

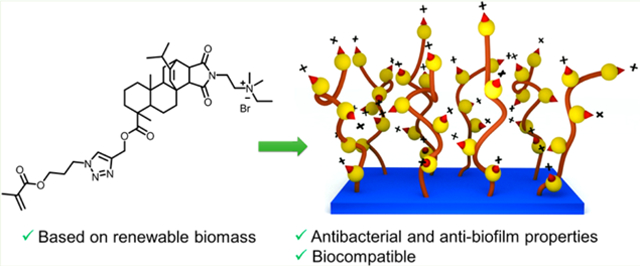
Abstract
We report antibacterial, antibiofilm, and biocompatible properties of surface-immobilized, quaternary ammonium-containing, resin acid-derived compounds and polycations that are known to be efficient antimicrobial agents with minimum toxicities to mammalian cells. Surface immobilization was carried out by the employment of two robust, efficient chemical methods: Copper-catalyzed azide–alkyne 1,3-dipolar cycloaddition click reaction, and surface-initiated atom transfer radical polymerization. Antibacterial and antibiofilm activities against Gram-positive Staphylococcus aureus and Gram-negative Escherichia coli were strong. Hemolysis assays and the growth of human dermal fibroblasts on the modified surfaces evidenced their biocompatibility. We demonstrate that the grafting of quaternary ammonium-decorated abietic acid compounds and polymers from surfaces enables the incorporation of renewable biomass in an effective manner to combat bacteria and biofilm formation in biomedical applications.
Graphical Abstract
INTRODUCTION
Microorganisms form biofilms on surfaces as a protective lifestyle in hostile environments.1–3 These sessile communities often develop from single planktonic cells into a three-dimensionally organized and remarkably complex community of microorganisms encapsulated within an emergent polymeric matrix.4–7 Depending on the type of microorganisms and the environment in which they live, the biofilms are characterized by structural heterogeneity, genetic diversity, and complex community interactions.5,8,9
Biofilms have become problematic in agricultural, industrial, environmental, and clinical settings.10 Microbes in these resilient structures are inherently resistant to antimicrobial agents and have become a major cause of infectious diseases.11–14 Many human diseases, such as dental caries, otitis, necrotizing fasciitis, and cystic fibrosis-related pneumonias, are related to bacterial biofilms.13 Bacterial biofilms produced on the surfaces of medical implants including catheters, heart valves, pacemakers, stents, prosthetic joints, and contact lenses are a significant issue that poses a considerable threat to patient morbidity and mortality and incurs substantial increases in healthcare costs each year.12
Despite the presence of advanced sterilization techniques, it is still difficult to eradicate biofilms and maintain sterility without frequent use of disinfectants. Therefore, there is an ever-growing demand for surfaces with inherent antimicrobial properties that can prevent bacterial colonization on them. Many methods for the antimicrobial modification of surfaces have been developed.15,16 Such methods include impregnating antimicrobial nanoparticles,17 grafting small molecules having antimicrobial properties18 or antimicrobial polymers on surfaces,19–22 coatings with antiquorum sensing molecules,23 loading antibiotics into surface matrices for slow release,24 photoactivated surfaces,25 changing hydrophobicity,26 and modifying surface nanotopography.27
Contact-active coatings, which kill bacteria upon contact, can be fabricated with chemically immobilized quaternary ammonium (QA) antimicrobial agents. Cationic surfaces made up of covalently attached materials such as QA organosilanes,28–30 antimicrobial peptides,19,31 and QA containing alkylated polymers such as polyvinylpyridines,32 polyethylenimines,33 poly(2-(dimethylamino)ethyl methacrylate),34,35 hyperbranced polyurea,36 and many others37,38 are known to be bactericidal. These materials are being studied extensively due to their ease of synthesis, self-sterilization properties, and long-lasting activities compared to other methods. Widespread utilization of such surfaces has necessitated the integration of sustainable materials for low-cost, noncytotoxic, and environmentally friendly antimicrobial surfaces.
In earlier efforts, we found that the modification of natural resin acids (from gum rosin) into quaternary ammonium compounds is a promising method for the preparation of highly effective antimicrobial agents, which kill bacteria via selective bacterial membrane lysis while exhibiting high biocompatibility.39–41 Herein we report the surface modification using low-cost resin acid derived cationic compounds and polymers to obtain effective antimicrobial and antibiofilm surfaces (Figure 1).
Figure 1.
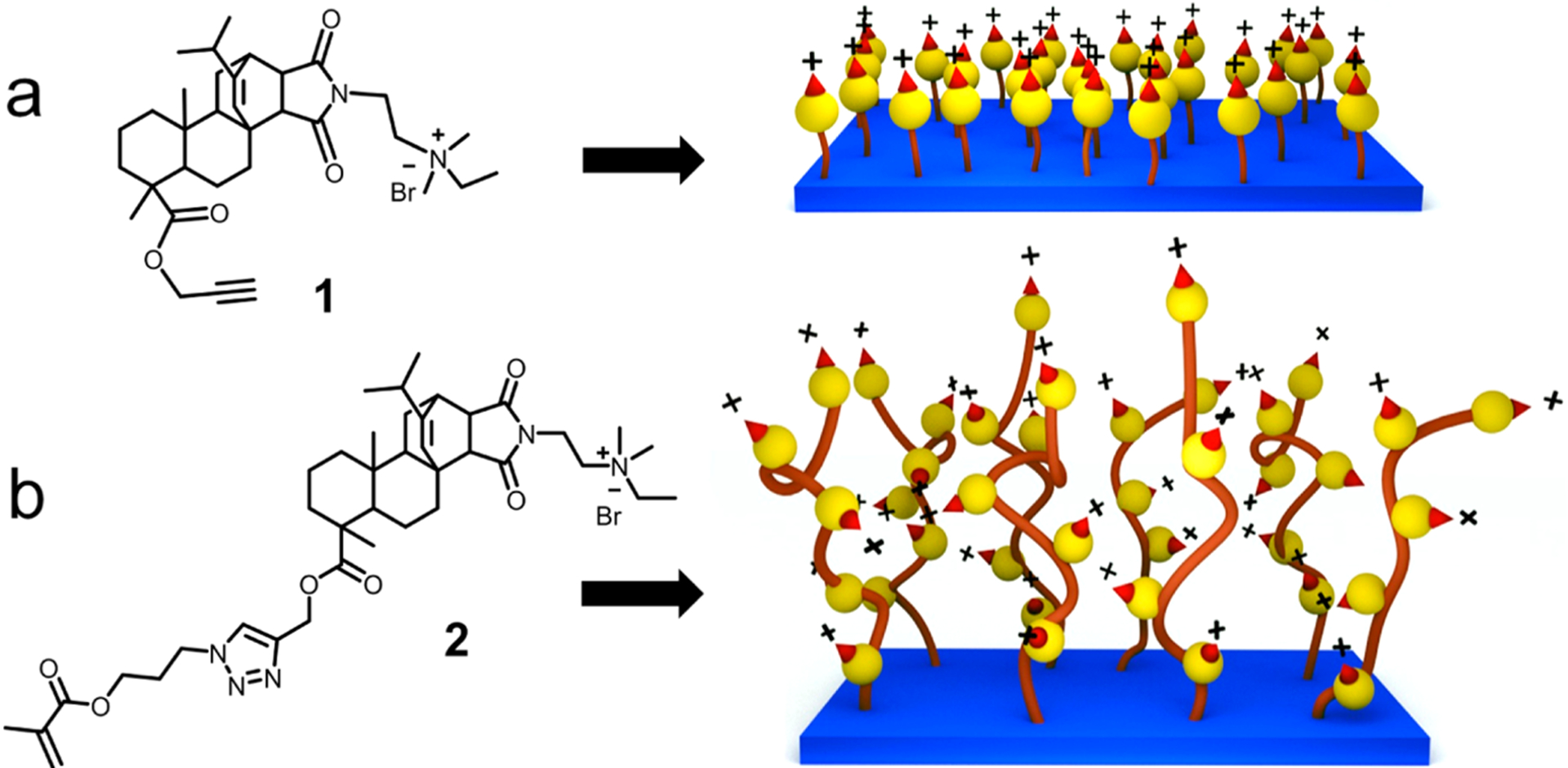
Illustration of resin acid derived cationic compounds and surfaces. (a) Compound 1 containing an alkyne functionality and the monolayer surface. (b) Methacrylate functionalized resin acid compound 2 and the polymer grafted surface.
The copper-catalyzed azide–alkyne 1,3-dipolar cycloaddition (CuAAC) click reaction was used to graft the cationic molecules to the substrate surfaces. Surface-initiated atom transfer radical polymerization (SI-ATRP) was used to functionalize substrate surfaces with cationic polymers. Although architecturally simple, these novel materials demonstrated promising antimicrobial properties with an increased resilience to bacterial biofilm formation against both Gram-positive and Gram-negative bacteria. Additional investigations revealed high levels of biocompatibility, illustrated by enhanced human dermal fibroblast (HDF) growth and low hemolysis of red blood cells.
EXPERIMENTAL SECTION
Materials.
Abietic acid (85%), maleic anhydride, N,N-dimethyla-minoethylamine (DAEA), propargyl alcohol, triethylamine (TEA), p-toluene sulfonic acid (PTSA), bromoethane, α-bromoisobutyryl bromide (BIBB), (3-aminopropyl)triethoxysilane (APTES), sodium bicarbonate, calcium chloride, 3-chloroprapanol, sodium azide, methacryloyl chloride, copper(I) bromide, N,N,N′,N′,N″-pentame-thyldiethylenetriamine (PMDETA), sulfuric acid, hydrogen peroxide, ethyl acetate, copper sulfate pentahydrate, and sodium ascorbate were purchased from Sigma-Aldrich, VWR, or Fisher Scientific and used as received. According to procedures reported in literature, (3-azidopropyl)trimethoxysilane (AzPTMS),42 bromotriethylorthosilicate (BrTEOS),43 and 3-azidopropyl methacrylate44 were prepared. QA containing resin acid derived compound 1 was synthesized, following our recent report.41 Acetic acid, dichloromethane (DCM), ethyl acetate, ethanol, hexane, diethyl ether, methanol, toluene, tetrahydrofuran (THF), and N,N-dimethylformamide (DMF) were obtained as ACS grade solvents. Standard protocols were followed to dry solvents or reagents. All other chemicals used for biological assays will be mentioned in the respective sections.
