

Summary
Efforts to probe the role of the gut microbiota in disease would benefit from a system in which patient-derived bacterial communities can be studied at scale. We addressed this by validating a strategy to propagate phylogenetically complex, diverse, stable, and highly reproducible stool-derived communities in vitro. We generated hundreds of in vitro communities cultured from diverse stool samples in various media; certain media generally preserved inoculum composition, and inocula from different subjects yielded source-specific community compositions. Upon colonization of germ-free mice, community composition was maintained and host proteome resembled the host from which the community was derived. Treatment with ciprofloxacin in vivo increased susceptibility to Salmonella invasion in vitro and the in vitro response to ciprofloxacin was predictive of compositional changes observed in vivo, including the resilience and sensitivity of each Bacteroides species. These findings demonstrate that stool-derived in vitro communities can serve as a powerful system for microbiota research.
Keywords: gut microbiota, ciprofloxacin, antibiotics, microbial ecology, culturomics, ex vivo, synthetic communities, microbiota perturbations, ecological stability
Introduction
The gut microbiota is a diverse community that performs functions important for host physiology (Hooper et al., 2012; Mayer et al., 2014; Sonnenburg and Backhed, 2016), and a wide range of environmental perturbations (Kashyap et al., 2013; Schubert et al., 2015; Tropini et al., 2018) lead to changes in community composition and function. Human and animal models have enabled some mechanistic understanding of these responses (Faith et al., 2011; Mark Welch et al., 2017; Reyes et al., 2013; Rezzonico et al., 2011), yet insight into microbiota recovery in even the most well-studied case (antibiotics) is limited. Antibiotics disrupt colonization resistance and lead to pathogen expansion and fecal shedding (Barthel et al., 2003; Stecher et al., 2007), and can dramatically affect host physiology, including changes in adiposity, insulin resistance, and cognitive function (Cho et al., 2012; Cox et al., 2014; Frohlich et al., 2016; Hwang et al., 2015). Understanding antibiotic effects across microbiotas, doses, and treatment regimens will assist in mitigating these adverse effects.
We recently showed, using mice colonized with human feces, that the microbiota shifts into a new, lower-diversity steady state after ciprofloxacin treatment, although certain taxa displayed a high level of resilience during recovery (Ng et al., 2019). It is unclear to what extent the response of each taxon can be predicted from its behavior in isolation; microbial interactions and drug modification can impact the efficacy of antibiotic treatment (Adamowicz et al., 2018; Aranda-Díaz et al., 2019; de Vos et al., 2017; Nicoloff and Andersson, 2016), which may underlie our current inability to explain individualized responses to fluoroquinolone treatment in humans (Dethlefsen and Relman, 2011) or why some antibiotics negatively impact taxa known to be resistant to the drug in vitro (Ivanov et al., 2008). To address these questions requires microbiota models that are scalable and high-throughput, and in which the magnitude of perturbations can be quantitatively tuned; in vivo investigations are limited in these regards.
In vitro co-culturing of collections of isolated species has revealed key interspecies interactions and metabolic roles (de Vos et al., 2017; Gutierrez and Garrido, 2019; Kehe et al., 2019; Venturelli et al., 2018). However, such synthetic communities generally lack the complexity of gut microbiotas; further, microbes within a host coevolve (Barroso-Batista et al., 2020), which could underlie individualized responses to antibiotics (Dethlefsen and Relman, 2011). It is also difficult to select which set of species best model the mammalian gut without knowledge of interspecies interactions. Sophisticated continuous-flow culture models (Macfarlane et al., 1998; Minekus et al., 1999) permit long time-scale experimentation and the manipulation of key environmental variables, but are also limited in throughput (Auchtung et al., 2016). Resuspension of stool in liquid media has shown promise for high-throughput analysis of drug metabolism by highly complex communities (Zimmermann et al., 2019), but the extent to which these approaches recapitulate gut microbiota behaviors is unclear. Passaging of diverse microbiotas from leaf and soil samples in minimal media led to the selection of simple communities whose family-level but not species-level compositions were governed by nutrient availability (Goldford et al., 2018); whether these findings also apply to human-relevant microbiotas and more complex media is unknown.
Here, we establish that stool-derived in vitro communities (SICs) are powerful tools for modeling responses of the gut microbiota to antibiotics in vivo. We demonstrate that hundreds of SICs generated from distinct initial inocula in diverse media can be phylogenetically complex and diverse, and stably preserve the composition of the initial inoculum. SIC composition was maintained during colonization of gnotobiotic mice, and the host proteome was mostly reprogrammed in the same manner as by the original humanization. SICs derived from the feces of ciprofloxacin-treated mice exhibited increased invasion by the pathogen Salmonella Typhimurium in vitro, consistent with previously observed decreases in colonization resistance after antibiotic treament. Ciprofloxacin caused similar compositional changes in SICs and humanized mice, highlighting the utility of SICs for predicting and interpreting the results of in vivo experiments in high throughput.
Results
Anaerobic batch culturing of humanized mice feces results in stable and reproducible in vitro communities
To generate SICs in a straightforward, scalable, and reproducible manner, we focused on ex-germ-free mice colonized with human feces (humanized mice). These humanized mice facilitate controlled experimental manipulation, enabling comparisons between in vitro and in vivo behaviors of communities of human commensals. We inoculated humanized mouse fecal samples into various media and repeatedly passaged in anaerobic batch cultures in high throughput (Fig. 1A).
Figure 1: High-throughput cultivation of feces from humanized mice yields stable, complex, and reproducible microbial communities.
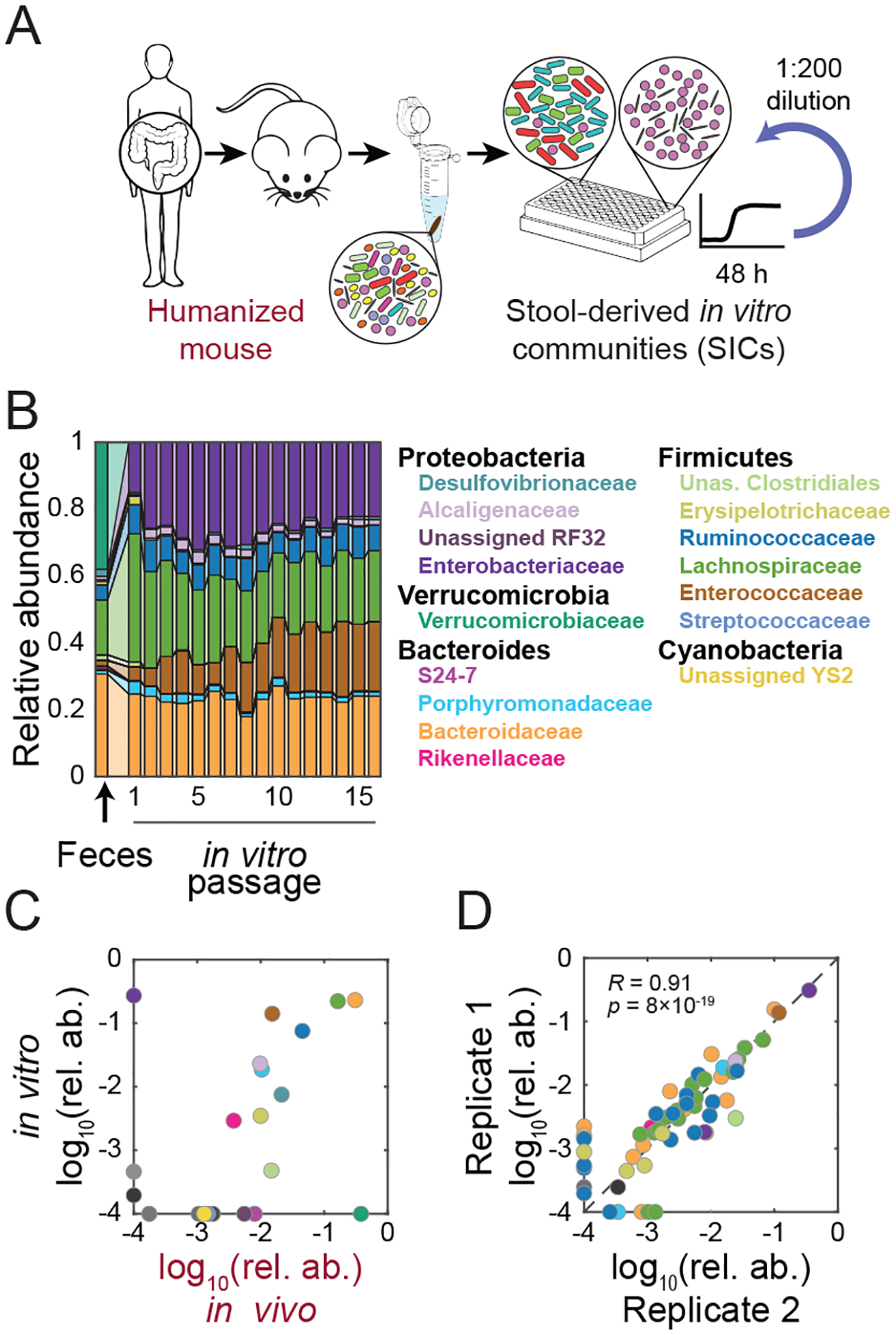
A) Experimental setup. germ-free mice were colonized with feces from a single human donor (“humanized”). Fecal samples from humanized mice were inoculated into anaerobic batch culture and passaged with dilution every 48 h to derive SICs.
B) In vitro passaging produces stable and complex SICs. Family-level compositions of a representative SIC derived from the feces of MD mouse 1 during 16 rounds of in vitro passaging in BHI.
C) In vitro passaging can produce an SIC that resembles the fecal inoculum. Family-level relative abundances (mean of passages 4–16) for the SIC in (B) compared with the fecal inoculum from which it originated.
D) In vitro-passaged SICs are highly reproducible. ASV-level relative abundances for two technical replicates of the SIC in (B) after 7 passages. R and p were computed using only ASVs present in both samples.
Relative abundances <10−4 in (C,D) were set to 10−4 for visualization.
To determine whether SICs reached stable compositions, we performed 16S sequencing on SICs derived from feces of two mice fed a diet deficient in microbiota-accessible carbohydrates (MACs) and grown in the complex medium BHI. At the family level, SICs stabilized by the second passage (Fig. 1B) and retained most families from the original feces (Fig. 1C). Of the families present in the inoculum but not in the passaged SIC, most were at low abundance in vivo (Fig. 1C); with the exception of the Verrucomicrobiaceae (Fig. 1B,C). In turn, the sole family to emerge from undetectable levels was the Enterobacteriaceae (Fig. 1B,C), whose members can be associated with dysbiosis. The fraction of amplicon sequence variants (ASVs, a proxy for strains) detectable in the fecal sample that were detectable on average beyond the fourth passage was 39.1±2.6% (23/58 ASVs). Of the lost ASVs, their median log10(relative abundance) in the inoculum was lower than that of the retained ASVs (−2.46 and −2.05, respectively; p=0.006 Wilcoxon test) and they constituted 55.1% of the inoculum, of which 38.0% corresponded to A. muciniphila, which has been shown to be fastidious (Tramontano et al., 2018). Lost ASVs were lost early in the culturing experiment, in 1.6±0.7 passages. Of the 37 ASVs lost in at least one replicate, 92% were lost in all three replicates, suggesting that ASV loss was not due to bottleneck effects of dilution during passaging. Thus, even though family-level composition is preserved for families conducive to culturing in these conditions, passaging leads to deterministic decreases to levels below the limit of detection biased toward species that start at lower abundance.
