distant from the sites of deamidation. Forsythe et al. showed that deamidation of two surface Asn residues in the N-terminal domain of γS caused long-range disruption of conformational dynamics in the C-terminal domain (Forsythe et al., 2019). Their work suggested that deamidation induced both local and global transient unfolding and that thermally-induced aggregation was enhanced for the deamidation variants in the presence of oxidized glutathione. Vetter et al. followed up on this study, demonstrating that with three deamidation mutations, γS again became more susceptible to oxidation-induced aggregation (Vetter et al., 2020). These previous studies and the results described here support allosteric conformational effects that destabilize protein structure and increase conformational dynamics, leading to increased susceptibility to oxidation.
The mutated residues (glutamine to glutamic acid and asparagine to aspartic acid) are structurally highly similar; therefore, the difference in charge after mutation is the likely basis for the altered structural dynamics. Upon deamidation, a negative charge is introduced at each deamidated site as the carboxylic acid moiety is ionized at the relevant pH values. These charges not only lower the pI of the protein significantly, but also induce regions of high negative charge on the surface of the protein (Figure S7). At neutral pH, γS-wt has an expected net charge of −1. With each additional deamidation mutation, the net charge decreases, reaching −10 for γS-9. A complex picture emerges from the evaluation of the benefits and drawbacks of introducing negative charges. Chong et al. proposed that additional negative charges increase the solubility for proteins that already have a net negative charge (Chong and Ham, 2014). However, the introduction of charged sites can also lead to an increase in fluctuating conformational transitions, or transient misfolding, which can drive aggregation through exposure of hydrophobic portions of the protein (Chong and Ham, 2015). Particularly for γ-crystallins, the drastic changes to surface charge might cause further disruption in the lens via the impairment of α-crystallin chaperone activity, as demonstrated previously for a deamidated variant of γS (Vetter et al., 2020).
Our findings support the hypothesis of the competing roles of net charge and conformational dynamics in governing aggregation propensity. As the deamidated variants gained negative charge, their structural stability decreased, as indicated by the observed unfolding temperatures (Figure 2B). Lower stability likely promotes transient misfolding, prompting an earlier onset of thermally induced aggregation in moderately charged variants (γS-3 and γS-5); however, as the proteins reached high negative charge (γS-7 and γS-9), the greater electrostatic repulsion, as evidenced by their A2 values, between molecules raises the energic barrier to aggregation (Figure 2D). Further studies into the local and global dynamic effects of charged residues are warranted.
γS serves as an oxidation sink in the lens
Previously, disulfide bonding in γS was thought to be solely a detrimental byproduct of oxidation. Recent work and the results of this study suggest that it may also be a functional adaptation that helps to sustain the redox balance in the lens (Roskamp et al., 2020; Serebryany et al., 2018). Here, we demonstrate that deamidation accelerates the oxidation of the C23, C25, and C27. Due to the unchanged global fold across the variants characterized in this study, we propose that we may extrapolate the behavior of the deamidated variants to the wild-type protein observed over a longer time period.
We suggest that the positioning of a highly solvent-exposed cysteine, C25, may be advantageous in γS due to its proximity to other cysteines that can form a stable internal disulfide bond. A possible mechanism for γS oxidoreductase activity emerges from the observations of the crystal structures determined in this study (Figure 7). Fully reduced, monomeric γS (1) contains the highly solvent-exposed C25, predisposing it to dimerization with a second molecule of γS. Although this schematic diagrams the proposed in vitro process with only γS available for crosslinking, in the lens this may also occur with a different crystallin species. We propose a transient dimer (2) forms with a crosslinking disulfide bond between C25 and a C25 of another crystallin monomer. We hypothesize this intermediate with only a single dimerizing disulfide forms preferentially due to the high solvent exposure of C25 and its known propensity for dimerization at this site, but suggest that this intermediate is quickly lost to disulfide exchange. This dimerizing bond exchanges with other nearby cysteines, forming an intramolecular disulfide bond between C23-C27 (3). This process removes a crosslinking disulfide bond and releases a recovered, reduced monomer of crystallin. After the formation of the C23-C27 bond, C25 occupies an orientation with reduced solvent exposure, as evidenced by the crystal structure of γS-9B. Further oxidation eventually leads to the formation of a fully oxidized and aggregation-prone dimer (Thorn et al., 2019), but this conversion may be slowed by the reduced solvent exposure of C25. The C23-C27 bond is apparently not highly disruptive to the protein fold in this region, indicating that this bond might serve to rescue γS or other crystallins from other, more deleterious disulfide bonds. If this hypothesis is correct, γS plays not only a structural role, but also a chemical role in the lens, acting as a final buffer against oxidative damage, even after depletion of glutathione.
Figure 7. Potential mechanism for the role of γS-crystallin as an oxidation sink in the eye lens.
