

Abstract
Perinatal inflammatory stress is associated with early life morbidity and lifelong consequences for pulmonary health. Chorioamnionitis, an inflammatory condition impacting the placenta and fluid surrounding the developing fetus, impacts 25–40% of preterm births. Severe chorioamnionitis with preterm birth is associated with significantly increased risk of pulmonary disease and secondary infections in childhood, suggesting that fetal inflammation may dramatically alter the development of the lung. Here we used intra-amniotic lipopolysaccharide (LPS) challenge to induce experimental chorioamnionitis in a prenatal rhesus macaque (Macaca mulatta) model mirrors structural and temporal aspects of human lung development. Inflammatory injury directly disrupted the developing gas exchange surface of the primate lung, with extensive damage to alveolar structure, particularly the close association and coordinated differentiation of alveolar type 1 pneumocytes and specialized alveolar capillary endothelium. Single cell RNA sequencing analysis defined a multicellular alveolar signaling niche driving alveologenesis that was extensively disrupted by perinatal inflammation, leading to a loss of gas exchange surface and alveolar simplification, with striking resemblance to chronic lung disease in newborns. Blockade of the inflammatory cytokines IL-1β and TNFα ameliorated LPS-induced inflammatory lung injury by blunting stromal responses to inflammation and modulating innate immune activation in myeloid cells, restoring structural integrity and key signaling networks in the developing alveolus. These data provide new insight into the pathophysiology of developmental lung injury and suggest that modulating inflammation is a promising therapeutic approach to prevent fetal consequences of chorioamnionitis.
One-Sentence Summary:
Rhesus macaque, a clinically relevant primate model, demonstrates that anti-inflammatory therapies can prevent perinatal inflammatory lung injury.
INTRODUCTION
The development of a functional gas exchange surface in the fetal lung is required for survival after birth. Mammalian lung development begins with budding of the lung from the anterior foregut endoderm, followed by stereotypic branching of lung tubules that generates the complex airway tree(1, 2). Proliferation and differentiation of epithelial cells in the distal lung generates alveoli, comprising the gas exchange surface of the mature lung(3). In humans and other primates, cellular differentiation and alveolar formation begins in utero, during the third trimester of pregnancy, and continues until adolescence(4–6). Alveolar type 2 (AT2) epithelial cells generate surfactant proteins and lipids to modulate alveolar surface tension(7), and alveolar type 1 (AT1) epithelial cells associate closely with alveolar endothelial cells to form a large surface for the exchange of oxygen and carbon dioxide(3, 8). Together, these processes are termed alveologenesis. At birth, fluid is reabsorbed and cleared from the alveolar space, and ventilation begins with air entry into the alveolus, accompanied by a reduction of pulmonary vascular resistance and increasing capillary blood flow to match ventilation to allow for adequate oxygenation at birth(9). When these carefully coordinated systems fail, neonatal respiratory distress results, which can be rapidly fatal without intensive care.
Cellular and genetic mechanisms directing lung development have been elucidated by studies of murine lung, but there are important structural and temporal differences in development between mouse and human lungs that limit the applicability of murine studies to human disease(10, 11). Recent studies demonstrate key differences in the cell-specific expression of transcription factors (TFs) and signaling molecules in humans compared to mice, during development(12, 13) and adulthood(11, 14). In addition, while humans and other primates have a distinct secretory epithelium lining respiratory bronchioles and alveolar ducts(15), common sites of damage in pulmonary diseases (Figure S1A–B) (16), rodents have a discrete bronchoalveolar duct junction (BADJ) (Figure S1C). Most notably, while murine alveologenesis occurs between postnatal day (PND) 3 and PND14, human alveologenesis begins in utero(4–6).
Defects in lung morphogenesis and lack of pulmonary surfactant are common causes of respiratory failure in the perinatal period, requiring intensive care to support ventilation(7). Incomplete AT2 cell differentiation causes surfactant deficiency and respiratory distress syndrome (RDS) (17), marked by ventilatory failure and hypoxemia requiring respiratory support. Premature infants who require intensive care for pulmonary pathologies are susceptible to developing chronic lung disease of prematurity (CLDP) or bronchopulmonary dysplasia (BPD) (18). These disorders are characterized by alveolar simplification, reduced gas exchange surface, and the need for long-term ventilatory support or supplemental oxygen.
Chorioamnionitis is defined as inflammation of the chorion and/or the amnion of the placenta and complicates 25–40% of preterm births(19–21). Importantly, chorioamnionitis is associated with increased risk of multiple respiratory comorbidities following delivery(22, 23), and is considered a distinct risk factor for the development of respiratory disease in childhood and later life(24). Numerous studies have demonstrated that chorioamnionitis is associated with increased pro-inflammatory cytokines in the amniotic fluid that contact the developing lung in utero(25, 26). The inflammatory milieu of chorioamnionitis is hypothesized to cause tissue injury and remodeling leading to lung disease in survivors.
We sought to investigate how inflammatory injury to the developing fetus alters the trajectory of lung development in the clinically relevant and developmentally appropriate non-human primate rhesus macaque (Macaca mulatta) model. Owing to continued scientific and clinical advances, most 26-week and later premature human infants can be successfully resuscitated after birth and supported in the neonatal intensive care unit, so human tissue obtained at these developmental times comes from small numbers of infants with severe non-pulmonary disease or after medical intervention(27). Given these factors, rhesus provides a unique opportunity to perform controlled experiments focused on understanding mechanisms of developmental lung injury. We previously showed that exposure of developing rhesus to intra-amniotic LPS causes intra-uterine inflammation and histological changes in the placenta consistent with chorioamnionitis(28). LPS robustly induces IL-1β, TNFα, and IL-6 after 16h in both lung tissue and alveolar wash in rhesus(28–30) and leads to rapid infiltration and activation of neutrophils and myeloid cells in the fetal rhesus lung(29). These findings mirror those from murine models of prenatal LPS, which induces dramatic in utero inflammation(31), exacerbates response to postnatal hyperoxia(32, 33), and causes long term changes to lung structure similar to CLDP/BPD (34). Given the striking differences between primate and murine lung development, the applicability of murine findings to human health and impacts of intra-uterine inflammation on gestational primate alveologenesis remain unclear.
Here, we used a combination of histological analysis, high content confocal imaging, and single cell RNA sequencing (scRNAseq) to show that rhesus lung development recapitulates critical aspects of human alveologenesis in the third trimester of gestation, with serial differentiation of epithelial, endothelial, and mesenchymal lineages required for morphological and physiological maturation of the distal lung. Developmental inflammation caused by intra-amniotic LPS leads to extensive lung injury, with significant disruption of alveolar structure, effacement of septa, injury to progenitor lineages, and loss of gas exchange surface. LPS exposure alters developmental signaling between alveolar cells, with substantial changes to signaling in both mesenchymal and lung innate immune lineages. In combination, these insults lead to alveolar simplification histologically resembling human CLDP/BPD. Blockade of major chorioamnionitis-associated cytokines IL-1β and TNFα (22, 25, 26, 28, 29) protects the developing lung from inflammatory injury, blunting histological disruption, and restoring the cellular niche and signaling networks of the alveolus. Together, these data provide new insight into key mechanisms of developmental lung injury and highlight targeted inflammatory blockade as a potential therapeutic approach to ameliorate lung injury in the vulnerable neonatal population.
RESULTS
Inflammatory injury disrupts the developing alveolar gas exchange surface
To evaluate dynamics of the development of the gas exchange surface in late gestation primates, we examined rhesus lung at multiple late prenatal time points (Figure 1A–J; S1A–F; S2A–O). Rhesus trimesters are approximately 55 days, compared to 90 days in humans; therefore, gestational day (GD) 105 equates to ~25–26 weeks of human gestation, GD130 to ~30–32 weeks, and GD150 to ~35–36 weeks(35) (Figure 1A). Morphologically, GD105 represents the transition from late canalicular to early saccular lung, while GD130 represents late saccular to early alveolar stage, and extensive alveolarization is evident by GD150 (Figure 1B–J; S1D–F). During alveolarization, alveolar septal structures (AS) expand as the gas exchange surface develops; epithelial and endothelial lineages form close interactions along the extending AS, preparing the lung for air breathing (Figure 1H–J). As alveologenesis proceeds, AT1 and AT2 cells (Figure 1E–G) are progressively specified while alveolar structural complexity increases with septal maturation. Recent mouse and human studies defined EDNRB+ endothelial cells as specialized alveolar capillaries (or aerocytes) which are critical for gas exchange(36–38). We detect similar maturation of the alveolar capillary network (Figure 1H–L) with increasingly close association and co-localization with AT1 cells during development of the primate gas exchange surface. Quantification (Figure 1K–L; S3A–D) demonstrated progressive increase in EDNRB+ cell surface volume and AT1 number, with close association seen by IHC (Figure 1H–J). Histological analysis (Figure S2J–P) and bulk RNA sequencing (Figure S4A–H) demonstrate high concordance of proliferative and global TF expression dynamics with murine models of lung development, but with structural and temporal similarities to human lung development (Figure S1A–C; S2Q). Therefore, rhesus macaque is a translationally relevant model of lung development, and GD130 is a period of active morphological and cellular maturation of alveolar septa and capillary networks.
Figure 1. Alveolar simplification and loss of gas exchange surface after perinatal inflammatory injury.
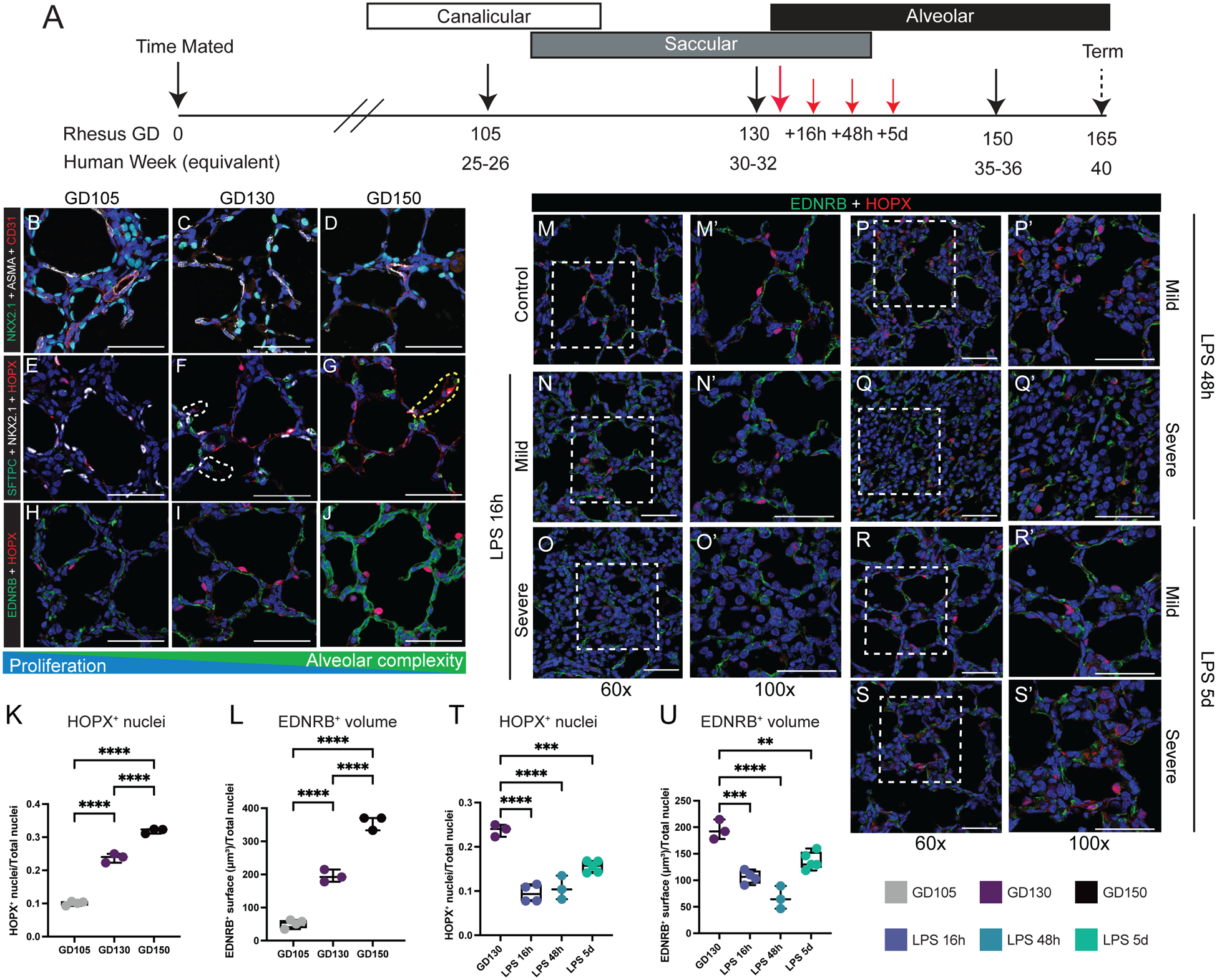
(A) Experimental design. Control animals obtained at at GD105 [canalicular; n=3], GD130 [saccular; n=11], and GD150 [alveolar; n=3] (black arrows). For injury, rhesus macaque dams received intra-amniotic LPS injection at GD130, with subsequent fetal delivery by C-section at 16h (n=8), 48h (n=3), or 5d (n=7) after injury (red arrows). (B-D) NKX2.1+ epithelial cells (green) form close interactions with both ASMA+ myofibroblasts (white) and CD31+ endothelial cells (red). (E-G) Epithelial differentiation of HOPX+ AT1 cells (red) and SFTPC+ AT2 cells (green) increase as development progresses. (H-J) Interactions of HOPX+ AT1 cells (red) and EDNRB+ alveolar capillary endothelial cells (ACs; green) increase during patterning of the developing gas exchange surface. K-L) Quantification of HOPX+ nuclei (L) and EDNRB+ surfaces (L) at each stage of lung development. (M-S) Extensive disruption of gas exchange surface, shown through the disordered patterning and interactions of HOPX+ AT1 cells (red) and EDNRB+ ACs (green) in both mild and severely injured regions up to 5d following LPS. T-U) Quantification of HOPX+ nuclei (T) and EDNRB+ surfaces (U) during injury. Scale bars = 50 μm. Full list of samples and treatment groups in Table S1. ** = p <0.01, *** = p <0.001, **** = p < 0.0001 by Kruskal-Wallace test.