A previous soil and leaf microbiota passaging study reported that despite convergence at the family level, genus-level composition was highly divergent across technical replicates (Goldford et al., 2018). In our experiments, family-level dynamics were similar (Fig. S1A) and the relative abundances of ASVs were highly correlated across all replicates (Fig. 1D, S1B). The fractions of families and genera that were detectable after passaging were higher than the fraction of ASVs that remained detectable (39.1%) and similar to each other (52.9% and 56.0%, respectively), suggesting that loss of a genus often meant the loss of an entire family. Three technical replicates of SICs derived from one mouse contained 50 ASVs at >0.1% (a conservative measure of detectability), and all ASVs present in only one replicate had relative abundance <1%; higher variation in low-abundance ASVs was expected due to counting noise. SIC composition was also maintained in higher-volume culturing (Fig. S1C) and after freezing and thawing (Fig. S1D). Thus, top-down in vitro cultivation from fecal samples can deterministically select complex SICs that are stable under passaging for several weeks.
Colonization of gnotobiotic mice with an SIC re-establishes gut microbiota composition and the host proteome
To determine whether and how SIC composition would be altered by colonization of a mammalian host, we introduced an SIC into germ-free mice (Fig. 2A). Family-level abundances were similar to those of the humanized mice from which the SIC was derived (Fig. 2B, S1E), and the Enterobacteriaceae that were overrepresented in vitro receded in vivo. To evaluate effects on host physiology, we quantified host and microbial proteins in the feces of humanized mice, germ-free mice, and SIC-colonized mice. Host proteomes of SIC-colonized mice were more similar to humanized mice than germ-free mice, with immunoglobulins upregulated in SIC-colonized mice (Fig. 2C–E, S1F). Therefore, re-introduction of SICs into a host largely re-establishes gut microbiota composition and the host proteome.
Figure 2: Re-introduction of SICs into a host re-establishes gut microbiota composition and promotes host homeostasis.
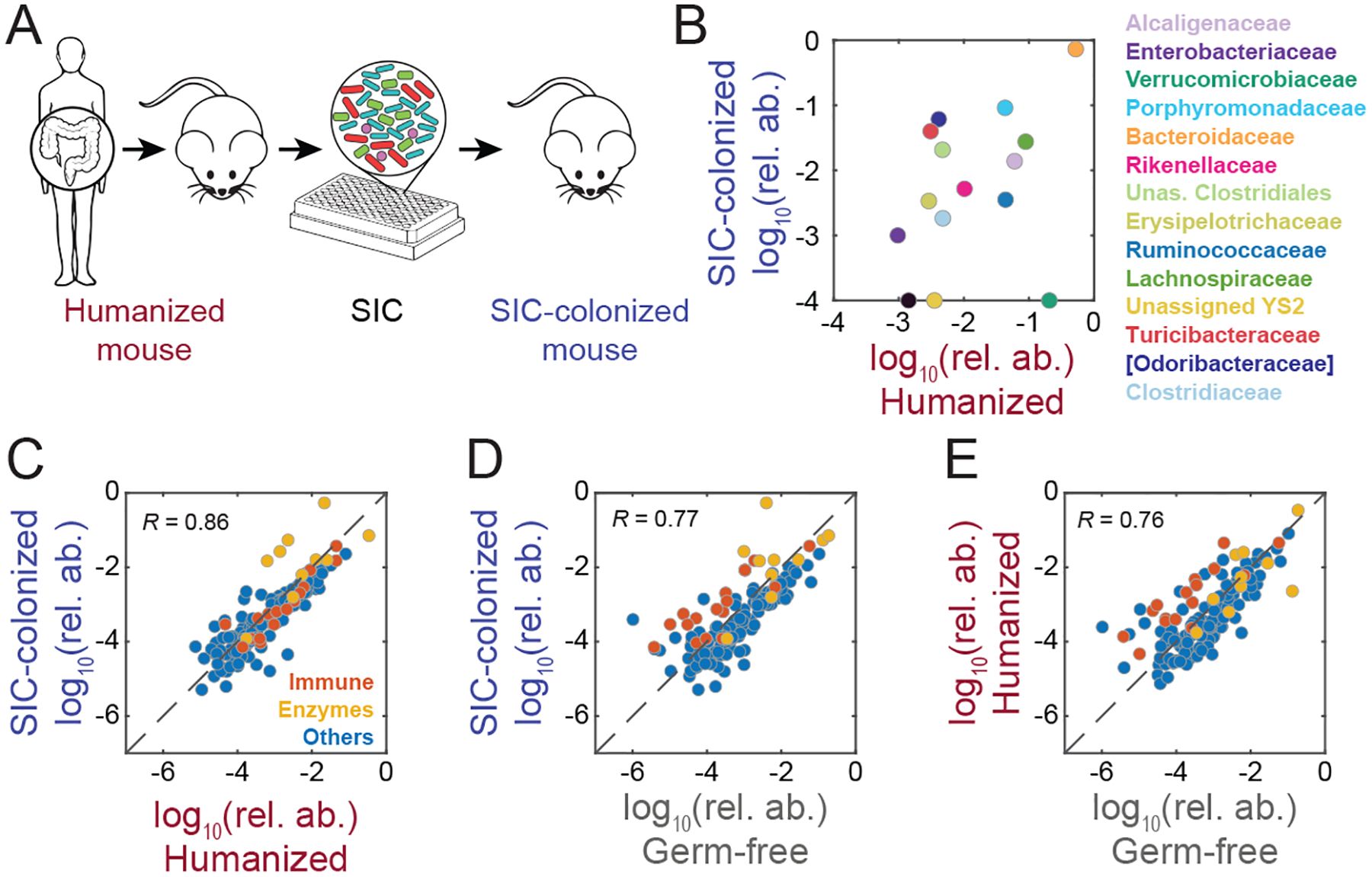
A) An SIC generated from a humanized mouse was used to colonize germ-free mice to determine whether SICs can re-establish the original microbiota.
B) SIC colonization of germ-free mice restored families overrepresented in vitro back to levels similar to those in the humanized mice from which the SIC was derived. Comparison of the family-level relative abundances of a humanized-mouse fecal inoculum and the mean family-level relative abundance of germ-free mice colonized with an SIC derived from the humanized-mouse fecal inoculum (inoculum n=1, SIC-colonized n=3). Relative abundances <10−4 were set to 10−4 for visualization.
C-E) C-E) The host proteomes in humanized and SIC-colonized mice were more highly correlated (C) than either set of mice with germ-free mice (D and E). Immune-related proteins (red) were similarly up-regulated in humanized and SIC-colonized mice relative to germ-free mice.
Initial inoculum composition and nutrient conditions affect SIC composition
To probe the determinants of SIC composition, we inoculated multiple media with a set of fecal samples that varied widely in composition, yet were composed of a common set of members so that they could be readily compared. We previously discovered that diet and antibiotics have large, distinct, interacting effects on the composition of the microbiota (Ng et al., 2013; Sonnenburg et al., 2016), thus we treated mice fed a standard diet (SD) or a MAC-deficient diet (MD) with ciprofloxacin for 5 days. We quantified microbiota composition before treatment (Pre), at the peak of treatment (Peak), during residual treatment (Res), and after (Post) treatment (Fig. 3A). As expected, treatment induced distinct microbiotas for the two diets (Fig. 3B). Nonetheless, mice generally exhibited reductions in Bacteroidetes and Proteobacteria and expansions in Firmicutes at the peak of treatment (Fig. S2A,B), and the number of uniquely detected ASVs (richness) dropped steadily from day 1 to day 5, followed by partial recovery (Fig. S2C). Thus, these fecal samples yielded qualitatively different inocula yet with the potential for overlapping strains.
Figure 3: High-throughput cultivation of humanized-mouse feces preserves inoculum composition and richness.
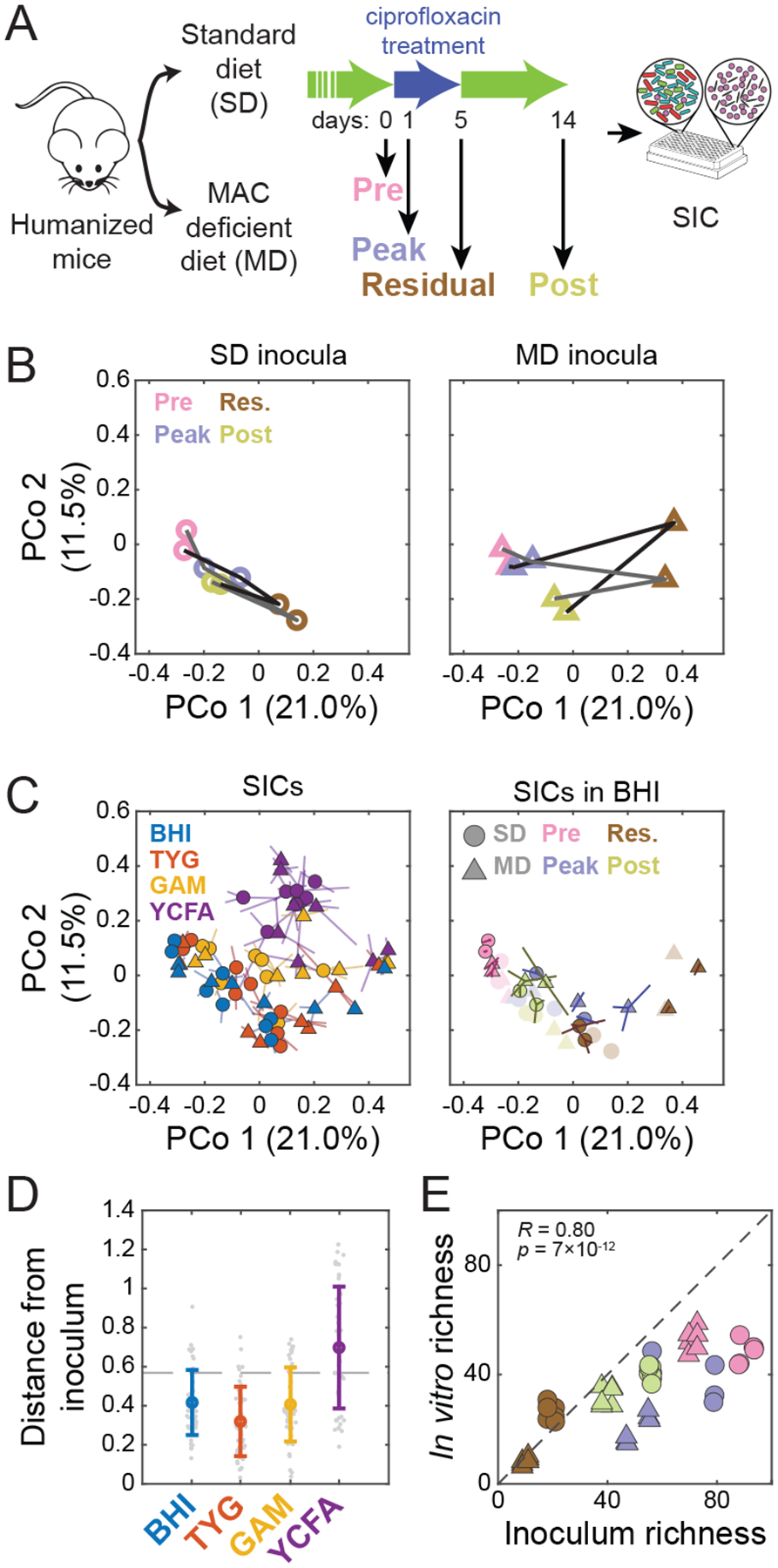
A) Experimental setup. Germ-free mice colonized with feces from a single human donor (“humanized”) were fed an SD or an MD and treated with ciprofloxacin for 5 days. Fecal samples from two mice on each diet were collected on four days (0, 1, 5, 14) before, during, at the end of, and after treatment, and were inoculated into anaerobic batch culture and passaged with dilution every 48 h to derive SICs. Sixteen samples (2 diets, 2 mice, 4 time points during ciprofloxacin treatment) were inoculated into 4 media (BHI, TYG, GAM, and YCFA) in triplicate.
B) Diet alters the trajectory of microbiota reorganization during ciprofloxacin treatment in vivo. PCoA of community composition of the fecal inocula using unweighted Unifrac distance computed on all in vivo and in vitro samples at the ASV level. Lines (black and grey) correspond to two different mice.