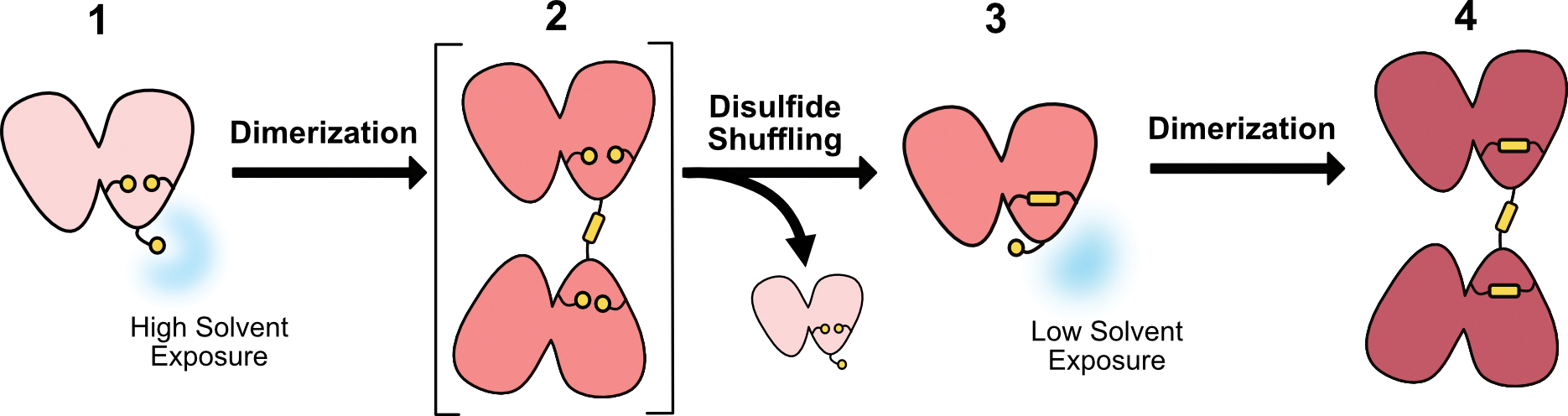
Monomeric, reduced γS-crystallin (1) with a highly solvent exposed C25 accepts an intermolecular disulfide bond from another γS-crystallin or another oxidized crystallin in the eye lens. This partially oxidized dimer (2) undergoes disulfide shuffling and the disulfide bond exchanges to C23-C27 forming an intramolecular disulfide bonded monomer (3) and releasing a recovered, reduced crystallin monomer. The reduced solvent exposure of C25 in the single disulfide bond intermediate (3) slows the formation of the fully oxidized disulfide-bonded dimer (4) that has been shown to be prone to aggregation.
In other proteins with disulfide reactivity, a CXXC motif has been reported, notably in thioredoxins and disulfide isomerases. The XX is often proline and glycine, both of which support a tight turn and the formation of a disulfide between the two cysteines (Oka et al., 2015). The same activity is recapitulated with the CXC motif, where X is usually glycine (Wilkinson and Gilbert, 2004). The activity of a CXC motif has been previously reported for human γD-crystallin, in a study showing disulfide exchange between wild-type γD and an aggregation prone variant, W42Q (Serebryany et al., 2018). In this case, X is serine, a highly flexible amino acid that is often present in flexible linker regions (Huang and Nau, 2003; Van Rosmalen et al., 2017); however, the disulfide bond was still postulated to cause conformational strain in the C-terminal domain. Upon disulfide exchange between the wild-type and W42Q variant, γS-W42Q was more destabilized by the disulfide and more rapidly aggregated out of solution (Serebryany et al., 2018). γS contains a similar, extended CXCXC motif. However, instead of glycine or proline that allow for tight turns, here the cysteines are separated by aspartic acid residues. As the structures reported here show, the favored disulfide forms between the first and last C in the motif (C23 and C27); the 3 separating residues likely allow for a less strained disulfide bond formation. Aspartic acid is, however, among the most flexible amino acids (Huang and Nau, 2003), lessening any structural disruption as well as supporting disulfide exchange between C25 and C23/27.
The presence of aspartic acid in this motif raises additional questions; is there a fitness benefit from proximal acidic residues? Repulsive electrostatic interactions between deprotonated cysteine and neighboring negatively charged residues have been suggested to destabilize the thiolate state and thereby raise cysteine pKa (Awoonor-Williams and Rowley, 2016). As thiolate is a much stronger nucleophile than the protonated thiol, an increase in cysteine pKa would likely be inhibitory to thiol-disulfide exchange (Bulaj et al., 1998; Nagy, 2013). Free cysteine in solution has a reported thiol pKa of 8.6 and most noncatalytic cysteine pKas fall within 7.4 – 9.1 (Awoonor-Williams and Rowley, 2016); however, dramatic shifts can been seen for catalytic cysteines, with pKa values as low as 2.88 reported (Pinitglang et al., 1997). The predicted pKa for C25 in γS is 9.35, calculated with PROPKA using the γS-wt crystal structure, 7N36 (Olsson et al., 2011; Søndergaard et al., 2011). C25 is therefore predicted to be slightly more basic than glutathione, the major antioxidant in the lens with a reported thiol pKa of 8.92 (Schafer and Buettner, 2001). In addition to similar pKa values, C25 in the CDCDC motif of γS and the structure of glutathione also share structural similarities. Glutathione is a tripeptide (L-γ-glutamyl-L-cysteinyl-glycine) with the amide bond formed with the γ-carboxyl rather than the α-carboxyl of glutamic acid. Like C25 in γS, the cysteine in glutathione is flanked by adjacent carboxylates. We plan to further investigate the role of γS and its interplay with glutathione in maintaining redox homeostasis in the eye lens.
Deamidation as a basis for rational mutagenesis for crystallization
On a more methodological note, deamidation may serve as a new target for rational mutagenesis to promote crystallization. Excellent work has been done in this area, with much of the effort focused on surface entropy reduction (SER) (Goldschmidt et al., 2007). In SER, surface residues with high conformational energy, such as Lys and Glu, are mutated to Ala or other small amino acids. This method is often successful but may sometimes lead to undesired structural changes. The deamidated variants reported here did not appear to change the overall fold of the protein but did more readily and reproducibly form crystals. For challenging crystallization targets, deamidation of surface Gln and Asn to Glu and Asp may provide less disruption to protein fold than other mutations, as the side chains are highly similar in size.