To model fetal chorioamnionitis, we treated rhesus dams at GD130 with intra-amniotic injection of endotoxin/lipopolysaccharide (LPS), an injury shown to mimic the inflammation present during chorioamnionitis in prenatal mouse, rabbit, pig, and sheep(25, 26, 30, 39), as well as rhesus macaque in prior studies from our group(26, 28, 30, 39). Here, we evaluated the hypothesis that this inflammatory milieu directly impairs the developmental progression of the rhesus lung. By 16h after LPS treatment (Figure 1M), extensive lung injury is evident (Figure 1M–U; 2A-J) with areas of both mild and severe disruption of tissue architecture. Inflammation impairs close opposition of AT1 cells and alveolar capillaries in both mildly and severely injured regions and causes loss of AS, most obvious at 48h after injury (Figure 1P–Q; 2C, F). Overall, in both mildly and severely injured regions, there is alveolar simplification following inflammatory injury (Figure 2A–D) as evidenced by significant decreases in mean linear intercept (Lm, p<0.001) and surface density of airspaces (Svair, p<0.001) (Figure 2G–J). More alveolar septa are detectable at 5d post-LPS (Figure 1R–S; 2D, F), primarily in mildly injured regions (Figure 2P, W). In severely injured regions, we also observed loss of differentiated epithelial cells (Figure 2K–O) and multiple endothelial lineages (Figure 2R–V, Y-CC) with partial recovery at 5d (Figure 2P–Q, W-X, DD-EE). Since chorioamnionitis and prematurity are known to increase risk of secondary viral infection(23), we assessed expression of the SARS-CoV-2 priming protease TMPRSS2 in control and injured lungs (Figure S5), and found no significant expression, even with inflammatory stress, concordant with recent mouse and human data(40). Together, our findings suggest LPS-mediated inflammation causes dramatic and persistent injury to the developing primate lung, with major alterations in cell quorum and tissue architecture resulting in pathological findings analogous to those found in premature infants after lung injury(18).
Figure 2. Extensive disruption of developing compartments of the primate lung after LPS-induced inflammation leading to alveolar simplification.
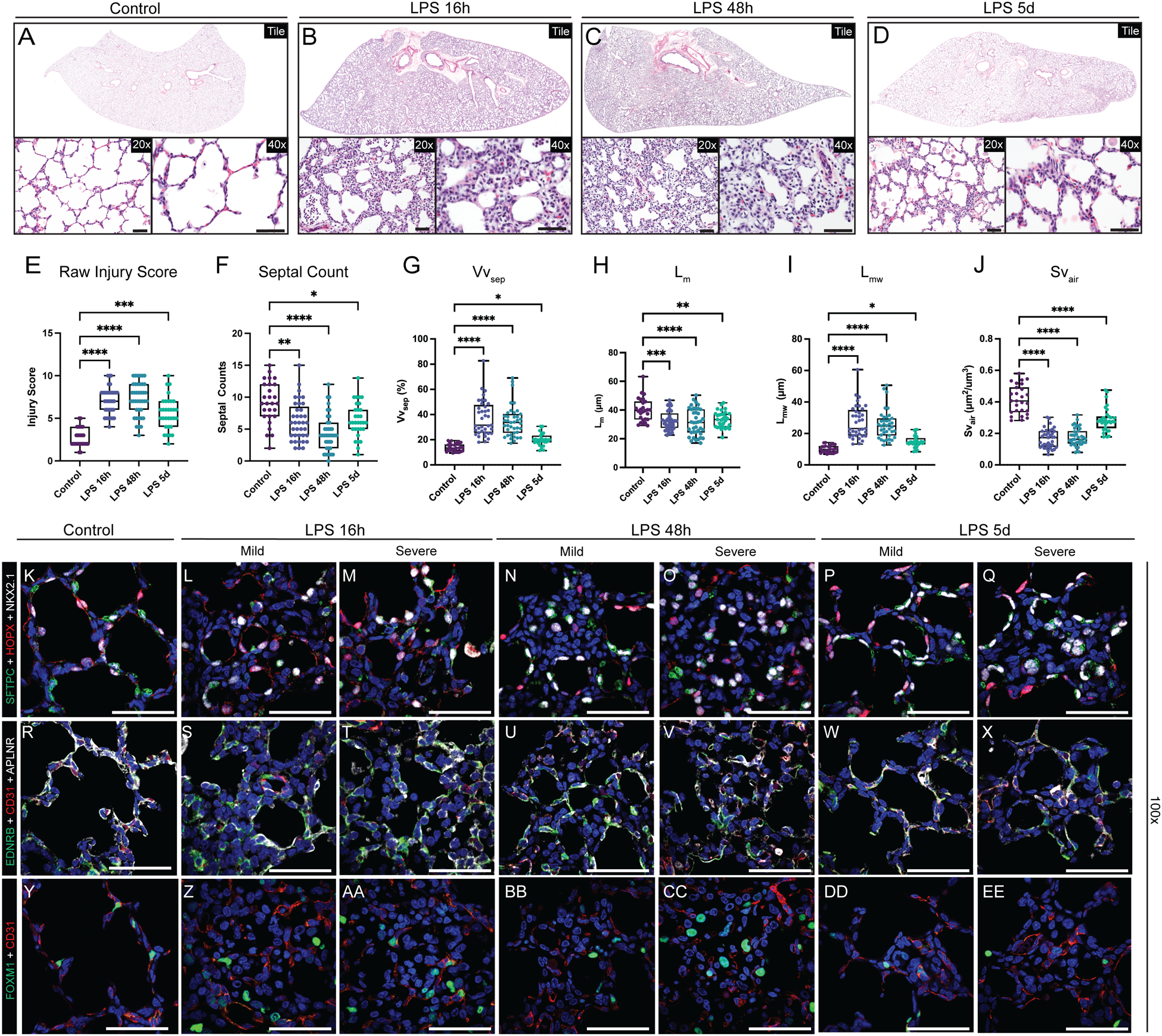
A-D) Histological analysis of LPS-induced lung injury at 16h, 48h, and 5d post-LPS. (E-J) Quantification of GD130 controls, LPS 16h, LPS 48h, LPS 5d. (E) Lung injury score calculated by ATS criteria (range = 0–10). (E) Counts of alveolar septa per field. G-J) Lung morphometry (VVsep [volume density of alveolar septa], Lm [mean linear intercept of air spaces], Lmw [mean transsectional wall length], and SVair [surface area density of air spaces]) demonstrating alveolar simplification. K-EE) Evaluation of epithelial (K-Q), differentiated capillary endothelial (R-X), and endothelial progenitor (Y-EE) populations after LPS-induced lung injury. Severe injury observed at 16h and 48h, with mild morphological improvement by 5d post-LPS. (K-Q) Destruction of organized alveolar epithelium, specifically loss of SFTPC+ AT2 cells (green) and severe damage to/disorganization of HOPX+ AT1 cells (red), with the most severe injury noted at 16h and 48h. (R-X) Loss of EDNRB+ alveolar capillaries (green) and disorganization of APLNR+ general capillaries (white) following LPS injury. (Y-EE) Nearly all FOXM1+ (green) cells lack expression of endothelial CD31, signifying loss of the CD31+/FOXM1+ proliferative endothelial progenitors. * = p <0.05, ** = p <0.01, *** = p <0.001, **** = p < 0.0001 by Kruskal-Wallace test. Scale bars = 50 μm.
A single cell atlas of developmental lung injury
To further evaluate pulmonary tissue remodeling and develop insight into the pathophysiology of perinatal lung injury, we performed single cell RNA sequencing (scRNAseq) on GD130 fetal lungs from control (IA saline) and experimental animals (16h after IA LPS) (Figure 3A; S6A–I). To specifically evaluate injury response in the developing primate gas exchange surface, we performed a focused analysis of epithelial, endothelial, and mesenchymal cell populations (Figure 3B–C). Clusters for subtypes of rhesus epithelium (Figure 3D), endothelium (Figure 3E), and mesenchyme (Figure 3F) were identified using Seurat v3(41), with parameters set to identify major cell populations rather than multiple similar subclusters. We used LungMAP(27, 42) and Human Cell Atlas(43) annotations from human lung to determine cell type identity on the basis of enriched expression of key marker genes. We identified major cell types described in humans with highest similarity to datasets from younger patients(27, 42).
Figure 3. Single cell interrogation of developmental lung injury.
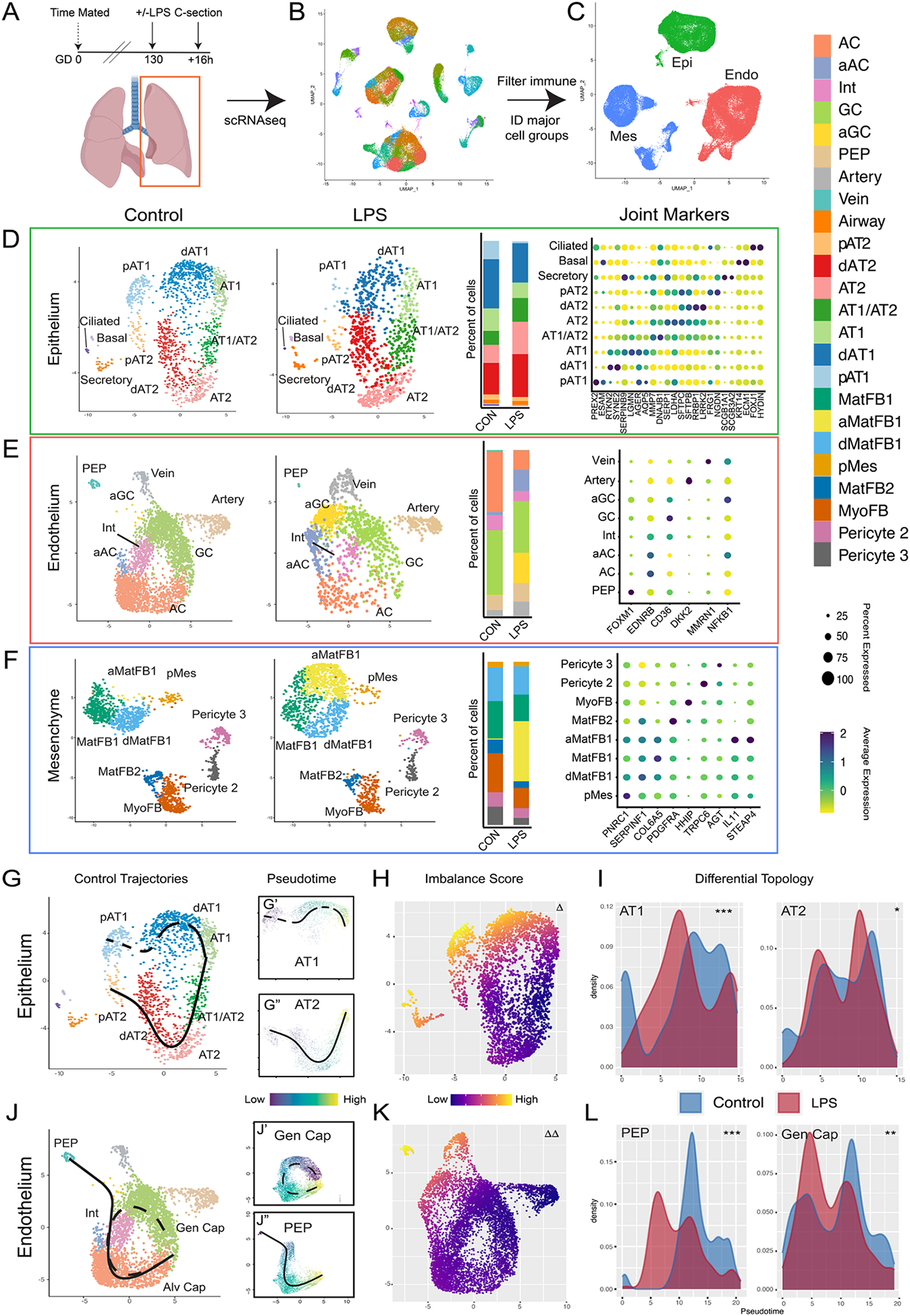
A-C) scRNAseq of control (n=2) and LPS-treated (n=2) primate lungs. UMAP projection of all cells (B) used to identify cells expressing PECAM1/CD31 (Endo), CDH1 (Epi), or COL1A1 (Mes) (C). D-F) Control and LPS UMAPs, cell type proportion per condition, and expression of canonical cell marker genes from epithelial (D), endothelial (E), and mesenchymal (F) cells. G-I) Cellular trajectories from Slingshot show AT1 and AT2 trajectories arising from separable progenitor lineages (G). LPS causes significant transcriptional imbalance (H) in AT1 cells and disrupts development of AT1 but not AT2 cells (I). J-L) Alveolar capillaries arise from both PEP and general capillaries in control lungs (J). LPS disrupts both AC trajectories (K, L). Δ = p <1e-10; Δ Δ = p <1e-13 for differential trajectory topology. *=p<1e-3, **=p<1e-10, ***= p<2.2e-16 for differential trajectory progression. AC = alveolar capillary, aAC = activated AC, Int = intermediate capillary, GC = general capillary, aGC = activated GC, PEP = proliferative endothelial progenitor, MatFB = matrix fibroblast, aMatFB = activated matrix fibroblast, dMatFB = differentiating matrix fibroblast, MyoFB = myofibroblast, pMes = proliferative mesenchyme, AT2 = alveolar type 2, pAT2 = progenitor AT2, dAT2 = differentiating AT2, AT1 = alveolar type 1, pAT1= progenitor AT1, dAT1 = differentiating AT1, CON = control.
