

Abstract
Single cell RNA-sequencing has accurately identified cell types within the human airway that express the Cystic Fibrosis Transmembrane Conductance regulator (CFTR) gene. Low abundance CFTR transcripts are seen in many secretory cells, while high levels are restricted to rare pulmonary ionocytes. Here we focus on the mechanisms coordinating basal CFTR expression in the secretory compartment. Cell-selective regulation of CFTR is achieved within its invariant topologically associating domain by the recruitment of cis-regulatory elements (CREs). CRE activity is coordinated by cell-type-selective transcription factors. One such factor, Krüppel-Like Factor 5 (KLF5), profoundly represses CFTR transcript and protein in primary human airway epithelial cells and airway cell lines. Here we reveal the mechanism of action of KLF5 upon the CFTR gene. We find that depletion or ablation of KLF5 from airway epithelial cells changes higher order chromatin structure at the CFTR locus. Critical looping interactions that are required for normal gene expression are altered, the H3K27ac active chromatin mark is redistributed, and CTCF occupancy is modified. However, mutation of a single KLF5 binding site within a pivotal airway cell CRE abolishes CFTR expression. Hence, KLF5 has both direct activating and indirect repressive effects, which together coordinate CFTR expression in the airway.
Graphical Abstract
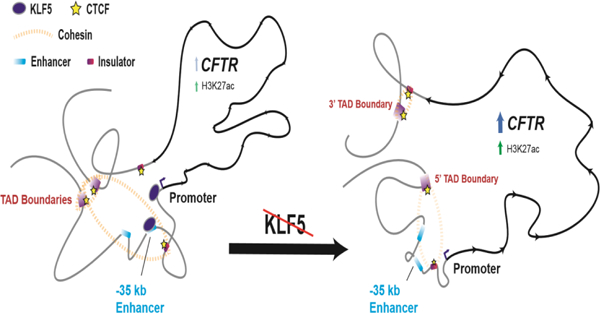
Introduction
Cystic fibrosis (CF) is an inherited, life-limiting disease caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Although lung pathology is the primary cause of mortality in CF patients [1], most lung epithelial cells express low levels of CFTR compared to other tissues affected by the disease [2–4]. Single-cell RNA-seq data identified a subset of secretory epithelial cells in the lung with detectable CFTR transcript in addition to a rare cell type (ionocytes), in which CFTR is abundant [5–7]. These divergent expression levels suggest distinct regulatory mechanisms between cell types. The role of cis-regulatory elements (CREs) in the cell type-selective regulation of CFTR has been a subject of extensive investigation [8]. Cooperation between intronic enhancers coordinates expression of the gene in intestinal epithelium [9]. CREs upstream of the promoter are essential for maintaining the higher order chromatin structure of the locus in airway epithelial cells [10], including sites at −44kb and −35kb [11, 12] that are required for gene expression [13]. Within the topologically associating domain (TAD) containing CFTR, between invariant boundaries at −80.1kb and +48.9kb [14], are enhancers and enhancer-blocking insulator elements that contribute differently to gene expression depending upon cell-type context [15]. CREs may contain binding sites for transcription factors (TFs) that control gene expression in a cell-specific context. Using a CRE-focused approach we identified multiple TFs with a pivotal role in regulating CFTR expression. Hepatocyte nuclear factor 1 (HNF1) and caudal-type homeobox 2 (CDX2) contribute to the function of the intestine-selective enhancers in intron 1 and 11 (legacy nomenclature) [9]. Furthermore, HNF1 interacts directly with several intronic CREs and the promoter [16, 17]. Forkhead box A1/2 are also enriched at regulatory sites within introns 10 and 11 [18] and are required for the looping and histone landscapes contributing to CFTR expression in the intestinal epithelium [19]. In airway epithelial cells, the −44kb CRE contains an antioxidant response element (ARE) involved in CFTR function. In normoxic conditions, the ARE at −44kb is occupied by the TF BTB and CNC homology 1 (BACH1) and v-Maf avian musculoaponeurotic fibrosarcoma oncogene homolog K (MAFK) heterodimers. Antioxidant treatment results in displacement of these repressive factors by Nrf2 and activation of CFTR expression [20]. BACH1, however, may play both an activating and repressive role on CFTR under different oxidative conditions [21]. The −35kb CRE was shown to be a potent enhancer of the CFTR promoter, both by enhancer assays in vitro [11] and by CRISPR/Cas9-mediated deletion [13]. This element may be regulated, in part, by the immune mediators interferon regulatory factors (IRF1/2) and nuclear factor Y (NF-Y). Targeting the complex network of TFs controlling CFTR in the airway epithelium may provide new therapeutic opportunities.
To identify TFs with either direct or indirect impacts on airway CFTR expression de novo, an siRNA depletion screen was performed in Calu-3 cells, targeting ~1500 human TFs and chromatin remodeling proteins. A subset of factors that activated or repressed CFTR in Calu-3 cells were also validated in primary human bronchial epithelial (HBE) cells [22]. Among the most potent repressors of CFTR was Krüppel-Like Factor 5 (KLF5). Depletion of KLF5 increased normalized CFTR mRNA levels more than 2.5-fold in Calu-3 cells and greater than 5-fold in HBE cells. Other members of the Krüppel-Like family have been implicated in CFTR regulation and CF pathogenesis. KLF4 was a key factor in the impaired Cx26-regulated wound repair process in CF airway basal cells [23]. It was also found to be upregulated in both CF lung and CF airway epithelial cell lines and to have a repressive effect on CFTR in wild-type (WT) cells [24]. KLF4 also has a key role in epithelial barrier function that is partially dependent on CFTR status [25]. Absence of KLF2 expression was observed in CF airways, correlating with a proinflammatory state [26]. KLF5 was previously found to be part of a transcriptional network in primary human tracheal epithelial cells and was predicted to occupy sites of open chromatin (CREs) at multiple CF-relevant loci [27]. Furthermore, KLF5 was a direct downstream target repressed by ETS Homologous Factor (EHF), a TF with a critical role in the human airway epithelium [28].
We recently showed that KLF5 is a potent regulator of genes with important biological functions in the human airway epithelium, including the proinflammatory response and wound repair processes [29]. In this context we generated cistrome data for KLF5 in the human airway epithelial cell lines Calu-3 and 16HBE14o- and found its occupancy at the promoter and the −35kb CRE of the CFTR gene. The Calu-3 and 16HBE14o- cell lines were chosen as they robustly express CFTR and are commonly used for CFTR functional studies.. Furthermore, they are effective models of the human airway epithelium with the latter able to be differentiated on air-liquid-interface (ALI). This observation of KLF5 occupancy suggested a direct role for the factor in regulating CFTR expression, which we investigate further here.
First, we use circular chromosome conformation capture with sequencing (4C-seq) to determine whether KLF5 is necessary for maintaining the higher order chromatin structure at the locus. We show that upon depletion or CRISPR/Cas9-mediated loss of detectable KLF5, substantial changes occur in the looping of CREs across the locus. To interrogate the mechanism, we assay the potential role of CCCTC binding factor (CTCF) and the cohesin complex in KLF5-mediated repression of CFTR. Next, we examine the active chromatin landscape across the CFTR locus in control cells and those deficient for KLF5, by assaying histone H3K27 acetylation, and show that loss of KLF5 alters H3K27ac enrichment particularly at CREs 5’ to the gene promoter. Lastly, we mutate the conserved KLF5 DNA-binding motif at the −35kb CRE and observe reduction of CFTR expression and alterations in locus architecture. These data suggest that KLF5 acts as a key regulator of CFTR expression with dual capabilities as a transcriptional activator and repressor, depending upon the context of its interactions with the gene.
Materials and Methods
Cell culture and siRNA transfection:
Calu-3 [30] and 16HBE14o- [31] cells were cultured in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum (FBS) using standard methods. TF depletion experiments were performed as previously described [29] for the negative control #2 (Dharmacon, D-001206–14-05), KLF5 (Dharmacon, M-013571–01-0005), CTCF (Ambion-s20968), and/or RAD21 (Ambion-s11725). 16HBE14o- cells were transfected using the same siRNA concentrations 24 h after seeding. All lysis was performed 72 h after transfection.
Chromatin immunoprecipitation and quantitative PCR (ChIP-qPCR):
ChIP was performed by standard protocols [9, 32]. Antibodies were specific for H3K27ac (Millipore 07–360), CTCF (07–729), or RNAPII (Cell Signaling Technology 14958S). ChIP-qPCR was performed using SYBR Green Master Mix (Thermo Fischer) on the QuantStudio 6 Flex System with results calculated using the percent input method with 2.5% starting chromatin input. Primer sequences are shown in Suppl.Table S1. All primers were previously published by the Harris group except for the mutagenesis primers and CRISPR-HDR primers/guides.