In conclusion, crystallins are among a unique class of extremely long-lived proteins (ELLPs). The half-lives of ELLPs far exceed those of average proteins, persisting for years or even the entire lifetime of an organism (Toyama et al., 2013). In metabolically inactive cells such as the eye lens, the cells’ resources to safeguard against protein degradation and aggregation are finite. Multiple and redundant mechanisms for maintaining lens transparency are likely necessary and the γ-crystallins appear to have a multidimensional role in the lens. γS-crystallin demonstrates resistance to significant structural change from deamidation mutations; however, these mutations predispose the protein to accumulate oxidative modifications. Furthermore, although glutathione is the main species responsible for maintaining redox balance in the lens, here we provide further evidence that γS-crystallin may also aid in reducing oxidized species by shuffling disulfide bonds internally to non-crosslinking positions. By this mechanism, γS may act as a last line of defense against oxidation. The proteins of the eye lens appear to employ methods to both counteract and exploit PTMs; the full role of crystallins and other lens molecules in slowing the irreversible aggregation that leads to cataract remains to be fully explored.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Rachel Martin (rwmartin@uci.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Coordinate data have been deposited at RCSB PDB and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bacterial and virus strains | ||
| Escherichia coli Rosetta(DE3) | Millipore Sigma | Cat #69450–3 |
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| HEPES | Fisher | BP310 |
| TRIS | Fisher | BP154 |
| NaCl | RPI | S23030 |
| NaN3 | Fisher | S227I |
| Dithiothreitol (DTT) | Fisher | R0861 |
| SYPRO orange dye | Sigma | S5692 |
| 5,5-dithiobis(2-nitrobenzoic acid) (DTNB) | Fisher | AC117540010 |
| Urea | Fisher | U15 |
| Ammonium bicarbonate | Fisher | A643 |
| Ammonium acetate | Fisher | A637 |
| Ammonium sulfate | Sigma | S5636 |
| Morpheus screen | Molecular Dimensions | MD1–124 |
| Hampton index screen | Hampton | HR2–134 |
| Critical commercial assays | ||
| Deposited data | ||
| Crystal structure of γS-wt | This paper | PDB: 7N36 |
| Crystal structure of γS-3 | This paper | PDB: 7N37 |
| Crystal structure of γS-5 | This paper | PDB: 7N38 |
| Crystal structure of γS-7 | This paper | PDB: 7N39 |
| Crystal structure of γS-9A | This paper | PDB: 7N3A |
| Crystal structure of γS-9B | This paper | PDB: 7N3B |
| NMR structure of γS-wt | (Kingsley et al., 2013) | PDB: 2M3T |
| Crystal structure of γS dimer | (Thorn et al., 2019) | PDB: 6FD8 |
| Crystal structure of γS C-terminal domain | (Purkiss et al., 2002) | PDB: 1HA4 |
| Crystal structure of γS-9C | (Norton-Baker et al., 2021) | PDB: 7NJE |
| Experimental models: Cell lines | ||
| Experimental models: Organisms/strains | ||
| Oligonucleotides | ||
| Recombinant DNA | ||
| pET28a(+) | (Brubaker et al., 2011) | N/A |
| Modifed pET28a(+) vectors | This paper | N/A |
| Software and algorithms | ||
| XDS | (Kabsch, 2010) | N/A |
| Phaser | (McCoy et al., 2007) | N/A |
| Phenix version 1.18.2 | (Liebschner et al., 2019) | N/A |
| Coot version 0.9.4 | (Emsley et al., 2010) | N/A |
| PyMol version 2.4.1 | Schrödinger | https://pymol.org/2/ |
| PROPKA | (Olsson et al., 2011) | propka.org |
| Prot pi | Roland Josuran, Zurich University of Applied Sciences | https://www.protpi.ch/Calculator/ProteinTool |
| Zetasizer | Malvern | N/A |
| OmniSEC | Malvern | N/A |
| Prism | GraphPad Software | http://www.graphpad.com/ |
| Other | ||
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial cell culture
Novagen Rosetta™(DE3) cells were transformed with plasmids encoding for γS-wt plasmid or the deamidation variants and used for protein expression in LB broth or autoinduction media (ZYM-5052) at 37 or 25 °C.
METHOD DETAILS
Expression and purification
Sites for deamidation were selected based on mass spectrometry data reported by Lapko et al. (Lapko et al., 2002), prioritizing sites with the highest reported incidence of deamidation and those with the highest calculated solvent-accessible surface area. Progressively more sites were mutated, starting with N15D, Q121E, N144D (3-site variant, γS-3), then N54D and Q93E (5-site variant, γS-5), then Q64E and Q17E (7-site variant, γS-7), and finally Q107E and Q71E (9-site variant, γS-9). Site-specific mutagenesis PCR was used to create the deamidated variants from the wild-type construct with an N-terminal 6x-His tag and a tobacco etch virus (TEV) protease cleavage sequence. The oligonucleotides used in the mutagenesis are listed in Table S1. Expression and purification of wild-type γS-crystallin (γS-wt) and the deamidated variants was performed as described previously (Brubaker et al., 2011). The constructs in pET28a(+) vector were used to transform Escherichia coli Rosetta (DE3) cells. Overexpression was achieved using Studier’s autoinduction method (ZYM-5052 media) for 1 h at 37 °C followed by 20−24 h at 25 °C incubation (Studier, 2005). The cells were lysed by sonication and the supernatant loaded onto a Ni-NTA column. An imidazole gradient was used to elute the tagged protein, then TEV protease (produced in-house) was added to cleave the 6x-His tag. Reapplication to the Ni-NTA column separated the TEV protease. The samples were reduced with 10 mM dithiothreitol (DTT), concentrated, and loaded on to a HiLoad 16/600 Superdex 75 pg column. The purified proteins were lyophilized for storage at −80 °C.
Differential scanning fluorimetry (DSF)
Differential scanning fluorimetry (DSF) analysis was performed in a Stratagene Mx3005P RT-PCR instrument (Agilent, Santa Clara, CA, USA) in thin-walled 96-well PCR plates with transparent Microseal® ‘B’ seals. The temperature ramp was between 25 °C and 94 °C with a 0.5 °C increase per cycle for 140 cycles and a cycle duration of 30 seconds. Each well contained 0.5 mg/ml protein and 2.5× SYPRO orange dye in 10 mM HEPES pH 7.0, 50 mM NaCl, 0.05% sodium azide. Midpoint of the unfolding temperatures (Tm) were calculated as described by Wright et al. (Wright et al., 2017). DSF data up to the fluorescence maxima were fitted in Prism (Graphpad) using a Boltzmann function:
Fmin is the minimum fluorescence value. Fmax is the maximum fluorescence value. T is the temperature. m characterizes the slope of the transition. The fraction of unfolded protein, Pu, was calculated as:
Dynamic light scattering (DLS)
Thermally-induced aggregation was assessed with dynamic light scattering (DLS) using a Zetasizer Nano-ZS (Malvern Analytical, Malvern, United Kingdom). Protein concentration was 1 mg/ml in 10 mM HEPES pH 7.0, 50 mM NaCl, 0.05% sodium azide. At each temperature, the sample was equilibrated for 120 seconds before the scattering measurements were performed in triplicate. Data was processed using the Zetasizer software. The autocorrelation function was used to derive the intensity of scattering as a function of particle size. A number distribution is then calculated to estimate the relative concentrations of particles of different sizes, accounting for their proportional light scattering. The reported apparent size is the number mean.