In the alveolar epithelium, we detect multiple distinct cellular states of AT1 and AT2 cells, and a cell population expressing markers of both cell types similar to the AT1/AT2 cell state described during murine lung development(44) and in humans(27). Recent lineage tracing suggested that AT1 and AT2 progenitor populations diverge early in mouse lung development, with distinct AT1- and AT2-defined clones emerging by E13.5 and becoming increasingly common as development proceeds; scRNAseq enabled separation of AT1 and AT2 progenitors from more fully differentiated distal epithelium by E17.5(45). Similarly, we identified distinct, separable progenitor AT1 and AT2 lineage clusters in prenatal rhesus. Pseudotemporal lineage inference(46) (Figure 3G) demonstrated separable trajectories supporting AT1 and AT2 cell evolution from these progenitors to fully differentiated cells through distinct differentiation intermediates. Lineage inference suggested that the AT1/AT2 cell state derives preferentially from the AT2 lineage branch, and predicts some AT1 differentiation through this state, in accordance with murine studies showing AT1 cells can arise from AT2 progenitors(47–49).
Within the endothelial population, in addition to arterial and venous endothelial cells, we identified three alveolar capillary endothelial lineages, which correspond to alveolar (AC/aerocyte) and general capillary (GC) populations described in mouse and human(36–38), as well as an intermediate population expressing markers for both AC and GC (Figure 3E). We also identified a separate endothelial population, characterized by a proliferative signature (despite cell cycle gene normalization) and distinct progenitor gene expression (Figure 3E). Pseudotime analysis predicted that this population functions as a progenitor lineage for both alveolar and general capillaries (Figure 3J). Comparison of the gene signature and pseudotime trajectory of these cells to previously reported single cell data from mouse development(50, 51) and early human development(52) suggested that this population, marked in our studies by FOXM1, represents a conserved proliferative endothelial progenitor (PEP) lineage for the lung (Figure S7A–K). Finally, we observed mesenchymal heterogeneity (Figure 3G) similar to that reported in both mouse and human, with multiple populations of pericytes, matrix fibroblasts, and myofibroblasts, although we do not detect a distinct lineage of smooth muscle cells, possibly owing to relatively lower representation of proximal airway in our dataset.
Disrupted patterning of the primate gas exchange surface during prenatal injury
We evaluated the impact of LPS injury on developmental trajectories of key epithelial and endothelial progenitor populations during alveolar patterning. We noted loss of progenitor lineages in both the epithelium and endothelium (Figure 3D–E), with reduction in quorum of AT1 and AT2 progenitors (Figure 3D) as well as PEPs (Figure 3E) in injured lungs. We combined pseudotime inference(46) and differential trajectory analysis(53) to directly evaluate the impact of LPS injury on developmental trajectories. In the epithelium, the AT1 differentiation trajectory is strikingly disrupted by LPS, with loss of AT1 progenitors and fully differentiated AT1 cells (Figure 3H–I). Conversely, the AT2 trajectory is only mildly disrupted, with loss of AT2 progenitors but compensatory differentiation along the baseline trajectory. In the endothelium, AC differentiation is reduced from both WT trajectories, with loss of FOXM1+ PEPs (Figure 2Y–EE; S7G–L) and decreased differentiation of AC along the trajectory from GCs, accompanied by preferential accumulation of EDNRB+/APLNR+ intermediates in both trajectories (Figure 3K–L). There was also a clear transition of both ACs and GC to an activated, IL-1- and TNF-responsive state (Figure 3E; S6G). Therefore, concordant with histological data, trajectory analysis confirms that a predominant effect of LPS on the developing lung is disorganization of critical AT1-AC interactions.
Immune response and loss of developmental signaling networks in the injured alveolus
Global analysis of differential gene expression (Figure S6F) of injured versus control lungs (Figure 4A) demonstrated extensive inflammatory pathway activation in all cell populations, with generalized activation of NFKB1 and RELA targets consistent with active NFκB signaling throughout lung structural cells (Figure S6G). Gene expression differences were common among related cell types and were consistent with previous observations that NFκB is an important signaling mechanism regulating the timing of mouse lung maturation(54). Markers of AT2 cell maturity increased, especially secretory and ER-related gene ontology (GO) terms (Figure 4A), consistent with prior reports that inflammation promotes AT2 maturation(39, 55). ACs and GCs both showed significant inflammatory activation with a shift to an activated transcriptional state characterized by expression of HIF1α-mediated hypoxia target genes (Figure S6H) and loss of angiogenic factors (Figure S6I). Matrix fibroblasts, particularly type 1 matrix fibroblasts, also showed response to LPS, taking on an inflammatory state marked by high level expression of IL-1 and TNF target genes.
Figure 4. Inflammatory injury disrupts cellular and developmental niche signaling in the alveolus.
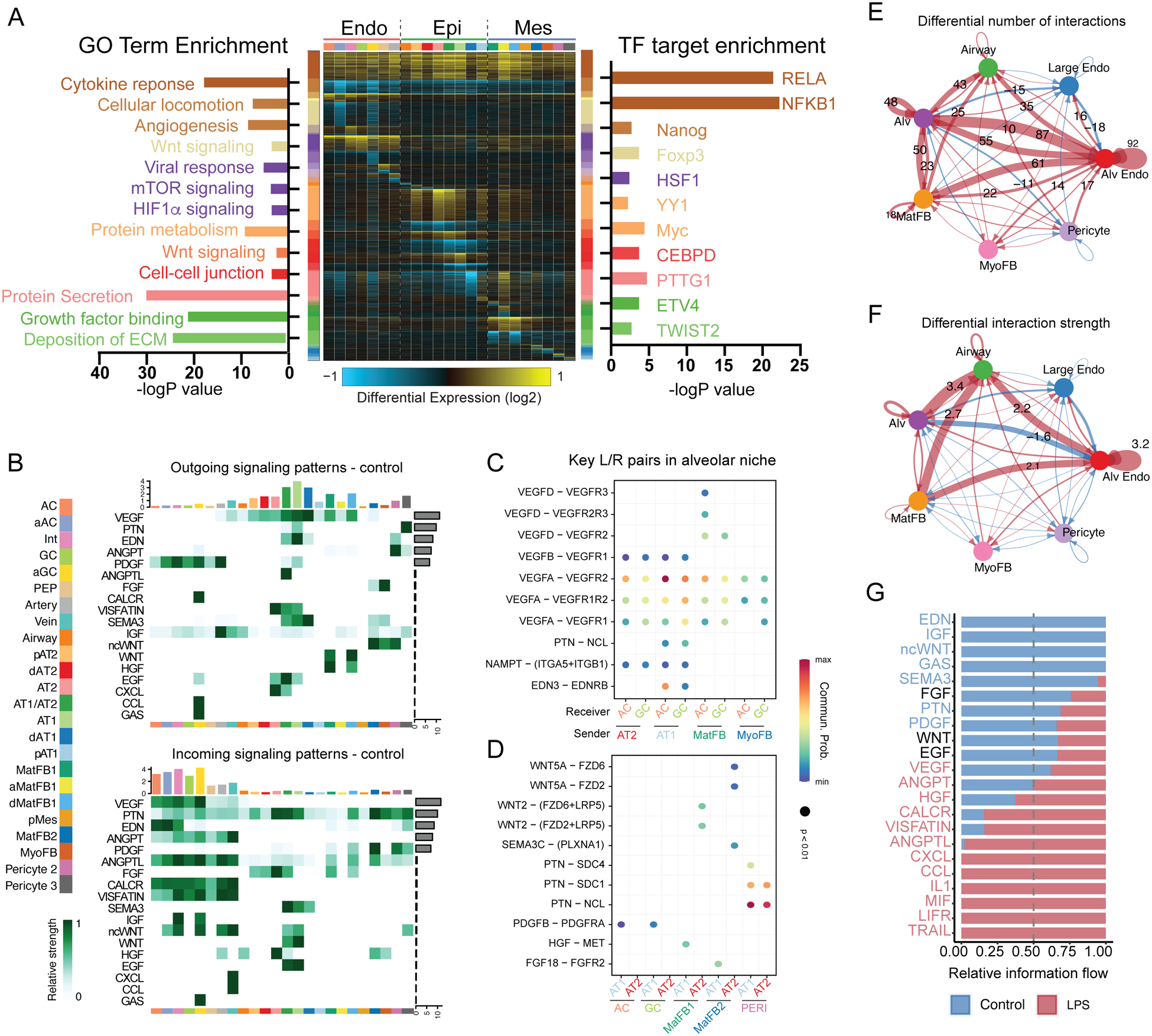
A) cellHarmony heatmap of LPS vs. control – differentially expressed genes (rows) for each cell population (columns), highlighting GO terms enriched in each gene set (left), and experimentally observed TF-target gene-module enrichment (right). Cell types colored as indicated in the sidebar. Y axis bar colors are based on differentially expressed gene sets, and bar size is the −log(p-value) for enrichment in that gene set. B-G) CellChat analysis of signaling milieu of the developing alveolus. B) Major ligand/receptor (L/R) relationships displayed as heatmap showing outgoing ligands (top) and incoming receptivity (bottom), with overrepresented signaling pathways (left Y axis), cell identity (bottom X axis, colors corresponding to sidebar cell identity), relative strength of the predicted signaling pathway in the overall network (right Y axis), and relative contribution of the cell population to the overall signaling milieu (top X axis). C-D) Key L/R interactions (L/R pair [Y axis]; sender/receiver cells [X Axis]) targeting ACs and general capillaries (C) or AT1 and AT2 cells (D). E-G) Changes in signaling milieu of LPS-treated lung - increased signaling in control (blue); increased signaling with LPS (red). Significant increases in number (E) and strength (F) of interactions occur with LPS, with shift from developmental signaling pathways enriched in the baseline alveolar niche to increased inflammatory signaling in the damaged alveolar niche (G). Cell identity abbreviations same as Figure 3. ECM = extracellular matrix.
We applied CellChat(56), which uses cell-specific expression of ligands and receptors to generate a network identifying key overrepresented pathways in single cell data, to decipher the signaling niche of the developing alveolus and define how inflammatory injury disrupts these communication patterns (Figure 4B–G). In general, major signaling pathways known to pattern murine distal lung development are recapitulated in rhesus, with similar cellular expression and receptivity patterns. For example, the WNT signaling pathway is defined by WNT2 ligand expression from a subset of matrix fibroblasts, while WNT5A is expressed primarily in myofibroblasts and pericytes; the response to both signals is prominent in rhesus alveolar epithelial and endothelial lineages, as expected from data in mouse lung and human precision-cut lung slices(47, 57, 58). We identified vascular endothelial growth factor (VEGF), pleiotrophin (PTN), endothelin (EDN), and platelet-derived growth factor (PDGF) signaling as major contributors to alveolar maturation (Figure 4B). Specifically, VEGFA and EDN3 ligand from developing AT1 cells signal to developing alveolar capillaries, likely contributing to the patterning of the alveolar gas exchange surface (Figure 4C). Contemporaneously, WNT, PTN, FGF, and HGF signals from mesenchymal cells promote epithelial maturation (Figure 4D). Matrix fibroblasts function as a major mesenchymal signaling hub, while myofibroblasts, which appear to be mechanically important in dynamic maturation of alveoli(59) (Figure 1B–D), are less prominent in the enriched signaling network. Together, these data define a baseline alveolar signaling niche, containing both conserved signaling interactions (e.g., WNT, FGF) and newly identified interactions (e.g., EDN), driving the patterning of the alveolar gas exchange surface in primates.
This alveolar signaling niche was dramatically disrupted by LPS-induced inflammatory injury. Differential interaction analysis demonstrated increases in both the number and strength of signaling interactions after LPS injury, with more interactions between alveolar epithelium, endothelium, and mesenchymal lineages (Figure 4E–F). Growth factors and maturation signals dominated the control signaling milieu, while LPS induced increases in CCL, CXCL, IL1, and TNF signaling pathways, consistent with a generalized inflammatory injury to the lung (Figure 4G). Corresponding loss of major developmental signals, including EDN, IGF, PTN, and WNT, implied damage to the developing AT1-AC network (Figure 4E–G). These changes were concordant with the observed histological disruption of the gas exchange surface, emphasizing that developing ACs are a key target of injury during late gestation. In the mesenchyme, the large, LPS-specific population of activated matrix fibroblasts (Figure 3F) appeared to elaborate pro-inflammatory signaling in the injured lung (Figure S8A–B), with robust expression of CCL and CXCL chemokines and decreased expression of WNT and HGF ligands. The net effect was a signaling milieu dominated by inflammatory ligands with reductions in major developmental pathways. These findings imply that direct inflammatory injury to the lung signaling niche and disruption of the developmental trajectory of key progenitors combine to underlie the phenotype of alveolar simplification evident at 5d post LPS exposure.