Methacrylate Monomer (Compound 2) Synthesis.
Compound 1 (8.97 g, 14 mmol, 1.0 equiv) and 3-azidopropyl methacrylate (3.07 g, 18 mmol, 1.25 equiv) were charged to a 100 mL round-bottom flask and dissolved in 60 mL of dry DMF. The flask was placed in an ice bath and stirred while bubbling nitrogen for 20 min. Cu(I)Br (80 mg, 0.558 mmol, 0.04 equiv) and PMDETA (125 mg, 0.721 mmol, 0.05 equiv) were dissolved in 2 mL of dry DMF under a nitrogen atmosphere and added to the reaction vessel. The flask was removed from the ice bath and allowed to heat to room temperature and react overnight. Upon completion of the reaction, the crude product was diluted in DCM and washed several times with aqueous/brine solution. The organic layer was then concentrated by rotoevaporation, precipitated twice in cold diethyl ether, and vacuum-dried. Yield: 70%. 1H and 13C NMR were recorded on a Bruker Avance III HD 300 spectrometer. 1H NMR (300 MHz, CDCl3) δ (ppm): δ 7.53 (s, 1H, C=CHN); 6.09 (s, 1H, CHH=CC=O); 5.60 (s, 1H, CHH= CCH3); 5.33 (s, 1H, CH=C); 5.21 (m, 2H, CCH2OC=O); 4.50 (t, 2H, CH2CH2ONN); 4.20 (t, 2H, CH2CH2OC=O); 3.79 (t, 2H, O=CNCH2CH2); 3.75 (t, 2H, O=CNCH2CH2); 3.60 (m, 2H, N + CH2CH3); 3.39 (s, 6H, N + (CH3)2); 2.89 (m, 1H, CHCHC=O); 2.56 (d, 1H, CH2CCHC=O); 2.36 (s, 3H, CH3C=CH2); 2.33 (m, 1H, CH2CHC=CH). 13C NMR (300 MHz, CDCl3): δ (ppm) 178.5 (CH2CC=OO); 178.2−177.1 (O=CNC=O); 167.1 (CH3CC=OO); 147.3 (CHC=CHC); 143.1 (NCH=CCH2); 136.0 (CH3C=CH2); 126.1 (NCH=CCH2); 124.6 (CHC=CHC); 124.0 (CH3C=CH2); 61.2−59.9 (CH2CH2N+ and CH3CH2 N+). Mass spectrometry was conducted on a Waters Micromass Q-Tof mass spectrometer, and the ionization source was positive ion electrospray. ES-MS: m/z 706.4540 (theoretical m/z: 706.4544 + H+ without Br−).
Preparation of Antimicrobial Surfaces.
Scheme 1 illustrates the synthetic route used for the preparation of the antimicrobial surfaces. As a fundamental study, glass substrates were used for the surface modifications. Glass slides were cut to the desired size (1.2 mm × 10 mm × 25 mm) and immersed in a solution of Piranha (H2SO4/H2O2, 3:1) at 50 °C for 3 h with occasional swirling. The surfaces were then carefully and thoroughly washed with DI water until the pH reached 7.0 and placed in an oven (120 °C) for 2 h. Later, they were transferred into a homemade reactor containing dry solutions of AzPTMS or BrTEOS (10 mM) in dry toluene. After assuring that all surfaces were completely immersed, the flask was heated at 110 °C for 24 h. The surfaces were purified by washing them thrice with toluene, ethanol, and acetone, consecutively.
Scheme 1. Grafting of Antimicrobial Materials on Surfacesa.
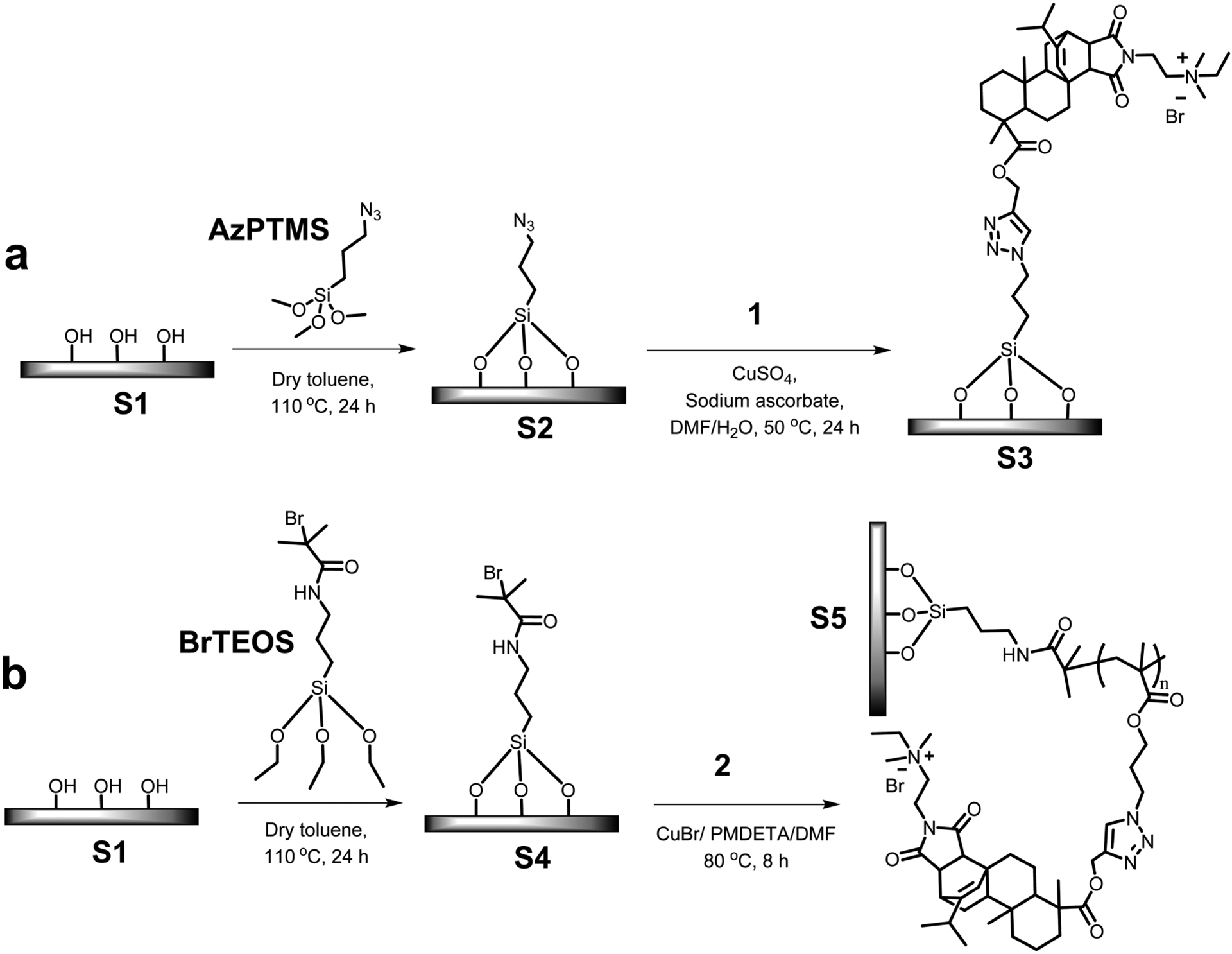
a(a) grafting a monolayer of compound 1 and (b) grafting the polymer with pendant cationic abietic acid derivative.
A solution of compound 1 was prepared by dissolving it (318 mg, 0.5 mmol, 1.0 equiv) in DMF (40 mL) and stirring for 10 min. Then azide-grafted surfaces (S2) were placed in the homemade reactor containing the solution along with CuSO4·5H2O (12.6 mg, 0.05 mmol, 0.1 equiv) in water (5 mL). The flask was covered and purged with nitrogen for 15 min. Sodium ascorbate (20 mg, 0.1 mmol, 0.2 equiv) in water (5 mL) was transferred using a needle. This flask was kept at 50 °C for 24 h. Compound 1 immobilized substrates were recovered from the reaction mixture and washed thoroughly first with DMF, ethanol, and acetone, consecutively, and finally with DI water. After drying under a stream of nitrogen, the modified glass slides were stored in a dry container.
ATRP initiator grafted substrates (S4) were kept in the homemade reactor, monomer 2 (4.716 g, 6 mmol, 1.0 equiv) was added along with 18 mL of dry DMF, and purged with N2 for 15 min. Then a mixture of CuBr (214.5 mg, 1.5 mmol, 0.25 equiv) and PMDETA (259.5 mg, 1.5 mmol, 0.25 equiv) in 12 mL of dry DMF was transferred. The reactor was immersed in a preheated oil bath at 80 °C and kept stirring for 8 h. After that, the substrates were carefully washed with DMF, ethanol, and acetone, consecutively, and finally with DI water. After drying under a stream of nitrogen, the surface-modified substrates were stored in a dry container.
Surface Characterizations.
The chemical compositions of the surfaces were determined by X-ray photoelectron spectroscopy (XPS) using a Kratos AXIS Ultra DLD XPS system equipped with a monochromatic Al Kα source. Static water contact angles were measured using a VCA Optima (AST Products, Inc.) system with a manual controller capable of casting 1 μL of Milli-Q water droplets. Static contact angles were recorded 5 s after placing the water drop on the surface. At least five replicate measurements were taken to calculate the average contact angle. Following reported procedures,32,45 the surface charge density was evaluated. Modified surfaces were dipped in a 1 wt % solution of fluorescein (Na salt) in deionized water for 10 min while shaking. Then the slides were thoroughly rinsed with deionized water, placed in 3 mL of 0.1 wt % cetyltrimethylammonium chloride in deionized water, and shaken for 20 min to desorb the dye from the surface. The absorbance of the resultant aqueous solution was measured at 501 nm, after adding 10 vol % of 100 mM aqueous phosphate buffer at pH 8.0. The surface charge density was calculated using the surface area of the substrate and the extinction coefficient (77 mM−1 cm−1).32,46,47 This staining method was also used qualitatively to observe the presence of active material on the surface using the confocal laser scanning microscopy (CLSM). For comparison, the same polymerization procedure was conducted using glass beads instead of glass slides. The grafted polymer was cleaved from the surface using HF acid, following a procedure reported in the literature.48 Then the molecular weight was determined by gel permeation chromatography (GPC). It was performed with the eluent DMF at a flow rate of 1.0 mL/min at 50 °C on a Varian system equipped with a ProStar 210 pump and a Varian 356-LC RI detector and three phenogel columns (Phenomenex Co.) calibrated with narrow dispersed polystyrene standards.