Small angle X-ray scattering (SAXS) data collection
Samples in 10 mM HEPES pH 7.0, 50 mM NaCl, 0.05% sodium azide, 2 mM DTT were concentrated to ~40 mg/mL in 0.5 mL Amicon-Ultra centrifugal concentrators and the flow through used as a buffer blank. A dilution series was prepared and the concentrations measured in triplicate on a Thermo Scientific™ NanoDrop™ OneC Microvolume UV-Vis Spectrophotometer (Fisher Scientific, Waltham, MA, USA). Data were collected at the SIBYLS beamline (12.3.1) at the Advanced Light Source in Berkeley, California using the high throughput mail-in SAXS service (Dyer et al., 2014). The concentration series for each protein was collected in triplicate. For each sample, a dose series was collected with a total exposure time of 10 s, framed at 0.3 s for a total for 33 frames. The sample oscillated during the exposure to reduce radiation damage. Data were collected using a PILATUS 2M detector. The monochromatic X-ray energy was 10 keV and detector distance was 2 m, resulting in scattering vectors, q, ranging from 0.0094 Å−1 to 0.4354 Å−1. Samples were at 20 °C during data collection. Data were initially processed as described (Hura et al., 2009). Additional processing was performed using BioXTAS RAW (Jeffries et al., 2016).
Calculation of the second virial coefficient (A2)
Static structure factor of a monodisperse solution at the origin S(c,0) related to the osmotic pressure Π is described by:
R is the gas constant 8.31 J mol−1 K−1. M is the molecular mass of the solute in g/mol. c is the concentration in g/mL. The osmotic pressure can be approximated by the series expansion:
For weakly interacting molecules at relatively low concentrations, 2MA2c ⋘ 1, the structure factor at the origin can be expressed as:
S(c,0) is evaluated by:
The slope of the linear fit from following gives 2MA2:
In an ideal solution, S(c,0) = 1. If A2 is positive, the osmotic pressure is higher and the interactions are repulsive, leading to particles that are evenly distributed. If A2 is negative, the osmotic pressure is lower and attractive interactions cause fluctuations in particle distribution (Koch et al., 2003; Vivarès and Bonneté, 2002).
Ellman’s assay
The lyophilized powders for γS-wt and the deamidation variants were reconstituted from the same stock of buffer and aged at room temperature stored out of direct light. Prior to each measurement, samples were vortexed and an aliquot was diluted in 2% SDS, 100 mM TRIS pH 8.0 to reach a final protein concentration 0.125 mg/mL and 5,5-dithiobis(2-nitrobenzoic acid) (DTNB) was added to reach a final concentration of 0.1 mM. Absorbance measurements were collected in an Epoch™ Microplate Spectrophotometer (BioTek, Winooski, VT, USA) at 412 nm in a 96-well plate. Each sample was made in triplicate and measurements taken in 5-minute intervals over 2 hours. The maximum value was used to calculate the thiol content. An extinction coefficient of 12968 M−1cm−1 was calculated from the calibration curve in 2% SDS, 100 mM TRIS pH 8.0 buffer using DTT as the thiol source (Figure S4) and used for all thiol concentration calculations.
Size-exclusion chromatography multi-angle light scattering (SEC-MALS)
SEC-MALS was performed on a Viskotek 305 TDA system (Malvern Analytical, Malvern, United Kingdom) equipped with a P2500 column. The running buffer used was 10 mM HEPES pH 7.0, 200 mM NaCl, 0.05% sodium azide.
Mass spectrometry
Proteins samples were denatured in 8 M urea, 1 M ammonium bicarbonate pH 8.0 for 1 h then iodoacetamide (IAM) was added to reach 50 mM. The reaction progressed at room temperature away from light for 2.5 h. DTT was added to reach 100 mM and incubated for 1 h. Mass spectra were collected on a Waters Xevo XS-QToF equipped with a phenyl column. Buffer A was 0.1% formic acid in water. Buffer B was 100% acetonitrile. For the protein intact mass spectra, the flow rate was 0.2 mL/min with a gradient of 0% to 97% B over 1.5 min then 97% B for 0.5 min. Spectra were processed using MassLynx. Deconvolution was performed by MaxEnt1.