Combined blockade of IL-1 and TNF signaling prevents inflammatory lung injury
We proceeded to test the hypothesis that direct blockade of inflammatory pathways driving experimental chorioamnionitis could protect the developing lung from injury. We treated pregnant rhesus dams with anakinra (100mg, Kineret, SOBI), a potent IL-1 receptor antagonist, and adalimumab (40mg, Humira, AbbVie Inc), an anti-TNF monoclonal antibody, by subcutaneous injection 3h prior to LPS, and by intra-amniotic injection (anakinra 50mg and adalimumab 40mg) 1h prior to LPS (Figure 5A). This combination blockade did not reduce myeloid or neutrophil accumulation in the lung compared to LPS, but modestly reduced BAL inflammatory cytokine levels and the expression of TLR, IL1, and TNF response pathways in several immune lineages of LPS-treated animals (see detailed analysis in our companion report (29)). In spite of these modest changes, inflammatory blockade provided remarkable protection to lung alveoli, reducing lung injury severity with many regions nearly indistinguishable from uninjured lung (Figure 5B–D). Histological quantification demonstrated normalization of septal numbers, injury score, and lung morphometry (Figure 5E–J). IHC demonstrated improvement in both lung morphology and cellular relationships. AT1 and AC structural organization improved (Figure 5K–S), with normalization of AT1 cell number (Figure 5CC) and EDNRB+ surface volume (Figure 5DD).
Figure 5. Combined IL-1 and TNF blockade protects the developing lung from inflammatory injury.
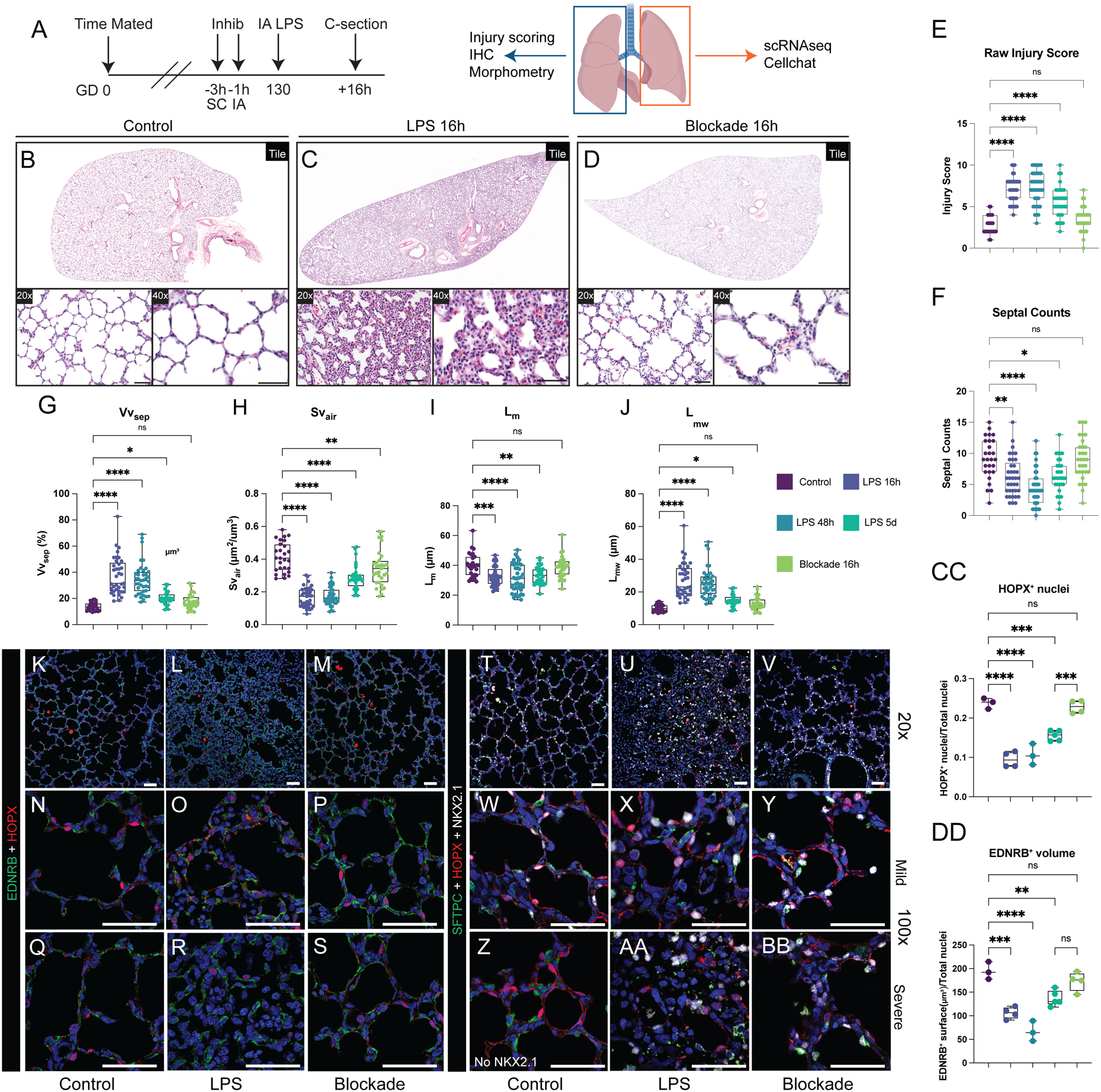
A-D) Experimental design and histological analysis of LPS-induced lung injury following combination blockade. E-J) Quantification of lung injury score (E), septal number (F), and lung morphometry (G-J) show near normalization compared to control and significant improvement compared to LPS. Quantification data reproduced from Figure 2 for comparison. K-S) Combination blockade prevents disruption of AT1/AC interactions. T-BB) Maintenance of differentiated epithelial cells in alveoli after combination blockade. CC-DD) Quantification of EDNRB+ surface volume (CC) and HOPX+ nuclei (DD) following combination blockade. Concordant with morphometry, AT1 cell numbers and EDNRB+ volume improve with blockade. Quantification data reproduced from Figure 1 for control and LPS comparison. * = p <0.05, ** = p <0.01, *** = p <0.001, **** = p < 0.0001 by Kruskal-Wallace test. Scale bars = 50μm.
We compared scRNAseq findings from LPS treatment, control, and combined cytokine blockade (Figure 6). Following blockade, cellular quorum by scRNAseq returned to near baseline levels, with both epithelial (Figure 6B–C) and endothelial progenitors (Figure 6D–E) observed at control levels. Nearly 80% of differentially regulated genes in LPS lungs were normalized after blockade, with extensive improvement in expression of genes associated with angiogenesis, cell cycle, and developmental pathways (Figure 6F–G). Signaling analysis with CellChat demonstrated improvement in major signaling pathways including EDN, PTN, FGF, and HGF after TNF and IL-1 blockade (Figure 6F–G). Despite these changes, a distinct inflammatory signal remained in blockade lungs compared to control, suggesting that the inflammatory blockade provided by anakinra and adalimumab was partial (Figure 6G). Strikingly, AT1 and AC cells showed improvements in immune response pathways and upregulated reciprocal ligand/receptor pairs in the EDN, PTN, and IGF pathways (Figure 6H–J). These results demonstrate that the combination of IL-1 and TNF blockade is sufficient to prevent severe lung injury in experimental chorioamnionitis and imply that protection of progenitor populations and maintenance of development signaling networks are major effects of combination blockade in protecting the developing alveolus from injury.
Figure 6. Combined IL-1 and TNF blockade maintains cellular quorum and alveolar niche signaling during LPS-mediated inflammatory injury.
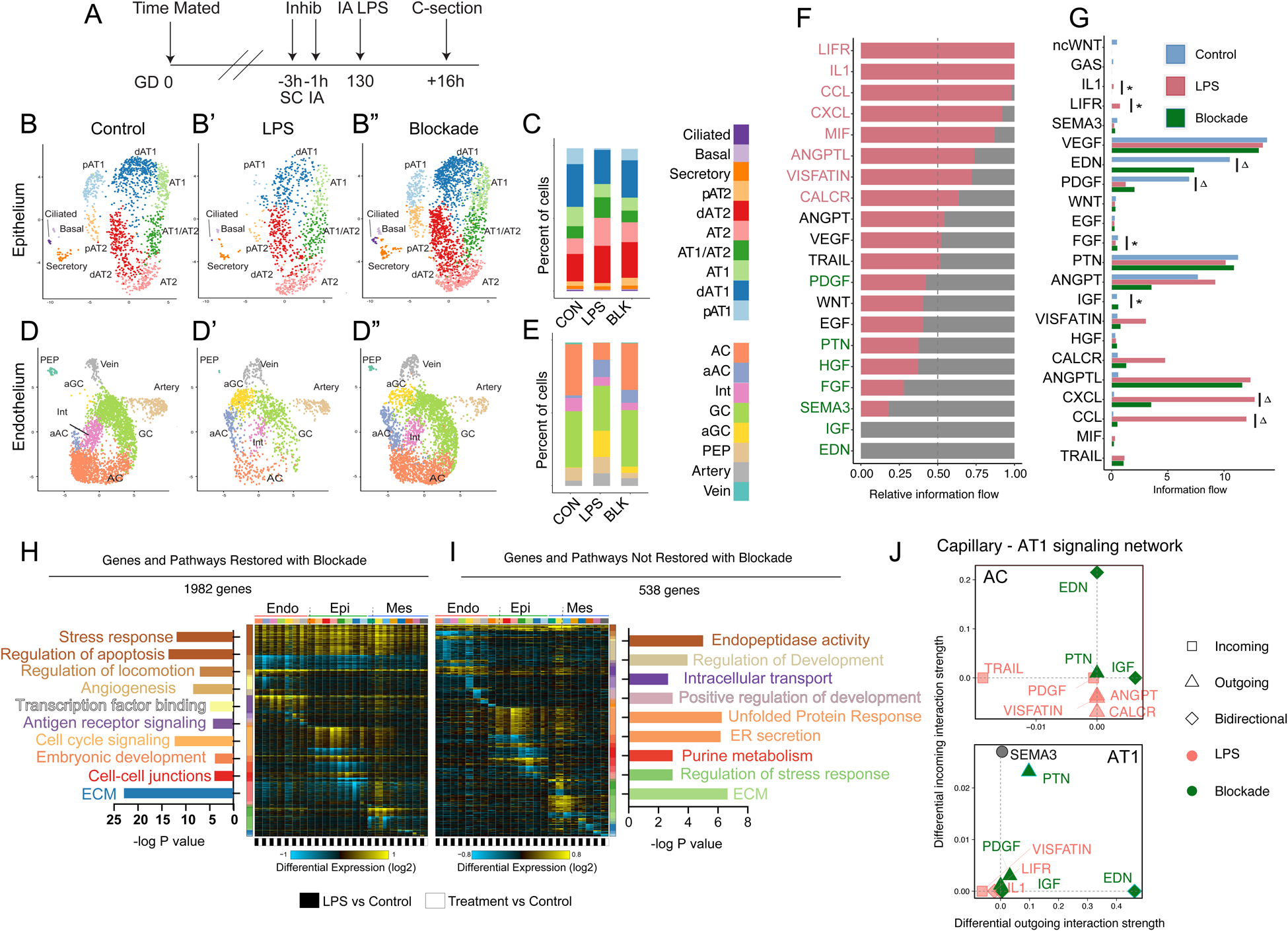
A) Experimental design, as in Figure 5. B-E) UMAP of epithelial (B) and endothelial (C) cell clusters from control (n=2, B, D, as in Fig 3), LPS-treated (n=2, B’, D’, as in Fig 3) and combination blockade (n=3, B”, D”) animals. C, E) Relative proportions of each cell type. F) CellChat analysis of the alveolar signaling milieu after blockade compared to LPS – enriched signals in LPS (red text); enriched signals in combined blockade (green text). Lung stromal signaling milieu reverts markedly compared to LPS treatment. FGF, HFG, pleiotrophin, and endothelin signaling improve with blockade. G) Comparison of intensity of major signaling pathways in control, LPS, and blockade conditions demonstrates normalization of some pathways (* = not significantly different than control, p>0.05 for control vs blockade) while others improve but do not normalize (Δ = significantly improved from LPS, but significantly elevated compared to control, p<0.05 for comparison to LPS and comparison to control), including most inflammatory pathways. H-I) GO term and pathway analysis of differentially regulated genes by cellHarmony, delineating the global effect of combination blockade. Genes and pathways are highlighted which revert (H) or do not revert (I) to near baseline expression with blockade. J) CellChat evaluation of AT1-AC interactions with LPS and following blockade.
Perinatal inflammation damages the developing alveolar signaling niche.
We hypothesized that blockade may reduce inflammatory activation of resident lung stromal and immune cells, preventing secondary injury to lung structure. To identify immune-stromal signaling relationships in LPS and blockade conditions, we used our unfiltered scRNAseq data set, containing both structural and immune cell lineages, and integrated cells from all captures to identify the predominant populations (Figure 7A). Signaling analysis identified myeloid immune lineages, including monocytes, macrophages, and dendritic cells, as a major signaling partner with structural cells in both LPS and blockade conditions (Figure 7B–C).
Figure 7. Combined IL-1 and TNF blockade prevents inflammatory injury to the alveolar signaling niche via modulation of CCL and CXCL cytokines in matrix fibroblasts and innate myeloid cells.