C) Medium and inoculum determine the final composition of passaged SICs. The 7th passage of all 192 SICs is shown in a PCoA of SIC composition using unweighted Unifrac distance computed on all in vivo and in vitro samples at the ASV level. Left: samples are colored by media, with shapes representing the diet in the mice from which the inocula were taken. Symbols are the centroid of three replicates, with lines connecting the replicates to the centroid. Right: SICs derived in BHI with colors and shapes representing the timepoint during ciprofloxacin treatment and diet, respectively, in the mice from which the inocula were taken. Symbols are the centroid of three replicates, with lines connecting the replicates to the centroid. Original fecal inocula are plotted in light colors.
D) Most steady-state SICs are similar to the fecal samples from which they were derived, as shown by weighted Unifrac distance of the 7th passage of each SIC to the corresponding fecal inoculum. Colored circles, mean distance for each medium; individual SICs in gray. Error bars, standard deviations; n=48. Dashed line, mean distance between fecal samples.
E) SIC diversity correlates with inoculum diversity in BHI. Richness (number of ASVs in rarefied data) was compared for the 7th passage of each SIC and the corresponding fecal inocula, and separated and colored/shaped as in (C). R and p are for Pearson coefficient; n=48.
To determine the extent to which the abiotic environment and inoculum dictated steady-state SIC composition, we inoculated the 16 fecal samples into four media (BHI, TYG, GAM and YCFA) and grew them in technical triplicates for ≥7 passages of anaerobic batch culturing. SICs were generally stable and reproducible in BHI and TYG (Fig. S3A,B); in GAM and YCFA, SICs were prone to fluctuations (Fig. S3A–C). All BHI SICs were robust to freezing and to growth in higher volumes (Fig. S4). In a Principal Coordinates Analysis of 16S data of all communities (Fig. 3C), the first coordinate was largely determined by the presence or absence of several co-occurring families, including Enterobacteriaceae (Fig. S5A). Most of these families were present in pre-treatment inocula and were undetectable in residual treatment (day 5) inocula (Fig. S5B), suggesting that the variation is mostly explained by the fecal inocula, not the culture media. Our data approximately clustered according to culture media; BHI and TYG SICs overlapped but were well separated from GAM and even more so from YCFA (Fig. 3C). BHI, TYG, and GAM SICs were more similar to their inocula than YCFA SICs when using a weighted approach that accounts for taxonomical relative abundances (Fig. 3D). Using presence and absence of taxa as a measure of similarity (thus giving more weight to low-abundance taxa), BHI and TYG are reasonable mimics of the conditions that produced the fecal pellet (Fig. 3C, S5C–E), regardless of inoculating composition. The data also clustered according to the time during antibiotic treatment when the inocula were sampled (Fig. 3C, S5C). The direction separating clusters of sampling time was approximately orthogonal to the direction separating SICs by medium (Fig. 3C, S5C), indicating that both the availability of taxa in the inoculum and the ability of those taxa to grow in a given medium dictate the composition of SICs.
We computed Pearson correlation coefficients of ASV abundances among pairs of technical replicates for all combinations of inoculum and medium, and found that 90% were >0.68 (Fig. S3A), with a mean and standard deviation of 0.86±0.22 (n=192). Across all pairwise comparisons, the mean number of unique ASVs (>0.1% in one replicate and not detectable in the other) was very low (3.0±3.3, n=384), with a mean log10(relative abundance) of −2.24±0.62 (n=1171), suggesting that many instances of unique ASVs were due to abundances lower than the limit of detection rather than due to stochasticity in passaging. SICs cultured in YCFA had the largest proportion of replicates with low correlation coefficients (<0.6; Fig. S3A,B), while every pair of replicates cultured in BHI had a correlation coefficient >0.6 (Fig. S3B,C). Half of the technical replicates shared members that constituted >99% of the total abundance, while only ~1% had shared members accounting for <5%. In general, the lowest replicability was observed in YCFA and in SICs cultured from inocula with low diversity (Fig. S2C, S3A–C), suggesting that higher diversity promotes deterministic behavior during passaging. Intriguingly, these findings indicate that SICs are not generally stable in GAM and YCFA, despite the ability of these media to promote the growth of many commensals in isolation (Browne et al., 2016; Rettedal et al., 2014).
SIC richness is highly correlated with that of the fecal sample due to maintenance of abundant species and within-family replacement
The presence or absence of multiple families dictating the first principal coordinate (Fig. S5A), and the observation that this coordinate mainly separated SICs by their inocula (Fig. 3C), suggested that SIC richness depends on the richness of the inoculum. Indeed, the richness of SICs grown in BHI, TYG, and GAM was highly correlated with that of the inoculum, with some SICs having >50 detectable ASVs (Fig. 3E, S5F).
Since BHI-passaged SICs were more compositionally similar to their inocula than SICs derived in other media (Fig. 3C,D), we focused on these SICs and queried the extent to which their richness was due to species that were detectable in the inoculum as opposed to emergent species that increased above the limit of detection due to passaging. While almost 40% of the ASVs detected in the inoculum were maintained, there were also 22.0±3.0 ASVs detected in SICs that were undetectable in the inoculum, which could be due to contamination or to a jump in abundance during passaging from below to above the limit of detection. The lack of contamination in control wells in our plates, along with the observation that replicates exhibited many of the same emergent ASVs (63% of the 27 emergent ASVs were present in all three replicates), suggested that contamination was generally low. These observations are reminiscent of the emergence of A. muciniphila within mice switched from a standard to MAC-deficient diet, even though these bacteria were often undetectable before the dietary switch (Earle et al., 2015). Most emergence in our SICs was due to within-family replacement; the only emergent family was the sole Gammaproteobacteria in the SIC (an Enterobacteriaceae) that had no detectable members in the inoculum. The ASVs that emerged typically did so in all three replicates, further underscoring the deterministic nature of SIC dynamics even at low abundance. As a whole, emergent ASVs accounted for 44.6±0.9% of the SIC, with 29.1±2.5% due to the Enterobacteriaceae ASV, and their median log10(relative abundance) in the SICs was similar to that of the ASVs that were retained (−2.13 and −2.14, respectively; p=0.35 Wilcoxon test). Emergent ASVs reached stable levels, defined as within 2 standard deviations of their mean log10(relative abundance) beyond the fourth passage, in 2.05±1.22 passages. These data suggest that the emergence of an ASV does not predict its abundance in the SIC and that restructuring of SICs (both loss and emergence) primarily occurs in the first two passages.
The total number of ASVs present in at least one inoculum (fecal gamma diversity) was 158, of which 68 were present in at least one BHI SIC and 79 in any medium. The total number of ASVs present in at least one SIC was 117. Of these, two appeared in at least one technical replicate of all BHI SICs, an Enterococcus species and a member of the Lachnospiraceae family; four more Lachnospiraceae were included when we ignored the low-diversity SIC cultured from residual treatment inocula. Intriguingly, these ASVs were present in the core microbiota of almost all mice at the point of minimum alpha diversity during antibiotic treatment (Ng et al., 2019), suggesting their general ability to persist through perturbations.
Species loss and emergence were ubiquitous across all SICs in all media. The fraction of ASVs detectable in the fecal sample that remained detectable in the seventh passage was negatively correlated with the diversity of the inoculum (for BHI, R=−0.89, p=10−17; Fig. S6A), consistent with our observation that less-abundant members are more likely to disappear. Conversely, the fraction of emergent ASVs was positively correlated with the diversity of the inoculum (R=0.61, p=10−6, Fig. S6B). However, the fraction of families lost was weakly anticorrelated with inoculum diversity (Fig. S6C), and the fraction of families that emerged was also weakly anticorrelated with inoculum diversity (Fig. S6D), indicating general species replacement within families. Almost 90% of the ASVs that were lost or gained did so within the first four passages (Fig. S6E,F). More than 66% of lost ASVs went undetectable from all the inocula in which they were present, indicating that most of the disappearance dynamics are deterministic. While A. muciniphila was present in all inocula, it was maintained in 16.7% of all SICs and in 31.3% of BHI SICs, albeit at decreased abundance relative to the inoculum.
Exposure to ciprofloxacin in vivo increases Salmonella invasion in vitro
The compositions of SICs derived during and after antibiotic treatment resembled the compositions of the inocula (Fig. 3B,C). Thus, we next asked whether the SICs recapitulated an important feature observed in vivo: decreased resistance to colonization by the pathogen Salmonella enterica serovar Typhimurium (Barthel et al., 2003; Lawley et al., 2008). Specifically, we asked whether exposure to ciprofloxacin led to compositional changes that reduce colonization resistance in the absence of the drug in vitro. We grew SICs derived from SD mice inocula gathered before, during the peak of, during residual, and after treatment in vivo (Pre-SD, Peak-SD, Res-SD, and Post-SD, respectively) in BHI. We challenged these SICs with S. Typhimurium for 48 h in BHI, diluted them, spotted them onto LB-streptomycin agar plates, and incubated them in aerobic conditions to select for S. Typhimurium (Fig. 4A). Pre-SD SICs had >10-fold less S. Typhimurium than communities derived from feces of ciprofloxacin-treated mice (Fig. 4B, S7A). Single-cell quantification of fluorescently tagged S. Typhimurium revealed an even larger difference in colonization efficiency between the Pre-SD and Res-SD SICs (Fig. 4C, S7B). Thus, changes in composition driven by antibiotic treatment in vivo lead to differences in SICs that mimic the resilience of the microbiota to S. Typhimurium invasion in vivo.
Figure 4: Pre-exposure of the gut microbiota to ciprofloxacin in vivo results in differential invasion of S. Typhimurium in vitro.
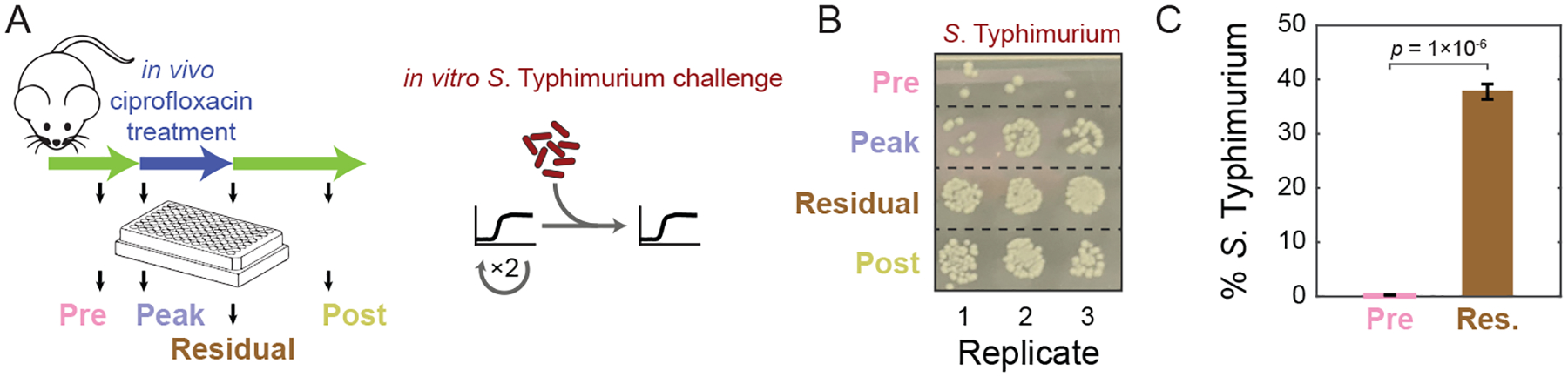
A) Experimental setup for in vitro challenge with S. Typhimurium. SICs passaged in BHI from pre-, peak, residual, and post-treatment humanized mouse fecal inocula were revived after freezing and passaged twice in BHI. SICs were mixed with S. Typhimurium and S. Typhimurium levels were quantified after 48 h of growth.