γS-crystallin crystallization, data collection, and structure refinement
Crystallization conditions were found via sparse matrix screening. Crystallization occurred at 20 °C and the protein:precipitant ratio was 1:1. For all protein samples, the lyophilized powder was resolubilized and buffer exchanged into 50 mM HEPES pH 6.8 with 5 mM DTT. The addition of DTT allowed for a uniform starting condition for all samples with cysteines reduced. For γS-wt, γS-3, and γS-5, the samples were aged overnight at 4 °C before crystallization. For γS-wt, crystals were grown in the Morpheus screen G5 (0.1 M Carboxylic Acids Mix, 0.1 M Buffer System 2 pH 7.5, 30 % v/v Precipitant Mix 1). The Carboxylic Acids Mix contains 0.2 M sodium formate, 0.2 M ammonium acetate, 0.2 M trisodium citrate, 0.2 M sodium potassium l-tartrate, and 0.2 M sodium oxamate. Buffer system 3 contains 0.1 M MOPS/HEPES-Na pH 7.5. Precipitant Mix 1 contains 20% w/v PEG 20 000 and 40% v/v PEG MME 550 (Gorrec, 2009). Protein concentration was 12 mg/mL. Crystals appeared between 14–28 days and were harvested after 1.5 months. For γS-3 and γS-5, crystals were grown in the Hampton Index screen condition G10 (0.2 M MgCl2, 0.1 M BIS-TRIS pH 5.5, 25 % w/v Polyethylene glycol 3350). Protein concentration was 18 mg/mL. Crystals appeared between 14–28 days and were harvested after 2 months. For γS-7, the protein sample was aged at 4 °C for 1–2 weeks before being crystallized in 0.1 M sodium acetate pH 5.0, 0.1 M ammonium sulfate, 28% PEG 3350. Crystals appeared after 1 day and were harvested after 1 week. For γS-9A, the protein sample was aged overnight at 4 °C before being crystallized in 0.1 M sodium acetate pH 5.45, 21% PEG 3350. Crystals appeared after 1 day and were harvested after 2–3 days. For γS-9B, the protein sample was aged at 4 °C for 1–2 weeks before being crystallized in 0.1 M sodium acetate pH 5.45, 0.1 M ammonium sulfate, 21% PEG 3350. Crystals appeared after 1 day and were harvested after 1 week. Crystals were cryoprotected with Paratone-N and flash-frozen by plunging into liquid nitrogen. X-ray diffraction data were collected at both the P13 or P14 beamline operated by the European Molecular Biology Laboratory (EMBL) at the PETRA-III synchrotron at DESY, Hamburg, Germany (https://www.embl-hamburg.de/services/mx/P13/ or https://www.embl-hamburg.de/services/mx/P14/). Diffraction was recorded on a PILATUS 6M or EIGER 16M detector. Diffraction data was processed using XDS (Kabsch, 2010). Phasing was performed using Phaser molecular replacement with chain A of PDB model 6FD8 as a search model (McCoy et al., 2007). Refinement was performed using the Phenix software suite with iterative manual improvement in Coot (Emsley et al., 2010; Liebschner et al., 2019). Structure images were generated in PyMOL (Schrödinger).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details are reported in the figure legends and the method details. For DSF (Figure S1), DLS (Figure 2C), SAXS (Figure S2F), calibration for Ellman’s assay (Figure S4), and Ellman’s assay on aged protein samples (Figure 5 and S5), data were analyzed in Prism (Graphpad) and are presented as mean ± SD (n=3). The X-ray data collection, structure determination, and validation were carried out as described in the method details section “γS-crystallin crystallization, data collection, and structure refinement,” using XDS, Phaser, Phenix, and Coot, as listed in the key resources table.
Supplementary Material
Highlights:
Crystal structures of cataract-associated variants of γS-crystallin reported
Increasing deamidation decreases stability and affects aggregation propensity
Overall fold of γS maintained among deamidated and disulfide-bonded variants
Deamidated γS variants form disulfide bonds more rapidly than wild-type γS
ACKNOWLEDGMENTS
This investigation was supported by NIH grants 2R01EY021514 and S10OD021594 to R.W.M. and CIFAR (R.W.M and R.J.D.M). B.N.-B. acknowledges support from the NSF GRFP and the Fulbright Fellowship. P.M. acknowledges support from the Joachim Herz Stiftung Add-on Fellowship and the Deutsche Forschungsgemeinschaft via grant No. 451079909. M.A.S.-P. acknowledges support from the HHMI Gilliam Fellowship. K.W.R. acknowledges support from the MAPS Training Grant, NSF DGE-1633631. The authors thank Dmitry Fishman and Felix Grun and Ben Katz for excellent management of the UCI Laser Spectroscopy Laboratories and Mass Spectrometry Facility, respectively. We also thank Kirsten Lampi and Larry David for advice about identifying free thiols using mass spectrometry. SAXS data collection at the SIBYLS beamline, Advanced Light Source, Lawrence Berkeley National Laboratory is funded by Department of Energy, Office of Basic Energy Sciences (DE-AC02-05CH11231) and Office of Biological and Environmental Research’s Integrated Diffraction Analysis Technologies (IDAT) program along with National Institute of Health project ALS-ENABLE (P30 GM124169).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Andley UP (2007). Crystallins in the eye: function and pathology. Prog. Retin. Eye Res. 26, 78–98. [DOI] [PubMed] [Google Scholar]
- Awoonor-Williams E, and Rowley CN (2016). Evaluation of methods for the calculation of the pKa of cysteine residues in proteins. J. Chem. Theory Comput. 12, 4662–4673. [DOI] [PubMed] [Google Scholar]
- Bloemendal H, De Jong W, Jaenicke R, Lubsen NH, Slingsby C, and Tardieu A (2004). Ageing and vision: structure, stability and function of lens crystallins. Prog. Biophys. Mol. Biol. 86, 407–485. [DOI] [PubMed] [Google Scholar]
- Brubaker WD, Freites JA, Golchert KJ, Shapiro RA, Morikis V, Tobias DJ, and Martin RW (2011). Separating instability from aggregation propensity in γS-crystallin variants. Biophys. J. 100, 498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulaj G, Kortemme T, and Goldenberg DP (1998). Ionization-reactivity relationships for cysteine thiols in polypeptides. Biochemistry 37, 8965–8972. [DOI] [PubMed] [Google Scholar]
- Chong SH, and Ham S (2014). Interaction with the surrounding water plays a key role in determining the aggregation propensity of proteins. Angew. Chemie - Int. Ed. 53, 3961–3964. [DOI] [PubMed] [Google Scholar]