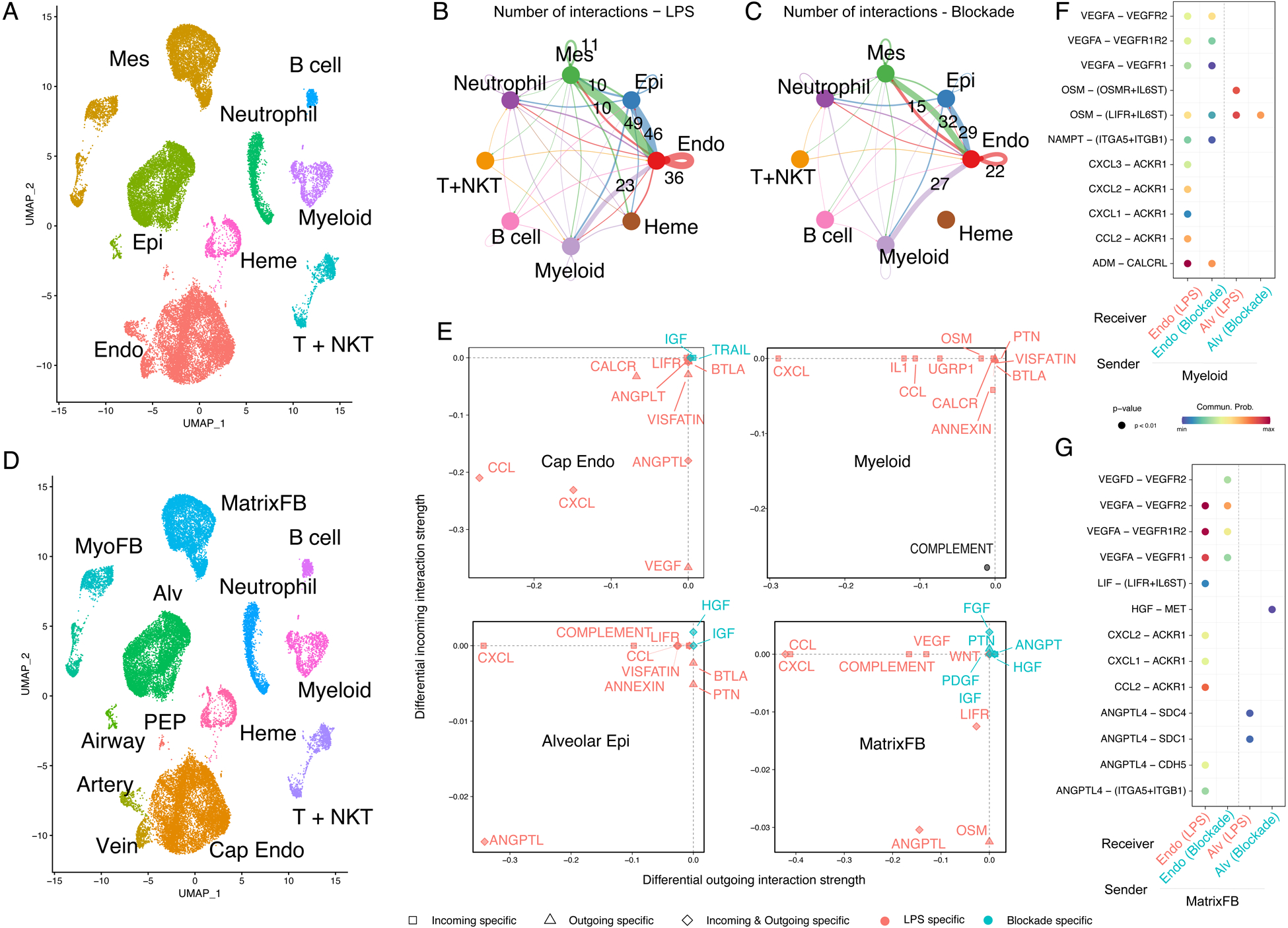
A) UMAP of major cell populations including immune cells used to identify key interactors. Myeloid immune lineages signal extensively to epithelium, endothelium, and mesenchyme in both LPS (B) and blockade (C) conditions. D) UMAP of more specific cell populations used for analysis in (E-G). E) CellChat differential secretion and receptivity analysis for major signaling modulators enriched in LPS (red) and after combination blockade (blue). Notably, blockade increases expression of key developmental niche signals. F-G) Major differential ligand and receptor pairs involved in myeloid signaling (F) or activated matrix fibroblast signaling (G) to endothelial and epithelial cells, showing intensity with either LPS (red) or blockade (blue). Mes = mesenchyme, Epi = epithelium, Endo = endothelium, Heme = hematopoietic, T + NKT = T cells and Natural Killer (NKT) cells, MyoFB = myofibroblast, MatrixFB = matrix fibroblasts, PEP = proliferative endothelial progenitor, Alv = alveolar epithelium, Cap endo = capillary endothelium (general capillaries and ACs).
Next, we subdivided structural cells into major subcategories (Figure 7D) and found that myeloid cells and activated matrix fibroblasts produced extensive inflammatory signaling modulators, including CXCL and CCL ligands, following LPS treatment (Figure 7E–G). The CXCL and CCL pathway response is predicted in alveolar endothelial and epithelial cells via atypical chemokine receptor 1 (ACKR1)(60) after LPS, likely contributing to the significant disruption of epithelial and endothelial patterning seen in these animals. ACKR family receptors are important modulators of cellular inflammatory response, and global knockout of ACKR1 in mice dramatically reduces lung inflammation following systemic LPS challenge(61). Adrenomedullin (ADM) signaling from myeloid cells to alveolar endothelium via CALCRL signaling also increased after LPS, and recent murine data has implicated ADM-CALCRL signaling in repair of the neonatal lung after hyperoxic injury(62, 63). Blockade of IL-1 and TNF signaling dramatically blunted CCL and CXCL cytokine expression in both matrix fibroblasts and myeloid cells (Figure 7F–G), and responses to these ligands correspondingly decreased throughout alveolar cells. ADM-CALCRL signaling decreased to near control levels in blockade (Figure 7F). Remarkably, matrix fibroblasts elaborated FGF, HGF, and IGF ligands (Figure 7E, G) preferentially after inflammatory blockade, similar to the milieu of the control alveolar niche, restoring a developmental pattern of signaling response in the epithelium and endothelium.
Taken together, these data support a model of developmental lung injury (Figure 8A–B) in fetal primates whereby LPS-induced inflammatory cytokine signaling directly injures the structural and signaling niches of the developing alveolus. This altered signaling milieu causes widespread activation of inflammatory pathways, promoting a pro-inflammatory state of myeloid immune lineages resident in or recruited to the lung. Together, these inflammatory signals potentiate injury to the alveolar niche through elaboration of CCL and CXCL ligands, ultimately leading to failed alveologenesis and alveolar simplification. Blockade of IL-1 and TNF signaling is sufficient to blunt this pro-inflammatory state, decreasing cell-intrinsic inflammatory response in structural cells and promoting a more tolerogenic phenotype in myeloid cells, leading to protection of the developing alveolus, maintenance of key developmental signaling relationships, and functional development of the gas exchange surface in preparation for birth and postnatal respiration (Figure 8C).
Figure 8. Model of pathogenesis and blockade of inflammatory lung injury in macaque.
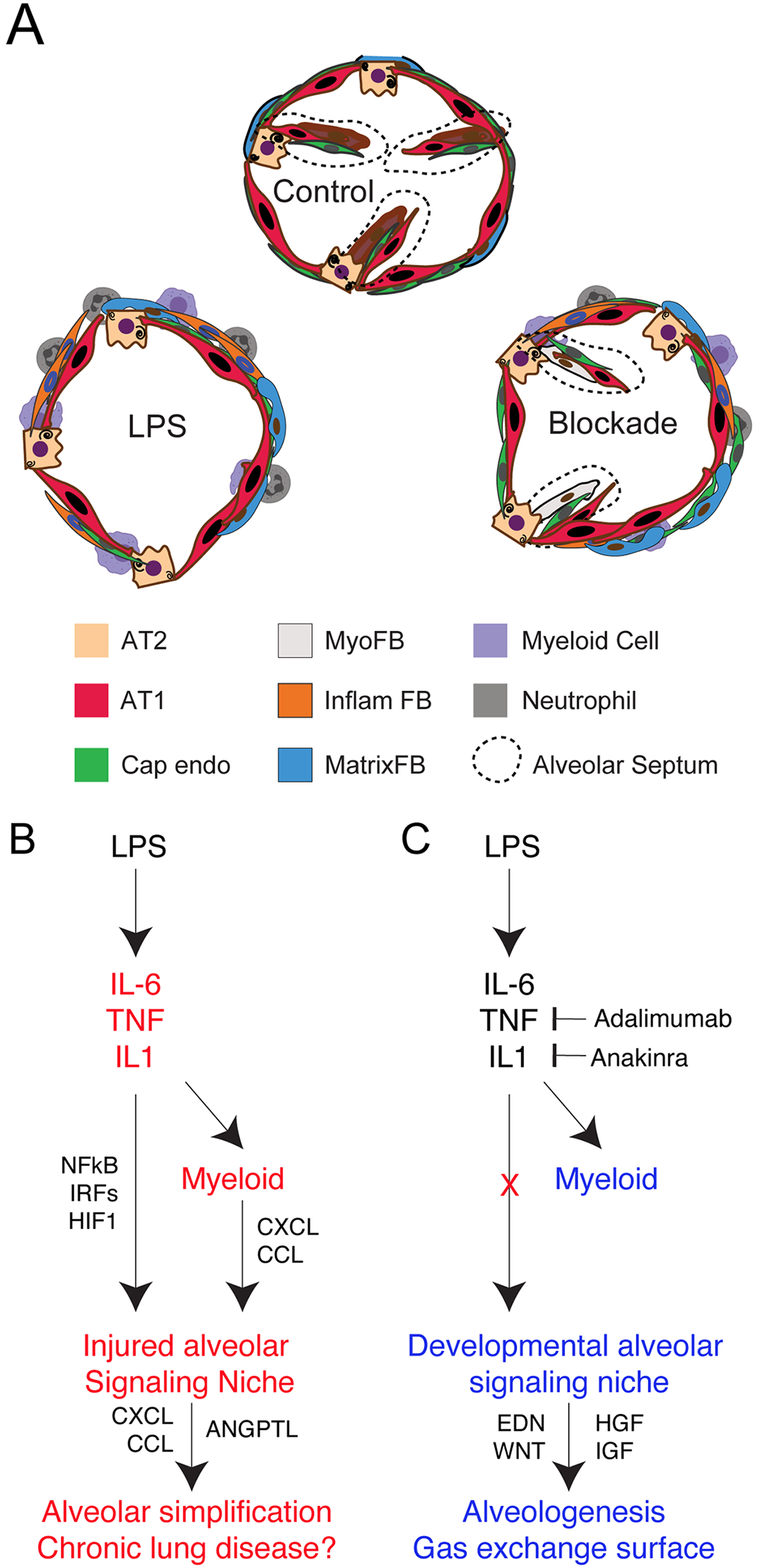
A-C) LPS causes significant alveolar simplification via activation of global inflammatory cytokines including IL-1, IL-6, and TNF, leading to activation of these pathways across the alveolar niche. Myeloid cells potentiate this injury via expression of CXCL and CCL cytokines, which are also elaborated from injured matrix fibroblasts, thus worsening injury. Inflammatory blockade blunts this pro-inflammatory state, reduces CCL and CXCL cytokine expression, and protects the alveolar signaling milieu dominated by developmental growth factors and morphogens, thereby promoting continued alveologenesis.
DISCUSSION
A conserved niche for mammalian alveologenesis
Successful adaption to postnatal life is entirely dependent upon the formation and function of pulmonary alveoli which mediate the efficient exchange of oxygen and carbon dioxide. Much of what we know about alveologenesis comes from decades of controlled mouse experimentation, which has generated detailed knowledge of signaling interactions and transcriptional regulators active during lung morphogenesis(3). Nonetheless, key differences between mouse and human lung limit what can be learned from purely murine studies. Nonhuman primates can be used for controlled experimentation, as in the current study, to directly assess causality; such causal studies are rarely possible in humans. Here, we utilized the rhesus macaque model to define the primate alveolar niche. Rhesus shares key morphological features and conserved developmental timing with human lung development, and our data demonstrate conservation in key signaling factors and cell signatures. Signature markers of alveolar epithelial cells(44, 45, 47), mesenchymal lineages(64–67), and endothelial specialization(36, 37, 51) are similar to those defined in mouse development and human lung datasets(14, 27, 42, 43). Here we present some candidate confirmation of RNA data with IHC in several cell populations here, but recent reports(68) have suggested variable concordance between RNA expression and protein expression in single cells, and future unbiased proteomic experiments will be needed to confirm that conservation at the RNA level is mirrored by protein expression across lung lineages. Nonetheless, the observed high-level conservation between mouse, primate, and human lung emphasizes the requirement for alveologenesis in mammalian physiology.
With strong connections to both mouse and human physiology and expression, the rhesus macaque model provides a valuable opportunity to bridge murine and human studies. Recent data has suggested drift in adult lung expression patterns and signaling between mouse and human(14), but our study suggests important evolutionary conservation during alveologenesis and may provide a framework for understanding perturbations in alveolar development beyond inflammatory injury. Of particular interest is the persistent disruption of developmental progenitors and differentiation trajectories up to 5d following injury, which may have important implications for neonatal CLDP/BPD pathophysiology. Extended evaluation of this model will be crucial to understanding whether intra-uterine inflammation causes developmental delay or true developmental arrest in the alveolus. Given that endothelial-AT1 interactions are central to alveolar development, and are directly injured by LPS, identifying determinants of this co-development is a high priority that may allow development of new therapeutics for both pediatric and adult lung diseases.