Contact Active Antibacterial Properties.
To rule out the presence of any physically adsorbed material on the surfaces, a standard diffusion assay was conducted using bacterial lawns of E. coli (ATCC 25922) and S. aureus (ATCC 25423) grown on tryptic soy agar. These bacterial strains were used throughout the study. To determine the antimicrobial activities of immobilized antimicrobial agent under dynamic contact conditions a modified test method was used.49 Sterile conical tubes (15 mL) containing 5 mL of tryptic soy broth (TSB) were inoculated with 15 μL of log-phase cultures of either E. coli or S. aureus. Modified substrates were sterilized with ethanol. After drying, they were immersed vertically in the tubes containing the cultures and incubated at 37 °C with continuous shaking at 150 rpm. After 24 h, 1.0 mL from each well was serially diluted with phosphate buffered saline (PBS) and plated on tryptic soy agar. After incubating 24 h at 37 °C the bacterial colonies were counted to obtain the colony forming units (CFU). The glass slides in the conical tubes were carefully rinsed with PBS and stained with propidium iodide and SYTO 9 (Live/Dead BacLight viability kit, Life Technology, Carlsbad, CA, U.S.A.). These fluorescent nucleic acid stains indicate cell viability as a function of membrane integrity. Healthy cells with intact membranes stain green, while dead or dying cells with compromised membranes stain red. CLSM was used to visualize the samples. ImageJ software was used to calculate the number cells and CFUs observed from these experiments.
Antibiofilm Activity in Solution.
E. coli and S. aureus were subcultured for purity from a frozen glycerol stock on TSA at 37 °C and subsequently used to inoculate 50 mL of 10% TSB. The culture was incubated overnight at 37 °C with gentle shaking (150 rpm). Sterile MBEC Biofilm Assay plates with 96 well bases (Innovotech, Edmonton, Canada) were used to test for biofilm formation. In a typical procedure, 50 mL of the overnight culture was diluted into sterile 10% TSB for an approximate cell density of 105 CFU/mL to create the inoculum. Then 150 μL of the inoculum was added to each well, and the peg lid was securely placed on the plate and sealed with parafilm. The plate was incubated for 24 h at 37 °C, 110 rpm. A sterile 96-well plate was seeded with various concentrations of 1 (0–500 μg/mL) in 10% TSB. The MBEC peg lid was rinsed in deionized water three times and inserted onto the antimicrobial challenge plate. The plate was incubated at 37 °C and 110 rpm for 24 h. After 24 h, the biofilm was rinsed in deionized water for 1 min and allowed to dry for 10 min at room temperature. The MBEC peg lid was then stained in 1% crystal violet for 1 min to stain the biofilm and rinsed three times in fresh deionized water. The peg lid was then treated with 100% methanol (1 min) to dissolve the crystal violet. Dissolved crystal violet was then measured at 600 nm with a Power Wave 200 Microplate Scanning Spectrophotometer (Bio-Tek Instruments, Winooski, VT).
Antibiofilm Activity of the Surfaces.
Biofilms were grown on treated and untreated substrates separately within a flow-through CDC Biofilm Reactor (CBR, BioSurface Technol., Bozeman, MT, U.S.A.). CBRs are chemostat reactors that allow a gradual but constant refreshment of nutrients to facilitate continuous growth of attached biofilm communities.50,51 The bacteria E. coli was subcultured from a glycerol stock in 3 g/L tryptic soy broth (TSB, Sigma-Aldrich, St. Louis, Missouri) at 37 °C. A total of 3 mL of an overnight culture were used to inoculate the sterile CBR containing 400 mL of 3 g/L TSB. The CBR was maintained at room temperature with a constant stir speed of 120 rpm. The flow rate of sterile 3 g/L TSB into the CBR was maintained at 0.1 mL/min. Biofilms were quantified using a crystal violet (CV) staining assay. The substrate surfaces were extracted in duplicate at 1, 2, 4 and 8 days postinoculation. The slides were gently rinsed with PBS and treated with 1% crystal violet to stain the biofilm cells. Excess stain was rinsed with deionized water and crystal violet was extracted with methanol. Cell density of the biofilm was quantified by measuring the absorbance of crystal violet at 595 nm on a UV-2450PC UV–vis Spectrophotometer (Shimadzu Corp., Kyoto, Japan).
Evaluation of Biocompatibility.
Surface-modified substrates were equilibrated in centrifuge tubes containing 10 mL of PBS and 0.2 mL of diluted mouse blood (800 μL of blood diluted with 1000 μL of PBS), following incubation at 37 °C for 1 h. PBS + 0.5% triton X100 and PBS were used as positive and negative controls, respectively. The tubes were centrifuged at 1500 rpm for 10 min and the optical absorbance of the supernatant was measured at 545 nm on a microplate reader. The hemolysis rate (HR) was calculated as follows: HR = (AS – AN)/(AP – AN), here AS, AN, and AP are the optical absorbance of the supernatant of the solution containing modified glass, the negative control, and the positive control, respectively. Additionally, in vitro biocompatibility studies were carried out by utilizing a HDF cell line that was obtained from Instrumentation Resources Facility, School of Medicine, University of South Carolina. HDF was propagated using Dulbecco’s modified Eagle medium (DMEM, Sigma) supplemented with 7.5 mM of l-glutamine, 1% of penicillin/streptomycin solution, 10% of fetal bovine serum (complete DMEM). HDF (passage 30) was seeded in 12 wells of tissue culture polystyrene plates containing different surfaces at a density of 1 × 104 cells/cm2 in a 1 mL volume of medium and allowed to proliferate for 96 h at 37 °C in a 10% CO2-modified atmosphere until 60–70% confluence was reached, as previously described.52 Morphological observations of the HDF after 4 days of in vitro culture on the different surfaces were made by phase contrast microscopy and analyzed with Lumenera’s INFINITY ANALYZE software.
The HDF cells grown on different surfaces were fixed with 2% para-formaldehyde for exactly 10 min. The fixed cells on the different surfaces were washed twice with PBS and then incubated with Alexa Fluor 488 Phalloidin antibody (Life Technology) for actin staining (green) for 60 min at room temperature. After three times of PBS wash, the surfaces were stained for DNA with 4’,6-diamidino-2-phenylindole (DAPI; blue) for 15 min at room temperature. After rinsing thoroughly with PBS, the different surfaces were mounted on glass slides in Dabco (Sigma-Aldrich) and stored at 4 °C. The images from different surfaces were taken using CLSM.
RESULTS AND DISCUSSION
Grafting Antimicrobial Agents on Substrate Surfaces.
Current efforts toward the utilization of renewable resources to generate useful and functional materials are steadily increasing.53,54 Diterpene resin acids produced largely by pine and conifer trees constitute a large bulk of biomass that can be effectively used to make novel materials. QA containing cationic compounds and polymers derived from these resin acids can act as highly effective antibacterial agents with remarkable biocompatibility (i.e., minimal cytotoxicity) toward mammalian cells.39,41,55 Unlike other small QA compounds, this unusual activity of the QA abietic acid derivatives is related to the optimum balance between the hydrophenanthrene ring skeleton and the quaternary ammonium group. In the current study, we prepared antimicrobial surfaces using these resin acid-derived QA cationic compounds. Typically, QA compounds and polymers have been covalently attached onto various substrate surfaces such as glass, polymers, paper and metals.56 In the present study, glass slides were used as the substrate since they are well-known to be inert and biocompatible. However, it is possible to use the same chemistry for any substrate with hydroxyl functionality. Two strategies were followed to develop antimicrobial cationic surface systems. The surfaces with a monolayer of compound 1 were prepared via click chemistry between the surface-immobilized azide groups and the alkyne moiety on compound 1 (Scheme 1a). Efficiency, versatility, and selectivity of the CuAAC click reaction are strongly demonstrated in a broad range of fields such as polymer and materials chemistry,57–59 bioconjugate chemistry,60 medicinal chemistry,61 and many other organic syntheses.62 Furthermore, atom transfer radical polymerization (ATRP) is a widely utilized and versatile method for controlled radical polymerization that allows a diverse range of polymers and architectures to be prepared.63 Surface-initiated ATRP (SI-ATRP) is commonly used to prepare polymer brushes on surfaces.64 This method is well explored to prepare antibacterial surfaces with high control, tunability, and long-term effectiveness.49 Therefore, SI-ATRP was used in this work to graft cationic polymers from surfaces, which were modified with the ATRP initiator (Scheme 1b).
Progress of the reactions were confirmed by determining the static water contact angle measurements (Figure 2a). Piranha-treated glass surfaces (S1) were highly hydrophilic and had a water contact angle of 17°. When AzPTMS was immobilized (S2), the surface hydrophobicity increased, which was evident from the contact angle increasing to 85°. These results were in good agreement with literature.42,45 Subsequent CuAAC click modification with the QA compound 1 amplified the surface hydrophilicity, leading to a final contact angle of 68°. Although there were positive charges on the compound 1 grafted surface (S3), the contact angle was much higher than that for pristine glass, which may be attributed to the hydrophilic/hydrophobic balance between the bulky hydrophenanthrene ring and the cationic groups. Surface attachment of ATRP initiator resulted in a surface (S4) with a contact angle of 60°, which was later increased to 82° (S5) after the SI-ATRP of the compound 2.
Figure 2.