- Chong SH, and Ham S (2015). Distinct role of hydration ater in protein misfolding and aggregation revealed by fluctuating thermodynamics analysis. Acc. Chem. Res. 48, 956–965. [DOI] [PubMed] [Google Scholar]
- Dasari S, Wilmarth PA, Reddy AP, Robertson LJG, Nagalla SR, and David LL (2009). Quantification of isotopically overlapping deamidated and 18O-labeled peptides using isotopic envelope mixture modeling. J. Proteome Res. 8, 1263–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer KN, Hammel M, Rambo RP, Tsutakawa SE, Rodic I, Classen S, Tainer JA, and Hura GL (2014). High-throughput SAXS for the characterization of biomolecules in solution: A practical approach. Methods Mol. Biol. 1091, 245–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Silva A, French-Pacheco L, Rivillas-Acevedo L, and Amero C (2020). Aggregation pathways of human γ D crystallin induced by metal ions revealed by time dependent methods. PeerJ 2020, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaugh SL, Mills IA, and King J (2006). Glutamine deamidation destabilizes human γD-crystallin and lowers the kinetic barrier to unfolding. J. Biol. Chem. 281, 30782–30793. [DOI] [PubMed] [Google Scholar]
- Forsythe HM, Vetter CJ, Jara KA, Reardon PN, David LL, Barbar EJ, and Lampi KJ (2019). Altered protein dynamics and increased aggregation of human γS-crystallin due to cataract-associated deamidations. Biochemistry 58, 4112–4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich MG, Wang Z, Schey KL, and Truscott RJW (2021a). Spontaneous Cleavage at Glu and Gln Residues in Long-Lived Proteins. ACS Chem. Biol. 16, 2244–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich MG, Wang Z, Schey KL, and Truscott RJW (2021b). Spontaneous protein–protein crosslinking at glutamine and glutamic acid residues in long-lived proteins. Biochem. J. 478, 327–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii N, Takata T, Fujii N, and Aki K (2016). Isomerization of aspartyl residues in crystallins and its influence upon cataract. Biochim. Biophys. Acta - Gen. Subj. 1860, 183–191. [DOI] [PubMed] [Google Scholar]
- Geiger T, and Clarke S (1987). Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides. Succinimide-linked reactions that contribute to protein degradation. J. Biol. Chem. 262, 785–794. [PubMed] [Google Scholar]
- Goldschmidt L, Cooper DR, Derewenda ZS, and Eisenberg D (2007). Toward rational protein crystallization: A web server for the design of crystallizable protein variants. Protein Sci. 16, 1569–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorrec F (2009). The MORPHEUS protein crystallization screen. J. Appl. Crystallogr. 42, 1035–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guseman AJ, Whitley MJ, González JJ, Rathi N, Ambarian M, and Gronenborn AM (2021). Assessing the structures and interactions of γD-crystallin deamidation variants. Structure 29, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SRA, Smith DL, and Smith JB (1998). Deamidation and disulfide bonding in human lens γ-crystallins. Exp. Eye Res. 67, 301–312. [DOI] [PubMed] [Google Scholar]
- Hooi MYS, Raftery MJ, and Truscott RJW (2012). Age-dependent deamidation of glutamine residues in human γS crystallin: deamidation and unstructured regions. Protein Sci. 21, 1074–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz J (1992). Alpha-crystallin can function as a molecular chaperone. Proc. Natl. Acad. Sci. U. S. A. 89, 10449–10453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F, and Nau WM (2003). A conformational flexibility scale for amino acids in peptides. Angew. Chemie - Int. Ed. 42, 2269–2272. [DOI] [PubMed] [Google Scholar]
- Hura GL, Menon AL, Hammel M, Rambo RP, Poole FL, Tsutakawa SE, Jenney FE, Classen S, Frankel KA, Hopkins RC, et al. (2009). Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS). Nat. Methods 6, 606–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffries CM, Graewert MA, Blanchet CE, Langley DB, Whitten AE, and Svergun DI (2016). Preparing monodisperse macromolecular samples for successful biological small-angle X-ray and neutron-scattering experiments. [DOI] [PMC free article] [PubMed]
- Kabsch W (2010). XDS. Acta Crystallogr. Sect. D D66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsley CN, Brubaker WD, Markovic S, Diehl A, Brindley AJ, Oschkinat H, and Martin RW (2013). Preferential and specific binding of human αb-crystallin to a cataract-related variant of γs-crystallin. Structure 21, 2221–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch MHJ, Vachette P, and Svergun DI (2003). Small-angle scattering: A view on the properties, structures and structural changes of biological macromolecules in solution. Q. Rev. Biophys. 36, 147–227. [DOI] [PubMed] [Google Scholar]
- Lampi KJ, Ma Z, Hanson SRA, Azuma M, Shih M, Shearer TR, Smith DL, Smith JB, and David LL (1998). Age-related changes in human lens crystallins identified by two-dimensional electrophoresis and mass spectrometry. Exp. Eye Res. 67, 31–43. [DOI] [PubMed] [Google Scholar]
- Lampi KJ, Oxford JT, Bachinger HP, Shearer TR, David LL, and Kapfer DM (2001). Deamidation of human βB1 alters the elongated structure of the dimer. Exp. Eye Res. 72, 279–288. [DOI] [PubMed] [Google Scholar]
- Lampi KJ, Amyx KK, Ahmann P, and Steel EA (2006). Deamidation in human lens βB2-crystallin destabilizes the dimer. Biochemistry 45, 3146–3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampi KJ, Wilmarth PA, Murray MR, and David LL (2014). Lens β-crystallins: The role of deamidation and related modifications in aging and cataract. Prog. Biophys. Mol. Biol. 115, 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapko VN, Purkiss AG, Smith DL, and Smith JB (2002). Deamidation in human γS-crystallin from cataractous lenses is influenced by surface exposure. Biochemistry 41, 8638–8648. [DOI] [PubMed] [Google Scholar]
- Lee B, and Richards FM (1971). The interpretation of protein structures: estimation of static accessibility. J. Mol. Biol. 55, 379–400. [DOI] [PubMed] [Google Scholar]