Beyond cellular and structural conservation, our data define a dedicated niche of signaling factors and gene networks that control the growth and patterning of the developing primate alveolus. WNT(47, 57, 58), FGF(69–71), EGF(72), and VEGF(36) signaling, all of which are critical in mouse alveolar development, function in multiple signaling loops during normal rhesus lung development and are heavily disrupted during injury, implying important functional conservation. We observed centrality of several less studied pathways in the primate alveolar niche; EDN and PTN signaling in AT1-AC interactions are particularly prominent pathways that merit further study in rhesus and other systems. Adaptation of techniques successfully used in human and mouse development, including organoids(12, 48) and precision-cut lung slices(57, 73) will be important in future studies to provide additional specificity to our understanding of these pathways, and to identify key signaling interactions that differ between mouse and primate.
Immune signals as important regulators of lung development and repair
A provocative aspect of our study is the finding that inflammatory blockade is sufficient to protect the developing lung from significant injury following LPS treatment. These data add to an evolving understanding of the role of immune cells in both lung development and lung repair. Recent data in adult mice demonstrated that inflammatory cytokine pathways, particularly IL-1(74, 75), functionally promote lung regeneration after multiple types of injury, driving AT2 proliferation(74) and AT1 differentiation through a regenerative transition state(75). Myeloid lineages have been reported to promote regeneration after pneumonectomy via CCR2/CCL2 signaling(76). We observe the opposite effect in the developing primate lung, where IL1 and TNF blockade protects the lung from injury, and CXCL/CCL chemokine signaling is a major hallmark of LPS-treated rhesus lung that is suppressed by IL1 and TNF blockade.
Our data suggest several possibilities to explain these differences. First, relating to tissue and immune maturity, recent reports demonstrate persuasively that the perinatal immune system differs dramatically from adult immunity(77, 78). Fetal immunity is carefully balanced to provide protection from pathogens while also allowing development of tolerance and colonization of microbiota. Extensive data from human studies, and our report on rhesus immune ontogeny(29), shows that chorioamnionitis disrupts this immune development(25, 26). Together, these data imply that signals for lung regeneration may be age-dependent, emphasizing the need to consider adult and pediatric regeneration as separable processes. Second, it is possible that the fetal immune system is less well positioned to generate the “correct” level of regenerative signaling in the alveolus after injury, or that the developing alveolus is more susceptible to injury from inflammatory signaling. Our data imply a central role of CXCL and CCL ligands signaling through atypical chemokine receptors in the fetal lung response to LPS; the ACKR family has been implicated in diverse processes in nonimmune cells(60), especially in directing cancer-associated endothelial biology(79). ACKR blockade is an area of active anti-cancer drug development(79), providing pharmaceuticals which could be considered as future therapeutics for chorioamnionitis. Taken together, our data emphasize that immune cells are a crucial component of the lung alveolar niche, participating in growth, maintenance, and regeneration of the distal lung throughout life.
Nonhuman primates bridge a gap in understanding and treating human lung disease
Defining actionable pathophysiology underlying complex human diseases remains a challenge, even in the era of unprecedented large patient data sets. Rhesus is an ideal model organism to address this challenge with translationally focused projects to model human pathophysiology. Our findings that IL-1 and TNF blockade protects the lung from injury, and that stromal and innate immune activation elaborates injurious signals that are suppressed with cytokine blockade, provide proof of principle that anti-inflammatory therapies protect the lung from severe injury. These data imply that future therapies targeting the immune system may hold promise for treatment of perinatal inflammation. The quality and flexibility of the rhesus model opens avenues for biomarker discovery, therapeutic development, and longitudinal analysis for chorioamnionitis and other complex diseases. Rhesus also provides a unique future opportunity to directly assess causal connections between perinatal inflammation and long-term respiratory health outcomes. Coordination of targeted primate studies with large human disease and organ consortia should be a future priority, as such efforts will enrich our combined knowledge of primate biology, maximize the value of human reference data, and provide opportunities for translational biology by assisting in development of pathophysiologically sophisticated, accurate models and new therapies for challenging and complex diseases.
MATERIALS AND METHODS
Study design/Ethics:
Between 2014 and 2019 adult female rhesus macaques (Macaca mulatta; total n=40) were time mated for planned hysterotomies at ~GD 105, ~GD 130 (~80% term gestation), or ~GD150 (see Table S1). For LPS treatment, Rhesus dams at ~GD 130 were randomized to receive 1 mL of saline ± LPS (derived from E. coli 055:B5, Sigma-Aldrich, St. Louis, MO cat #L2880) at 1 mg/mL, a dose shown in prior studies to generate intra-uterine inflammation similar to that observed in human chorioamnionitis patients (27), via ultrasound-guided intraamniotic (IA) injection. Hysterotomies were performed 16h, 48h, or 5d post-LPS with delivery of the fetus at approximately GD130. For animals in the combined blockade (anakinra + adalimumab) treatment group, the pregnant females simultaneously received both adalimumab (40 mg subcutaneously [SQ] 3h prior to LPS and 40 mg IA 1h prior to LPS) and anakinra (100 mg SQ 3h prior to LPS and 50 mg IA 1h prior to LPS), were given IA LPS as above, and sacrificed 16h post LPS for analysis. All animals were obtained within 2d before or after the target gestational age when possible. There were no instances of spontaneous death or preterm labor in the experimental animals. At delivery, fetuses were euthanized with pentobarbital and underwent necropsy immediately. The right upper lung lobe was fixed for histologic analysis, and fresh fetal lung tissue was sent by overnight delivery for next day processing of the left lung for single cell isolation. Samples spanning different years were assayed and analyzed in the same experimental replicates in our laboratory to minimize technical variability. Characteristics and techniques of evaluation for all animals are listed in Table S1. All animal procedures were approved by the Institutional Animal Care and Use Committees (IACUC) at Cincinnati Children’s Hospital and the University of California Davis, and all studies were conducted in accordance with CCHMC biosafety protocols.
Tissue Fixation and Processing:
The right upper lung lobe was perfused with saline and inflation fixed with 10% formalin at 30 cm H2O pressure for at least 24h. Tissue was dehydrated through an ethanol gradient, paraffin-embedded, and sectioned at a thickness of 5 μm for hematoxylin and eosin staining (for morphological examination and injury scoring), immunofluorescence, and RNAscope.
Imaging:
Images were taken using a Nikon Eclipse Ti A1R LUN-V Inverted Confocal Microscope (with Plan Apo λ 20x, Plan Apo IR 60x WI DIC N2, and Plan Apo λ 100x Oil objectives, using NIS-Elements AR v5.20.02) and Nikon Eclipse NiE Upright Widefield Microscope (Nikon DS-Fi3 Camera with Plan Fluor 4x, Plan Apo 10x, Plan Apo VC 20x DIC N2, and Plan Apo 40x DIC M N2 objectives, using NIS-Elements AR v5.20.01), respectively.
Histological Analysis, Morphometrics, and Quantification:
Representative 40x H&E images from n=3 animals in each experimental group (control GD 130 [27 fields], LPS 16h [37 fields], LPS 48h [39 fields], LPS 5d [31 fields], and LPS + combination blockade [31 fields]) were randomly selected for analysis, including lung morphometry, injury scoring, and septal counts. Images were deidentified and scored independently, using parameters established by (80), by two individuals blinded to the treatment condition of the samples. Septa were counted manually, considering any multicellular extensions into the alveolar space that were not part of the alveolar wall. For lung morphometric analysis, the same 40x image fields were evaluated using the FIJI-macro (run in FIJI/ImageJ v1.53) established by (81) with minor modifications to report volume density of alveolar septa (Vvsep), mean linear intercept of the airspaces (Lm), mean transsectional wall length (Lmw), and surface area density of the air spaces (Svair). Input images were converted to the size/pixel density specified by (81), and calculation constants were adjusted based on image width/height (μm) for 40x images. The same 165 40x magnification images were used for injury scoring, septal counts, and morphometric analysis. Statistical analyses were compared among experimental conditions using ANOVA (Kruskal-Wallace test which does not assume equal distribution of data in all groups) with prespecified multiple comparisons in GraphPad Prism 9.0.
Immunofluorescence:
Following deparaffinization, rehydration, and sodium citrate (10 mM, pH 6.0) antigen retrieval, immunofluorescence was used to detect protein expression on paraffin sections. Antibodies listed in Table S2 were detected using ImmPress HRP Universal antibody polymer detection kit (Horse Anti-mouse/rabbit IgG, Vector Labs, MP-7500), as previously described for mouse (48), with TSA plus fluorophores (1:100); antibodies listed in Table S3 were detected using standard immunofluorescence protocols, as previously described (50). Sections were stained with DAPI (Invitrogen, D1306, 1:1000) and mounted using Prolong Gold antifade mounting medium (Invitrogen, P36930). TMPRSS2 IHC and FOXJ1 RNAscope were performed as previously reported (40).
Immunofluorescence Quantification:
Confocal 60x z-stacks of representative 5 μm tissue sections from each timepoint and experimental group (control GD 105, control GD 130, control GD 150, LPS 16h, LPS 48h, LPS 5d, LPS + combination blockade) were converted from .nd2 (exported from NIS-Elements AR acquisition software) to .ims (Imaris input file) using Imaris File Converter x64 v9.8.0 (Oxford Instruments) for analysis in Imaris x64 v9.8.0 (Oxford Instruments). All .ims files for each condition were loaded into Imaris for batch analysis for the following parameters: DAPI+ nuclei (405 channel+ spots), EDNRB+/fluorescein+ surfaces (488 channel+ surfaces), HOPX+/Cy3.5+ nuclei (561 channel+ spots). Following batch addition/quantification of the elaborated parameters, total counts of HOPX+ nuclei and total volume of EDNRB+ surfaces (μm3) were recorded and normalized to counts of total nuclei (DAPI+ nuclei) per field/z-stack. Statistical analyses were compared among developmental time points and experimental conditions using ANOVA (Kruskal-Wallace test which does not assume equal distribution of data in all groups) with prespecified multiple comparisons in GraphPad Prism 9.0. See Figure S3 for an example of this process.
Lung Single Cell Suspension:
After manually dissecting out gross airways, fresh rhesus fetal lung tissue (~500 mg/tube) was cut into small pieces and transferred into a gentleMACS C-tube (Miltenyi Biotec, 130–093-237) containing 5 mL of digestion buffer (9 mL of PBS [Gibco, 10010–023] combined with 1 mL of Dispase [stock: 50 U/mL; final concentration: 5 U/mL, Corning, 354235], 50 μL of DNase [stock: 5 mg/mL; final concentration: 0.025 mg/mL or 50 U/mL, GoldBio, D-301], and 100 μL Collagenase Type I [stock: 48000 U/mL; final concentration of 480 U/mL, Gibco, 17100–017]). C-tubes were processed using the gentleMACS Octo Dissociator with Heaters (Miltenyi Biotec, 130–096-427), running programs “m_lung_01_02” (36 sec) twice, “37C_m_LIDK_1” (36 min 12 sec) once, and “m_lung_01_02” (36 sec) once. Resulting tissue suspension was filtered through 100 μm cell strainer (Greiner Bio-One, 542000) into a 50 mL conical and resuspended in 50 mL PBS. Following centrifugation (1000g for 5 min at 4°C), cells were resuspended and incubated in 5 mL of RBC lysis buffer (Invitrogen, 00–4333-57) for 5 min on ice. Following another centrifugation (1000g for 5 min at 4°C), cells were resuspended in 20 mL of DMEM/F-12 media (Gibco, 11320–033) (without serum) and filtered through a 40 μm cell strainer (Greiner Bio-One, 542040). Cell counts and viability were determined manually using a hemocytometer.
scRNA sequencing:
From this suspension, 16,000 cells per animal were loaded into one channel of the Chromium system using the v3 single cell reagent kit (10x Genomics, Pleasanton, CA) by the CCHMC Gene Expression Core and these libraries were sequenced as a pool on a Novaseq 6000 in the CCHMC DNA Sequencing Core. Details of scRNAseq analysis pipeline, including packages used and parameters, are found in the Supplemental Materials.
Statistical Tests:
All data met the assumptions of the statistical tests used (details reported in each figure legend). Statistical tests used for single cell analyses are described in the Supplemental Materials and figure legends. For comparing differences between groups, we used Graphpad Prism 9.0 to perform either unpaired two-tailed Student’s t-test for two groups or ANOVA (Kruskal-Wallace test which does not assume equal distribution of data in all groups) with prespecified multiple comparison testing for groups of three or more.
Supplementary Material
Figure S1. Rhesus macaque lung development models key aspects of human lung organogenesis.
Figure S2. Progressive cellular maturation of the rhesus lung during third trimester gestation.
Figure S3. Method for quantification of gas exchange surface development.
Figure S4. Bulk RNA sequencing of murine and rhesus lung during development.
Figure S5. Low level expression of SARS-CoV-2 protease TMPRSS2 in fetal primate lung.
Figure S6. Power calculations and subset analysis of scRNAseq data.
Figure S7. Conservation of the proliferative endothelial progenitor lineage across mammalian evolution.
Figure S8. Major signaling patterns in LPS-treated lungs.