B) SICs derived from mice treated with ciprofloxacin are more susceptible to S. Typhimurium. Colonies of S. Typhimurium SL1344 after 48 h of growth with SICs diluted 1:104 and grown aerobically on LB+streptomycin.
C) Single-cell quantification of mCherry-tagged S. Typhimurium 14028s after co-culture with SICs derived from pre- and residual-treatment mice fecal inocula. p-value is from a Student’s two-sided t-test; n=3.
Ciprofloxacin alters SICs in a manner consistent with isolate sensitivities and in vivo dynamics
We isolated 15 strains from a Pre-MD SIC and measured their MICs (Table S1). The Lachnospiraceae exhibited the highest MICs (Fig. 5A); consistent with the high levels of Lachnospiraceae and Ruminococcaceae in humanized mice at the peak of ciprofloxacin treatment (Fig. S2B). Most sensitive families (MIC <32 μg/mL) decreased in abundance in vivo (Fig. 5A); the lone exception was the Bacteroidaceae, which increased in three of four mice (Fig. 5A), consistent with our previous observation that a highly resistant Bacteroides vulgatus strain (MIC >512 μg/mL) can be selected in vivo (Ng et al., 2019). These results suggest that ciprofloxacin sensitivities are generally similar in vitro and in vivo.
Figure 5: In vitro treatment of an SIC with ciprofloxacin results in changes in community composition consistent with strain sensitivity.
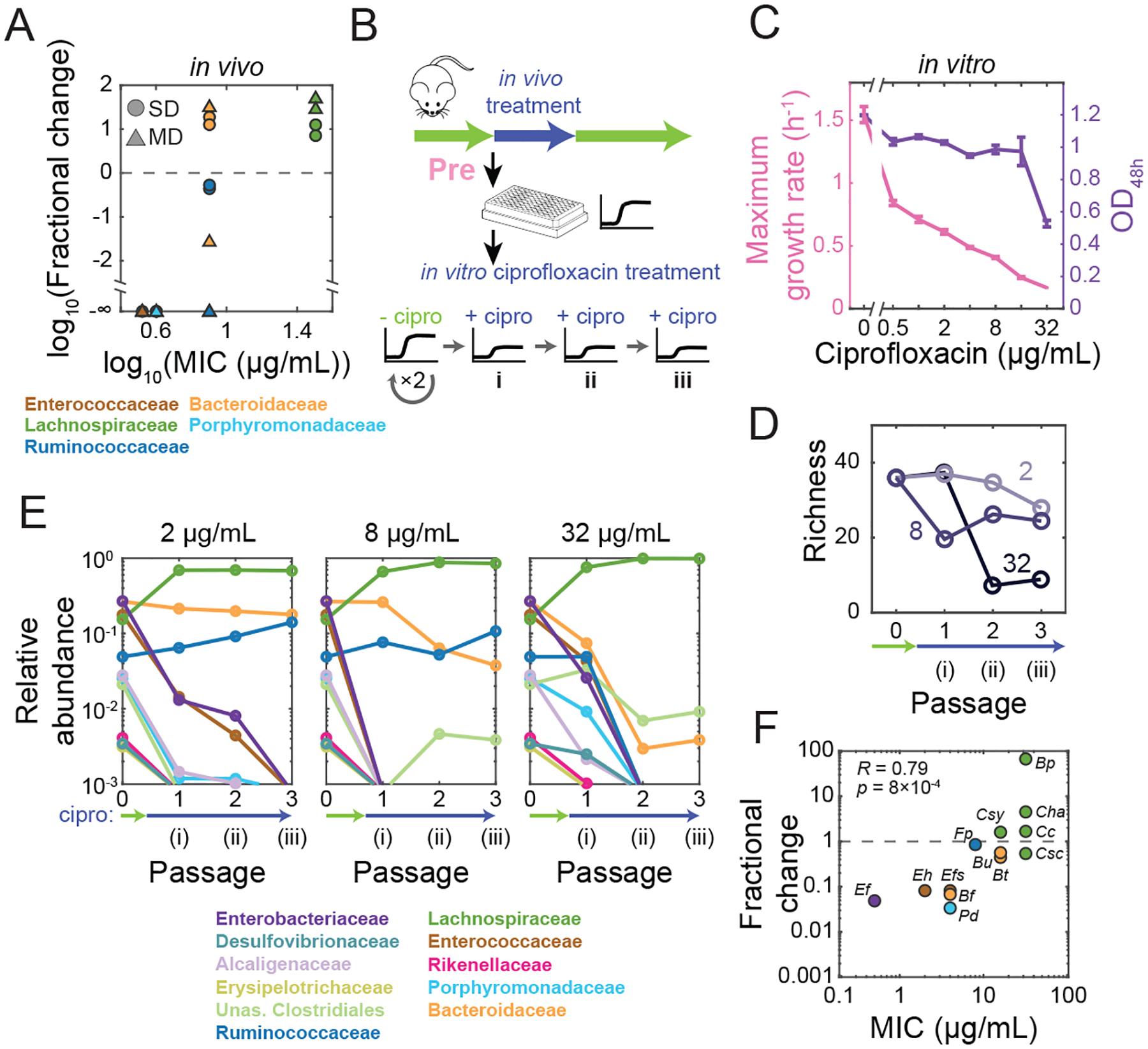
A) Changes in family-level abundances during ciprofloxacin treatment in vivo can be predicted by strain-level sensitivities in vitro. Fractional changes were computed as the ratio of residual and pre-treatment time-point abundances in vivo. MIC is the mean value across strains in a given family determined from growth in isolation in BHI (Table S1).
B) Experimental setup. An SIC passaged in BHI from a pre-treatment humanized mouse fecal inoculum (Pre-SD) was passaged in ciprofloxacin three times (i,ii,iii).
C) Pre-SD SIC yield and growth rate decreased with increasing concentrations of ciprofloxacin. OD was measured after 48 h of growth with ciprofloxacin. Lines, means of triplicate growth curves; error bars, standard deviations.
D) Richness of the Pre-SD SIC decreased in a dose-dependent manner. Data are means of two technical replicates.
E) Ciprofloxacin treatment in vitro selects for a few families. Family-level composition of the Pre-SD SIC across passages in ciprofloxacin. The limit of detection was ~10−3. Data are means of two technical replicates.
F) Growth of ASVs in the Pre-SD SIC under ciprofloxacin treatment can be predicted by their sensitivity in isolation. Fractional-abundance changes of ASVs corresponding to the 15 isolates in (A) were computed after one passage in 2 μg/mL ciprofloxacin.
We next queried the responses of a Pre-SD SIC to ciprofloxacin (Fig. 5B). The maximum growth rate of the SIC decreased monotonically with drug concentration (Fig. 5C) and the collective MIC for the SIC was >32 μg/mL (Fig. 5C). To assess the effects of long-term exposure to ciprofloxacin on SIC composition, we passaged the Pre-SD SIC in BHI with ciprofloxacin twice more (Fig. 5Bii,iii). The maximum growth rate of the SICs remained low, suggesting that community remodeling took place during the first ciprofloxacin passage (Fig. S7C). At 2 μg/mL, diversity decreased steadily (Fig. 5D); at 32 μg/mL, diversity declined to lower than untreated SICs derived from the residual-treatment inoculum (Fig. 5D, S2C). For all doses, many families decreased to undetectable levels, including the abundant Enterobacteriaceae and Enterococcaceae (Fig. 5E), while members of the Lachnospiraceae, Ruminococcaceae, and Bacteroidaceae persisted after three rounds in 2 and 8 μg/mL ciprofloxacin (Fig. 5E). At 32 μg/mL, the Ruminococcaceae were undetectable by the second passage and the Bacteroidaceae decreased to very low levels (Fig. 5E), likely explaining the large decrease in maximum OD relative to 16 μg/mL (Fig. 5C).
Antibiotic pressure selected for strains that emerged from below the detection limit due to the decrease of other, more sensitive species: at 2 μg/mL the Lachnospiraceae increased from 12 to 16 detectable ASVs while the Bacteroidaceae decreased from 12 to 4 detectable ASVs. This emergence was not due to contamination: at every concentration, changes were highly reproducible between replicates (>89% and >69% of ASVs that became undetectable or detectable, respectively, did so in both replicates). Moreover, the MICs of the 15 isolates were highly correlated with the fractional change in abundance upon 2 μg/mL ciprofloxacin treatment of the Pre-SD SIC (Fig. 5F). Hence, the compositional changes observed in SICs can largely be predicted by individual sensitivities in isolation.
SICs derived from pre-exposed inocula show increased resilience to ciprofloxacin treatment
To ask whether pre-exposed SICs show increased resilience to ciprofloxacin treatment in vitro, we selected a low diversity SIC derived from an SD mouse inoculum during residual treatment in vivo (Res-SD, Fig. S7D) and grew it in BHI with ciprofloxacin either continuously (Fig. S7Di,ii,ii) or transiently (Fig. S7Di,iv,v). The Res-SD SIC was less sensitive than the Pre-SD SIC in terms of growth rate (Fig. 5C, S7E), and the final OD was largely unaffected (Fig. S7E). These data suggest that the Res-SD SIC derived from the feces of antibiotic-treated mice is less susceptible to ciprofloxacin.
We next asked whether the robustness of Res-SD SIC growth behaviors during ciprofloxacin treatment could be explained by particular compositional differences with the Pre-SD SIC. The Res-SD SIC displayed distinct responses compared to the Pre-SD SIC, maintaining its diversity even at the highest ciprofloxacin concentrations (Fig. 5D, S7F). At all concentrations, Bacteroidaceae and Lachnospiraceae dominated even after three passages (Fig. S7G), in contrast to the decrease of most Bacteroidaceae members in the Pre-SD SIC (Fig. 5E). The most abundant lost family from Res-SD SIC was the Enterococcaceae (Fig. S7G); the Ruminococcaceae and Verrucomicrobiaceae were lost at 32 and 2 μg/mL, respectively. These losses were counterbalanced by emergent ASVs within the resistant families. The composition was quantitatively similar at the ASV level between the two replicates at low concentrations (Fig. S7H). At 32 μg/mL, the two replicates were more similar to each other than the Pre-SD SIC after treatment (Fig. S7I), suggesting that pre-exposure to the drug in mice decreases the stochastic nature of species elimination upon subsequent exposure in vitro. The composition of the Res-SD SIC before treatment and after two recovery passages were highly similar at low doses (Fig. S7J). These data imply that the members of the Res-SD SIC are more resistant to the drug, in line with their pre-selection in vivo.
SICs successfully predict in vivo compositional changes due to ciprofloxacin
Drug concentration is typically undetermined along the gastrointestinal tract and potentially heterogeneous in space and time. Other variables are also difficult to measure and control, such as environmental reservoirs that play a role in bacterial migration and repopulation. The SICs we generated from antibiotic-treated mice provide the opportunity to study antibiotic perturbations in a precisely controlled and high-throughput manner and to assess similarity to in vivo behavior.
In vivo, the microbiota undergoes a complex recovery that likely involves differential susceptibilities, intrinsic recovery capacities, the emergence of resistant mutants, and metabolic interactions (Ng et al., 2019). To investigate the response of SICs in conditions mimicking recovery, after one round in ciprofloxacin we diluted the Pre-SD SIC into BHI without ciprofloxacin (Fig. 6Ai,iv,v). Maximum growth rate increased after one round of treatment at all doses (Fig. S7K); at low doses, growth rate rebounded to values similar to those before treatment, while at >8 μg/mL it only partially recovered (Fig. S7K), likely due to extinction of the Enterococcaceae (Fig. S7L, Table S1). Diversity increased slightly after drug removal (Fig. 6B), suggesting the recovery of certain species from below the limit of detection. Thus, diversity recovery in vitro (Fig. 6B) can be partially explained by re-emergence of ASVs that became undetectable but not extinct during treatment.