Reverse transcription and quantitative PCR (RT-qPCR):
Total RNA from confluent cultures was extracted with TRIzol (Invitrogen) and cDNA prepared with the TaqMan reverse transcription kit (Invitrogen). CFTR mRNA levels were assayed using Taqman fast advanced master mix for cDNA diluted 1:4 [16]. Results were normalized to the housekeeping control beta-2-microglobulin (β2M). Primer sequences are shown in Suppl.Table S1.
Western blot:
Cells were lysed in NET buffer (10 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 1X Sigma Protease Inhibitor), and protein concentration measured by Bradford protein assay (Bio-Rad). Proteins were separated by standard PAGE protocols, and western blots were probed with antibodies specific for CFTR (CFF-596) KLF5 (sc-398470), CTCF (Millipore-07–729), RAD21 (Millipore-05–908), and β-tubulin (T4026, Sigma-Aldrich).
Transient reporter gene (luciferase) assays
The pGL3B vector (Promega), pGL3B245 containing the CFTR minimal promoter, and the 350bp core sequence of the −35kb CFTR enhancer together with the promoter were described previously [11]. The Regulatory Sequence Analysis Tools (RSAT) matrix scan tool was used to predict KLF5 binding sites [33]. Site-directed mutagenesis was performed on both of the two most significant KLF5 motifs found in the −35kb 350bp core element using the Agilent QuikChange Lightning Site-Directed Mutagenesis Kit. 16HBE14o- cells were co-transfected with each luciferase vector and a modified pRL Renilla luciferase positive control vector at a 1:10 ratio using Lipofectamine 3000 (Thermo Fischer Scientific). Cells were lysed after 48 h and lysates assayed for firefly and Renilla luciferase activity using the Dual-Luciferase Reporter Assay Kit (Promega). Transfections (n=2) were performed using two replicate mutagenesis constructs in triplicate.
4C-seq
4C-seq libraries were generated from Calu-3 and 16HBE14o- as described previously [34]. Chromatin from ~ 1×107 cells was digested using NlaIII and DpnII or Csp6I as the primary or secondary restriction enzymes, respectively. All 4C experiments were done at least twice for every condition and clonal cell line. Enzyme pairs and primer sequences used for each viewpoint are shown in Supplementary Table S1. Two-nucleotide barcode sequences were added to the P5 linker to enable library multiplexing on the same sequencing flow cell. Domainogram visualizations were generated using the 4Cseqpipe pipeline [35] with default parameters on the hg19 genome. Quantifications of 4C-seq reads were generated using the pipe4C pipeline v1.1 [34] with default parameters. The deepTools bigwigCompare tool was used to subtract read density tracks [36].
CRISPR KLF5-null 16HBE14o- and homology directed repair (HDR)
Generation of the KLF5-null 16HBE14o- cells was described previously [29]. KLF5-motif 16HBE14o- mutants were generated using a single-guide RNA designed to target 24 bases directly downstream of the 3’ KLF5 motif in the −35kb CRE and cloned into pBlueScript (pBS). A single stranded DNA template was generated with 96 bp homology arms flanking the KLF5 motifs and a SacI restriction enzyme site to facilitate screening (Suppl. Fig. S5). 16HBE14o- cells were co-transfected with pMJ920 (wild-type Cas9 plasmid tagged with GFP) (Addgene, plasmid #42234), pBS containing the −35kb gRNA, and the HDR template at a 1:1:5 ratio using Lipofectamine 3000 (Life Technologies). GFP-positive cells were sorted by fluorescence-activated cell sorting and single cells were manually diluted to 96-well plates. Clones were expanded and screened for homozygous motif conversion by PCR amplification of the site followed by SacI digest and sequencing. Guide, template, and screening primers are shown in Supplementary Table S1.
Accession Numbers
Gene Expression Omnibus IDs: Calu-3 and 16HBE14o- H3K27ac ChIP-seq (GSE132808), Calu-3 and 16HBE14o- KLF5 ChIP-seq (GSE164853).
Results
Depletion of KLF5 alters the looping interactions between the CFTR TAD boundaries and upstream cis-regulatory elements
The CFTR locus is contained within a highly conserved TAD with boundaries predicted at sites of CTCF occupancy at −80.1kb 5’ and +48.9kb 3’ to the coding region [32]. We showed previously that the direct looping interactions of CREs encompassing enhancers at −44kb and −35kb, and a CTCF-binding insulator at −20.9kb, were critical for maintaining higher order chromatin structure and expression of CFTR in airway epithelial cells. Since transcription factors are known to be directly recruited to CREs and contribute to chromatin looping, we hypothesized that the mechanism of CFTR repression by KLF5 might similarly involve regulation of the 3D architecture of the CFTR locus. To test this hypothesis, we performed 4C-seq in Calu-3 cells treated with a pool of four negative control siRNAs or four siRNA targeting KLF5. By using viewpoints at the −20.9kb insulator element [37] (Fig 1A), +48.9kb 3’ TAD boundary (Fig 1B), CFTR promoter (Fig S1A), and −80.1kb 5’ TAD boundary (Fig S1B), we assayed the impact of KLF5 depletion on looping interactions across the locus. For each viewpoint, 4C-seq read quantification in the KLF5-depleted cells was subtracted from the NC-treated cells to identify regions of substantial change between them. We found that depletion of KLF5 markedly affects the looping between several intergenic CREs, the TAD boundaries, and intronic elements. Depletion of KLF5 resulted in a significant loss of interactions between a viewpoint at the −20.9kb site, the intron 10c element, sites close to the 3’ end of the gene including the CRE at +6.8kb, and the 3’ TAD boundary (Fig. 1A, red arrows and bars). The +6.8kb CRE is a DNase I hypersensitive site (DHS) and a CTCF binding insulator element [37]. We also observed a reduction in interactions over a region between the +15.6kb insulator (non-CTCF binding) [9, 10] and the 3’ TAD boundary that encompasses the open chromatin peak at 36.6 kb [9, 38]. Reorganization is also evident around the −80.1kb DHS within and beyond the 5’ TAD boundary. The diminished interaction between the 5’ and 3’ elements is also observed in the reciprocal comparison using the +48.9kb 3’ TAD boundary as a viewpoint (Fig. 1B, red bar and arrows). The −80.1kb 5’ TAD boundary viewpoint exhibits a marked gain in interactions across the whole region 5’ to the promoter (Fig. S1A, red bars). It also confirms observations with the −20.9kb viewpoint showing a rearrangement of the 3D architecture both within and outside the TAD, and a significant loss of interaction with the +48.9kb 3’ TAD boundary (Fig. 1A, red arrow). The promoter interactions are also consistent with data from the other viewpoints, exhibiting substantial gain in interaction with the ~40kb around the −80.1kb 5’ TAD boundary and diminished looping with the middle of the locus (intron 10c) and the 3’ end of the gene. Very few significant changes were observed between the viewpoints and other intronic elements. Changes detected within ~20kb of the viewpoints are not significant due to the disproportionate read density in the windows, which enhance relatively minor changes compared to regions beyond the viewpoint.
Figure 1. KLF5 depletion alters the 3D structure of the CFTR locus in Calu-3 cells.