Table S1. Rhesus Sample Information
Table S2. ImmPRESS HRP + TSA Antibodies and Reagents
Table S3. Standard Immunofluorescence Antibodies
Table S4. scRNAseq Metrics
Acknowledgments:
The authors would like to thank the Gene Expression Core (especially Kelly Rangel and Shawn Smith), Confocal Imaging Core (especially director Matt Kofron), and DNA Sequencing Core of the Cincinnati Children’s Research Foundation for extensive technical support. This work has been in extensive collaboration with the California National Primate Research Center, and we would especially like to thank Paul-Michael Sosa, Jennifer Kendrick, and Sarah Lockwood for invaluable help in animal management and care.
Funding:
National Institutes of Health grant HL00752 (AT)
National Institutes of Health grant HL135258 (M Gu)
National Institutes of Health grant AI138553 (LM)
National Institutes of Health grant OD011107 (LM)
National Institutes of Health grant HL142485 (LM)
National Institutes of Health grant HD98389 (SGK)
National Institutes of Health grant HL148865 (JAW, NS)
National Institutes of Health grant AI150748 (ERM, WJZ)
National Institutes of Health grant HL153045 (YX)
National Institutes of Health grant HL122642 (YX)
National Institutes of Health grant HL141174 (VVK)
National Institutes of Health grant HL149631 (VVK)
National Institutes of Health grant HL152973 (VVK)
National Institutes of Health grant HL149366 (IL)
National Institutes of Health grant HL134745 (JAW)
National Institutes of Health grant ES029234 (CAC)
National Institutes of Health grant HD084686 (HD)
National Institutes of Health grant HL155611 (HD)
National Institutes of Health grant HL142708 (HD)
National Institutes of Health grant HL140178 (WJZ)
National Institutes of Health LungMAP2 Pilot Program (WJZ, M Guo)
LAM Foundation (YX, M Guo)
Francis Family Foundation (HD)
Cincinnati Children’s Hospital Procter Scholar Award (WJZ)
Cincinnati Children’s Hospital ARC Award (CAC, JAW, SK)
Footnotes
Publisher's Disclaimer: * This manuscript has been accepted for publication in Science Translational Medicine. This version has not undergone final editing. Please refer to the complete version of record at www.sciencetranslationalmedicine.org/. The manuscript may not be reproduced or used in any manner that does not fall within the fair use provisions of the Copyright Act without the prior written permission of AAAS.
Competing interests:
The authors declare that they have no competing interests for the current work, including patents, financial holdings, advisory positions, or other interests.
Data and materials availability:
Extensive additional confocal and histological imaging from animals reported here are available at https://research.cchmc.org/lungimage/). All mouse and primate RNA sequencing data has been deposited at GEO (GSE178680 and GSE169390) and is available at the LungMAP portal (bit.ly/rhesuslung). Explorable versions of the single cell RNAseq data are available in a Shiny portal (bit.ly/rhesuslung_explore) and a CZI CellxGene portal (bit.ly/rhesuslung_cellxgene).
References and Notes
- 1.Zorn AM, Wells JM, Vertebrate endoderm development and organ formation. Annual Review of Cell and Developmental 25, 221–251 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Warburton D, Schwarz M, Tefft D, Flores-Delgado G, Anderson KD, Cardoso WV, The molecular basis of lung morphogenesis. Mechanisms of development 92, 55–81 (2000). [DOI] [PubMed] [Google Scholar]
- 3.Whitsett JA, Kalin TV, Xu Y, Kalinichenko VV, Building and Regenerating the Lung Cell by Cell. Physiol Rev 99, 513–554 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hyde DM, Blozis SA, Avdalovic MV, Putney LF, Dettorre R, Quesenberry NJ, Singh P, Tyler NK, Alveoli increase in number but not size from birth to adulthood in rhesus monkeys. American Journal of Physiology-Lung Cellular and Molecular Physiology 293, L570–L579 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Dunnill M, Postnatal growth of the lung. Thorax 17, 329 (1962). [Google Scholar]
- 6.Cooney TP, Thurlbeck WM, The radial alveolar count method of Emery and Mithal: a reappraisal 1--postnatal lung growth. Thorax 37, 572–579 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whitsett JA, Weaver TE, Hydrophobic surfactant proteins in lung function and disease. New England Journal of Medicine 347, 2141–2148 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Hsia CC, Hyde DM, Weibel ER, Lung structure and the intrinsic challenges of gas exchange. Comprehensive physiology 6, 827–895 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hillman NH, Kallapur SG, Jobe AH, Physiology of Transition from Intrauterine to Extrauterine Life. Clin Perinatol 39, 769–783 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Basil MC, Katzen J, Engler AE, Guo M, Herriges MJ, Kathiriya JJ, Windmueller R, Ysasi AB, Zacharias WJ, Chapman HA, Kotton DN, Rock JR, Snoeck H-W, Vunjak-Novakovic G, Whitsett JA, Morrisey EE, The Cellular and Physiological Basis for Lung Repair and Regeneration: Past, Present, and Future. Cell Stem Cell 26, 482–502 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Basil MC, Morrisey EE, Lung regeneration: a tale of mice and men. Seminars in Cell & Developmental Biology 100, 88–100 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nikolić MZ, Caritg O, Jeng Q, Johnson J-AA, Sun D, Howell KJ, Brady JL, Laresgoiti U, Allen G, Butler R, Zilbauer M, Giangreco A, Rawlins EL, Human embryonic lung epithelial tips are multipotent progenitors that can be expanded in vitro as long-term self-renewing organoids. eLife 6, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller AJ, Hill DR, Nagy MS, Aoki Y, Dye BR, Chin AM, Huang S, Zhu F, White ES, Lama V, Spence JR, In Vitro Induction and In Vivo Engraftment of Lung Bud Tip Progenitor Cells Derived from Human Pluripotent Stem Cells. Stem Cell Rep 10, 101–119 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Travaglini KJ, Nabhan AN, Penland L, Sinha R, Gillich A, Sit RV, Chang S, Conley SD, Mori Y, Seita J, Berry GJ, Shrager JB, Metzger RJ, Kuo CS, Neff N, Weissman IL, Quake SR, Krasnow MA, A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature, 1–7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pan H, Deutsch GH, Wert SE, Ambalavanan N, Ansong C, Ardini-Poleske ME, Bagwell J, Chan C, Deutsch GH, Frevert C, Gabriel D, Hagood JS, Hill CB, Holden-Wiltse J, Jegga AG, Mariani TJ, Masci AM, Pan H, Shi W, Warburton D, Wert SE, K. A, Comprehensive anatomic ontologies for lung development: A comparison of alveolar formation and maturation within mouse and human lung. J Biomed Semant 10, 18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koo H-KK, Vasilescu DMM, Booth S, Hsieh A, Katsamenis OL, Fishbane N, Elliott WM, Kirby M, Lackie P, Sinclair I, Warner JA, Cooper JD, Coxson HO, Paré PD, Hogg JC, Hackett T-LL, Small airways disease in mild and moderate chronic obstructive pulmonary disease: a cross-sectional study. Lancet Respir Medicine 6, 591–602 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Whitsett JA, Wert SE, Weaver TE, Diseases of pulmonary surfactant homeostasis. Annu Rev Pathol 10, 371–393 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thebaud B, Goss KN, Laughon M, Whitsett JA, Abman SH, Steinhorn RH, Aschner JL, Davis PG, McGrath-Morrow SA, Soll RF, Jobe AH, Bronchopulmonary dysplasia. Nat Rev Dis Primers 5, 78 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goldenberg RL, Culhane JF, Iams JD, Romero R, Epidemiology and causes of preterm birth. Lancet 371, 75–84 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldenberg RL, Hauth JC, Andrews WW, Intrauterine infection and preterm delivery. N Engl J Med 342, 1500–1507 (2000). [DOI] [PubMed] [Google Scholar]
- 21.Kim CJ, Romero R, Chaemsaithong P, Chaiyasit N, Yoon BH, Kim YM, Acute chorioamnionitis and funisitis: definition, pathologic features, and clinical significance. Am J Obstet Gynecol 213, S29–S52 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McDowell KM, Jobe AH, Fenchel M, Hardie WD, Gisslen T, Young LR, Chougnet CA, Davis SD, Kallapur SG, Pulmonary Morbidity in Infancy after Exposure to Chorioamnionitis in Late Preterm Infants. Ann Am Thorac Soc 13, 867–876 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Montgomery S, Bahmanyar S, Brus O, Hussein O, Kosma P, Palme-Kilander C, Respiratory infections in preterm infants and subsequent asthma: a cohort study. BMJ Open 3, e004034–e004034 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Massaro D, Massaro GD, Critical period for alveologenesis and early determinants of adult pulmonary disease. American Journal of Physiology-Lung Cellular and Molecular Physiology 287, L715–L717 (2004). [DOI] [PubMed] [Google Scholar]
- 25.Kallapur S, Presicce P, Rueda C, Jobe A, Chougnet C, Fetal Immune Response to Chorioamnionitis. Semin Reprod Med 32, 056–067 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jackson CM, Mukherjee S, Wilburn AN, Cates C, Lewkowich IP, Deshmukh H, Zacharias WJ, Chougnet CA, Pulmonary Consequences of Prenatal Inflammatory Exposures: Clinical Perspective and Review of Basic Immunological Mechanisms. Front Immunol 11, 1285 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang A, Chiou J, Poirion OB, Buchanan J, Valdez MJ, Verheyden JM, Hou X, Kudtarkar P, Narendra S, Newsome JM, Guo M, Faddah DA, Zhang K, Young RE, Barr J, Sajti E, Misra R, Huyck H, Rogers L, Poole C, Whitsett JA, Pryhuber G, Xu Y, Gaulton KJ, Preissl S, Sun X, Single cell multiomic profiling of human lung reveals cell type-specific and age-dynamic control of SARS-CoV2 host genes. eLife 9, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Presicce P, Park C-W, Senthamaraikannan P, Bhattacharyya S, Jackson C, Kong F, Rueda CM, DeFranco E, Miller LA, Hildeman DA, Salomonis N, Chougnet CA, Jobe AH, Kallapur SG, IL-1 signaling mediates intrauterine inflammation and chorio-decidua neutrophil recruitment and activation. Jci Insight 3, e98306 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jackson CM, Demmert M, Mukherjee S, Isaacs T, Gray J, Senthamaraikannan P, Presicce P, Chetal K, Salomonis N, Miller LA, Jobe AH, Kallapur SG, Zacharias WJ, Lewkowich IP, Deshmukh H, Chougnet CA, A potent myeloid response is rapidly activated in the lungs of premature Rhesus macaques exposed to intra-uterine inflammation. bioRxiv, 2021.2005.2028.444219 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Presicce P, Cappelletti M, Senthamaraikannan P, Ma F, Morselli M, Jackson CM, Mukherjee S, Miller LA, Pellegrini M, Jobe AH, Chougnet CA, Kallapur SG, TNF-Signaling Modulates Neutrophil-Mediated Immunity at the Feto-Maternal Interface During LPS-Induced Intrauterine Inflammation. Front Immunol 11, 558 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salminen A, Paananen R, Vuolteenaho R, Metsola J, Ojaniemi M, Autio-Harmainen H, Hallman M, Maternal Endotoxin-Induced Preterm Birth in Mice: Fetal Responses in Toll-Like Receptors, Collectins, and Cytokines. Pediatric Research 63, 280–286 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Velten M, Britt RD Jr., Heyob KM, Welty SE, Eiberger B, Tipple TE, Rogers LK, Prenatal inflammation exacerbates hyperoxia-induced functional and structural changes in adult mice. Am J Physiol Regul Integr Comp Physiol 303, R279–290 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Faksh A, Britt RD, Vogel ER, Kuipers I, Thompson MA, Sieck GC, Pabelick CM, Martin RJ, Prakash YS, Effects of antenatal lipopolysaccharide and postnatal hyperoxia on airway reactivity and remodeling in a neonatal mouse model. Pediatric Research 79, 391–400 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berger J, Bhandari V, Animal models of bronchopulmonary dysplasia. The term mouse models. Am J Physiol Lung Cell Mol Physiol 307, L936–947 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barry PA, Lockridge KM, Salamat S, Tinling SP, Yue Y, Zhou SS, Gospe SM, Britt WJ, Tarantal AF, Nonhuman Primate Models of Intrauterine Cytomegalovirus Infection. Ilar J 47, 49–64 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Ellis LV, Cain MP, Hutchison V, Flodby P, Crandall ED, Borok Z, Zhou B, Ostrin EJ, Wythe JD, Chen J, Epithelial Vegfa Specifies a Distinct Endothelial Population in the Mouse Lung. Dev Cell 52, 617–630.e616 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gillich A, Zhang F, Farmer CG, Travaglini KJ, Tan SY, Gu M, Zhou B, Feinstein JA, Krasnow MA, Metzger RJ, Capillary cell-type specialization in the alveolus. Nature 586, 785–789 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Niethamer TK, Stabler CT, Leach JP, Zepp JA, Morley MP, Babu A, Zhou S, Morrisey EE, Defining the role of pulmonary endothelial cell heterogeneity in the response to acute lung injury. eLife 9, e53072 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schmidt AF, Kannan PS, Bridges J, Presicce P, Jackson CM, Miller LA, Kallapur SG, Chougnet CA, Jobe AH, Prenatal inflammation enhances antenatal corticosteroid-induced fetal lung maturation. Jci Insight 5, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schuler BA, Habermann AC, Plosa EJ, Taylor CJ, Jetter C, Negretti NM, Kapp ME, Benjamin