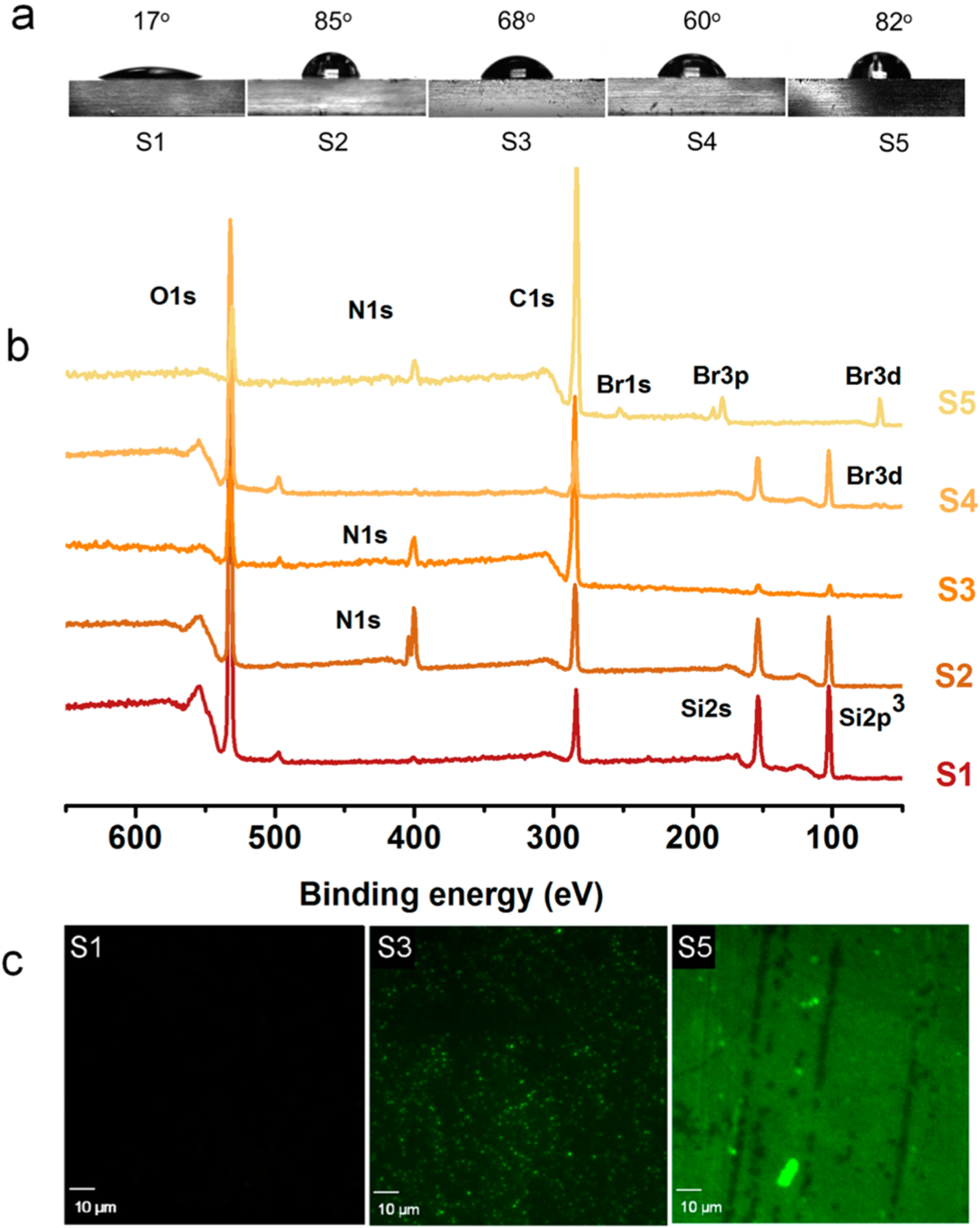
Surface characterizations. (a) Static water contact angles on various modified surfaces. (b) XPS survey spectra. (c) CLSM images of surfaces stained using fluorescein. S1, pristine glass; S2, azide-grafted; S3, compound 1-grafted; S4, ATRP initiator-grafted; and S5, polymer-grafted.
Additionally, XPS revealed the change of surface composition and the progression of the chemical modifications. The XPS survey scans illustrated the increase of carbon content and the appearance of nitrogen on surface S2 and bromine on surface S5, compared to that of surface S1 (Figure 2b). After the completion of CuAAC reaction and the SI-ATRP, the carbon content increased significantly on both surfaces S3 and S5. Depletion of silane peak intensity also confirmed the formation of a layer of material on top of the glass surface. XPS did not detect copper on the surfaces, thus, nullifying the argument that residual catalysts from surface modifications may have an effect on bactericidal properties. Also, previous control studies using copper against different strains of bacteria have not shown significant amounts of toxicity at low concentrations, indicating that our resin-acid derived compounds and polymers were responsible for the antimicrobial properties of the surfaces.39 Grafting of cationic compound 1 and the cationic polymer resulted in surfaces that bear covalently attached cations. These cations can be stained with a negatively charged dye such as fluorescein. It is useful to qualitatively confirm the presence of cationic groups on surfaces. For the polymer grafted surface, the stain was visible even to the naked eye, indicating the increase of cationic groups on surface S5. Figure 2c shows the CLSM images of different surfaces stained with fluorescein.
The density of QA groups on the glass surfaces was determined by a colorimetric method which is based on fluorescent complexation between fluorescein disodium salt and QA groups, as reported earlier.32,45 It can be envisioned that not all the cations could be exposed to fluorescein due to steric crowding, especially on the polymer grafted surface. Therefore, this method quantifies the “solvent accessible” density of QA groups on the glass surfaces. Under the given conditions, fluorescein anions bind to QA groups via charge–charge interactions. It is assumed that one dye molecule binds with one QA group. A stronger detergent such as cetyltrimethylammonium chloride can be used to remove the bound fluorescein from the surface. The stain can then be quantified by measuring the absorbance at 501 nm, which is characteristic to fluorescein. There were approximately 0.5 cationic groups per nm2 on the surface S3. Surface S5 had a cationic group density of 6.3/nm2. Grafting density was approximated to 0.13/nm2, with respect to the molecular weight obtained from the cleaved polymer, which was Mn,GPC = 39400 g/mol.
Contact Active Antibacterial Activity of the Surfaces.
In this work, we observed strong bactericidal effects upon contact of bacteria with QA resin acid derived cationic surfaces. It was found that there was no observable leakage of materials from the surfaces demonstrated by standard diffusion assays. The CLSM images (Figure 3) show the development of S. aureus and E. coli cells on surfaces after 24 h incubation with surfaces S1, S3, and S5. The Live/Dead kit used for this viability assay consists of two stains: propidium iodide (PI) and SYTO 9, both of which stain nucleic acids. Green-fluorescing SYTO 9 is able to enter all cells live or dead and is used for assessing total cell counts, whereas red fluorecing PI enters only cells with damaged cytoplasmic membranes (e.g., dead cells). Therefore, bacteria observed from CLSM must result from pronounced growth on pristine glass surface.
Figure 3.
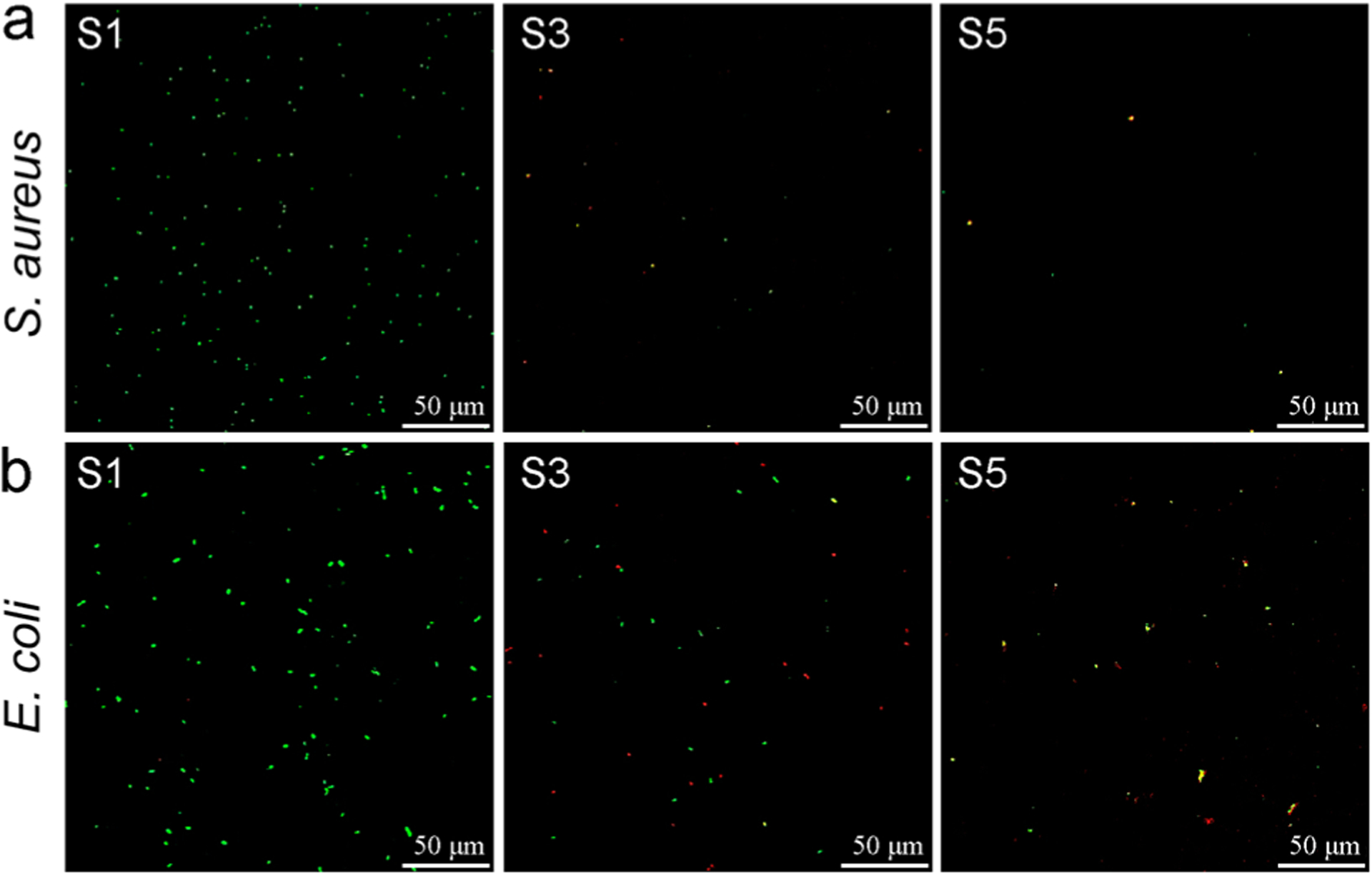
Stained (Live/Dead stain) surfaces after 24 h incubation with (a) S. aureus and (b) E. coli. Green cells indicate live bacteria colonizing the surface, while dead cells appear in red color. S1, pristine surface; S3, compound 1-grafted; and S5, polymer-grafted.