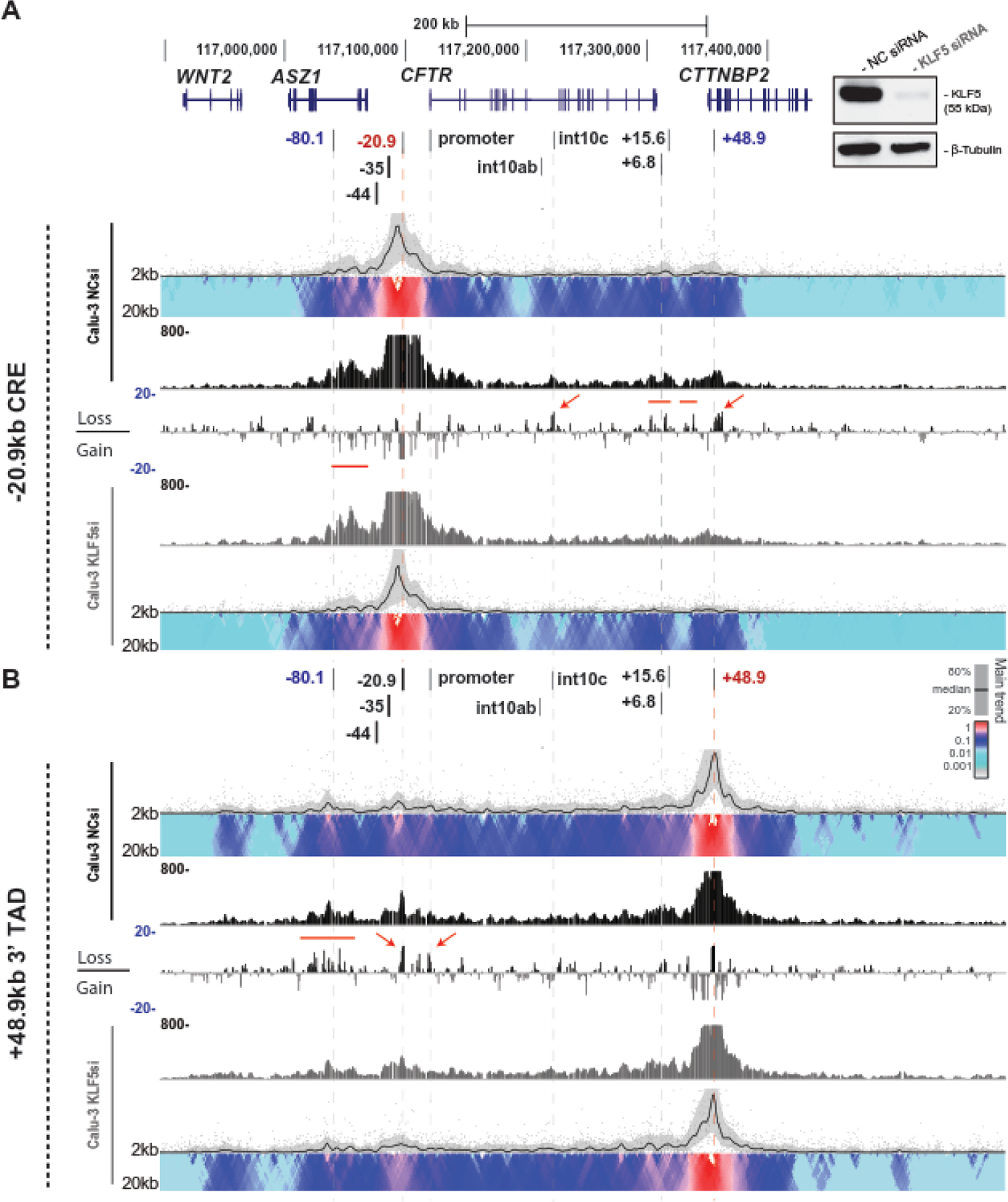
4C-seq data are shown for Calu-3 cells treated with negative control siRNA (NCsi) or siRNA targeting KLF5 (KLF5si). The genomic location of CFTR on chromosome 7 and its cis-regulatory elements (CREs) are shown at the top. 4C-seq data generated using the −20.9kb CRE (A) and +48.9kb 3’ TAD boundary (B) viewpoints are shown. Inset panel shows western blots probed with antibodies specific for KLF5 and β-Tubulin (normalizer) to illustrate the effective depletion of KLF5 in 4C experiments. For each viewpoint, domainograms at the top and bottom use an intensity gradient to show the relative interactions within a window size from 2 to 20kb. The corresponding trend in the contact profile using a 5kb window is shown above the domainograms. Relative interactions are normalized to read enrichment (set to 1) at the viewpoint, with a gradient of intensity from red (highest interaction) to blue to grey. Read quantification tracks in the IGV genome browser are shown below for NCsi or above for KLF5si. Between the conditions are read tracks of the subtraction of the KLF5-si 4C-seq from the NCsi cells in log2 scale.
KLF5-null cell lines show similarly altered looping interactions between the CFTR TAD boundaries and upstream cis-regulatory elements
We showed earlier that siRNA-mediated depletion of KLF5 in Calu-3 and primary HBE cells significantly upregulated the CFTR transcript [22]. To determine if the repressive role of KLF5 on CFTR expression was recapitulated in an orthogonal model, we used KLF5-null 16HBE14o- cell lines generated previously by CRISPR/Cas9 [29]. Consistent with the Calu-3 data, KLF5-null 16HBE14o- cells express ~2-fold more CFTR transcript than parental wildtype or nontargeted (in a CRISPR experiment) wildtype (Suppl. Fig S2A). The KLF5-null cells also showed substantially enhanced levels of CFTR protein compared to clonal WT (Suppl. Fig S2B). Next, we asked whether CFTR locus looping was also altered in the KLF5-null cells compared to non-targeted 16HBE14o- cells. Again, using 4C-seq we observed that, consistent with the results in Calu-3 cells, looping interactions between the CFTR promoter, the −80.1kb 5’ TAD boundary, and the upstream CREs such as −35kb and −20.9kb were significantly increased in all three homozygous KLF5-null clones (Fig 2, red arrows). A reduction in looping with the promoter viewpoint was also apparent around intron 10c, the 3’ end of the gene around the +6.8kb and +15.6kb CREs, and the intergenic region near the 3’ TAD boundary (Fig 2, red lines). Also as seen in Calu-3 cells, there is a marked reduction between the promoter and both the 48.9kb site and 3’ adjacent sequences. All three clones exhibited a significantly reduced interaction within a site in intron 16, which corresponds to a DHS in some airway and pancreatic cell lines [39, 40]. A gain of looping interactions between the −20.9kb CRE and the extended region within ~50kb of the 5’ TAD boundary confirmed findings with the promoter viewpoint (Fig S3, red line). Diminished −20.9kb interactions with 3’ end of the gene, proximal to the +6.8kb insulator, and both proximal and distal to the +48.9kb TAD boundary (Fig S3, red arrow and lines) are also consistent with the promoter viewpoint data. A viewpoint at the +48.9kb 3’ TAD boundary also shows diminished interactions with the −80.1kb TAD boundary and 5’ CREs including the −20.9kb site (Fig S4, red arrows) in the KLF5-null clones.
Figure 2. Loss of KLF5 causes substantial change in higher order chromatin structure at the CFTR locus in 16HBE14o- cells.
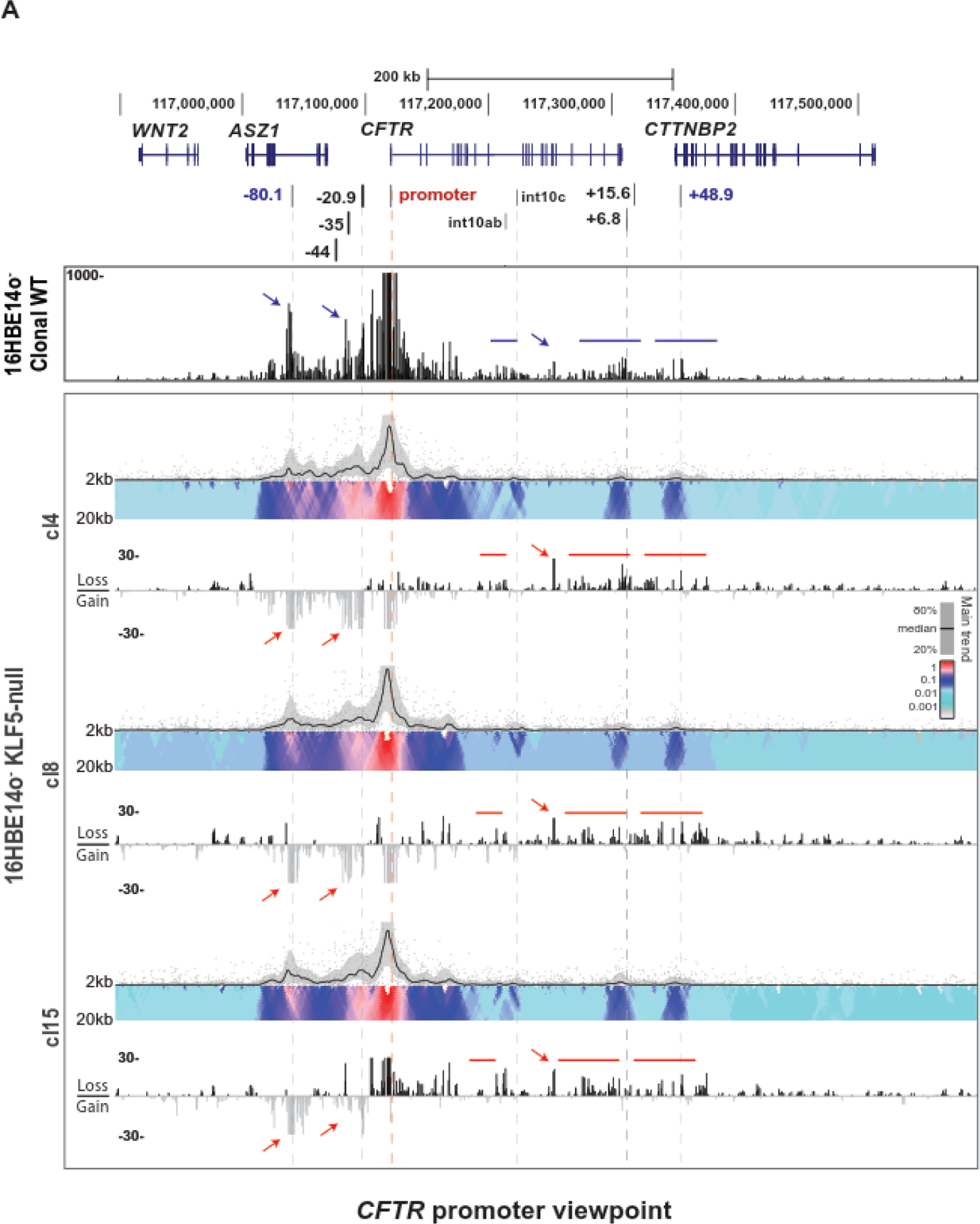
4C-seq data are shown for clonal WT (n=2) or KLF5-null (n=3) 16HBE14o- clones using a CFTR promoter viewpoint (for location see Table S1). The domainogram for each cell clone is shown above, and below is the subtraction of the read quantification tracks of each KLF5-null clone 4C-seq from the WT cells in log2 scale. Regions of specific interest are marked by bars and arrows in blue for the clonal WT track and red for the KLF5-null clones.