JT, Gulleman P, Nichols DS, Braunstein LZ, Hackett A, Koval M, Guttentag SH, Blackwell TS, Webber SA, Banovich NE, Cohort VC-C, Network HCAB, Kropski JA, Sucre JM, Age-determined expression of priming protease TMPRSS2 and localization of SARS-CoV-2 in lung epithelium. J Clin Invest 131, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, Satija R, Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e1821 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guo M, Du Y, Gokey JJ, Ray S, Bell SM, Adam M, Sudha P, Perl AK, Deshmukh H, Potter SS, Whitsett JA, Xu Y, Single cell RNA analysis identifies cellular heterogeneity and adaptive responses of the lung at birth. Nat Commun 10, 37 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schiller HB, Montoro DT, Simon LM, Rawlins EL, Meyer KB, Strunz M, Braga FAV, Timens W, Koppelman GH, Budinger GRS, Burgess JK, Waghray A, Berge M. v. d., Theis FJ, Regev A, Kaminski N, Rajagopal J, Teichmann SA, Misharin AV, Nawijn MC, The Human Lung Cell Atlas: A High-Resolution Reference Map of the Human Lung in Health and Disease. Am J Resp Cell Mol 61, 31–41 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Treutlein B, Brownfield DG, Wu AR, Neff NF, Mantalas GL, Espinoza FH, Desai TJ, Krasnow MA, Quake SR, Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature 509, 371–375 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frank DB, Penkala IJ, Zepp JA, Sivakumar A, Linares-Saldana R, Zacharias WJ, Stolz KG, Pankin J, Lu M, Wang Q, Babu A, Li L, Zhou S, Morley MP, Jain R, Morrisey EE, Early lineage specification defines alveolar epithelial ontogeny in the murine lung. Proc Natl Acad Sci U S A 116, 4362–4371 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Street K, Risso D, Fletcher RB, Das D, Ngai J, Yosef N, Purdom E, Dudoit S, Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 19, 477 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frank DB, Peng T, Zepp JA, Snitow M, Vincent TL, Penkala IJ, Cui Z, Herriges MJ, Morley MP, Zhou S, Lu MM, Morrisey EE, Emergence of a Wave of Wnt Signaling that Regulates Lung Alveologenesis by Controlling Epithelial Self-Renewal and Differentiation. Cell Reports 17, 2312–2325 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zacharias WJ, Frank DB, Zepp JA, Morley MP, Alkhaleel FA, Kong J, Zhou S, Cantu E, Morrisey EE, Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature 555, 251–255 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, Randell SH, Noble PW, Hogan BL, Type 2 alveolar cells are stem cells in adult lung. J Clin Invest 123, 3025–3036 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Du Y, Kitzmiller JA, Sridharan A, Perl AK, Bridges JP, Misra RS, Pryhuber GS, Mariani TJ, Bhattacharya S, Guo M, Potter SS, Dexheimer P, Aronow B, Jobe AH, Whitsett JA, Xu Y, Lung Gene Expression Analysis (LGEA): an integrative web portal for comprehensive gene expression data analysis in lung development. Thorax 72, 481 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ren X, Ustiyan V, Guo M, Wang G, Bolte C, Zhang Y, Xu Y, Whitsett JA, Kalin TV, Kalinichenko VV, Postnatal Alveologenesis Depends on FOXF1 Signaling in c-KIT+ Endothelial Progenitor Cells. Am J Resp Crit Care 200, 1164–1176 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cao J, O’Day DR, Pliner HA, Kingsley PD, Deng M, Daza RM, Zager MA, Aldinger KA, Blecher-Gonen R, Zhang F, Spielmann M, Palis J, Doherty D, Steemers FJ, Glass IA, Trapnell C, Shendure J, A human cell atlas of fetal gene expression. Science 370, eaba7721 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bézieux H. R. d., Berge K. V. d., Street K, Dudoit S, Trajectory inference across multiple conditions with condiments: differential topology, progression, differentiation, and expression. bioRxiv, 2021.2003.2009.433671 (2021). [Google Scholar]
- 54.Xu Y, Wang Y, Besnard V, Ikegami M, Wert SE, Heffner C, Murray SA, Donahue LR, Whitsett JA, Transcriptional programs controlling perinatal lung maturation. PLoS One 7, e37046 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bachurski CJ, Ross GF, Ikegami M, Kramer BW, Jobe AH, Intra-amniotic endotoxin increases pulmonary surfactant proteins and induces SP-B processing in fetal sheep. Am J Physiol Lung Cell Mol Physiol 280, L279–285 (2001). [DOI] [PubMed] [Google Scholar]
- 56.Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan C-H, Myung P, Plikus MV, Nie Q, Inference and analysis of cell-cell communication using CellChat. Nat Commun 12, 1088 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sucre JMS, Vickers KC, Benjamin JT, Plosa EJ, Jetter CS, Cutrone A, Ransom M, Anderson Z, Sheng Q, Fensterheim BA, Ambalavanan N, Millis B, Lee E, Zijlstra A, Königshoff M, Blackwell TS, Guttentag SH, Hyperoxia Injury in the Developing Lung is Mediated by Mesenchymal Expression of Wnt5A. Am J Resp Crit Care 0, 1249–1262 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shu W, Guttentag S, Wang Z, Andl T, Ballard P, Lu MM, Piccolo S, Birchmeier W, Whitsett JA, Millar SE, Morrisey EE, Wnt/beta-catenin signaling acts upstream of N-myc, BMP4, and FGF signaling to regulate proximal-distal patterning in the lung. Developmental biology 283, 226–239 (2005). [DOI] [PubMed] [Google Scholar]
- 59.Zepp JA, Morley MP, Loebel C, Kremp MM, Chaudhry FN, Basil MC, Leach JP, Liberti DC, Niethamer TK, Ying Y, Jayachandran S, Babu A, Zhou S, Frank DB, Burdick JA, Morrisey EE, Genomic, epigenomic, and biophysical cues controlling the emergence of the lung alveolus. Science 371, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bonecchi R, Graham GJ, Atypical Chemokine Receptors and Their Roles in the Resolution of the Inflammatory Response. Frontiers in Immunology 7, 224 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dawson TC, Lentsch AB, Wang Z, Cowhig JE, Rot A, Maeda N, Peiper SC, Exaggerated response to endotoxin in mice lacking the Duffy antigen/receptor for chemokines (DARC). Blood 96, 1681–1684 (2000). [PubMed] [Google Scholar]
- 62.Menon RT, Shrestha AK, Reynolds CL, Barrios R, Caron KM, Shivanna B, Adrenomedullin Is Necessary to Resolve Hyperoxia-Induced Experimental Bronchopulmonary Dysplasia and Pulmonary Hypertension in Mice. Am J Pathology 190, 711–722 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Menon RT, Shrestha AK, Shivanna B, Hyperoxia exposure disrupts adrenomedullin signaling in newborn mice: Implications for lung development in premature infants. Biochem Bioph Res Co 487, 666–671 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Endale M, Ahlfeld S, Bao E, Chen X, Green J, Bess Z, Weirauch MT, Xu Y, Perl AK, Temporal, spatial, and phenotypical changes of PDGFRalpha expressing fibroblasts during late lung development. Developmental biology 425, 161–175 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zepp JA, Zacharias WJ, Frank DB, Cavanaugh CA, Zhou S, Morley MP, Morrisey EE, Distinct Mesenchymal Lineages and Niches Promote Epithelial Self-Renewal and Myofibrogenesis in the Lung. Cell 170, 1134–1148 e1110 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Branchfield K, Li R, Lungova V, Verheyden JM, McCulley D, Sun X, A three-dimensional study of alveologenesis in mouse lung. Developmental biology 409, 429–441 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kumar ME, Bogard PE, Espinoza FH, Menke DB, Kingsley DM, Krasnow MA, Mesenchymal cells. Defining a mesenchymal progenitor niche at single-cell resolution. Science 346, 1258810 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mair F, Erickson JR, Voillet V, Simoni Y, Bi T, Tyznik AJ, Martin J, Gottardo R, Newell EW, Prlic M, A Targeted Multi-omic Analysis Approach Measures Protein Expression and Low-Abundance Transcripts on the Single-Cell Level. Cell Reports 31, 107499 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.White AC, Xu J, Yin Y, Smith C, Schmid G, Ornitz DM, FGF9 and SHH signaling coordinate lung growth and development through regulation of distinct mesenchymal domains. Development 133, 1507–1517 (2006). [DOI] [PubMed] [Google Scholar]
- 70.Beers MF, Mulugeta S, Surfactant protein C biosynthesis and its emerging role in conformational lung disease. Annu Rev Physiol 67, 663–696 (2005). [DOI] [PubMed] [Google Scholar]
- 71.Padela S, Yi M, Cabacungan J, Shek S, Belcastro R, Masood A, Jankov RP, Tanswell AK, A critical role for fibroblast growth factor-7 during early alveolar formation in the neonatal rat. Pediatr Res 63, 232–238 (2008). [DOI] [PubMed] [Google Scholar]
- 72.Li C, Lee MK, Gao F, Webster S, Di H, Duan J, Yang CY, Bhopal N, Peinado N, Pryhuber G, Smith SM, Borok Z, Bellusci S, Minoo P, Secondary crest myofibroblast PDGFRalpha controls the elastogenesis pathway via a secondary tier of signaling networks during alveologenesis. Development 146, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Akram KM, Yates LL, Mongey R, Rothery S, Gaboriau DCA, Sanderson J, Hind M, Griffiths M, Dean CH, Live imaging of alveologenesis in precision-cut lung slices reveals dynamic epithelial cell behaviour. Nat Commun 10, 1178 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Katsura H, Kobayashi Y, Tata PR, Hogan BLM, IL-1 and TNFalpha Contribute to the Inflammatory Niche to Enhance Alveolar Regeneration. Stem Cell Rep 12, 657–666 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Choi J, Park J-E, Tsagkogeorga G, Yanagita M, Koo B-K, Han N, Lee J-H, Inflammatory Signals Induce AT2 Cell-Derived Damage-Associated Transient Progenitors that Mediate Alveolar Regeneration. Cell Stem Cell 27, 366–382.e367 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lechner AJ, Driver IH, Lee J, Conroy CM, Nagle A, Locksley RM, Rock JR, Recruited Monocytes and Type 2 Immunity Promote Lung Regeneration following Pneumonectomy. Cell Stem Cell 21, 120–134 e127 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Park J-E, Jardine L, Gottgens B, Teichmann SA, Haniffa M, Prenatal development of human immunity. Science 368, 600–603 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang X, Zhivaki D, Lo-Man R, Unique aspects of the perinatal immune system. Nat Rev Immunol 17, 495–507 (2017). [DOI] [PubMed] [Google Scholar]
- 79.Massara M, Bonavita O, Mantovani A, Locati M, Bonecchi R, Atypical chemokine receptors in cancer: friends or foes? J Leukocyte Biol 99, 927–933 (2016). [DOI] [PubMed] [Google Scholar]
- 80.Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS, Kuebler WM, An Official American Thoracic Society Workshop Report: Features and Measurements of Experimental Acute Lung Injury in Animals. 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Salaets T, Tack B, Gie A, Pavie B, Sindhwani N, Jimenez J, Regin Y, Allegaert K, Deprest J, Toelen J, A semi-automated method for unbiased alveolar morphometry: Validation in a bronchopulmonary dysplasia model. PLOS ONE 15, e0239562 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen J, Bardes EE, Aronow BJ, Jegga AG, ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic acids research 37, W305–311 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McGinnis CS, Murrow LM, Gartner ZJ, DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst 8, 329–337.e324 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vieth B, Ziegenhain C, Parekh S, Enard W, Hellmann I, powsimR: power analysis for bulk and single cell RNA-seq experiments. Bioinformatics 33, 3486–3488 (2017). [DOI] [PubMed] [Google Scholar]
- 85.Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, Zhang F, Mundlos S, Christiansen L, Steemers FJ, Trapnell C, Shendure J, The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.DePasquale EAK, Schnell D, Dexheimer P, Ferchen K, Hay S, Chetal K, Valiente-Alandí Í, Blaxall BC, Grimes HL, Salomonis N, cellHarmony: cell-level matching and holistic comparison of single-cell transcriptomes. Nucleic acids research 47, e138–e138 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Rhesus macaque lung development models key aspects of human lung organogenesis.
Figure S2. Progressive cellular maturation of the rhesus lung during third trimester gestation.
Figure S3. Method for quantification of gas exchange surface development.
Figure S4. Bulk RNA sequencing of murine and rhesus lung during development.
Figure S5. Low level expression of SARS-CoV-2 protease TMPRSS2 in fetal primate lung.
Figure S6. Power calculations and subset analysis of scRNAseq data.
Figure S7. Conservation of the proliferative endothelial progenitor lineage across mammalian evolution.
Figure S8. Major signaling patterns in LPS-treated lungs.
Table S1. Rhesus Sample Information
Table S2. ImmPRESS HRP + TSA Antibodies and Reagents
Table S3. Standard Immunofluorescence Antibodies
Table S4. scRNAseq Metrics
Data Availability Statement
Extensive additional confocal and histological imaging from animals reported here are available at https://research.cchmc.org/lungimage/). All mouse and primate RNA sequencing data has been deposited at GEO (GSE178680 and GSE169390) and is available at the LungMAP portal (bit.ly/rhesuslung). Explorable versions of the single cell RNAseq data are available in a Shiny portal (bit.ly/rhesuslung_explore) and a CZI CellxGene portal (bit.ly/rhesuslung_cellxgene).