However, the number of cells attached to the surface remarkably reduced when the compound 1 was grafted (Figures 3 and 4a,b). This is possibly an antiadherence property of these QA surfaces. Compared to S1, there is an 86% reduction of attached S. aureus cells and 47% reduction of attached E. coli cells on S3. Interestingly, S3 showed prominent bactericidal properties, observed in red-fluorescent cells (i.e., having compromised cell membranes). The modification with QA compound 1 resulted in a 61% loss in S. aureus viability and 49% in E. coli viability (Figure 4c,d). Further, surfaces with QA polymer prepared by SI-ATRP also exhibited a similar trend. However, the antibacterial activity was much higher in terms of bactericidal as well as antibiofilm properties. There was a 94% reduction of S. aureus cells and a 59% reduction of E. coli cells attached on S5, compared to S1. After incubation, most of the cells located on S5 fluoresced red, indicating membrane damage with 70% of S. aureus cells and 69% of E. coli cells. In comparison with the results of S3, there was a notable increase in bactericidal activity of S5 against both S. aureus and E. coli. In addition, the modified glass surfaces were more bactericidal against Gram-positive S. aureus than they were against Gram-negative E. coli. This may be attributed to the bacterial cell envelope structural difference where Gram-negative E. coli has an additional outer membrane.
Figure 4.
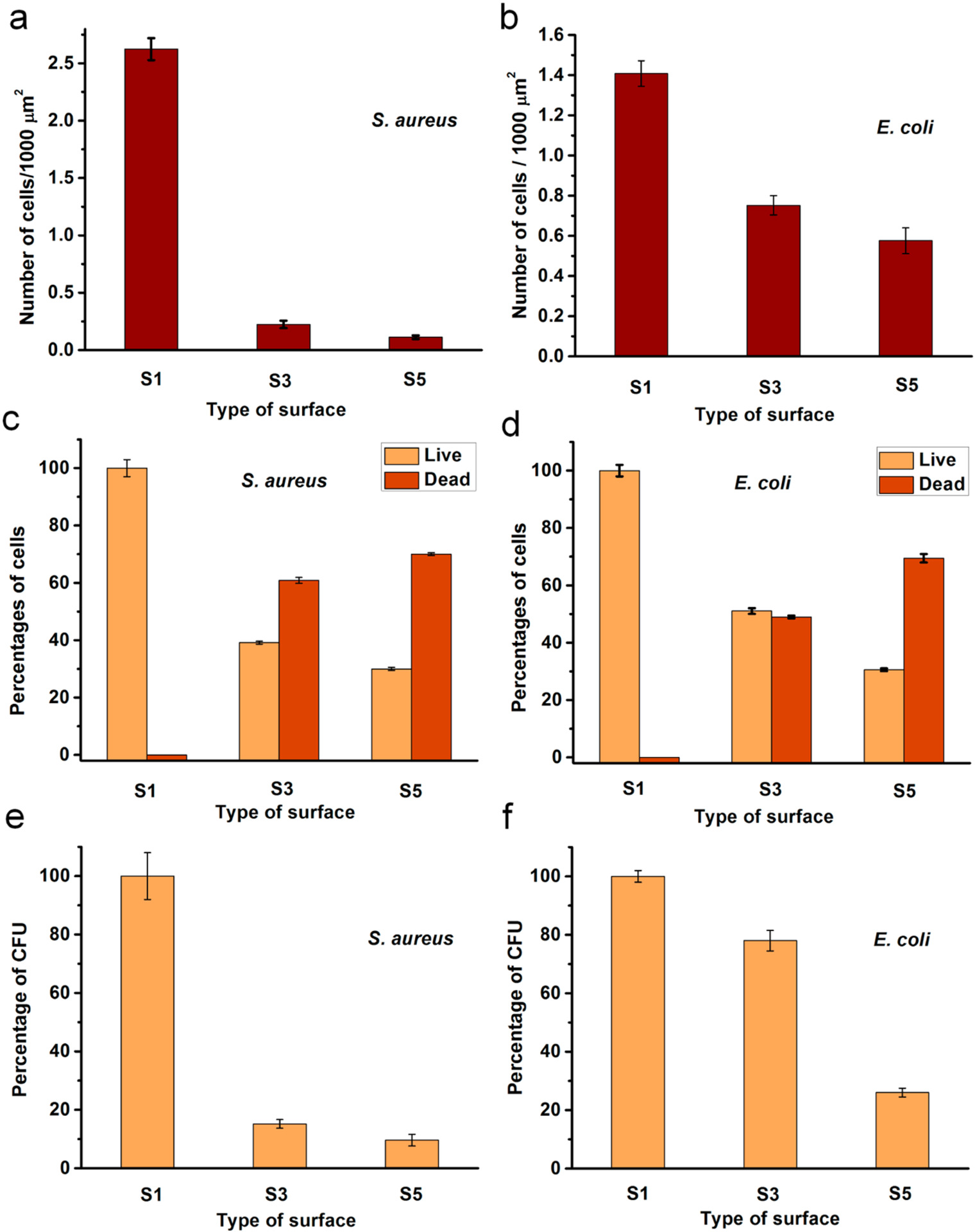
Antibacterial activity of the modified surfaces after 24 h: (a, b) Number of cells on the surface; (c, d) Percentages of live or dead cells on the surfaces; (e, f) Percentages of CFU obtained for the bacterial cultures exposed to surfaces. S1, pristine surface; S3, compound 1-grafted; and S5, polymer-grafted.
These cationic surfaces were able to kill planktonic bacterial cells that came in contact with them. This phenomena was probed from CFU counts of the liquid bacterial media incubated in contact with the modified surfaces. S. aureus showed more susceptibility against both surfaces S3 and S5 with 84% or more reduction of CFU (Figure 4e,f). However, E. coli cells were less affected by the cationic surfaces resulting with the highest inhibition of 74% CFU in contact with S5 relative to control S1.
Antibiofilm Properties.
It has been demonstrated that QA compounds can effectively eradicate bacterial biofilm formation.65 Since the material in this study showed strong antibacterial activities, a CDC bioreactor was used to study long-term antibiofilm activity. In most studies, relatively short periods of time (<1 day) are used to study biofilm growth on cationically modified surfaces. However, given that bacteria are dynamic and can adapt to their environment, we conducted experiments over longer periods of time. The results of the antimicrobial challenge assay indicated that compound 1 has strong antibiofilm activity. The lowest concentration of compound 1, which prevented the growth of previously established S. aureus or E. coli biofilms on the pegs of the antimicrobial challenge plate, was 250 μg/mL. This was determined by measuring the mass of biofilm on each peg indirectly with dissolved crystal violet. The results for antibiofilm activity of modified surfaces conducted with the CDC bioreactor are summarized in Figure 5. It should be noted that the model systems used for surface antimicrobial activity and antibiofilm properties are quite different, hence a direct comparison would not be meaningful. It is obvious that the unmodified surface S1 showed prominent and gradual growth of bacterial biofilm on the surface. However, QA-attached surfaces exhibited reduced growth of biofilm on them. For example, S3 with a monolayer of compound 1 showed 77% less S. aureus biofilm biomass at 2 days of incubation and S5 with QA polymer demonstrated a 64% reduction of biofilm compared to the untreated glass S1. The cationic surfaces were more effective toward S. aureus biofilm growth compared to E. coli. This can be expected because the QA compounds have shown to be more antimicrobial toward Gram-positive bacteria. Over 8 days, S3 and S5 demonstrated slow growth of biofilms, which was significantly less than that of the pristine surfaces. A similar or higher amount of biofilm on QA polymer-coated surface may be attributed to the fact that the increased surface area/roughness provided by the grafted polymer may have more attachment sites.
Figure 5.