Diverse roles of CTCF in the KLF5-mediated regulation of CFTR
In earlier work, we showed that depletion of architectural proteins CTCF and RAD21, a component of the cohesin complex, disrupted the 3D structure, histone landscape, and TF occupancy across the CFTR locus in the colon carcinoma cell line Caco-2, and increased CFTR expression [32]. We also showed above that the CTCF-binding insulator elements at −20.9kb and +6.8kb exhibited noteworthy changes in looping interactions measured by 4C-seq, following loss of KLF5. To investigate whether the mechanism of KLF5-mediated regulation of CFTR involves these architectural proteins, we first separately depleted KLF5, CTCF, or RAD21 and then combinations of these factors in Calu-3, and assayed CFTR transcript (normalized to β2M) and protein (Fig 3A). As expected, loss KLF5 alone increased CFTR expression, whereas depletion of CTCF, RAD21, or both, significantly reduced expression, in contrast with the previous observations in Caco-2 cells, However, when both KLF5 and CTCF, or KLF5 and RAD21 were depleted simultaneously, the effect of KLF5 depletion was dominant and CFTR transcript and protein were significantly increased, albeit at a lower level than when the architectural proteins were not depleted. Next, to determine whether the localization of architectural proteins at the CFTR locus, rather than their abundance at the same sites, was critical in controlling gene expression we performed ChIP-qPCR for CTCF in the clonal WT and KLF5-null 16HBE14o- cells (Fig 3B). Consistent with the alterations in higher order chromatin organization, we observed a significant loss of CTCF enrichment at the +48.9kb 3’ TAD boundary and also at the −20.9kb insulator in the KLF5-null cells. We also observed a slight reduction in CTCF occupancy at the −80.1kb 5’ TAD boundary and the +6.8kb insulator. Together, these results suggest that loss of KLF5 impairs the normal recruitment of architectural proteins to critical structural elements at the locus.
Figure 3: The relationship of KLF5 and architectural proteins at the CFTR locus.
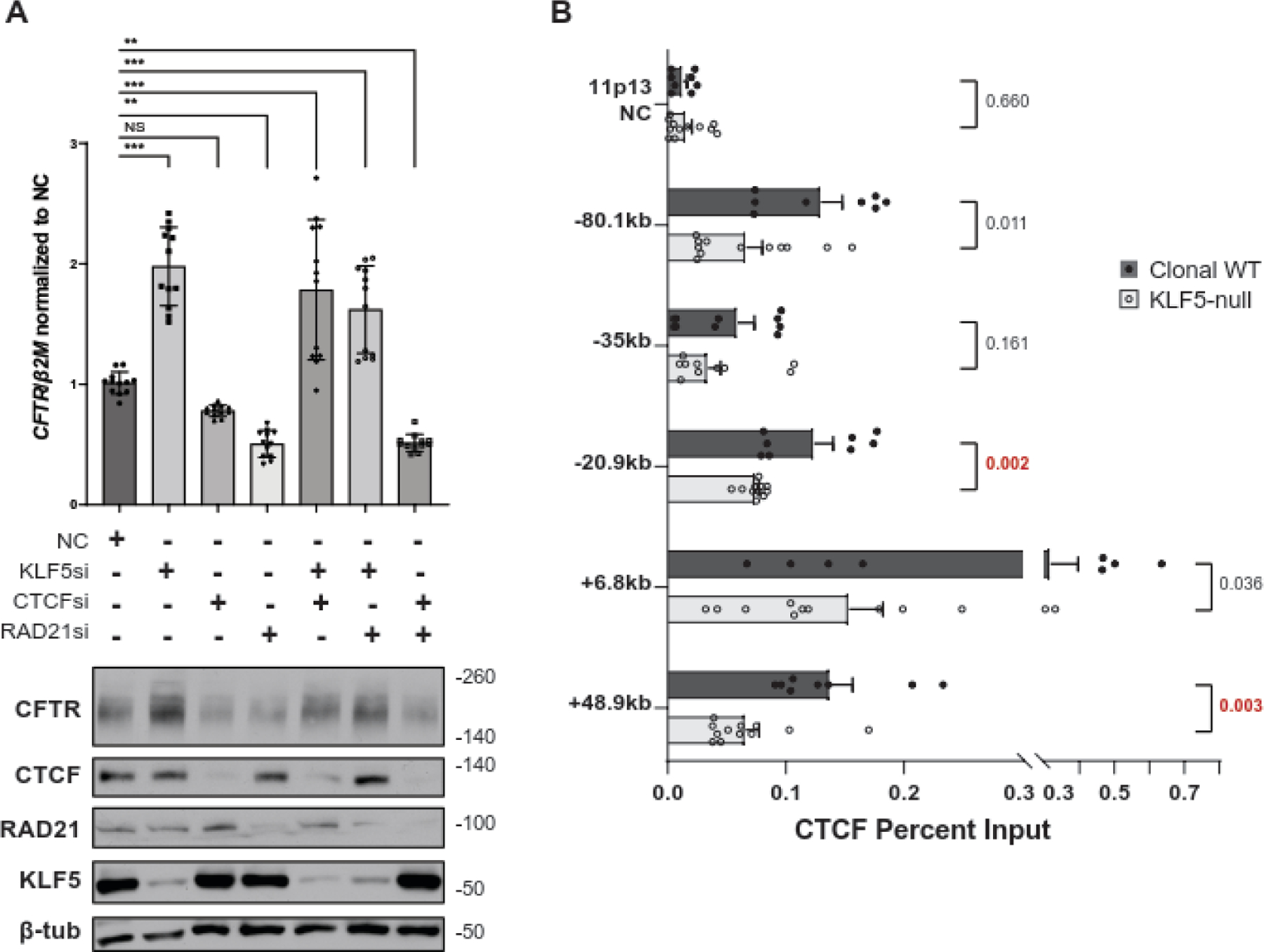
A. Loss of KLF5 overrides the impact of CTCF/Rad21 depletion on CFTR expression. Change in CFTR transcript in Calu-3 treated with NC siRNA or siRNA targeting KLF5, CTCF, and/or RAD21 using RT-qPCR (above) and corresponding change in CFTR protein by western blot (below). CFTR transcript abundance is normalized to beta-2-microglobulin (β2M). *p < 0.01, **p < 0.001, ***p < 0.0001 using a two-way analysis of variance plus multiple comparisons test. B. Absence of KLF5 decreases CTCF occupancy at key sites in the CFTR locus. ChIP-qPCR for CTCF in clonal WT (n=12) or KLF5-null 16HBE14o- cells (n=12) at CFTR locus CREs or a negative control site at chromosome 11p13. The p-value from unpaired T-tests are shown next to each comparison.
Depletion or loss of KLF5 alters H3K27 acetylation across the CFTR locus
Krüppel-Like factors are known to interact with histone deacetylases in diverse cell types [41–43]. We documented the histone modification landscape across CFTR in airway epithelial cells previously [13], and aligned histone marks with regions of open chromatin at CREs (Fig. 4A). In both Calu-3 and 16HBE14o- cells, the −44kb and −35kb enhancers, and CFTR promoter, exhibit strong peaks of H3K27ac enrichment (GEO: GSE132808). The −35 kb site and the promoter also have peaks of KLF5 occupancy (GEO: GSE164853) (Fig 4A). Hence, we next asked whether loss of KLF5 altered the distribution of H3K27ac across the locus and particularly at specific CREs, which might be pivotal in the observed changes in CFTR expression. We performed ChIP-qPCR for H3K27ac in Calu-3 cells depleted for KLF5 (Fig 4B), and in the KLF5-null 16HBE14o- cells (Fig. 4C), using primers at these critical CREs. No significant changes in H3K27ac occupancy were seen in either cell type at the −44kb CRE, which has a very high basal signal, nor at a negative control site (DHS intron 10ab). Similarly, there was no significant change in abundance of this histone mark at the +48.9 kb TAD boundary, a site that is enriched for H3K27ac in Calu-3 but not 16HBE14o-. However, at the −35kb DHS, where KLF5 binds directly, H3K27ac was significantly increased following depletion (Fig. 4B) or loss (Fig. 4C) of KLF5. Depletion of KLF5 was also associated with significant de novo H3K27ac deposition at both the −20.9kb and +6.8 kb enhancer blocking insulator elements in Calu-3 cells (Fig. 4B), while a similar change upon loss of KLF5 in 16HBE14o- cells was only seen at the −20.9 kb site (Fig. 4C).