Figure 6: In vitro treatment of an SIC with ciprofloxacin results in similar responses as in vivo.
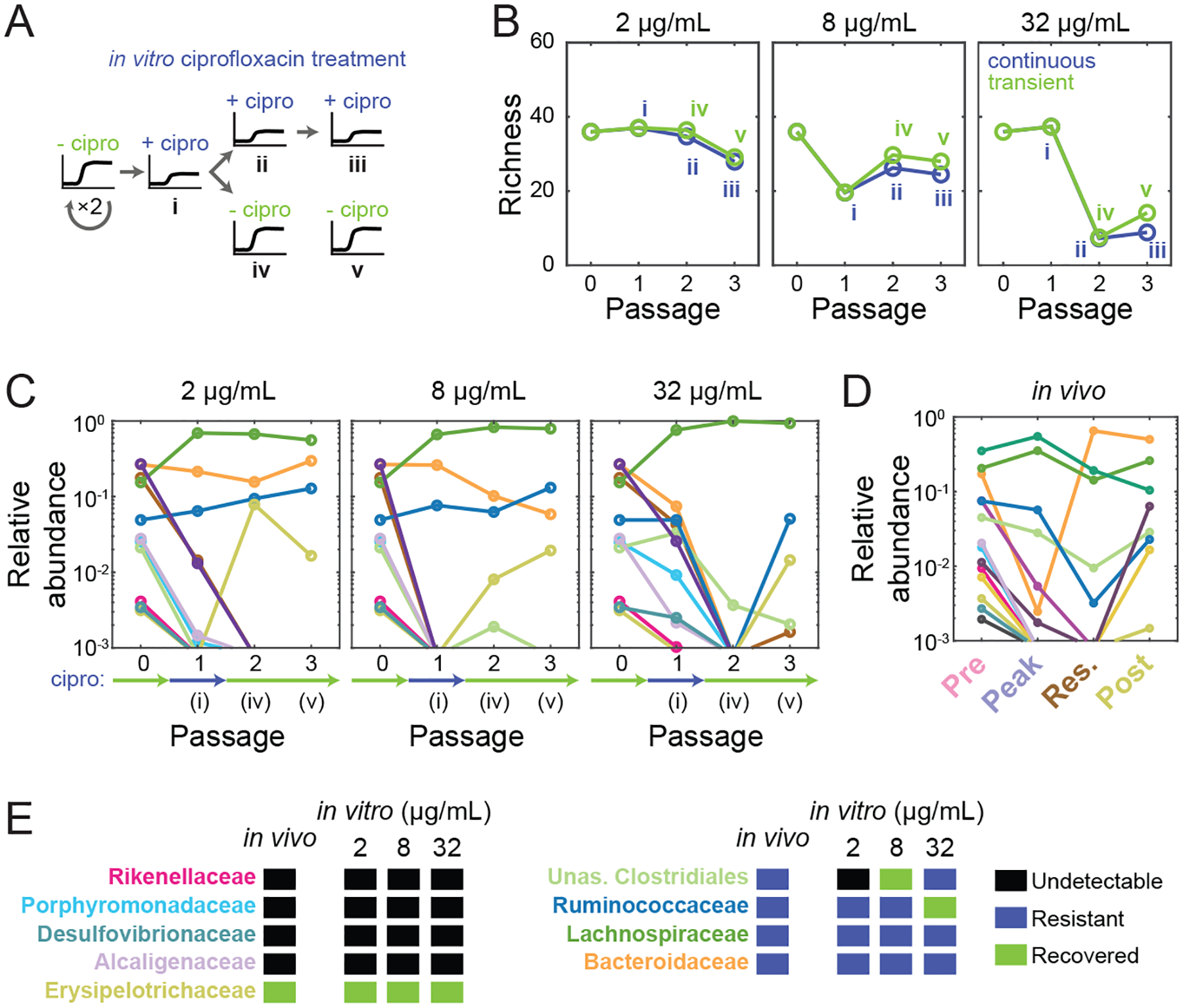
A) Experimental setup mimicking transient treatment in vivo. An SIC passaged in BHI from a pre-treatment humanized mouse fecal inoculum (Pre-SD) was passaged in ciprofloxacin three times (i,ii,iii); or in ciprofloxacin once and then twice without the drug (i,iv,v).
B) Richness of the Pre-SD SIC recovered only partially after removal of ciprofloxacin (green) versus continuous exposure (blue). Data are means of two technical replicates.
C) Some families (e.g. Erysipelotrichaceae and Ruminococcaceae) recover after transient ciprofloxacin treatment. Data are the mean log10(relative abundance) at the family level of two replicates during one round of ciprofloxacin treatment and two rounds of recovery.
D) Ciprofloxacin-induced changes to family-level abundances in SD mice in vivo resemble changes in vitro in (C). Data are the mean log10(relative abundance) at the family level of two mice.
E) Most families display similar qualitative abundance changes in vivo and in vitro.
The small increase in SIC diversity after ciprofloxacin removal (Fig. 6B) involved recovery of Erysipelotrichaceae members from below the limit of detection (Fig. 6C), consistent with its recovery in vivo (Fig. 6D,E). A second treatment after one passage without the drug again made the Erysipelotrichaceae undetectable (Fig. S7M), indicating that sensitivity to ciprofloxacin was unchanged. The Ruminococcaceae, exhibited recovery (Fig. 6C), unsurprisingly since they also survived continued treatment at intermediate concentrations (Fig. 5E). The other families did not recover from undetectable levels after removing the drug in vitro or in vivo (Fig. 6D,E). Taken together, these results suggest that in vitro perturbations can be used as a proxy for many aspects of in vivo dynamics.
Bacteroides recovery in vitro is due to a combination of resistance and resilience
The unexpected recovery of the Bacteroidaceae in vivo motivated us to probe the dynamics of these ASVs. In vivo, although most Bacteroides ASVs were undetectable at the peak and residual time points, most reappeared post-treatment (Fig. 7A). Only B. vulgatus increased in relative abundance to become dominant during residual treatment (Fig. 7B); this strain was likely highly resistant to ciprofloxacin based on our assessment of Bacteroides isolates from the residual fecal sample (Ng et al., 2019). Consistent with this observation, B. vulgatus was the only Bacteroidaceae ASV present in vitro after 3 passages with 32 μg/mL ciprofloxacin, reaching a stable abundance in the Pre-SD (Fig. 7B) and Res-SD SICs (Fig. S7N). B. caccae and B. uniformis were present after 3 passages of the Pre-SD SIC at 8 μg/mL, and B. thetaiotaomicron was present after 3 passages at 2 μg/mL (Fig. 7B); all three species recovered in vivo post-treatment (Fig. 7A), which our data suggests is due to their intrinsic resistance. Other Bacteroides species, such as B. fragilis, became undetectable after one passage at 2 μg/mL in both SICs (Fig. 7B, S7N). These trends are consistent with the higher MICs of B. uniformis and B. thetaiotaomicron compared to B. fragilis (Table S1). Thus, sensitivities of isolates are predictive of Bacteroides resistance during continuous ciprofloxacin treatment.
Figure 7: In vitro treatment of an SIC reveals resistance and resilience in the Bacteroides genus.
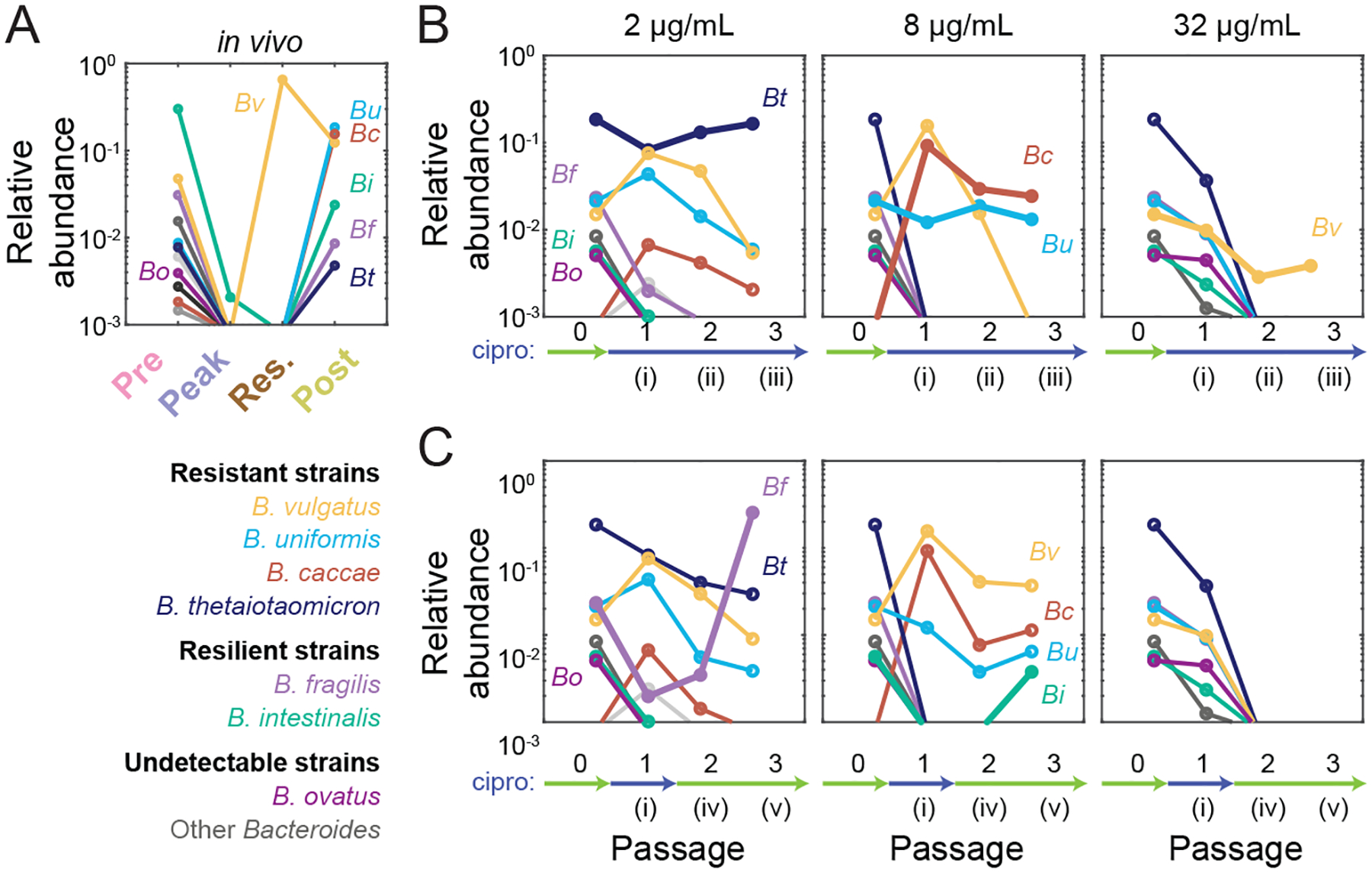
A) Bacteroides dynamics in vivo consist of B. vulgatus dominance during treatment and the recovery of several species after treatment. Data are the mean log10(relative abundance) at the ASV level across two SD mice: Bacteroides ovatus (Bo), B. vulgatus (Bv), B. uniformis (Bu), B. caccae (Bc), B. intestinalis (Bi), B. fragilis (Bf), and B. thetaiotaomicron (Bt). Other Bacteroides are shown in shades of grey.
B,C) Bacteroides survival and recovery in vivo can be respectively explained by resistance and resilience characteristics of the Pre-SD SIC in vitro during continuous treatment (B) or one round of treatment followed by two rounds of recovery (C). Data are the mean log10(relative abundance) of two replicates during one round of ciprofloxacin treatment and two rounds of recovery at the ASV level. Thick lines highlight Bacteroides ASVs that show resistance (B) or resilience (C) at the highest concentration that they display the behavior.