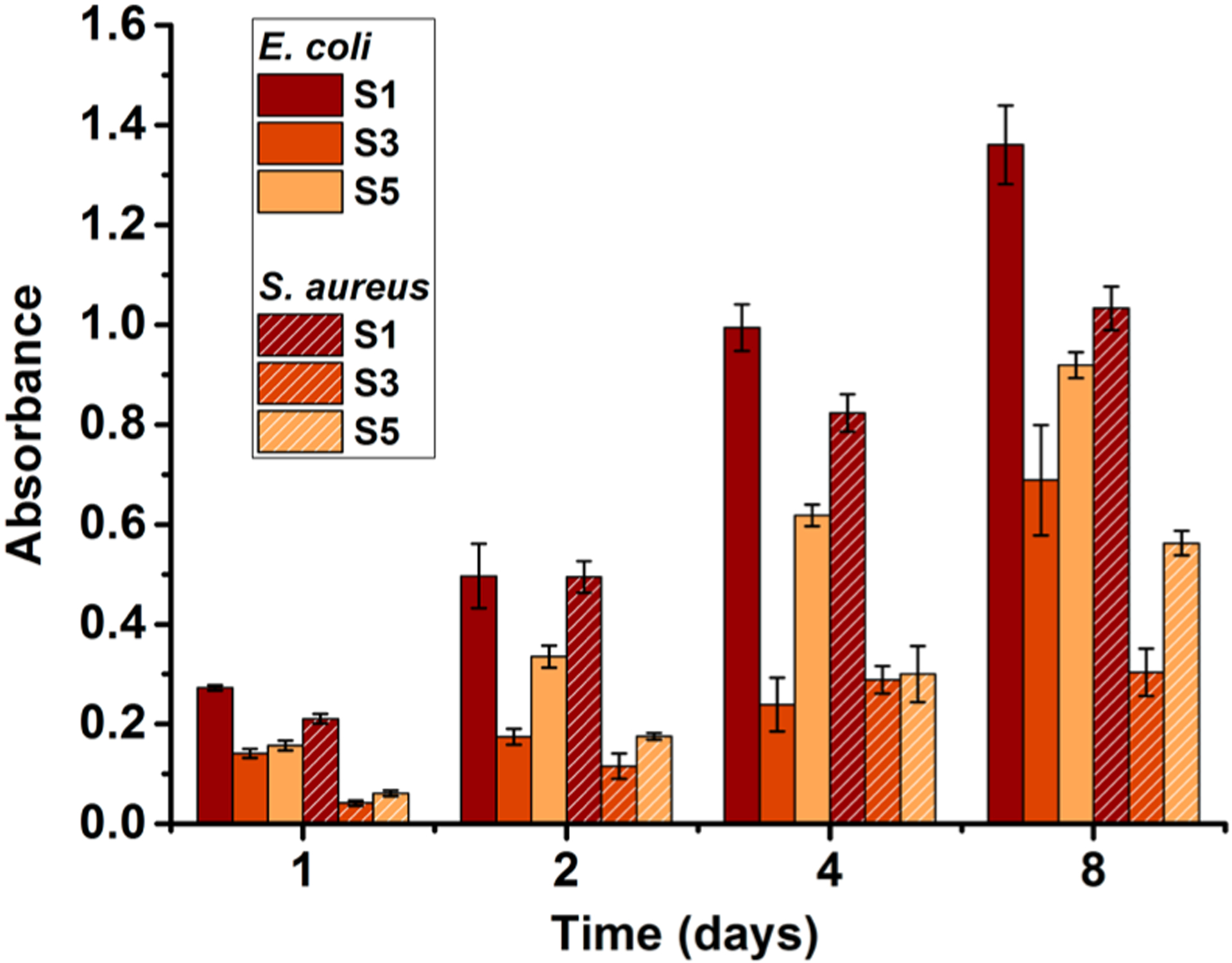
Amounts of E. coli and S. aureus biofilm biomass accumulated on the surfaces after incubating in the CDC bioreactor. S1, pristine surface; S3, compound 1-grafted; and S5, polymer-grafted.
The complicated process of biofilm formation is initially governed by physiochemical interactions between the bacterial cells and the target surface. Therefore, surface chemical composition, charge, hydrophobicity and contour act as primary factors in the success or failure of colony initiation on a surface.21,22 Antibacterial surfaces may deactivate any planktonic cells that successfully interact with the surface, hence, preventing the next stages of biofilm formation. We postulated that the reduction in bacterial growth was the result of two possible mechanisms (Figure 6). First, the cells are killed upon contact with the polymer and, therefore, cannot readily establish a biofilm. Second, the attachment of bacterial cells on the surface is reduced due to unfavorable surface properties such as charge and hydrophilic/hydrophobic balance. The antibacterial and antibiofilm assay results of this study indicated that the surface modification procedure induces both mechanisms.
Figure 6.
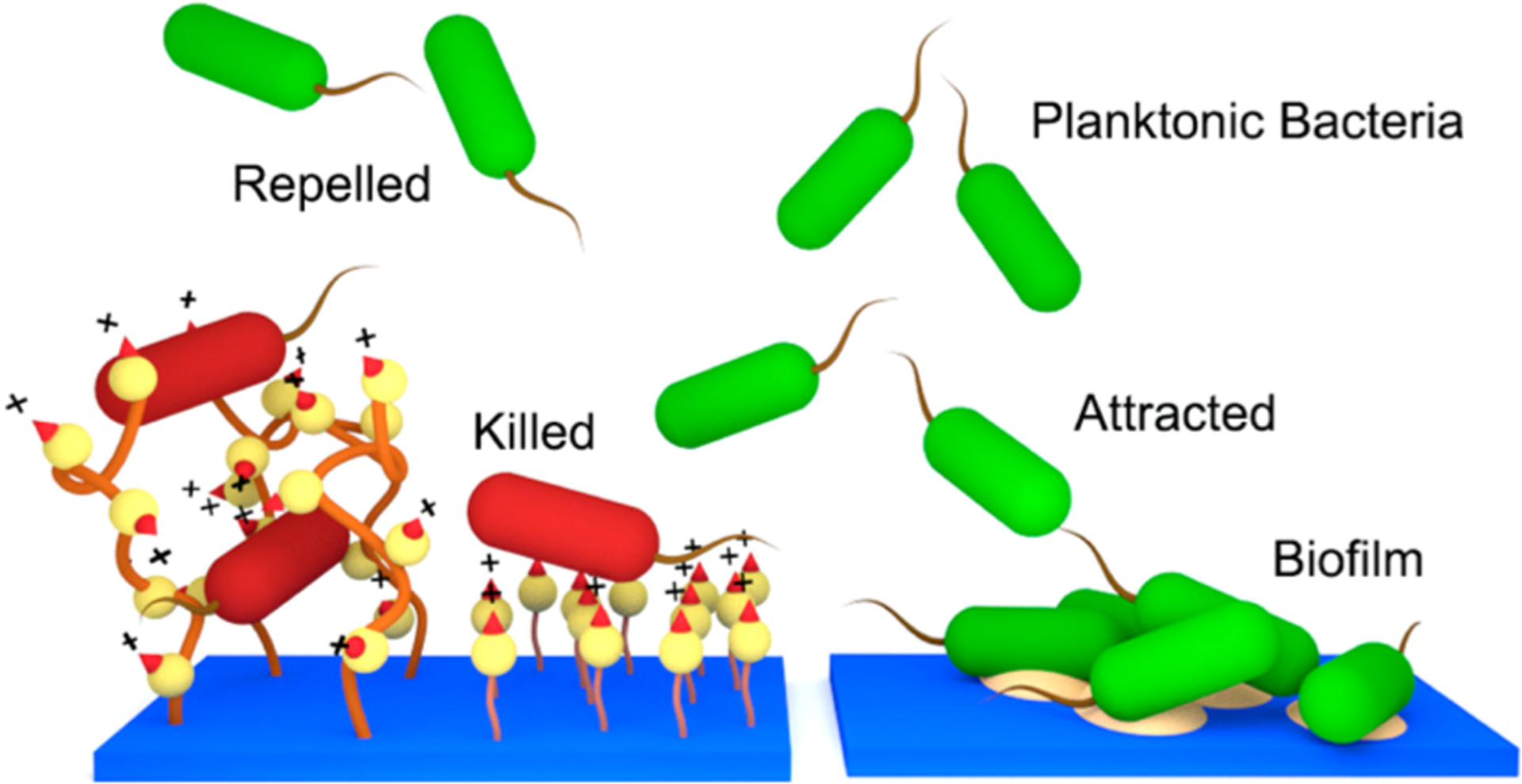
Comparison of antimicrobial mechanisms of the QA surfaces (left) and pristine surface (right). Resin acid containing cationic surfaces may act as a bactericidal and bacterial repelling coating.
Biocompatibility of the Surfaces.
In our earlier report, it was demonstrated that QA decorated abietic acid compounds and polymers are highly biocompatible while showing strong antimicrobial properties.41 Here, additional biocompatibility evaluations were carried out for the modified surfaces utilizing hemolysis activity assays and surface cell growth assays. The hemolysis on chemically modified glass surfaces indicated a high degree of biocompatibility (i.e., noncytotoxicity). Unmodified glass is known to have a negligible hemolytic effect. The hemolysis level of S3 was found to be <4%. The polymer-grafted glass S5 resulted in insignificant hemolysis (<0.1%). Interestingly, the cationic surfaces showed improved proliferation of fibroblast cells. HDF cell proliferation over 4 days (Figure 7) showed continuous increase of cell density on the cationic surfaces.
Figure 7.