Figure 4. Loss of KLF5 enhances H3K27ac occupancy at CFTR CREs.
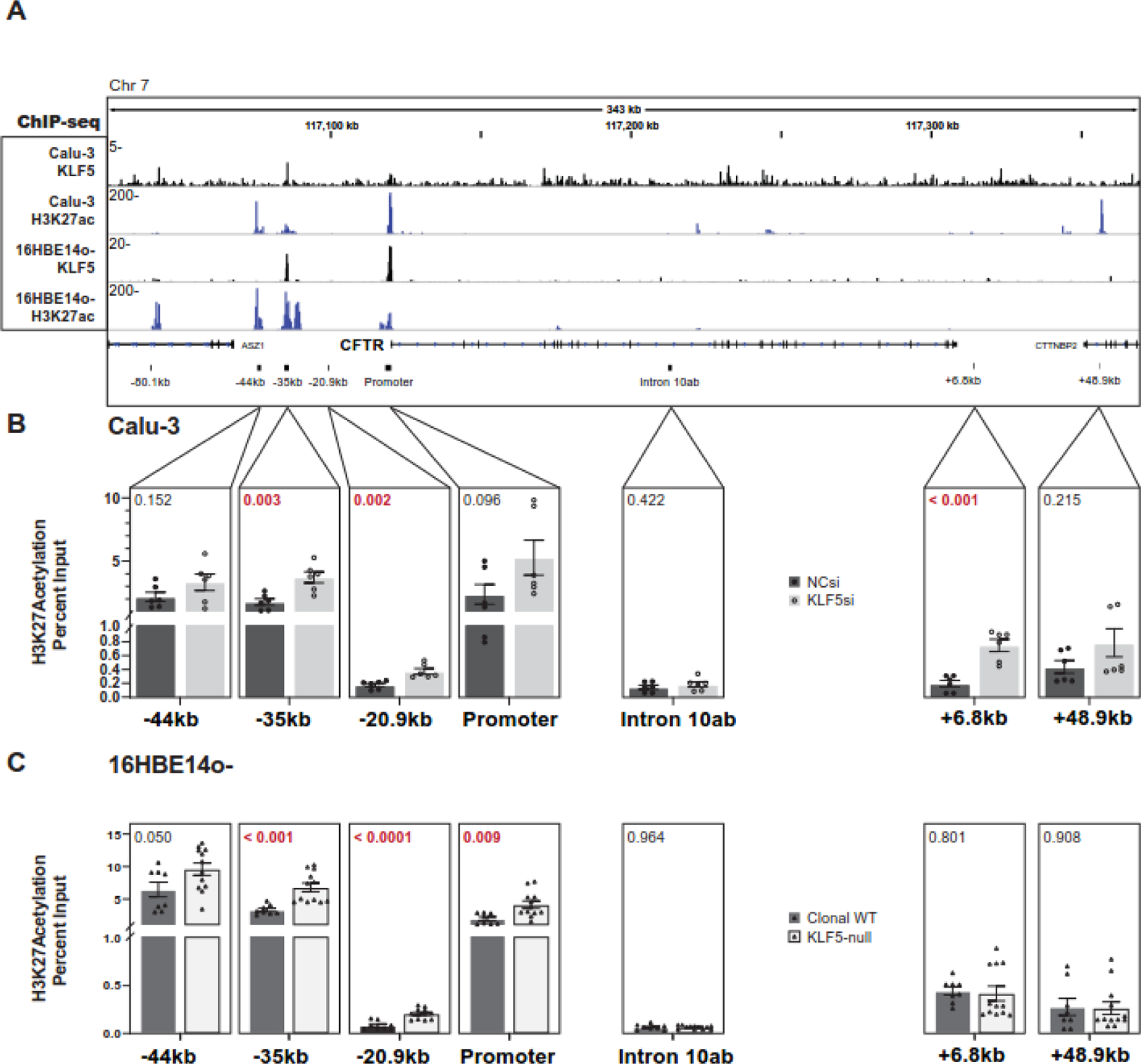
A. KLF5 and H3K27ac ChIP-seq IDR signal across the CFTR locus in Calu-3 and 16HBE14o- cells. CFTR CRE sites are shown below. B. ChIP-qPCR for H3K27ac in Calu-3 cells treated with NC siRNA or siRNA targeting KLF5 at key CREs (n= 3 experiments; n=2 technical replicates). C. ChIP-qPCR for H3K27ac in clonal WT (n=2) or KLF5-null 16HBE14o- (n=3) cells, n=2 experiments). The p-values from unpaired T-tests are shown above each comparison.
Disruption of the KLF5-binding motif at the −35kb 5’ enhancer element perturbs CFTR expression
KLF5 occupies the −35kb 5’ enhancer in both Calu-3 and 16HBE14o- cells as shown by the ChIP-seq peak in Fig. 4A and data in GEO:GSE164853. To ascertain the importance of this direct binding of KLF5, we used in silico tools to predict its binding motif(s) in the 350 bp core enhancer of the −35 kb element [11]. Regulatory Sequence Analysis Tools (RSAT) matrix scan identified two probable KLF5 binding sites. Each of these sites was mutated in constructs containing the 350bp enhancer sequence and a minimal CFTR promoter driving luciferase expression (Fig 5A). (The CRISPR-HDR template containing the homology arms and the mutated KLF5 sequence are shown in Suppl. Table S1). Mutant and control plasmids were co-transfected into 16HBE14o- cells with a renilla normalization control vector (Fig 5B). The relative luciferase expression was significantly lower from both mutant constructs compared to the parental core enhancer plasmid, though neither reduced expression to the promoter-only construct levels. The 3’ mutant KLF5 site which caused a greater reduction in luciferase activity was chosen as the target for CRISPR homology-directed repair (HDR) in the same cell line (Fig. S5). 16HBE14o- cells were transfected with a plasmid expressing Cas9, a gRNA targeting the region immediately downstream of the HDR site, and a 200bp HDR template encompassing the mutation. Transfected cells were manually diluted to single-cell density to ensure the isolation of clones that were homozygous for the conversion. Three homozygous −35kb KLF5-site mutant HDR clones (cl4, cl33 and cl57) were generated, which expressed significantly lower amounts of CFTR transcript (Fig. 5C) and protein (Fig. 5D) than clonal WT 16HBE14o-cells. Since we observed a consistent reduction in transcription in the three mutant clones, we next assayed the occupancy of RNA polymerase II (RNAPII) at the CFTR promoter and key CREs in mutant and clonal WT cells (Fig. 5E). No change in RNAPII binding was observed in mutant cells at the negative control DHS 10ab site, nor at the −20.9kb insulator. However, RNAPII enrichment was significantly reduced in cl4 and cl33 at the promoter and both upstream enhancers at −44kb and −35kb. 4C-seq was then performed using the same viewpoints as above to determine whether locus architecture was changed upon loss of this critical KLF5 binding site (Fig. 5E). A viewpoint at the −80.1kb 5’ TAD boundary gained 5’ looping interactions outside the TAD in HDR cells, and lost interactions both within the gene and at its 3’ end when compared to parental WT cells. This is consistent with the observations in the KLF5-depleted Calu-3 and KLF5-null 16HBE14o- cells. However, in contrast to those cell lines, the −35kb KLF5 site mutation is associated with specific structural changes, including both a gain in −80.1 kb looping adjacent (5’) to the −35kb CRE and with the promoter (Fig 5F, dashed orange arrows), and a loss with other elements (red line). Looping between the −80.1kb and +48.9kb TAD boundaries was also significantly increased (red arrows).
Figure 5: Mutation of the KLF5 binding site in the −35kb CFTR CRE impairs gene expression and alters chromatin architecture.