Like B. fragilis, B. intestinalis was undetectable by the second passage even at 2 μg/mL (Fig. 7B). Nonetheless, both species recovered in vivo in SD mice (Fig. 7A). Remarkably, these species also re-emerged in vitro upon further passage without drug in the Pre-SD SIC (Fig. 7C), as did B. fragilis in the Res-SD SIC (Fig. S7O). In fact, B. fragilis became dominant after recovery from treatment with 2 μg/mL in the Pre-SD SIC (Fig. 7C), similar to the replacement of resistant B. vulgatus in vivo by other Bacteroides (Ng et al., 2019) and consistent with dominance of B. fragilis in the Res-SD SIC (Fig. S7N). B. intestinalis was detected in the Pre-SD SIC only after recovery from treatment with 8 μg/mL (Fig. 7C), suggesting that different ciprofloxacin concentrations can alter the competitive landscape of Bacteroides species. We refer to the ability of strains to recover from undetectable levels during treatment as “resilience”. Interestingly, B. vulgatus was only detected when the SIC was continuously exposed (Fig. 7B) and not after ciprofloxacin was removed (Fig. 7C), suggesting that recovery of other families can drive resistant members to undetectable levels. Species that did not recover in vivo, such as B. ovatus, did not recover in vitro (Fig. 7A,C). In sum, fecal-derived SICs can distinguish between resistance and resilience.
Discussion
In vitro co-culturing has proven instrumental in enhancing our understanding of microbial ecology, affording the ability to design consortia through combinatorial culturing and providing the potential to capitalize on bacterial genetics for functional modifications of individual members. Here, we have shown that in vitro passaging of fecal samples is a complementary and powerful tool for generating stable, reproducible, complex SICs. Our approach yields complex SICs with >50 species, and it is likely possible to further increase SIC diversity in media supplemented with specific nutrients that promote the growth of fastidious organisms (Tramontano et al., 2018). While YCFA supports the growth of a wide range of gut commensals in isolation, in SICs the Enterococcaceae take over (Fig. S5A,C), indicating that media that support isolate growth do not necessarily support diverse SICs. Improved understanding of the contributions of interspecies interactions and nutrient availability to community assembly will be instrumental to engineering future gut models, and SICs represent a major step toward this goal.
Our study shows that SICs can shed light on behaviors observed in vivo. SICs derived from mice during and after antibiotic treatment were more susceptible to invasion by S. Typhimurium than SICs derived before treatment (Fig. 4). While previous studies reported that ciprofloxacin treatment can increase the colonization susceptibility of chemostat-grown microbial communities (Carman et al., 2004), our experiments were conducted in the absence of ciprofloxacin at the time of challenge. Thus, the compositional changes elicited by antibiotics in vivo are maintained in vitro, leading to different functional outcomes. Our quantitative comparison between the ciprofloxacin response of the gut microbiota in vivo and in vitro revealed remarkable similarities in taxon-specific changes in diversity (Fig. 6E) despite differences in spatial structure, duration of treatment, and population sizes between SICs and the gut. Thus, SICs have the potential to shed light on many aspects of the dynamics that are confounded by multiple factors in vivo. The observed correlation between abundance changes upon continuous ciprofloxacin treatment and strain sensitivities (Fig. 5F) suggests that interspecies interactions are not strong modifiers of ciprofloxacin sensitivity in these communities. Nevertheless, the predicted emergence of resistant strains led to a subsequent out-competition by close relatives upon removal of the drug (SI), highlighting the role of ecological interactions on community structure. Further studies using SICs can elucidate the role of ecology on antibiotic sensitivity and community structure, especially in the case of antibiotics where indirect resistance can benefit multiple species (Brook, 2009; Sorg et al., 2016). The lack of substantial recovery in richness in vitro after treatment (Fig. 6B) suggests that the ~4-fold increase in vivo post-treatment (Fig. S2C) could come from environmental reseeding, supporting the physiological relevance of reservoirs suggested in vivo by comparisons of singly housed and co-housed mice (Ng et al., 2019). Nonetheless, the recovery in vitro of some species such as B. intestinalis from undetectable levels did not require reseeding (Fig. 7C), indicating built-in resilience; such findings point to the ability of SICs to elucidate aspects of community ecology that can be uncoupled from host dependence. Taken together, these findings indicate that dissecting how a gut community is remodeled by a perturbation in vitro can serve as a pre-screening tool before expensive animal experiments. Many other fascinating questions should be tackled in the future using our in vitro approach, which can easily be scaled to thousands of communities.
SIC colonization of germ-free mice largely resulted in the same composition and shifted the host proteome of germ-free mice toward that of humanized mice (Fig. 2B–E). Moreover, the observation that fast-growing species expand in SICs (Fig. 1B,C) but recede in SIC-colonized mice (Fig. 2B) reveals the few bacterial taxa whose growth is controlled by the host, highlighting further importance of in vitro models. These results suggest the potential of personalized SICs to study community function and its relationship with perturbations and the host, as well as to develop scalable and safe therapeutic alternatives to fecal microbiota transplants (Khoruts and Weingarden, 2014). A combination of top-down and bottom-up stool-derived communities should serve as powerful, complementary resources for the microbiota field. More generally, SICs may provide a bridge between mechanistic studies of microbial ecology and human-relevant in vivo systems.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Kerwyn Casey Huang (kchuang@stanford.edu).
Materials availability
Bacterial communities described in this study can be requested from the lead contact. This study did not generate any other new unique reagents.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request. Custom code used in this paper is available at doi:10.5281/zenodo.570655410.5281/zenodo.5706554. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse strains
All mouse experiments were conducted in accordance with the Administrative Panel on Laboratory Animal Care, Stanford University’s IACUC. Germ-free, Swiss-Webster mice were bred from an in-house colony and maintained in sterile incubators with food and water ad libitum. Females 6–12 weeks of age were cohoused (3–5 per cage). At the end of the experiments, mice were euthanized with CO2, and death was confirmed via cervical dislocation.
Human subjects
Fecal samples were obtained from an anonymous donor (~40 years old healthy white male).
Bacterial strains
Strains isolated from SICs and characterized in this study are listed in Table S1. S. Typhimurium strains SL1344 and mCherry-expressing 14028s were obtained from the Monack lab at Stanford University.
METHODS DETAILS
Mouse humanization and fecal sampling
20 germ-free mice (5 per cage) were gavaged with a human fecal sample obtained from a healthy male donor as previously described (Kashyap et al., 2013). The microbiota was allowed to equilibrate for 6–8 weeks before perturbation experiments commenced to allow for mucus normalization (Johansson et al., 2015). For experiments involving a dietary switch, mice were first fed a standard diet (Purina LabDiet 5010) rich in MACs, and then a defined low-MAC diet (Harlan TD.86489) in which the sole carbohydrates are sucrose (31% w/w), cornstarch (31% w/w), and cellulose (5% w/w) for two weeks to allow the microbiota to re-equilibrate. We then gavaged all mice (2 cages SD, 2 cages MD; n=20 total) for 5 days twice daily with 3 mg ciprofloxacin (Sigma Aldrich) dissolved in 200 μL water. Since we previously showed that the responses of cage-mates to antibiotics are highly reproducible and stereotypical (Ng et al., 2019), we collected fecal samples from two randomly selected mice in different cages from each dietary condition on day 0 (pre-treatment) and on day 1 (peak of treatment, when the culturable load reached a maximum decrease of 10- to 100-fold (Ng et al., 2019)), day 5 (residual treatment, when ciprofloxacin was not being administered but was still detected in feces (Ng et al., 2019)), and day 14 (post-treatment). Fecal pellets were transported on ice and moved into an anaerobic chamber within 30 min.
SIC passaging, storage, and revival
Approximately 100 mg of stool samples from humanized mice or human donors were resuspended into 200 μL phosphate-buffered saline (PBS) in 1.5-mL microcentrifuge tubes. Samples were incubated in PBS for 10–15 min at room temperature to encourage large pieces of fecal matter to disintegrate. Samples were vortexed for 5 min and allowed to sit for 1 min to let food particles settle. One microliter of this resuspension was inoculated into 200 μL medium in 96-well polystyrene microplates (Greiner Bio-One). Each resuspension was inoculated into four media (Table S2) in triplicate. The plate was covered with optical film, with a small (~0.5 mm) hole poked above each well, outside of the plate reader’s light path, to allow gas exchange.
Incubation and optical density (OD) measurements were performed with an Epoch 2 plate reader (BioTek) in an anaerobic chamber at 37 °C with continuous shaking and OD600 measured at 7.5-min intervals. Since some commensals require >24 h to reach saturation (Tramontano et al., 2018), plates were incubated in the plate reader for 48 h and then 1 μL of this saturated culture was transferred into 200 μL of fresh medium in a new plate. We refer to a 48-h cycle from inoculation to saturation as one passage. Samples used in the passaging experiments in Fig. 3 were sometimes taken in consecutive days, hence the plate was removed from the plate reader every 24 h, to inoculate and/or passage corresponding SICs. On a given day, if the communities were not passaged, then the whole volume was transferred into a new plate.
After passaging, 50 μL of the cultures were mixed with 50 μL sterile 50% glycerol (v/v in water) in crimp vials, sealed, and stored at −80 °C for long-term storage. The remaining volume was stored in the 96-well plate at −80 °C with an aluminum seal until DNA was extracted. BHI SICs were revived for experimentation by inoculation of frozen glycerol stocks directly into 3 mL BHI. Revived communities were incubated at 37 °C for 48 h and passaged twice before experimentation by diluting 1:200 into fresh medium every 48 h.
SIC colonization
Mice were humanized with a human fecal sample obtained from the same healthy anonymous donor as was used for the ciprofloxacin-treatment experiment (Fig. 1A). Four weeks after humanization, fecal pellets were collected from 11 mice housed across 3 cages. A mixture of the 11 fecal pellets was used to inoculate 3 mL BHI to derive an SIC as described above. The SIC was passaged every 48 h with a 1:200 dilution into fresh medium. After 5 passages, a frozen stock was made by mixing equal volumes of a saturated culture and 50% glycerol (v/v in water). The SIC was revived by inoculating the frozen stock into 3 mL BHI and passaging twice before using 200 μL of the saturated culture to gavage three co-housed germ-free, female, Swiss-Webster mice. Fecal pellets were sampled from the germ-free mice on the day that they were gavaged, on day 24 after colonization for proteome analysis, and on day 28 for 16S sequencing.
Strains and growth media
Growth media are listed in Table S2. These media were chosen because of their ability to grow diverse microbes: the nutrient-rich Brain Heart Infusion (BHI) is widely used to culture fastidious microorganisms; Tryptone-Yeast extract-Glucose (TYG), which is often used to culture members of the Bacteroides genus that includes many prevalent human commensals (Whitaker et al., 2017); Gifu Anaerobic Medium (GAM; a mixture of animal, plant, and yeast digests and extracts supplemented with glucose and starch), which cultures individual species with relative yields similar to their abundances in human microbiotas (Rettedal et al., 2014); and Yeast extract, Casitone, and Fatty Acids (YCFA), which has been used to culture a wide range of human gut commensals (Browne et al., 2016). Isolated strains are listed in Table S1. All media and materials were made anaerobic by incubating in a custom anerobic chamber (Coy Labs) for 48 h before use.
16S rRNA sequencing
DNA was extracted from whole fecal pellets or 50 μL of culture with a DNeasy PowerSoil HTP 96 kit (Qiagen 12955–4). 16S rRNA amplicons were generated using Earth Microbiome Project-recommended 515F/806R primer pairs using the 5PRIME HotMasterMix (Quantabio 2200410) with the following program in a thermocycler: 94 °C for 3 min, 35 cycles of 94 °C for 45 s, 50 °C for 60 s, and 72 °C for 90 s, followed by 72 °C for 10 min. PCR products were cleaned, quantified, and pooled using the UltraClean 96 PCR Cleanup kit (Qiagen 12596–4) and Quant-iT dsDNA High Sensitivity Assay kit (Invitrogen Q33120). Samples were sequenced with 250- or 300-bp reads on a MiSeq (Illumina).