- Lee CM, and Afshari NA (2017). The global state of cataract blindness. Curr. Opin. Ophthalmol. 28, 98–103. [DOI] [PubMed] [Google Scholar]
- Liebschner D, Afonine PV, Baker ML, Bunkoczi G, Chen VB, Croll TI, Hintze B, Hung LW, Jain S, McCoy AJ, et al. (2019). Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr. Sect. D Struct. Biol. 75, 861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YC, Wilkins M, Kim T, Malyugin B, and Mehta JS (2017). Cataracts. Lancet 390, 600–612. [DOI] [PubMed] [Google Scholar]
- Lynnerup N, Kjeldsen H, Heegaard S, Jacobsen C, and Heinemeier J (2008). Radiocarbon dating of the human eye lens crystallines reveal proteins without carbon turnover throughout life. PLoS One 3, 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael R, and Bron AJ (2011). The ageing lens and cataract: a model of normal and pathological ageing. Philos. Trans. R. Soc. B Biol. Sci. 366, 1278–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita H, and Mizushima N (2016). Autophagy in the lens. Exp. Eye Res. 144, 22–28. [DOI] [PubMed] [Google Scholar]
- Morishita H, Eguchi T, Tsukamoto S, Sakamaki Y, Takahashi S, Saito C, Koyama-Honda I, and Mizushima N (2021). Organelle degradation in the lens by PLAAT phospholipases. Nature 592, 1–5. [DOI] [PubMed] [Google Scholar]
- Nagy P (2013). Kinetics and mechanisms of thiol-disulfide exchange covering direct substitution and thiol oxidation-mediated pathways. Antioxidants Redox Signal. 18, 1623–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton-Baker B, Mehrabi P, Boger J, Schonherr R, von Stetten D, Schikora H, Kwok AO, Martin RW, Miller RJD, Redecke L, et al. (2021). A simple vapor-diffusion method enables protein crystallization inside the HARE serial crystallography chip. Acta Crystallogr. Sect. D Struct. Biol D77, 820–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka OBV, Yeoh HY, and Bulleid NJ (2015). Thiol-disulfide exchange between the PDI family of oxidoreductases negates the requirement for an oxidase or reductase for each enzyme. Biochem. J. 469, 279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson MHM, Søndergaard CR, Rostkowski M, and Jensen JH (2011). PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 7, 525–537. [DOI] [PubMed] [Google Scholar]
- Pande A, Pande J, Asherie N, Lomakin A, Ogun O, King J, and Benedek GB (2001). Crystal cataracts: Human genetic cataract caused by protein crystallization. Proc. Natl. Acad. Sci. U. S. A. 98, 6116–6120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pande A, Mokhor N, and Pande J (2015). Deamidation of human γS-Crystallin increases attractive protein interactions: implications for cataract. Biochemistry 54, 4890–4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinitglang S, Watts AB, Patel M, Reid JD, Noble MA, Gul S, Bokth A, Naeem A, Patel H, Thomas EW, et al. (1997). A classical enzyme active center motif lacks catalytic competence until modulated electrostatically. Biochemistry 36, 9968–9982. [DOI] [PubMed] [Google Scholar]
- Purkiss AG, Bateman OA, Goodfellow JM, Lubsen NH, and Slingsby C (2002). The X-ray crystal structure of human γS-crystallin C-terminal domain. J. Biol. Chem. 277, 4199–4205. [DOI] [PubMed] [Google Scholar]
- Qin S, and Zhou HX (2019). Calculation of Second Virial Coefficients of Atomistic Proteins Using Fast Fourier Transform. J. Phys. Chem. B 123, 8203–8215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintanar L, Domínguez-Calva JA, Serebryany E, Rivillas-Acevedo L, Haase-Pettingell C, Amero C, and King JA (2016). Copper and Zinc Ions Specifically Promote Nonamyloid Aggregation of the Highly Stable Human γ-D Crystallin. ACS Chem. Biol. 11, 263–272. [DOI] [PubMed] [Google Scholar]
- Ray NJ, Hall D, and Carver JA (2016). Deamidation of N76 in human γS-crystallin promotes dimer formation. Biochim. Biophys. Acta - Gen. Subj. 1860, 315–324. [DOI] [PubMed] [Google Scholar]
- Rocha MA, Sprague‐Piercy MA, Kwok AO, Roskamp KW, and Martin RW (2021). Chemical properties determine solubility and stability in βγ‐crystallins of the eye lens. ChemBioChem 22, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskamp KW, Kozlyuk N, Sengupta S, Bierma JC, and Martin RW (2019). Divalent cations and the divergence of βγ-crystallin function. Biochemistry 58, 4505–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskamp KW, Azim S, Kassier G, Norton-Baker B, Sprague-Piercy MA, Miller RJD, and Martin RW (2020). Human γS-crystallin–copper binding helps buffer against aggregation caused by oxidative damage. Biochemistry 59, 2371–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Rosmalen M, Krom M, and Merkx M (2017). Tuning the flexibility of glycine-serine linkers to allow rational design of multidomain proteins. Biochemistry 56, 6565–6574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer FQ, and Buettner GR (2001). Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 30, 1191–1212. [DOI] [PubMed] [Google Scholar]
- Searle BC, Dasari S, Wilmarth PA, Turner M, Reddy AP, David LL, and Nagalla SR (2005). Identification of protein modifications using MS/MS de novo sequencing and the OpenSea alignment algorithm. J. Proteome Res. 4, 546–554. [DOI] [PubMed] [Google Scholar]
- Serebryany E, and King JA (2014). The βγ-crystallins: native state stability and pathways to aggregation. Prog. Biophys. Mol. Biol. 115, 32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serebryany E, Yu S, Trauger SA, Budnik B, and Shakhnovich EI (2018). Dynamic disulfide exchange in a crystallin protein in the human eye lens promotes cataract-associated aggregation. J. Biol. Chem. 293, 17997–18009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra A, Gallart-Palau X, Wei J, and Sze SK (2016). Characterization of Glutamine Deamidation by Long-Length Electrostatic Repulsion-Hydrophilic Interaction Chromatography-Tandem Mass Spectrometry (LERLIC-MS/MS) in Shotgun Proteomics. Anal. Chem. 88, 10573–10582. [DOI] [PubMed] [Google Scholar]