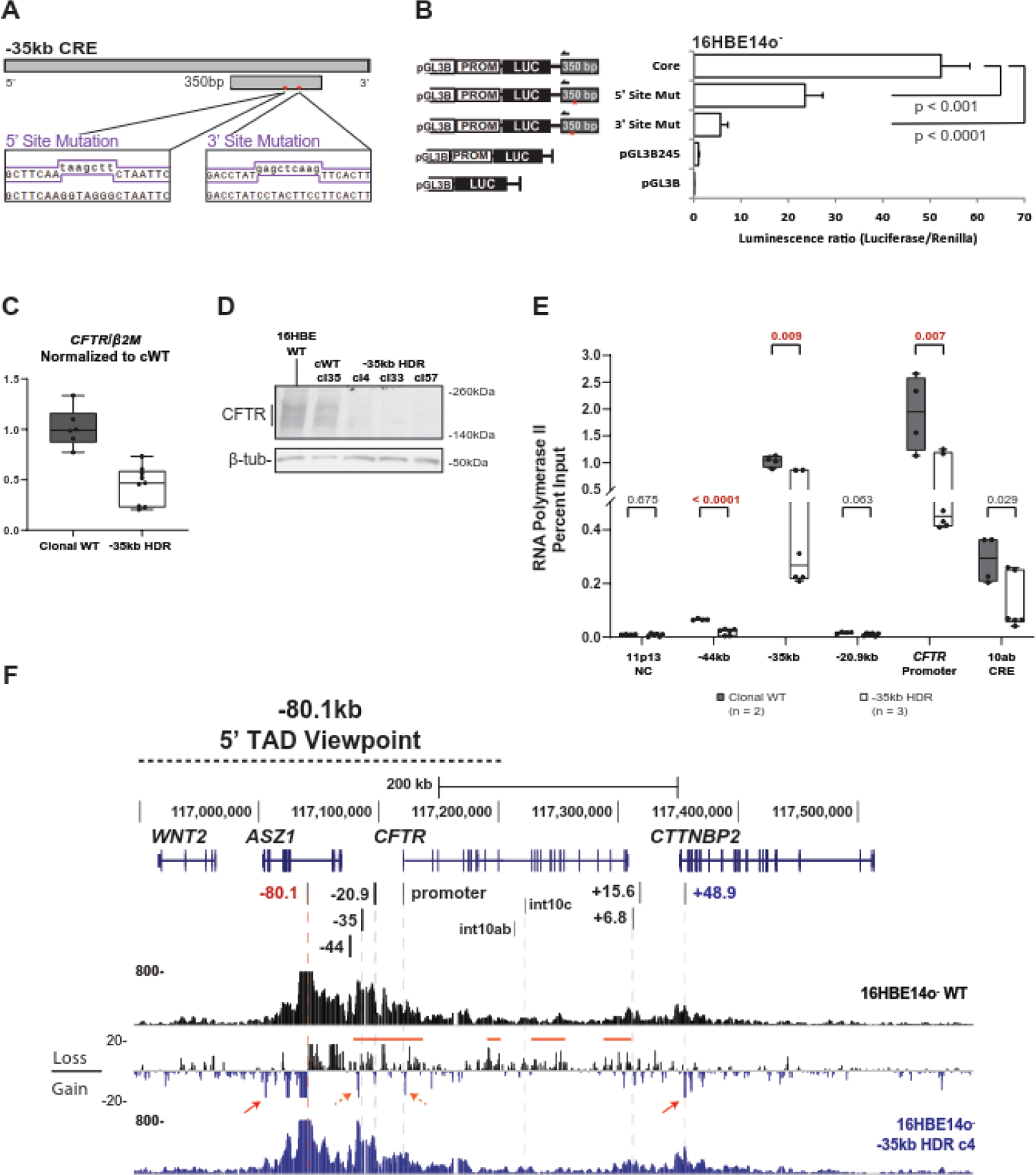
A. Schematic of the 5’ and 3’ KLF5-motif mutations generated in the 350bp core element of the −35kb CRE. Mutagenesis primer sequences are shown in the purple lines below. The 5’ and 3’ mutations contain a HindIII and SacI restriction enzyme site, respectively. B. Transient reporter gene assays in 16HBE14o- cells using luciferase vector pGL3B, pGL3B with the CFTR minimal promoter (pGL3B-245) [58], pGL3B-245 with the 350bp core of the −35kb enhancer [11], or the latter vector with mutation of one 6bp KLF5 motif. Luciferase activity was measured relative to Renilla 48h post-transfection. C. RT-qPCR analysis of CFTR expression in clonal WT 16HBE14o- cells (n=2) compared to −35kb KLF5-site mutant HDR clones (n=3), (n=3 experiments) D. Western blot showing CFTR protein levels in parental WT, clonal WT, or −35kb HDR 16HBE14o- cells. E. ChIP-qPCR with antibody specific for RNApolII (RPB1) comparing clonal WT and −35kb HDR clones. The p-value from unpaired T-tests of the RT-qPCR are shown above each comparison. F. 4C-seq data for WT 16HBE14o- or −35kb HDR clone 4 using the −80.1kb 5’ TAD boundary as the viewpoint. Read quantification for each cell line is shown, and between them the subtraction of HDR clone 4 from WT. Regions of interest are noted with the red bars and arrows.
Discussion
Higher order chromatin structure at the CFTR locus provide more global insights into the contributions of CREs and specific TFs to the cell type-selective regulation of large, complex genes. Here, by examining the balance of direct and indirect control of CFTR locus architecture, the histone landscape, and recruitment of RNAPll by a single key TF (KLF5), we enhance understanding of the mechanisms whereby perturbations of normal gene regulation may cause disease. Moreover, these mechanisms may contribute to cellular heterogeneity within tissues, such as the diversity of airway secretory epithelial cells, corresponding to variable levels of CFTR expression [44, 45]. First, considering indirect control, we investigate features of the CFTR locus that are altered upon depletion of the potent repressive factor KLF5. Next, by mutation of a critical KLF5 binding site within a strong airway cell-selective 5’ enhancer that is recruited to the gene promoter by chromatin looping, we address direct regulatory mechanisms. Of note, KLF5 occupancy at the gene promoter in airway cells may be predominantly the result of enhancer looping, since direct binding motifs in the promoter are relatively low affinity. Our data support a pivotal role for KLF5 in establishing the epigenetic landscape of CFTR in secretory epithelial cells of the human airway, document the importance of direct KLF5 recruitment to critical CREs within these cells, and illustrate the relationship between higher order chromatin structure and gene expression at this large and complex locus.
In these studies, KLF5 was found to be a potent repressor of CFTR expression in two human airway epithelial cell lines with diverse phenotypes, consistent with our earlier observations on primary HBE cells [22]. The 3D structure of the CFTR locus has features that are ubiquitous (the TAD boundaries), in addition to others that are cell type selective (the looping of CREs to the gene promoter). Since we showed earlier that CRISPR/Cas9-mediated deletion of specific CREs significantly altered locus architecture and/or CFTR expression [13, 15], we assayed changes in the 3D landscape in airway epithelial cells with reduced or absent KLF5 using 4C-seq. A substantial loss of association was observed between 5’ elements and the 3’ TAD-boundary, while few significant changes in long-range interactions were observed within the gene body. It may be relevant that intronic enhancers drive CFTR expression in the Caco2 cells, but 5’ enhancers outside the gene are critical in airway cells. The most consistent change in locus architecture upon loss of KLF5 involved the CTCF-binding insulator element at −20.9kb. In both cell lines, this site simultaneously showed stronger interactions with the promoter and the −80.1kb 5’ TAD boundary, while showing diminished interactions with the +48.9kb 3’ TAD boundary. These changes in interaction profiles suggest a profound change in locus structure and physical distance between CREs when KLF5 is lost. The close association of enhancers, and gene promoters optimizes the environment for recruitment of RNAPII [46]. CRISPR deletions of the −44kb or −35kb CREs, which resulted in loss of CFTR expression, reduced the interaction frequency of the −20.9kb site with the −80.1kb 5’ TAD boundary but increased downstream interactions [13]. This contrasts with the changes observed in cells deficient for KLF5. It is possible that the locus architecture may be held by specific anchor points at the TAD boundaries and CREs, mediated by KLF5, which enables low but detectable CFTR expression. Upon loss of KLF5 the anchor constraints are relieved between the upstream sites and the 3’ end of the locus, enhancing access to the promoter.
In order to show that the impact of KLF5 on locus architecture was not unique to CFTR, we performed 4C-seq experiments after KLF5 depletion at another genomic region. We chose the well-studied chromosome 11p13 region [47, 48] encompassing the E74 Like ETS Transcription Factor 5 (ELF5), EHF and APAF1 Interacting Protein (APIP) locus. Single-nucleotide polymorphisms (SNPs) in the intergenic region between EHF and APIP are associated with CF lung disease severity [49, 50] and CRISPR/Cas9-mediated removal of CREs altered 3D chromatin structure and the coordinate regulation of EHF and ELF5 [48]. KLF5 binds to several sites in this genomic interval including a region of open chromatin in EHF intron 6. We used 4C-seq to determine the impact of siRNA-mediated depletion of KLF5 on this region in Calu-3 cells (Fig S6). Loss of KLF5 significantly reduced the looping interactions of viewpoints on the 5’ side at DHS11.2516 and the 3’ TAD boundary in APIP intron 4, with this highly active site in EHF intron 6. ELF5 is not expressed in Calu-3 cells, while EHF and APIP are slightly upregulated after KLF5 depletion (GEO:GSE164853), with fold changes of 1.48 (p-adj = 0.007) and 1.35 (p-adj = 0.002), respectively. These data support a broader role for KLF5 in establishing and maintaining long-range chromatin 3D structures, particularly between CREs, at multiple loci.