Samples were de-multiplexed with Qiime v. 1.9.1 (Caporaso et al., 2010) using the commands “split_libraries_fastq.py --rev_comp_mapping_barcodes --rev_comp_barcode --store_demultiplexed_fastq --max_bad_run_length 999 --min_per_read_length_fraction .01 --sequence_max_n 999 --phred_quality_threshold 0” and “split_sequence_file_on_sample_ids.py”. Subsequent processing was performed using DADA2 as previously described (Callahan et al., 2016). truncLenF and truncLenR parameters were set to 240 and 180, respectively. Resulting ASV sequences were assigned taxonomy with Qiime, using the commands “assign_taxonomy.py”, “align_seqs.py”, and “make_philogeny.py
Isolation of strains
SICs were resuspended in PBS and plated onto BHI, BHI-S, or GAM plates. Plates were incubated at 37 °C in an anaerobic chamber. After 2 days, colonies were grown in the medium corresponding to the plate from which they were isolated for 2 days, and glycerol stocked. Strains were identified via high-throughput MALDI-TOF mass spectrometry of whole colonies, and subsequent matching of spectra to a reference library with a MALDI Biotyper System (Bruker), following manufacturer’s instructions and including formic acid lysis. 15 distinct strains were obtained and their taxonomy was checked by Sanger sequencing. Genomic DNA was extracted from pure cultures using a DNeasy UltraClean 96 Microbial Kit (Qiagen 10196–4) or DNeasy Blood and Tissue Kit (Qiagen 69504) kit. The 16S gene was amplified using primers 5’AGAGTTTGATCCTGGCTCAG and 5’GACGGGCGGTGWGTRCA, and the amplicon was Sanger sequenced. Taxonomic assignment was performed by alignment using BLAST against the 16S ribosomal RNA sequences (Bacterial and Archaea) database.
Antibiotic sensitivity estimation
Frozen stocks of isolated strains (Table S1) were streaked onto BHI 5% sheep blood agar plates and incubated for 48 h at 37 °C in anaerobic conditions. A single colony of each strain was inoculated into a 3 mL monoculture in BHI, and incubated for 48 h at 37 °C. Cultures were then diluted 1:200 into fresh BHI containing 0.5–32 μg/mL ciprofloxacin. The minimum inhibitory concentration (MIC) for each strain was calculated as the minimum dose necessary to completely inhibit growth as measured by OD after 48 h of incubation.
In vitro S. Typhimurium challenge
SICs were revived in 200 μL BHI in microplates and passaged twice by diluting 1 μL into 200 μL fresh medium every 48 h. A frozen stock of S. Typhimurium strain SL1344 was grown anaerobically at 37 °C for 24 h in BHI. One microliter of the SICs and 1 μL of S. Typhimurium adjusted to OD600=0.1 was diluted into 200 μL fresh BHI. Saturated co-cultures were serially diluted into sterile PBS after 48 h of incubation at 37 °C. Three microliters of a 1:104 dilution were spotted onto LB agar with 50 μg/mL streptomycin to select for S. Typhimurium. Pictures of colonies (Fig. 4B, S8A) were taken after 48 h of incubation at 37 °C in aerobic conditions.
For single-cell quantification of S. Typhimurium levels, we followed the protocol described above to grow SICs and an S. Typhimurium 14028s strain expressing cytoplasmic mCherry (Table S1) prior to co-culturing. SICs and S. Typhimurium were mixed at various ratios and incubated anaerobically for 48 h at 37 °C. Saturated cultures were removed from the anaerobic chamber and spotted onto PBS agar plates to perform high-throughput phase and fluorescence microscopy (Shi et al., 2017).
In vitro antibiotic treatment
SICs were revived in 3 mL BHI and passaged twice before experimentation by diluting 15 μL into 3 mL fresh medium every 48 h. One microliter of the SICs was then diluted into 200 μL fresh BHI containing 0.5–32 μg/mL ciprofloxacin. Growth was measured in a plate reader as described above, and 48 h-old cultures were stored at −80 °C until DNA was extracted.
Isolation of stool proteins and peptides
Isolation of stool proteins broadly followed the protocol in (Gonzalez et al., 2020). Briefly, ~50 mg mouse pellets or pelleted cells from 1 mL saturated cultures were aliquoted into a 96-well plate along with ~600 mg of 0.1-mm ceramic beads (Omni International, #27–6006). To each well, 750 μL lysis buffer (6 M urea, 5% sodium dodecyl sulfate, and 50 mM Tris, pH 8.1) was added and plates were sealed with sealing mats (Omni International, #27–530). Sealed plates were subjected to 10 min of bead beating at 20 Hz using a Qiagen Tissuelyser II. After bead beating, each plate was centrifuged at 3000 rcf at 4 °C for 10 min. Five hundred microliters of the resulting supernatant were transferred to a new 2-mL 96-well plate (Waters, 186002482), sealed with a sealing mat, spun again at 3000 rcf at 4 °C for 10 min, then transferred to a fresh 2-mL plate. Samples were reduced with 10 μL of 500 mM dithiothreitol (Sigma-Aldrich) for 30 min at 47 °C and alkylated with 30 μL of 500 mM iodoacetamide (Sigma-Aldrich) for 1 h at room temperature in the dark. Fifty microliters of the reduced and alkylated supernatant were transferred to a new 2-mL 96-well plate for further processing, while the remaining material was stored at −80 °C.
Supernatant-resident stool proteins were washed, digested, and eluted as described in the Protifi S-trap protocol (http://www.protifi.com/wp-content/uploads/2018/08/S-Trap-96-well-plate-long-1.4.pdf). Briefly, the 50 μL supernatant was acidified with 5 μL of 12% phosphoric acid to which 300 μL S-trap binding buffer was added. Each resulting mixture was loaded into a single well. Positive pressure was used to load the proteins into each well with a Positive Pressure-96 Processor (Waters) with pressure set at 6–9 psi on the “Low-Flow” setting. Loaded proteins were washed with 300 μL Binding Buffer (90% methanol and 10% triethylammonium bicarbonate buffer (TEAB, Sigma-Aldrich, #T7408), adjusted to pH 7.1 using phosphoric acid) five times. After washing, 125 μL digestion buffer (100 mM TEAB and 5 μg trypsin) were added and proteins were digested for 3 h at 47 °C. Peptides were eluted with 100 μL TEAB, followed by 100 μL of 0.2% formic acid, followed by 100 μL of 50% acetonitrile, 0.2% formic acid. These peptides were captured in a 1-mL 96-well plate (Thermo Scientific, #AB-1127) and the volume was dried down in a Centrivap speedvac (Model 7810016). Plated samples were desalted using RP-S cartridges on an Agilent Bravo AssayMAP using the built-in desalting protocol, eluted with 50% acetontrile, and dried down. Plated peptide concentrations were normalized using a Nanodrop ND-1000.
Mass spectrometry
Peptide samples were diluted to 0.5 μg/μL. Subsequently, 1 μL was loaded onto an in-house, laser-pulled 100-μm inner diameter nanospray column packed to ~220 mm with 3-μm 2A C18 beads (Reprosil). Peptides were separated by reversed-phase chromatography on a Dionex Ultimate 3000 HPLC. Buffer A of the mobile phase contained 0.1% formic acid in HPLC-grade water, and buffer B contained 0.1% formic acid in acetonitrile. An initial 2-min isocratic gradient flowing 3% B was followed by a linear increase to 25% B for 115 min, then increased to 45% B over 15 min, and a final increase to 95% B over 15 min, whereupon B was held for 6 min and returned back to baseline in 2 min and held for 10 min, for a total of 183 min. The HPLC flow rate was 0.400 μL/min. Samples were run on a Thermo Fusion Lumos mass spectrometer that collected mass spectrometry data in positive-ion mode within the 400–1500 m/z range with an initial Orbitrap scan resolution of 120,000, followed by high-energy collision-induced dissociation and analysis in the orbitrap using “Top Speed” dynamic identification with dynamic exclusion enabled (repeat count of 1, exclusion duration of 90 s). The automatic gain control was set to 4×105 for FT full mass spectrometry and to 104 for ITMSn.
Peptide/protein database searching
Mass spectra were searched using Proteome Discoverer 2.2 using the built-in SEQUEST search algorithm. Two FASTA databases were searched: Uniprot Swiss-Prot Mus musculus (taxon ID 10090, downloaded January 2017) and a database containing common preparatory contaminants. Target-decoy searching at both the peptide and protein level was employed with a strict false discovery-rate cutoff of 0.05 using the Percolator algorithm built into Proteome Discoverer 2.2. Enzyme specificity was set to tryptic, with static peptide modifications set to cambamidomethylation (+57.0214 Da). Dynamic modifications were set to oxidation (+15.995 Da) and N-terminal protein acetylation (+42.011 Da). Only high-confidence proteins (q < 0.01) were used for analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
16S data analysis
Custom MATLAB R2018a (MathWorks) scripts were used for 16S data analysis.
ASVs were classified as contaminants and removed from further analysis if they appeared with significantly higher relative abundances (p<0.001, t-test) in control samples (water added to DNA extraction step).
Alpha diversity (richness) was measured by rarefying all samples to 5000 reads and calculating the number of taxa with relative abundance >0. Shannon diversity was calculated using , where pi is the relative abundance of the ith taxon and N is the total number of taxa with pi>0. Evenness was calculated using the Pielou index by normalizing Shannon diversity by the maximum Shannon index for the number of taxa: .
Weighted and unweighted Unifrac distances between samples for beta diversity were calculated using custom MATLAB code. Unifrac was calculated as described in (Lozupone and Knight, 2005). Weighted Unifrac was calculated as described in (Lozupone et al., 2007): for samples A and B, , where n is the total number of branches in the tree, bi is the length of branch i, and Ai and Bi are the relative abundances of the taxa that descend from branch i in samples A and B, respectively. No further normalization was performed. Only taxa present at >0.01% in more than two samples were used to calculate distances. Principal coordinate analyses were performed on unweighted Unifrac distances between samples with >5000 reads.
Growth analyses
Maximal growth rate was calculated as the maximal slope of ln(OD) with respect to time (calculated from a linear regression of a sliding window of 11 timepoints) using custom Matlab R2018b (Mathworks) code.
Proteome analysis
Custom MATLAB R2018a (MathWorks) scripts were used to analyze protein abundances. M. musculus analyses were restricted to proteins that were 10 times more abundant in germ-free, humanized, or SIC-colonized mice samples than in any in vitro bacterial cultures in our dataset.
Bacterial invasion
Percentages of S. Typhimurium (Fig. 4C, S8B) were estimated by calculating the proportion of pixels within cell contours segmented from phase images whose fluorescence was above a threshold that clearly separated mCherry-positive and mCherry-negative cells using custom Matlab code.
Statistical analyses
For two-condition comparisons, two-sample t tests were applied. Pearson correlation coefficients and the p-values for testing the hypothesis that there is no relationship between the observed phenomena were calculated on log10(relative abundance data).
Supplementary Material
Acknowledgments
The authors thank Rebecca Culver, Carolina Tropini, Kali Pruss, Alice Cheng, Manohary Rajendram, Surya Tripathi, and Myles Bartholomew for technical assistance; and Rita Oliveira, Lisa Willis, Handuo Shi, and Po-Yi Ho for valuable comments on the manuscript. Some reference strains used in this study were kindly provided by Denise Monack. A.A.-D. is a Howard Hughes Medical Institute International Student Research fellow, a Stanford Bio-X Bowes fellow, and a Siebel Scholar. The authors acknowledge funding from the Allen Discovery Center at Stanford on Systems Modeling of Infection (to K.M.N. and K.C.H.). J.L.S. and K.C.H. are Chan Zuckerberg Investigators.
Footnotes
Declaration of Interests
The authors declare no competing interests.