- Slingsby C, Wistow GJ, and Clark AR (2013). Evolution of crystallins for a role in the vertebrate eye lens. Protein Sci. 22, 367–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Søndergaard CR, Olsson MHM, Rostkowski M, and Jensen JH (2011). Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J. Chem. Theory Comput. 7, 2284–2295. [DOI] [PubMed] [Google Scholar]
- Srivastava OP, Srivastava K, Chaves JM, and Gill AK (2017). Post-translationally modified human lens crystallin fragments show aggregation in vitro. Biochem. Biophys. Reports 10, 94–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens R, Stevens L, and Price NC (1959). The Stabilities of Various Thiol Compounds used in Protein Purifications. Science (80-. ). 1959–1959. [Google Scholar]
- Studier FW (2005). Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234. [DOI] [PubMed] [Google Scholar]
- Takata T, Oxford JT, Brandon TR, and Lampi KJ (2007). Deamidation alters the structure and decreases the stability of human lens βA3-crystallin. Biochemistry 46, 8861–8871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata T, Oxford JT, Demeler B, and Lampi KJ (2008). Deamidation destabilizes and triggers aggregation of a lens protein, βA3-crystallin. Protein Sci. 17, 1565–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemoto L, and Boyle D (2000). Increased deamidation of asparagine during human senile cataractogenesis. Mol. Vis. 6, 164–168. [PubMed] [Google Scholar]
- Tao Y, and Julian RR (2014). Identification of amino acid epimerization and isomerization in crystallin proteins by tandem LC-MS. Anal. Chem. 86, 9733–9741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorn DC, Grosas AB, Mabbitt PD, Ray NJ, Jackson CJ, and Carver JA (2019). The structure and stability of the disulfide-linked γS-crystallin dimer provide insight into oxidation products associated with lens cataract formation. J. Mol. Biol. 431, 483–497. [DOI] [PubMed] [Google Scholar]
- Toyama BH, Savas JN, Park SK, Harris MS, Ingolia NT, Yates JR, and Hetzer MW (2013). Identification of long-lived proteins reveals exceptional stability of essential cellular structures. Cell 154, 971–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendra VPR, Khan I, Chandani S, Muniyandi A, and Balasubramanian D (2016). Gamma crystallins of the human eye lens. Biochim. Biophys. Acta - Gen. Subj. 1860, 333–343. [DOI] [PubMed] [Google Scholar]
- Vetter CJ, Thorn DC, Wheeler SG, Mundorff CC, Halverson KA, Wales TE, Shinde UP, Engen JR, David LL, Carver JA, et al. (2020). Cumulative deamidations of the major lens protein γS-crystallin increase its aggregation during unfolding and oxidation. Protein Sci. 29, 1945–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivarès D, and Bonneté F (2002). X-ray scattering studies of Aspergillus flavus urate oxidase: Towards a better understanding of PEG effects on the crystallization of large proteins. Acta Crystallogr. Sect. D Biol. Crystallogr. 58, 472–479. [DOI] [PubMed] [Google Scholar]
- Wilkinson B, and Gilbert HF (2004). Protein disulfide isomerase. Biochim. Biophys. Acta 1699, 35–44. [DOI] [PubMed] [Google Scholar]
- Wilmarth PA, Tanner S, Dasari S, Nagalla SR, Riviere MA, Bafha V, Pevzner PA, and David LL (2006). Age-related changes in human crystalline determined from comparative analysis of post-translational modifications in young and aged lens: Does deamidation contribute to crystallin insolubility? J. Proteome Res. 5, 2554–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, et al. (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wistow GJ; Piatigorsky J (1988). Lens crystallins: The evolution and expression of proteins for a highly specialized tissue. Ann. Rev. Biochem. 1988. 470–504. [DOI] [PubMed] [Google Scholar]
- Wright TA, Stewart JM, Page RC, and Konkolewicz D (2017). Extraction of thermodynamic parameters of protein unfolding using parallelized differential scanning fluorimetry. J. Phys. Chem. Lett. 8, 553–558. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coordinate data have been deposited at RCSB PDB and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bacterial and virus strains | ||
| Escherichia coli Rosetta(DE3) | Millipore Sigma | Cat #69450–3 |
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| HEPES | Fisher | BP310 |
| TRIS | Fisher | BP154 |
| NaCl | RPI | S23030 |
| NaN3 | Fisher | S227I |
| Dithiothreitol (DTT) | Fisher | R0861 |
| SYPRO orange dye | Sigma | S5692 |
| 5,5-dithiobis(2-nitrobenzoic acid) (DTNB) | Fisher | AC117540010 |
| Urea | Fisher | U15 |
| Ammonium bicarbonate | Fisher | A643 |
| Ammonium acetate | Fisher | A637 |
| Ammonium sulfate | Sigma | S5636 |
| Morpheus screen | Molecular Dimensions | MD1–124 |
| Hampton index screen | Hampton | HR2–134 |
| Critical commercial assays | ||
| Deposited data | ||
| Crystal structure of γS-wt | This paper | PDB: 7N36 |
| Crystal structure of γS-3 | This paper | PDB: 7N37 |
| Crystal structure of γS-5 | This paper | PDB: 7N38 |
| Crystal structure of γS-7 | This paper | PDB: 7N39 |
| Crystal structure of γS-9A | This paper | PDB: 7N3A |
| Crystal structure of γS-9B | This paper | PDB: 7N3B |
| NMR structure of γS-wt | (Kingsley et al., 2013) | PDB: 2M3T |
| Crystal structure of γS dimer | (Thorn et al., 2019) | PDB: 6FD8 |
| Crystal structure of γS C-terminal domain | (Purkiss et al., 2002) | PDB: 1HA4 |
| Crystal structure of γS-9C | (Norton-Baker et al., 2021) | PDB: 7NJE |
| Experimental models: Cell lines | ||
| Experimental models: Organisms/strains | ||
| Oligonucleotides | ||
| Recombinant DNA | ||
| pET28a(+) | (Brubaker et al., 2011) | N/A |
| Modifed pET28a(+) vectors | This paper | N/A |
| Software and algorithms | ||
| XDS | (Kabsch, 2010) | N/A |
| Phaser | (McCoy et al., 2007) | N/A |
| Phenix version 1.18.2 | (Liebschner et al., 2019) | N/A |
| Coot version 0.9.4 | (Emsley et al., 2010) | N/A |
| PyMol version 2.4.1 | Schrödinger | https://pymol.org/2/ |
| PROPKA | (Olsson et al., 2011) | propka.org |
| Prot pi | Roland Josuran, Zurich University of Applied Sciences | https://www.protpi.ch/Calculator/ProteinTool |
| Zetasizer | Malvern | N/A |
| OmniSEC | Malvern | N/A |
| Prism | GraphPad Software | http://www.graphpad.com/ |
| Other | ||
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.