Since we observed KLF5 depletion-associated changes in looping interactions between the CTCF-binding elements at −20.9kb, +6.8kb and +48.9kb, we asked whether the mechanism of KLF5 repression of CFTR involved the architectural proteins CTCF and cohesin. In Calu-3 cells, depletion of RAD21 alone or both CTCF and RAD21 downregulated CFTR expression in contrast, when depletion of these factors was coupled with that of KLF5, CFTR was upregulated to levels similar to those seen with loss of KLF5 alone, indicating that the KLF5-mediated repression of the gene is somewhat independent of the architectural protein complex. Loss of KLF5 was not only associated with a reduction in looping interactions, but also with a significant loss of CTCF binding specifically at the −20.9kb and +48kb sites (while overall abundance of the CTCF and RAD21 did not change (Fig. 5B). This redistribution may cause relaxation of the 3D structure mediated by the CTCF-cohesin complex between these sites and thus increased accessibility to the CFTR promoter.
The well-established H3K27ac histone landscape of CFTR marks the airway-selective enhancer elements and sites of transcription factor binding. We observed greater enrichment of the active histone mark at both of the sites of KLF5 occupancy, at −35kb and the promoter, which may correlate with increased enhancer activity. Alternatively, following loss of KLF5, this CRE gains a minor enhancer signature which has been found at other CTCF-binding sites in a TF-dependent manner [51]. As H3K27ac enrichment does not significantly change at the other enhancer at −44kb nor at +48.9kb, upon KLF5 loss, this TF may maintain levels of the active mark at the CFTR locus in concert with the looping structure in the upstream region containing its binding sites.
Krüppel-like factors have triplicate zinc-finger DNA binding domains and well-characterized binding motifs [52]. Direct binding of KLF5 to the −35kb, as shown in both Calu-3 and 16HBE14o- by ChIP-seq, likely activates CFTR expression. Mutation of the conserved binding motifs for KLF5, either in a luciferase reporter gene construct or in the endogenous CFTR locus, significantly reduced gene expression. This may result from impaired recruitment of the transcriptional complex, as suggested by the reduction in RNA polymerase II occupancy. Mutation of the KLF5 motif also disrupts the looping structure of the locus between the −80.1kb 5’ TAD boundary, upstream CREs, and across the gene body. In combination these data suggest KLF5 has dual and opposite functions in CFTR regulation. In isolation, it is an activating TF at the −35kb enhancer. Disruption of binding at the upstream enhancer may impact KLF5-mediated mechanisms at the promoter where we observe lower KLF5 affinity, restricting access to RNAPII and other activating transcription factors. However, in the context of the larger CFTR locus it has an additional repressive function in controlling accessibility of the gene, specifically between CTCF-binding CREs. Members of the Krüppel-like family of TFs are extensively studied for both activating and repressive functions in different cellular contexts (reviewed in [53]). Consistent with our model for CFTR regulation by KLF5, this factor may directly promote recruitment of the transcriptional complex and establish the 3D conformation of gene loci, such as at enhancers in cancer driver events [54]. Our results on the CFTR locus may enable novel therapeutic approaches to modulate CFTR expression, using chemical biology approaches drawn from other diseases. The synthetic retinoic acid receptor agonist Am80 was found to inhibit expression and function of KLF5 [55–57]. Targeted, drug-mediated depletion of KLF5 in tissues with low levels of CFTR expression could enhance efforts to restore normal CFTR protein function.
Supplementary Material
KLF5 is a key transcription factor (TF) in coordinating airway CFTR gene expression
KLF5 represses CFTR by modulating chromatin looping and active histone mark occupancy
KLF5 also augments CFTR expression through direct binding at a potent enhancer
We reveal a mechanism for coordinate activation and repression of a gene by one TF
Acknowledgements
We thank the Case Western Reserve University genomics core for deep sequencing.
Funding
This work was supported by the National Institutes of Health (R01 HL094585; R01 HL117843; R01 HD068901 [A. H.]; T32 GM008056 [A. P.]) and the Cystic Fibrosis Foundation (Harris 16G0, 15/17XX0 and 18P0 [A. H.]). The funders had no role in the design of the study, in the collection, analyses, or interpretation of the data, in the writing of the manuscript, or in the decision to publish the results.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declare no conflict of interest.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data Availability
To review GEO accession GSE194288:
Go to https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE194288
Enter token ufcfwwkorhyzdsf into the box
References.
- [1].Elborn JS. Cystic fibrosis. Lancet 2016;388:2519–31. [DOI] [PubMed] [Google Scholar]
- [2].Harris A, Chalkley G, Goodman S, Coleman L. Expression of the cystic fibrosis gene in human development. Development 1991;113:305–10. [DOI] [PubMed] [Google Scholar]
- [3].Crawford I, Maloney PC, Zeitlin PL, Guggino WB, Hyde SC, Turley H, et al. Immunocytochemical localization of the cystic fibrosis gene product CFTR. Proc Natl Acad Sci U S A 1991;88:9262–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hyde K, Reid CJ, Tebbutt SJ, Weide L, Hollingsworth MA, Harris A. The cystic fibrosis transmembrane conductance regulator as a marker of human pancreatic duct development. Gastroenterology 1997;113:914–9. [DOI] [PubMed] [Google Scholar]
- [5].Plasschaert LW, Zilionis R, Choo-Wing R, Savova V, Knehr J, Roma G, et al. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 2018;560:377–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Montoro DT, Haber AL, Biton M, Vinarsky V, Lin B, Birket SE, et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 2018;560:319–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Okuda K, Dang H, Kobayashi Y, Carraro G, Nakano S, Chen G, et al. Secretory Cells Dominate Airway CFTR Expression and Function in Human Airway Superficial Epithelia. Am J Respir Crit Care Med 2021;203:1275–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Harris A Human Molecular Genetics and the long road to treating cystic fibrosis. Hum Mol Genet 2021;30:R264–R73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ott CJ, Blackledge NP, Kerschner JL, Leir SH, Crawford GE, Cotton CU, et al. Intronic enhancers coordinate epithelial-specific looping of the active CFTR locus. Proc Natl Acad Sci U S A 2009;106:19934–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gheldof N, Smith EM, Tabuchi TM, Koch CM, Dunham I, Stamatoyannopoulos JA, et al. Cell-type-specific long-range looping interactions identify distant regulatory elements of the CFTR gene. Nucleic Acids Res 2010;38:4325–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhang Z, Leir SH, Harris A. Immune mediators regulate CFTR expression through a bifunctional airway-selective enhancer. Mol Cell Biol 2013;33:2843–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gosalia N, Harris A. Chromatin Dynamics in the Regulation of CFTR Expression. Genes (Basel) 2015;6:543–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].NandyMazumdar M, Yin S, Paranjapye A, Kerschner JL, Swahn H, Ge A, et al. Looping of upstream cis-regulatory elements is required for CFTR expression in human airway epithelial cells. Nucleic Acids Res 2020;48:3513–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Smith EM, Lajoie BR, Jain G, Dekker J. Invariant TAD Boundaries Constrain Cell-Type-Specific Looping Interactions between Promoters and Distal Elements around the CFTR Locus. Am J Hum Genet 2016;98:185–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yang R, Kerschner JL, Gosalia N, Neems D, Gorsic LK, Safi A, et al. Differential contribution of cis-regulatory elements to higher order chromatin structure and expression of the CFTR locus. Nucleic Acids Res 2016;44:3082–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mouchel N, Henstra SA, McCarthy VA, Williams SH, Phylactides M, Harris A. HNF1alpha is involved in tissue-specific regulation of CFTR gene expression. Biochem J 2004;378:909–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ott CJ, Suszko M, Blackledge NP, Wright JE, Crawford GE, Harris A. A complex intronic enhancer regulates expression of the CFTR gene by direct interaction with the promoter. J Cell Mol Med 2009;13:680–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kerschner JL, Harris A. Transcriptional networks driving enhancer function in the CFTR gene. Biochem J 2012;446:203–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kerschner JL, Gosalia N, Leir SH, Harris A. Chromatin remodeling mediated by the FOXA1/A2 transcription factors activates CFTR expression in intestinal epithelial cells. Epigenetics 2014;9:557–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zhang Z, Leir SH, Harris A. Oxidative stress regulates CFTR gene expression in human airway epithelial cells through a distal antioxidant response element. Am J Respir Cell Mol Biol 2015;52:387–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].NandyMazumdar M, Paranjapye A, Browne J, Yin S, Leir SH, Harris A. BACH1, the master regulator of oxidative stress, has a dual effect on CFTR expression. Biochem J 2021;478:3741–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Mutolo MJ, Leir SH, Fossum SL, Browne JA, Harris A. A transcription factor network represses CFTR gene expression in airway epithelial cells. Biochem J 2018;475:1323–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Crespin S, Bacchetta M, Bou Saab J, Tantilipikorn P, Bellec J, Dudez T, et al. Cx26 regulates proliferation of repairing basal airway epithelial cells. Int J Biochem Cell Biol 2014;52:152–60. [DOI] [PubMed] [Google Scholar]