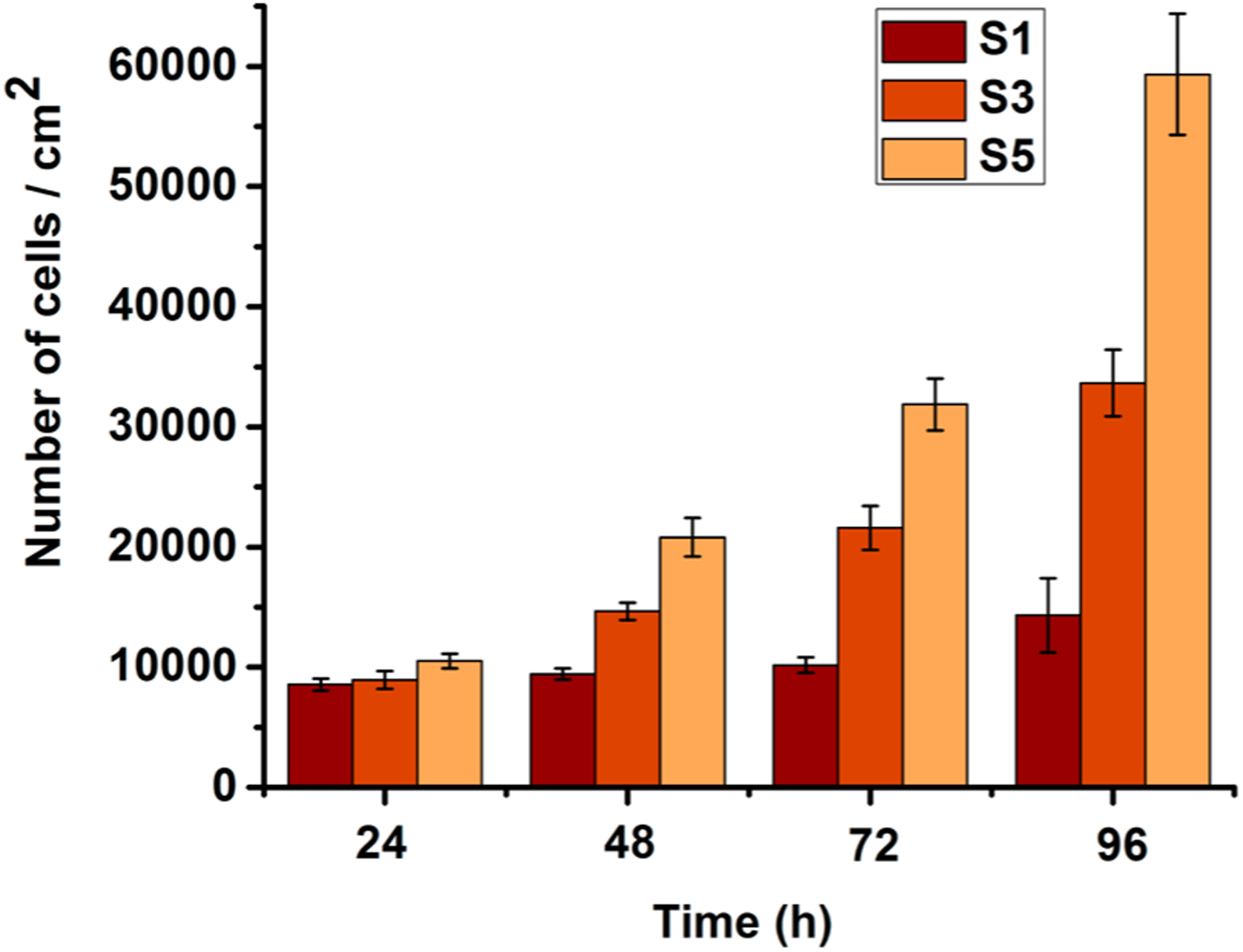
Amount of HDF cells proliferated on the surfaces. S1, pristine surface; S3, compound 1-grafted; and S5, polymer-grafted.
Figure 8 shows the fluorescence microscopy and phase contrast images of the fibroblast cells grown on surfaces S1, S3, and S5. HDF cells were attached and spread well, with typical cell morphologies on surfaces S3 and S5. However, surface S1 did not show pronounced growth when compared to surface S3 or S5.
Figure 8.
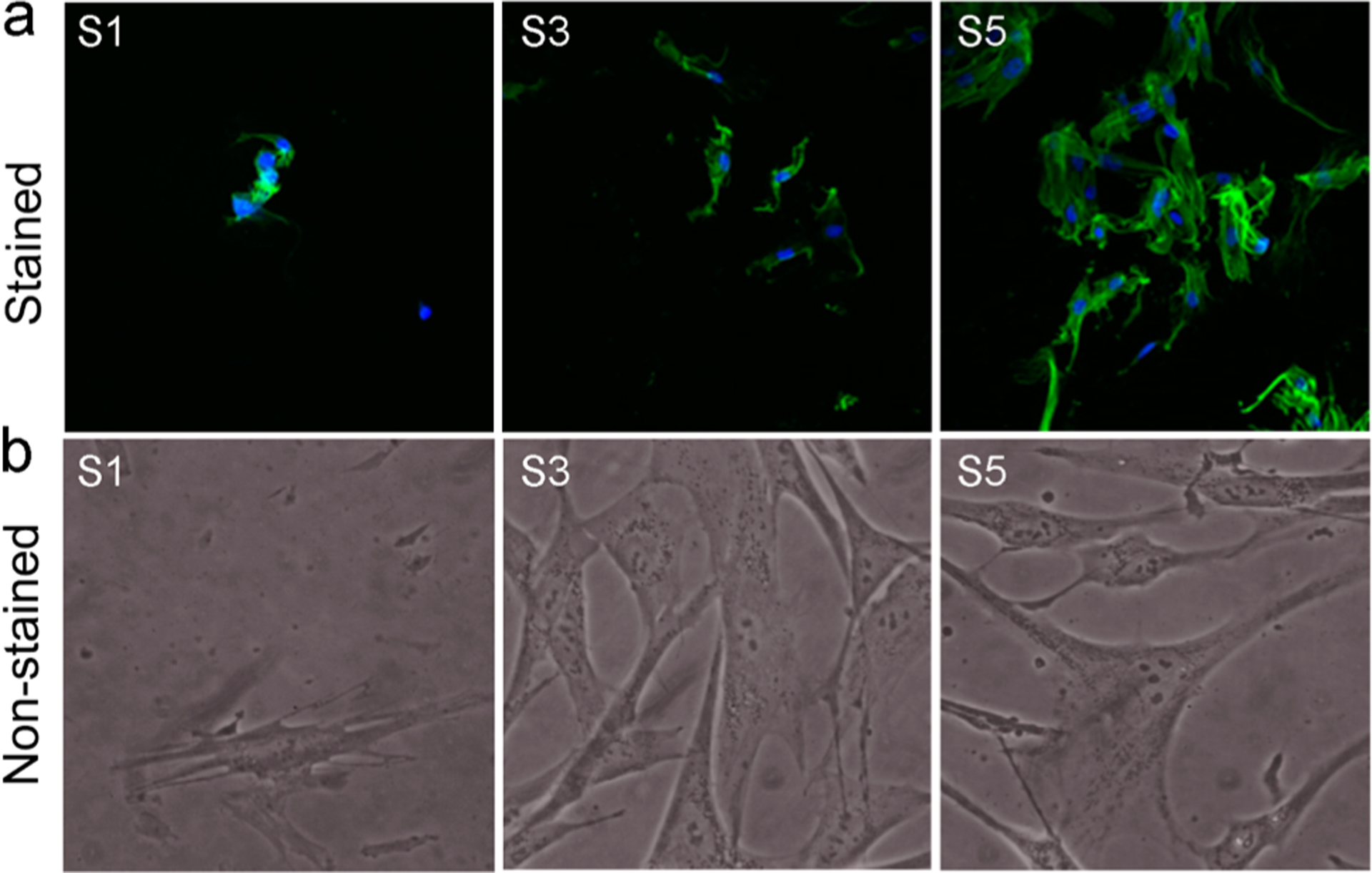
Morphology of HDF cells proliferated on the surfaces after 4 days. (a) Fluorescence microscopy images after staining. (b) Images under the phase contrast microscope. S1, pristine surface; S3, compound 1-grafted; and S5, polymer-grafted.
After 2 days of seeding, cells changed their shape and began to spread. Cells on surfaces S3 and S5 had stretched out many filopodia, and most of them had further extended to be triangles and polygons, while the cells on surface S1 spread to be flat with no or less ejection of filopodia. There was a difference in reaching confluence of the HDF cells within the different surfaces. Confluence of the HDF cells was observed on surfaces S3 and S5. There was a difference in cell proliferation on the surfaces, which was indicated by the varying extents of cell spreading observed on different surfaces. The surface S5 was highly cell-adhesive. HDF, when attached to S5, proliferated to mature fibroblasts by extending their filopodia and having a branched cytoplasm surrounding their speckled nucleus. It was interesting to observe an improved proliferation of human fibroblast cells on the cationic surfaces, which have been similarly reported in earlier studies,66,67 stating that cationic surfaces promote fibroblast spreading, proliferation, and extracellular matrix production.
5. CONCLUSIONS
In conclusion, we have developed a simple and effective method to prepare antimicrobial surfaces using resin acid derived cationic compounds and polymers. The cationic compound 1 and polymer grafted surfaces showed strong antimicrobial activity against the bacteria, S. aureus and E. coli. The surfaces were more active against Gram-positive (i.e., S. aureus) bacteria. The surfaces were resistant to biofilm growth for longer durations of time, as demonstrated by the CDC bioreactor assay. In addition, biocompatibility of surfaces was proven to be excellent, as demonstrated by hemolysis assays and HDF growth assays. These contact active coatings may be used to modify relevant surfaces to actively eliminate infectious microorganisms by destroying planktonic cells as well as controlling their biofilm growth. Therefore, this work presents a promising approach for the incorporation of renewable resources for developing surfaces that can control the spread of infectious diseases.