References
- Adamowicz EM, Flynn J, Hunter RC, and Harcombe WR (2018). Cross-feeding modulates antibiotic tolerance in bacterial communities. ISME J 12, 2723–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranda-Díaz A, Obadia B, Thomsen T, Hallberg ZF, Güvener ZT, Ludington WB, and Huang KC (2021). Bacterial interspecies interactions modulate pH-mediated antibiotic tolerance. Elife 9, e51493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auchtung JM, Robinson CD, Farrell K, and Britton RA (2016). MiniBioReactor Arrays (MBRAs) as a Tool for Studying C. difficile Physiology in the Presence of a Complex Community. Methods Mol Biol 1476, 235–258. [DOI] [PubMed] [Google Scholar]
- Barroso-Batista J, Pedro MF, Sales-Dias J, Pinto CJG, Thompson JA, Pereira H, Demengeot J, Gordo I, and Xavier KB (2020). Specific Eco-evolutionary Contexts in the Mouse Gut Reveal Escherichia coli Metabolic Versatility. Curr Biol 30, 1049–1062. [DOI] [PubMed] [Google Scholar]
- Barthel M, Hapfelmeier S, Quintanilla-Martinez L, Kremer M, Rohde M, Hogardt M, Pfeffer K, Russmann H, and Hardt WD (2003). Pretreatment of mice with streptomycin provides a Salmonella enterica serovar Typhimurium colitis model that allows analysis of both pathogen and host. Infect Immun 71, 2839–2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brook I (2009). The role of beta-lactamase-producing-bacteria in mixed infections. BMC Infect Dis 9, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne HP, Forster SC, Anonye BO, Kumar N, Neville BA, Stares MD, Goulding D, and Lawley TD (2016). Culturing of ‘unculturable’ human microbiota reveals novel taxa and extensive sporulation. Nature 533, 543–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, and Holmes SP (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods 13, 581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7, 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carman RJ, Simon MA, Fernandez H, Miller MA, and Bartholomew MJ (2004). Ciprofloxacin at low levels disrupts colonization resistance of human fecal microflora growing in chemostats. Regul Toxicol Pharmacol 40, 319–326. [DOI] [PubMed] [Google Scholar]
- Cho I, Yamanishi S, Cox L, Methe BA, Zavadil J, Li K, Gao Z, Mahana D, Raju K, Teitler I, et al. (2012). Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature 488, 621–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox LM, Yamanishi S, Sohn J, Alekseyenko AV, Leung JM, Cho I, Kim SG, Li H, Gao Z, Mahana D, et al. (2014). Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell 158, 705–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vos MGJ, Zagorski M, McNally A, and Bollenbach T (2017). Interaction networks, ecological stability, and collective antibiotic tolerance in polymicrobial infections. Proc Natl Acad Sci U S A 114, 10666–10671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethlefsen L, and Relman DA (2011). Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci U S A 108 Suppl 1, 4554–4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan SH, Hold GL, Harmsen HJ, Stewart CS, and Flint HJ (2002). Growth requirements and fermentation products of Fusobacterium prausnitzii, and a proposal to reclassify it as Faecalibacterium prausnitzii gen. nov., comb. nov. Int J Syst Evol Microbiol 52, 2141–2146. [DOI] [PubMed] [Google Scholar]
- Earle KA, Billings G, Sigal M, Lichtman JS, Hansson GC, Elias JE, Amieva MR, Huang KC, and Sonnenburg JL (2015). Quantitative Imaging of Gut Microbiota Spatial Organization. Cell Host Microbe 18, 478–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith JJ, McNulty NP, Rey FE, and Gordon JI (2011). Predicting a human gut microbiota’s response to diet in gnotobiotic mice. Science 333, 101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohlich EE, Farzi A, Mayerhofer R, Reichmann F, Jacan A, Wagner B, Zinser E, Bordag N, Magnes C, Frohlich E, et al. (2016). Cognitive impairment by antibiotic-induced gut dysbiosis: Analysis of gut microbiota-brain communication. Brain Behav Immun 56, 140–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldford JE, Lu N, Bajic D, Estrela S, Tikhonov M, Sanchez-Gorostiaga A, Segre D, Mehta P, and Sanchez A (2018). Emergent simplicity in microbial community assembly. Science 361, 469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez CG, Wastyk HC, Topf M, Gardner CD, Sonnenburg JL, and Elias JE (2020). High-Throughput Stool Metaproteomics: Method and Application to Human Specimens. mSystems 5, e00200–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez N, and Garrido D (2019). Species Deletions from Microbiome Consortia Reveal Key Metabolic Interactions between Gut Microbes. mSystems 4, e00185–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper LV, Littman DR, and Macpherson AJ (2012). Interactions between the microbiota and the immune system. Science 336, 1268–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang I, Park YJ, Kim YR, Kim YN, Ka S, Lee HY, Seong JK, Seok YJ, and Kim JB (2015). Alteration of gut microbiota by vancomycin and bacitracin improves insulin resistance via glucagon-like peptide 1 in diet-induced obesity. FASEB J 29, 2397–2411. [DOI] [PubMed] [Google Scholar]
- Ivanov II, Frutos Rde L, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, Finlay BB, and Littman DR (2008). Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe 4, 337–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson ME, Jakobsson HE, Holmen-Larsson J, Schutte A, Ermund A, Rodriguez-Pineiro AM, Arike L, Wising C, Svensson F, Backhed F, and Hansson GC (2015). Normalization of Host Intestinal Mucus Layers Requires Long-Term Microbial Colonization. Cell Host Microbe 18, 582–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashyap PC, Marcobal A, Ursell LK, Larauche M, Duboc H, Earle KA, Sonnenburg ED, Ferreyra JA, Higginbottom SK, Million M, et al. (2013). Complex interactions among diet, gastrointestinal transit, and gut microbiota in humanized mice. Gastroenterology 144, 967–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehe J, Kulesa A, Ortiz A, Ackerman CM, Thakku SG, Sellers D, Kuehn S, Gore J, Friedman J, and Blainey PC (2019). Massively parallel screening of synthetic microbial communities. Proc Natl Acad Sci U S A 116, 12804–12809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoruts A, and Weingarden AR (2014). Emergence of fecal microbiota transplantation as an approach to repair disrupted microbial gut ecology. Immunol Lett 162, 77–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawley TD, Bouley DM, Hoy YE, Gerke C, Relman DA, and Monack DM (2008). Host transmission of Salmonella enterica serovar Typhimurium is controlled by virulence factors and indigenous intestinal microbiota. Infect Immun 76, 403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone C, and Knight R (2005). UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71, 8228–8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone CA, Hamady M, Kelley ST, and Knight R (2007). Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol 73, 1576–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlane GT, Macfarlane S, and Gibson GR (1998). Validation of a Three-Stage Compound Continuous Culture System for Investigating the Effect of Retention Time on the Ecology and Metabolism of Bacteria in the Human Colon. Microb Ecol 35, 180–187. [DOI] [PubMed] [Google Scholar]
- Mark Welch JL, Hasegawa Y, McNulty NP, Gordon JI, and Borisy GG (2017). Spatial organization of a model 15-member human gut microbiota established in gnotobiotic mice. Proc Natl Acad Sci U S A 114, E9105–E9114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer EA, Knight R, Mazmanian SK, Cryan JF, and Tillisch K (2014). Gut microbes and the brain: paradigm shift in neuroscience. J Neurosci 34, 15490–15496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minekus M, Smeets-Peeters M, Bernalier A, Marol-Bonnin S, Havenaar R, Marteau P, Alric M, Fonty G, and Huis in’t Veld JH (1999). A computer-controlled system to simulate conditions of the large intestine with peristaltic mixing, water absorption and absorption of fermentation products. Appl Microbiol Biotechnol 53, 108–114. [DOI] [PubMed] [Google Scholar]
- Ng KM, Aranda-Diaz A, Tropini C, Frankel MR, Van Treuren WW, O’Laughlin C, Merrill BD, Yu FB, Pruss KM, Oliveira RA, et al. (2019). Recovery of the gut microbiota after antibiotics depends on host diet, community context, and environmental reservoirs. Cell Host Microbe 26, 650–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng KM, Ferreyra JA, Higginbottom SK, Lynch JB, Kashyap PC, Gopinath S, Naidu N, Choudhury B, Weimer BC, Monack DM, and Sonnenburg JL (2013). Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature 502, 96–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoloff H, and Andersson DI (2016). Indirect resistance to several classes of antibiotics in cocultures with resistant bacteria expressing antibiotic-modifying or -degrading enzymes. J Antimicrob Chemother 71, 100–110. [DOI] [PubMed] [Google Scholar]
- Rettedal EA, Gumpert H, and Sommer MO (2014). Cultivation-based multiplex phenotyping of human gut microbiota allows targeted recovery of previously uncultured bacteria. Nat Commun 5, 4714. [DOI] [PubMed] [Google Scholar]
- Reyes A, Wu M, McNulty NP, Rohwer FL, and Gordon JI (2013). Gnotobiotic mouse model of phage-bacterial host dynamics in the human gut. Proc Natl Acad Sci U S A 110, 20236–20241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezzonico E, Mestdagh R, Delley M, Combremont S, Dumas ME, Holmes E, Nicholson J, and Bibiloni R (2011). Bacterial adaptation to the gut environment favors successful colonization: microbial and metabonomic characterization of a simplified microbiota mouse model. Gut Microbes 2, 307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert AM, Sinani H, and Schloss PD (2015). Antibiotic-Induced Alterations of the Murine Gut Microbiota and Subsequent Effects on Colonization Resistance against Clostridium difficile. MBio 6, e00974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Colavin A, Lee TK, and Huang KC (2017). Strain Library Imaging Protocol for high-throughput, automated single-cell microscopy of large bacterial collections arrayed on multiwell plates. Nat Protoc 12, 429–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg ED, Smits SA, Tikhonov M, Higginbottom SK, Wingreen NS, and Sonnenburg JL (2016). Diet-induced extinctions in the gut microbiota compound over generations. Nature 529, 212–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg JL, and Backhed F (2016). Diet-microbiota interactions as moderators of human metabolism. Nature 535, 56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorg RA, Lin L, van Doorn GS, Sorg M, Olson J, Nizet V, and Veening JW (2016). Collective Resistance in Microbial Communities by Intracellular Antibiotic Deactivation. PLoS Biol 14, e2000631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stecher B, Robbiani R, Walker AW, Westendorf AM, Barthel M, Kremer M, Chaffron S, Macpherson AJ, Buer J, Parkhill J, et al. (2007). Salmonella enterica serovar Typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol 5, 2177–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tramontano M, Andrejev S, Pruteanu M, Klunemann M, Kuhn M, Galardini M, Jouhten P, Zelezniak A, Zeller G, Bork P, et al. (2018). Nutritional preferences of human gut bacteria reveal their metabolic idiosyncrasies. Nat Microbiol 3, 514–522. [DOI] [PubMed] [Google Scholar]
- Tropini C, Moss EL, Merrill BD, Ng KM, Higginbottom SK, Casavant EP, Gonzalez CG, Fremin B, Bouley DM, Elias JE, et al. (2018). Transient Osmotic Perturbation Causes Long-Term Alteration to the Gut Microbiota. Cell 173, 1742–1754 e1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venturelli OS, Carr AC, Fisher G, Hsu RH, Lau R, Bowen BP, Hromada S, Northen T, and Arkin AP (2018). Deciphering microbial interactions in synthetic human gut microbiome communities. Mol Syst Biol 14, e8157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker WR, Shepherd ES, and Sonnenburg JL (2017). Tunable Expression Tools Enable Single-Cell Strain Distinction in the Gut Microbiome. Cell 169, 538–546 e512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, Zimmermann-Kogadeeva M, Wegmann R, and Goodman AL (2019). Separating host and microbiome contributions to drug pharmacokinetics and toxicity. Science 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request. Custom code used in this paper is available at doi:10.5281/zenodo.570655410.5281/zenodo.5706554. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.