- [24].Sousa L, Pankonien I, Clarke LA, Silva I, Kunzelmann K, Amaral MD. KLF4 Acts as a wt-CFTR Suppressor through an AKT-Mediated Pathway. Cells 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sousa L, Pankonien I, Simoes FB, Chanson M, Amaral MD. Impact of KLF4 on Cell Proliferation and Epithelial Differentiation in the Context of Cystic Fibrosis. Int J Mol Sci 2020;21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Saavedra MT, Patterson AD, West J, Randell SH, Riches DW, Malcolm KC, et al. Abrogation of anti-inflammatory transcription factor LKLF in neutrophil-dominated airways. Am J Respir Cell Mol Biol 2008;38:679–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bischof JM, Ott CJ, Leir SH, Gosalia N, Song L, London D, et al. A genome-wide analysis of open chromatin in human tracheal epithelial cells reveals novel candidate regulatory elements for lung function. Thorax 2012;67:385–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Fossum SL, Mutolo MJ, Tugores A, Ghosh S, Randell SH, Jones LC, et al. Ets homologous factor (EHF) has critical roles in epithelial dysfunction in airway disease. J Biol Chem 2017;292:10938–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Paranjapye A, NandyMazumdar M, Browne JA, Leir SH, Harris A. Kruppel-like factor 5 regulates wound repair and the innate immune response in human airway epithelial cells. J Biol Chem 2021;297:100932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Shen BQ, Finkbeiner WE, Wine JJ, Mrsny RJ, Widdicombe JH. Calu-3: a human airway epithelial cell line that shows cAMP-dependent Cl- secretion. Am J Physiol 1994;266:L493–501. [DOI] [PubMed] [Google Scholar]
- [31].Cozens AL, Yezzi MJ, Kunzelmann K, Ohrui T, Chin L, Eng K, et al. CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am J Respir Cell Mol Biol 1994;10:38–47. [DOI] [PubMed] [Google Scholar]
- [32].Gosalia N, Neems D, Kerschner JL, Kosak ST, Harris A. Architectural proteins CTCF and cohesin have distinct roles in modulating the higher order structure and expression of the CFTR locus. Nucleic Acids Res 2014;42:9612–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Nguyen NTT, Contreras-Moreira B, Castro-Mondragon JA, Santana-Garcia W, Ossio R, Robles-Espinoza CD, et al. RSAT 2018: regulatory sequence analysis tools 20th anniversary. Nucleic Acids Res 2018;46:W209–W14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Krijger PHL, Geeven G, Bianchi V, Hilvering CRE, de Laat W. 4C-seq from beginning to end: A detailed protocol for sample preparation and data analysis. Methods 2020;170:17–32. [DOI] [PubMed] [Google Scholar]
- [35].van de Werken HJ, Landan G, Holwerda SJ, Hoichman M, Klous P, Chachik R, et al. Robust 4C-seq data analysis to screen for regulatory DNA interactions. Nat Methods 2012;9:969–72. [DOI] [PubMed] [Google Scholar]
- [36].Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 2016;44:W160–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Blackledge NP, Ott CJ, Gillen AE, Harris A. An insulator element 3’ to the CFTR gene binds CTCF and reveals an active chromatin hub in primary cells. Nucleic Acids Res 2009;37:1086–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zhang Z, Ott CJ, Lewandowska MA, Leir SH, Harris A. Molecular mechanisms controlling CFTR gene expression in the airway. J Cell Mol Med 2012;16:1321–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Smith DJ, Nuthall HN, Majetti ME, Harris A. Multiple potential intragenic regulatory elements in the CFTR gene. Genomics 2000;64:90–6. [DOI] [PubMed] [Google Scholar]
- [40].McCarthy VA, Ott CJ, Phylactides M, Harris A. Interaction of intestinal and pancreatic transcription factors in the regulation of CFTR gene expression. Biochim Biophys Acta 2009;1789:709–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Munemasa Y, Suzuki T, Aizawa K, Miyamoto S, Imai Y, Matsumura T, et al. Promoter region-specific histone incorporation by the novel histone chaperone ANP32B and DNA-binding factor KLF5. Mol Cell Biol 2008;28:1171–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Seo S, Lomberk G, Mathison A, Buttar N, Podratz J, Calvo E, et al. Kruppel-like factor 11 differentially couples to histone acetyltransferase and histone methyltransferase chromatin remodeling pathways to transcriptionally regulate dopamine D2 receptor in neuronal cells. J Biol Chem 2012;287:12723–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].He M, Zheng B, Zhang Y, Zhang XH, Wang C, Yang Z, et al. KLF4 mediates the link between TGF-beta1-induced gene transcription and H3 acetylation in vascular smooth muscle cells. FASEB J 2015;29:4059–70. [DOI] [PubMed] [Google Scholar]
- [44].Deprez M, Zaragosi LE, Truchi M, Becavin C, Ruiz Garcia S, Arguel MJ, et al. A Single-Cell Atlas of the Human Healthy Airways. Am J Respir Crit Care Med 2020;202:1636–45. [DOI] [PubMed] [Google Scholar]
- [45].Travaglini KJ, Nabhan AN, Penland L, Sinha R, Gillich A, Sit RV, et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 2020;587:619–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Meng H, Bartholomew B. Emerging roles of transcriptional enhancers in chromatin looping and promoter-proximal pausing of RNA polymerase II. J Biol Chem 2018;293:13786–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Stolzenburg LR, Yang R, Kerschner JL, Fossum S, Xu M, Hoffmann A, et al. Regulatory dynamics of 11p13 suggest a role for EHF in modifying CF lung disease severity. Nucleic Acids Res 2017;45:8773–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Swahn H, Sabith Ebron J, Lamar KM, Yin S, Kerschner JL, NandyMazumdar M, et al. Coordinate regulation of ELF5 and EHF at the chr11p13 CF modifier region. J Cell Mol Med 2019:Nov;23(11):7726–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Wright FA, Strug LJ, Doshi VK, Commander CW, Blackman SM, Sun L, et al. Genome-wide association and linkage identify modifier loci of lung disease severity in cystic fibrosis at 11p13 and 20q13.2. Nat Genet 2011;43:539–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Corvol H, Blackman SM, Boelle PY, Gallins PJ, Pace RG, Stonebraker JR, et al. Genome-wide association meta-analysis identifies five modifier loci of lung disease severity in cystic fibrosis. Nat Commun 2015;6:8382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kim YW, Kang Y, Kang J, Kim A. GATA-1-dependent histone H3K27 acetylation mediates erythroid cell-specific chromatin interaction between CTCF sites. FASEB J 2020;34:14736–49. [DOI] [PubMed] [Google Scholar]
- [52].Anderson KP, Kern CB, Crable SC, Lingrel JB. Isolation of a gene encoding a functional zinc finger protein homologous to erythroid Kruppel-like factor: identification of a new multigene family. Mol Cell Biol 1995;15:5957–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Luo Y, Chen C. The roles and regulation of the KLF5 transcription factor in cancers. Cancer Sci 2021;112:2097–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Liu Y, Guo B, Aguilera-Jimenez E, Chu VS, Zhou J, Wu Z, et al. Chromatin Looping Shapes KLF5-Dependent Transcriptional Programs in Human Epithelial Cancers. Cancer Res 2020;80:5464–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zhang XH, Zheng B, Han M, Miao SB, Wen JK. Synthetic retinoid Am80 inhibits interaction of KLF5 with RAR alpha through inducing KLF5 dephosphorylation mediated by the PI3K/Akt signaling in vascular smooth muscle cells. FEBS Lett 2009;583:1231–6. [DOI] [PubMed] [Google Scholar]
- [56].Muta K, Nakazawa Y, Obata Y, Inoue H, Torigoe K, Nakazawa M, et al. An inhibitor of Kruppel-like factor 5 suppresses peritoneal fibrosis in mice. Perit Dial Int 2021;41:394–403. [DOI] [PubMed] [Google Scholar]
- [57].Liu L, Koike H, Ono T, Hayashi S, Kudo F, Kaneda A, et al. Identification of a KLF5-dependent program and drug development for skeletal muscle atrophy. Proc Natl Acad Sci U S A 2021;118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Smith AN, Barth ML, McDowell TL, Moulin DS, Nuthall HN, Hollingsworth MA, et al. A regulatory element in intron 1 of the cystic fibrosis transmembrane conductance regulator gene. J Biol Chem 1996;271:9947–54. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
To review GEO accession GSE194288:
Go to https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE194288
Enter token ufcfwwkorhyzdsf into the box