ACKNOWLEDGMENTS
This work was supported by National Science Foundation (DMR-1206072) and in part by NIH P20 GM103641. The authors would like to thank Dr. Shuguo Ma at the USC XPS Facility for the help to obtain XPS data.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Costerton JW; Cheng KJ; Geesey GG; Ladd TI; Nickel JC; Dasgupta M; Marrie TJ Annu. Rev. Microbiol 1987, 41, 435–464. [DOI] [PubMed] [Google Scholar]
- (2).Branda SS; Vik Å; Friedman L; Kolter R Trends Microbiol. 2005, 13, 20–26. [DOI] [PubMed] [Google Scholar]
- (3).Donlan RM Clin. Infect. Dis 2001, 33, 1387–1392. [DOI] [PubMed] [Google Scholar]
- (4).Berk V; Fong JCN; Dempsey GT; Develioglu ON; Zhuang X; Liphardt J; Yildiz FH; Chu S Science 2012, 337, 236–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Davies DG; Parsek MR; Pearson JP; Iglewski BH; Costerton JW; Greenberg EP Science 1998, 280, 295–298. [DOI] [PubMed] [Google Scholar]
- (6).Tuson HH; Weibel DB Soft Matter 2013, 9, 4368–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Flemming H-C; Wingender J Nat. Rev. Microbiol 2010, 8, 623–633. [DOI] [PubMed] [Google Scholar]
- (8).Decho AW; Frey RL; Ferry JL Chem. Rev 2011, 111, 86–99. [DOI] [PubMed] [Google Scholar]
- (9).Stewart PS; Franklin MJ Nat. Rev. Microbiol 2008, 6, 199–210. [DOI] [PubMed] [Google Scholar]
- (10).Fletcher MH; Jennings MC; Wuest WM Tetrahedron 2014, 70, 6373–6383. [Google Scholar]
- (11).Hall-Stoodley L; Costerton JW; Stoodley P Nat. Rev. Microbiol 2004, 2, 95–108. [DOI] [PubMed] [Google Scholar]
- (12).Høiby N; Bjarnsholt T; Givskov M; Molin S; Ciofu O Int. J. Antimicrob. Agents 2010, 35, 322–332. [DOI] [PubMed] [Google Scholar]
- (13).Costerton JW; Stewart PS; Greenberg EP Science 1999, 284, 1318–1322. [DOI] [PubMed] [Google Scholar]
- (14).Hoyle B; Costerton JW Prog. Drug Res 1991, 37, 91–105. [DOI] [PubMed] [Google Scholar]
- (15).Hasan J; Crawford RJ; Ivanova EP Trends Biotechnol. 2013, 31, 295–304. [DOI] [PubMed] [Google Scholar]
- (16).Campoccia D; Montanaro L; Arciola CR Biomaterials 2013, 34, 8533–8554. [DOI] [PubMed] [Google Scholar]
- (17).Taglietti A; Arciola CR; D’Agostino A; Dacarro G; Montanaro L; Campoccia D; Cucca L; Vercellino M; Poggi A; Pallavicini P; Visai L Biomaterials 2014, 35, 1779–1788. [DOI] [PubMed] [Google Scholar]
- (18).Swanson TE; Cheng X; Friedrich CJ Biomed. Mater. Res., Part A 2011, 97, 167–176. [DOI] [PubMed] [Google Scholar]
- (19).Gao G; Lange D; Hilpert K; Kindrachuk J; Zou Y; Cheng JTJ; Kazemzadeh-Narbat M; Yu K; Wang R; Straus SK; Brooks DE; Chew BH; Hancock REW; Kizhakkedathu JN Biomaterials 2011, 32, 3899–3909. [DOI] [PubMed] [Google Scholar]
- (20).Muñoz-Bonilla A; Fernández-García M Prog. Polym. Sci 2012, 37, 281–339. [Google Scholar]
- (21).Muñoz-Bonilla A; Fernández-García M Eur. Polym. J 2015, 65, 46–62. [Google Scholar]
- (22).Siedenbiedel F; Tiller JC Polymers 2012, 4, 46–71. [Google Scholar]
- (23).Ho KKK; Chen R; Willcox MDP; Rice SA; Cole N; Iskander G; Kumar N Biomaterials 2014, 35, 2336–2345. [DOI] [PubMed] [Google Scholar]
- (24).Brohede U; Forsgren J; Roos S; Mihranyan A; Engqvist H; Strømme MJ Mater. Sci.: Mater. Med 2009, 20, 1859–1867. [DOI] [PubMed] [Google Scholar]
- (25).Foster H; Ditta I; Varghese S; Steele A Appl. Microbiol. Biotechnol 2011, 90, 1847–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Zhang X; Wang L; Levanen E RSC Adv. 2013, 3, 12003–12020. [Google Scholar]
- (27).Ivanova EP; Hasan J; Webb HK; Gervinskas G; Juodkazis S; Truong VK; Wu AHF; Lamb RN; Baulin VA; Watson GS; Watson JA; Mainwaring DE; Crawford RJ Nat. Commun 2013, 4, 2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Isquith AJ; Abbott EA; Walters PA Appl. Microbiol 1972, 24, 859–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Gottenbos B; van der Mei HC; Klatter F; Nieuwenhuis P; Busscher HJ Biomaterials 2002, 23, 1417–23. [DOI] [PubMed] [Google Scholar]
- (30).Li Z; Lee D; Sheng X; Cohen RE; Rubner MF Langmuir 2006, 22, 9820–9823. [DOI] [PubMed] [Google Scholar]
- (31).Gabriel M; Nazmi K; Veerman EC; Nieuw Amerongen AV; Zentner A Bioconjugate Chem. 2006, 17, 548–550. [DOI] [PubMed] [Google Scholar]
- (32).Tiller JC; Liao C-J; Lewis K; Klibanov AM Proc. Natl. Acad. Sci. U. S. A 2001, 98, 5981–5985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Lin J; Qiu S; Lewis K; Klibanov AM Biotechnol. Bioeng 2003, 83, 168–172. [DOI] [PubMed] [Google Scholar]
- (34).Huang J; Murata H; Koepsel RR; Russell AJ; Matyjaszewski K Biomacromolecules 2007, 8, 1396–1399. [DOI] [PubMed] [Google Scholar]
- (35).Dong H; Huang J; Koepsel RR; Ye P; Russell AJ; Matyjaszewski K Biomacromolecules 2011, 12, 1305–1311. [DOI] [PubMed] [Google Scholar]
- (36).Asri LATW; Crismaru M; Roest S; Chen Y; Ivashenko O; Rudolf P; Tiller JC; van der Mei HC; Loontjens TJA; Busscher HJ Adv. Funct. Mater 2014, 24, 346–355. [Google Scholar]
- (37).Klibanov AM J. Mater. Chem 2007, 17, 2479–2482. [Google Scholar]
- (38).Ferreira L; Zumbuehl AJ Mater. Chem 2009, 19, 7796–7806. [Google Scholar]
- (39).Wang J; Chen YP; Yao K; Wilbon PA; Zhang W; Ren L; Zhou J; Nagarkatti M; Wang C; Chu F; He X; Decho AW; Tang C Chem. Commun 2012, 48, 916–918. [DOI] [PubMed] [Google Scholar]
- (40).Chen Y; Wilbon PA; Chen YP; Zhou J; Nagarkatti M; Wang C; Chu F; Decho AW; Tang C RSC Adv. 2012, 2, 10275–10282. [Google Scholar]
- (41).Ganewatta MS; Chen YP; Wang J; Zhou J; Ebalunode J; Nagarkatti M; Decho AW; Tang C Chem. Sci 2014, 5, 2011–2016. [Google Scholar]
- (42).Godula K; Rabuka D; Nam KT; Bertozzi CR Angew. Chem., Int. Ed 2009, 48, 4973–4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Beltrán-Osuna ÁA; Cao B; Cheng G; Jana SC; Espe MP; Lama B Langmuir 2012, 28, 9700–9706. [DOI] [PubMed] [Google Scholar]
- (44).Yan Y; Zhang J; Qiao Y; Tang C Macromol. Rapid Commun 2014, 35, 254–259. [DOI] [PubMed] [Google Scholar]
- (45).Huang J; Koepsel RR; Murata H; Wu W; Lee SB; Kowalewski T; Russell AJ; Matyjaszewski K Langmuir 2008, 24, 6785–6795. [DOI] [PubMed] [Google Scholar]
- (46).Sjöback R; Nygren J; Kubista M Spectrochim. Acta, Part A 1995, 51, L7–L21. [Google Scholar]
- (47).Murata H; Koepsel RR; Matyjaszewski K; Russell AJ Biomaterials 2007, 28, 4870–4879. [DOI] [PubMed] [Google Scholar]
- (48).Li Y; Benicewicz BC Macromolecules 2008, 41, 7986–7992. [Google Scholar]
- (49).Lee SB; Koepsel RR; Morley SW; Matyjaszewski K; Sun Y; Russell AJ Biomacromolecules 2004, 5, 877–82. [DOI] [PubMed] [Google Scholar]
- (50).Donlan RM; Piede JA; Heyes CD; Sanii L; Murga R; Edmonds P; El-Sayed I; El-Sayed MA Appl. Environ. Microbiol 2004, 70, 4980–4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Goeres DM; Loetterle LR; Hamilton MA; Murga R; Kirby DW; Donlan RM Microbiology 2005, 151, 757–762. [DOI] [PubMed] [Google Scholar]
- (52).Zhang YZ; Venugopal J; Huang ZM; Lim CT; Ramakrishna S Biomacromolecules 2005, 6, 2583–9. [DOI] [PubMed] [Google Scholar]
- (53).Yao K; Tang C Macromolecules 2013, 46, 1689–1712. [Google Scholar]
- (54).Chen Y; Wilbon PA; Zhou J; Nagarkatti M; Wang C; Chu F; Tang C Chem. Commun 2013, 49, 297–299. [DOI] [PubMed] [Google Scholar]
- (55).Ganewatta MS; Tang C Polymer 2015, 63, A1–A29. [Google Scholar]
- (56).Madkour AE; Dabkowski JM; Nüsslein K; Tew GN Langmuir 2009, 25, 1060–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Fournier D; Hoogenboom R; Schubert US Chem. Soc. Rev 2007, 36, 1369–80. [DOI] [PubMed] [Google Scholar]
- (58).Binder WH; Sachsenhofer R Macromol. Rapid Commun 2008, 29, 952–981. [Google Scholar]
- (59).Nandivada H; Jiang X; Lahann J Adv. Mater 2007, 19, 2197–2208. [Google Scholar]
- (60).Breinbauer R; Köhn M ChemBioChem 2003, 4, 1147–1149. [DOI] [PubMed] [Google Scholar]
- (61).Tron GC; Pirali T; Billington RA; Canonico PL; Sorba G; Genazzani AA Med. Res. Rev 2008, 28, 278–308. [DOI] [PubMed] [Google Scholar]
- (62).Kolb HC; Finn MG; Sharpless KB Angew. Chem., Int. Ed 2001, 40, 2004–2021. [DOI] [PubMed] [Google Scholar]
- (63).Matyjaszewski K; Tsarevsky NV J. Am. Chem. Soc 2014, 136, 6513–6533. [DOI] [PubMed] [Google Scholar]
- (64).Pyun J; Kowalewski T; Matyjaszewski K Macromol. Rapid Commun 2003, 24, 1043–1059. [Google Scholar]
- (65).Jennings MC; Ator LE; Paniak TJ; Minbiole KPC; Wuest WM ChemBioChem 2014, 15, 2211–2215. [DOI] [PubMed] [Google Scholar]
- (66).Kishida A; Iwata H; Tamada Y; Ikada Y Biomaterials 1991, 12, 786–92. [DOI] [PubMed] [Google Scholar]
- (67).De Rosa M; Carteni M; Petillo O; Calarco A; Margarucci S; Rosso F; De Rosa A; Farina E; Grippo P; Peluso GJ Cell. Physiol 2004, 198, 133–143. [DOI] [PubMed] [Google Scholar]